
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Meglimin hydrochloride
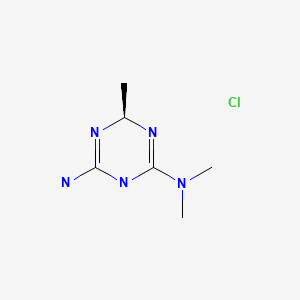

Meglimin hydrochloride
Imeglimin
hydrochloride
Twymeeg
| Formula | C6H13N5. HCl |
|---|---|
| CAS | 775351-61-6 (HCl). , C6H14ClN5 191.66CAS 775351-65-0, FREEFORM 155.20 |
| Mol weight | 191.6619 |
AntidiabeticAPPROVED PMDA JAPAN2021/6/23, イメグリミン塩酸塩
(4R)-6-N,6-N,4-trimethyl-1,4-dihydro-1,3,5-triazine-2,6-diamine
1,3,5-Triazine-2,4-diamine,1,6-dihydro-N,N,6-trimethyl-,(+)-(9CI)
(4R)-6-N,6-N,4-trimethyl-1,4-dihydro-1,3,5-triazine-2,6-diamine
JAPAN
Twymeeg Tablets 500 mg
(Sumitomo Dainippon Pharma Co., Ltd.)

Imeglimin is an experimental drug being developed as an oral anti-diabetic.[1][2] It is an oxidative phosphoryl
Imeglimin (brand name Twymeeg) is an oral anti-diabetic medication.[1][2] It was approved for use in Japan in June 2021.[3]
It is an oxidative phosphorylation blocker that acts to inhibit hepatic gluconeogenesis, increase muscle glucose uptake, and restore normal insulin secretion. It is the first approved drug of this class of anti-diabetic medication.


PATENT
https://patents.google.com/patent/WO2012072663A1/enEXAMPLESExample 1 : Synthesis and isolation of (+)-2-amino-3,6-dihydro-4-dimethylamino-6- methyl-l,3,5-triazine hydrochloride by the process according to the invention
Preliminary step: Synthesis of racemic 2-amino-3,6-dihydro-4-dimethylamino- 6-methyl-l,3,5-triazine hydrochloride:
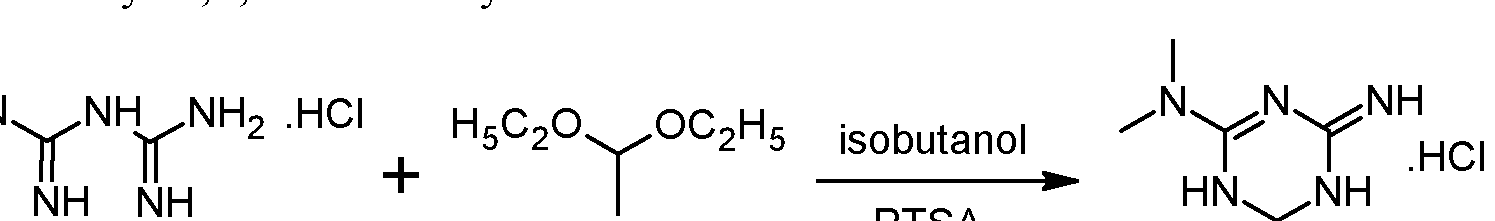
Metformin hydrochloride is suspended in 4 volumes of isobutanol. Acetaldehyde diethylacetal (1.2 eq.) and para-toluenesulfonic acid (PTSA) (0.05 eq) are added and the resulting suspension is heated to reflux until a clear solution is obtained. Then 2 volumes of the solvent are removed via distillation and the resulting suspension is cooled to 20°C. The formed crystals are isolated on a filter dryer and washed with isobutanol (0.55 volumes). Drying is not necessary and the wet product can be directly used for the next step.Acetaldehyde diethylacetal can be replaced with 2,4,6-trimethyl-l,3,5-trioxane (paraldehyde).- Steps 1 and 2: formation of the diastereoisomeric salt and isolation of the desired diastereoisomer

Racemic 2-amino-3,6-dihydro-4-dimethylamino-6-methyl-l,3,5-triazine hydrochloride wet with isobutanol (obtained as crude product from preliminary step without drying) and L-(+)-Tartaric acid (1 eq.) are dissolved in 2.2 volumes of methanol at 20-40°C. The obtained clear solution is filtered and then 1 equivalent of triethylamine (TEA) is added while keeping the temperature below 30°C. The suspension is heated to reflux, stirred at that temperature for 10 minutes and then cooled down to 55°C. The temperature is maintained at 55°C for 2 hours and the suspension is then cooled to 5- 10°C. After additional stirring for 2 hours at 5-10°C the white crystals are isolated on a filter dryer, washed with methanol (2 x 0.5 Vol) and dried under vacuum at 50°C. The yield after drying is typically in the range of 40-45%
– Steps 3 and 4: transformation of the isolated diastereoisomer of the tartrate salt into the hydrochloride salt and recovery of the salt

γ ethanol HN^NH(+) 2-amino-3,6-dihydro-4-dimethylamino-6-methyl-l,3,5-triazine tartrate salt is suspended in 2 volumes of ethanol and 1.02 equivalents of HCl-gas are added under vacuum (-500 mbar). The suspension is heated to reflux under atmospheric pressure (N2) and 5% of the solvent is removed via distillation. Subsequent filtration of the clear colourless solution into a second reactor is followed by a cooling crystallization, the temperature is lowered to 2°C. The obtained suspension is stirred at 2°C for 3 hours and afterwards the white crystals are isolated with a horizontal centrifuge. The crystal cake is washed with ethanol and dried under vacuum at 40°C. The typical yield is 50-55% and the mother liquors can be used for the recovery of about 25-30%) of (+)-2-amino- 3,6-dihydro-4-dimethylamino-6-methyl-l,3,5-triazine tartrate.Example 2: Modification of the solvent of steps 3 and 4
– Steps 3 and 4: transformation of the isolated diastereoisomer of the tartrate salt into the hydrochloride salt and recovery of the salt
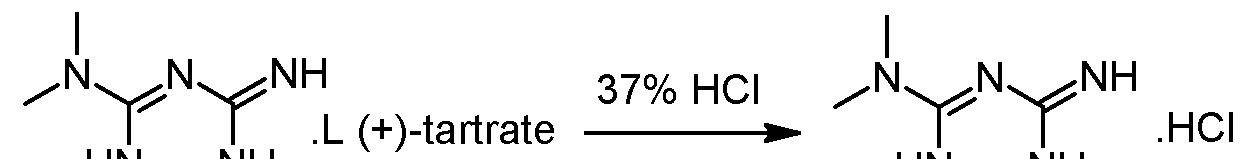
HN^NH acetone HN^NH(+) 2-amino-3,6-dihydro-4-dimethylamino-6-methyl-l,3,5-triazine tartrate salt synthesized according to steps 1 and 2 of example 1 is suspended in 1 volume (based on total amount of (+) 2-amino-3,6-dihydro-4-dimethylamino-6-methyl-l,3,5-triazine tartrate salt) of acetone at 20°C. To this suspension 1.01 equivalents of 37% Hydrochloric acid are added. The suspension is heated to reflux under atmospheric pressure (N2) and water is added until a clear solution is obtained. 1.5 vol of acetone are added at reflux temperature. The compound starts crystallising and the obtained suspension is kept at reflux for 2 hours followed by a cooling crystallization to 0°C. The obtained suspension is stirred at 0°C for 2 hours and the white crystals are isolated by centrifugation. The crystal cake is washed with isopropanol and dried under vacuum at 40°C in a continuous drying oven.
References
- ^ Vuylsteke V, Chastain LM, Maggu GA, Brown C (September 2015). “Imeglimin: A Potential New Multi-Target Drug for Type 2 Diabetes”. Drugs in R&D. 15 (3): 227–32. doi:10.1007/s40268-015-0099-3. PMC 4561051. PMID 26254210.
- ^ Dubourg J, Fouqueray P, Thang C, Grouin JM, Ueki K (April 2021). “Efficacy and Safety of Imeglimin Monotherapy Versus Placebo in Japanese Patients With Type 2 Diabetes (TIMES 1): A Double-Blind, Randomized, Placebo-Controlled, Parallel-Group, Multicenter Phase 3 Trial”. Diabetes Care. 44 (4): 952–959. doi:10.2337/dc20-0763. PMID 33574125.
- ^ Poxel SA (June 23, 2021). “Poxel and Sumitomo Dainippon Pharma Announce the Approval of TWYMEEG® (Imeglimin hydrochloride) for the Treatment of Type 2 Diabetes in Japan” (Press release).
| Clinical data | |
|---|---|
| Trade names | Twymeeg |
| Legal status | |
| Legal status | Rx-only in Japan |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 775351-65-0 |
| PubChem CID | 24812808 |
| ChemSpider | 26232690 |
| UNII | UU226QGU97 |
| CompTox Dashboard (EPA) | DTXSID50228237 |
| Chemical and physical data | |
| Formula | C6H13N5 |
| Molar mass | 155.205 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
/////////Imeglimin hydrochloride, Twymeeg, JAPAN 2021, APPROVALS 2021, Antidiabetic, イメグリミン塩酸塩, ATI DIABETES, DIABETES, Imeglimin
CC1N=C(NC(=N1)N(C)C)N.Cl
NEW DRUG APPROVALS
ONE TIME
$10.00
CVnCoV, zorecimeran, CureVac COVID-19 vaccine
CVnCoV
cas 2541470-90-8
An optimized, non-chemical modified mRNA encoding the prefusion-stabilized full-length spike protein of SARS-CoV-2 virus (Curevac)
zorecimeran, CureVac COVID-19 vaccine
CureVac/Bayer
GSK
NCT04674189 NCT04449276 NCT04515147 NCT04652102
EudraCT-2020-004066-19
mRNA-based vaccine
PHASE 3
| CVnCoV | Humoral and cellular responses | CD4+ T-cells, CD8+ T-cells | N/A | N/A | Rhesus macaque | [124] |
124. Rauch S, Gooch K, Hall Y, Salguero FJ, Dennis MJ, Gleeson FV. et al. mRNA vaccine CVnCoV protects non-human primates from SARS-CoV-2 challenge infection. bioRxiv. 2020. 2020 12.23.424138
The CureVac COVID-19 vaccine is a COVID-19 vaccine candidate developed by CureVac N.V. and the Coalition for Epidemic Preparedness Innovations (CEPI).[1] The vaccine showed inadequate results in its Phase III trials with only 47% efficacy.[2] The European Medicines Agency stated that: “(…) medicine developers should design studies to demonstrate a rate of efficacy of at least 50%.”[3].
The CVnCov Vaccine (or CV07050101) is in development by CureVac AG. The vaccine uses mRNA technology to create a protein associated with SARS-CoV2, and upon administration and replication, to initiate subsequent immune responses in the body. As of June 2020, the company received regulatory approval from German and Belgian Authorities to commence Phase 1 clinical trials of this vaccine (NCT04449276).
Efficacy
On 16 June 2021,[4] CureVac said its vaccine showed 47% efficacy from its Phase III trial. This was based on interim analysis of 134 COVID cases in its Phase III study conducted in Europe and Latin America. The final analysis for the trials requires a minimum of 80 additional cases.[2]
Pharmacology
CVnCoV is an mRNA vaccine that encodes the full-length, pre-fusion stabilized coronavirus spike protein, and activates the immune system against it.[5][6][7] CVnCoV technology does not interact with the human genome.[6] CVnCoV uses unmodified RNA,[8] unlike the Pfizer–BioNTech COVID-19 vaccine and Moderna COVID-19 vaccine, which both use nucleoside-modified RNA.[9]
Manufacturing
Manufacturing of mRNA vaccines can be performed rapidly in high volume,[10] including use of portable, automated printers (“RNA microfactories”) for which CureVac has a joint development partnership with Tesla.[11]
mRNA vaccines require stringent cold chain refrigeration throughout manufacturing, distribution and storage.[12][13] The CureVac technology for CVnCoV uses a non-modified, more natural mRNA less affected by hydrolysis, enabling storage at 5 °C (41 °F) and relatively simplified cold chain requirements that facilitate up to three months of storage and distribution to world regions that do not have specialized ultracold equipment.[6][10]
CureVac has a European-based network to accelerate manufacturing of CVnCoV, if proven safe and effective, for production of up to 300 million doses in 2021 and 600 million doses in 2022.[10][14] An estimated 405 million doses will be provided to EU states.[14]
Clinical trials
In November 2020, CureVac reported results of a Phase I-II clinical trial that CVnCoV (active ingredient zorecimeran) was well-tolerated, safe, and produced a robust immune response.[15][16]
In December 2020, CureVac began a Phase III clinical trial of CVnCoV with 36,500 participants.[17][18] Bayer will provide clinical trial support and international logistics for the Phase III trial, and may be involved in eventual manufacturing should the vaccine prove to be safe and effective.[19][20] In February 2021, the EU’s CHMP started a rolling review of CVnCoV.[21][22] In April 2021, the same procedure began in Switzerland.[23]
Brand names
The manufacturer currently markets the vaccine under the name CVnCoV.[24] Zorecimeran is the proposed international nonproprietary name (pINN).[25]
References
- ^ “CureVac focuses on the development of mRNA-based coronavirus vaccine to protect people worldwide”. CureVac(Press release). 15 March 2020. Retrieved 17 February 2021.
- ^ Jump up to:a b Burger, Ludwig (16 June 2021). “CureVac fails in pivotal COVID-19 vaccine trial with 47% efficacy”. Reuters. Retrieved 17 June 2021.
- ^ https://www.ema.europa.eu/en/human-regulatory/overview/public-health-threats/coronavirus-disease-covid-19/treatments-vaccines/vaccines-covid-19/covid-19-vaccines-studies-approval#what-is-the-level-of-efficacy-that-can-be-accepted-for-approval?-section
- ^ “CureVac Provides Update on Phase 2b/3 Trial of First-Generation COVID-19 Vaccine Candidate, CVnCoV”. 16 June 2021.
- ^ https://www.curevac.com/wp-content/uploads/2020/10/20201023-CureVac-Manuscript-draft-preclinical-data.pdf
- ^ Jump up to:a b c Schlake T, Thess A, Fotin-Mleczek M, Kallen KJ (November 2012). “Developing mRNA-vaccine technologies”. RNA Biology. 9(11): 1319–30. doi:10.4161/rna.22269. PMC 3597572. PMID 23064118.
- ^ “Understanding mRNA COVID-19 vaccines”. US Centers for Disease Control and Prevention. 18 December 2020. Retrieved 5 January 2021.
- ^ “COVID-19”. CureVac. Retrieved 21 December 2020.
- ^ Dolgin, Elie (25 November 2020). “COVID-19 vaccines poised for launch, but impact on pandemic unclear”. Nature Biotechnology: d41587–020–00022-y. doi:10.1038/d41587-020-00022-y. PMID 33239758. S2CID 227176634.
- ^ Jump up to:a b c Nawrat A (3 December 2020). “Q&A with CureVac: resolving the ultra-cold chain logistics of Covid-19 mRNA vaccines”. Pharmaceutical Technology. Retrieved 5 January 2021.
- ^ “Tesla to make molecule printers for German COVID-19 vaccine developer CureVac”. Reuters. 2 July 2020. Retrieved 19 December 2020.
- ^ Kartoglu U, Milstien J (July 2014). “Tools and approaches to ensure quality of vaccines throughout the cold chain”. Expert Review of Vaccines. 13 (7): 843–54. doi:10.1586/14760584.2014.923761. PMC 4743593. PMID 24865112.
- ^ Hanson CM, George AM, Sawadogo A, Schreiber B (April 2017). “Is freezing in the vaccine cold chain an ongoing issue? A literature review”. Vaccine. 35 (17): 2127–2133. doi:10.1016/j.vaccine.2016.09.070. PMID 28364920.
- ^ Jump up to:a b Kansteiner F (17 November 2020). “CureVac, armed with COVID-19 vaccine deal, plots ‘pandemic-scale’ Euro manufacturing expansion”. FiercePharma, Questex LLC. Retrieved 5 January2021.
- ^ “CureVac’s Covid-19 vaccine induces immune response in study”. Clinical Trials Arena. 3 November 2020. Retrieved 5 January 2021.
- ^ “CureVac’s COVID-19 vaccine triggers immune response in Phase I trial”. Reuters. 2 November 2020. Retrieved 5 January2021.
- ^ “Multicenter Clinical Study Evaluating the Efficacy and Safety of Investigational SARS-CoV-2 mRNA Vaccine CVnCoV in Adults 18 Years of Age and Older”. EU Clinical Trials Register. 19 November 2020. Retrieved 5 January 2021.
Proposed INN: zorecimeran
- ^ “A Study to Determine the Safety and Efficacy of SARS-CoV-2 mRNA Vaccine CVnCoV in Adults”. ClinicalTrials.gov. 8 December 2020. NCT04652102. Retrieved 19 December 2020.
- ^ Burger L (7 January 2021). “CureVac strikes COVID-19 vaccine alliance with Bayer”. Reuters. Retrieved 17 February 2021.
- ^ “CureVac and Bayer join forces on COVID-19 vaccine candidate CVnCoV”. CureVac (Press release). 7 January 2021. Retrieved 17 February 2021.
- ^ “EMA starts rolling review of CureVac’s COVID-19 vaccine (CVnCoV)”. European Medicines Agency (EMA) (Press release). 11 February 2021. Retrieved 12 February 2021.
- ^ “CureVac Initiates Rolling Submission With European Medicines Agency for COVID-19 Vaccine Candidate, CVnCoV”. CureVac(Press release).
- ^ “CureVac starts review process in Switzerland for COVID-19 vaccine hopeful”. Reuters. 19 April 2021. Retrieved 19 April 2021.
- ^ “Celonic and CureVac Announce Agreement to Manufacture over 100 Million Doses of CureVac’s COVID-19 Vaccine Candidate, CVnCoV”. CureVac (Press release). 30 March 2021. Retrieved 14 April 2021.
- ^ World Health Organization (October 2020). “International Nonproprietary Names for Pharmaceutical Substances (INN). Proposed INN: List 124 – COVID-19 (special edition)” (PDF). WHO Drug Information. 34 (3): 668–69. Archived (PDF) from the original on 27 November 2020.
External links
| Scholia has a profile for zorecimeran (Q97154239). |
- “Zorecimeran”. Drug Information Portal. U.S. National Library of Medicine.
| Vaccine description | |
|---|---|
| Target | SARS-CoV-2 |
| Vaccine type | mRNA |
| Clinical data | |
| Other names | CVnCoV, CV07050101 |
| Routes of administration | Intramuscular |
| ATC code | None |
| Identifiers | |
| DrugBank | DB15844 |
| UNII | 5TP24STD1S |
| Part of a series on the |
| COVID-19 pandemic |
|---|
| COVID-19 (disease)SARS-CoV-2 (virus) |
| showTimeline |
| showLocations |
| showInternational response |
| showMedical response |
| showImpact |
| COVID-19 portal |
- Rego GNA, Nucci MP, Alves AH, Oliveira FA, Marti LC, Nucci LP, Mamani JB, Gamarra LF: Current Clinical Trials Protocols and the Global Effort for Immunization against SARS-CoV-2. Vaccines (Basel). 2020 Aug 25;8(3). pii: vaccines8030474. doi: 10.3390/vaccines8030474. [Article]
- Speiser DE, Bachmann MF: COVID-19: Mechanisms of Vaccination and Immunity. Vaccines (Basel). 2020 Jul 22;8(3). pii: vaccines8030404. doi: 10.3390/vaccines8030404. [Article]
- CureVac & Covid-19 [Link]
- Smart Patients [Link]
- Regulatory News [Link]
////////////zorecimeran, CVnCoV, CV07050101, CORONA VACCINE, COVID 19, VACCINE, CUREVAC, SARS-CoV-2, CV07050101, SARS-CoV-2 mRNA vaccine
NEW DRUG APPROVALS
one time
$10.00
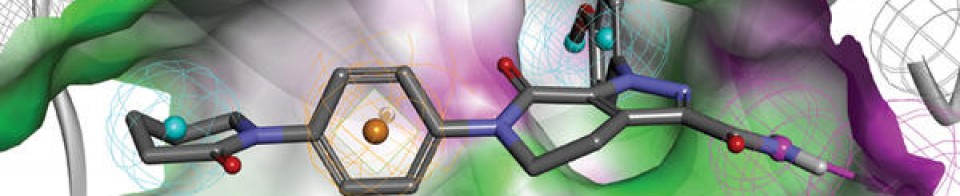
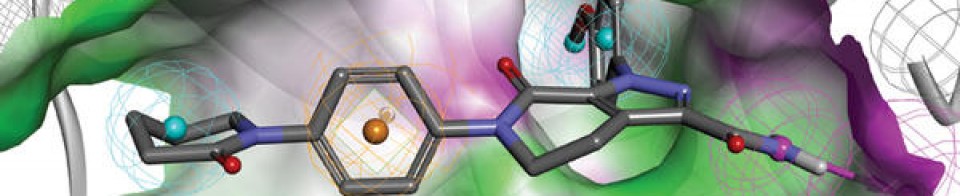
Tucidinostat, Chidamide

Tucidinostat, Chidamide
ツシジノスタット
2021/6/23 PMDA JAPAN APPROVED,
| Hiyasta |
| Formula | C22H19FN4O2 |
|---|---|
| CAS | 1616493-44-7 |
| Mol weight | 390.4103 |
Antineoplastic, Histone deacetylase inhibitor
Chidamide (Epidaza) is a histone deacetylase inhibitor (HDI) developed in China.[1] It was also known as HBI-8000.[2] It is a benzamide HDI and inhibits Class I HDAC1, HDAC2, HDAC3, as well as Class IIb HDAC10.[3]
Chidamide is approved by the Chinese FDA for relapsed or refractory peripheral T-cell lymphoma (PTCL), and has orphan drug status in Japan.[2][better source needed] As of April 2015 it is only approved in China.[1]
Chidamide is being researched as a treatment for pancreatic cancer.[4][5][6] However, it is not US FDA approved for the treatment of pancreatic cancer.
Chidamide (Epidaza®), a class I HDAC inhibitor, was discovered and developed by ChipScreen and approved by the CFDA in December 2014 for the treatment of recurrent of refractory peripheral T-cell lymphoma. Chidamide, also known as CS055 and HBI- 8000, is an orally bioavailable benzamide type inhibitor of HDAC isoenzymes class I 1–3, as well as class IIb 10, with potential antineoplastic activity. It selectively binds to and inhibits HDAC, leading to an increase in acetylation levels of histone protein H3.74 This agent also inhibits the expression of signaling kinases in the PI3K/ Akt and MAPK/Ras pathways and may result in cell cycle arrest and the induction of tumor cell apoptosis. Currently, phases I and II clinical trials are underway for the treatment of non-small cell lung cancer and for the treatment of breast cancer, respectively.
Chemical Synthesis
The scalable synthetic approach to chidamide very closely follows the discovery route. The sequence began with the condensation of commercial nicotinaldehyde (52) and malonic acid (53) in a mixture of pyridine and piperidine. Next, activation of acid 54 with N,N0-carbonyldiimidazole (CDI) and subsequent reaction with 4-aminomethyl benzoic acid (55) under basic conditions afforded amide 56 in 82% yield. Finally, activation of 56 with CDI prior to treatment with 4-fluorobenzene- 1,2-diamine (57) and subsequent treatment with TFA and THF yielded chidamide (VIII) in 38% overall yield from 52. However, no publication reported that mono-N-Boc-protected bis-aniline was used to approach Chidamide.
References
- ^ Jump up to:a b Lowe D (April 2015). “China’s First Homegrown Pharma”. Seeking Alpha.
- ^ Jump up to:a b “Chipscreen Biosciences Announces CFDA Approval of Chidamide (Epidaza) for PTCLs in China”. PR Newswire Association LLC.
- ^ “HUYA Bioscience International Grants An Exclusive License For HBI-8000 In Japan And Other Asian Countries To Eisai”. PR Newswire Association LLC. February 2016.
- ^ Qiao Z, Ren S, Li W, Wang X, He M, Guo Y, et al. (April 2013). “Chidamide, a novel histone deacetylase inhibitor, synergistically enhances gemcitabine cytotoxicity in pancreatic cancer cells”. Biochemical and Biophysical Research Communications. 434 (1): 95–101. doi:10.1016/j.bbrc.2013.03.059. PMID 23541946.
- ^ Guha M (April 2015). “HDAC inhibitors still need a home run, despite recent approval”. Nature Reviews. Drug Discovery. 14 (4): 225–6. doi:10.1038/nrd4583. PMID 25829268. S2CID 36758974.
- ^ Wang SS (2015-04-02). “A New Cancer Drug, Made in China”. The Wall Street Journal. Retrieved 13 April 2015.
| Clinical data | |
|---|---|
| Trade names | Epidaza |
| Other names | Tucidinostat |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1616493-44-7 |
| PubChem CID | 9800555 |
| ChemSpider | 7976319 |
| UNII | 87CIC980Y0 |
| Chemical and physical data | |
| Formula | C22H19FN4O2 |
| Molar mass | 390.418 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
/////Tucidinostat, Antineoplastic, Histone deacetylase inhibitor, ツシジノスタット , Epidaza, Chidamide, APPROVALS 2021, JAPAN 2021
NEW DRUG APPROVALS
one time
$10.00
Isotretinoin

Isotretinoin
Title: Isotretinoin
CAS Registry Number: 4759-48-2
CAS Name: 13-cis-Retinoic acid
Additional Names: 2-cis-vitamin A acid; neovitamin A acid
Manufacturers’ Codes: Ro-4-3780Trademarks: Accutane (Roche); Isotrex (Stiefel); Oratane (Douglas); Roaccutane (Roche)
Molecular Formula: C20H28O2Molecular Weight: 300.44Percent Composition: C 79.95%, H 9.39%, O 10.65%
Literature References: Naturally occurring metabolite of vitamin A, q.v.; inhibits sebum production. Prepn: C. D. Robeson et al.,J. Am. Chem. Soc.77, 4111 (1955). Stereoselective process: R. Lucci, EP111325; idem,US4556518 (1984, 1985 both to Hoffmann-La Roche). Toxicology and teratogenicity study: J. J. Kamm, J. Am. Acad. Dermatol.6, 652 (1982). Identification as endogenous metabolite of all-trans-retinoic acid: M. E. Cullum, M. H. Zile, J. Biol. Chem.260, 10590 (1985). HPLC determn in serum: G. Tang, R. M. Russell, J. Lipid Res.31, 175 (1990). Review of pharmacology and clinical efficacy in acne: A. R. Shalita et al.,Cutis42, Suppl. 6A, 1-19 (1988). Symposium on clinical experience: Dermatology195, Suppl. 1, 1-37 (1997).
Properties: Reddish-orange plates from isopropyl alcohol, mp 174-175°. uv max: 354 nm (e 39800). LD50 (20 day) in mice, rats (mg/kg): 904, 901 i.p.; 3389, >4000 orally (Kamm).
Melting point: mp 174-175°Absorption maximum: uv max: 354 nm (e 39800)Toxicity data: LD50 (20 day) in mice, rats (mg/kg): 904, 901 i.p.; 3389, >4000 orally (Kamm)Therap-Cat: Antiacne.Keywords: Antiacne.
Isotretinoin, also known as 13-cis-retinoic acid and sold under the brand name Accutane among others, is a medication primarily used to treat severe acne. It is also used to prevent certain skin cancers (squamous-cell carcinoma), and in the treatment of other cancers. It is used to treat harlequin-type ichthyosis, a usually lethal skin disease, and lamellar ichthyosis. It is a retinoid, meaning it is related to vitamin A, and is found in small quantities naturally in the body. Its isomer, tretinoin, is also an acne drug.
The most common adverse effects are a transient worsening of acne (lasting 1–4 months), dry lips (cheilitis), dry and fragile skin, and an increased susceptibility to sunburn. Uncommon and rare side effects include muscle aches and pains (myalgias), and headaches. Isotretinoin is known to cause birth defects due to in-utero exposure because of the molecule’s close resemblance to retinoic acid, a natural vitamin A derivative which controls normal embryonic development. It is also associated with psychiatric side effects, most commonly depression but also, more rarely, psychosis and unusual behaviours. Other rare side effects include hyperostosis, and premature epiphyseal closure, have been reported to be persistent.
In the United States, a special procedure is required to obtain the pharmaceutical. In most other countries, a consent form is required which explains these risks. In other countries, such as Israel, it is prescribed like any other medicine from a dermatologist (after proper blood tests).
Women taking isotretinoin must not get pregnant during and for one month after the discontinuation of isotretinoin therapy. Sexual abstinence or effective contraception is mandatory during this period. Barrier methods by themselves (e.g., condoms) are not considered adequate due to the unacceptable failure rates of approximately 3%. Women who become pregnant while taking isotretinoin therapy are generally counseled to have an abortion.
It was patented in 1969 and approved for medical use in 1982.[2] It sold well, but in 2009, Roche decided to discontinue manufacturing due to diminishing market share due to the availability of the many generic versions and the settling of multiple lawsuits over side effects. It continues to be manufactured as of 2019 by Absorica, Amnesteem, Claravis, Myorisan, Sotret, and Zenatane.[3]
Medical uses
Isotretinoin is used primarily for severe cystic acne and acne that has not responded to other treatments.[4][5][6][7] Many dermatologists also support its use for treatment of lesser degrees of acne that prove resistant to other treatments, or that produce physical or psychological scarring.[8] Isotretinoin is not indicated for treatment of prepubertal acne and is not recommended in children less than 12 years of age.[9]
It is also somewhat effective for hidradenitis suppurativa and some cases of severe rosacea.[10] It can also be used to help treat harlequin ichthyosis, lamellar ichthyosis and is used in xeroderma pigmentosum cases to relieve keratoses. Isotretinoin has been used to treat the extremely rare condition fibrodysplasia ossificans progressiva. It is also used for treatment of neuroblastoma, a form of nerve cancer.
Isotretinoin therapy has furthermore proven effective against genital warts in experimental use, but is rarely used for this indication as there are more effective treatments. Isotretinoin may represent an efficacious and safe alternative systemic form of therapy for recalcitrant condylomata acuminata (RCA) of the cervix. In most countries this therapy is currently unapproved and only used if other therapies failed.[11][12]
Prescribing restrictions
Isotretinoin is a teratogen; there is about a 20–35% risk for congenital defects in infants exposed to the drug in utero, and about 30–60% of children exposed to isotretinoin prenatally have been reported to show neurocognitive impairment.[13] Because of this, there are strict controls on prescribing isotretinoin to women who may become pregnant and women who become pregnant while taking isotretinoin are strongly advised to terminate their pregnancies.[13]
In most countries, isotretinoin can only be prescribed by dermatologists or specialist physicians; some countries also allow limited prescription by general practitioners and family doctors. In the United Kingdom[14] and Australia,[15][16] isotretinoin may be prescribed only by or under the supervision of a consultant dermatologist. Because severe cystic acne has the potential to cause permanent scarring over a short period, restrictions on its more immediate availability have proved contentious.[17] In New Zealand, isotretinoin can be prescribed by any doctor but subsidised only when prescribed by a vocationally-registered general practitioner, dermatologist or nurse practitioner.[18]
In the United States, since March 2006 the dispensing of isotretinoin is run through a website called iPLEDGE. The FDA required the companies marketing the drug in the US, which at the time that iPLEDGE was launched were Roche, Mylan, Barr, and Ranbaxy, to put this website in place as a risk evaluation and mitigation strategy. These companies formed a group called the Isotretinoin Products Manufacturing Group, and it hired Covance to run the website.[19][20] Prescribers, pharmacists, and all people to whom the drug is prescribed need to register on the site and log information into it. Women with child-bearing potential must commit to using two forms of effective contraception simultaneously for the duration of isotretinoin therapy and for a month immediately preceding and a month immediately following therapy. Additionally they must have two negative pregnancy tests 30 days apart and have negative pregnancy tests before each prescription is written.[21][22]
History[edit]
The compound 13-cis retinoic acid was first studied in the 1960s at Roche Laboratories in Switzerland by Werner Bollag as a treatment for skin cancer. Experiments completed in 1971 showed that the compound was likely to be ineffective for cancer and, surprisingly, that it could be useful to treat acne. However, they also showed that the compound was likely to cause birth defects, so in light of the events around thalidomide, Roche abandoned the product. In 1975, Gary Peck and Frank Yoder independently rediscovered the drug’s use as a treatment of cystic acne while studying it as a treatment for lamellar ichthyosis, and published that work. Roche resumed work on the drug. In clinical trials, subjects were carefully screened to avoid including women who were or might become pregnant. Roche’s New Drug Application for isotretinoin for the treatment of acne included data showing that the drug caused birth defects in rabbits. The FDA approved the application in 1982.
Scientists involved in the clinical trials published articles warning of birth defects at the same time the drug was launched in the US, but nonetheless isotretinoin was taken up quickly and widely, both among dermatologists and general practitioners. Cases of birth defects showed up in the first year, leading the FDA to begin publishing case reports and to Roche sending warning letters to doctors and placing warning stickers on drug bottles, and including stronger warnings on the label. Lawsuits against Roche started to be filed. In 1983 the FDA’s advisory committee was convened and recommended stronger measures, which the FDA took and were that time unprecedented: warning blood banks not to accept blood from people taking the drug, and adding a warning to the label advising women to start taking contraceptives a month before starting the drug. However use of the drug continued to grow, as did the number of babies born with birth defects. In 1985 the label was updated to include a boxed warning. In early 1988 the FDA called for another advisory committee, and FDA employees prepared an internal memo estimating that around 1,000 babies had been born with birth defects due to isotretinoin, that up to around 1,000 miscarriages had been caused, and that between 5,000 and 7,000 women had had abortions due to isotretinoin. The memo was leaked to the New York Times[77] a few days before the meeting, leading to a storm of media attention. In the committee meeting, dermatologists and Roche each argued to keep the drug on the market but to increase education efforts; pediatricians and the CDC argued to withdraw the drug from the market. The committee recommended to restrict physicians who could prescribe the drug and to require a second opinion before it could be prescribed. The FDA, believing it did not have authority under the law to restrict who had the right to prescribe the drug, kept the drug on the market but took further unprecedented measures: it required to Roche to make warnings yet more visible and graphic, provide doctors with informed consent forms to be used when prescribing the drug, and to conduct follow up studies to test whether the measures were reducing exposure of pregnant women to the drug. Roche implemented those measures, and offered to pay for contraception counseling and pregnancy testing for women prescribed the drug; the program was called the “Pregnancy Prevention Program”.
A CDC report published in 2000[78] showed problems with the Pregnancy Prevention Program and showed that the increase in prescriptions was from off-label use, and prompted Roche to revamp its program, renaming it the “Targeted Pregnancy Prevention Program” and adding label changes like requirements for two pregnancy tests, two kinds of contraception, and for doctors to provide pharmacists with prescriptions directly; providing additional educational materials, and providing free pregnancy tests. The FDA had another advisory meeting in late 2000 that again debated how to prevent pregnant women from being exposed to the drug; dermatologists testified about the remarkable efficacy of the drug, the psychological impact of acne, and demanded autonomy to prescribe the drug; others argued that the drug be withdrawn or much stricter measures be taken. In 2001 the FDA announced a new regulatory scheme called SMART (the System to Manage Accutane Related Teratogenicity) that required Roche to provide defined training materials to doctors, and for doctors to sign and return a letter to Roche acknowledging that they had reviewed the training materials, for Roche to then send stickers to doctors, which doctors would have to place on prescriptions they give people after they have confirmed a negative pregnancy test; prescriptions could only be written for 30 days and could not be renewed, thus requiring a new pregnancy test for each prescription.[citation needed]
In February 2002, Roche’s patents for isotretinoin expired, and there are now many other companies selling cheaper generic versions of the drug. On June 29, 2009, Roche Pharmaceuticals, the original creator and distributor of isotretinoin, officially discontinued both the manufacture and distribution of their Accutane brand in the United States due to what the company described as business reasons related to low market share (below 5%), coupled with the high cost of defending personal-injury lawsuits brought by some people who took the drug.[79] Generic isotretinoin will remain available in the United States through various manufacturers. Roche USA continues to defend Accutane and claims to have treated over 13 million people since its introduction in 1982. F. Hoffmann-La Roche Ltd. apparently will continue to manufacture and distribute Roaccutane outside of the United States.[80]
Among others, actor James Marshall sued Roche over allegedly Accutane-related disease that resulted in removal of his colon.[81] The jury, however, decided that James Marshall had a pre-existing bowel disease.[82]
Several trials over inflammatory bowel disease claims have been held in the United States thus far, with many of them resulting in multimillion-dollar judgments against the makers of isotretinoin.[83]
Society and culture
Brands
As of 2017 isotretinoin was marketed under many brand names worldwide: A-Cnotren, Absorica, Accuran, Accutane, Accutin, Acne Free, Acnecutan, Acnegen, Acnemin, Acneone, Acneral, Acnestar, Acnetane, Acnetin A, Acnetrait, Acnetrex, Acnogen, Acnotin, Acnotren, Acretin, Actaven, Acugen, Acutret, Acutrex, Ai Si Jie, Aisoskin, Aknal, Aknefug Iso, Aknenormin, Aknesil, Aknetrent, Amnesteem, Atlacne, Atretin, Axotret, Casius, Ciscutan, Claravis, Contracné, Curacne, Curacné, Curakne, Curatane, Cuticilin, Decutan, Dercutane, Effederm, Epuris, Eudyna, Farmacne, Flexresan, Flitrion, I-Ret, Inerta, Inflader, Inotrin, Isac, Isdiben, Isoacne, Isobest, Isocural, Isoderm, Isoface, IsoGalen, Isogeril, Isolve, Isoprotil, Isoriac, Isosupra, Isosupra Lidose, Isotane, Isotina, Isotinon, Isotren, Isotret, Isotretinoin, Isotretinoina, Isotretinoína, Isotretinoine, Isotretinoïne, Isotrétinoïne, Isotretinoinum, Isotrex, Isotrin, Isotroin, Izotek, Izotziaja, Lisacne, Locatret, Mayesta, Myorisan, Neotrex, Netlook, Nimegen, Noitron, Noroseptan, Novacne, Oralne, Oraret, Oratane, Piplex, Policano, Procuta, Reducar, Retin A, Roaccutan, Roaccutane, Roacnetan, Roacta, Roacutan, Rocne, Rocta, Sotret, Stiefotrex, Tai Er Si, Teweisi, Tretin, Tretinac, Tretinex, Tretiva, Tufacne, Zenatane, Zerocutan, Zonatian ME, and Zoretanin.[1]
As of 2017 it was marketed as a topical combination drug with erythromycin under the brand names Isotrex Eritromicina, Isotrexin, and Munderm.[1]
Research
While excessive bone growth has been raised a possible side effect, a 2006 review found little evidence for this.[84]
syn

C. D. Robeson et al., J. Am. Chem. Soc. 77, 4111 (1955). Stereoselective process: R. Lucci, EP 111325; idem, US 4556518 (1984, 1985 both to Hoffmann-La Roche). doi:10.1021/jo00349a001.
syn
J Chem Soc 1968,(16),1982-83
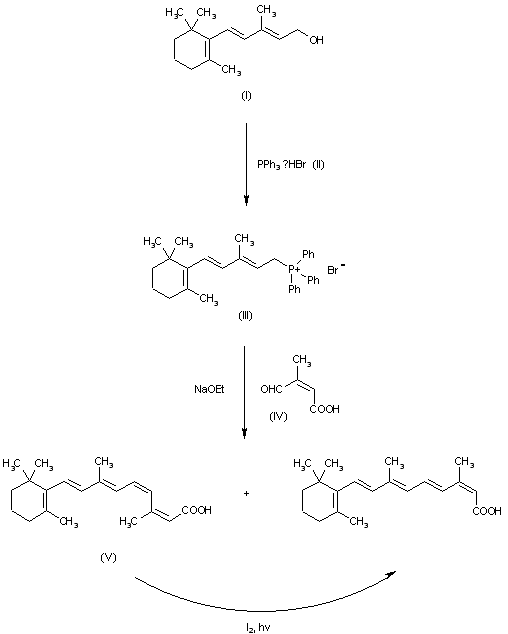
The reaction of vinyl-beta-ionol (I) with triphenylphosphonium bromide (II) in ethanol gives the corresponding phosphonium salt (III), which is condensed through a Wittig reaction with cis-beta-formylcrotonic acid (IV) by means of sodium ethoxide in ethanol to afford a mixture of cis-2-cis-4-vitamin A acid (V) and the desired product. Finally, compound (V) is isomerized bv irradiation with diffuse light in ether in the presence of iodine.
syn
Tetrahedron 2000,56(37),7211
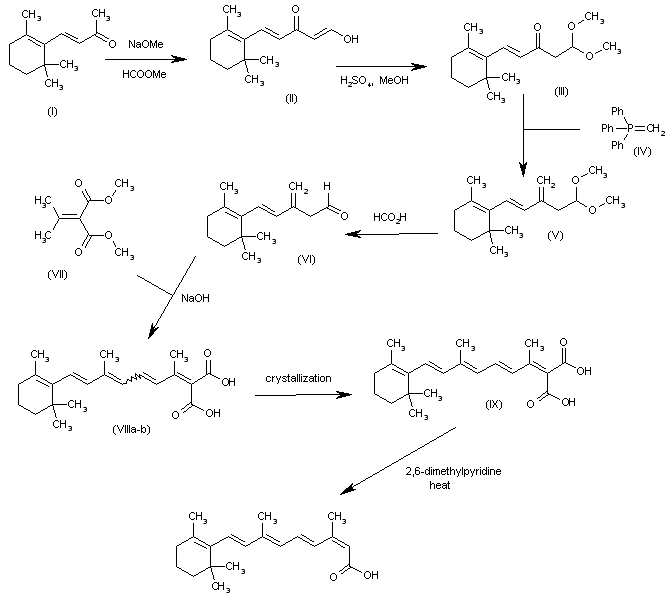
The formylation of the beta-ionone (I) with methyl formate and NaOMe gives the enol (II), which by reaction with methanol and H2SO4 yields the dimethylacetal (III). The reaction of (III) with methylenetriphenylphosphorane (IV) affords the methylene compound (V), which is treated with formic acid to provide the aldehyde (VI). The condensation of (VI) with isopropylidenemalonic acid dimethyl ester (VII) by means of NaOH gives the polyenic malonic acid (VIII) as a mixture of isomers that is separated by crystallization in ethyl ether to yield the desired all-trans-isomer (IX). Finally, this malonic acid is selectively monodecarboxylated by means of refluxing 2,6-dimethylpyridine to afford the target (E,E,E,Z)-isomer.
References
- ^ Jump up to:a b c “Isotretinoin international brands”. Drugs.com. Retrieved 1 June 2017.
- ^ Fischer J, Ganellin CR (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 476. ISBN 978-3-527-60749-5.
- ^ “Isotretinoin (Oral Route) Description and Brand Names – Mayo Clinic”.
- ^ Merritt B, Burkhart CN, Morrell DS (June 2009). “Use of isotretinoin for acne vulgaris”. Pediatric Annals. 38 (6): 311–20. doi:10.3928/00904481-20090512-01. PMID 19588674.
- ^ Jump up to:a b Layton A (May 2009). “The use of isotretinoin in acne”. Dermato-Endocrinology. 1(3): 162–9. doi:10.4161/derm.1.3.9364. PMC 2835909. PMID 20436884.
- ^ Jump up to:a b c “Roaccutane 20mg Soft Capsules – Summary of Product Characteristics”. UK Electronic Medicines Compendium. 1 July 2015.
- ^ Jump up to:a b c US Label (PDF) (Report). FDA. 22 October 2010 [January 2010]. Retrieved 1 June2017. See FDA Index page for NDA 018662 for updates
- ^ Strauss JS, Krowchuk DP, Leyden JJ, Lucky AW, Shalita AR, Siegfried EC, Thiboutot DM, Van Voorhees AS, Beutner KA, Sieck CK, Bhushan R (April 2007). “Guidelines of care for acne vulgaris management”. Journal of the American Academy of Dermatology. 56 (4): 651–63. doi:10.1016/j.jaad.2006.08.048. PMID 17276540.
- ^ Jump up to:a b c d “Isotretinoin (oral formulations): CMDH scientific conclusions – Scientific conclusions and grounds for the variation to the terms of the Marketing Authorisation(s)”(PDF). European Medicines Agency. August 2017. Retrieved 17 May 2019.
- ^ Jump up to:a b Klasco RK, editor. Drugdex system, vol. 128. Greenwood Village (CO): Thomson Micromedex; 2006.[page needed]
- ^ Georgala S, Katoulis AC, Georgala C, Bozi E, Mortakis A (June 2004). “Oral isotretinoin in the treatment of recalcitrant condylomata acuminata of the cervix: a randomised placebo controlled trial”. Sexually Transmitted Infections. 80 (3): 216–8. doi:10.1136/sti.2003.006841. PMC 1744851. PMID 15170007.
- ^ Sehgal VN, Srivastava G, Sardana K (June 2006). “Isotretinoin–unapproved indications/uses and dosage: a physician’s reference”. International Journal of Dermatology. 45 (6): 772–7. doi:10.1111/j.1365-4632.2006.02830.x. PMID 16796650.
- ^ Jump up to:a b Choi JS, Koren G, Nulman I (March 2013). “Pregnancy and isotretinoin therapy”. Canadian Medical Association Journal. 185 (5): 411–3. doi:10.1503/cmaj.120729. PMC 3602257. PMID 23296582.
- ^ Joint Formulary Committee. British National Formulary (47th ed.). London: British Medical Association and Royal Pharmaceutical Society of Great Britain. ISBN 978-0-85369-584-4.[page needed]
- ^ “Fresh call for GPs to prescribe Roaccutane”. AustralianDoctor. 19 June 2012.
- ^ Specifically, doctors who are fellows of the Australasian College of Dermatologists (FACD); cf. Pharmaceutical Services Branch, Guide to poisons and therapeutic goods legislation for medical practitioners and dentists, Sydney: NSW Department of Health; 2006.[page needed]
- ^ James M (June 1996). “Isotretinoin for severe acne”. Lancet. 347 (9017): 1749–50. doi:10.1016/S0140-6736(96)90814-4. PMID 8656912. S2CID 28756302.
- ^ “Acne, Isotretinoin, and Depression”. MEDSAFE (New Zealand Ministry of Health). June 2013 [June 2005]. Retrieved 7 February 2014.
- ^ Thiboutot, D. M.; Cockerell, C. J. (1 August 2006). “iPLEDGE: A Report from the Front Lines of Dermatologic Practice”. AMA Journal of Ethics. 8 (8): 524–528. doi:10.1001/virtualmentor.2006.8.8.pfor1-0608. ISSN 1937-7010. PMID 23234692.
- ^ Darves, Bonnie (March 9, 2006). “Dermatologists Frustrated With Problematic iPledge Program”. Medscape.
- ^ “iPledge (About iPledge)”.
- ^ “Isotretinoin (marketed as Accutane) Capsule Information”. U.S. Food and Drug Administration (FDA). 2018-11-03.
- ^ Jump up to:a b c “Isotretinoin 20mg capsules – – (eMC)”. http://www.medicines.org.uk. Retrieved 2017-12-27.
- ^ “Isotretinoin 20mg capsules – – (eMC)”. http://www.medicines.org.uk. Retrieved 2018-01-10.
- ^ David M, Hodak E, Lowe NJ (1988). “Adverse effects of retinoids”. Medical Toxicology and Adverse Drug Experience. 3 (4): 273–88. doi:10.1007/bf03259940. PMID 3054426. S2CID 12432684.
- ^ DiGiovanna JJ (November 2001). “Isotretinoin effects on bone”. Journal of the American Academy of Dermatology. 45 (5): S176-82. doi:10.1067/mjd.2001.113721. PMID 11606950.
- ^ Ellis CN, Madison KC, Pennes DR, Martel W, Voorhees JJ (1984). “Isotretinoin therapy is associated with early skeletal radiographic changes”. Journal of the American Academy of Dermatology. 10 (6): 1024–9. doi:10.1016/S0190-9622(84)80329-1. PMID 6588057.
- ^ “Isotretinoin risks in acne treatment: Page 3 of 4”. October 2014.
- ^ Jump up to:a b Moy A, McNamara NA, Lin MC (September 2015). “Effects of Isotretinoin on Meibomian Glands”. Optometry and Vision Science. 92 (9): 925–30. doi:10.1097/OPX.0000000000000656. PMID 26154692. S2CID 205905994.
- ^ Jump up to:a b Lambert RW, Smith RE (March 1989). “Effects of 13-cis-retinoic acid on the hamster meibomian gland”. The Journal of Investigative Dermatology. 92 (3): 321–5. doi:10.1111/1523-1747.ep12277122. PMID 2918239.
- ^ Fraunfelder FT, Fraunfelder FW, Edwards R (September 2001). “Ocular side effects possibly associated with isotretinoin usage”. American Journal of Ophthalmology. 132 (3): 299–305. doi:10.1016/S0002-9394(01)01024-8. PMID 11530040.
- ^ Jump up to:a b c d e f g h Brelsford M, Beute TC (September 2008). “Preventing and managing the side effects of isotretinoin”. Seminars in Cutaneous Medicine and Surgery. 27 (3): 197–206. doi:10.1016/j.sder.2008.07.002. PMID 18786498.
- ^ Scheinfeld N, Bangalore S (May 2006). “Facial edema induced by isotretinoin use: a case and a review of the side effects of isotretinoin”. Journal of Drugs in Dermatology. 5 (5): 467–8. PMID 16703787.
- ^ Jump up to:a b “Updated measures for pregnancy prevention during retinoid use”. European Medicines Agency. 21 June 2018.
- ^ Roche Products Pty Ltd. Roaccutane (Australian Approved Product Information). Dee Why (NSW): Roche; 2005.[page needed]
- ^ Leyden JJ, Del Rosso JQ, Baum EW (February 2014). “The use of isotretinoin in the treatment of acne vulgaris: clinical considerations and future directions”. The Journal of Clinical and Aesthetic Dermatology. 7 (2 Suppl): S3–S21. PMC 3970835. PMID 24688620.
- ^ BNF, edition 57[page needed]
- ^ Jump up to:a b c d e f g h i j k l m Bremner JD, Shearer KD, McCaffery PJ (January 2012). “Retinoic acid and affective disorders: the evidence for an association”. The Journal of Clinical Psychiatry (Systematic Review). 73 (1): 37–50. doi:10.4088/JCP.10r05993. PMC 3276716. PMID 21903028.
- ^ Jump up to:a b c Kontaxakis VP, Skourides D, Ferentinos P, Havaki-Kontaxaki BJ, Papadimitriou GN (January 2009). “Isotretinoin and psychopathology: a review”. Annals of General Psychiatry. 8: 2. doi:10.1186/1744-859X-8-2. PMC 2637283. PMID 19154613.
- ^ Jump up to:a b c d Borovaya A, Olisova O, Ruzicka T, Sárdy M (September 2013). “Does isotretinoin therapy of acne cure or cause depression?”. International Journal of Dermatology. 52 (9): 1040–52. doi:10.1111/ijd.12169. PMID 23962262.
- ^ Jump up to:a b “Interactive Drug Analysis Profile – Isotretinoin”. mhra.gov.uk. Medicines & Healthcare Products Regulatory Agency. 31 March 2017.
- ^ Jump up to:a b Goodfield MJ, Cox NH, Bowser A, McMillan JC, Millard LG, Simpson NB, Ormerod AD (June 2010). “Advice on the safe introduction and continued use of isotretinoin in acne in the U.K. 2010”. The British Journal of Dermatology. 162 (6): 1172–9. doi:10.1111/j.1365-2133.2010.09836.x. PMID 21250961.
- ^ Jump up to:a b Ludot M, Mouchabac S, Ferreri F (June 2015). “Inter-relationships between isotretinoin treatment and psychiatric disorders: Depression, bipolar disorder, anxiety, psychosis and suicide risks”. World Journal of Psychiatry. 5 (2): 222–7. doi:10.5498/wjp.v5.i2.222. PMC 4473493. PMID 26110123.
- ^ Wysowski DK, Pitts M, Beitz J (October 2001). “An analysis of reports of depression and suicide in patients treated with isotretinoin”. Journal of the American Academy of Dermatology. 45 (4): 515–9. doi:10.1067/mjd.2001.117730. PMID 11568740.
- ^ Jump up to:a b Rowe C, Spelman L, Oziemski M, Ryan A, Manoharan S, Wilson P, Daubney M, Scott J (May 2014). “Isotretinoin and mental health in adolescents: Australian consensus”. The Australasian Journal of Dermatology (Review). 55 (2): 162–7. doi:10.1111/ajd.12117. PMID 24283385. S2CID 29178483.
- ^ Palha JA, Goodman AB (June 2006). “Thyroid hormones and retinoids: a possible link between genes and environment in schizophrenia” (PDF). Brain Research Reviews. 51(1): 61–71. doi:10.1016/j.brainresrev.2005.10.001. hdl:1822/3943. PMID 16325258. S2CID 30773986.
- ^ Jump up to:a b c d Goodman AB (March 1994). “Retinoid dysregulation as a cause of schizophrenia”. The American Journal of Psychiatry. 151 (3): 452–3. doi:10.1176/ajp.151.3.452b. PMID 8109664.
- ^ Goodman AB (May 1996). “Congenital anomalies in relatives of schizophrenic probands may indicate a retinoid pathology”. Schizophrenia Research. 19 (2–3): 163–70. doi:10.1016/0920-9964(96)88523-9. PMID 8789914. S2CID 12089905.
- ^ Goodman AB (July 2005). “Microarray results suggest altered transport and lowered synthesis of retinoic acid in schizophrenia”. Molecular Psychiatry. 10 (7): 620–1. doi:10.1038/sj.mp.4001668. PMID 15838536.
- ^ Samad TA, Krezel W, Chambon P, Borrelli E (December 1997). “Regulation of dopaminergic pathways by retinoids: activation of the D2 receptor promoter by members of the retinoic acid receptor-retinoid X receptor family”. Proceedings of the National Academy of Sciences of the United States of America. 94 (26): 14349–54. Bibcode:1997PNAS…9414349S. doi:10.1073/pnas.94.26.14349. PMC 24972. PMID 9405615.
- ^ Crockett SD, Porter CQ, Martin CF, Sandler RS, Kappelman MD (September 2010). “Isotretinoin use and the risk of inflammatory bowel disease: a case-control study”. The American Journal of Gastroenterology. 105 (9): 1986–93. doi:10.1038/ajg.2010.124. PMC 3073620. PMID 20354506.
- ^ Lowenstein EB, Lowenstein EJ (2011). “Isotretinoin systemic therapy and the shadow cast upon dermatology’s downtrodden hero”. Clinics in Dermatology. 29 (6): 652–61. doi:10.1016/j.clindermatol.2011.08.026. PMID 22014987.
- ^ “Drug Safety Update – Latest advice for medicines users – October 2017” (PDF). Medicines and Healthcare products Regulatory Agency. 3 October 2017. Retrieved 17 May2019.
- ^ “Pharmacovigilance Risk Assessment Committee (PRAC) – Minutes for the meeting on 3–6 July 2017” (PDF). European Medicines Agency. 1 September 2017. p. 44. Retrieved 17 May 2019.
- ^ Kremer I, Gaton DD, David M, Gaton E, Shapiro A (1994). “Toxic effects of systemic retinoids on meibomian glands”. Ophthalmic Research. 26 (2): 124–8. doi:10.1159/000267402. PMID 8196934.
- ^ Griffin JN, Pinali D, Olds K, Lu N, Appleby L, Doan L, Lane MA (November 2010). “13-Cis-retinoic acid decreases hypothalamic cell number in vitro”. Neuroscience Research. 68 (3): 185–90. doi:10.1016/j.neures.2010.08.003. PMID 20708044. S2CID 207152111.
- ^ Crandall J, Sakai Y, Zhang J, Koul O, Mineur Y, Crusio WE, McCaffery P (April 2004). “13-cis-retinoic acid suppresses hippocampal cell division and hippocampal-dependent learning in mice”. Proceedings of the National Academy of Sciences of the United States of America. 101 (14): 5111–6. Bibcode:2004PNAS..101.5111C. doi:10.1073/pnas.0306336101. JSTOR 3371827. PMC 387382. PMID 15051884.
- ^ Sakai Y, Crandall JE, Brodsky J, McCaffery P (June 2004). “13-cis Retinoic acid (accutane) suppresses hippocampal cell survival in mice”. Annals of the New York Academy of Sciences. 1021 (1): 436–40. Bibcode:2004NYASA1021..436S. doi:10.1196/annals.1308.059. PMID 15251924.
- ^ Nelson AM, Cong Z, Gilliland KL, Thiboutot DM (September 2011). “TRAIL contributes to the apoptotic effect of 13-cis retinoic acid in human sebaceous gland cells”. The British Journal of Dermatology. 165 (3): 526–33. doi:10.1111/j.1365-2133.2011.10392.x. PMC 3166444. PMID 21564055.
- ^ Nelson AM, Gilliland KL, Cong Z, Thiboutot DM (October 2006). “13-cis Retinoic acid induces apoptosis and cell cycle arrest in human SEB-1 sebocytes”. The Journal of Investigative Dermatology. 126 (10): 2178–89. doi:10.1038/sj.jid.5700289. PMID 16575387.
- ^ Wachter K (2009). “Isotretinoin’s Mechanism of Action Explored”. Skin & Allergy News. 40(11): 32. doi:10.1016/S0037-6337(09)70553-4.
- ^ Isotretinoin’s Mechanism of Action Elucidated Archived 2010-04-04 at the Wayback Machine. Medconnect (2009-08-28). Retrieved on 2010-11-13.
- ^ Nelson AM, Zhao W, Gilliland KL, Zaenglein AL, Liu W, Thiboutot DM (April 2008). “Neutrophil gelatinase-associated lipocalin mediates 13-cis retinoic acid-induced apoptosis of human sebaceous gland cells”. The Journal of Clinical Investigation. 118 (4): 1468–78. doi:10.1172/JCI33869. PMC 2262030. PMID 18317594.
- ^ Jump up to:a b Peck GL, Olsen TG, Yoder FW, Strauss JS, Downing DT, Pandya M, Butkus D, Arnaud-Battandier J (February 1979). “Prolonged remissions of cystic and conglobate acne with 13-cis-retinoic acid”. The New England Journal of Medicine. 300 (7): 329–33. doi:10.1056/NEJM197902153000701. PMID 153472.
- ^ Shalita A (2001). “The integral role of topical and oral retinoids in the early treatment of acne”. Journal of the European Academy of Dermatology and Venereology. 15: 43–9. doi:10.1046/j.0926-9959.2001.00012.x. PMID 11843233.
- ^ [unreliable medical source?]Farrell LN, Strauss JS, Stranieri AM (December 1980). “The treatment of severe cystic acne with 13-cis-retinoic acid. Evaluation of sebum production and the clinical response in a multiple-dose trial”. Journal of the American Academy of Dermatology. 3 (6): 602–11. doi:10.1016/S0190-9622(80)80074-0. PMID 6451637.
- ^ [unreliable medical source?]Jones H, Blanc D, Cunliffe WJ (November 1980). “13-cis retinoic acid and acne”. Lancet. 2 (8203): 1048–9. doi:10.1016/S0140-6736(80)92273-4. PMID 6107678. S2CID 40877032.
- ^ Pendino F, Flexor M, Delhommeau F, Buet D, Lanotte M, Segal-Bendirdjian E (June 2001). “Retinoids down-regulate telomerase and telomere length in a pathway distinct from leukemia cell differentiation”. Proceedings of the National Academy of Sciences of the United States of America. 98 (12): 6662–7. Bibcode:2001PNAS…98.6662P. doi:10.1073/pnas.111464998. JSTOR 3055868. PMC 34517. PMID 11371621.
- ^ Φαχαντίδης, Παναγιώτης Ε. (2007). Η επίδραση της ισοτρετινοϊνης και των αναστολέων της 5α-αναγωγάσης στις μεταλλοπρωτεάσες του συνδετικού ιστού σε ασθενείς με ακμή[The influence of isotretinoin and 5-a reductase inhibitors in metaloproteases of connective tissue in patients with ance] (in Greek). Aristotle University of Thessaloniki.[unreliable medical source?]
- ^ Toyoda M, Nakamura M, Makino T, Kagoura M, Morohashi M (June 2002). “Sebaceous glands in acne patients express high levels of neutral endopeptidase”. Experimental Dermatology. 11 (3): 241–7. doi:10.1034/j.1600-0625.2002.110307.x. PMID 12102663. S2CID 23468315.
- ^ Wysowski DK, Swartz L (May 2005). “Relationship between headache and depression in users of isotretinoin”. Archives of Dermatology. 141 (5): 640–1. doi:10.1001/archderm.141.5.640. PMID 15897395.
- ^ Magin P, Pond D, Smith W (February 2005). “Isotretinoin, depression and suicide: a review of the evidence”. The British Journal of General Practice. 55 (511): 134–8. PMC 1463189. PMID 15720936.
- ^ Ng CH, Schweitzer I (February 2003). “The association between depression and isotretinoin use in acne”. The Australian and New Zealand Journal of Psychiatry. 37 (1): 78–84. doi:10.1046/j.1440-1614.2003.01111.x. PMID 12534661. S2CID 8475675.
- ^ Jump up to:a b c d e “FDA information, side effects, and uses / Accutane (isotretinoin)”. U. S. Food and Drug Administration (FDA). Retrieved 20 January 2014.
- ^ “FDA information, side effects, and uses / Accutane (isotretinoin) : Table 2 Pharmacokinetic Parameters of Isotretinoin Mean (%CV), N=74“. U. S. Food and Drug Administration (FDA). Retrieved 20 January 2014.
- ^ “FDA information, side effects, and uses / Accutane (isotretinoin) : Drug Interactions“. U. S. Food and Drug Administration (FDA). Retrieved 20 January 2014.
- ^ Gina Kolata for the New York Times. April 22, 1988 Anti-Acne Drug Faulted in Birth
- ^ CDC. January 21, 2000 Accutane®-Exposed Pregnancies — California, 1999 MMWR Weekly 49(02);28-31
- ^ Shari Roan (7 November 2009). “New study may deal final blow to acne drug Accutane”. LA Times.
- ^ “Roche Discontinues and Plans to Delist Accutane in the U.S.” (Press release). Genentech. 2009-06-29. Archived from the original on 2009-11-08. Retrieved 2010-11-12.
- ^ Feeley J (2011-03-11). “Roche Accutane Acne Drug Caused ‘Tragedy’ for Actor, Brian Dennehy Says”. Bloomberg.
- ^ Silverman E (2011-11-04). “It’s Curtains On Actor’s Accutane Lawsuit”. Pharmalot. UBM Canon.
- ^ Voreacos D (May 30, 2007). “Roche Found Liable in First Of 400 Suits Over Accutane”. The Washington Post. Bloomberg News. Retrieved April 30, 2012.
- ^ Halverstam CP, Zeichner J, Lebwohl M (2006). “Lack of significant skeletal changes after long-term, low-dose retinoid therapy: case report and review of the literature”. Journal of Cutaneous Medicine and Surgery. 10 (6): 291–9. doi:10.2310/7750.2006.00065. PMID 17241599. S2CID 36785828.
External links
////////////Antiacne, 13-cis-Retinoic acid, 2-cis-vitamin A acid, neovitamin A acid, Isotretinoin
NEW DRUG APPROVALS
ONE TIME
$10.00
COVAX-19
Vaxine Pty Ltd company logo

Vaxine’s promising new COVID-19 vaccine candidate
A new multivalent COVID-19 vaccine developed by Australian company Vaxine to tackle the new virus variants could be game-changer in the fight against COVID-19
The world desperately needs a vaccine that blocks virus transmission and protects against all the variants. Covax-19 vaccine may soon change history”— Sharen Pringle, Vaxine Business Mananager
ADELAIDE, SA, AUSTRALIA, May 16, 2021 /EINPresswire.com/ — Professor Petrovsky, who is the Chairman and Research Director of Australian-based Vaxine Pty Ltd, explains that the two biggest challenges to tackling the COVID-19 pandemic are to develop a vaccine that completely prevents virus transmission something other COVID-19 have not been completely successful in achieving, and the second being to find a vaccine that protects equally against all the evolving immune-escape variants.
Professor Petrovsky has been researching coronavirus vaccines for the last 17 years, having previously published scientific papers on vaccines against both the SARS and MERS coronaviruses, which were highly protective in relevant animal models. He also recently published data from a collaboration with the US Army on development of a promising Ebola vaccine that protected mice against this most lethal disease after just a single vaccine dose. He has now successfully taken the same approach to design a protein-based vaccine against COVID-19.
Studies in a broad range of animal models including mice, hamsters, ferrets and monkeys, have recently revealed the high potential of this vaccine that is currently known as Covax-19(TM), but which likely will be soon rebranded as in its latest iteration it moves into late stage human trials in a number of countries.
Recent breakthrough data generated by Vaxine’s partner, Professor Kaissar Tabynov who leads the International Center for Vaccinology at the Kazakh National Agrarian University has shown that Vaxine’s unique spike protein antigen which is produced using insect cells in culture, was unique in that it not only totally protected hamsters from infection themselves but also prevented them from transmitting the virus to unvaccinated animals that were placed in the same cage two days after the vaccinated animals had been challenged with virus. Protection against transmission was not seen in hamsters given other vaccines making this finding unique to Vaxine’s spike protein antigen.
This hamster data reinforced findings in hamster, ferret and monkey challenge study performed by collaborating US Universities, who showed that two doses of Vaxine’s Covax-19 vaccine provided complete clearance of recoverable virus from the lungs and nose of animals when sampled just days after an infectious challenge.
“COVAX-19 vaccine has now been shown to be highly protective against the original Wuhan strain of the virus in hamster, ferret and monkey infection models performed by independent academic institutions in multiple countries, attesting to the strength of our protein-based vaccine approach”, says Prof. Petrovsky.
“A key element in the success of Covax-19 vaccine is the inclusion of Vaxine’s Advax adjuvant technology which acts as a turbocharger to drive an optimal immune response against the virus” explains Prof. Petrovsky who has been working on this promising vaccine adjuvant technology for the last 20 years with funding support from the US National Institutes of Health.
“We have now shown that our COVAX-19 vaccine can provide effective immunity including an ability to block nasal virus replication and this in turn successfully prevents transmission of the virus to vaccine-naïve animals,” he explains.
Follow on studies to confirm and expand upon these initial findings are currently underway at several US universities as well as Kazakh National Agrarian University, with a manuscript describing some of the initial animal data currently under review at a leading vaccine journal.
In another major breakthrough the team has now developed the vaccine into a multivariant format designed to protect against all the recently described variant strains of COVID-19, with work also underway on the most recently described Indian strains.
While the data is still preliminary says Prof. Petrovsky, the immune responses to the multivalent vaccine in mice are generating equally strong antibody binding activity against all the major virus variants. “This is extremely exciting as the world desperately needs vaccines able to protect against all the new strains of the virus including the UK, South African and Brazilian strains. By contrast , the currently available vaccines are clearly not as strong against some of these variants as they are against the original Wuhan strain” he explains.
Already there have been multiple confirmed cases of vaccine breakthrough where otherwise healthy individuals who have received mRNA, adenovirus or inactivated whole virus vaccines have become infected generally with either the South African or Brazilian variants.
This problem of immune-escape will only get worse over time as more complex variants emerge which is why Vaxine has been putting all its energy into finding a robust solution to this issue before proceeding with Phase 3 clinical trials of its Covax-19 vaccine.
Dr. Petrovsky went on to conclude “Now we have a multivalent formulation of Covax-19 vaccine that is showing high promise in animal studies, we plan to work as fast as we can to advance this new vaccine formulation in human trials, while expanding manufacturing capacity to ensure we are able to produce enough vaccine to meet the enormous global demand that will be attracted by such a successful vaccine.”
“To help us in this task Vaxine is looking to assemble a global network of partner organisations in countries around the world to assist Vaxine with vaccine development, clinical trials, manufacturing, distribution and sales. This is going to be a mammoth effort as we go to war against this insidious virus that continues to wreak havoc around the globe, with WHO recently predicting that the second year of the pandemic is likely to be much worse even than the first, an ominous warning for many countries that still remain poorly prepared and lacking in local vaccine manufacturing capability.
Vaxine wishes to help developing countries to establish their own local state-of-the-art vaccine manufacturing facilities, providing advice on appropriate facility design and undertaking technology transfer of its state of the art protein production technology to such facilities.
Countries in the developing world can no longer afford to sit and wait for outside organisations like COVAX to solve their vaccine supply problems, instead Vaxine proposes to help such countries find their own local solutions to the vaccine supply bottleneck for this.
Sharen Pringle
Vaxine Pty Ltd
437 033 400
email us here……..https://www.einnews.com/pr_news/541113168/covid-19-vaccine-breakthrough
Currently, the Australian influenza vaccine and adjuvant specialist and the Polish protein drug maker have just inked a memorandum of understanding, so the terms of a future contract remain to be defined. However, the technology behind is interesting.
The partners intent to utilize an insect cell-based recombinant spike protein of SARS-CoV–2 in combination with Vaxine’s proprietary Advax™ adjuvant and have already started Phase I testing in Australia with first result expected later this month. The company announced it will use artificial intelligence to evalutate clinical data in real time and announced the ambition to complete Phase II and III trials at the end of this year. “Supported by Microsoft technology, we aim to collect and analyse the COVAX-19™ trial data in real time, rather than waiting until the end of the trial before seeing if the vaccine is working, which is the traditional process,” said Vaxine’s Research Director Professor Nikolai Petrovsky from Flinders University in Adelaide.
Preclinically, Vaxine Pty Ltd’s syntetic spike protein with the company’s non-inflammatory Advax™ adjuvant, induced antibody and T-cell immune responses against the co-administered antigen. In various animal models, Covax-19 vaccination provided robust protection against an infection with the novel coronavirus.
The Phase I of Vaxine Pty Ltd in running since July in 40 healthy volunteers. If results are positive, the Australian vaccine maker is to expand studies and manufacturing to Europe. Under a future agreement Mabion SA would lead clinical development, manufacturing, regulatory negotiations and could exclusively market the vaccine in the EU and – optionally – in the US……..https://european-biotechnology.com/up-to-date/latest-news/news/mabion-to-licence-covid-19-jab-from-vaxine-pty-ltd.html
////////////////COVAX-19, corona virus, covid 19, Vaxine, australia, vaccine
NEW DRUG APPROVALS
ONE TIME
$10.00
Uprifosbuvir

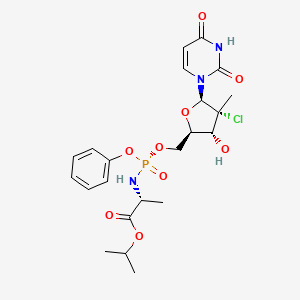
Uprifosbuvir
MK 3682, IDX 21437
ウプリホスブビル;
| Formula | C22H29ClN3O9P |
|---|---|
| CAS | 1496551-77-9 |
| Mol weight | 545.9071 |
уприфосбувир [Russian] [INN]أوبريفوسبوفير [Arabic] [INN]乌磷布韦 [Chinese] [INN]
propan-2-yl (2R)-2-[[[(2R,3R,4R,5R)-4-chloro-5-(2,4-dioxopyrimidin-1-yl)-3-hydroxy-4-methyloxolan-2-yl]methoxy-phenoxyphosphoryl]amino]propanoate
Isopropyl (2R)-2-{[(R)-{[(2R,3R,4R,5R)-4-chloro-5-(2,4-dioxo-3,4-dihydro-1(2H)-pyrimidinyl)-3-hydroxy-4-methyltetrahydro-2-furanyl]methoxy}(phenoxy)phosphoryl]amino}propanoate
IDX-21437, DB15206, SB18784, D10996, Q27281714
Uprifosbuvir (MK-3682) is an antiviral drug developed for the treatment of Hepatitis C. It is a nucleotide analogue which acts as an NS5B RNA polymerase inhibitor. It is currently in Phase III human clinical trials.[1][2][3]
Uprifosbuvir is under investigation in clinical trial NCT02332707 (Efficacy and Safety of Grazoprevir (MK-5172) and Uprifosbuvir (MK-3682) With Elbasvir (MK-8742) or Ruzasvir (MK-8408) for Chronic Hepatitis C Genotype (GT)1 and GT2 Infection (MK-3682-011)).Hepatitis C viruss (HCV) have the newly-increased patients of 3-4 million every year, and World Health Organization (WHO) is estimated in global sense More than 200,000,000, in China more than 10,000,000 patients, HCV belongs to flaviviridae hepatovirus virus to dye person.Long-term hepatitis C virus Gently to inflammation, weight is to liver cirrhosis, hepatocarcinoma for poison infection.And during hepatitis C cirrhosis patients in decompensation, can there are various complication, such as abdomen Water abdominal cavity infection, upper gastrointestinal hemorrhage, hepatic encephalopathy, hepatorenal syndrome, liver failure etc. are showed.The side of HCV infection is treated initially Method is interferon and interferon and ribavirin combination therapy, and only 50% therapist has reaction, and interferon to the method With obvious side effect, such as flu-like symptoms, body weight lower and fatigue and weak, and interferon and ribavirin Conjoint therapy then produces sizable side effect, including haemolysis, anemia and tired etc..U.S. FDA have approved multiple HCV medicines, including the polymerization of protease inhibitor, ucleosides and non-nucleoside in recent years Enzyme inhibitor and NS5A inhibitor etc..The protease inhibitor class medicine of FDA approvals has three:VX‐950 (Telaprevir), SCH-503034 (Boceprevir) and TMC435 (Simeprevir), the shortcoming of protease inhibitor is It is also easy to produce that mutation, toxicity is big, poor bioavailability, it is effective to individual other gene type.Eggs of the Telaprevir as the first generation White enzyme inhibitor has logged out market.The second filial generation and third generation protease inhibitor of high activity and wide spectrum is mainly used as and other One of component of drug combination of hepatitis C medicine.NS5A inhibitor is the highly active anti-HCV medicament of a class.The most representative Daclatasive for having BMS, The Ombitasvir of the Ledipasvir and AbbVie of Gilead, as this kind of medicine independent medication is easy to produce drug resistance, They treat one of drug component of HCV primarily as drug combination.The AG14361 of hepatitis C is generally divided into two kinds of ucleosides and non-nucleoside.At present, clinically only Suo Feibu One ucleosides hepatitis C medicine of Wei is listed by FDA approvals, and other are still in the anti-hepatitis C virus medicine of ucleosides of clinical experimental stage Thing also has the MK-3682 (IDX21437) of Mo Shadong, the AL-335 of the ACH-3422 and Alios of Achillion drugmakers.Third Hepatitis virus have the features such as Multi-genotype and fast variation, and single medicine treatment hepatitis C has generation drug resistance fast, to part Genotype cure rate is low and the various defects such as course for the treatment of length.In order to overcome these defects, the treatment of drug combination is primarily now taken Scheme, in order to overcome these defects, primarily now takes the therapeutic scheme of drug combination, the Sovaldi conducts of FDA approval listings The key component of drug combination, for the patient of 4 type of 1 type of gene and gene be Suo Feibuwei, profit Ba Wei woodss and Polyethylene Glycol-α- The drug combination of interferon three, the course for the treatment of are 12 weeks;For 1 type of gene and the patient of 3 types, the big woods joints of Suo Feibuwei and Li Ba Medication, the course for the treatment of are respectively 12 weeks and 24 weeks.- 2016 years 2013, FDA ratified Suo Feibuwei and NS3 protein inhibitors again in succession Simeprevir shares the patient of 1 type of therapeutic gene;The NS5A inhibitor Daclatavir therapeutic genes 1 of Suo Feibuwei and BMS With the patient of 3 types.Harvoni is the patient that Suo Feibuweijia NS5A inhibitor Ledipasvir is used for 1 type of gene.Even if using Same nucleoside, the NS5A inhibitor and/or NS3 protease inhibitor for sharing varying strength can effectively extend composition of medicine Clinical application range and Shorten the Treatment Process.In June, 2016, FDA have approved Suo Feibuwei and more potent secondary NS5A inhibitor Velpatasvir shares the hepatitis C patient suitable for all gene types, it is not necessary to carry out genetic test.Just in three phases clinic Suo Feibuwei, NS5A inhibitor Velpatasvir and NS3 protease inhibitor Voxilaprevir goes for all of disease People, is try to the course for the treatment of and shortened to 8 weeks from 12 weeks.Suo Feibuwei just in clinical trial target spots different with hepatitis C virus are directed to Drug regimen (such as Suo Feibuweijia new type NS 5A inhibitor Velpatasvir and/or protease inhibitor GS5816), its knot Fruit show than single drug more wide spectrum, effectively, and can be with Shorten the Treatment Process.MSD Corp. is by MK-3682 and NS5A inhibitor Grazoprevir and/or protease inhibitor Elbasvir is used as new drug regimen, effective for all genotype of HCV, And further shorten to the course for the treatment of of 8 weeks.New deuterated nucleoside phosphoric acid ester compound disclosed in patent of the present invention, especially The double deuterated compound such as VI-1b2 in 5 ‘-position, shows than the more preferable bioavailability of former compound MK-3682 and longer partly declines Phase.In addition, this kind of novel nucleoside phosphoramidate is significantly superior to the Suo Feibuwei of clinical practice in terms of anti-hepatitis C activity, On sugared ring, chlorine atom replaces fluorine atom, and cytotoxicity is significantly reduced in surveyed cell line.By to base, sugared ring With the transformation and optimization of prodrug moiety system, the anti-hepatitis C activity of partial synthesis compound is higher than Suo Feibuwei 2-10 times, meanwhile, In the optimization of metabolism key position, synthesis compound shows that in blood plasma the higher metabolic stabilities of peso Fei Buwei and chemistry are steady It is qualitative.Therefore this kind of new deuterated nucleotide phosphate and NS5A inhibitor and/or egg as shown in formula a, a1, a2, b, b1, b2 The newtype drug combination constituted by white enzyme inhibitor is with extremely wide application prospect.Deuterium is the naturally occurring hydrogen isotope of nature, the deuterated isotopic body in common drug all containing trace.Deuterium without It is malicious, “dead”, it is safe to human body, C-D keys are more stable (6-9 times) than c h bond, hydrogen is replaced with after deuterium, can extend medicine Half-life, while pharmacologically active (shape difference of H and D is little, J Med Chem.2011,54,2529-2591) is not affected, in addition Deuterated medicine usually shows more preferable bioavailability and less toxicity, and the active ribonucleoside triphosphote of its metabolism is more stable, So deuterated nucleoside phosphoramidate will be better than corresponding nucleoside medicine in the curative effect of clinical practice.For example, 2013 It is exactly a deuterated compound that the nucleoside anti hepatitis C virus drug ACH-3422 of clinical trial is in the approval of year FDA, with non-deuterium (WO2014169278, WO are 2014169280) than having higher bioavailability and longer half-life for the former compound phase in generation.
Based on above-mentioned present Research, we design and are prepared for the new deuterated nucleoside that compound VI-1b2 is representative Phosphoramidate.Below we will be described in the architectural feature of deuterated nucleoside phosphoramidate of our inventions, preparation method, Antiviral activity experimental result and it as anti-hepatitis c virus drug combination key component and NS5A inhibitor and/ Or the drug regimen of protease inhibitor is in the application of anti-virus aspect.
The EPA awarded the greener reaction conditions to the pharmaceutical company Merck & Co. for building a prodrug synthesis that eliminated the use of toxic reagents. Prodrugs are molecules that get metabolized by our bodies into an active pharmaceutical. Some hepatitis C and HIV medications are prodrugs and get synthesized through a method call pronucleotide (ProTide) synthesis. The method uses toxic and corrosive thionyl chloride, plus an excess of expensive pentafluorophenol that generates a lot of waste. Merck’s new method creates their target compounds in 90 to 92% yields without these reagents and eliminates the need for halogenated solvents entirely through strategic catalyst loading and the use of different starting materials from the traditional route.
The design of greener chemicals award went to the development of more environmentally friendly versions of chemicals called thermoset binders, which can serve as carpet adhesives and are involved in the manufacture of mineral and fiberglass products. Generally, these chemicals are based on formaldehyde or polycarboxylic acids, and they can give off toxic formaldehyde and often use small amounts of sulfuric and hypophosphorous acid as catalysts to activate them. The insulation and commercial roofing company Johns Manville created a new binder based on the reaction between renewable dextrose, fructose, and other simple sugars, bound together by the α-carbon-containing cross-linking agent glyoxal. The reaction also uses a biodegradable acid in water as a catalyst. The binder can be made in just one step instead of the traditional multistep synthesis. Also, the synthesis can be done directly at the manufacturing site, instead of beforehand like with the traditional approach, meaning this new binder creates fewer of the health and environmental hazards that come from storage and transportation.
NEW DRUG APPROVALS
ONE TIME
$10.00
SYN
US 20170226146,

Paper
Organic Process Research & Development (2021), 25(3), 661-667.
https://pubs.acs.org/doi/10.1021/acs.oprd.0c00487

A novel application of the synthesis of pronucleotide (ProTide) 5′-phosphoramidate monoesters promoted by aluminum-based Lewis acids is described. In the multikilogram synthesis of uprifosbuvir (MK-3682, 1), a clinical candidate for the treatment of hepatitis C, this methodology provided >100:1 diastereoselectivity at the phosphorus stereocenter and >100:1 selectivity for the 5′-mono phosphorylation over undesired bisphosphorylation side products. The high diastereoselectivity and mono/bis ratio achieved enabled elimination of the tedious workup associated with the tert-butyl magnesium chloride protocol commonly used to install this functionality in similar nucleotide prodrugs, achieving a near doubling of the isolated yield from 45% to 81%. The process development and purity control strategy of MK-3682, as well as handling of the pyrophoric reagent on scale, will also be discussed.
PAPER
Science (Washington, DC, United States) (2020), 369(6504), 725-730.
Science (Washington, DC, United States) (2017), 356(6336), 426-430.
Chemical Science (2017), 8(4), 2804-2810.
PATENT
CN 106543253
https://patents.google.com/patent/CN106543253A/zh
PATENT
WO 2014058801
https://patents.google.com/patent/WO2014058801A1/enExample 1Preparation of 2′-Chloro Nucleoside Analogs
Scheme 1


Ethyl (3R)-2-chloro-3-[(4R)-2,2-dimethyl-l,3-dioxolan-4-yl]-3-hydroxy-2- methylpropanoate (A2):

[00273] A 5 L flange flask was fitted with a thermometer, nitrogen inlet, pressure equalizing dropping funnel, bubbler, and a suba»seal. Methyl lithium solution (1.06 L, 1.6 M in diethylether, 1.7 equiv.) was added, and the solution was cooled to about -25 °C.Diisopropyl amine (238 ml, 1.7 equiv.) was added using the dropping funnel over about 40 minutes. The reaction was left stirring, allowing to warm to ambient temperature overnight. C02(s)/acetone cooling was applied to the LDA solution, cooling to about -70 °C.[00274] i?-Glyceraldehyde dimethylacetal solution (50% in DCM) was evaporated down to -100 mbar at a bath temp of 35 °C, to remove the DCM, then azeotroped with anhydrous hexane (200 ml), under the same Buchi conditions. 1H NMR was used to confirm that all but a trace of DCM remained.[00275] The fresh aldehyde (130 g, 1 mol) and ethyl 2-chloropropionionate (191 ml, 1.5 equiv.) were placed in a 1 L round bottom flask, which was filled with toluene (800 ml). This solution was cooled in a C02(s)/acetone bath, and added via cannula to the LDA solution over about 50 minutes, keeping the internal temperature of the reaction mixture cooler than -60 °C. The mixture was stirred with cooling (internal temp, slowly fell to ~ -72 °C) for 90 min, then warmed to room temperature over 30 minutes using a water bath. This solution was added to a sodium dihydrogen phosphate solution equivalent to 360 g of NaH2P04 in 1.5 L of ice/water, over about 10 minutes, with ice-bath cooling. The mixture was stirred for 20 minutes, then transferred to a sep. funnel, and partitioned. The aqueous layer was further extracted with EtOAc (2 x 1 L), and the combined organic extracts were dried over sodium sulfate. The volatiles were removed in vacuo (down to 20 mbar). The resultant oil was hydrolyzed crude.
(3R,4R,5R)-3-chIoro-4-hydroxy-5-(hydroxymethyI)-3-methyIoxoIan-2-one (A4):

H O CI[00276] The crude oil A2 was taken up in acetic acid (1.5 L, 66% in water) and heated to 90 °C over one hour, then at held at that temperature for one hour. Once the mixture had cooled to room temperature, the volatiles were removed in vacuo, and azeotroped with toluene (500 ml). The resultant oil was combined with some mixed material from an earlier synthesis and columned in two portions (each -1.25 L of silica, 38→ 75% EtOAc in DCM). The lower of the two main spots is the desired material; fractions containing this material as the major component were combined and the solvent removed in vacuo to give 82 g of orange solid whose 1 H NMR showed the material to be of about 57% purity (of the remainder 29% was the indicated epimer). This material was recrystallized fromtoluene/butanone (600 ml / -185 ml), the butanone being the ‘good’ solvent. The resultant solid was filtered washing with toluene and hexane, and dried in vacuo to give product of about 92% purity (30 g).(2R,3R,4R)-2-[(benzoyIoxy)methyI]-4-chIoro-4-methyI-5-oxooxoIan-3-yI benzoate(A5):

[00277] A 2 L 3 -neck round bottom flask was fitted with an overhead stirrer, thermometer and pressure equalizing dropping funnel (→N2). The intermediate A4 (160 mmol) in acetonitrile (1 L) was added, followed by 4-dimethylaminopyridine (3.2 mmol) and benzoyl chloride (352 mmol). Finally triethylamine (384 mmol) was added over 10 minutes using the dropping funnel. The addition of the triethylamine is accompanied by a mild exotherm, which obviated the addition of a cold water bath to keep the internal temperature below 25 °C. The reaction was stirred at ambient temperature for 2.5 hours. The reaction mixture was transferred to a sep. funnel with EtOAc (2 L) and half saturated brine (2 L), and partitioned. The aqueous layer was re-extracted with EtOAc (1 L). The combined organic layers were washed with 50%> sodium bicarbonate/25%) brine (1.5 L) and dried over sodium sulfate, to give 62 g of solid. This was recrystallized from 1.8 L of 1 : 1 toluene/trimethylpentane (95 °C), to give 52.4 g of product.[00278] 1H NMR (CDCls, 400 MHz): δ (ppm) 1.91 (s, 3H), 4.57 (dd, J= 5.12Hz and J = 12.57Hz, 1H), 4.77 (dd, J= 3.29Hz and J= 12.68Hz, 1H), 4.92-4.96 (m, 1H), 5.60 (d, J = 8.36Hz, 1H), 7.38-7.66 (m, 6H), 7.97-7.99 (m, 2H), 8.08-8.10 (m, 2H); MS (ESI) m/z= 411.1(MNa ).
3,5-Di-0-benzoyl-2-C-chloro-2-C-methyl-D-ribofuranose (A6):

[00279] To a solution of A5 (14.48 mmol) in anhydrous tetrahydrofurane (70 ml) was added under inert atmosphere at -35°C, LiAlH(OtBu)3 (1M in tetrahydrofurane, 21.7 mmol) over a 30 min period. The reaction mixture was stirred for 1 hour at -20 °C and quenched by addition of a saturated NH4C1 solution, keeping the temperature bellow 0 °C. Ethyl acetate was added and the white suspension was filtered through a pad of celite and washed with ethyl acetate. The filtrate was extracted with ethyl acetate twice. The combined organic layers were dried over anhydrous sodium sulfate, filtered and evaporated under reduced pressure. The residue was purified by chromatography on silica gel (eluent: petroleum ether/ethyl acetate 0 to 20%). The product was dried in vacuum (50 °C) overnight to afford expected intermediate as a colorless oil in 96% yield (mixture α/β: 45/55).[00280] 1H NMR (CDC13, 400 MHz): δ (ppm) 1.74 (s, 1.75HP), 1.76 (s, 1.25Ha), 4.42-4.69 (m, 3H), 5.30 (d, J= 12.8Hz, 0.55HP), 5.43-5.47 (m, 0.45Ha), 5.60 (d, J= 7.0Hz, 0.55HP), 5.78 (d, J= 7.0Hz , 0.45Ha), 7.35-7.41 (m, 2H), 7.45-7.56 (m, 3H), 7.59-7.65 (m, 1H), 7.96- 8.04 (m, 2H), 8.06-8.14 (m, 2H); MS (ESI) m/z= 413 (MNa+).3,5-Di-0-benzoyl-2-C-chloro-2-C-methyl-D-arabinofuranosyl bromide (A7):

[00281] To a solution of A6 (12.80 mmol) in anhydrous dichloromethane (80 ml) was added under inert atmosphere at -20 °C, triphenylphosphine (18.0 mmol). The reaction mixture was stirred for 15 minutes at -20 °C and CBr4 (19.20 mmol) was added. The reaction mixture was then stirred for 1 hour at -20 °C. The crude was partially concentrated under reduced pressure (bath temperature bellow 30 °C) and directly purified by chromatography on silica gel (eluent: petroleum ether/ethyl acetate 0 to 30%) to afford a mixture of β sugar A7a (1.67 g) and a sugar A7b (2.15 g) as a colorless gum in 66%> global yield.[00282] 1H NMR (CDC13, 400 MHz): β sugar δ (ppm) 1.93 (s, 3H), 4.60-4.88 (m, 3H), 6.08 (d, J= 7.9 Hz, 1H), 6.62 (s, 1H), 7.31-7.38 (m, 2H), 7.41-7.55 (m, 3H), 7.59-7.65 (m, 1H), 8.00-8.05 (m, 2H), 8.06-8.12 (m, 2H); a sugar δ (ppm) 1.88 (s, 3H), 4.66-4.89 (m, 3H), 5.37 (d, J= 4.88Hz, 1H), 6.44 (s, 1H), 7.41-7.55 (m, 4H), 7.54-7.65 (m, 2H), 8.00-8.05 (m, 2H), 8.14-8.20 (m, 2H); MS (ESI) m/z= 476/478 (MNa+).3 ,5′-Di-0-benzoyl-2′-C-chloro-2′-C-methyl-4-benzoyl-cytidine (A8):

[00283] To a suspension of N-benzoyl cytosine (9.48 mmol), and a catalytic amount of ammonium sulfate in 4-chlorobenzene (24 ml) was added HMDS (28.44 mmol). The reaction mixture was heated during 2 hours at 140 °C. The solvent was removed under inert atmosphere and the residue was taken in 4-chlorobenzene (15 ml). Then, A7b (4.74 mmol) in chlorobenzene (10 ml) was added dropwise to the reaction mixture followed by SnCl4 (14.22 mmol) dropwise. The reaction mixture was stirred at 70 °C overnight, cooled to room temperature and diluted with dichloromethane and a saturated NaHC03 solution. The white suspension was filtered through a pad of celite and washed with dichloromethane. The filtrate was extracted with dichloromethane twice. The combined organic layers were dried over anhydrous Na2S04, filtered and evaporated under reduced pressure to afford expected intermediate as a white solid in 89% yield.[00284] 1H NMR (DMSO, 400 MHz): δ (ppm) 1.58 (s, 3H), 4.68-4.81 (m, 3H), 5.68 (brs, 1H), 6.55 (brs, 1H), 7.36 (d, J= 7.84 Hz, 1H), 7.39-7.76 (m, 9H), 7.88-8.07 (m, 6H), 8.30 (d, J= 7.84 Hz, 1H); MS (ESI) m/z= 588 (MH+).3′,5′-Di-0-benzoyl-2,-C-chloro-2,-C-methyluridine (A9):

[00285] A suspension of A8 (4.19 mmol) in an acetic acid/water mixture (67 ml/17 ml, v/v), was heated at 110 °C for 3 hours. The reaction mixture was evaporated to dryness and co-evaporated with toluene (three times) to afford expected intermediate in quantitative yield as an oil which was directly used for the next step; MS (ESI) m/z= 485 (MH+). 2 -C-Chloro-2 -C-methyluridine (301):

H O CI[00286] Intermediate A9 (4.19 mmol) in 7 N methanolic ammonia (80 ml) was stirred at room temperature for 24 hours. The mixture was evaporated to dryness, diluted with water and transferred into a separatory funnel. The aqueous layer was extracted withdichloromethane and water was removed under reduced pressure. The residue was purified by flash RP18 gel chromatography (eluent: water/acetonitrile 0 to 40%) to afford pure expected compound as a white foam in 79% yield.[00287] 1H NMR (DMSO, 400 MHz): δ (ppm) 1.44 (s, 3H), 3.60-3.68 (m, 1H), 3.80-3.94 (m, 3H), 5.39 (t, J= 4.45 Hz, 1H), 5.63 (d, J= 8.26 Hz, 1H), 5.93 (d, J= 5.72 Hz, 1H), 6.21 (s, 1H), 8.16 (d, J= 8.90 Hz, 1H), 11.44 (m, 1H); MS (ESI) m/z= 277 (MH+).2′-C-Chloro-2′-C-methyl-3-benzyloxymethyluridine (Al 1):

H O CI[00288] To a solution of 301 (0.361 mmol) in anhydrous DMF (4 ml) was added at -5 °C, DBU (0.723 mmol) followed by benzyloxymethylchloride (0.542 mmol). The reaction mixture was stirred for 45 minutes between -5 °C and 5 °C. The solvent was evaporated under reduced pressure and the residue was purified by chromatography on silica gel (eluent: dichloromethane/methanol 0 to 10%) to afford pure expected intermediate as a white solid in 80% yield.[00289] 1H NMR (DMSO, 400 MHz): δ (ppm) 1.41 (s, 3H), 3.61-3.69 (m, 1H), 3.82-3.95 (m, 3H), 4.57 (s, 2H), 5.32 (s, 2H), 5.43 (t, J= 4.46Hz, 1H), 5.80 (d, J= 8.08Hz, 1H), 5.96 (d, J= 4.46 Hz, 1H), 6.23 (s, 1H), 7.22-7.36 (m, 5H), 8.25 (d, J= 8.22Hz, 1H); MS (ESI) m/z= 397 (MH+). Isopropyl (2S)-2-[[chloro(phenoxy)phosphoryl]amino]propanoate (A12a):

2,2-Dimethylpropyl (2S)-2-[[chloro(phenoxy)phosphoryl]amino]propanoate (A12b):

[00290] To a solution of aminoester, HC1 salt (0.434 mmol) in anhydrous dichloromethane (or acetonitrile) (4 ml) (3 times vacuo/nitrogen) under nitrogen was added at -30°C phenyldichlorophosphate (0.434 mmol) followed by N-methylimidazole (2.90 mmol)(or only 1.45 mmol for A12b). The reaction mixture was stirred at -30°C during 1 hour. The reaction was monitored by LC/MS (the sample was quenched by methanol or water) to check the complete formation of expected intermediate A12a [MS (ESI) m/z= 302 (MH+)(-OMe compounder A12b [MS (ESI) m/z= 314 (MH~)].Compound (A13a), (A13b) or (83ii):[00291] To the previous reaction mixture containing A12 was added All (or 302) (0.29 mmol) at -25°C under nitrogen. The reaction mixture was allowed to warm up slowly to room temperature overnight, and then diluted with dichloromethane and water (or with NaHCC”3 and EtOAc). The organic layer was extracted, dried, filtered and evaporated under reduced pressure. The crude residue was purified by chromatography on silica gel (eluent: dichloromethane/methanol 0 to 10%) (followed by preparative HPLC for A29).Compound (A13a):

[00292] Mixture of diastereoisomers; MS (ESI) m/z= 666 (MH+). Compound (A13b):

[00293] Mixture of diastereoisomers; MS (ESI) m/z= 692.3 (MH ).Compound (83ii):

[00294] Glassy solid; 1H NMR (CDCI3, 400MHz): δ (ppm) 1.19-1.24 (m, 9H), 1.35 (d, J = 7.1Hz, 3H), 3.95-4.05 (m, 1H), 4.31 (d, J= 8.1Hz, 2H), 4.41 (d, J= 9.0Hz, 1H), 4.59 (d, J = 7.1Hz, 2H), 4.98 (heptuplet, J= 6.28Hz, 1H), 6.38 (brs, 1H), 6.52 (s, 1H), 7.08-7.15 (m, 1H), 7.23-7.30 (m, 4H), 8.07 (s, 1H), 8.31 (s, 1H); 31P NMR (CDC13, 161.98 MHz): δ (ppm) 3.96 (s, IP); MS (ESI) m/z= 569.20 (MH+).Compounds (40iia) and (40iib):

[00295] To a solution of A13 (0.29 mmol) in anhydrous ethanol (6 ml) was added trifluoroacetic acid (2.9 mmol) dropwise (then 3 times vacuo/nitrogen purges), followed by Palladium hydroxide (20% on Carbon). The reaction mixture was purged 3 timesvacuo/nitrogen, and 3 times vacuo/hydrogen and then stirred under hydrogen for 5 hours. The reaction mixture was diluted with ethyl acetate and filtered through a pad of celite. The filtrate was evaporated under reduced pressure, and the crude compound was purified by preparative MS/HP LC to afford two pure compounds in 48% global yield.[00296] Compound 40ii (diastereoisomer 1): white solid; 1H NMR (CDC13, 400 MHz): δ (ppm) 1.22-1.26 (m, 6H), 1.37 (d, J= 7.08 Hz, 3H), 1.51 (s, 3H), 3.71-3.88 (m, 2H), 3.97- 4.06 (m, 1H), 4.16-4.18 (m, 1H), 4.45-4.57 (m, 2H), 4.97-5.07 (m, 1H), 5.57 (d, J= 8.20 Hz, 1H), 6.39 (s, 1H), 7.18-7.37 (m, 5H), 7.44 (d, J= 8.20 Hz, 1H), 8.40 (s, 1H); 31P NMR (CDC13, 161.98 MHz): δ (ppm) 4.20 (s, IP); MS (ESI, El+) m/z= 546 (MH+).[00297] Compound 40ii (diastereoisomer 2): white solid; 1H NMR (CDC13, 400 MHz): δ (ppm) 1.24-1.26 (m, 6H), 1.36 (d, J= 7.04 Hz, 3H), 1.59 (s, 3H), 3.69-3.77 (m, 1H), 3.91- 3.99 (m, 2H), 4.17-4.19 (m, 1H), 4.43-4.59 (m, 2H), 5.01-5.06 (m, 1H), 5.68 (d, J= 8.20 Hz, 1H), 6.42 (s, 1H), 7.21-7.39 (m, 5H), 7.60 (d, J=8.20 Hz, 1H), 8.14 (s, 1H); 31P NMR (CDC13, 161.98 MHz): δ (ppm) 3.47 (s, IP); MS (ESI) m/z= 546 (MH+).Compound 42ii:

[00298] Compound 42ii was synthesized from compound A13b (0.144 mmol) as described for compound 40ii.[00299] White solid; 1H NMR (MeOD, 400 MHz) δ (ppm) 0.94 (s, 9H), 1.40 (d, J= 7.10 Hz, 3H), 1.53 (s, 3H), 3.76 (d, J= 10.43 H, 1H), 3.86 (d, J= 10.44 H, 1H), 3.98-4.06 (m, 2H), 4.18-4.22 (m, 1H), 4.39-4.44 (m, 1H), 4.52-4.57 (m, 1H), 5.62 (d, J= 8.18 Hz, 1H), 6.40 (s, 1H), 7.20-7.29 (m, 3H), 7.36-7.41 (m, 2H), 7.74 (d, J= 8.18 Hz, 1H); 31P NMR (MeOD, 161.98 MHz) δ (ppm) 3.68 (s, IP); MS (ESI) m/z = 574.08 (MH+).
PAPER
US 20170226146
https://patents.google.com/patent/US20170226146A1/en
- [0250]
- [0251]
A 3-neck 100 mL jacketed round bottom flask with nitrogen inlet and mechanical stirrer was charged with compound 4 (3.0 g, 10.8 mmol), compound 13 (0.484 g, 2.17 mmol, 0.20 equiv), 2-butanone (21 mL), and 2,6-lutidine (2.53 mL, 21.7 mmol, 2.0 equiv). The resulting slurry was cooled to −15° C., then a solution of compound 12 (7.96 g, 13.0 mmol) in 2-butanone (3 mL) was added over 14 hours. The reaction mixture was allowed to stir at −15° C. for an additional 25 hours and then warmed to 20° C. n-Heptane (16 mL) was added with stirring over a 1 hour period then the mixture was allowed to stir at 25° C. for 3 hours, then filtered through a fitted funnel. The filter cake was slurry-washed with a 3:2 mixture of 2-butanone and n-heptane (10 mL and then 15 mL), then dried by pulling nitrogen stream through the fritted funnel. The filter cake was slurried in a 10:1 mixture of water and 2-butanone (21 mL) and then filtered. This slurrying and filtration sequence was repeated two more times. The resulting filter cake was dried with nitrogen stream through the fritted funnel to provide compound 6.
Example 21Alternate Preparation of Compound A
- [0252]
- [0253]
Compound 6 (0.072 mmol, 1 equiv), K2HPO4 (63.0 mg, 0.361 mmol) and compound 14 (5.45 mg, 0.018 mmol) were added to a 1 dram vial with 4 A mol sieves (40 mg). To the resulting mixture was added DCM (800 μl), then the resulting reaction was allowed to stir for 5 minutes. To the reaction mixture was then added compound 14 (28.7 mg, 0.094 mmol, 1.3 equiv) and the resulting reaction was allowed to stir for about 15 hours at room temperature to provide Compound A.
- [0256]
- [0257]
A 100 mL reactor with nitrogen inlet and mechanical stirrer was charged with compound 4 (7.00 g, 25.3 mmol), compound 15 (0.225 g, 0.506 mmol, 0.020 equiv), 1,3-dioxolane (42 mL), and 2,6-lutidine (4.42 mL, 38.0 mmol, 1.5 equiv). The mixture was cooled to −10° C. and a 33 wt % solution of compound 12 in isopropyl acetate (29 mL, 30 mmol) was added over 1 hour. The reaction mixture was allowed to stir at −10° C. for additional 40 hours, then isopropyl acetate (28 mL) was added, and the resulting mixture was warmed to 0° C. A 10 wt % aqueous NaHSO4 solution was added (14 mL), and the mixture was allowed to stir at 30° C. for 30 minutes, then the layers were separated. To the organic layer was added an aqueous solution containing 5 wt % NaHCO3 and 5 wt % Na2SO4 (21 mL). The mixture was allowed to stir at 50° C. for 6 h. The layers were separated. To the organic layer was added 10 wt % aqueous NaCl solution (21 mL). The mixture was allowed to stir at 50° C. for 30 min. The organic layer was separated, combined with isopropyl acetate (5 mL) and concentrated in vacuo to half volume at 20000 pa in a 50° C. bath. The resulting solution was solvent-switched with isopropanol (4×35 mL) to 60 g weight. The mixture was seeded with 100 mg of compound A at 60° C. The resulting slurry was allowed to stir at 55° C. for 30 minutes, then n-Heptane (35 mL) was added over 1 hour at 55° C. The resulting slurry was allowed to stir for an additional 1 hour at 55° C., then cooled to room temperature and filtered. The filter cake was washed with a 1:1 mixture of isopropanol and n-heptane (3×14 mL), followed by n-heptane (14 mL), then dried under nitrogen to provide Compound A.
PAPER
https://pubs.rsc.org/en/content/articlelanding/2021/sc/d1sc01978c#!divAbstract
Uprifosbuvir is an antiviral agent developed for treatment of chronic hepatitis C infections. Its original synthesis route requires twelve steps with an overall yield of only 1 %. Such a difficult and time-consuming synthesis approach is acceptable for the early trial phase of a new drug, but impractical for broad application as hepatitis C treatment or for repurposing against novel viral diseases.
Artis Klapars, John Y. L. Chung, and colleagues, Merck & Co., Inc., Rahway, NJ, USA, and WuXi STA, Shanghai, China, have developed a synthesis route for uprifosbuvir requiring only five steps and starting from readily available uridine. Initially, uridine is selectively oxidized after OH-acylation with pivaloyl chloride in an acyl migration/oxidation process driven by complexation with the Lewis acid BF3*OEt2 in toluene. In the second step, methylation is achieved by MeMgBr/MgCl2 in a toluene/anisole mixture where a more reactive methyl-manganese species is formed in-situ from the Grignard reagent, providing high yield and a good diastereomeric ratio (dr). Subsequently, the tertiary chloride group is introduced. Due to the high functional-group density, a cyclodehydration step is required before chlorination to avoid side reactions. The chlorination is carried out using dichlorodimethylsilane with FeCl3*6H2O and tetramethyldisiloxane as additives which avoids the hazardous use of HCl gas under pressure required in the initial synthesis. In the final step, the regioselective phosphoramidation is achieved using a chlorophosphoramidate precursor and a dimeric chiral imidazole carbamate catalyst which led to a dr of 97:3 starting from a 1:1 diastereomeric mixture of the chlorophosphoramidate reagent.
Uprifosbuvir was synthesized with an overall yield of 50 %, a vast improvement compared to the 1 % of the original synthesis route. Additionally, the newly developed synthesis steps have the potential to provide easier access to other nucleoside-based antiviral agents.
- Efficient Synthesis of Antiviral Agent Uprifosbuvir Enabled by New Synthetic Methods,
Artis Klapars, John Chung, John Limanto, Ralph Calabria, Louis-Charles Campeau, Kevin Campos, Wenyong Chen, Stephen M Dalby, Tyler A Davis, Daniel DiRocco, Alan Hyde, Amude M Kassim, Mona Utne Larsen, Guiquan Liu, Peter Maligres, Aaron Moment, Feng Peng, Rebecca Ruck, Michael Shevlin, Bryon L Simmons, Zhiguo Jake Song, Lushi Tan, Timothy J Wright, Susan Zultanski,
Chemical Science 2021.
https://doi.org/10.1039/D1SC01978C
Efficient synthesis of antiviral agent uprifosbuvir enabled by new synthetic methods†
Artis Klapars, *a
This article is Open Access

All publication charges for this article have been paid for by the Royal Society of Chemistry
Abstract
An efficient route to the HCV antiviral agent uprifosbuvir was developed in 5 steps from readily available uridine in 50% overall yield. This concise synthesis was achieved by development of several synthetic methods: (1) complexation-driven selective acyl migration/oxidation; (2) BSA-mediated cyclization to anhydrouridine; (3) hydrochlorination using FeCl3/TMDSO; (4) dynamic stereoselective phosphoramidation using a chiral nucleophilic catalyst. The new route improves the yield of uprifosbuvir 50-fold over the previous manufacturing process and expands the tool set available for synthesis of antiviral nucleotides.

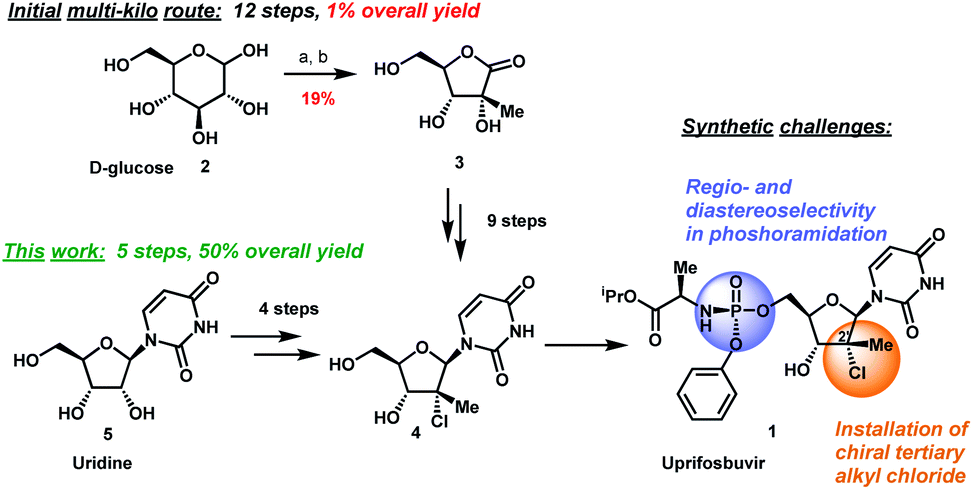
Scheme 1 Synthetic approaches to uprifosbuvir 1 with the two main challenges highlighted. (a) Me2NH, AcOH, EtOH/MeOH, 80 °C, 1.5 h; (b) Ca(OH)2, water, 70 °C, 24 h, 19% over 2 steps.9
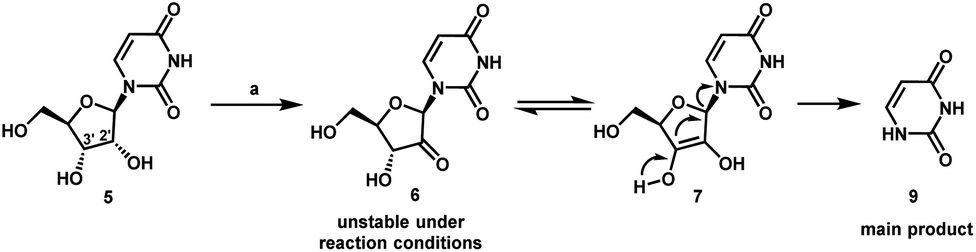
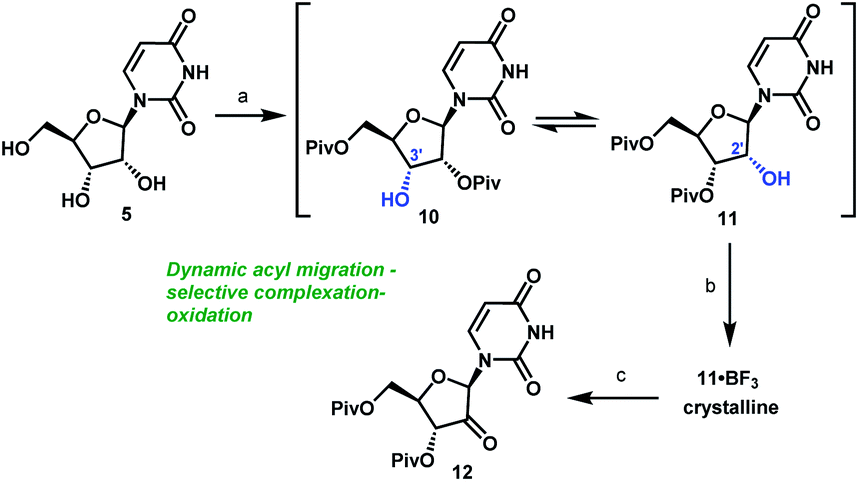
Scheme 3 Complexation-driven selective acyl migration/oxidation to access 12. (a) PivCl, pyridine, 0 °C, 16 h; (b) BF3·OEt2, PhMe, 40 °C, 10 h; (c) TEMPO, Bu4NBr, AcOOH, dioctyl sulphide, PhMe, −10 °C to 20 °C, 24 h, 83% from 5.
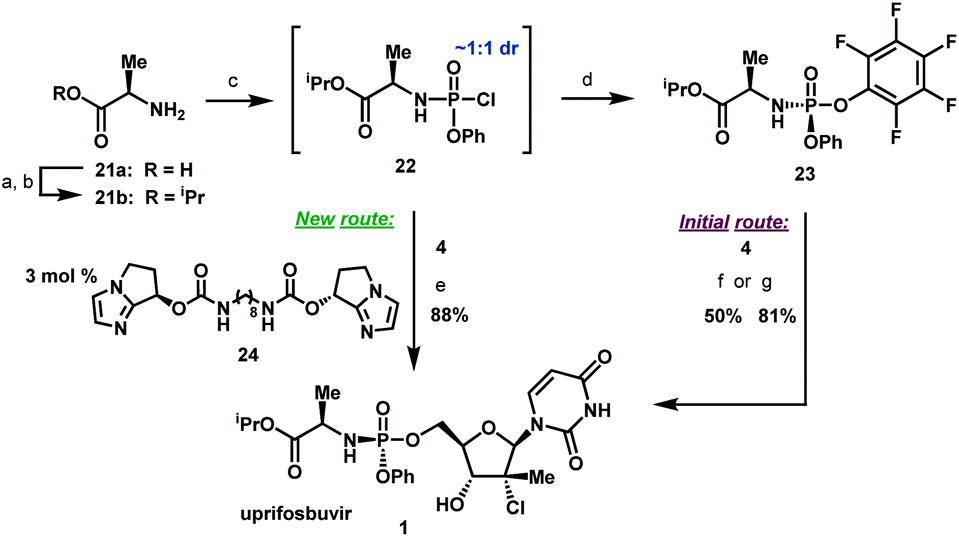
Scheme 6 Completion of uprifosbuvir synthesis. (a) TMS-Cl, iPrOH, 70 °C, 12 h; (b) NEt3, iPrOAc, wiped film evaporation, 80%; (c) PhOP(O)Cl2, NEt3, iPrOAc, −20 °C, 2 h, 90%; (d) C6F5OH, NEt3, iPrOAc, −5 °C to 10 °C, 18 h, 76%;26 (e) 4, 3 mol% 24, 2,6-lutidine, 1,3-dioxolane, −10 °C, 24 h, 88%; (f) 4, tBuMgCl, THF, −5 °C to 5 °C, 15 h, 50%;27 (g) 4, Me2AlCl, 2,6-lutidine, THF, 35 °C, 16 h, 81%.27
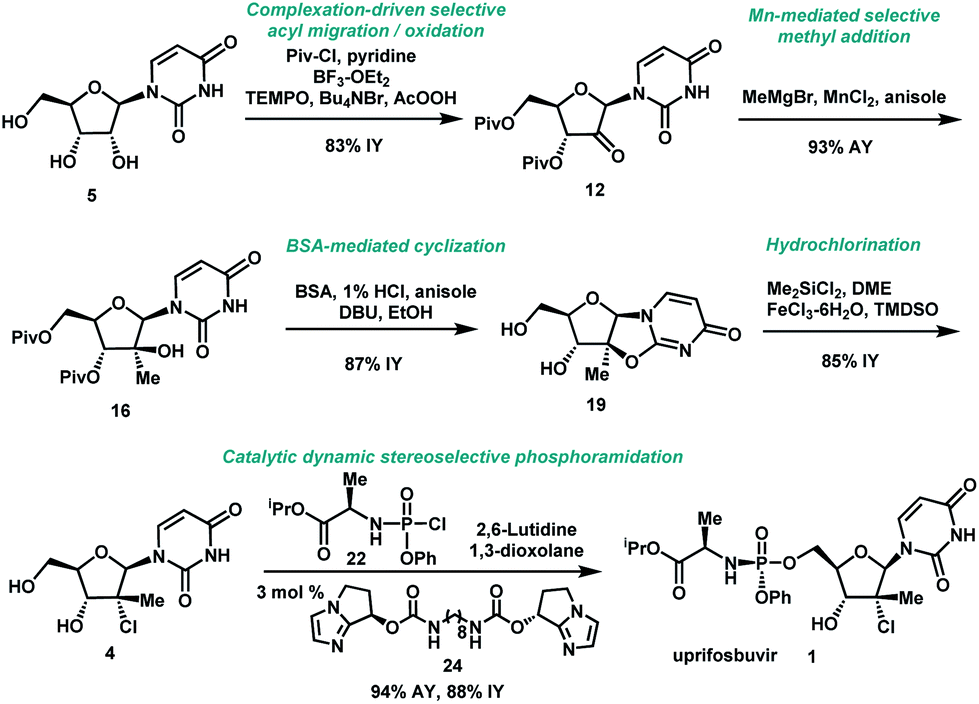
Scheme 7 Summary of uprifosbuvir synthesis. AY = assay yield; IY = isolated yield.
https://www.rsc.org/suppdata/d1/sc/d1sc01978c/d1sc01978c1.pdf
PAPERhttps://www.sciencedirect.com/science/article/abs/pii/S0960894X17308314
References
- ^ Soriano V, Fernandez-Montero JV, de Mendoza C, Benitez-Gutierrez L, Peña JM, Arias A, Barreiro P (August 2017). “Treatment of hepatitis C with new fixed dose combinations”. Expert Opinion on Pharmacotherapy. 18 (12): 1235–1242. doi:10.1080/14656566.2017.1346609. PMID 28644739. S2CID 205819421.
- ^ Borgia G, Maraolo AE, Nappa S, Gentile I, Buonomo AR (March 2018). “NS5B polymerase inhibitors in phase II clinical trials for HCV infection”. Expert Opinion on Investigational Drugs. 27 (3): 243–250. doi:10.1080/13543784.2018.1420780. PMID 29271672. S2CID 3672885.
- ^ Lawitz E, Gane E, Feld JJ, Buti M, Foster GR, Rabinovitz M, et al. (September 2019). “Efficacy and safety of a two-drug direct-acting antiviral agent regimen ruzasvir 180 mg and uprifosbuvir 450 mg for 12 weeks in adults with chronic hepatitis C virus genotype 1, 2, 3, 4, 5 or 6”. Journal of Viral Hepatitis. 26 (9): 1127–1138. doi:10.1111/jvh.13132. PMID 31108015. S2CID 160014275.
| Clinical data | |
|---|---|
| Trade names | Uprifosbuvir |
| Legal status | |
| Legal status | US: Investigational New Drug |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1496551-77-9 |
| PubChem CID | 90055716 |
| DrugBank | DB15206 |
| ChemSpider | 57427403 |
| UNII | JW31KPS26S |
| KEGG | D10996 |
| ChEMBL | ChEMBL3833371 |
| Chemical and physical data | |
| Formula | C22H29ClN3O9P |
| Molar mass | 545.9 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
Uprifosbuvir (MK-3682) is an antiviral drug developed for the treatment of Hepatitis C. It is a nucleotide analogue which acts as an NS5B RNA polymerase inhibitor. It is currently in Phase III human clinical trials.[1][2][3]
References
- ^ Soriano V, Fernandez-Montero JV, de Mendoza C, Benitez-Gutierrez L, Peña JM, Arias A, Barreiro P (August 2017). “Treatment of hepatitis C with new fixed dose combinations”. Expert Opinion on Pharmacotherapy. 18 (12): 1235–1242. doi:10.1080/14656566.2017.1346609. PMID 28644739. S2CID 205819421.
- ^ Borgia G, Maraolo AE, Nappa S, Gentile I, Buonomo AR (March 2018). “NS5B polymerase inhibitors in phase II clinical trials for HCV infection”. Expert Opinion on Investigational Drugs. 27 (3): 243–250. doi:10.1080/13543784.2018.1420780. PMID 29271672. S2CID 3672885.
- ^ Lawitz E, Gane E, Feld JJ, Buti M, Foster GR, Rabinovitz M, et al. (September 2019). “Efficacy and safety of a two-drug direct-acting antiviral agent regimen ruzasvir 180 mg and uprifosbuvir 450 mg for 12 weeks in adults with chronic hepatitis C virus genotype 1, 2, 3, 4, 5 or 6”. Journal of Viral Hepatitis. 26 (9): 1127–1138. doi:10.1111/jvh.13132. PMID 31108015. S2CID 160014275.
//////////uprifosbuvir, MK 3682, ウプリホスブビル, уприфосбувир, أوبريفوسبوفير , 乌磷布韦 , IDX-21437, DB15206, SB18784, D10996, Q27281714, IDX 21437, PHASE 3
CC(C)OC(=O)C(C)NP(=O)(OCC1C(C(C(O1)N2C=CC(=O)NC2=O)(C)Cl)O)OC3=CC=CC=C3
NEW DRUG APPROVALS
ONE TIME
$10.00
ARCT-021 (LUNAR-COV19)
ARCT-021 (LUNAR-COV19)
cas 2541451-24-3
A lipid-enabled and UnlockedNucleomonomer Agent modified RNA (LUNAR) of self-replicating RNA for vaccination against spike protein of SARS-CoV-2 (Arcturus)
Self-replicating RNA vaccine
| Arcturus Therapeutics and Duke-NUS Medical School, Singapore |
- OriginatorArcturus Therapeutics
- ClassCOVID-19 vaccines; RNA vaccines; Viral vaccines
- Mechanism of ActionImmunostimulants
- Orphan Drug StatusNo
- New Molecular EntityNo
- Available For LicensingYes – COVID 2019 infections
- Phase IICOVID 2019 infections
- 01 Mar 2021Arcturus Therapeutics has patent pending for STARR platform in USA
- 01 Mar 2021Immunogenicity data from a preclinical studies in COVID-2019 infections released by Arcturus Therapeutics
- 01 Mar 2021Arcturus Therapeutics completes a phase I/II trial in COVID-2019 infection in the Singapore
ref International Journal of Biological Sciences (2021), 17(6), 1446-1460. https://www.ijbs.com/v17p1446.htm
| LUNAR-COV19 | T7 | m7GpppNmN | Yes | VEEV-FL-S | N1-methyl pseudouridine | Silicon column | |
| protein | [54] |
ARCT-021: Currently undergoing phase 1/2 clinical trials, it combines two technologies, i.e., saRNA STARR™ and LUNAR® lipid-mediated delivery method. It was designed to enhance and extend antigen expression, enabling vaccination at lower doses [87]. In addition, LUNAR® lipids are pH-sensitive and biodegradable, causing minimal lipid accumulation in cells after multiple dosing [87]The Arcturus COVID-19 vaccine, commonly known as ARCT-021 and LUNAR-COV19, is a COVID-19 vaccine candidate developed by Arcturus Therapeutics.
| LUNAR- COV19 | 1 | Day 0 | 0.2 μg and 10 μg (Preclinical) | IM | Arcturus Therapeutics | N/A | Phase 2 | NCT04668339 NCT04480957 | [54] |
54. de Alwis R, Gan ES, Chen S, Leong YS, Tan HC, Zhang SL. et al. A Single Dose of Self-Transcribing and Replicating RNA Based SARS-CoV-2 Vaccine Produces Protective Adaptive Immunity In Mice. bioRxiv. 2020. 2020 09.03.280446
Development
Arcturus Therapeutics partnered with Singapore’s Duke–NUS Medical School to develop a COVID-19 vaccine.[1] The company also partnered with Catalent, a contract development and manufacturing organization, to manufacture multiple batches of Arcturus’ COVID-19 mRNA vaccine candidate.[2]
Clinical research
Phase I-II
LUNAR-COV19 clinical trials in humans began in July 2020.[3] On 4 January 2021, Arcturus Therapeutics started phase-2 clinical trials.[4]
Deployment
Arcturus has entered into development and supply agreements with the Economic Development Board of Singapore and supply agreements with the Israel Ministry of Health for LUNAR-COV19.[5][6]
Arcturus Therapeutics Receives FDA Allowance to Proceed with Phase 2 Study of ARCT-021 (LUNAR-COV19) Vaccine Candidate in the United States
Phase 2 study to be conducted in the U.S. and Singapore, and will evaluate both single dose and two dose priming regimens of ARCT-021 in up to 600 participants
Anticipate interim Phase 2 data in early 2021; targeting global Phase 3 study start in Q2 2021 which could allow application for emergency use authorization/conditional approval in H2 2021January 04, 2021 07:01 AM Eastern Standard Time
SAN DIEGO–(BUSINESS WIRE)–Arcturus Therapeutics Holdings Inc. (the “Company”, “Arcturus”, Nasdaq: ARCT), a leading clinical-stage messenger RNA medicines company focused on the development of infectious disease vaccines and significant opportunities within liver and respiratory rare diseases, today announced that the Company has received allowance of the Investigational New Drug (IND) application from the U.S. Food and Drug Administration (FDA) for the Phase 2 clinical study of its vaccine candidate ARCT-021 following review of data from the Phase 1/2 study.
Arcturus Therapeutics Receives FDA Allowance to Proceed with Phase 2 Study of ARCT-021 (LUNAR-COV19) Vaccine Candidate in the United States
Arcturus previously announced that the ARCT-021 Phase 2 study had been approved to proceed by the Singapore Health Sciences Authority (HSA), who reviewed the same data as reviewed by the FDA. These Phase 1/2 study results demonstrated favorable tolerability and both humoral and cellular immunogenicity following administration of ARCT-021.
The Phase 2 study will enroll 600 participants, with 450 receiving ARCT-021 and 150 receiving placebo. Both older and younger adult participants will be included. Early interim analyses of safety and immunogenicity will be performed to inform dose selection for a Phase 3 study, which is targeted to start in Q2 2021, if the Phase 2 study is successful.
“Allowance of the IND for our ARCT-021 Phase 2 clinical study represents an important milestone for the program and we look forward to starting to screen study participants at U.S. and Singapore clinical sites very soon,” said Steve Hughes, M.D., Chief Medical Officer of Arcturus. “We have advanced ARCT-021 to Phase 2 based on promising interim results from our Phase 1/2 study and extensive preclinical data. Our prior clinical results show that ARCT-021 administration results in humoral and cellular immunogenicity, and we are encouraged by an increasing body of evidence highlighting the potential importance of T cells in providing protection against SARS-CoV-2 infection and COVID-19. We believe that ARCT-021 holds promise to be a highly effective vaccine with a differentiated clinical profile, including the potential to only require a single dose for protection.”
About Arcturus Therapeutics
Founded in 2013 and based in San Diego, California, Arcturus Therapeutics Holdings Inc. (Nasdaq: ARCT) is a clinical-stage mRNA medicines and vaccines company with enabling technologies: (i) LUNAR® lipid-mediated delivery, (ii) STARR™ mRNA Technology and (iii) mRNA drug substance along with drug product manufacturing expertise. Arcturus’ diverse pipeline of RNA therapeutic and vaccine candidates includes self-replicating mRNA vaccine programs for SARS-CoV-2 (COVID-19) and Influenza, and other programs to potentially treat Ornithine Transcarbamylase (OTC) Deficiency, Cystic Fibrosis, and Cardiovascular Disease along with partnered programs including Glycogen Storage Disease Type 3, Hepatitis B Virus, and non-alcoholic steatohepatitis (NASH). Arcturus’ versatile RNA therapeutics platforms can be applied toward multiple types of nucleic acid medicines including messenger RNA, small interfering RNA, replicon RNA, antisense RNA, microRNA, DNA, and gene editing therapeutics. Arcturus’ technologies are covered by its extensive patent portfolio (205 patents and patent applications, issued in the U.S., Europe, Japan, China and other countries). Arcturus’ commitment to the development of novel RNA therapeutics has led to collaborations with Janssen Pharmaceuticals, Inc., part of the Janssen Pharmaceutical Companies of Johnson & Johnson, Ultragenyx Pharmaceutical, Inc., Takeda Pharmaceutical Company Limited, CureVac AG, Synthetic Genomics Inc., Duke-NUS Medical School, and the Cystic Fibrosis Foundation. For more information visit www.ArcturusRx.com. In addition, please connect with us on Twitter and LinkedIn.
References
- ^ Teo J (15 April 2020). “Coronavirus: Clinical trials for Singapore’s vaccine project could start in August”. The Straits Times. Retrieved 27 April 2020.
- ^ Stanton D (6 May 2020). “With Arcturus, Catalent bags another COVID project”. Bioprocess Insider. Retrieved 8 May 2020.
- ^ Clinical trial number NCT04480957 for “Phase 1/2 Ascending Dose Study of Investigational SARS-CoV-2 Vaccine ARCT-021 in Healthy Adult Subjects” at ClinicalTrials.gov
- ^ “Arcturus Therapeutics Receives FDA Allowance to Proceed with Phase 2 Study of ARCT-021 (LUNAR-COV19) Vaccine Candidate in the”. Bloomberg. 4 January 2021. Retrieved 17 January 2021.
- ^ Anwar N (26 November 2020). “Singapore’s co-developed vaccine candidate is in ‘good shape’ for delivery in 2021”. CNBC. Retrieved 18 March 2021.
- ^ Cheok M, Mookerjee I (5 August 2020). “Singapore Will Get First Claim to Any Successful Arcturus Vaccine”. Bloomberg. Retrieved 18 March 2021.
External links
| Scholia has a profile for Lunar-COV19 (Q98713328). |
| Vaccine description | |
|---|---|
| Target | SARS-CoV-2 |
| Vaccine type | mRNA |
| Clinical data | |
| Other names | ARCT-021, LUNAR-COV19 |
| Routes of administration | Intramuscular |
| Part of a series on the |
| COVID-19 pandemic |
|---|
| COVID-19 (disease)SARS-CoV-2 (virus) |
| showTimeline |
| showLocations |
| showInternational response |
| showMedical response |
| showImpact |
| COVID-19 portal |
/////////COVID-19, SARS-CoV-2, corona virus, singapore, ARCT 021, LUNAR-COV19
NEW DRUG APPROVALS
one time
$10.00
DAPAGLIFLOZIN

DAPAGLIFLOZIN, BMS-512148
ダパグリフロジン;
(2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol,
Cas 461432-26-8
| Molecular Formula: C21H25ClO6 |
| Molecular Weight: 408.87 |
Dapagliflozin propandiol monohydrate; 960404-48-2
| Molecular Weight | 502.98 |
| Formula | C21H25ClO6•C3H8O2•H2O |
Bristol-Myers Squibb (Originator)
AstraZeneca
TYPE 2 DIABETES,SGLT-2 Inhibitors
launched 2012, as forxiga in EU, FDA 2014, JAPAN PMDA 2014
Dapagliflozin propanediol monohydrate was first approved by European Medicine Agency (EMA) on November 12, 2012, then approved by the U.S. Food and Drug Administration (FDA) on January 8, 2014, and approved by Pharmaceuticals and Medical Devices Agency of Japan (PMDA) on March 24, 2014. It was co-developed and co-marketed as Forxiga® by Bristol-Myers Squibb and AstraZeneca in EU.
Dapagliflozin propanediol monohydrate is a sodium-glucose co-transporter 2 (SGLT2) inhibitor indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus.
Forxiga® is available as tablet for oral use, containing 5 mg or 10 mg of free Dapagliflozin. The recommended starting dose is 5 mg once daily in the morning.

Dapagliflozin propanediol is a solvate containing 1:1:1 ratio of the dapagliflozin, (S)-(+)-1,2-propanediol, and water.
US——-In 2011, the product was not recommended for approval by the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee. In 2011, the FDA assigned a complete response letter to the application. A new application was resubmitted in 2013 by Bristol-Myers Squibb and AstraZeneca in the U.S
WILMINGTON, Del. & PRINCETON, N.J.--(BUSINESS WIRE)--December 12, 2013-- USFDA



Sales:$518.7 Million (Y2015); 
$235.8 Million (Y2014);
$33 Million (Y2013);ATC Code:A10BX09
Approved Countries or AreaUpdate Date:2015-07-29
- US
- EU
- JP
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2014-01-08 | Marketing approval | Farxiga | Type 2 diabetes | Tablet | 5 mg/10 mg | AstraZeneca |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2012-11-12 | Marketing approval | Forxiga | Type 2 diabetes | Tablet, Film coated | Eq. 5 mg/10 mg Dapagliflozin | Bristol-Myers Squibb, AstraZeneca |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2014-03-24 | Marketing approval | Forxiga | Type 2 diabetes | Tablet, Film coated | 5 mg/10 mg | Bristol-Myers Squibb, AstraZeneca, Ono |
MoreChemical Structure
AstraZeneca (NYSE:AZN) and Bristol-Myers Squibb Company (NYSE:BMY) today announced the U.S. Food and Drug Administration’s (FDA) Endocrinologic and Metabolic Drugs Advisory Committee (EMDAC) voted 13-1 that the benefits of dapagliflozin use outweigh identified risks and support marketing of dapagliflozin as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus. The Advisory Committee also voted 10-4 that the data provided sufficient evidence that dapagliflozin, relative to comparators, has an acceptable cardiovascular risk profile.
The FDA is not bound by the Advisory Committee’s recommendation but takes its advice into consideration when reviewing the application for an investigational agent. The Prescription Drug User Fee Act (PDUFA) goal date for dapagliflozin is Jan. 11, 2014.

Dapagliflozin is being reviewed by the FDA for use as monotherapy, and in combination with other antidiabetic agents, as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes. It is a selective and reversible inhibitor of sodium-glucose cotransporter 2 (SGLT2) that works independently of insulin to help remove excess glucose from the body. Dapagliflozin, an investigational compound in the U.S., was the first SGLT2 inhibitor to be approved anywhere in the world. Dapagliflozin is currently approved under the trade name [Forxiga](TM) for the treatment of adults with type 2 diabetes, along with diet and exercise, in 38 countries, including the European Union and Australia.
http://online.wsj.com/article/PR-CO-20131212-910828.html?dsk=y
Reference:1. WO03099836A1 / US6515117B2.
2. WO2010048358.
3. J. Med. Chem. 2008, 51, 1145–1149.
4. WO2004063209A2 / US7375213B2.
5. WO2008002824A1 / US7919598B2.Route 2
Reference:1. WO2010022313 / US8283454B2.Route 3
Reference:1. WO2013068850.Route 4
Reference:1. Org. Lett. 2012, 14, 1480-1483.
PAPER
https://www.future-science.com/doi/10.4155/fmc-2020-0154
Patent
https://patents.google.com/patent/WO2016178148A1/en
1. A process for the preparation of dapagliflozin in amorphous form, the process comprising:
(a) reducing a compound of formula II to a compound of formula ΠΙ in the presence of a Lewis acid;

(b) silylating a compound of formula IV with hexamethyldisilazane to form a compound of formula V;

(c) reacting the compound of formula III with the compound of formula V in the presence of a strong base followed by treatment with an acid in the presence of an alcohol to prepare a compound of formula VII, wherein R is an alkyl group selected from C1-5 alkyl;

(d) converting the compound of formula VII to dapagliflozin;
(e) acetylating dapagliflozin to give D-glucitol, l ,5-anhydro-l -C-[4-chloro-3-[(4- ethoxyphenyl)methyl] phenyl]-, 2,3,4,6-tetraacetate, (IS)-, a compound of formula VIII;

(f) optionally, purifying the compound of formula VIII with a solvent selected from halogenated hydrocarbons, alcohols, ethers, or mixtures thereof; (g) hydrolyzing the compound of formula VIII obtained in step (f) to give dapagliflozin;
EXAMPLE 1: Preparation of 5-bromo-2-chlorobenzoyl chloride
To a suspension of 5-bromo-2-chiorobenzoic acid (lOg) in methylene di chloride (40niL), dimethylformamide (0.2g) and thionyl chloride were added and the reaction mixture was refluxed for about 2h. After completion of reaction, the solvent was distilled out. The mass obtained was degassed under vacuum followed by stripping with cyclohexane to give crude 5-bromo-2-chlorobenzoyl chloride (10.8g).
[0189] EXAMPLE 2: Preparation of 5-bromo-2-chloro-4′-ethoxybenzophenone (compound of Formula II)
5-bromo-2-chlorobenzoyl chloride (10.7g) was dissolved in methylene dichloride (40mL) and the reaction mixture was cooled to about -8°C to about -12°C under inert atmosphere. Aluminum chloride (5.65g) was added to the reaction mixture followed by addition of a solution of ethoxybenzene in methylene dichloride. The reaction mixture was stirred for about lh at about -8°C to -12°C and then quenched in dilute hydrochloric acid followed by extraction with methylene dichloride. The organic layer was washed with sodium bicarbonate solution and concentrated. The residue obtained was crystallized from methanol to give 5-bromo-2-chloro-4′-ethoxybenzophenone (8.5g). HPLC purity: 99.34%
[0190] EXAMPLE 3: Preparation of 5-bromo-2-chloro-4′-ethoxydiphenylmethane (compound of formula III)
To a mixture of 5-bromo-2-chloro-4′-ethoxybenzophenone (lOg) and methylene dichloride (50mL), cooled to about 0°C to about 5°C, triethylsilane (11.98g) and titanium chloride (22.3g) were added. The reaction mixture was stirred for about 3h at about 10°C to about 15°C. The reaction mixture was quenched into chilled water. The organic layer was separated, washed with water and sodium bicarbonate solution and concentrated under vacuum followed by stripping with toluene. The residue obtained was stirred with methanol, filtered and dried to give 5-bromo-2-chloro-4′-ethoxydiphenylmethane (9g). HPLC purity: 99.4%
[0191] EXAMPLE 4: Preparation of 2,3,4,6-tetra-0-(trimethylsilyl)-D-glucono-l,5- lactone (compound of Formula V)
To a mixture of D-glucono- 1,5 -lactone (lOg) and iodine (0.28g) in methylene dichloride (80mL), hexamethyldisilazane (36.1g) was added and the reaction mixture was refluxed. After completion of reaction, the reaction mixture was concentrated and degassed to give 2,3,4,6-tetra-0-(trimethylsilyl)-D-glucono-l,5-lactone as liquid (25g). HPLC purity: 95%
[0192] EXAMPLE 5: Preparation of D-glucopyranoside, methyl l-C-[4-chloro-3-[(4- ethoxyphenyl)methyl] phenyl] (compound of Formula VII wherein R is methyl)
To a mixture of 2,3,4,6-tetra-0-(trimethylsilyl)-D-glucono-l ,5-lactone (25g) and 5- bromo-2-chloro-4′-ethoxydiphenylmethane (8.7g) in tetrahydrofuran (174mL), cooled to about -75°C to about -88 °C under nitrogen atmosphere, n-butyl lithium in hexane (50mL) was slowly added. The reaction mixture was stirred at about the same temperature and then mixture of methanol and methanesulphonic acid was added to it. The reaction mixture was quenched into sodium bicarbonate solution and extracted with ethyl acetate. The organic layer was separated, washed with saturated sodium chloride solution and concentrated under vacuum to obtain a residue. The residue was purified with a mixture of toluene and cyclohexane. Yield: 1 lg as thick mass with 80-85% HPLC purity.
reacting the compound of formula III with the compound of formula V in the presence of a strong base followed by treatment with an acid in the presence of an alcohol to prepare a compound of formula VII, wherein R is an alkyl group selected from C1-5 alkyl;
[0193] EXAMPLE 6: Preparation of D-glucitol, l,5-anhydro-l-C-[4-chloro-3-[(4- ethoxyphenyl) methyl] phenyl]
To a mixture of D-glucopyranoside, methyl l -C-[4-chloro-3-[(4- ethoxyphenyl)methyl] phenyl] in methylene di chloride (40mL) and acetonitrile (40mL), cooled to about -40°C to about -45°C, triethylsilane (8.74g) was added followed by addition of boron trifluoride etherate (10.67g) maintaining the temperature at about -40°C to about -45°C. The reaction mixture was quenched in sodium bicarbonate solution and extracted with ethyl acetate. The organic layer was separated, concentrated and degassed under vacuum to give title compound (1 lg) as thick residue with 80-85% HPLC purity.
[0194] EXAMPLE 7: Preparation of D-Glucitol, l,5-anhydro-l-C-[4-chloro-3-[(4- ethoxyphenyl)methyl]phenyl]-, 2,3,4,6-tetraacetate, (lS)-
To a cooled solution of D-glucitol, l,5-anhydro-l -C-[4-chloro-3-[(4-ethoxyphenyl) methyl] phenyl]- (l lg) in methylene dichloride (55mL) at about 0°C to about 5°C, diisopropylethylamme, dmiethylaminopyridine and acetic anhydride were added and the reaction mixture was stirred. After completion of reaction, the reaction mixture was quenched by adding water. The aqueous layer was separated and extracted with methylene dichloride. The organic layer was separated, washed with sodium bicarbonate solution and concentrated under vacuum to obtain residue which was stripped out with methanol. The residue was purified with methanol and charcoal, followed by diisopropyl ether and methanol crystallization. Yield: lOg; HPLC purity: 99.6%
acetylating dapagliflozin to give D-glucitol, l ,5-anhydro-l -C-[4-chloro-3-[(4- ethoxyphenyl)methyl] phenyl]-, 2,3,4,6-tetraacetate, (IS)-, a compound of formula VIII;
[0195] EXAMPLE 8: Preparation of D-glucitol, l,5-anhydro-l-C-[4-chloro-3-[(4- ethoxyphenyl)methyl] phenyl] (Dapagliflozin)
To a stirred solution of D-glucitol, l,5-anhydro-l-C-[4-chloro-3-[(4- ethoxyphenyl)methyl] phenyl]-, 2,3,4,6-tetraacetate, (IS)-, (lOg) in THF: methanol: water mixture (50mL: 50mL:30mL), sodium hydroxide was added and the reaction mixture was stirred. After completion of reaction, the solvents were distilled out under vacuum and the residue obtained was dissolved in methylene dichloride and washed with water and brine and dried over sodium sulfate. The reaction mixture was concentrated and degassed to give off- white to white solids of D-glucitol, l,5-anhydro-l-C-[4-chloro-3-[(4- ethoxyphenyl)methyl] phenyl]- (dapagliflozin) Yield: 7g (XRD matches with amorphous form) HPLC purity: 99.8%
[0197] EXAMPLE 10: Preparation of D-Glucitol, l,5-anhydro-l-C-[4-chloro-3-[(4- ethoxyphenyl)methyl]phenyl]-, 2,3,4,6-tetraacetate, (IS)- from D-glucono-1,5- lactone (One-pot Synthesis)
To a mixture of D-glucono-l,5-lactone (lOg) in methylene dichloride (80mL), hexamethyldisilazane (36. lg) was added and the reaction mixture was refluxed. After completion of reaction, the reaction mixture was concentrated and degassed. The residue obtained was dissolved in tetrahydrofuran. 5-Bromo-2-chloro-4′-ethoxydiphenylmethane (8.7g) was added to the reaction mixture which was cooled to about -75°C to about-85°C under nitrogen atmosphere. n-Butyl lithium in hexane (50mL) was slowly added to the reaction mixture maintaining the temperature between -75°C to about -85°C. The reaction mixture was stirred at about the same temperature and then mixture of methanol and methanesulphonic acid was added to it. The reaction mixture was quenched into sodium bicarbonate solution and extracted with ethyl acetate. The organic layer was separated, washed with saturated sodium chloride solution and concentrated under vacuum to obtain a residue. This residue was purified by a mixture of toluene and cyclohexane. To the product obtained, methylene dichloride and acetonitrile were added and the reaction mixture was cooled to about -40°C to about -45°C. Triethylsilane (8.74g) was added to the reaction mixture followed by addition of boron trifluoride etherate (10.67g) maintaining temperature at about -40°C to about -45°C. The reaction mixture was quenched in sodium bicarbonate solution. The aqueous layer was separated and extracted with ethyl acetate. The organic layer was separated, concentrated and degassed under vacuum. The thick residue obtained was dissolved in methylene dichloride and cooled to about 0°C to about 5°C. Diisopropylethylamine, dimethylaminopyridine and acetic anhydride were added to the reaction mixture which was stirred. After completion of reaction, the reaction mixture was quenched by adding water. The aqueous layer was separated and extracted with methylene dichloride. The organic layer was separated, washed with sodium bicarbonate solution and concentrated under vacuum to obtain residue which was stripped out with methanol The residue obtained was recrystallized with methanol and charcoal to give title compound (iOg) with 99.7% HPLC purity.
PAPER
Bioorganic & Medicinal Chemistry, 26(14), 3947-3952; 2018
https://www.sciencedirect.com/science/article/abs/pii/S0968089618309386?
Abstract
The cardiovascular complications were highly prevalent in type 2 diabetes mellitus (T2DM), even at the early stage of T2DM or the state of intensive glycemic control. Therefore, there is an urgent need for the intervention of cardiovascular complications in T2DM. Herein, the new hybrids of NO donor and SGLT2 inhibitor were design to achieve dual effects of anti-hyperglycemic and anti-thrombosis. As expected, the preferred hybrid 2 exhibited moderate SGLT2 inhibitory effects and anti-platelet aggregation activities, and its anti-platelet effect mediated by NO was also confirmed in the presence of NO scavenger. Moreover, compound 2 revealed significantly hypoglycemic effects and excretion of urinary glucose during an oral glucose tolerance test in mice. Potent and multifunctional hybrid, such as compound 2, is expected as a potential candidate for the intervention of cardiovascular complications in T2DM.
Graphical abstract

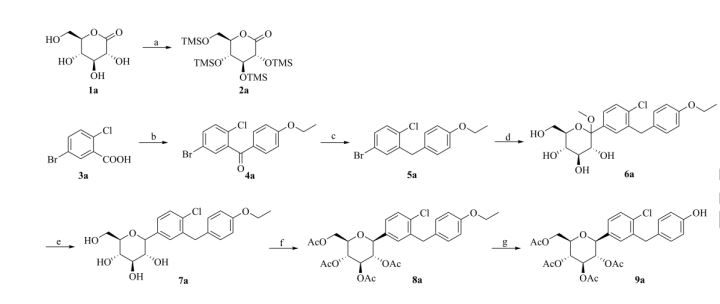
Scheme 1. Synthesis of target compounds 1-3. Reagents and conditions: (a) TMSCl, NMM, THF, 35 °C; (b) (COCl)2, CH2Cl2, DMF, then phenetole, AlCl3, 0 °C; (c) Et3SiH, BF3·OEt2, CH2Cl2, CH3CN, 25 °C; (d) n-BuLi, THF, toluene, -78 °C, then 2a followed by MeOH, CH3SO3H; (e) Et3SiH, BF3·OEt2, CH2Cl2, CH3CN, -10 °C; (f) Ac2O, pyridine, CH2Cl2, DMAP;
PATENT
Indian Pat. Appl., 2014MU03972,
PATENT
| Dapagliflozin, also known as SGLT2 inhibitor, chemical name is (2S,3R,4R,5S,6R)-2-[3-(4-ethoxybenzyl)-4-chlorophenyl]-6- Hydroxymethyltetrahydro-2H-pyran-3,4,5-triol, a sodium-glucose cotransporter 2 inhibitor, announced by the U.S. Food and Drug Administration (FDA) on January 8, 2014 , approved the use of dapagliflozin for the treatment of type 2 diabetes, the specific structural formula is as follows: |
| |
| Dapagliflozin works by inhibiting sodium-glucose transporter 2 (SGLT2), a protein in the kidney that allows glucose to be reabsorbed into the blood. This allows excess glucose to be excreted through the urine, thereby improving blood sugar control without increasing insulin secretion. |
| At present, there are two main methods for synthesizing dapagliflozin. One uses 5-bromo-2-chlorobenzoic acid as the starting material, which is chlorinated, Falk acylated, reduced, and then combined with 2,3,4 ,6-tetra-0-trimethylsilyl-D-glucopyranosic acid 1,5-lactone is condensed, methyl etherified, and demethoxylated to obtain dapagliflozin. The specific process route is as follows: |
| |
| The method has expensive starting materials and too many process steps, so it is not suitable for industrial production, and dangerous n-butyllithium needs to be used in the reaction process, so the requirements for experimental conditions are too high; |
| Another method is to use o-toluidine as the starting material, undergo bromination, diazotization, chlorination, and alkylation reactions, and then react with 2,3,4,6-tetra-0-trimethylsilyl -D-glucopyranosic acid 1,5-lactone is condensed, then methyl etherified and demethoxylated to obtain dapagliflozin. The specific process route is as follows: |
| |
| AIBN will be used in this reaction, which will produce highly toxic cyanide, which will seriously pollute the environment, and also requires the use of n-butyllithium, which requires high experimental conditions and is dangerous to operate, and is not suitable for large-scale production. |
| Example 1 |
| Weigh 16g (0.8mol) of activated magnesium, add two iodine pellets, heat to 35°C under nitrogen protection, then add 500mL of anhydrous THF to it, without magnesium, and then add 200mL of 10% iodobenzene to it 2000mL of THF solution (total 2000mL: 20g iodobenzene (0.53mol)+2000mL THF), after the color of the solution subsided, add the remaining iodobenzene solution dropwise, react for 5h, and filter to obtain the Grignard reagent THF solution of iodobenzene. |
| 433.4g (1.1mol) of peracetyl sugar and 1500mL of dichloromethane were added, cooled to 0°C in an ice-water bath, 545.5mL of 33% hydrogen bromide in acetic acid solution (2.2mol) was added dropwise to the reaction flask, and the mixture was gradually heated up The mixture was heated to 25° C. and stirred for 1.5 h. Saturated sodium bicarbonate solution was added to quench the reaction. The aqueous solution was extracted with dichloromethane, dried, and concentrated by rotary evaporation. 423.6 g of white solid were obtained, which was 2,3,4,6-tetraacetyl bromoglucose, and the yield was 93%. |
| Weigh 217.83g of 2,3,4,6-tetraacetyl bromoglucose (0.53mol) and dissolve it in 2500mL of toluene, then add 5.6g of europium oxide (3mol%) to it, under nitrogen protection, reduce the temperature to – 20°C, dropwise add the prepared iodobenzene Grignard reagent to it, and control the temperature at -20~-15°C. After the dropwise addition, the temperature is raised to 5°C for reaction for 2h, vacuum concentrated to an oily substance, and then added to it. 2000mL of toluene was dissolved, extracted twice with saturated aqueous sodium chloride solution, the organic phase was dried, concentrated in vacuo to an oily substance, and 1000mL of 3:1 n-hexane/ethyl acetate solution was added to it, heated to dissolve, cooled, and a solid was precipitated out, filtered , and dried to obtain 259.49 g of solid with a yield of 85%. |
| The above-mentioned solid was dissolved in 1000mL 2:3:1 tetrahydrofuran/ethanol/water solution, then 40g sodium hydroxide solid (1mol) was added to it, stirred overnight, vacuum concentrated to obtain oil after the reaction was finished, and then 1000mL of ethyl acetate was added thereto. The ester was dissolved, extracted twice with saturated aqueous sodium chloride solution and twice with saturated aqueous sodium thiosulfate solution, dried the organic phase, concentrated in vacuo to an oil, to which was added 200 mL of 1:3 ethyl acetate/acetonitrile solution , heated to dissolve, cooled and recrystallized to obtain 147.1 g of white rice-like crystals with a yield of 80.2% and a purity of 99.2% (determined by high performance liquid chromatography, external standard method). |
| IR(cm-1):1689,1612,1588,1523,1455,1253,1089,820。1H NMR(500MHz,CDCl 3 ):δ:7.36(d,J=8.2Hz,1H),7.32(d,J=1.9Hz,1H),7.23(dd,J=8.3,2.0Hz,1H),7.09(d,J=8.6Hz,2H),6.82(d,J=8.6Hz,2H),4.85(s,1H),4.41(s,3H),3.93~4.02(m,5H),3.70(dd,J=11.7,1.3Hz,1H),3.44(dd,J=11.7,5.6Hz,1H),3.26~3.28(m,1H)。LC-MS,m/z:[M+Na]+=431。 |
| Example 2 |
| Weigh 26.7g (1.33mol) of activated magnesium, add two iodine pellets, heat to 35°C under nitrogen protection, then add 500mL of anhydrous THF to it, without magnesium, and then add 200mL of 10% iodine to it The THF solution of benzene (2000mL in total: 20g iodobenzene (0.53mol)+2000mL THF), after the color of the solution subsides, add the remaining iodobenzene solution dropwise, react for 5h, and filter to obtain the Grignard reagent THF solution of iodobenzene . |
| 433.4g (1.1mol) of peracetyl sugar and 1500mL of dichloromethane were added, cooled to 0°C in an ice-water bath, 272.75mL of 33% hydrogen bromide in acetic acid solution (1.1mol) was added dropwise to the reaction flask, and the mixture was gradually heated up The mixture was heated to 25° C. and stirred for 1.5 h. Saturated sodium bicarbonate solution was added to quench the reaction. The aqueous solution was extracted with dichloromethane, dried, and concentrated by rotary evaporation. 381.5 g of white solid were obtained, which was 2,3,4,6-tetraacetyl bromoglucose, and the yield was 83.8%. |
| Weigh 217.83g of 2,3,4,6-tetraacetyl bromoglucose (0.53mol) and dissolve it in 5000mL of toluene, then add 5.76g of gadolinium oxide (3mol%) to it, under nitrogen protection, reduce the temperature to – 20°C, dropwise add the prepared iodobenzene Grignard reagent to it, and control the temperature at -20~-15°C. After the dropwise addition, the temperature is raised to 5°C for reaction for 2h, vacuum concentrated to an oily substance, and then added to it. 2000mL of toluene was dissolved, extracted twice with saturated aqueous sodium chloride solution, the organic phase was dried, concentrated in vacuo to an oily substance, and 1000mL of 3:1 n-hexane/ethyl acetate solution was added to it, heated to dissolve, cooled, and a solid was precipitated out, filtered , dried to obtain solid 281.2g, yield 92.1%. |
| The above-mentioned solid was dissolved in 1000mL 2:3:1 tetrahydrofuran/ethanol/water solution, then 40g sodium hydroxide solid (1mol) was added to it, stirred overnight, vacuum concentrated to obtain oil after the reaction was finished, and then 1000mL of ethyl acetate was added thereto. The ester was dissolved, extracted twice with saturated aqueous sodium chloride solution and twice with saturated aqueous sodium thiosulfate solution, dried the organic phase, concentrated in vacuo to an oil, to which was added 200 mL of 1:4 ethyl acetate/acetonitrile solution , heated to dissolve, cooled and recrystallized to obtain 150.9 g of white rice-like crystals with a yield of 88.2% and a purity of 99.5% (determined by high performance liquid chromatography, external standard method). |
| Example 3 |
| Weigh 37.4g (1.862mol) of activated magnesium, add two iodine pellets, heat to 35°C under nitrogen protection, then add 500mL of anhydrous THF to it, without magnesium, and then add 200mL of 10% iodine to it The THF solution of benzene (2000mL in total: 20g iodobenzene (0.53mol)+2000mL THF), after the color of the solution subsides, add the remaining iodobenzene solution dropwise, react for 5h, and filter to obtain the Grignard reagent THF solution of iodobenzene . |
| 433.4g (1.1mol) of peracetyl sugar and 1500mL of dichloromethane were added, cooled to 0°C in an ice-water bath, 818.25mL of 33% hydrogen bromide in acetic acid solution (3.3mol) was added dropwise to the reaction flask, and the mixture was gradually heated up The mixture was heated to 25° C. and stirred for 1.5 h. Saturated sodium bicarbonate solution was added to quench the reaction. The aqueous solution was extracted with dichloromethane, dried, and concentrated by rotary evaporation. 375.3 g of white solid were obtained, which was 2,3,4,6-tetraacetyl bromoglucose, and the yield was 82.4%. |
| Weigh 217.83g 2,3,4,6-tetraacetyl bromoglucose (0.53mol) and dissolve it in 7500mL toluene, then add 5.6g europium oxide (3mol%) to it, under nitrogen protection, reduce the temperature to – 20°C, dropwise add the prepared iodobenzene Grignard reagent to it, and control the temperature at -20~-15°C. After the dropwise addition, the temperature is raised to 5°C for reaction for 2h, vacuum concentrated to an oily substance, and then added to it. 2000mL of toluene was dissolved, extracted twice with saturated aqueous sodium chloride solution, the organic phase was dried, concentrated in vacuo to an oily substance, and 1000mL of 3:1 n-hexane/ethyl acetate solution was added to it, heated to dissolve, cooled, and a solid was precipitated out, filtered , and dried to obtain 273.25 g of solid with a yield of 89.5%. |
| The above-mentioned solid was dissolved in 1000mL 2:3:1 tetrahydrofuran/ethanol/water solution, then 40g sodium hydroxide solid (1mol) was added to it, stirred overnight, vacuum concentrated to obtain oil after the reaction was finished, and then 1000mL of ethyl acetate was added thereto. The ester was dissolved, extracted twice with saturated aqueous sodium chloride solution and twice with saturated aqueous sodium thiosulfate solution, the organic phase was dried, concentrated in vacuo to an oil, to which was added 200 mL of 1:5 ethyl acetate/acetonitrile solution , heated to dissolve, cooled and recrystallized to obtain 152.7 g of white rice-like crystals with a yield of 82.9% and a purity of 99.4% (determined by high performance liquid chromatography, external standard method). |
| Example 4 |
| Weigh 26.7g (1.33mol) of activated magnesium, add two iodine pellets, heat to 35°C under nitrogen protection, then add 500mL of anhydrous THF to it, without magnesium, and then add 200mL of 10% iodine to it The THF solution of benzene (2000mL in total: 20g iodobenzene (0.53mol)+2000mL THF), after the color of the solution subsides, add the remaining iodobenzene solution dropwise, react for 5h, and filter to obtain the Grignard reagent THF solution of iodobenzene . |
| 433.4g (1.1mol) of peracetyl sugar and 1500mL of dichloromethane were added, cooled to 0°C in an ice-water bath, 545.5mL of 33% hydrogen bromide in acetic acid solution (2.2mol) was added dropwise to the reaction flask, and the mixture was gradually heated up The mixture was heated to 25° C. and stirred for 1.5 h. Saturated sodium bicarbonate solution was added to quench the reaction. The aqueous solution was extracted with dichloromethane, dried, and concentrated by rotary evaporation. 425.1 g of white solid were obtained, which was 2,3,4,6-tetraacetyl bromoglucose in 94% yield. |
| Weigh 217.83g of 2,3,4,6-tetraacetyl bromoglucose (0.53mol) and dissolve it in 5000mL of toluene, then add 5.76g of gadolinium oxide (3mol%) to it, under nitrogen protection, reduce the temperature to – 20°C, dropwise add the prepared iodobenzene Grignard reagent to it, control the temperature at -20~-15°C, after the dropwise addition, raise the temperature to 5°C for 2 hours, concentrate in vacuo to an oily substance, then add to it 2000mL of toluene was dissolved, extracted twice with saturated aqueous sodium chloride solution, the organic phase was dried, concentrated in vacuo to an oily substance, then 1000mL of 3:1 n-hexane/ethyl acetate solution was added to it, heated to dissolve, cooled, and a solid was precipitated out, filtered , and dried to obtain 278.6 g of solid with a yield of 91.2%. |
| The above-mentioned solid was dissolved in 1000mL 2:3:1 tetrahydrofuran/ethanol/water solution, then 40g sodium hydroxide solid (1mol) was added to it, stirred overnight, vacuum concentrated to obtain oil after the reaction was completed, and 1000mL of ethyl acetate was added thereto. The ester was dissolved, extracted twice with saturated aqueous sodium chloride solution and twice with saturated aqueous sodium thiosulfate solution, the organic phase was dried, concentrated in vacuo to an oil, to which was added 200 mL of 1:4 ethyl acetate/acetonitrile solution , heated to dissolve, cooled and recrystallized to obtain 155.62 g of white rice-like crystals with a yield of 84.5% and a purity of 99.7% (determined by high performance liquid chromatography, external standard method). |
Patent
Sodium-glucose co-transporter-2 (SGLT2) inhibitors are a group of oral medicines used for treating diabetes that have been approved since 2013. SGLT2 inhibitors prevent the kidneys from re-absorbing glucose back into the blood by passing into the bladder. Glucose is re-absorbed back into the blood via the renal proximal tubules. SGLT2 is a protein predominantly expressed in the renal proximal tubules and is likely to be major transporter responsible for this uptake. Glucose-lowering effect of SGLT-2 inhibitors occurs via an insulin-independent mechanism mostly through glucosuria by increasing the urinary excretion of glucose.
It has been shown that the treatment with SGLT2 inhibitors in patients with type II diabetes lowers HbAlc, reduces body weight, lowers systemic blood pressure (BP) and induces a small increase in LDL-C and HDL-C levels.
SGLT2 inhibitors inhibit the reabsorption of sodium and glucose from the tubule and hence, more sodium is delivered in the macula densa causing arteriole dilation, reduced intraglomerular pressure and decreased hyperfiltration. SGLT2 inhibitors cause natriuresis and volume depletion, and an increase in circulating levels of renin, angiotensin and aldosterone. They also reduce albuminuria and slow GFR loss through mechanisms that appear independent of glycemia.
Dapagliflozin is a highly potent and reversible SGLT2 inhibitor, which increases the amount of glucose excreted in the urine and improves both fasting and post-prandial plasma glucose levels in patients with type 2 diabetes. Dapagliflozin has also been shown to tend to reduce liver fat content in some studies in a diabetic population.
Dapagliflozin is available on the market in the form of dapagliflozin propanediol monohydrate and is sold under trade name Forxiga or Farxiga in the form of fdm-coated tablets. Further it is available on the market as a combination product with metformin hydrochloride which is sold under trade name Xigduo IR or Xigduo XR in the form of film-coated tablets. In addition, it is available on the market as a combination product with saxagliptin hydrochloride which is sold under trade name Qtem in the form of film-coated tablets. Moreover, it is available on the market as a combination product with saxagliptin hydrochloride and metformin hydrochloride which is sold under trade name Qtemmet XR in the form of film-coated tablets.
Dapagliflozin as a monotherapy and in a combination with other active substances has demonstrated its efficacy in improving glycaemic control and reducing body weight and blood pressure in a broad spectrum of patients with type II diabetes, including those with high baseline HbAlc and the elderly. A sustained reduction in serum uric acid concentration was also observed. Dapagliflozin provides significant improvement in HbAlc, reduction in insulin dose and reduction in body weight in patients with type 1 diabetes as adjunct therapy to adjustable insulin.
Dapagliflozin can be in its free form or any stereoisomer or any pharmaceutically acceptable salt or co crystal complex or a hydrate or a solvate thereof and in any polymorphic forms and any mixtures thereof.
Dapagliflozin as a substance was first disclosed in US 6,515,117. The process for the preparation of dapagliflozin involves the reaction of 4-bromo- 1 -chloro-2-(4-ethoxybenyl)benzene with 2,3,4,6-tetra-O-trimethyl silyl -D-gluconolactone, the obtained compound 3 on demethoxylation yields diastereomeric mixture of Dapagliflozin. Hie diastereomeric mixture of dapagliflozin is further acetylated with acetic anhydride in the presence of pyridine and dimethylaminopyridine yields, then recrystallized from absolute ethanol to yield the desired tetra-acetyJated b-C-glucoside as a white solid. Compound tetra-acetylated b-C-glucoside is treated with lithium hydroxide hydrate which undergoes deprotection to yield the compound dapagliflozin.
Several other documents, patents and applications disclose the process for the preparation of dapagliflozin such as for example WOO 127128, WO03099836, W02004063209, W02006034489,
W02010022313, WO2012019496, W02013064909, W02013068850, W02013079501, WO2014094544, WO2014159151, WO2014206299, W02015040571, WO2015044849, WO2015063726, WO2015132803, WO2015155739, W02016098016, WO2016128995, WO2016178148, WO2017042683, WO2017063617, W02018029611, WO2018029264, WO2018142422.
Prior art documents already provided some compositions of SGLT2 inhibitor dapagliflozin.
W02008116179 discloses immediate release formulation in the form of a stock granulation or in the form of a capsule or a tablet which comprises dapagliflozin propylene glycol hydrate, one or more bulking agent, one or more binder and one or more disintegrant.
WO2011060256 describes the bilayer tablet comprising dapagliflozin having sustained release profde in one layer and metformin in another layer while WO2011060290 describes immediate release formulation of dapagliflozin and metformin.
WO2012163546 discloses the pharmaceutical composition comprising cyclodextrin and dapagliflozin.
Co-crystals of dapagliflozin with lactose are described in WO2014178040.
Solid dispersion compositions comprising amorphous dapagliflozin and at least one polymer are disclosed in W02015011113 and in WO2015128853.
CN103721261 discloses the combination of SGLT2 inhibitor with vitamins such as vitamin B.
Pharmaceutical composition preparation comprising dapagliflozin L-proline and metformin and/or DPP-IV inhibitor is disclosed in WO2018124497.
EP2252289A1 provides a combination of SGLT inhibitor with DPP4 inhibitor showing synergistic effect in increasing plasma active GLP-1 level in a patient over that provided by administration of the SGLT inhibitor or the DPP4 inhibitor alone.
EP2395983A1 relates to a pharmaceutical composition comprising a SGLT2 inhibitor, a DPP4 inhibitor and a third antidiabetic agent which is suitable in the treatment or prevention of one or more conditions selected from type 1 diabetes mellitus, type 2 diabetes mellitus, impaired glucose tolerance and hyperglycemia.
Example A: HPLC method
The purity of Dapagliflozine in general may be determined with the following HPLC method: column: XBridge C18, 150×4.6mm, 3.5; flow-rate: 0.9ml/min; column temperature: 50°C, wavelength: UV 225 nm; mobile phase: eluent A: 0.1% H3PO4, Eluent B: methanol; gradient:
Sample preparation: Accurately weigh about 40mg of sample and dissolve in 50 ml of solvent. Calculation: Use area per cent method. Do not integrate solvent peaks.
Example 1: Preparation of 5-bromo-2-chlorobenzoyl chloride
5-bromo-2-chlorobenzoic acid (450 g) was suspended in dichloromethane (2.25 L) and dimethylformamide (0.74 ml). At 15 – 30°C oxalyl chloride (180.3 ml) was slowly added. During addition gas evolution of HC1 and CO2 occurred. The reaction was performed at 20-30°C. The reaction was considered to be complete if 2-chloro-5-bromobenzoic acid was below 1% (area percent purity). The mixture was concentrated at elevated temperature until oily residue was obtained.
Example 2: Preparation of (5-bromo-2-chlorophenyl)(4-ethoxyphenyl)methanone
Dichloromethane (900 ml) was charged into reactor and then aluminum chloride (267.6 g) was added. The reaction mixture was cooled below 5°C and ethoxybenzene (256.1 ml) was slowly added. After complete addition, the mixture was gradually cooled below -5°C. In a separate reactor, 5-bromo-2-chlorobenzoyl chloride (485g) was dissolved in dichloromethane (900 ml). This solution was slowly added to the mixture of aluminum chloride and ethoxybenzene with such rate that temperature was kept below -5°C. After complete addition the mixture was stirred below -5°C until reaction was finished. The reaction was considered to be complete if methyl ester was below 1 % (the reaction mixture is sampled in methanol). After reaction was completed the reaction mixture was slowly added into cooled 1M HC1 solution and flushed with of dichloromethane (450 ml). The organic phase was separated and water phase was washed again with dichloromethane. Organic phases were combined and washed with water and NaHC03 solution. So obtained organic phase was concentrated to oily residue and dissolved in methanol ethyl acetate mixture in 10 to 1 ratio at reflux temperature. The clear solution was gradually cooled down to 35-45 °C and seeded with pure 5-bromo-2-chlorophenyl(4-ethoxyphenyl)methanone. The reaction mass was gradually cooled down to 0-10°C and stirred at that temperature up to 4 hours. The precipitate was isolated and washed with precooled methanol. The product was dried to a final LOD (Loss on drying) content of less than 1.0% with a yield of 564g (87% mass yield).
Example 3: Preparation of 4-bromo-l-chloro-2-(4-ethoxybenzyl)benzene
5-bromo-2-chlorophenyl(4-ethoxyphenyl)methanone (400g) was dissolved in 1.62L tetrahydrofuran. Into solution NaBTL (53.5g ) was added. After addition, the mixture was stirred at ambient temperature for 30-60 min followed by cooling of reaction mixture below -5°C. Aluminum chloride (314g) was added in portion and reaction mixture maintained below 5°C. After addition, the reaction mixture was gradually heated to reflux temperature and stirred until reaction was complete. Reaction mixture was cooled to ambient temperature and mixture of THF/water was slowly added into reaction mixture followed by addition of water and stirred at ambient temperature. Organic phase was collected and washed with saturated NaCl solution. Organic phase was concentrated to oily residue and dissolved in ethanol (800ml) at elevated temperature. Solution was cooled to 25-30°C and seeded with pure 4-bromo-l-chloro-2-(4-ethoxybenzyl)benzene. The reaction mass was gradually cooled to -2 to 10°C and stirred at that temperature. The product was isolated and washed with precooled ethanol and dried until final LOD (Loss on drying) content was less than 1.0%. Yield was 322 g (89%).
Example 4: Preparation of 3R,4S,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6- (hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol
4-bromo-l-chloro-2-(4-ethoxybenzyl)benzene (97.5g ) and toluene (1.46L) was charge into reactor. Solution was heated to reflux temperature and approximately half of the solvent was distilled out. Tetrahydrofuran (195 mL) was charged into the solution and mixture was cooled below -70°C. Solution of 15% «-Buli in hexane (227.5 ml) was slowly added and temperature was kept below -70°C. After complete addition solution was stirred at temperature below -70°C to complete reaction. Solution of 2,3,4,6-tetra-O-trimethylsilyl-D-gluconolactone (182 g) in toluene (243 mL) was added into reaction mixture at temperature below -70°C. After complete addition, the mixture was stirred below -70°C, warmed to approximately -65°C and then mixture of 57.6 g methanesulfonic acid in 488 ml methanol was added. After addition, the mixture was gradually warmed to ambient temperature and stirred until reaction was complete. After reaction was finished reaction mixture was slowly added into saturated NaHCCL solution (630ml) and stirred. Into quenched mixture 975 ml of heptane and 585 ml methanol was added. The mixture was stirred for additional 15 min. Organic phase was washed with water/methanol mixture several times. Water phases were combined and distilled to remove organic solvents. Into the residual water phase, toluene was added to perform extraction. Organic phases were combined and washed with water. Organic phase was distillated at elevated temperature until oily residue was obtained.
Example 5: Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(hydroxymethyl) tetrahydro2H-pyran-3,4,5-triol – Dapagliflozin
Dichloromethane (656 mL) was charged into oily residue from step 4 and stirred at ambient temperature until clear solution was obtained. Triethysilane (122 mL) was added into the so obtained solution. Reaction mixture was cooled below -30°C and 94.2 mL of boron trifluoride etherate was slowly added at temperatures below -30°C. After complete addition, the mixture was stirred below -30°C for one hour and gradually warmed to -5 to 0°C until reaction was completed. After reaction was finished saturated NaHCCL solution (468 mL) was slowly added. Reaction mixture was distilled to remove organic solvents followed by addition ethyl acetate into the residue. Organic phase was collected and washed again with saturated NaHC03 and water. So obtained organic phase was distillated at elevated temperature until oily residue is obtained.
Example 6: Preparation of (2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(4-chloro-3-(4-ethoxybenzyl)phenyl) tetrahydro-2H-pyran-3,4,5-triyl triacetate.
Oily residual from example 5 was dissolved in dichloromethane (602 mL) at ambient temperature followed by addition of DMAP (6.22g). Reaction mixture was cooled to 0 – 10°C and 144.3 mL of acetic anhydride was added at temperatures below 10°C. Reaction mixture was gradually warmed to ambient temperature and stirred until reaction was completed. Reaction mass was washed with water with saturated NaHC03. Organic phase was collected and concentrated to oily residue to which ethanol (1.68L) was charged and approximately 300 ml ethanol was removed by distillation. The clear solution was gradually cooled to 60-65 °C and seeded. The reaction mass was gradually cooled to 20-25 °C and product was isolated. The product was dried at 50°C in vacuum until LOD (Loss on drying) below 1.0% 123g of product was obtained with yield 71%. HPLC purity: 99.97 %.
Formula 9 Formula 2
(2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(4-chloro-3-(4-ethoxybenzyl)phenyl) tetrahydro-2H-pyran-3,4,5-triyl triacetate (740 g) as prepared according to process described in examples 1 to 6 was charged into solution of methanol (2.27L), water (0.74L) and NaOH (23 lg) at 35-45°C and stirred at 35-45°C until reaction was completed. After reaction was finished 1M HC1 (1.63L) was slowly added. Reaction mass was distilled to remove organic solvents and product was extracted by tert-butyl methyl ether. Combined organic phases were washed with water and concentrated at elevated temperature until oily residue was obtained. Content of impurity IMP A was below 0.02%.
Example 7a: Preparation of amorphous dapagliflozin.
Oily residue as prepared according to example 7 comprising approximately 262 g of dapagliflozin was dissolved in toluene (2.5L) at temperature 60-70 °C. Solution of dapagliflozin in toluene was slowly added into heptane (5.3L) at temperature between 10 to 15°C and stirring rate with P/V at 4 W/m3. After complete addition, the suspension was cooled to 0°C and stirred with unchanged stirring rate. Suspension was isolated and washed with precooled heptane. Filtration rate was 14 · 104 m/s. Isolated product was dried in vacuum dryer at temperatures between 25 °C to 50 °C. Content of impurity IMP A was below 0.02% and residual heptane and toluene were 1673 ppm and below 89 ppm.
(2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(4-chloro-3-(4-ethoxybenzyl)phenyl) tetrahydro-2H-pyran-3,4,5-triyl triacetate (30 g) of 4 different qualities obtained by the process known in the prior art was charged into solution of methanol (90 mL), water (30 mL) and NaOH (9.36 g) and stirred at 35-45°C until reaction was completed and sampled for HPLC analysis (Sample 1).
After reaction was finished 1M HC1 (66 mL) was slowly added. Reaction mass was distilled to remove organic solvents and product was extracted by tert-butyl methyl ether. To the combined organic phases 68ml of 1M NaOH was added and pH was set to 12.5 to 13.5. Phases were separated and organic phase is washed again with 68ml of water without pH correction. So obtained organic phase was sampled for HPLC analysis (Sample 2) and concentrated at elevated temperature until oily residue was obtained.
Oily residue comprising approximately 21 g of dapagliflozin was dissolved in toluene (210 mL) at temperature 60-70 °C. Solution of dapagliflozin in toluene was slowly added into heptane (420 mL) at temperature between 10 to 15°C and stirring rate with P/V as defined in Table 1. After complete addition, the suspension was cooled to 0°C and stirred with unchanged stirring rate. Suspension was isolated and washed with precooled heptane. Filtration rate was as defined in Table 1. Isolated product was dried in vacuum dryer at temperatures between 25 °C to 50 °C. Amorphous dapagliflozin with content of impurity IMP A as shown in Table 1 and residual heptane and toluene as shown in Table 1 was obtained for each cases.
Table 1 : Process parameters used in the preparation of four different starting materials (cases).
As it is evident from Table 2 the final amorphous dapagliflozin prepared by the extraction process according to the present invention contains less than 0.02% of impurity IMP A irrespective of the level of impurity IMP A present in the starting material.
Table 2: Content of impurity IMP A in the final amorphous dapagliflozin obtained with and without extraction.
Example 9
Oily residue, as obtained by the procedure described in example 8 case 1, containing approximately 2 g of dapagliflozin was dissolved in 1.5ml of isopropyl acetate and 6ml of tert-butyl methyl ether at temperature 50-55 °C. So prepared solution was charged into 25 mL of heptane at 0°C. After complete addition the suspension was stirred at -10 to 0°C. Suspension was isolated and washed with precooled heptane at temperatures between 25°C to 50 °C. 1.5 g of dapagliflozin was obtained with content of impurity IMP A was below 0.02%.
Example 10
Oily residue, as obtained by the procedure described in example 8 case 1, containing approximately 2 g of dapagliflozin was dissolved in 1.5ml of isopropyl acetate and 6ml of tert-butyl methyl ether at temperature 50-55 °C. So prepared solution was charged into 40 mL of heptane at 0°C. After complete addition the suspension was stirred at -10 to 0°C. Suspension was isolated and washed with precooled heptane at temperatures between 25°C to 50 °C. 1.5 g of dapagliflozin was obtained with content of impurity IMP A was below 0.02%.
Example 11
Dapagliflozin (30g) was dissolved in toluene (285mL) at temperature 60-70 °C. Solution of dapagliflozin in toluene was slowly added into heptane (600mL) at 5°C and stirring rate with P/V at 16 W/m3. After complete addition, the suspension was cooled to -10°C and stirred with unchanged stirring rate. Suspension was isolated and washed with precooled heptane. Isolated product was dried in vacuum dryer at temperatures between 25 °C to 50 °C. Content of residual heptane and toluene were 1480 ppm and 732 ppm.
Example 12
Dapagliflozin (30g) was dissolved in toluene (285mL) at temperature 60-70 °C. Solution of dapagliflozin in toluene was slowly added into heptane (600mL) at 20°C and stirring rate with P/V at 16 W/m3. After complete addition, the suspension was cooled to 5°C and stirred with unchanged stirring rate. Suspension was isolated and washed with precooled hcptanc Isolated product was dried in vacuum dryer at temperatures between 25 °C to 50 °C. Content of residual heptane and toluene were 2873 ppm and 639 ppm.
Comparative Example 1
Dapagliflozin (30g) was dissolved in toluene (285mL) at temperature 60-70 °C. Solution of dapagliflozin in toluene was slowly added into heptane (600mL) at -5°C and stirring rate with P/V at 16 W/m3. After complete addition, the suspension was cooled to -15°C and stirred with unchanged stirring rate. Suspension was isolated and washed with precooled heptane. Isolated product was dried in vacuum dryer at temperatures between 25 °C to 50 °C. Content of residual heptane and toluene were 1940 ppm and 1557 ppm.
Comparative Example 2
Dapagliflozin (30g) was dissolved in toluene (285mL) at temperature 60-70 °C. Solution of dapagliflozin in toluene was slowly added into heptane (600mL) at 25°C and stirring rate with P V at 16 W/m3. After complete addition, the suspension was cooled to 20°C and stirred with unchanged stirring rate. Suspension was isolated and washed with precooled heptane. Isolated product was dried in vacuum dryer at temperatures between 25 °C to 50 °C. Content of residual heptane and toluene were 3663 ppm and 2047 ppm.
Comparative Example 3
Dapagliflozin (30g) was dissolved in toluene (285mL) at temperature 60-70 °C. Solution of dapagliflozin in toluene was slowly added into heptane (600mL) at 30°C and stirring rate with P/V at 16 W/m3. After complete addition, the suspension was cooled to 15°C and stirred with unchanged stirring rate. Suspension was isolated and washed with precooled heptane. Isolated product was dried in vacuum dryer at temperatures between 25 °C to 50 °C. Content of residual heptane and toluene were 2425 ppm and 1812 ppm.
///////////////////////////////////////////
POST YOUR INTERMEDIATES LIST HERE FOR WORLDWIDE REACH

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
PATENT
https://patents.google.com/patent/WO2017206808A1/en
Daggliflozin (English name: Dapagliflozin) is a new Sodium glucose co-transporters 2 (SGLT-2) inhibitor developed by Bristol-Myers Squibb and AstraZeneca. Approved by the European Commission on November 14, 2012, and marketed in the United States on January 8, 2014, to improve glycemic control in adult patients with type 2 diabetes by combining diet and exercise; the trade name is Farxiga, currently offering 5 mg and 10 mg tablets. At the same time, a combination of dapagliflozin and metformin hydrochloride has also been marketed.The chemical name of dapagliflozin is (2S,3R,4R,5S,6R)-2-(3-(4-ethoxybenzyl)-4-chlorophenyl)-6-hydroxymethyltetrahydro-2H – pyran-3,4,5-triol, the chemical formula is C 21 H 25 ClO 6 , CAS No. 461432-26-8, the structural formula is shown as 2, clinically used as a pharmaceutical for dapagliflozin (S) -1,2-propanediol monohydrate, the structural formula is as shown in 1.

The synthesis of β-type C-aryl glycosidic bonds is a key point in the synthetic route during the preparation of dapagliflozin. At present, there are four synthetic methods for the synthesis of dapagliflozin reported in the literature and patents.Route 1: The synthetic route of dapagliflozin reported in patent WO03099836A1 is as follows:

The route uses 2-chloro-5-bromobenzoic acid (12) as raw material to react with phenethyl ether to form intermediate 11 and then triethylsilane to obtain intermediate 10; intermediate 10 and n-butyl The lithium is reacted at -78 ° C, and then subjected to a nucleophilic addition reaction with the intermediate 9, and then methoxylated to obtain the intermediate 8; the intermediate 8 is subjected to acylation reduction and deprotection to obtain the intermediate 2. The disadvantage of this method is that the β-type C-aryl glycosidic bond synthesis of the compound is carried out at a low temperature of -78 ° C, which is obviously difficult to meet the needs of industrial production; and, through nucleophilic addition, methoxylation, The five-step reaction of acetylation, reduction and hydrolysis can synthesize the β-type C-aryl glycosidic bond. The procedure is relatively long, and the purity of the intermediate 2 is only 94%.Route 2: The synthetic route of dapagliflozin reported in the literature OrgLett.2012, 14, 1480 is as follows:

The intermediate 14 of the route is reacted with di-n-butyl-n-hexylmagnesium for 48 hours at 0 ° C, and then reacted with zinc bromide to prepare an organozinc reagent by Br/Mg/Zn exchange reaction, and then with intermediate 4 Intermediate 3 was prepared by nucleophilic substitution reaction; finally, intermediate 2 was obtained by deprotection with sodium methoxide. The synthesis method is relatively novel, and the synthesis step is short. However, the research experiment is conducted only as a synthesis method, and the post treatment of the intermediate 3 is performed by column chromatography. The purity of the intermediate 2 produced was not reported. Moreover, the di-n-butyl-n-hexylmagnesium reagent used in the route is not a commonly used reagent, and is not commercially available in China. It can only be prepared by reacting dibutylmagnesium with n-hexyllithium reagent before the test, and the operation is cumbersome and difficult to mass. use.Route 3: The synthetic route of dapagliflozin reported in patent WO2013068850A2 is as follows:

The route uses 1,6-anhydroglucose (20) as a raw material, protects the 2,4-hydroxyl group by tert-butyldiphenylchlorosilane, and then protects the 3-position hydroxyl group with phenylmagnesium bromide. Intermediate 18. The intermediate 14 is subjected to an Br/Mg/Al exchange reaction to prepare an organoaluminum reagent 16, which is reacted with an intermediate 18 to form an intermediate 15, and finally, deprotected to obtain an intermediate 2. The synthesis method is very novel and is also used as a synthetic methodological study. The purification of the intermediates is carried out by column chromatography. The 1,6-anhydroglucose (20) used in the route is very expensive; and the multi-step reaction in the route uses a format reagent, a preparation format reagent or an organoaluminum reagent, which is cumbersome and cumbersome to perform, and is difficult to scale synthesis. The purity of the intermediate 2 produced was not reported.Route 4: The synthetic route of dapagliflozin reported in patent WO2013152476A1 is as follows:

The route uses 2-chloro-5-iodobenzoic acid (24) as raw material to form intermediate 22 by Friedel acylation and reduction reaction, and exchange with I-Mg at -5 ° C with isopropyl magnesium chloride lithium chloride. The intermediate 8 is obtained by nucleophilic addition and methoxylation with the intermediate 9, and then the intermediate 2 is obtained by reduction with triethylsilane, and the intermediate 2 is further purified by co-crystallizing with L-valine. Finally, The pure intermediate 2 was obtained by removing L-valine. This route is a modified route of Route 1, which replaces n-butyllithium with isopropylmagnesium chloride chloride to raise the reaction temperature of the reaction from -78 °C to -5 °C. However, the problem of a long step of synthesizing a β-type C-aryl glycosidic bond still exists. The obtained intermediate 2 is not optically pure, and needs to be purified by co-crystallizing with L-valine, and the work amount of post-treatment is increased, and finally the purity of the intermediate 2 is 99.3%.Among the four synthetic routes described above for dapagliflozin, route one and route four are commonly used synthetic methods for β-type C-aryl glycosidic bonds, and the route is long, and the optical purity of the obtained product is not high, and further purification is required. Post processing is cumbersome. Moreover, the reaction required at -78 °C in Route 1 requires high equipment and high energy consumption, which undoubtedly increases the cost. Although both Route 2 and Route 3 are new methods, most of the purification of intermediates used is column chromatography. Such a process is not suitable for scale production in factories; and some of the synthetic routes are used. Reagents are not commercially available or expensive, and there is no advantage in such route costs. Therefore, there is an urgent need to find a new method for the synthesis of dapagliflozin, and to enable industrial production, and the route has a cost advantage.Repeating the procedure reported in the literature in Equation 2, the yield of Intermediate 3 was only 46%. The organic zinc reagent is prepared by Br/Mg/Zn exchange reaction, and the exchange reaction yield is 78%; and the raw material is prepared by X/Li/Zn exchange reaction to prepare an organic zinc reagent, and the exchange reaction yield is 98.5%, which is also the two Different reaction pathways lead to the essential reason for the different yields of intermediate 3. Moreover, the price of commercially available 1.0 mol/L di-n-butyl magnesium n-heptane solution 500 mL is 1380 yuan, and the price of 1.6 mol/L n-hexyl lithium n-hexane solution 500 mL is 950 yuan, and 2.5 mol/L n-butyl lithium. The price of 500 mL of n-hexane solution is only 145 yuan. Therefore, the method for preparing dapagliflozin by preparing an organozinc reagent by X/Li/Zn and then synthesizing the β-type C-aryl glycosidic bond designed by the invention has the advantages of cost, ease of operation and industrialization. Very obvious advantage.In order to solve this problem, the original compound company uses a eutectic method in the production of dapagliflozin to make dapagliflozin together with a solvent or an amino acid compound, since the compound 2 sugar ring structure contains four hydroxyl groups and is easy to absorb moisture and deteriorate. The crystal is made into a relatively stable solid, easy to store, stable and controllable in quality, and easy to prepare. Among them, the marketed dapagliflozin forms a stable eutectic with (S)-1,2-propanediol and water (1). The original crystal form patent (CN101479287B, CN103145773B) reported that all 11 crystal forms are dapagliflozin solvate or dapagliflozin. Crystal. Among them, there are two preparation methods for the da forme (S)-1,2-propanediol monohydrate (1) having a crystal structure of type Ia:Method 1: The preparation method is as follows:

Compound 7 is deprotected with sodium hydroxide to obtain compound 2, then compound 2 is extracted with isopropyl acetate, (S)-1,2-propanediol ((S)-PG) is added, and seed crystal of compound 1 is added. Then, cyclohexane was added to crystallize and separated to obtain a eutectic of the compound (1) of the type Ia.Method 2: The preparation method is as follows:

Compound 8 is subjected to reduction of methoxy group by triethylsilane and boron trifluoride diethyl ether complex, and then the reaction solution is extracted with methyl tert-butyl ether (MTBE), and (S)-1,2-propanediol ( (S)-PG), a seed crystal of the compound 1 is added, and then cyclohexane is added to crystallize, and the mixture is separated and dried to obtain a eutectic of the compound (1) of the type Ia.The above two methods for preparing the eutectic are all used in the cyclohexane solvent, which is listed in the appendix of the 2015 edition of the Pharmacopoeia (four parts) as the second type of solvent that should be restricted, with a residual limit of 0.388%. The solvent residue of the final product obtained must reach the specified limit, and the post-treatment process is complicated, time-consuming and labor-intensive, and the production cost is correspondingly increased. The invention finds a suitable solvent on the basis of the synthetic route to prepare a medicinal crystal form, and has obvious advantages in both the method and the process operation steps.The synthetic route is as follows:

Comparative Example 1, (1S)-2,3,4,6-tetra-O-pivaloyl-1,5-anhydro-1-[3-(4-ethoxyphenylmethyl)-4- Preparation of chlorophenyl]glucosamine (Compound 3)Under nitrogen protection, 1.0 mol/L di-n-butylmagnesium-n-heptane solution (16 mL) was cooled to 0 ° C, and 1.6 mol/L n-hexane lithium n-hexane solution (10 mL) was slowly added dropwise. After the addition was completed, 0 ° C After stirring for 15 h, dry n-butyl ether (2.5 mL) was added to prepare a solution of di-n-butyl-n-hexylmagnesium lithium solution, which was calibrated with iodine and stored for use.Zinc bromide (2.7 g) and lithium bromide (1.04 g) were added with n-butyl ether (20 mL), heated to 50 ° C for 4 h, and cooled for use. 4-(2-Chloro-5-bromo-benzyl) phenyl ether (6.513 g) was added with toluene (8 mL) and n-butyl ether (5 mL) under nitrogen, cooled to 0 ° C, and 0.61 mol/L was added dropwise. n-Butyl-n-hexylmagnesium lithium solution (13.1 mL), after the addition is completed, the reaction was kept at 0 ° C for 48 h, and the above-mentioned alternate zinc bromide and lithium bromide n-butyl ether solution were added, and the reaction was kept at 0 ° C for 1 h, and added 2 , 3,4,6-tetra-O-pivaloyl-α-D-bromoglucopyranose (14.49 g) in toluene (25 mL), heated to 100 ° C to stir the reaction, after TLC detection reaction, add 1 mol / L diluted hydrochloric acid (60 mL), taken after stirring extraction, the organic phase was washed with water (40 mL), then washed with saturated brine (40 mL), dried over anhydrous Na 2 SO 4, concentrated under reduced pressure, column chromatography (petroleum ether / Ethyl acetate = 20:1) 10.38 g of Compound 3 as a pale yellow oil. Yield: 46%. Purity: 99.02%. The organozinc reagent prepared by the method has an iodine calibration yield of 78%.The calibration method of the concentration of the prepared organic zinc reagent: accurately weighed iodine (1 mmol), placed in a three-necked flask, replaced nitrogen, and added anhydrous 0.5 mol/L LiCl tetrahydrofuran solution (5 mL), stirred and dissolved, and cooled to 0 ° C. The prepared organozinc reagent was slowly added dropwise until the color of the brownish yellow solution disappeared.Example 2 (1S)-2,3,4,6-tetra-O-pivaloyl-1,5-anhydro-1-[3-(4-ethoxyphenylmethyl)-4-chloro Preparation of phenyl]glucitol (compound 3)Zinc bromide (2.25 g) and lithium bromide (0.87 g) were added with n-butyl ether (30 mL), heated to 50 ° C for 2 h, and cooled for use. 4-(2-Chloro-5-iodo-benzyl) phenyl ether (7.45 g) was added with toluene (10 mL) and n-butyl ether (10 mL) under nitrogen, cooled to -20 ° C, and slowly added dropwise 1.6 mol / L-n-hexyl lithium n-hexane solution (14mL), control the internal temperature does not exceed -10 ° C, after the completion of the addition, the temperature is incubated at -20 ° C for 0.5 h, adding the above-mentioned spare zinc bromide and lithium bromide n-butyl ether solution, The reaction was stirred at 20 ° C for 3 h. Add 2,3,4,6-tetra-O-pivaloyl-α-D-bromoglucopyranose (11.59g) toluene (50mL) solution, heat to 120 ° C and stir the reaction for 4h, after TLC detection reaction, was added 1mol / L diluted hydrochloric acid (40 mL), water (20 mL), and extracted, the organic phase was washed with water (40 mL), dried over anhydrous Na 2 SO 4, concentrated with n-heptane (15mL) and methanol (60 mL) and recrystallized 10.8 g of Compound 3 as a white solid was obtained in a yield: 72.42%. Purity: 99.47%. Melting point: 99.5 to 101.6 °C. (The organic zinc reagent prepared by this method was iodine-calibrated in a yield of 98.5%.) ESI-MS (m/z): 767.30 [M+Na] + . 1 H-NMR (400 MHz, CDCl 3 ): δ 7.33 (1H, d), 7.14-7.17 (2H, m), 7.05 (2H, d), 6.79-6.81 (2H, dd), 5.39 (1H, t ), 5.21-5.31 (2H, m), 4.33 (1H, d), 4.17-4.20 (1H, dd), 3.94-4.11 (5H, m), 3.79-3.83 (1H, m), 1.39 (3H, t ), 1.20 (9H, s), 1.16 (9H, s), 1.11 (9H, s), 0.86 (9H, s).Example 3, (1S)-2,3,4,6-tetra-O-pivaloyl-1,5-anhydro-1-[3-(4-ethoxyphenylmethyl)-4-chloro Preparation of phenyl]glucitol (compound 3) PrepareZinc bromide (3.38 g) and lithium bromide (1.3 g) were added with n-butyl ether (40 mL), heated to 50 ° C for 2 h, and cooled for use. 4-(2-Chloro-5-iodo-benzyl) phenyl ether (7.45 g) was added with toluene (20 mL) and n-butyl ether (5 mL) under nitrogen, cooled to -50 ° C, and slowly added dropwise 2.5 mol / L-butyllithium hexane solution (8mL), control the internal temperature does not exceed -30 ° C, after the addition is completed, the reaction is kept at -50 ° C for 10 h, adding the above-mentioned alternate zinc bromide and lithium bromide n-butyl ether solution, The reaction was stirred at -20 ° C for 10 h. Add 2,3,4,6-tetra-O-pivaloyl-α-D-bromoglucopyranose (34.77g) toluene (80mL) solution, heat to 100 ° C and stir the reaction for 24h, after TLC detection reaction, was added 1mol / L diluted hydrochloric acid (60 mL), water (50 mL), and extracted, the organic phase was washed with water (40 mL), dried over anhydrous Na 2 SO 4, concentrated with n-heptane (15mL) and methanol (60 mL) and recrystallized 10.854 g of Compound 3 as a white solid. Yield: 72.81%. Purity: 99.53%.Example 4, (1S)-2,3,4,6-tetra-O-pivaloyl-1,5-anhydro-1-[3-(4-ethoxyphenylmethyl)-4-chloro Preparation of phenyl]glucitol (compound 3)N-butyl ether (50 mL) was added to zinc iodide (3.19 g) and lithium iodide (1.34 g), and the mixture was heated to 50 ° C for 1.5 h, and cooled for use. 4-(2-Chloro-5-iodo-benzyl) phenyl ether (7.45 g) was added with toluene (15 mL) and n-butyl ether (5 mL) under nitrogen, cooled to -60 ° C, and slowly added dropwise 1.6 mol / L-n-hexyl lithium n-hexane solution (13.8mL), control the internal temperature does not exceed -20 ° C, after the addition is completed, the reaction is kept at -60 ° C for 5 h, and the above-mentioned alternate zinc iodide and lithium iodide n-butyl ether solution is added. The reaction was stirred at 25 ° C for 1 h. Add 2,3,4,6-tetra-O-pivaloyl-α-D-bromoglucopyranose (23.2g) toluene (50mL) solution, heat to 140 ° C reflux reaction for 0.5h, after TLC detection reaction was added 1mol / L diluted hydrochloric acid (50 mL), water (50 mL), and extracted, the organic phase was washed with water (40 mL), dried over anhydrous SO 4 Na 2, concentrated by weight of n-heptane (15mL) and methanol (60 mL) Crystallization gave 10.51 g of Compound 3 as a white solid, yield 70.5%. Purity: 99.41%.Example 5, (1S)-2,3,4,6-tetra-O-pivaloyl-1,5-anhydro-1-[3-(4-ethoxyphenylmethyl)-4-chloro Preparation of phenyl]glucitol (compound 3)To the zinc bromide (2.25 g) and lithium bromide (0.87 g), cyclopentyl methyl ether (30 mL) was added, and the mixture was heated to 50 ° C for 3 hours, and cooled for use. 4-(2-Chloro-5-iodo-benzyl) phenyl ether (7.45 g) was added with toluene (10 mL) and cyclopentyl methyl ether (10 mL) under nitrogen, cooled to -5 ° C, and slowly added dropwise. Mol / L n-hexyl lithium n-hexane solution (12.5mL), control the internal temperature does not exceed 0 ° C, after the addition is completed, the reaction is kept at -5 ° C for 3 h, adding the above-mentioned spare zinc bromide and lithium bromide cyclopentyl methyl ether The solution was incubated at -5 ° C for 4 h, and a solution of 2,3,4,6-tetra-O-pivaloyl-α-D-bromoglucopyranose (17.39 g) in toluene (40 mL) was added and heated to 80 ℃ reaction was stirred 6h, after completion of the reaction by TLC, was added 1mol / L diluted hydrochloric acid (50 mL), water (50 mL), and extracted, the organic phase was washed with water (40 mL), dried over anhydrous 2 SO 4 Na, and concentrated under reduced pressure, Recrystallization of n-heptane (15 mL) and methanol (60 mL) gave 8.15 g of Compound 3 as a white solid. Purity: 99.39%.Example 6, (1S)-2,3,4,6-tetra-O-pivaloyl-1,5-anhydro-1-[3-(4-ethoxyphenylmethyl)-4-chloro Preparation of phenyl]glucitol (compound 3)Zinc bromide (4.5 g) and lithium bromide (1.74 g) were added with n-butyl ether (60 mL), heated to 50 ° C for 3 h, and cooled for use. 4-(2-Chloro-5-bromo-benzyl) phenyl ether (6.513 g) was added with toluene (15 mL) and n-butyl ether (5 mL) under nitrogen, cooled to -30 ° C, and slowly added dropwise 2.5 mol / L-butyllithium n-hexane solution (8.4mL), control the internal temperature does not exceed -20 ° C, after the addition is completed, the reaction is kept at -30 ° C for 3 h, and the above-mentioned alternate zinc bromide and lithium bromide n-butyl ether solution is added. The reaction was incubated at -5 ° C for 4 h, and a solution of 2,3,4,6-tetra-O-pivaloyl-α-D-bromoglucopyranose (14.49 g) in toluene (50 mL) was added and heated to 120 ° C for stirring. the reaction 4h, after completion of the reaction by TLC, was added 1mol / L diluted hydrochloric acid (50 mL), water (40 mL), and extracted, the organic phase was washed with water (40 mL), dried over anhydrous Na 2 SO 4, and concentrated under reduced pressure, n-heptyl Recrystallization of the alkane (15 mL) and methanol (60 mL) gave 10.38 g of Compound 3 as a white solid. Purity: 99.54%.Example 7, (1S)-2,3,4,6-tetra-O-pivaloyl-1,5-anhydro-1-[3-(4-ethoxyphenylmethyl)-4-chloro Preparation of phenyl]glucitol (compound 3)Methyl bromide (40 mL) was added to zinc bromide (2.25 g) and lithium bromide (0.87 g), and the mixture was heated to 50 ° C for 3 h, and cooled for use. 4-(2-Chloro-5-iodo-benzyl) phenyl ether (7.45 g) was added with toluene (15 mL), methyl tert-butyl ether (15 mL), cooled to -40 ° C, and slowly added dropwise. 1.6mol/L n-hexyl lithium n-hexane solution (13.8mL), control the internal temperature does not exceed -30 ° C, after the addition is completed, the reaction is kept at -40 ° C for 4 h, and the above-mentioned alternate zinc bromide and lithium bromide are added. The butyl ether solution was incubated at 5 ° C for 7 h, and a solution of 2,3,4,6-tetra-O-pivaloyl-α-D-bromoglucopyranose (17.39 g) in toluene (50 mL) was added and heated. to 90 deg.] C the reaction was stirred 8h, after completion of the reaction by TLC, was added 1mol / L diluted hydrochloric acid (40 mL), water (40 mL), and extracted, the organic phase was washed with water (40 mL), dried over anhydrous Na 2 SO 4, and concentrated under reduced pressure Recrystallization from n-heptane (15 mL) and methanol (60 mL) gave 9.41 g of Compound 3 as a white solid. Purity: 99.42%. Example 8. Preparation of dapagliflozin (S)-1,2-propanediol monohydrate eutectic (Compound 1)To the compound 3 (37.27 g), methanol (190 mL) was added, and sodium methoxide (10.8 g) was added thereto, and the mixture was heated under reflux for 3 hours. After the TLC reaction was completed, methanol was concentrated, and isopropyl acetate (100 mL) was added to the residue, and water was added. (60 mL), extracted with stirring and the organic phase washed with water (50 mL). (S)-1,2-propanediol (3.8g) and water (0.9g) were added to the organic phase, stirred until it was dissolved, and n-heptane (200 mL) was added, and the mixture was stirred for 2 hours under ice-cooling, suction filtration, filter cake Washing with n-heptane and drying at 30 ° C gave 23.89 g of Compound 1 as a white solid. Yield: 95%. Purity: 99.79%. Melting point: 69.1 to 75.6 °C. The product obtained was subjected to KF = 3.74% (theoretical value: 3.58%). ESI-MS (m/z): 431.22 [M+Na] + . 1 H-NMR (400 MHz, CD 3 OD): δ 7.33 – 7.37 (2H, m), 7.28-7.30 (1H, dd), 7.11 (2H, d), 6.80-6.83 (2H, dd), 4.1 ( 1H, d), 3.98-4.05 (4H, m), 3.88-3.91 (1H, dd), 3.74-3.82 (1H, m), 3.68-3.73 (1H, m), 3.37-3.49 (5H, m), 3.28-3.34 (1H, m), 1.37 (1H, t), 1.15 (3H, d).The crystal form of the obtained product was subjected to thermogravimetric analysis (TGA) by a Universal V4.7A TA instrument, and the TGA curve (Fig. 1) showed a weight loss of about 18.52% from about room temperature to about 240 ° C. The original form Ia crystal form The TGA plot shows a value of 18.7%.The crystal form of the obtained product was subjected to differential scanning calorimetry (DSC) by a Universal V4.7A TA instrument, and the DSC curve (Fig. 2) showed endotherm in the range of about 60 ° C to 85 ° C. The DSC plot shows a range of approximately 50 ° C to 78 ° C.
The crystal form of the obtained product was examined by a Bruker D8advance instrument for powder X-ray diffraction (PXRD), and the 2X value of the PXRD pattern (Fig. 3) (CuKα).

There are characteristic peaks at 3.749°, 7.52°, 7.995°, 8.664°, 15.134°, 15.708°, 17.069°, 18.946°, 20.049°, which are completely consistent with the characteristic peaks of the PXRD pattern of the Ia crystal form in the original patent.In combination with the nuclear magnetic data and melting point of the prepared crystal form, the crystal form of the product (Compound 1) obtained by the present invention is consistent with the pharmaceutically acceptable crystalline form Ia reported in the original patent.
Patent Citations
Publication numberPriority datePublication dateAssigneeTitleCN101479287A *2006-06-282009-07-08布里斯托尔-迈尔斯斯奎布公司Crystalline solvates and complexes of (is) -1, 5-anhydro-l-c- (3- ( (phenyl) methyl) phenyl) -d-glucitol derivatives with amino acids as sglt2 inhibitors for the treatment of diabetesCN104496952A *2014-11-282015-04-08深圳翰宇药业股份有限公司Synthesis method of dapagliflozinCN105153137A *2015-09-172015-12-16上海应用技术学院Preparation method of empagliflozinFamily To Family CitationsCN104829572B *2014-02-102019-01-04江苏豪森药业集团有限公司Dapagliflozin novel crystal forms and preparation method thereofCN105399735A *2015-12-292016-03-16上海应用技术学院Empagliflozin intermediate, and preparation method and application thereof* Cited by examiner, † Cited by third party
Non-Patent Citations
TitleCHEN DEJIN ET AL., CHINA MASTER’S THESES FULL-TEXT DATABASE, ENGINEERING TECHNOLOGY I, vol. B016-731, no. 3, 15 March 2016 (2016-03-15) *LEMAIRE S. ET AL.: “Stereoselective C-glycosylation of furanosyl halides with arylzinc reagents”, PURE APPL. CHEM., vol. 86, no. 3, 4 March 2014 (2014-03-04), pages 329 – 333 *LEMAIRE S. ET AL.: “Stereoselective C-Glycosylation Reactions with Arylzinc Reagents”, ORGANIC LETTERS, vol. 14, no. 6, 2 March 2012 (2012-03-02), pages 1480 – 1483, XP055069093 ** Cited by examiner, † Cited by third partyCLIP
Chemical Synthesis
Dapagliflozin propanediol hydrate, an orally active sodium glucose cotransporter type 2 (SGLT-2) inhibitor, was developed by Bristol-Myers Squibb (BMS) and AstraZeneca for the once-daily treatment of type 2 diabetes. As opposed to competitor SGLT-2 inhibitors, dapagliflozin was not associated with renal toxicity or long-term deterioration of renal function in phase III clinical trials. The drug exhibits excellent SGLT2 potency with more than 1200 fold selectivity over the SGLT1 enzyme.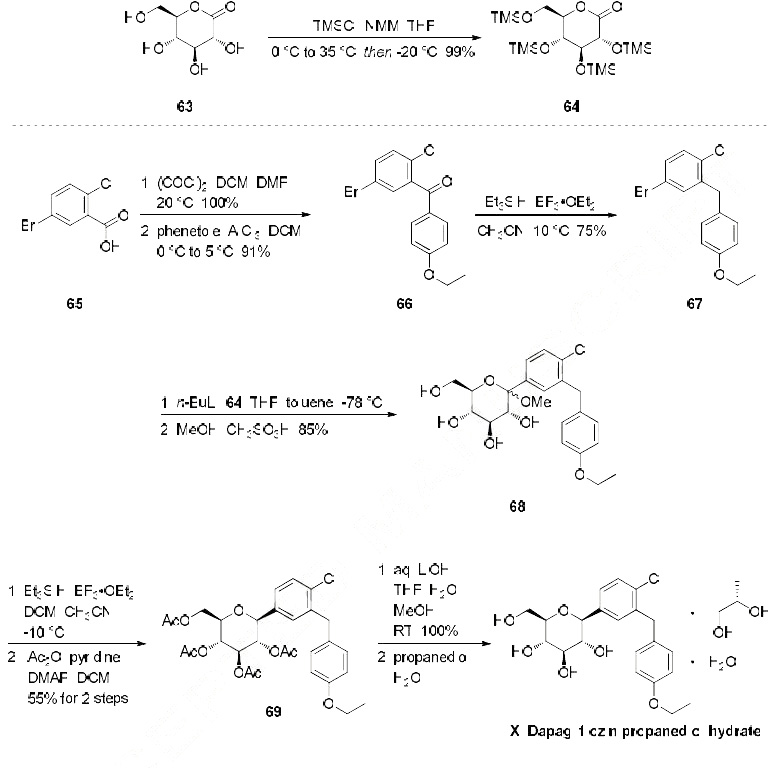
Dapagliflozin propanediol monohydrate
PAPER
https://link.springer.com/article/10.1007/s12039-020-1747-x
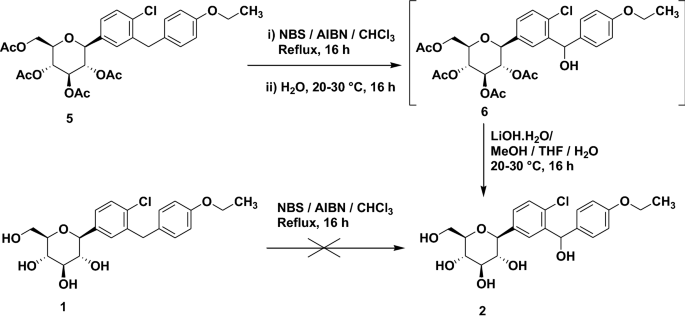
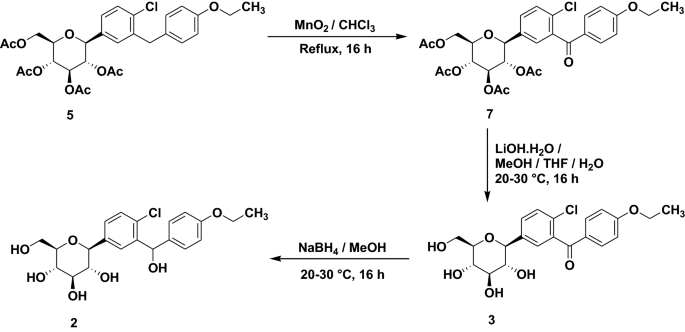

PATENTS
WO 2010138535
WO 2011060256
WO 2012041898
WO 2012163990
WO 2013068850
WO 2012163546
WO 2013068850
WO 2013079501

The IC50 for SGLT2 is less than one thousandth of the IC50 for SGLT1 (1.1 versus 1390 nmol/l), so that the drug does not interfere with the intestinal glucose absorption.[7

dapagliflozin being an inhibitor of sodiumdependent glucose transporters found in the intestine and kidney (SGLT2) and to a method for treating diabetes, especially type II diabetes, as well as hyperglycemia, hyperinsulinemia, obesity, hypertriglyceridemia, Syndrome X, diabetic
complications, atherosclerosis and related diseases, employing such C-aryl glucosides alone or in combination with one, two or more other type antidiabetic agent and/or one, two or more other type therapeutic agents such as hypolipidemic agents.
Approximately 100 million people worldwide suffer from type II diabetes (NIDDM – non-insulin-dependent diabetes mellitus), which is characterized by hyperglycemia due to excessive hepatic glucose production and peripheral insulin resistance, the root causes for which are as yet unknown. Hyperglycemia is considered to be the major risk factor for the development of diabetic complications, and is likely to contribute directly to the impairment of insulin secretion seen in advanced NIDDM. Normalization of plasma glucose in NIDDM patients would be predicted to improve insulin action, and to offset the development of diabetic complications. An inhibitor of the sodium-dependent glucose transporter SGLT2 in the kidney would be expected to aid in the normalization of plasma glucose levels, and perhaps body weight, by enhancing glucose excretion.
Dapagliflozin can be prepared using similar procedures as described in U.S. Pat. No. 6,515,117 or international published applications no. WO 03/099836 and WO 2008/116179
WO 03/099836 A1 refers to dapagliflozin having the structure according to formula 1 .

formula 1
WO 03/099836 A1 discloses a route of synthesis on pages 8-10, whereby one major step is the purification of a compound of formula 2

formula 2
The compound of formula 2 provides a means of purification for providing a compound of formula 1 since it crystallizes. Subsequently the crystalline form of the compound of formula 2 can be deprotected and converted to dapagliflozin. Using this process, dapagliflozin is obtained as an amorphous glassy off-white solid containing 0.1 1 mol% of EtOAc. Crystallization of a pharmaceutical drug is usually advantageous as it provides means for purification also suitable for industrial scale preparation. However, for providing an active pharmaceutical drug a very high purity is required. In particular, organic impurities such as EtOAc either need to be avoided or further purification steps are needed to provide the drug in a
pharmaceutically acceptable form, i.e. substantially free of organic solvents. Thus, there is the need in the art to obtain pure and crystalline dapagliflozinwhich is substantially free of organic solvents.
WO 2008/002824 A1 discloses several alternative solid forms of dapagliflozin, such as e.g. solvates containing organic alcohols or co-crystals with amino acids such as proline and phenylalanine. For instance, the document discloses crystalline
dapagliflozin solvates which additionally contain water molecules (see e.g.
Examples 3-6), but is silent about solid forms of dapagliflozin which do not contain impurities such as organic alcohols. As described above, it is desirable to provide the pharmaceutical active drug in a substantially pure form, otherwise triggering further expensive and time-consuming purification steps. In contrast, the document relates to dapagliflozin solvates where an alcohol and water are both incorporated into the crystal lattice. Hence, there is the need in the art to obtain pure and crystalline dapagliflozin suitable for pharmaceutical production.
WO 2008/1 16179 A1 refers to an immediate release pharmaceutical composition comprising dapagliflozin and propylene glycol. Propylene glycol is a chiral
substance and (S)-propylene glycol used is very expensive. Consequently, also the immediate release pharmaceutical composition is more expensive.
Crystalline forms (in comparision to the amorphous form) often show desired different physical and/or biological characteristics which may assist in the manufacture or formulation of the active compound, to the purity levels and uniformity required for regulatory approval. As described above, it is desirable to provide the pharmaceutical active drug in a substantially pure form, otherwise triggering further expensive and time-consuming purification steps.
PATENT
WO 2008/ 1 16179 Al seems to disclose an immediate release formulation comprising dapagliflozin and propylene glycol hydrate. WO 2008/ 116195 A2 refers to the use of an SLGT2 inhibitor in the treatment of obesity
http://www.google.com/patents/US20120282336
http://www.tga.gov.au/pdf/auspar/auspar-dapagliflozin-propanediol-monohydrate-130114.pdf
Example 2 Dapagliflozin (S) PGS—(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (S)-propane-1,2-diol hydrate (1:1:1)
Dapagliflozin (S) propylene glycol hydrate (1:1:1) can be prepared using similar procedures as described in published applications WO 08/002824 and WO 2008/116179, the disclosures of which are herein incorporated by reference in their entirety for any purpose. SGLT2 EC50=1.1 nM.
Example 3 Dapagliflozin (R) PGS—(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (R)-propane-1,2-diol hydrate (1:1:1)
Dapagliflozin (R) propylene glycol hydrate (1:1:1) can be prepared using similar procedures as described in WO 08/002824 and WO 2008/116179, the disclosures of which are herein incorporated by reference in their entirety for any purpose. SGLT2 EC50=1.1 nM.
WO 2008/002824 A1 discloses several alternative solid forms of dapagliflozin, such as e.g. solvates containing organic alcohols or co-crystals with amino acids such as proline and phenylalanine. For instance, the document discloses crystalline
dapagliflozin solvates which additionally contain water molecules (see e.g.
Examples 3-6), but is silent about solid forms of dapagliflozin which do not contain impurities such as organic alcohols. As described above, it is desirable to provide the pharmaceutical active drug in a substantially pure form, otherwise triggering further expensive and time-consuming purification steps. In contrast, the document relates to dapagliflozin solvates where an alcohol and water are both incorporated into the crystal lattice. Hence, there is the need in the art to obtain pure and crystalline dapagliflozin suitable for pharmaceutical production.
WO 2008/1 16179 A1 refers to an immediate release pharmaceutical composition comprising dapagliflozin and propylene glycol. Propylene glycol is a chiral
substance and (S)-propylene glycol used is very expensive. Consequently, also the immediate release pharmaceutical composition is more expensive.
Surprisingly, amorphous dapagliflozin can be purified with the process of the present invention. For instance amorphous dapagliflozin having a purity of 99,0% can be converted to crystalline dapagliflozin hydrate having a purity of 100% (see examples of the present application). Moreover, said crystalline dapagliflozin hydrate does not contain any additional solvent which is desirable. Thus, the process of purifying dapagliflozin according to the present invention is superior compared with the process of WO 03/099836 A1 .
Additionally, the dapagliflozin hydrate obtained is crystalline which is advantageous with respect to the formulation of a pharmaceutical composition. The use of expensive diols such as (S)-propanediol for obtaining an immediate release pharmaceutical composition as disclosed in WO 2008/1 16179 A1 can be avoided
PAPER
In Vitro Characterization and Pharmacokinetics of Dapagliflozin …
dmd.aspetjournals.org/content/…/DMD29165_supplemental_data_.doc
Dapagliflozin (BMS-512148), (2S,3R,4R,5S,6R)-2-(3-(4-Ethoxybenzyl)-4-chlorophenyl)
-6-hydroxymethyl-tetrahydro-2H-pyran-3,4,5-triol. 1H NMR (500 MHz, CD3OD) δ 7.33
(d, J = 6.0, 1H), 7.31 (d, J = 2.2, 1H), 7.31 (dd, J = 2.2, 6.0, 1H), 7.07 (d, J = 8.8, 2H),
6.78 (d, J = 8.8, 2H), 4.07-3.90 (m, 7H), 3.85 (d, J = 10.6, 1H), 3.69 (dd, J = 5.3, 10.6,
1H), 3.42-3.25 (m, 4H), 1.34 (t, J = 7.0, 3H). 13C NMR (125 MHz, CD3OD) δ 158.8,
140.0, 139.9, 134.4, 132.9, 131.9, 130.8, 130.1, 128.2, 115.5, 82.9, 82.2, 79.7, 76.4, 71.9,
64.5, 63.1, 39.2, 15.2.
HRMS calculated for C21H25ClNaO6 (M+Na)+
For C21H25ClO6: C, 61.68; H, 6.16. Found: C, 61.16; H, 6.58.
: 431.1237; found 431.1234. Anal. Calcd
SECOND SETJ. Med. Chem., 2008, 51 (5), pp 1145–1149DOI: 10.1021/jm701272q
1H NMR (500 MHz, CD3OD) δ 7.33 (d, J = 6.0, 1H), 7.31 (d, J = 2.2, 1H), 7.31 (dd, J = 2.2, 6.0, 1H), 7.07 (d, J = 8.8, 2H), 6.78 (d, J = 8.8, 2H), 4.07–3.90 (m, 7H), 3.85 (d, J = 10.6, 1H), 3.69 (dd, J = 5.3, 10.6, 1H), 3.42–3.25 (m, 4H), 1.34 (t, J = 7.0, 3H);
13C NMR (125 MHz, CD3OD) δ 158.8, 140.0, 139.9, 134.4, 132.9, 131.9, 130.8, 130.1, 128.2, 115.5, 82.9, 82.2, 79.7, 76.4, 71.9, 64.5, 63.1, 39.2, 15.2;
HRMS calcd for C21H25ClNaO6 (M + Na)+ 431.1237, found 431.1234. Anal. Calcd for C21H25ClO6: C, 61.68; H, 6.16. Found: C, 61.16; H, 6.58.
HPLC
- HPLC measurements were performed with an Agilent 1100 series instrument equipped with a UV-vis detector set to 240 nm according to the following method:
Column: Ascentis Express RP-Amide 4.6 x 150 mm, 2.7 mm;
Column temperature: 25 °C
– Eluent A: 0.1 % formic acid in water
– Eluent B: 0.1 % formic acid in acetonitrile
– Injection volume: 3 mL
– Flow: 0.7 mL/min
– Gradient:Time [min][%] B0.02525.06526.07029.07029.52535.025……………………..
PATENT
http://www.google.com/patents/WO2013068850A2?cl=en
EXAMPLE 24 – Synthesis of 2,4-di-6>-ieri-butyldiphenylsilyl-l-C-(4-chloro-3-(4- ethoxybenzyl)phenyl)- -D-glucopyranoside 2,4-di-6>-TBDPS-dapagliflozin; (IVj”))

[0229] l-(5-Bromo-2-chlorobenzyl)-4-ethoxybenzene (1.5 g, 4.6 mmol) and magnesium powder (0.54 g, 22.2 mmol) were placed in a suitable reactor, followed by THF (12 mL) and 1,2- dibromoethane (0.16 mL). The mixture was heated to reflux. After the reaction had initiated, a solution of l-(5-bromo-2-chlorobenzyl)-4-ethoxybenzene (4.5 g, 13.8 mmol) in THF (28 mL) was added dropwise. The mixture was allowed to stir for another hour under reflux, and was then cooled to ambient temperature, and then titrated to determine the concentration. The above prepared 4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl magnesium bromide (31 mL, 10 mmol, 0.32 M in THF) and A1C13 (0.5 M in THF, 8.0 mL, 4.0 mmol) were mixed at ambient temperature to give a black solution, which was stirred at ambient temperature for 1 hour. To a solution of
I, 6-anhydro-2,4-di-6>-ieri-butyldiphenylsilyl- -D-glucopyranose (0.64 g, 1.0 mmol) in PhOMe (3.0 mL) at ambient temperature was added phenylmagnesium bromide (0.38 mL, 1.0 mmol, 2.6 M solution in Et20). After stirring for about 5 min the solution was then added into the above prepared aluminum mixture via syringe, followed by additional PhOMe (1.0 mL) to rinse the flask. The mixture was concentrated under reduced pressure (50 torr) at 60 °C (external bath temperature) to remove low-boiling point ethereal solvents and then PhOMe (6mL) was added. The reaction mixture was heated at 130 °C (external bath temperature) for 8 hours at which time HPLC assay analysis indicated a 51% yield of 2,4-di-6>-ieri-butyldiphenylsilyl-l-C-(4-chloro-3- (4-ethoxybenzyl)phenyl)- -D-glucopyranoside. After cooling to ambient temperature, the reaction was treated with 10% aqueous NaOH (1 mL), THF (10 mL) and diatomaceous earth at ambient temperature, then the mixture was filtered and the filter cake was washed with THF. The combined filtrates were concentrated and the crude product was purified by silica gel column chromatography (eluting with 1:30 EtOAc/77-heptane) affording the product 2,4-di-6>- ieri-butyldiphenylsilyl- 1 – -(4-chloro-3 -(4-ethoxybenzyl)phenyl)- β-D-glucopyranoside (0.30 g, 34%) as a white powder.
1H NMR (400 MHz, CDC13) δ 7.56-7.54 (m, 2H), 7.43-7.31 (m, 13H), 7.29-7.22 (m, 6H), 7.07- 7.04 (m, 2H), 7.00 (d, J= 2.0 Hz, IH), 6.87 (dd, J= 8.4, 2.0 Hz, IH), 6.83-6.81 (m, 2H), 4.18 (d, J= 9.6 Hz, IH), 4.02 (q, J= 6.9 Hz, 2H), 3.96 (d, J= 10.8 Hz, 2H), 3.86 (ddd, J= 11.3, 7.7, 1.1 Hz, IH), 3.76 (ddd, J= 8.4, 8.4, 4.8 Hz, IH), 3.56 (ddd, J= 9.0, 6.4, 2.4 Hz, IH), 3.50 (dd, J=
I I.4, 5.4 Hz, IH), 3.44 (dd, J= 9.4, 8.6 Hz, IH), 3.38 (dd, J= 8.8, 8.8 Hz, IH), 1.70 (dd, J= 7.8, 5.4 Hz, IH, OH), 1.42 (t, J= 6.8 Hz, 3H), 1.21 (d, J= 5.2 Hz, IH, OH), 1.00 (s, 9H), 0.64 (s, 9H); 13C NMR (100 MHz, CDC13) δ 157.4 (C), 138.8 (C), 137.4 (C), 136.3 (CH x2), 136.1 (CH x2), 135.2 (CH x2), 135.0 (C), 134.9 (CH x2), 134.8 (C), 134.2 (C), 132.8 (C), 132.0 (C), 131.6 (CH), 131.1 (C), 129.9 (CH x2), 129.7 (CH), 129.6 (CH), 129.5 (CH), 129.4 (CH), 129.2 (CH), 127.58 (CH x2), 127.57 (CH x2), 127.54 (CH x2), 127.31 (CH), 127.28 (CH x2), 114.4 (CH x2), 82.2 (CH), 80.5 (CH), 79.3 (CH), 76.3 (CH), 72.7 (CH), 63.4 (CH2), 62.7 (CH2), 38.2 (CH2), 27.2 (CH3 x3), 26.6 (CH3 x3), 19.6 (C), 19.2 (C), 14.9 (CH3). EXAMPLE 25 -Synthesis of dapagliflozin ((25,3R,4R,55,6/?)-2-[4-chloro-3-(4- ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol; (Ij))

IVj’ U
[0230] A solution of the 2,4-di-6>-ieri-butyldiphenylsilyl-l-C-(4-chloro-3-(4- ethoxybenzyl)phenyl)- -D-glucopyranoside (60 mg, 0.068 mmol) in THF (3.0 mL) and TBAF (3.0 mL, 3.0 mmol, 1.0 M in THF) was stirred at ambient temperature for 15 hours. CaC03 (0.62 g), Dowex^ 50WX8-400 ion exchange resin (1.86 g) and MeOH (5mL) were added to the product mixture and the suspension was stirred at ambient temperature for 1 hour and then the mixture was filtrated through a pad of diatomaceous earth. The filter cake was rinsed with MeOH and the combined filtrates was evaporated under vacuum and the resulting residue was purified by column chromatography (eluting with 1 : 10 MeOH/DCM) affording dapagliflozin (30 mg).
1H NMR (400 MHz, CD3OD) δ 7.37-7.34 (m, 2H), 7.29 (dd, J= 8.2, 2.2 Hz, 1H), 7.12-7.10 (m, 2H), 6.82-6.80 (m, 2H), 4.10 (d, J= 9.6 Hz, 2H), 4.04 (d, J= 9.2 Hz, 2H), 4.00 (q, J= 7.1 Hz, 2H), 3.91-3.87 (m, 1H), 3.73-3.67(m, 1H), 3.47-3.40 (m, 3H), 3.31-3.23 (m, 2H), 1.37 (t, J= 7.0 Hz, 3H);
13C NMR (100 MHz, CD3OD) δ 157.4 (C), 138.6 (C), 138.5 (C), 133.1 (C), 131.5 (C), 130.5 (CH), 129.4 (CH x2), 128.7 (CH), 126.8 (CH), 114.0 (CH x2), 80.5 (CH), 80.8 (CH), 78.3 (CH), 75.0 (CH), 70.4 (CH), 63.0 (CH2), 61.7 (CH2), 37.8 (CH2), 13.8 (CH3);
LCMS (ESI) m/z 426 (100, [M+NH4]+), 428 (36, [M+NH4+2]+), 447 (33, [M+K]+).
Example 1 – Synthesis of l,6-anhydro-2,4-di-6>-ieri-butyldiphenylsilyl- -D-glucopyranose (II”)

III II”
[0206] To a suspension solution of l,6-anhydro- -D-glucopyranose (1.83 g, 11.3 mmol) and imidazole (3.07 g, 45.2 mmol) in THF (10 mL) at 0 °C was added dropwise a solution of TBDPSC1 (11.6 mL, 45.2 mmol) in THF (10 mL). After the l,6-anhydro-P-D-gJucopyranose was consumed, water (10 mL) was added and the mixture was extracted twice with EtOAc (20 mL each), washed with brine (10 mL), dried (Na2S04) and concentrated. Column
chromatography (eluting with 1 :20 EtOAc/rc-heptane) afforded 2,4-di-6>-ieri-butyldiphenylsilyl- l,6-anhydro- “D-glucopyranose (5.89 g, 81%).
1H NMR (400 MHz, CDC13) δ 7.82-7.70 (m, 8H), 7.49-7.36 (m, 12H), 5.17 (s, IH), 4.22 (d, J= 4.8 Hz, IH), 3.88-3.85 (m, IH), 3.583-3.579 (m, IH), 3.492-3.486 (m, IH), 3.47-3.45 (m, IH), 3.30 (dd, J= 7.4, 5.4 Hz, IH), 1.71 (d, J= 6.0 Hz, IH), 1.142 (s, 9H), 1.139 (s, 9H); 13C NMR (100 MHz, CDCI3) δ 135.89 (CH x2), 135.87 (CH x2), 135.85 (CH x2), 135.83 (CH x2), 133.8 (C), 133.5 (C), 133.3 (C), 133.2 (C), 129.94 (CH), 129.92 (CH), 129.90 (CH), 129.88 (CH), 127.84 (CH2 x2), 127.82 (CH2 x2), 127.77 (CH2 x4), 102.4 (CH), 76.9 (CH), 75.3 (CH), 73.9 (CH), 73.5 (CH), 65.4 (CH2), 27.0 (CH3 x6), 19.3 (C x2).
PATENT
WO 2016147197, DAPAGLIFLOZIN, NEW PATENT, HARMAN FINOCHEM LIMITED
LINK>>> (WO2016147197) A NOVEL PROCESS FOR PREPARING (2S,3R,4R,5S,6R)-2-[4-CHLORO-3-(4-ETHOXYBENZYL)PHENY 1] -6-(HY DROXY METHYL)TETRAHYDRO-2H-PY RAN-3,4,5-TRIOL AND ITS AMORPHOUS FORM
PATENT
PATENT
WO2016018024, CRYSTALLINE COMPOSITE COMPRISING DAPAGLIFLOZIN AND METHOD FOR PREPARING SAME
HANMI FINE CHEMICAL CO., LTD. [KR/KR]; 59, Gyeongje-ro, Siheung-si, Gyeonggi-do 429-848 (KR)
Dapagliflozin, sold under the brand name Farxiga among others, is a medication used to treat type 2 diabetes and, with certain restrictions, type 1 diabetes.[2] It is also used to treat adults with certain kinds of heart failure.[3][4][5]
Common side effects include hypoglycaemia (low blood sugar), urinary tract infections, genital infections, and volume depletion (reduced amount of water in the body).[6] Diabetic ketoacidosis is a common side effect in type 1 diabetic patients.[7] Serious but rare side effects include Fournier gangrene.[8] Dapagliflozin is a sodium-glucose co-transporter-2 (SGLT-2) inhibitor and works by removing sugar from the body with the urine.[9]
It was developed by Bristol-Myers Squibb in partnership with AstraZeneca. In 2018, it was the 227th most commonly prescribed medication in the United States, with more than 2 million prescriptions.[10][11]
Medical uses
Dapagliflozin is used along with diet and exercise to improve glycemic control in adults with type 2 diabetes and to reduce the risk of hospitalization for heart failure among adults with type 2 diabetes and known cardiovascular disease or other risk factors.[12][3] It appears more useful than empagliflozin.[13][verification needed]
In addition, dapagliflozin is indicated for the treatment of adults with heart failure with reduced ejection fraction to reduce the risk of cardiovascular death and hospitalization for heart failure.[3][4][5] It is also indicated to reduce the risk of kidney function decline, kidney failure, cardiovascular death and hospitalization for heart failure in adults with chronic kidney disease who are at risk of disease progression.[14]
In the European Union it is indicated in adults:
- for the treatment of insufficiently controlled type 2 diabetes mellitus as an adjunct to diet and exercise:
- as monotherapy when metformin is considered inappropriate due to intolerance;
- in addition to other medicinal products for the treatment of type 2 diabetes;
- for the treatment of insufficiently controlled type 1 diabetes mellitus as an adjunct to insulin in patients with BMI ≥ 27 kg/m2, when insulin alone does not provide adequate glycaemic control despite optimal insulin therapy; and
- for the treatment of heart failure with reduced ejection fraction.[5]
Adverse effects
Since dapagliflozin leads to heavy glycosuria (sometimes up to about 70 grams per day) it can lead to rapid weight loss and tiredness. The glucose acts as an osmotic diuretic (this effect is the cause of polyuria in diabetes) which can lead to dehydration. The increased amount of glucose in the urine can also worsen the infections already associated with diabetes, particularly urinary tract infections and thrush (candidiasis). Rarely, use of an SGLT2 drug, including dapagliflozin, is associated with necrotizing fasciitis of the perineum, also called Fournier gangrene.[15]
Dapagliflozin is also associated with hypotensive reactions. There are concerns it may increase the risk of diabetic ketoacidosis.[16]
Dapagliflozin can cause dehydration, serious urinary tract infections and genital yeast infections.[3] Elderly people, people with kidney problems, those with low blood pressure, and people on diuretics should be assessed for their volume status and kidney function.[3] People with signs and symptoms of metabolic acidosis or ketoacidosis (acid buildup in the blood) should also be assessed.[3] Dapagliflozin can cause serious cases of necrotizing fasciitis of the perineum (Fournier gangrene) in people with diabetes and low blood sugar when combined with insulin.[3]
To lessen the risk of developing ketoacidosis (a serious condition in which the body produces high levels of blood acids called ketones) after surgery, the FDA has approved changes to the prescribing information for SGLT2 inhibitor diabetes medicines to recommend they be stopped temporarily before scheduled surgery. Canagliflozin, dapagliflozin, and empagliflozin should each be stopped at least three days before, and ertugliflozin should be stopped at least four days before scheduled surgery.[17]
Symptoms of ketoacidosis include nausea, vomiting, abdominal pain, tiredness, and trouble breathing.[17]
Use is not recommended in patients with eGFR < 45ml/min/1.73m2, though data from 2021 shows the reduction in the kidney failure risks in people with chronic kidney disease using dapagliflozin.[18]
Mechanism of action
Dapagliflozin inhibits subtype 2 of the sodium-glucose transport proteins (SGLT2) which are responsible for at least 90% of the glucose reabsorption in the kidney. Blocking this transporter mechanism causes blood glucose to be eliminated through the urine.[19] In clinical trials, dapagliflozin lowered HbA1c by 0.6 versus placebo percentage points when added to metformin.[20]
Regarding its protective effects in heart failure, this is attributed primarily to haemodynamic effects, where SGLT2 inhibitors potently reduce intravascular volume through osmotic diuresis and natriuresis. This consequently may lead to a reduction in preload and afterload, thereby alleviating cardiac workload and improving left ventricular function.[21]
Selectivity
The IC50 for SGLT2 is less than one thousandth of the IC50 for SGLT1 (1.1 versus 1390 nmol/L), so that the drug does not interfere with intestinal glucose absorption.[22]
Names
Dapagliflozin is the International nonproprietary name (INN),[23] and the United States Adopted Name (USAN).[24]
There is a fixed-dose combination product dapagliflozin/metformin extended-release, called Xigduo XR.[25][26][27]
In July 2016, the fixed-dose combination of saxagliptin and dapagliflozin was approved for medical use in the European Union and is sold under the brand name Qtern.[28] The combination drug was approved for medical use in the United States in February 2017, where it is sold under the brand name Qtern.[29][30]
In May 2019, the fixed-dose combination of dapagliflozin, saxagliptin, and metformin hydrochloride as extended-release tablets was approved in the United States to improve glycemic control in adults with type 2 diabetes when used in combination with diet and exercise. The FDA granted the approval of Qternmet XR to AstraZeneca.[31] The combination drug was approved for use in the European Union in November 2019, and is sold under the brand name Qtrilmet.[32]
History
In 2012, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) issued a positive opinion on the drug.[5]
Dapagliflozin was found effective in several studies in participants with type 2 and type 1 diabetes.[5] The main measure of effectiveness was the level of glycosylated haemoglobin (HbA1c), which gives an indication of how well blood glucose is controlled.[5]
In two studies involving 840 participants with type 2 diabetes, dapagliflozin when used alone decreased HbA1c levels by 0.66 percentage points more than placebo (a dummy treatment) after 24 weeks.[5] In four other studies involving 2,370 participants, adding dapagliflozin to other diabetes medicines decreased HbA1c levels by 0.54-0.68 percentage points more than adding placebo after 24 weeks.[5]
In a study involving 814 participants with type 2 diabetes, dapagliflozin used in combination with metformin was at least as effective as a sulphonylurea (another type of diabetes medicines) used with metformin.[5] Both combinations reduced HbA1c levels by 0.52 percentage points after 52 weeks.[5]
A long-term study, involving over 17,000 participants with type 2 diabetes, looked at the effects of dapagliflozin on cardiovascular (heart and circulation) disease.[5] The study indicated that dapagliflozin’s effects were in line with those of other diabetes medicines that also work by blocking SGLT2.[5]
In two studies involving 1,648 participants with type 1 diabetes whose blood sugar was not controlled well enough on insulin alone, adding dapagliflozin 5 mg decreased HbA1c levels after 24 hours by 0.37% and by 0.42% more than adding placebo.[5]
Dapagliflozin was approved for medical use in the European Union in November 2012.[5] It is marketed in a number of European countries.[33]
Dapagliflozin was approved for medical use in the United States in January 2014.[34][14]
In 2020, the U.S. Food and Drug Administration (FDA) expanded the indications for dapagliflozin to include treatment for adults with heart failure with reduced ejection fraction to reduce the risk of cardiovascular death and hospitalization for heart failure.[3] It is the first in this particular drug class, sodium-glucose co-transporter 2 (SGLT2) inhibitors, to be approved to treat adults with New York Heart Association’s functional class II-IV heart failure with reduced ejection fraction.[3]
Dapagliflozin was shown in a clinical trial to improve survival and reduce the need for hospitalization in adults with heart failure with reduced ejection fraction.[3] The safety and effectiveness of dapagliflozin were evaluated in a randomized, double-blind, placebo-controlled study of 4,744 participants.[3] The average age of participants was 66 years and more participants were male (77%) than female.[3] To determine the drug’s effectiveness, investigators examined the occurrence of cardiovascular death, hospitalization for heart failure, and urgent heart failure visits.[3] Participants were randomly assigned to receive a once-daily dose of either 10 milligrams of dapagliflozin or a placebo (inactive treatment).[3] After about 18 months, people who received dapagliflozin had fewer cardiovascular deaths, hospitalizations for heart failure, and urgent heart failure visits than those receiving the placebo.[3]
In July 2020, the FDA granted AstraZeneca a Fast Track Designation in the US for the development of dapagliflozin to reduce the risk of hospitalisation for heart failure or cardiovascular death in adults following a heart attack.[35]
In August 2020, it was reported that detailed results from the Phase III DAPA-CKD trial showed that AstraZeneca’s FARXIGA® (dapagliflozin) on top of standard of care reduced the composite measure of worsening of renal function or risk of cardiovascular (CV) or renal death by 39% compared to placebo (p<0.0001) in patients with chronic kidney disease (CKD) Stages 2-4 and elevated urinary albumin excretion. The results were consistent in patients both with and without type 2 diabetes (T2D)[36]
In April 2021, the FDA expanded the indications for dapagliflozin (Farxiga) to include reducing the risk of kidney function decline, kidney failure, cardiovascular death and hospitalization for heart failure in adults with chronic kidney disease who are at risk of disease progression.[14] The efficacy of dapagliflozin to improve kidney outcomes and reduce cardiovascular death in people with chronic kidney disease was evaluated in a multicenter, double-blind study of 4,304 participants.[14]
///////////////////////////////////////////
POST YOUR INTERMEDIATES LIST HERE FOR WORLDWIDE REACH
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
SYN
https://pharmacia.pensoft.net/article/70626/
Synthesis
Dapagliflozin is an approved drug by U.S. Food and Drug Administration (FDA). Dapagliflozin is a representative of SGLT-2 inhibitors, actively considered to cure diabetes type 2. Thus, methodology of dapagliflozin synthesis has rarely published (Ellsworth et al. 2002; Meng 2008). Scheme 1 have shown the general synthetic route for the synthesis of dapagliflozin. Gluconolactone 3 which was protected by trimethylsilyl TMS was treated with aryl lithium. Aryl lithium was obtained by reacting aryl bromide 2 (exchange of Li/Br) with n-BuLi. Methyl C-aryl glucoside 4 was produced by treatment of resulting mixture with methane sulfonic acid in the presence of methanol. Compound 4 was subjected to acetylation in the presence of Ac2O, resulted in the formation of 5 followed by reduction of 5 to 6 with the help of Et3SiH and BF3.OEt2. Finally, dapagliflozin 1 was produced via hydrolysis of 6 by LiOH (Deshpande et al. 2008; Meng 2008).
Ellsworth B, Washburn WN, Sher PM, Wu G, Meng W (2002) C-Aryl glucoside SGLT2 inhibitors and method, Google Patents. https://patents.google.com/patent/US6515117B2/en
Meng W, Ellsworth BA, Nirschl AA, McCann PJ, Patel M, Girotra RN, Wu G, Sher PM, Morrison EP, Biller SA, Zahler R, Deshpande PP, Pullockaran A, Hagan DL, Morgan N, Taylor JR, Obermeier MT, Humphreys WG, Khanna A, Discenza L, Robertson JG, Wang A, Han S, Wetterau JR, Janovitz EB, Flint OP, Whaley JM, Washburn WN (2008) “Discovery of dapagliflozin: a potent, selective renal sodium-dependent glucose cotransporter 2 (SGLT2) inhibitor for the treatment of type 2 diabetes. ” Journal of Medicinal Chemistry 51(5): 1145–1149. https://doi.org/10.1021/jm701272q
Deshpande PP, Ellsworth BA, Singh J, Denzel TW, Lai C, Crispino G, Randazzo ME, Gougoutas JZ (2008) Methods of producing C-aryl glucoside SGLT2 inhibitors, Google Patents. https://patents.google.com/patent/US7375213B2/en

Jun et al. has reported a few improvements to the scheme 1. In the improved methodology scheme 2, trimethylsilyl chloride was added to gluconolactone 7 in the presence of N-methylmorpholine and tetrahydrofuran THF (Horton et al. 1981), followed by the formation of persilyated lactone 3. After completing the reaction of aryl bromide 2 with n-BuLi, added to persilyated lactone 3. Intermediate lactol 8 was produced by treating resulting reaction mixture with trifluoroacetic acid in aqueous form. Then ethyl C-aryl glycoside 9 yielded when subsequently compound 8 was subjected to methane-sulfonic acid in ethyl alcohol. Crude product 9 in the form of oil was secured after the screening of solvents. Jun et al. proposed that more than 98% pure 9 was collected as crystalline solvate after the crystallization of crude oil from n-propanol and n-heptane mixture (Yu et al. 2019). Moreover, Wang et al. proposed that a high extent of diastereoselectivity obtained after the reduction of tetra-O-unprotected methyl C-aryl glucoside by utilizing Et3SiH and BF3.Et2O (Wang et al. 2014). The nature of active pharmaceutical ingredient is amorphous foam which is isolated after the reduction of 9. Production of cocrystalline complex facilitate the isolation and purification of API (Deng et al. 2017). It is concluded that more than 99.7% pure dapagliflozin produced in overall 79% yield, after the crystallization of a mixture consists of n-heptane and ethyl acetate (Yu et al. 2019).
Zheng et al. designed the production methodology of dapagliflozin by introducing NO donor group at the last steps of general route of dapagliflozin synthesis (scheme 1). Novel hybrids achieved by the combination of dapagliflozin and NO donor, having excellent dual characteristics of anti-hyperglycemic and anti-thrombosis. The figure 2 represent the modifiable site (4-position) of dapagliflozin.
Horton D, Priebe W (1981) “Synthetic routes to higher-carbon sugars. Reaction of lactones with 2-lithio-, 3-dithiane. ” Carbohydrate Research 94(1): 27–41. https://doi.org/10.1016/S0008-6215(00)85593-7
Yu J, Cao Y, Yu H, Wang JJ (2019) “A Concise and Efficient Synthesis of Dapagliflozin. ” Organic Process Research & Development 23(7): 1458–1461. https://doi.org/10.1021/acs.oprd.9b00141
Wang X-j, Zhang L, Byrne D, Nummy L, Weber D, Krishnamurthy D, Yee N, Senanayake CH (2014) “Efficient synthesis of empagliflozin, an inhibitor of SGLT-2, utilizing an AlCl3-promoted silane reduction of a β-glycopyranoside. ” Organic Letters 16(16): 4090–4093. https://doi.org/10.1021/ol501755h
Deng J-H, Lu T-B, Sun CC, J-M Chen (2017) “Dapagliflozin-citric acid cocrystal showing better solid state properties than dapagliflozin. ” European Journal of Pharmaceutical Sciences 104: 255–261. https://doi.org/10.1016/j.ejps.2017.04.008

Scheme 3 has shown the formation strategy of new hybrids of nitric oxide with dapagliflozin. During the synthesis process, the compound 6 was treated with BBr3 that result in the formation of phenol 10, which was further subjected to condensation with bromoalkane and then undergo hydrolysis and produce 11 intermediates. Target compound was obtained by the reacting 11 with silver nitrate in acetonitrile (Li et al. 2018).
Li Z, Xu X, Deng L, Liao R, Liang R, Zhang B, Zhang LJB (2018) Design, synthesis and biological evaluation of nitric oxide releasing derivatives of dapagliflozin as potential anti-diabetic and anti-thrombotic agents. Bioorganic & Medicinal Chemistry 26(14): 3947–3952. https://doi.org/10.1016/j.bmc.2018.06.017

Lin et al. fabricated green route (scheme 4) for the production of dapagliflozin. 5-bromo-2-chlorobenzoic acid 12 and gluconolactone were utilized to initiate the synthesis. By taking BF3.Et2O in catalytic amount to produce 13, overall yield of 76% was obtained in one-pot way via considering the Friedel-Craft acylation and ketallization. There was no need to do work-up operations to separate the diaryl ketal 13 as it was easily crystallized from the mixture. Compound 14 was produce as a result of condensation between 13 and 3. Overall yield of 68% of compound 15 was produced by the deprotection of silyl group in ethyl alcohol media. In THF presence, single crystals of 15 was achieved and characterized by XRD-analysis. High yield of dapagliflozin was obtained after the reduction of 15 that was carried out by triethylsilane in the presence of boron trifluoride diethyl etherate in dichloromethane. Upon crystallization from the mixture having heptane and ethyl acetate, greater than 98% pure dapagliflozin was produced by green synthetic pathway (Hu et al. 2019).
Hu L, Zou P, Wei W, Yuan X-M, Qiu X-L, Gou S-H (2019) “Facile and green synthesis of dapagliflozin. ” Synthetic Communications 49(23): 3373–3379. https://doi.org/10.1080/00397911.2019.1666283
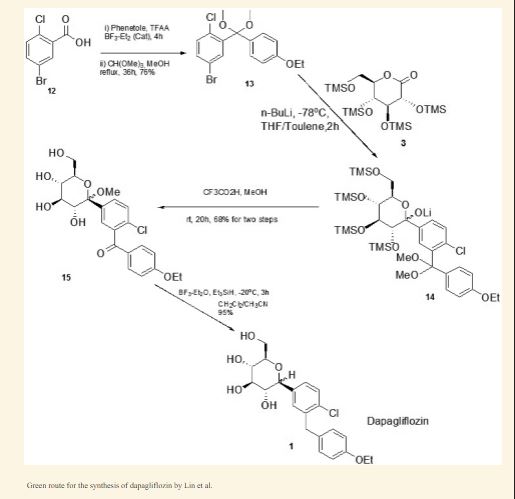
PAPER
A Concise and Efficient Synthesis of Dapagliflozin
Cite this: Org. Process Res. Dev.2019, 23, 7, 1458–1461
Publication Date:June 27, 2019
https://doi.org/10.1021/acs.oprd.9b00141
https://pubs.acs.org/doi/10.1021/acs.oprd.9b00141
file:///C:/Users/Inspiron/Downloads/op9b00141_si_001.pdf SUPP
Abstract

A concise and efficient synthesis of the SGLT-2 inhibitor dapagliflozin (1) has been developed. This route involves ethyl C-aryl glycoside 9 as the key intermediate, which is easily crystallized and purified as the crystalline n-propanol solvate with high purity (>98.5%). The tetra-O-unprotected compound 9 could be directly reduced to crude dapagliflozin with high diastereoselectivity. The final pure API product 1 was isolated and purified with high purity (>99.7%). The process has been implemented on a multikilogram scale.
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(hydroxymethyl)tetra-hydro-2Hpyran-3,4,5-triol (1)
Compound 1 has a melting point of 88.3oC.
The MsOH solution should be used immediately to avoid sulfonate formation and we have no data on the presence of sulfonates in the API.
1HNMR (400 MHz, DMSO-d6) δ (ppm) 7.32-7.37 (m, 2H), 7.22-7.24 (m, 1H), 7.08-7.10 (m, 2H), 6.80-6.84 (m, 2H), 4.94-4.96 (m, 2H), 4.82-4.83 (d, J=5.6 Hz, 1H), 4.43-4.46 (m, 1H), 3.93-4.02 (m, 5H), 3.68-3.73 (m, 1H), 3.42-3.48 (m, 1H), 3.10-3.28 (m, 4H), 1.27-1.30 (m, 3H).
13C NMR (100 MHz, DMSOd6) δ(ppm) 157.38, 140.14, 138.27, 132.40, 131.69, 131.27, 130.03, 129.13, 127.81, 114.78, 81.67, 81.18, 78.80, 75.19, 70.80, 63.37, 61.86, 38.14, 15.15.
IR(KBr): 3415, 2979, 2918, 1617, 1512, 1475, 1391, 1242, 1103, 1045, 913, 825 cm-1.
MS(m/z):431.12[M + Na]+ .
1H NMR and 13C NMR spectra for Compound
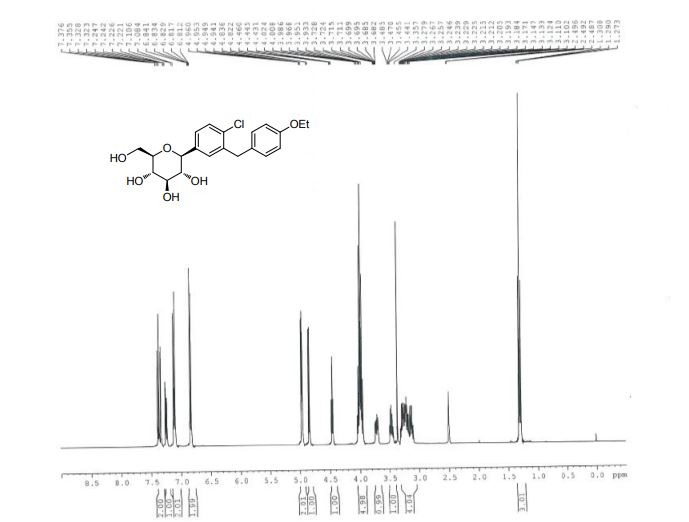
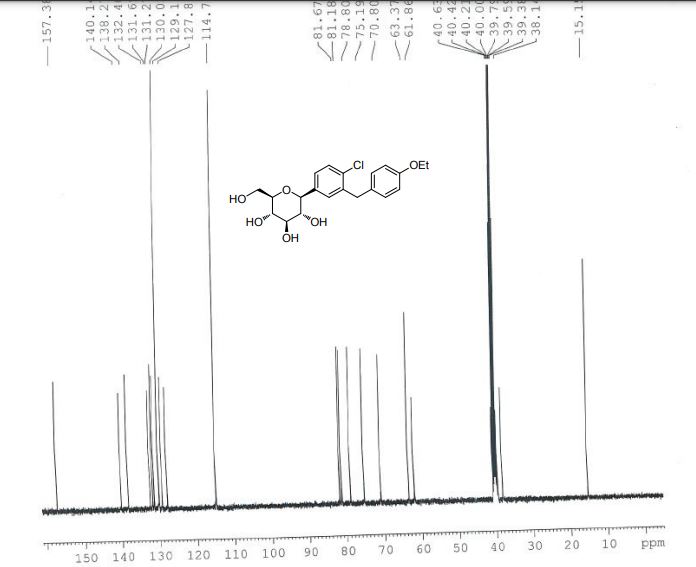
IR and Mass spectra for Compound
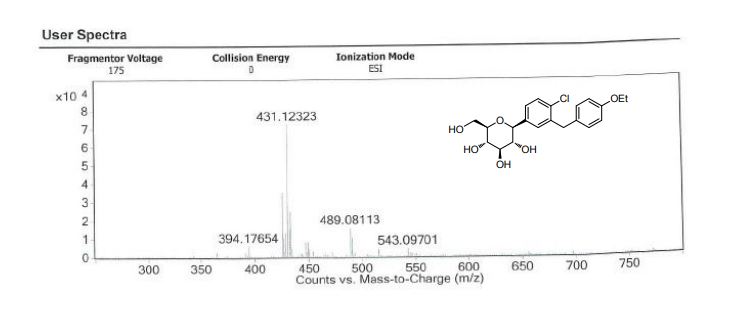
DSC spectra for Compound
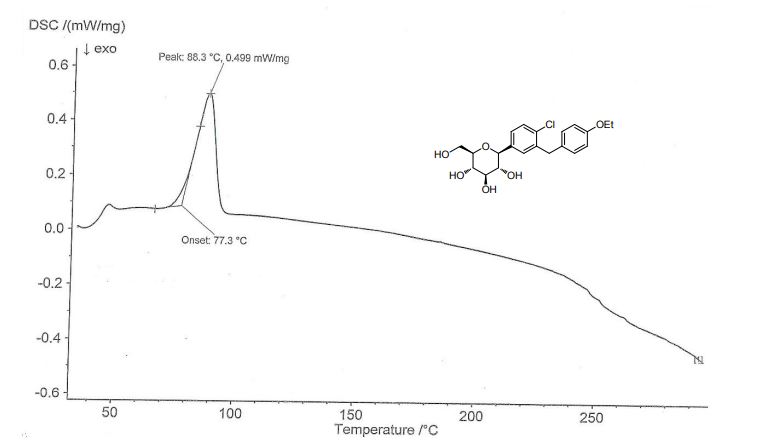
PAPER
https://pubs.acs.org/doi/10.1021/ol300220p
rg. Lett.2012, 14, 6, 1480–1483
Publication Date:March 2, 2012
https://doi.org/10.1021/ol300220p
Abstract

A general, transition-metal-free, highly stereoselective cross-coupling reaction between glycosyl bromides and various arylzinc reagents leading to β-arylated glycosides is reported. The stereoselectivity of the reaction is explained by invoking anchimeric assistance via a bicyclic intermediate. Stereochemical probes confirm the participation of the 2-pivaloyloxy group. Finally, this new method was applied to a short and efficient stereoselective synthesis of Dapagliflozin and Canagliflozin.
CROSS REF J. Med. Chem 2008, 51, 1145
H NMR (360 MHz, MeOD): δ 7.35‐7.28 (m, 3H); 7.09 (d, J=8.4Hz, 2H), 6.80 (d, J=8.8Hz, 2H), 4.09 (d, J=9.5Hz, 1H); 4.03‐3.96 (m, 4H); 3.89‐3.85 (m, 1H); 3.71‐3.66 (m, 1H); 3.45‐3.36 (m, 4H), 3.28‐3.26 (m, 2H); 1.36 (t, J=6.9Hz, 3H).
13CNMR (90 MHz, MeOD): δ 14.80, 38.31, 61.80, 63.38, 69.88, 74.67, 77.93, 79.35, 81.08, 114.48, 126.35, 128.20, 129.01, 129.72, 130.62, 131.14, 134.15, 137.04, 139.01, 157.31.
PAPER
https://pubs.acs.org/doi/10.1021/ja00199a028
Research
One study found that it had no benefit on heart disease risk or overall risk of death in people with diabetes.[37] Another study found that in heart failure with a reduced ejection fraction, dapagliflozin reduced the risk of worsening of heart failure or progression to death from cardiovascular causes, irrespective of diabetic status.[38]
References
- ^ Jump up to:a b “Dapagliflozin (Farxiga) Use During Pregnancy”. Drugs.com. 30 August 2018. Retrieved 5 May 2020.
- ^ Jump up to:a b “Farxiga- dapagliflozin tablet, film coated”. DailyMed. 3 February 2020. Retrieved 5 May 2020.
- ^ Jump up to:a b c d e f g h i j k l m n o “FDA approves new treatment for a type of heart failure”. U.S. Food and Drug Administration (FDA) (Press release). 5 May 2020. Retrieved 5 May 2020.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b National Institute for Health and Care Excellence (24 February 2021). “Dapagliflozin for treating chronic heart failure with reduced ejection fraction”. NICE Technology Appraisal Auidance [TA679]. NICE. Retrieved 9 May 2021.
- ^ Jump up to:a b c d e f g h i j k l m n “Forxiga EPAR”. European Medicines Agency (EMA). Retrieved 17 February 2020. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Ptaszynska, Agata; Johnsson, Kristina M.; Parikh, Shamik J.; De Bruin, Tjerk W. A.; Apanovitch, Anne Marie; List, James F. (2014). “Safety Profile of Dapagliflozin for Type 2 Diabetes: Pooled Analysis of Clinical Studies for Overall Safety and Rare Events”. Drug Safety. 37 (10): 815–829. doi:10.1007/s40264-014-0213-4. PMID 25096959. S2CID 24064402.
- ^ Dandona, Paresh; Mathieu, Chantal; Phillip, Moshe; Hansen, Lars; Tschöpe, Diethelm; Thorén, Fredrik; Xu, John; Langkilde, Anna Maria; DEPICT-1 Investigators (2018). “Efficacy and Safety of Dapagliflozin in Patients with Inadequately Controlled Type 1 Diabetes: The DEPICT-1 52-Week Study”. Diabetes Care. 41(12): 2552–2559. doi:10.2337/dc18-1087. PMID 30352894. S2CID 53027785.
- ^ Hu, Yang; Bai, Ziyu; Tang, Yan; Liu, Rongji; Zhao, Bin; Gong, Jian; Mei, Dan (2020). “Fournier Gangrene Associated with Sodium-Glucose Cotransporter-2 Inhibitors: A Pharmacovigilance Study with Data from the U.S. FDA Adverse Event Reporting System”. Journal of Diabetes Research. 2020: 1–8. doi:10.1155/2020/3695101. PMC 7368210. PMID 32695827.
- ^ FARXIGA- dapagliflozin tablet, film coated. DailyMed. Retrieved 6 May 2021.
- ^ “The Top 300 of 2021”. ClinCalc. Retrieved 18 February 2021.
- ^ “Dapagliflozin – Drug Usage Statistics”. ClinCalc. Retrieved 18 February 2021.
- ^ “FDA Approves Farxiga to Treat Type 2 Diabetes” (Press release). U.S. Food and Drug Administration (FDA). 8 January 2014. Archived from the original on 9 January 2014. Retrieved 15 November 2016. This article incorporates text from this source, which is in the public domain.
- ^ Zelniker TA, Wiviott SD, Raz I, et al. (January 2019). “SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: a systematic review and meta-analysis of cardiovascular outcome trials”. Lancet. 393(10166): 31–9. doi:10.1016/S0140-6736(18)32590-X. PMID 30424892. S2CID 53277899.
However, in patients with atherosclerotic cardiovascular disease, the effect of empagliflozin on cardiovascular death was more pro-nounced than that of canagliflozin or dapagliflozin
- ^ Jump up to:a b c d “FDA Approves Treatment for Chronic Kidney Disease”. U.S. Food and Drug Administration (FDA) (Press release). 30 April 2021. Retrieved 30 April 2021. This article incorporates text from this source, which is in the public domain.
- ^ “FDA warns about rare occurrences of a serious infection of the genital area with SGLT2 inhibitors for diabetes”. U.S. Food and Drug Administration (FDA). 9 February 2019. This article incorporates text from this source, which is in the public domain.
- ^ “SGLT2 inhibitors: Drug Safety Communication – FDA Warns Medicines May Result in a Serious Condition of Too Much Acid in the Blood”. U.S. Food and Drug Administration (FDA). 15 May 2015. Archived from the original on 27 October 2016. Retrieved 15 November 2016. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b “FDA revises labels of SGLT2 inhibitors for diabetes to include warning”. U.S. Food and Drug Administration. 19 March 2020. Retrieved 6 June 2020. This article incorporates text from this source, which is in the public domain.
- ^ McMurray, John J.V.; Wheeler, David C.; Stefánsson, Bergur V.; Jongs, Niels; Postmus, Douwe; Correa-Rotter, Ricardo; Chertow, Glenn M.; Greene, Tom; Held, Claes; Hou, Fan-Fan; Mann, Johannes F.E.; Rossing, Peter; Sjöström, C. David; Toto, Roberto D.; Langkilde, Anna Maria; Heerspink, Hiddo J.L.; DAPA-CKD Trial Committees Investigators (2021). “Effect of Dapagliflozin on Clinical Outcomes in Patients with Chronic Kidney Disease, with and Without Cardiovascular Disease” (PDF). Circulation. 143 (5): 438–448. doi:10.1161/CIRCULATIONAHA.120.051675. PMID 33186054. S2CID 226948086.
- ^ “Life Sciences – Clarivate”. Clarivate. Archived from the original on 5 November 2007.
- ^ “UEndocrine: Internet Endocrinology Community”. uendocrine.com. Archived from the original on 5 February 2013.
- ^ Lan NS, Fegan PG, Yeap BB, Dwivedi G (October 2019). “The effects of sodium-glucose cotransporter 2 inhibitors on left ventricular function: current evidence and future directions”. ESC Heart Fail. 6 (5): 927–935. doi:10.1002/ehf2.12505. PMC 6816235. PMID 31400090.
- ^ Schubert-Zsilavecz, M, Wurglics, M, Neue Arzneimittel 2008/2009
- ^ “International Nonproprietary Names for Pharmaceutical Substances (INN). Recommended International Nonproprietary Names: List 59” (PDF). World Health Organization. 2008. p. 50. Retrieved 15 November 2016.
- ^ “Statement on a Nonproprietary Name Adopted by the USAN Council” (PDF). American Medical Association. Archived from the original (PDF) on 7 February 2012. Retrieved 15 November2016.
- ^ “US FDA Approves Once-Daily Xigduo XR Tablets for Adults with Type 2 Diabetes”. AstraZeneca. 30 October 2014.
- ^ “Drug Approval Package: Xigduo XR (dapagliflozin and metformin HCl) Extended-Release Tablets”. U.S. Food and Drug Administration (FDA). 7 April 2015. Retrieved 5 May 2020.
- ^ “Xigduo XR- dapagliflozin and metformin hydrochloride tablet, film coated, extended release”. DailyMed. 3 February 2020. Retrieved 5 May 2020.
- ^ “Qtern EPAR”. European Medicines Agency (EMA). Retrieved 7 May 2020.
- ^ “Drug Approval Package: Qtern (dapagliflozin and saxagliptin)”. U.S. Food and Drug Administration (FDA). 10 October 2018. Retrieved 8 May 2020.
- ^ “Qtern- dapagliflozin and saxagliptin tablet, film coated”. DailyMed. 24 January 2020. Retrieved 17 February 2020.
- ^ “Drug Approval Package: Qternmet XR”. U.S. Food and Drug Administration (FDA). 27 January 2020. Retrieved 17 February2020.
- ^ “Qtrilmet EPAR”. European Medicines Agency (EMA). Retrieved 30 March 2020.
- ^ “Forxiga”. Drugs.com. 4 May 2020. Retrieved 5 May 2020.
- ^ “Drug Approval Package: Farxiga (dapagliflozin) Tablets NDA #202293”. U.S. Food and Drug Administration (FDA). 24 December 1999. Retrieved 5 May 2020.
- ^ “FARXIGA Granted Fast Track Designation in the US for Heart Failure Following Acute Myocardial Infarction Leveraging an Innovative Registry-Based Trial Design”. http://www.businesswire.com. 16 July 2020. Retrieved 20 July 2020.
- ^https://www.businesswire.com/news/home/20200830005009/en/FARXIGA-Demonstrated-Unprecedented-Reduction-Risk-Kidney-Failure
- ^ “Type 2 diabetes. Cardiovascular assessment of dapagliflozin: no advance”. Prescrire International. 29 (211): 23. January 2020. Retrieved 2 February 2020.
- ^ McMurray JJ, Solomon SD, Inzucchi SE, et al. (November 2019). “Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction”. New England Journal of Medicine. 381 (21): 1995–2008. doi:10.1056/NEJMoa1911303. PMID 31535829.
External links
- “Dapagliflozin”. Drug Information Portal. U.S. National Library of Medicine.
- “Dapagliflozin mixture with metformin hydrochloride”. Drug Information Portal. U.S. National Library of Medicine.
- “Dapagliflozin mixture with saxagliptin”. Drug Information Portal. U.S. National Library of Medicine.
Clinical trials
- Clinical trial number NCT00528372 for “A Phase III Study of BMS-512148 (Dapagliflozin) in Patients With Type 2 Diabetes Who Are Not Well Controlled With Diet and Exercise” at ClinicalTrials.gov
- Clinical trial number NCT00643851 for “An Efficacy & Safety Study of BMS-512148 in Combination With Metformin Extended Release Tablets” at ClinicalTrials.gov
- Clinical trial number NCT00859898 for “Study of Dapagliflozin in Combination With Metformin XR to Initiate the Treatment of Type 2 Diabetes” at ClinicalTrials.gov
- Clinical trial number NCT00528879 for “A Phase III Study of BMS-512148 (Dapagliflozin) in Patients With Type 2 Diabetes Who Are Not Well Controlled on Metformin Alone” at ClinicalTrials.gov
- Clinical trial number NCT00660907 for “Efficacy and Safety of Dapagliflozin in Combination With Metformin in Type 2 Diabetes Patients” at ClinicalTrials.gov
- Clinical trial number NCT00680745 for “Efficacy and Safety of Dapagliflozin in Combination With Glimepiride (a Sulphonylurea) in Type 2 Diabetes Patients” at ClinicalTrials.gov
- Clinical trial number NCT01392677 for “Evaluation of Safety and Efficacy of Dapagliflozin in Subjects With Type 2 Diabetes Who Have Inadequate Glycaemic Control on Background Combination of Metformin and Sulfonylurea” at ClinicalTrials.gov
- Clinical trial number NCT00683878 for “Add-on to Thiazolidinedione (TZD) Failures” at ClinicalTrials.gov
- Clinical trial number NCT00984867 for “Dapagliflozin DPPIV Inhibitor add-on Study” at ClinicalTrials.gov
- Clinical trial number NCT00673231 for “Efficacy and Safety of Dapagliflozin, Added to Therapy of Patients With Type 2 Diabetes With Inadequate Glycemic Control on Insulin” at ClinicalTrials.gov
- Clinical trial number NCT02229396 for “Phase 3 28-Week Study With 24-Week and 52-week Extension Phases to Evaluate Efficacy and Safety of Exenatide Once Weekly and Dapagliflozin Versus Exenatide and Dapagliflozin Matching Placebo” at ClinicalTrials.gov
- Clinical trial number NCT02413398 for “A Study to Evaluate the Effect of Dapagliflozin on Blood Glucose Level and Renal Safety in Patients With Type 2 Diabetes (DERIVE)” at ClinicalTrials.gov
- Clinical trial number NCT01730534 for “Multicenter Trial to Evaluate the Effect of Dapagliflozin on the Incidence of Cardiovascular Events (DECLARE-TIMI58)” at ClinicalTrials.gov
- Clinical trial number NCT03036124 for “Study to Evaluate the Effect of Dapagliflozin on the Incidence of Worsening Heart Failure or Cardiovascular Death in Patients With Chronic Heart Failure (DAPA-HF)” at ClinicalTrials.gov
///////////DAPAGLIFLOZIN, ダパグリフロジン, BMS 512148, TYPE 2 DIABETES, SGLT-2 Inhibitors, EU 2012, forxiga, FDA 2014, JAPAN 2014, DIABETES
- Statement on a nonproprietory name adopted by the USAN council
- Efficacy and Safety of Dapagliflozin, Added to Therapy of Patients With Type 2 Diabetes With Inadequate Glycemic Control on Insulin, ClinicalTrials.gov, April 2009
- Trial Details for Trial MB102-020, Bristol-Myers Squibb, May 2009
- “FDA panel advises against approval of dapagliflozin”. 19 July 2011.
- Prous Science: Molecule of the Month November 2007
- UEndocrine: Internet Endocrinology Community
- Schubert-Zsilavecz, M, Wurglics, M, Neue Arzneimittel 2008/2009
- more1) Pal, Manojit et al; Improved Process for the preparation of SGLT2 inhibitor dapagliflozin via glycosylation of 5-bromo-2-Chloro-4′-ethoxydiphenylmethane with Gluconolactone ;. Indian Pat Appl,. 2010CH03942 , 19 Oct 20122) Lemaire, Sebastien et al; Stereoselective C-Glycosylation Reactions with Arylzinc Reagents ;
- Organic Letters , 2012, 14 (6), 1480-1483;3) Zhuo, Biqin and Xing, Xijuan; Process for preparation of Dapagliflozin amino acid cocrystals ;
- Faming Zhuanli Shenqing , 102 167 715, 31 Aug 20114) Shao, Hua et al; Total synthesis of SGLT2 inhibitor Dapagliflozin ;
- Hecheng Huaxue , 18 (3), 389-392; 20105) Liou, Jason et al; Processes for the preparation of C-Aryl glycoside amino acid complexes as potential SGLT2 Inhibitors ;. PCT Int Appl,.
- WO20100223136) Seed, Brian et al; Preparation of Deuterated benzyl-benzene glycosides having an inhibitory Effect on sodium-dependent glucose co-transporter; . PCT Int Appl,.
- WO20100092437) Song, Yanli et al; Preparation of benzylbenzene glycoside Derivatives as antidiabetic Agents ;. PCT Int Appl,.
- WO20090265378) Meng, Wei et al; D iscovery of Dapagliflozin: A Potent, Selective Renal Sodium-Dependent Glucose cotransporter 2 (SGLT2) Inhibitor for the Treatment of Type 2 Diabetes ;
- Journal of Medicinal chemistr y, 2008, 51 (5), 1145 -1149;9) Gougoutas, Jack Z. et al; Solvates Crystalline complexes of amino acid with (1S)-1 ,5-anhydro-LC (3 – ((phenyl) methyl) phenyl)-D-glucitol were prepared as for SGLT2 Inhibitors the treatment of Diabetes ;. PCT Int Appl,.
- WO200800282410) Deshpande, Prashant P. et al; Methods of producing C-Aryl glucoside SGLT2 Inhibitors ;..
- U.S. Pat Appl Publ,. 20,040,138,439
NEW DRUG APPROVALS
one time
$10.00
IIBR-100

IIBR-100
Brilife
Recombinant vesicular stomatitis virus (rVSV) vaccine
Israel Institute for Biological Research
Hadassah Medical Center; Sheba Medical Center Hospital
The SARS-CoV-2 virus is responsible for the COVID-19 pandemic. The pandemic emerged from Wuhan Province in China in December 2019 and was declared by the WHO Director-General a Public Health Emergency of International Concern on 30 January 2020.
In this study, a vaccine developed by IIBR for SARS-CoV-2 virus will be assessed for its safety and potential efficacy in volunteers. The study is comprised of two phases, a dose-escalation phase (phase I) during which subjects (18-55 years old) will be randomly allocated to receive a single administration of IIBR-100 100 at low, mid or high dose or saline or two administrations of IIBR-100 at low dose, or saline, 28 days apart.
Based on results obtained during phase I, and cumulative phase I data review, the expansion phase (phase II) has begun, during which larger cohorts as well as elderly age subjects will be randomly allocated to receive a single administration of IIBR-100 at low, mid or high dose or saline, or two administrations of IIBR-100 at low, mid or high dose (prime-boost) or saline, 28 days apart. Additional top-dose (prime-boost) may be implemented when immunogenicity of any prime-boost arm is considered insufficient.
Based on immunogenicity preliminary data and DSMB recommendations, the two administrations of mid, high and top dose (prime-boost) or saline will continue.
The subjects will be followed for a period of up to 12 months post last vaccine administration to assess the safety and efficacy of the vaccine.
https://clinicaltrials.gov/ct2/show/NCT04608305
IIBR-100 also known as Brilife is a COVID-19 vaccine candidate developed by The Israel Institute for Biological Research.[1][2]
References
- ^ Clinical trial number NCT04608305 for “Phase I/II Randomized, Multi-Center, Placebo-Controlled, Dose-Escalation Study to Evaluate the Safety, Immunogenicity and Potential Efficacy of an rVSV-SARS-CoV-2-S Vaccine (IIBR-100) in Adults” at ClinicalTrials.gov
- ^ Jeffay N (29 December 2020). “As Israel goes vaccine-wild, will the homegrown version lose its shot?”. The Times of Israel. Retrieved 1 January 2021.
candidate developed by The Israel Institute for Biological Research.[1][2]
Israeli institute’s COVID vaccine candidate said very effective in animal trials
Secretive Israeli research center’s shot shows near 100% efficacy in non-human trials, is on par with US company Moderna’s candidate, TV report says
Israeli researchers at a top secret research center have made progress on a coronavirus vaccine that shows a high level of effectiveness in animals, according to a Friday TV report.
However, there is no guarantee that the vaccine under development will be effective in humans, or will be available soon.
The Israel Institute for Biological Research (IIBR), a secretive unit that works under the Prime Minister’s Office, developed a vaccine that shows close to 100 percent protection against the virus in lab animals, the Channel 12 report said, citing “a security source.”
The vaccine under development is on par in effectiveness with a vaccine being developed by US biotechnology company Moderna, the report said.
Unlike vaccines developed abroad, the domestic vaccine will first be delivered to Israeli citizens, it added. If successful, it was expected to provide protection against the disease with a single dose.
The institute has not started human trials but was preparing to manufacture 10 to 15 million doses, report said.
Hebrew media have reported on potential breakthroughs at the shadowy institute several times before, starting in mid-March, with the Defense Ministry pushing back on some of the claims to tamper expectations.

Magen David Adom medical workers test Israelis for the coronavirus at a drive-through site in Lod, on July 10, 2020. (Yossi Aloni/Flash90)
IIBR said last month that it had completed successful coronavirus vaccine trials on rodents, paving the way for further testing on other animals and then possibly human trials.
In a paper published on the website of bioRxiv, an online repository for papers that haven’t yet been peer-reviewed, the institute, which is based in Ness Ziona, said it hopes to have a finished vaccine in a year, or possibly even earlier.
In the abstract of the report, the researchers say their vaccine, which they tested on hamsters, “results in rapid and potent induction of neutralizing antibodies against SARS-CoV-2,” the virus that causes COVID-19.
Earlier this month a vaccine adviser to the government cautioned that there was no guarantee that the shots being developed will prove widely effective.
In May, the institute confirmed that it had isolated an antibody it believed could be used to develop treatments against the virus. The development would not be useful in the creation of a vaccine, but would rather be a move toward a drug treatment for those who have already contracted the disease.
Tal Zaks, Moderna’s Israeli chief medical officer, described to Channel 12 on Friday the company’s push into Phase 3 testing of its vaccine candidate, which was developed with the National Institutes of Health, and began its first injections Monday.
The trial, the world’s largest vaccine study, plans to test the vaccine on 30,000 volunteers.
There’s still no guarantee that the experimental vaccine, developed by the National Institutes of Health and Moderna Inc., will really offer protection.
“The first time we saw the first model, that the vaccine, even if it’s just in mice, successfully stimulated the immune system to identify the virus and neutralize it, I knew that we hadn’t missed anything, that we had the correct vaccine,” he said.
“And of course the second ‘ah-ha’ moment was when we saw the first clinical results, when it was clear that in humans we weren’t just getting to antibody levels we were seeing in sick people, which is what we aspired to, but we were getting to even higher levels,” Zaks said.
A Nurse gives a volunteer an injection, as the world’s biggest study of a possible COVID-19 vaccine, developed by the US National Institutes of Health and Moderna Inc., gets underway on July 27, 2020, in Binghamton, NY. (AP Photo/Hans Pennink)
Last month Israel signed a deal with Moderna for the potential purchase of its coronavirus vaccine if it ends up proving effective.
Moderna said the vaccination was administered in Savannah, Georgia, the first site to get underway among more than seven dozen trial sites scattered around the country.
Several other vaccines made by China and by Britain’s Oxford University earlier this month began smaller final-stage tests in Brazil and other hard-hit countries.
The massive studies aren’t just to test if the shots work — they’re needed to check each potential vaccine’s safety. And following the same study rules will let scientists eventually compare all the shots.
It normally takes years to create a new vaccine from scratch, but scientists are setting speed records this time around, spurred by knowledge that vaccination is the world’s best hope against the pandemic.
If everything goes right with the final studies, it still will take months for the first data to trickle in from the Moderna test, followed by the Oxford one.
Governments around the world are trying to stockpile millions of doses of those leading candidates so if and when regulators approve one or more vaccines, immunizations can begin immediately. But the first available doses will be rationed, presumably reserved for people at highest risk from the virus.
Coronavirus cases in Israel rose by 1,791 in 24 hours on Friday and the national death toll hit 512, according to the latest Health Ministry figures.
The total case count stood at 70,970, with 320 patients in serious condition, including 98 on ventilators. The number of recovered patients reached 43,850.
Israel has the fifth-highest number of new coronavirus infections per capita in the world, overtaking the United States, according to data compiled by a scientific publication based at Oxford University.
And while Israel has seen the number of new coronavirus cases rocket to more than 2,000 a day in recent weeks, a new Hebrew University report published on Thursday asserted that Israel has managed to gain control of the second wave of the coronavirus, thanks to a recent stabilization in the number of seriously and moderately ill patients.
The curve for seriously and moderately ill patients began to spike in late June before stabilizing in recent days, the researchers reported. They credited the restrictions imposed by the government in recent weeks to limit crowding for helping to flatten the curve.
According to the report, the death toll will climb by roughly 200 in the coming three weeks as a result of the high infection rate over the past month.
Experts have blamed a too-speedy reopening and the lack of an effective contact-tracing program as main factors in the virus resurgence, which has come as new daily coronavirus cases around the world have also reached record highs.
| Vaccine description | |
|---|---|
| Target | SARS-CoV-2 |
| Vaccine type | Viral vector |
| Clinical data | |
| Other names | Brilife |
| Routes of administration | Intramuscular |
| Part of a series on the |
| COVID-19 pandemic |
|---|
| COVID-19 (disease)SARS-CoV-2 virus (variants) |
| showTimeline |
| showLocations |
| showInternational response |
| showMedical response |
| showImpact |
| COVID-19 portal |
//////IIBR-100, Brilife, COVID-19, vaccine, israel, corona virus, covid 19, SARS-CoV-2
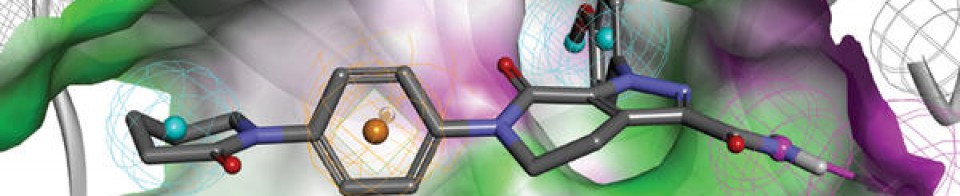
NEW DRUG APPROVALS
one time
$10.00
ABDALA, CIGB-66

ABDALA
CIGB-66, is a COVID-19 vaccine
Cuba says Abdala vaccine 92.28% effective against coronavirus
The announcement came just days after the government said another homegrown vaccine, Soberana 2, has proved to be 62% effective with just two of its three doses.
- June 22, 2021, 10:03 IST
Havana: Cuba said on Monday that its three-shot Abdala vaccine against the coronavirus has been proved 92.28% effective in last-stage clinical trials.
The announcement came just days after the government said another homegrown vaccine, Soberana 2, has proved to be 62% effective with just two of its three doses.
“Hit by the pandemic, our scientists at the Finlay Institute and Center for Genetic Engineering and Biotechnology have risen above all the obstacles and given us two very effective vaccines,” President Miguel Diaz-Canel tweeted.
The announcement came from state-run biopharmaceutical corporation BioCubaFarma, which oversees Finlay, the maker of Soberana 2, and the Center for Genetic Engineering and Biotechnology, the producer of Abdala.
Both vaccines are expected to be granted emergency authority by local regulators shortly.
Cuba, whose biotech sector has exported vaccines for decades, has five coronavirus vaccine candidates.
The Caribbean’s largest island is facing its worst Covid-19 outbreak since the start of the pandemic following the arrival of more contagious variants, setting new records for daily coronavirus cases.
The Communist-run country has opted not to import foreign vaccines but to rely on its own. Some experts said it was a risky bet but it appears to have paid off, putting Cuba in position to burnish its scientific reputation, generate much-needed hard currency through exports and strengthen the vaccination drive worldwide.
Several countries from Argentina and Jamaica to Mexico, Vietnam and Venezuela have expressed an interest in buying Cuba’s vaccines. Iran started producing Soberana 2 earlier this year as part of late-phase clinical trials.
Cuba’s authorities have already started administering the experimental vaccines en masse as part of “intervention studies” they hope will slow the spread of the virus.
About a million of the country’s 11.2 million residents have been fully vaccinated to date.
Daily cases have halved in the capital, Havana, since the start of the vaccination campaign a month ago, using Abdala, according to official data.
Cuba has reported a total of 169,365 Covid-19 cases and 1,170 deaths.
ABDALA, technical name CIGB-66, is a COVID-19 vaccine candidate developed by the Center for Genetic Engineering and Biotechnology in Cuba.[1][2] This vaccine candidate, named after a patriotic drama by Cuban independence hero José Martí, is a protein subunit vaccine containing COVID-derived proteins that trigger an immune response.[3] However, none of the clinical trial full results have been published. This candidate followed a previous one called CIGB-669 (MAMBISA).[4]
The vaccine is one of two Cuba-developed COVID-19 vaccines in Phase III trials.[5][6][7]
Clinical research
Phase I/II
In July 2020, CIGB-66 commenced phase I/II clinical trials.[8]
Phase III
The Phase III trial compares 3 doses of the vaccine administered at 0, 14 and 28 days against a placebo, with the primary outcome measuring the proportion of cases reported for each group 14 days after the third dose.
The trial was registered on 18 March 2021. The first dose was administered on 22 March and by April 4, the 48,000 participants had received their first dose,[9][10] and second doses started being administered from April 5.[11][12] Third doses have started being administered on 19 April[13][14][15] and on May 1, 97% of the original participants had received their 3 doses, the others 3% were lost in the process.
Intervention study
124,000 people aged 19 to 80 received 3 doses of the vaccine as part of an intervention study, with the primary outcome measuring the proportion of cases and deaths for the vaccinated compared to the unvaccinated population.[16]
A wider intervention study with the 1.7 million inhabitants of Havana is expected to start in May with the ABDALA and Soberana 2 vaccine.[17]
Efficacy
From May 3, the efficacy of the vaccine will start being evaluated.[18][19][20]
The “first evaluation of efficacy” can begin when there is 50 cases, then there is a second evaluation at 100 cases and a definitive efficacy can “finally be demonstrated” at 150 cases, Cuban Center for Genetic Engineering and Biotechnology director said.[21]
Production outside Cuba
Venezuela has claimed that it will manufacture the vaccine[22] but this claim has not yet materialised.[23] State-owned EspromedBIO will manufacture the vaccine but some “arrangements” are needed to start production.[24] In April, Nicolás Maduro said that a capacity of 2 Million doses per month is hoped to be reach by “August, September approximately”.[25
In June 2021, Vietnam’s Ministry of Health announced that negotiations were ongoing between Cuba and Vietnam for Abdala vaccine production. The Institute of Vaccines and Medical Biologicals (IVAC) was named as the focal point for receiving technology transfer.[26]
References
- ^ “ABDALA Clinical Study – Phase III”. rpcec.sld.cu. Registro Público Cubano de Ensayos Clínicos. Retrieved 22 March 2021.
- ^ “ABDALA Clinical Study”. rpcec.sld.cu. Registro Público Cubano de Ensayos Clínicos. Retrieved 22 March 2021.
- ^ Yaffe H (31 March 2021). “Cuba’s five COVID-19 vaccines: the full story on Soberana 01/02/Plus, Abdala, and Mambisa”. LSE Latin America and Caribbean blog. Retrieved 31 March 2021.
- ^ “MAMBISA Study”. rpcec.sld.cu. Registro Público Cubano de Ensayos Clínicos. Retrieved 22 March 2021.
- ^ “Three-shot Cuban COVID-19 vaccine candidate moves forward in phase III”. http://www.bioworld.com. Retrieved 10 April 2021.
- ^ “Cuba’s Abdala COVID-19 vaccine enters phase 3 clinical trial – Xinhua | English.news.cn”. http://www.xinhuanet.com. Retrieved 10 April 2021.
- ^ Zimmer C, Corum J, Wee SL. “Coronavirus Vaccine Tracker”. The New York Times. ISSN 0362-4331. Retrieved 10 April 2021.
- ^ “ABDALA Clinical Study”. rpcec.sld.cu. Registro Público Cubano de Ensayos Clínicos. Retrieved 21 March 2021.
- ^ BioCubaFarma (4 April 2021). “[Translated] “The application of the 1st dose of #Abdala, in volunteer 48 thousand, of the Phase III Clinical Trial. Next Monday, April 5, the application of the 2nd dose of this vaccine candidate begins. #VcaunasCubanasCovid19 .””. Twitter. Retrieved 10 April 2021.
- ^ “Covid Check-in: Cuba’s Homegrown Vaccines”. AS/COA. Retrieved 10 April 2021.
- ^ BioCubaFarma (5 April 2021). “[Translated] “The application of the 2nd dose of the vaccine candidates begins today #Abdala and #Soberana02 , as part of the 3rd phase of the clinical trial. Workers of @Emcomed1 in Havana and eastern provinces, from very early hours they carry out their distribution until the vaccination centers””. Twitter (in Spanish). Retrieved 10 April 2021.
- ^ “Two Cuban Vaccines Start Second Dose Phase III Trials”. Kawsachun News. 5 April 2021. Retrieved 10 April 2021.
- ^ “Abdala: Comienza tercera dosis en el Oriente cubano”. http://www.cuba.cu (in Spanish). Retrieved 21 April 2021.
- ^ BioCubaFarma. “[Translated] “Application of the 3rd dose of the vaccine candidate begins #Abdala in the provinces of Granma, Santiago de Cuba and Guantánamo. The application of the 2nd dose of #Soberana02 within the framework of the EC Phase III.#VacunasCubanasCovid19”. Twitter. Retrieved 21 April 2021.
- ^ Noticias, Agencia Cubana de. “Convergen múltiples voluntades para éxito de estudio Abdala en Bayamo”. ACN (in Spanish). Retrieved 21 April 2021.
- ^ “ABDALA-Intervention | Registro Público Cubano de Ensayos Clínicos”. rpcec.sld.cu. Retrieved 10 April 2021.
- ^ Ministerio de Salud Pública en Cuba. “Sitio oficial de gobierno del Ministerio de Salud Pública en Cuba”. Sitio oficial de gobierno del Ministerio de Salud Pública en Cuba (in Spanish). Retrieved 23 April 2021.
- ^ “Scientists announce Abdala’s administration of 3rd dose will finish”. http://www.plenglish.com/index.php?o=rn&id=66941&SEO=scientists-announce-abdalas-administration-of-3rd-dose-will-finish (in Spanish). Retrieved 2 May 2021.
- ^ Noticias, Agencia Cubana de. “Concluye aplicación de vacuna Abdala en Oriente de Cuba”. ACN (in Spanish). Retrieved 2 May2021.
- ^ “Cuba conclui ensaios clínicos de candidata a vacina contra covid-19”. R7.com (in Portuguese). 2 May 2021. Retrieved 2 May2021.
- ^ “Abdala cerca de concluir la fase III de ensayos clínicos; Mambisa se alista para avanzar a nueva fase (+Video)”. Granma.cu (in Spanish). Retrieved 3 May 2021.
- ^ “Cuba says it’s ‘betting it safe’ with its own Covid vaccine”. NBC News. Retrieved 10 April 2021.
- ^ “Maduro struggles to make his grand vaccine promise”. Eminetra.co.uk. 2 May 2021. Retrieved 3 May 2021.
- ^ “Venezuela producirá la vacuna cubana anticovid Abdala”. http://www.efe.com (in Spanish). Retrieved 3 May 2021.
- ^ Apr 11, Reuters /; 2021; Ist, 16:27. “Indonesian President orders Java rescue efforts after quake kills 8 – Times of India”. The Times of India. Retrieved 3 May 2021.
- ^ Ministry of Health Vietnam (16 June 2021). “Bộ trưởng Bộ Y tế đàm phán với Cuba về hợp tác sản xuất vaccine”. giadinh.net.vn(in Vietnamese). Retrieved 17 June 2021.
External links
| Scholia has a profile for Abdala (Q106390652). |
| Vaccine description | |
|---|---|
| Target | SARS-CoV-2 |
| Vaccine type | Protein subunit |
| Clinical data | |
| Other names | ABDALA |
| Routes of administration | Intramuscular |
| Part of a series on the |
| COVID-19 pandemic |
|---|
| COVID-19 (disease)SARS-CoV-2 virus (variants) |
| showTimeline |
| showLocations |
| showInternational response |
| showMedical response |
| showImpact |
| COVID-19 portal |
//ABDALA, CUBA, CIGB-66, COVID-19, vaccine, CORONA VIRUS, SARS-CoV-2,
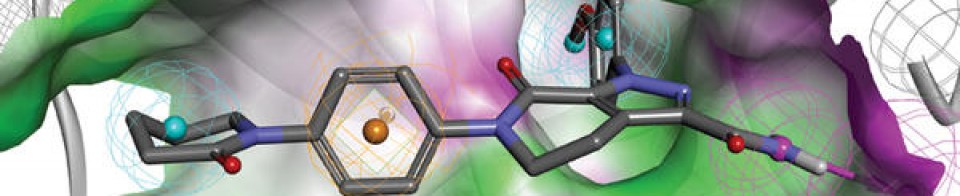
NEW DRUG APPROVALS
ONE TIME
$10.00

{kind=link}
{kind=link}
.svg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}