
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

Telotristat ethyl
Molecular Formula, C27-H26-Cl-F3-N6-O3,
Molecular Weight, 574.9884,
RN: 1033805-22-9
UNII: 8G388563M
LX 1032
(2S)-2-Amino-3-[4-[2-amino-6-[[(1R)-1-[4-chloro-2-(3-methylpyrazol-1-yl)phenyl]-2,2,2-trifluoroethyl]oxy]pyrimidin-4-yl]phenyl]propionic acid ethyl ester
Ethyl-4-(2-amino-6-{(1R)-1-[4-chlor-2-(3-methyl-1H-pyrazol-1-yl)phenyl]-2,2,2-trifluorethoxy}-4-pyrimidinyl)-L-phenylalaninat

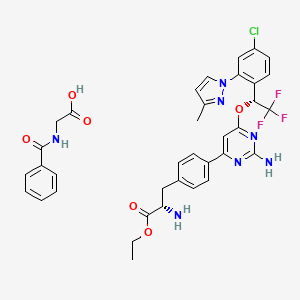
CAS: 1137608-69-5 (etiprate), LX 1606
Chemical Formula: C36H35ClF3N7O6
Molecular Weight: 754.16
- LX 1032 hippurate
- LX 1606



Carcinoid syndrome is a cluster of symptoms sometimes seen in people with carcinoid tumors. These tumors are rare, and often slow-growing. Most carcinoid tumors are found in the gastrointestinal tract. Carcinoid syndrome occurs in less than 10 percent of patients with carcinoid tumors, usually after the tumor has spread to the liver. The tumors in these patients release excess amounts of the hormone serotonin, resulting in diarrhea. Complications of uncontrolled diarrhea include weight loss, malnutrition, dehydration, and electrolyte imbalance.
“Today’s approval will provide patients whose carcinoid syndrome diarrhea is not adequately controlled with another treatment option,” said Julie Beitz, M.D., director of the Office of Drug Evaluation III in the FDA’s Center for Drug Evaluation and Research.
Xermelo, in a regimen with SSA therapy, is approved in tablet form to be taken orally three times daily with food. Xermelo inhibits the production of serotonin by carcinoid tumors and reduces the frequency of carcinoid syndrome diarrhea.
The safety and efficacy of Xermelo were established in a 12-week, double-blind, placebo-controlled trial in 90 adult participants with well-differentiated metastatic neuroendocrine tumors and carcinoid syndrome diarrhea. These patients were having between four to 12 daily bowel movements despite the use of SSA at a stable dose for at least three months. Participants remained on their SSA treatment, and were randomized to add placebo or treatment with Xermelo three times daily. Those receiving Xermelo added on to their SSA treatment experienced a greater reduction in average bowel movement frequency than those on SSA and placebo. Specifically, 33 percent of participants randomized to add Xermelo on to SSA experienced an average reduction of two bowel movements per day compared to 4 percent of patients randomized to add placebo on to SSA.
The most common side effects of Xermelo include nausea, headache, increased levels of the liver enzyme gamma-glutamyl transferase, depression, accumulation of fluid causing swelling (peripheral edema), flatulence, decreased appetite and fever. Xermelo may cause constipation, and the risk of developing constipation may be increased in patients whose bowel movement frequency is less than four bowel movements per day. Patients treated with a higher than recommended dosage of Xermelo developed severe constipation in clinical trials. One patient required hospitalization and two other patients developed complications of either intestinal perforation or intestinal obstruction. Patients should be monitored for severe constipation. If a patient experiences severe constipation or severe, persistent or worsening abdominal pain, they should discontinue Xermelo and contact their healthcare provider.
The FDA granted this application fast track designation and priority review. The drug also received orphan drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
Xermelo is manufactured by Woodlands, Texas-based Lexicon Pharmaceuticals, Inc.
SYNTHESIS…….WO 2011100285

5.67. Synthesis of (S)-2-Amino-3-[4-(2-amino-6-{R-l-[4-chloro-2-(3-methyl-pyrazol-l-yll- phenyll-2,2,2-trifluoro-ethoxy)-pyrimidin-4-yl)-phenyll-propionic acid ethyl ester

The title compound was prepared stepwise, as described below:
Step 1: Synthesis of l-(2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-ethanone. To a 500 ml 2 necked RB flask containing anhydrous methanol (300 ml) was added thionyl chloride (29.2 ml, 400 mmol) dropwise at 0-5°C (ice water bath) over 10 minutes. The ice water bath was removed, and 2-bromo-4-chloro-benzoic acid (25 g, 106 mmol) was added. The mixture was heated to mild reflux for 12h. Progress of the reaction was monitored by TLC and LCMS. After completion of the reaction, the reaction mixture was concentrated. Crude product was dissolved in dichloromethane (DCM, 250 ml), washed with water (50 ml), sat. aq. NaHC03 (50 ml), brine (50 ml), dried over sodium sulfate, and concentrated to give the 2- bromo-4-chloro-benzoic acid methyl ester (26 g, 99 %), which was directly used in the following step.
2-Bromo-4-chloro-benzoic acid methyl ester (12.4 g, 50 mmol) in toluene (200 ml) was cooled to -70°C, and trifluoromethyl trimethyl silane (13 ml, 70 mmol) was added.
Tetrabutylamonium fluoride (1M, 2.5 ml) was added dropwise, and the mixture was allowed to warm to room temperature over 4h, after which it was stirred for 10 hours at room temperature. The reaction mixture was concentrated to give the crude [l-(2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-l-methoxy-ethoxy]-trimethyl-silane. The crude intermediate was dissolved in methanol (100 ml) and 6N HCI (100 ml) was added. The mixture was kept at 45-50°C for 12h. Methanol was removed, and the crude was extracted with dichloromethane (200 ml). The combined DCM layer was washed with water (50 ml), NaHC03 (50 ml), brine (50 ml), and dried over sodium sulfate. Removal of solvent gave a crude product, which was purified by ISCO column chromatography, using 1-2% ethyl acetate in hexane as solvent, to afford l-(2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-ethanone (10 g, 70%). !H-NMR (300 MHz, CDC ): δ (ppm) 7.50 (d,lH), 7.65(d,lH), 7.80(s,lH).
Step 2: Synthesis of R-l-(2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-ethanol. To catechol borane (1M in THF 280 ml, 280 mmol) in a 2L 3-necked RB flask was added S-2-methyl-CBS oxazaborolidine (7.76 g, 28 mmol) under nitrogen, and the resulting mixture was stirred at room temperature for 20 min. The reaction mixture was cooled to -78°C (dry ice/acetone bath), and 1-(2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-ethanone (40 g, 139 mmol) in THF (400 ml) was added dropwise over 2 hours. The reaction mixture was allowed to warm to -36°C, and was stirred at that temperature for 24 hours, and further stirred at -32 °C for another 24h. 3N NaOH (250 ml) was added, and the cooling bath was replaced by ice-water bath. Then 30 % hydrogen peroxide in water (250 ml) was added dropwise over 30 minutes. The ice water bath was removed, and the mixture was stirred at room temperature for 4 hours. The organic layer was separated, concentrated and re-dissolved in ether (200 ml). The aqueous layer was extracted with ether (2 x 200 ml). The combined organic layers were washed with IN aq. NaOH (4 x 100 ml), brine, and dried over sodium sulfate. Removal of solvent gave crude product which was purified by column chromatography using 2 to 5% ethyl acetate in hexane as solvent to give desired alcohol 36.2 g (90 %, e.e. >95%). The alcohol (36.2 g) was crystallized from hexane (80 ml) to obtain R-l-(2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-ethanol 28.2 g (70 %; 99-100 % e.e.). !H-NMR (400 MHz, CDCIs) δ (ppm) 5.48 (m, 1H), 7.40 (d, 1H), 7.61 (d, 2H).
Step 3: Synthesis of R-l-[4-chloro-2-(3-methyl-pyrazol-l-yl)-phenyll-2.2.2-trifluoro-ethanol. R-l-(2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-ethanol (15.65 g, 54.06 mmol), 3-methylpyrazole (5.33 g, 65 mmol), Cul (2.06 g, 10.8 mmol), 2CO3 (15.7 g, 113.5 mmol), (lR,2R)-N,N’-dimethyl-cyclohexane-l,2-diamine (1.54 g, 10.8 mmol) and toluene (80 ml) were combined in a 250 ml pressure tube and heated to 130°C (oil bath temperature) for 12 hours. The reaction mixture was diluted with ethyl acetate and washed with H2O (4 x 100 ml), brine, and dried over sodium sulfate. Removal of solvent gave a crude product, which was purified by ISCO column chromatography using 5-10 % ethyl acetate in hexane as solvent to get R-l-[4-chloro-2-(3-methyl-pyrazol-l-yl)-phenyl]-2,2,2-trifluoro-ethanol (13.5 g; 86 %). i-H-NMR (400 MHz, CDC ): δ (ppm) 2.30(s, 3H), 4.90(m, 1H), 6.20(s, 1H), 6.84(d, 1H), 7.20(s, 1H), 7.30(d, 1H), 7.50(d, 1H).
Step 4: Synthesis of (S)-2-Amino-3- 4-(2-amino-6-fR-l-[4-chloro-2-(3-methyl-pyrazol-l-yl)-phenyll^^^-trifluoro-ethoxyl-pyrimidin^-yll-phenvD-propionic acid ethyl ester. R-l-[4-chloro-2-(3-methyl-pyrazol-l-yl)-phenyl]-2,2,2-trifluoro-ethanol (17.78 g, 61.17 mmol), (S)-3-[4-(2-amino-6-chloro-pyrimidine-4-yl)-phenyl]-2-tert-butoxycarbonylamino-propionic acid (20.03 g, 51 mmol), 1,4-dioxane (250 ml), and CS2CO3 (79.5 g, 244 mmol) were combined in a 3-necked 500 ml RB flask and heated to 100°C (oil bath temperature) for 12-24 hours. The progress of reaction was monitored by LCMS. After the completion of the reaction, the mixture was cooled to 60°C, and water (250 ml) and THF (400 ml) were added. The organic layer was separated and washed with brine (150 ml). The solvent was removed to give crude BOC protected product, which was taken in THF (400 ml), 3N HCI (200 ml). The mixture was heated at 35-40 °C for 12 hours. THF was removed in vacuo. The remaining aqueous layer was extracted with isopropyl acetate (2x 100 ml) and concentrated separately to recover the unreacted alcohol (3.5 g). Traces of remaining organic solvent were removed from the aqueous fraction under vacuum.
To a 1L beaker equipped with a temperature controller and pH meter, was added H3PO4 (40 ml, 85 % in water) and water (300 ml) then 50 % NaOH in water to adjust pH to 6.15. The temperature was raised to 58 °C and the above acidic aqueous solution was added dropwise into the buffer with simultaneous addition of 50 % NaOH solution in water so that the pH was maintained between 6.1 to 6.3. Upon completion of addition, precipitated solid was filtered and washed with hot water (50-60°C) (2 x 200 ml) and dried to give crude (S)-2-amino-3-[4-(2-amino-6-[R-l-[4-chloro-2-(3-methyl-pyrazol-l-yl)-phenyl]-2,2,2-trifluoro-ethoxy}-pyrimidin-4-yl)-phenyl}^ propionic acid (26.8 g; 95 %). LCMS and HPLC analysis indicated the compound purity was about 96-97 %.
To anhydrous ethanol (400 ml) was added SOC (22 ml, 306 mmol) dropwise at 0-5°C.
Crude acid (26.8 ) from the above reaction was added. The ice water bath was removed, and the reaction mixture was heated at 40-45°C for 6-12 hours. After the reaction was completed, ethanol was removed in vacuo. To the residue was added ice water (300 ml), and extracted with isopropyl acetate (2 x 100 ml). The aqueous solution was neutralized with saturated Na2C03 to adjust the pH to 6.5. The solution was extracted with ethyl acetate (2 x 300 ml). The combined ethyl acetate layer was washed with brine and concentrated to give 24 g of crude ester (HPLC purity of 96-97 %). The crude ester was then purified by ISCO column chromatography using 5 % ethanol in DCM as solvent to give (S)-2-amino-3-[4-(2-amino-6-{R-l-[4-chloro-2-(3-methyl-pyrazol-l-yl)-phenyl]-2,2,2-trifluoro-ethoxy}-pyrimidin-4-yl)-phenyl}-propionic acid ethyl ester (20.5g; 70 %; HPLC purity of 98 %). LCMS M+l = 575. !H-NMR (400 MHz, CDsOD): δ (ppm) 1.10 (t, 3H), 2.25 (s, 3H), 2.85 (m, 2H), 3.65 (m, IH), 4.00 (q, 2H), 6.35 (s, IH), 6.60 (s, IH), 6.90 (m, IH), 7.18 (d, 2H), 7.45 (m, 2H), 7.70 (d, IH), 7.85 (m, 3H).
SYNTHESIS OF INTERMEDIATE
WO 2009048864

https://google.com/patents/WO2009048864A1?cl=en
6.15. Preparation of 6SV3-(4-(2-Amino-6-chloropyrimidin-4-yl)phenyl)-2- (fert-butoxycarbonylamino)propanoic Acid Using the Lithium Salt of (S)-2-(te^-butoxycarbonylamino)-3-(4-(4,4,5,5-tetramethyl-l,3,2- dioxaborolan-2-yl)phenyl)propanoic Acid

During preparation of compound 7, the isolation of the free acid can be optionally omitted. Thus, an aqueous solution of the lithium salt of compound 7 in 100 ml water, prepared from 5.0 g of Boc-Tyr-OMe (4, 17 mmol), was mixed 2-amino-4,6- dichloropyrimidine (3.3 g, 1.2 eq), potassium bicarbonate (5.0 g, 3 eq), bis(triphenylphosphine)palladium(II) dichloride (60 mg, 0.5 mol%), and 100 ml ethanol. The resulting mixture was heated at 700C for 5 hours. Additional 2-amino-4,6- dichloropyrimidine (1.1 g, 0.4 eq) was added and heating was continued at 7O0C for an additional 2 hours. HPLC analysis showed about 94% conversion. Upon cooling and filtration, the filtrate was analyzed by HPLC against a standard solution of compound 8. The assay indicated 3.9 g compound 8 was contained in the solution (59% yield from compound 4).
6.16. Alternative Procedure for Preparation of (S)-3-(4-f2-Amino-6- chloropyrimidin-4-yl)phenyl)-2-(fe^-butoxycarbonylamino)propanoic Acid Using Potassium Carbonate as Base

The boronic acid compound 11 (Ryscor Science, Inc., North Carolina, 1.0 g, 4.8 mmol) and potassium carbonate (1.32 g, 2 eq) were mixed in aqueous ethanol (15 ml ethanol and 8 ml water). Di-ter£-butyldicarbonate (1.25 g, 1.2 eq) was added in one portion. After 30 minutes agitation at room temperature, HPLC analysis showed complete consumption of the starting compound 11. The 2-amino-4,6- dichloropyrimidine (1.18 g, 1.5 eq) and the catalyst bis(triphenylphosphine)palladium(II) dichloride (34 mg, 1 mol%) were added and the resulting mixture was heated at 65-700C for 3 hours. HPLC analysis showed complete consumption of compound 12. After concentration and filtration, HPLC analysis of the resulting aqueous solution against a standard solution of compound 8 showed 1.26 g compound 8 (67% yield).
6.17. Alternative procedure for preparation of (5)-3-(4-(2-Amino-6-

The boronic acid compound 11 (10 g, 48 mmol) and potassium bicarbonate (14.4 g, 3 eq) were mixed in aqueous ethanol (250 ml ethanol and 50 ml water). Oi-tert- butyldicarbonate (12.5 g, 1.2 eq) was added in one portion. HPLC analysis indicated that the reaction was not complete after overnight stirring at room temperature. Potassium carbonate (6.6 g, 1.0 eq) and additional di-te/t-butyldicarbonate (3.1 g, 0.3 eq) were added. After 2.5 hours agitation at room temperature, HPLC analysis showed complete consumption of the starting compound 11. The 2-amino-4,6-dichloropyrimidine (11.8 g, 1.5 eq) and the catalyst bis(triphenylphosphine)-palladium(II) dichloride (0.34 g, 1 mol%” were added and the resulting mixture was heated at 75-8O0C for 2 hours. HPLC analysis showed complete consumption of compound 12. The mixture was concentrated under reduced pressure and filtered. The filtrate was washed with ethyl acetate (200 ml) and diluted with 3 : 1 THF/MTBE (120 ml). This mixture was acidified to pH about 2.4 by 6 N hydrochloric acid. The organic layer was washed with brine and concentrated under reduced pressure. The residue was precipitated in isopropanol, filtered, and dried at 500C under vacuum to give compound 8 as an off-white solid (9.0 g, 48% yield). Purity: 92.9% by HPLC analysis. Concentration of the mother liquor yielded and additional 2.2 g off-white powder (12% yield). Purity: 93.6% by HPLC analysis
PATENT
https://www.google.com/patents/WO2013059146A1?cl=en
This invention is directed to solid pharmaceutical dosage forms in which an active pharmaceutical ingredient (API) is (S)-ethyl 2-amino-3-(4-(2-amino-6-((R)-l-(4-chloro-2-(3- methyl-lH-pyrazol-l-yl)phenyl)-2,2,2-trifluoroethoxy)pyrimidin-4-yl)phenyl)propanoate
(telotristat):

or a pharmaceutically acceptable salt thereof. The compound, its salts and crystalline forms can be obtained by methods known in the art. See, e.g., U.S. patent no. 7,709,493.
PATENT
http://www.google.co.in/patents/WO2008073933A2?cl=en
6.19. Synthesis of (S)-2-Amino-3-r4-q-amino-6-{R-l-r4-chloro-2-(3-methyl- Pyrazol-l-yl)-phenyll-2,2,2-trifluoro-ethoxy}-pyrimidin-4-yl)-phenyll- propionic acid ethyl ester

The title compound was prepared stepwise, as described below: Step 1 : Synthesis of l-(2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-ethanone. To a 500 ml 2 necked RB flask containing anhydrous methanol (300 ml) was added thionyl chloride (29.2 ml, 400 mmol) dropwise at 0-50C (ice water bath) over 10 min. The ice water bath was removed, and 2-bromo-4-chloro-benzoic acid (25 g, 106 mmol) was added. The mixture was heated to mild reflux for 12h. Progress of the reaction was monitored by TLC and LCMS. After completion of the reaction, the reaction mixture was concentrated. Crude product was dissolved in dichloromethane (DCM, 250 ml), washed with water (50 ml), sat. aq. NaHCO3 (50 ml), brine (50 ml), dried over sodium sulfate, and concentrated to give the 2- bromo-4-chloro-benzoic acid methyl ester (26 g, 99 %), which was directly used in the following step.
2-Bromo-4-chloro-benzoic acid methyl ester (12.4 g, 50 mmol) in toluene (200 ml) was cooled to -700C, and trifluoromethyl trimethyl silane (13 ml, 70 mmol) was added. Tetrabutylamonium fluoride (IM, 2.5 ml) was added dropwise, and the mixture was allowed to warm to room temperature over 4h, after which it was stirred for 1Oh at room temperature. The reaction mixture was concentrated to give the crude [l-(2-bromo-4-chloro-phenyl)-2,2,2- trifluoro-l-methoxy-ethoxy]-trimethyl-silane. The crude intermediate was dissolved in methanol (100 ml) and 6N HCl (100 ml) was added. The mixture was kept at 45-500C for 12h. Methanol was removed, and the crude was extracted with dichloromethane (200 ml). The combined DCM layer was washed with water (50 ml), NaHCO3 (50 ml), brine (50 ml), and dried over sodium sulfate. Removal of solvent gave a crude product, which was purified by ISCO column chromatography, using 1-2% ethyl acetate in hexane as solvent, to afford 1- (2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-ethanone (10 g, 70%). 1H-NMR (300 MHz, CDCl3): δ (ppm) 7.50 (d,lH), 7.65(d,lH), 7.80(s,lH).
Step 2: Synthesis of R-l-(2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-ethanol. To catechol borane (IM in THF 280 ml, 280 mmol) in a 2L 3-necked RB flask was added S-2- methyl-CBS oxazaborolidine (7.76 g, 28 mmol) under nitrogen, and the resulting mixture was stirred at room temperature for 20 min. The reaction mixture was cooled to -78°C (dry ice/acetone bath), and l-(2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-ethanone (40 g, 139 mmol) in THF (400 ml) was added dropwise over 2h. The reaction mixture was allowed to warm to -36°C, and was stirred at that temperature for 24 h, and further stirred at -32°C for another 24h. 3N NaOH (250 ml) was added, and the cooling bath was replaced by ice-water bath. Then 30 % hydrogen peroxide in water (250 ml) was added dropwise over 30 minutes. The ice water bath was removed, and the mixture was stirred at room temperature for 4h. The organic layer was separated, concentrated and re-dissolved in ether (200 ml). The aqueous layer was extracted with ether (2 x 200 ml). The combined organic layers were washed with IN aq. NaOH (4 x 100 ml), brine, and dried over sodium sulfate. Removal of solvent gave crude product which was purified by column chromatography using 2 to 5% ethyl acetate in hexane as solvent to give desired alcohol 36.2 g (90 %, e.e. >95%). The alcohol (36.2 g) was crystallized from hexane (80 ml) to obtain R-l-(2-bromo-4-chloro- phenyl)-2,2,2-trifiuoro-ethanol 28.2 g (70 %; 99-100 % e.e.). 1H-NMR (400 MHz, CDCl3) δ (ppm) 5.48 (m, IH), 7.40 (d, IH), 7.61 (d, 2H). Step 3: Synthesis of R-l-r4-chloro-2-(3-methyl-pyrazol-l-vπ-phenyl1-2.2.2-trifluoro- ethanol. R-l-(2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-ethanol (15.65g, 54.06 mmol), 3- methylpyrazole (5.33 g, 65 mmol), CuI (2.06 g, 10.8 mmol), K2CO3 (15.7 g, 113.5 mmol), (lR,2R)-N,N’-dimethyl-cyclohexane-l,2-diamine (1.54 g, 10.8 mmol) and toluene (80 ml) were combined in a 250 ml pressure tube and heated to 1300C (oil bath temperature) for 12 h. The reaction mixture was diluted with ethyl acetate and washed with H2O (4 x 100 ml), brine, and dried over sodium sulfate. Removal of solvent gave a crude product, which was purified by ISCO column chromatography using 5-10 % ethyl acetate in hexane as solvent to get R-I- [4-chloro-2-(3-methyl-pyrazol-l-yl)-phenyl]-2,2,2-trifluoro-ethanol (13.5 g; 86 %). 1H-NMR (400 MHz, CDCl3): δ (ppm) 2.30(s, 3H), 4.90(m, IH), 6.20(s, IH), 6.84(d, IH), 7.20(s, IH), 7.30(d, IH), 7.50(d, IH).
Step 4: Synthesis of (S)-2-Amino-3- r4-(2-amino-6- (R-I- r4-chloro-2-(3-methyl- pyrazol- 1 -ylVphenyl~|-2,2.,2-trifluoro-ethoxy| -pyrimidin-4-yl)-phenyU -propionic acid ethyl ester. R-l-[4-chloro-2-(3-methyl-pyrazol-l-yl)-phenyl]-2,2,2-trifluoro-ethanol (17.78 g, 61.17 mmol), (S)-3-[4-(2-amino-6-chloro-pyrimidine-4-yl)-phenyl]-2-tert- butoxycarbonylamino-propionic acid (20.03 g, 51 mmol), 1,4-dioxane (250 ml), and Cs2CO3 (79.5 g, 244 mmol) were combined in a 3-necked 500 ml RB flask and heated to 1000C (oil bath temperature) for 12-24 h. The progress of reaction was monitored by LCMS. After the completion of the reaction, the mixture was cooled to 600C, and water (250 ml) and THF (400 ml) were added. The organic layer was separated and washed with brine (150 ml). The solvent was removed to give crude BOC protected product, which was taken in THF (400 ml), 3N HCl (200 ml). The mixture was heated at 35-400C for 12h. THF was removed in vacuo. The remaining aqueous layer was extracted with isopropyl acetate (2x 100 ml) and concentrated separately to recover the unreacted alcohol (3.5 g). Traces of remaining organic solvent were removed from the aqueous fraction under vacuum.
To a IL beaker equipped with a temperature controller and pH meter, was added H3PO4 (40 ml, 85 % in water) and water (300 ml) then 50 % NaOH in water to adjust pH to 6.15. The temperature was raised to 58°C and the above acidic aqueous solution was added dropwise into the buffer with simultaneous addition of 50 % NaOH solution in water so that the pH was maintained between 6.1 to 6.3. Upon completion of addition, precipitated solid was filtered and washed with hot water (50-600C) (2 x 200 ml) and dried to give crude (S)-2- amino-3-[4-(2-amino-6-{R-l-[4-chloro-2-(3-methyl-pyrazol-l-yl)-phenyl]-2,2,2-trifluoro- ethoxy}-pyrimidin-4-yl)-phenyl} -propionic acid (26.8 g; 95 %). LCMS and HPLC analysis indicated the compound purity was about 96-97 %. To anhydrous ethanol (400 ml) was added SOCl2 (22 ml, 306 mmol) dropwise at 0-
5°C. Crude acid (26.8 g ) from the above reaction was added. The ice water bath was removed, and the reaction mixture was heated at 40-450C for 6-12h. After the reaction was completed, ethanol was removed in vacuo. To the residue was added ice water (300 ml), and extracted with isopropyl acetate (2 x 100 ml). The aqueous solution was neutralized with saturated Na2CO3 to adjust the pH to 6.5. The solution was extracted with ethyl acetate (2 x 300 ml). The combined ethyl acetate layer was washed with brine and concentrated to give 24 g of crude ester (HPLC purity of 96-97 %). The crude ester was then purified by ISCO column chromatography using 5 % ethanol in DCM as solvent to give (S)-2-amino-3-[4-(2- amino-6- (R- 1 -[4-chloro-2-(3-methyl-pyrazol- 1 -yl)-phenyl]-2,2,2-trifluoro-ethoxy} – pyrimidin-4-yl)-phenyl} -propionic acid ethyl ester (20.5g; 70 %; HPLC purity of 98 %). LCMS M+l = 575. 1H-NMR (400 MHz, CD3OD): δ (ppm) 1.10 (t, 3H), 2.25 (s, 3H), 2.85 (m, 2H), 3.65 (m, IH), 4.00 (q, 2H), 6.35 (s, IH), 6.60 (s, IH), 6.90 (m, IH), 7.18 (d, 2H), 7.45 (m, 2H), 7.70 (d, IH), 7.85 (m, 3H).
PATENT
WO 2011056916
https://www.google.com/patents/WO2011056916A1?cl=en
PATENT
WO 2010065333
CLIP,……..PL CHECK ERROR



REFERENCES
Kulke, M.H.; Hoersch, D.; Caplin, M.E.; et al.
Telotristat ethyl, a tryptophan hydroxylase inhibitor for the treatment of carcinoid syndrome
J Clin Oncol 2017, 35(1): 14
| WO2010056992A1 * | Nov 13, 2009 | May 20, 2010 | The Trustees Of Columbia University In The City Of New York | Methods of preventing and treating low bone mass diseases |
| US7709493 | May 20, 2009 | May 4, 2010 | Lexicon Pharmaceuticals, Inc. | 4-phenyl-6-(2,2,2-trifluoro-1-phenylethoxy)pyrimidine-based compounds and methods of their use |
| US20090088447 * | Sep 25, 2008 | Apr 2, 2009 | Bednarz Mark S | Solid forms of (s)-ethyl 2-amino-3-(4-(2-amino-6-((r)-1-(4-chloro-2-(3-methyl-1h-pyrazol-1-yl)phenyl)-2,2,2-trifluoroethoxy)-pyrimidin-4-yl)phenyl)propanoate and methods of their use |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US9199994 | Sep 5, 2014 | Dec 1, 2015 | Karos Pharmaceuticals, Inc. | Spirocyclic compounds as tryptophan hydroxylase inhibitors |
| US9512122 | Sep 1, 2015 | Dec 6, 2016 | Karos Pharmaceuticals, Inc. | Spirocyclic compounds as tryptophan hydroxylase inhibitors |
///////////telotristat ethyl, fast track designation,priority review,orphan drug designation, Xermelo , Woodlands, Texas-based, Lexicon Pharmaceuticals, Inc, fda 2017, LX 1606, LX 1032
O=C(OCC)[C@@H](N)Cc1ccc(cc1)c2cc(nc(N)n2)O[C@H](c3ccc(Cl)cc3n4ccc(C)n4)C(F)(F)F
O=C(OCC)[C@@H](N)CC1=CC=C(C2=NC(N)=NC(O[C@H](C3=CC=C(Cl)C=C3N4N=C(C)C=C4)C(F)(F)F)=C2)C=C1.O=C(O)CNC(C5=CC=CC=C5)=O


[…] TELOTRISTAT ETHYL […]
LikeLike
[…] TELOTRISTAT ETHYL […]
LikeLike