




Graphical abstract

PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
Home » sex arousal
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |


Bremelanotide is a compound that is currently under investigation for its potential uses in managing reperfusion injury, female sexual dysfunction or hemorrhagic shock. The chemical may also see success in managing modulate inflammation or limiting the effects of ischemia.
N-Acetyl-L-norleucyl-L-alpha-aspartyl-L-histidyl-D-phenylalanyl-L-arginyl-L-tryptophyl-L-lysine (2-7)-lactam
Bremelanotide, PT 141, CAS NO.: 189691-06-3
Bremelanotide ![]() i/ˌbrɛmɨˈlænətaɪd/ (formerly PT-141) is a compound under drug development by Palatin Technologies as a treatment for female sexual dysfunction, hemorrhagic shock and reperfusion injury. It functions by activating the melanocortin receptors MC1R and MC4R, to modulate inflammation and limiting ischemia.[2] It was originally tested for intranasal administration in treating female sexual dysfunction but this application was temporarily discontinued in 2008 after concerns were raised over adverse side effects of increased blood pressure. As of December 2014, Palatin is conducting a human Phase 3 study[3] using a subcutaneous drug delivery system that appears to have little effect on blood pressure.
i/ˌbrɛmɨˈlænətaɪd/ (formerly PT-141) is a compound under drug development by Palatin Technologies as a treatment for female sexual dysfunction, hemorrhagic shock and reperfusion injury. It functions by activating the melanocortin receptors MC1R and MC4R, to modulate inflammation and limiting ischemia.[2] It was originally tested for intranasal administration in treating female sexual dysfunction but this application was temporarily discontinued in 2008 after concerns were raised over adverse side effects of increased blood pressure. As of December 2014, Palatin is conducting a human Phase 3 study[3] using a subcutaneous drug delivery system that appears to have little effect on blood pressure.
Palatin, in collaboration with European licensee Gedeon Richter, is developing an sc formulation of the synthetic peptide bremelanotide (PT-141; BMT), a melanocortin MCR-4 agonist and a synthetically modified analog of PT-14, also analogous to alpha-melanocyte-stimulating hormone (alpha-MSH), for the potential treatment of female sexual dysfunction (FSD) including hypoactive sexual desire disorder (HSDD)

The Bremelanotide or PT-141 is a mean that explains the revolution caused by the medical world in a silent but attractive manner in the human health related study. Bremelanotide is the latest arrival from the company called Palatin Technologies which forms the basic treatment for the hemorrhagic shock and reperfusion injury.( In short about the company, the Palatin Technologies is the owner of this research and is located in New Jersey. Hence this medicine is a Jersey based Product. And regarding the product under research, is waiting for the approval from the Food and Drug Association. Once this is done, the company has targeted to reach those customers, whom the Viagra has approached. This has the effect of helping the male patients suffering with an erectile dysfunction syndrome. Also if it gets the approval as a treatment measure for the female sexual dysfunction, then this medicine is expected to bring a relief to the post-menopausal and also supports or provides their sexual happiness and also they are checking regarding thehyposexual desire disorder. This is expected to be a blockbuster, if released. So this medicine is waiting for a confirmation as well as an approval.
In February 2015, a randomized, double-blind, placebo-controlled, open-label extension, phase III trial (NCT02338960; BMT-302, Reconnect Study) was initiated in the US in premenopausal women (expected n = 550) with hypoactive sexual desire disorder to evaluate the efficacy and safety of bremelanotide. At that time, the trial was expected to complete in July 2017
Study – Potential Use Erectile Dysfunction
One study has explored the potential use of bremelanotide as a replacement for natural peptide melanocyte stimulating hormones for the sake of treating erectile dysfunction.
Additional study is necessary to determine the extent of the effects bremelanotide has on the brain and natural stimulating chemicals.


This is an advanced research involved even now. This functions by activating the Melanocortin, which is a group of peptide hormones which includes the adrenocorticotropic hormone and also the different forms of the melanocyte stimulating hormones. These melanocortins are produced or prepared from the proopiomelanocortin in the pituitary glands. The melanocortin releases or exert their effects by making a bind with the melanocortin and thereby activating it).The Bremelanotide functions by activating the melanocortin receptors and thereby makes a modulation in the inflammation. This is actually produced for making use in treating the sexual dysfunction. Due to certain reasons; the process of researching was kept under hold in recently, since it created some adverse side effects of increased blood pressure. In the chemistry of the preparation of the bremelanotide, the Peptide Melanaton II forms the basic compound. This compound is tested using a sunless tanning agent.
The actual information about the peptide melanaton has the effect of making sexual arousal and speed as well as sudden erections and some other side effects. However, there are several other measures taken to test the property of the same under several other health situations to make a detailed study about the chemical compound structure to make a change in the combination of the chemical structure. This medicine has made a revolution in the field of science of the human structure. When made a deep verification of the compound structure of the chemical study showed the following information. The structural design has an appearance of white colored powder like material, which has an accurate purity of nearly 98%. The actual molecular weight of the compound formed is around 1025.2. This compound has the collective share of Amino acids in the composition, peptide and acetate contents also.
The study of the compound structure PT-141 has an enhanced support of making a recombination that produces a different profile of the same medicine but in a different standard with different properties that may support the human requirement.
Bremelanotide PT-141 is known for its aphrodisiac properties
![]()
Bremelanotide was developed from the peptide hormone Melanotan II which underwent testing as a sunless tanningagent. In initial testing, Melanotan II did induce tanning but additionally caused sexual arousal and spontaneous erections as unexpected side effects in nine out of the ten original male volunteer test subjects.[4]
In studies, bremelanotide was shown to induce lordosis in an animal model[5] and was also effective in treating sexual dysfunction in both men (erectile dysfunction or impotence) and women (sexual arousal disorder). Unlike Viagra and other related medications, it does not act upon the vascular system, but directly increases sexual desire via the nervous system.[6]
A Phase III clinical trial was scheduled to begin in the first half of 2007, but was delayed until August 2007. On August 30, Palatin announced that the U.S. Food and Drug Administration had expressed serious concerns regarding therisk/benefit ratio of bremelanotide with regards to the side effect of increased blood pressure. The FDA stated that it would consider alternate uses for bremelanotide, including as a treatment for individuals who do not respond to more established ED treatments. However, On May 13, 2008, Palatin Technologies announced it had “discontinued development of Bremelanotide for the treatment of male and female sexual dysfunction” while concurrently announcing plans to develop it as a treatment for hemorrhagic shock instead.[7] The company additionally announced intentions to focus its attention on another compound, PL-6983, that causes lower blood pressure in animal models.[8]Palatin has since re-initiated Bremelanotide studies for ED and FSD using a subcutaneous delivery method. On August 12, 2009, the company announced that in a double-blind study of 54 volunteers bremelanotide failed to evoke the hypertensive side effects seen with the nasal delivery system used in prior studies, concluding that “variability of uptake” inherent in intranasal administration of the drug resulted in “increases in blood pressure and gastrointestinal events…primarily related to high plasma levels in [only] a subset of patients” and that subcutaneous administration of the drug circumvented the potential for this side effect.[8] Palatin has completed a human Phase 2B study utilizing subcutaneous administration and reported positive results.[9]

Bremelanotide is a cyclic hepta-peptide lactam analog of alpha-melanocyte-stimulating hormone (alpha-MSH) that activates the melanocortin receptors MC3-R and MC4-R in thecentral nervous system. It has the amino acid sequence Ac-Nle-cyclo[Asp-His-D-Phe-Arg-Trp-Lys]-OH or cyclo-[Nle4, Asp5, D-Phe7, Lys10]alpha-MSH-(4-10). It is a metabolite of Melanotan II that lacks the C-terminal amide function.
 |
|
| Systematic (IUPAC) name | |
|---|---|
| (3S,6S,9R,12S,15S,23S)-15-[(N-acetyl-L-norleucyl)amino]-9-benzyl-6-{3-[(diaminomethylidene)amino]propyl}-12-(1H-imidazol-5-ylmethyl)-3-(1H-indol-3-ylmethyl)-2,5,8,11,14,17-hexaoxo-1,4,7,10,13,18-hexaa zacyclotricosane-23-carboxylic acid | |
| Clinical data | |
| Legal status |
|
| Pharmacokinetic data | |
| Half-life | 120 minutes[1] |
| Identifiers | |
| CAS number | 189691-06-3 |
| ATC code | None |
| PubChem | CID 9941379 |
| ChemSpider | 8116997 |
| UNII | 6Y24O4F92S |
| KEGG | D06569 |
| ChEMBL | CHEMBL2070241 |
| Chemical data | |
| Formula | C50H68N14O10 |
| Molecular mass | 1025.2 g/mol |
Sexual dysfunction, including both penile erectile dysfunction or impotence and female sexual dysfunction, are common medical problems. Significant effort has been devoted over the last twenty or more years to develop methods, devices and compounds for treatment of sexual dysfunction. While more effort has been undertaken for treatment of penile erectile dysfunction, female sexual dysfunction is also an area to which significant research and effort has been devoted.
At present, one commonly used orally administered drug for treatment of sexual dysfunction in the male is Viagra®, a brand of sildenafil, which is a phosphodiesterase 5 inhibitor, increasing the persistence of cyclic guanosine monophosphate and thereby enhancing erectile response. There are several other medical treatment alternatives currently available depending on the nature and cause of the impotence problem. Some men have abnormally low levels of the male hormone testosterone, and treatment with testosterone injections or pills may be beneficial. However, comparatively few impotent men have low testosterone levels. For many forms of erectile dysfunction, treatment may be undertaken with drugs injected directly into the penis, including drugs such as papaverin, prostaglandin E1, phenoxybenzamine or phentolamine. These all work primarily by dilating the arterial blood vessels and decreasing the venous drainage. Urethral inserts, such as with suppositories containing prostaglandin, may also be employed. In addition, a variety of mechanical aids are employed, including constriction devices and penile implants.
A variety of treatments have also been explored for female sexual dysfunction, including use of sildenafil, although the Food and Drug Administration has not specifically approved such use. Testosterone propionate has also been employed to increase or augment female libido.
Melanocortin receptor-specific compounds have been explored for use of treatment of sexual dysfunction. In one report, a cyclic α-melanocyte-stimulating hormone (“α-MSH”) analog, called Melanotan-II, was evaluated for erectogenic properties for treatment of men with psychogenic erectile dysfunction. Wessells H. et al., J Urology 160:389-393 (1998); see also U.S. Pat. No. 5,576,290, issued Nov. 19, 1996 to M. E. Hadley, entitled Compositions and Methods for the Diagnosis and Treatment of Psychogenic Erectile Dysfunction and U.S. Pat. No. 6,051,555, issued Apr. 18, 2000, also to M. E. Hadley, entitled Stimulating Sexual Response in Females. The peptides used in U.S. Pat. Nos. 5,576,290 and 6,051,555 are also described in U.S. Pat. No. 5,674,839, issued Oct. 7, 1997, to V. J. Hruby, M. E. Hadley and F. Al-Obeidi, entitled Cyclic Analogs of Alpha–MSH Fragments, and in U.S. Pat. No. 5,714,576, issued Feb. 3, 1998, to V. J. Hruby, M. E. Hadley and F. Al-Obeidi, entitled Linear Analogs of Alpha–MSH Fragments. Melanotan-II is a peptide of the following formula:

Additional related peptides are disclosed in U.S. Pat. Nos. 5,576,290, 5,674,839, 5,714,576 and 6,051,555. These peptides are described as being useful for both the diagnosis and treatment of psychogenic sexual dysfunction in males and females. These peptides are related to the structure of melanocortins.
In use of Melanotan-II, significant erectile responses were observed, with 8 of 10 treated men developing clinically apparent erections, and with a mean duration of tip rigidity greater than 80% for 38 minutes with Melanotan-II compared to 3.0 minutes with a placebo (p=0.0045). The drug was administered by subcutaneous abdominal wall injection, at doses ranging from 0.025 to 0.157 mg/kg body weight. Transient side effects were observed, including nausea, stretching and yawning, and decreased appetite.
The minimum peptide fragment of native α-MSH needed for erectile response is the central tetrapeptide sequence, His6-Phe7-Arg8-Trp9 (SEQ ID NO:1). In general, all melanocortin peptides share the same active core sequence, His-Phe-Arg-Trp (SEQ ID NO:1), including melanotropin neuropeptides and adrenocorticotropin. Five distinct melanocortin receptor subtypes have been identified, called MC1-R through MC5-R, and of these MC3-R and MC4-R are believed to be expressed in the human brain. MC3-R has the highest expression in the arcuate nucleus of the hypothalamus, while MC4-R is more widely expressed in the thalamus, hypothalamus and hippocampus. A central nervous system mechanism for melanocortins in the induction of penile erection has been suggested by experiments demonstrating penile erection resulting from central intracerebroventricular administration of melanocortins in rats. While the mechanism of His-Phe-Arg-Trp (SEQ ID NO:1) induction of erectile response has not been fully elucidated, it has been hypothesized that it involves the central nervous system, and probably binding to MC3-R and/or MC4-R.
Other peptides and constructs have been proposed which are ligands that alter or regulate the activity of one or more melanocortin receptors. For example, International Patent Application No. PCT/US99/09216, entitled Isoquinoline Compound Melanocortin Receptor Ligands and Methods of Using Same, discloses two compounds that induce penile erections in rats. However, these compounds were administered by injection at doses of 1.8 mg/kg and 3.6 mg/kg, respectively, and at least one compound resulted in observable side effects, including yawning and stretching. Other melanocortin receptor-specific compounds with claimed application for treatment of sexual dysfunction are disclosed in International Patent Application No. PCT/US99/13252, entitled Spiropiperidine Derivatives as Melanocortin Receptor Agonists.
Both cyclic and linear α-MSH peptides have been studied; however, the peptides heretofore evaluated have had an amide or —NH2 group at the carboxyl terminus. See, for example, Wessells H. et al., J Urology, cited above; Haskell-Luevano C. et al., J Med Chem 40:2133-39 (1997); Schiöth H. B. et al., Brit J Pharmacol 124:75-82 (1998); Schiöth H. B. et al., Eur J Pharmacol 349:359-66 (1998); Hadley M. E. et al., Pigment Cell Res 9:213-34 (1996); Bednarek M. A. et al., Peptides20:401-09 (1999); U.S. Pat. Nos. 6,054,556, 6,051,555 and 5,576,290; and, International Patent Applications PCT/US99/04111 and PCT/US98/03298. While significant research has been conducted in an effort to determine the optimal structure of α-MSH peptides, including a variety of structure-function, agonist-antagonist, molecular modeling and pharmacophore studies, such studies have relied upon peptides with an art conventional —NH2 group at the carboxyl terminus. Further, it has long been believed that biologically active neuropeptides, including α-MSH peptides, are amidated, with an —NH2 group at the carboxyl terminus, and that such amidation is required both for biological activity and stability. See, for example, Metabolism of Brain Peptides, Ed. G. O’Cuinn, CRC Press, New York, 1995, pp. 1-9 and 99-101.
…………………………………………….
Bioorganic and Medicinal Chemistry Letters, 2005 , vol. 15, 4 pg. 1065 – 1068
http://www.sciencedirect.com/science/article/pii/S0960894X04014842

Figure 2.
NMR structural analysis on compound 3.

Figure 4.
NMR structural analysis of compound 1.
……………………………………………….
In a preferred embodiment, the invention provides the peptide
Ac-Nle-cyclo(-Asp-His-D-Phe-Arg-Trp-Lys)-OH Compound 1
The peptide of Compound 1 has a formula of C50H68N14O10, and a net molecular weight of 1025.18. This peptide may be synthesized by solid-phase means and purified to greater than 96% purity by HPLC, yielding a white powder that is a clear, colorless solution in water. The structure of Compound 1 is:

In general, the peptide compounds of this invention may be synthesized by solid-phase synthesis and purified according to methods known in the art. Any of a number of well-known procedures utilizing a variety of resins and reagents may be used to prepare the compounds of this invention.
The peptides of this invention may be in the form of any pharmaceutically acceptable salt. Acid addition salts of the compounds of this invention are prepared in a suitable solvent from the peptide and an excess of an acid, such as hydrochloric, hydrobromic, sulfuric, phosphoric, acetic, trifluoroacetic, maleic, succinic or methanesulfonic. The acetate salt form is especially useful. Where the compounds of this invention include an acidic moiety, suitable pharmaceutically acceptable salts may include alkali metal salts, such as sodium or potassium salts, or alkaline earth metal salts, such as calcium or magnesium salts.
The invention provides a pharmaceutical composition that includes a peptide of this invention and a pharmaceutically acceptable carrier. The carrier may be a liquid formulation, and is preferably a buffered, isotonic, aqueous solution. Pharmaceutically acceptable carriers also include excipients, such as diluents, carriers and the like, and additives, such as stabilizing agents, preservatives, solubilizing agents, buffers and the like, as hereafter described.
EXAMPLE 1
Peptide Synthesis
The peptide Ac-Nle-cyclo(-Asp-His-D-Phe-Arg-Trp-Lys)-OH was synthesized by standard solid phase peptide synthesis methods, and is a cyclic heptapeptide melanocortin peptide analog with a free acid at the carboxyl terminus and an acetylated amino group at the amino terminus, with the structure:

The peptide has a net molecular weight of 1025.18, and is supplied in an acetate salt form. The peptide is a white, odorless amorphous hygroscopic powder, soluble in 0.9% saline, composed of C50H68N14O10. For synthesis, an Fmoc-Lys(R3)-p-alkoxybenzyl alcohol resin was transferred to a solid phase peptide synthesizer reactor with a mechanical stirrer. The R3group, such as 1-(1′-adamantyl)-1-methyl-ethoxycarbonyl (Adpoc), allyloxycarbonyl (Aloc) or 4-methyltrityl (Mtt), was removed and the next Fmoc-protected amino acid (Fmoc-Trp(Boc)-OH) was added to the resin through standard coupling procedures. The Fmoc protective group was removed and the remaining amino acids added individually in the correct sequence, by repeating coupling and deprotection procedures until the amino acid sequence was completed. After completion of coupling with the last Fmoc-amino acid derivative, Fmoc-Nle-OH, and cleavage of the Fmoc protective group, the exposed terminal amino group was acetylated with acetic anhydride and pyridine in N,N-dimethylformamide (DMF). The peptide-resin was dried and the Lys and Asp protective groups cleaved. The Lys and Asp deprotected peptide resin was suspended in a suitable solvent, such as DMF, dichloromethane (DCM) or 1-methyl-2-pyrrolidone (NMP), a suitable cyclic coupling reagent, such as 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium tetrafluoroborate (TBTU), 2-(7-aza-1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium tetrafluoroborate (TATU), 2-(2-oxo-1(2H)-pyridyl)-1,1,3,3-tetramethyluronium tetrafluoroborate (TBTU) or N,N′-dicyclohexylcarbodiimide/1-hydroxybenzotriazole (DCCl/HOBt) was added, and coupling initiated by use of a suitable base, such as N,N-diispropylethylamine (DIPEA), sym-collidine or N-methylmorpholine (NMM). After cyclization, the peptide-resin was washed and the peptide cleaved from the resin and any remaining protective groups using trifluoroacetic acid (TFA) in the presence of water and 1,2-ethanedithiol (EDT). The final product was precipitated by adding cold ether and collected by filtration. Final purification was by reversed phase HPLC using a C18 column. The purified peptide was converted to acetate salt by passage through an ion-exchange column.
…………………………………………..

Compounds of the Invention.
in a preferred embodiment of the present invention, fie rneianocortin receptor agonist is;
Ac-Nie”Cyc/o{-Asp-His–D–Phe-Arg–Trp»Lys)–OH (bremeianotide)
The peptide of bremeianotide has a formula of CsaHesN< C½, and a net mofecufar weight of 1025.18, This peptide may be synthesized by conventional means, including either solid-phase or Squid-phase techniques, and purified to greater than 99% purity by HPLC, yielding a white powder that is a clear, colorless solution in water. The structure of bremeianotide is:

Bremeianotide may be in the form of any pharmaceutically acceptable salt. Acid addition salts of the compounds of this invention are prepared in a suitable solvent from the peptide and an excess of an acid, such as hydrochloric, hydrobromic, sulfuric, phosphoric, acetic, trifluoroacefie, maieic, citric, tartaric, oxalic, succinic or methanesu!fonic acid. The acetate salt form is especially useful.
in a preferred embodiment, bremelanotide is an acetate salt form, and is formulated in a buffered aqueous solution including giycerin, and prepackaged in a syringe and auto-injector device. In alternative embodiments, bremelanotide is any pharmaceutically acceptable salt form, and is formulated in any pharmaceutically acceptable aqueous solution, the aqueous solution optionally including one or more salts, such as sodium chloride, one or more acids, such as citric acid, and one or more additional ingredients, including cellulose or derivatives thereof, saccharides o
polysaccharides such as dextrose, and any of a wide variety of surfactants, chelating agents and preservatives.
………………………………………….
In yet another embodiment of the present invention, the melanocortin receptor agonist is:
Ac–Nle-cyclo(-Asp–His–D–Phe–Arg–Trp–Lys)-OH PT-141
The peptide of PT-141 has a formula of C50H68N14O10, and a net molecular weight of 1025.18. This peptide may be synthesized by conventional means, including either solid-phase or liquid-phase techniques, and purified to greater than 99% purity by HPLC, yielding a white powder that is a clear, colorless solution in water. The structure of PT-141 is:

In one embodiment of the invention, PT-141 is synthesized by solid-phase synthesis and purified according to methods known in the art. Any of a number of well-known procedures utilizing a variety of resins and reagents may be used to prepare PT-141.
PT-141 may be in the form of any pharmaceutically acceptable salt. Acid addition salts of the compounds of this invention are prepared in a suitable solvent from the peptide and an excess of an acid, such as hydrochloric, hydrobromic, sulfuric, phosphoric, acetic, trifluoroacetic, maleic, citric, tartaric, oxalic, succinic or methanesulfonic acid. The acetate salt form is especially useful. Where the compounds of this invention include an acidic moiety, suitable pharmaceutically acceptable salts may include alkali metal salts, such as sodium or potassium salts, or alkaline earth metal salts, such as calcium or magnesium salts.
In a preferred embodiment, PT-141 is an acetate salt form, and is formulated in a buffered aqueous solution including glycerin, prepackaged in a metered unit dose intranasal delivery device. In alternative embodiments, PT-141 is any pharmaceutically acceptable salt form, and is formulated in any pharmaceutically acceptable aqueous solution, the aqueous solution optionally including one or more salts, such as sodium chloride, one or more acids, such as citric acid, and one or more additional ingredients, including cellulose or derivatives thereof, saccharides or polysaccharides such as dextrose, and any of a wide variety of surfactants, chelating agents and preservatives. In one preferred embodiment, PT-141 is administered to patients in volumes of 100 μL, with the quantity of PT-141 delivered determined by the concentration thereof. As described hereafter, in one preferred embodiment a metered unit dose contains 7.5 mg of PT-141.
While certain embodiments of the present invention are described primarily in the context of PT-141, it is to be understood that other melanocortin receptor agonists may be employed. For example, the metallopeptide melanocortin receptor agonists disclosed in WO 02/064091, filed on Feb. 13, 2001, and U.S. Ser. No. 10/640,755, filed on Aug. 13, 2003, both entitled Melanocortin Metallopeptides for Treatment of Sexual Dysfunction; and WO 01/13112, filed on Jun. 14, 2000, entitled Melanocortin Metallopeptide Constructs, Combinatorial Libraries and Applications, may be employed. In addition, the peptidomimetic melanocortin receptor agonists disclosed in U.S. Ser. No. 10/776,419, filed on Feb. 10, 2004, entitled Peptidomimetics of Biologically Active Metallopeptides; the pyrrolidine melanocortin receptor agonists disclosed in U.S. Ser. No. 10/766,657, filed on Feb. 10, 2004, entitled Pyrrolidine Melanocortin-Specific Compounds; and the bicyclic melanocortin receptor agonists disclosed in PCT/US04/01505, filed on Jan. 20, 2004, entitled Bicyclic Melanocortin-Specific Compounds, may also be employed. Also particular preferred are the piperazine melanocortin agonists disclosed in PCT/US04/01462, filed on Jan. 20, 2004 and U.S. Ser. No. 10/762,079, filed on Jan. 20, 2004, both entitled piperazine Melanocortin-Specific Compounds; the melanocortin agonists disclosed in WO 03/006620, filed on Jul. 11, 2002, entitled Linear and Cyclic Melanocortin Receptor-Specific Peptides; WO 04/005324, filed on Jul. 9, 2003, entitled Peptide Compositions for Treatment of Sexual Dysfunction; PCT/US00/18217, filed on Jun. 29, 2000 and U.S. Ser. No. 10/040,547, filed on Jan. 4, 2002, entitled Compositions and Methods for Treatment of Sexual Dysfunction; and U.S. Ser. No. 10/638,071, filed on Aug. 8, 2003, entitled Cyclic Peptide Compositions and Methods for Treatment of Sexual Dysfunction. The entire disclosure of each of the foregoing are incorporated here by reference. It is to be understood that the foregoing listing of patent applications disclosing melanocortin receptor agonists is intended to only be exemplary, and that other melanocortin receptor agonists, whether heretofore known or hereafter developed, may similarly be used in the practice of this invention.
…………………….
NMR prediction


| PALATIN TECHNOLOGIES, INC.: ‘Bremelanotide in Premenopausal Women With Female Sexual Arousal Disorder and/or Hypoactive Sexual Desire Disorder‘ CLINICALTRIALS.GOV (NCT01382719, [Online] 20 March 2012, page 1 Retrieved from the Internet: <URL:http://clinicaltrials.gov/archive/NCT0 1382719/ 2012-03 20> [retrieved on 2014-02-10] | ||
| 2 | * | PALATIN TECHNOLOGIES, INC.: ‘Reports Positive Bremelanotide Study; Improved Safety Profile with Subcutaneous Administration‘ PR NEWSWIRE., [Online] 12 August 2009, Retrieved from the Internet: <URL:http://www.thefreelibrary.com/Palatin +Technolo9ies,+Inc.+Reports+Positive+Bremel anotide+Study%38…-a020561 3302> [retrieved on 2014-02-10] |
| 3 | * | SAFARINEJAD, MR.: ‘Evaluation of the Safety and Efficacy of Bremelanotide, a Melanocortin Receptor Agonist, in Female Subjects with Arousal Disorder: A Double-Blind Placebo-Controlled, Fixed Dose, Randomized Study”.‘ INTERNATIONAL SOCIETY FOR SEXUAL MEDICINE. vol. 5, 2008, pages 887 – 897 |
| US8455617 | Jun 7, 2010 | Jun 4, 2013 | Astrazeneca Ab | Melanocortin receptor-specific peptides |
| US8455618 | Oct 26, 2011 | Jun 4, 2013 | Astrazeneca Ab | Melanocortin receptor-specific peptides |
| US8487073 | Nov 23, 2010 | Jul 16, 2013 | Palatin Technologies, Inc. | Melanocortin receptor-specific peptides for treatment of sexual dysfunction |
| US8729224 | Jun 5, 2013 | May 20, 2014 | Palatin Technologies, Inc. | Melanocortin receptor-specific peptides for treatment of female sexual dysfunction |
| EP2266567A1 | May 26, 2009 | Dec 29, 2010 | Æterna Zentaris GmbH | Use of cetrorelix in combination with PDE V inhibitors for the treatment of sex hormone dependent disorders |
| EP2266568A1 | May 26, 2009 | Dec 29, 2010 | Æterna Zentaris GmbH | Use of LHRH antagonists in combination with PDE V inhibitors for the treatment of sex hormone dependent disorders |
| WO2013067309A1 | Nov 2, 2012 | May 10, 2013 | Xion Pharmaceutical Corporation | Methods and compositions for oral administration of melanocortin receptor agonist compounds |
| WO2014071339A2 * | Nov 5, 2013 | May 8, 2014 | Palatin Technologies, Inc. | Uses of bremelanotide in therapy for female sexual dysfunction |
| WO2009151714A2 * | Mar 24, 2009 | Dec 17, 2009 | Palatin Technologies, Inc. | Therapeutic for treatment of circulatory shock, ischemia, inflammatory disease and related conditions |
| US6794489 * | Jan 4, 2002 | Sep 21, 2004 | Palatin Technologies, Inc. | Peptide sequence ac-nle-cyclo(-asp-his-d-phe-arg-trp-lys)-oh derived from a melanocyte-stimulating hormone (? alpha -msh?) analog, called melanotan-ii |
| US20050222014 * | May 26, 2005 | Oct 6, 2005 | Palatin Technologies, Inc. | Administering phosphodiestarase inhibitors and melanocortin receptor antagonist: synergistic mixture |
| US20110065652 * | Nov 23, 2010 | Mar 17, 2011 | Palatin Technologies, Inc. | Melanocortin Receptor-Specific Peptides for Treatment of Sexual Dysfunction |
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE

 Googleplus
Googleplus
 Location in Madhya Pradesh
Location in Madhya Pradesh
en.wikipedia.org/wiki/Khajuraho_Group_of_Monuments
The Khajuraho Group of Monuments are a group of Hindu and Jain temples in Madhya Pradesh, India. About 620 kilometres (385 mi) southeast of New Delhi, …








Hotel Chandela – A Taj Leisure Hotel


LATUR, MAHARASHTRA, INDIA
http://en.wikipedia.org/wiki/Latur
| Latur लातूर Lattalur, Ratnapur |
|
|---|---|
| City | |
|
Latur
Location in Maharashtra, India |
|
| Coordinates: 18.40°N 76.56°ECoordinates: 18.40°N 76.56°E | |
| Country | |
| State | Maharashtra |
| Region | Aurangabad Division |
| District | Latur |
| Settled | Possibly 7th century AD |
| Government | |
| • Body | Latur Municipal Corporation |
| • Mayor | Akhtar Shaikh |
| Area[1] | |
| • Total | 117.78 km2(45.48 sq mi) |
| Area rank | 89 |
| Elevation | 515 m (1,690 ft) |
| Population (2011) | |
| • Total | 382,754 |
| • Rank | 89th |
| • Density | 3,200/km2(8,400/sq mi) |
| Demonym | Laturkar |
| Languages | |
| • Official | Marathi |
| Time zone | IST (UTC+5:30) |
| PIN |
|
| Telephone code | 91-2382 |
| Vehicle registration | MH-24 |
| Sex ratio | 923.54 ♀/1000 ♂ |
| Literacy | 89.67 |
| Distance from Mumbai | 497 kilometres (309 mi) E (land) |
| Distance fromHyderabad | 337 kilometres (209 mi) NW (land) |
| Distance fromAurangabad, Maharashtra | 294 kilometres (183 mi) SE (land) |
| Climate | BSh (Köppen) |
| Precipitation | 666 millimetres (26.2 in) |
| Avg. summer temperature | 41 °C (106 °F) |
| Avg. winter temperature | 13 °C (55 °F) |
| http://www.citypopulation.de/world/Agglomerations.html | |



his Is The Famous ‘Ganj-Golai’ As The Central Place Of The Latur City. There Are 16 Roads Connecting To This Place And Seperate Markets i.e. Jewellers …

लातूर जिल्हयातील चित्र संग्रह
LATUR AIRPORT
 LATUR AIRPORT
LATUR AIRPORT


2012 Navratri Mahotsav in Latur

SOS Children’s Village Latur


Latur, India: Carnival Resort


Ausa Near Latur

Chakur near Latur

Vilasrao Deshmukh’s ancestral home at Babhalgaon village in Latur. Machindra Amle



UDGIR: Udgir is one of the most important towns of Latur district. Udgir has a great historical significance. It has witnessed the war between the Marathas …
 The city of Latur is located in India’s welathiest state, Maharashtra. Together with many of the surrounding villages, Latur was all but destroyed in the
The city of Latur is located in India’s welathiest state, Maharashtra. Together with many of the surrounding villages, Latur was all but destroyed in the

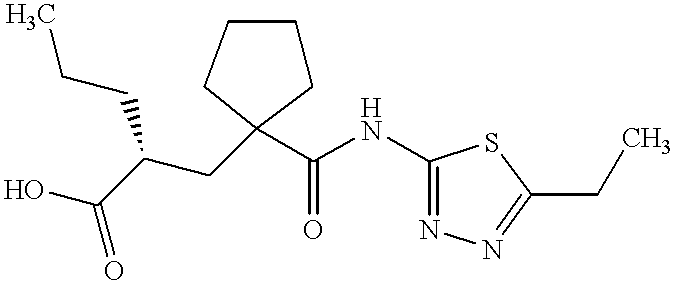
UK-414,495
Molecular Formula: C16H25N3O3S
Molecular Weight: 339.453
UK 414495
CAS 388630-36-2
OF
(-)-(2R)-2-[[1-[[(5-Ethyl-1,3,4-thiadiazol-2-yl)amino]carbonyl]cyclopentyl]methyl]pentanoic acid;
AND
Cyclopentanepropanoic acid, 1-[[(5-ethyl-1,3,4-thiadiazol-2-yl)amino]carbonyl]-α-propyl-, (αR)-
((R)-2-({1-[(5-ethyl-1,3,4-thiadiazol-2-yl) carbamoyl]cyclopentyl}methyl) valeric acid)
(2R)-2-[(1-{[(5-Ethyl-1,3,4-thiadiazol-2-yl)amino]carbonyl}cyclopentyl) methyl]pentanoic acid
…………………………………………………
Cas 337962-93-3 RACEMIC…………2-[[1-[[(5-Ethyl-1,3,4-thiadiazol-2-yl)amino]carbonyl]cyclopentyl]methyl]pentanoic acid

…………………………………………………………………..
ITS ENANTIOMER
(+)-(2S)-2-[[1-[[(5-Ethyl-1,3,4-thiadiazol-2-yl)amino]carbonyl]cyclopentyl]methyl]pentanoic acid……………337962-74-0
CAS SUMMARY
| Cas number | 388630-36-2 337962-74-0 (enantiomer) 337962-93-3 (racemate) 388630-59-9 (sodium salt) |

 desired
desired
UK-414,495 is a drug developed by Pfizer for the treatment of female sexual arousal disorder.[1] UK-414,495 acts as a potent, selective inhibitor of the enzyme neutral endopeptidase, which normally serves to break down the neuropeptide VIP. The consequent increase in VIP activity alters blood flow to the genital region leading to increased lubrication and muscle relaxation.[2][3][4]

A female equivalent of Viagra could soon be available to help women increase their sexual arousal, scientists claim.
For years they have endeavoured to create an alternative for women that mimics the effects of the male Viagra pill.
Now, the pharmaceutical company behind the original pill has created a prototype which increases blood flow to the genitalia in a similar way to Viagra.

Pfizer have come up with a prototype version of the female equivalent of Viagra
More than half of women experience sexual dysfunction at some point in their lives.
They may suffer a lack of desire, emotional or mental health problems and physical problems that mean they avoid having sex.
Pharmaceutical giant Pfizer has developed a drug, so far called only UK-414,495, which is supposed to increase sexual arousal, but will not affect desire, mood or emotional problems.
Some women take Viagra with mixed results and the drug has been used in fertility treatment to increase blood flow to the pelvis and encourage an embryo to implant in the womb.
But this is the first pill that claims to be an equivalent of the male Viagra.
The research, which involved animals, is published by the British Journal of Pharmacology, though Pfizer say they won’t develop the drug and warn that the chemical may not work the same way in humans, according to the Telegraph.
Chris Wayman, the lead researcher, said: ‘Before this work, we knew surprisingly little about the processes that control all of these changes.

Pfizer claim the tablets may help overcome female sexual arousal disorder
‘Now that we are beginning to establish the pathways involved in sexual arousal, scientists may be able to find ways of helping women who would like to overcome female sexual arousal disorder.
‘While the particular chemical compound in this research did not prove appropriate for further developments, the implications of the research could lead to the development of a product in the future.’
Viagra was originally developed as a treatment for high blood pressure and the heart condition angina, but men who took part in early trials realised the drug had an interesting side effect.
Clinical trials suggested the drug had little effect on angina and instead induced erections in men.
The drug first went on sale in 1998 and has since been prescribed to 25million men, creating a multi-billion pound global market.
The name Viagra has become so associated with men’s erectile problems that many cures are marketed as ‘herbal viagra’.
It is known by many nicknames, including Vitamin V and the Blue Pill.
Read more: http://www.dailymail.co.uk/health/article-1265842/Female-Viagra-help-women-increase-sexual-arousal.html#ixzz39lkmpSik
…………………………………

scheme
http://www.google.com/patents/US20020052370




| Ex | Prec | n | Y | Data |
| 43 | Prep 37 | 0 |
 |
1H NMR (CDCl3, 400 MHz) δ: 0.92 (t, 3H), 1.35 (t, 3H), 1.25-1.80 (m, 11H), 2.20-2.50 (m, 4H), 2.95 (q, 2H), 12.10 (bs, 1H); LRMS: m/z 339.8 (MH+) Anal. Found: C, 56.46; H, 7.46; N, 12.36. C16H25N3O3S requires C, 56.62; H, 7.44; N, 12.37%. |
Example 29 (2R)-2-[(1-{[(5-Ethyl-1,3,4-thiadiazol-2-yl)amino]carbonyl}cyclopentyl) methyl]pentanoic acid
[0354]
desired[0355] and
Example 30 (2S)-2-[(1-{[(5-Ethyl-1,3,4-thiadiazol-2-yl)amino]carbonyl}cyclopentyl) methyl]pentanoic acid
[0356]
 undesired
undesired[0357] The acid from Example 4 (824 mg) was further purified by HPLC using an AD column and using hexane:iso-propanol:trifluoroacetic acid (85:15:0.2) as eluant to give the title compound of example 29 as a white foam, 400 mg, 99.5% ee, 1H NMR (CDCl3, 400 MHz) δ: 0.90 (t, 3H), 1.36 (m, 6H), 1.50-1.80 (m, 9H), 2.19 (m, 1H), 2.30 (m, 1H), 2.44 (m, 1H), 2.60 (m, 1H), 2.98 (q, 2H), 12.10-12.30 (bs, 1H), LRMS: m/z 338 (MH−), [α]D=−9.0°(c=0.1, methanol),
and
the title compound of example 30 as a white foam, 386 mg, 99% ee, 1H NMR (CDCl3, 400 MHz) δ: 0.90 (t, 3H), 1.38 (m, 6H), 1.50-1.79 (m, 9H), 2.19 (m, 1H), 2.30 (m, 1H), 2.44 (m, 1H), 2.60 (m, 1H), 2.98 (q, 2H), 12.10-12.27 (bs, 1H);
[0358] LRMS: m/z 338 (MH−); and [α]D=+3.8°(c=0.1, methanol).
[0359] Alternatively, Example 29 may be prepared as follows:
[0360] To a solution of the product from Preparation 51a (574 g, 1.45 mol) in dichloromethane (2.87 L) was added trifluoroacetic acid (1.15 L) over a period of 50 minutes with cooling at 10° C. After addition was complete, the reaction was allowed to warm to ambient temperature with stirring under a nitrogen atmosphere for 24 hours. Deionised water (2.6 L) was then added. The reaction mixture was then washed with deionised water (3×2.6 L). The dichloromethane layer was concentrated to a volume of approximately 1 L to give the crude title compound (439 g, 1.29 mol, 96% yield) as a solution in dichloromethane. A purified sample of the title compound was obtained using the following procedure. To a dichloromethane solution (2.34 L) of the crude product, that had been filtered to remove any particulate contamination, was added isopropyl acetate (1.38 L). The resultant mixture was distilled at atmospheric pressure whilst being simultaneously replaced with isopropyl acetate until the solution temperature reached 87° C. The heating was stopped and the solution was allowed to cool to ambient temperature with stirring for 14 hours to give a cloudy brown solution. The agitation rate was then increased and crystallisation commenced. The suspension was then allowed to granulate for 12 hours at ambient temperature. The resultant suspension was then cooled to 0° C. for 3.5 hours and the solid was then collected by filtration. The filter cake was then washed with isopropyl acetate (2×185 ml, then 2×90 ml) and the solid was dried under vacuum at 40-45° C. for 18 hours to give the title compound (602 g, 0.18 mol, 70% yield) as a cream coloured, crystalline solid;
m.p.: 130-136° C.;
LRMS (negative APCI): m/z [M−H]− 338;
1H-NMR (CDCl3, 300 MHz) δ: 0.92 (t, 3H), 1.27-1.52 (m, 7H), 1.52-1.89 (m, 8H), 2.11-2.27 (m, 1H), 2.27-2.37 (m, 1H), 2.42-2.55 (m, 1H), 2.65 (dd, 2H), 3.00 (q, 2H), 12.25 (bs, 1H).
[0361] Example 29 may be purified as follows:
[0362] The title product from Example 29 was disolved in methanol. To this solution was added sodium methoxide (1 equivalent) in methanol (1 ml/g of Example 29) and the mixture was stirred at room temperature for 20 minutes. The solvent was removed in vacuo and the residue was azeotoped with ethyl acetate to give a brown residue. Ethyl acetate was added and the solution filtered to give a brown solid which was washed with tert-butylmethyl ether to give the crude sodium salt of Example 29. This crude product (35 g) was partitioned between water (200 ml) and ethyl acetate (350 ml). Concentrated hydrochloric acid (˜7 ml) was added until the pH of the aqueous layer was pH2. The aqueous phase was washed with ethyl acetate (2×100 ml). The combined layers were dried using magnesium sulphate. The solvent was removed in vacuo to give a light brown solid (31 g). Ethyl acetate (64 ml, 2 ml/g) and diisopropyl ether (155 ml, 5 ml/g) were added and the mixture heated to 68° C. until a clear solution was obtained (˜30 min). Upon cooling to room temperature, crystallisation of the free acid occurred. After 30 minutes stirring at room temperature the product was collected by filtration and washed with diisopropyl ether. The product was dried in a vacuum oven at 50° C. overnight. (20.2 g, 61% recovery from the sodium salt.); m.p. 135 degC (determined using a Perkin Elmer DSC7 at a heating rate of 20° C./minute).
[0372] The title compound of Example 29 metabolysed to form (2R)-1-(2-{[(5-ethyl-1,3,4-thiadiazol-2-yl)amino]carbonyl}pentyl)cyclopentanecarboxylic acid.

[0373] This compound was prepared as follows:
[0374] The product from Preparation 102 (430 mg, 1 mmol) was taken up in ethanol (5 mls) and methanol (1 ml) and hydrogenated at 30 psi hydrogen pressure at room temperature for 2 h. The mixture was then filtered through a plug of Arbocel®) and evaporated to a yellow oil. This oil was purified by column chromatography using firstly 19:1, then 9:1 DCM:MeOH as eluant to provide the product as a clear oil (120 mg, 35%); 1HNMR (400 MHz, CDCl3) 0.88 (t, 3H), 1.20-1.88 (m, 13H), 1.90-2.03 (m, 1H), 2.24-2.38 (m, 1H), 2.43-2.72 (m, 2H), 2.95 (q, 2H); LRMS m/z 340.2 (M+H).
Example 31 (R)-2-{[1-({[2-(Hydroxymethyl)-2,3-dihydro-1H-inden-2-yl]amino}carbonyl)-cyclopentyl]methyl}pentanoic acid
[0375] and

Example 32 (S)-2-{[1-({[2-(Hydroxymethyl)-2.3-dihydro-1H-inden-2-yl]amino}carbonyl)-cyclopentyl]methyl}pentanoic acid
[0376]

[0377] 2-{[1-({[2-(Hydroxymethyl)-2,3-dihydro-1H-inden-2-yl]amino}carbonyl)-cyclopentyl]methyl}pentanoic acid (WO 9110644, Example 8) was further purified by HPLC using an AD column and hexane:isopropanol:trifluoroacetic acid (90:10:0.1) as eluant, to give the title compound of Example 31, 99% ee, [α]D=+10.40 (c=0.067, ethanol) and the title compound of Example 32, 99% ee, [α]D=−10.9° (c=0.046, ethanol).
………………..
http://www.google.com/patents/US6734186
Example 7 (+)-2-[(1-{[(5-Ethyl-1,3,4-thiadiazol-2-yl)amino]carbonyl}cyclopentyl)methyl]pentanoic Acid (F63)

The acid from Preparation 18 (18/ex4) (824 mg) was further purified by HPLC using an AD column and using hexane:iso-propanol:trifluoroacetic acid (85:15:0.2) as eluant to give the title compound of example 7 as a white foam, 386 mg, 99% ee,1H NMR (CDCl3, 400 MHz) δ: 0.90 (t, 3H), 1.38 (m, 6H), 1.50-1.79 (m, 9H), 2.19 (m, 1H), 2.30 (m, 1H), 2.44 (m, 1H), 2.60 (m, 1H), 2.98 (q, 2H), 12.10-12.27 (bs, 1H); LRMS: m/z 338 (MH-); and [α]D=+3.80°(c=0.1, methanol)
Novel selective inhibitors of neutral endopeptidase for the treatment of female sexual arousal disorder. Synthesis and activity of functionalized glutaramides
J Med Chem 2006, 49(14): 4409

Female sexual arousal disorder (FSAD) is a highly prevalent sexual disorder affecting up to 40% of women. We describe herein our efforts to identify a selective neutral endopeptidase (NEP) inhibitor as a potential treatment for FSAD. The rationale for this approach, together with a description of the medicinal chemistry strategy, lead compounds, and SAR investigations are detailed. In particular, the strategy of starting with the clinically precedented selective NEP inhibitor, Candoxatrilat, and targeting low molecular weight and relatively polar mono-carboxylic acids is described. This led ultimately to the prototype development candidate R–13, for which detailed pharmacology and pharmacokinetic parameters are presented.
ACID ENTRY 13
…………………………………………..
WO 2002002513
http://www.google.com/patents/WO2002002513A1?cl=en
…………………..
WO 2002003995
http://www.google.com/patents/WO2002003995A2?cl=en
Scheme 12
LiAIHψ THF, 6hr at reflux




Example 1
( f?)-2-r(1 r(5-ethyl-1.3.4-thiadiazol-2-yl)aminolcarbonyl)cvclopentyl) methyllpentanoic acid
and
Example 2
( S)-2-r(1-fr(5-Ethyl-1.3.4-thiadiazol-2-vnaminolcarbonyl)cvclopentyl)- methyllpentanoic acid
The title product from stage c) below (824mg) was further purified by HPLC using an AD column and using hexane:/sσ-propanol:trifluoroacetic acid (85:15:0.2) as elutant to give the title product from Example 1 , 400mg, 99.5% ee, 1H NMR (CDCI3, 400MHz) δ: 0.90 (t, 3H), 1.36 (m, 6H), 1.50-1.80 (m, 9H), 2.19 (m, 1 H), 2.30 (m, 1 H), 2.44 (m, 1 H), 2.60 (m, 1 H), 2.98 (q, 2H), 12.10-12.30 (bs, 1 H), LRMS : m/z 338 (MH“ ), [α]D = -9.0° (c = 0.1 , methanol), and the title product from Example 2, 386mg, 99% ee, 1H NMR (CDCl3, 400MHz) δ: 0.90 (t, 3H), 1.38 (m, 6H), 1.50-1.79 (m, 9H), 2.19 ( , 1 H), 2.30 ( , 1H), 2.44 (m, 1 H), 2.60 (m, 1 H), 2.98 (q, 2H), 12.10-12.27 (bs, 1H); LRMS: m/z 338 (MH“); and [α]D = +3.8° (c = 0.1 , methanol)
Preparation of Starting Materials a) 1 -r2-(tø/t-Butoxycarbonyl)-4-pentvπ-cvclopentane carboxylic acid
A mixture of 1 -[2-(tø t-butoxycarbonyl)-4-pentenyl]-cyclopentane carboxylic acid (EP 274234) (23g, 81.5mmol) and 10% palladium on charcoal (2g) in dry ethanol (200ml) was hydrogenated at 30psi and room temperature for 18 hours. The reaction mixture was filtered through Arbocel®, and the filtrate evaporated under reduced pressure to give a yellow oil. The crude product was purified by column chromatography on silica gel, using ethyl acetate:pentane (40:60) as the eluant, to provide the desired product as a clear oil, 21 g, 91%; 1H NMR (CDCI3, 0.86 (t, 3H), 1.22-1.58 (m, 15H), 1.64 (m, 4H), 1.78 (dd, 1H), 2.00-2.18 ( , 3H), 2.24 ( , 1H); LRMS : m/z 283 (M-HV b) tert-Butyl 2-1Ϊ1 -flT5-ethyl-1.3.4-thiadiazol-2-vnaminolcarbonyl)- cvclopentvDmethyllpentanoate.
1 -(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (0.21 mmol), 1 – hydroxybenzotriazole hydrate (0.2mmol), N-methylmorpholine (0.31 mmol) and 2-amino-5-ethyl-1 ,3,4-thiadiazole (0.22mmol) were added to a solution of the product from stage a) above (150mg, 0.53mmol) in N,N- dimethylformamide (3ml), and the reaction stirred at 90°C for 18 hours. The cooled solution was diluted with ethyl acetate (90ml), washed with water
(3x25ml), and brine (25ml), then dried (MgSO ) and evaporated under reduced pressure. The crude product was purified by chromatography on silica gel, using ethyl acetate:pentane (30:70) as the eluant to afford the title compound, 92%; 1H NMR (CDCI3, 300MHz) δ: 0.82 (t, 3H), 1.20-1.80 (m, 22H), 1.84 (m, 1 H), 2.20 (m, 4H), 3.04 (q, 2H), 9.10 (bs, 1 H); LRMS : m/z
396.2 (MH+).
c) . 2-r(1-H,(5-ethyl-1.3.4-thiadiazol-2-yl)amino1carbonyl)cvclopentyl) methyllpentanoic acid.
Trifluoroacetic acid (5ml) was added to a solution of the title product from stage b) above (0.31 mmol) in dichloromethane (5ml), and the solution stirred at room temperature for 4 hours. The reaction mixture was concentrated under reduced pressure and the residue azeotroped with toluene and dichloromethane to afford the title compound as a clear oil, 81 %, 1H NMR
(CDCI3, 400MHz) δ: 0.92 (t, 3H), 1.35 (t, 3H), 1.25-1.80 (m, 11 H), 2.20-2.50 (m, 4H), 2.95 (q, 2H), 12.10 (bs, 1 H); LRMS : m/z 339.8 (MH+); Anal. Found: C, 56.46; H, 7.46; N, 12.36. Cι6H25N3O3S requires C, 56.62; H, 7.44; N, 12.37%.
………………………………………

………………………………..
Original Research Article
SEE
The discovery of small molecule inhibitors of neutral endopeptidase. Structure-activity studies on functionalized glutaramides
Chem Biol Drug Des 2006, 67(1): 74
Optimization of oral pharmacokinetics in the discovery of clinical candidates for the treatment of sexual dysfunction
237th ACS Natl Meet (March 22-26, Salt Lake City) 2009, Abst MEDI 173
Novel selective inhibitors of neutral endopeptidase for the treatment of female sexual arousal disorder. Synthesis and activity of functionalized glutaramides
J Med Chem 2006, 49(14): 4409
Bioorganic & Medicinal Chemistry (2007), 15(1), 142-159
Journal of Medicinal Chemistry (2007), 50(24), 6165-6176.
|
5-7-2004
|
Treatment of sexual dysfunction
|
|
|
11-15-2002
|
Treatment of sexual dysfunction
|
|
|
5-3-2002
|
Cyclopentyl-substituted glutaramide derivatives as inhibitors of neutral endopeptidase
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
| (R)-2-({1-[(5-ethyl-1,3,4-thiadiazol-2-yl)carbamoyl]cyclopentyl}methyl)valeric acid | |
| Clinical data | |
| Legal status | ? |
| Identifiers | |
| CAS number | 337962-93-3 |
| ATC code | ? |
| PubChem | CID 9949799 |
| Chemical data | |
| Formula | C16H25N3O3S |
| Mol. mass | 339.452 g/mol |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US6734186 * | Nov 8, 2000 | May 11, 2004 | Pfizer Inc. | Phosphodiesterase 2 inhibitor |
| US7956195 * | Dec 21, 2007 | Jun 7, 2011 | Abbott Laboratories | reacting arylboronic acids with a cycloalkanone, in the presence of a rhodium catalyst or BINAP, to form a substituted arylcycloalkanone, then formin of a hydantoin, alkylation of the hydantoin, resolution, hydrolysis of the hydantoin to the amino acids and esterification of acids; chemical intermediates |
| WO2005007166A1 * | Jul 12, 2004 | Jan 27, 2005 | Alasdair Mark Naylor | Treatment of sexual dysfunction |
Female Sexual Response
The female sexual response phase of arousal is not easily distinguished from the phase of desire until physiological changes begin to take place in the vagina and clitoris as well as other sexual organs. Sexual excitement and pleasure are accompanied by a combination of vascular and neuromuscular events which lead to engorgement of the clitoris, labia and vaginal wall, increased vaginal lubrication and dilatation of the vaginal lumen (Levin, 1980; Ottesen, 1983; Levin, 1991; Levin, 1992; Sjoberg, 1992; Wagner, 1992; Schiavi et al., 1995; Masters et al., 1996; Berman et al., 1999).
Vaginal engorgement enables transudation to occur and this process is responsible for increased vaginal lubrication. Transudation allows a flow of plasma through the epithelium and onto the vaginal surface, the driving force for which is increased blood flow in the vaginal capillary bed during the aroused state. In addition engorgement leads to an increase in vaginal length and luminal diameter, especially in the distal ⅔ of the vaginal canal. The luminal dilatation of the vagina is due to a combination of smooth muscle relaxation of its wall and skeletal muscle relaxation of the pelvic floor muscles. Some sexual pain disorders such as vaginismus are thought to be due, at least in part, by inadequate relaxation preventing dilatation of the vagina; it has yet to be ascertained if this is primarily a smooth or skeletal muscle problem. (Levin, 1980; Oltesen, 1983; Levin, 1991; Levin, 1992; Sjoberg, 1992; Wagner, 1992; Schiavi et al., 1995; Master et al., 1996; Berman et al., 1999).
The vasculature and micro vasculature of the vagina are innervated by nerves containing neuropeptides and other neurotransmitter candidates. These include calcitonin gene-related peptide (CGRP), neuropeptide Y (NPY; Sequence No. 4), nitric oxide synthase (NOS), substance P and vasoactive intestinal peptide (VIP; Sequence No. 8) (Hoyle et al., 1996). Peptides that are present in the clitoris are discussed infra. Nitric oxide synthase, which is often colocalised with VIP (Sequence No. 8), displays a greater expression, immunologically, in the deep arteries and veins rather than in the blood vessels of the propria (Hoyle et al., 1996).
Female Sexual Dysfunction
It is known that some individuals can suffer from female sexual dysfunction (FSD). FSD is best defined as the difficulty or inability of a woman to find satisfaction in sexual expression. FSD is a collective term for several diverse female sexual disorders (Leiblum, 1998, Berman et al., 1999). The woman may have lack of desire, difficulty with arousal or orgasm, pain with intercourse or a combination of these problems. Several types of disease, medications, injuries or psychological problems can cause FSD.
Studies investigating sexual dysfunction in couples reveals that up to 76% of women have complaints of sexual dysfunction and that 30-50% of women in the USA experience FSD.
Sub-types of FSD include hypoactive sexual desire disorder, female sexual arousal disorder, orgasmic disorder and sexual desire disorder.
Treatments in development are targeted to treat specific subtypes of FSD, predominantly desire and arousal disorders.
The categories of FSD are best defined by contrasting them to the phases of normal female sexual response: desire, arousal and orgasm (Leiblum 1998). Desire or libido is the drive for sexual expression—and manifestations often include sexual thoughts either when in the company of an interested partner or when exposed to other erotic stimuli. In contrast, sexual arousal is the vascular response to sexual stimulation, an important component of which is vaginal lubrication and elongation of the vagina. Thus, sexual arousal, in contrast to sexual desire, is a response relating to genital (e.g. vaginal and clitoral) blood flow and not necessarily sensitivity. Orgasm is the release of sexual tension that has culminated during arousal. Hence, FSD typically occurs when a woman has an inadequate or unsatisfactory response in any of these phases, usually desire, arousal or orgasm. FSD categories include hypoactive sexual desire disorder, sexual arousal disorder, orgasmic disorders and sexual pain disorders.
Hypoactive sexual desire disorder is present if a woman has no or little desire to be sexual, and has no or few sexual thoughts or fantasies. This type of FSD can be caused by low testosterone levels, due either to natural menopause or to surgical menopause. Other causes include illness, medications, fatigue, depression and anxiety.
Female sexual arousal disorder (FSAD) is characterised by inadequate genital response to sexual stimulation. The genitalia (e.g. the vagina and/or the clitoris) do not undergo the engorgement that characterises normal sexual arousal. The vaginal walls are poorly lubricated, so that intercourse is painful. Orgasms may be impeded. Arousal disorder can be caused by reduced oestrogen at menopause or after childbirth and during lactation, as well as by illnesses, with vascular components such as diabetes and atherosclerosis. Other causes result from treatment with diuretics, antihistamines, antidepressants eg SSRIs or antihypertensive agents. FSAD is discussed in more detail infra.
Sexual pain disorders (which include dyspareunia and vaginismus) are characterised by pain resulting from penetration and may be caused by medications which reduce lubrication, endometriosis, pelvic inflammatory disease, inflammatory bowel disease or urinary tract problems.
The prevalence of FSD is difficult to gauge because the term covers several types of problem, some of which are difficult to measure, and because the interest in treating FSD is relatively recent. Many women’s sexual problems are associated either directly with the female ageing process or with chronic illnesses such as diabetes and hypertension.
There are wide variations in the reported incidence and prevalence of FSD, in part explained by the use of differing evaluation criteria, but most investigators report that a significant proportion of otherwise healthy women have symptoms of one or more of the FSD subgroups. By way of example, studies comparing sexual dysfunction in couples reveal that 63% of women had arousal or orgasmic dysfunction compared with 40% of men have erectile or ejaculatory dysfunction (Frank et al., 1978).
However, the prevalence of female sexual arousal disorder varies considerably from survey to survey. In a recent National Health and Social Life Survey 19% of women reported lubrication difficulties whereas 14% of women in an outpatient gynaecological clinic reported similar difficulties with lubrication (Rosen et al., 1993).
Several studies have also reported dysfunction with sexual arousal in diabetic women (up to 47%), this included reduced vaginal lubrication (Wincze et al., 1993). There was no association between neuropathy and sexual dysfunction.
Numerous studies have also shown that between 11-48% of women overall may have reduced sexual desire with age. Similarly, between 11-50% of women report problems with arousal and lubrication, and therefore experience pain with intercourse. Vaginismus is far less common, affecting approximately 1% of women.
Studies of sexually experienced women have detailed that 5-10% have primary anorgasmia. Another 10% have infrequent orgasms and a further 10% experience them inconsistently (Spector et al., 1990).
Because FSD consists of several subtypes that express symptoms in separate phases of the sexual response cycle, there is not a single therapy. Current treatment of FSD focuses principally on psychological or relationship issues. Treatment of FSD is gradually evolving as more clinical and basic science studies are dedicated to the investigation of this medical problem. Female sexual complaints are not all psychological in pathophysiology, especially for those individuals who may have a component of vasculogenic dysfunction (eg FSAD) contributing to the overall female sexual complaint. There are at present no drugs licensed for the treatment of FSD. Empirical drug therapy includes oestrogen administration (topically or as hormone replacement therapy), androgens or mood-altering drugs such as buspirone or trazodone. These treatment options are often unsatisfactory due to low efficacy or unacceptable side effects.
Since interest is relatively recent in treating FSD pharmacologically, therapy consists of the following:- psychological counselling, over-the-counter sexual lubricants, and investigational candidates, including drugs approved for other conditions. These medications consist of hormonal agents, either testosterone or combinations of oestrogen and testosterone and more recently vascular drugs, that have proved effective in male erectile dysfunction. None of these agents has been demonstrated to be very effective in treating FSD.
Female Sexual Arousal Disorder (FSAD)
The sexual arousal response consists of vasocongestion in the pelvis, vaginal lubrication and expansion and swelling of the external genitalia. The disturbance causes marked distress and/or interpersonal difficulty. Studies investigating sexual dysfunction in couples reveals that there is a large number of females who suffer from sexual arousal dysfunction; otherwise known as female sexual arousal disorder (FSAD).
The Diagnostic and Statistical Manual (DSM) IV of the American Psychiatric Association defines Female Sexual Arousal Disorder (FSAD) as being:
“a persistent or recurrent inability to attain or to maintain until completion of the sexual activity adequate lubrication-swelling response of sexual excitement. The disturbance must cause marked distress or interpersonal difficulty.”
FSAD is a highly prevalent sexual disorder affecting pre-, peri- and post menopausal (±HRT) women. It is associated with concomitant disorders such as depression, cardiovascular diseases, diabetes and UG disorders.
The primary consequences of FSAD are lack of engorgement/swelling, lack of lubrication and lack of pleasurable genital sensation. The secondary consequences of FSAD are reduced sexual desire, pain during intercourse and difficulty in achieving an orgasm.
It has recently been hypothesised that there is a vascular basis for at least a proportion of patients with symptoms of FSAD (Goldstein et al., 1998) with animal data supporting this view (Park et al., 1997).
Drug candidates for treating FSAD, which are under investigation for efficacy, are primarily erectile dysfunction therapies that promote circulation to the male genitalia. They consist of two types of formulation, oral or sublingual medications (Apomorphine, Phentolamine, Sildenafil), and prostaglandin (PGE1-Alprostadil) that are injected or administered transurethrally in men, and topically to the genitalia in women.
The present invention seeks to provide an effective means of treating FSD, and in particular FSAD.
SUMMARY
The present invention is based on findings that FSAD is associated with reduced genital blood flow—in particular reduced blood flow in the vagina and/or the clitoris. Hence, treatment of women with FSAD can be achieved by enhancement of genital blood flow with vasoactive agents. In our studies, we have shown that cAMP mediates vaginal and clitoral vasorelaxation and that genital (e.g. vaginal and clitoral) blood flow can be enhanced/potentiated by elevation of cAMP levels. This is a seminal finding.
In this respect, no one has previously proposed that FSAD can be treated in such a way—i.e. by direct or indirect elevation of cAMP levels. Moreover, there are no teachings in the art to suggest that FSAD was associated with a detrimental modulation of cAMP activity and/or levels or that cAMP is responsible for mediating vaginal and clitoral vasorelaxation. Hence, the present invention is even further surprising.
In addition, we have found that by using agents of the present invention it is possible to increase genital engorgement and treat FSAD—e.g. increased lubrication in the vagina and increased sensitivity in the vagina and clitoris. Thus, in a broad aspect, the present invention relates to the use of a cAMP potentiator to treat FSD, in particular FSAD.
The present invention is advantageous as it provides a means for restoring a normal sexual arousal response—namely increased genital blood flow leading to vaginal, clitoral and labial engorgement. This will result in increased vaginal lubrication via plasma transudation, increased vaginal compliance and increased genital (e.g. vaginal and clitoral) sensitivity. Hence, the present invention provides a means to restore, or potentiate, the normal sexual arousal response.
More particularly, the present invention relates to:
A pharmaceutical composition for use (or when in use) in the treatment of FSD, in particular FSAD; the pharmaceutical composition comprising an agent capable of potentiating cAMP in the sexual genitalia of a female suffering from FSD, in particular FSAD; wherein the agent is optionally admixed with a pharmaceutically acceptable carrier, diluent or excipient.
The use of an agent in the manufacture of a medicament (such as a pharmaceutical composition) for the treatment of FSD, in particular FSAD; wherein the agent is capable of potentiating cAMP in the sexual genitalia of a female suffering from FSD, in particular FSAD.
A method of treating a female suffering from FSD, in particular FSAD; the method comprising delivering to the female an agent that is capable of potentiating cAMP in the sexual genitalia; wherein the agent is in an amount to cause potentiation of cAMP in the sexual genitalia of the female; wherein the agent is optionally admixed with a pharmaceutically acceptable carrier, diluent or excipient.
An assay method for identifying an agent that can be used to treat FSD, in particular FSAD, the assay method comprising: determining whether an agent can directly or indirectly potentiate cAMP; wherein a potentiation of cAMP in the presence of the agent is indicative that the agent may be useful in the treatment of FSD, in particular FSAD.
