
Home » Antineoplastic
Category Archives: Antineoplastic
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Perzebertinib, Bizrolertinib
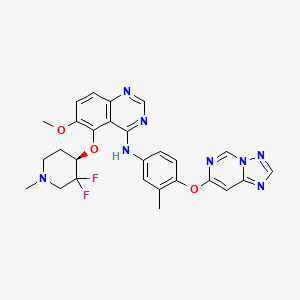
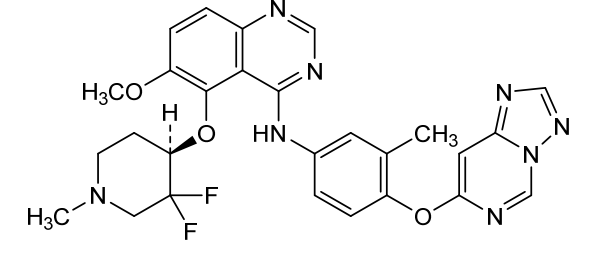
Perzebertinib, Bizrolertinib
CAS 2414056-31-6
MFC27H26F2N8O3 MW548.5 g/mol
5-[(4R)-3,3-difluoro-1-methylpiperidin-4-yl]oxy-6-methoxy-N-[3-methyl-4-([1,2,4]triazolo[1,5-c]pyrimidin-7-yloxy)phenyl]quinazolin-4-amine
- 4-Quinazolinamine, 5-[[(4R)-3,3-difluoro-1-methyl-4-piperidinyl]oxy]-6-methoxy-N-[3-methyl-4-([1,2,4]triazolo[1,5-c]pyrimidin-7-yloxy)phenyl]-
- 5-[[(4R)-3,3-Difluoro-1-methyl-4-piperidinyl]oxy]-6-methoxy-N-[3-methyl-4-([1,2,4]triazolo[1,5-c]pyrimidin-7-yloxy)phenyl]-4-quinazolinamine
5-{[(4R)-3,3-difluoro-1-methylpiperidin-4-yl]oxy}-6-methoxy-N-{3-methyl-4-[([1,2,4]triazolo[1,5-c]pyrimidin-7-yl)oxy]phenyl}quinazolin4-amine
epidermal growth factor receptor tyrosine kinase inhibitor, antineoplastic, ZN-A-1041, ZN 1041, RG 6596, Bizrolertinib, UN8TM5120C
Perzebertinib (also known as bizrolertinib or by developmental codes ZN-A-1041, ZN-1041, and RG6596) is an orally active, potent, and highly selective HER2 (ERBB2) tyrosine kinase inhibitor (TKI) designed to treat advanced solid tumors, primarily HER2-positive breast cancer.
Mechanism of Action
Perzebertinib functions as a selective, irreversible inhibitor of the HER2 tyrosine kinase. It blocks the ATP-binding site of the receptor to stop autophosphorylation. This action shuts down downstream signaling via the PI3K/AKT and MAPK pathways, successfully suppressing the growth, survival, and migration of tumor cells overexpressing HER2.
Key Clinical Advantages
- Blood-Brain Barrier (BBB) Penetration: The drug is designed to cross the blood-brain barrier effectively. This makes it highly valuable for treating brain metastases, a common and aggressive complication in advanced HER2-positive breast cancers.
- EGFR Sparing: Unlike older pan-EGFR/HER2 inhibitors, perzebertinib is engineered to spare wild-type EGFR. Sparing EGFR helps minimise common on-target side effects like severe skin rash and diarrhea.
- Efflux Resistance: It is not a substrate for P-gp or BCRP efflux pumps, allowing it to maintain high concentrations within central nervous system (CNS) tissues.
Development and Clinical Status
Initially discovered and developed by Suzhou Zanrong Pharmaceutical Technology (Zion Pharma), the asset is being co-developed in partnership with Roche and Genentech.
The drug has progressed through Phase 1 clinical studies evaluating its safety and pharmacokinetics in advanced solid tumors, moving forward into Phase 2/3 evaluations for HER2-positive advanced or locally advanced metastatic breast cancer. It is frequently evaluated as a monotherapy or in combination regimens alongside established therapies like capecitabine, trastuzumab, or pertuzumab
PAT
US11723908, Example 37, EG 77
https://patentscope.wipo.int/search/en/detail.jsf?docId=US344952565&_cid=P10-MS9QYW-06569-1
PAT
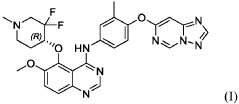
International Patent Publication No. WO 2020/057511 A1, which is incorporated herein by reference in its entirety, discloses quinazoline compounds that inhibit type I receptor tyrosine kinases, demonstrate good brain penetration in animals, and possess favorable toxicity profiles (for example a decreased activity against hERG), and thus particularly useful in the treatment of type I receptor tyrosine kinases mediated diseases or conditions, in particular ErbB2-associated disease or conditions, including cancer (e.g., metastatic cancer, such as brain metastases). A specific compound, which is identified as (R)-N-(4-([1,2,4]triazolo[1,5-c]pyrimidin-7-yloxy)-3-methylphenyl)-5-((3,3-difluoro-1-methylpiperidin-4-yl)oxy)-6-methoxyquinazolin-4-amine (also referred to as compound (I) herein),
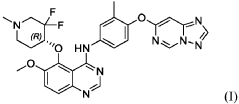
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020057511&_cid=P10-MS9R3W-09545-1
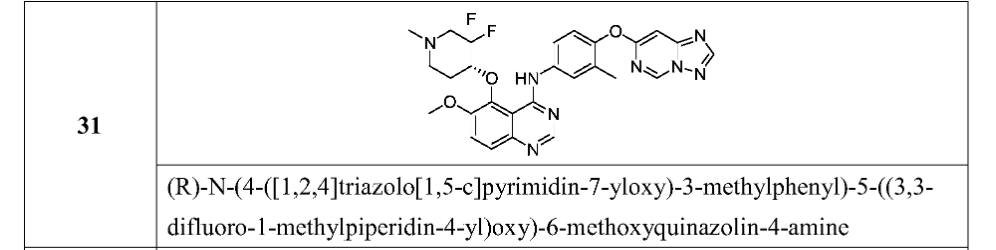

[0681]
(S) -N- (4- ( [1, 2, 4] triazolo [1, 5-c] pyrimidin-7-yloxy) -3-methylphenyl) -5- ( (3, 3-difluoro-1-methylpiperidin-4-yl) oxy) -6-methoxyquinazolin-4-amine

Step 5: (R) -N- (4- ( [1, 2, 4] triazolo [1, 5-c] pyrimidin-7-yloxy) -3-methylphenyl) -5- ( (3, 3-difluoro-1-methylpiperidin-4-yl) oxy) -6-methoxyquinazolin-4-amine and
[0696]
(S) -N- (4- ( [1, 2, 4] triazolo [1, 5-c] pyrimidin-7-yloxy) -3-methylphenyl) -5- ( (3, 3-difluoro-1-methylpiperidin-4-yl) oxy) -6-methoxyquinazolin-4-amine
[0697]
[0698]
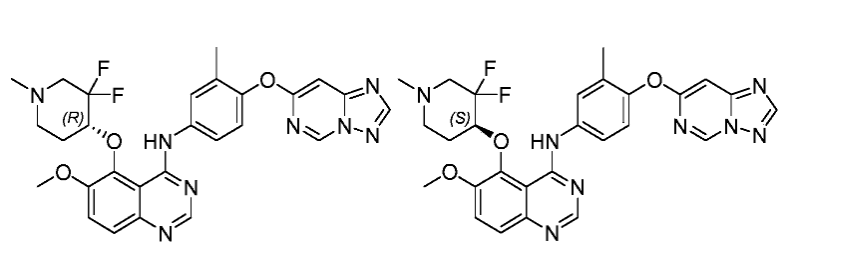
To a solution of 4-chloro-5- ( (3, 3-difluoro-1-methylpiperidin-4-yl) oxy) -6-methoxyquinazoline (410 mg, 1.19 mmol) in Propan-2-ol (60 mL) was added TsOH. H 2O (68 mg, 0.36 mmol) and 4- ( [1, 2, 4] triazolo [1, 5-c] pyrimidin-7-yloxy) -3-methylaniline (259 mg, 1.07 mmol) . The resulting mixture was stirred at 100℃ under Ar 2protection and concentrated. The residue was dissolved in H 2O (100 mL) , basified with aq. NaHCO 3to pH =7-8, extracted with DCM: MeOH = 20: 1 (100 mLx3) . The combined organic layers were dried over anhydrous Na 2SO 4, filtered and concentrated. The residue was purified by column chromatography (DCM/MeOH=30/1) to give product (300 mg, 46%yield) as white solid. The racemic material was subsequently separated by chiral SFC to give two isomers:
[0699]
(R) -N- (4- ( [1, 2, 4] triazolo [1, 5-c] pyrimidin-7-yloxy) -3-methylphenyl) -5- ( (3, 3-difluoro-1-methylpiperidin-4-yl) oxy) -6-methoxyquinazolin-4-amine (Peak 1, retention time 6.241 min, ee: >99%) (100 mg, 67%) as a white solid. MS (ESI) m/z: 549.2 (M+H) +. 1H NMR (400 MHz, CDCl 3) δ 10.04 (s, 1H) , 9.20 (s, 1H) , 8.61 (s, 1H) , 8.33 (s, 1H) , 7.88 (d, J = 2.0 Hz, 1H) , 7.79-7.76 (m, 1H) 7.69 (d, J = 9.2 Hz, 1H) , 7.53 (d, J = 9.2 Hz, 1H) , 7.11 (d, J = 8.8 Hz, 1H) , 6.90 (s, 1H) , 4.84-4.79 (m, 1H) , 4.03 (s, 3H) , 3.22-3.21 (m, 1H) , 2.93 (d, J = 7.2 Hz, 1H) , 2.38 (s, 3H) , 2.41-2.34 (m, 1H) , 2.34-2.27 (m, 1H) , 2.19 (s, 3H) , 2.16-2.10 (m, 2H) .
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma



AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

References
- Quinazoline derivatives as antitumor agentsPublication Number:US-2023293533-A1Priority Date:2018-09-18
- Quinazoline derivatives as antitumor agentsPublication Number:ES-2971927-T3Priority Date:2018-09-18Grant Date:2024-06-10
- Quinazoline derivatives as antitumor agentsPublication Number:EP-4360713-B1Priority Date:2018-09-18Grant Date:2024-10-30
- Quinazoline derivatives as antitumor agentsPublication Number:ES-3007082-T3Priority Date:2018-09-18Grant Date:2025-03-19
- Quinazoline derivatives as antitumor agentsPublication Number:EP-4360713-A2Priority Date:2018-09-18
- Quinazoline derivatives as antitumor agentsPublication Number:EP-3853220-B1Priority Date:2018-09-18Grant Date:2024-01-03
- Quinazoline derivatives as antitumor agentsPublication Number:EP-3853220-A1Priority Date:2018-09-18
- Quinazoline derivatives as antitumor agentsPublication Number:US-2021386742-A1Priority Date:2018-09-18
- Quinazoline derivatives as antitumor agentsPublication Number:US-11723908-B2Priority Date:2018-09-18Grant Date:2023-08-15
- Crystalline forms of quinazoline derivatives, preparation, compositions and uses thereofPublication Number:CN-120623183-APriority Date:2021-10-20
- Crystalline forms of quinazoline derivatives, preparation, compositions and uses thereofPublication Number:CN-120309621-APriority Date:2021-10-20
- Quinazoline derivatives as antitumor agentsPublication Number:WO-2020057511-A1Priority Date:2018-09-18
- Quinazoline Derivatives as Antitumor AgentsPublication Number:JP-7546550-B2Priority Date:2018-09-18Grant Date:2024-09-06
- Quinazoline derivatives as antitumor agentsPublication Number:CA-3099776-A1Priority Date:2018-09-18
/////////perzebertinib, anax labs, epidermal growth factor receptor tyrosine kinase inhibitor, antineoplastic, ZN-A-1041, ZN 1041, RG 6596, Bizrolertinib, UN8TM5120C
Pasodacigib
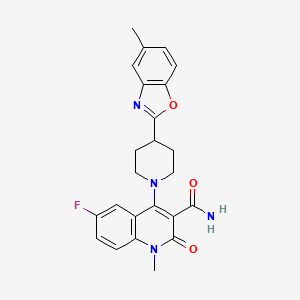
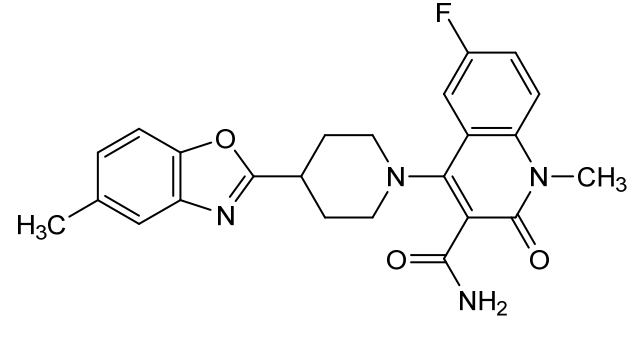
Pasodacigib
Cas 2648721-77-9
MFC24H23FN4O3 mw 434.5 g/mol
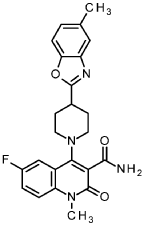
6-fluoro-1-methyl-4-[4-(5-methyl-1,3-benzoxazol-2-yl)piperidin-1-yl]-2-oxo-1,2-dihydroquinoline-3-carboxamide
6-fluoro-1-methyl-4-[4-(5-methyl-1,3-benzoxazol-2-yl)piperidin-1-yl]-2-oxo-1,2-dihydroquinoline-3-carboxamide
diacylglycerol kinase inhibitor, antineoplastic, BAY 2862789, BAY-2862789, XM6U88YE6H
Pasodacigib (also known by its developmental code BAY 2862789 or BAY-2862789) is an investigational small-molecule drug developed by Bayer AG. It functions as a potent and selective diacylglycerol kinase alpha (DGKα) inhibitor designed for cancer immunotherapy.
🧪 Mechanism of Action
- Targeting DGKα: Diacylglycerol kinase alpha (DGKα) is an enzyme that converts diacylglycerol (DAG) into phosphatidic acid (PA) within cells.
- T-Cell Reactivation: In the tumor microenvironment, overactive DGKα metabolises DAG, which depletes the signaling required for T-cell activation. By blocking this enzyme, pasodacigib aims to restore DAG levels, reactivating the patient’s own T-cells to mount a clinically beneficial anti-tumor immune response.
📋 Key Details & Chemical Properties
- Developer: Bayer AG
- CAS Registry Number: 2648721-77-9
- Molecular Formula: C₂₄H₂₃FN₄O₃
- Molecular Weight: 434.46 g/mol
- Research Status: It is an investigational drug that has entered clinical trial assessment (such as the Bayer-led study NCT05858164) to evaluate its safety and efficacy in treating advanced malignancies.
⚠️ Important Medical Disclaimer
Pasodacigib is strictly an investigational compound undergoing clinical development and laboratory research. It is not approved by the FDA or any other global regulatory authority for prescription, public medical use, or patient purchase.
If you are looking into this molecule for scientific research or a clinical study, please let me know if you need specific details regarding its chemical structure, information on related DGK inhibitors, or updates on active oncology clinical trials
Pat
https://patentscope.wipo.int/search/en/detail.jsf?docId=US400268899&_cid=P22-MS6W4B-66581-1
Example 298
6-fluoro-1-methyl-4-[4-(5-methyl-1,3-benzoxazol-2-yl)piperidin-1-yl]-2-oxo-1,2-dihydroquinoline-3-carboxamide
| 1H NMR (400 MHz, DMSO-d 6) δ ppm 2.02-2.15 (m, 2H) 2.17-2.26 (m, 2H) 2.44 (s, 3H) 3.13-3.25 (m, 3H) 3.35-3.43 (m, 2H) 3.59 (s, 3H) 7.18 (d, 1H) 7.47-7.63 (m, 6H) 7.73 (br s, 1H). |
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021105117&_cid=P22-MS6W2Y-65422-1
Intermediate 101
6-fluoro-1-methyl-2H-3,1-benzoxazine-2,4(1 H)-dione
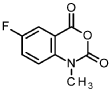
To a solution of 5.00 g 6-fluoro-2H-3,1-benzoxazine-2,4(1 H)-dione (26.8 mmol, CAS 321-69-7) and 9.3 ml. N,N-diisopropylethylamine (54 mmol) in 40 ml. dimethylformamide was added 5.1 ml. iodomethane (80 mmol) at rt and the mixture was stirred overnight. The reaction mixture was diluted with 1000 ml. water and the resulting solid was collected by filtration, the filter cake was washed with water and dried in vacuum to give 4.93 g of the title compound (99 % purity, 93 % yield).
1H NMR (400 MHz, DMSO-cfe) d ppm 3.47 (s, 3H), 7.40-7.61 (m, 1 H), 7.69-7.93 (m, 2H).Intermediate 102
6-fluoro-4-hydroxy-1-methyl-2-oxo-1 ,2-dihydroquinoline-3-carbonitrile
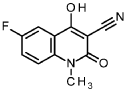
4.85 g 6-fluoro-1 -methyl-2H-3,1 -benzoxazine-2,4(1 H)-dione (intermediate 101 , 24.6 mmol,) was solubilised in 50 ml. tetrahydrofurane, 34 ml. triethylamine (250 mmol) and then 15.1 ml. ethyl cyanoacetate (133 mmol) were added carefully and the suspension was stirred 72 h at 900. The reaction mixture was cooled down to rt, concentrated under reduced pressure, the residue was diluted with water and ethyl acetate (1 :1) and the mixture was adjusted to pH = 1 with hydrogen chloride solution (2 M in water). The resulting solid was filtered and the filter cake was washed with less water and ethyl acetate to give 4.40 g of the title compound (100 % purity, 82 % yield).
1H NMR (400 MHz, DMSO-cfe) d ppm 3.34 (br s, 3H), 7.01 -7.44 (m, 2H), 7.51 -7.70 (m, 1 H).
Intermediate 103
4-chloro-6-fluoro-1-methyl-2-oxo-1,2-dihydroquinoline-3-carbonitrile
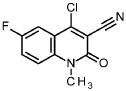
A mixture of 4.40 g 6-fluoro-4-hydroxy-1-methyl-2-oxo-1 ,2-dihydroquinoline-3-carbonitrile (intermediate 102, 20.0 mmol) and 19 ml. phosphoric trichloride (200 mmol) was stirred overnight at 900. The reaction mixture was cooled down to rt, diluted with hexane and the resulting solid was filtered. The filter cake was carefully added to a half saturated solution of sodium bicarbonate, the resulting suspension was filtered, the solid was washed with water, ethyl acetate and then with ethanol and dried in vacuum to give 4.17 g of the title compound (100 % purity, 88 % yield).
1H NMR (400 MHz, DMSO-cfe) d ppm 3.67 (s, 3H); 7.58 – 8.09 (m, 3H).
Example 217
6-fluoro-1-methyl-4-[4-(5-methyl-1,3-benzoxazol-2-yl)pipendin-1-yl]-2-oxo-1,2-dihydroquinoline-3-carbonitrile
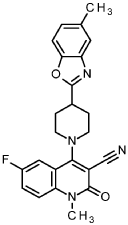
80 mg 4-chloro-6-fluoro-1-methyl-2-oxo-1 ,2-dihydroquinoline-3-carbonitrile (338 pmol, intermediate 103) was suspended in 2.5 ml. 2-propanol, 180 mI_ N,N-diisopropylethylamine (1.0 mmol) and 87.7 mg 5-methyl-2-(piperidin-4-yl)-1 ,3-benzoxazole (406 pmol, CAS 199292-77-8) were added and the mixture was stirred for 2 h at 900. The reaction mixture was cooled down to rt and the suspension was diluted with water and stirred for 15 min. The solid was filtered off and washed with water and ethanol to give 127 mg of the title compound (98 % purity, 88 % yield).
1H NMR (400 MHz, DMSO-cfe) d ppm 2.07 – 2.19 (m, 2 H) 2.27 – 2.35 (m, 2 H) 2.44 (s, 3 H) 3.43 (tt, 1 H) 3.55 – 3.64 (m, 5 H) 3.81 (br d, 2 H) 7.18 (dd, 1 H) 7.53 (d, 1 H) 7.54 – 7.61 (m, 2 H) 7.61 – 7.70 (m, 2 H).
LC-MS (Method 2): R, = 1.32 min; MS (ESIpos): m/z = 417.4 [M+H]+
Example 298
6-fluoro-1-methyl-4-[4-(5-methyl-1,3-benzoxazol-2-yl)pipendin-1-yl]-2-oxo-1,2-dihydroquinoline-3-carboxamide
68 mg 6-fluoro-1 -methyl-4-[4-(5-methyl-1 ,3-benzoxazol-2-yl)piperidin-1-yl]-2-oxo-1 ,2-dihydroquinoline-3-carbonitrile (160 pmol, example 217), 9 mg palladium(ll)acetate (40 pmol) and 142 mg acetaldoxime (2.4 mmol) were stirred in 1 .5 ml. ethanol for 5 h at 80TT The reaction mixture was diluted with water, extracted with ethyl acetate two times, the combined organic layers were filtered through a waterresistant filter and the filtrate was concentrated under reduced pressure. The residue was purified by RP-HPLC (column: X-Bridge C18 5pm 100x30mm, mobile phase: acetonitrile / water (0.2 vol. % ammonia 32 %)-gradient) to give 41 .4 mg of the title compound (100 % purity, 60 % yield).
1H NMR (400 MHz, DMSO-cfe) d ppm 2.02 – 2.15 (m, 2 H) 2.17 – 2.26 (m, 2 H) 2.44 (s, 3 H) 3.13 – 3.25 (m, 3 H) 3.35 – 3.43 (m, 2 H) 3.59 (s, 3 H) 7.18 (d, 1 H) 7.47 – 7.63 (m, 6 H) 7.73 (br s, 1 H).
LC-MS (Method 2): R, = 1 .17 min; MS (ESIpos): m/z = 435.4 [M+H]
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- DGK alpha inhibitors as immune activation substituted aminoquinolones of (2)Publication Number:CN-119523983-APriority Date:2019-11-28
- Cyclopropyl modulators of p2y12 receptorPublication Number:US-2017002015-A1Priority Date:2009-07-27
- Substituted aminoquinolones as dgkalpha inhibitors for immune activationPublication Number:EP-4065574-A1Priority Date:2019-11-28
- Substituted aminoquinolones as dgkalpha inhibitors for immune activationPublication Number:WO-2021105117-A1Priority Date:2019-11-28
- Substituted aminoquinolones as immunoactive DGK alpha inhibitorsPublication Number:CN-115003665-APriority Date:2019-11-28
- Substituted aminoquinolones as inhibitors of immune-activated DGKαPublication Number:CN-119564686-APriority Date:2019-11-28
- DGK alpha inhibitors as immune activation substituted aminoquinolones of (2)Publication Number:CN-119424429-APriority Date:2019-11-28
- Combinations of dgk (diacylglycerol kinase) inhibitors and immune checkpoint inhibitors and modulatorsPublication Number:TW-202448461-APriority Date:2023-02-06
- Substituted aminoquinolones as DGKalpha inhibitors for immune activationPublication Number:US-11998539-B2Priority Date:2019-11-28Grant Date:2024-06-04
- Substituted aminoquinolones as dgkalpha inhibitors for immune activationPublication Number:US-2023148194-A1Priority Date:2019-11-28
- Substituted aminoquinolones as dgkalpha inhibitors for immune activationPublication Number:US-2023201186-A1Priority Date:2019-11-28
- DGK alpha inhibitors as immune activation substituted aminoquinolones of (2)Publication Number:CN-115003665-BPriority Date:2019-11-28Grant Date:2024-11-08
- Combination of ccr8 antibodies with dgk inhibitors in the treatment of cancerPublication Number:WO-2024165468-A1Priority Date:2023-02-06
- Combination of ccr8 antibodies with dgk inhibitorsPublication Number:TW-202436351-APriority Date:2023-02-06
- Combinations of dgk (diacylglycerol kinase) inhibitorsPublication Number:WO-2024165470-A1Priority Date:2023-02-06
- Combinations of dgk (diacylglycerol kinase) inhibitors and immune checkpoint inhibitors and modulatorsPublication Number:WO-2024165469-A1Priority Date:2023-02-06
- Combinations of dgk (diacylglycerol kinase) inhibitorsPublication Number:TW-202448460-APriority Date:2023-02-06
/////////pasodacigib, anax labs, diacylglycerol kinase inhibitor, antineoplastic, BAY 2862789, BAY-2862789, XM6U88YE6H
Palacaparib
Palacaparib
CAS 2756333-39-6
MFC21H22F2N6O2 MW428.4 g/mol
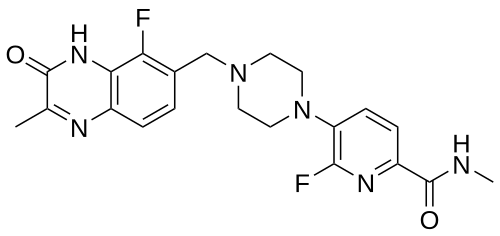
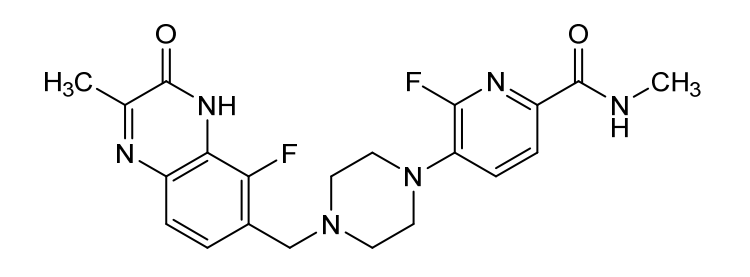
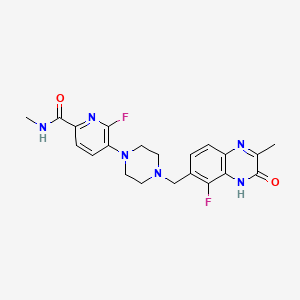
6-fluoro-5-[4-[(5-fluoro-2-methyl-3-oxo-4H-quinoxalin-6-yl)methyl]piperazin-1-yl]-N-methylpyridine-2-carboxamide
2-Pyridinecarboxamide, 6-fluoro-5-(4-((5-fluoro-3,4-dihydro-2-methyl-3-oxo-6-quinoxalinyl)methyl)-1-piperazinyl)-N-methyl-
6-fluoro-5-{4-[(5-fluoro-2-methyl-3-oxo-3,4-dihydroquinoxalin-6-yl)methyl]piperazin-1-yl}-N-methylpyridine-2-carboxamide
poly (ADP-ribose) polymerase (PARP) inhibitor, antineoplastic, AZD 9574, 9UG32UQW48
Palacaparib is an investigational new drug that is being evaluated by AstraZeneca for the treatment of prostate cancer.[1] It is a selective PARP1 inhibitor.[2][3]
Palacaparib (also known as AZD9574) is an investigational, orally bioavailable cancer drug developed by AstraZeneca. It belongs to a class of medications called PARP inhibitors.
Key Characteristics
- High Selectivity: It selectively targets the PARP1 enzyme. It features an 8,000-fold greater selectivity for PARP1 over other PARP forms like PARP2. This targeted approach aims to lower typical bone marrow toxicities linked with older, non-selective PARP inhibitors.
- Brain Penetrance: Unlike many earlier options, it successfully crosses the blood-brain barrier. This trait makes it a prime candidate for managing primary brain tumors and central nervous system (CNS) metastases.
Mechanism of Action
- Enzyme Binding: Palacaparib tightly binds to the PARP1 enzyme at single-strand DNA break locations.
- DNA Trapping: It traps the enzyme on the damaged DNA, blocking the base excision repair pathway.
- Synthetic Lethality: This stalling stalls replication forks and forces single-strand breaks to progress into double-strand breaks.
- Cell Death: In tumors with homologous recombination deficiency (HRD)—such as BRCA1/2 mutations—cells cannot fix these severe breaks, triggering apoptosis (programmed cell death).
Clinical Research and Targets
According to active registries from the National Cancer Institute (NCI), Palacaparib is undergoing early-phase human trials both as a single agent and alongside other treatments. Researchers are testing its efficacy across several oncological areas:
- Advanced Solid Malignancies: Studies focus heavily on HRD-positive tumors, including specific types of breast, ovarian, and pancreatic cancers.
- Prostate Cancer: Active monotherapy and combination trials target metastatic prostate cancer.
- CNS Malignancies: Preclinical designs show promise in treating gliomas when paired with radiation or alkylating agents like temozolomide.
- OriginatorAstraZeneca
- ClassAntineoplastics; Carbamates; Fluorinated hydrocarbons; Ketones; Piperazines; Pyridines; Quinoxalines; Small molecules
- Mechanism of ActionPoly(ADP-ribose) polymerase-1 inhibitors
- Phase I/IIProstate cancer; Solid tumours
- 03 Jun 2026Phase-I/II clinical trials in Prostate cancer (Combination therapy, Hormone refractory, Second-line therapy or greater, Metastatic disease) in USA (PO) (NCT07590934)
- 14 May 2026AstraZeneca plans a phase I/II trial for Prostate cancer (Metastatic disease, Combination therapy, Second-line therapy or greater, Hormone-refractory) in USA, Australia, Germany, Italy, South Korea, Spain, United Kingdom (PO) in May 2026 (NCT07590934) (EudraCT2025-524920-23)
- 19 Feb 2026Chemical structure information added.
Palacaparib is an orally bioavailable central nervous system (CNS) penetrant and inhibitor of nuclear enzyme poly(ADP-ribose) polymerase (PARP) 1, with potential antineoplastic activity. Upon oral administration, palacaparib selectively binds to PARP1, thereby preventing repair of damaged DNA via the base excision repair (BER) pathway. This agent enhances the accumulation of DNA strand breaks and promotes genomic instability eventually leading to apoptosis. Palacaparib may enhance the cytotoxicity of DNA-damaging agents and reverse tumor cell chemo- and radioresistance. PARP1 catalyzes post-translational ADP-ribosylation of nuclear proteins that signal and recruit other proteins to repair damaged DNA and plays a key role in the repair of single strand DNA (ssDNA) breaks and double-strand break (DSBs). Palacaparib is able to penetrate the blood-brain barrier (BBB).
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021260092&_cid=P22-MS178J-58931-1
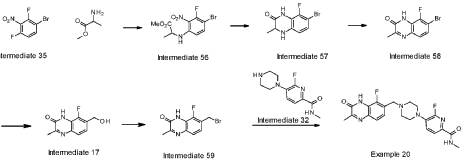
Example 20: 6-fluoro-5-[4-[(5-fluoro-2-methyl-3-oxo-4H-ciuinoxalin-6-yl)methvnDiDerazin-1-vn-N-methyl-pyridine-2-carboxamide
Triethylphosphane (20.90 ml, 145.06 mmol) was added dropwise with an addition funnel to a stirred suspension of 8-fluoro-7-(hydroxymethyl)-3-methyl-1 H-quinoxalin-2-one (intermediate 17) (15.1 g, 72.53 mmol) and 1 ,2-dibromo-1 ,1 ,2,2-tetrachloroethane (52.0 g, 159.56 mmol) in DCM (400 mL) at 0°C under nitrogen. The mixture was stirred at r.t for 3 h gave a light-yellow suspension. Crude LCMS indicated full conversion. DCM was removed under vacuum; the residue was slurry in 300 mL diethyl ether at rt and the light yellow ppt was filtered and washed with 200 ml ether. The solid was taken into 300 ml of water, stirred at rt for 10 min, the solid was collected by filtration, thorough wash (200 ml) with water to remove the salts. The solid was dried under vacuum for overnight (no heat). The solid was washed with hexanes and dried in vacuum in a bushel funnel to give 7-(bromomethyl)-8-fluoro-3-methylquinoxalin-2(1 H)-one (intermediate 59) (22.76 g, 116 %, likely contains some inorganic salts) as an off white solid. Used as such for next reaction. 1 H NMR (500 MHz, DMSO-c/6) 2.42 (3H, s), 4.65 – 4.93 (2H, m), 7.28 – 7.42 (1 H, m), 7.51 (1 H, d), 12.53 (1 H, br s); m/z (ES+) [M+H]+ = 271 , 273.
To a flask charged with 7-(bromomethyl)-8-fluoro-3-methylquinoxalin-2(1 H)-one (intermediate 59) (22.76 g) and 6-fluoro-N-methyl-5-(piperazin-1-yl)picolinamide, 2HCI ( intermediate 32) (24.24 g, 77.9 mmol) in acetonitrile (350 ml), was added DIPEA (38.0 ml, 217.59 mmol) at rt and the resulting mixture was stirred at 70°C for 4 h. Reaction was not complete. To the mixture was added 5 g of Kl and 2 g of Nal and the mixture was stirred at 50°C for 20 h. More 540 mgs (~0.03eq) of 6-fluoro-N-methyl-5-(piperazin-l-yl)picolinamide, 2HCI (intermediate 32) was added to the mixture and the stirring continued at 50°C for 2 h. The solid from the reaction suspension was collected by filtration, washed with acetonitrile and dried. The resulting material was then suspended in water (~400 ml), slurred at rt for 20 min, filtered and dried (97% purity by LCMS). The solid was then dissolved into a mixture of DCM/MeOH (3/1) (about 1.5 L) at reflux, filtered through a pad of silica gel, removed most of the DCM until solid precipitate out and the mixture was kept at rt for 20 min. The solid was collected by filtration and repeated the procedure for the filtrate, and the solids were combined to yield the product 6-fluoro-5-[4-[(5-fluoro-2-methyl-3-oxo-4H-quinoxalin-6-yl)methyl]piperazin-1-yl]-N-methyl-pyridine-2-carboxamide (example 20) (26 g, 84%) as a light yellow solid. 1 H NMR (500 MHz, DMSO-c/6) 2.41 (3H, s), 2.57 – 2.69 (4H, m), 2.76 (3H, d), 3.16 (4H, br s), 3.70 (2H, s), 7.29 (1 H, br t), 7.40 – 7.60 (2H, m), 7.83 (1 H, d), 8.38 (1 H, br d), 12.44 (1 H, br s); m/z (ES+) [M+H]+ = 429.
PAT
- Chemical compoundsPublication Number:US-12421208-B2Priority Date:2020-06-25Grant Date:2025-09-23
- Chemical compoundsPublication Number:US-11795158-B2Priority Date:2020-06-25Grant Date:2023-10-24
- Chemical compoundsPublication Number:US-2022009901-A1Priority Date:2020-06-25
- Combining ATR inhibitors and PARP inhibitors for cancer treatmentPublication Number:IL-309388-APriority Date:2021-06-16
- Use of ATR inhibitors in combination with PARP inhibitors to treat cancerPublication Number:KR-20240021884-APriority Date:2021-06-16
- Quinoxaline derivatives as anticancer drugsPublication Number:CN-115768760-APriority Date:2020-06-25
- Quinoxaline derivatives as anti-cancer drugsPublication Number:WO-2021260092-A1Priority Date:2020-06-25
- Quinoxaline derivatives as anti-cancer drugsPublication Number:EP-4172152-A1Priority Date:2020-06-25
SYN
- Discovery of 6-Fluoro-5-{4-[(5-fluoro-2-methyl-3-oxo-3,4-dihydroquinoxalin-6-yl)methyl]piperazin-1-yl}-N-methylpyridine-2-carboxamide (AZD9574): A CNS-Penetrant, PARP1-Selective InhibitorPublication Name:Journal of Medicinal ChemistryPublication Date:2024-12-10PMID:39655996DOI:10.1021/acs.jmedchem.4c01725
- New Horizons of Synthetic Lethality in Cancer: Current Development and Future PerspectivesPublication Name:Journal of Medicinal ChemistryPublication Date:2024-07-02PMCID:PMC11284803PMID:38955347DOI:10.1021/acs.jmedchem.4c00113
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
 | |
| Clinical data | |
|---|---|
| Other names | AZD-9574 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2756333-39-6 |
| PubChem CID | 162524593 |
| IUPHAR/BPS | 11946 |
| ChemSpider | 115008044 |
| UNII | 9UG32UQW48 |
| ChEMBL | ChEMBL5095223 |
| PDB ligand | A1H64 (PDBe, RCSB PDB) |
| Chemical and physical data | |
| Formula | C21H22F2N6O2 |
| Molar mass | 428.444 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- “Palacaparib”. AdisInsight. Springer Nature Switzerland AG. Retrieved 5 July 2026.
- Johannes JW, Balazs AY, Barratt D, Bista M, Chuba MD, Cosulich S, et al. (December 2024). “Discovery of 6-Fluoro-5-{4-[(5-fluoro-2-methyl-3-oxo-3,4-dihydroquinoxalin-6-yl)methyl]piperazin-1-yl}-N-methylpyridine-2-carboxamide (AZD9574): A CNS-Penetrant, PARP1-Selective Inhibitor”. Journal of Medicinal Chemistry. 67 (24): 21717–21728. doi:10.1021/acs.jmedchem.4c01725. PMID 39655996.
- Shkil DO, Chesnokova NA, Ivashchenko AA, Petersen EV, Maximov PY (May 2026). “Structural Determinants of PARP1 Selectivity from Molecular Dynamics Analysis of PARP1 and PARP2 Complexes”. Molecules. 31 (10). Basel, Switzerland: 1592. doi:10.3390/molecules31101592. PMC 13210057. PMID 42197145.
///////////palacaparib, ANAX LABS, poly (ADP-ribose) polymerase (PARP) inhibitor, antineoplastic, AZD 9574, 9UG32UQW48
Navlimetostat
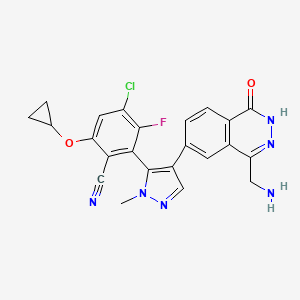
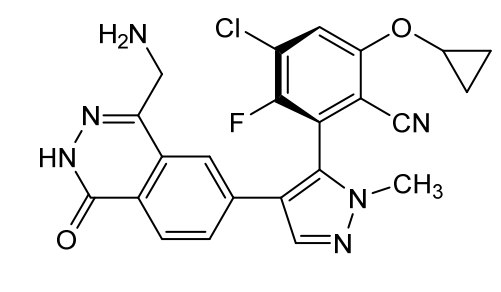
Navlimetostat
CAS 2630904-45-7
ALSO 2630904-44-6
MF C23H18ClFN6O2 MW464.9 g/mol
- Benzonitrile, 2-(4-(4-(aminomethyl)-1,2-dihydro-1-oxo-6-phthalazinyl)-1-methyl-1H-pyrazol-5-yl)-4-chloro-6-(cyclopropyloxy)-3-fluoro-, (2S)-
- (M)-27
- 2-[4-[4-(aminomethyl)-1-oxo-2H-phthalazin-6-yl]-2-methylpyrazol-3-yl]-4-chloro-6-cyclopropyloxy-3-fluorobenzonitrile
(2M)-2-{4-[4-(aminomethyl)-1-oxo-1,2-dihydrophthalazin-6-yl] -1-methyl-1H-pyrazol-5-yl}-4-chloro-6-(cyclopropyloxy)-3-fluorobenzonitrile
antineoplastic, MRTX-1719, BMS-986504, MRTX 1719, BMS 986504
Navlimetostat (also known as MRTX-1719 or BMS-986504) is an investigational, first-in-class oral targeted cancer therapy being developed by Bristol-Myers Squibb. It works by selectively binding to the PRMT5-MTA complex, exploiting synthetic lethality to kill cancer cells with MTAP gene deletions while sparing healthy cells.
Navlimetostat is currently in Phase 1/2 clinical trials for advanced solid tumors, including MTAP-deficient non-small cell lung cancer (NSCLC), pancreatic cancer, and glioblastoma.
Key highlights and ongoing research:
- Mechanism: In MTAP-deleted cancer cells, a metabolite called MTA accumulates and binds to PRMT5. Navlimetostat targets and inhibits this specific PRMT5-MTA complex, leading to tumor cell death.
- Clinical Trials: It is currently being investigated as a monotherapy (e.g., in MTAP-deleted advanced solid tumors) and in combination with other agents like pumitamig
- OriginatorMirati Therapeutics
- DeveloperBristol-Myers Squibb; Mirati Therapeutics
- ClassAntineoplastics; Small molecules
- Mechanism of ActionPRMT5 protein inhibitors
- Phase II/IIIAdenocarcinoma; Non-small cell lung cancer
- Phase I/IIMesothelioma; Neurilemmoma; Pancreatic cancer; Solid tumours
- 22 May 2026University of Southampton in collaboration with Bristol-Myers Squibb plans a phase II SELECTmeso1 trial for Malignant mesothelioma (Second-line therapy or greater) in United Kingdom in May 2026 (PO, Tablet) (NCT07602946)
- 13 May 2026Northwestern University plans a phase Ib/II trial for Solid tumours (Metastatic disease, Second-line therapy or greater, Combination therapy) in USA(PO) in December 2027 (NCT07594626)
- 12 May 2026M.D. Anderson Cancer Center plans a phase I/II trial for Non-small cell lung cancer (Combination therapy, Late-stage disease, Metastatic disease, Second-line therapy or greater) in USA (PO), in November 2026 (NCT07579221)
PRMT5 Inhibitor BMS-986504 is an orally bioavailable methylthioadenosine (MTA)-selective inhibitor of the protein arginine methyltransferase 5 (PRMT5), with potential antineoplastic activity. Upon oral administration, PRMT5 inhibitor BMS-986504 targets, binds to and inhibits PRMT5 that is bound to MTA, a complex that is elevated in methylthioadenosine phosphorylase (MTAP)-deleted cancer cells, thereby specifically inhibiting the function of PRMT5 solely within MTAP-deleted cancer cells and not in normal, healthy cells. By inhibiting the methyltransferase activity of PRMT5, levels of both monomethylated and dimethylated arginine residues in histones H2A, H3 and H4 are decreased. This modulates the expression of genes involved in several cellular processes, including cellular proliferation. This may increase the expression of antiproliferative genes and/or decrease the expression of genes that promote cell proliferation, which may lead to decreased growth of rapidly proliferating cancer cells. BMS-986504 also causes dysregulated RNA splicing and decreased retinoblastoma protein (pRb). Together, this decreases proliferation and increases apoptosis specifically in MTAP-deleted cancer cells. PRMT5, a type II methyltransferase that catalyzes the formation of both omega-N monomethylarginine (MMA) and symmetric dimethylarginine (sDMA) on histones and a variety of other protein substrates involved in signal transduction and cellular transcription, is essential for the viability of cancer and normal cells. It is overexpressed in several neoplasms. Elevated levels are associated with decreased patient survival. MTAP is deleted in certain cancer cells leading to an accumulation of the metabolite MTA; MTA binds to and partially inhibits the activity of PRMT5.
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021050915&_cid=P12-MR76CL-04796-1
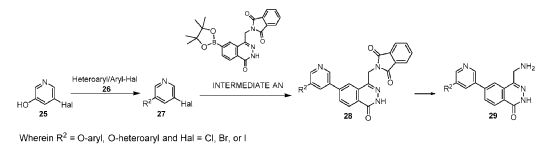
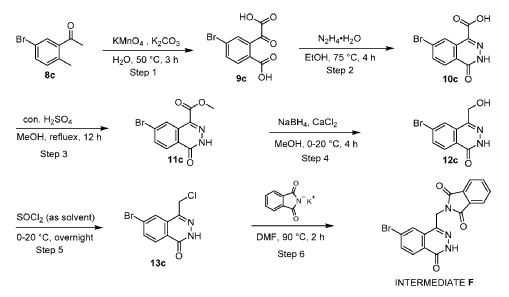
[0186] Step 6: To a solution of 6-bromo-4-(chloromethyl)-2H-phthalazin-1-one 13c (148 g, crude) in DMF (1.5 L) was added (1,3-dioxoisoindolin-2-yl)potassium (121 g, 653 mmol). The reaction mixture was stirred at 90 °C for 2 hours and then cooled to 25 °C. The formed precipitate was filtered and washed with DMF (200 mL x 2) and the filter cake triturated with water (1.00 L), filtered and dried to give Intermediate F, 2-[(7-bromo-4-oxo-3H-phthalazin-1-yl)methyl]isoindoline-1,3-dione (162 g, 413 mmol, 76% yield) as a white solid.1H NMR (400 MHz, DMSO-d6) d = 12.59 (s, 1H), 8.43 (d, J = 1.2 Hz, 1H), 8.18 (d, J = 8.4 Hz, 1H), 8.07 (dd, J = 1.6, 8.4 Hz, 1H), 7.97 – 7.93 (m, 2H), 7.92 – 7.86 (m, 2H), 5.19 (s, 2H). LCMS [M+1]: 383.9.
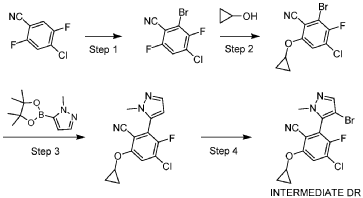
[0327] Step 4: A mixture of 4-chloro-6-(cyclopropoxy)-3-fluoro-2-(2-methylpyrazol-3-yl)benzonitrile (180 mg, 0.617 mmol, 1.00 eq) and N-bromosuccinimide (220 mg, 1.23 mmol, 2.00 eq.) in acetonitrile (10 mL) was stirred at 40 °C for 2 hours under a nitrogen atmosphere. After such time the mixture was concentrated and the residue was purified by prep-TLC (SiO2, petroleum ether: ethyl acetate 20%) to give 2-(4-bromo-2-methyl-pyrazol-3-yl)-4-chloro-6-(cyclopropoxy)-3-fluoro-benzonitrile (170 mg, 0.455 mmol, 74% yield) as a white solid. LCMS [M+1] + = 371.8; 1H NMR (400 MHz, CDCl3) d = 7.61 (s, 1H), 7.55 (d, J = 6.0 Hz, 1H), 3.93 – 3.85 (m, 1H), 3.80 (s, 4H), 0.97 – 0.94 (m, 4H).
EXAMPLE 16-7 and 16-8
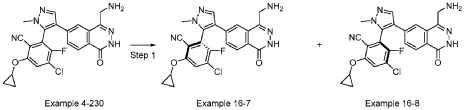
[0590] Example 4-230, 2-(4-(4-(aminomethyl)-1-oxo-1,2-dihydrophthalazin-6-yl)-1-methyl-1H-pyrazol-5-yl)-4-chloro-6-cyclopropoxy-3-fluorobenzonitrile (30 mg, 0.065 mmol) separated by SFC (DAICEL CHIRALPAK IC (250 mm × 30 mm x 10 mm); mobile phase:
[0.1% NH3H2O isopropanol]; B%: 40% isocratic, 4.1 min cycle; 120 min total ) to give example 16-7 (ee > 99%, 13 mg, 0.026 mmol, 25% yield) as a yellow solid and example 16-8 (8 mg, ee = 84% ). Example 16-8 was then then further separated by SFC (DAICEL CHIRALPAK IC (250 mm × 30 mm,10 mm); mobile phase: [0.1% NH3H2O EtOH]; B%: 60% isocratic, 3.1 min cycle; total 50 min) to give Example 16-8 (ee > 99%, 4 mg, 0.007 mmol, 7% yield) as a yellow gum. Spectra data for Example 16-7: LCMS [M+1] + = 465.1; 1H NMR (400 MHz, DMSO-d6) d = 12.59 – 12.44 (s, 1H), 8.29 (s, 1H), 8.15 (d, J = 8.4 Hz, 1H), 8.01 (d, J = 6.0 Hz, 1H), 7.75 (s, 1H), 7.67 (br d, J = 7.6 Hz, 1H), 4.23 – 4.17 (m, 1H), 3.86 (br s, 2H), 3.78 (s, 3H), 0.94 – 0.88 (m, 2H), 0.84 – 0.79 (m, 2H). Spectra data for Example 16-8: LCMS [M+1] + = 465.1; 1H NMR (400 MHz, DMSO-d6) d = 12.49 – 12.37 (s, 1H), 8.26 (s, 1H), 8.15 (d, J = 8.4 Hz, 1H), 8.00 (d, J = 6.0 Hz, 1H), 7.73 (d, J = 1.6 Hz, 1H), 7.72 – 7.68 (m, 1H), 4.19 (m, 1H), 3.80 (s, 2H), 3.77 (s, 3H), 0.93 – 0.87 (m, 2H), 0.83 – 0.78 (m, 2H).
PAT
- MTA-synergistic PRMT5 inhibitorsPublication Number: CN-114728912-APriority Date: 2019-09-12
- Mta-cooperative prmt5 inhibitorsPublication Number: WO-2021050915-A1Priority Date: 2019-09-12
- MTA-Cooperative PRMT5 InhibitorsPublication Number: US-2021078994-A1Priority Date: 2019-09-12
- MTA-Cooperative PRMT5 InhibitorsPublication Number: US-2021079003-A1Priority Date: 2019-09-12
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- Dynamic Kinetic Resolution of Axially Chiral MRTX1719Publication Name: SynfactsPublication Date: 2022-10-18DOI: 10.1055/s-0041-1738738
- Design and evaluation of achiral, non-atropisomeric 4-(aminomethyl)phthalazin-1(2H)-one derivatives as novel PRMT5/MTA inhibitorsPublication Name: Bioorganic & Medicinal ChemistryPublication Date: 2022-10-01PMID: 35926325DOI: 10.1016/j.bmc.2022.116947
- Synthesis of MRTX1719Publication Name: SynfactsPublication Date: 2022-03-18DOI: 10.1055/s-0041-1737934
- Fragment-Based Discovery of MRTX1719, a Synthetic Lethal Inhibitor of the PRMT5•MTA Complex for the Treatment of MTAP -Deleted CancersPublication Name: Journal of Medicinal ChemistryPublication Date: 2022-01-18PMID: 35041419DOI: 10.1021/acs.jmedchem.1c01900
- Disordered methionine metabolism in MTAP/CDKN2A-deleted cancers leads to dependence on PRMT5Publication Name: Science (New York, N.Y.)Publication Date: 2016-03-11PMID: 26912361DOI: 10.1126/science.aad5944
- Synthesis of MRTX1719DOI: 10.1055/s-0041-1737934Publication Date: 2022Publication Name: Synfacts
- Dynamic Kinetic Resolution of Axially Chiral MRTX1719DOI: 10.1055/s-0041-1738738Publication Date: 2022Publication Name: Synfacts
////////navlimetostat, anax labs, antineoplastic, MRTX-1719, BMS-986504, MRTX 1719, BMS 986504
Lonitoclax
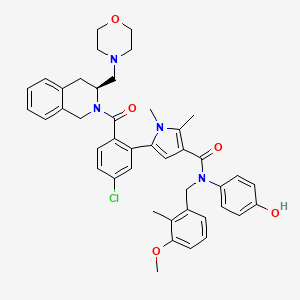
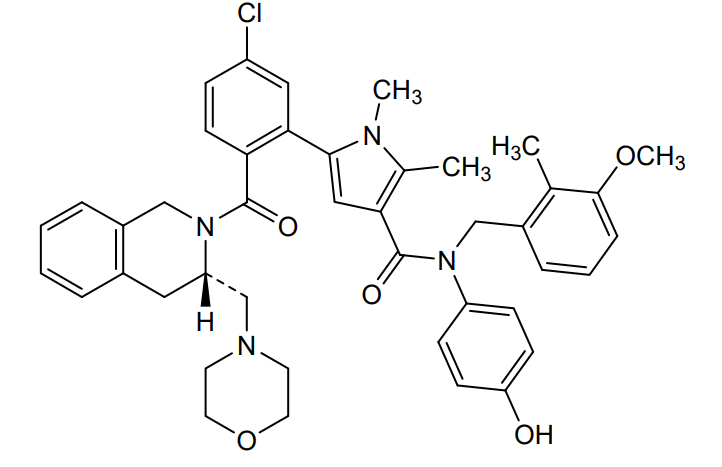
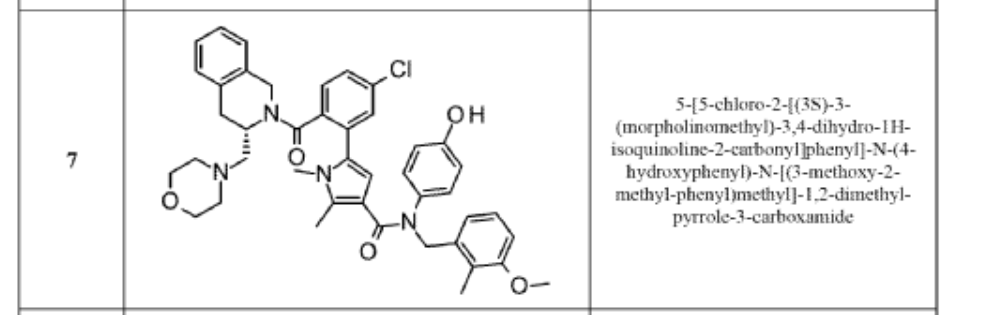
Lonitoclax
CAS 2952589-57-8
MF C43H45ClN4O5 MW733.3 g/mol
5-[5-chloro-2-[(3S)-3-(morpholin-4-ylmethyl)-3,4-dihydro-1H-isoquinoline-2-carbonyl]phenyl]-N-(4-hydroxyphenyl)-N-[(3-methoxy-2-methylphenyl)methyl]-1,2-dimethylpyrrole-3-carboxamide
- 1H-Pyrrole-3-carboxamide, 5-[5-chloro-2-[[(3S)-3,4-dihydro-3-(4-morpholinylmethyl)-2(1H)-isoquinolinyl]carbonyl]phenyl]-N-(4-hydroxyphenyl)-N-[(3-methoxy-2-methylphenyl)methyl]-1,2-dimethyl-
- 5-(5-Chloro-2-(((3S)-3-(morpholin-4-ylmethyl)-3,4-dihydroisoquinolin-2-(1-H)-yl)carbonyl)phenyl)-N-4-hydroxyphenyl)-N-(3-methoxy-2-methylbenzyl)-1,2-dimethyl-1H-pyrrole-3-carboxamide
- 5-[5-chloro-2-[(3S)-3-(morpholinomethyl)- 3,4-dihydro-1H-isoquinoline-2- carbonyl]phenyl]-N-(4-hydroxyphenyl)-N- [(3-methoxy-2-methyl-phenyl)methyl]-1,2- dimethyl-pyrrole-3-carboxamide
- 5-{5-Chloro-2-[(3S)-3-[(morpholin-4-yl)methyl]-3,4-dihydroisoquinoline-2(1H)-carbonyl]phenyl}-N-(4-hydroxyphenyl)-N-[(3-methoxy-2-methylphenyl)methyl]-1,2-dimethyl-1H-pyrrole-3-carboxamide
5-(5-chloro-2-{(3S)-3-[(morpholin-4-yl)methyl]-3,4-dihydroisoquinoline-2(1H)-carbonyl}phenyl)-N-(4-
hydroxyphenyl)-N-[(3-methoxy-2-methylphenyl)methyl]-1,2-dimethyl-1H-pyrrole-3-carboxamide
B-cell lymphoma 2 (Bcl-2) inhibitor, antineoplastic, ZE50-0134, ZE50 0134, Lomond Therapeutics, CANCER, 76NBC3X6A3
Lonitoclax (also known as ZE50-0134) is an investigational, next-generation, orally administered B-cell lymphoma 2 (Bcl-2) inhibitor being developed for the treatment of hematologic malignancies like Acute Myeloid Leukemia (AML) and Chronic Lymphocytic Leukemia (CLL). Developed by Lomond Therapeutics, the drug is engineered as a highly selective option to improve upon existing first-generation Bcl-2 inhibitors like venetoclax.
Mechanism and Advantages Over Venetoclax
Unlike earlier therapies, lonitoclax features a unique binding mode and a structurally distinct chemotype. Its design yields several pharmacology advantages:
- Higher Selectivity: It binds tightly to Bcl-2 while demonstrating exceptional selectivity over Bcl-xL, which helps lower hematologic toxicities.
- Limited Immune Suppression: In preclinical data, lonitoclax spared healthy non-malignant immune cells (B cells, CD8 T cells, and NK cells), a major shift from the immunosuppressive profile of venetoclax.
- Reduced Drug Interaction & Accumulation: It features a shorter half-life (~9–10 hours) and minimal CYP3A4 (P4503A4) inhibition. This prevents the drug from building up dangerously and mitigates the risk of Tumor Lysis Syndrome (TLS), potentially enabling safer outpatient treatments.
Clinical Development Status
Lonitoclax is currently advancing through early-phase clinical trials:
- IND Clearances: The U.S. FDA cleared Investigational New Drug (IND) applications evaluating lonitoclax for CLL/SLL and as a combination treatment for relapsed or refractory AML.
- Healthy Volunteer Studies: Phase 1 single ascending dose (SAD) studies in healthy adults confirmed that the drug is well tolerated with linear pharmacokinetics and no significant safety issues. Target engagement was confirmed through plasma apoptosis assays.
- Combination Trials: Active Phase 1b multicenter trials are underway evaluating the safety, efficacy, and synergy of lonitoclax when combined with hypomethylating agents like azacitidine in AML patients.
SYN
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023129553&_cid=P11-MQVQMH-93381-1
ADVERISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- Bcl-2 inhibitorsPublication Number: WO-2023129553-A1Priority Date: 2021-12-29
- BCL-2 InhibitorsPublication Number: US-2025115577-A1Priority Date: 2021-12-29
- Bcl-2 inhibitorsPublication Number: EP-4457223-A1Priority Date: 2021-12-29
///////Lonitoclax, ANAX LABS, B-cell lymphoma 2 (Bcl-2) inhibitor, antineoplastic, ZE50-0134, ZE50 0134, Lomond Therapeutics, CANCER, 76NBC3X6A3
Lomonitinib
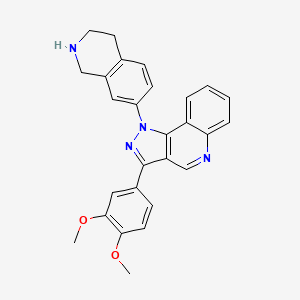
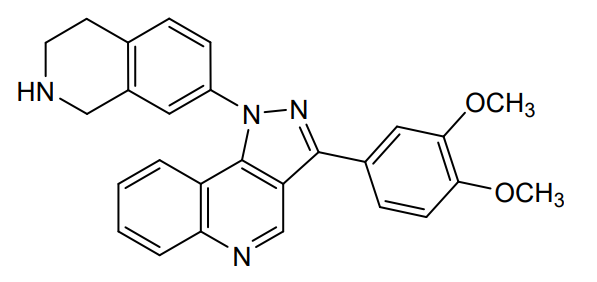
Lomonitinib
CAS 2923221-56-9
MF C27H24N4O2 MW436.5 g/mol
3-(3,4-dimethoxyphenyl)-1-(1,2,3,4-tetrahydroisoquinolin-7-yl)pyrazolo[4,5-c]quinoline
- 3-(3,4-dimethoxyphenyl)-1-(1,2,3,4-tetrahydroisoquinolin-7-yl)pyrazolo[4,3-c]quinoline
- 1H-Pyrazolo[4,3-c]quinoline, 3-(3,4-dimethoxyphenyl)-1-(1,2,3,4-tetrahydro-7-isoquinolinyl)-
- 3-(3,4-dimethoxyphenyl)-1-(1,2,3,4-tetrahydroisoquinolin-7-yl)-1H-pyrazolo[4,3-c]quinoline
- 7-[3-(3,4-dimethoxyphenyl)-1H- pyrazolo[4,3-c]quinolin-1-yl]-1,2,3,4- tetrahydroisoquinoline
- 7-[3-(3,4-Dimethoxyphenyl)-1H-pyrazolo[4,3-c]quinolin-1-yl]-1,2,3,4-tetrahydroisoquinoline
3-(3,4-dimethoxyphenyl)-1-(1,2,3,4-tetrahydroisoquinolin-7-yl)-1H-pyrazolo[4,3-c]quinoline
tyrosine kinase inhibitor, antineoplastic, ZE46-0134, Eilean Therapeutics, U4DPU7W7QU
Lomonitinib (also known as ZE46-0134) is a highly potent, selective, orally bioavailable pan-FLT3 and IRAK4 small molecule inhibitor being developed for the treatment of Acute Myeloid Leukemia (AML). Developed by Eilean Therapeutics in collaboration with Expert Systems, it uniquely targets both primary mutations and the major drug-resistance pathways that cause other AML therapies to fail.
Mechanism of Action
Lomonitinib utilizes a dual-targeting framework to bypass conventional drug resistance:
- Pan-FLT3 Inhibition: It binds to and blocks FMS-like tyrosine kinase 3 (FLT3) mutations. This includes the challenging FLT3-ITD-F691L “gatekeeper” mutation, which typically confers resistance to all currently approved standard FLT3 inhibitors like gilteritinib.
- IRAK4 Inhibition: It simultaneously targets interleukin-1 receptor-associated kinase 4 (IRAK4). IRAK4 activation acts as a key “escape pathway” that cancer cells use to survive and build adaptive resistance to standalone FLT3 therapy.
Key Clinical Advantages
According to preclinical models and clinical data presented at the American Society of Hematology (ASH), lomonitinib offers unique benefits:
- Superior Efficacy: In vivo models demonstrate stronger anti-tumor activity and deeper responses in gatekeeper mutation-dependent disease compared to gilteritinib.
- Favorable Loading Strategy: Because of its wide therapeutic index and low toxicity, clinicians can administer a high loading dose on Day 1 followed by a smaller maintenance dose. This achieves effective therapeutic drug levels by Day 4, a rapid target engagement not possible with older long-half-life FLT3 inhibitors.
- Low Drug Interactions: Clinical profiles show minimal pharmacokinetic interference from proton pump inhibitors (PPIs) or CYP3A4 inhibitors like itraconazole.
Development Status
Lomonitinib is currently classified as an investigational new drug:
- Clinical Trials: It is undergoing open-label, dose-escalation Phase 1/1b trials in both Australia and the United States (such as trial NCT06366789) evaluating adults with FLT3-mutated relapsed or refractory AML.
- Partnerships: The drug is being studied in the US in collaboration with The Leukemia & Lymphoma Society as part of their Beat AML master clinical trial portfolio
Lomonitinib is an orally bioavailable inhibitor of FMS-like tyrosine kinase 3 (FLT3; CD135; STK1; FLK2) mutations and interleukin-1 receptor-associated kinase 4 (IRAK4), with potential antineoplastic activity. Upon oral administration, lomonitinib targets, binds to and inhibits the activity of FLT3 mutations, including the FLT3-ITD-F691L gatekeeper mutation, while sparing the wild-type form of FLT3. This inhibits the proliferation of FLT3 mutant-expressing cancer cells. In addition, lomonitinib targets, binds to, and inhibits the kinase activity of IRAK4. This inhibits IRAK4-mediated signaling and may reduce adaptive resistance to FLT3 inhibition as toll-like receptor (TLR) activation plays an important role in resistance to FLT3 inhibition. FLT3, a class III receptor tyrosine kinase (RTK), is overexpressed or mutated in most B-lineage neoplasms and in acute myeloid leukemias. IRAK4, a serine/threonine-protein kinase, plays a key role in both the TLR and IL-1R signaling pathways.
- Dose Escalation and Expansion Study to Evaluate the Safety, PK, PD and Efficacy of ZE46-0134 in Adults With FLT3 Mutated or Spliceosome Mutated Relapsed or Refractory Acute Myeloid LeukemiaCTID: NCT06366789Phase: Phase 1Status: RecruitingDate: 2025-12-31
- Study of Biomarker-Based Treatment of Acute Myeloid LeukemiaCTID: NCT03013998Phase: Phase 1/Phase 2Status: RecruitingDate: 2025-12-17
- Study of Single and Multiple Ascending Doses of ZE46-0134 in Healthy VolunteersCTID: NCT06399315Phase: Phase 1Status: CompletedDate: 2025-12-09
SYN
PAT
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US445571045&_cid=P12-MQUBPX-10324-1
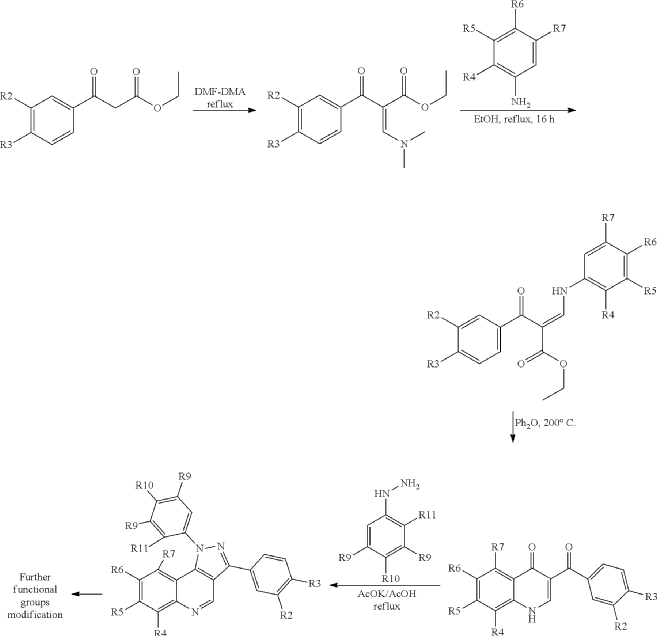

Example 34: 3-(3,4-dimethoxyphenyl)-1-(1,2,3,4-tetrahydroisoquinolin-7-yl)-1H-pyrazolo[4,3-c]quinoline dihydrochloride (1.31)
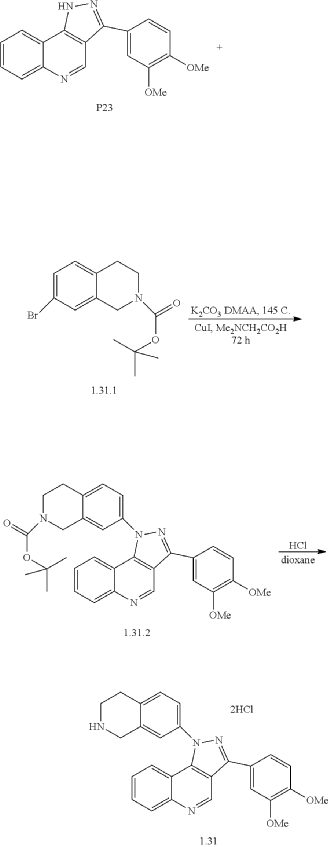
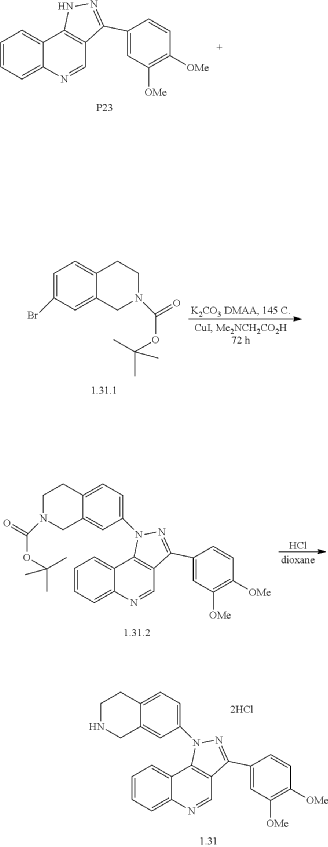
| A mixture of 3-(3,4-dimethoxyphenyl)-1H-pyrazolo[4,3-c]quinoline (P23) (153 mg, 0.5 mmol), tert-butyl 7-bromo-3,4-dihydroisoquinoline-2(1H)-carboxylate (1.31.1) (172 mg, 0.55 mmol), K 2CO 3 (83 mg, 0.6 mmol), CuI (10 mg, 0.05 mmol), N,N-dimethylglycine (11 mg, 0.1 mmol), and DMAA (2 mL) was stirred under Ar at 145° C. for 72 h, cooled to ambient temperature, diluted with CHCl 3, washed with 1% aq. solution of Na 2EDTA, and concentrated under reduced pressure. The residue was subjected to HPLC to afford 87 mg (33%) of tert-Butyl 7-(3-(3,4-dimethoxyphenyl)-1H-pyrazolo[4,3-c]quinolin-1-yl)-3,4-dihydroisoquinoline-2(1H)-carboxylate (1.31.2). 1H NMR (400 MHz, DMSO-d 6): δ 9.57 (s, 1H), 8.19 (d, J=8.4 Hz, 1H), 7.77 (m, 1H), 7.69 (dd, J 1=8.0 Hz, J 2=1.6 Hz, 1H), 7.63 (s, 1H), 7.57 (m, 3H), 7.51 (m, 2H), 7.17 (d, J=8.4 Hz, 1H), 4.63 (s, 2H), 3.88 (s, 3H), 3.86 (s, 3H), 3.68 (m, 2H), 2.98 (m, 2H), 1.45 (s, 9H). LCMS (ESI) m/z 538 [MH] +. |
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- Substituted 1H-pyrazolo [4,3-c ] quinolines, methods of preparation and uses thereofPublication Number: CN-118076605-APriority Date: 2021-10-15
- SUBSTITUTED 1H-PYRAZOLO [4,3-c] QUINOLINES, METHODS OF PREPARATION, AND USE THEREOFPublication Number: US-2025011319-A1Priority Date: 2021-10-15
////////lomonitinib, anax labs, tyrosine kinase inhibitor, antineoplastic, ZE46-0134, Eilean Therapeutics, U4DPU7W7QU
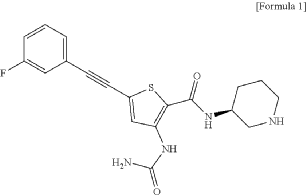
Lasmotinib
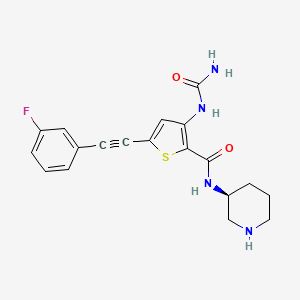
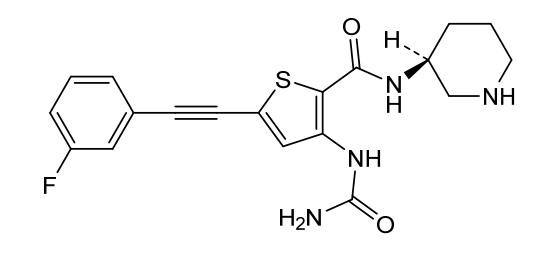
Lasmotinib
CAS 2127107-15-5
MF C19H19FN4O2S MW386.4 g/mol
3-(carbamoylamino)-5-[2-(3-fluorophenyl)ethynyl]-N-[(3S)-piperidin-3-yl]thiophene-2-carboxamide
- N-(2-(N-((3S)(3-Piperidyl))carbamoyl)-5-(2-(3-fluorophenyl)ethynyl)(3-thienyl))aminamide
- 2-Thiophenecarboxamide, 3-((aminocarbonyl)amino)-5-(2-(3-fluorophenyl)ethynyl)-N-(3S)-3-piperidinyl-
- 3-(carbamoylamino)-5-[2-(3-fluorophenyl)ethynyl]-N-[(3S)-piperidin-3-yl]thiophene-2-carboxamide
- 2-Thiophenecarboxamide, 3-[(aminocarbonyl)amino]-5-[2-(3-fluorophenyl)ethynyl]-N-(3S)-3-piperidinyl-
- 3-(carbamoylamino)-5-(2-(3-fluorophenyl)ethynyl)-N-((3S)-piperidin-3-yl)thiophene-2-carboxamide
3-(carbamoylamino)-5-[(3-fluorophenyl)ethynyl]-N-[(3S)-piperidin-3-yl]thiophene-2-carboxamide
tyrosine kinase inhibitor, antineoplastic, PHI-101, PHI 101, U2UY9TBQ8Z
Lasmotinib (also known by its research code PHI-101) is a next-generation, orally bioavailable targeted cancer therapy. It functions as a dual FLT3 and CHK2 inhibitor. It is primarily being investigated to treat Acute Myeloid Leukemia (AML) and ovarian cancer.
How It Works
- FLT3 Inhibition: It targets FMS-like tyrosine kinase 3 (FLT3), an enzyme that is often mutated in AML. Lasmotinib is designed to attack not just single activating mutations (ITD or TKD), but also difficult-to-treat double and triple-resistant mutations.
- CHK2 Inhibition: It also inhibits Checkpoint Kinase 2 (CHK2), preventing cancer cells from repairing DNA damage. This causes the cancer cells to undergo apoptosis (programmed cell death).
Key Clinical Advantages
- High Efficacy: In relapsed or refractory AML patients who have previously failed other FLT3 inhibitors, lasmotinib has demonstrated high rates of composite complete remission.
- Safety Profile: Preclinical and early-stage trials indicate a promising safety profile with a very low or 0% occurrence rate of cardiotoxicity (heart damage), which is a common hurdle for some other FLT3-targeting drugs.
Current Development & Combinations
- Developer: Discovered by Seoul National University Hospital and being developed by Pharos iBio.
- Synergistic Therapies: Lasmotinib is currently moving into global clinical trials as a powerful combination therapy. Research shows it synergizes strongly with existing treatments like Venetoclax or Azacytidine, as well as with emerging Menin inhibitors (such as bleximenib) to achieve deep tumor growth inhibition
Lasmotinib is an orally bioavailable inhibitor of checkpoint kinase 2 (chk2), with potential antineoplastic and chemopotentiating activities. Upon oral administration, lasmotinib binds to and inhibits the activity of chk2, which may prevent the repair of DNA damage caused by DNA-damaging agents. This may result in tumor cell apoptosis and potentiate the antitumor efficacies of various chemotherapeutic agents. Chk2, an ATP-dependent serine–threonine kinase, is a key component in the DNA replication-monitoring checkpoint system and is activated by double-stranded breaks (DSBs); activated chk2 is overexpressed by a variety of cancer cell types.
- Chk2 Inhibitor for Recurrent EpitheliAl periToneal, fallopIan or oVarian cancEr (CREATIVE Phase IA Trial)CTID: NCT04678102Phase: Phase 1Status: Unknown statusDate: 2023-06-26
- Evaluation of the Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of PHI 101 for the Treatment of AMLCTID: NCT04842370Phase: Phase 1Status: Unknown statusDate: 2021-04-20
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=JP405710409&_cid=P21-MQIVJB-43702-2
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US465154324&_cid=P21-MQIVJB-43702-2
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2024015484&_cid=P21-MQIVJB-43702-2
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2025210599&_cid=P21-MQIVJB-43702-2
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
PAT
- Inhibitors of brutons tyrosine kinasePublication Number: US-2021070748-A1Priority Date: 2015-06-02
- New, substituted quinoline compounds as inhibitors of S-nitrosoglutathion reductasePublication Number: HU-E025653-T2Priority Date: 2010-10-08
- New hybrid oligomers. Their preparation process and pharmaceutical compositions containing themPublication Number: AU-2006256439-A1Priority Date: 2005-03-18
- NEW THIOPHENE COMPOUND SUBSTITUTED IN POSITIONS 2,3,5, USED AS A PROTEIN KINASE INHIBITORPublication Number: BR-112018016729-B1Priority Date: 2016-02-16
- 2, 3, 5-substituted thiophene compounds as protein kinase inhibitorsPublication Number: CN-108884066-BPriority Date: 2016-02-16Grant Date: 2021-08-24
- 2,3,5-substituted thiophene compound as protein kinase inhibitorPublication Number: US-10442796-B2Priority Date: 2016-02-16Grant Date: 2019-10-15
- Novel compound of 2,3,5-substituted thiophene as a protein kinase inhibitorPublication Number: RU-2724957-C2Priority Date: 2016-02-16Grant Date: 2020-06-29
- Novel 2,3,5-substituted thiophene compounds as protein kinase inhibitorsPublication Number: KR-101965326-B1Priority Date: 2016-02-16Grant Date: 2019-04-03
- Novel 2,3,5-substituted thiophene compound as protein kinase inhibitorPublication Number: WO-2017142325-A1Priority Date: 2016-02-16
- Novel 2,3,5-substituted thiophene compound as protein kinase inhibitorPublication Number: US-2019047993-A1Priority Date: 2016-02-16
- Novel 2,3,5-substituted thiophene compounds as protein kinase inhibitorsPublication Number: KR-20180136425-APriority Date: 2016-02-16
- Novel 2,3,5-substituted thiophene compound as protein kinase inhibitorPublication Number: EP-3418275-B1Priority Date: 2016-02-16Grant Date: 2021-03-17
- Novel 2,3,5-substituted thiophene compounds that are protein kinase inhibitorsPublication Number: JP-2019504900-APriority Date: 2016-02-16
- Use of 2,3,5-substituted thiophene compound for enhancement of radiotherapyPublication Number: EP-3804719-A1Priority Date: 2018-05-30
- Use of 2,3,5-substituted thiophene compound to prevent, ameliorate, or treat breast cancersPublication Number: EP-3804718-A1Priority Date: 2018-05-30
- Novel 2,3,5-substituted thiophene compound as protein kinase inhibitorPublication Number: EP-3418275-A1Priority Date: 2016-02-16
- New 2,3,5-substituted thiophene compound as a protein kinase inhibitorPublication Number: RU-2018130703-APriority Date: 2016-02-16
- Novel 2,3,5-substituted thiophene compounds as protein kinase inhibitorsPublication Number: KR-20190035671-APriority Date: 2016-02-16
- Radiotherapy-enhancing applications of 2,3,5-substituted thiophene compoundsPublication Number: JP-2021525285-APriority Date: 2018-05-30
- Use of 2,3,5-substituted thiophene compound for enhancement of radiotherapyPublication Number: US-2021205289-A1Priority Date: 2018-05-30
- Use of 2,3,5-Substituted Thiophene Compound for Prevention, Improvement or Treatment of Breast CancerPublication Number: KR-102227117-B1Priority Date: 2018-05-30Grant Date: 2021-03-15
- Use of 2,3,5-Substituted Thiophene Compound for Prevention, Improvement or Treatment of Breast CancerPublication Number: KR-20190136976-APriority Date: 2018-05-30
- Use of 2,3,5-substituted thiophene compound to prevent, ameliorate, or treat breast cancersPublication Number: US-2021205290-A1Priority Date: 2018-05-30
////////lasmotinib, anax labs, tyrosine kinase inhibitor, antineoplastic, PHI-101, PHI 101, U2UY9TBQ8Z
Lanisidenib
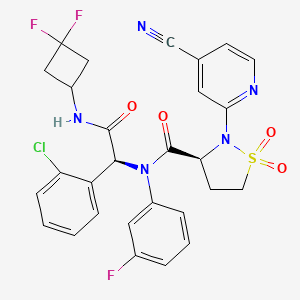
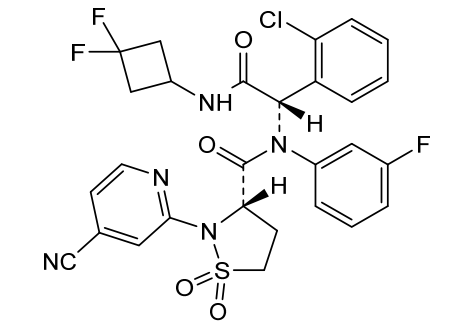
Lanisidenib
Cas 2135537-20-9
MF C28H23ClF3N5O4S MW618.03 g/mol
(3S)-N-[(1S)-1-(2-chlorophenyl)-2-[(3,3-difluorocyclobutyl)amino]-2-oxoethyl]-2-(4-cyano-2-pyridinyl)-N-(3-fluorophenyl)-1,1-dioxo-1,2-thiazolidine-3-carboxamide
IUPAC Name: (3S)-N-{(1S)-1-(2-chlorophenyl)-2-[(3, 3-difluorocyclobutyl)amino]-2-oxoethyl}-2-(4-cyanopyridin-2-yl)-N-(3-fluorophenyl)-1,1-dioxo-1λ⁶,2-thiazolidine-3-carboxamide
(3S)-N-{(1S)-1-(2-chlorophenyl)-2-[(3,3-difluorocyclobutyl)amino]-2-oxoethyl}-2-(4-cyanopyridin-2-yl)-N-(3-fluorophenyl)-1,1-dioxo1λ6,2-thiazolidine-3-carboxamide
isocitrate dehydrogenase inhibitor, antineoplastic, G5J396CG5J
Lanisidenib is a potent, selective isocitrate dehydrogenase (IDH) inhibitor that exhibits antineoplastic (anti-cancer) activity. It works by targeting abnormal IDH enzymes, which are frequently mutated in various malignancies, such as certain myeloid leukemias and solid tumours. By blocking these mutant enzymes, it halts the production of oncometabolites that drive cancer progression
Research and Availability
The compound is primarily utilized in biochemical research and preclinical drug screening platforms. Specialty chemical suppliers, such as MedChemExpress and AdooQ BioScience, distribute it exclusively for laboratory research
SYN
Inhibitors of Mutant Isocitrate Dehydrogenases 1 and 2 (mIDH1/2): An Update and Perspective
Publication Name: Journal of Medicinal Chemistry
Publication Date: 2018-05-31
PMID: 29847930
DOI: 10.1021/acs.jmedchem.8b00159
PAT
| Step F: (S)—N—((S)-1-(2-chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-2-(4-cyanopyridin-2-yl-N-(3-fluorophenyl)-isothiazolidine-3-carboxamide 1,1-dioxide |
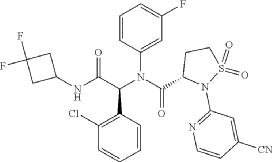
| At room temperature, 3-amino-5-Fluorouridine (57 mg, 0.508 mmol) and o-chlorobenzaldehyde (72 mg, 0.512 mmol) were dissolved in methanol, and stirred for 30 min. (S)-2-(4-cyanopyridin-2-yl)isothiazolidine-3-carboxylic acid 1,1-dioxide (136 mg, 0.508 mmol) was then added into the mixed solution, stirred for 10 min, then added with 1,1-difluoro-3-isocyanocyclobutane (prepared according to the method described in patent CN103097340, 60 mg, 0.508 mmol), and stirred overnight. The solvent was removed and the residue was separated by thin layer chromatography, to give the title compound (S)—N—((S)-1-(2-chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-2-(4-cyanopyridin-2-yl-N-(3-fluorophenyl)-isothiazolidine-3-carboxamide 1,1-dioxide (the compound of formula I). |
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019057142&_cid=P20-MQBQHW-03190-1
A sulfonamide compound with the structure shown in Formula I has the chemical name: (S)-N-((S)-1-(2-chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-2-(4-cyanopyridin-2-yl)-N-(3-fluorophenyl)-isothiazolidin-3-carboxamide 1,1-dioxide.
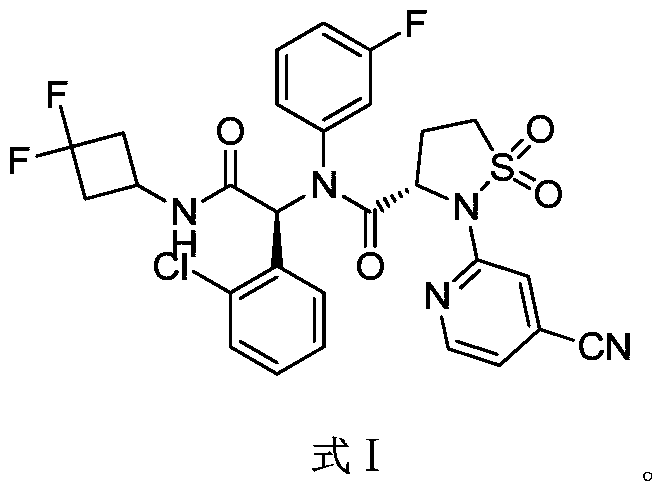
Step F: (S)-N-((S)-1-(2-chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-2-(4-cyanopyridin-2-yl)-N-(3-fluorophenyl)-isothiazolidin-3-carboxamide 1,1-dioxide
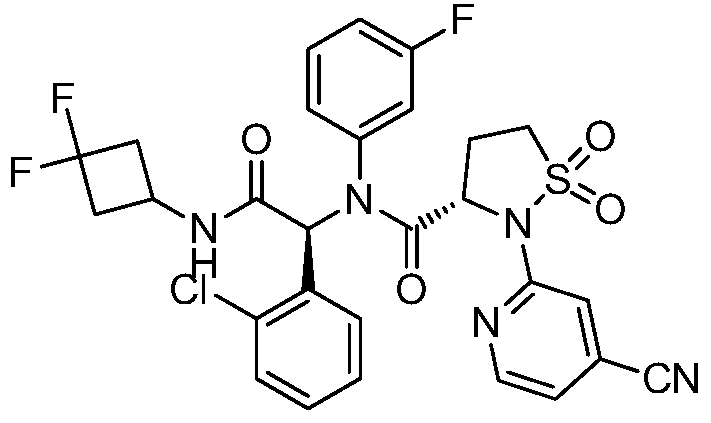
At room temperature, 3-amino-5-fluoropyridine (57 mg, 0.508 mmol) and o-chlorobenzaldehyde (72 mg, 0.512 mmol) were dissolved in methanol and stirred for 30 minutes. Then, (S)-2-(4-cyanopyridin-2-yl)isothiazolidin-3-carboxylic acid 1,1-dioxide (136 mg, 0.508 mmol) was added to the mixture and stirred for 10 minutes. Finally, 1,1-difluoro-3-isocyanocyclobutane (refer to the patent) was added. Prepared by the method described in CN103097340, 60 mg (0.508 mmol), stirred overnight, solvent removed, and separated by thin-layer chromatography to obtain the title compound (S)-N-((S)-1-(2-chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-2-(4-cyanopyridin-2-yl)-N-(3-fluorophenyl)-isothiazolidin-3-carboxamide 1,1-dioxide (compound of formula I).
[0134]
1H-NMR(400MHz,CDCl 3):δ=8.46(m,1H),7.67(d,J=8.8Hz,1H),7.63(s,1H),7.22-6.84(m,8H),6.47(d,J=3.6,1H),6.08(s,1H),4.82(d,J=6.1Hz,1H),4.33(m,1H),3.68-3.60(m,1H),3.40-3.28(m,1H),3.10-2.98(m,2H),2.68-2.38(m,4H)。
[0135]
m/z=618[M+H] +。
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
PAT
- Sultam compound and application method thereofPublication Number: US-11111240-B2Priority Date: 2016-03-22Grant Date: 2021-09-07
- Sultam Compound And Application Method ThereofPublication Number: US-2021047314-A1Priority Date: 2016-03-22
- Lactam compounds and methods of using the samePublication Number: CN-113666922-APriority Date: 2016-03-22
- Endosulfonamide compound and method of use thereofPublication Number: TW-201736354-APriority Date: 2016-03-22
- Sultam compound and application method thereofPublication Number: EP-3434671-B1Priority Date: 2016-03-22Grant Date: 2020-10-21
- Internal sulfonamide compounds and methods of use thereofPublication Number: CN-109071471-BPriority Date: 2016-03-22Grant Date: 2021-05-07
- Sultam compounds and methods of use thereofPublication Number: KR-102389985-B1Priority Date: 2016-03-22Grant Date: 2022-04-22
- Endosulfonamide compounds and methods of usePublication Number: TW-I729094-BPriority Date: 2016-03-22Grant Date: 2021-06-01
- Crystalline sulfamide compoundsPublication Number: KR-102707847-B1Priority Date: 2017-09-22Grant Date: 2024-09-23
- Crystalline sulfamide compoundPublication Number: KR-20200057049-APriority Date: 2017-09-22
- Crystalline sulfamide compoundPublication Number: CA-3076405-A1Priority Date: 2017-09-22
- Crystalline sulfamide compoundPublication Number: US-2020291012-A1Priority Date: 2017-09-22
- Crystalline sulfamide compoundPublication Number: US-11254665-B2Priority Date: 2017-09-22Grant Date: 2022-02-22
- Preparation method of lactam compoundPublication Number: CN-118580235-APriority Date: 2023-03-03
- A kind of internal sulfonamide compound crystalPublication Number: CN-111065630-APriority Date: 2017-09-22
- Crystalline sulfamide compoundPublication Number: EP-3686191-A1Priority Date: 2017-09-22
- A kind of internal sulfonamide compound crystallizationPublication Number: CN-111065630-BPriority Date: 2017-09-22Grant Date: 2022-12-30
- Crystalline sulfamide compoundPublication Number: EP-3686191-B1Priority Date: 2017-09-22Grant Date: 2022-12-14
///////////lanisidenib, anax labs, isocitrate dehydrogenase inhibitor, antineoplastic, G5J396CG5J
Itareparib
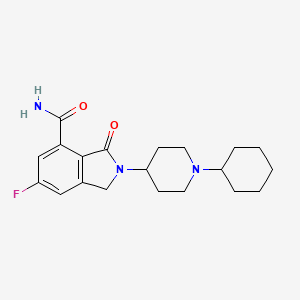
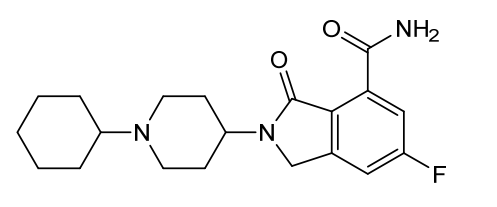
Itareparib
CAS 1606995-47-4
MF C20H26FN3O2 MW359.4 g/mol
2-(1-Cyclohexyl-4-piperidinyl)-6-fluoro-2,3-dihydro-3-oxo-1H-isoindole-4-carboxamide
1H-ISOINDOLE-4-CARBOXAMIDE, 2-(1-CYCLOHEXYL-4-PIPERIDINYL)-6-FLUORO-2,3-DIHYDRO-3-OXO-
2-(1-cyclohexylpiperidin-4-yl)-6-fluoro-3-oxo-2,3-dihydro-1H-isoindole4-carboxamide
poly (ADP-ribose) polymerase (PARP) inhibitor, antineoplastic, NMS-03305293, NMS-293, NMS 03305293, NMS 293, KFI1190L8L, NV 578,
Itareparib is the inhibitor for PARP and exhibits antineoplastic activity.
Itareparib (development code NMS-03305293 or NMS-293) is an experimental, next-generation PARP1-selective oral inhibitor being developed by the biopharmaceutical company Nerviano Medical Sciences for the treatment of various advanced solid tumors and brain cancers
Key Characteristics & Mechanism
Unlike first-generation poly(ADP-ribose) polymerase (PARP) inhibitors, itareparib features a highly specialized mechanism designed to improve clinical safety and versatility:
- Non-Trapping Profile: Traditional PARP inhibitors trap the PARP enzyme onto DNA, forming PARP-DNA complexes. This trapping causes significant bone marrow toxicity (myelosuppression), leading to severe side effects like anemia, neutropenia, and thrombocytopenia. Itareparib is engineered to be “non-trapping,” avoiding these complexes to protect healthy blood cells.
- High Brain Penetrance: It crosses the blood-brain barrier effectively, making it uniquely suitable for treating primary and secondary central nervous system (CNS) malignancies.
- Ideal Combinability: Because it does not cause overlapping bone marrow toxicity, it can be safely paired with other DNA-damaging therapies like traditional chemotherapies and antibody-drug conjugates (ADCs).
Clinical Development & Target Indications
Itareparib is currently advancing through Phase I and Phase II clinical trials. It is being investigated across several oncology settings:
- Glioblastoma (GBM): Evaluated in Phase II clinical studies for relapsed, IDH wild-type glioblastoma in combination with the chemotherapy drug temozolomide (TMZ).
- Ovarian Cancer: Evaluated in Phase Ia/Ib trials (such as trial NCT06930755) in combination with topotecan for patients with recurrent, platinum-resistant ovarian, fallopian tube, or peritoneal cancers. [1]
- Small Cell Lung Cancer (SCLC) & Astrocytoma: Explored in ongoing combination trials targeting highly aggressive tumors where conventional PARP inhibitors are limited by overlapping toxicity.
- Study of NMS-03305293 in Adult Patients With Relapsed Ovarian CancerCTID: NCT06930755Phase: Phase 1Status: RecruitingDate: 2026-05-28
- Study of NMS-03305293 in Adult Patient With Relapsed Small Cell Lung CancerCTID: NCT06931626Phase: Phase 1Status: RecruitingDate: 2025-11-12
- Ph I/II Study of NMS-03305293+TMZ in Adult Patients With Recurrent GlioblastomaCTID: NCT04910022Phase: Phase 1/Phase 2Status: Active, not recruitingDate: 2025-08-19
- Study of NMS-03305293 in Pts with Selected Advanced/Metastatic Solid TumorsCTID: NCT04182516Phase: Phase 1Status: TerminatedDate: 2024-09-19
A Phase I/II Combination Study of NMS-03305293 and Temozolomide in Adult Patients with Recurrent Glioblastoma
EudraCT: 2020-003417-35
Phase: Phase 1, Phase 2
Status: Trial now transitioned
Date: 2021-11-10
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US275481284&_cid=P10-MQA9O8-42416-1
2-(1-Cyclohexyl-piperidin-4-yl)-6-fluoro-3-oz-2,3-dihydro-1H-isoindole-4-carboxylic Acid Amide (I), cpd 29 [R═F; n=m=0; R1=piperidin-4-yl; R2=1-cyclohexyl]
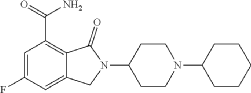
| To a stirred solution of 2-(1-cyclohexyl-piperidin-4-yl)-6-fluoro-3-oxo-2,3-dihydro-1H-isoindole-4-carbonitrile (IV) (100 mg, 0.3 mmol) in acetic acid (5 mL), concentrated sulfuric acid (2.7 mL) was added dropwise during 30 min. The reaction was then warmed at 80° C. for 9 h, cooled at room temperature and poured into cold water (10 mL). The aqueous phase was then made basic by adding concentrated aqueous ammonia and extracted with dichloromethane (3×10 mL). The combined organic phases were washed with 2N aqueous sodium hydroxide (2×12 mL) and brine, dried over Na 2SO 4 and evaporated to dryness in vacuo. The title compound was obtained as a white solid (43 mg, 40%) after purification through column chromatography ((dichloromethane/methanol/ammonia solution, 7N in methanol:97/2/1). |
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014064149&_cid=P10-MQA9K8-39764-1
2-(1-Cyclohexyl-piperidin-4-yl)-6-fluoro-3-oxo-2,3-dihydro-1 H-isoindole-4-carboxylic acid amide (I), cpd 29
[R = F; n = m = 0; R1 = piperidin-4-yl; R2 = 1-cyclohexyl]
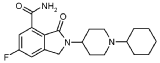
To a stirred solution of 2-(1-cyclohexyl-piperidin-4-yl)-6-fluoro-3-oxo-2,3-dihydro-1 H-isoindole-4-carbonitrile (IV) (100 mg, 0.3 mmol) in acetic acid (5 mL), concentrated sulfuric acid (2.7 mL) was added dropwise during 30 min. The reaction was then warmed at 80 °C for 9 h, cooled at room temperature and poured into cold water (10 mL). The aqueous phase was then made basic by adding concentrated aqueous ammonia and extracted with dichloromethane (3 x 10 mL). The combined organic phases were washed with 2N aqueous sodium hydroxide (2 X 12 mL) and brine, dried over Na2S04 and evaporated to dryness in vacuo. The title compound was obtained as a white solid (43 mg, 40%) after purification through column chromatography ((dichloromethane/methanol/ammonia solution, 7N in methanol: 97/2/1).
1H NMR (400.5 MHz, DMSO- cfe) δ ppm 1.00 – 1.14 (m, 1 H), 1.14 – 1.28 (m, 4 H), 1.53 – 1.61 (m, 1 H), 1.67 – 1.80 (m, 6 H), 2.25 – 2.36 (m, 3 H), 2.88 – 2.95 (m, 2 H), 3.94 – 4.03 (m, 1 H), 4.55 (s, 2 H), 7.66 (dd, JHF = 7.7, JHH = 2.6 Hz, 1 H), 7.85 (br. s., 1 H), 7.89 (dd, JHF = 10.9, JHH = 2.6 Hz, 1 H), 10.78 (br. s., 1 H).
HRMS (ESI+): calcd. for C20H27FN3O2 [M + H]+ 360.2082; found 360.2098
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
- 4-carboxamido-isoindolinone derivatives as selective parp-1 inhibitorsPublication Number: US-2020407314-A1Priority Date: 2012-10-26
- 4-carboxamido-isoindolinone derivatives as selective PARP-1 inhibitorsPublication Number: US-11773064-B2Priority Date: 2012-10-26Grant Date: 2023-10-03
- 4-Carboxamide-isoindolinone derivatives as selective PARP-1 inhibitorsPublication Number: JP-6314147-B2Priority Date: 2012-10-26Grant Date: 2018-04-18
- 4-carboxamido-isoindolinone derivatives as selective PARP-1 inhibitorsPublication Number: ES-2813530-T3Priority Date: 2012-10-26Grant Date: 2021-03-24
- 4-carboxamido-isoindolinone derivatives as selective PARP-1 inhibitorsPublication Number: US-10385018-B2Priority Date: 2012-10-26Grant Date: 2019-08-20
- 4-carboxamido-isoindolinone derivatives as selective parp-1 inhibitorsPublication Number: US-2015274662-A1Priority Date: 2012-10-26
- 4-carboxamido-isoindolinone derivatives as selective parp-1 inhibitorsPublication Number: WO-2014064149-A1Priority Date: 2012-10-26
- 4-carboxamido-isoindolinone derivatives as selective parp-1 inhibitorsPublication Number: US-11420940-B2Priority Date: 2012-10-26Grant Date: 2022-08-23
- DERIVATIVES OF 4-CARBOXAMIDO-ISOINDOLINONA AS SELECTIVE INHIBITORS OF PARP-1.Publication Number: MX-2015005245-APriority Date: 2012-10-26
- 4-carboxamido-isoindolinone derivatives as selective parp-1 inhibitorsPublication Number: US-2019330151-A1Priority Date: 2012-10-26
- COMPOUNDS DERIVED FROM 4-CARBOXAMIDO-ISOINDOLINONE, PROCESS OF PREPARATION OF THESE, IN VITRO METHOD TO SELECTIVELY INHIBIT PARP-1 PROTEIN ACTIVITY, PHARMACEUTICAL COMPOSITION AND USE OF THE REFERRED COMPOUNDSPublication Number: BR-112015009130-B1Priority Date: 2012-10-26
- 4-carboxamido-isoindolinone derivatives as selective parp-1 inhibitorsPublication Number: US-2022363636-A1Priority Date: 2012-10-26
- 4-carboxamido-isoindolinone derivatives as selective parp-1 inhibitorsPublication Number: CA-2889581-A1Priority Date: 2012-10-26
- 4-carboxamido-isoindolinone derivatives as selective PARP-1 inhibitorsPublication Number: US-10800739-B2Priority Date: 2012-10-26Grant Date: 2020-10-13
- 4-carboxamido-isoindolinone derivatives as selective parp-1 inhibitorsPublication Number: EP-2912032-A1Priority Date: 2012-10-26
- 4-carboxamido-isoindolinone derivatives as selective parp-1 inhibitorsPublication Number: EP-2912032-B1Priority Date: 2012-10-26Grant Date: 2020-05-27
- DERIVATIVES 4-CARBOXAMIDO-ISOINDOLINONE AS PARP-1 SELECTIVE INHIBITORS, METHOD FOR THEIR PRODUCTION AND APPLICATIONPublication Number: EA-028506-B1Priority Date: 2012-10-26
- 4-Formylamino-isoindolinone derivatives as selective PARP-1 inhibitorsPublication Number: CN-104768948-APriority Date: 2012-10-26
- 4-carboxamido-isoindolinone derivatives as selective parp-1 inhibitorsPublication Number: CA-2889581-CPriority Date: 2012-10-26Grant Date: 2021-06-29
////////itareparib, ANAX LABS, poly (ADP-ribose) polymerase (PARP) inhibitor, antineoplastic, NMS-03305293, NMS-293, NMS 03305293, NMS 293, KFI1190L8L, NV 578,
Imofinostat
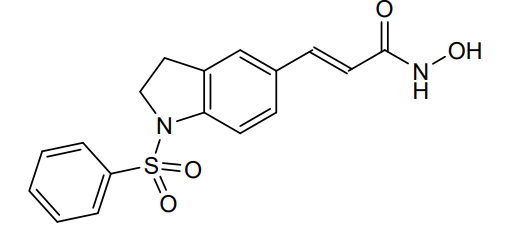
Imofinostat
CAS 1338320-94-7
MF C17H16N2O4S MW 344.4 g/mol
- 3-(1-(Benzenesulfonyl)-2,3-dihydro-1H-indol-5-yl)-N-hydroxyacrylamide
- (E)-3-[1-(benzenesulfonyl)-2,3-dihydroindol-5-yl]-N-hydroxyprop-2-enamide
(2E)-3-[1-(benzenesulfonyl)-2,3-dihydro-1H-indol-5-yl]-N-hydroxyprop2-enamide
histone deacetylase inhibitor, antineoplastic, ABT-301, MPT0E028, ABT 301, MPT0E 028, T65L58FI65
Imofinostat (also known as ABT-301 or MPT0E028) is an orally bioavailable, small-molecule histone deacetylase (HDAC) inhibitor primarily being developed as an innovative precision oncology treatment. Developed by companies like AnBogen Therapeutics and Formosa Pharmaceuticals, it is designed to reactivate tumor suppressor genes that cancer cells have silenced, thereby triggering cancer cell death (apoptosis) and stopping tumor growth.
Mechanism of Action
Imofinostat works through a distinct multi-modality approach to fight cancer cells:
- HDAC Inhibition: It acts as a potent inhibitor of human pan-histone deacetylase enzymes, showing preferential selectivity for Class I HDACs (especially HDAC3). This blocks the deacetylation of histone proteins, causing chromatin to remodel and forcing cancer cells to express tumor-suppressor genes.
- Akt Pathway Targeting: Independent of its epigenetic effects, it can directly target and reduce the activation (phosphorylation) of the Akt protein kinase, a major pathway that cancer cells use to survive and multiply.
- Microenvironment Modulation: Preclinical data shows it alters the tumor microenvironment by converting “cold tumors” (invisible to the immune system) into “hot tumors” by promoting the infiltration of CD8+ cytotoxic T cells.
Current Clinical Status & Indications
Imofinostat is actively moving through clinical trial pipelines, focusing heavily on combination therapies to overcome treatment resistance:
- Colorectal Cancer (CRC): It is currently being evaluated in a global Phase 1/2 clinical trial (NCT07244705). It is combined with the immune checkpoint inhibitor tislelizumab (Tevimbra®) and the anti-angiogenic drug bevacizumab to treat advanced, metastatic colorectal cancer.
- Pancreatic Cancer: Recent data presented at the 2026 American Association for Cancer Research (AACR) Annual Meeting demonstrates that imofinostat disrupts the HDAC3-NRF2 pathway. This action breaks down chemotherapy resistance in highly aggressive KRAS-mutant pancreatic ductal adenocarcinoma, making tumors much more sensitive to treatments like gemcitabine.
- Other Solid Tumors: Phase 1 monotherapy trials have confirmed that the drug possesses a highly competitive safety profile across a broad variety of advanced solid tumors.
Imofinostat is an orally bioavailable N-hydroxyacrylamide-derived inhibitor of both human pan-histone deacetylase (HDAC) enzymes and the serine/threonine protein kinase Akt (protein kinase B), with potential antineoplastic activity. Upon administration, imofinostat selectively binds to and inhibits HDACs, which inhibits deacetylation of histone proteins and leads to the accumulation of highly acetylated histones. This may result in both an induction of chromatin remodeling, and the selective transcription of tumor suppressor genes. This prevents cell division and induces both cell cycle arrest and apoptosis, which may inhibit the proliferation of susceptible tumor cells. In addition, imofinostat inhibits the phosphorylation and activation of Akt, which prevents the activation of downstream signaling pathways, independent of its HDAC inhibitory activity. HDACs, upregulated in many tumor cell types, are a family of enzymes that deacetylate histone proteins. Akt, overexpressed in many tumor cell types, plays a key role in tumor cell proliferation and survival.
Dose-Seeking Study of MPT0E028 in Subjects With Advanced Solid Malignancies Without Standard Treatment
CTID: NCT02350868
Phase: Phase 1
Status: Completed
Date: 2019-04-11
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2011126821&_cid=P11-MQ4LAI-84972-1
COMD 12
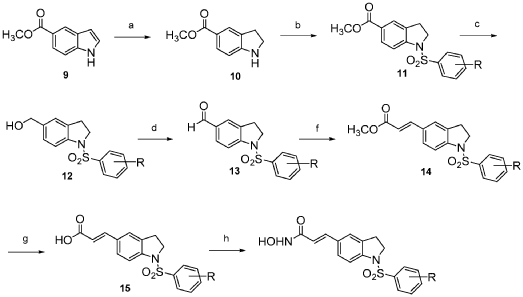
Compound 12 was synthesized via the route as shown in Scheme 3 above (reagents and conditions: (a) NaBH3CN, AcOH; (b) Benzenesulfonyl chloride, 4-methoxybenzenesulfonyl chloride, 3,4-dimethoxybenzenesulfonyl chloride, 4-fluorobenzenesulfonyl chloride, or 4-nitrobenzenesulfonyl chloride, pyridine; (c) L1AIH4, THF; (d) PDC, MS, CH2C12; f) Ph3P = CH-COOCH3, CH2C12; (g) 1M LiOH(aq), dioxane; (h) (i) NH2OTHP, PyBOP, NEt3, DMF; (ii) TFA, MeOH; (i) Fe, NH4C1, Isopropanol, H20).
2,3-Dihydro-lH-indole-5-carboxylic acid methyl ester (10): sodium cyanoborohydride (0.16 g, 2.57 mmol) was added to a solution of methyl indole-5-carboxylate (9) (0.30 g, 1.71 mmol) in AcOH (2 mL) at 0 °C. The reaction mixture was warmed to room temperature and stirred for 2 h before it was quenched with water at 0 °C. Concentrated NaOH was added to reach pH=10. The aqueous layer was extracted with CH2CI2 (15 mL x 3). The combined organic layer was dried over anhydrous MgS04 and concentrated under reduced pressure to give a yellow residue, which was purified by silica gel chromatography (EtOAc: n-hexane = 1 : 2) to afford 10 (0.28 g). 1H NMR (500MHz, CDC13): δ 3.06 (t, J= 8.5 Hz, 2H), 3.65 (t, J= 8.5 Hz, 2H), 3.84 (s, 3H), 6.53-6.55 (m, 1H), 7.75-7.76 (m, 2H).
l-Benzenesulfonyl-2,3-dihydro-lH-indole-5-carboxylic acid methyl ester (11): To a solution of 10 (0.28 g, 1.58 mmol) in pyridine (2 mL), benzenesulfonyl chloride (0.40 ml, 3.16 mmol) was added. The reaction mixture was refluxed overnight. The mixture was then purified by silica gel chromatography (EtOAc: n-hexane = 1 : 3) to afford 11 (0.40 g). 1H NMR (500MHz, CDCI3): δ 2.99 (t, J= 8.6 Hz, 2H), 3.87 (s, 3H), 3.97 (t, J= 8.6 Hz, 2H), 7.45-7.48 (m, 2H), 7.56-7.59 (m, 1H), 7.66 (d, J= 8.5 Hz, 1H), 7.75 (s, 1H), 7.82 (d, J= 7.7 Hz, 2H), 7.90 (d, J= 7.9 Hz, 1H).
(l-Benzenesulfonyl-2,3-dihydro-lH-indol-5-yl)-methanol (12): LAH (0.10 g, 2.52 mmol) was added to a solution of 11 (0.40 g, 1.26 mmol) in THF (10 mL) at 0 °C. The reaction mixture was warmed to room temperature and stirred for 2 h before it was quenched with water and then extracted with CH2CI2 (15 mL x 3). The combined organic layer was dried over anhydrous MgS04 and concentrated under reduced pressure. The reaction mixture was purified by silica gel chromatography (EtOAc: n-hexane = 1 : 1) to afford 12 (0.24 g). 1H NMR (500MHz, CDC13): δ 2.83 (t, J= 8.4 Hz, 2H), 3.92 (t, J= 8.5 Hz, 2H), 4.49 (s, 2H), 7.09 (s, 1H), 7.16 (d, J= 8.2 Hz, 1H), 7.46-7.49 (m, 2H), 7.53 (d, J= 8.2 Hz, 1H), 7.60 (t, J= 7.5 Hz, 1H), 7.76 (d, J= 7.7 Hz, 2H).
l-Benzenesulfonyl-2,3-dihydro-lH-indole-5-carbaldehyde (13): molecular sieves (0.63g) were added to a solution of 12 (0.24 g, 0.83 mmol) in CH2C12 (10 mL), PDC (0.63 g, 1.66 mmol). The mixture was stirred at room temperature overnight before it was filtered through celite. The organic layer was concentrated under reduced pressure then purified by silica gel chromatography (EtOAc: n-hexane = 1 : 2) to afford 13 (0.19 g). 1H NMR (500MHz, CDC13): δ 3.05 (t, J= 8.6 Hz, 2H), 4.01 (t, J= 8.7 Hz, 2H), 7.46-7,49 (m, 2H), 7.58-7.62 (m, 2H), 7.71 (d, J= 8.3 Hz, 1H), 7.75 (d, J= 8.3 Hz, 1H), 7.84 (d, J= 7.8 Hz, 2H), 9.85 (s, 1H).
3-(l-Benzenesulfonyl-2,3-dihydro-lH-indol-5-yl)-acrylic acid methyl ester (14): Methyl (triphenylphosphoranylidene) acetate (0.27 g, 0.79 mmol) was added to a solution of 13 (0.19g,
0.66 mmol) in CH2CI2 (10 mL). The mixture was stirred at room temperature for 3h before it was
quenched with water and then extracted with CH2CI2 (15 mL x 3). The combined organic layer was dried over anhydrous MgS04 and concentrated under reduced pressure to give a yellow residue, which was then purified by silica gel chromatography (EtOAc: n-hexane = 1 : 3) to afford 14
(0.20 g).
3-(l-Benzenesulfonyl-2,3-dihydro-lH-indol-5-yl)-acrylic acid (15): 1M LiOH aqueous solution (1.16 ml, 1.16 mmol) was added to a solution of 14 (0.20g, 0.58 mmol) in dioxane
(15 mL). The reaction mixture was stirred at 40 °C overnight before it was concentrated under reduced pressure. The residue was dissolved in water and concentrated HCl was added up to acidic pH to give the precipitation, which was dried by vacuum to afford 15 (0.16 g). 1H NMR (500MHz, CD3OD): δ 2.92 (t, J= 8.5 Hz, 2H), 3.96 (t, J= 8.5 Hz, 2H), 6.33 (d, J= 15.9 Hz, 1H), 7.38 (s, 1H), 7.41 (d, J= 8.5 Hz, 1H), 7.50-7.53 (m, 2H), 7.55 (d, J= 16.1 Hz, 1H), 7.58-7.64 (m, 2H), 7.82 (d, J = 7.6 Hz, 2H).
3-(l-Benzenesulfonyl-2,3-dihydro-lH-indol-5-yl)-N-hydroxy-acrylamide
(Compound 12): NH2OTHP (0.05 g, 0.44 mmol) was added to a solution of 15 (0.12 g, 0.37 mmol), PyBOP (0.20 g, 0.39 mmol), triethylamine (0.12 ml, 0.88 mmol) in DMF (1.5 mL). The reaction mixture was stirred at room temperature for 1 h before it was quenched with water, followed by extraction with EtOAc (15 mL x 3). The combined organic layer was dried over anhydrous MgS04 and concentrated under reduced pressure. The residue was purified by silica gel chromatography (CH2C12: CH3OH = 30 : 1 : l%NH3(aq)) to give a white solid, which was treated with TFA (1.13 ml, 15.21 mmol) in the presence of CH3OH (25 mL) and stirred overnight at room temperature. The reaction mixture was concentrated under reduced pressure to give a white residue, which was recrystallized by CH3OH to afford Compound 12 (0.12 g). 1H NMR (500MHz,
CD3OD): δ 2.91 (t, J= 8.5 Hz, 2H), 3.96 (t, J= 8.4 Hz, 2H), 6.32 (d, J= 15.8 Hz, 1H), 7.32 (s, 1H), 7.37-7.39 (m, 1H), 7.46 (d, J= 15.7 Hz, 1H), 7.50-7.53 (m, 2H), 7.58-7.64 (m, 2H), 7.82 (d, J= 7.8 Hz, 2H). MS (EI) mlz: 170 (100%), 344 (M+, 3.21%). HRMS (EI) for Ci7Hi6N204S (M+): calcd, 344.0831; found, 344.0829.
PAT
US20150368195
https://patentscope.wipo.int/search/en/detail.jsf?docId=US154007904&_cid=P11-MQ4M0P-01888-1
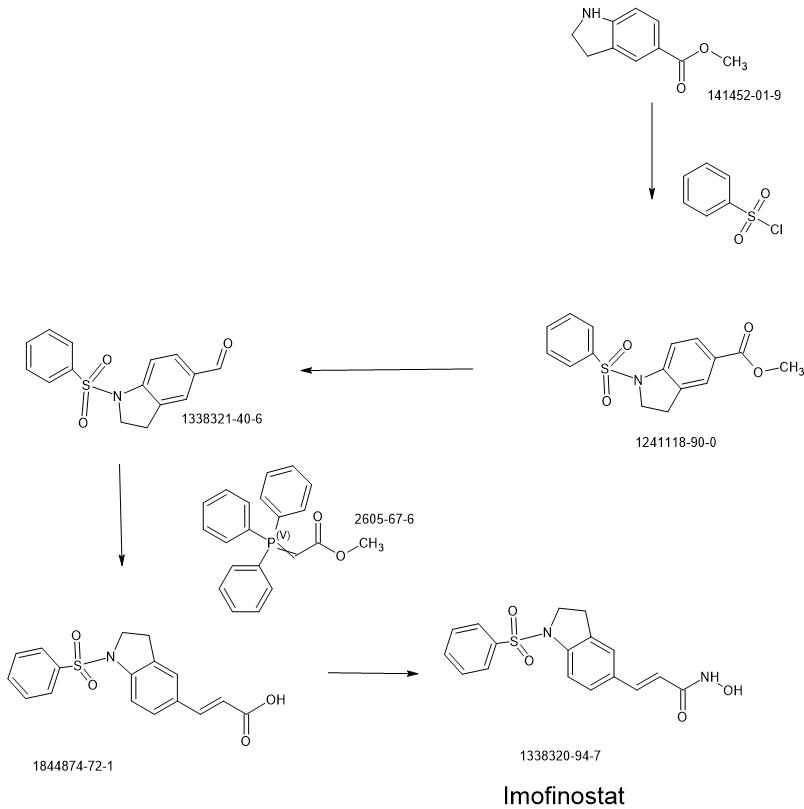
PAT
- Indolyl or indolinyl hydroxamate compoundsPublication Number: US-8846748-B2Priority Date: 2010-03-29Grant Date: 2014-09-30
- Indolyl or indolinyl hydroxamate compoundsPublication Number: US-9598364-B2Priority Date: 2010-03-29Grant Date: 2017-03-21
- Indolyl or indolinyl hydroxamate compoundsPublication Number: WO-2011126821-A2Priority Date: 2010-03-29
- Indolyl or indolinyl hydroxamate compoundsPublication Number: EP-2552887-A2Priority Date: 2010-03-29
- Indolyl or indolinyl hydroxamate compoundsPublication Number: US-2011245315-A1Priority Date: 2010-03-29
- Indolyl or indolinyl hydroxamate compoundsPublication Number: US-2014364477-A1Priority Date: 2010-03-29
- Indolyl or indolinyl hydroxamate compoundsPublication Number: EP-2552887-B1Priority Date: 2010-03-29Grant Date: 2018-10-24
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
//////////imofinostat, anax labs, histone deacetylase inhibitor, antineoplastic, ABT-301, MPT0E028, ABT 301, MPT0E 028, T65L58FI65

{kind=link}