
Home » japan 2017
Category Archives: japan 2017
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Pralatrexate プララトレキサート

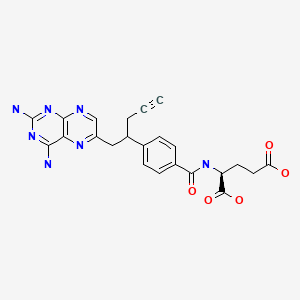
Pralatrexate (JAN/USAN/INN);
10-Propargyl-10-deazaaminopterin;
Folotyn (TN)
Antineoplastic
| Product
CAS:
|
FOLOTYN (Allos Therapeutics)
146464-95-1
|
|---|---|
| Formula |
C23H23N7O5
|
| Exact mass |
477.1761
|

- (2S)-2-((4-((1RS)-1-((2,4-diaminopteridin-6-yl)methyl)but-3-ynyl)benzoyl)amino)pentanedioic acid
- (2S)-2-({4-[1-(2,4-diaminopteridin-6-yl)pent-4-yn-2-yl]benzoyl}amino)pentanedioic acid
- 10-Propargyl-10-deazaaminopterin
- N-(4-(1-((2,4-Diamino-6-pteridinyl)methyl)-3-butynyl)benzoyl)-L-glutamic acid
- PDX
- UNII:A8Q8I19Q20
Japan approved 2017
| 2017/7/3 | PMDA | JAPAN | Pralatrexate | Difolta | Mundipharma | NME |

EMA 2012
The molecule contains two asymmetric carbon centres (C10) and (C19). The C10 position exists in the RS-configuration (approx. 50:50 ratio) on the link between the two aryl groups. The C19 position is contained in the glutamic acid moiety and predominantly exists in the S-configuration. Pralatrexate is an off-white to yellow crystalline material, soluble in aqueous solutions at pH 6.5 or higher and practically insoluble in chloroform, and ethanol. It predominantly exists as a single polymorph (form A).
Pralatrexate, chemically known as “(2S)-2-[[4-[(1RS)-1-[(2,4-diaminopteridin-6-yl)methyl]but-3-ynyl]benzoyl- ]-amino]pentanedioic acid”, also known as “10-Propargyl-10-deazaminopterin” or “PDX”, is a compound which has been tested and found useful in the treatment of cancer. In its racemic form, 2S)-2-[[4-[(1RS)-1-[(2,4-diaminopteridin-6-yl)methyl]but-3-ynyl]benzoyl]a- mino]-pentanedioic acid has been approved by the U.S. Food and Drug Administration (FDA) as a treatment for relapsed and refractory peripheral T-cell lymphoma.
Pralatrexate, was first disclosed in Journal of Medicinal Chemistry. 36: 2228-2231 (1993) by DeGraw et al., and subsequently in U.S. Pat. No. 5,374,726 and U.S. Pat. No. 5,354,741.
Pralatrexate is an antimetabolite for the treatment of relapsed or refractory peripheral T-cell lymphoma. It is more efficiently retained in cancer cells than methotrexate. FDA approved on September 24, 2009.
Pralatrexate (brand name Folotyn) is an anti-cancer therapy.[1] It is the first drug approved as a treatment for patients with relapsed or refractory peripheral T-cell lymphoma, or PTCL[2] — a biologically diverse group of aggressive blood cancers that have a poor prognosis.[2]

Approval
Folotyn was approved by the U.S. Food and Drug Administration (FDA) in September 2009 under the FDA’s accelerated approval,[2] which allows for earlier approval of drugs that meet unmet medical needs.[3] Pralatrexate injection is marketed in the U.S. under the name Folotyn by Spectrum Pharmaceuticals.[2] Clinical trials are currently underway to explore the potential of Folotyn in other blood related cancers and solid tumors.[4]

Mechanism
Pralatrexate is an antifolate (a folate analogue metabolic inhibitor) designed to accumulate preferentially in cancer cells.[1] Based on preclinical studies, researchers believe that pralatrexate selectively enters cells expressing reduced folate carrier type 1 (RFC-1), a protein that is overexpressed on certain cancer cells compared to normal cells.[1]
Antifolates, such as pralatrexate, are part of a group of compounds known as antimetabolites with structural similarity to naturally occurring molecules involved in DNA synthesis.[5] Cancer cells mistake antimetabolites for normal metabolites[5] allowing the compound to stop or slow critical enzymes involved in DNA synthesis which then triggers cell death.[1] Because of their primary effect on DNA synthesis, the antimetabolites are most effective against actively dividing cells and are largely cell-cycle phase specific.[5]
The selectivity of pralatrexate for cancer cells is based upon the observation that cancer cells generally have an overexpression of reduced folate carrier protein-1 (RTC-1) compared to normal somatic cells. This carrier protein allows the entrance of pralatrexate into the cell. Upon entering the cell, folypolyglutamate synthase FPGS catalyzes the polyglutamination of pralatrexate so that it is retained inside the cell.
Once inside, pralatrexate competitively inhibits dihydrofolate reductase (DHFR) and thymidylate synthase. Subsequent depletion of thymidine monophosphate (TMP) occurs so that the cancer cell is unable to synthesize DNA and RNA. As a result, the cancer cell cannot proliferate and is forced to undergo apoptosis. Pralatrexate is more effective against cells that are actively dividing.
Biological Activity
Pralatrexate (Folotyn) is an antifolate, and structurally a folate analog. It acts as an inhibitor of dihydrofolate reductase. It is selective for the reduced folate carrier type 1. Its IC50 is < 300 nM in some cell lines.
Conversion of different model animals based on BSA (Value based on data from FDA Draft Guidelines)
| Species | Mouse | Rat | Rabbit | Guinea pig | Hamster | Dog |
| Weight (kg) | 0.02 | 0.15 | 1.8 | 0.4 | 0.08 | 10 |
| Body Surface Area (m2) | 0.007 | 0.025 | 0.15 | 0.05 | 0.02 | 0.5 |
| Km factor | 3 | 6 | 12 | 8 | 5 | 20 |
| Animal A (mg/kg) = Animal B (mg/kg) multiplied by | Animal B Km |
| Animal A Km |
For example, to modify the dose of resveratrol used for a mouse (22.4 mg/kg) to a dose based on the BSA for a rat, multiply 22.4 mg/kg by the Km factor for a mouse and then divide by the Km factor for a rat. This calculation results in a rat equivalent dose for resveratrol of 11.2 mg/kg.
Discovery
Research on this class of drugs began in the 1950s at SRI International, where scientists were focused on developing new chemotherapies and antifolates that would be effective against tumor cells.[1]
In the late 1970s, researchers at Memorial Sloan Kettering Cancer Center discovered that cancerous cells take in natural folate through a protein identified as plasma membrane transporter (now referred to as “reduced folate carrier type 1” or “RFC-1”). Further research showed that when normal cells evolve into cancerous cells they often overproduce RFC-1 to ensure they get enough folate.[6]
A subsequent scientific collaboration was ultimately formed among SRI International, Memorial Sloan Kettering Cancer Center, and the Southern Research Institute with the intention of developing an antifolate with greater therapeutic selectivity – an agent that could be more effectively internalized into tumors (transported into the cells through RFC-1) and would be more toxic to cancer cells than normal cells.[6]
This collaboration, supported by the National Cancer Institute,[7] led to the identification of pralatrexate in the mid-1990s. Pralatrexate was later licensed to Allos Therapeutics in 2002 for further development.[8] Allos Therapeutics, Inc. was acquired by Spectrum Pharmaceuticals, Inc. on September 5, 2012. Allos is now a wholly owned subsidiary of Spectrum.[9]
Pralatrexate, is a 10-deazaaminopterin derivative which has been developed for the potential treatment of malignancies. Pralatrexate is an antifolate, structurally a folate analog inhibitor of dihydrofolate reductase (DHF ) exhibiting high affinity for reduced folate carrier- 1 (RFC- 1) and iolylpolyglutamate synthetase (FPGS). with antineoplastic and immunosuppressive activities, resulting in extensive internalization and accumulation in tumour cells. Pralatrexate selectively enters cells expressing RFC- 1. Intracellularly, this agent is highly polyglutamylated and competes for the folate binding site of DHFR, blocking tetrahydrofolate synthesis, which may result in depletion of nucleotide precursors; inhibition of DNA. RNA and protein synthesis; and apoptotic tumor cell death. Efficient intracellular polyglutamylation of pralatrexate results in higher intracellular concentrations compared to non-polyglutamylated pralatrexate, which is more readily effuxed by the MRP (multidrug resistance protein) drug efflux pump. RFC- 1, an oncofetal protein expressed at highest levels during embryonic development, may be over expressed on the cell surfaces of various cancer cell types. Pralatrexate is the first and only drug approved by the Food and Drug Administration as a treatment for relapsed or refractory peripheral T-cell lymphoma, demonstrating the ability to reduce tumor size, but not to prolong life.
Pralatrexate is a folate analog metabolic inhibitor that competitively inhibits dihydrofolate reductase. It is also a competitive inhibitor for polyglutamylation by the enzyme folylpolyglutamyl synthetase. This inhibition results in the depletion of thymidine and other biological molecules the synthesis of which depends on single carbon transfer.
US 200510267117 discloses that T cell lymphoma is treated by administering to a patient suffering from T cell lymphoma a therapeutically effective amount of IO-propargyl-10- deazaaminopterin. Remission is observed in human patients, even with drug resistant T cell lymphoma at weekly dosages levels as low as 30 mg/m2. In general, the 10-propargyl-lO- deazaaminopterin is administered in an amount of from 30 to 275 mg/m2 per dose.
US 2011/0190305 discloses diastereomers of 10-propargyl-l 0-deazaminopterin, compositions comprising optically pure diastereomers of 10-propargyl-l 0-deazaminopterin, in particular the two (R,S) diastereomers about the C 10 position, method of preparation of the diastereomers and method of treatment of conditions related to inflammatory disorders and cancer.
US005354751 discloses heteroaroyl-10-deazaaminopterins and 10-alkenyl or 10-alkynyl-lO- deazaaminopterins having pronounced anti-inflammatory activity, anti-leukemic and anti- tumorigenic activity, as well as a method for treatment of inflammatory diseases, leukemia and tumors. Pharmaceutical compositions containing these heteroaroyl-lO-deazaaminopterin compounds are also disclosed. The invention further concerns a process for preparation of these compounds. A method for preparation of I0-propargyl-10-deazaaminopterin compound is also disclosed in this document.
Journal publication Bioorganic and Medicinal Chemistry (19) 2011, page 1151, synthetic approaches to the 2009 new drugs, also discloses a method for synthesis of Pralatrexate. The method comprises alkylating dimethyl homotrephthalate with propargyl bromide in the presence of KH in THF and then with 2,4-diamino-6-(bromomethyl)pteridine hydrobromide in the presence of KH in D F to afford crude product. Subsequent hydrolysis of the diester with aqueous NaOH, followed by acidification with acetic acid to give crude carboxylic acid, followed by thermally induced decarboxylation in D SO to give 10-deazapteroic acid derivative. Activation of carboxylic acid as a mixed anhydride using t-butyl chioroformate prior to coupling with diethyl L-glutamate hydrochloride in the presence of Et3 in DMF to give lO-propargyl-IO-deaza-aminopterin diethyl ester. Finally, saponification of diethyl ester with aqueous NaOH in 2-methoxyethanol, followed by acidifying with AcOH giving Pralatrexate.
Methods of preparing Pralatrexate known in the prior art are not only complicated but preparation of Pralatrexate using the methods disclosed in the prior art also result in very high manufacturing cost. Therefore, there is a need for an improved, simple and cost effective method for preparation of Pralatrexate which can be used for industrial scale preparation of this compound.
PATENT
scheme- 1 .

Scheme-1

Scheme 2

Scheme 3
scheme 4.

https://patents.google.com/patent/WO2013164856A1/en
PATENT
https://patents.google.com/patent/WO2014016740A2/en
Pralatrexate, chemically known as “(25)-2-[[4-[(lR5)-l-[(2,4-diaminopteridin-6- yl)methyl]but-3-ynyl]benzoyl]- amino] pentanedioic acid”, also known as “10-Propargyl- 10-deazaminopterin” or “PDX”, is a compound which has been tested and found useful in the treatment of cancer. In its racemic form, 2S)-2-[[4-[(lRS)-l-[(2,4-diaminopteridin-6- yl)methyl]but-3-ynyl]benzoyl]amino]- pentanedioic acid has been approved by the U.S. Food and Drug Administration (FDA) as a treatment for relapsed and refractory peripheral T-cell lymphoma.
Pralatrexate, represented by Formula (I), was first disclosed in Journal of Medicinal Chemistry. 36: 2228-2231 (1993) by DeGraw et al., and subsequently in US 5374726 and US 5354741.

DeGraw et al, publication, US 5374726 and US 5354741 disclose method for the synthesis of Pralatrexate of Formula (I), comprising alkylation of homoterephthalic acid dimethyl ester with propargyl bromide using Potassium Hydride, which is further coupled with 2,4-diamino-6-bromomethylpteridine in presence of Potassium Hydride followed by hydrolysis in presence of NaOH in 2-methoxyethanol-water mixture and decarboxylation at high temperature in DMSO and subsequent coupling with L-glutamic acid diethyl ester using t-butyl chloroformate and a base, and finally hydrolysis of the product with NaOH in 2-methoxyethanol-water mixture to give Pralatrexate of Formula (I). The process is outlined below as synthetic Scheme- 1.


Scheme- 1
The methods disclosed in DeGraw et al., publication, US 5374726 and US 5354741 suffer from the following disadvantages, which are outlined below:
(i) Use of pyrophoric Potassium hydride in the initial alkylation step and the subsequent coupling step.
(ii) Amide formation in the penultimate step by use of a hazardous chloroformate reagent.
(iii) The final product has a purity of -95% and is contaminated with the 10- deazaminopterin impurity to the level of 4%, which affects the final quality of Active Pharmaceutical ingredient (API) and does not meet the Pharmacopeial specifications. Use of 2-methoxyethanol in the last step which is classified under guideline of International Conference on Harmonisation of Pharmaceutical for Human USE (ICH) as a Class-2 solvent, with a maximum daily exposure limit of 50 ppm. Extensive use of column chromatography during the method adding to the cost of manufacture.
(vi) Low yield of the final Pralatrexate (-5.5 %).
US 6028071 discloses a process for the preparation of Pralatrexate of Formula (I) comprising coupling of homoterephthalic acid dimethyl ester with propargyl bromide using NaH in THF, further coupling of the product with 2,4-diamino-6- bromomethylpteridine using NaH in DMF, followed by hydrolysis with a base in 2- methoxyethanol-water mixture, and decarboxylation at elevated temperatures at 115- 120°C in DMSO, and finally coupling of the product with L-glutamic acid dimethyl ester using benzotriazole-l-yloxytris(dimethylamino) phosphonium hexafluorophosphate (BOP) and triethylamine in DMF, and finally hydrolysis with NaOH in methanol-water mixture to yield Pralatrexate. The process is outlined below as synthetic Scheme-2.

3, R = CH3
4 ■■ H

Scheme-2 The process disclosed in US 6028071 suffer from the following disadvantages outlined below
(i) Use of sodium hydride in the initial alkylation step and the subsequent coupling step.
(ii) Using benzotriazole-l-yloxytris(dimethylamino) phosphonium hexafluoro phosphate (BOP) in coupling reaction that liberates Hexamethylphosphoramide (HMPA), which is carcinogenic
(iii) Extensive column chromatography during the process adding to the cost of manufacture
(iv) Quality of the API obtained by this process is only -98%.
(v) Low yield of Pralatrexate is obtained (2.06%).
(vi) In the propargylation step the ratio of oc-monopropargyl homoterephthalic acid dimethyl ester to oc-monopropargyl homoterephthalic acid dimethyl ester is not less than 75:25.
US 20110190305 relates to optically pure diastereomers of 10-propargyl-lO- deazaminopterin, in particular the two ( ,S) diastereomers about the CIO position. None of the prior art discloses a process for preparing substantially pure Pralatrexate. When the present inventors practiced the invention disclosed in US 6028071 to ascertain the purity of Pralatrexate, they found the content of individual diastereomers at the CIO position to be 50+3.66%.
Example- 11
10-Propar gyl- 10-deazaminopterin (Pralatrexate)
To aqueous NaOH (11.6 g NaOH in 472 mL DM water) and Methanol (944 mL), 10- Propargyl-10-deazaminopterin Dimethyl Ester (59.0 g) was added at 20-25°C and stirred the reaction mass for 8 hours. After completion of reaction which was monitored by HPLC, pH of the reaction mass was adjusted to 6.6 with acetic acid. Excess methanol was evaporated under reduced pressure below 40° C and DM water (1298mL) was added to the residual solution. The pH of the residual solution was adjusted to 4.5 with dilute acetic acid. The reaction mass was stirred for 30 minutes at 20-25° C and filtered the solid precipitated. The solid was furthered purified with DM water (590 mL) by stirring at 20- 25°C for 30-35 minutes. The solid was filtered and dried under vacuum at 35-40° C to give 39 g (70 %) of the title compound.
Purity: 99.56 %
Water content = 4.8 % (w/w)
*H NMR (DMSO-d6; 400MHz): δ 1.91 (m, 1H), 2.05 (m, 1H), 2.33 (t, J=7.2 Hz, 2H), 2.59 (bm, 2H), 2.78 (s, 1H), 3.14-3.20 (bm, 1H), 3.28 (dd, J=14.4 Hz & 6.4 Hz, 1H), 3.64 (quintet, J=7.2 Hz), 4.35 (bm, 1H), 6.30 (bs, 2H, NH2), 7.39 (d, J=8.0 Hz, 2H), 7.61 & 7.63 (2xbs, 2H, NH2), 7.73 (d, J=8.0Hz, 2H), 8.39 (bs, 1H), 8.50 (d, J=7.6 Hz, 1H, NH), 12.20 (bs, 2H, 2xC02H).
13C NMR (DMSO-d6; 100MHz): δ 24.84 (CH2), 25.94 (CH2), 30.46 (CH2), 39.08 (CH2), 43.05 (CH), 51.93 (CH), 72.90 (CH), 82.57 (C), 121.51 (C), 127.35 (2xCH), 127.35 (2xCH), 132.22 (C), 146.69 (C), 147.20 (C), 150.56 (CH), 154.17 (C), 162.41 (C), 162.77 (C), 166.42 & 166.46 (CONH), 173.54 (C02H), 173.94 (C02H).
MS (ES+) m/z: 478 [M+H]+.
IR (KBr, cm-1): 1540, 1557, 1639, 1704, 3300, 3420.
XRD (°2Theta; Cu): 8.47, 10.85, 12.28, 14.34, 15.00, 15.78, 18.90, 21.79, 24.20, 27.5, 28.92, 34.28.
Example- 12 discloses the preparation of Pralatrexate according to US 6028071.
Example-12
To 10-Propargyl-lO-deazaminopterin dimethyl ester (3.0 g) in methanol (181.8 mL), aqueous sodium hydroxide (0.52 g of sodium hydroxide in 13.1 mL demineralized water) was added at 20-25°C accompanied by stirring. The reaction mixture was stirred for 2h at 20-25°C, kept for further 8 hours at the same temperature and diluted with demineralized water (181.8 mL). methanol was recovered under vacuum below 40°C and the residue was left at 20-25°C for 24 hrs. The reaction was monitored by HPLC and acidified with acetic acid (7.5 mL). The solid obtained was filtered, washed with demineralized water (15 mL) and suck-dried for 2-3 hrs. The product was dried under vacuum at 50-55°C for 12 hours.
Weight : 2.5
Yield (%) : 89.2
Purity by HPLC (%) : 99.61
PATENT
Pralatrexate, (2S)-2-[[4-[(1 RS)-1-[(2,4-diaminopteridin-6-yl)methyl]but-3-ynyl]benzoyl]amino]pentandioic acid, also referred to as 10-propargyl-10-deaza-aminopterin, is an anti-cancer drug having the following formula:

Pralatrexate is approved for a treatment for patients with relapsed or refractory peripheral T-cell lymphoma. It is an antifolate and acts as an inhibitor of
dihydrofolate reductase.
[0004] Pralatrexate is disclosed in several documents such as DeGraw et al., (J. Med. Chem, 1993, 36, 2228), US 6,028,071, US 5,354,751, EP 0944389, EP 1891957 and WO 98/02163. US 6,028,071 discloses Pralatrexate and a preparation thereof, as described in the following reaction scheme:

EXAMPLES
Reference examples:
[0099] Pt-MADES (compound 5) and Pt-MADAC (compound 6) may be prepared according to procedures disclosed in US 6,028,071, example 1.
Example 1 : Decarboxylation of Pt-MADAC (compound 6)

[00100] Pt-MADAC (compound 6) (17 g, 43.3 mmol, containing 1.4% of impurity hydro-Pt-MADAC (compound 6a) according to HPLC analysis) was added to N-methyl-2-pyrrolidone (170 mL, 10 Vol.) pre-heated at 120°C. Upon dissolution of the solid, N, N-diisopropylethyl amine (5.6 mL, 32.1 mmol) was added. The reaction mixture was stirred at 120 °C for 0.5 h, then cooled down to room temperature and poured into water (1700 mL, 100 Vol.). The pH was adjusted to 4.5 by addition of aq. HCl (16% w/w). A precipitate formed and was isolated by filtration. The collected solid was dried in a drying oven at 45 °C for 18 h to provide Pt-MADEC (compound 7) as a yellow solid (14.6 g, 97% yield, purity 87.1%), containing 7.5% of Pt-lactone (compound 7b) and 1.4% of hydro-Pt-MADEC (compound 7a) according to HPLC analysis.
Example 2: Decarboxylation of Pt-MADAC (Compound 6) without use of a base
[00101] Pt-MADAC (compound 6) (2 g, 5.10 mmol, containing 1.4% of impurity hydro-Pt-MADAC (compound 6a) according to HPLC analysis) was added to N-methyl-2-pyrrolidone (20 mL, 10 Vol.) pre-heated at 120 °C. The reaction mixture was stirred at 120 °C for 1 h, then cooled down to room temperature and poured into water (200 mL, 100 Vol.). The pH was adjusted to 4.5 by addition of aq. HCl (16% w/w) and the precipitate that formed was isolated by filtration. Drying in a drying oven at 45 °C for 18 h furnished Pt-MADEC (compound 7) as a yellow solid (1.65 g, 93% yield, purity 85.8%), containing 6.9% of Pt-lactone (compound 7b) and 1.4% of hydro-Pt-MADEC (compound 7a) according to HPLC analysis.
Example 3: Purification of Pt-MADEC 7 by precipitation of the corresponding potassium salt

[00102] Pt-MADEC (compound 7) (4 g, corresponding to 9.9 mmol of product considering the residual solvent content, prepared according to example 1) was added to aqueous KOH (12.9 mmol of KOH in 40 mL of water, 10 Vol.). The solid dissolved rapidly, and after 0.5 h, the formation of a precipitate started. After 0.5 h at room temperature the reaction mixture was cooled to 0 °C. After 1 h at 0 °C, the precipitate that had formed was isolated by filtration. Drying in a drying oven at 45 °C for 18 h furnished K-Pt-MADEC (compound 11) as a pale yellow solid (2.5 g, 65% yield, purity 99.5%), containing 0.3% of K-Pt-lactone-open (compound 11b) and 0.1% of hydro-K-Pt-MADEC (compound 11a) according to HPLC analysis.
Example 4: Purification of Pt-MADEC 7 derived from Pt-MADAC containing Pt-NADAC (5%) and Pt-lactone as impurities

[00103] Pt-MADAC (compound 6) (10 g) containing 5% of impurity Pt-NADAC (compound 6c) (according to HPLC analysis) was subjected to the decarboxylation conditions described in Example 1. The isolated Pt-MADEC (compound 7) (8.7 g, 87% yield, purity 83.4%) contained 6.2% of Pt-lactone (compound 7b), 1.6% of Pt- NADEC (compound 7c) and 3.4% of unreacted Pt-NADAC (compound 6c) according to HPLC analysis.
[00104] The compound was subjected to the purification conditions described in Example 3, furnishing K-Pt-MADEC (compound 11) (5.3 g, 55% yield, purity 98.5%), containing 0.3% of K-Pt-lactone-open (compound lib), 0.88% of K-Pt-NADEC (compound 11c) and 0.18% of K-Pt-NADAC (compound 1 Id) according to HPLC analysis.
Example 5: Purification of Pt-MADEC 7 by precipitation of the corresponding sodium salt

[00105] Pt-MADEC (compound 7) (2 g, corresponding to 4.95 mmol of product considering the residual solvent content) was added to aqueous NaOH (6.44 mmol of NaOH in 20 mL of water, 10 Vol.). The solid dissolved rapidly, then after 5 minutes the formation of a precipitate started. After 0.5 h at room temperature, the reaction mixture was cooled to 0 °C. After 1 h at 0 °C the precipitate that had formed was isolated by filtration. The collected solid was then dried in a drying oven at 45 °C for 18 h to provide the Na-Pt-MADEC (compound 12) as a pale yellow solid (1.6 g, 75% yield, purity 96.6%), containing 1.8% of Na-Pt-lactone-open (compound 12b) and 0.3% of hydro-Na-Pt-MADEC (compound 12a) according to HPLC analysis.
Example 6: Purification of Pt-MADEC 7 by precipitation of the corresponding lithium salt

[00106] Pt-MADEC (compound 7) (2 g, corresponding to 4.27 mmol of product, considering the residual solvent content), was added to aqueous LiOH (5.55 mmol of LiOH, in 20 mL of water, 10 Vol.). The solid dissolved rapidly, and after 15 minutes the formation of a precipitate started. After 0.5 h at room temperature the reaction mixture was cooled to 0 °C. After 1 h at 0 °C the precipitate that had formed was isolated by filtration. The collected solid was then dried in a drying oven at 45 °C for 18 h to provide Li-Pt-MADEC (compound 13) as a pale yellow solid (1.1 g, 73% yield, purity 97.7%), containing 0.6% of Li-Pt-lactone-open (compound 13b) and 0.9% of hydro-Li-Pt-MADEC (compound 13a) according to HPLC analysis.
Example 7: Synthesis of Pt-lactone
[00107] To a suspension of Pt-MAD AC (compound 6) (12.2 g, 31.2 mmol) in acetonitrile (122 mL, 10 Vol.), copper (I) iodide (595 mg, 3.12 mmol) and N,N-diisopropylethyl amine (10.9 mL, 62.2 mmol) were added. The reaction mixture was heated to reflux and stirred at reflux for 48 h., and then cooled to room temperature. The resulting precipitate was filtered and washed with acetonitrile (24 mL, 2 Vol.). The collected solid was then dried in a drying oven at 45 °C for 18 h to provide Pt-lactone (compound 7b) as a brown solid (12.3 g, >100% yield, purity 94.1%).
Example 8: Purification of Pt-MADES by formation of the corresponding DMF solvate
[00108] Pt-MADES (compound 5) (40 g, purity 94.7%) was added to DMF (400 mL, 10 Vol.), pre-heated at 120 °C. After dissolution of the product, the reaction mixture was kept at 120°C for 0.5 h, and then cooled to room temperature. The
resulting precipitate was filtered and washed with acetone (2 x 200 mL, 2 x 5 Vol.). Drying in a drying oven at 45 °C for 18 h furnished Pt-MADES (compound 5) as a pale yellow solid (36.4 g, 91% yield, purity 97.5%).
Example 9: purification of Pt-MADES
[00109] Pt-MADES (compound 5) (30 g, purity 94.7%) was suspended in a mixture of MeOH (300 mL, 10 Vol.) and formamide (150 mL, 5 Vol.). The suspension was heated at 60-65 °C for 2 h, then cooled to room temperature. Upon stirring at room temperature for 15 h, the resulting precipitate was filtered and washed with methanol (2 x 30 mL, 2 x 1 Vol.). Drying in a drying oven at 50 °C for 18 h furnished Pt-MADES (compound 5) as a pale yellow solid (22.5 g, 75% yield, purity 98.0%).
Example 10: purification of Pt-MADAC
[00110] Pt-MADAC (compound 6) (50 g, purity 94.0%) was suspended in a mixture of MeOH (150 mL, 3 Vol.) and formamide (50 mL, 1 Vol.). The suspension was heated at 60-65 °C for 2 h, and then cooled to room temperature. Upon stirring at room temperature for 15 h, a precipitate formed and was filtered and washed with methanol (2 x 50 mL, 2 x 1 Vol.). The collected precipitate was then dried in a drying oven at 50°C for 18 h. The thus-produced Pt-MADES was then suspended in MeOH (500 mL, 10 Vol.). This suspension was heated at reflux for 1 h, and then cooled to 0°C. The resulting precipitate was filtered and washed with methanol (2 x 50 mL, 2 x 1 Vol.). Drying in a drying oven at 50 °C for 18 h furnished Pt-MADAC (compound 6) as a pale yellow solid (41 g, 82% yield, purity 96.3%).
Example 11: Preparation of Pralatrexate, sodium salt (PLT-Na) (Compound 10a) by hydrolysis of Pralatrexate ethyl ester (PLT-ES) (Compound 9B)

[00111] A reactor was charged with EtOH (195 mL) and aqueous NaOH (3.75 M, 19.5 mL, 73 mmol). The mixture was then cooled down to 10 °C. 10-Propargyl-10-
deazaaminopterin ethyl ester (PLT-ES, Compound 9B, 13 g, 24.4 mmol) was added, and the temperature was increased to 25 °C over 0.5 h. The resulting suspension was then stirred at 25 °C for 17 h. The solid in the suspension was then isolated by filtration and washed with EtOH (65 mL). The collected solid was then dried in a drying oven at 45 °C for 18 h to provide (2S)-2-[[4-[(1 RS)-1-[(2,4-diaminopteridin-6-yl)methyl]but-3-ynyl]benzoyl]amino]pentanedioic acid disodium salt (Na-PLT; compound 10a) as a pale yellow solid (10.9 g, 86% yield, purity 99.6%).
Example 12: Preparation of Pralatrexate (Compound 10)

[00112] (2S)-2-[[4-[(1 RS)-1-[(2,4-Diaminopteridin-6-yl)methyl]but-3-ynyl]benzo-yl]amino]pentanedioic acid disodium salt (Na-PLT, Compound 10a, 10.9 g, 20.9 mmol) was dissolved in water (109 mL). The pH of the solution was adjusted to 4.5 by addition of aqueous HCl 1N. A precipitate formed and was isolated by filtration and washed with water (54 mL). The collected solid was then dried in a drying oven at 45 °C for 17 h to provide Pralatrexate (Compound 10) as a white solid (9.4 g, 81% yield, and purity 99.7%).
Example 13: Preparation of Pralatrexate ethyl ester (PLT-ES) (compound 9B)

[00113] A reactor was charged with potassium 10-propargyl-4-deoxy-4-amino-10-deazapteroate (K-Pt-MADEC, compound 11, 11.1 g, 28.7 mmol). DCM (82 ml) and 1-methyl-2-pyrrolidinone (16.7 ml) were added to obtain a suspension. 1-Hydroxy-benzotriazole hydrate (HOBt, 0.77 g, 5.74 mmol), N-(3-dimethylaminopropyl)-N’-ethylcarbodiimide hydrochloride (EDC, 6.60 g, 34.4 mmol) and (L)-glutamic acid
diethyl ester hydrochloride (7.98 g, 34.4 mmol) were sequentially added. The resulting mixture was stirred at room temperature until HPLC analysis showed reaction completion. DCM was removed under reduced pressure and MeOH (22.2 mL) was added. The reaction mixture was poured into water pre-acidified with aqueous HCl 16% (w/w, 16.9 mL). The pH was adjusted to 4.5 by addition of aqueous NaOH, resulting in the precipitation of the PLT-ES. The solid precipitate was isolated by filtration and dried in oven at 55 °C for 18 h to provide PLT-ES as a pale yellow solid (13 g, 85% yield, and purity 99.0%).
Example 14: Preparation of crystalline Pralatrexate ethyl ester (compound 9B)
[00114] PLT-ES (Compound 9B, lg) was dissolved in EtOH (15 ml). After a few minutes a precipitate started to form, and the precipitate was isolated by filtration after 45 minutes (0.5g, yellow solid, amorphous form). The mother liquor was left at room temperature overnight, resulting in the precipitation of a yellow solid which was isolated by filtration (0.3g, crystalline form, PXRD is shown on Figure 7).
Example 15: Hydrolysis of Pralatrexate ethyl ester (compound 9B)

[00115] A reactor was charged with MeOH (48 mL), aqueous NaOH (3.75 M, 18.0 mL, 67.5 mmol), and water (6 mL), and the mixture was cooled down to 10 °C. PLT-ES 9B (12 g, 22.5 mmol) was added and the temperature was increased to 25 °C over 1 h. The resulting suspension was stirred at 25 °C for 1 h, and then EtOH (168 mL) was added, resulting in the formation of a precipitate. After stirring for an additional 24 h the solid precipitate was isolated by filtration and washed with EtOH (120 mL).
The collected solid was then dried in a drying oven at 60 °C for 18 h to provide Na-PLT 10a (10.5 g, 90% yield, purity 99.8%) as a pale yellow solid.
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014068599
Pralatrexate is chemically, N-(4-{ 1-[(2,4-diaminopteridin-6-yl)methyl]but-3-yn-1 -yl}benzoyl)-L-glutamic acid and has the structural formula:

Pralatrexate is an anti-cancer therapy. It is the first drug approved as a treatment for patients with relapsed or refractory peripheral T-cell lymphoma, or PTCL – a biologically diverse group of aggressive blood cancers. Pralatrexate is currently marketed under the trade name FOLOTYN® by Alios.
Pralatrexate was disclosed in U.S. patent nos. 5,354,751 and 6,028,071.
According to the ‘071 patent, alpha-propargylhomoterephthalic acid dimethyl ester substantially free of homoterephthalic acid dimethyl ester was obtained by chromatographing alpha-propargylhomoterephthalic acid dimethyl ester residue obtained as part of the reaction between homoterephthalic acid dimethyl ester and propargyl bromide in the presence of
tetrahydrofuran and sodium hydride on silica gel using cyclohexane and ethyl acetate (8:1) for the elution.
Pralatrexate was also reported in J. Med. Chem, 1993, 36, 2228-2231. According to the paper, pralatrexate is prepared by crystallizing pralatrexate diethyl ester in a mixture of 2-methoxyethanol and water in the presence of sodium hydroxide.
International patent application publication no. WO 2012/061469 (‘469 patent) disclosed crystalline Form A, Form B and Form C of pralatrexate. According to the ‘469 patent, crystalline pralatrexate Form A can be prepared by crystallizing amorphous pralatrexate in formamide.
According to the ‘469 patent, crystalline pralatrexate Form B can be prepared by crystallizing amorphous pralatrexate in methanol or water.
According to the ‘469 patent, crystalline pralatrexate Form C can be prepared by crystallizing amorphous pralatrexate in a mixture of methanol and water.
Alpha-propargylhomoterephthalic acid dimethyl ester is a key staring material for the preparation of pralatrexate.
Example 1 :
Preparation of Alpha-propargylhomoterephthalic acid dimethyl ester
Sodium hydride (60 gm; 60%) was added to tetrahydrofuran (1500 ml) at room temperature and then cooled to 10 to 15°C. To the solution was added a solution of homoterephthalic acid dimethyl ester (250 gm) in tetrahydrofuran (250 ml) slowly for 15 minutes. The reaction mass was then cooled to 0 to -5°C and then added propargyl bromide (130 gm) in tetrahydrofuran (125 ml) slowly for 15 minutes at 0 to -5°C. The reaction mass was maintained for 2 hours at 0 to -5°C and then added methanol (50 ml). The temperature of the reaction mass was raised to room temperature and then added water (1500 ml) and diisopropyl ether (2500 ml), and then the layers were separated. The organic layer were dried with sodium sulfate and then concentrated to obtain 275 gm of alpha-propargylhomoterephthalic acid dimethyl ester.
Chromatographic purity of alpha-propargylhomoterephthalic acid dimethyl ester: 62.0%; Content of homoterephthalic acid dimethyl ester: 12.0%.
Example 2:
Purification of Alpha-propargylhomoterephthalic acid dimethyl ester
Alpha-propargylhomoterephthalic acid dimethyl ester (275 gm; HPLC Purity: 62.0%) as obtained in example 1 was dissolved in a mixture of hexane (1200 ml) and diisopropyl ether (65 ml) at room temperature. The solution was stirred for 15 hours at room temperature and filtered. The solid obtained was dried to obtain 175 gm of alpha-propargylhomoterephthalic acid dimethyl ester.
Chromatographic purity of alpha-propargylhomoterephthalic acid dimethyl ester: 74.6%; Content of homoterephthalic acid dimethyl ester: 0.4%.
Example 3:
Preparation of 10-propargyl-10-deazaminopterin diethyl ester
Step-I: Preparation oƒ 10-proparsyl-10-carbomethoxy-4-deoxy-4-amino-10-deazapteroic acid methyl ester
Sodium hydride (120 gm; 60%) was added to dimethylformamide (750 ml) at room temperature and then cooled to 0 to -5°C. To the solution was added a solution of alpha-propargylhomoterephthalic acid dimethyl ester (250 gm) in dimethylformamide (750 ml)
slowly for 15 minutes. The reaction mixture was maintained for 30 minutes at 0 to -5 C and then cooled to -20 to -25°C. To the reaction mixture was added 6-bromomethyl-pteridine-2,4-diamine (300 gm) in dimethylformamide (1500 ml) slowly for 30 minutes. The reaction mass was maintained for 2 hours at -20 to -25°C and then added methanol (300 ml). The temperature of the reaction mass was raised to room temperature and then added water (15000 ml) and diisopropyl ether (1500 ml). The contents were stirred for 2 hours at room temperature and filtered. The solid obtained was dried to obtain 198 gm of 10-propargyl-10-carbomethoxy-4-deoxy-4-amino-10-deazapteroic acid methyl ester.
Step-II: Preparation oƒ 10-proparsyl-10-carboxy-4-deoxy-4-amino-10-deazapteroic acid
2-Methoxyethanol (775 gm) was added to 10-propargyI-10-carbomethoxy-4-deoxy-4-amino-10-deazapteroic acid methyl ester (155 gm) at room temperature and then cooled to 15 to 20°C. To the reaction mixture was added a solution of sodium hydroxide (120 gm) in water (930 ml) and maintained for 4 hours at room temperature. The pH of the reaction mass was adjusted to 4.5 to 4.6 with acetic acid (50%) and then added water (3100 ml). The reaction mass was stirred for 2 hours, filtered and then dried to obtain 125 gm of 10-propargyl-10-carboxy-4-deoxy-4-amino-10-deazapteroic acid.
Step-III: Preparation oƒ 10-proparsyl-4-deoxy-4-amino-10-deazapteroic acid
10-Propargyl-10-carboxy-4-deoxy-4-amino-10-deazapteroic acid (135 gm) was added to dimethyl sulfoxide (1350 ml) at 120 to 125°C and maintained for 45 minutes at 120 to 125°C. The reaction mass was poured into water (3000 ml), maintained for 24 hours at room temperature and filtered to obtain a wet solid. To the wet solid was basified and then acetified, and maintained for 2 hours at room temperature. The separated solid was filtered and then dried to obtain 56 gm of 10-propargyl-4-deoxy-4-amino-10-deazapteroic acid.
Step-IV: Preparation oƒ 10-propargyl-10-deazaminopterin diethyl ester
Dimethylformamide (1 12 ml) was added to 10-propargyl-4-deoxy-4-amino-10-deazapteroic acid (14 gm) and stirred for 15 minutes. To the reaction mixture was added triethylamine (14 ml) and then cooled to 0 to -5°C. A solution of (benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate (21 gm) in dimethylformamide (28 ml) was added to the reaction mixture and maintained for 1 hour at 0 to -5°C. To the
reaction mixture was added L-glutamic acid diethyl ester (10 gm) in dimethylformamide (28 ml) slowly, maintained for 2 hours at -10 to -15°C and filtered. The pH of the filtrate obtained was adjusted with sodium hydroxide solution and then added water (700 ml) slowly for 45 minutes. The reaction mass was maintained for 2 hours at room temperature, filtered and then dried to obtain 14 gm of 10-propargyl-10-deazaminopterin diethyl ester.
Example 4:
Preparation of pralatrexate
10-Propargyl-10-deazaminopterin diethyl ester (40 gm) was dissolved in tetrahydrofuran (320 ml) at room temperature. The solution was then cooled to 15 to 20°C and added a solution of sodium hydroxide (24 gm) in water (400 ml) slowly for 15 minutes. The reaction mass was maintained for 45 minutes at 15 to 20°C and then added a mixture of tetrahydrofuran (200 ml) and ethyl acetate (200 ml). The layers were separated and to the aqueous layer was added water (80 ml). The separated aqueous layer was then concentrated and pH was adjusted to 4.7 to 4.8 with acetic acid (10%). The contents were stirred for 1 hour at room temperature and filtered. The solid obtained was then dried to obtain 28 gm of pralatrexate.
Chromatographic purity of pralatrexate: 98.5%;
Content of 10-propargyl-4-deoxy-4-amino-10-dezapteroic acid: 0.3%;
Content of 10-deazaaminopterin: 0.5%;
Example 5:
Purification of pralatrexate
The pralatrexate (28 gm: HPLC Purity: 98.5%) as obtained in example 4 was dissolved in tetrahydrofuran (400 ml) and then heated to 60°C. To the contents were added water (200 ml) at 60°C and then cooled to 5 to 10°C. The contents were stirred for 2 hours 30 minutes at 5 to 10°C, filtered and then dried to obtain a solid. The solid was dissolved in dimethyl sulfoxide (138 ml) and then stirred to obtain a clear solution. The solution was filtered through celite bed and then added ethanol (690 ml) slowly for 1 hour. The contents were stirred for 1 hour at room temperature, filtered and then dried to obtain 20 gm of pure pralatrexate.
Chromatographic purity of pralatrexate: 99.5%;
Content of 10-propargyl-4-deoxy-4-amino-10-dezapteroic acid: 0.06%; Content of 10-deazaaminopterin: 0.08%.
PAPER
Nonpolyglutamatable Antifolate N-alpha-(4 amino-4-deoxypteroyl)-Ndelta-hemiphthaloyl-L-ornithine”, JOURNAL OF MEDICINAL CHEMISTRY, AMERICAN CHEMICAL SOCIETY, US, vol. 45, no. 8, 1 January 2002 (2002-01-01), pages 1690 – 1696, XP002291409, ISSN: 0022-2623, DOI: 10.1021/JM010518T *

Details are disclosed for the synthesis of Nα-[4-[2-(2,4-diaminoquinazolin-6-yl)ethyl]benzoyl]-Nδ-hemiphthaloyl-l-ornithine (2) and Nα-[4-[5-(2,4-diaminoteridin-6-yl)pent-1-yn-4-yl]benzoyl]-Nδ-hemiphthaloyl-l-ornithine (6) as analogues of Nα-(4-amino-4-deoxypteroyl)-Nδ-hemiphthaloyl-l-ornithine (1, PT523), a nonpolyglutamatable antifolate currently in advanced preclinical development. In a 72 h growth inhibition assay against cultures of CCRF-CEM human leukemic lymphoblasts, the IC50 of 2 and 6 was 0.69 ± 0.044 nM and 1.3 ± 0.35 nM, respectively, as compared with previously reported values 4.4 ± 0.10 nM for aminopterin (AMT) and 1.5 ± 0.39 nM for PT523. In a spectrophotometric assay of dihydrofolate reductase (DHFR) inhibition using dihydrofolate and NADPH as the cosubstrates, the previously unreported compounds 2 and the mixed 10R and 10S diastereomers of 6 had Ki values of 0.21 ± 0.05 pM and 0.60 ± 0.02 pM, respectively, as compared with previously reported values of 3.70 ± 0.35 pM for AMT and 0.33 ± 0.04 pM for PT523. Thus, while they were comparable to 1 and several of its previously studied analogues in their ability to bind to DHFR and inhibit the growth of CCRF-CEM cells, 2 and the mixed diastereomers of 6 were several times more active than AMT despite the fact that they cannot form γ-polyglutamylated metabolites of the type formed in cells from AMT and other classical antifolates with a glutamate side chain.
Synthesis and In Vitro Antitumor Activity of New Deaza Analogues of the Nonpolyglutamatable Antifolate Nα-(4-Amino-4-deoxypteroyl)-Nδ-hemiphthaloyl-l-ornithine (PT523)

PATENT
https://patents.google.com/patent/CN107488112A/zh
PATENT
US 20150183789
https://patents.google.com/patent/US9440979B2/en
PATENT
Publication numberPriority datePublication dateAssigneeTitle
References
- ^ Jump up to:a b c d e [1], Allos Therapeutics Press Release, “Allos Therapeutics’ Pralatrexate Demonstrates Anticancer Activity in Multiple Cancer Cell Lines”.
- ^ Jump up to:a b c d [2], Allos Therapeutics Press Release, “Allos Therapeutics’ FOLOTYN(TM) First and Only FDA-Approved Therapy for Relapsed or Refractory Peripheral T-cell Lymphoma”.
- Jump up^ [3], FDA, “Fast Track, Accelerated Approval and Priority Review”.
- Jump up^ [4], Allos Therapeutics, “Allos Therapeutics, Inc. Q1 2010 Earnings Call Transcript”.
- ^ Jump up to:a b c [5], Psychiatric Times, “Principles of Oncologic Pharmacotherapy”.
- ^ Jump up to:a b [6], Memorial Sloan Kettering Cancer Center Press Release, “FDA Approves Lymphoma Drug Developed at Memorial Sloan Kettering”.
- Jump up^ [7], National Cancer Institute “NCI Cancer Bulletin: The Next Steps in Drug Development at NCI”.
- Jump up^ “FDA Approves Pralatrexate for Treatment of Peripheral T-Cell Lymphoma” (Press release). SRI International. 2009-09-25. Retrieved 2013-07-10.
- Jump up^ Avery, Greg (2012-09-07). “Purchase of Allos Therapeutics is completed”. Denver Business Journal. Retrieved 2013-07-10.
External links
- Pralatrexate Development Information
- Clinical trials of pralatrexate at ClinicalTrials.gov
- Folotyn website
- FocusonPTCL website
- ASAP Support For Assisting Patients
 |
|
 |
|
| Clinical data | |
|---|---|
| Trade names | Folotyn |
| AHFS/Drugs.com | Monograph |
| License data |
|
| Pregnancy category |
|
| Routes of administration |
Intravenous |
| ATC code | |
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| ChemSpider | |
| UNII | |
| ChEBI | |
| ChEMBL | |
| ECHA InfoCard | 100.205.791 |
| Chemical and physical data | |
| Formula | C23H23N7O5 |
| Molar mass | 477.47 g/mol |
| 3D model (JSmol) | |
////////////プララトレキサート , japan 2017, Pralatrexate
NC1=NC2=NC=C(CC(CC#C)C3=CC=C(C=C3)C(=O)N[C@@H](CCC(O)=O)C(O)=O)N=C2C(N)=N1
Amenamevir アメナメビル
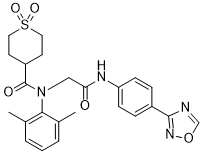
Amenamevir アメナメビル
M-5220
CAS 841301-32-4
Chemical Formula: C24H26N4O5S
Molecular Weight: 482.555
N-(2-((4-(1,2,4-oxadiazol-3-yl)phenyl)amino)-2-oxoethyl)-N-(2,6-dimethylphenyl)tetrahydro-2H-thiopyran-4-carboxamide 1,1-dioxide
PMDA

| 2017/7/3 | PMDA | APPROVED | JAPAN | Amenamevir | Amenalief
BRAND |
Maruho
COMPANY |

![]()
Amenamevir, also known as ASP2151, is a herpes virus helicase-primase inhibitor. ASP2151 had significantly better anti-HSV activity against herpes simplex keratitis than valacyclovir and acyclovir after systemic or topical use.
| アメナメビル Amenamevir  C24H26N4O5S : 482.55 [841301-32-4] |
Amenamevir is an oral helicase-primase inhibitor launched in 2017 in Japan for the treatment of herpes zoster (shingles). The product is being marketed by Maruho.
Amenamevir had been in phase III clinical trials for herpes simplex virus;
In August 2012, Astellas Pharma granted Maruho development and commercialization rights in Japan.
US 20050032855
WO 2006082822
WO 2006082820
WO 2006082821
WO 2009123169
WO 2010047295
JP 2006241144
Patent
The publicly known crystal (following and alpha type crystal) of the compound A of disclosure to the aforementioned Patent document 2 is obtained by re-crystallizing from an ethanol water mixed solvent, and has the melting point of about 220 to 222 degree C. The present invention relates to multi-form crystals other than the alpha form crystal concerned, and relates to beta, gamma, delta, and epsilon type crystal specifically. In a surprising thing, each of these multi-form crystals is crystals stable to a degree usable as a medicinal manufacture field object, and has a preferable property in the surface of solubility, absorbency, stability, and/or a handling property
PATENT
US20050032855, EP1844776A1.

REFERENCES
1: Ohtsu Y, Otsuka S, Nakamura T, Noguchi K. Regulated bioanalysis of conformers – A case study with ASP2151 in dog plasma and urine. J Chromatogr B Analyt Technol Biomed Life Sci. 2015 Aug 1;997:56-63. doi: 10.1016/j.jchromb.2015.05.028. Epub 2015 May 28. PubMed PMID: 26093120.
2: James SH, Larson KB, Acosta EP, Prichard MN. Helicase-primase as a target of new therapies for herpes simplex virus infections. Clin Pharmacol Ther. 2015 Jan;97(1):66-78. doi: 10.1002/cpt.3. Epub 2014 Nov 18. Review. PubMed PMID: 25670384.
3: Muylaert I, Zhao Z, Elias P. UL52 primase interactions in the herpes simplex virus 1 helicase-primase are affected by antiviral compounds and mutations causing drug resistance. J Biol Chem. 2014 Nov 21;289(47):32583-92. doi: 10.1074/jbc.M114.609453. Epub 2014 Oct 2. PubMed PMID: 25278021; PubMed Central PMCID: PMC4239612.
4: Biswas S, Sukla S, Field HJ. Helicase-primase inhibitors for herpes simplex virus: looking to the future of non-nucleoside inhibitors for treating herpes virus infections. Future Med Chem. 2014 Jan;6(1):45-55. doi: 10.4155/fmc.13.192. Review. PubMed PMID: 24358947.
5: Andrei G, Snoeck R. Advances in the treatment of varicella-zoster virus infections. Adv Pharmacol. 2013;67:107-68. doi: 10.1016/B978-0-12-405880-4.00004-4. Review. PubMed PMID: 23886000.
6: Sasaki S, Miyazaki D, Haruki T, Yamamoto Y, Kandori M, Yakura K, Suzuki H, Inoue Y. Efficacy of herpes virus helicase-primase inhibitor, ASP2151, for treating herpes simplex keratitis in mouse model. Br J Ophthalmol. 2013 Apr;97(4):498-503. doi: 10.1136/bjophthalmol-2012-302062. Epub 2013 Jan 29. PubMed PMID: 23361434.
7: Katsumata K, Chono K, Kato K, Ohtsu Y, Takakura S, Kontani T, Suzuki H. Pharmacokinetics and pharmacodynamics of ASP2151, a helicase-primase inhibitor, in a murine model of herpes simplex virus infection. Antimicrob Agents Chemother. 2013 Mar;57(3):1339-46. doi: 10.1128/AAC.01803-12. Epub 2012 Dec 28. PubMed PMID: 23274658; PubMed Central PMCID: PMC3591930.
8: Chono K, Katsumata K, Suzuki H, Shiraki K. Synergistic activity of amenamevir (ASP2151) with nucleoside analogs against herpes simplex virus types 1 and 2 and varicella-zoster virus. Antiviral Res. 2013 Feb;97(2):154-60. doi: 10.1016/j.antiviral.2012.12.006. Epub 2012 Dec 20. PubMed PMID: 23261844.
9: Chono K, Katsumata K, Kontani T, Shiraki K, Suzuki H. Characterization of virus strains resistant to the herpes virus helicase-primase inhibitor ASP2151 (Amenamevir). Biochem Pharmacol. 2012 Aug 15;84(4):459-67. doi: 10.1016/j.bcp.2012.05.020. Epub 2012 Jun 9. PubMed PMID: 22687623.
10: Katsumata K, Weinberg A, Chono K, Takakura S, Kontani T, Suzuki H. Susceptibility of herpes simplex virus isolated from genital herpes lesions to ASP2151, a novel helicase-primase inhibitor. Antimicrob Agents Chemother. 2012 Jul;56(7):3587-91. doi: 10.1128/AAC.00133-12. Epub 2012 Apr 23. PubMed PMID: 22526302; PubMed Central PMCID: PMC3393391.
11: Tyring S, Wald A, Zadeikis N, Dhadda S, Takenouchi K, Rorig R. ASP2151 for the treatment of genital herpes: a randomized, double-blind, placebo- and valacyclovir-controlled, dose-finding study. J Infect Dis. 2012 Apr 1;205(7):1100-10. doi: 10.1093/infdis/jis019. Epub 2012 Feb 20. PubMed PMID: 22351940.
12: Himaki T, Masui Y, Chono K, Daikoku T, Takemoto M, Haixia B, Okuda T, Suzuki H, Shiraki K. Efficacy of ASP2151, a helicase-primase inhibitor, against thymidine kinase-deficient herpes simplex virus type 2 infection in vitro and in vivo. Antiviral Res. 2012 Feb;93(2):301-4. doi: 10.1016/j.antiviral.2011.11.015. Epub 2011 Dec 4. PubMed PMID: 22155691.
13: Katsumata K, Chono K, Sudo K, Shimizu Y, Kontani T, Suzuki H. Effect of ASP2151, a herpesvirus helicase-primase inhibitor, in a guinea pig model of genital herpes. Molecules. 2011 Aug 25;16(9):7210-23. doi: 10.3390/molecules16097210. PubMed PMID: 21869749.
14: Andrei G, Snoeck R. Emerging drugs for varicella-zoster virus infections. Expert Opin Emerg Drugs. 2011 Sep;16(3):507-35. doi: 10.1517/14728214.2011.591786. Epub 2011 Jun 24. Review. PubMed PMID: 21699441.
15: Chono K, Katsumata K, Kontani T, Kobayashi M, Sudo K, Yokota T, Konno K, Shimizu Y, Suzuki H. ASP2151, a novel helicase-primase inhibitor, possesses antiviral activity against varicella-zoster virus and herpes simplex virus types 1 and 2. J Antimicrob Chemother. 2010 Aug;65(8):1733-41. doi: 10.1093/jac/dkq198. Epub 2010 Jun 9. PubMed PMID: 20534624
///////////Amenamevir, アメナメビル, japan 2017, ASP2151, ASP 2151, M-5220, MARUHO, Amenalief
O=C(C(CC1)CCS1(=O)=O)N(C2=C(C)C=CC=C2C)CC(NC3=CC=C(C4=NOC=N4)C=C3)=O
Forodesine Hydrochloride

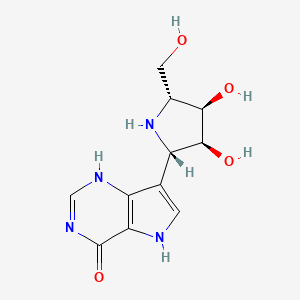
Forodesine
- Molecular FormulaC11H14N4O4
- Average mass266.253 Da

CAS No. : 284490-13-7
Molecular Formula: C11H15ClN4O4
Average Mass: 302.72 g/mol
Forodesine (INN; also known as Immucillin H; trade names Mundesine and Fodosine) is a transition-state analog inhibitor of purine nucleoside phosphorylase[1] studied for the treatment of patients with T-cell acute lymphoblastic leukemia (T-ALL) and for treatment of B-cell acute lymphocytic leukemia (B-ALL).
Forodesine was originally discovered by Vern Schramm‘s laboratory at the Albert Einstein College of Medicine in New York and Industrial Research Limited in New Zealand.
Forodesine is being developed by BioCryst Pharmaceuticals. As of 2008, it is currently in phase II clinical trials.[2].
In 2006, BioCryst entered into a licensing agreement with Mundipharma International Holdings Limited to develop and commercialize forodesine in markets across Europe, Asia, and Australasia for use in oncology.[3]
In April 2017, forodesine was approved in Japan for the treatment of relapsed/refractory peripheral T-cell lymphoma.[4]
ema
On 20 September 2010, orphan designation (EU/3/10/780) was granted by the European Commission to Mundipharma Research Limited, United Kingdom, for forodesine for the treatment of chronic lymphocytic leukaemia
| Active substance | Forodesine hydrochloride |
|---|---|
| Decision number | P/69/2010 |
| PIP number | EMEA-000785-PIP01-09 |
| Pharmaceutical form(s) | Hard capsule |
| Condition(s)/indication(s) | Cutaneous T-cell lymphoma (CTCL) |
| Route(s) of administration | Oral use |
| PIP applicant | Applicant: Mundipharma Research Ltd E-mail: paediatric@mundipharma-rd.eu Country: United Kingdom Phone: +44 1223424900 Fax: +44 1223426054 |
| Decision type | W: decision granting a waiver in all age groups for the listed condition(s) |
On 20 September 2010, orphan designation (EU/3/10/780) was granted by the European Commission to Mundipharma Research Limited, United Kingdom, for forodesine for the treatment of chronic lymphocytic leukaemia.
What is chronic lymphocytic leukaemia?
Chronic lymphocytic leukaemia (CLL) is cancer of a type of white blood cell called B lymphocytes. In this disease, the lymphocytes multiply too quickly and live for too long, so that there are too many of them circulating in the blood. The cancerous lymphocytes look normal, but they are not fully developed and do not work properly. Over a period of time, the abnormal cells replace the normal white blood cells, red blood cells and platelets (components that help the blood to clot) in the bone marrow (the spongy tissue inside the large bones in the body). CLL is the most common type of leukaemia and mainly affects older people. It is rare in people under the age of 40 years. CLL is a long-term debilitating and life-threatening disease because some patients develop severe infections. What is the estimated number of patients affected by the condition? At the time of designation, CLL affected approximately 3 in 10,000 people in the European Union (EU)*. This is equivalent to a total of around 152,000 people, and is below the threshold for orphan designation, which is 5 people in 10,000. This is based on the information provided by the sponsor and the knowledge of the Committee for Orphan Medicinal Products (COMP).
What treatments are available? Treatment for CLL is complex and depends on a number of factors, including the extent of the disease, whether it has been treated before, and the patient’s age, symptoms and general state of health. Patients whose CLL is not causing any symptoms or is only getting worse very slowly may not need
Forodesine Hydrochloride was originally developed by BioCryst Pharmaceuticals and then licensed to Mundipharma and in particular is marketed in Japan under the trade name Mundesine®. Forodesine Hydrochloride is a transitional analogue inhibitor of purine nucleoside phosphorylase (PNP). Mundesine® is approved for the treatment of peripheral T-cell lymphoma (PTCL).
Mundesine® is a capsule that contains 100mg of free Forodesine per capsule. The recommended dose is 300mg orally, twice daily.
In 2004, the compound was eligible for orphan drug treatment for non-Hodgkin’s lymphoma (NHL), chronic myelogenous leukemia (CLL) and hairy cell leukemia, respectively. In 2007, the compound was eligible for the EU orphan drug for the treatment of acute lymphoblastic leukemia (ALL) and cutaneous T-cell lymphoma (CTCL). In 2010, the compound was eligible for EU orphan drug for treatment of CLL. In 2006, the compound obtained Japanese orphan drug eligibility for CTCL treatment.
Forodesine, or 7-[(2S,3S,4R,5R)-3,4-dihydroxy-5-(hydroxymethyl)-2-pyrrolidinyl]-l,5-dihydropyrrolo[2,3-e]pyrimidin-4-one, is an inhibitor of purine nucleoside phosphorylase. It is currently in development as a treatment for peripheral T-Cell Lymphoma .
W099/19338 describes a compound genus as a new class of inhibitors of nucleoside metabolism, including Forodesine. The compounds effect as inhibitors of purine nucleoside phosphorylase is taught as efficacious to suppress T-cell function and to treat infections caused by protozoan parasites.
WO00/61783 describes a number of processes for preparing molecules described in W099/19338. Reaction scheme 3 on page 23 of the published application describes a synthesis of Forodesine, characterised by the removal of two acid labile protecting groups in the final step to yield the hydrochloride salt.
Forodesine is a particularly difficult molecule to make on a commercial scale. The current process for manufacture requires a coupling reaction under cryogenic temperature conditions of -55C. Subsequent steps involve the use of a high pressure hydrogenation reaction. Such extreme reaction conditions provide for safety concerns, particularly when conducted on a bulk scale. Further the products of the reaction were extremely challenging to purify. The effect of all this is to require more sophisticated and expensive equipment at the manufacturing plant; all of which add up to an increased cost of goods for patients. Accordingly a new manufacturing process was sought.
Surprisingly a new route has been invented which is shorter, cheaper, less dangerous and provides an increased overall yield whilst still conforming to the required purity profile.
The current manufacturing process is described in Fig 1.
5C


, MeOH, reflux xchange
tallisation

Fig l
Within the diagram, the following acronyms are used, wherein NCS is N-Chlorosuccinimide, OTBDMS is t-butyldimethylsiloxy protecting group, MtBE is methyl t-butyl ether, (BOC)20 is di-t-butyldicarbonate and BOC is t-butyloxycarbonyl protecting group,
Particularly problematic in this process is the requirement to conduct the coupling of process step (iii) at exceptionally low temperature. Further challenges are provided by process step (v) the hydrogenation reaction to remove the benxylyoxymethyl (BOM) protecting group, before removing the other acid labile protecting groups.
Conducting hydrogenation reactions with their need for a high pressure environment requires specialist equipment. Such apparatus is expensive, adding to the cost of the materials produced. Despite the use of specialist equipment, safety concerns can never be eradicated. Whilst BOM can, in certain circumstances, be acid labile, treatment of analogues of the molecules described in Fig 1 with acid has always resulted in incomplete removal of the protecting group, leading to a large number of partially deprotected impurities. This makes purification exceptionally difficult as well as reducing the overall yield for the step.
A new improved process has been developed as described in Fig 2:
Toluene



Fig 2
The new route has a number of clear advantages. The coupling reaction (ix) is conducted at a warmer -15°C, rather than the challenging cryogenic conditions of -55°C required previously. It eradicates the hydrogenation step, avoiding the need for dangerous high pressure conditions. It also makes the overall process much quicker and cheaper; not only are the conditions challenging, but the reagents used in large quantities such as palladium are expensive and environmentally challenging.
The classical method to remove a BOM protecting group is by catalytic hydrogenation. It is however known to be unstable in acid conditions. For this reasons there have been previous attempts to remove BOM at the same time as the three acid labile protecting groups. This has always been unsuccessful as treatment with acid typically resulted in incomplete deprotection, leading to a mixture of products. This made for a tricky purification and a reduced yield. Surprisingly under the particular conditions described herein it has been possible to effect the transformation in greater yield and without a difficult purification. The final product is obtained in equal or greater purity than material obtained from the previous route.
PATENT
Scheme 13
HO OH 1 . HCI/Acetone, MeOH OCH,
2. PPh3, imidazole I
HO (EtO)2POCH2CN
OH O O
Ribose Λ 13a

References for preparation of compound 13a:
1. Mishra, Girija Prasad; Rao, Batchu Venkateswara; Tetrahedron: Asymmetry (2011), 22(7), 812-817.
2. Brock, E. Anne; Davies, Stephen G.; Lee, James A.; Roberts, Paul M.; Thomson,
James E; Organic Letters (2011), 13(7), 1594-1597.
3. WO 2010/085377 A2 (incorporated by reference).
4. Yadav, J. S.; Reddy, P. Narayana; Reddy, B. V. Subba; Synlett (2010), (3), 457- 461.
5. Song, Kai; Zheng, Guo-jun; Huaxue Shiji (2010), 32(2), 171-172.
6. Prabhakar, Peddikotla; Rajaram, Singanaboina; Reddy, Dorigondla Kumar;
Shekar, Vanam; Venkateswarlu, Yenamandra; Tetrahedron: Asymmetry (2010), 21(2), 216-221.
7. CN 101182342 A.
8. Baird, Lynton J.; Timmer, Mattie S. M.; Teesdale-Spittle, Paul H.; Harvey, Joanne
E; Journal of Organic Chemistry (2009), 74(6), 2271-2277.
9. Wang, Xiang-cheng; Wang, Gang; Qu, Gang-lian; Huaxue Shijie (2008), 49(4), 226-228.
10. Ivanova, N. A.; Valiullina, Z. R.; Shitikova, O. V.; Miftakhov, M. S; Russian
Journal of Organic Chemistry (2007), 43(5), 742-746.
11. Braga, Fernanda Gambogi; Coimbra, Elaine Soares; Matos, Magnum de Oliveira;
Lino Carmo, Arturene Maria; Cancio, Marisa Damato; da Silva, Adilson David; European Journal of Medicinal Chemistry (2007), 42(4), 530-537.
12. Wender, Paul A.; Bi, F. Christopher; Buschmann, Nicole; Gosselin, Francis; Kan, Cindy; Kee, Jung-Min; Ohmura, Hirofumi; Organic Letters (2006), 8(23), 5373- 5376.
13. Fei, Xiangshu; Wang, Ji-Quan; Miller, Kathy D.; Sledge, George W.; Hutchins, Gary D.; Zheng, Qi-Huang; Nuclear Medicine and Biology (2004), 31(8), 1033- 1041.
14. Abdel-Rahman, Adel A.-H.; Abdel-Megied, Ahmed E.-S.; Goda, Adel E.-S.; Zeid,
Ibrahim F.; El Ashry, El Sayed H; Nucleosides, Nucleotides & Nucleic Acids (2003), 22(11), 2027-2038.
15. Palmer, Andreas M.; Jager, Volker; European Journal of Organic Chemistry
(2001), (7), 1293-1308.
16. Paquette, Leo A.; Bailey, Simon; Journal of Organic Chemistry (1995), 60(24),
7849-56.
17. Classon, Bjoern; Liu, Zhengchun; Samuelsson, Bertil; Journal of Organic
Chemistry (1988), 53(26), 6126-30.
18. Kissman, Henry M.; Baker, B. R; Journal of the American Chemical Society
(1957), 79 5534-40.
References for cyclizations related to preparation of compounds of type 13d:
1. Davies, Stephen G.; Durbin, Matthew J.; Goddard, Euan C; Kelly, Peter M.;
Kurosawa, Wataru; Lee, James A.; Nicholson, Rebecca L.; Price, Paul D.;
Roberts, Paul M.; Russell, Angela J.; Scott, Philip M.; Smith, Andrew D; Organic & Biomolecular Chemistry (2009), 7(4), 761-776.
2. Davies, Stephen G.; Nicholson, Rebecca L.; Price, Paul D.; Roberts, Paul M.;
Russell, Angela J.; Savory, Edward D.; Smith, Andrew D.; Thomson, James E; Tetrahedron: Asymmetry (2009), 20(6-8), 758-772.
3. Davies, Stephen G.; Nicholson, Rebecca L.; Price, Paul D.; Roberts, Paul. M.;
Smith, Andrew D; Synlett (2004), (5), 901-903.
4. Brock, E. Anne; Davies, Stephen G.; Lee, James A.; Roberts, Paul M.; Thomson, James E; Organic Letters (2011), 13(7), 1594-1597.
5. Gary B. Evans, Richard H. Furneaux, Andrzej Lewandowicz, Vern L. Schramm, and Peter C. Tyler, Journal of Medicinal Chemistry (2003), 46, 3412-3423.
PATENT
WO 2016110527
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016110527
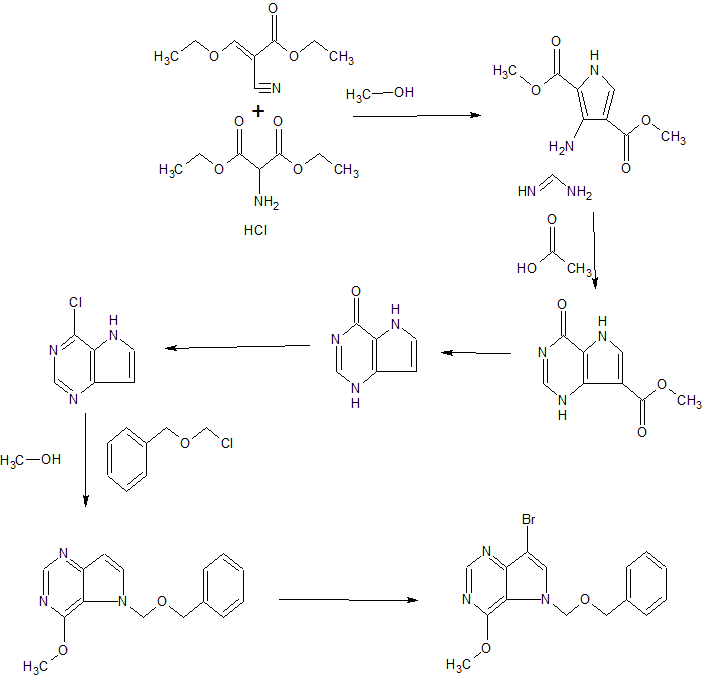

The invention also provides for the synthesis of a compound of formula (II)

By reacting a compound of Formula (VII)

With di-t-butyldicarbonate.
Preferably the reaction is conducted at -10 to -20°C, in methyl t-butyl ether & heptane
The invention also provides for the synthesis of a compound of formula (VII)

By reacting a compound of Formula (IV)

With a suitable base to form

Before reacting with a compound of Formula (III)

Example 1
Stage 1 Manufacture of (III)
Compound of formula (III) (approx. 130g) in toluene solution is added to a suspension of N-Chlorosuccinimide in toluene at 20°C over a period of 90min. The reaction mixture is stirred at 20°C for 1 hour then chilled to 0°C and stirred for a further hour. The precipitated succinimide by-product is removed by filtration and the filtered solution charged directly to a 45% potassium hydroxide solution (aq) containing
tetrabutylammonium bromide. The reaction mixture is stirred at 0°C and completion of reaction is confirmed by GC analysis. Water is then added to the two-phase mixture to dissolve inorganic precipitates and the toluene product solution is washed with a 28% ammonium hydroxide/acetic acid buffer mixture with sodium chloride added. After phase separation the organic phase solution is stabilised with triethylamine. Magnesium sulfate is added to dry the solution. After filtration, the yield of (III) is determined by R.O.E. and GC purity.
Stage 2 Manufacture of (II)
Stage 2a Lithiation
A suspension of compound of formula (IV) (approx. 200g) in MtBE is chilled to -15°C and treated with /7-Hexyl lithium (2.5M in hexanes) added over 2h, maintaining the reaction mixture at -15°C. The mixture is then stirred for 3h at -15°C.
Stage 2b Coupling with (IV)
After lithiation is complete, a compound formula (III) in toluene solution is added to the reaction mixture maintaining the contents at -15°C. The reaction mixture is then stirred at this temperature for 1.5h.
Stage 2c Boc anhydride quench
A solution of di-t-butyldicarbonate in MtBE is added to the above reaction mixture at -15°C. The solution is stirred for a further 30min.
Workup and Purification
The reaction mixture is quenched by addition of RO water, then filtered. The aqueous layer is separated and run to waste. The organic layer is again washed with water. The organic layer is concentrated to a low volume and solvent replaced by heptane. The mix is stirred for 16h and filtered again.
The solution is passed through a silica gel column and eluted with heptane. The resulting solution is treated with charcoal – stirred for 3h, then filtered. The product (II) is progressed as a solution in heptane to the next stage.
Stage 3 Manufacture of Crude Forodesine (la)
Stage 3 Deprotection with cone. HCI
Concentrated hydrochloric acid is added to (II) in heptane and the mixture stirred. The acid phase is separated off and stirred for 16h at ambient temperature. The solution is then heated to 40°C for 6h. The water is then distilled off under reduced pressure to a minimum volume.
Ethanol is then added to precipitate the crude Forodesine (la) which is isolated by filtration after cooling 0-5°C. It is washed with ethanol and dried in a vacuum oven at 75°C to a constant weight.
Stage 4a Decolourization of crude Forodesine (la) using Ion-Exchange Column
Crude Forodesine (la) is dissolved in water and loaded onto a freshly prepared ion-exchange column containing Dowex 50WX4 resin in the Na+ form activated with 30% sodium hydroxide solution. The ion-exchange column is eluted with 4 x lOOmL water followed by 4 x lOOmL 2M HCI. The HCI fractions are collected separately as they contain the desired product. The 2M HCI fractions are combined and concentrated under vacuum with minimum RO water added to dissolve the residue. 1,4-Dioxane is added to the aqueous solution to precipitate the product. The mixture is stirred at 20°C for 1.5h. The product is filtered, washed with 1,4-dioxane and dried in a vacuum oven at 35°C to a constant weight to give decolourised BCX1777.
Stage 4b Recrystallization of Forodesine
Decolourised Forodesine is added to in 0.6M dilute hydrochloric acid and heated to 45°C to dissolve. The resulting solution is hot filtered and washed through with some RO Water. The solution is cooled to 20°C and ethanol added over at least lh. The mixture is then seeded with Forodesine HCI. The resulting slurry is stirred for 8h at 20°C, then cooled to 2°C for a further 1.5h. The product is isolated by filtration, washed twice with cold ethanol then dried in a vacuum oven at 75°C to a constant weight to give a white crystalline Forodesine HCI (approx. 50g).
While the invention has been described in detail and with reference to specific embodiments thereof, it will be apparent to one of skill in the art that various changes and modifications can be made therein without departing from the spirit and scope thereof. Moreover, all embodiments described herein are considered to be broadly applicable and combinable with any and all other consistent embodiments, as appropriate.
PAPER
Journal of Medicinal Chemistry (2009), 52(4), 1126-1143.
Third-Generation Immucillins: Syntheses and Bioactivities of Acyclic Immucillin Inhibitors of Human Purine Nucleoside Phosphorylase
* To whom correspondence should be addressed. Phone: +64-4-9313040. Fax: +64-4-9313055. E-mail: g.evans@irl.cri.nz., †
Carbohydrate Chemistry Team, Industrial Research Limited.
, ‡
Department of Biochemistry, Albert Einstein College of Medicine of Yeshiva University.

References
- Jump up^ Kicska GA, Long L, Hörig H, et al. (April 2001). “Immucillin H, a powerful transition-state analog inhibitor of purine nucleoside phosphorylase, selectively inhibits human T lymphocytes”. Proc. Natl. Acad. Sci. U.S.A. 98 (8): 4593–8. Bibcode:2001PNAS…98.4593K. doi:10.1073/pnas.071050798. PMC 31879
 . PMID 11287638.
. PMID 11287638. - Jump up^ “Complete list of clinical trials for forodesine (BCX-1777) (ClinicalTrials.gov)”. Retrieved 2008-07-22.
- Jump up^ “Biocryst Initiates Pivotal Fodosine Phase IIb Clinical Trial In Patients With Relapsed/Refractory T-Lymphoblastic Leukemia/Lymphoma”(Press release). January 16, 2007.
- Jump up^ “BioCryst Announces Mundipharma Receives Approval for Mundesine® in Japan” (Press release). April 3, 2017.
External links
- “From cell biology to therapy: forodesine”. Hematology Meeting Reports. 2 (5): 106–111. 2008.
- Gore, L; Stelljes, M; Quinones, R (2007). “Forodesine treatment and post-transplant graft-versus-host disease in two patients with acute leukemia: Facilitation of graft-versus-leukemia effect?”. Seminars in Oncology. 34 (6 Suppl 5): S35–9. doi:10.1053/j.seminoncol.2007.11.005. PMID 18086346.
- 18 December 2006 Fodosine orphan designation by the European Commission for acute lymphoblastic leukaemia.
- BioCryst Pharmaceuticals, Inc. have entered into an exclusive license agreement with Mundipharma for develop and commercialize BioCryst’s lead compound, Forodesine.
- Birmingham, Alabama – February 2, 2006 Mundipharma will obtain rights in markets across Europe, Asia and Australasia to Forodesine™ in the field of oncology in exchange for a $10 million up-front payment. Furthermore, Mundipharma will commit up to an additional $15 million to assist in the evaluation of Forodesine’s™ therapeutic safety and efficacy profile. BioCryst may also receive future event payments totalling $155 million in addition to royalties on product sales of Forodesine™ by Mundipharma.
- News BioCryst provides Fodosine update March 27, 2007. “Voluntarily Placed on Hold by BioCryst (…) we don’t think the final response rate will be as high as 18%”.
- The European Commission granted a marketing authorisation valid throughout the European Union for Atriance on 22 August 2007 for acute lymphoblastic leukaemia. What benefit has Atriance shown during the studies? Atriance was shown to be effective in a proportion of the patients in both studies. In the first study, among the 39 children and young adults who se cancer had not responded to two or more previous treatments, five (13%) had a complete response to treatment after a month, with no evidence of disease and normal blood counts. In the second study, among the 28 adults and adolescents with cancer that had not responded to two or more previous tre atments, five (18%) had a complete response to treatment. In both studies, more patients had a partial response to Atriance treatment, with blood counts returning towards normal levels.
- Lino Berton collects all the information on Forodesine in www.linoberton.com site, putting them in a row. In 2014 he published the book Qualcosa che non muore where he tells his incredible experience in the closed trial early in 2007.
- Il Giornale.it (in Italian). “Come si boicotta un farmaco che funziona”. Dated 08-01-2016.
 |
|
| Clinical data | |
|---|---|
| Trade names | Mundesine and Fodosine |
| Routes of administration |
oral |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C11H14N4O4 |
| Molar mass | 266.26 g·mol−1 |
| 3D model (JSmol) | |
/////////Forodesine Hydrochloride, Forodesine, BCX 1777, Immucillin-H, FOSODINE, JAPAN 2017
Novel Drug Approvals for 2017, A Review/Compilation
CDSCO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO, Novel Drug Approvals for 2017, A Review Compilation (USFDA, EMA, PMDA, CDSCO).
Any errors in this compilation, email amcrasto@gmail.com, Call +919323115463
Some gaps will be filled up soon keep watching……………..
INDEX, NAME (click on the title, it contains link)
SECTION A; USFDA Approvals
6 BENRALIZUMAB
17 DURVALUMAB
24 GUSELKUMAB
36 OZENOXACIN
40 SARILUMAB
41 SECNIDAZOLE
INDEX, FORMULATION NAME
USFDA
•Aliqopa (COPANLISIB) to treat adults with relapsed follicular lymphoma — a slow-growing type of nonHodgkin lymphoma (a cancer of the lymph system) — who have received at least two prior systemic therapies;
• ALUNBRIG, BRIGATINIB, To treat patients with anaplastic lymphoma kinase (ALK)-positive metastatic non-small cell lung cancer (NSCLC) who have progressed on or are intolerant to crizotinib
• Austedo, Deutetrabenazine For the treatment of chorea associated with Huntington’s disease
• Bavencio (avelumab) for the treatment of patients 12 years and older with a rare and aggressive form of cancer called metastatic Merkel cell carcinoma, including those who have not received prior chemotherapy;
•BAXDELLA, Delafloxacin, BACTERIAL INFECTIONS
• Benznidazole to treat children ages 2 to 12 years with Chagas disease, a parasitic infection that can cause serious heart illness after years of infection, and can also affect swallowing and digestion. This is the first treatment approved in the United States for this rare disease;
• Besponsa (inotuzumab ozogamicin) for the treatment of adults with a type of cancer of the blood called relapsed or refractory B-cell precursor acute lymphoblastic leukemia;
•BEVYXXA, BETRIXABAN, For the prophylaxis of venous thromboembolism (VTE) in adult patients hospitalized for an acute medical illness
• BRINEURA, CERLIPONASE ALFA, To treat a specific form of Batten disease
• Calquence (ACALABRUTINIB) to treat adults with mantle cell lymphoma who have received at least one prior therapy. Mantle cell lymphoma is a particularly aggressive cancer;
• DUPIXENT, (DUPILUMAB) To treat adults with moderate-to-severe eczema (atopic dermatitis)
• Emflaza (deflazacort) to treat patients age 5 years and older with Duchenne muscular dystrophy, a rare genetic disorder that causes progressive muscle deterioration and weakness;
• FASENRA, BENRALIZUMAB, For add-on maintenance treatment of patients with severe asthma aged 12 years and older, and with an eosinophilic phenotype
• Giapreza (angiotensin II), for the treatment of hypotension in adults with distributive or vasodilatory shock (dangerously low blood pressure despite adequate heart function) whose blood pressure remains low despite receiving fluids and treatment with drugs called vasopressors;
• HEMLIBRA EMICIZUMAB To prevent or reduce the frequency of bleeding episodes in adult and pediatric patients with hemophilia A who have developed antibodies called Factor VIII (FVIII) inhibitors.
• Idhifa (enasidenib) for the treatment of adults with relapsed or refractory acute myeloid leukemia, a form of blood cancer, who have a specific genetic mutation;
• IMFINZI, DURVALUMAB To treat patients with locally advanced or metastatic urothelial carcinoma
• Ingrezza (valbenazine) to treat adults with tardive dyskinesia, a side effect of some antipsychotic medications whereby patients can experience uncontrollable stiff, jerky movements of their face and body, and other uncontrolled movements such as eye-blinking, sticking out the tongue, and arm-waving;
• KEVZARA SARILUMAB, RHEUMATOID ARTHRITIS
• KISQALI, RIBOCICLIB, To treat postmenopausal women with a type of advanced breast cancer
• Macrilen macimorelin acetate, For the diagnosis of adult growth hormone deficiency
• Mavyret (glecaprevir and pibrentasvir) to treat adults with chronic hepatitis C virus genotypes 1-6 without cirrhosis (liver disease) or with mild cirrhosis, including patients with moderate to severe kidney disease, as well as those who are on hemodialysis;
• Mepsevii (vestronidase alfa-vjbk) to treat patients with Sly syndrome or mucopolysaccharidosis type 7 – a rare genetic disorder where an enzyme deficiency results in skeletal abnormalities, developmental delay, enlarged liver and spleen, and narrowed airways, which can lead to respiratory infections;
• Nerlynx (neratinib) for the extended adjuvant treatment — a form of therapy administered after an initial treatment to further lower the risk of the cancer coming back — of early-stage, human epidermal growth factor receptor 2 (HER2)-positive breast cancer;
• OCREVUS, OCRELIZUMAB, To treat patients with relapsing and primary progressive forms of multiple sclerosis
• OZEMPIC SEMAGLUTIDE To improve glycemic control in adults with type 2 diabetes mellitus
•PARSABIV, ETELCALCETIDE, To treat secondary hyperparathyroidism in adult patients with chronic kidney disease undergoing dialysis
• Prevymis (letermovir) for prevention of an infection called cytomegalovirus (CMV) in patients who are receiving a bone marrow transplant. CMV disease can cause serious health issues in these patients;
• Radicava (edaravone) to treat patients with amyotrophic lateral sclerosis, commonly referred to as Lou Gehrig’s disease, a rare disease that attacks and kills the nerve cells that control voluntary muscles;
• RHOPRESSA, NETARSUDIL, To treat glaucoma or ocular hypertension
• Rydapt (midostaurin) to treat adults newly diagnosed with a form of blood cancer known as acute myeloid leukemia who have a specific genetic mutation called FLT3, in combination with chemotherapy;
• Siliq (brodalumab) to treat adults with moderate-to-severe plaque psoriasis, a chronic disorder in which the body’s immune system sends out faulty signals that speed growth of skin cells that then accumulate, causing red, flaky patches that can appear anywhere on the body;
•SOLOSEC, SECNIDAZOLE To treat bacterial vaginosis
• STEGLATRO ERTUGLIFLOZIN To improve glycemic control in adults with type 2 diabetes mellitus
• Symproic (Naldemedine) for the treatment of opioid-induced constipation in adults with chronic noncancer pain; • Tremfya (guselkumab) for the treatment of adults with moderate-to-severe plaque psoriasis;
• Trulance (plecanatide) to treat adults with chronic idiopathic constipation, which is a persistent condition of constipation due to unknown origin;
• TYMLOS, Abaloparatide, To treat osteoporosis in postmenopausal women at high risk of fracture or those who have failed other therapies
• Vabomere (vaborbactam and meropenem) for treatment of adults with complicated urinary tract infections, including pyelonephritis (kidney infection) caused by bacteria;
• Verzenio (abemaciclib) to treat adults who have hormone receptor (HR)-positive, HER2-negative advanced or metastatic breast cancer that has progressed after taking therapy that alters a patient’s hormones (endocrine therapy);
• Vosevi (sofosbuvir/velpatasvir/voxilaprevir) to treat adults with chronic hepatitis C virus genotypes 1-6 without cirrhosis (liver disease) or with mild cirrhosis;
• Xadago (safinamide) as an add-on treatment for patients with Parkinson’s disease who are currently taking levodopa/carbidopa and experiencing “off” episodes;
• XERMELO, TELOTRISTAT ETHYL combined with somatostatin analog (SSA) therapy to treat adults with carcinoid syndrome diarrhea that SSA therapy alone has inadequately controlled, and;
• XEPI OZENOXACIN TO TREAT IMPETIGO
•XERMELO, TELOTRISTAT ETHYL, To treat carcinoid syndrome diarrhea
• Zejula (niraparib) for the maintenance treatment (intended to delay cancer growth) of adults with recurrent epithelial ovarian, fallopian tube or primary peritoneal cancer, whose tumors have completely or partially shrunk (complete or partial response, respectively) in response to platinum-based chemotherapy
USFDA
| No. | Drug Name |
Active Ingredient | Approval Date | FDA-approved use on approval date |
|---|---|---|---|---|
| 46. | Giapreza | angiotensin II | 12/21/2017 | To increase blood pressure in adults with septic or other distributive shock |
| 45. | Macrilen | macimorelin acetate | 12/20/2017 | For the diagnosis of adult growth hormone deficiency |
| 44. | Steglatro | ertugliflozin | 12/19/2017 | To improve glycemic control in adults with type 2 diabetes mellitus |
| 43. | Rhopressa | netarsudil | 12/18/2017 | To treat glaucoma or ocular hypertension |
| 42. | Xepi | ozenoxacin | 12/11/2017 | To treat impetigo Drug Trials Snapshot |
| 41. | Ozempic | semaglutide | 12/5/2017 | To improve glycemic control in adults with type 2 diabetes mellitus |
| 40. | Hemlibra | emicizumab | 11/16/2017 | To prevent or reduce the frequency of bleeding episodes in adult and pediatric patients with hemophilia A who have developed antibodies called Factor VIII (FVIII) inhibitors. |
| 39. | Mepsevii | vestronidase alfa-vjbk | 11/15/2017 | To treat pediatric and adult patients with an inherited metabolic condition called mucopolysaccharidosis type VII (MPS VII), also known as Sly syndrome. |
| 38. | Fasenra | benralizumab | 11/14/2017 | For add-on maintenance treatment of patients with severe asthma aged 12 years and older, and with an eosinophilic phenotype Drug Trials Snapshot |
| 37. | Prevymis | letermovir | 11/8/2017 | To prevent infection after bone marrow transplant Drug Trials Snapshot |
| 36. | Vyzulta | latanoprostene bunod ophthalmic solution | 11/2/2017 | To treat intraocular pressure in patients with open-angle glaucoma or ocular hypertension. Drug Trials Snapshot |
| 35. | Calquence | acalabrutinib | 10/31/2017 | To treat adults with mantle cell lymphoma Press Release Drug Trials Snapshot |
| 34. | Verzenio | abemaciclib | 9/28/2017 | To treat certain advanced or metastatic breast cancers Press Release Drug Trials Snapshot |
| 33. | Solosec | secnidazole | 9/15/2017 | To treat bacterial vaginosis Drug Trials Snapshot |
| 32. | Aliqopa | copanlisib | 9/14/2017 | To treat adults with relapsed follicular lymphoma Press Release Drug Trials Snapshot |
| 31. | benznidazole | benznidazole | 8/29/2017 | To treat children ages 2 to 12 years old with Chagas disease Press Release Drug Trials Snapshot |
| 30. | Vabomere | meropenem and vaborbactam | 8/29/2017 | To treat adults with complicated urinary tract infections Press Release Drug Trials Snapshot |
| 29. | Besponsa | inotuzumab ozogamicin | 8/17/2017 | To treat adults with relapsed or refractory acute lymphoblastic leukemia Press Release Drug Trials Snapshot |
| 28. | Mavyret | glecaprevir and pibrentasvir | 8/3/2017 | To treat adults with chronic hepatitis C virus Press Release Drug Trials Snapshot |
| 27. | Idhifa | enasidenib | 8/1/2017 | To treat relapsed or refractory acute myeloid leukemia Press Release Drug Trials Snapshot |
| 26. | Vosevi | sofosbuvir, velpatasvir and voxilaprevir | 7/18/2017 | To treat adults with chronic hepatitis C virus Press Release Drug Trials Snapshot |
| 25. | Nerlynx | neratinib maleate | 7/17/2017 | To reduce the risk of breast cancer returning Press Release Drug Trials Snapshot |
| 24. | Tremfya | guselkumab | 7/13/2017 | For the treatment of adult patients with moderate-to-severe plaque psoriasis Drug Trials Snapshot |
| 23. | Bevyxxa | betrixaban | 6/23/2017 | For the prophylaxis of venous thromboembolism (VTE) in adult patients hospitalized for an acute medical illness Drug Trials Snapshot |
| 22. | Baxdela | delafloxacin | 6/19/2017 | To treat patients with acute bacterial skin infections Drug Trials Snapshot |
| 21. | Kevzara | sarilumab | 5/22/2017 | To treat adult rheumatoid arthritis Drug Trials Snapshot |
| 20. | Radicava | edaravone | 5/5/2017 | To treat patients with amyotrophic lateral sclerosis (ALS) Press Release Drug Trials Snapshot |
| 19. | Imfinzi | durvalumab | 5/1/2017 | To treat patients with locally advanced or metastatic urothelial carcinoma Web Post Drug Trials Snapshot |
| 18. | Tymlos | abaloparatide | 4/28/2017 | To treat osteoporosis in postmenopausal women at high risk of fracture or those who have failed other therapies Drug Trials Snapshot |
| 17. | Rydapt | midostaurin | 4/28/2017 | To treat acute myeloid leukemia Press Release Chemistry Review(s) (PDF) Drug Trials Snapshot |
| 16. | Alunbrig | brigatinib | 4/28/2017 | To treat patients with anaplastic lymphoma kinase (ALK)-positive metastatic non-small cell lung cancer (NSCLC) who have progressed on or are intolerant to crizotinib Drug Trials Snapshot |
| 15. | Brineura | cerliponase alfa | 4/27/2017 | To treat a specific form of Batten disease Press Release Drug Trials Snapshot |
| 14. | Ingrezza | valbenazine | 4/11/2017 | To treat adults with tardive dyskinesia Press Release Chemistry Review(s) (PDF)Drug Trials Snapshot |
| 13. | Austedo | deutetrabenazine | 4/3/2017 | For the treatment of chorea associated with Huntington’s disease Drug Trials Snapshot, Chemistry Review(s) (PDF) |
| 12. | Ocrevus | ocrelizumab | 3/28/2017 | To treat patients with relapsing and primary progressive forms of multiple sclerosis Press Release Drug Trials Snapshot |
| 11. | Dupixent | dupilumab | 3/28/2017 | To treat adults with moderate-to-severe eczema (atopic dermatitis) Press Release Drug Trials Snapshot |
| 10. | Zejula | niraparib | 3/27/2017 | For the maintenance treatment for recurrent epithelial ovarian, fallopian tube or primary peritoneal cancers Press Release Drug Trials Snapshot |
| 9. | Symproic | naldemedine | 3/23/2017 |
For the treatment of opioid-induced constipation |
| 8. | Bavencio | avelumab | 3/23/2017 | To treat metastatic Merkel cell carcinoma Press Release Drug Trials Snapshot |
| 7. | Xadago | safinamide | 3/21/2017 | To treat Parkinson’s disease Press Release Drug Trials SnapshotChemistry Review(s) (PDF) |
| 6. | Kisqali | ribociclib | 3/13/2017 | To treat postmenopausal women with a type of advanced breast cancer Drug Trials Snapshot |
| 5. | Xermelo | telotristat ethyl | 2/28/2017 | To treat carcinoid syndrome diarrhea Press Release Drug Trials Snapshot |
| 4. | Siliq | brodalumab | 2/15/2017 | To treat adults with moderate-to-severe plaque psoriasis Press Release Drug Trials Snapshot |
| 3. | Emflaza | deflazacort | 2/9/2017 | To treat patients age 5 years and older with Duchenne muscular dystrophy (DMD) Press Release Drug Trials Snapshot |
| 2. | Parsabiv | etelcalcetide | 2/7/2017 | To treat secondary hyperparathyroidism in adult patients with chronic kidney disease undergoing dialysis Drug Trials Snapshot |
| 1. | Trulance | plecanatide | 1/19/2017 | To treat Chronic Idiopathic Constipation (CIC) in adult patients. Press Release Drug Trials Snapshot |
* This information is currently accurate. In rare instances, it may be necessary for FDA to change a drug’s new molecular entity (NME) designation or the status of its application as a novel new biologics license application (BLA). For instance, new information may become available which could lead to a reconsideration of the original designation or status. If changes must be made to a drug’s designation or the status of an application as a novel BLA, the Agency intends to communicate the nature of, and the reason for, any revisions as appropriate.
| USFDA 2017 | ||||||
| 2017/12/21 | Angiotensin II | Giapreza | La Jolla Pharmaceutical | |||
| 2017/12/20 | Ertugliflozin | Steglatro | Merck Sharp Dohme | |||
| 2017/12/20 | Macimorelin acetate | Macrilen | Aeterna Zentaris GmbH | |||
| 2017/12/18 | Netarsudil mesylate | Rhopressa | Aerie Pharmaceuticals | |||
| 2017/12/11 | Ozenoxacin | Xepi | Ferrer Internacional S.A. | |||
| 2017/12/5 | Semaglutide | Ozempic | Novo Nordisk Inc | |||
| 2017/11/16 | Emicizumab | Hemlibra | Genentech | BLA | ||
| 2017/11/15 | Vestronidase alfa | Mepsevii | Ultragenyx Pharmaceutical | BLA | ||
| 2017/11/14 | Benralizumab | Fasenra | AstraZeneca AB | BLA | ||
| 2017/11/8 | Letermovir | Prevymis | Merck Sharp Dohme | |||
| 2017/11/2 | Latanoprostene bunod | Vyzulta | Bausch & Lomb Incorporated | |||
| 2017/10/31 | Acalabrutinib | Calquence | AstraZeneca Pharmaceuticals LP | |||
| 2017/9/28 | Abemaciclib | Verzenio | Eli Lilly | |||
| 2017/9/15 | Secnidazole | Solosec | Symbiomix Therapeutics | |||
| 2017/9/14 | Copanlisib | Aliqopa | Bayer Healthcare Pharmaceuticals | |||
| 2017/8/29 | Benznidazole | Chemo Research | ||||
| 2017/8/29 | Meropenem – Vaborbactam | Vabomere | Rempex Pharmaceuticals | |||
| 2017/8/17 | Inotuzumab ozogamicin | Besponsa | Wyeth Pharmaceuticals | BLA | ||
| 2017/8/3 | Glecaprevir – Pibrentasvir | Mavyret | AbbVie | |||
| 2017/8/1 | Enasidenib | Idhifa | Celgene Corporation | |||
| 2017/7/18 | Sofosbuvir – Velpatasvir – Voxilaprevir | Vosevi | Gilead Sciences | |||
| 2017/7/17 | Neratinib maleate | Nerlynx | Puma Biotechnology | |||
| 2017/7/13 | Guselkumab | Tremfya | Janssen Biotech | BLA | ||
| 2017/6/23 | Betrixaban | Bevyxxa | Portola Pharmaceuticals | |||
| 2017/6/19 | Delafloxacin meglumine | Baxdela | Melinta Therapeutics | |||
| 2017/5/22 | Sarilumab | Kevzara | Sanofi Synthelabo | BLA | ||
| 2017/5/5 | Edaravone | Radicava | Mitsubishi Tanabe Pharma America | |||
| 2017/5/1 | Durvalumab | Imfinzi | AstraZeneca UK | BLA | ||
| 2017/4/28 | Abaloparatide | Tymlos | Radius Health | |||
| 2017/4/28 | Midostaurin | Rydapt | Novartis Pharmaceuticals | |||
| 2017/4/28 | Brigatinib | Alunbrig | Ariad Pharmaceuticals | |||
| 2017/4/27 | Cerliponase alfa | Brineura | BioMarin Pharmaceutical | BLA | ||
| 2017/4/11 | Valbenazine | Ingrezza | Neurocrine Biosciences | |||
| 2017/4/3 | Deutetrabenazine | Austedo | Teva Pharmaceuticals | |||
| 2017/3/28 | Ocrelizumab | Ocrevus | Genentech | BLA | ||
| 2017/3/28 | Dupilumab | Dupixent | Regeneron Pharmaceuticals | BLA | ||
| 2017/3/27 | Niraparib | Zejula | Tesaro | |||
| 2017/3/23 | Naldemedine tosylate | Symproic | Shionogi | |||
| 2017/3/23 | Avelumab | Bavencio | EMD Serono | BLA | ||
| 2017/3/23 | Safinamide mesylate | Xadago | Newron Pharmaceuticals | |||
| 2017/3/21 | Ribociclib | Kisqali | Novartis Pharmaceuticals | |||
| 2017/2/28 | Telotristat ethyl | Xermelo | Lexicon Pharmaceuticals | |||
| 2017/2/15 | Brodalumab | Siliq | Valeant Pharmaceuticals | BLA | ||
| 2017/2/9 | Deflazacort | Emflaza | Marathon Pharmaceuticals | |||
| 2017/2/8 | Etelcalcetide hydrochloride | Parsavib | KAI Pharmaceuticals | |||
| 2017/1/19 | Plecanatide | Trulance | Synergy Pharmaceuticals |
1 Abaloparatide
RADIUS

FDA 4/28/2017
To treat osteoporosis in postmenopausal women at high risk of fracture or those who have failed other therapies
Drug Trials Snapshot

2 Abemaciclib
ELI LILLY
| Verzenio | abemaciclib | FDA 9/28/2017 | To treat certain advanced or metastatic breast cancers Press Release Drug Trials Snapshot |
LINK https://newdrugapprovals.org/2015/10/19/abemaciclib-bemaciclib/
3 Acalabrutinib
| Calquence | FDA APPROVED
10/31/2017 |
To treat adults with mantle cell lymphoma Press Release Drug Trials Snapshot |

-Facebook.png)
4 Angiotensin II
LA JOLLA
| Giapreza | angiotensin II | 12/21/2017 | To increase blood pressure in adults with septic or other distributive shock Press Release Drug Trials Snapshot |


5 AVELUMAB
MERCK

| Bavencio | FDA 3/23/2017 | To treat metastatic Merkel cell carcinoma Press Release Drug Trials Snapshot |
6 BENRALIZUMAB
ASTRA ZENECA
Fasenra benralizumab
FDA 11/14/2017
For add-on maintenance treatment of patients with severe asthma aged 12 years and older, and with an eosinophilic phenotype
Drug Trials Snapshot


7 Benznidazole
CHEMO RESEARCH


![]()
| benznidazole | FDA
8/29/2017 |
To treat children ages 2 to 12 years old with Chagas disease Press Release Drug Trials Snapshot |
8 BETRIXABAN
PORTOLA PHARMA

| Bevyxxa | FDA
6/23/2017 |
For the prophylaxis of venous thromboembolism (VTE) in adult patients hospitalized for an acute medical illness Drug Trials Snapshot
|
![]()

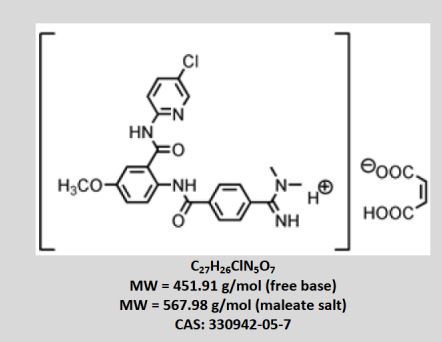
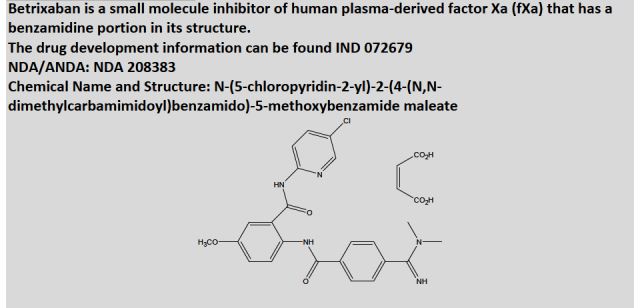
9 BRIGATINIB
TAKEDA
![]()

| Alunbrig | FDA
4/28/2017 |
To treat patients with anaplastic lymphoma kinase (ALK)-positive metastatic non-small cell lung cancer (NSCLC) who have progressed on or are intolerant to crizotinib Drug Trials Snapshot |
10 BRODALUMAB
VALEANT PHARMA
| Siliq | FDA
2/15/2017 |
To treat adults with moderate-to-severe plaque psoriasis Press Release Drug Trials Snapshot |

LINK ,,,,https://newdrugapprovals.org/2017/02/16/fda-approves-new-psoriasis-drug-siliq-brodalumab/
11 CERLIPONASE ALFA
![]()

| Brineura | FDA 4/27/2017 | To treat a specific form of Batten disease Press Release Drug Trials Snapshot |
12 Copanlisib
| Aliqopa | FDA APPROVED
9/14/2017 |
To treat adults with relapsed follicular lymphoma Press Release Drug Trials Snapshot |
![]()
LINK…..https://newdrugapprovals.org/2017/11/20/copanlisib/
13 DEFLAZACORT
MARATHON PHARMA
![]()
| Emflaza | FDA 2/9/2017 | To treat patients age 5 years and older with Duchenne muscular dystrophy (DMD) Press Release Drug Trials Snapshot |
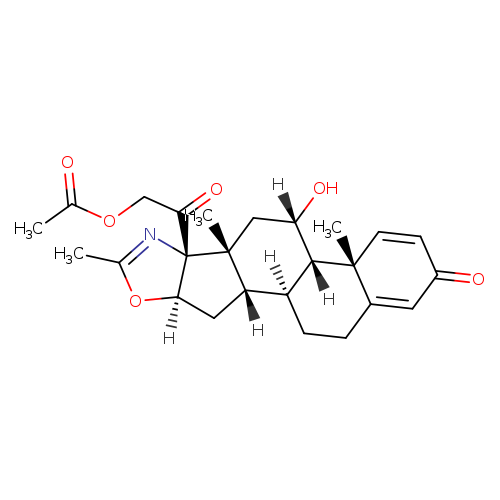
LINK……https://newdrugapprovals.org/2017/02/17/deflazacort/
14 DELAFLOXACIN
| Baxdela | FDA APPROVED
6/19/2017 |
To treat patients with acute bacterial skin infections |
![]()
![]()
LINK……..https://newdrugapprovals.org/2018/01/25/delafloxacin/
15 Deutetrabenazine
TEVA

LINK……………https://newdrugapprovals.org/2015/08/15/sd-809-deutetrabenazine-nda-submitted-by-teva/
| Austedo | FDA 4/3/2017 | For the treatment of chorea associated with Huntington’s disease Drug Trials Snapshot Chemistry Review(s) (PDF) |
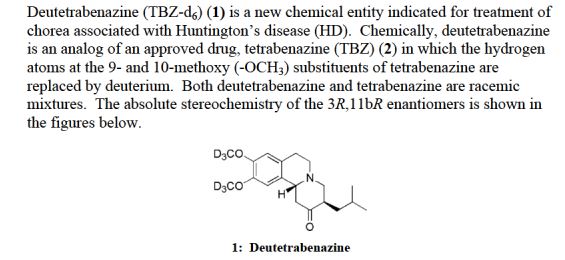

16 DUPILUMAB
SANOFI/REGENERON
![]()

| Dupixent | FDA | 3/28/2017 | To treat adults with moderate-to-severe eczema (atopic dermatitis) Press Release Drug Trials Snapshot |
LINK…….https://newdrugapprovals.org/2017/03/29/fda-approves-new-eczema-drug-dupixent-dupilumab/
17 DURVALUMAB
ASTRA ZENECA
durvalumab FDA 5/1/2017To treat patients with locally advanced or metastatic urothelial carcinoma
Web Post
Drug Trials Snapshot
18 EDAVARONE

MITSUBISHI TANABE
| Radicava | FDA 5/5/2017 | To treat patients with amyotrophic lateral sclerosis (ALS) Press Release Drug Trials Snapshot |


19 EMICIZUMAB
ROCHE

| Hemlibra | emicizumab | FDA 11/16/2017 | To prevent or reduce the frequency of bleeding episodes in adult and pediatric patients with hemophilia A who have developed antibodies called Factor VIII (FVIII) inhibitors. Press Release Drug Trials Snapshot |

20 Enasidenib

![]()
| Idhifa | FDA
8/1/2017 |
To treat relapsed or refractory acute myeloid leukemia Press Release Drug Trials Snapshot |

21 Ertugliflozin
MERCK

| Steglatro | ertugliflozin | FDA
12/19/2017 |
To improve glycemic control in adults with type 2 diabetes mellitus Drug Trials Snapshot |
LINK https://newdrugapprovals.org/2014/02/10/ertugliflozin/

22 ETELCALCETIDE
Amgen
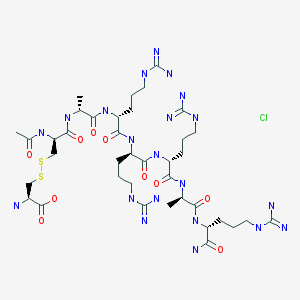
| Parsabiv | FDA APPROVED
2/7/2017 |
To treat secondary hyperparathyroidism in adult patients with chronic kidney disease undergoing dialysis Drug Trials SnapshotSYNTHESIS LINK……..https://cen.acs.org/articles/96/i4/the-year-in-new-drugs-2018.html |

SYNTHESIS LINK……..https://cen.acs.org/articles/96/i4/the-year-in-new-drugs-2018.html
23 GLECAPREVIR
ABBVIE

| Mavyret | glecaprevir and pibrentasvir | FDA 8/3/2017 | To treat adults with chronic hepatitis C virus Press Release Drug Trials Snapshot |
LINK https://newdrugapprovals.org/2016/10/05/glecaprevir-abt-493/

24 GUSELKUMAB
JOHNSON AND JOHNSON
guselkumab
FDA 7/13/2017
For the treatment of adult patients with moderate-to-severe plaque psoriasis
Drug Trials Snapshot
![]()

25 Inotuzumab ozogamicin
PFIZER

![]()

| Besponsa | FDA
8/17/2017 |
To treat adults with relapsed or refractory acute lymphoblastic leukemia Press Release Drug Trials Snapshot |
26 LATANOPROSTENE
VALEANT
latanoprostene bunod ophthalmic solution
FDA 11/2/2017
To treat intraocular pressure in patients with open-angle glaucoma or ocular hypertension.
Drug Trials Snapshot
![]()
27 LETERMOVIR
MERCK
| Prevymis | FDA 11/8/2017 | To prevent infection after bone marrow transplant Drug Trials Snapshot |
LINK https://newdrugapprovals.org/2016/05/16/letermovir-aic-246/

28 Macimorelin acetate
AETERNA ZENTARIS

| Macrilen | macimorelin acetate | FDA
12/20/2017 |
For the diagnosis of adult growth hormone deficiency Drug Trials Snapshot |
29 MEROPENEM

30 MIDOSTAURIN
NOVARTIS
- Chemistry Review(s) (PDF)

| Rydapt | FDA
4/28/2017 |
To treat acute myeloid leukemia Press Release Drug Trials Snapshot |

31 Naldemedine
FDA 3/23/2017, Symproic, For the treatment of opioid-induced constipation


32 NERATINIB MALEATE
PUMA BIOTECH
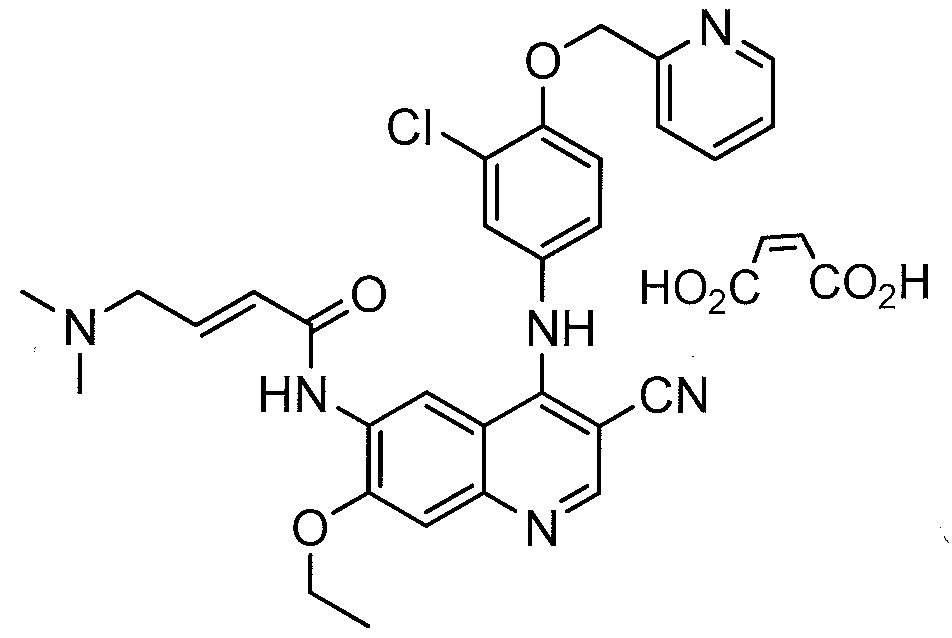


| Nerlynx | FDA | 7/17/2017 | To reduce the risk of breast cancer returning Press Release Drug Trials Snapshot |
33 NETARSUDIL
| Rhopressa | FDA APPROVED
12/18/2017 |
To treat glaucoma or ocular hypertension |


LINK……https://newdrugapprovals.org/2018/01/29/netarsudil/
34 NIRAPARIB
TESARO
| Zejula | FDA | 3/27/2017 | For the maintenance treatment for recurrent epithelial ovarian, fallopian tube or primary peritoneal cancers Press Release Drug Trials Snapshot |

![]()
![]()
LINK…https://newdrugapprovals.org/2016/12/22/niraparib-mk-4827/
35 OCRELIZUMAB
ROCHE
| Ocrevus | FDA | 3/28/2017 | To treat patients with relapsing and primary progressive forms of multiple sclerosis Press Release Drug Trials Snapshot |
![]()

36 OZENOXACIN
MEDIMETRIX

| Xepi | ozenoxacin | FDA
12/11/2017 |
To treat impetigo Drug Trials Snapshot |
37 Pibrentasvir
ABBVIE

| Mavyret | glecaprevir and pibrentasvir | FDA 8/3/2017 | To treat adults with chronic hepatitis C virus Press Release Drug Trials Snapshot |
LINK https://newdrugapprovals.org/2016/06/08/abt-530-pibrentasvir/
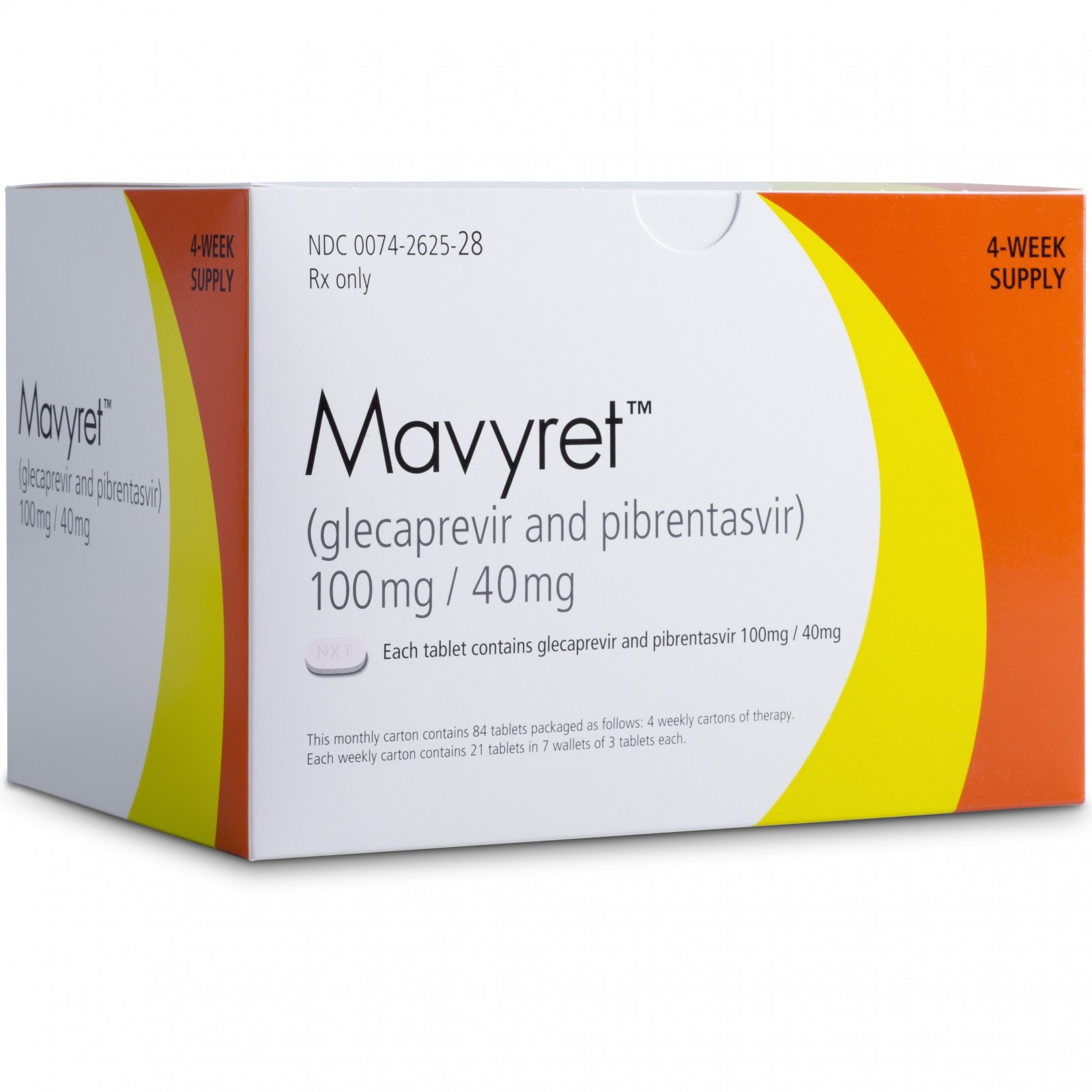

38 PLECANATIDE
Plecanatide 普卡那肽 ليكاناتيد плеканатид
SYNERGY PHARMA


| Trulance | FDA APPROVED
1/19/2017 |
To treat Chronic Idiopathic Constipation (CIC) in adult patients. Press Release Drug Trials Snapshot |
39 RIBOCICLIB
NOVARTIS
Structure..link for correct structure
| Kisqali | FDA 3/13/2017 | To treat postmenopausal women with a type of advanced breast cancer Drug Trials Snapshot |

LINK https://newdrugapprovals.org/2015/10/19/ribociclib/
40 SARILUMAB
SANOFI /REGENERON

| Kevzara | sarilumab | FDA 5/22/2017 | To treat adult rheumatoid arthritis Drug Trials Snapshot |
LINK https://newdrugapprovals.org/2013/11/25/late-stage-success-for-sanofiregeneron-ra-drug-sarilumab/

![]()
41 SECNIDAZOLE
SYMBIOMIX
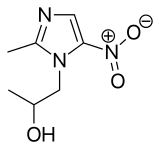
| Solosec | FDA | 9/15/2017 | To treat bacterial vaginosis Drug Trials Snapshot |

42 SAFINAMIDE
NEWRON PHARMA

![]()
![]()
- Chemistry Review(s) (PDF) for correct structure
| Xadago | FDA 3/21/2017 | To treat Parkinson’s disease Press Release Drug Trials Snapshot |
43 Semaglutide
NOVO NORDISK
| Ozempic | semaglutide | FDA
12/5/2017 |
To improve glycemic control in adults with type 2 diabetes mellitus Drug Trials Snapshot |
LINK https://newdrugapprovals.org/2013/07/22/a-survey-of-promising-late-stage-diabetes-drugs/

44 SOFOSBUVIR
45 TELOTRISTAT ETHYL
LEXICON
| Xermelo | FDA
2/28/2017 |
To treat carcinoid syndrome diarrhea Press Release Drug Trials Snapshot |


46 VABORBACTAM
THE MEDICINES CO

| Vabomere | meropenem and vaborbactam | FDA
8/29/2017 |
To treat adults with complicated urinary tract infections Press Release Drug Trials Snapshot |

47 VALBENAZINE
NEUROCRINE
- Chemistry Review(s) (PDF)
![]()
| Ingrezza | FDA
4/11/2017 |
To treat adults with tardive dyskinesia Press Release Drug Trials Snapshot |
48 Vestronidase alfa-vjbk
ULTRAGENYX
| Mepsevii | vestronidase alfa-vjbk | FDA 11/15/2017 | To treat pediatric and adult patients with an inherited metabolic condition called mucopolysaccharidosis type VII (MPS VII), also known as Sly syndrome. Press Release Drug Trials Snapshot |
![]()
49 VELPATASVIR
50 VOXILAPREVIR
GILEAD
| Vosevi | sofosbuvir, velpatasvir and voxilaprevir | FDA 7/18/2017 | To treat adults with chronic hepatitis C virus Press Release Drug Trials Snapshot |
SECTION B; EMA approvals
European Medicines Agency’s – Human medicines: Highlights of 2017
Advances in medicines authorizations are essential for public health as they have the potential to improve treatment of diseases. In 2017, EMA recommended 92 medicines for marketing authorization. Of these, 35 had a new active substance, which has never been authorized in the European Union (EU) before. Many of these medicines represent a significant improvement in their therapeutic areas; they include medicines for children, for rare diseases and advanced therapies42. Amongst the 35 new active substances (NAS) that EMA recommended, 11 were new drugs and biologics to treat cancer, 05 to treat neurological disorders, 04 for infectious diseases, 04 for immunology/rheumatology, 03 for endocrinology, 02 each for Uro-nephrology, haematology, and dermatology, 01 for Pneumonology, and 01 for hepatology/gastroenterology class of drugs.



EUROPE
| 2017/11/16 | Niraparib | Zejula | Tesaro UK Limited | O | NME | ||
| 2017/11/10 | Adalimumab | Cyltezo | Boehringer Ingelheim International GmbH | B | |||
| 2017/11/10 | Miglustat | Miglustat Gen.Orph | Gen.Orph | G | |||
| 2017/11/10 | Ritonavir | Ritonavir Mylan | MYLAN S.A.S | G | |||
| 2017/11/10 | Padeliporfin | Tookad | STEBA Biotech S.A | ||||
| 2017/11/10 | Guselkumab | Tremfya | Janssen-Cilag International N.V. | BLA | |||
| 2017/9/27 | Dupilumab | Dupixent | sanofi-aventis groupe | BLA | |||
| 2017/9/21 | Darunavir / Cobicistat / Emtricitabine / Tenofovir alafenamide | Symtuza | Janssen-Cilag International N.V. | ||||
| 2017/9/21 | Atezolizumab | Tecentriq | Roche Registration Limited | BLA | |||
| 2017/9/18 | Avelumab | Bavencio | Merck Serono Europe Limited | O | BLA | ||
| 2017/9/18 | Entecavir | Entecavir Mylan | Mylan S.A.S | G | |||
| 2017/9/18 | Lacosamide | Lacosamide Accord | Accord Healthcare Ltd | G | |||
| 2017/9/18 | Midostaurin | Rydapt | Novartis Europharm Ltd | O | NME | ||
| 2017/9/18 | Telotristat ethyl | Xermelo | Ipsen Pharma | O | NME | ||
| 2017/9/5 | Trientine | Cuprior | GMP-Orphan SA | ||||
| 2017/9/5 | Efavirenz / Emtricitabine / Tenofovir disoproxil | Efavirenz/Emtricitabine/Tenofovir disoproxil Mylan | Mylan S.A.S | G | |||
| 2017/8/24 | Tivozanib hydrochloride monohydrate | Fotivda | EUSA Pharma (UK) Limited | NME | |||
| 2017/8/24 | Adalimumab | Imraldi | Samsung Bioepis UK Limited (SBUK) | B | |||
| 2017/8/24 | Nitisinone | Nitisinone MDK (previously Nitisinone MendeliKABS) | MendeliKABS Europe Ltd | G | |||
| 2017/8/22 | Ribociclib | Kisqali | Novartis Europharm Ltd | NME | |||
| 2017/8/22 | Cladribine | Mavenclad | Merck Serono Europe Limited | ||||
| 2017/7/26 | Glecaprevir / Pibrentasvir | Maviret | AbbVie Limited | NME | |||
| 2017/7/26 | Sofosbuvir / Velpatasvir / Voxilaprevi | Vosevi | Gilead Sciences International Ltd | NME | |||
| 2017/7/19 | Insulin lispro | Insulin lispro Sanofi | sanofi-aventis groupe | B | |||
| 2017/7/19 | Patiromer sorbitex calcium | Veltassa | Vifor Fresenius Medical Care Renal Pharma France | NME | |||
| 2017/7/17 | Efavirenz / Emtricitabine / Tenofovir disoproxil | Efavirenz/Emtricitabine/Tenofovir disoproxil Zentiva | Zentiva k.s. | G | |||
| 2017/7/17 | Brodalumab | Kyntheum | LEO Pharma A/S | BLA | |||
| 2017/7/17 | beclometasone / formoterol / glycopyrronium bromide | Trimbow | Chiesi Farmaceutici S.p.A. | ||||
| 2017/7/13 | Rituximab | Blitzima | Celltrion Healthcare Hungary Kft. | B | |||
| 2017/7/13 | Cariprazine | Reagila | Gedeon Richter | ||||
| 2017/7/10 | Spheroids of human autologous matrix-associated chondrocytes | Spherox | CO.DON AG | ||||
| 2017/7/6 | Cenegermin | Oxervate | Dompe farmaceutici s.p.a. | O | BLA | ||
| 2017/6/29 | Inotuzumab ozogamicin | Besponsa | Pfizer Limited | O | BLA | ||
| 2017/6/23 | Etanercept | Erelzi | Sandoz GmbH | ||||
| 2017/6/23 | Sarilumab | Kevzara | Sanofi-Aventis Groupe | NME | |||
| 2017/6/23 | Dimethyl fumarate | Skilarence | Almirall S.A | ||||
| 2017/6/23 | Carglumic acid | Ucedane | Lucane Pharma | G | |||
| 2017/6/15 | Rituximab | Rixathon, Riximyo B | Sandoz GmbH | ||||
| 2017/6/2 | Pentosan polysulfate sodium | Elmiron | bene-Arzneimittel GmbH | ||||
| 2017/6/2 | Nonacog beta pegol | Refixia | Novo Nordisk A/S | BLA | |||
| 2017/5/30 | Cerliponase alfa | Brineura | BioMarin International Limited | O E | BLA | ||
| 2017/5/30 | Nusinersen | Spinraza | Biogen Idec Ltd | O | NME | ||
| 2017/5/24 | Meningococcal group b vaccine (recombinant, adsorbed) | Trumenba | Pfizer Limited | ||||
| 2017/5/22 | Ivabradine | Ivabradine Accord | Accord Healthcare Ltd | G | |||
| 2017/5/8 | Dinutuximab beta | Dinutuximab beta Apeiron | Apeiron Biologics AG | O E | |||
| 2017/4/28 | Emtricitabine – tenofovir disoproxil mixt | Emtricitabine/Tenofovir disoproxil Krka d.d. | KRKA, d.d., Novo mesto | G | |||
| 2017/4/24 | Parathyroid hormone | Natpar | Shire Pharmaceuticals Ireland Ltd | O C | BLA | ||
| 2017/4/20 | Edoxaban | Roteas | Daiichi Sankyo Europe GmbH | ||||
| 2017/3/22 | Tofacitinib citrate | Xeljanz | Pfizer Limited | NME | |||
| 2017/3/20 | Umeclidinium | Rolufta | GlaxoSmithKline Trading Services Limited | ||||
| 2017/3/3 | Chlormethine | Ledaga | Actelion Registration Ltd. | O | |||
| 2017/2/27 | Pregabalin | Pregabalin Zentiva | Zentiva k.s. | G | |||
| 2017/2/17 | Rituximab | Truxima | Celltrion Healthcare Hungary Kft. | B | |||
| 2017/2/13 | Etanercept | Lifmior | Pfizer Limited | ||||
| 2017/2/13 | Baricitinib | Olumiant | Eli Lilly Nederland B.V. | NME | |||
| 2017/1/19 | Mercaptamine | Cystadrops | Orphan Europe S.A.R.L. | O | |||
| 2017/1/18 | Bezlotoxumab | Zinplava | Merck Sharp & Dohme Limited | NME | |||
| 2017/1/11 | Teriparatide | Movymia | STADA Arzneimittel AG | B | |||
| 2017/1/11 | Insulin glargine / lixisenatide | Suliqua | Sanofi-Aventis Groupe | ||||
| 2017/1/9 | Insulin aspart | Fiasp | Novo Nordisk A/S | ||||
| 2017/1/9 | Tadalafil | Tadalafil | Mylan S.A.S | G | |||
| 2017/1/9 | Tenofovir alafenamide | Vemlidy | Gilead Sciences International Ltd | ||||
| 2017/1/4 | Lonoctocog alfa | Afstyla | CSL Behring GmbH | BLA | |||
| 2017/1/4 | Darunavir | Darunavir Mylan | Mylan S.A.S. | G | |||
| 2017/1/4 | Insulin glargine | Lusduna | Merck Sharp & Dohme Limited | B | |||
| 2017/1/4 | Teriparatide | Terrosa | Gedeon Richter Plc. | B |
SECTION B; EMA Approvals
Combined drugs USFDA+EMA +PMDA list are listed below. trying to simplify search
1 Abaloparatide USFDA
2 Abemaciclib USFDA
3 ACALABRUTINIB USFDA
3A ALOFISEL EMA
3B AMENAMEVIR JAPAN
4 ANGIOTENSIN II USFDA
4A Atezolizumab EMA
5 AVELUMAB USFDA+EMA
6 BENRALIZUMAB USFDA+EMA
6A BARICITINIB JAPAN
7 BENZNIDAZOLE USFDA
8 BETRIXABAN USFDA
9 BRIGATINIB USFDA
10 BRODALUMAB USFDA+EMA
10A BUROSUMAB EMA
10B CARIPRAZINE HYDROCHLORIDE EMA
11 CERLIPONASE ALPA USFDA+EMA
12 COPANLISIB USFDA
13 DEFLAZACORT USFDA
14 Delafloxacin USFDA
15 Deutetrabenazine USFDA
16DUPILUMAB USFDA+EMA
17 DURVALUMAB USFDA
18 EDAVARONE USFDA
19 EMICIZUMAB USFDA
20 Enasidenib USFDA
21 ERTUGLIFLOZIN USFDA
22 ETELCALCETIDE USFDA
22A FORODESINE JAPAN
22B FLUCICLOVINE EMA
23 GLECAPREVIR USFDA+EMA
24 GUSELKUMAB USFDA+EMA
25 INOTUZUMAB OZOGAMICIN USFDA+EMA
26 LATANOPROSTENE USFDA
27 LETERMOVIR USFDA+EMA
27A Utetium lu 177 dotatate EMA
28 MACIMORELIN ACETATE USFDA
29 MEROPENEM USFDA
30 MIDOSTAURIN USFDA+EMA
31 NALDEMEDINE USFDA
32 NERATINIB USFDA
33 NETARSUDIL USFDA
34A NONACOG EMA
34B NUCINERSEN EMA +Japan
35 Ocrelizumab USFDA+EMA
35A OXERVATE EMA
36 OZENOXACIN USFDA
36A PATIROMER EMA
36B PADELIPORFIN EMA
36C PEMAFIBRATE JAPAN
37 PIBRENTASVIR USFDA+EMA
38 PLECANATIDE USFDA
39A ROLAPITANT EMA
39BRURLOCTOCOG EMA
40 SARILUMAB USFDA+EMA
41 SECNIDAZOLE USFDA
42 SAFINAMIDE USFDA
43 SEMAGLUTIDE USFDA+EMA
43A SODIUM ZIRCONIUM CYCLOCYLICATE EMA
44 SOFOSBUVIR USFDA+EMA
44A SPHEROX EMA
45 TELOTRISTAT ETHYL USFDA+EMA
45A TIVOZANIB EMA
45B TOFACITINIB EMA
45C TRUMENBA EMA
46 VABORBACTAM USFDA
47 VALBENAZINE USFDA
48 VESTRONIDASE ALFA-VJBK USFDA
49 VELPATASVIR USFDA+EMA
50 VOXILAPREVIR USFDA+EMA
Drugs EMA list missed out in usfda list
3A ALOFISEL
link………https://newdrugapprovals.org/2018/03/02/alofisel-darvadstrocel-cx-601/
4A Atezolizumab
WILL BE UPDATED
10A BUROSUMAB
WILL BE UPDATED
10B CARIPRAZINE HYDROCHLORIDE
WILL BE UPDATED
22B FLUCICLOVINE
![]()
SEE EMA
| Axumin : EPAR – Summary for the public | EN = English | 06/07/2017 |
27A Lutetium lu 177 dotatate
WILL BE UPDATED
34A NONACOG
WILL BE UPDATED
34B NUCINERSEN
EMA AND JAPAN 2017 APPROVED


35A OXERVATE
WILL BE UPDATED
36A PATIROMER
WILL BE UPDATED
36B PADELIPORFIN

| NAME | Tookad |
|---|---|
| AGENCY PRODUCT NUMBER | EMEA/H/C/004182 |
| ACTIVE SUBSTANCE | padeliporfin di-potassium |
| INTERNATIONAL NON-PROPRIETARY NAME(INN) OR COMMON NAME | padeliporfin |
| THERAPEUTIC AREA | Prostatic Neoplasms |
| ANATOMICAL THERAPEUTIC CHEMICAL (ATC) CODE | L01XD07 |
| ADDITIONAL MONITORING | This medicine is under additional monitoring. This means that it is being monitored even more intensively than other medicines. For more information, see medicines under additional monitoring. |
| MARKETING-AUTHORISATION HOLDER | STEBA Biotech S.A |
|---|---|
| REVISION | 0 |
| DATE OF ISSUE OF MARKETING AUTHORISATION VALID THROUGHOUT THE EUROPEAN UNION | 10/11/2017 |
Contact address:
STEBA Biotech S.A
7 place du theatre
L-2613 Luxembourg
Luxembourg
38A PRALATREXATE
Japan approved 2017
| 2017/7/3 | PMDA | JAPAN | Pralatrexate | Difolta | Mundipharma | NME |
39A ROLAPITANT
WILL BE UPDATED
39B RURLOCTOCOG
WILL BE UPDATED
43A SODIUM ZIRCONIUM
WILL BE UPDATED
44A SPHEROX
WILL BE UPDATED
45A TIVOZANIB

Pharmacotherapeutic group
Antineoplastic agents
Therapeutic indication
Fotivda is indicated for the first line treatment of adult patients with advanced renal cell carcinoma (RCC) and for adult patients who are VEGFR and mTOR pathway inhibitor-naïve following disease progression after one prior treatment with cytokine therapy for advanced RCC.
Treatment of advanced renal cell carcinoma
Fotivda : EPAR -Product Information

Tivozanib is synthesized in three main steps using well defined starting materials with acceptable
specifications.
Adequate in-process controls are applied during the synthesis. The specifications and control methods for
intermediate products, starting materials and reagents have been presented. The critical process
parameters are duly justified, methodology is presented and control is adequate.
The characterisation of the active substance and its impurities are in accordance with the EU guideline on
chemistry of new active substances. Potential and actual impurities were well discussed with regards to
their origin and characterised.
The active substance is packaged in a low-density polyethylene (LDPE) bag which complies with the EC
directive 2002/72/EC and EC 10/2011 as amended.
Product details
| Name | Fotivda |
|---|---|
| Agency product number | EMEA/H/C/004131 |
| Active substance | tivozanib |
| International non-proprietary name(INN) or common name | tivozanib hydrochloride monohydrate |
| Therapeutic area | Carcinoma, Renal Cell |
| Anatomical therapeutic chemical (ATC) code | L01XE |
Publication details
| Marketing-authorisation holder | EUSA Pharma (UK) Limited |
|---|---|
| Revision | 0 |
| Date of issue of marketing authorisation valid throughout the European Union | 24/08/2017 |
Contact address:
EUSA Pharma (UK) Limited
Breakspear Park, Breakspear Way
Hemel Hempstead, HP2 4TZ
United Kingdom
45B TOFACITINIB
WILL BE UPDATED
45C TRUMENBA
WILL BE UPDATED
SECTION C JAPANFORODOS

SECTION C New Drugs JAPAN
https://www.pmda.go.jp/english/review-services/reviews/approved-information/drugs/0002.html
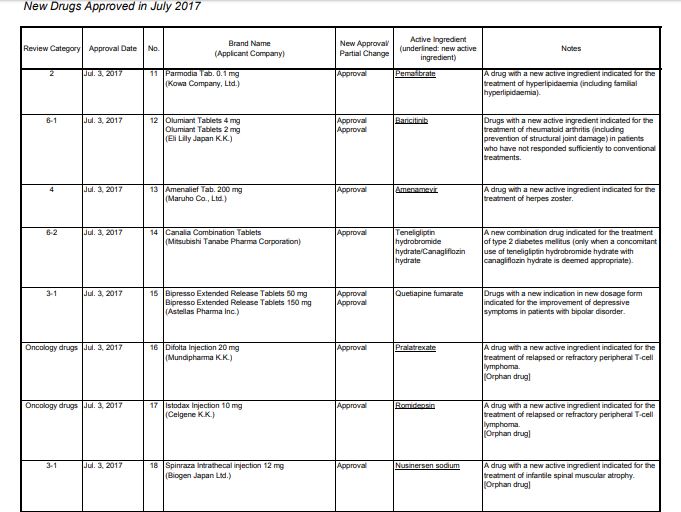
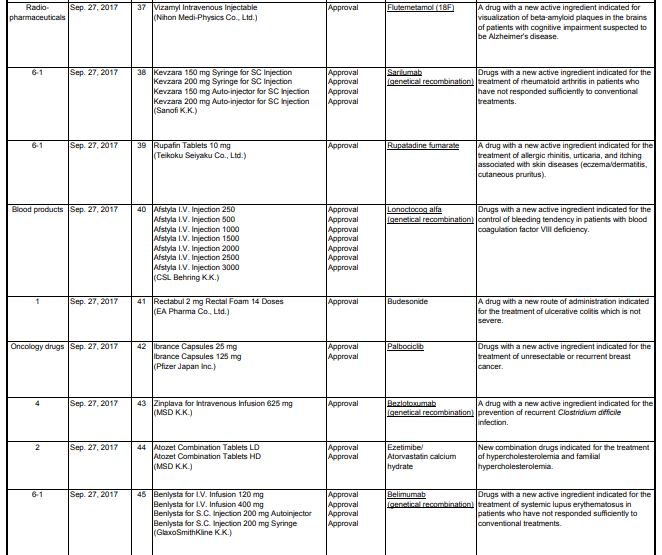
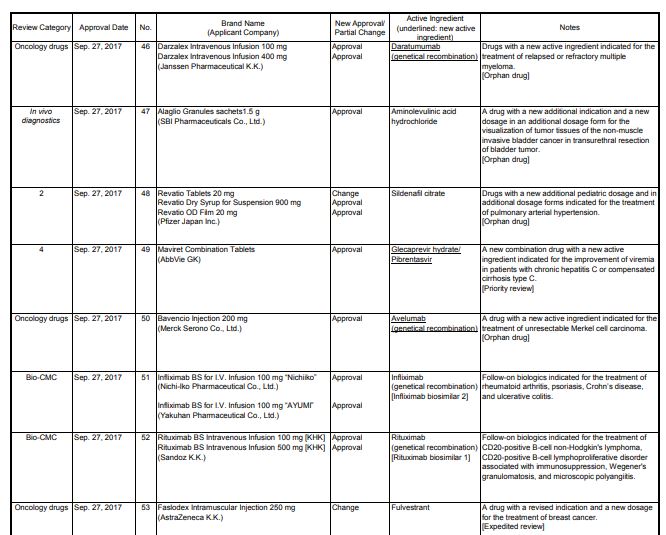
JAPAN 2017
| 2017/9/27 | Avelumab (genetical recombination) | Bavencio | Merck Serono | BLA |
| 2017/9/27 | Glecaprevir – pibrentasvir mixt | Maviret | Abbvie | NME |
| 2017/9/27 | Daratumumab (genetical recombination) | Darzalex | Janssen Pharmaceutical | BLA |
| 2017/9/27 | Belimumab (genetical recombination) | Benlysta | GlaxoSmithKline | BLA |
| 2017/9/27 | Bezlotoxumab (genetical recombination) | Zinplava | MDS | BLA |
| 2017/9/27 | Palbociclib | Ibrance | Pfizer | NME |
| 2017/9/27 | Lonoctocog alfa (genetical recombination) | Afstyla | CSL Behring | BLA |
| 2017/9/27 | Rupatadine fumarate | Rupafin | Teikoku seiyaku | NME |
| 2017/9/27 | Sarilumab (genetical receombination) | Kevzara | Sanofi | BLA |
| 2017/9/27 | Flutemetamol (18F) | Vizamyl | Nihon Medi-Physics | NME |
| 2017/7/3 | Nusinersen sodium | Spinraza | Biogen Japan | |
| 2017/7/3 | Romidepsin | Istodax | Celgene | NME |
| 2017/7/3 | Pralatrexate | Difolta | Mundipharma | NME |
| 2017/7/3 | Amenamevir | Amenalief | Maruho | NME |
| 2017/7/3 | Baricitinib | Olumiant | Lilly | NME |
| 2017/7/3 | Pemafibrate | Parmodia | Kowa | NME |
| 2017/3/30 | Human prothrombin complex, freeze-dried concentrated | Kcentra | CSL Behring | |
| 2017/3/30 | Ixazomib citrate | Ninlaro | Takeda | NME |
| 2017/3/30 | Forodesine hydrochloride | Mundesine | Mundipharma | |
| 2017/3/30 | Aflibercept beta (genetical recombination) | Zaltrap | Sanofi | |
| 2017/3/30 | Hydromorphone hydrochloride | Narusus, Narurapid | DaiichiSankyo-pp | |
| 2017/3/30 | Naldemedine tosylate | Symproic | Shionogi | NME |
| 2017/3/30 | Guanfacine hydrochloride | Intuniv | Shionogi |
3B AMENAMEVIR

Originally developed by Astellas, the drug was licensed to Maruho. Amenamevir treats herpes zoster by inhibiting the activity of the helicase-primer enzyme during viral DNA replication and blocking the virus’s proliferation.
Amenalief® is an oral film-coated tablet containing 200 mg of amenamevir per tablet. Recommended dose of 1 day, 400mg each time, after meals.
22A FORODESINE HYDROCHLORIDE

LINK https://newdrugapprovals.org/2018/03/06/forodesine-hydrochloride/
6A BARICITINIB JAPAN

Originally developed by Incyte, Baricitinib was later licensed to and for sale by Lilly under the trade name Olumiant®. Baricitinib is an irreversible inhibitor of Janus kinase 1 (JAK1) and Janus kinase 2 (JAK2). Olumiant® is approved for the treatment of mild to moderate rheumatoid arthritis in adult patients who are not responsive or intolerant to other anti-arthritic drugs. This product can be used alone or in combination with methotrexate.
Olumiant® is a film-coated tablet containing 2 mg or 4 mg per tablet. Recommended oral dose is 4mg daily, with meals or fasting food, you can take any time period.
2017/7/3PMDA Baricitinib Olumiant Lilly
36C PEMAFIBRATE

LINK https://newdrugapprovals.org/2016/04/24/pemafibrate/
SECTION D
CDSCO INDIA

http://www.cdsco.nic.in/forms/list.aspx?lid=2034&Id=11 http://www.cdsco.nic.in/forms/list.aspx?lid=2034&Id=11
![]()







KEEP WATCHING UNDER CONSTRUCTION AND WILL BE PASTED SOON………………………………………..
KEEP WATCHING UNDER CONSTRUCTION AND WILL BE PASTED SOON………………………………………..
KEEP WATCHING UNDER CONSTRUCTION AND WILL BE PASTED SOON………………………………………..
KEEP WATCHING UNDER CONSTRUCTION AND WILL BE PASTED SOON………………………………………..
REFERENCES
2 http://www.ema.europa.eu/docs/en_GB/document_library/Report/2018/01/WC500242079.pdf
“NEW DRUG APPROVALS” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This is a compilation for educational purposes only. P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent



 amcrasto@gmail.com
amcrasto@gmail.com
I , Dr A.M.Crasto is writing this blog to share the knowledge/views, after reading Scientific Journals/Articles/News Articles/Wikipedia. My views/comments are based on the results /conclusions by the authors(researchers). I do mention either the link or reference of the article(s) in my blog and hope those interested can read for details. I am briefly summarising the remarks or conclusions of the authors (researchers). If one believe that their intellectual property right /copyright is infringed by any content on this blog, please contact or leave message at below email address amcrasto@gmail.com. It will be removed ASAP
////////EMA APPROVALS, USFDA Approvals, ACALABRUTINIB, AVELUMAB, BETRIXABAN, BRODALUMAB, COPANLISIB, DEFLAZACORT, Delafloxacin, Deutetrabenazine, DUPILUMAB, ETELCALCETIDE, Naldemedine, NETARSUDIL, NIRAPARIB, Ocrelizumab, PLECANATIDE, RIBOCICLIB, SAFINAMIDE, TELOTRISTAT ETHYL, VALBENAZINE, CERLIPONASE, BRIGATINIB, MIDOSTAURIN, Abaloparatide, BENZNIDAZOLE, NERATINIB, inotuzumab ozogamicin, Enasidenib, LETERMOVIR, GLECAPREVIR, PIBRENTASVIR, VOXILAPREVIR, SOFOSBUVIR, EDAVARONE, abemaciclib, ANGIOTENSIN II, VESTRONIDASE, macimorelin acetate, ERTUGLIFLOZIN, SEMAGLUTIDE, EMICIZUMAB, eu 2017, fda 2017, BENRALIZUMAB, DURVALUMAB, GUSELKUMAB, LATANOPROSTENE, OZENOXACIN, SARILUMAB, SECNIDAZOLE, BENRALIZUMAB, TIVOZANIB, SARILUMAB, FLUCICLOVINE,
Pemafibrate, Пемафибрат , بيرمافيبرات , 佩玛贝特 , ペマフィブラート ,


Pemafibrate
NDA Filing Japan, Phase 2 in EU, US
A PPAR-α agonist potentially for the treatment of dyslipidemia.

K-877, K-13675, (R)-
CAS No. 848259-27-8,
Molecular Formula,C28-H30-N2-O6,Molecular Weight,490.553
- Originator Kowa Pharmaceutical
- Class Antihyperlipidaemics
- Mechanism of Action Peroxisome proliferator-activated receptor alpha agonists
- Preregistration Dyslipidaemias
Most Recent Events
- 01 Feb 2016 Kowa Research Institute completes a phase I drug-interaction trial in Healthy volunteers in USA (PO) (NCT02719431)
- 12 Jan 2016 Kowa Research Institute plans the phase III PROMINENT trial for Dyslipidaemia (In patients with diabetes mellitus) in countries worldwide
- 01 Jan 2016 Kowa Research Institute initiates a phase I drug-interaction trial in Healthy volunteers in USA (PO) (NCT02719431)

UPDATE ADDED ON MARCH 2017
Pemafibrate
- Molecular FormulaC28H30N2O6
- Average mass490.548 Da
|
Antihyperlipidemic, Triglyceride synthesis inhibitor, Peroxisome proliferator-activated receptor (PPAR) alpha agonist
|
Pemafibrate, marketed as Parmodia, is a peroxisome proliferator-activated receptor alpha (PPARα) agonist. It is developed and marketed by Kowa Pharmaceuticals.
In 3 July 2017, Pharmaceuticals and Medical Devices Agency approved it in Japan. It is available in 0.1 mg tablets.[1]
References
- Pemafibrate, pharmacodia.com
| ペマフィブラート Pemafibrate C28H30N2O6 : 490.55 [848259-27-8] |
 |
|
| Clinical data | |
|---|---|
| Trade names | Parmodia |
| Synonyms | K-13675 |
| Routes of administration |
Oral |
| Identifiers | |
| CAS Number | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C28H30N2O6 |
| Molar mass | 490.56 g·mol−1 |
| 3D model (JSmol) | |
////////////Pemafibrate, Пемафибрат , بيرمافيبرات , 佩玛贝特 , ペマフィブラート ,
Pemafibrate, also known as K-877 and (R)-K 13675, is a PPAR alpha agonist. (R)-K-13675 decreases the secretion of inflammatory markers without affecting cell proliferation or tube formation. Peroxisome proliferator-activated receptor-alpha (PPAR-alpha) is a key regulator of lipid and glucose metabolism and has been implicated in inflammation. (R)-K-13675 was associated with the inhibition of inflammatory responses without affecting cell proliferation or angiogenesis, and subsequently may induce an anti-atherosclerotic effect.
Pemafibrate had been filed NDA by Kowa for the treatment of dyslipidemia in the Japan in 2015.
Pemafibrate is in phase II clinical trials for the treatment of dyslipidemia in the US and EU.

Reference:1. US2009023944A1.
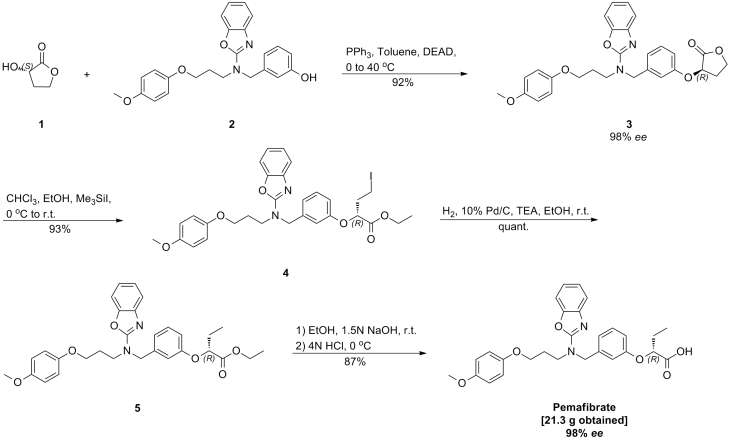
Reference:1. US2009076280A1.
http://www.google.com/patents/US20090076280
Example 5 Synthesis of (R)-2-{3-[N-(benzoxazole-2-yl)-N-(3-(4-methoxyphenoxy)propyl)aminomethyl]phenyloxy}butyric acid (Compound (6))
-
Ethyl (R)-2-{3-[N-(benzoxazole-2-yl)-N-(3-(4-methoxyphenoxy)propyl)aminomethyl]phenyloxy}butylate (26.0 g) was dissolved in ethanol (200 mL), and 1.5N NaOH (50 mL) was added to the solution, followed by stirring for 1 hour at room temperature. The reaction mixture was washed with diethyl ether, and the formed aqueous layer was acidified with 4N HCl under ice cooling. The thus-treated aqueous layer was extracted with ethyl acetate, and the extract was washed sequentially with water and saturated brine. The washed extract was dried over sodium sulfate anhydrate and concentrated under reduced pressure. The residue was purified through silica gel column chromatography (chloroform/methanol=10/1), to thereby yield the target product (21.3 g, 87%, 98% ee).
Optical Purity:
-
Measurement conditions: HPLC
-
Column: CHIRALPAK AD
-
Solvent: n-hexane/IPA/TFA=100/30/0.1
-
Flow rate: 2 mL/min
-
Retention time: 4.19 min (S-form; 3.68 min)
-
1H-NMR (400 MHz, CD3OD) δ ppm: 0.94 (t, J=7 Hz, 3H), 1.81 (m, 2H), 1.99 (quintet, J=6 Hz, 2H), 3.60 (t, J=7 Hz, 2H), 3.61 (s, 3H), 3.85 (t, J=6 Hz, 2H), 4.40 (t, J=6 Hz, 1H), 4.65 (s, 2H), 6.69-6.80 (m, 7H), 6.91 (dt, J=7, 1 Hz, 1H), 7.05 (dt, J=7, 1 Hz, 1H), 7.12-7.18 (m, 4H).

Reference:1. Bioorg. Med. Chem. Lett. 2007, 17, 4689-4693.
Landmark Trial Entitled “PROMINENT” To Explore The Prevention Of Heart Disease In Diabetic Patients With High Triglycerides And Low HDL-C
Trial will evaluate if lowering triglycerides and increasing functional HDL with Kowa’s potent selective peroxisome proliferator activator receptor-alpha (PPAR-alpha) modulator, K-877 (pemafibrate) can reduce the elevated risk of cardiovascular disease in high-risk diabetic patients who are already taking statins
Jan 12, 2016, 09:00 ET from Kowa Research Institute, Inc.
RESEARCH TRIANGLE PARK, N.C., Jan. 12, 2016 /PRNewswire/ — Kowa Research Institute, Inc., announced plans to conduct an international, multi-center cardiovascular outcomes trial evaluating triglyceride reduction and increasing functional HDL with K-877 (pemafibrate), in high-risk diabetic patients with high triglyceride and low HDL-C levels who are already taking statins. K-877 is a highly potent and selective peroxisome proliferator activator receptor-alpha (PPAR-alpha) modulator (SPPARMalpha), a promising category of metabolic therapy.
Paul Ridker, MD, director of the Center for Cardiovascular Disease Prevention (CCVDP) at Brigham and Women’s Hospital (BWH), a teaching affiliate of Harvard Medical School, and Aruna Pradhan, MD, a cardiologist at BWH, will be co-Principal Investigators of the planned trial.
“This trial is unprecedented,” said Gary Gordon, MD, President, Kowa Research Institute, Inc. “Statins are effective in lowering cardiovascular risk among patients with high cholesterol, but residual risk remains, particularly in patients with high triglyceride levels and low HDL-C levels. Kowa will be the first company to run a major, randomized clinical trial investigating whether modulating PPAR-alpha to lower triglycerides and increase functional HDL in diabetic patients can reduce cardiovascular risk when added to statin therapy.”
Evidence supports a role for triglyceride-rich lipoproteins and low HDL-C as important contributors to atherosclerosis. Kowa specifically set out to create the most potent and selective PPAR-alpha modulator ever developed, and succeeded with K-877, which is at least 1,000 times as potent and selective as other drugs. Kowa has completed clinical development of K-877 for hyperlipidemia in Japan, and has submitted it to the PMDA for approval as a new drug. Kowa’s clinical studies have shown K-877 significantly reduces triglycerides, ApoC3, and remnant cholesterol and increases functional HDL and FGF21.
The Pemafibrate to Reduce cardiovascular OutcoMes by reducing triglycerides IN diabetic patiENTs (PROMINENT) Phase 3 K-877 cardiovascular outcomes trial will recruit an estimated 10,000 high-risk diabetic patients worldwide. All participants will receive aggressive, standard of care management of cardiovascular risk factors including treatment with high-intensity statins. In addition, patients will receive either K-877 or placebo. The trial will include diabetic patients with and without established cardiovascular disease and will test whether K-877 reduces the occurrence of heart attacks, hospitalizations for unstable angina requiring unplanned revascularization, stroke, or death from cardiovascular causes.
“Cardiovascular disease remains the number one cause of death worldwide,” said Dr. Gordon. “Reducing residual cardiovascular risk with K-877 would be valuable to physicians managing patients’ cardiovascular disease.”
About Kowa Company, Ltd. and Kowa Research Institute, Inc.
Kowa Company, Ltd. (Kowa) is a privately held multinational company headquartered in Nagoya, Japan. Established in 1894, Kowa is actively engaged in various manufacturing and trading activities in the fields of pharmaceuticals, life science, information technology, textiles, machinery and various consumer products. Kowa’s pharmaceutical division is focused on research and development for cardiovascular therapeutics (dyslipidemia, type 2 diabetes and atherosclerosis), ophthalmology and anti-inflammatory agents. The company’s flagship product, LIVALO® (pitavastatin), is approved in 45 countries around the world.
Kowa Research Institute, Inc., headquartered in Research Triangle Park, NC, is the division of Kowa responsible for the clinical development of Kowa’s new drugs in the United States. Kowa Research Institute was established in 1997 in California and began operations at the current location in 2003. For more information about Kowa Research Institute, visit www.kowaus.com.
| 1 | NCT00610441 | Dose Finding Study in Adults With Attention-Deficit/Hyperactivity Disorder (ADHD)(174007/P05805/MK-8777-003) | Completed | Drug: MK-8777|Drug: Placebo | Phase 2 | Merck Sharp & Dohme Corp. |
| 2 | NCT00610649 | Trial to Determine the Maximum Tolerated Dose (MTD) Based on Safety and Tolerability, of Org 26576 in Participants With Major Depressive Disorder (174001/P05704/MK-8777-001) | Completed | Drug: MK-8777|Drug: Placebo | Phase 2 | Merck Sharp & Dohme Corp. |
| 3 | NCT02073084 | A Thorough Corrected QT Interval Trial | Completed | Drug: K-877 Low Dose|Drug: Moxifloxacin|Other: Placebo|Drug: K-877 High Dose | Phase 1 | Kowa Research Institute, Inc. |
| 4 | NCT02273986 | Drug-Drug Interaction Study in Health Adult Volunteers | Completed | Drug: Digoxin|Drug: K-877 | Phase 1 | Kowa Research Institute, Inc. |
| 5 | NCT02275962 | Drug-Drug Interaction Study in Healthy Adult Volunteers | Active, not recruiting | Drug: K-877|Drug: Rifampin | Phase 1 | Kowa Research Institute, Inc. |
| 6 | NCT02275975 | Drug-Drug Interaction Study in Healthy Adult Volunteers | Completed | Drug: K-877|Drug: Fluconazole | Phase 1 | Kowa Research Institute, Inc. |
| 7 | NCT02275988 | Drug-Drug Interaction Study in Healthy Adult Volunteers | Completed | Drug: K-877|Drug: Clarithromycin | Phase 1 | Kowa Research Institute, Inc. |
| 8 | NCT02276001 | Drug-Drug Interaction Study in Healthy Adult Volunteers | Completed | Drug: K-877|Drug: Cyclosporine | Phase 1 | Kowa Research Institute, Inc. |
| US6653334 * | Dec 27, 2002 | Nov 25, 2003 | Kowa Co., Ltd. | Benzoxazole compound and pharmaceutical composition containing the same |
| US7109226 * | Sep 3, 2004 | Sep 19, 2006 | Kowa Co., Ltd. | PPAR-activating compound and pharmaceutical composition comprising the compound |
| US7183295 * | Apr 20, 2006 | Feb 27, 2007 | Kowa Co., Ltd. | PPAR-activating compound and pharmaceutical composition comprising the compound |
///////Pemafibrate, NDA, Kowa, dyslipidemia, Japan, 2015, phase II clinical trials, US and EU, K-877, K-13675, (R)-
CC[C@H](C(=O)O)Oc1cccc(c1)CN(CCCOc2ccc(cc2)OC)c3nc4ccccc4o3
CC[C@@H](OC1=CC=CC(CN(C2=NC3=CC=CC=C3O2)CCCOC4=CC=C(OC)C=C4)=C1)C(O)=O
Baricitinib

Baricitinib
NDA submitted jan 2016
2-[1-ethylsulfonyl-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]azetidin-3-yl]acetonitrile,
3-Azetidineacetonitrile, 1-(ethylsulfonyl)-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1Hpyrazol-1-yl]-
2-(3-(4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-1-(ethylsulfonyl)azetidin-3-yl)acetonitrile
3-Azetidineacetonitrile, 1-(ethylsulfonyl)-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl]-
2-(3-(4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-1-(ethylsulfonyl)azetidin-3-yl)acetonitrile
For the treatment of rheumatoid arthritis and diabetic kidney disease
Incyte Corporation INNOVATOR
http://www.ama-assn.org/resources/doc/usan/baricitinib.pdf
MF C16H17N7O2S
MW 371.4
SPONSOR Eli Lilly and Company
CODE LY3009104, INCB028050
CAS 1187594-09-7
UPDATE……..APPROVED PMDA 2017
WO 2009114512
2-[3-(4-{7H-pyrrolo[2,3-d]pyrimidin-4-yl}-1H-pyrazol- 1-yl)-1-(ethylsulfonyl)azetidin-3-yl]acetonitrile (baricitinib) FREE FORM
m.p. 193–195 °C;
IR: 3203, 3113, 2998, 2847, 2363, 1584, 1328, 1137 cm–1.
Anal. calcd for C16H17N7 O2 S: C, 51.74; H, 4.61; N, 26.40; found: C, 51.91; H, 4.49; N, 26.57%. MS (m/z): 372 [M + H]+;
1 H NMR (300 MHz, DMSO-d6 ): δ 1.25 (t, J = 7.3 Hz, 3H), 3.23 (m, J = 7.3 Hz, 2H), 3.69 (s, 2H), 4.24 (d, J = 9.0 Hz, 2H), 4.61 (d, J = 9.0 Hz, 2H), 7.08 (s, 1H), 7.62 (s, 1H), 8.47 (s, 1H), 8.71 (s, 1H), 8.92 (s, 1H), 12.12 (s, 1H);
13C NMR (125 MHz, DMSO-d6 ): δ 7.4, 24.9, 39.3, 43.4, 58.5, 99.9, 113.0, 116.6, 126.9, 129.5, 139.9, 149.3, 150.9, 152.2.
REF Journal of Chemical Research, Volume 40, Number 4, April 2016, pp. 205-208(4)

Baricitinib phosphate
{1-(Ethylsulfonyl)-3-[4-(1H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl]-3-azetidinyl}acetonitrile phosphate (1:1)
Cas 1187595-84-1, C16H20N7O6PS, 469.41
- Originator Incyte Corporation
- Developer Eli Lilly; Incyte Corporation
- Class Acetonitriles; Antipsoriatics; Antirheumatics; Azetidines; Pyrazoles; Pyrimidines; Pyrroles; Small molecules
- Mechanism of Action Janus kinase 1 inhibitors; Janus kinase-2 inhibitors
Highest Development Phases
- Preregistration Rheumatoid arthritis
- Phase II Atopic dermatitis; Diabetic nephropathies; Psoriasis; Systemic lupus erythematosus
Most Recent Events
- 01 Jul 2016 Eli Lilly completes a phase I trial in Healthy volunteers in China (PO) (NCT02758613)
- 09 Jun 2016 Efficacy and adverse events data from the RA-BEYOND phase III trial in Rheumatoid arthritis presented at the 17thAnnual Congress of the European League Against Rheumatism (EULAR-2016)
- 09 Jun 2016 Final efficacy and safety data from the phase III trials, RA-BEAM and RA-BEGIN in Rheumatoid arthritis were presented at the 17th Annual Congress of the European League Against Rheumatism (EULAR – 2016)
The Janus kinase (JAK) is a family of four tyrosine receptor kinases that play a pivotal role in cytokine receptor signalling pathways via their interaction with signal transducers and
activators of transcription proteins. The four JAK family members are Janus kinase 1 (JAK1), Janus kinase 2 (JAK2), Janus kinase 3 (JAK3) and tyrosine kinase (TYK2), whose
lengths range from 120 to 140 kDa. It has been shown that JAK2 activation may be critical for tumour growth and progression,indicating its selection as a therapeutic target. Moreover, since JAK3 is required for immune cell development, targeting JAK3 could be a useful strategy for generating a novel class of immunosuppressant drugs. JAK1 and TYK2 have been implicated in disease and immune suppression.
Over the past decade there have been extensive efforts to identify and design novel small-molecule JAK inhibitors with varied profiles of subtype selectivity to address unmet medical needs Ruxolitinib is a Janus kinase inhibitor with selectivity for subtypes JAK1 and JAK2. It was approved by the U.S. Food and Drug Administration (FDA) for the treatment
of intermediate or high-risk myelofibrosis in November 2011.
Selective inhibitors of JAK are viewed as having considerable potential as disease-modifying anti-inflammatory drugs for the treatment of rheumatoid arthritis. Tofacitinib, which was the first oral non-biological disease-modifying antirheumatic drug, was approved for the management of rheumatoid arthritis (RA) at the end of 2012. Baricitinib and filgotinib are beingmevaluated in phase III and phase II clinical trials respectively for
the treatment of rheumatoid arthritis. Baricitinib (also known as LY3009104 or INCB028050) is a novel and potent small molecule inhibitor of the Janus kinase family of enzymes with selectivity for JAK1 and JAK2. In in vitro studies baricitinib inhibited JAK1 and JAK2 in the low nanomolar range, while it demonstrated low inhibitory activity for JAK3 and moderate activity for TYK2.9–13 The data from two phase III studies showed that baricitinib can achieve impressive responses in RA patients who have not responded well to established therapies.
Therefore, improvement in the preparation of baricitinib is of practical significance.
1 L. Tan, K. Akahane, R. McNally, K.M.S.E. Reyskens, S.B. Ficarro, S. Liu,
G.S. Herter-Sprie, S. Koyama, M.J. Pattison, K. Labella, L. Johannessen,
E.A. Akbay, K. Wong, D.A. Frank, J.A. Marto, T.A. Look, J.S.C. Arthur,
M.J. Eck and N.S. Gray, J. Med. Chem., 2015, 58, 6589.
2 J.J. Kulagowski, W. Blair, R.J. Bull, C. Chang, G. Deshmukh, H.J. Dyke,
C. Eigenbrot, N. Ghilardi, P. Gibbons, T.K. Harrison, P.R. Hewitt, M.
Liimatta, C.A. Hurley, A. Johnson, T. Johnson, J.R. Kenny, P.B. Kohli, R.J.
Maxey, R. Mendonca, K. Mortara, J. Murray, R. Narukulla, S. Shia, M.
Steffek, S. Ubhayakar, M. Ultsch, A. Abbema, S.I. Ward, B. Waszkowycz
and M. Zak, J. Med. Chem., 2012, 55, 5901.
3 J.F. Kadow, Y. Ueda, N.A. Meanwell, T.P. Connolly, T. Wang, C. Chen, K.
Yeung, J. Zhu, J.A. Bender, Z. Yang, D. Parker, P. Lin, R.J. Colonno, M.
Mathew, D. Morgan, M. Zheng, C. Chien and D. Grasela, J. Med. Chem.,
2012, 55, 2048.
4 Q. Su, S. Ioannidis, C. Chuaqui, L. Almeida, M. Alimzhanov, G. Bebernitz,
K. Bell, M. Block, T. Howard, S. Huang, D. Huszar, J.A. Read, C.R. Costa,
J. Shi, M. Su, M. Ye and M. Zinda, J. Med. Chem., 2014, 57, 144.
5 J.D. Clark, M.E. Flanagan and J. Telliez, J. Med. Chem., 2014, 57, 5023.
6 P. Norman, Expert. Opin. Investig. Drugs, 2014, 23, 1067.
7 L.J. Farmer, M.W. Ledeboer, T. Hoock, M.J. Arnost, R.S. Bethiel, Y.L.
Bennani, J.J. Black, C.L. Brummel, A. Chakilam, W.A. Dorsch, B. Fan,
J.E. Cochran, S. Halas, E.M. Harrington, J.K. Hogan, D. Howe, H. Huang,
D.H. Jacobs, L.M. Laitinen, S. Liao, S. Mahajan, V. Marone, G. Martinez-
Botella, P. McCarthy, D. Messersmith, M. Namchuk, L. Oh, M.S. Penney,
A.C. Pierce, S.A. Raybuck, A. Rugg, F.G. Salituro, K. Saxena, D. Shannon,
D. Shlyakter, L. Swenson, S. Tian, C. Town, J. Wang, T. Wang, M.W.
Wannamaker, R.J. Winquist and H.J. Zuccola, J. Med. Chem., 2015, 58,
7195.
8 S.C. Meyer and R.L. Levine, Clin. Cancer. Res., 2014, 20, 2051.
Baricitinib (formerly INCB28050, LY3009104)[1] is an oral JAK1 and JAK2 inhibitor.
Baricitinib is in Phase III development by Eli Lilly and Incyte as a potential treatment for rheumatoid arthritis.[2] It is in Phase II development as a potential treatment for psoriasis and diabetic nephropathy. The related compound in JAK inhibitor is Tofacitinib, currently approved for the treatment of rheumatoid arthritis (RA) in the United States.
The companies announced 52-week data from a Phase IIb study of baricitinib at the European Congress of Rheumatology meeting in Madrid which showed that clinical improvements previously observed at week 24 were sustained for the full year in RA patients. Specifically, 49% of patients were ACR50 responders (ie a 50% improvement in their condition) after 52 weeks compared to 41% at week 24. For the full year, 21% reached ACR70 compared with 27% after 24 weeks.
To date, baricitinib, an orally administered selective JAK1 and JAK2 inhibitor, has demonstrated “an acceptable safety profile and side effects have generally been straightforward to manage”, said Oxford University’s Peter Taylor. He added that “these encouraging findings support further investigation of this new drug in RA”.
Baricitinib is already in Phase III for RA and in Phase II for psoriasis and diabetic nephropathy.
WO2009114512, also to Incyte, discloses azetidine and cyclobutane derivatives of the general structure shown below as JAK inhibitors.

Baricitinib (also known as LY3009104 or INCB28050) is in phase II clinical trials for the treatment of rheumatoid arthritis and diabetic kidney disease. Baricitinib is shown below.

About Baricitinib
Baricitinib is a once-daily, oral, selective JAK1 and JAK2 inhibitor. There are four known JAK enzymes: JAK1, JAK2, JAK3 and TYK2. JAK-dependent cytokines have been implicated in the pathogenesis of a number of inflammatory and autoimmune diseases, suggesting that JAK inhibitors may be useful for the treatment of a broad range of inflammatory conditions. Baricitinib demonstrates approximately 100-fold greater potency of inhibition against JAK1 and JAK2 than JAK 3 in kinase assays.
In December 2009, Lilly and Incyte announced an exclusive worldwide license and collaboration agreement for the development and commercialization of baricitinib and certain follow-on compounds for patients with inflammatory and autoimmune diseases. Baricitinib is currently in Phase 3 clinical development for rheumatoid arthritis and Phase 2 development for psoriasis and diabetic nephropathy.
About Rheumatoid Arthritis
Rheumatoid arthritis is an autoimmune diseasei characterized by inflammation and progressive destruction of joints.ii More than 23 million people worldwide suffer from RA.iii Approximately three times as many women as men have the disease. Patients and physicians indicate there remains an important opportunity to improve patient care. Current treatment of RA includes the use of non-steroidal anti-inflammatory drugs, oral disease-modifying anti-rheumatic drugs such as methotrexate, and injectable biological response modifiers that target selected mediators implicated in the pathogenesis of RA.iv
About Baricitinib Phase 3 Trials
Lilly and Incyte have conducted four pivotal Phase 3 clinical trials of baricitinib in patients with moderately-to-severely active rheumatoid arthritis to support regulatory submission in most countries. An additional Phase 3 study was initiated to support clinical development in China and remains ongoing. The clinical trial program includes a wide range of patients including those who are methotrexate naïve, inadequate responders to methotrexate, inadequate responders to conventional disease-modifying anti-rheumatic drugs, or inadequate responders to TNF inhibitors. Patients completing any of the five Phase 3 studies can enroll in a long-term extension study. For additional information on this clinical trial program, please visit http://www.clinicaltrials.gov.
About Incyte
Incyte Corporation is a Wilmington, Delaware-based biopharmaceutical company focused on the discovery, development and commercialization of proprietary therapeutics for oncology and inflammation. For additional information on Incyte, please visit the Company’s web site at http://www.incyte.com.
About Eli Lilly and Company
Lilly is a global healthcare leader that unites caring with discovery to make life better for people around the world. We were founded more than a century ago by a man committed to creating high-quality medicines that meet real needs, and today we remain true to that mission in all our work. Across the globe, Lilly employees work to discover and bring life-changing medicines to those who need them, improve the understanding and management of disease, and give back to communities through philanthropy and volunteerism. To learn more about Lilly, please visit us at http://www.lilly.com and newsroom.lilly.com/social-channels.
SOURCE: Eli Lilly
Biological Activity
Baricitinib (formerly INCB28050, LY3009104) is a selective orally bioavailable JAK1/JAK2 inhibitor. Baricitinib preferentially inhibits JAK1 and JAK2, with 10-fold selectivity over Tyk2 and 100-fold over JAK3. INCB-28050 (baricitinib) inhibits intracellular signaling of multiple proinflammatory cytokines including IL-6 at concentrations <50 nM. Baricitinib also inhibits pSTAT3 stimulated by IL-23 with IC50 of 20 nM in isolated naive T-cells. Baricitinib (INCB028050) was also effective in multiple murine models of arthritis, with no evidence of suppression of humoral immunity or adverse hematologic effects. Baricitinib reduces levels of pSTAT3 in a dose- and time-dependent manner in the peripheral blood of rAIA animals. INCB28050 (Baricitinib) (10 mg/mL, p.o.) improves a composite score of joint damage by 47% in the murine CIA model.
Conversion of different model animals based on BSA (Value based on data from FDA Draft Guidelines)
| Species | Mouse | Rat | Rabbit | Guinea pig | Hamster | Dog |
| Weight (kg) | 0.02 | 0.15 | 1.8 | 0.4 | 0.08 | 10 |
| Body Surface Area (m2) | 0.007 | 0.025 | 0.15 | 0.05 | 0.02 | 0.5 |
| Km factor | 3 | 6 | 12 | 8 | 5 | 20 |
| Animal A (mg/kg) = Animal B (mg/kg) multiplied by | Animal B Km |
| Animal A Km |
For example, to modify the dose of resveratrol used for a mouse (22.4 mg/kg) to a dose based on the BSA for a rat, multiply 22.4 mg/kg by the Km factor for a mouse and then divide by the Km factor for a rat. This calculation results in a rat equivalent dose for resveratrol of 11.2 mg/kg.


As shown in Scheme 1, Rodgers et al. have reported the first synthetic route to baricitinib.
ref J.D. Rodgers, S. Shepard, T.P. Maduskuie, H. Wang, N. Falahatpisheh, M. Rafalski, A.G. Arvanitis, L. Sorace, R.K. Jalluri, J.S. Fridman and K. Vaddi, 2007, US20070135461.
tert-Butyl 3-oxoazetidine- 1-carboxylate (1) was employed as the starting material. This was transformed to compound 2 by a Horner–Emmons reaction, followed by deprotection of the N-Boc group in acidic conditions. The intermediate 4 was obtained by the sulfonamidation reaction of compound 3 with ethanesulfonyl chloride. The other part of baricitinib was acquired by utilising 4-chloro-7H-pyrrolo[2,3-d]pyrimidine (5) as the starting material. Compound 5 reacted with [2-(chloromethoxy)ethyl] trimethylsilane (SEM-Cl) to afford the intermediate 6, which was converted by reaction with 7 via the intermediate 8 to 4-(1H-pyrazol-4-yl)-7-{[2-(trimethylsilyl)ethoxy]methyl}-7Hpyrrolo[2,3-d]pyrimidine (9) via a Suzuki coupling reaction and a hydrolysis reaction. After the nucleophilic addition reaction and deprotection of the SEM group, baricitinib was obtained through eight steps. This synthetic route had drawbacks of high cost, low overall yield and the requirement of strict operating conditions.
PATENT
WO2009114512
EXAMPLES
Example 1. {l-(ethylsulfonyl)-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-lH-pyrazol-l- yl]azetidin-3-yl}acetonitrile trifluoroacetic acid salt

Step 1. tert-butyl 3-(cyanomethylene)azetidine-l-carboxylate
0I \t
To a suspension of sodium hydride (60% dispersion in mineral oil, 0.257 g, 6.42 mmol) in tetrahydrofuran (32 mL) at 0 0C under a nitrogen atmosphere was added diethyl cyanomethylphosphonate (1.19 g, 6.72 mmol) (purchased from Aldrich). The reaction was then stirred for 45 minutes at room temperature. A solution of tert-butyl 3-oxoazetidine-l- carboxylate (1.00 g, 5.84 mmol) (purchased from Alfa Aesar) in tetrahydrofuran (8.8 mL) was introduced dropwise and the mixture was stirred for 16 hours. Brine and ethyl acetate were added and the layers separated. The aqueous layer was extracted with three portions of ethyl acetate. The combined extracts were dried over sodium sulfate, filtered and concentrated to afford product, used without further purification in Step 2 (1.12 g, 99%). 1H NMR (300 MHz, CDCl3): δ 5.38 (p, IH), 4.73-4.68 (m, 2H), 4.64-4.59 (m, 2H), 1.46 (s, 9H).
Step 2. tert-butyl 3-(cyanomethyl)’3-[4-(7-[2-(trimethylsilyl)ethoxy]methyl-7H-pyrrolo[2,3- djpyrim idin-4-yl) – 1 H-pyrazol-1 -yl]azetidine-l -carboxylate

To a solution of 4-(lH-pyrazol-4-yl)-7-[2-(trimethylsilyl)ethoxy]methyl-7H- pyrrolo[2,3-d]pyrimidine (4.61 g, 14.6 mmol) (prepared according to the method of WO 2007/070514 in Example 65, Step 2) and tert-butyl 3-(cyanomethylene)azetidine-l- carboxylate (2.84 g, 14.6 mmol) in acetonitrile (100 mL) was added 1,8- diazabicyclo[5.4.0]undec-7-ene (2.19 mL, 14.6 mmol). The reaction was stirred at room temperature for 16 hours. The acetonitrile was removed in vacuo and the residue was dissolved in ethyl acetate. This solution was sequentially washed with IN HCl and brine, dried over sodium sulfate, filtered and concentrated. The residue was purified by flash column chromatography, eluting with 80% ethyl acetate/hexanes to afford desired product (5.36 g, 72%).
1H NMR (300 MHz, CDCl3): δ 8.86 (s, IH), 8.44 (s, IH), 8.34 (s, IH), 7.42 (d, IH), 6.80 (d, IH), 5.68 (s, 2H), 4.54 (d, 2H), 4.29 (d, 2H), 3.59-3.51 (m, 2H), 3.33 (s, 2H), 1.47 (s, 9H), 0.96-0.89 (m, 2H), -0.06 (s, 9H); LCMS (M+H)+: 510.2.
Step 3. 3-[4-(7-[2-(trimethylsilyl)ethoxy]methyl-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-lH- pyrazol- 1 -yl] azetidin-3-ylacetonitrile

To a solution of tert-butyl 3-(cyanomethyl)-3-[4-(7-[2-(trimethylsilyl)ethoxy]methyl- 7H-pyrrolo[2,3-d]pyrimidin-4-yl)-lH-pyrazol-l-yl]azetidine-l-carboxylate (5.36 g, 10.5 mmol) in 1,4-dioxane (100 mL) was added 4.00 M of hydrogen chloride in 1,4-dioxane (40 mL, 160 mmol) and the mixture was stirred at room temperature for 16 hours. The reaction was poured into saturated sodium bicarbonate solution sufficient to neutralize. The product was extracted with three portions of ethyl acetate. The combined extracts were washed with brine, dried over sodium sulfate, filtered and concentrated to afford product which was used without further purification (3.0 g, 69%). 1H NMR (400 MHz, CDCl3): δ 8.85 (s, IH), 8.42 (s, IH), 8.32 (s, IH), 7.41 (d, IH), 6.80 (d, IH), 5.68 (s, 2H), 4.30 (d, 2H), 3.88 (d, 2H), 3.58-3.51 (m, 2H), 3.42 (s, 2H), 0.96-0.89 (m, 2H), -0.06 (s, 9H); LCMS (M+H)+: 410.2. Step 4. l-(ethylsulfonyl)-3-[4-(7-[2-(trimethylsilyl)ethoxy]methyl-7H-pyrrolo[2,3- d]pyritnidin-4-yl)-lH-pyrazol-l-yl]azetidin-3-ylacetonitrile

To a solution of 3-[4-(7-[2-(trimethylsilyl)ethoxy]methyl-7H-pyrrolo[2,3- d]pyrimidin-4-yl)-lH-pyrazol-l-yl]azetidin-3-ylacetonitrile (0.100 g, 0.244 mmol) in tetrahydrofuran (2 mL) containing N,N-diisopropylethylamine (0.085 mL, 0.49 mmol) was added ethanesulfonyl chloride (0.023 mL, 0.24 mmol). After stirring for 1.5 hours, the reaction mixture was poured into dilute HCl and extracted with three portions of ethyl acetate. The combined extracts were washed with brine, dried over sodium sulfate, decanted and concentrated to afford product, used without further purification in Step 5 (111 mg, 91%).
1H NMR (300 MHz, CDCl3): δ 8.86 (s, IH), 8.63 (s, IH), 8.35 (s, IH), 7.45 (d, IH), 6.83 (d, IH), 5.68 (s, 2H), 4.63 (d, 2H), 4.26 (d, 2H), 3.54 (t, 2H), 3.42 (s, 2H), 3.09 (q, 2H), 1.41 (t, 3H), 0.92 (t, 2H), -0.06 (s, 9H); LCMS (M+H)+: 502.1.
Step 5. l-(ethylsulfonyl)-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-lH-pyrazol-l-yl]azetidin-3- ylacetonitrile trifluoroacetate salt
To a solution of l-(ethylsulfonyl)-3-[4-(7-[2-(trimethylsilyl)ethoxy]methyl-7H- pyrrolo[2,3-d]pyrimidin-4-yl)-lH-pyrazol-l-yl]azetidin-3-ylacetonitrile (0.111 g, 0.22 mmol) in methylene chloride (3 mL) was added trifluoroacetic acid (2 mL) and the solution was stirred for 1.5 hours. The solvents were removed in vacuo and the residue was dissolved in methanol (3 mL) and ethylenediamine (0.1 mL) was added. After stirring for 3 hours, the volume was reduced in vacuo and the product was purified by preparative-HPLC/MS, (SunFire Cl 8 column, eluting with a gradient Of MeCNZH2O containing 0.1% TFA) to afford the product as the trifluoroacetic acid salt (50 mg, 47%). 1H NMR (400 MHz, d6-dmso): δ 12.55 (br d, IH), 9.03 (s, IH), 8.83 (s, IH), 8.56 (s, IH), 7.79-7.75 (m, IH), 7.24-7.19 (m, IH), 4.59 (d, 2H), 4.26 (d, 2H), 3.71 (s, 2H), 3.25 (q, 2H), 1.24 (t, 3H); LCMS (M+H)+: 372.1.
Alternatively, the deprotection and sulfonylation steps could be performed in the reverse order, as in Example 2.
Example 66. tert-Butyl 3-oxoazetidine-l-carboxylate (7).
A solution of tert-buty\ 3-hydroxyazetidine-l-carboxylate (24, 50 g, 289 mmol) in ethyl acetate (400 mL) was cooled to 0 0C. The resulting solution was then treated with solid TEMPO (0.5 g, 3.2 mmol, 0.011 equiv) and a solution of potassium bromide (KBr, 3.9 g, 33.2 mmol, 0.115 equiv) in water (60 mL) at 0 – 5 0C. While keeping the reaction temperature between 0 – 5 0C a solution of saturated aqueous sodium bicarbonate (NaHCO3, 450 mL) and an aqueous sodium hypochlorite solution (NaClO, 10 – 13 % available chlorine, 450 mL) were added. Once the solution of sodium hypochlorite was added, the color of the reaction mixture was changed immediately. When additional amount of sodium hypochlorite solution was added, the color of the reaction mixture was gradually faded. When TLC showed that all of the starting material was consumed, the color of the reaction mixture was no longer changed. The reaction mixture was then diluted with ethyl acetate (EtOAc, 500 mL) and two layers were separated. The organic layer was washed with water (500 rnL) and the saturated aqueous sodium chloride solution (500 mL) and dried over sodium sulfate (Na2SO4). The solvent was then removed under reduced pressure to give the crude product, tert-butyl 3-oxoazetidine-l-carboxylate (7, 48 g, 49.47 g theoretical, 97% yield), which was found to be sufficiently pure and was used directly in the subsequent reaction without further purification. For crude 7: 1H NMR (CDCl3, 300 MHz), δ 4.65 (s, 4H), 1.42 (s, 9H) ppm.
Example 67. tert-Buty\ 3-(cyanomethylene)azetidine-l-carboxylate (9). Diethyl cyanomethyl phosphonate (8, 745 g, 4.20 mol, 1.20 equiv) and anhydrous tetrahydrofuran (THF, 9 L) was added to a four-neck flask equipped with a thermowell, an addition funnel and the nitrogen protection tube at room temperature. The solution was cooled with an ice-methanol bath to -14 0C and a 1.0 M solution of potassium tert-butoxide (^-BuOK) in anhydrous tetrahydrofuran (THF, 3.85 L, 3.85 mol, 1.1 equiv) was added over 20 min while keeping the reaction temperature below -5 0C. The resulting reaction mixture was stirred for 3 h at -10 0C and a solution of l-terf-butoxycarbonyl-3-azetidinone (7, 600 g, 3.50 mol) in anhydrous tetrahydrofuran (THF, 2 L) was added over 2 h while keeping the internal temperature below -5 0C. The reaction mixture was stirred at -5 to -10 0C over 1 h and then slowly warmed up to room temperature and stirred at room temperature for overnight. The reaction mixture was then diluted with water (4.5 L) and saturated aqueous sodium chloride solution (NaCl, 4.5 L) and extracted with ethyl acetate (EtOAc, 2 x 9 L). The combined organic layers were washed with brine (6 L) and dried over anhydrous sodium sulfate (Na2SO4). The organic solvent was removed under reduced pressure and the residue was diluted with dichloromethane (CH2Cl2, 4 L) before being absorbed onto silica gel (Siθ2, 1.5 Kg). The crude product, which was absorbed on silica gel, was purified by flash column chromatography (SiO2, 3.5 Kg, 0 – 25% EtOAc/hexanes gradient elution) to afford tert-butyl 3-(cyanomethylene)azetidine-l-carboxylate (9, 414.7 g, 679.8 g theoretical, 61% yield) as white solid. For 9: 1H NMR (CDCl3, 300MHz), δ 5.40 (m, IH), 4.70 (m, 2H), 4.61 (m, 2H), 1.46 (s, 9H) ppm; Ci0H14N2O2 (MW, 194.23), LCMS (EI) mle 217 (M+ + Na).
C8H13NO3 MoI Wt 171 19

2

11 step 3 10
C7H10N2O2S C5H7CIN2 MoI Wt 186 23 MoI Wt 130 58
Example 68. 2-(l-(Ethylsulfonyl)azetidin-3-ylidene)acetonitrile (11).
A solution of tert-buty\ 3-(cyanomethylene)azetidine-l-carboxylate (9, 100Og, 5.2 mol) in acetonitrile (7 L) and a 3 N aqueous HCl solution (7 L) was stirred at room temperature for 18 h. When HPLC showed that all the starting material (9) was consumed, the reaction mixture was concentrated under reduced pressure to dryness. The residue, which contains the crude desired deprotection product (10), was then suspended in acetonitrile (12 L) and the resulting suspension was cooled to O – 5 0C. Diisopropyethylamine (DIEA, 3.14 L, 18.03 mol, 3.5 equiv) was then slowly added while keeping the internal temperature below 5 0C. The resulting homogeneous solution was allowed to cool down to O 0C and ethane sulfonyl chloride (EtSO2Cl, 730 mL, 7.73 mol, 1.5 equiv) was added over 1 h while keeping the internal temperature below 5 0C. The resulting reaction mixture was allowed to gradually warm to room temperature and stirred at room temperature for overnight. When HPLC showed that the reaction was complete, the reaction mixture was concentrated under reduced pressure to a volume of approximately 2 L. The bath temperature of the rotary evaporator is set to not exceed 45 0C. The concentrated residue was then diluted with dichloromethane (CH2CI2, 10 L) and the resulting dichloromethane solution was washed with aqueous sodium chloride solution (10 L). The aqueous phase was back extracted with dichloromethane (CH2CI2, 5 L). The combined organic layers were dried over anhydrous sodium sulfate
(Na2SO^ and the residue was absorbed onto silica gel (SiO2, 1 Kg) under reduced pressure. The bath temperature of the rotary evaporator was set to not exceed 45 0C. The material was then loaded onto a silica gel column (SiO2, 2.5 Kg) and eluted with 20 – 60 % ethyl acetate in heptane to afford 2-(l-(ethylsulfonyl)azetidin-3-ylidene)acetonitrile (11, 882 g, 968.4 g theoretical, 91 % yield) as off-white solids. For 11: 1H NMR (CDCl3, 300 MHz) δ 5.46 (m, IH), 4.77 (m, 2H), 4.70 (m, 2H), 3.05 (q, 2H), 1.39 (t, 3H) ppm; C7Hi0N2O2S (MW, 186.23), LCMS (EI) mle 187 (M+ + H).
Example 69. 2-(l-(Ethylsulfonyl)-3-(4-(7-((2-(trimethyIsiIyl)ethoxy)methyl)-7H- pyrrolo[2,3-</]pyrimidin-4-yl)-lH-pyrazol-l-yl)azetidin-3-yl)acetonitrile (12).
Method A. To a suspension of 4-(lH-pyrazol-4-yl)-7-((2-
(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-^pyrimidine (5, 440 g, 1.395 mol) and 2-(l- (ethylsulfonyl)azetidin-3-ylidene)acetonitrile (11, 312.4 g, 1.68 mol, 1.2 equiv) in acetonitrile (4.4 L) was added DBU (249.8 mL, 1.67 mol, 1.2 equiv) drop wise to keep the reaction temperature between 15 – 25 0C. After adding DBU, the reaction mixture became homogeneous, but a precipitate appeared in 30 min. The reaction mixture was stirred for 3 h at room temperature. When ΗPLC showed that the reaction was deemed complete, the reaction mixture was quenched with water (11 L). The resulting mixture was stirred at room temperature for additional 30 min and then filtered. The solid cake was washed with water (4 L), MTBE (2 L) and dried in vacuum oven at 35 0C for 24 h to afford crude 2-(l –
(ethylsulfonyl)-3-(4-(7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-(i]pyrimidin-4-yl)- lH-pyrazol-l-yl)azetidin-3-yl)acetonitrile (12, 681 g, 699.8 g theoretical, 97.3 % yield) as white solids, which was found to be sufficiently pure for the subsequent reaction without further purification. For 12: 1HNMR (CDCl3, 300 MHz), δ 8.86 (s, IH), 8.45 (s, IH), 8.35 (s, IH), 7.43 (d, IH), 6.80 (d, IH), 5.68 (s, 2H), 4.65 (d, 2H), 4.27 (d, 2H), 3.55 (s, 2H), 3.4 (t, 2H), 3.07 (m, 2H), 1.42 (m, 3H), 0.92 (m, 2H), -0.05 (s, 9H) ppm; C22H3IN7O3SSi (MW, 501.68), LCMS (EI) mle 502 (M+ + H).

12
C15H21N5OSi C22H31N7O3SSi MoI Wt 315 45 MoI Wt 501 68

Phosphate salt
C16H17N7O2S C16H20N7O6PS
MoI Wt 371 42 MoI Wt 469 41
Example 72. tert-Butyl 3-(cyanomethyl)-3-(4-(7-((2-(trimethylsilyl)ethoxy)methyl)-7H- pyrroIo[2,3-rf]pyrimidin-4-yl)-lH-pyrazol-l-yl)azetidine-l-carboxyIate (15).
To a suspension of tert-butyl 3-(cyanomethylene)azetidine-l-carboxylate (9, 417.2 g, 2.15 mol, 1.05 equiv) and 4-(lH-pyrazol-4-yl)-7-((2-(trimethylsilyl)ethoxy)methyl)-7H- pyrrolo[2,3-<f]pyrimidine (5, 645 g, 2.04 mol) in acetonitrile (4.9 L) was added DBU (30.5 mL, 0.204 mol, 0.1 equiv) drop wise at room temperature. The resulting reaction mixture was then stirred at room temperature for 3 h. After about 1 h, a clear, brown solution was obtained. When LCMS showed that no starting material remained, silica gel (SiO2, 1 Kg) was added and the mixture was concentrated to dryness under reduced pressure. This material, which contains the crude desired product (15), was then loaded onto a pre-packed silica column (Siθ2, 2.5 Kg) and the column was eluted with 60 – 80% of ethyl acetate/heptane. The fractions containing the pure desired product (15) were combined and concentrated under reduced pressure to give the desired product as thick oil which was then stirred in heptane at room temperature until crystallization occurred. The solids were collected by filtration and washed with heptane to afford tert-buty\ 3-(cyanomethyl)-3-(4-(7-((2- (trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-ύ(]pyrimidin-4-yl)-lH-pyrazol-l-yl)azetidine- 1-carboxylate (15, 1014.9 g, 1039.7 g theoretical, 97.6% yield) as white solids. For 15: 1H NMR (DMSO-^6, 300 MHz) δ 8.93 (s, IH), 8.77 (s, IH), 8.47 (s, IH), 7.80 (d, IH, J= 3.8 Hz), 7.20 (d, IH, J = 3.7 Hz), 5.63 (s, 2H), 4.50 (d, 2H, J= 9.3 Hz), 4.21 (d, 2H, J= 9.3 Hz), 3.66 (s, 2H), 3.52 (t, 2H, J= 7.8 Hz), 1.40 (s, 9H), 0.82 (t, 2H, J= 8.1 Hz), -0.12 (s, 9H) ppm; C25H35N7O3Si (MW, 509.68), LCMS (EI) m/e 510 (M+ + H) and 532 (M+ + Na).

15
C15H21N5OSi C25H35N7O3Si MoI Wt 31545 MoI Wt 509 68

16 12
C20H27N7OSi C22H31N7O3SSi MoI Wt 409 56 MoI Wt 501 68

14 phosphate
C16H17N7O2S C16H20N7O6PS

MoI Wt 371 42 MoI Wt 46941
Example 77. (4-(l-(3-(Cyanomethyl)-l-(ethylsulfonyl)azetidin-3-yl)-lH-pyrazol-4-yl)- 7H-pyrrolo[2,3-</]pyrimidin-7-yl)methyl pivalate (20).
To a suspension of [4-(lH-pyrazol-4-yl)-7H-pyrrolo[2,3-</|pyrimidin-7-yl]methyl pivalate (19, 10.0 g, 33.4 mmol) and 2-(l-(ethylsulfonyl)azetidin-3-ylidene)acetonitrile (11, 6.22 g, 33.4 mmol, 1.0 equiv) in N,N-dimethylformamide (DMF, 20 mL) was added DBU (254 mg, 1.67 mmol, 0.05 equiv) drop wise to keep the reaction temperature between 15 – 25 0C. After adding DBU, the reaction mixture became homogeneous within 90 min. The reaction mixture was stirred for 3 h at room temperature. When ΗPLC showed that the reaction was deemed complete, the reaction mixture was quenched with water (120 mL) and acetonitrile (80 mL). The resulting mixture was stirred at room temperature for an additional 30 min. The solids were collected by filtration, washed with a mixture of acetonitrile and water (2/3 by volume, 2 x 20 mL), and dried in vacuum oven at 40 – 45 0C for 24 h to afford crude (4-(l-(3-(cyanomethyl)-l-(ethylsulfonyl)azetidin-3-yl)-lH-pyrazol-4-yl)-7H- pyrrolo[2,3-</)pyrimidin-7-yl)methyl pivalate (20, 14.5 g, 16.2 g theoretical, 89.5 % yield) as white solids, which was found to be sufficiently pure (> 98.0% by ΗPLC) for the subsequent reaction without further purification. For 20: 1FTNMR (CDCl3, 300 MHz), δ 8.87 (s, IH), 8.43 (s, IH), 8.37 (s, IH), 7.51 (d, IH, J= 3.6 Hz), 6.76 (d, IH, J= 3.6 Hz), 6.26 (s, 2H),
4.64 (d, 2H, J = 9.6 Hz), 4.25 (d, 2H, J = 9.6 Hz), 3.41 (s, 2H), 3.09 (q, 2H, J= 7.6 Hz), 1.42 (t, 3H, J= 7.6 Hz), 1.17 (s, 9H) ppm; C22H27N7O4S (MW, 485.56), LCMS (EI) mle 486 (M+ + H).

C15H17N5O2 C22H27N7O4S MoI Wl 299 33 MoI Wt 48556

14 phosphate
C16H17N7O2S C16H20N7O6PS MoI Wt 371 42 MoI Wt 46941
PAPER
Authors: Xu, Jiaojiao; Cai, Jin; Chen, Junqing; Zong, Xi; Wu, Xuan; Ji, Min; Wang, Peng
Source: Journal of Chemical Research, Volume 40, Number 4, April 2016, pp. 205-208(4), http://dx.doi.org/10.3184/174751916X14569294811333
A highly efficient method for the synthesis of baricitinib was developed. The starting material tert-butyl 3-oxoazetidine-1-carboxylate was converted to intermediate 2-(1-(ethylsulfonyl)azetidin-3-ylidene)acetonitrile via the Horner–Emmons reaction, deprotection of the N-Boc-group and a final sulfonamidation reaction. Then the nucleophilic addition reaction was carried out smoothly to afford the borate intermediate in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene under reflux. Finally, the desired compound baricitinib was obtained by the Suzuki coupling reaction of 4-chloro-7-H-pyrrolo[2,3-d]pyrimidine with the above borate intermediate. All compounds were characterised by IR, MS, 1H NMR and 13C NMR. The overall yield in this synthetic route was as high as 49%. Moreover, this procedure is straightforward to carry out, has low cost and is suitable for industrial production.

In order to improve the procedure, we designed a novel synthetic route for the synthesis of baricitinib (Scheme 2). Similarly, we also applied tert-butyl 3-oxoazetidine-1-carboxylate (1) as the starting material. First, we optimised the preparation of compound 4. In the Horner–Emmons reaction, NaH was used as the base instead of t-BuOK, which led to a yield as high as 84%. Then the deprotection of the N-Boc group was carried out smoothly under trifluoroacetic acid (TFA) cleavage conditions to afford compound 3, which was reacted with ethanesulfonyl chloride without further purification. Next, the nucleophilic addition reaction between compound 4 and 4-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)-lH-pyrazole (11) proceeded successfully in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU). With the intermediate compound 12 in hand, we optimised the conditions of the Suzuki coupling reaction with compound 5. Several coupling systems were evaluated, such as Pd(PPh3 )4 – K2 CO3 –t-butanol/H2 O, Pd(PPh3 )4 –Na2 CO3 –t-butanol/H2 O and Pd(OAc)2 –K2 CO3 –dioxane/H2 O. The Pd(PPh3 )4 –CsF–t-butanol/ toluene/H2 O system afforded the most satisfactory yield. Finally, baricitinib was obtained efficiently and the overall yield was as high as 49% based on tert-butyl 3-oxoazetidine-1-carboxylate (1)
PATENT
Baricitinib is a Janus kinase (JAK) inhibitor. It is chemically designated as { 1 (ethylsulfonyl)-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-lH-pyrazol-l-yl]azetidin-3-yl}acetonitrile, having the structure as depicted in Formula I.

Formula I
U.S. Patent No. 8,158,616 discloses processes for the preparation of baricitinib of Formula I and [4-(lH-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl]methyl pivalate of Formula II.

Formula II
U.S. Patent No. 8, 158,616 involves a three-step process for the preparation of [4- (lH-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl]methyl pivalate of Formula II as depicted in Scheme 1 below:
Scheme 1

Formula V

Formula VI

Formula II Formula VII
The process disclosed in U.S. Patent No. 8, 158,616 involves the use of sodium hydride as a base for reacting 4-chloro-7H-pyrrolo[2,3-d]pyrimidine of Formula III with chloromethyl pivalate of Formula IV, and the use of a protected pyrazole borolane derivative of Formula VI for the conversion of (4-chloro-7H-pyrrolo[2,3-d]pyrimidin-7- yl)methyl 2,2-dimethylpropanoate of Formula V into [4-(lH-pyrazol-4-yl)-7H- pyrrolo[2,3-d]pyrimidin-7-yl]methyl pivalate of Formula II.
The use of sodium hydride is not suitable on an industrial scale due to its inflammable and hazardous nature. The use of a protected pyrazole borolane derivative of Formula VI increases the cost of the manufacturing process, as an additional deprotection step is required for obtaining [4-(lH-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl]methyl pivalate of Formula II.
Thus, there exists a need for the development of an economical and industrially advantageous process for the preparation of [4-(lH-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl]methyl pivalate of Formula II that avoids the use of sodium hydride and involves a lesser number of steps.
The present invention provides a convenient, economical, and industrially advantageous two-step process for the preparation of [4-(lH-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl]methyl pivalate of Formula II. The process of the present invention involves the use of an alkali or alkaline earth metal hydroxide, carbonate, or bicarbonate as a base for reacting 4-chloro-7H-pyrrolo[2,3-d]pyrimidine of Formula III with chloromethyl pivalate of Formula IV, and the use of an unprotected pyrazole borolane of Formula VIII for the conversion of (4-chloro-7H-pyrrolo[2,3-d]pyrimidin-7-yl)methyl 2,2-dimethylpropanoate of Formula V into [4-(lH-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl]methyl pivalate of Formula II. The process of the present invention provides [4-(lH-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl]methyl pivalate of Formula II in high yield.
A first aspect of the present invention provides a process for the preparation of [4-(lH-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl]methyl pivalate of Formula II,

Formula II
comprising the steps of:
i) reacting 4-chloro-7H-pyrrolo[2,3-d]pyrimidine of Formula III

Formula III
with chloromethyl pivalate of Formula IV

Formula IV
in the presence of an alkali or alkaline earth metal hydroxide, carbonate, bicarbonate as a base to obtain (4-chloro-7H-pyrrolo[2,3-d]pyrimidin-7- yl)methyl 2,2-dimethylpropanoate of Formula V; and

ii) reacting the (4-chloro-7H-pyrrolo[2,3-d]pyrimidin-7-yl)methyl 2,2- dimethylpropanoate of Formula V with 4-(4,4,5,5-tetramethyl-l,3,2 dioxaborolan-2-yl)-lH-pyrazole of Formula VIII

Formula VIII
to obtain the [4-(lH-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl]methyl pivalate of Formula II.
A second aspect of the present invention provides a process for the preparation of baricitinib of Formula I,

Formula I
comprising the steps of:
i) reacting 4-chloro-7H-pyrrolo[2,3-d]pyrimidine of Formula III

Formula III
with chloromethyl pivalate of Formula IV

Formula IV
in the presence of an alkali or alkaline earth metal hydroxide, carbonate, or bicarbonate base to obtain (4-chloro-7H-pyrrolo[2,3-d]pyrimidin-7-yl)methyl 2,2-dimethylpropanoate of Formula V;

Formula V
ii) reacting the (4-chloro-7H-pyrrolo[2,3-d]pyrimidin-7-yl)methyl 2,2- dimethylpropanoate of Formula V with 4-(4,4,5,5-tetramethyl-l,3,2- dioxaborolan-2-yl)-lH-pyrazole of Formula VIII

Formula VIII
to obtain [4-( lH-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl]methyl pivalate of Formula II; and

Formula II
iii) reacting the [4-(lH-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl]methyl pivalate of Formula II with [l-(ethylsulfonyl)azetidin-3-ylidene]acetonitrile of Formula IX

Formula IX
to obtain baricitinib of Formula I.
EXAMPLES
Example 1 : Preparation of (4-chloro-7H-pyrrolor2.3-dlpyrimidin-7-yl)methyl 2.2-dimethylpropanoate (Formula V)
4-Chloro-7H-pyrrolo[2,3-d]pyrimidine (25 g; Formula III), potassium carbonate (27 g), and chloromethyl pivalate (27 g; Formula IV) were added to a reaction vessel containing N,N-dimethylformamide (100 mL) at ambient temperature. The reaction mixture was stirred for 14 hours. The progress of the reaction was monitored by thin layer chromatography. Water (250 mL) was added to the reaction mixture, and then the mixture was stirred for 2 hours. The reaction mixture was filtered, then washed with water (50 mL), and then dried under reduced pressure at 40°C to 45°C for 12 hours to obtain (4-chloro-7H-pyrrolo[2,3-d]pyrimidin-7-yl)methyl 2,2-dimethylpropanoate.
Yield: 98.85%
Example 2: Preparation of r4-(lH-pyrazol-4-yl)-7H-pyrrolor2.3-dlpyrimidin-7-yllmethyl pivalate (Formula II)
(4-Chloro-7H-pyrrolo[2,3-d]pyrimidin-7-yl)methyl 2,2-dimethylpropanoate (10 g; Formula V), water (50 mL), and potassium carbonate (15.5 g) were added into a reaction vessel at ambient temperature. 4-(4,4,5,5-Tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (8.7 g; Formula VIII), 1,4-dioxane (100 mL), and
tetrakis(triphenylphosphine)palladium(0) (0.08 g) were added to the reaction mixture. The reaction mixture was heated to a temperature of 80°C to 85°C, and then stirred at the same temperature for 14 hours. The progress of the reaction was monitored by thin layer chromatography. On completion, ethyl acetate (100 mL) was added to the reaction mixture. The contents were stirred for 1 hour, then filtered through a Hyflo®, and then washed with ethyl acetate (40 mL). The organic layer was separated, and then concentrated under reduced pressure to obtain [4-(lH-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl]methyl pivalate.
Yield: 82.27%
PATENT
PATENT
https://www.google.com/patents/CN105566332A?cl=en
PATENT
The present invention provides processes for the preparation of baricitinib of Formula I and an intermediate of Formula V. The present invention also provides the of the intermediate of Formula V for the preparation of baricitinib.

Formula V
Background of the Invention
Baricitinib is a Janus kinase (JAK) inhibitor. It is chemically designated as (ethylsulfonyl)-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-lH-pyrazol-l-yl]azetidin-3 yl}acetonitrile, having the structure as depicted in Formula I.

Formula I
U.S. Patent No. 8,158,616 discloses a process for the preparation of baricitinib comprising the reaction of 2-(l-(ethylsulfonyl)azetidin-3-ylidene)acetonitrile of Formula II with 4-(lH-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl methyl pivalate of Formula
III to provide an intermediate of Formula IV, followed by deprotection of the intermediate of Formula IV to obtain baricitinib of Formula I, as depicted in Scheme I below:
Scheme I

Formula IV
The process disclosed in U.S. Patent No. 8, 158,616 requires a deprotection step in the last stage of the synthesis, which adds to the cost of the overall synthesis.
Thus, there exists a need for an alternate, cost-effective, and industrially advantageous process for the preparation of baricitinib.
EXAMPLES
Example 1 : Preparation of 3-(cvanomethylene)azetidine hydrochloride (Formula VIP
Aqueous hydrochloric acid (6N, 10 mL) and montmorillonite K-10 (2 g) were added into a reaction vessel at ambient temperature. The contents were stirred for 1 hour, and then filtered under reduced pressure to obtain activated montmorillonite K-10. The activated montmorillonite K-10 was added into another reaction vessel containing tert-butyl 3-(cyanomethylidene)azetidine-l-carboxylate (2 g; Formula VI) and methanol (20
mL) at ambient temperature. The reaction mixture was refluxed for about 12 hours to about 15 hours. On completion, the reaction mixture was filtered under reduced pressure followed by recovery of methanol under reduced pressure at about 40°C to about 45°C to obtain 3-(cyanomethylene)azetidine hydrochloride.
Yield: 75%
Example 2: Preparation of 2-(l-(ethylsulfonyl)azetidin-3-ylidene)acetonitrile (Formula ID
N,N-Diisopropylethylamine (4.5 mL) was added into a reaction vessel containing acetonitrile (50 mL) and 3-(cyanomethylene)azetidine hydrochloride (1.5 g; Formula VII) at about 0°C to about 10°C. The reaction mixture was stirred for about 10 minutes.
Ethanesulfonyl chloride (2.22 g) was added into the reaction mixture at about 0°C to about 5°C over about 5 minutes. The temperature of the reaction mixture was raised to about 20°C to about 25 °C, and then the reaction mixture was stirred for about 16 hours. On completion of the reaction, acetonitrile was recovered from the reaction mixture under reduced pressure at about 40°C to about 45°C to obtain an oily residue. Dichloromethane (50 mL) was added into the residue. The contents were washed with a saturated sodium chloride solution (30 mL), followed by complete recovery of dichloromethane under reduced pressure at about 40°C to obtain 2-(l-(ethylsulfonyl)azetidin-3-ylidene)acetonitrile .
Yield: 98.59%
Example 3: Preparation of { l-(ethylsulfonyl)-3-[4-(4.4.5.5-tetramethyl-1.3.2-dioxaborolan-2-yl)-lH-pyrazol-l-yllazetidin-3-yl}acetonitrile (Formula V)
1,4-Dioxane (20 mL) was added into a reaction vessel containing a solution of potassium carbonate (4.5 g) in water (30 mL) at about 20°C to about 25 °C. 2-(l-(Ethylsulfonyl)azetidin-3-ylidene)acetonitrile (2 g; Formula II) and 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (2.30 g; Formula VIII) were added into the reaction mixture at about 20°C to about 25 °C. The reaction mixture was stirred at about 20°C to about 25 °C for about 16 hours to about 18 hours. On completion of the reaction, 1,4-dioxane was recovered from the reaction mixture under reduced pressure at about 45 °C to obtain a residue. Ethyl acetate (20 mL) was added into the residue, and the contents were stirred for about 5 minutes. The organic and aqueous layers were separated. The organic layer was concentrated under reduced pressure at about 45 °C to obtain { l-(ethylsulfonyl)- 3-[4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazol-l-yl]azetidin-3-yl}acetonitrile.
Yield: 85.78%
Mass: 381.4 [M + H]+
Example 4: Preparation of baricitinib (Formula I)
4-Chloro-7H-pyrrolo[2,3-d]pyrimidine (0.8 g; Formula IX) was added into a reaction vessel containing a solution of potassium carbonate (2.1 g) in water (30 mL) at about 20°C to about 25°C. A solution of { l-(ethylsulfonyl)-3-[4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazol-l-yl]azetidin-3-yl}acetonitrile (2.0 g; Formula V) in 1,4-dioxane (30 mL) was added into the reaction mixture at about 20°C to about 25 °C, followed by the addition of tetrakis(triphenylphosphine)palladium(0) (0.1 g). The reaction mixture was stirred at about 80°C to about 85°C for about 5 hours. On completion of the reaction, 1,4-dioxane was recovered from the reaction mixture under reduced pressure at about 45°C to obtain a residue. Ethyl acetate (50 mL) was added into the residue, and then the contents were stirred for about 5 minutes. The organic and aqueous layers were separated. The organic layer was concentrated under reduced pressure at about 45°C to obtain baricitinib.
Yield: 99.0%
Patent
PATENT
Figure 4: Infra-red (IR) spectrum of the crystalline form of baricitinib.
Example: Preparation of crystalline form of baricitinib
(4-( 1 -(3-(Cyanomethyl)- 1 -(ethylsulfonyl)azetidin-3-yl)- lH-pyrazol-4-yl)-7H-pyrrolo [2,3-d]pyrimidin-7-yl)methyl pivalate (8 g), methanol (40 mL), tetrahydrofuran (160 mL), and 1M sodium hydroxide (18.4 mL) were added into a reaction vessel at 20°C to 25°C. The reaction mixture was stirred for 3 hours. Progress of the reaction was monitored by thin layer chromatography. On completion, the reaction mixture was quenched with water (80 mL). The pH was adjusted to 7.0 to 7.5 by adding IN hydrochloric acid. Half of the solvent was recovered at a temperature of 40°C to 50°C. The reaction mixture was stirred at 20°C to 25°C for 18 hours, and then cooled to 5°C to 10°C. The solids were filtered, washed with a mixture of acetonitrile (50 mL) and water (100 mL), and then dried at 40°C to 50°C under reduced pressure for 24 hours to obtain the crystalline form of baricitinib.
Yield: 70%
PATENT
Figure 1 : X-ray Powder Diffraction (XRPD) pattern of the crystalline form of baricitinib. BELOW

Figure 2: Differential Scanning Calorimetry (DSC) thermogram of the crystalline form of baricitinib.
Figure 3 : Thermogravimetric Analysis (TGA) of the crystalline form of baricitinib.
Figure 4: Infra-red (IR) spectrum of the crystalline form of baricitinib.
The crystalline form of baricitinib is further characterized by a DSC having endotherms at about 180.63°C and about 207.98°C.
The crystalline form of baricitinib has a water content of about 3%, as determined by TGA.
The crystalline form of baricitinib is also characterized by an XRPD pattern as depicted in Figure 1, a DSC thermogram as depicted in Figure 2, a TGA as depicted in Figure 3, and an IR spectrum as depicted in Figure 4.
The preparation of the crystalline form of baricitinib is carried out by reacting (4-(l-(3-(cyanomethyl)-l-(ethylsulfonyl)azetidin-3-yl)-lH-pyrazol-4-yl)-7H-pyrrolo [2,3-d]pyrimidin-7-yl)methyl pivalate with a base in the presence of one or more solvents at a temperature of about 15°C to 50°C, stirring the reaction mixture for about 30 minutes to about 10 hours, partially recovering the solvent(s) from the reaction mixture at a temperature of about 35°C to about 60°C under reduced pressure, stirring the contents at about 15°C to 35°C for about 5 hours to about 24 hours, filtering the solid, washing the solid with a mixture of acetonitrile and water, and drying.
The (4-( 1 -(3 -(cyanomethyl)- 1 -(ethylsulfonyl)azetidin-3 -yl)- lH-pyrazol-4-yl)-7H-pyrrolo [2,3-d]pyrimidin-7-yl)methyl pivalate may be obtained by following the process disclosed in U.S. Patent No. 8, 158,616.
Example: Preparation of crystalline form of baricitinib
(4-( 1 -(3-(Cyanomethyl)- 1 -(ethylsulfonyl)azetidin-3-yl)- lH-pyrazol-4-yl)-7H-pyrrolo [2,3-d]pyrimidin-7-yl)methyl pivalate (8 g), methanol (40 mL), tetrahydrofuran (160 mL), and 1M sodium hydroxide (18.4 mL) were added into a reaction vessel at 20°C to 25°C. The reaction mixture was stirred for 3 hours. Progress of the reaction was monitored by thin layer chromatography. On completion, the reaction mixture was quenched with water (80 mL). The pH was adjusted to 7.0 to 7.5 by adding IN hydrochloric acid. Half of the solvent was recovered at a temperature of 40°C to 50°C. The reaction mixture was stirred at 20°C to 25°C for 18 hours, and then cooled to 5°C to 10°C. The solids were filtered, washed with a mixture of acetonitrile (50 mL) and water (100 mL), and then dried at 40°C to 50°C under reduced pressure for 24 hours to obtain the crystalline form of baricitinib.
Yield: 70%
PATENT
https://www.google.com/patents/WO2015145286A1?cl=en
EXAMPLES
Comparative Examples
Example 1 : Repetition of the process according to Example 78. Method B of U.S. Patent No. 8.158.616
4-( 1 -(3 -(Cyanomethyl)- 1 -(ethylsulfonyl)azetidin-3 -yl)- lH-pyrazol-4-yl)-7H- pyrrolo[2,3-d]pyrimidin-7-yl)methyl pivalate (1 g), methanol (5 mL), tetrahydrofuran (20 mL), and 1M sodium hydroxide (2.3 mL) were added into a reaction vessel at 20°C to 25 °C. The reaction mixture was stirred for 3 hours. Progress of the reaction was monitored by thin layer chromatography. On completion, the reaction mixture was quenched by adding water (20 mL). The pH was adjusted to 7.0 to 7.5 by adding IN hydrochloric acid, and the contents were stirred for 1.5 hours. No solid material was obtained. Example 2: Repetition of the process according to Example 78. Method C of U.S. Patent No. 8.158.616
4-( 1 -(3 -(Cyanomethyl)- 1 -(ethylsulfonyl)azetidin-3 -yl)- lH-pyrazol-4-yl)-7H- pyrrolo[2,3-d]pyrimidin-7-yl)methyl pivalate (2 g), lithium hydroxide monohydrate (0.51 g), acetonitrile (8 mL), and 2-propanol (2 mL) were added into a reaction vessel at 20°C to 25°C. The reaction mixture was stirred at 45°C to 50°C for 6 hours. Progress of the reaction was monitored by thin layer chromatography. On completion, the reaction mixture was cooled to 20°C to 25°C. The pH was adjusted to 6.0 to 7.0 by adding IN hydrochloric acid, and the contents were stirred overnight. No solid material was obtained.
Working Example:
Preparation of an amorphous form of baricitinib
4-( 1 -(3 -(Cyanomethyl)- 1 -(ethylsulfonyl)azetidin-3 -yl)- lH-pyrazol-4-yl)-7H- pyrrolo[2,3-d]pyrimidin-7-yl)methyl pivalate (1 g), methanol (5 mL), tetrahydrofuran (20 mL), and 1M sodium hydroxide (2.3 mL) were added into a reaction vessel at 20°C to 25 °C. The reaction mixture was stirred for 3 hours. Progress of the reaction was monitored by thin layer chromatography. On completion, the reaction mixture was quenched by adding water (20 mL). The pH was adjusted to 7.0 to 7.5 by adding IN hydrochloric acid, followed by completely recovering the solvent under reduced pressure at 40°C to 50°C. A sticky material was obtained. Water (10 mL) was added to the sticky material at 20°C to 25°C. The contents were stirred for 10 minutes. A solid material was precipitated out. The solid material was filtered, washed with water (20 mL), and then dried under reduced pressure at 40°C to 45°C for 24 hours to obtain the amorphous form of baricitinib.
Yield: 81%.
The amorphous form of baricitinib may be used in a pharmaceutical composition with one or more pharmaceutically acceptable carriers, diluents, or excipients, and optionally other therapeutic ingredients. The pharmaceutical composition may be used for the treatment of JAK-associated diseases.
PATENT
CN 105693731
https://www.google.com/patents/CN105693731A?cl=en
Baricitinib crystal form A and preparation method
Crystalline forms of 1 ethylsulfonyl 3 4 7H pyrrolo 2 3 d pyrimidin 4 yl …
priorart.ip.com/IPCOM/000244270
Nov 27, 2015 – Crystalline forms of baricitinib were found and are described … on their appearance temperature As follows the polymorph observed at room …
IR (KBr): 3203, 3116, 2256, 1583, 1328, 1138 cm-1.
(d, J=3.2 Hz, 1H), 7.10 (d, J=3.4 Hz, 1H), 4.62 (d, J=9.0 Hz, 2H), 4.26 (d, J=9.1 Hz,
2H), 3.72 (s, 2H), 3.26 (q, J=7.3 Hz, 2H), 1.26 (t, J=7.3 Hz, 3H) ppm.
116.86, 113.25, 100.14, 58.74, 56.26, 43.50, 27.03, 7.63 ppm.
7.10-100.14, 4.62-58.74, 4.26-58.74, 3.72-27.03, 3.26-43.50, 1.26-7.63.
56.26), 8.73-(152.39, 149.55, 113.25), 8.50-(129.80, 122.42), 7.64-(152.39, 113.25,
100.14), 7.10-(152.39, 127.13, 113.25), (4.62, 4.26)-(58.74, 56.26, 27.03), 3.72-
(116.86, 58.74, 56.26), 3.26-7.63, 1.26-43.50.
C 51.74%; H 4.61%; N 26.40%; S 8.63%.
Found C 51.62%; H 4.59%; N 26.28%; S 8.78%.
References
- “Baricitinib” (pdf). Statement on a nonproprietary name adopted by the USAN council. American Medical Association.
- “Lilly, Incyte Treatment Shows Positive Results”. http://www.insideindianabusiness.com. 9 Dec 2014. Retrieved 2 Mar 2015.
| Patent | Submitted | Granted |
|---|---|---|
| AZETIDINE AND CYCLOBUTANE DERIVATIVES AS JAK INHIBITORS [US8158616] | 2009-09-17 | 2012-04-17 |
| AZETIDINE AND CYCLOBUTANE DERIVATIVES AS JAK INHIBITORS [US2013225556] | 2013-03-29 | 2013-08-29 |
| JANUS KINASE INHIBITORS FOR TREATMENT OF DRY EYE AND OTHER EYE RELATED DISEASES [US2010113416] | 2010-05-06 | |
| METHOD OF TREATING MUSCULAR DEGRADATION [US2013310340] | 2013-05-15 | 2013-11-21 |
| METHOD OF SELECTING THERAPEUTIC INDICATIONS [US2014170157] | 2012-06-15 | 2014-06-19 |
| CYCLODEXTRIN-BASED POLYMERS FOR THERAPEUTIC DELIVERY [US2014357557] | 2014-05-30 | 2014-12-04 |
| Azetidine and cyclobutane derivatives as JAK inhibitors [US8420629] | 2011-12-09 | 2013-04-16 |
| BIOMARKERS AND COMBINATION THERAPIES USING ONCOLYTIC VIRUS AND IMMUNOMODULATION [US2014377221] | 2013-01-25 | 2014-12-25 |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2010039939A1 * | Oct 1, 2009 | Apr 8, 2010 | Incyte Corporation | Janus kinase inhibitors for treatment of dry eye and other eye related diseases |
| WO2011028685A1 | Aug 31, 2010 | Mar 10, 2011 | Incyte Corporation | Heterocyclic derivatives of pyrazol-4-yl-pyrrolo[2,3-d]pyrimidines as janus kinase inhibitors |
| WO2011103423A1 * | Feb 18, 2011 | Aug 25, 2011 | Incyte Corporation | Cyclobutane and methylcyclobutane derivatives as janus kinase inhibitors |
| WO2012068450A1 * | Nov 18, 2011 | May 24, 2012 | Incyte Corporation | Cyclobutyl substituted pyrrolopyridine and pyrrolopyrimidine derivatives as jak inhibitors |
| WO2012177606A1 | Jun 19, 2012 | Dec 27, 2012 | Incyte Corporation | Azetidinyl phenyl, pyridyl or pyrazinyl carboxamide derivatives as jak inhibitors |
| WO2013026025A1 | Aug 17, 2012 | Feb 21, 2013 | Incyte Corporation | Cyclohexyl azetidine derivatives as jak inhibitors |
| WO2013173506A2 | May 15, 2013 | Nov 21, 2013 | Rigel Pharmaceuticals, Inc. | Method of treating muscular degradation |
| WO2014028756A1 * | Aug 15, 2013 | Feb 20, 2014 | Concert Pharmaceuticals, Inc. | Deuterated baricitinib |
| WO2014138168A1 * | Mar 5, 2014 | Sep 12, 2014 | Incyte Corporation | Processes and intermediates for making a jak inhibitor |
| WO2015095492A1 | Dec 18, 2014 | Jun 25, 2015 | Incyte Corporation | Tricyclic heterocycles as bet protein inhibitors |
| WO2015123424A1 | Feb 12, 2015 | Aug 20, 2015 | Incyte Corporation | Cyclopropylamines as lsd1 inhibitors |
| WO2015131031A1 | Feb 27, 2015 | Sep 3, 2015 | Incyte Corporation | Jak1 inhibitors for the treatment of myelodysplastic syndromes |
| CN102844317B * | Feb 18, 2011 | Jun 3, 2015 | 因西特公司 | Cyclobutane and methylcyclobutane derivatives as janus kinase inhibitors |
| CN103415515B * | Nov 18, 2011 | Aug 26, 2015 | 因塞特公司 | 作为jak抑制剂的环丁基取代的吡咯并吡啶和吡咯并嘧啶衍生物 |
| CN103797010A * | Jun 19, 2012 | May 14, 2014 | 因塞特公司 | Azetidinyl phenyl, pyridyl or pyrazinyl carboxamide derivatives as JAK inhibitors |
| CN104024256A * | Sep 6, 2012 | Sep 3, 2014 | 因塞特公司 | Processes and intermediates for making a JAK inhibitor |
| EP2647629A1 * | Dec 1, 2011 | Oct 9, 2013 | Nissan Chemical Industries, Ltd. | Pyrazole compound having therapeutic effect on multiple myeloma |
| EP2647629A4 * | Dec 1, 2011 | Apr 23, 2014 | Nissan Chemical Ind Ltd | Pyrazole compound having therapeutic effect on multiple myeloma |
| US8415362 | Jun 12, 2008 | Apr 9, 2013 | Incyte Corporation | Pyrazolyl substituted pyrrolo[2,3-b]pyrimidines as Janus kinase inhibitors |
| US8722693 | Dec 5, 2013 | May 13, 2014 | Incyte Corporation | Salts of the Janus kinase inhibitor (R)-3-(4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile |
| US8822481 | Apr 18, 2014 | Sep 2, 2014 | Incyte Corporation | Salts of the janus kinase inhibitor (R)-3-(4-(7H-pyrrolo[2,3-d] pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile |
| US8829013 | Apr 18, 2014 | Sep 9, 2014 | Incyte Corporation | Salts of the Janus kinase inhibitor (R)-3-(4-(7H-pyrrolo[2,3-D]pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile |
| US8933085 | Nov 18, 2011 | Jan 13, 2015 | Incyte Corporation | Cyclobutyl substituted pyrrolopyridine and pyrrolopyrimidine derivatives as JAK inhibitors |
| US8933086 | Sep 20, 2013 | Jan 13, 2015 | Incyte Corporation | Heteroaryl substituted pyrrolo[2,3-B]pyridines and pyrrolo[2,3-B]pyrimidines as Janus kinase inhibitors |
| US8946245 | Mar 30, 2011 | Feb 3, 2015 | Incyte Corporation | Heteroaryl substituted pyrrolo[2,3-b]pyridines and pyrrolo[2,3-b]pyrimidines as Janus kinase inhibitors |
| US8987443 | Mar 5, 2014 | Mar 24, 2015 | Incyte Corporation | Processes and intermediates for making a JAK inhibitor |
| US9023840 | Feb 21, 2014 | May 5, 2015 | Incyte Corporation | Azetidinyl phenyl, pyridyl or pyrazinyl carboxamide derivatives as JAK inhibitors |
| US9034884 | Nov 18, 2011 | May 19, 2015 | Incyte Corporation | Heterocyclic-substituted pyrrolopyridines and pyrrolopyrimidines as JAK inhibitors |
| US9079912 | May 12, 2014 | Jul 14, 2015 | Incyte Corporation | Heteroaryl substituted pyrrolo[2,3-B] pyridines and pyrrolo[2,3-B] pyrimidines as Janus kinase inhibitors |
| US9193733 | May 17, 2013 | Nov 24, 2015 | Incyte Holdings Corporation | Piperidinylcyclobutyl substituted pyrrolopyridine and pyrrolopyrimidine derivatives as JAK inhibitors |
| US9206187 | Sep 6, 2013 | Dec 8, 2015 | Incyte Holdings Corporation | Heteroaryl substituted pyrrolo[2,3-B] pyridines and pyrrolo[2,3-B] pyrimidines as Janus kinase |
| US9216984 | Nov 8, 2013 | Dec 22, 2015 | Incyte Corporation | 3-[4-(7H-pyrrolo[2,3-D]pyrimidin-4-yl)-1H-pyrazol-1-yl]octane—or heptane-nitrile as JAK inhibitors |
| US9221845 | Mar 11, 2015 | Dec 29, 2015 | Incyte Holdings Corporation | Processes and intermediates for making a JAK inhibitor |

PHOSHATE SEE……http://www.medchemexpress.com/product_pdf/HY-15315A/Baricitinib%20phosphate-NMR-HY-15315A-08874-2013.pdf
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
2-[1-ethylsulfonyl-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]azetidin-3-yl]acetonitrile
|
|
| Clinical data | |
| Legal status |
|
| Identifiers | |
| CAS Number | 1187594-09-7 |
| ATC code | None |
| PubChem | CID: 44205240 |
| ChemSpider | 26373084 |
| ChEMBL | CHEMBL2105759 |
| PDB ligand ID | 3JW (PDBe, RCSB PDB) |
| Chemical data | |
| Formula | C16H17N7O2S |
| Molecular mass | 371.42 g/mol |
SEE………http://apisynthesisint.blogspot.in/2016/01/baricitinib.html
//////////LY3009104, INCB028050, LY 3009104, INCB 028050, nda, baricitinib
CCS(=O)(=O)N1CC(C1)(CC#N)N2C=C(C=N2)C3=C4C=CNC4=NC=N3
Review of literature
Almost all the synthetic methods (WO2009114512A1, CN201510880931.X, CN201610080433.1, WO2016088094A1, WO2016125080A2, WO2016205487A1, CN201610903498.1, WO2017109524A1, CN201710181322.4, CN201710165830.3) reported for the preparation of baricitinib employed important intermediates 2-(1-(ethylsulfonyl)azetidin-3-ylidene)acetonitrile(2) and tert-butyl 3-(cyanomethylene)azetidine-1-carboxylate(3), for which the development of a green and facile synthetic method for intermediates 2 and 3 has a strong demand. However, several reported research-scale synthetic methods for the preparation of intermediates 2 and 3
1
In Scheme 1, compounds 2-(chloromethyl)oxirane (I-1) and diphenylmethanamine (I-2) were used as the starting material (WO2009114512A1). Intermediate 2 was obtained through reduction reaction, boc-protecting reaction, oxidizing reaction, and wittig reaction, which was then employed to afford intermediate 3 by deprotect and hinsber reactions
Synthesis of intermediate 2 and 3 using 2-(chloromethyl)oxirane (I-1) and diphenylmethanamine (I-2) as starting material
2
In Scheme 2, compound azetidin-3-ol hydrochloride (II-1) was used as start material, which was employed to afford intermediate 3 through hinsber reaction, oxidizing reaction, and wittig reaction (WO2016205487A1) Besides, another patent reported that the start material 1-amino-3-chloropropan-2-ol hydrochloride (III-1) was first reacted with ethanesulfonyl chloride to afford compound N-(3-chloro-2-hydroxypropyl)ethanesulfonamide (III-2), which was then converted to the same intermediate 1-(ethylsulfonyl)azetidin-3-ol (III-3, II-2) after cyclization
Synthesis of intermediate 3 with II-1 as starting material
3
Key intermediate 3 was obtained by the same method as that of Scheme 2 (Scheme 3, CN201710165830.3)
Synthesis of intermediate 3 with III-1 as starting material
4
In Scheme 4, compound azetidin-3-one hydrochloride (IV-1) was used as raw start material, which was converted to intermediate 3 through hinsber reaction and aldol condensation reaction (CN201610903498.1).
Synthesis of intermediate 3 with IV-1 as starting material
van Vollenhoven R, Helt C, Arora V, Zhong J, Correia AP, de la Torre I, Muram D (2018) Safety and efficacy of baricitinib in patients receiving conventional synthetic disease-modifying antirheumatic drugs or corticosteroids. Rheumatol Ther 5(2):525–536
https://www.mdpi.com/1424-8247/12/1/37/htm
Baricitinib (2, Figure 2) is the active ingredient of Olumiant®, commercialized by Eli Lilly and Co. Its IUPAC name is: 2-[1-(ethanesulfonyl)-3-(4-{7H-pyrrolo[2,3-d]pyrimidin-4-yl}-1H-pyrazol-1-yl)azetidin-3-yl] acetonitrile, CAS 1187594-09-7.
After a rejection in April 2017, baricitinib (2 mg tablets) has been approved on May 31, 2018 for treatment of rheumatoid arthritis. [3] Noticeably, it had been approved, for the same purpose, in the European Union (EU) in February 2017. [4]
There are essentially two routes for the preparation of baricitinib 2. As depicted in Scheme 1, they can be distinguished by introducing central pyrazole ring in the molecule. In the original procedure [20,21], the pyrazole ring was linked to the pyrrolo[2,3-d]pyrimidine system (to afford 6) and then coupled to the azetidine moiety 7 to give the intermediate 8. In an alternative route [22,23], the bound between the pyrazole and the azetidine was formed (to yield 10) before reaction with the fused system 9.
Scheme 2. Preparation of baricitinib 2 from a 4-pyrazolyl-7H-pyrrolo[2,3-d]pyrimidine
Thus (Scheme 2), 4-chloro-7H– pyrrolo[2,3-d]pyrimidine was protected on position 7 by reaction with 2-(trimethylsilyl)ethoxymethyl chloride. The protected fused system was then coupled with 4-pyrazoleboronic acid pinacol ester 12 by a Suzuli-Miyaura reaction, giving 6. Parallelly, 7 was obtained from 1-Boc-3-azetidinone 13 and diethyl cyanomethylphosphonate. Reaction between 6 and 7 in the presence of DBU afforded the ester 8. Subsequent hydrolysis, decarboxylation, sulfonation, and finally deprotection of the pyrrolopyrimidine moiety yielded the targeted derivative 2. In a variant [21], also used to prepare deuterated samples of 2 [24], the azetidine derivative 7 has been deprotected and sulfonated before coupling with 6.
Scheme 3. Preparation of baricitinib 2 from a 4-chloro-7H-pyrrolo[2,3-d]pyrimidine.
In a more recent patent [22], the sulfonated azetidine 14 (Scheme 3) was prepared from azetidine-3-ol by a sequence including a sulfonation, an oxidation, and introduction of the cyanomethylene moiety. Interestingly, there is no need to protect any position in that sequence. Additionally, let us emphasize that the oxidation step could be performed both in batch or under flow conditions. [22,25]. Then, 14 was reacted with 4-pyrazoleboronic acid pinacol ester 12 to yield 10. The bound between the azetidinylpyrazole group and the pyrrolo[2,3-d]pyrimidine system was then created through a Suzuki-Miyaura reaction involving 7-Boc-4-chloro-7H-pyrrolo[2,3-d]pyrimidine 9 or even the unprotected 4-chloro-7H-pyrrolo[2,3-d]pyrimidine.
- 20 Rodgers, J.; Shepard, S.; Maduskuie, T.; Wang, H.; Falahatpisheh, N.; Rafalski, M.; Arvanitis, A.; Storace, L.; Jalluri, R.; Fridman, J.; et al. Heteroaryl Substituted Pyrrolo[2,3-b]pyridines and Pyrrolo[2,3-b]pyrimidines as Janus Kinase Inhibitors. US20070135461A1, 14 June 2007.
- 21 Rodgers, J.D.; Shepard, S.; Li, Y.-L.; Zhou, J.; Liu, P.; Meloni, D.; Xia, M. Azetidine and Cyclobutane Derivatives as JAK Inhibitors. U.S. Patent US20090233903A1, 17 September 2009.
- 22 Kobierski, M.E.; Kopach, M.E.; Martinelli, J.R.; Varie, D.L.; Wilson, T.M. Processes and Intermediates for the Preparation of {1(Ethylsulfonyl)-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1yl]azetidin-3-yl}acetonitrile. WO 2016/ 205487, 22 December 2016]
- 23 Xu, J.; Cai, J.; Chen, J.; Zong, X.; Wu, X.; Ji, M.; Wang, P. An Efficient Synthesis of Baricitinib. Chem. Res.2016, 40, 205–208.
- 24 Tung, R.D. Deuterated Baricitinib. U.S. Patent US20180221374A1, 9 August 201
- 25 Hughes, D.L. Applications of Flow Chemistry in Drug Development: Highlights of Recent Patent Literature. Process Res. Dev.2018, 22, 13–20

Reference:1. WO2009114512A1 / US2009233903A1.



: J. Med. Chem. 2019, 62, 7340−7382
Baricitinib (Olumiant). Baricitinib is an inhibitor of Janus family tyrosine kinase (JAK)-1 and -2 approved by the USFDA in 2017 as monotherapy or in combination with methotrexate for the treatment of adults with moderate to severe active rheumatoid arthritis.83 Baricitinib was discovered by Incyte and codeveloped with Eli Lilly. It is also in clinical trials for the treatment of atopic dermatitis, systemic lupus erythematosus, and giant cell arteritis. Numerous synthetic routes to baricitinib have been reported in the patent literature.28 The largest scale synthesis was reported by Incyte and is described in Schemes 35−37. 28a Horner−Emmons reaction between tert-butyl 3-oxoazetidine1-carboxylate (156) and diethyl cyanomethyl phosphonate (157) gave cyanomethylene azetidine 158 in 61% yield (Scheme 35). Acidic removal of the Boc protecting group was followed by treatment with ethanesulfonyl chloride to give the sulfonamide subunit of baricitinib 159 in 91% yield. The synthesis of baricitinib was completed as described in Scheme 36. Deprotonation of chloropyrrolopyrimidine 160 with sodium hydride followed by treatment with trimethylsilylethoxymethyl chloride gave the SEM protected chloropyrrolopyrimidine 161 in 89% yield. Suzuki coupling with commercially available boronic ester 162 followed by acidic removal of the ethoxyethyl protecting group gave pyrazole 163 in 87% yield. 1,4-Addition of pyrazole 163 to cyanomethylazetidine 159 was accomplished in the presence of DBU to give SEM-protected baricitinib 164 in high yield. Finally, treatment of 164 with lithium tetrafluoroborate followed by ammonium hydroxide gave baricitinib (XVII) in 81% yield. An alternative synthesis of baricitinib that avoids the use of the ethoxylethyl and SEM protecting groups and changes the order of the 1,4- addition and Suzuki coupling steps has also been reported on large scale and is described in Scheme 37. 28b,84
(28) (a) Rodgers, J. D.; Shepard, S.; Li, Y.-L.; Zhou, J.; Liu, P.; Meloni, D.; Xia, M. Preparation of Azetidine and Cyclobutane Derivatives as Jak Inhibitors. WO 2009114512, 2009. (b) Kobierski, M. E.; Kopach, M. E.; Martinelli, J. R.; Varie, D. L.; Wilson, T. M. Processes and Intermediates for the Preparation of {1-(Ethylsulfonyl)- 3-[4-(7H-pyrrolo[2,3-D]pyrimidin-4-yl)-1H-pyrazol-1-yl]azetidin-3- yl}acetonitrile. WO 2016205487, 2016.
(84) Xu, J.; Cai, J.; Chen, J.; Zong, X.; Wu, X.; Ji, M.; Wang, P. An Efficient Synthesis of Baricitinib. J. Chem. Res. 2016, 40, 205−208.
///////////////





Formulations f.c. tablet 2 mg, 4 mg (as phosphate) References Jiaojiao, X. et al., Journal of Chem. Research, (2016) 40 (4), 205. a WO 2009 114512 (Incyte; 17.09.2009; US-prior. 11.03.2008). US 8 158 616 (Incyte; 17.04.2012; US-prior. 11.03.2008). b WO 2016 205487 (Eli Lilly & Co.; 22.12.2016; US-prior. 19.06.2015). Preparation of I Qiyan, L. et al., Organic Letters, (2009) 11(9), 1999-2002
