
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Difelikefalin acetate
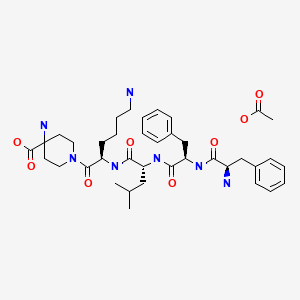
Difelikefalin acetate
ジフェリケファリン酢酸塩
CAS 1024829-44-4
| Formula | C36H53N7O6. (C2H4O2)x |
|---|
D-Phe-D-Phe-D-Leu-D-Lys-[ω(4-aminopiperidine-4- carboxylic acid)]-OH
FDA APPROVED, 2021/8/23, FORSUVA
Analgesic, Antipruritic, Opioid receptor agonist
Treatment of moderate-to-severe pruritus associated with chronic kidney disease in adults undergoing hemodialysis
Difelikefalin, CR-845; MR-13A-9; MR-13A9
4-amino-1- (D-phenylalanyl-D-phenylalanyl-D-leucyl-D-lysyl) piperidine-4-carboxylic acid
C36H53N7O6, 679.40573
| ORIGINATOR | Ferring Pharmaceuticals |
|---|---|
| DEVELOPER | Cara Therapeutics |
| CLASS | Analgesic drugs (peptides) |
| MECHANISM OF ACTION | Opioid kappa receptor agonists |
| WHO ATC CODES | D04A-X (Other antipruritics), N02A (Opioids) |
| EPHMRA CODES | D4A (Anti-Pruritics, Including Topical Antihistamines, Anaesthetics, etc), N2A (Narcotics) |
| INDICATION | Pain, Osteoarthritis, Pruritus |
Difelikefalin, sold under the brand name Korsuva , is an analgesic opioid peptide used for the treatment of moderate-to-severe pruritus. It acts as a peripherally specific, highly selective agonist of the κ-opioid receptor (KOR).[3][4][5][6]
Difelikefalin was approved for medical use in the United States in August 2021.[2][7][8]
Difelikefalin acts as an analgesic by activating KORs on peripheral nerve terminals and KORs expressed by certain immune system cells.[3] Activation of KORs on peripheral nerve terminals results in the inhibition of ion channels responsible for afferent nerve activity, causing reduced transmission of pain signals, while activation of KORs expressed by immune system cells results in reduced release of proinflammatory, nerve-sensitizing mediators (e.g., prostaglandins).[3]

NEW DRUG APPROVALS
ONE TIME
$10.00
Research
It is under development by Cara Therapeutics as an intravenous agent for the treatment of postoperative pain.[3][4][6] An oral formulation has also been developed.[6] Due to its peripheral selectivity, difelikefalin lacks the central side effects like sedation, dysphoria, and hallucinations of previous KOR-acting analgesics such as pentazocine and phenazocine.[3][4] In addition to use as an analgesic, difelikefalin is also being investigated for the treatment of pruritus (itching).[3][4][5] Difelikefalin has completed phase II clinical trials for postoperative pain and has demonstrated significant and “robust” clinical efficacy, along with being safe and well tolerated.[4][6] It has also completed a phase III clinical trial for uremic pruritus in hemodialysis patients.[9]Kappa opioid receptors have been suggested as targets for intervention for treatment or prevention of a wide array of diseases and conditions by administration of kappa opioid receptor agonists. See for example, Jolivalt et al., Diabetologia, 49(11):2775-85; Epub Aug. 19, 2006), describing efficacy of asimadoline, a kappa receptor agonist in rodent diabetic neuropathy; and Bileviciute-Ljungar et al., Eur. J. Pharm. 494:139-46 (2004) describing the efficacy of kappa agonist U-50,488 in the rat chronic constriction injury (CCI) model of neuropathic pain and the blocking of its effects by the opioid antagonist, naloxone. These observations support the use of kappa opioid receptor agonists for treatment of diabetic, viral and chemotherapy- induced neuropathic pain. The use of kappa receptor agonists for treatment or prevention of visceral pain including gynecological conditions such as dysmenorrheal cramps and endometriosis has also been reviewed. See for instance, Riviere, Br. J. Pharmacol. 141:1331-4 (2004).[0004] Kappa opioid receptor agonists have also been proposed for the treatment of pain, including hyperalgesia. Hyperalgesia is believed to be caused by changes in the milieu of the peripheral sensory terminal occur secondary to local tissue damage. Tissue damage (e.g., abrasions, burns) and inflammation can produce significant increases in the excitability of polymodal nociceptors (C fibers) and high threshold mechanoreceptors (Handwerker et al. (1991) Proceeding of the VIth World Congress on Pain, Bond et al., eds., Elsevier Science Publishers BV, pp. 59-70; Schaible et al. (1993) Pain 55:5-54). This increased excitability and exaggerated responses of sensory afferents is believed to underlie hyperalgesia, where the pain response is the result of an exaggerated response to a stimulus. The importance of the hyperalgesic state in the post-injury pain state has been repeatedly demonstrated and appears to account for a major proportion of the post-injury/inflammatory pain state. See for example, Woold et al. (1993) Anesthesia and Analgesia 77:362-79; Dubner et al.(1994) In, Textbook of Pain, Melzack et al., eds., Churchill-Livingstone, London, pp. 225-242.[0005] Kappa opioid receptors have been suggested as targets for the prevention and treatment of cardiovascular disease. See for example, Wu et al. “Cardioprotection of Preconditioning by Metabolic Inhibition in the Rat Ventricular Myocyte – Involvement of kappa Opioid Receptor” (1999) Circulation Res vol. 84: pp. 1388-1395. See also Yu et al. “Anti-Arrhythmic Effect of kappa Opioid Receptor Stimulation in the Perfused Rat Heart: Involvement of a cAMP-Dependent Pathway”(1999) JMoI Cell Cardiol, vol. 31(10): pp. 1809-1819.[0006] It has also been found that development or progression of these diseases and conditions involving neurodegeneration or neuronal cell death can be prevented, or at least slowed, by treatment with kappa opioid receptor agonists. This improved outcome is believed to be due to neuroprotection by the kappa opioid receptor agonists. See for instance, Kaushik et al. “Neuroprotection in Glaucoma” (2003) J. Postgraduate Medicine vol. 49 (1): pp. 90-95. [0007] The presence of kappa opioid receptors on immune cells (Bidlak et al.,(2000) Clin. Diag. Lab. Immunol. 7(5):719-723) has been implicated in the inhibitory • action of a kappa opioid receptor agonist, which has been shown to suppress HIV-I expression. See Peterson PK et al, Biochem Pharmacol 2001, 61(19):1145-51. [0008] Walker, Adv. Exp. Med. Biol. 521: 148-60 (2003) appraised the antiinflammatory properties of kappa agonists for treatment of osteoarthritis, rheumatoid arthritis, inflammatory bowel disease and eczema. Bileviciute-Ljungar et al., Rheumatology 45:295-302 (2006) describe the reduction of pain and degeneration in Freund’s adjuvant-induced arthritis by the kappa agonist U-50,488.[0009] Wikstrom et al, J. Am. Soc. Nephrol. 16:3742-7 (2005) describes the use of the kappa agonist, TRK-820 for treatment of uremic and opiate-induced pruritis, and Ko et al., J. Pharmacol. Exp. Ther. 305: 173-9 (2003) describe the efficacy of U- 50,488 in morphine-induced pruritis in the monkey. [0010] Application of peripheral opioids including kappa agonists for treatment of gastrointestinal diseases has also been extensively reviewed. See for example, Lembo, Diges. Dis. 24:91-8 (2006) for a discussion of use of opioids in treatment of digestive disorders, including irritable bowel syndrome (IBS), ileus, and functional dyspepsia.[0011] Ophthalmic disorders, including ocular inflammation and glaucoma have also been shown to be addressable by kappa opioids. See Potter et ah, J. Pharmacol. Exp. Ther. 309:548-53 (2004), describing the role of the potent kappa opioid receptor agonist, bremazocine, in reduction of intraocular pressure and blocking of this effect by norbinaltorphimine (norBNI), the prototypical kappa opioid receptor antagonist; and Dortch-Carnes et al, CNS Drug Rev. 11(2): 195-212 (2005). U.S. Patent 6,191,126 to Gamache discloses the use of kappa opioid agonists to treat ocular pain. Otic pain has also been shown to be treatable by administration of kappa opioid agonists. See U.S. Patent 6,174,878 also to Gamache.[0012] Kappa opioid agonists increase the renal excretion of water and decrease urinary sodium excretion (i.e., produces a selective water diuresis, also referred to as aquaresis). Many, but not all, investigators attribute this effect to a suppression of vasopressin secretion from the pituitary. Studies comparing centrally acting and purportedly peripherally selective kappa opioids have led to the conclusion that kappa opioid receptors within the blood-brain barrier are responsible for mediating this effect. Other investigators have proposed to treat hyponatremia with nociceptin peptides or charged peptide conjugates that act peripherally at the nociceptin receptor, which is related to but distinct from the kappa opioid receptor (D. R. Kapusta, Life ScL, 60: 15-21, 1997) (U.S. Pat. No. 5,840,696). U.S. Pat Appl. 20060052284.
PATENTJpn. Tokkyo Koho, 5807140US 20090156508WO 2008057608
PATENTUS 20100075910https://patents.google.com/patent/US8236766B2/en


Example 2Synthesis of Compound (2): D-Phe-D-Phe-D-Leu-D-Lys-[ω(4-aminopiperidine-4-carboxylic acid)]-OHSee the scheme of FIG. 3 and Biron et al., Optimized selective N-methylation of peptides on solid support. J. Peptide Science 12: 213-219 (2006). The amino acid derivatives used were Boc-D-Phe-OH, Fmoc-D-Phe-OH, Fmoc-D-Leu-OH, Fmoc-D-Lys(Dde)-OH, and N-Boc-amino-(4-N-Fmoc-piperidinyl)carboxylic acid. HPLC and MS analyses were performed as described in the synthesis of compound (1) described above.The fully protected resin-bound peptide was synthesized manually starting from 2-Chlorotrityl chloride resin (1.8 g, 0.9 mmol; Peptide International). Attachment of N-Boc-amino-(4-N-Fmoc-piperidinyl)carboxylic acid followed by peptide chain elongation and deprotection of Dde in D-Lys(Dde) at Xaa4 was carried out according to the procedure described in the synthesis of compound (1). See above. The resulting peptide resin (0.9 mmol; Boc-D-Phe-D-Phe-D-Leu-D-Lys-(N-Boc-amino-4-piperidinylcarboxylic acid)-[2-Cl-Trt resin]) was split and a portion of 0.3 mmol was used for subsequent cleavage. The peptide resin (0.3 mmol) was then treated with a mixture of TFA/TIS/H2O (15 ml, v/v/v=95:2.5:2.5) at room temperature for 90 minutes. The resin was then filtered and washed with TFA. The filtrate was evaporated in vacuo and the crude synthetic peptide amide (0.3 mmol; D-Phe-D-Phe-D-Leu-D-Lys-[ω(4-aminopiperidine-4-carboxylic acid)]-OH) was precipitated from diethyl ether.For purification, the crude synthetic peptide amide (0.3 mmol) was dissolved in 2% acetic acid in H2O (50 ml) and the solution was loaded onto an HPLC column and purified using TEAP buffer system with a pH 5.2 (buffers A=TEAP 5.2 and B=20% TEAP 5.2 in 80% ACN). The compound was eluted with a linear gradient of buffer B, 7% B to 37% B over 60 minutes. Fractions with purity exceeding 95% were pooled and the resulting solution was diluted with two volumes of water. The diluted solution was then loaded onto an HPLC column for salt exchange and further purification with a TFA buffer system (buffers A=0.1% TFA in H2O and B=0.1% TFA in 80% ACN/20% H2O) and a linear gradient of buffer B, 2% B to 75% B over 25 minutes. Fractions with purity exceeding 97% were pooled, frozen, and dried on a lyophilizer to yield the purified synthetic peptide amide as white amorphous powder (93 mg). HPLC analysis: tR=16.43 min, purity 99.2%, gradient 5% B to 25% B over 20 min; MS (MH+): expected molecular ion mass 680.4, observed 680.3.Compound (2) was also prepared using a reaction scheme analogous to that shown in FIG. 3 with the following amino acid derivatives: Fmoc-D-Phe-OH, Fmoc-D-Leu-OH, Fmoc-D-Lys(Boc)-OH, and Boc-4-amino-1-Fmoc-(piperidine)-4-carboxylic acid.The fully protected resin-bound peptide was synthesized manually starting from 2-Chlorotrityl chloride resin (PS 1% DVB, 500 g, 1 meq/g). The resin was treated with Boc-4-amino-1-Fmoc-4-(piperidine)-4-carboxylic acid (280 g, 600 mmol) in a mixture of DMF, DCM and DIEA (260 mL of each) was added. The mixture was stirred for 4 hours and then the resin was capped for 1 h by the addition of MeOH (258 mL) and DIEA (258 mL).The resin was isolated and washed with DMF (3×3 L). The resin containing the first amino acid was treated with piperidine in DMF (3×3 L of 35%), washed with DMF (9×3 L) and Fmoc-D-Lys(Boc)-OH (472 g) was coupled using PyBOP (519 g) in the presence of HOBt (153 g) and DIEA (516 mL) and in DCM/DMF (500 mL/500 mL) with stiffing for 2.25 hours. The dipeptide containing resin was isolated and washed with DMF (3×3.6 L). The Fmoc group was removed by treatment with piperidine in DMF(3×3.6 L of 35%) and the resin was washed with DMF (9×3.6 L) and treated with Fmoc-D-Leu-OH (354 g), DIC (157 mL) and HOBt (154 g) in DCM/DMF (500 mL/500 mL) and stirred for 1 hour. Subsequent washing with DMF (3×4.1 L) followed by cleavage of the Fmoc group with piperidine in DMF (3×4.2 L of 35%) and then washing of the resin with DMF (9×4.2 L) provided the resin bound tripeptide. This material was treated with Fmoc-D-Phe-OH (387 g), DIC (157 mL) and HOBt (153 g) in DCM/DMF (500 mL/500 mL) and stirred overnight. The resin was isolated, washed with DMF (3×4.7 L) and then treated with piperidine in DMF (3×4.7 L of 35%) to cleave the Fmoc group and then washed again with DMF (9×4.7 L). The tetrapeptide loaded resin was treated with Fmoc-D-Phe-OH (389 g), DIC (157 mL) and HOBt (154 g) in DCM/DMF (500 mL/500 mL) and stirred for 2.25 hours. The resin was isolated, washed with DMF (3×5.2 L) and then treated piperidine (3×5.2 L of 35%) in DMF. The resin was isolated, and washed sequentially with DMF (9×5.2 L) then DCM (5×5.2 L). It was dried to provide a 90.4% yield of protected peptide bound to the resin. The peptide was cleaved from the resin using TFA/water (4.5 L, 95/5), which also served to remove the Boc protecting groups. The mixture was filtered, concentrated (⅓) and then precipitated by addition to MTBE (42 L). The solid was collected by filtration and dried under reduced pressure to give crude synthetic peptide amide.For purification, the crude synthetic peptide amide was dissolved in 0.1% TFA in H2O and purified by preparative reverse phase HPLC (C18) using 0.1% TFA/water—ACN gradient as the mobile phase. Fractions with purity exceeding 95% were pooled, concentrated and lyophilized to provide pure synthetic peptide amide (>95.5% pure). Ion exchange was conducted using a Dowex ion exchange resin, eluting with water. The aqueous phase was filtered (0.22 μm filter capsule) and freeze-dried to give the acetate salt of the synthetic peptide amide (2) with overall yield, 71.3%, >99% purity.Hydrochloride, hydrobromide and fumarate counterions were evaluated for their ability to form crystalline salts of synthetic peptide amide (2). Approximately 1 or 2 equivalents (depending on desired stoichiometry) of hydrochloric acid, hydrobromic acid or fumaric acid, as a dilute solution in methanol (0.2-0.3 g) was added to synthetic peptide amide (2) (50-70 mg) dissolved in methanol (0.2-0.3 g). Each individual salt solution was added to isopropyl acetate (3-5 mL) and the resulting amorphous precipitate was collected by filtration and dried at ambient temperature and pressure. Crystallization experiments were carried out by dissolving the 10-20 mg of the specific amorphous salt obtained above in 70:30 ethanol-water mixture (0.1-0.2 g) followed by the addition of ethanol to adjust the ratio to 90:10 (˜0.6-0.8 mL). Each solution was then seeded with solid particles of the respective precipitated salt. Each sample tube was equipped with a magnetic stir bar and the sample was gently stirred at ambient temperature. The samples were periodically examined by plane-polarized light microscopy. Under these conditions, the mono- and di-hydrochloride salts, the di-hydrobromide salt and the mono-fumarate salt crystallized as needles of 20 to 50 μm in length with a thickness of about 1 μm.PATENT
WO 2008057608
https://patents.google.com/patent/WO2008057608A2/en Compound (2): D-Phe-D-Phe-D-Leu-D-Lys-[ω(4-aminopiperidine-4- carboxylic acid)]-OH (SEQ ID NO: 2):

EXAMPLE 2: Synthesis of compound (2)[00288] D-Phe-D-Phe-D-Leu-D-Lys-[ω(4-aminopiperidine-4-carboxylic acid)]-OH (SEQ ID NO: 2):[00289] See the scheme of Figure 2 and B iron et al., Optimized selective N- methylation of peptides on solid support. J. Peptide Science 12: 213-219 (2006). The amino acid derivatives used were Boc-D-Phe-OH, Fmoc-D-Phe-OH, Fmoc-D-Leu- OH, Fmoc-D-Lys(Dde)-OH, and N-Boc-amino-(4-N-Fmoc-piperidinyl) carboxylic acid. HPLC and MS analyses were performed as described in the synthesis of compound (1) described above.[00290] The fully protected resin-bound peptide was synthesized manually starting from 2-Chlorotrityl chloride resin (1.8 g, 0.9 mmol; Peptide International). Attachment of N-Boc-amino-(4-N-Fmoc-piperidinyl) carboxylic acid followed by peptide chain elongation and deprotection of Dde in D-Lys(Dde) at Xa^ was carried out according to the procedure described in the synthesis of compound (1). See above. The resulting peptide resin (0.9 mmol; Boc-D-Phe-D-Phe-D-Leu-D-Lys-(N- Boc-amino-4-piperidinylcarboxylic acid)-[2-Cl-Trt resin]) was split and a portion of 0.3 mmol was used for subsequent cleavage. The peptide resin (0.3 mmol) was then treated with a mixture of TFA/TIS/H2O (15 ml, v/v/v = 95:2.5:2.5) at room temperature for 90 min. The resin was then filtered and washed with TFA. The filtrate was evaporated in vacuo and the crude peptide (0.3 mmol; D-Phe-D-Phe-D- Leu-D-Lys-[ω(4-aminopiperidine-4-carboxylic acid)]-OH) was precipitated from diethyl ether.[00291] For purification, the crude peptide (0.3 mmol) was dissolved in 2% acetic acid in H2O (50 ml) and the solution was loaded onto an HPLC column and purified using TEAP buffer system with a pH 5.2 (buffers A = TEAP 5.2 and B = 20% TEAP 5.2 in 80% ACN). The compound was eluted with a linear gradient of buffer B, 7%B to 37%B over 60 min. Fractions with purity exceeding 95% were pooled and the resulting solution was diluted with two volumes of water. The diluted solution was then loaded onto an HPLC column for salt exchange and further purification with a TFA buffer system (buffers A = 0.1% TFA in H2O and B = 0.1% TFA in 80% ACN/20% H2O) and a linear gradient of buffer B, 2%B to 75%B over 25 min. Fractions with purity exceeding 97% were pooled, frozen, and dried on a lyophilizer to yield the purified peptide as white amorphous powder (93 mg). HPLC analysis: tR = 16.43 min, purity 99.2%, gradient 5%B to 25%B over 20 min; MS (M+H+): expected molecular ion mass 680.4, observed 680.3.[00292] Compound (2) was also prepared using a reaction scheme analogous to that shown in figure 2 with the following amino acid derivatives: Fmoc-D-Phe-OH, Fmoc-D-Leu-OH, Fmoc-D-Lys(Boc)-OH, and Boc-4-amino-l-Fmoc-(piperidine)-4- carboxylic acid.[00293] The fully protected resin-bound peptide was synthesized manually starting from 2-Chlorotrityl chloride resin (PS 1%DVB, 500 g, 1 meq/g). The resin was treated with Boc-4-amino-l-Fmoc-4-(piperidine)-4-carboxylic acid (280 g, 600 mmol) in a mixture of DMF, DCM and DIEA (260 mL of each) was added. The mixture was stirred for 4 hours and then the resin was capped for Ih by the addition of MeOH (258 mL) and DIEA[00294] (258 mL). The resin was isolated and washed with DMF (3 x 3 L). The resin containing the first amino acid was treated with piperidine in DMF (3 x 3 L of 35%), washed with DMF (9 x 3 L) and Fmoc-D-Lys(Boc)-OH (472 g) was coupled using PyBOP (519 g) in the presence of HOBt (153 g) and DIEA (516 mL) and in DCM/DMF (500 mL/ 500 mL) with stirring for 2.25 hours. The dipeptide containing resin was isolated and washed with DMF (3 x 3.6 L). The Fmoc group was removed by treatment with piperidine in DMF [00295] , (3 x 3.6 L of 35%) and the resin was washed with DMF (9 x 3.6 L) and treated with Fmoc-D-Leu-OH (354 g), DIC (157 mL) and HOBt (154 g) in DCM/DMF (500 mL / 500 mL) and stirred for 1 hour. Subsequent washing with DMF (3 x 4.1 L) followed by cleavage of the Fmoc group with piperidine in DMF (3 x 4.2 L of 35%) and then washing of the resin with DMF (9 x 4.2 L) provided the resin bound tripeptide. This material was treated with Fmoc-D-Phe-OH (387 g), DIC (157 mL) and HOBt (153 g) in DCM/DMF (500 mL / 500 mL) and stirred overnight. The resin was isolated, washed with DMF (3 x 4.7 L) and then treated with piperidine in DMF (3 x 4.7 L of 35%) to cleave the Fmoc group and then washed again with DMF (9 x 4.7 L). The tetrapeptide loaded resin was treated with Fmoc-D-Phe-OH (389 g), DIC (157 mL) and HOBt (154 g) in DCM/DMF (500 mL / 500 mL) and stirred for 2.25 hours. The resin was isolated, washed with DMF (3 x 5.2 L) and then treated piperidine (3 x 5.2 L of 35%) in DMF. The resin was isolated, and washed sequentially with DMF (9 x 5.2 L) then DCM (5 x 5.2 L). It was dried to provide a 90.4% yield of protected peptide bound to the resin. The peptide was cleaved from the resin using TFA/ water (4.5 L, 95/5), which also served to remove the Boc protecting groups. The mixture was filtered, concentrated (1/3) and then precipitated by addition to MTBE (42 L). The solid was collected by filtration and dried under reduced pressure to give crude peptide.[00296] For purification, the crude peptide was dissolved in 0.1% TFA in H2O and purified by preparative reverse phase HPLC (C 18) using 0.1% TF A/water – ACN gradient as the mobile phase. Fractions with purity exceeding 95% were pooled, concentrated and lyophilized to provide pure peptide (> 95.5% pure). Ion exchange was conducted using a Dowex ion exchange resin, eluting with water. The aqueous phase was filtered (0.22 μm filter capsule) and freeze-dried to give the acetate salt of the peptide (overall yield, 71.3%, >99% pure).
PATENT
κ opioid receptor agonists are known to be useful as therapeutic agents for various pain. Among, kappa opioid receptor agonist with high selectivity for peripheral kappa opioid receptors, are expected as a medicament which does not cause the central side effects. Such as peripherally selective κ opioid receptor agonist, a synthetic pentapeptide has been reported (Patent Documents 1 and 2). The following formula among the synthetic pentapeptide (A)
[Formula 1] Being Represented By Compounds Are Useful As Pain Therapeutics. The Preparation Of This Compound, Solid Phase Peptide Synthesis Methods In Patent Documents 1 And 2 Have Been Described.Document 1 Patent: Kohyo 2010-510966 JP
Patent Document 2: Japanese Unexamined Patent Publication No. 2013-241447 Compound (1) or a salt thereof and compound (A), for example as shown in the following reaction formula, 4-aminopiperidine-4-carboxylic acid, D- lysine (D-Lys), D- leucine (D-Leu) , it can be prepared by D- phenylalanine (D-Phe) and D- phenylalanine (D-Phe) sequentially solution phase peptide synthesis methods condensation.[Of 4]The present invention will next to examples will be described in further detail.Example
1 (1) Synthesis of Cbz-D-Lys (Boc) -α-Boc-Pic-OMe (3)
to the four-necked flask of 2L, α-Boc-Pic- OMe · HCl [α-Boc-4 – aminopiperidine-4-carboxylic acid methyl hydrochloride] were charged (2) 43.7g (148mmol), was suspended in EtOAc 656mL (15v / w). To the suspension of 1-hydroxybenzotriazole (HOBt) 27.2g (178mmol), while cooling with Cbz-D-Lys (Boc) -OH 59.2g (156mmol) was added an ice-bath 1-ethyl -3 – (3-dimethylcarbamoyl amino propyl) was added to the carbodiimide · HCl (EDC · HCl) 34.1g (178mmol). After 20 minutes, stirring was heated 12 hours at room temperature. After completion of the reaction, it was added and the organic layer was 1 N HCl 218 mL of (5.0v / w). NaHCO to the resulting organic layer 3 Aq. 218ML (5.0V / W), Et 3 N 33.0 g of (326Mmol) was stirred for 30 minutes, and the mixture was separated. The organic layer HCl 218ML 1N (5.0V / W), NaHCO 3 Aq. 218mL (5.0v / w), NaClaq . Was washed successively with 218ML (5.0V / W), Na 2 SO 4 dried addition of 8.74g (0.2w / w). Subjected to vacuum filtration, was concentrated under reduced pressure resulting filtrate by an evaporator, and pump up in the vacuum pump, the Cbz-D-Lys (Boc) -α-Boc-Pic-OMe (3) 88.9g as a white solid obtained (96.5% yield, HPLC purity 96.5%).[0033](2) D-Lys (Boc) Synthesis Of -Arufa-Boc-Pic-OMe (4)
In An Eggplant-Shaped Flask Of 2L, Cbz-D-Lys (Boc) -Arufa-Boc-Pic-OMe (3) 88.3g (142mmol) were charged, it was added and dissolved 441mL (5.0v / w) the EtOAc. The 5% Pd / C to the reaction solution 17.7g (0.2w / w) was added, After three nitrogen substitution reduced pressure Atmosphere, Was Performed Three Times A Hydrogen Substituent. The Reaction Solution Was 18 Hours With Vigorous Stirring At Room Temperature To Remove The Pd / C And After The Completion Of The Reaction Vacuum Filtration. NaHCO The Resulting Filtrate 3 Aq. 441ML And (5.0V / W) Were Added For Liquid Separation, And The Organic Layer Was Extracted By The Addition Of EtOAc 200ML (2.3V / W) In The Aqueous Layer. NaHCO The Combined Organic Layer 3 Aq. 441ML And (5.0V / W) Were Added for liquid separation, and the organic layer was extracted addition of EtOAc 200mL (2.3v / w) in the aqueous layer. NaClaq the combined organic layers. 441mL and (5.0v / w) is added to liquid separation, was extracted by the addition EtOAc 200ML Of (2.3V / W) In The Aqueous Layer. The Combined Organic Layer On The Na 2 SO 4 Dried Addition Of 17.7 g of (0.2W / W), Then The Filtrate Was Concentrated Under Reduced Pressure Obtained Subjected To Vacuum Filtration By an evaporator, and pump up in the vacuum pump, D-Lys (Boc) -α-Boc-Pic- OMe (4) to give 62.7g (90.5% yield, HPLC purity 93.6%).(3) Cbz-D-Leu -D-Lys (Boc) -α-Boc-Pic-OMe synthesis of (5)
in the four-necked flask of 2L, D-Lys (Boc) -α-Boc-Pic-OMe (4) was charged 57.7 g (120 mmol), was suspended in EtOAc 576mL (10v / w). HOBt 19.3g (126mmol) to this suspension, was added EDC · HCl 24.2g (126mmol) while cooling in an ice bath added Cbz-D-Leu-OH 33.4g (126mmol). After 20 minutes, after stirring the temperature was raised 5 hours at room temperature, further the EDC · HCl and stirred 1.15 g (6.00 mmol) was added 16 h. After completion of the reaction, it was added liquid separation 1N HCl 576mL (10v / w) . NaHCO to the resulting organic layer 3 Aq. 576ML (10V / W), Et 3 N 24.3 g of (240Mmol) was stirred for 30 minutes, and the mixture was separated. The organic layer HCl 576ML 1N (10V / W), NaHCO 3 Aq. 576mL (10v / w), NaClaq . Was washed successively with 576ML (10V / W), Na 2 SO 4 dried addition of 11.5g (0.2w / w). After the filtrate was concentrated under reduced pressure obtained subjected to vacuum filtration by an evaporator, and pump up in the vacuum pump, the Cbz-D-Leu-D- Lys (Boc) -α-Boc-Pic-OMe (5) 85.8g It was obtained as a white solid (98.7% yield, HPLC purity 96.9%).(4) D-Leu-D -Lys (Boc) -α-Boc-Pic-OMe synthesis of (6)
in an eggplant-shaped flask of 1L, Cbz-D-Leu- D-Lys (Boc) -α-Boc-Pic -OMe the (5) 91.9g (125mmol) were charged, was added and dissolved 459mL (5.0v / w) the EtOAc. The 5% Pd / C to the reaction solution 18.4g (0.2w / w) was added, After three nitrogen substitution reduced pressure atmosphere, was performed three times a hydrogen substituent. The reaction solution was subjected to 8 hours with vigorous stirring at room temperature to remove the Pd / C and after the completion of the reaction vacuum filtration. NaHCO the resulting filtrate 3 Aq. 200mL (2.2v / w) were added to separate liquid, NaHCO to the organic layer 3 Aq. 200mL (2.2v / w), NaClaq . It was sequentially added washed 200mL (2.2v / w). To the resulting organic layer Na 2 SO 4 dried added 18.4g (0.2w / w), to the filtrate concentrated under reduced pressure obtained subjected to vacuum filtration by an evaporator, and a pump-up with a vacuum pump. The resulting amorphous solid was dissolved adding EtOAc 200mL (2.2v / w), was crystallized by the addition of heptane 50mL (1.8v / w). Was filtered off precipitated crystals by vacuum filtration, the crystals were washed with a mixed solvent of EtOAc 120mL (1.3v / w), heptane 50mL (0.3v / w). The resulting crystal 46.1g to added to and dissolved EtOAc 480mL (5.2v / w), was crystallized added to the cyclohexane 660mL (7.2v / w). Was filtered off under reduced pressure filtered to precipitate crystals, cyclohexane 120mL (1.3v / w), and washed with a mixed solvent of EtOAc 20mL (0.2v / w), and 30 ° C. vacuum dried, D-Leu- as a white solid D-Lys (Boc) -α- Boc-Pic-OMe (6) to give 36.6 g (48.7% yield, HPLC purity 99.9%).(5) Synthesis of Cbz-D-Phe-D- Leu-D-Lys (Boc) -α-Boc-Pic-OMe (7)
to the four-necked flask of 1L, D-Leu-D- Lys (Boc) -α-Boc-Pic-OMe with (6) 35.8g (59.6mmol) was charged, it was suspended in EtOAc 358mL (10v / w). To this suspension HOBt 9.59g (62.6mmol), Cbz- D-Phe-OH 18.7g was cooled in an ice bath is added (62.6mmol) while EDC · HCl 12.0g (62.6mmol) It was added. After 20 minutes, a further EDC · HCl After stirring the temperature was raised 16 hours was added 3.09 g (16.1 mmol) to room temperature. After completion of the reaction, it was added and the organic layer was 1N HCl 358mL of (10v / w). NaHCO to the resulting organic layer 3 Aq. 358ML (10V / W), Et 3 N 12.1 g of (119Mmol) was stirred for 30 minutes, and the mixture was separated. The organic layer HCl 358ML 1N (10V / W), NaHCO 3 Aq. 358mL (10v / w), NaClaq . Was washed successively with 358ML (10V / W), Na 2 SO 4 dried addition of 7.16g (0.2w / w). After the filtrate was concentrated under reduced pressure obtained subjected to vacuum filtration by an evaporator, and pump up in the vacuum pump, Cbz-D-Phe-D -Leu-D-Lys (Boc) -α-Boc-Pic-OMe (7) was obtained 52.5g as a white solid (yield quant, HPLC purity 97.6%).(6) D-Phe-D -Leu-D-Lys (Boc) synthesis of -α-Boc-Pic-OMe ( 8)
in an eggplant-shaped flask of 2L, Cbz-D-Phe- D-Leu-D-Lys ( Boc) -α-Boc-Pic- OMe (7) the 46.9g (53.3mmol) were charged, the 840ML EtOAc (18V / W), H 2 added to and dissolved O 93.8mL (2.0v / w) It was. The 5% Pd / C to the reaction mixture 9.38g (0.2w / w) was added, After three nitrogen substitution reduced pressure atmosphere, was performed three times a hydrogen substituent. The reaction solution was subjected to 10 hours with vigorous stirring at room temperature to remove the Pd / C and after the completion of the reaction vacuum filtration. NaHCO the resulting filtrate 3 Aq. 235mL (5.0v / w) were added to separate liquid, NaHCO to the organic layer 3 Aq. 235mL (5.0v / w), NaClaq . It was added sequentially cleaning 235mL (5.0v / w). To the resulting organic layer Na 2 SO 4 dried addition of 9.38g (0.2w / w), then the filtrate was concentrated under reduced pressure obtained subjected to vacuum filtration by an evaporator, pump up with a vacuum pump to D-Phe -D-Leu-D-Lys ( Boc) -α-Boc-Pic-OMe (7) was obtained 39.7g (yield quant, HPLC purity 97.3%).351mL was suspended in (10v / w). To this suspension HOBt 7.92g (51.7mmol), Boc-D-Phe-OH HCl HCl(8) D-Phe-D -Phe-D-Leu-D-Lys-Pic-OMe Synthesis Of Hydrochloric Acid Salt (1)
In An Eggplant-Shaped Flask Of 20ML Boc-D-Phe-D -Phe-D- Leu-D- lys (Boc) -α -Boc- Pic-OMe (9) and 2.00gg, IPA 3.3mL (1.65v / w), was suspended by addition of PhMe 10mL (5v / w). It was stirred at room temperature for 19 hours by addition of 6N HCl / IPA 6.7mL (3.35v / w). The precipitated solid was filtered off by vacuum filtration and dried under reduced pressure to a white solid of D-Phe-D-Phe- D- Leu-D-Lys-Pic- OMe 1.59ghydrochloride (1) (yield: 99 .0%, HPLC purity 98.2%) was obtained.(9) D-Phe-D -Phe-D-Leu-D-Lys-Pic-OMe Purification Of The Hydrochloric Acid Salt (1)
In An Eggplant-Shaped Flask Of 20ML-D-Phe-D- Phe D-Leu -D-Lys- pic-OMe hydrochloride crude crystals (1) were charged 200mg, EtOH: MeCN = 1: after stirring for 1 hour then heated in a mixed solvent 4.0 mL (20v / w) was added 40 ° C. of 5 , further at room temperature for 2 was time stirring slurry. Was filtered off by vacuum filtration, the resulting solid was dried under reduced pressure a white solid ((1) Purification crystals) was obtained 161 mg (80% yield, HPLC purity 99.2% ).(10) D-Phe-D -Phe-D-Leu-D-Lys-Pic Synthesis (Using Purified
(1)) Of (A) To A Round-Bottomed Flask Of 10ML D-Phe-D-Phe-D- -D-Lys Leu-Pic-OMe Hydrochloride Salt (1) Was Charged With Purified Crystal 38.5Mg (0.0488Mmol), H 2 Was Added And Dissolved O 0.2ML (5.2V / W). 1.5H Was Stirred Dropwise 1N NaOH 197MyuL (0.197mmol) at room temperature. After completion of the reaction, concentrated under reduced pressure by an evaporator added 1N HCl 48.8μL (0.0488mmol), to obtain a D-Phe-D-Phe- D-Leu-D-Lys- Pic (A) (yield: quant , HPLC purity 99.7%).
D-Phe-D-Phe- D-Leu-D-Lys-Pic-OMe (1) physical properties 1 H NMR (400 MHz, 1M DCl) [delta] ppm by: 0.85-1.02 (yd,. 6 H), 1.34-1.63 ( m, 5 H), 1.65-2.12 ( m, 5 H), 2.23-2.45 (m, 2 H), 2.96-3.12 (m, 4 H), 3.19 (ddt, J = 5.0 & 5.0 & 10.0 Hz), 3.33-3.62 (m, 1 H), 3.68-3.82 (m, 1 H), 3.82-3.95 (m, 4 H), 3.95-4.18 (m, 1 H), 4.25-4.37 (m, 2 H), 4.61-4.77 (M, 2 H), 7.21-7.44 (M, 10 H) 13 C NMR (400MHz, 1M DCl) Deruta Ppm: 21.8, 22.5, 24.8, 27.0, 30.5, 30.8, 31.0, 31.2, 31.7, 37.2 , 37.8, 38.4, 39.0, 39.8, 40.4, 40.6, 41.8, 42.3, 49.8, 50.2, 52.2, 52.6, 54.6, 55.2, 57.7, 57.9, 127.6, 128.4, 129.2, 129.6, 129.7, 129.8 dp 209.5 ℃Example 2
(Trifluoroacetic Acid (TFA)
Use) (1) D-Phe-D-Phe-D-Leu-D-Lys-Pic-OMe TFA Synthesis Of Salt (1)
TFA 18ML Eggplant Flask Of 50ML (18V / W) , 1- Dodecanethiol 1.6ML (1.6V / W), Triisopropylsilane 0.2ML (0.2V / W), H 2 Sequentially Added Stirring The O 0.2ML (0.2V / W) Did. The Solution To The Boc-D-Phe- D- Phe-D-Leu-D -Lys (Boc) -α-Boc-Pic-OMe the (9) 1.00g (1.01mmol) was added in small portions with a spatula. After completion of the reaction, concentrated under reduced pressure by an evaporator, it was added dropwise the resulting residue in IPE 20mL (20v / w). The precipitated solid was filtered off, the resulting solid was obtained and dried under reduced pressure to D-Phe-D-Phe- D-Leu -D-Lys-Pic-OMe · TFA salt as a white solid (1) (Osamu rate 93.0%, HPLC purity 95.2%).(2) D-Phe-D -Phe-D-Leu-D-Lys-Pic synthesis of (A)
to a round-bottomed flask of 10mL D-Phe-D-Phe -D-Leu-D-Lys-Pic-OMe TFA were charged salt (1) 83mg (0.0843mmol), was added and dissolved H2O 431μL (5.2v / w). Was 12h stirring dropwise 1N NaOH 345μL (0.345mmol) at room temperature. After completion of the reaction, concentrated under reduced pressure by an evaporator added 1N HCl 84.3μL (0.0843mmol), to obtain a D-Phe-D-Phe- D-Leu-D-Lys-Pic (A) ( yield: quant, HPLC purity 95.4%).Example
3 (HCl / EtOAc
Use) (1) In An Eggplant-Shaped Flask Of 30ML Boc-D-Phe-D -Phe-D-Leu-D-Lys (Boc) -Arufa-Boc-Pic-OMe (9) 1. It was charged with 00g (1.01mmol ), was added and dissolved EtOAc7.0mL (7.0v / w). 4N HCl / EtOAc 5.0mL (5.0v / w) was added after 24h stirring at room temperature, the precipitated solid was filtered off by vacuum filtration, washed with EtOAc 2mL (2.0v / w). The resulting solid D-Phe-D-Phe- D-Leu-D-Lys-Pic-OMe hydrochloride (1) was obtained 781mg of a white solid was dried under reduced pressure (the 96.7% yield, HPLC purity 95.4%).(2) D-Phe-D -Phe-D-Leu-D-Lys-Pic (A) Synthesis of
eggplant flask of 10mL D-Phe-D-Phe -D-Leu-D-Lys-Pic-OMe hydrochloride were charged salt (1) 90 mg (0.112 mmol), H 2 was added and dissolved O 0.47mL (5.2v / w). Was 12h stirring dropwise 1N NaOH 459μL (0.459mmol) at room temperature. After completion of the reaction, concentrated under reduced pressure by an evaporator added 1N HCl 0.112μL (0.112mmol), was obtained D-Phe-D-Phe- D-Leu-D-Lys-Pic (A) ( yield: quant, HPLC purity 93.1%).4 Example
Compound (1) Of The Compound By Hydrolysis Synthesis Of (The A) (Compound (1) Without
Purification) Eggplant Flask 10ML D-Phe-D-Phe -D-Leu-D-Lys-Pic-OMe (1) Charged Hydrochloride Were (Without Pre-Step Purification) 114.5Mg (0.142Mmol), H 2 Was Added And Dissolved O 595MyuL (5.2V / W). Was 14H Stirring Dropwise 1N NaOH 586MyuL (0.586Mmol) At Room Temperature. After Completion Of the reaction, concentrated under reduced pressure by an evaporator added 1N HCl 0.15μL (0.150mmol), was obtained D-Phe-D-Phe- D-Leu-D-Lys-Pic (A) (yield: quant, HPLC purity 95.2 %).Example 1 Comparative
Path Not Via The Compound (1) (Using Whole Guard Boc-D-Phe-D-Phe-D-Leu-D-Lys (Boc) -Alpha-Boc-Pic-OMe
(A)) (1) D–Boc Phe- D-Phe-D-Leu-D-Lys (Boc) -Arufa-Boc-Pic-OH Synthesis Of
Eggplant Flask Of 30ML Boc-D-Phe-D -Phe-D-Leu-D- Lys (Boc) -α- Boc-Pic -OMe (9) were charged 1.00g (1.00mmol), was added and dissolved MeOH 5.0mL (5.0v / w). After stirring for four days by the addition of 1N NaOH 1.1 mL (1.10mmol) at room temperature, further MeOH 5.0mL (5.0v / w), 1N NaOH 2.0mL the (2.0mmol) at 35 ℃ in addition 3h and the mixture was stirred. After completion of the reaction, 1 N HCl 6.1 mL was added, After distilling off the solvent was concentrated under reduced pressure was separated and the organic layer was added EtOAc 5.0mL (5.0mL) .NaClaq. 5.0mL (5.0v / w) Wash the organic layer was added, the organic layer as a white solid was concentrated under reduced pressure to Boc-D-Phe-D- Phe-D-Leu-D-Lys (Boc) – α-Boc-Pic-OH 975.1mg (99.3% yield, HPLC purity 80.8% )(2) D-Phe-D -Phe-D-Leu-D-Lys-Pic synthesis of (A)
to a round-bottomed flask of 20mL Boc-D-Phe-D -Phe-D-Leu-D-Lys (Boc) It was charged -α-Boc-Pic-OH ( 10) 959mg (0.978mmol), was added and dissolved EtOAc 4.9mL (5.0v / w). And 4h stirring at room temperature was added dropwise 4N HCl / EtOAc 4.9mL (5.0mL) at room temperature. After completion of the reaction, it was filtered under reduced pressure, a white solid as to give D-Phe-D-Phe- D-Leu-D-Lys-Pic the (A) (96.4% yield, HPLC purity 79.2%) . If not via the compound of the present invention (1), the purity of the compound obtained (A) was less than 80%.
PATENThttp://www.google.com/patents/US20110212882
References
- ^ Janecka A, Perlikowska R, Gach K, Wyrebska A, Fichna J (2010). “Development of opioid peptide analogs for pain relief”. Curr. Pharm. Des. 16 (9): 1126–35. doi:10.2174/138161210790963869. PMID 20030621.
- ^ Jump up to:a b https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/214916s000lbl.pdf
- ^ Jump up to:a b c d e f g h i j Raymond S. Sinatra; Jonathan S. Jahr; J. Michael Watkins-Pitchford (14 October 2010). The Essence of Analgesia and Analgesics. Cambridge University Press. pp. 490–491. ISBN 978-1-139-49198-3.
- ^ Jump up to:a b c d e Jeffrey Apfelbaum (8 September 2014). Ambulatory Anesthesia, An Issue of Anesthesiology Clinics. Elsevier Health Sciences. pp. 190–. ISBN 978-0-323-29934-3.
- ^ Jump up to:a b Alan Cowan; Gil Yosipovitch (10 April 2015). Pharmacology of Itch. Springer. pp. 307–. ISBN 978-3-662-44605-8.
- ^ Jump up to:a b c d Charlotte Allerton (2013). Pain Therapeutics: Current and Future Treatment Paradigms. Royal Society of Chemistry. pp. 56–. ISBN 978-1-84973-645-9.
- ^ “Korsuva: FDA-Approved Drugs”. U.S. Food and Drug Administration. Retrieved 24 August 2021.
- ^ “Vifor Pharma and Cara Therapeutics announce U.S. FDA approval of Korsuva injection for the treatment of moderate-to-severe pruritus in hemodialysis patients” (Press release). Vifor Pharma. 24 August 2021. Retrieved 24 August 2021 – via Business Wire.
- ^ Fishbane S, Jamal A, Munera C, Wen W, Menzaghi F (2020). “A phase 3 trial of difelikefalin in hemodialysis patients with pruritus”. N Engl J Med. 382 (3): 222–232. doi:10.1056/NEJMoa1912770. PMID 31702883.
External links
- “Difelikefalin”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03422653 for “A Study to Evaluate the Safety and Efficacy of CR845 in Hemodialysis Patients With Moderate-to-Severe Pruritus (KALM-1)” at ClinicalTrials.gov
- Clinical trial number NCT03636269 for “CR845-CLIN3103: A Global Study to Evaluate the Safety and Efficacy of CR845 in Hemodialysis Patients With Moderate-to-Severe Pruritus (KALM-2)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Korsuva |
| Other names | CR845, FE-202845, D-Phe-D-Phe-D-Leu-D-Lys-[γ-(4-N-piperidinyl)amino carboxylic acid][1] |
| License data | US DailyMed: Difelikefalin |
| Routes of administration | Intravenous |
| Drug class | Kappa opioid receptor agonist |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [2] |
| Pharmacokinetic data | |
| Bioavailability | 100% (IV)[3] |
| Metabolism | Not metabolized[3] |
| Elimination half-life | 2 hours[3] |
| Excretion | Excreted as unchanged drug via bile and urine[3] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1024828-77-0 |
| PubChem CID | 24794466 |
| ChemSpider | 44208824 |
| UNII | NA1U919MRO |
| KEGG | D11111 |
| Chemical and physical data | |
| Formula | C36H53N7O6 |
| Molar mass | 679.863 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
//////////Difelikefalin acetate, FDA 2021, APPROVALS 2021, FORSUVA, ジフェリケファリン酢酸塩 , Difelikefalin, CR 845, MR 13A-9, MR-13A9, PEPTIDE
Lonapegsomatropin
FPTIPLSRLF DNAMLRAHRL HQLAFDTYQE FEEAYIPKEQ KYSFLQNPQT SLCFSESIPT
PSNREETQQK SNLELLRISL LLIQSWLEPV QFLRSVFANS LVYGASDSNV YDLLKDLEEG
IQTLMGRLED GSPRTGQIFK QTYSKFDTNS HNDDALLKNY GLLYCFRKDM DKVETFLRIV
QCRSVEGSCG F
(Disulfide bridge: 53-165, 182-189)

Lonapegsomatropin, ロナペグソマトロピン
FDA APPROVED, 25/8/21, Skytrofa, Treatment of growth hormone deficiency
To treat short stature due to inadequate secretion of endogenous growth hormone
1934255-39-6 CAS, UNII: OP35X9610Y
Molecular Formula, C1051-H1627-N269-O317-S9[-C2-H4-O]4n
ACP 001; ACP 011; lonapegsomatropin-tcgd; SKYTROFA; TransCon; TransCon growth hormone; TransCon hGH; TransCon PEG growth hormone; TransCon PEG hGH; TransCon PEG somatropin,
WHO 10598
PEPTIDE
Biologic License Application (BLA): 761177
Company: ACENDIS PHARMA ENDOCRINOLOGY DIV A/S
SKYTROFA is a human growth hormone indicated for the treatment of pediatric patients 1 year and older who weigh at least 11.5 kg and have growth failure due to inadequate secretion of endogenous growth hormone (GH) (1).
- OriginatorAscendis Pharma
- DeveloperAscendis Pharma; VISEN Pharmaceuticals
- ClassGrowth hormones; Hormonal replacements; Polyethylene glycols
- Mechanism of ActionSomatotropin receptor agonists
- Orphan Drug StatusYes – Somatotropin deficiency
- RegisteredSomatotropin deficiency
- 25 Aug 2021Registered for Somatotropin deficiency (In children, In infants) in USA (SC)
- 27 May 2021Ascendis Pharma expects European Commission decision on the Marketing Authorisation Application (MAA) for Somatotropin deficiency (In children, In infants, In neonates) in fourth quarter of 2021
- 27 May 2021Phase-III clinical trials in Somatotropin deficiency (In children, Treatment-naive) in Japan (SC)
Ascendis Pharma A/S Announces U.S. Food and Drug Administration Approval of SKYTROFA® (lonapegsomatropin-tcgd), the First Once-weekly Treatment for Pediatric Growth Hormone Deficiency
SKYTROFA, the first FDA approved treatment utilizing TransCon™ technology, is a long-acting prodrug of somatropin that releases the same somatropin used in daily therapies –
– Once weekly SKYTROFA demonstrated higher annualized height velocity (AHV) at week 52 compared to a daily growth hormone with similar safety and tolerability –
– Availability in the U.S. expected shortly supported by a full suite of patient support programs –
– Ascendis Pharma to host investor conference call today, Wednesday, August 25 at 4:30 p.m. E.T. –
COPENHAGEN, Denmark, Aug. 25, 2021 (GLOBE NEWSWIRE) — Ascendis Pharma A/S (Nasdaq: ASND), a biopharmaceutical company that utilizes its innovative TransCon technologies to potentially create new treatments that make a meaningful difference in patients’ lives, today announced that the U.S. Food and Drug Administration (FDA) has approved SKYTROFA (lonapegsomatropin-tcgd) for the treatment of pediatric patients one year and older who weigh at least 11.5 kg (25.4 lb) and have growth failure due to inadequate secretion of endogenous growth hormone (GH).
As a once-weekly injection, SKYTROFA is the first FDA approved product that delivers somatropin (growth hormone) by sustained release over one week.
“Today’s approval represents an important new choice for children with GHD and their families, who will now have a once-weekly treatment option. In the pivotal head-to-head clinical trial, once-weekly SKYTROFA demonstrated higher annualized height velocity at week 52 compared to somatropini,” said Paul Thornton, M.B. B.Ch., MRCPI, a clinical investigator and pediatric endocrinologist in Fort Worth, Texas. “This once-weekly treatment could reduce treatment burden and potentially replace the daily somatropin therapies, which have been the standard of care for over 30 years.”
Growth hormone deficiency is a serious orphan disease characterized by short stature and metabolic complications. In GHD, the pituitary gland does not produce sufficient growth hormone, which is important not only for height but also for a child’s overall endocrine health and development.
The approval includes the new SKYTROFA® Auto-Injector and cartridges which, after first removed from a refrigerator, allow families to store the medicine at room temperature for up to six months. With a weekly injection, patients switching from injections every day can experience up to 86 percent fewer injection days per year.
“SKYTROFA is the first product using our innovative TransCon technology platform that we have developed from design phase through non-clinical and clinical development, manufacturing and device optimization, and out to the patients. It reflects our commitment and dedication to addressing unmet medical needs by developing a pipeline of highly differentiated proprietary products across multiple therapeutic areas,” said Jan Mikkelsen, Ascendis Pharma’s President and Chief Executive Officer. “We are grateful to the patients, caregivers, clinicians, clinical investigators, and our employees, who have all contributed to bringing this new treatment option to children in the U.S. with GHD.”
In connection with the commercialization of SKYTROFA, the company is committed to offering a full suite of patient support programs, including educating families on proper injection procedures for SKYTROFA as the first once-weekly treatment for children with GHD.
“It is wonderful that patients and their families now have the option of a once-weekly growth hormone therapy,” said Mary Andrews, Chief Executive Officer and co-founder of the MAGIC Foundation, a global leader in endocrine health, advocacy, education, and support. “GHD is often overlooked and undertreated in our children and managing it can be challenging for families. We are excited about this news as treating GHD is important, and children have a short time to grow.”
The FDA approval of SKYTROFA was based on results from the phase 3 heiGHt Trial, a 52-week, global, randomized, open-label, active-controlled, parallel-group trial that compared once-weekly SKYTROFA to daily somatropin (Genotropin®) in 161 treatment-naïve children with GHDii. The primary endpoint was, AHV at 52 weeks for weekly SKYTROFA and daily hGH treatment groups. Other endpoints included adverse events, injection-site reactions, incidence of anti-hGH antibodies, annualized height velocity, change in height SDS, proportion of subjects with IGF-1 SDS (0.0 to +2.0), PK/PD in subjects < 3 years, and preference for and satisfaction with SKYTROFA.
At week 52, the treatment difference in AHV was 0.9 cm/year (11.2 cm/year for SKYTROFA compared with 10.3 cm/year for daily somatropin) with a 95 percent confidence interval [0.2, 1.5] cm/year. The primary objective of non-inferiority in AHV was met for SKYTROFA in this trial and further demonstrated a higher AHV at week 52 for lonapegsomatropin compared to daily somatropin, with similar safety, in treatment-naïve children with GHD.
No serious adverse events or discontinuations related to SKYTROFA were reported. Most common adverse reactions (≥ 5%) in pediatric patients include: infection, viral (15%), pyrexia (15%), cough (11%), nausea and vomiting (11%), hemorrhage (7%), diarrhea (6%), abdominal pain (6%), and arthralgia and arthritis (6%)ii. In addition, both arms of the study reported low incidences of transient, non-neutralizing anti-hGH binding antibodies and no cases of persistent antibodies.
Conference Call and Webcast Information
| Date | Wednesday, August 25, 2021 |
| Time | 4:30 p.m. ET/1:30 p.m. Pacific Time |
| Dial In (U.S.) | 844-290-3904 |
| Dial In (International) | 574-990-1036 |
| Access Code | 8553236 |
A live webcast of the conference call will be available on the Investors and News section of the Ascendis Pharma website at www.ascendispharma.com. A webcast replay will be available on this website shortly after conclusion of the event for 30 days.
The Following Information is Intended for the U.S. Audience Only
INDICATION
SKYTROFA® is a human growth hormone indicated for the treatment of pediatric patients 1 year and older who weigh at least 11.5 kg and have growth failure due to inadequate secretion of endogenous growth hormone (GH).
IMPORTANT SAFETY INFORMATION
- SKYTROFA is contraindicated in patients with:
- Acute critical illness after open heart surgery, abdominal surgery or multiple accidental trauma, or if you have acute respiratory failure due to the risk of increased mortality with use of pharmacologic doses of somatropin.
- Hypersensitivity to somatropin or any of the excipients in SKYTROFA. Systemic hypersensitivity reactions have been reported with post-marketing use of somatropin products.
- Closed epiphyses for growth promotion.
- Active malignancy.
- Active proliferative or severe non-proliferative diabetic retinopathy.
- Prader-Willi syndrome who are severely obese, have a history of upper airway obstruction or sleep apnea or have severe respiratory impairment due to the risk of sudden death.
- Increased mortality in patients with acute critical illness due to complications following open heart surgery, abdominal surgery or multiple accidental trauma, or those with acute respiratory failure has been reported after treatment with pharmacologic doses of somatropin. Safety of continuing SKYTROFA treatment in patients receiving replacement doses for the approved indication who concurrently develop these illnesses has not been established.
- Serious systemic hypersensitivity reactions including anaphylactic reactions and angioedema have been reported with post-marketing use of somatropin products. Do not use SKYTROFA in patients with known hypersensitivity to somatropin or any of the excipients in SKYTROFA.
- There is an increased risk of malignancy progression with somatropin treatment in patients with active malignancy. Preexisting malignancy should be inactive with treatment completed prior to starting SKYTROFA. Discontinue SKYTROFA if there is evidence of recurrent activity.
- In childhood cancer survivors who were treated with radiation to the brain/head for their first neoplasm and who developed subsequent growth hormone deficiency (GHD) and were treated with somatropin, an increased risk of a second neoplasm has been reported. Intracranial tumors, in particular meningiomas, were the most common of these second neoplasms. Monitor all patients with a history of GHD secondary to an intracranial neoplasm routinely while on somatropin therapy for progression or recurrence of the tumor.
- Because children with certain rare genetic causes of short stature have an increased risk of developing malignancies, practitioners should thoroughly consider the risks and benefits of starting somatropin in these patients. If treatment with somatropin is initiated, carefully monitor these patients for development of neoplasms. Monitor patients on somatropin therapy carefully for increased growth, or potential malignant changes of preexisting nevi. Advise patients/caregivers to report marked changes in behavior, onset of headaches, vision disturbances and/or changes in skin pigmentation or changes in the appearance of preexisting nevi.
- Treatment with somatropin may decrease insulin sensitivity, particularly at higher doses. New onset type 2 diabetes mellitus has been reported in patients taking somatropin. Undiagnosed impaired glucose tolerance and overt diabetes mellitus may be unmasked. Monitor glucose levels periodically in all patients receiving SKYTROFA. Adjust the doses of antihyperglycemic drugs as needed when SKYTROFA is initiated in patients.
- Intracranial hypertension (IH) with papilledema, visual changes, headache, nausea, and/or vomiting has been reported in a small number of patients treated with somatropin. Symptoms usually occurred within the first 8 weeks after the initiation of somatropin and resolved rapidly after cessation or reduction in dose in all reported cases. Fundoscopic exam should be performed before initiation of therapy and periodically thereafter. If somatropin-induced IH is diagnosed, restart treatment with SKYTROFA at a lower dose after IH-associated signs and symptoms have resolved.
- Fluid retention during somatropin therapy may occur and is usually transient and dose dependent.
- Patients receiving somatropin therapy who have or are at risk for pituitary hormone deficiency(s) may be at risk for reduced serum cortisol levels and/or unmasking of central (secondary) hypoadrenalism. Patients treated with glucocorticoid replacement for previously diagnosed hypoadrenalism may require an increase in their maintenance or stress doses following initiation of SKYTROFA therapy. Monitor patients for reduced serum cortisol levels and/or need for glucocorticoid dose increases in those with known hypoadrenalism.
- Undiagnosed or untreated hypothyroidism may prevent response to SKYTROFA. In patients with GHD, central (secondary) hypothyroidism may first become evident or worsen during SKYTROFA treatment. Perform thyroid function tests periodically and consider thyroid hormone replacement.
- Slipped capital femoral epiphysis may occur more frequently in patients undergoing rapid growth. Evaluate pediatric patients with the onset of a limp or complaints of persistent hip or knee pain.
- Somatropin increases the growth rate and progression of existing scoliosis can occur in patients who experience rapid growth. Somatropin has not been shown to increase the occurrence of scoliosis. Monitor patients with a history of scoliosis for disease progression.
- Cases of pancreatitis have been reported in pediatric patients receiving somatropin. The risk may be greater in pediatric patients compared with adults. Consider pancreatitis in patients who develop persistent severe abdominal pain.
- When SKYTROFA is administered subcutaneously at the same site over a long period of time, lipoatrophy may result. Rotate injection sites when administering SKYTROFA to reduce this risk.
- There have been reports of fatalities after initiating therapy with somatropin in pediatric patients with Prader-Willi syndrome who had one or more of the following risk factors: severe obesity, history of upper airway obstruction or sleep apnea, or unidentified respiratory infection. Male patients with one or more of these factors may be at greater risk than females. SKYTROFA is not indicated for the treatment of pediatric patients who have growth failure due to genetically confirmed Prader-Willi syndrome.
- Serum levels of inorganic phosphorus, alkaline phosphatase, and parathyroid hormone may increase after somatropin treatment.
- The most common adverse reactions (≥5%) in patients treated with SKYTROFA were: viral infection (15%), pyrexia (15%), cough (11%), nausea and vomiting (11%), hemorrhage (7%), diarrhea (6%), abdominal pain (6%), and arthralgia and arthritis (6%).
- SKYTROFA can interact with the following drugs:
- Glucocorticoids: SKYTROFA may reduce serum cortisol concentrations which may require an increase in the dose of glucocorticoids.
- Oral Estrogen: Oral estrogens may reduce the response to SKYTROFA. Higher doses of SKYTROFA may be required.
- Insulin and/or Other Hypoglycemic Agents: SKYTROFA may decrease insulin sensitivity. Patients with diabetes mellitus may require adjustment of insulin or hypoglycemic agents.
- Cytochrome P450-Metabolized Drugs: Somatropin may increase cytochrome P450 (CYP450)-mediated antipyrine clearance. Carefully monitor patients using drugs metabolized by CYP450 liver enzymes in combination with SKYTROFA.
You are encouraged to report side effects to FDA at (800) FDA-1088 or www.fda.gov/medwatch. You may also report side effects to Ascendis Pharma at 1-844-442-7236.
Please click here for full Prescribing Information for SKYTROFA.
About SKYTROFA® (lonapegsomatropin-tcgd)
SKYTROFA® is a once-weekly prodrug designed to deliver somatropin over a one-week period. The released somatropin has the same 191 amino acid sequence as daily somatropin.
SKYTROFA single-use, prefilled cartridges are available in nine dosage strengths, allowing for convenient dosing flexibility. They are designed for use only with the SKYTROFA® Auto-Injector and may be stored at room temperature for up to six months. The recommended dose of SKYTROFA for treatment-naïve patients and patients switching from daily somatropin is 0.24 mg/kg body weight, administered once weekly. The dose may be adjusted based on the child’s weight and insulin-like growth factor-1 (IGF-1) SDS.
SKYTROFA has been studied in over 300 children with GHD across the Phase 3 program which consists of the heiGHt Trial (for treatment-naïve patients), the fliGHt Trial (for treatment-experienced patients), and the enliGHten Trial (an ongoing long-term extension trial). Patients who completed the heiGHt Trial or the fliGHt Trial were able to continue into the enliGHten Trial and some have been on SKYTROFA for over four years.
SKYTROFA is being evaluated for pediatric GHD in Phase 3 trials in Japan and Greater China, including the People’s Republic of China, Hong Kong, Macau and Taiwan. Ascendis Pharma is also conducting the global Phase 3 foresiGHt Trial in adults with GHD. SKYTROFA has been granted orphan designation for GHD in both the U.S. and Europe.
About TransCon™ Technologies
TransCon refers to “transient conjugation.” The proprietary TransCon platform is an innovative technology to create new therapies that are designed to potentially optimize therapeutic effect, including efficacy, safety and dosing frequency. TransCon molecules have three components: an unmodified parent drug, an inert carrier that protects it, and a linker that temporarily binds the two. When bound, the carrier inactivates and shields the parent drug from clearance. When injected into the body, physiologic conditions (e.g., pH and temperature) initiate the release of the active, unmodified parent drug in a predictable manner. Because the parent drug is unmodified, its original mode of action is expected to be maintained. TransCon technology can be applied broadly to a protein, peptide or small molecule in multiple therapeutic areas, and can be used systemically or locally.
About Ascendis Pharma A/S
Ascendis Pharma is applying its innovative platform technology to build a leading, fully integrated biopharma company focused on making a meaningful difference in patients’ lives. Guided by its core values of patients, science and passion, the company utilizes its TransCon technologies to create new and potentially best-in-class therapies.
Ascendis Pharma currently has a pipeline of multiple independent endocrinology rare disease and oncology product candidates in development. The company continues to expand into additional therapeutic areas to address unmet patient needs.
Ascendis is headquartered in Copenhagen, Denmark, with additional facilities in Heidelberg and Berlin, Germany, in Palo Alto and Redwood City, California, and in Princeton, New Jersey.
Please visit www.ascendispharma.com (for global information) or www.ascendispharma.us (for U.S. information).

NEW DRUG APPROVALS
ONE TIME
$10.00
///////////Lonapegsomatropin, Skytrofa, APPROVALS 2021, FDA 2021, PEPTIDE, ロナペグソマトロピン , ACP 00, ACP 011, lonapegsomatropin-tcgd, TransCon, TransCon growth hormone, TransCon hGH, TransCon PEG growth hormone, TransCon PEG hGH, TransCon PEG somatropin, ORPHAN DRUG
MVC COVID-19 vaccine, Taiwan’s covid vaccine
Medigen vaccine
MVC COVID-19 vaccine
- MVC-COV1901
track it https://covid19.trackvaccines.org/vaccines/24/
MVC-COV1901 is a vaccine candidate developed and commercialized by Medigen Vaccine Biologics Corporation. The vaccine candidate contains a perfusion form of the SARS-Cov2 recombinant spike protein. Medigen has combined forces with Dynavax, which offers an advanced adjuvant, CpG 1018 (also known as ISS-1018), for use with its vaccine. As of September 2020, the vaccine candidate is in Phase 1 clinical trials to assess its safety and immunogenicity (NCT04487210).
The MVC COVID-19 vaccine, designated MVC-COV1901 and also known as the Medigen COVID-19 vaccine, is a protein subunit COVID-19 vaccine developed by Medigen Vaccine Biologics Corporation [zh] in Taiwan, American company Dynavax Technologies and the U.S. National Institute of Health.[1][2]
This vaccine is made by the recombinant S-2P spike protein adjuvanted with CpG 1018 supplied by Dynavax.[3] Preliminary results from Phase I trials on 77 participants were published in June 2021, indicating what the authors described as “robust” immune system response elicited by the vaccine.[4]
The study authors have assessed the humoral immune response by measuring quantities of binding IgG to S protein, and also the cellular immune response by measuring the quantities of IFN-γ and IL-4 secreting T cells.[4]
Taiwan-based Medigen Vaccine Biologics Corporation (MVC) and Dynavax Technologies Corporation, in the US, have announced the rollout of its COVID-19 vaccine, MVC-COV1901. Approximately 600,000 people are anticipated to receive the Medigen vaccine this week.
Ryan Spencer, Chief Executive Officer of Dynavax commented, “We are pleased that Medigen’s vaccine is now available for the people of Taiwan. We are very excited for this first, of hopefully multiple, EUAs and approvals for COVID-19 vaccines that include CpG 1018 adjuvant. Considering the limitations of current vaccines and the global vaccine shortage, we believe adjuvanted vaccines can contribute significantly to current vaccination efforts.”
In July, MVC received Taiwan Emergency Use Authorization and approval for inclusion in Taiwan’s COVID-19 vaccine immunization program, MVC-COV1901.
MVC COVID-19 vaccine is indicated for adults over 20 years old and is administered in two doses 28 days apart for prevention of COVID-19.
The Advisory Committee recommended that MVC should submit safety monitoring report monthly during the declared EUA period and should submit a vaccine effectiveness report within one year after obtaining EUA approval.
(CNN)Taiwan’s President Tsai Ing-wen received her first shot of the island’s homegrown Covid-19 vaccine on Monday, a public show of support for the new drug which is central to plans for inoculation self sufficiency amid low immunization rates and struggles to obtain vaccines from overseas.Monday’s island-wide rollout of the Medigen Covid-19 vaccine, developed by Taipei-based Medigen Vaccine Biologics Corporation, comes after the drug was approved for emergency use last month by Taiwanese authorities for anyone above 20 years old, with at least 28 days between the two doses.The vaccine has yet to complete phase 3 clinical trials and no efficacy data is available. Paul Torkehagen, Medigen’s director of overseas business development, told CNN in May that the company designed a “very large” phase 2 clinical trial to ensure the vaccine’s safety and effectiveness, with 3,800 participants. Normally, a stage 2 clinical trial only involves several hundred people. Data from the trials showed that 99.8% of participants were able to form antibodies against Covid-19 after taking two doses of the vaccine, Medigen’s CEO Charles Chen said. 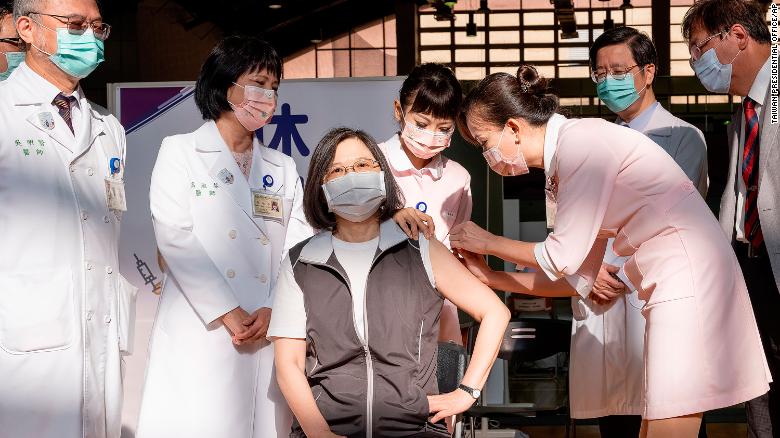
Taiwanese President Tsai Ing-wen, center, receives her first shot of the island’s first domestically developed coronavirus vaccine at the Taiwan University Hospital in Taipei, Taiwan on Monday, August 23.
Taiwan’s Centers for Disease Control said in a July 19 statement that the vaccine posed no serious health effects. Taiwan has ordered 5 million doses of the vaccine from Medigen and more than 700,000 people have already signed up to receive it, according to Reuters.In a Facebook post after receiving the vaccine at a hospital in Taipei, Tsai said she hadn’t suffered from any post-vaccination pain and thanked the health care workers who had administered the shot.”Taking the vaccine can protect yourself, your family, as well as medical staff,” Tsai wrote. “Let’s do our part in boosting Taiwan’s collective defense against the virus!”With its borders sealed to most travelers and strict measures enacted to contain local outbreaks, Taiwan has so far been largely successful in containing Covid-19, reporting fewer than 16,000 total confirmed infections and 828 deaths. But the island has struggled to vaccinate its more than 23 million population, partly due to difficulties obtaining doses from international suppliers.Taiwan’s government has only managed to import around 10 million Covid-19 vaccines, according to Reuters. In July it ordered another 36 million doses of the Moderna shot.Fewer than 5% of Taiwan’s population has received both doses of their Covid-19 vaccine, according to Reuters, as the island delays second dose vaccinations so more people can receive a first shot.On Monday, Taiwan reported four new Covid-19 cases, according to the Central Epidemic Command Center (CECC). Authorities announced on the weekend they would ease virus prevention measures to allow for larger gatherings and the opening of study centers and indoor amusement parks.But Health and Welfare Minister Chen Shih-chung said current Covid-19 restrictions — which include the closure of bars and nightclubs — would remain in place until at least September 6, with the possibility of an extension if the global outbreak continued to grow.Taiwan could become increasingly isolated if it keeps pursuing its “Covid zero” strategy, with both Australia and New Zealand hinting they might abandon the approach once vaccinations reach a certain level.In an opinion piece published on Sunday, Australian Prime Minister Scott Morrison said that while lockdowns to prevent Covid-19 transmission were “sadly necessary for now,” they may not be once vaccination rates increased to the targets of 70% and 80%.”This is what living with Covid is all about. The case numbers will likely rise when we soon begin to open up. That is inevitable,” he said.In neighboring New Zealand, which has also attempted to eliminate the virus within its borders, Covid-19 response minister Chris Hipkins told local media the highly-contagious Delta variant raised “some pretty big questions about what the long-term future of our plans are.”“At some point we will have to start to be more open in the future,” he said.
History
On 16 February 2020, Medigen Vaccine Biologics Corp. (MVC) signed a collaboration agreement with National Institutes of Health (NIH) for COVID-19 vaccine development. The partnership will allow MVC to obtain NIH’s COVID-19 vaccine and related biological materials to conduct animal studies in Taiwan.[5]
On 23 July 2020, Medigen Vaccine Biologics (MVC) announced collaboration with Dynavax Technologies to develop COVID-19 vaccine. The COVID-19 candidate vaccine will have the combination of SARS-CoV2 spike protein created by MVC and Dynavax’s vaccine adjuvant CpG 1018, which was used in a previously FDA-approved adult hepatitis B vaccine.[6][7]
Clinical trials
On 13 October 2020, Medigen Vaccine Biologics received Taiwan’s government subsidies for the initiation of Phase 1 Clinical Trial in Taiwan starting early October. The Phase 1 Clinical Trial was held at National Taiwan University Hospital with 45 participants ranging the age of 20-50.[8][9]
On 25 January 2021, Medigen Vaccine Biologics initiated Phase 2 Clinical Trial for its COVID-19 vaccine candidate MVC-COV1901 with the first participant being dosed. The Phase 2 Clinical Trial for the MVC COVID-19 vaccine was a randomized, double-blinded, and multi-center clinical trial, planned to enroll 3,700 participants of any age 20 above.[3][10][11]
On 10 June 2021, Medigen Vaccine Biologics released its COVID-19 vaccine Phase 2 interim analysis results, which demonstrates good safety profile in participants. The Phase 2 Clinical Trial in the end included 3,800 participants with all participants receiving second dose by 28 April 2021. Medigen Vaccine Biologics announced that it will request Emergency Use Authorization (EUA) with the concluding of the Phase 2 Clinical Trial.[12]
On 20 July 2021, Medigen Vaccine Biologics filed a Phase 3 Clinical Trial IND application with Paraguay’s regulatory authority, which was later approved. The Phase 3 Clinical Trial, however, was different from regular Phase 3 Clinical Trial, which uses immune-bridging trial to compare the performance of MVC COVID-19 vaccine with the Oxford-AstraZeneca COVID-19 vaccine.[13] The decision was a controversial announcement as immune-bridging trials were not fully approved or widely accepted by health authorities. In addition, the accuracy of immune-bridging trials were also been questioned for years.[citation needed]
Adolescents trial
In July 2021, Medigen commenced phase II trials for adolescents aged 12-18.[14]
Authorization
| Full authorization Emergency authorization |
See also: List of COVID-19 vaccine authorizations § Medigen
On July 19, 2021, MVC COVID-19 vaccine obtained Emergency Use Authorization (EUA) approval from the Taiwanese government after fulfilling EUA requirements set by Taiwanese authority.[15] The EUA, however, was met with controversy due to the lack of efficacy data and Phase 3 Clinical Trial. On August 23, 2021, President Tsai Ing-Wen was among the first Taiwanese to receive a dose of the vaccine. [16]
References
- ^ “Dynavax and Medigen Announce Collaboration to Develop a Novel Adjuvanted COVID-19 Vaccine Candidate”. GlobeNewswire. 23 July 2020. Retrieved 7 June 2021.
- ^ 黃驛淵 (10 June 2021). “【獨家】【國產疫苗解盲1】高端實體疫苗針劑首曝光 「每天9萬劑」生產基地直擊” (in Chinese). Mirror Media.
- ^ Jump up to:a b “Medigen Vaccine Biologics COVID-19 Vaccine Adjuvanted with Dynavax’s CpG 1018 Announces First Participant Dosed in Phase 2 Clinical Trial in Taiwan”. http://www.medigenvac.com. Retrieved 7 August 2021.
- ^ Jump up to:a b Hsieh SM, Liu WD, Huang YS, Lin YJ, Hsieh EF, Lian WC, Chen C, Janssen R, Shih SR, Huang CG, Tai IC, Chang SC (25 June 2021). “Safety and immunogenicity of a Recombinant Stabilized Prefusion SARS-CoV-2 Spike Protein Vaccine (MVCCOV1901) Adjuvanted with CpG 1018 and Aluminum Hydroxide in healthy adults: A Phase 1, dose-escalation study”. EClinicalMedicine: 100989. doi:10.1016/j.eclinm.2021.100989. ISSN 2589-5370. PMC 8233066. PMID 34222848.
- ^ “MVC and NIH Collaborate to Develop COVID-19 Vaccine”. http://www.medigenvac.com. Retrieved 7 August 2021.
- ^ “Medigen Collaborates with Dynavax to Develop Novel Adjuvanted COVID-19 Vaccine Candidate”. http://www.medigenvac.com. Retrieved 7 August 2021.
- ^ “MVC Signed an License Agreement with NIH on COVID-19 Vaccine”. Medigen. 5 May 2020. Retrieved 27 July 2021.
- ^ “Medigen’s COVID-19 Vaccine Combined with Dynavax’s CpG 1018 Adjuvant Receives Taiwan Government Subsidy with First Participant Dosed in Early October”. http://www.medigenvac.com. Retrieved 7 August 2021.
- ^ “A Study to Evaluate MVC-COV1901 Vaccine Against COVID-19 in Adult (COVID-19)”. clinicaltrials.gov. United States National Library of Medicine. Retrieved 11 March 2021.
- ^ “A Study to Evaluate the Safety and Immunogenicity of MVC-COV1901 Against COVID-19”. clinicaltrials.gov. United States National Library of Medicine. Retrieved 11 March 2021.
- ^ “A Study to Evaluate MVC-COV1901 Vaccine Against COVID-19 in Elderly Adults”. clinicaltrials.gov. United States National Library of Medicine. 28 March 2021. Retrieved 3 April 2021.
- ^ “MVC Released COVID-19 Vaccine Phase 2 Interim Analysis Result”. http://www.medigenvac.com. Retrieved 7 August 2021.
- ^ “MVC Announces Paraguay Approval of IND Application for Phase 3 Clinical Trial”. http://www.medigenvac.com. Retrieved 7 August 2021.
- ^ “A Study to Evaluate MVC-COV1901 Vaccine Against COVID-19 in Adolescents”. clinicaltrials.gov. United States National Library of Medicine. 6 July 2021. Retrieved 6 July 2021.
- ^ “MVC COVID-19 Vaccine Obtains Taiwan EUA Approval”. http://www.medigenvac.com. Retrieved 7 August 2021.
- ^ Taiwan begins contested rollout of new Medigen domestic vaccine, Nikkei Asia, Erin Hale, August 23, 2021
| Vaccine description | |
|---|---|
| Target | SARS-CoV-2 |
| Vaccine type | Protein subunit |
| Clinical data | |
| Other names | MVC-COV1901 |
| Routes of administration | Intramuscular |
| Legal status | |
| Legal status | Full and Emergency Authorizations: List of MVC COVID-19 vaccine authorizations |
| Identifiers | |
| DrugBank | DB15854 |
| Part of a series on the |
| COVID-19 pandemic |
|---|
| COVID-19 (disease)SARS-CoV-2 (virus)CasesDeaths |
| showTimeline |
| showLocations |
| showInternational response |
| showMedical response |
| showEconomic impact and recession |
| showImpacts |
| COVID-19 portal |
////////Medigen vaccine, MVC COVID-19 vaccine, SARS-CoV-2, covid 19, corona virus, taiwan, approvals 2021, iss 1018, CpG 1018, MVC-COV1901
NEW DRUG APPROVALS
one time
$10.00
Pepinemab, VX 15
(Heavy chain)
QVQLVQSGAE VKKPGSSVKV SCKASGYSFS DYYMHWVRQA PGQGLEWMGQ INPTTGGASY
NQKFKGKATI TVDKSTSTAY MELSSLRSED TAVYYCARYY YGRHFDVWGQ GTTVTVSSAS
TKGPSVFPLA PCSRSTSEST AALGCLVKDY FPEPVTVSWN SGALTSGVHT FPAVLQSSGL
YSLSSVVTVP SSSLGTKTYT CNVDHKPSNT KVDKRVESKY GPPCPPCPAP EFLGGPSVFL
FPPKPKDTLM ISRTPEVTCV VVDVSQEDPE VQFNWYVDGV EVHNAKTKPR EEQFNSTYRV
VSVLTVLHQD WLNGKEYKCK VSNKGLPSSI EKTISKAKGQ PREPQVYTLP PSQEEMTKNQ
VSLTCLVKGF YPSDIAVEWE SNGQPENNYK TTPPVLDSDG SFFLYSRLTV DKSRWQEGNV
FSCSVMHEAL HNHYTQKSLS LSLGK
(Light chain)
DIVMTQSPDS LAVSLGERAT INCKASQSVD YDGDSYMNWY QQKPGQPPKL LIYAASNLES
GVPDRFSGSG SGTDFTLTIS SLQAEDVAVY YCQQSNEDPY TFGQGTKLEI KRTVAAPSVF
IFPPSDEQLK SGTASVVCLL NNFYPREAKV QWKVDNALQS GNSQESVTEQ DSKDSTYSLS
STLTLSKADY EKHKVYACEV THQGLSSPVT KSFNRGEC
(Disulfide bridge: H22-H96, H132-L218, H145-H201, H224-H’224, H227-H’227, H259-H319, H365-H423, H’22-H’96, H’132-L’218, H’145-H’201, H’259-H’319, H’365-H’423, L23-L92, L138-L198, L’23-L’92, L’138-L’198)
Pepinemab
VX15/2503
Antineoplastic, Anti-human semaphorin 4D antibody
Monoclonal antibody
Treatment of solid tumors, multiple sclerosis and Huntington’s disease
| Formula | C6442H9910N1702O2052S48 |
|---|---|
| MOL WGT | 145481.0022 |
- Moab VX15/2503
- Pepinemab
- UNII-BPZ4A29SYE
- VX-15
- VX15
- VX15/2503
| Product name | Pepinemab Biosimilar – Anti-SEMA4D mAb – Research Grade |
|---|---|
| Source | CAS 2097151-87-4 |
| Species | Chimeric,Humanized |
| Expression system | Mammalian cells |
- OriginatorVaccinex
- DeveloperBristol-Myers Squibb; Children’s Oncology Group; Emory University; Merck KGaA; National Cancer Institute (USA); Teva Pharmaceutical Industries; UCLAs Jonsson Comprehensive Cancer Center; Vaccinex
- ClassAntibodies; Antidementias; Antineoplastics; Immunotherapies; Monoclonal antibodies
- Mechanism of ActionCD100 antigen inhibitors
- Orphan Drug StatusYes – Huntington’s disease
- New Molecular EntityYes
- Phase IIHuntington’s disease
- Phase I/IIAlzheimer’s disease; Non-small cell lung cancer; Osteosarcoma; Solid tumours; Squamous cell cancer
- Phase IColorectal cancer; Malignant melanoma; Pancreatic cancer
- No development reportedMultiple sclerosis
- 22 May 2021Pepinemab is still in phase I trials for Colorectal cancer and Pancreatic cancer in USA (NCT03373188)
- 17 May 2021Phase-I/II clinical trials in Squamous cell cancer (Combination therapy, Late-stage disease, Metastatic disease, Recurrent, Second-line therapy or greater) in USA (IV) (NCT04815720)
- 17 May 2021Vaccinex plans a phase I/II trial for Alzheimer’s disease (In volunteers), in H2 2021
Semaphorin 4D (SEMA4D) plays a role in multiple cellular processes that contribute to the pathophysiology of neuroinflammatory/neurodegenerative diseases. SEMA4D is, therefore, a uniquely promising target for therapeutic development.
Pepinemab is a novel monoclonal antibody that blocks the activity of SEMA4D, and preclinical testing has demonstrated the beneficial effects of anti-SEMA4D treatment in a variety of neurodegenerative disease models. Vaccinex is committed to the development of this potentially important antibody that has the potential to help people with different neurodegenerative disorders that share common mechanisms of pathology.
Note: Pepinemab (VX15/2503) is an investigational drug currently in clinical studies. It has not been demonstrated to be safe and effective for any disease indication. There is no guarantee that pepinemab (VX15/2503) will be approved for the treatment of any disease by the U.S. Food and Drug Administration or by any other health authority worldwide.
////////////////////Pepinemab, VX15/2503, vx 15, Antineoplastic, Anti-human semaphorin 4D antibody, Monoclonal antibody, solid tumors, multiple sclerosis, Huntington’s disease, PEPTIDES
NEW DRUG APPROVALS
ONE TIME
$10.00
AVASOPASEM MANGANESE
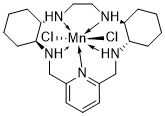
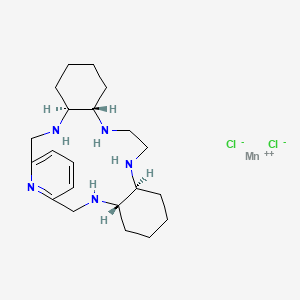
AVASOPASEM
Average: 518.83
Monoisotopic: 517.134397
Chemical FormulaC21H35Cl3MnN5
manganese(2+);(4S,9S,14S,19S)-3,10,13,20,26-pentazatetracyclo[20.3.1.04,9.014,19]hexacosa-1(26),22,24-triene;dichloride
- Manganese, dichloro((4aS,13aS,17aS,21aS)-1,2,3,4,4a,5,6,12,13,13a,14,15,16,17,17a,18,19,20,21,21a-eicosahydro-7,11-nitrilo-7H-dibenzo(b,H)-5,13,18,21-tetraazacycloheptadecine-kappaN5,kappaN13,kappaN18,kappaN21,kappaN22)-, (pb-7-11-2344’3′)-
CAS 435327-40-5
- A superoxide dismutase mimetic.
- GC 4419
- M-40419
- SC-72325A
- For the Reduction of The Severity and Incidence of Radiation and Chemotherapy-Induced Oral Mucositis
Avasopasem manganese, also known as GC4419, is a highly-selective small molecule mimetic of superoxide dismutase (SOD) being investigated for the reduction of radiation-induced severe oral mucositis.1,2 This drug has potential application for radiation-induced esophagitis and oral mucositis, in addition to being currently tested against COVID-19.
Avasopasem manganese is a superoxide dismutase mimetic that rapidly and selectively converts superoxide to hydrogen peroxide and oxygen in order to protect normal tissue from radiation therapy-induced damage.1 This drug is currently being investigated against oral mucositis, esophagitis, and COVID-19.
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018152353
Transition metal pentaaza 15-membered macrocyclic ring complexes having the macrocyclic ring system corresponding to Formula A have been shown to be effective in a number of animal and cell models of human disease, as well as in treatment of conditions afflicting human patients.
For example, in a rodent model of colitis, one such compound, GC4403, has been reported when administered by intraperitoneal (ip) injection to significantly reduce the injury to the colon of rats subjected to an experimental model of colitis (see Cuzzocrea et al., Europ. J. Pharmacol., 432, 79-89 (2001)).
GC4403 administered ip has also been reported to attenuate the radiation damage arising both in a clinically relevant hamster model of acute, radiation-induced oral mucositis (Murphy et al., Clin. Can. Res., 74(13), 4292 (2008)), and lethal total body irradiation of adult mice (Thompson et al., Free Radical Res., 44(5), 529-40 (2010)).
Similarly, another such compound, GC4419, administered ip has been shown to attenuate VEGFr inhibitor-induced pulmonary disease in a rat model (Tuder, et al., Am. J. Respir. Cell Mol. Biol., 29, 88-97 (2003)), and to increase the anti-tumor activity of anti-metabolite and anti-mitotic agents in mouse cancer models (see, e.g., WO2009/143454). In other studies, GC4419 and GC4403 have been shown to be similarly potent in various animal models of disease. Additionally, another such compound, GC4401, administered ip has been shown to provide protective effects in animal models of septic shock (S. Cuzzocrea, et. al., Crit. Care Med., 32(1 ), 157 (2004)) and pancreatitis (S. Cuzzocrea, et. al., Shock, 22(3), 254-61 (2004)).
[0003] Certain of these compounds have also been shown to possess potent anti-inflammatory activity and prevent oxidative damage in vivo. For example, GC4403 administered ip has been reported to inhibit inflammation in a rat model of inflammation (Salvemini, et.al., Science, 286, 304 (1999)), and prevent joint disease in a rat model of collagen-induced arthritis (Salvemini et al., Arthritis & Rheumatism, 44(12), 2009-2021 (2001)). In addition, these compounds have been reported to possess analgesic activity and to reduce inflammation and edema by systemic administration in the rat-paw carrageenan hyperalgesia model, see, e.g., U.S. Pat. No. 6,180,620.
[0004] Compounds of the class comprising GC4419 have also been shown to be safe and effective in the prevention and treatment of disease in human subjects. For example, GC4419 administered by intravenous (iV) infusion has been shown to reduce oral mucositis in head-and-neck cancer patients undergoing chemoradiation therapy (Anderson, C, Phase 1 Trial of Superoxide Dismutase (SOD) Mimetic GC4419 to Reduce Chemoradiotherapy (CRT)-lnduced Mucositis (OM) in Patients (pts) with Mouth or Oropharyngeal Carcinoma (OCC), Oral Mucositis Research Workshop,
MASCC/ISOO Annual Meeting on Supportive Care in Cancer, Copenhagen, Denmark (June 25, 2015)).
[0005] However, the administered dose when delivered systemically, for example by a parenteral route, can be limited in animal models and particularly in humans by systemic exposure and resulting toxicity that appears to be similar in nature among the pentaaza 15-membered macrocyclic ring dismutase mimetics of Formula A, particularly GC4403, GC4419, GC4401 and related compounds sharing the dicyclohexyl and pyridine motif in the macrocycle ring (e.g., compounds sharing the dicyclohexyl and pyridine motif generally include compounds according to Formula (I) below herein having W as an unsubstituted pyridine moiety, and wherein U and V are transcyclohexanyl fused rings) . For example, the maximum tolerated dose of GC4403 delivered as a 30-minute iv infusion in humans is 25 mg, or roughly 0.35 mg/kg in a 70-kg subject, and similar limitations exist for animal parenteral dosing. Thus, the efficacy of treatment of conditions such as local inflammatory disease or tissue damage of the alimentary canal may be limited when using systemic delivery of GC4403 and similar compounds.
[0006] In each of these compounds comprising the pentaaza 15-membered macrocyclic ring of Formula A, the five nitrogens contained in the macrocyclic ring each form a coordinate covalent bond with the manganese (or other transition metal coordinated by the macrocycle) at the center of the molecule. Additionally, manganese (or other appropriate transition metal coordinated with the macrocycle) forms coordinate covalent bonds with “axial ligands” in positions perpendicular to the roughly planar macrocycle. Such coordinate covalent bonds are characterized by an available “free” electron pair on a ligand forming a bond to a transition metal via donation and sharing of the electron pair thus forming a two-electron bond between the metal and the donor atom of the ligand (Cotton, F.A. & G. Wilkinson, Advanced Inorganic Chemistry, Chapter 5, “Coordination Compounds”, 2nd revised edn., Interscience Publishers, p.139 (1966); lUPAC Gold Book, online version http://goldbook.iupac.org/C01329.html). The coordinate covalent nature of the bonds between manganese (or other such appropriate transition metal) and the five macrocyclic ring nitrogens and between manganese (or other such transition metal) and each of the two chloro axial ligands is evidenced, for example, by the “single crystal” X-ray crystal structure of GC4403 (Fig. 11 ) and GC4419 (Fig. 12).
[0007] Coordination compounds contrast with ionic compounds, for example, salts, where in the solid state the forces between anions and cations are strictly coulombic electrostatic forces of attraction between ions of opposite charge. Thus, in salts, discrete cations and anions provide the force to maintain the solid state structure; e.g., such as the chloride ion and the sodium ion in a typical salt such as sodium chloride (Cotton, F.A. & G. Wilkinson, Advanced Inorganic Chemistry, Chapter 5, “The Nature of Ionic Substances”, 2nd revised edn., Interscience Publishers, pp. 35-36, 45-49 (1966).
[0008] Although pentaaza 15-membered macrocyclic ring complexes have been disclosed in the literature for a number of anti-inflammatory indications, the representative disclosures discussed above illustrate that such compounds are generally administered by intraperitoneal (ip) or intravenous (iv) injection to potentiate systemic bioavailability. Local (e.g. topical) administration has been reported as ineffective in animal models of inflammatory disease, particularly when measured against the efficacy of systemic administration methods (Murphy et al., Clin. Can. Res., 74(13), 4292 (2008); WO 2008/045559). One research group has reported inhibition of colonic tissue injury and neutrophil accumulation by intracolonic administration of a prototype pentaaza macrocycle superoxide dismutase mimetic (MnPAM) (having a different structure from GC4403), though that disclosure neither addresses systemic bioavailability of the compounds described therein, nor explore limitations resulting from systemic bioavailability impacting safety and/or efficacy of that specific compound (Weiss et al., J. Biol. Chem., 271(42): 26149-26156 (1996); Weiss, R. and Riley, D., Drugs Future, 21 (4): 383-389 (1996)).
[0009] Aspects of the present disclosure provide for formulations of pentaaza macrocyclic ring complexes of the class comprising GC4419, GC4403, and GC4401 that exhibit limited systemic bioavailability when administered orally (e.g. less than 20%, less than 15%, and even less than 10% bioavailability when dosed in appropriate oil-based formulations; see Table 1 and when combined with other formulations even less than 5%, and even less than 1%; see Example 28). In general, drug absorption from the gastrointestinal tract occurs via passive uptake so that absorption is favored when the drug is in a non-ionized (neutral) and lipophilic form. See, e.g., Goodman & Gilman’s: The Pharmacological Basis of Therapeutics, Ninth Edition, p. 5-9 (1996). Without wishing to be limited to any particular theory, this is also believed to be the case for this class of compounds, as exemplified by GC4403, where the axial ligands are both chloro moieties forming a coordinate covalent bond to the manganese and a neutral complex results:
The Mn(ll) pentaaza macrocyclic ring dichloro complexes, such as GC4419, GC4401, GC4444, and GC4403 (structures shown below) were synthesized using literature procedures. For GC4403 the chiral R,R-diaminocyclohexane is utilized as starting material,2 whereas for GC4419, the mirror-image enantiomer of GC4403, the chiral S,S-diaminocyclohexane is utilized instead.3,4 The remainder of the synthesis of GC4419 can be identical in all respects to the method published for GC4403.2 The synthesis of the GC4401 complex was reported previously in reference 5.
[00213] The synthesis of GC4444 which contains the additional 11-R-Methyl substituent generating a fifth chiral center on carbon (and is also derived from R,R-diaminocyclohexane) is made from the corresponding chiral tetraamine whose synthesis was published in reference 6 as Example 5C.
Syntheses of Axial Ligand Derivatives
[00214] The same Mn(II) pentaaza macrocyclic ring dichloro complexes (GC4419, GC4403, GC4444 and GC4401 ) were also used as the starting material precursors for the syntheses of other axial ligand bound derivatives using a generic synthesis scheme in which a large excess of a salt of an anion is used to displace the chloro ligand thereby generating the new compound.
EXAMPLE 2
[00215] Synthesis of Manganese(ll)bis-acetato[(4aS,13aS,17aS,21aS)-1,2,3,4,48,5,6,12,13,13a,14,15,16,17, 17a,18,19,20,21,21a- Eicosahydro-11,7-nitrilo-7H-dibenzo[b,h][1,4,7,10] tetraazacycloheptadecine-KN5, κΝ13, κΝ18, κΝ21, κΝ22]-, [bis-Acetato (GC4419)]. GC4701
[00216] Using a 500-mL Erlenmeyer, 100 mL of deionized (“DI”) water was added to 5.3 g of GC4419; the mixture was stirred vigorously for 15-20 min, then sonicated for 5 min. The resulting light brownish suspension was filtered through a 10-20 μ fritted funnel (ca. 0.3 g of solid material remained in the funnel). The resulting clear solution was added into a sodium acetate solution (ca. 429 mmol, 21 equiv in 100 mL DI water) as a stream in one portion. No solid separated and the yellowish solution was stirred for 5 additional min. The solution was transferred to a separatory funnel and extracted (3 X 50 mL) with dichloromethane. The organic layers were separated, combined, and transferred back into a separatory funnel. The dichloromethane solution was back-extracted (2 X 50 mL) with aqueous sodium acetate (32 g/100 mL). The dichloromethane layer was dried over MgSO4 (ca. 10 g) for 30 min (w/stirring), filtered using a 10-20 μ fritted funnel, and the solution taken to dryness using a rotavap. To the yellow oily solid resulting from taking the solution to dryness was added methanol (50 mL). This solution was then again taken to dryness on the rotovap to yield a light yellow foam/glass. This material was dried in vacuo at room temperature for two days.
[00217] The isolated yellowish brittle (4.11 g, 75% yield based on GC4419) was analyzed by HPLC and showed a purity of 99.7% and elemental analysis showed 0.98 wt. % residual chlorine. The elemental analysis is consistent with the expected bis-(acetato) structure C25H41MnN5O4●2H2O. Anal Cal’d: C, 53.00% ; H, 8.01 %; N, 12.36%, and Mn, 9.70%. Anal Found: C, 53.10% ; H, 8.34% ; Mn, 9.86%, N, 12.56%, and CI (as total halogen content), 0.98 wt. %.
Patent
WO 2002071054
https://patents.google.com/patent/WO2002071054A1/enSuperoxide dismutase (SOD) enzymes are enzymes that catalyze the dismutation of the free radical superoxide, the one-electron reduction product of molecular oxygen. The dismutation of the free radical superoxide involves the conversion of this one-electron reduction product of molecular oxygen to the nonradical molecular oxygen. Superoxide dismutase enzymes are a class of oxidoreductases which contain either Cu/Zn, Fe, or Mn at the active site. Superoxide dismutase (SOD) mimetic compounds are low molecular weight catalysts which mimic the natural enzyme function of the superoxide dismutase enzymes. Thus, superoxide dismutase mimetic compounds also catalyze the conversion of superoxide into oxygen and hydrogen peroxide, rapidly eliminating the harmful biologically generated superoxide species that are believed to contribute to tissue pathology in a number of diseases and disorders. These diseases and disorders include reperfusion diseases, such as those following myocardial infarct or stroke, inflammatory disorders such as arthritis, and neurological disorders such as Parkinson’s disease. Chem Reviews, 1999 vol 99, No. 9, 2573-2587.Superoxide dismutase mimetic compounds possess several advantages over the superoxide dismutase enzymes themselves in that their chemical properties can be altered to enhance stability, activity and biodistribution while still possessing the ability to dismutase the harmful superoxide. Superoxide dismutase mimetic compounds have generated intense interest and have been the focus of considerable efforts to develop them as a therapeutic agent for the treatment of a wide range of diseases and disorders, including reperfusion injury, ischemic myocardium post-ischemic neuropathies, inflammation, organ transplantation and radiation induced injury. Most of the superoxide dismutase mimics currently being developed as therapeutic agents are synthetic low molecular weight manganese-based superoxide dismutase mimetic compounds. Chem Reviews, 2576. Superoxide dismutase mimetic compounds are metal complexes in which the metal can coordinate axial ligands. Examples of such metal complexes include, but are not limited to, complexes of the metals Mn and Fe. Many of the complexes of the metals Mn and Fe do not possess superoxide dismutase activity but possess properties that enable them to be put to other therapeutic and diagnostic uses. These therapeutic and diagnostic uses include MRI imaging enhancement agents, peroxynitrite decomposition catalysts, and catalase mimics. These metal complexes, however, share the structural similarity of possessing a metal that can coordinate exchangeable ligands. These metal complexes exist in water as a mixture of species in which various ligands are possible. An illustration of such a mixture is provided by M40403 , a Mn(π) complex of a nitrogen-containing fifteen membered macrocyclic ligand, shown in Scheme 1. One of the forms for this metal complex is the dichloro complex, which when dissolved in water another form is generated where one of the chloride anions immediately dissociates from the metal generating the [Mn(Cl)(aquo)]+ complex. The problem in aqueous solvent systems or any solvent which has a potential donor atom is that there are a variety of potential ligands available to coordinate axially to the Mn(π) ion of the complex, hi conducting an analysis of a sample containing a metal complex by high performance liquid chromatography (HPLC) the chromatogram tends to be very broad and unresolved due to the presence of the various species of complexes, as shown in Scheme 1. This phenomena makes the identification and quantification of metal complexes by standard HPLC techniques quite difficult. Therefore, in light of the developing roles of metal complexes as therapeutics in the treatment of various disorders and diagnostic agents, a substantial need exists for an effective and workable high performance liquid chromatography method for analyzing metal complexes.

Scheme 1An additional complication which exists is the issue of the acid stability of the metal complex. As the pH decreases, the rate at which the complex becomes protonated and experiences instability increases. This presents particular problems for the use of HPLC as a method of detection and quantification of the metal complexes because the mobile phase used for reverse phase HPLC frequently contains mixtures of organic solvents and water in various combinations with trifluoroacetic acid. The trifluoroacetic acid is commonly present between about 0.1 to about 0.5% by weight. The presence of the trifluoroacetic acid causes the complex to dissociate. This dissociation destroys the potential of any such method to be used for release testing for purity. Furthermore, the trifluoroacetate anion causes the formation of some of the trifluoroacetato complex which could possess a different retention time from the chloro complexes thus, confusing the chromatography. Thus, the phenomenon of ligand exchange, coupled with the acid instability of the metal complexes, provides considerable challenges to the effort to detect and quantify metal complexes using HPLC. These challenges and needs have surprisingly been met by the invention described below.Analytical HPLC is a powerful method to obtain information about a sample compound including information regarding identification, quantification and resolution of a compound. HPLC has been used particularly for the analysis of larger compounds and for the analysis of inorganic ions for which liquid chromatography is unsuitable. Skoog, D.A., West, M.A., Analytical Chemistry, 1986, p. 520. As an analytical tool HPLC takes advantage of the differences in affinity that a particular compound of interest has for the stationary phase and the mobile phase (the solvent being continuously applied to the column). Those compounds having stronger interactions with the mobile phase than with the stationary phase will elute from the column faster and thus have a shorter retention time. The mobile phase can be altered in order to manipulate the interactions of the target compound and the stationary phase. In normal-phase HPLC the stationary phase is polar, such as silica, and the mobile phase is a nonpolar solvent such as hexane or isopropyl ether. In reversed- phase HPLC the stationary phase is non-polar, often a hydrocarbon, and the mobile phase is a relatively polar solvent. Since 1974 when reversed-phase packing materials became commercially available, the number of applications for reversed- phase HPLC has grown, and reversed- phase HPLC is now the most widely used type of HPLC. Reversed-phase HPLC’s popularity can be attributed to its ability to separate a wide variety of organic compounds. Reversed-phase chromatography is especially useful in separating the related components of reaction mixtures, and therefore is a useful analytical tool for determining the various compounds produced by reactions. To create a non-polar stationary phase silica or synthetic polymer based adsorbents are modified with hydrocarbons. The most popular bonded phases are Cl, C4, C8 and C18. Silica based adsorbents modified with trimethylchlorosilane (Cl) and butyldimethylchlorosilane (C4) have a few applications in HPLC, mainly for protein separation or purification. These adsorbents show significant polar interactions. Octyl (C8) and octadecyl (C18) modified adsorbents are the most widely used silica based adsorbents, with almost 80% of all HPLC separations being developed with these adsorbents.The most important parameter in reversed-phase HPLC is the mobile phase. The type of mobile phase employed in the HPLC will have a significant effect on the retention of the analytes in the sample, and varying the composition of the mobile phase allows the chromatographer to adjust the retention times of target components in the mixture to desired values. This ability provides the HPLC method with flexibility. The mobile phase in reversed-phase chromatography has to be polar and it also has to provide reasonable competition for the adsorption sites for the analyte molecules. Solvents that are commonly employed as eluent components in reversed-phase HPLC are acetonitrile, dioxane, ethanol, methanol, isopropanol, tetrahydrofuran, and water. In reversed phase HPLC of high molecular weight biological compounds, the solvents acetonitrile, isopropanol or propanol are most frequently used. Popular additives to the mobile phase for the improvement of resolution include mixtures of phosphoric acid and amines and periϊuorinated carboxylic acids, especially trifluoroacetic acid (TFA). HPLC exploits the differences in affinity that a particular compound of interest has for the stationary phase and the mobile phase. This phenomenon can be utilized to separate compounds based on the differences in their physical properties. Thus, HPLC can be used to separate stereoisomers, diastereomers, enantiomers, mirror image stereoisomers, and impurities. Stereoisomers are those molecules which differ from each other only in the way their atoms are oriented in space. The particular arrangement of atoms that characterize a particular stereoisomer is known as its optical configuration, specified by known sequencing rules as, for example, either + or – (also D or L) and/or R or S. Stereoisomers are generally classified as two types, enantiomers or diastereomers. Enantiomers are stereoisomers which are mirror-images of each other. Enantiomers can be further classified as mirror-image stereoisomers that cannot be superimposed on each other and mirror-image stereoisomers that can be superimposed on each other. Mirror- image stereoisomers that can be superimposed on each other are known as meso compounds. Diastereomers are stereoisomers that are not mirror images of each other. Diastereomers have different physical properties such as melting points, boiling points, solubilities in a given solvent, densities, refractive indices, etc. Diastereomers can usually be readily separated from each other by conventional methods, such as fractional distillation, fractional crystallization, or chromatography, including HPLC.Enantiomers, however, present special challenges because their physical properties are identical. They generally cannot be separated by conventional methods, especially if they are in the form of a racemic mixture. Thus, they cannot be separated by fractional distillation because their boiling points are identical and they cannot be separated by fractional crystallization because their solubilites are identical (unless the solvent is optically active). They also cannot be separated by conventional chromatography such as HPLC because (unless the adsorbent is optically active) they are held equally onto the adsorbent. HPLC methods employing chiral stationary phases are a very common approach to the separation of enantiomers. To be able to separate racemic mixtures of stereoisomers, the chiral phase has to form a diastereomeric complex with one of the isomers, or has to have some other type of stereospecific interaction. The exact mechanism of chiral recognition is not yet completely understood. In reversed-phaseHPLC a common type of chiral bonded phase is chiral cavity phases.The ability to be able to separate diastereomers and enantiomers by HPLC is a useful ability in evaluating the success of synthetic schemes. It is often desirable to separate stereoisomers as a means of evaluating the enantiomeric purity of production samples. All references listed herein are hereby incorporated by reference in their entiretyExamples 1 (traditional mobile phase) and 2 (mobile phase containing excess of salt of a coordinating anion).


+X“

Scheme 2 Any metal complex possessing a metal that is capable of coordinating a monodentate ligand can be used in the present invention. Examples of such metal complexes include, but are not limited to, complexes of the metals Mn and Fe. The metal complexes of the invention preferably have therapeutic and diagnostic utilities. These therapeutic and diagnostic utilities include, but are not limited to, use as superoxide dismutase mimetic compounds, MRI imaging enhancement agents, peroxynitrite decomposition catalysts, and catalase mimics. The preferred metal complexes for use in the invention are superoxide dismutase mimetic compounds. Examples of such superoxide dismutase mimetic compounds include, but are not limited to, the following complexes of the metals Mn and Fe. Iron based superoxide dismutase mimetics include, but are not limited to, Fera(salen) complexes, Fera(l,4,7,10,13-pentaazacyclopentadecane) derivatives and Feffl(porphyrinato) complexes. Manganese based superoxide dismutase mimetic compounds include, but are not limited to, metal complexes containing manganese(π) or manganese(m). Examples of manganese based superoxide dismutase mimetic compounds include Mnm(porphyrinato) complexes, Mnffl(salen) complexes, and Mnπ(l ,4,7, 10, 13-pentaazacyclopentadecane) derivatives. Mnπ(l ,4,7, 10,13- pentaazacyclopentadecane) derivatives are more preferred for use in the invention. Examples of Mnπ(l,4,7,10,13-pentaazacyclopentadecane) derivatives preferred for use in the invention include, but are not limited to, M40403 and M40401, as shown in Scheme 3 below.Furthermore, stereoisomers of all of the above metal complexes can be used in the process of the present invention. Diastereomers of the same metal complexes can also be detected and separated by the method of the present invention. As it is often desirable to separate stereoisomers as a means of evaluating the chemical and optical purity of production samples, the metal complexes can also comprise products of a reaction stream. Enantiomers of any of the metal complexes referenced above can be used in the chiral HPLC method of the invention for the separation of enantiomers of a metal complex.
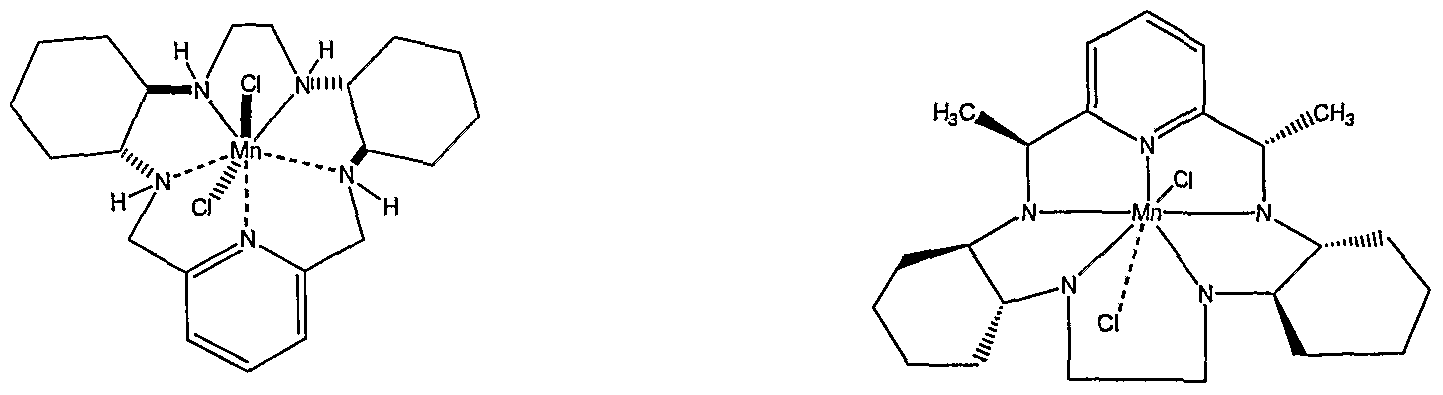
M40403 M40401

M40484Scheme 3The ligand is a coordinating anion that binds to the metal cation of the metal complex. The coordinating anion can serve as an axial ligand for a superoxide dismutase mimetic compound. Examples of such anions include, but are not limited to, chloride anions, thiocyanate anions, stearate anions, acetate anions, trifluoroacetate anions, carboxylate anions, formate anions, or azide anions. Preferred anions include chloride anions, thiocyanate anions, and formate anions. More preferred anions are chloride anions. The more preferred anions in the chiral HPLC embodiment of the invention are thiocyanate anions. When present in an excess, the thiocyanate anions bind to the coordinating metal of the complexes preferentially to the chloride anions. An excess of thiocyanate anions will produce the bis(thiocyanato) complexes of M40403 and M40419 as shown in Scheme 4.

M40403 M40403-(SCN)2

M40419 M40419-(SCN)2Scheme 4An example of the use of the acetate anion as the coordinating anion with M40403 is shown in Scheme 5 below. Scheme 6 illustrates the use of the formate anion as the coordinating anion with M40403.

M40403 M40403-(OAc)2Scheme 5

M40403 M40403-(Formate)2Scheme 6The coordinating anion is supplied by a salt of the coordinating anion. Salts of the chloride anion include, but are not limited to, sodium chloride, lithium chloride, potassium chloride, ammonium chloride, or tetraalkylammonium chloride. Preferred salts of the chloride anion include sodium chloride, lithium chloride and tetrabutylammonium chloride. Salts of the thiocyanate anion include, but are not limited to, sodium thiocyanate, potassium thiocyanate, ammonium thiocyanate, or lithium thiocyanate. Preferred salts of the thiocyanate anion include sodium thiocyanate and potassium thiocyanate. Salts of the acetate anion include, but are not limited to, potassium acetate, sodium acetate, ammonium acetate, ammonium trifluoroacetate and lithium acetate. Preferred salts of the acetate anion include ammonium acetate. Salts of the formate anion include, but are not limited to, potassium formate, sodium formate, ammonium formate and lithium formate. Preferred salts of the formate anion include ammonium formate. Salts of the cyanate anion include but are not limited to, sodium cyanate, potassium cyanate, or ammonium cyanate. Salts of the carboxylate anion include, but are not limited to, potassium carboxylate, ammonium carboxylate and sodium carboxylate. Salts of the stearate anion include, but are not limited to, lithium stearate and sodium stearate. Salts of the azide anion include, but are not limited to, sodium azide, potassium azide, and lithium azide. The salt added to the mobile phase can also be a mixture of any of these salts. Examples include a mixture of tetrabutylammonium chloride and lithium chloride.EXAMPLESExperimental For Examples 1-8 Chemicals, Solvents and MaterialsAll solvents used in the study were HPLC grade or equivalent. All chemicals were ACS reagent grade or equivalent.HPLC System and Data AnalysisThe HPLC chromatography was performed using a Gilson system (Model 306 pump, Model 155 UN-V detector, Model 215 liquid handler, Unipoint Software,Win98), a Narian system (Model 310 pump, Model 340 UN-N detector, Model 410 autosampler Star Workstation, Win98) or SSI system (Acuflow Series IN pump, Acutect 500 UV-N detector, Alcott Model 718 autosampler, HP Model 3395 integrator).Example 1HPLC Analysis of M40403 using Method 1

M40403 Method 1: Analytical Column: Waters YMC ODS-AQ S5 120A (4.6 x 50 mm); System A: 0.1% trifluoroacetic acid in H2O; System B: 0.08% trifluoroacetic acid in acetonitrile; Gradient: 10-50% system B over 10 min; Flow rate: 3ml/min; Detector wavelength: 265. Injected 20 μl of stock solution of M40403 prepared by dissolving 1 mg in 1 ml of water and diluting with 1 ml of system A. The HPLC chromatogram of M40403 using method 1 is shown in Figure 1. Example 2 HPLC Analysis of M40403 using Method 2Method 2: Analytical Column: Waters YMC 9DS-AQ S5 12θΛ (4.6 x 50 MM); System A: 0.5 N aqueous NaCl; System B: 1 :4 water/CH3CN; Gradient: 10-50% system B over 9 min; Flow rate: 3mL/min; Detector wavelength: 265 nm. Injected 20 μl of stock solution of M40403 prepared by dissolving 1 mg in 1 ml of system A. The HPLC chromatogram of M40403 using method 2 is shown in Figure 2.Example 3 HPLC Analysis of M40403 using Method 3Method 3: Analytical Column: Waters Symmetry Shield RP18, 5 μm, 250 x 4.6 mm;Mobile Phase: Acetonitrile: 0.125 M Tetrabutylammonium Chloride in water (pH 6.5), 5%: 95% H20(v/v); Flow rate: 1 mL/min; Detection wavelength: 265nm. Injected 20 μl of stock solution of M40403 prepared by dissolving 1 mg in 1 ml of mobile phase. The HPLC chromatogram of M40403 using method 3 is shown in Figure 3.The HPLC chromatogram of M40403 and related compounds using method 3 is shown in Figure 3a. Method 3 allows a separation of M40402 (bisimine of M40403), M40414 (monoimine of M40403) and M40475 (free ligand of M40403) (see chromatogram in Figure 3a).Example 4HPLC Analysis of M40403 using Method 4Method 4: Analytical Column: Waters Symmetry Shield RP18, 5 μm, 250 x 4.6 mm; Mobile Phase: Acetonitrile: 0.125 M Tetrabutylammonium Chloride and 0.5 M LiCl in water (pH 6.5), 5%: 95% H20 (v/v); Flow rate: lmL/min; Detection wavelength: 265 nm. Injected 20 μl of stock solution of M40403 prepared by dissolving 1 mg in 1 ml of system A. The HPLC chromatogram of M40403 using method 4 is shown in Figure 4.The HPLC chromatogram of M40403 and related compounds using method 4 is shown in Figure 4a. Method 4 allows a separation of M40402 (bisimine of M40403), M40414 (monoimine of M40403) and M40475 (free ligand of M40403) and all diastereomers of M40403 (see chromatogram in Figure 4a).Example 5 HPLC Analysis of M40401 using Method 1

M40401 Method 1: Analytical Column: Waters YMC ODS-AQ S5 120A (4.6 x 50 mm); System A: 0.1 % trifluoroacetic acid in H2O; System B: 0.08% trifluoroacetic acid in acetonitrile; Gradient: 10-50% system B over 10 min; Flow rate: 3ml/min; Detector wavelength: 265. Injected 20 μl of stock solution of M40401 prepared by dissolving 1 mg in 1 ml of water and diluting with 1 ml of system A. The HPLC chromatogram of M40401 using method 1 is shown in Figure 5.Example 6 HPLC with various NaCl concentrations:An HPLC was taken of M40401 with various concentrations of NaCl.Analytical Column: Waters YMC 9DS-AQ S5 120 A (4.6 x 50 mm);System A: (A) H2O (no NaCl) ; (B) 0.01 M NaCl in water; (C) 0.5 M NaCl in water;System B: acetonitrile; Gradient: 0-100% system B over 10 min; Flow: 3 ml/min;Detector wavelength: 265 nm. Injected 20 μl of stock solution of M40401 prepared by dissolving 1 mg in 1 ml of system A. The HPLC chromatogram of M40401 using various NaCl concentrations is shown in Figure 6. Example 7 HPLC Analysis of M40401 using Method 2Method 2: Analytical Column: Waters YMC ODS-AQ S5 12θΛ (4.6 x 50 MM); System A: 0.5 N aqueous NaCl; System B: 1 :4 water/CH3CN; Gradient 1 : 10-50% system B over 9 min; Flow rate: 3 mL/min; Detector wavelength: 265 nm. Injected 20 μl of stock solution of M40403 prepared by dissolving 1 mg in 1 ml of system A.The HPLC chromatogram of M40401 using method 2 is shown in Figure 7. Method 2 allows a separation of M40472 (bisimine of M40401), M40473 (monoimine of M40401), free ligand of M40403 and two isomers of M40401 (M40406, M40404).Example 8HPLC Analysis of M40401 using Method 3Method 3: Analytical Column: Waters Symmetry Shield RP18, 5 m, 250 4.6 mm; Mobile Phase: Acetonitrile: 0.125 M Tetrabutylammom‘um Chloride in H20 (pH 6.5), 5: 95%) H20 (v/v); Flow rate: lmL/min; Detection wavelength: 265 nm. The HPLC chromatogram of M40401 using method 3 is shown in Figure 8.Method 3 allows a separation of M40472 (bisimine of M40401), M40473 (monoimine of M40401), free ligand of M40403 and two isomers of M40401 (M40406, M40404).Example 9 HPLC Analysis of M40401 using Method 4Method 4: Analytical Column: Waters Symmetry Shield RP18, 5 μm, 250 x 4.6 mm;Mobile Phase: Acetonitrile: 0.125 M Tetrabutylammonium Chloride and 0.5 M LiCl in water (pH 6.5), 5: 95%> H2O (v/v); Flow rate: 1 mL/min; Detection wavelength: 265 nm; Injected 20 μl of stock solution of M40401 prepared by dissolving 1 mg in 1 ml of a mobile phase. The HPLC chromatogram of M40401 using method 4 is shown in Figure 9.The HPLC chromatogram of M40401 and related compounds using method 4 is shown in Figure 9a. Method 4 allows a separation of M40472 (bisimine of M40401), M40473 (monoimine of M40401), free ligand of M40403 and two isomers of M40401 (M40406, M40404). Example 10HPLC of M40403-(HCOO“)2 Using Formate AnionAn HPLC of M40403 employing the formate anion was taken. Analytical Column: Waters YMC 9DS-AQ S5 120 A (4.6 x 50 mm); System A: 0.025 M ammonium formate in water; System B: 1 : 4 = 0.125 M ammonium formate in water/ acetonitrile; Gradient: 0-100% system B over 10 min; Flow: 3 ml/min;Detector wavelength: 265 nm. Injected 20 μl of stock solution of M40403-(Formate)2 prepared by dissolving 1 mg in 1 ml of system A. The HPLC chromatogram of M40403-(HCOO“)2 is shown in Figure 10.Example 11 HPLC of M40403-(OAc)2 Using Acetate AnionAn HPLC of M40403 employing the acetate anion was taken.Analytical Column: Waters YMC 9DS-AQ S5 120 A (4.6 x 50 mm); System A: 0.025 M ammonium acetate in water; System B: 1: 4 = 0.125 M ammonium acetate in water/ acetonitrile; Gradient: 0-100% system B over 10 min; Flow: 3 ml/min;Detector wavelength: 265 nm. Injected 20 μl of stock solution of M40403-(OAc)2 prepared by dissolving 1 mg in 1 ml of system A. The HPLC chromatogram of M40403 -(OAc)2 is shown in Figure 11.Example 12An HPLC method to separate the diastereomers of superoxide dismutase mimetic compound M40403. Four stereoisomer mixtures were prepared (Part A) as shown in Schemes 5-9 and then separated (Part B) via reversed-phase high performance liquid chromatography. Part A: Synthesis of Stereoisomers Of M40403M40403 is synthesized from its single-isomer, tetra-amine precursor M40400 in the reaction shown in Scheme 7.

M40400 M40402

M40403Scheme 7The various stereoisomers of M40403 are synthesized from the various isomers of 1,2-diaminocyclohexane which provides the chiral carbon centers in M40403. The 1,2-diaminocyclohexane isomers used to prepare the R,R+R,S) M40403 stereoisomer mixture of Set 1 are shown in Scheme 6. Similarly, the 1,2-diaminocyclohexane isomers used to prepare the (R,R+S,S) M40403 stereoisomer mixture of Set 2 are shown in Scheme 7. The 1,2-diaminocyclohexane isomers used to prepare the (R,S+R,S) M40403 stereoisomer mixture of Set 3 are shown in Scheme 8. The 1,2- diaminocyclohexane isomers used to prepare the (S,S+R,S) M40403 stereoisomer mixture of Set 4 are shown in Scheme 9. As shown in Schemes 6-9 the M40403 diastereomers are prepared by template cyclization, followed by reduction with sodium borohydride.

Scheme 8

(S.S.S.S)Scheme 9

(S.R.R.S)Scheme 10

Scheme 11Table 1


Part B: Separation of Stereoisomer MixturesChemicals, Materials, and MethodsTetrabutylammonium chloride hydrate (98%, 34,585-7) was purchased from Aldrich Chemical Company. Sodium chloride (99.6%, S-9888) was purchased from Sigma Chemical Company. All other solvents (HPLC-grade unless otherwise indicated) and reagents were purchased from Fisher Scientific and were of the finest grade available. The SymmetryShield® RP18 column (4.6 mm x 250 mm, 5 μm particle size) and its corresponding guard column were purchased from Waters Corporation. Reversed-Phase HPLC ExperimentsPreparation of Standard SolutionsHPLC Mobile phased was an aqueous solution consisting of 0.125 M tetrabutylammonium chloride (TBAC) and 0.5 M LiCl, prepared by adding tetrabutylammonium chloride hydrate (36.99 g) and solid LiCl (21.2 g) to a 1 L volumetric flask, diluting to volume with Millipore water, and inverting the flask several times to obtain a homogeneous solution. The resulting solution was filtered through a 0.45 μm nylon filter prior to use. Mobile phase B was HPLC-grade acetonitrile. Samples of each diastereoisomer set for HPLC-UN analysis were prepared at concentrations of ~ 3.0 mg/mL in a 50:50 mixture of 0.5 M LiCl in MeOH:
PATENT
SOLID STATE FORMS OF AVASOPASEM MANGANESE AND PROCESS FOR PREPARATION THEREOF
Avasopasem manganese (GC4419), has the following chemical structure:
[0003] Avasopasem manganese is a highly selective small molecule superoxide dismutase (SOD) mimetic which is being developed for the reduction of radiation-induced severe oral mucositis (SOM). The compound is described in U.S. Patent No. 8,263,568.
[0004] Polymorphism, the occurrence of different crystalline forms, is a property of some molecules and molecular complexes. A single molecule may give rise to a variety of polymorphs having distinct crystal structures and physical properties like melting point, thermal behaviors (e.g., measured by thermogravimetric analysis (“TGA”), or differential scanning calorimetry (“DSC”)), X-ray diffraction (XRD) pattern, infrared absorption fingerprint, and solid state (13C) NMR spectrum. One or more of these techniques may be used to distinguish different polymorphic forms of a compound.
[0005] Different salts and solid state forms (including solvated forms) of an active pharmaceutical ingredient may possess different properties. Such variations in the properties of different salts and solid state forms and solvates may provide a basis for improving formulation, for example, by facilitating better processing or handling characteristics, changing the
dissolution profile in a favorable direction, or improving stability (polymorph as well as chemical stability) and shelf-life. These variations in the properties of different salts and solid state forms may also offer improvements to the final dosage form, for instance, if they serve to improve bioavailability. Different salts and solid state forms and solvates of an active pharmaceutical ingredient may also give rise to a variety of polymorphs or crystalline forms, which may in turn provide additional opportunities to assess variations in the properties and characteristics of a solid active pharmaceutical ingredient.
[0006] Discovering new solid state forms and solvates of a pharmaceutical product may yield materials having desirable processing properties, such as ease of handling, ease of processing, storage stability, and ease of purification or as desirable intermediate crystal forms that facilitate conversion to other polymorphic forms. New solid state forms of a pharmaceutically useful compound can also provide an opportunity to improve the performance characteristics of a pharmaceutical product. It enlarges the repertoire of materials that a formulation scientist has available for formulation optimization, for example by providing a product with different properties, including a different crystal habit, higher crystallinity, or polymorphic stability, which may offer better processing or handling characteristics, improved dissolution profile, or improved shelf-life (chemical/physical stability). For at least these reasons, there is a need for additional solid state forms (including solvated forms) of Avasopasem manganese.
EXAMPLES
Preparation of starting materials
[00119] Avasopasem manganese can be prepared according to methods known from the literature, for example U.S. Patent No. 8,263,568. Alternatively, Avasopasem manganese can be prepared by the template method reported for the enantiomeric analogue GC4403, which has the formula:
GC4403 is disclosed in International Appl. No. WO 98/58636 (as compound SC-72325) and Riley, D.P, and Schall, O.F., Advances in Inorganic Chemistry (2007), 59, 233-263. Thus, GC4403 can be synthesized via the template route described in the literature using the chiral R,R-l,2-diamminocyclohexane [Salvemini, D., et ah, Science (1999), 286, 304-6 , and Aston, K, et al., Inorg. Chem. (2001), 40(8), 1779-89] Avasopasem manganese (GC4419) can be prepared by the same method except that the chiral R,R-l,2-diamminocyclohexane is replaced with S,S-1 ,2-diamminocyclohexane.
Example 1: Preparation of Avasopasem manganese Form AMI
[00120] Avasopasem manganese (0.1 grams) was dissolved in dichloromethane (0.5 ml) at 25-30°C in a test tube. The solution was filtered through 0.45 micron filter and the clear solution was subjected to slow solvent evaporation at 25°C by covering the tube with paraffin film with a pin hole. After, 2 days, the obtained solid was analyzed by XRD- Form AMI; as shown in Figure 1
- GlobeNewswire: Galera Therapeutics Announces Avasopasem Manganese Improved Markers of Chronic Kidney Disease in Patients Receiving Cisplatin [Link]
- Galera Therapeutics: AVASOPASEM (GC4419) [Link]
///////////AVASOPASEM, Avasopasem manganese, GC-4419, GC4419, GC 4419, M 40419, M40419; M-40419, SC 72325A, SC-72325A, SC72325A,
[Cl-].[Cl-].[Mn++].C1CC[C@@H]2NCC3=CC=CC(CN[C@H]4CCCC[C@@H]4NCCN[C@H]2C1)=N3

NEW DRUG APPROVALS
ONE TIME
$10.00
Belzutifan
Belzutifan
CAS 1672668-24-4
383.34 g·mol−1 C17H12F3NO4S
3-[[(1S,2S,3R)-2,3-difluoro-1-hydroxy-7-methylsulfonyl-2,3-dihydro-1H-inden-4-yl]oxy]-5-fluorobenzonitrile
MK-6482, PT-2977, UNII-7K28NB895L, 7K28NB895L
3-[(1S,2S,3R)-2,3-Difluoro-1-hydroxy-7-methylsulfonylindan-4-yl]oxy-5-fluorobenzonitrile
GTPL11251, PT 2977 [WHO-DD], BDBM373040
FDA APPROVED 8/13/2021, Welireg
To treat von Hippel-Lindau disease under certain conditions
EMA Drug Information
| Disease/Condition | Treatment of von Hippel-Lindau disease |
|---|---|
| Active Substance | 3-(((1S,2S,3R)-2,3-difluoro-1-hydroxy-7-(methylsulfonyl)-2,3-dihydro-1H-inden-4-yl)oxy)-5-fluorobenzonitrile |
| Status of Orphan Designation | Positive |
| Decision Date | 2020-08-21 |
FDA approves belzutifan for cancers associated with von Hippel-Lindau disease
On August 13, 2021, the Food and Drug Administration approved belzutifan (Welireg, Merck), a hypoxia-inducible factor inhibitor for adult patients with von Hippel-Lindau (VHL) disease who require therapy for associated renal cell carcinoma (RCC), central nervous system (CNS) hemangioblastomas, or pancreatic neuroendocrine tumors (pNET), not requiring immediate surgery.
Belzutifan was investigated in the ongoing Study 004 (NCT03401788), an open-label clinical trial in 61 patients with VHL-associated RCC (VHL-RCC) diagnosed based on a VHL germline alteration and with at least one measurable solid tumor localized to the kidney. Enrolled patients had other VHL-associated tumors, including CNS hemangioblastomas and pNET. Patients received belzutifan 120 mg once daily until disease progression or unacceptable toxicity.
The primary efficacy endpoint was overall response rate (ORR) measured by radiology assessment, as assessed by an independent review committee using RECIST v1.1. Additional efficacy endpoints included duration of response (DoR), and time- to- response (TTR). An ORR of 49% (95% CI:36, 62) was reported in patients with VHL-associated RCC. All patients with VHL-RCC with a response were followed for a minimum of 18 months from the start of treatment. The median DoR was not reached; 56% of responders had DoR ≥ 12 months and a median TTR of 8 months. In patients with other VHL-associated non-RCC tumors, 24 patients with measurable CNS hemangioblastomas had an ORR of 63% and 12 patients with measurable pNET had an ORR of 83%. Median DoR was not reached, with 73% and 50% of patients having response durations ≥ 12 months for CNS hemangioblastomas and pNET, respectively.
The most common adverse reactions, including laboratory abnormalities, reported in ≥ 20% of patients who received belzutifan were decreased hemoglobin, anemia, fatigue, increased creatinine, headache, dizziness, increased glucose, and nausea. Anemia and hypoxia from belzutifan use can be severe. In Study 004, anemia occurred in 90% of patients and 7% had Grade 3 anemia. Patients should be transfused as clinically indicated. The use of erythropoiesis stimulating agents for treatment of anemia is not recommended in patients treated with belzutifan. In Study 004, hypoxia occurred in 1.6% of patients. Belzutifan can render some hormonal contraceptives ineffective, and belzutifan exposure during pregnancy can cause embryo-fetal harm.
The recommended belzutifan dosage is 120 mg administered orally once daily with or without food.
View full prescribing information for Welireg.
This review was conducted under Project Orbis, an initiative of the FDA Oncology Center of Excellence. Project Orbis provides a framework for concurrent submission and review of oncology drugs among international partners. For this review, FDA collaborated with the Australian Therapeutic Goods Administration (TGA), Health Canada, and the Medicines and Healthcare products Regulatory Agency (MHRA) of the United Kingdom. The application reviews are ongoing at the other regulatory agencies.
This review used the Real-Time Oncology Review (RTOR) pilot program, which streamlined data submission prior to the filing of the entire clinical application, as well as the Assessment Aid and the Product Quality Assessment Aid, voluntary submissions from the applicant to facilitate the FDA’s assessment. The FDA approved this application approximately 1 month ahead of the FDA goal date.
This application was granted priority review for this indication. A description of FDA expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.
Belzutifan, sold under the brand name Welireg, is a medication used for the treatment of von Hippel–Lindau disease-associated renal cell carcinoma.[1][2][3][4][5][6] It is taken by mouth.[1]
The most common side effects include decreased hemoglobin, anemia, fatigue, increased creatinine, headache, dizziness, increased glucose, and nausea.[2]
Belzutifan is an hypoxia-inducible factor-2 alpha (HIF-2α) inhibitor.[1][2][7]
Belzutifan is the first drug to be awarded an “innovation passport” from the UK Medicines and Healthcare products Regulatory Agency (MHRA).[8][4] Belzutifan was approved for medical use in the United States in August 2021.[2][9] Belzutifan is the first hypoxia-inducible factor-2 alpha inhibitor therapy approved in the U.S.[9]
Medical uses
Belzutifan is indicated for treatment of adults with von Hippel-Lindau (VHL) disease who require therapy for associated renal cell carcinoma (RCC), central nervous system (CNS) hemangioblastomas, or pancreatic neuroendocrine tumors (pNET), not requiring immediate surgery.[2]
PATENT
WO 2019191227
https://patents.google.com/patent/WO2019191227A1/en

PATENT
WO 2015035223
https://patents.google.com/patent/WO2015035223A1/enScheme 9


[01237] 3-r(15,25.3 ?‘)-2.3-difluoro-l-hvdroxy-7-methylsulfonyl-indan-4- νΠοχν-5-fluoro-benzonitrile (Compound 289)[01238] Step A: r(15.2/?V4- -cvano-5-fluoro-phenoxy)-2-fluoro-7- methylsulfonyl-indan-l -vH acetate: To a stirred solution of 3-fluoro-5-[(15,27?)-2-fluoro-l – hydroxy-7-methylsulfonyl-indan-4-yl]oxy-benzonitrile (2.00 g, 5.47 mmol) in DCM (27 mL) was added 4-(dimethylamino)pyridine (0.2 g, 1.64 mmol) and triethylamine (1.53 mL, 10.9 mmol). Acetic anhydride (1.00 mL, 10.9 mmol) was added dropwise at 0 °C under nitrogen. The reaction mixture was stirred at ambient temperature overnight. The reaction mixture was diluted with DCM, washed with saturated aqueous NaHC03 and brine, dried andconcentrated. The residue was purified by flash chromatography on silica gel (20-40% EtOAc/hexane) to give [(lS,2/?)-4-(3-cyano-5-fluoro-phenoxy)-2-fluoro-7-methylsulfonyl- indan-l-yl] acetate (1.95 g, 87%). LCMS ESI (+) m/z 408 (M+H).[01239] Step B: Γ( 1 .25.35)-3-bromo-4-(3-cvano-5-fluoro-Dhenoxy)-2-fluoro- 7-methylsulfonyl-indan-l-yll acetate and f(15.25,3/?)-3-bromo-4-(3-cyano-5-fluoro- phenoxy)-2-fluoro-7-methylsulfonyl-indan-l -yl1 acetate: To a stirred solution of [(15,2/?)-4- (3-cyano-5-fluoro-phenoxy)-2-fluoro-7-methylsulfonyl-indan-] -yl] acetate (1.95 g, 4.79 mmol) in 1 ,2-dichloroethane (24 mL) was added N-bromosuccinimide (0.94 g, 5.27 mmol) and 2,2′-azobisisobutyronitrile (8 mg, 0.05 mmol). The reaction mixture was heated at 80 °C for 3 hours. After cooling, the reaction mixture was diluted with DCM, washed with saturated aqueous NaHC03 and brine, dried and concentrated. The residue was purified by column chromatography on silica gel (20-30% EtOAc hexane) to give [(lS,2S,3S)-3-bromo- 4-(3-cyano-5-fluoro-phenoxy)-2-fluoro-7-methylsulfonyl-indan-l-yl] acetate (1 .52 g, 65%). LCMS ESI (+) m/z 486, 488 (M+H). Further elution with 30-50% EtOAc/hexane gave the more polar product [(lS,2S,3/?)-3-bromo-4-(3-cyano-5-fluoro-phenoxy)-2-fluoro-7- methylsulfonyl-indan-l -yl] acetate (0.583 g, 25%). LCMS ESI (+) m/z 486, 488 (M+H). [01240] Step C: rd5.2^.3 V4-(3-cvano-5-fluoro-phenoxy)-2-fluoro-3- hvdroxy-7-methylsulfonyl-indan- 1 -yll acetate: To a combined mixture of [(1 ,25,35)-3- bromo-4-(3-cyano-5-fluoro-phenoxy)-2-fluoro-7-methylsulfonyl-indan-l -yl] acetate and [( 15,2S,3/?)-3-bromo-4-(3-cyano-5-fluoro-phenoxy)-2-fluoro-7-methylsulfonyl-indan- 1 -yl] acetate prepared in Step B (2.05 g, 4.22 mmol) were added 1 ,2-dimethoxyethane (28 mL) and water (0.050 mL) followed by silver perchlorate hydrate (1.42 g, 6.32 mmol). The reaction mixture was heated at 70 °C for 2 hours. After cooling, the reaction mixture was diluted with EtOAc and filtered through Celite. The filtrate was washed with water and brine, dried and concentrated. The residue was purified by flash chromatography on silica gel (20-50%) to give [(15,2/?,35)-4-(3-cyano-5-fluoro-phenoxy)-2-fluoro-3-hydroxy-7-methylsulfonyl-indan- 1 -yl] acetate (0.416 g, 23%) as the less polar product. LCMS ESI (+) m/z 441 (M+NH4+). Further elution with 60% EtOAc/hexane gave [(15,2/?,3R)-4-(3-cyano-5-fluoro-phenoxy)-2- fluoro-3-hydroxy-7-methylsulfonyl-indan-l-yl] acetate (0.58 g, 32 %). LCMS ESI (+) m/z 441 (M+NH4+).[01241] Step D: r(15.25.3/? -4-(3-cvano-5-fluoro-phenoxyV2.3-difluoro-7- methylsulfonyl-indan-l-vH acetate: To a stirred solution of [(15,2/?,35)-4-(3-cyano-5-fluoro- phenoxy)-2-fluoro-3-hydroxy-7-methylsulfonyl-indan-l-yl] acetate (416 mg, 0.98 mmol) in DCM (10 mL) was added (diethylamino)sulfur trifluoride (DAST) (0.26 mL, 2.0 mmol) at – 78 °C under nitrogen. The reaction mixture was allowed to warm to 0 °C and stirred for 15 minutes. The reaction was quenched by saturated aqueous NaHC03. The mixture was partitioned between EtOAc and water. The aqueous layer was extracted with EtOAc. The combined organic layers were washed with brine, dried and concentrated. The residue was purified by flash chromatography on silica gel (20-40% EtOAc/hexane) to give [(15,25,3/?)- 4-(3-cyano-5-fluoro-phenoxy)-2,3-difluoro-7-methylsulfonyl-indan-l -yl] acetate (310 mg, 74%). LCMS ESI (+) m/z 426 (M+H).[01242] Step E: 3-r(15.25.3^)-2.3-difluoro-l-hvdroxy-7-methylsulfonyl-indan-4-vnoxy-5-fluoro-benzonitrile (Compound 289): Prepared as described in Example 288 Step F substituting [(l ?)-4-(3-cyano-5-fluoro-phenoxy)-3,3-difluoro-7-methylsulfonyl-indan- 1-yl] acetate with [(15,25,3/?)-4-(3-cyano-5-fluoro-phenoxy)-2,3-difluoro-7-methylsulfonyl- indan-l-yl] acetate. LCMS ESI (+) m/z 384 (M+H); Ή NMR (400 MHz, CDC13): δ 8.13 (d, 1H), 7.31-7.25 (m, 1 H), 7.23-7.19 (m, 1 H), 7.14-7.09 (m, 1H), 7.04 (d, 1H), 6.09-5.91 (m, 1 H), 5.87-5.80 (m, 1 H), 5.25-5.05 (m, 1H), 3.32 (s, 3H), 2.95 (d, 1H).
PatentWO 2016145032https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016145032&tab=PCTDESCRIPTIONCOMPD 289
PATENTWO 2016145045WO 2016168510WO 2016057242WO 2019191227
| PMID | Publication Date | Title | Journal |
|---|---|---|---|
| 31282155 | 2019-08-08 | 3-[(1S,2S,3R)-2,3-Difluoro-1-hydroxy-7-methylsulfonylindan-4-yl]oxy-5-fluorobenzonitrile (PT2977), a Hypoxia-Inducible Factor 2α (HIF-2α) Inhibitor for the Treatment of Clear Cell Renal Cell Carcinoma | Journal of medicinal chemistry |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| WO-2020146758-A1 | Methods to treat mitochondrial-associated dysfunctions or diseases | 2019-01-10 | |
| WO-2020092100-A1 | Solid dispersions and pharmaceutical compositions comprising a substituted indane and methods for the preparation and use thereof | 2018-10-30 | |
| TW-202003430-A | Methods of reducing inflammation of the digestive system with inhibitors of HIF-2-alpha | 2018-03-28 | |
| WO-2019191227-A1 | Methods of reducing inflammation of the digestive system with inhibitors of hif-2-alpha | 2018-03-28 | |
| US-2019151347-A1 | Compositions and methods of modulating hif-2a; to improve muscle generation and repair | 2017-11-20 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| US-2019048421-A1 | Biomarkers of response to hif-2-alpha inhibition in cancer and methods for the use thereof | 2015-09-21 | |
| WO-2017053192-A1 | Biomarkers of response to hif-2-alpha inhibition in cancer and methods for the use thereof | 2015-09-21 | |
| US-10335388-B2 | Combination therapy of a HIF-2-alpha inhibitor and an immunotherapeutic agent and uses thereof | 2015-04-17 | 2019-07-02 |
| US-2018140569-A1 | Combination therapy of a hif-2-alpha inhibitor and an immunotherapeutic agent and uses thereof | 2015-04-17 | |
| US-2019282535-A1 | Combination therapy of a hif-2-alpha inhibitor and an immunotherapeutic agent and uses thereof | 2015-04-17 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| WO-2016168510-A1 | Combination therapy of a hif-2-alpha inhibitor and an immunotherapeutic agent and uses thereof | 2015-04-17 | |
| US-10786480-B2 | Combination therapy of a HIF-2-α inhibitor and an immunotherapeutic agent and uses thereof | 2015-04-17 | 2020-09-29 |
| US-10278942-B2 | Compositions for use in treating pulmonary arterial hypertension | 2015-03-11 | 2019-05-07 |
| US-10512626-B2 | Compositions for use in treating glioblastoma | 2015-03-11 | 2019-12-24 |
| US-2018042884-A1 | Compositions for use in treating glioblastoma | 2015-03-11 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| US-2018177754-A1 | Compositions for use in treating pulmonary arterial hypertension | 2015-03-11 | |
| US-2019015377-A1 | Compositions for Use in Treating Pulmonary Arterial Hypertension | 2015-03-11 | |
| WO-2016145032-A1 | Compositions for use in treating pulmonary arterial hypertension | 2015-03-11 | |
| WO-2016145045-A1 | Compositions for use in treating glioblastoma | 2015-03-11 | |
| US-10098878-B2 | HIF-2α inhibitors for treating iron overload disorders | 2014-10-10 | 2018-10-16 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| US-2020190031-A1 | Aryl ethers and uses thereof | 2013-09-09 | |
| US-9896418-B2 | Aryl ethers and uses thereof | 2013-09-09 | 2018-02-20 |
| US-9908845-B2 | Aryl ethers and uses thereof | 2013-09-09 | 2018-03-06 |
| US-9969689-B2 | Aryl ethers and uses thereof | 2013-09-09 | 2018-05-15 |
| WO-2015035223-A1 | Aryl ethers and uses thereof | 2013-09-09 |
Merck Team Wins 2021 Pete Dunn Award
05-17-2021 10:52 AM

The ACS Green Chemistry Institute (GCI) Pharmaceutical Roundtable honors the work of Stephen Dalby, François Lévesque, Cecilia Bottecchia and Jonathan McMullen at Merck with the 2021 Peter J. Dunn Award for Green Chemistry & Engineering Impact in the Pharmaceutical Industry. The team’s innovation is titled, “Greener Manufacturing of Belzutifan (MK-6482) Featuring a Photo-Flow Bromination.”
Belzutifan is an important new drug used in the treatment of cancer and other non-oncology diseases. Acquired by Merck in 2019 through the purchase of Peloton Therapeutics, a new, greener manufacturing process for its synthesis was needed. Over the next 18 months, the team developed a more direct route from commodity chemical to API, employed new reaction conditions, particularly in the oxidation sequence, and incorporated new technology, photo-flow.
Despite this accelerated timeline, the team achieved a five-fold improvement in overall yield with a commensurate 73% reduction in process mass intensity (PMI) compared to the original route. Notably, the Merck team also developed a visible light-initiated radical bromination performed in flow. According to the L.-C. Campeau, Executive Director and Head of Process Chemistry and Discovery Process Chemistry at Merck, this is the “first example of a photo-flow reaction run on manufacturing scale at Merck and represents the linchpin of the synthesis.”
The improved process for Belzutifan, which is expected to launch this year, will reduce the waste associated with its manufacture and is aligned with Merck’s corporate sustainability goals.
“The Merck team delivered an excellent example of the application of innovative technologies to develop a more sustainable synthesis of the pharmaceutically-active compound, Belzutifan,” comments Paul Richardson, Director of Oncology and Chemical Synthesis at Pfizer and Co-Chair of the ACS GCI Pharmaceutical Roundtable. “Using the guiding principles of green chemistry, for example, in the use of catalysis and a relatively benign reaction media, further illustrate the Merck team’s work as worthy of recognition for the 2021 Peter Dunn Award.”
The award will be presented at the June 11 GC&E Friday, part of the 25th Annual Green Chemistry & Engineering Conference. During this session from 10 a.m. – 1 p.m., Stephen Dalby & Jon MacMullen will be discussing the details of this innovative process.
References
- ^ Jump up to:a b c d https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/215383s000lbl.pdf
- ^ Jump up to:a b c d e f “FDA approves belzutifan for cancers associated with von Hippel-Lindau”. U.S. Food and Drug Administration (FDA). 13 August 2021. Retrieved 13 August 2021.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “Belzutifan”. SPS – Specialist Pharmacy Service. 18 March 2021. Retrieved 25 April 2021.
- ^ Jump up to:a b “MHRA awards first ‘innovation passport’ under new pathway”. RAPS (Press release). Retrieved 25 April 2021.
- ^ “Merck Receives Priority Review From FDA for New Drug Application for HIF-2α Inhibitor Belzutifan (MK-6482)” (Press release). Merck. 16 March 2016. Retrieved 25 April 2021 – via Business Wire.
- ^ “FDA Grants Priority Review to Belzutifan for von Hippel-Lindau Disease–Associated RCC”. Cancer Network. Retrieved 26 April 2021.
- ^ {{cite journal |vauthors=Choueiri TK, Bauer TM, Papadopoulos KP, Plimack ER, Merchan JR, McDermott DF, Michaelson MD, Appleman LJ, Thamake S, Perini RF, Zojwalla NJ, Jonasch E | display-authors=6 |title=Inhibition of hypoxia-inducible factor-2α in renal cell carcinoma with belzutifan: a phase 1 trial and biomarker analysis |journal=Nat Med |volume= |issue= |pages= |date=April 2021 |pmid=33888901 |doi=10.1038/s41591-021-01324-7 }
- ^ “First Innovation Passport awarded to help support development and access to cutting-edge medicines”. Medicines and Healthcare products Regulatory Agency (MHRA) (Press release). 26 February 2021. Retrieved 14 August 2021.
- ^ Jump up to:a b “FDA Approves Merck’s Hypoxia-Inducible Factor-2 Alpha (HIF-2α) Inhibitor Welireg (belzutifan) for the Treatment of Patients With Certain Types of Von Hippel-Lindau (VHL) Disease-Associated Tumors” (Press release). Merck. 13 August 2021. Retrieved 13 August 2021 – via Business Wire.
External links
- “Belzutifan”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT04195750 for “A Study of Belzutifan (MK-6482) Versus Everolimus in Participants With Advanced Renal Cell Carcinoma (MK-6482-005)” at ClinicalTrials.gov
- Clinical trial number NCT03401788 for “A Phase 2 Study of Belzutifan (PT2977, MK-6482) for the Treatment of Von Hippel Lindau (VHL) Disease-Associated Renal Cell Carcinoma (RCC) (MK-6482-004)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Pronunciation | bell-ZOO-ti-fan |
| Trade names | Welireg |
| Other names | MK-6482, PT2977 |
| License data | US DailyMed: Belzutifan |
| Routes of administration | By mouth |
| Drug class | Antineoplastic |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1][2] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1672668-24-4 [KEGG] |
| PubChem CID | 117947097 |
| ChemSpider | 59053536 |
| UNII | 7K28NB895L |
| KEGG | D11954 |
| ChEMBL | ChEMBL4585668 |
| PDB ligand | 72Q (PDBe, RCSB PDB) |
| Chemical and physical data | |
| Formula | C17H12F3NO4S |
| Molar mass | 383.34 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
/////////Belzutifan, Welireg, FDA 2021, APPROVALS 2021, MK 6482, PT 977, Antineoplastic
CS(=O)(=O)C1=C2C(C(C(C2=C(C=C1)OC3=CC(=CC(=C3)C#N)F)F)F)O
NEW DRUG APPROVALS
ONE TIME
$10.00
Naftopidil, KT 611
Naftopidil
1-[4-(2-methoxyphenyl)piperazin-1-yl]-3-naphthalen-1-yloxypropan-2-ol
C24H28N2O3, 392.49
CAS 57149-07-2
1-(4-(2-methoxyphenyl)piperazin-1-yl)-3-(naphthalen-1-yloxy)propan-2-ol
Naftopidil (Flivas), BM-15275NaftopidilCAS Registry Number: 57149-07-2
CAS Name: 4-(2-Methoxyphenyl)-a-[(1-naphthalenyloxy)methyl]-1-piperazineethanolAdditional Names:RS-1-[4-(2-methoxyphenyl)-1-piperazinyl]-3-(1-naphthoxy)-2-propanol; 1-(2-methoxyphenyl)-4-[3-(naphth-1-yloxy)-2-hydroxypropyl]-piperazine
Manufacturers’ Codes: KT-611Trademarks: Avishot (Kanebo); Flivas (Asahi)Molecular Formula: C24H28N2O3Molecular Weight: 392.49Percent Composition: C 73.44%, H 7.19%, N 7.14%, O 12.23%Literature References: a1-Adrenergic blocker and serotonin (5HT1A) receptor agonist. Prepn: E. C. Witte et al.,DE2408804; eidem,US3997666 (1975, 1976 both to Boehringer Mann.). Clinical pharmacodynamics: R. Kirsten et al.,Eur. J. Clin. Pharmacol.46, 271 (1994). Clinical pharmacokinetics: M. J. G. Farthing et al.,Postgrad. Med. J.70, 363 (1994). HPLC determn in human plasma: G. Niebch et al.,J. Chromatogr.534, 247 (1990). Clinical evaluation in BPH: K. Yasuda et al.,Prostate25, 46 (1994). Review of pharmacology and clinical experience: H. M. Himmel, Cardiovasc. Drug Rev.12, 32-47 (1994).
Properties: Crystals from isopropanol, mp 125-126°; also reported as colorless crystals, mp 125-129°. Insol in water. Partition coefficient (octanol/water): 75. LD50 in mice, rats (g/kg): 1.3, 6.4 orally (Himmel).Melting point: mp 125-126°; mp 125-129°Log P: Partition coefficient (octanol/water): 75Toxicity data: LD50 in mice, rats (g/kg): 1.3, 6.4 orally (Himmel)
Derivative Type: DihydrochlorideCAS Registry Number: 57149-08-3Molecular Formula: C24H28N2O3.2HClMolecular Weight: 465.41Percent Composition: C 61.94%, H 6.50%, N 6.02%, O 10.31%, Cl 15.24%Properties: Crystals from methanol/ethanol (1:2), mp 212-213°.Melting point: mp 212-213°
Therap-Cat: Antihypertensive; a-blocker in treatment of symptomatic benign prostate hypertrophy.Keywords: a-Adrenergic Blocker; Antihypertensive.
Naftopidil (INN, marketed under the brand name Flivas) is a drug used in benign prostatic hypertrophy which acts as a selective α1-adrenergic receptor antagonist or alpha blocker.[1]
PATENT
DE 2408804
CN 101671317
CN 102816136
JP 2013023467
JP 2014118360
IN 2011CH00466
US 20150353473
CN 104744405
IN 2013CH06042
IN 2012DE02071
JP 2016044182
PAPER
ChemMedChem (2009), 4(3), 393-9.
The Journal of organic chemistry (2013), 78(18), 9076-84.
e-EROS Encyclopedia of Reagents for Organic Synthesis (2014), 1-5
European journal of medicinal chemistry (2015), 96, 83-91.
Bioorganic & medicinal chemistry letters (2018), 28(9), 1534-1539.
ChemistrySelect (2019), 4(26), 7745-7750.
Green Chemistry (2019), 21(16), 4422-4433. |
PAPER
https://www.scielo.br/j/jbchs/a/q5qDxfT9mSwtL9hhQYxyhgs/?lang=en#

(S)-1-(4-(2-Methoxyphenyl)piperazin-1-yl)-3-(naphthalene1-yloxy)propan-2-ol (2b) To a solution of epoxide 8b (0.1 g, 0.5 mmol) in anhydrous 2-propanol (10 mL) was added 1-(2-methoxyphenyl) piperazine (0.096 g, 0.5 mmol) and the reaction mixture was refluxed for 32 h. After completion of reaction, the solvent was removed under reduced pressure and purification was carried out by flash column chromatography (230-400 mesh silica). The EtOAc:petroleum ether (60:40) was used as solvent system for elution, it afforded the (S)-(+)-naftopidil 2b as a yellow solid (0.156 g, 80%); mp 126-127°C; [α]D 25 +4.3o (c 1.55, MeOH);3 [α]D 25 +4.5o (c 1.5, MeOH); IR (CHCl3) νmax/cm-1 3403, 3031, 2977, 2907, 1261, 1225; 1 H NMR (300 MHz, CDCl3) d 2.58-2.70 (m, 4H, N-CH2), 2.80-2.85 (m, 2H, CH2N), 3.03-3.51 (m, 4H, NCH2), 3.51 (bs, 1H, OH), 3.75 (s, 3H, OCH3), 4.02-4.10 (m, 2H, OCH2), 4.19-4.23 (m, 1H, CH), 6.72-6.85 (m, 2H, Ar-H), 6.83-6.85 (d, 2H, J 3.9 Hz, Ar-H), 6.87-6.95 (1H, m, Ar-H), 7.14-7.29 (1H, m, Ar-H), 7.33-7.42 (3H, m, Ar-H), 7.69-7.72 (m, 1H, Ar-H), 8.19-8.22 (m, 1H, Ar-H); 13C NMR (75 MHz, CDCl3) d 50.44 (NCH2), 53.43 (NCH2), 55.17 (OCH3), 60.85 (CH2N), 65.47 (CH), 70.36 (OCH2), 104.73 (Ar), 111.03 (Ar), 118.05 (Ar), 120.39 (Ar), 120.83 (Ar), 121.78 (Ar), 122.91 (Ar), 125.07 (Ar), 125.41 (Ar), 125.67 (Ar), 126.26 (Ar), 127.32 (Ar), 134.31 (Ar), 140.87 (Ar), 152.04 (Ar), 154.21 (Ar); LC-MS m/z 393.36 (M+ + 1), 415.36 (M+ + Na); For compound 2a: [α]D 25 -10.6o (c 1, MeOH,);6 [α]D 25 -11.7o (c 1, MeOH).
PATENT
CN 1473820
PATENT
WO 2018026371
https://patents.google.com/patent/WO2018026371A1/en
PATENT
JP-2021104982
Naftopidil monohydrochloride dihydrate and its use for the preparation of naftopidil , which is known as an ameliorating agent for dysuria associated with benign prostatic hyperplasia.Naftopidil is known as an ameliorating agent for dysuria associated with benign prostatic hyperplasia. Since naftopidil is administered as a free form, there is a need for a method for preparing the free form that can be obtained efficiently and with high purity.
Japanese Unexamined Patent Publication No. 50-12186 (Patent Document 1) discloses a method for preparing naftopidil, and states that naftopidil was obtained in a yield of 29% to 79% in the examples thereof. In particular, in Example 3, naftopidil is obtained via naftopidil hydrochloride anhydride, but the yield is 49%, and the purity is not described.
Japanese Patent Application Laid-Open No. 2013-23467 (Patent Document 2) reacts 1- (2-methoxyphenyl) piperazin with 2-[(1-naphthyloxy) methyl] oxylane to obtain crude naftopidil, which is obtained as toluene. Discloses a method for obtaining purified naftopidil from water and water, as well as a mixed solvent of toluene and methanol. In this method, the yield of crude naftopidil did not reach 80%, and the purity after two purification operations using toluene and water, and then toluene and methanol was said to be 99.62% at the highest. ing. In this method, crude naftopidil is not chlorinated with hydrochloric acid.
In Indian patent application 466 / CHE / 2011 (Patent Document 3), crude naftopidil was recrystallized from ethyl acetate to obtain naftopidil in a yield of 79% and a purity of 99.90%, and further recrystallized from methanol to obtain purity. It discloses a method of obtaining 99.99% naftopidil. Even with this method, crude naftopidil is not chlorinated with hydrochloric acid.
Indian Patent Application 2071 / DEL / 2012 (Patent Document 4) discloses a method for producing green chemical naftopidil using metal nanoparticles. Here, naftopidil is purified by column chromatography using silica gel to obtain naftopidil in a yield of 63%, but the purity is not disclosed.patcit 1: Japanese Patent Application
Laid-Open No. 50-12186 patcit 2: Japanese Patent Application Laid-Open No.
2013-23467 patcit 3: Indian Patent Application 466 / CHE / 2011
patcit 4: Indian Patent Application 2071 / DEL / 2012Production
of Naftopidil Monohydrochloride Dihydrate The naftopidil monohydrochloride dihydrate according to the present invention is preferably prepared according to the following scheme.
[Chem. 2]
That is, it can be obtained by reacting 2-[(1-naphthyloxy) methyl] oxylane with 1- (2-methoxyphenyl) piperazine by adding a solvent such as toluene, and then adding / presenting hydrochloric acid. ..The present invention will be described in more detail with reference to the following examples. The reactions in the examples below, and the numbers given to the compounds, are as shown in the scheme below.
[Chem. 3]
Example 1
100 g of 1 -naphthol [1] was dissolved in chloromethyloxylan [2], and a sodium methoxide methanol solution was added dropwise. After completion of the reaction, the reaction was washed with water and the organic layer was concentrated to obtain 2-[(1-naphthyloxy) methyl] oxylan [3] (yield 89%).
Example 2
A toluene solution of 1- (2-methoxyphenyl) piperazin [4] was added dropwise to a toluene solution of 5.0 g of 2-[(1-naphthyloxy) methyl] oxylan [3]. After completion of the reaction, the mixture was washed with water and cooled by adding hydrochloric acid. After the suspension is filtered off, it is dried and (2RS) -1- [4- (2-methoxyphenyl) piperazin-1-yl] -3- (naphthalene-1-yloxy) propan-2-ol. Hydrochloride dihydrate [5] was obtained (yield 95%).
Example 3
A toluene solution of 1- (2-methoxyphenyl) piperazin [4] was added dropwise to a toluene solution of 5.0 g of 2-[(1-naphthyloxy) methyl] oxylan [3]. After completion of the reaction, the mixture was washed with water, methanol and hydrochloric acid were added to separate the liquids, and the mixture was cooled. After the suspension is filtered off, it is dried and (2RS) -1- [4- (2-methoxyphenyl) piperazin-1-yl] -3- (naphthalene-1-yloxy) propan-2-ol. Hydrochloride dihydrate [5] was obtained (yield 81%).
Example 4
A toluene solution of 1- (2-methoxyphenyl) piperazin [4] was added dropwise to a toluene solution of 5.0 g of 2-[(1-naphthyloxy) methyl] oxylan [3]. After completion of the reaction, the mixture was washed with water, methanol and hydrochloric acid were added, and the mixture was cooled. After the suspension is filtered off, it is dried and (2RS) -1- [4- (2-methoxyphenyl) piperazin-1-yl] -3- (naphthalene-1-yloxy) propan-2-ol. Hydrochloride dihydrate [5] was obtained (yield 86%).
Example 5
A toluene solution of 1- (2-methoxyphenyl) piperazin [4] was added dropwise to a toluene solution of 100 g of 2-[(1-naphthyloxy) methyl] oxylan [3]. After completion of the reaction, the mixture was washed with water, methanol and hydrochloric acid were added, and the mixture was cooled. After the suspension is filtered off, it is dried and (2RS) -1- [4- (2-methoxyphenyl) piperazin-1-yl] -3- (naphthalene-1-yloxy) propan-2-ol. Hydrochloride dihydrate [5] was obtained (yield 92%).
Example 6
(2RS) -1- [4- (2-methoxyphenyl) piperazin-1-yl] -3- (naphthalene-1-yloxy) propan-2-ol monohydrochloride dihydrate [5 ] Toluene and sodium hydroxide aqueous solution were added to 7.0 g. The organic layer was washed with water and concentrated, and then metall and acetonitrile were added and cooled. After the suspension is filtered off, it is dried and (2RS) -1- [4- (2-methoxyphenyl) piperazin-1-yl] -3- (naphthalene-1-yloxy) propan-2-ol [ 6] was obtained (yield 82%, chemical purity 99.98%).
Example 7
(2RS) -1- [4- (2-methoxyphenyl) piperazin-1-yl] -3- (naphthalene-1-yloxy) propan-2-ol monohydrochloride dihydrate [5 ] Toluene and an aqueous sodium hydroxide solution were added to 12.0 g. The organic layer was washed with water and concentrated, then metall was added and cooled. After the suspension is filtered off, it is dried and (2RS) -1- [4- (2-methoxyphenyl) piperazin-1-yl] -3- (naphthalene-1-yloxy) propan-2-ol [ 6] was obtained (yield 90%, chemical purity 99.99%).
Example 8
(2RS) -1- [4- (2-Methoxyphenyl) piperazin-1-yl] -3- (naphthalene-1-yloxy) propan-2-ol monohydrochloride dihydrate [5 ] Toluene, methanol, and potassium hydroxide aqueous solution were added to 116 g. The organic layer was washed with water and concentrated, then 2-propanol was added and cooled. After the suspension is filtered off, it is dried and (2RS) -1- [4- (2-methoxyphenyl) piperazin-1-yl] -3- (naphthalene-1-yloxy) propan-2-ol [ 6] was obtained (yield 90%, chemical purity 99.98%).
Comparative Example 1
A toluene solution of 1- (2-methoxyphenyl) piperazin [4] was added dropwise to a 10.0 g toluene solution of 2-[(1-naphthyloxy) methyl] oxylan [3]. After completion of the reaction, the mixture was washed with water and cooled. After the suspension is filtered off, it is dried and (2RS) -1- [4- (2-methoxyphenyl) piperazin-1-yl] -3- (naphthalene-1-yloxy) propan-2-ol [ 6] Crude crystals were obtained (yield 89%).
Comparative Example 2
(2RS) -1- [4- (2-methoxyphenyl) piperazin-1-yl] -3- (naphthalene-1-yloxy) propan-2-ol [6] obtained in Comparative Example 1. Methoxyol and acetonitrile were added to 6.0 g of the crude crystals of the above, and the mixture was cooled. After the suspension is filtered off, it is dried and (2RS) -1- [4- (2-methoxyphenyl) piperazin-1-yl] -3- (naphthalene-1-yloxy) propan-2-ol [ 6] was obtained (yield 85%, chemical purity 99.96%).
Naftopidil one identification hydrochloride dihydrate
(1) water and HCl content
mosquito – Le Fischer – water content value measured by the law was 7.3% to 7.5%. The amount of HCl measured by neutralization titration was 8.0% to 8.1%. Determined from these naftopidil: HCl: H 2 When calculating these molar ratios from O weight ratio of approximately 1: 1: 2. From this, it was judged that naftopidil monohydrochloride dihydrate was obtained.
(2) Powder X-ray Diffraction
The chart of the results of powder X-ray diffraction (Cu-Kα) of naftopidil monohydrochloride dihydrate was as shown in FIG. For reference, a chart of naftopidil is shown as FIG.
(3) Differential Thermal Analysis / Thermogravimetric Analysis
(TG / DTA) The chart of the results of differential thermal analysis / thermogravimetric analysis (TG / DTA) of naphthopidyl monohydrochloride dihydrate is as shown in FIG. rice field. Here, the measurement conditions were such that the heating rate was 5 ° C./min. For reference, a chart of naftopidil is shown as FIG.
PAPERShivani; Journal of Organic Chemistry 2007, V72(10), P3713-3722 https://pubs.acs.org/doi/10.1021/jo062674j
References
- ^ Sakai H, Igawa T, Onita T, Furukawa M, Hakariya T, Hayashi M, Matsuya F, Shida Y, Nishimura N, Yogi Y, Tsurusaki T, Takehara K, Nomata K, Shiraishi K, Shono T, Aoki D, Kanetake H (2011). “Efficacy of naftopidil in patients with overactive bladder associated with benign prostatic hyperplasia: prospective randomized controlled study to compare differences in efficacy between morning and evening medication”. Hinyokika Kiyo. 57 (1): 7–13. PMID 21304253.
| Clinical data | |
|---|---|
| Trade names | ertv |
| AHFS/Drugs.com | International Drug Names |
| Routes of administration | Oral |
| ATC code | none |
| Legal status | |
| Legal status | In general: ℞ (Prescription only) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 57149-07-2 |
| PubChem CID | 4418 |
| ChemSpider | 4265 |
| UNII | R9PHW59SFN |
| CompTox Dashboard (EPA) | DTXSID5045176 |
| ECHA InfoCard | 100.220.557 |
| Chemical and physical data | |
| Formula | C24H28N2O3 |
| Molar mass | 392.499 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (verify) |
/////////////////Naftopidil, KT 611, a-Adrenergic Blocker, Antihypertensive.
COC1=CC=CC=C1N2CCN(CC2)CC(COC3=CC=CC4=CC=CC=C43)O
NEW DRUG APPROVALS
ONE TIME
$10.00
Pralnacasan
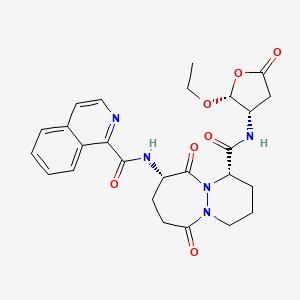
Pralnacasan
VX 740
cas 192755-52-5
(4S,7S)-N-[(2R,3S)-2-ethoxy-5-oxooxolan-3-yl]-7-(isoquinoline-1-carbonylamino)-6,10-dioxo-2,3,4,7,8,9-hexahydro-1H-pyridazino[1,2-a]diazepine-4-carboxamide
N-[(4S,7S)-4-{[(2R,3S)-2-ethoxy-5-oxooxolan-3-yl]carbamoyl}-6,10-dioxo-octahydro-1H-pyridazino[1,2-a][1,2]diazepin-7-yl]isoquinoline-1-carboxamide
(1S,9S)-N-((2R,3S)-2-Ethoxy-5-oxotetrahydrofuran-3-yl)-9-((isoquinolin-1-ylcarbonyl)amino)-6,10-dioxooctahydro-6-H-pyridazino(1,2-a)(1,2)diazepine-1-carboxamide
6H-Pyridazino(1,2-a)(1,2)diazepine-1-carboxamide, N-((2R,3S)-2-ethoxytetrahydro-5-oxo-3-furanyl)octahydro-9-((1-isoquinolinylcarbonyl)amino)-6,10-dioxo-, (1S,9S)-
- HMR 3480
- HMR3480
- HMR3480/VX-740
- Pralnacasan
- UNII-N986NI319S
- VX 470
- VX-740
C26H29N5O7, 523.543
NSAID, ICE inhibitor & metastasis inhibitor.пралнаказан [Russian] [INN]برالناكاسان [Arabic] [INN]普那卡生 [Chinese] [INN]

Pralnacasan is an orally bioavailable pro-drug of a potent, non-peptide inhibitor of interleukin-1beta converting enzyme (ICE).Pralnacasan is a potent, non-peptide inhibitor of interleukin-1beta converting enzyme (ICE, aka Caspase-1). It was originally discovered by Vertex Pharmaceuticals and licensed for development to Aventis Pharma. In 2003 Aventis and Vertex Pharmaceuticals agreed to voluntarily discontinue development based on results from a 9-month animal toxicity trial that showed liver abnormalities due to chronic high doses of pralnacasan. Pralnacasan has also been investigated for the treatment of Partial Epilepsy; advancing to Phase II clinical trials.Pralnacasan is a potent, non-peptide inhibitor of interleukin-1beta converting enzyme (ICE). Pralnacasan is an oral, anti-cytokine drug candidate licensed for development by Aventis Pharma from Vertex Pharmaceuticals. In November 2003, Aventis and Vertex Pharmaceuticals announced that they had voluntarily suspended the phase II clinical trials of pralnacasan due to results from an animal toxicity study that demonstrated liver abnormalities after a nine-month exposure to pralnacasan at high doses. While no similar liver toxicity has been seen to date in human trials, the companies will evaluate the animal toxicity results before proceeding with the phase II clinical program.Pralnacasan inhibits interleukin-1beta converting enzyme (ICE), an enzyme that regulates the production of IL-1 and IFN gamma – intercellular mediators that initiate and sustain the process of inflammation. Inhibiting ICE may be an effective strategy for curtailing damaging inflammatory processes common to a number of acute and chronic conditions, such as rheumatoid arthritis (RA) and osteoarthritis.
PAPERhttps://pubs.rsc.org/en/content/articlelanding/2017/ob/c7ob01403a/unauth
IDrugs (2003), 6(2), 154-158.
Chemistry (Weinheim an der Bergstrasse, Germany) (2017), 23(2), 360-369PAPER
Bioorganic & Medicinal Chemistry Letters (2006), 16(16), 4233-4236.https://www.sciencedirect.com/science/article/abs/pii/S0960894X06006184?
Abstract
Novel 1-(2-acylhydrazinocarbonyl)cycloalkyl carboxamides were designed as peptidomimetic inhibitors of interleukin-1β converting enzyme (ICE). A short synthesis was developed and moderately potent ICE inhibitors were identified (IC50 values <100 nM). Most of the synthesized examples were selective for ICE versus the related cysteine proteases caspase-3 and caspase-8, although several dual-acting inhibitors of ICE and caspase-8 were identified. Several of the more potent ICE inhibitors were also shown to inhibit IL-1β production in a whole cell assay (IC50 < 500 nM).
Graphical abstract
Novel 1-(2-acylhydrazinocarbonyl)cycloalkyl carboxamides were designed and synthesized as selective peptidomimetic inhibitors of interleukin-1β converting enzyme (ICE IC50 values <100 nM).

PAPEROrganic letters (2014), 16(13), 3488-91.https://pubs.acs.org/doi/10.1021/ol501425b
Abstract

Peptides containing N2-acyl piperazic or 1,6-dehydropiperazic acids can be formed efficiently via a novel multicomponent reaction of 1,4,5,6-tetrahydropyridazines, isocyanides, and carboxylic acids. Remarkably, the reaction’s induced intramolecularity can enable the regiospecific formation of products with N2-acyl piperazic acid, which counters the intrinsic and troublesome propensity for piperazic acids to react at N1 in acylations. The utility of the methodology is demonstrated in the synthesis of the bicyclic core of the interleukin-1β converting enzyme inhibitor, Pralnacasan.
PatentWO 9722619WO 9903852WO 9952935
PATENTWO 2000042061https://patents.google.com/patent/WO2000042061A1/enThe invention particularly relates to the process as defined above in which the compound of formula (I) is 9- (1, 3-dihydro-1,3, dioxo-2H-isoindol-2-yl) -3 ,, 7, 8, 9, 10-hexahydro-6, 10-dioxo-6H-pyridazino- [1,2- a] [1, 2] ethyl diazepine-1-carboxylate:

The invention particularly relates to the process as defined above in which the compound of formula (Iopt) is- (lS-cis) -9- (1, 3-dihydro-l, 3-dioxo-2H-isoindol-2-yl) – 3,4,7,8,9, 10-hexahydro-β, 10-dioxo -6H-pyridazino- [1,2- a] [1, 2] ethyl diazeρine-1-carboxylate:

The compounds of formula (I) can be generally used for the synthesis of medicaments as indicated in patent EP 94095. The compounds of formulas (II) and (III) and (F) are known and can be prepared according to the experimental method described below.The invention also relates to the application of the process as defined above as an intermediate step for the preparation of a compound of formula (V)

via the compound of formula (Iopt) as defined above, characterized in that this process comprises the steps of the process for the preparation of the compounds of formula (Iopt) from the compounds of formula (II) as defined above.The subject of the invention is also the application as defined above, characterized in that the compound of formula (Iopt) is (lS-cis) -9- (1, 3-dihydro-l, 3-dioxo -2H- isoindol-2-yl) -3,4,7,8,9, 10-hexahydro-6, 10-dioxo-6H- pyridazino- [1,2-a] [1, 2] diazepine-1- ethyl carboxylate

The subject of the invention is also the application of the process as defined above as an intermediate step in the overall process for preparing the compounds of formula (I) and (Iopt) as defined above. Finally, the subject of the invention is, as intermediate compound, the compound of formula (IA) as defined above.Preparation 1 Preparation of bis (phenylmethyl) 1,2-hydrazinecarboxylate1.5 liters of methanol and 25 g of 80% hydrazine monohydrate are placed under nitrogen. Cooled to 0 ° C and then introduced 75 g of benzyl chloroformate and a solution of 93 g of sodium carbonate in 1100 ml of demineralized water. Maintaining the reaction mixture for 1 hour at 0 ° C, drained and washed by displacement with a mixture of 100 ml of methanol and 100 ml of water, then washed by displacement with 500 ml of water at 0 C °. Dried and obtained 107.6 g of the desired product. Preparation 2Preparation of N-phthaloyl-L-glutamic anhydride D (+) 2-tetrahydro-2,6,6-dioxo-2H-pyran-3-yl-1H-isoindole-1,3 (2H) – dione (R)Stage a: N-phthaloyl-L-glutamic acid2- (1, 3-dihydro-1,3, dioxo-2H-isoindole-2-yl) acid – pentanedioic (2S)To a solution of 14.4 g of sodium carbonate in 180 ml of water is added 10 g of L-glutamic acid then 16 g of N-carbethoxyphthalimide (nefkens reagent, commercial). The mixture is stirred at ambient temperature for 2 hours and then extracted with ethyl acetate. The organic phase is evaporated under reduced pressure until a dry extract is obtained and 2.74 g of crude product is obtained. Washing is carried out with sodium bicarbonate, then after return to the acid and extraction with ethyl acetate, 370 mg of expected product and H 2 N-C0 2 Et are isolated. Furthermore, the aqueous phase is brought to pH = 2 with 36% hydrochloric acid at a temperature below 5 ° C and then extracted with ethyl acetate, washed with a saturated chloride solution. sodium, dry, filter and concentrate under reduced pressure until 22.7 g of expected product is obtained in the form of an oil.Mass spectrum (MH) “ = 276 “ Infrared (Nujol):1775 cm “1 (m), 1720 cm ” 1 (F, complex): CO 1611 cm “1 : Aromatic Stage b:To the product obtained in stage a), 160 ml of tetrahydrofuran are added and 18.6 g of DCC (1, 3-Dicyclohexyl-carbodiimide) dissolved in 55 ml of tetrahydrofuran are added dropwise over 30 minutes. Stirred for 1 hour at 15-17 ° C, then filtered, rinsed with tetrahydrofuran, evaporated under reduced pressure until a dry extract is obtained which is taken up in isopropyl ether. After 30 minutes of stirring, the filter is washed and dried. 14.98 g of expected product are obtained. α D = -52.63 λ H NMR (DMSO) 2.12 (m, 1H); 2.61 (m, 1H); 2.98 (dm, 1H); 3.16 (ddd, 1H); 5.48 (dd, 1H); 7.82 (m,> 4H)Example 1: (IS-cis) -9- (1, 3-dihydro-1,3, dioxo-2H-isoindol-2-yl) -3,4,7,8,9,10-hexahydro-6,10 -dioxo-6H-pyridazino- [1,2- a] [1,2] diazepine-1-ethyl carboxylate. Stage a: Preparation of 2,5-dibromopentanoic acid 39 ml of bromine are added to a mixture of 106 g of 5-bromopentanoic acid and 1 ml of phosphorus tribromide. The reaction mixture is brought to 70-80 ° C for 16 h 30. The reaction medium is brought to 100 ° C for 15 minutes and allowed to return to room temperature. 147 g of sought product is obtained.Stage b: Preparation of ethyl 2,5-dibromopentanoate24.37 g of oxalyl chloride are added to a mixture containing 50 g of the acid prepared in the preceding stage, 15 drops of dimethylformamide and 300 ml of dichloromethane. The reaction mixture is kept under stirring at at room temperature, until the reaction is complete. The reaction mixture is cooled to 10 ° C and 50 ml of ethyl alcohol are added. Stirred for 30 minutes at 10 ° C, allowed to return to room temperature and stirred for 3 hours at room temperature. It is brought to dryness and the desired product is obtained. Stage c: CyclizationPreparation of (S) -tetrahydro-1,2,3-pyridazinetricarboxylate of 3-ethyl-1,2-bis (phenylmethyl) and (R) -tetrahydro-1,2,3-pyridazinetricarboxylate of 1,2 -bis (phenylmethyl). A suspension of 12.1 g of ethyl 2,5-dibromopentanoate (stage b) in 50 ml of diglyme is introduced at 20-25 ° C. in a suspension containing 10.42 g of 1,2-hydrazine carboxylate of bis (phenylmethyl) (preparation 1), 65 ml of diglyme and 8.26 g of potassium carbonate. The suspension obtained is heated to 90 ° C. and stirring is continued for 48 hours. Cooled to 20 ° C, poured into a solution containing 50 ml of 2N hydrochloric acid and 150 ml of a mixture of water and ice. Extraction is carried out with ethyl acetate, washing with water and drying. It is filtered, rinsed with ethyl acetate and dried. Finally, the crude product is purified by chromatography on silica, eluting with a heptane / ethyl acetate mixture 40/20 and 10.71 g of sought product is obtained. Stage d: Acylation and hydrogenolysisPreparation of α, (IS) – [3-oxo-3- (tetrahydro-3-ethoxycarbonyl-1 (2H) -pyridazinyl) propyl] -1,3-dihydro-1,3-dioxo-2H-isoindole acid -2-aceticThe mixture consisting of 15g of tetrahydro-1,2,3-pyridazinetricarboxylate of 3-ethyl-1,2-bis (phenylmethyl) is placed under hydrogen pressure (1.3 bar) for 24 hours. R + S mixture as prepared in stage c 150 ml of tetrahydrofuran, 2.5 g of palladium on carbon (10%) and 9.08 g of phthaloylglutamic acid anhydride as prepared according to preparation 2. After filtration, we evaporated under reduced pressure until a dry extract is obtained which is taken up in 100 ml of ethyl acetate and 150 ml of a saturated solution of sodium bicarbonate. It is extracted 3 times and the bicarbonate solution is acidified to pH = 3 with 36% hydrochloric acid. It is extracted 3 times with dichloromethane and washed with water. 13.16 g of crude product are obtained, which product is purified by chromatography on silica, eluting with a toluene / ethyl acetate / acetic acid 20/80 / 1.5 mixture to obtain 12.7 g of the expected product.NMR (250MHz, CDC1 3 ): 1.24 (d, 3H, OCH 2 CH 3 ); 4.12 (q, 2H, OCH 2 CH 3 ); 4.36-4.40 (m, 1H, Hl in alpha or beta position); 4.69-4.92 (m, 1H, H9 in the alpha position); 7.70 – 7.86 H aromatic. Stage el: cyclization with POCl 3– (lS-cis) -9- (1, 3-dihydro-l, 3-dioxo-2H-isoindol-2-yl) – 3,4,7,8,9, 10-hexahydro-6, 10-dioxo -6H-pyridazino- [1,2- a] [1, 2] ethyl diazepine-1-carboxylate. – (lR-trans) -9- (1, 3-dihydro-1,3, dioxo-2H-isoindol-2-yl) – 3,4,7,8, 9, 10-hexahydro-6,10-dioxo -6H-pyridazino- [1,2-a] [1,2] diazepine-1-ethyl carboxylate.To a solution of 20 ml of dichloroethane heated beforehand to 75 ° C., the following solutions A and B are added over 3 hours: A: 417 mg of the ester prepared in stage d in 4 ml of dichloroethane to which 1 ml of a solution of 1.2 ml of 2,6-lutidine in 5 ml of dichloroethane. B: 1 ml of a solution of 1.9 ml of P0Cl 3 in 10 ml of dichloroethane, then the mixture is stirred for 1 hour at this temperature. Cool to 10 ° C., add demineralized water, extract with dichloromethane and evaporate under reduced pressure to obtain a crude product (0.415 g) which is purified by chromatography on silica eluting with the heptane / dichloromethane mixture. / ethyl acetate 1/1/1. 161.8 mg of the SS diastereoisomer, 126.7 mg of the SR diastereoisomer and 5.8 mg of the SS + SR mixture are isolated. Stage e2: cyclization with POBr 3– (lS-cis) -9- (1, 3-dihydro-l, 3-dioxo-2H-isoindol-2-yl) – 3,4,7, 8, 9, 10-hexahydro-6, 10-dioxo -6H-pyridazino- [1, 2- a] [1, 2] ethyl diazepine-1-carboxylate. – (lR-trans) -9- (1, 3-dihydro-l, 3-dioxo-2H-isoindol-2-yl) – 3,4,7, 8, 9, 10-hexahydro-6, 10-dioxo -6H-pyridazino- [1, 2- a] [1, 2] ethyl diazepine-1-carboxylate.To a solution of 20 ml of dichloroethane heated beforehand to 80 ° C., the following solutions A and B are added over 3 hours:A: 417 mg of the ester prepared in stage d in 4 ml of dichloroethane to which 1 ml of a solution of 2.4 ml of 2,6-lutidine in 10 ml of dichloroethane was added. B: 1 ml of a solution of 5.85 g of POBr 3 in 10 ml of dichloroethane, then the mixture is stirred for 1 hour at this temperature. Cool to 10 ° C, add demineralized water, extract with dichloromethane and evaporate under reduced pressure to obtain a crude product (0.419 g) which is purified by chromatography on silica eluting with the heptane / dichloromethane / mixture 1/1/1 ethyl acetate. 163 mg of the SS diastereoisomer, 143 mg of the SR diastereoisomer and 6.2 mg of the SS + SR mixture are isolated.Stage f: deracemization / epimerization – (lS-cis) -9- (1, 3-dihydro-l, 3-dioxo-2H-isoindol-2-yl) – 3,4,7,8, 9, 10-hexahydro -6,10-dioxo-6H-pyridazino- [1, 2- a] [1, 2] ethyl diazepine-1-carboxylate.Is introduced at a temperature of -45 / -48 ° C in one hour 30 minutes, a solution containing 0.029 g of potassium terbutylate and 0.3 ml of dimethylformamide in a mixture containing 0.194 g of the mixture SS + SR prepared in stage d , 1.5 ml of dimethylformamide and 0.75 ml of terbutanol. The mixture is kept stirring for 1 hour and, after cooling to -50 ° C., 0.4 g of powdered ammonium chloride is introduced. Stirred 10 minutes at -45 ° C, add 1 ml of ammonium chloride at 20 ° C and stirred again 10 minutes. 2 ml of water are added after 5 minutes demineralized. Extracted with ethyl acetate, washed with demineralized water, decanted, concentrated and dried. 0.166 g of expected SS diastereoisomer is obtained. ” D = -75.3 ° (1% in methanol) NMR (250MHz, CDC1 3 ): 1.73 (m, 3H, H-2alpha H-3alpha H-3beta; 1.24 (d, 3H, OCH 2 CH 3 ); 2.38 (m, 3H, H2beta, H7alpha, H8 alpha); 2.92 (m, 1H, H4alpha); 3.39 – 3.44 (m, 1H, H8beta); 3.62 (m, 1H, H7beta); 4.23 (m, 2H, OCH 2 CH 3 ); 4.66-4.71 (m, 1H, H4 in beta position); 5.26-5.41 (m, 2H, Hl and H9 in the alpha position); 7.72 – 7.88 H aromatics.
PATENT
WO 2000010979https://patents.google.com/patent/WO2000010979A1/en

formula II, said compound has the structure:
In the synthesis of these inhibitors, the terminal carbon of Ri adjacent the -COOH moiety contains a protecting substituent. Preferably that protecting
substituent is
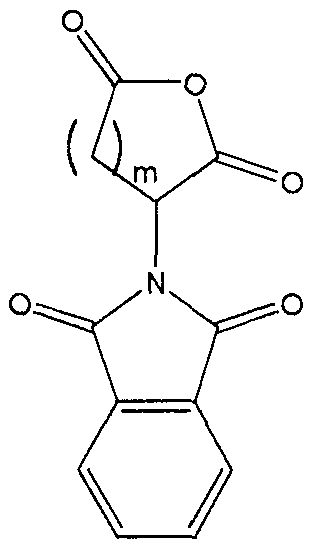
The synthesis steps from compound H to the inhibitors set forth above involve removal of the protecting substituent on Rx; coupling of the R5-NH- or R5′-NH- moiety in its place; hydrolysis of the R2 group;N .(CJ2)m.—Tand coupling of the amine ( (Ch,2)a Rs or -NH-Z)in its place. The removal of the protecting substituent on Ri is typically carried out with hydrazine. The subsequent coupling of the R5-NH- or R5′-NH- moiety is achieved with standard coupling reagents, such as EDC, DCC or acid chloride . Depending upon the nature of R2, its hydrolysis may be achieved with an acid (when R2 is t-butyl), a hydroxide (when R2 is any other alkyl, alkenyl or alkynyl or Ar) or hydrogenolysis (when R2 is an Ar-substituted alkyl, alkenyl or alkynyl) . This produces the corresponding acid from the ester.The acid is then coupled to the amine with standard coupling reagents, such as EDC, DCC or acid chloride .In order that this invention be more fully understood, the following examples are set forth. These examples are for the purpose of illustration only and are not to be construed as limiting the scope of the invention in any way. EXAMPLE 1Synthesis of a 7,6 Scaffold for a Caspase InhibitorA.

Compound A’ was dissolved m 5 equivalents of S0C12 and then heated to 80°C for 1 hour. The solution was then cooled to 50°C and 2 equivalents of bromine were added. The solution was incubated at 50°C for an additional 12 hours until the red color disappeared. We then cooled the solution to 10°C and added 4 volumes of water. The solution was then re-heated to 50°C for another hour. We then separated the organic and aqueous layer, washed the organic layer consecutively with water, Na2S0 and then brme, removing the aqueous layer after each washing. The final organic layer was then isolated, dried over Na2S0 and concentrated to produce compound B’ as an amber oil.B.

Compound B’ was treated with 1 equivalent of tert-butanol and 0.1 equivalents of 4- (dimethylammo) – pyπdme a solution of and the resulting solution cooled to 7°C. We then added a solution of 1 equivalent of DCC m toluene while maintaining reaction temperature at less than 22°C. The cooling bath was removed and the reaction was stirred at ambient temperature under a nitrogen atmosphere for 16 hours. The reaction mixture was then diluted with hexane and cooled to 9°C . The resulting solids were removed by filtration. The filtrate was washed consecutively with 0. IN HC1, water, and then sodium bicarbonate. The filtrate was then dried over sodium sulfate and concentrated in vacuo to afford compound C as a yellow oil.C.

Compound D’ was combined with 1.2 equivalents of compound C and dissolved in DMF at ambient temperature under nitrogen atmosphere. We then added granular sodium sulfate, 2.5 equivalents of LiOH monohydrate, and then 0.1 equivalents Bu4NI to the resulting solution. The reaction temperature was maintained at between 20°C and 30°C and allowed to stir for 16 hours. The reaction mixture was then diluted with ethyl acetate and water and the layers separated. The organic layer was washed with water and then brine, dried over sodium sulfate and concentrated in vacuo to produce an amber oil. This oil was then dissolved in 5 volumes of ethanol at ambient temperature. We then added 2.5 volumes of water. The resulting mixture was allowed to stir until a white solid formed (approximately 5 hours) . The crystallized product was isolated via filtration then dried in vacuo to afford compound E’ as a white solid.D.

We dissolved compound E’ in THF. We then added, at ambient temperature under a nitrogen atmosphere, 0.02 equivalents of triethylamine and 0.01 equivalents of Pd(OAc)2. A solution of 2.5 equivalents of triethylsilane (Et3SiH) in THF was then added and the resulting black solution was allowed to stir for 16 hours to complete the reaction. We then added a saturated, aqueous solution of sodium bicarbonate followed by a solution of compound F’ in THF. After 30 minutes, the layers were separated and the aqueous layer acidified to pH 4.5 with aqueous citric acid. The product in the aqueous layer was then extracted into ethyl acetate. The organic layer was isolated, washed with brine, dried over sodium sulfate and concentrated in vacuo to produce a white foam. This crude product was then recrystallized from MTBE to afford compound G’ as a white powder. E.
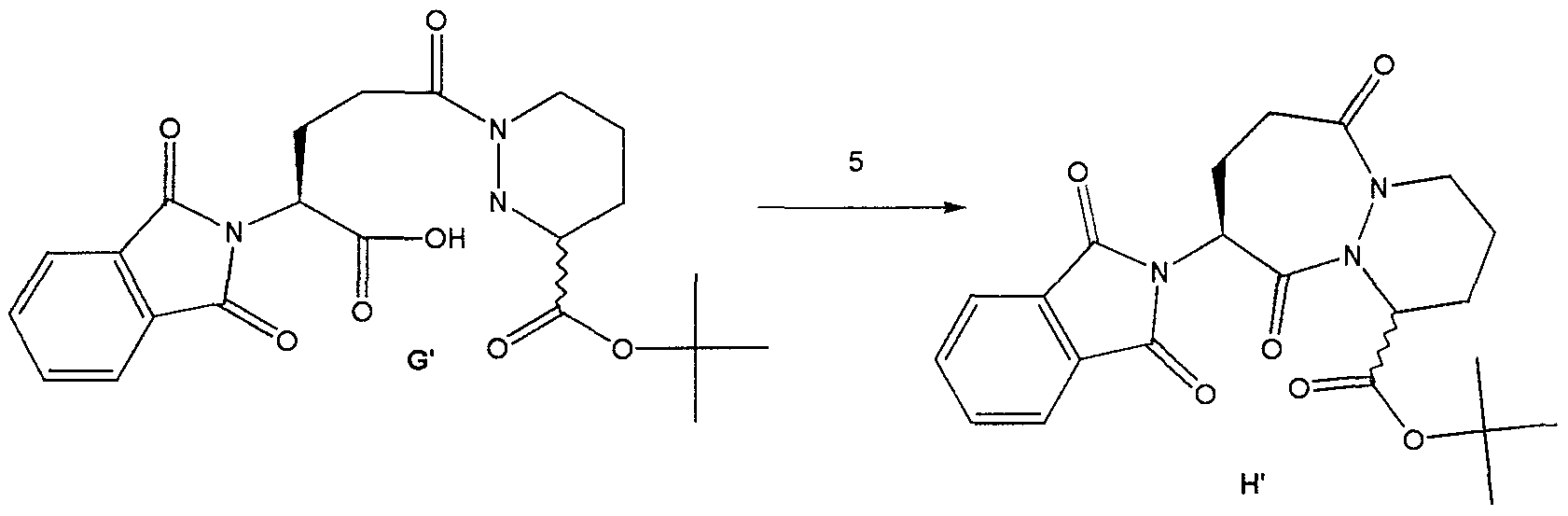
Method #1:To a suspension of compound G’ and 0.1 equivalents of DMF m dichloroethane, at 70°C we added 5 equivalents of 2, 6-lutιdme simultaneously with 2.5 equivalents of S0C12 over a period of 3 hours. The reaction was then diluted with toluene and washed consecutively with NaHC03 and br e. The solution was then dried over Na2S04 and concentrated in vacuo to afford compound H’ as a yellow solid.Method #2:To a suspension of compound G’ m dichloroethane, at 70°C, we added 4 equivalents of 2,6- lutid e followed by 2 equivalents of methanesulfonyl chloride. The resulting solution was stirred at 70°C for 12 hours. The reaction was then diluted with toluene and washed consecutively with NaHC03 and brme. The solution was then dried over Na2S04 and concentrated in vacuo to afford compound H’ as a white solid. Method #2 produced a significantly higher yield of H’ as compared to Method #1. EXAMPLE 2 Use of Intermediate H’ to Produce an Inhibitor of ICE A.

t-Butyl-9-amino-6 , 10-dioxo-l ,2,3,4,7,8,9, 10-octahydro-6- H-pyridazino [1 ,2-a] [1 ,2] diazepine-1-carboxylate (GB2,128,984) To a suspension of H’ (107 g, 0.25 mol) in ethanol (900 iriL) was added hydrazine (27 L, 0.55 mol) and the resulting mixture was allowed to stir at ambient temperature. After 4 hours, the reaction was concentrated in vacuo and the resulting white solid was suspended in acetic acid (IL of 2N) and allowed to stir at ambient temperature for 16 hours. The resulting white solid was filtered off and washed with water. The filtrate was made basic by the addition of solid sodium carbonate and the product extracted with dichloromethane. The organic layer was washed with brine, dried over magnesium sulfate and concentrated in vacuo to afford 79 mg of compound I’ as a yellow viscous oil.B.

t-Butyl-9- (isoquinolin-1-oylamino) -6, 10-dioxo- 1,2,3,4,7,8,9, 10-octahydro-6-H-pyridazino [ 1 , 2-a] [1,2] diazepine-1-carboxylate To a solution of the amine I’ (79 g, 0.265 mol) and isoquinolin-1-carboxylic acid (56g, 0.32 mol) in dichloromethane : DMF (400mL: 400mL) was added hydroxybenztriazole (54 g, 0.4 mol) and l-(3- dimethylaminopropyl) -3-ethylcarbodiimide hydrochloride (74 g, 0.39 mol) and the resulting mixture was allowed to stir at ambient temperature for 16 hours. The reaction mixture was poured into water and extracted with ethyl acetate. The ethyl acetate layer was washed with 0.5N sodium bisulfate, water, sodium bicarbonate, brine, dried over sodium sulfate and concentrated in vacuo to afford 122 g of compound J’ as an orange solid-foam.C.

9- (isoquinolin-1-oylamino) -6, 10-dioxo-l ,2 ,3 ,4 , 7 , 8 , 9 , 10- octahydro-6-H-pyridazino [1 ,2-a] [1,2] diazepine-1- carboxylate A solution of the ester J’ (122 g) in dichloromethane and trifluoroacetic acid (200 mL) was allowed to stir at ambient temperature for 16 hours. The reaction mixture was concentrated to a black oil which was then triturated with acetonitrile and ether to afford 98 g of compound K’ as a pale yellow solid. D .

K'[IS, 9S (2RS, 3S) ] N-(2-benzyloxγ-5-oxotetrahydrofuran-3- yl) -6 , 10-dιoxo-9- (ιsoquιnolιn-1-oγlamιno) -1,2,3,4,7,8,9, 10-octahydro-6-H-pyrιdazιno [ 1 , 2-a] [1,2] dιazepιne-l-carboxamιde To a solution of (3S, 2RS) 3- allyloxycarbonylammo-2- (4-chlorobenzyl) oxy-5- oxotetrahydrofuran [Bioorg. & Med. Chem. Lett., 2, pp. 615-618 (1992)] (4.4 g, 15.1 mmol) in dichloromethane was added N, N-dimethylbarbituric acid (5.9g, 3.8 mmol) then tetrakispalladium ( 0) tπphenyl phosphme (1.7 g, 1.5 mmol) and the resulting mixture was allowed to stir at ambient temperature for 15 minutes. To the resulting mixture was added the acid, compound K’ (5.0 g, 12.6 mmol), hydroxybenztπazole (2.0 g, 14.8 mmol) then and 1- (3-dιmethylammopropyl) -3-ethylcarbodιιmιde hydrochloride (2.7g, 14 mmol) and the reaction was allowed to stir for 3 hours at ambient temperature. The reaction mixture was then poured into water and extracted with ethyl acetate. The organics were washed with 0.5M sodium bisulfate, water, sodium bicarbonate, br e, dried over magnesium sulfate and concentrated m vacuo to afford 2.6 g of the crude product as a yellow foam. The crude material was purified by column chromatography (Sι02, dichloromethane : acetone 9:1 – 3:1) to afford 1.2 g of the compound L’ . Compound L’ and related compounds that may be synthesized using the method of this invention as an intermediate step are described in WO 97/22619, the disclosure of which is herein incorporated by reference. Those related compounds may be synthesized from the product of the method of this invention, H or H’ , through modifications of the procedure set forth in Example 2. Such modifications are well known in the art.
PATENTWO 2001083458https://patents.google.com/patent/WO2001083458A2/enScheme IV

C 2 5,> R’==OH (S)-VI-a *•* 6 6., R R”==<CI

Example 1

(S) -t-butyl- bis- (1,2-benzyloxycarbonyl) – hexahydropyridazine-3-carboxylate (>90% ee) : To a solution of bis-Cbz hydrazine and (R) -t-butyl-2, 5- dimesylvalerate (from the diol prepared by the method of Schmidt et al., Synthesis, p. 223 (1996)) in DMF was added Na2S04 then TBAF (2.5 equivalents). The resulting reaction mixture was allowed to stir at room temperature for 24 hrs. The reaction was then diluted with ethyl acetate. The organic layer was washed sequentially with 10% citric acid and brine, dried over anhydrous Na2S04 and concentrated in vacuo to afford the title compound. The optical purity of the title compound was greater than 90% ee as determined by HPLC using a ChiralPak® AD column and eluting with ethanol at 0.7 ml per minute.Example 2

(S) -t-butyl-bis- (1 ,2-benzyloxycarbonyl) – hexahydropyridazine-3-carboxylate (40% ee) : To a solution of bis-Cbz hydrazine and (R) -t-butyl-2, 5-dimesylvalerate(96.5% ee) in DMF was added Na2S04 then K2C03 (5 equivalents) and TBAI (0.1 equivalents). The resulting reaction mixture was heated at 80°C for 24 hrs. The reaction was allowed to cool and diluted with ethyl acetate. The organic layer was washed sequentially with 10% citric acid and brine, dried over anhydrous Na2S04 and concentrated in vacuo to afford the title compound as a 70:30 mixture of the S:R enantiomers (40% ee, as determined by HPLC using a ChiralPak® AD column, eluting with ethanol at 0.7 ml/min) .Example 3

Racemic t-butyl- bis- (1 ,2-benzyloxycarbonyl) – hexahydropyridazine-3-carboxylate: To a solution of bis- Cbz hydrazine and (R) -t-butyl-2, 5-dimesylvalerate (96.5% ee) in THF was added NaH (2 equivalents) . The resulting reaction mixture was stirred at room temperature. The reaction was quenched then diluted with ethyl acetate. The organic layer was washed sequentially with 10% citric acid and brine, dried over anhydrous Na2S04 and concentrated in vacuo to afford the title compound as a racemic mixture.Example 4 A. Deprotection and salt formation

Hexahydro-pyridazine-3-carboxylic acid tert-butyl ester , L-tartaric acid salt (B) : Compound A was combined with 10% Pd/C (10% w/w) in tetrahydrofuran. The resulting suspension was stirred at 60 °C under a hydrogen atmosphere until deprotection complete. The catalyst was removed via filtration, to the filtrate was added L- tartaric acid (1 equivalent) and the resulting solution concentrated in vacuo.B. Enantiomeric Enrichment
Hθ

The concentrate (B) was taken up in n-butanol(10 volumes), heated to reflux, then allowed to slowly cool to ambient temperature while stirring. The resulting solids were collected via filtration to afford(S) -piperazic acid, t-butyl ester as the tartrate salt (C) in 33% yield.C. Chiral AnalysisCompound (C) was suspended in water and DCM and cooled. We then added NaOH to basify the aqueous layer. The layers were then separated and to the organic layer we added two equivalents of benzyl chloroformate andNaOH. After stirring for 1 hour, the layers were again separated and the organic layer was washed with water.The organic layer was then dried over MgS04 and then concentrated in vacuo to produce the bis-Cbz piperazic acid, t-butyl ester for chiral HPLC analysis. The bis-Cbz piperazic acid, t-butyl ester was applied to a Chiralpak AD HPLC column (Chiral Technologies, Exton, PA) and eluted with ethanol at 0.8 ml/minute. Fractions from the column were quantitate by absorption at 210 nm. The results demonstrated that (S)- piperazic acid, t-butyl ester accounted for 94.5% of the piperazic acid, t-butyl ester present in the preparation.
Example 5 Conversion of Intermediate IV to Intermediate Vl-a Cbzy


IV’ C02t-Bu yi-a C02t-Bu Tetrahydro-pyridazine-l,3-dicarboxylic acid 1-benzyl ester 3-tert-butyl ester (Vl-a) : Compound IV (1 mmol) is combined with toluene and sodium hydroxide (aqueous, 2M, 3 equivalents) and the resulting mixture cooled to 1 °C. A solution of benzylchloroformate (1.05 equivalents) in toluene is added while maintaining the reaction pH at 10 or higher by the addition of sodium hydroxide, as needed. After stirring an additional 1 hour, allow the mixture to warm to room temperature then extract with ethyl acetate. The organic layer is washed with brine, dried over sodium sulfate and concentrated to afford Vl-a.Example 6 Conversion of Intermediate X to an Inhibitor of ICE
A. Phthalimide removal to form IX-b

X IX-b t-Butyl-9-amino-6 , 10-dioxo-l ,2,3,4,7,8,9, 10-octa ydro-6-H-pyridazino[l,2-a] [1,2] diazepine-1-carboxylate (GB 2,128,984): To a suspension of X (107 g, 0.25 mol) in ethanol (900 mL) was added hydrazine (27 mL, 0.55 mol) and the resulting mixture was allowed to stir at ambient temperature. After 4 hours, the reaction was concentrated in va cuo and the resulting white solid was suspended in acetic acid (1L of 2N) and allowed to stir at ambient temperature for 16 hours. The resulting white solid was filtered off and washed with water. The filtrate was made basic by the addition of solid sodium carbonate and the product extracted with dichloromethane. The organic layer was washed with brine, dried over magnesium sulfate and concentrated in va cuo to afford 79g of compound IX-b as a yellow viscous oil.B. Formation of compound XII

IX-b XII t-Butyl-9- (isoquinolin-1-oylamino) -6 , 10-dioxo- 1,2,3,4,7,8,9, 10-octahydro-6-H-pyridazino [1 , 2-a] [1,2] diazepine-1-carboxylate (XII) : To a solution of IX-b (79 g, 0.265 mol) and isoquinolin-1-carboxylic acid (56g, 0.32 mol) in dichloromethane and DMF (400mL: 00mL) was added hydroxybenzotriazole (54 g, 0.4 mol) and l-(3- dimethylaminopropyl) -3-ethylcarbodiimide hydrochloride (74 g, 0.39 mol) and the resulting mixture was allowed to stir at ambient temperature for 16 hours. The reaction mixture was poured into water and extracted with ethyl acetate. The ethyl acetate layer was washed with 0.5N sodium bisulfate, water, sodium bicarbonate, brine, dried over sodium sulfate and concentrated in vacuo to afford 122 g of compound XII as an orange solid-foam.t-Butyl ester hydrolysis to form compound XIII

XIII 9- (isoquinolin-1-oylamino) -6 , 10-dioxo-l ,2,3,4,7,8,9, 10- octahydro-6-H-pyridazino [1 , 2-a] [1 , 2] diazepine-1- carboxylate (XIII) : A solution of the ester XII (from step B) (122 g) in dichloromethane and trifluoroacetic acid (200 mL) was allowed to stir at ambient temperature for 16 hours. The reaction mixture was concentrated to a black oil which was then triturated with acetonitrile and ether to afford 98 g of compound XIII as a pale yellow solid.D. Formation of compound 4-b

[1S, 9S (2RS,3S) ]N- (2-benzyloxy-5-oxotetrahydrofuran-3- yl) -6,10-dioxo-9- (isoquinolin-1-oylamino) – 1,2,3,4,7,8,9, 10-octahydro-6-H-pyridazino [1 , 2-a] [1,2] diazepine-1-carboxamide (4-b) : To a solution of (3S, 2RS) 3-allyloxycarbonylamino-2-benzyloxy-5-oxotetrahydrofuran [Bioorq. & Med. Chem. Lett., 2, pp. 615-618 (1992)] (4.4 g, 15.1 mmol) in dichloromethane was added N,N- dimethylbarbituric acid (5.9g, 3.8 mmol) then tetrakispalladium(O) triphenyl phosphine (1.7 g, 1.5 mmol) and the resulting mixture was allowed to stir at ambient temperature for 15 minutes. To the resulting mixture was added the acid, compound XIII (from step C) (5.0 g, 12.6 mmol), hydroxybenzotriazole (2.0 g, 14.8 mmol), then 1- (3-dimethylaminopropyl) -3-ethylcarbodiimide hydrochloride (2.7g, 14 mmol) and the reaction was allowed to stir for 3 hours at ambient temperature. The reaction mixture was then poured into water and extracted with ethyl acetate. The organics were washed with 0.5M sodium bisulfate, water, sodium bicarbonate, brine, dried over magnesium sulfate and concentrated in vacuo to afford 2.6 g of the crude product as a yellow foam. The crude material was purified by column chromatography (Si02, dichloromethane: acetone 9:1 – 3:1) to afford 1.2 g of the compound 4-b. Compounds of formulae VII and VIII, and related compounds, that may be synthesized using the method of this invention as an intermediate step are described in WO 97/22619 and United States Patent 6,204,261 the disclosure of which is herein incorporated by reference. Those related compounds may be synthesized from the product of the method of this invention, I, IV, or V, through modifications of the procedure set forth in Examples 4 through 6. Such modifications are well known in the art.PATENTUS 6559304https://patents.google.com/patent/US6559304B1PATENTWO 2008074816https://patents.google.com/patent/WO2008074816A1/en
Patent
Publication numberPriority datePublication dateAssigneeTitleEP0094095A2 *1982-05-121983-11-16F. Hoffmann-La Roche AgBicyclic carboxylic acids and their alkyl and aralkyl estersUS4692438A *1984-08-241987-09-08Hoffmann-La Roche Inc.Pyridazo-diazepines, diazocines, and -triazepines having anti-hypertensive activityWO1993023403A1 *1992-05-151993-11-25Merrell Dow Pharmaceuticals Inc.NOVEL MERCAPTOACETYLAMIDO PYRIDAZO[1,2]PYRIDAZINE, PYRAZOLO[1,2]PYRIDAZINE, PYRIDAZO[1,2-a][1,2]DIAZEPINE AND PYRAZOLO[1,2-a][1,2]DIAZEPINE DERIVATIVES USEFUL AS INHIBITORS OF ENKEPHALINASE AND ACEWO1994011353A1 *1992-11-121994-05-26University College LondonProcess for the preparation of (3r)- and (3s)-piperazic acid derivativesWO1995035308A1 *1994-06-171995-12-28Vertex Pharmaceuticals IncorporatedINHIBITORS OF INTERLEUKIN-1β CONVERTING ENZYMEFamily To Family CitationsUS6204261B11995-12-202001-03-20Vertex Pharmaceuticals IncorporatedInhibitors of interleukin-1β Converting enzyme inhibitorsFR2777888B11998-04-272004-07-16Hoechst Marion Roussel IncNOVEL DERIVATIVES OF ACID (3,4,7,8,9,10-HEXAHYDRO-6,10- DIOXO-6H-PYRIDAZINO [1,2-A] [1,2] DIAZEPINE-1-CARBOXYLIC, THEIR PROCESS OF PREPARATION AND THEIR APPLICATION TO THE PREPARATION OF MEDICINESFR2777889B11998-04-272004-07-09Hoechst Marion Roussel IncNOVEL DERIVATIVES OF OCTAHYDRO-6,10-DIOXO-6H- PYRIDAZINO [1,2-A] [1,2] DIAZEPINE-1-CARBOXYLIC, THEIR PREPARATION PROCESS AND THEIR APPLICATION TO THE PREPARATION OF THERAPEUTICALLY ACTIVE COMPOUNDS
////////////////Pralnacasan, VX 740, VX 470, HMR 3480, пралнаказан , برالناكاسان , 普那卡生 ,
CCOC1C(CC(=O)O1)NC(=O)C2CCCN3N2C(=O)C(CCC3=O)NC(=O)C4=NC=CC5=CC=CC=C54
NEW DRUG APPROVALS
ONE TIME
$10.00
VX 148
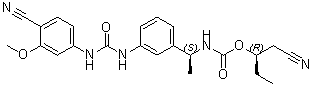
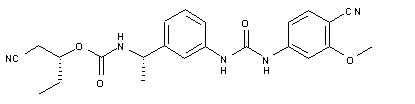
VX 148
297730-05-3
Name: VX-148
CAS#: 297730-05-3
Chemical Formula: C23H25N5O4
Exact Mass: 435.19065
Molecular Weight: 435.48
Elemental Analysis: C, 63.44; H, 5.79; N, 16.08; O, 14.70
| Molecular Weight | 435.48 |
| Formula | C23H25N5O4 |
| CAS No. | 297730-05-3 (VX 148); |
| Chemical Name | Carbamic acid, N-[(1S)-1-[3-[[[(4-cyano-3-methoxyphenyl)amino]carbonyl]amino]phenyl]ethyl]-, (1R)-1-(cyanomethyl)propyl ester |
- OriginatorVertex Pharmaceuticals
- ClassAntipsoriatics
- Mechanism of ActionInosine monophosphate dehydrogenase inhibitors
- DiscontinuedPsoriasis; Transplant rejection; Viral infections
- 13 Nov 2003Interim data from a media release have been added to the adverse events and Skin Disorders therapeutic trials sections
- 23 May 2003Vertex Pharmaceuticals has completed enrolment in a phase IIa trial for Psoriasis in Iceland
- 24 Dec 2002Phase-II clinical trials in Psoriasis in Iceland (unspecified route)
VX-148 is a second-generation, orally administered inhibitor of inosine monophosphate dehydrogenase (IMPDH). The IMPDH enzyme plays a key role in regulating immune response and proliferation of specific cell types, including lymphocytes. VX-148 is a developed for the treatment of autoimmune diseases.
Investigated for use/treatment in autoimmune diseases, psoriasis and psoriatic disorders, and viral infection.
VX-148 is a novel, uncompetitive IMPDH inhibitor with a K(i) value of 6 nM against IMPDH type II enzyme. VX-148 is slightly more potent than mycophenolic acid and VX-497 in inhibiting the proliferation of mitogen-stimulated primary human lymphocytes (IC(50) value of ~80 nM). The inhibitory activity of VX-148 is alleviated in the presence of exogenous guanosine. VX-148 does not inhibit proliferation of nonlymphoid cell types such as fibroblasts, indicating selectivity for inhibition of IMPDH activity. VX-148 is orally bioavailable in rats and mice; oral administration of VX-148 inhibits primary antibody response in mice in a dose-dependent manner with an ED(50) value of 38 mg/kg b.i.d. VX-148 significantly prolongs skin graft survival at 100 mg/kg b.i.d. in mice.
SYN
WO 0056331
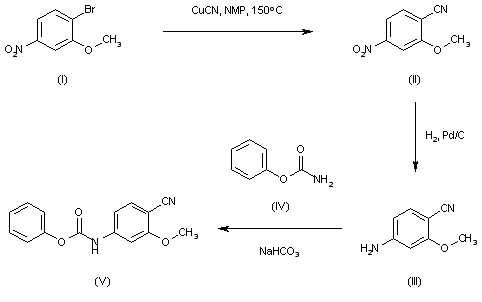
The intermediate carbamate (V) has been obtained as follows. The reaction of 4-bromo-3-methoxynitrobenzene (I) with CuCN in NMP at 150 C gives 2-methoxy-4-nitrobenzonitrile (II), which is reduced with H2 over Pd/C in ethyl acetate to yield 4-amino-2-methoxybenzonitrile (III). Finally, this compound is condensed with phenyl carbamate (IV) by means of NaHCO3 in ethyl acetate to afford the desired carbamate intermediate (V).
SYN
The reduction of 3-nitroacetophenone (VI) by means of NaBH4 in ethanol gives 1-(3-nitrophenyl)ethanol (VII), which is treated with DPPA and DBU in hot toluene to yield the azido derivative (VIII). The reduction of (VIII) with PPh3 in THF/water affords 1-(3-nitrophenyl)ethylamine (IX) as a racemic mixture that is submitted to optical resolution with L-(+)-tartaric acid to provide the desired (S)-isomer (X). The reduction of the nitro group of (X) by means of H2 over Pd/C in methanol gives 1(S)-(3-aminophenyl)ethylamine (XI), which is condensed with 2(R)-hydroxypentanenitrile (XII) and CDI to yield the carbamate (XIII). Finally, this compound is condensed with intermediate carbamate (V) by means of TEA in hot ethyl acetate to afford the target urea.
- Jain J, Almquist SJ, Heiser AD, Shlyakhter D, Leon E, Memmott C, Moody CS, Nimmesgern E, Decker C: Characterization of pharmacological efficacy of VX-148, a new, potent immunosuppressive inosine 5′-monophosphate dehydrogenase inhibitor. J Pharmacol Exp Ther. 2002 Sep;302(3):1272-7. [Article]
////////////VX 148, phase 2
O=C(O[C@H](CC)CC#N)N[C@H](C1=CC=CC(NC(NC2=CC=C(C#N)C(OC)=C2)=O)=C1)C

NEW DRUG APPROVALS
one time
$10.00
VX- ? (3-[2-(4-fluorophenyl)-lH-indol-3-yl]-N-[(3S,4R)-4-hydroxy-2- oxo-pyrrolidin-3-yl ] propanamide)
VX- ?
CAS 2446817-72-5
HYDRATE 2446818-26-2
Acetic acid, 1-methylethyl ester 2446818-27-3
C21 H20 F N3 O3, 381.4
1H-Indole-3-propanamide, 2-(4-fluorophenyl)-N-[(3S,4R)-4-hydroxy-2-oxo-3-pyrrolidinyl]-
3-[2-(4-fluorophenyl)-lH-indol-3-yl]-N-[(3S,4R)-4-hydroxy-2- oxo-pyrrolidin-3-yl ] propanamide
use in treating focal segmental glomerulosclerosis (FSGS) and/or non-diabetic kidney disease (NDKD).

NEW DRUG APPROVALS
one time
$10.00
PATENT
SOLID FORMS OF APOL1 INHIBITOR AND METHODS OF USING SAME
Compound I is disclosed as Compound 87 in U.S. Provisional Application No.62/780,667 filed on December 17, 2018, U.S. Application No. 16/717,099 filed onDecember 17, 2019, and PCT International Application No. PCT/US2019/066746 filed on December 17, 2019, the entire contents of each of which are incorporated herein by reference.
Compound I, which can be employed in the treatment of diseases mediated by APOLl, such as FSGS and NDKD
Example 1. Synthesis of Compound
Preparation of Compound I and Forms Thereof
Compound I Compound I /‘– PrOAc solvate Form A
n-pentanol/
n-heptane
Compound I
Form B
Step 1. Synthesis of 3-[2-(4-fluorophenyl)-lH-indol-3-yl]propanoic acid (C101)
[00156] To a mixture of C104 (100.0 g, 1.0 equiv) and phenyl hydrazine hydrochloride (72.2 g, 1.05 eqiv) was charged AcOH (800 mL, 8 vol). The mixture was agitated and heated to 85 °C for 16 hours. The batch was cooled to 22 °C. A vacuum was applied and the batch distill at <70 °C to ~3 total volumes. The batch was cooled to 19- 25 °C. The reactor was charged with iPrOAc (800 mL, 8 vol) and then charged with water (800 mL, 8 vol). The internal temperature was adjusted to 20 – 25 °C and the biphasic mixture was stirred for no less than 0.5 h. Stirring was stopped and the phases allowed to separate for no less than 0.5 h. The lower aqueous layer was removed. 1 N HC1 (500 mL, 5 vol) was charged to the reactor. The internal temperature was adjusted to 20 – 25 °C, and the
biphasic mixture was stirred for no less than 0.5 h. Stirring was stopped and phases were allowed to separate for no less than 0.5 h. The lower aqueous layer was removed. The reactor was charged with 1 N HC1 (500 mL, 5 vol). The internal temperature was adjusted to 20 – 25 °C, and the biphasic mixture was stirred for no less than 0.5 h. Stirring was stopped and phases were allowed to separate for no less than 0.5 h. The lower aqueous layer was removed. Water (500 mL, 5 vol) was charged to the reactor. The internal temperature was adjusted to 20 – 25 °C, and the biphasic mixture was stirred for no less than 0.5 h. Stirring was stopped and phases were allowed to separate for no less than 0.5 h. The lower aqueous layer was removed. Water (500 mL, 5 vol) was charged to the reactor. The internal temperature was adjusted to 20 – 25 °C, and the biphasic mixture was stirred for no less than 0.5 h. Stirring was stopped and phases were allowed to separate for no less than 0.5 h. The lower aqueous layer was removed. The organic phase was distilled under vacuum at <75 °C to 3 total volumes. The reactor was charged with toluene (1000 mL, 10 vol). The organic phase was distilled under vacuum at <75 °C to 5 total volumes. The reactor was charged with toluene (1000 mL, 10 vol). The organic phase was distilled under vacuum at <75 °C to 5 total volumes. The resulting slurry was heated to an internal temperature of 85 °C until complete dissolution of solids was achieved. The mixture was allowed to stir for 0.5 h at 85 °C and then cooled to an internal temperature of 19 – 25 °C over 5 h. The mixture was allowed to stir at 25 °C for no less than 2 h. The slurry was filtered. The filter cake was washed with toluene (1 x 2 vol (200 mL) and 1 x 1.5 vol (150 mL)). The solids were dried under vacuum with nitrogen bleed at 60 °C to afford product C101 (95.03 g, 70%).
Step 2. Synthesis of Compound I
[00157] A mixture of 3-[2-(4-fluorophenyl)-lH-indol-3-yl]propanoic acid C101 (50 g, 1.0 equiv), S2 hydrochloride (28.3 g, 1.05 equiv), and CDMT (34.1 g, 1.1 equiv) was charged with 2-MeTHF (200 mL, 4 vol) and DMF (50 mL, 1 vol) and the mixture was agitated. The internal temperature adjusted to <13 °C. The reactor was charged with NMM (64.5 g, 3.5 equiv) over 1 h, while maintaining internal temperature <20 °C. The internal temperature was adjusted to 25 °C and the batch was stirred at that temperature for 14 h. The batch was cooled to 10 °C and charged with water (250 mL, 5 vol) while keeping the internal temperature <20 °C. The batch was then warmed to 20 – 25 °C. Stirring was stopped, and the phases allowed to separate for 10 min. The lower aqueous phase was removed. The aqueous layer was back extracted with 2-MeTHF (2 x 200 mL, 2 x 4 vol) at
20 – 25 °C. The combined organic phases were washed with 1 N HC1 (500 mL, 10 vol) at 20 – 25 °C by mixing for 10 min and settling for 10 min. The lower aqueous phase was removed. The organic phases were washed with 0.25 N HC1 (2 x 250 mL, 2 x 5 vol) at 20 – 25 °C by mixing for 10 min and settling for 10 min for each wash. Lower aqueous phases were removed after each wash. The organic phase was washed with water (250 mL, 5 vol) at 20 – 25 °C by mixing for 10 min and settling for 10 min. The reactor was charged with 20 wt % Nuchar RGC® and stirred for 4 h. The reaction mixture was filtered through a pad of celite®. The reactor and celite® pad were rinsed with 2-MeTHF. The combined organics were distilled under vacuum at <50 °C to 5 total volumes. The reactor was charged with iPrOAc (500 mL, 10 vol). The organic phase was distilled under vacuum at <50 °C to 5 total volumes. The mixture was charged with additional iPrOAc (400 mL, 8 vol) and distillation under vacuum was repeated. The mixture was charged with additional iPrOAc (250 mL, 5 vol), heated to an internal temperature of 75 °C and stirred for 5 h. The slurry was cooled to 25 °C, over 5 h and stirred for no less than 12 h. The slurry was filtered and the filter cake washed with iPrOAc (2 x 50 mL, 2 x 1 vol). The solids were dried under vacuum with nitrogen bleed at 55 – 60 °C to afford Compound I as an iPrOAc solvate (60.38 g including 9.9% w/w iPrOAc, 80.8% yield).
Recrystallization to Form A of Compound I
[00158] Compound I as an iPrOAc solvate (17.16 g after correction for iPrOAc content, 1.0 equiv) was charged to a reactor. A mixture of IP A (77 mL, 4.5 vol) and water (137 mL, 8 vol) were charged to the reactor. The slurry was heated to an internal temperature of 75 °C. The batch was cooled to an internal temperature of 25 °C over 10 h and then stirred at 25 °C for at least 12 h. The slurry was filtered. The filter cake was washed with 36/64 IP A/water (2 x 52 mL, 2 x 3 vol). The solids were dried under vacuum with nitrogen bleed at 60 °C to afford Compound I as a neat, crystalline form (Form A, 15.35 g, 89%).
[00159] The X-ray powder diffractogram of Compound I Form A (FIG. 50) was acquired at room temperature using a PANalytical Empyrean diffractometer equipped with PIXcel ID detector. The peaks are listed in Table A below.
Table A. XRPD of Form A of Compound I
|
I
PATENT
- WO2020131807
Alternative Preparation I of Compound 87 (Indole preparation route C)
Step 1. Synthesis of 2-(4-fluorophenyl)-lH-indole (C98)
[00401] To a stirred suspension of indole (5 g, 42.7 mmol) and (4- fluorophenyl)boronic acid (8.96 g, 64.0 mmol) in AcOH (200 mL) was
added Pd(OAc)2.Trimer (1.44 g, 6.4 mmol) and the mixture stirred at room temperature for 16 h under 02-balloon pressure. Then the reaction mixture was filtered through a Celite® pad, washed with EtOAc (500 mL). The filtrates were washed with water, sat. NaHC03 solution, brine solution, then dried over Na2S04 and concentrated under reduced pressure. Purification by silica gel chromatography (Gradient: 0-10 % EtOAc in heptane) yielded the product afforded 2-(4-fluorophenyl)-lH-indole (5.5 g, 61 %). ‘H NMR (300 MHz, DMSO-de) 5 11.51 (s, 1H), 7.9 (t, J = 5.4 Hz, 2H), 7.52 (d, J = 7.8 Hz, 1H), 7.39 (d, J = 8.1 Hz, 1H), 7.30 (t, J = 8.7 Hz, 2H), 7.09 (t, J = 12 Hz, 1H), 6.99 (t, J = 7.5 Hz, 1H), 6.86 (s, 1H). LCMS m/z 212.4 [M+H]+.
Step 2. Synthesis of methyl (E)-3-[2-(4-fluorophenyl)-lH-indol-3-yl]prop-2-enoate (C99)
[00402] 2-(4-fluorophenyl)-lH-indole (1.0 g, 4.76 mmol) and methyl 3,3-dimethoxypropanoate (0.81 mL, 5.7 mmol) were suspended in dichloromethane (15 mL). Trifluoroacetic acid (2.00 mL, 26 mmol) was added rapidly via syringe, resulting in a clear brown solution. The reaction mixture was heated to 40 °C for three hours. The reaction was diluted with dichloromethane (15 mL) to give an amber solution which was washed with saturated aqueous NaHCCh (25 mL) to yield a bright yellow/light amber biphasic mixture. The phases were separated and the organic layer was washed with saturated NaHCCh (30 mL), then dried (MgSCh) and filtered. The mixture was concentrated under a nitrogen stream overnight. The crude product was obtained as a yellow powder. The product was dissolved in minimum 2-MeTHF and pentane added until the suspension became lightly cloudy. The suspension was allowed to stand overnight, and the precipitate was filtered off. The filter cake was washed with heptane (2 x 15 mL), and dried in vacuo at 40 °C to afford the product as a yellow powder. Methyl (E)-3-[2-(4-fluorophenyl)-lH-indol-3-yl]prop-2-enoate (1.30 g, 86 %). ¾ NMR (300 MHz, Chloroform -if) d 8.41 (s, 1H), 8.01 – 7.95 (m, 1H), 7.92 (d, J = 16.0 Hz,
1H), 7.58 – 7.50 (m, 2H), 7.46 – 7.41 (m, 1H), 7.33 – 7.27 (m, 2H), 7.22 (t, J = 8.6 Hz, 2H), 6.59 (d, J = 16.0 Hz, 1H), 3.79 (s, 3H). LCMS m/z 295.97 [M+H]+.
Step 3. Synthesis of methyl 3-[2-(4-fluorophenyl)-lH-indol-3-yl]propanoate (CIOO)
[00403] To a solution of methyl (E)-3-[2-(4-fluorophenyl)-lH-indol-3-yl]prop-2-enoate (7 g, 0.02 mol) in EtOAc (350 mL) was added Palladium on carbon (4 g, 10 %w/w, 0.004 mol) and stirred at room temperature for 2 h under an atmosphere of H2 (bladder pressure). The reaction mixture was filtered through a pad of Celite® and washed with EtOAc (400 mL). The filtrates was concentrated to afford methyl 3-[2-(4-fluorophenyl)-lH-indol-3-yl]propanoate (7.1 g, 100 %). 1H MR (300 MHz, DMSO-<fc) 5 11.2 (s, 1H), 7.65 (q, J = 5.4 Hz, 2H), 7.54 (d, J = 8.1 Hz, 1H), 7.36 (t, J = 9.0 Hz, 3H), 7.10 (t, J = 8.1 Hz, 1H), 7.02 (t, J = 7.8 Hz, 1H), 3.53 (s, 3H), 3.10 (t, J = 15.9 Hz, 2H), 2.63 (t, J = 15.9 Hz, 2H). LCMS m/z 298.21 [M+H]+. The product was used directly in the subsequent step without further purification.
Step 4. Synthesis of 3-[2-(4-fluorophenyl)-lH-indol-3-yl]propanoic acid (C101)
[00404] To stirred solution of methyl 3-[2-(4-fluorophenyl)-lH-indol-3-yl]propanoate (14.4 g, 0.05mol) in THF (300 mL), MeOH (300 mL) and H2O (250 mL) was cooled to -10°C. LiOH.H20 (10.1 g, 0.24 mol) was slowly added in a portion-wise manner. The reaction mixture was allowed to stir at room temperature for 16 h. The mixture was
evaporated and ice cold water (200 mL) was added, pH was adjusted to pH- 2 with 1M HC1 (400 mL, Cold solution). The mixture was stirred for 10 minutes, filtered and dried to afford 3-[2-(4-fhiorophenyl)-lH-indol-3-yl]propanoic acid (12.9 g, 94 %). ‘H NMR (400 MHz, DMSCMJ) 5 12.11 (s, 1H), 11.18 (s, 1H), 7.65 (q, J = 5.2 Hz, 2H), 7.56 (d, J = 7.6 Hz, 1H), 7.36 (t, J = 8.8 Hz, 3H), 7.10 (t, J = 8 Hz, 1H), 7.01 (t, J = 8 Hz, 1H), 3.06 (t, J = 16.4 Hz, 2H), 2.55 (t, J = 16 Hz, 2H). LCMS m/z 284.21 [M+H]+.
Step 5. Synthesis of 3-[2-(4-fluorophenyl)-lH-indol-3-yl]-N-[(3S,4R)-4-hydroxy-2- oxo-pyrrolidin-3-yl ] propanamide (87)
[00405] A mixture of 3-[2-(4-fluorophenyl)-lH-indol-3-yl]propanoic acid C101 (40 g, 120.0 mmol) and (3S,4R)-3-amino-4-hydroxy-pyrrolidin-2-one (Hydrochloride salt) S2 (23.8 g, 156.0 mmol) in DMF (270 mL) was stirred at room temperature for 5 minutes. CDMT (27.2 g, 154.9 mmol) and NMM (53 mL, 482.1 mmol) were added and the mixture was stirred at room temperature for 2 h. The mixture was poured into water (140 mL) and then stirred for 1 h at room temperature, then filtered and washing the solids with water (50 mL). The solids were dissolved in 1 : 1 IP A/water (-400 mL, until all solids dissolved) with heating (reflux) and stirring. The mixture was allowed to cool slowly to room temperature overnight. The mixture was cooled to 0 oC and stirred to break up crystals for filtration. The crystals were then filtered off, rinsed with cold 1 : 1 IP A/water to afford a tan solid (45 g). The solid was dissolved in IPA (200 mL) and heated to 80 °C to dissolve the solid. Activated charcoal (10 g) was added and the mixture was heated with stirring for 30 minutes. The mixture was filtered through Celite ® and solvent removed under reduced pressure. A mixture of 40:60 IP A/water (350 mL) was added to the solid and the mixture was heated until all solids dissolved. The mixture was cooled to room temperature over 5 h. Solids precipitated within the mixture. The mixture was then cooled to 0 °C and stirred for 1 h. The solids were filtered off and air dried on funnel for 1 h, then in a vacuum at 55 °C overnight to afford the product. 3-[2-(4-fluorophenyl)-lH-indol-3-yl]-N-[(3S,4R)-4-hydroxy-2-oxo-pyrrolidin-3-yl]propanamide (36.6 g, 79 %). ¾ NMR (300 MHz, Methanol-i¾) d 7.63 (ddt, J= 8.6, 5.1, 2.7 Hz, 3H), 7.35 (dt, J= 8.1, 1.0 Hz, 1H), 7.25 – 7.16 (m, 2H), 7.11 (ddd, J= 8.1, 7.0, 1.3 Hz, 1H), 7.03 (ddd, J = 8.0, 7.0, 1.2 Hz, 1H), 4.34 (td, J= 7.6, 6.8 Hz, 1H), 4.22 (d, J= 7.7 Hz, 1H), 3.55 (dd, J= 9.9, 7.5 Hz, 1H), 3.26 – 3.18 (m, 2H), 3.10 (dd, J= 9.9, 6.8 Hz, 1H), 2.69 – 2.59 (m, 2H). LCMS m/z 382.05 [M+H]+. The
product contained 0.23 % IPA by weight by NMR (1439 ppm IPA by residual solvent analysis). Purity is 99.5 % by (qNMR).
Alternative Preparation II of Compound 87 ( Indole Preparation route D)
Step 1. Synthesis of 5-(4-fluorophenyl)-5-oxo-pentanoic acid (Cl 04)
[00406] To a stirred suspension of AlCb(13.9 g, 0.10 mol) in dichloromethane (50 mL) was added a solution of tetrahydropyran-2,6-dione (5.93 g, 0.05
mol) in dichloromethane (100 mL) at 0 °C over a period of 15 minutes and stirred for 30 min. Then to the reaction mixture was added fluorobenzene (5 g, 0.05 mol) at 0 °C over a period of 15 min, gradually allowed to room temperature and stirred for 16 h. Then the reaction mixture was added to ice water (50 mL) under stirring. The resulting solid was filtered to afford a light yellow solid. The solid was diluted with 3 % NaOH solution (50 mL) and dichloromethane (50 mL). The aqueous layer was separated and acidified with IN HC1 at 0 °C. The mixture was then extracted with EtOAc (100 mL), dried over Na2SC>4, and concentrated under reduced pressure. The solid was then washed with pentane and dried to afford 5-(4-fluorophenyl)-5-oxo-pentanoic acid as an off white solid. (6 g, 53 %). ¾ NMR (300 MHz, DMSO-^) d 12.07 (s, 1H), 8.06 (d, J = 6 Hz, 1H), 8.02 (d, J = 5.4 Hz, 1H), 7.36 (t, J = 8.7 Hz, 2H), 3.06 (t, J = 12 Hz,
2H), 2.31 (t, J = 7.2 Hz, 2H), 1.86-1.78 (m, 2H). LCMS m/z 211.18 [M+H]+.
Step 2. Synthesis of 3-[2-(4-fluorophenyl)-lH-indol-3-yl]propanoic acid (Cl 01) [00407] Phenylhydrazine (Hydrochloride salt) (375.7 g, 2.6 mol) was combined with the 5-(4-fluorophenyl)-5-oxo-pentanoic acid (507.7 g, 2.4 mol) in a 12 L three-necked round-bottomed flask equipped with an overhead stirrer, temperature probe, and reflux condenser. AcOH (5 L) was added. The stirring was initiated and ZnCk (605 g, 4.44 mol) was added. The white suspension rapidly thickened after a few minutes (due to formation of the hydrazine intermediate). Approx. 500 mL of extra AcOH was added to aid stirring. The reaction was then heated to 100 °C for three hours. The reaction was cooled to room temperature and poured into water (approx. 6 L). The mixture was extracted with EtOAc (approx 8 L). The extract was washed with water, dried
(MgS04), filtered, and evaporated in vacuo to afford a golden yellow solid. The solid was triturated with approx. 4 L of 10 % EtOAc/DCM and filtered. The filter cake was washed with 50 % dichloromethane/heptane (approx 1 L). The filter cake was dissolved in 40 % EtOAc/dichloromethane (approx. 2L) and filtered over a plug of silica gel. The plug was eluted with 40 % EtOAc/ dichloromethane until the product had been eluted (checked by TLC (25 % EtOAc/ dichloromethane)). The filtrate was evaporated in vacuo to afford 382.6 g of an off-white solid (Crop 1). All filtrates were combined and evaporated in vacuo. The remaining solid was dissolved in 10 %
EtOAc/dichloromethane (approx. 1 L) and chromatographed on a 3 kg silica gel cartridge on the ISCO Torrent (isocratic gradient of 10 % EtOAc/dichloromethane). Product fractions were combined and evaporated in vacuo to afford a yellow solid that was slurried with dichloromethane, cooled under a stream of nitrogen, and filtered. The filter cake was washed with 50 % dichloromethane/heptane and dried in vacuo to afford 244.2 g of product (Crop 2). Altogether, both crops afforded 3-[2-(4-fluorophenyl)-lH-indol-3-yl]propanoic acid (626.8 g, 93 %). ¾ NMR (300 MHz, DMSO-i/e) d 12.15 (s, 1H), 11.20 (s, 1H), 7.74 – 7.62 (m, 2H), 7.57 (d, J = 7.8 Hz, 1H), 7.47 – 7.28 (m, 3H), 7.11 (ddd, J = 8.1, 7.0, 1.2 Hz, 1H), 7.02 (ddd, J = 7.9, 7.0, 1.1 Hz, 1H), 3.17 – 2.85 (m, 2H), 2.61 – 2.52 (m, 2H) ppm. 19F NMR (282 MHz, DMSO-i/e) d -114.53 ppm. LCMS m/z 284.15 [M+H]+.
Step 3. Synthesis of 3-[2-(4-fluorophenyl)-lH-indol-3-yl]-N-[(3S,4R)-4-hydroxy-2- oxo-pyrrolidin-3-yl ] propanamide (87)
[00408] A 3-L three neck RBF under nitrogen was equipped with a 150 mL addition funnel and thermocouple, then loaded with 3-[2-(4-fluorophenyl)-lH-indol-3-yl]propanoic acid (77.2 g, 228.6 mmol), (3S,4R)-3-amino-4-hydroxy-pyrrolidin-2-one
(Hydrochloride salt) (36.6 g, 239.9 mmol) and CDMT (44.2 g, 251.7 mmol). DMF (320 mL) was added and the orange slurry was cooled to -5 °C (acetone/brine/dry ice). NMM (88 mL, 800.4 mmol) was added via a funnel over 75 minutes to keep the internal temp <0 °C. The slurry was stirred at between -10 and 0 °C for 1 hour, then allowed to warm to ambient temperature progressively over 2 hours. Additional reagents were added (10 % of the initial quantities), and the mixture was stirred overnight at ambient temperature. Water (850 mL) was added over 60 minutes, maintaining the internal temperature at <25 °C (ice bath). This slow water addition allows for complete dissolution of any visible salt before precipitation of the product. The resulting thick slurry was stirred at ambient temperature overnight. The solid was recovered by filtration and washed with water (3 x 500 mL). The solid was dried under a stream of air at ambient temperature, then purified by crystallization.
Crystallization of 3- [2-( 4-fluorophenyl)-lH-indol-3-yl ]-N-[ ( 3S, 4R)-4-hydroxy-2-oxo- pyrrolidin-3-yl ] propanamide (87)
[00409] Under nitrogen atmosphere, a 2-L, 3 -neck flask equipped with addition funnel and thermocouple was charged with a light brown suspension of the crude 3-[2-(4-fluorophenyl)-lH-indol-3-yl]-N-[(3S,4R)-4-hydroxy-2-oxo-pyrrolidin-3-yljpropanamide (89.5 g) in IPA (225 mL, 2.5 vol). The slurry was heated to 50 °C and water (675 mL, 7.5 vol) was added until near-complete dissolution of solid was observed. The temperature was adjusted to 70 °C-to achieve full dissolution, yielding a clear amber solution. After 30 minutes, the heat source was removed and the mixture was cooled to ambient temperature over the weekend, stirring gently while maintaining the nitrogen atmosphere. The solid was recovered by filtration, washed with IPA:H20 = 1 :2 (2 x 300 mL, 2 x 3.3 vol) dried under a stream of air overnight to afford the product. 3-[2-(4-fluorophenyl)-lH-indol-3-yl]-N-[(3S,4R)-4-hydroxy-2-oxo-pyrrolidin-3-yl]propanamide (84.8 g, 92 %). ¾ NMR (300 MHz, DMSO-^) d 11.19 (s, 1H), 8.23 (d, J= 7.5 Hz, 1H), 7.77 (s, 1H), 7.72 – 7.63 (m, 2H), 7.60 (d, J= 7.8 Hz, 1H), 7.41 -7.31 (m, 3H), 7.12 (ddd, J= 8.1, 7.0, 1.2 Hz, 1H), 7.03 (ddd, J= 8.0, 7.0, 1.1 Hz, 1H), 5.49 (d, J= 5.0 Hz, 1H), 4.20 – 4.06 (m, 2H), 3.38 (s, 1H), 3.11 – 3.00 (m, 2H), 2.92 (dd, J= 9.4, 6.6 Hz, 1H). LCMS m/z 382.15 [M+H]+.
Crystallization of 3- [2-( 4-fluorophenyl)-lH-indol-3-yl J-N-[ ( 3S, 4R)-4-hydroxy-2-oxo- pyrrolidin-3-yl ] propanamide (87)
[00410] A 2-L, 3-neck flask equipped with addition funnel and thermocouple was charged with a light brown suspension of the crude 3-[2-(4-fluorophenyl)-lH-indol-3- yl]-N-[(3S,4R)-4-hydroxy-2-oxo-pyrrolidin-3-yl]propanamide in IPA (225 mL, 1 vol). The slurry was heated to 50 °C and water (675 mL, 3 vol) was added until near- complete dissolution of solid observed (mL). Temperature was increased to 70 °C under nitrogen (full dissolution, yielding a clear amber solution). After 30 minutes, the heat was removed and the mixture cooled to ambient temperature over the weekend, stirring gently under nitrogen atmosphere. The solid was recovered by filtration and washed with IPAiLLO = 1 :2 (2 x 300 mL).The solid was dried under a stream of air overnight to afford the product. 3-[2-(4-fluorophenyl)-lH-indol-3-yl]-N-[(3S,4R)-4-hydroxy-2-oxo- pyrrolidin-3-yl]propanamide (84.8 g, 92 %). ¾ NMR (300 MHz, DMSO-i/e) d 11.19 (s, 1H), 8.23 (d, J= 7.5 Hz, 1H), 7.77 (s, 1H), 7.72 – 7.63 (m, 2H), 7.60 (d, J= 7.8 Hz,
1H), 7.41 – 7.31 (m, 3H), 7.12 (ddd, J= 8.1, 7.0, 1.2 Hz, 1H), 7.03 (ddd, 7= 8.0, 7.0,
1.1 Hz, 1H), 5.49 (d, J= 5.0 Hz, 1H), 4.20 – 4.06 (m, 2H), 3.38 (s, 1H), 3.11 – 3.00 (m, 2H), 2.92 (dd, J= 9.4, 6.6 Hz, 1H). LCMS m/z 382.15 [M+H]+.
Large Scale Preparation of Compound 87
/- PrOAc solvate Form A
Step 1. Synthesis of 3-[2-(4-fluorophenyl)-lH-indol-3-yl]propanoic acid (C101)
[00411] To a mixture of C104 (100.0 g, 1.0 equiv) and phenyl hydrazine hydrochloride (72.2 g, 1.05 eqiv) was charged AcOH (800 mL, 8 vol). The mixture was agitated and heated to 85 °C for 16 hours. The batch was cooled to 22 °C. A vacuum was applied and the batch distill at <70°C to ~3 total volumes. The batch was cooled to 19- 25 °C. The reactor was charged with iPrOAc (800 mL, 8 vol) and then charged with water (800 mL, 8 vol). The internal temperature was adjusted to 20 – 25 °C and the biphasic mixture was stirred for no less than 0.5 h. Stirring was stopped and the phases allowed to separate for no less than 0.5 h. The lower aqueous layer was removed. 1 N HC1 (500 mL, 5 vol) was charged to the reactor. The internal temperature was adjusted to 20 – 25 °C, and the biphasic mixture was stirred for no less than 0.5 h. Stirring was stopped and phases were allowed to separate for no less than 0.5 h. The lower aqueous layer was removed. The reactor was charged with 1 N HC1 (500 mL, 5 vol). The internal temperature was adjusted to 20 – 25 °C, and the biphasic mixture was stirred for no less than 0.5 h.
Stirring was stopped and phases were allowed to separate for no less than 0.5 h. The lower aqueous layer was removed. Water (500 mL, 5 vol) was charged to the reactor.
The internal temperature was adjusted to 20 – 25 °C, and the biphasic mixture was stirred for no less than 0.5 h. Stirring was stopped and phases were allowed to separate for no less than 0.5 h. The lower aqueous layer was removed. Water (500 mL, 5 vol) was charged to the reactor. The internal temperature was adjusted to 20 – 25 °C, and the biphasic mixture was stirred for no less than 0.5 h. Stirring was stopped and phases were allowed to separate for no less than 0.5 h. The lower aqueous layer was removed. The organic phase was distilled under vacuum at <75 °C to 3 total volumes. The reactor was charged with toluene (1000 mL, 10 vol). The organic phase was distilled under vacuum at <75 °C to 5 total volumes. The reactor was charged with toluene (1000 mL, 10 vol). The organic phase was distilled under vacuum at <75 °C to 5 total volumes. The resulting slurry was heated to an internal temperature of 85 °C until complete dissolution of solids was achieved. The mixture was allowed to stir for 0.5 h at 85 °C and then cooled to an internal temperature of 19 – 25 °C over 5 h. The mixture was allowed to stir at 25 °C for no less than 2 h. The slurry was filtered. The filter cake was washed with toluene (1 x 2 vol (200 mL) and 1 x 1.5 vol (150 mL)). The solids were dried under vacuum with nitrogen bleed at 60 °C to afford product C101 (95.03 g, 70%).
Purification of Compound 87 by Recrystallization to Form A
[00412] Compound 87 as an iPrOAc solvate (17.16 g after correction for iPrOAc content, 1.0 equiv) was charged to a reactor. A mixture of IP A (77 mL, 4.5 vol) and water (137 mL, 8 vol) were charged to the reactor. The slurry was heated to an internal temperature of 75 °C. The batch was cooled to an internal temperature of 25 °C over 10 h and then stirred at 25 °C for at least 12 h. The slurry was filtered. The filter cake was washed with 36/64 IP A/water (2 x 52 mL, 2 x 3 vol). The solids were dried under vacuum with nitrogen bleed at 60 °C to afford Compound 87 as a neat, crystalline form (Form A, 15.35 g, 89%).
Synthetic Procedure
[00413] A mixture of 3-[2-(4-fluorophenyl)-lH-indol-3-yl]propanoic acid C101 (50 g, 1.0 equiv), S2 hydrochloride (28.3 g, 1.05 equiv), and CDMT (34.1 g, 1.1 equiv) was charged with 2-MeTHF (200 mL, 4 vol) and DMF (50 mL, 1 vol) and the mixture was agitated. The internal temperature adjusted to <13 °C. The reactor was charged with NMM (64.5 g, 3.5 equiv) over 1 h, while maintaining internal temperature <20 °C. The internal temperature was adjusted to 25 °C and the batch was stirred at that temperature for 14 h. The batch was cooled to 10 °C and charged with water (250 mL, 5 vol) while keeping the internal temperature <20 °C. The batch was then warmed to 20 – 25 °C. Stirring was stopped, and the phases allowed to separate for 10 min. The lower aqueous phase was removed. The aqueous layer was back extracted with 2-MeTHF (2 x 200 mL, 2 x 4 vol) at 20 – 25 °C. The combined organic phases were washed with 1 N HC1 (500 mL, 10 vol) at 20 – 25 °C by mixing for 10 min and settling for 10 min. The lower aqueous phase was removed. The organic phases were washed with 0.25 N HC1 (2 x 250 mL, 2 x 5 vol) at 20 – 25 °C by mixing for 10 min and settling for 10 min for each wash. Lower aqueous phases were removed after each wash. The organic phase was washed with water (250 mL, 5 vol) at 20 – 25 °C by mixing for 10 min and settling for 10 min. The reactor was charged with 20 wt % Nuchar RGC® and stirred for 4 h. The reaction mixture was filtered through a pad of celite®. The reactor and celite® pad were rinsed with 2-MeTHF. The combined organics were distilled under vacuum at <50 °C to 5 total volumes. The reactor was charged with iPrOAc (500 mL, 10 vol). The organic phase was distilled under vacuum at <50 °C to 5 total volumes. The mixture was charged with additional iPrOAc (400 mL, 8 vol) and distillation under vacuum was repeated. The mixture was charged with additional iPrOAc (250 mL, 5 vol), heated to an internal
temperature of 75 °C and stirred for 5 h. The slurry was cooled to 25 °C, over 5 h and stirred for no less than 12 h. The slurry was filtered and the filter cake washed with iPrOAc (2 x 50 mL, 2 x 1 vol). The solids were dried under vacuum with nitrogen bleed at 55 – 60 °C to afford Compound 87 as an iPrOAc solvate (60.38 g including 9.9% w/w iPrOAc, 80.8% yield).
Form A of Compound 87
[00414] Compound 87 hydrate form was converted to the dehydrated, neat crystalline form (Form A) after drying.
Hydrate Form A of Compound 87
[00415] A mixture of IP A (4.5 vol) and water (8 vol) was added to compound 87
(iPrOAc solvate containing ~2.5 – 11 wt% iPrOAc, 1.0 equiv). The slurry was heated to an internal temperature of 75 °C and filtered hot. The filtrate was cooled to 25 °C for at least 12 h. The slurry was filtered. The filter cake was washed with 36/64 IP A/water (2 x 3 vol). The solids were dried under vacuum with nitrogen bleed at 55 – 60 °C. The product was isolated as Hydrate form.
IPAC Solvate of Compound 87:
[00416] The large scale synthesis described above provided an iPrOAc solvate containing ~2.5 – 11 wt% iPrOAc after drying.
Amorphous Form of Compound 87
[00417] ~lg of compound 87 was dissolved in 22mL of acetone. The solution was evaporated using a Genevac. The resulted solid was dried at 60C under vacuum overnight. The dried solid was amorphous form.
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| WO-2020131807-A1 | Inhibitors of apol1 and methods of using same | 2018-12-17 | |
| US-2020377479-A1 | Inhibitors of apol1 and methods of using same | 2018-12-17 |
///////////
O=C(N[C@@H]1C(=O)NC[C@H]1O)CCc1c2ccccc2[NH]c1c1ccc(F)cc1
SIMILAR

predicted
VX 147
cas 2446816-88-0 predicted
O=C(N[C@@H]1C(=O)NC[C@H]1O)CCc1c2cc(F)cc(F)c2[NH]c1c1ccc(F)cc1
- OriginatorVertex Pharmaceuticals
- ClassSmall molecules; Urologics
- Mechanism of ActionApolipoprotein L1 inhibitors
- Orphan Drug StatusNo
- New Molecular EntityYes
Highest Development Phases
- Phase IIFocal segmental glomerulosclerosis
- Phase IKidney disorders
Most Recent Events
- 14 Apr 2020Phase-II clinical trials in Focal segmental glomerulosclerosis in USA (PO) (EudraCT2020-000185-42) (NCT04340362)
- 31 Dec 2019Vertex Pharmaceuticals completes phase I clinical trial in Focal segmental glomerulosclerosis and Kidney disorders (In volunteers) in USA (PO)
- 05 Aug 2019Vertex Pharmaceuticals plans a phase II proof-of-concept trial for focal segmental glomerulosclerosis in 2020
| NCT Number ICMJE | NCT04340362 |
|---|---|
| Other Study ID Numbers ICMJE | VX19-147-101 2020-000185-42 ( EudraCT Number ) |

{kind=link}
{kind=link}
.svg){kind=link}
{kind=link}
{kind=link}