

| Record ID | Title | Status | Phase |
|---|---|---|---|
| NCT03041311 | Carboplatin, Etoposide, and Atezolizumab With or Without Trilaciclib (G1T28), a CDK 4/6 Inhibitor, in Extensive Stage Small Cell Lung Cancer (SCLC) | Recruiting | 2 |
| NCT02978716 | Trilaciclib (G1T28), a CDK 4/6 Inhibitor, in Combination With Gemcitabineand Carboplatin in Metastatic Triple Negative Breast Cancer (mTNBC) | Recruiting | 2 |
| NCT02514447 | Trilaciclib (G1T28), a CDK 4/6 Inhibitor, in Patients With Previously Treated Extensive Stage SCLC Receiving Topotecan Chemotherapy | Recruiting | 2 |
| NCT02499770 | Trilaciclib (G1T28), a CDK 4/6 Inhibitor, in Combination With Etoposide and Carboplatin in Extensive Stage Small Cell Lung Cancer (SCLC) | Active, not recruiting | 2 |
Home » Posts tagged 'Inc.'
Tag Archives: Inc.
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
CIFORADENANT
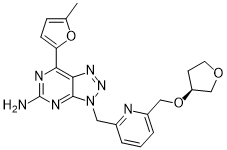

CIFORADENANT
1202402-40-1
Chemical Formula: C20H21N7O3
Molecular Weight: 407.434
CPI-444, CPI 444, CPI444, V81444, V-81444, V 81444,
UNII 8KFO2187CP
Corvus Pharmaceuticals, Inc. PHASE 1
(S)-7-(5-methylfuran-2-yl)-3-((6-(((tetrahydrofuran-3-yl)oxy)methyl)pyridin-2-yl)methyl)-3H-[1,2,3]triazolo[4,5-d]pyrimidin-5-amine
| 3H-1,2,3-TRIAZOLO(4,5-D)PYRIMIDIN-5-AMINE, 7-(5-METHYL-2-FURANYL)-3-((6-((((3S)-TETRAHYDRO-3-FURANYL)OXY)METHYL)-2-PYRIDINYL)METHYL)- |
(73 S)-15 -methyl-6-oxa-2(7,3)-[1,2,3]triazolo[4,5- d]pyrimidina-4(2,6)-pyridina-1(2)-furana-7(3)- oxolanaheptaphan-25 -amine adenosine receptor antagonist
Ciforadenant, also known as CPI-444 and V81444, is an orally administered antagonist of the adenosine A2A receptor. Upon oral administration, CPI-444 binds to adenosine A2A receptors expressed on the surface of immune cells, including T-lymphocytes, natural killer (NK) cells, macrophages and dendritic cells (DCs). This prevents tumor-released adenosine from interacting with the A2A receptors on these key immune surveillance cells, thereby abrogating adenosine-induced immunosuppression in the tumor microenvironment.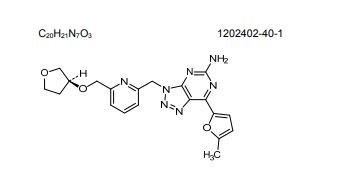
Ciforadenant is an antagonist of adenosine A2A being developed by Corvus , under license from Vernalis , for the oral treatment of advanced solid tumor; the company is also developing the drug in combination with atezolizumab , for non-small-cell lung cancer.
In 2015, Vernalis licensed the exclusive rights of the product for use of all therapeutic application to Corvus.
Synthesis
WO 2009156737

PATENT
WO 2009156737
US 8450328
WO2017112917
WO 2018175473
WO 2018009972
WO 2018049271
WO 2018022992
PATENT
PATENT
WO-2018183965
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018183965&redirectedID=true
EXAMPLES
Reaction Scheme 1
[0314] Referring to Reaction Scheme 1 , the process to manufacture triazolo[4,5]pyramidine derivatives and intermediates thereof in accordance with the present disclosure, such as the compound known as CPI-444, consists of three chemical steps and uses starting materials known as CP-55, CP-56 and CP-60. The intermediate known as CP-57 is formed at step la without isolation (telescoped) and taken to the next step to form the compound known as CP-58 at step lb. Suzuki coupling using CP-60 during step 2 generates crude CPI-444 which undergoes crystallization during step 3 to form CPI-444.
[0315] Previously described processes for making triazolo[4,5]pyramidine derivatives and intermediates thereof utilized a compound known as CP-59:
[0316] Moreover, such previously described process utilize triethylamine which takes a longer time for the layers to separate where excessive rag layer is observed during phase separation. [0317] The present inventors unexpectedly and surpisingly found that the replacement of CP-59 with CP-60 improved ease of handling and improved process efficiency. In addition, the present inventors unexpectedly and surpisingly found that the use of potassium carbonate (K2CO3) during step 2 improves the phase separation and minimizes rag layer formation upon reaction completion. Finally, Step 3 employs the use of thermocycler in order to facilitate the removal of residual solvents such as isopropyl alcohol.
[0318] Accordingly, the processes in accordance with the teachings of the present disclosure are an improvement over, and are more suitable for commercial scale-up, than processes previously described.
[0319] Starting material (C-55) is commercially available through Astatech, Inc., Keystone Business Park, 2525 Pearl Buck Road, Bristol, PA, 19007, USA; or Suven, SDE Serene Chambers, Road No.5, Avenue 7 Banjara Hills, Hyderabad, 500034, India.
[0320] CP-60 is commercially available through ARK Pharma, Inc., 3860 North Ventura Drive, Arlington Heights, IL, 60004, USA; or Boron Technology Institute, Road No. 2, Building No. 10, room No. 259, Haidian District, Beijing, China.
EXAMPLE 1. Preparation of CP-56
Reaction Scheme 1
Boc20, CbzCI
[0321] Preparation of Dimethyl pyridine-2,6-dicarboxylate:
Pyridine-2,6-dicarboxylic acid (900g, leq) is suspended in methanol(5 volume) and added H2SO4. (19g). The mixture is heated to reflux for approximately 4hr. After reaction completion, the mixture is cooled to 5- 10°C to allow the solids to precipitate. The solids are stirred for an additional hour. The solids are collected by filtration. The wet-cake is re-dissolved in DCM (3 volume) and extract in sequence with an aqueous saturated solution of NaHC03 (2 Volume) followed by with a 5% brine solution (2 Volume). The organic layer is concentrated to dryness to obtain dimethyl pyridine-2,6-dicarboxylate; 914.85g, purity 100%, yield 87.%.
[0322] Preparation of pyridine-2,6-diyldimethanol:
Dimethyl pyridine-2,6-dicarboxylate (885g, leq) is dissolved in EtOH (4425g, 5 Volume) at room temperature. The NaBH4 (341 g, 2eq) is added slowly to the reaction while keeping the internal temperature below 30°C using an ice bath. The reaction is heated to 35°C for approximately 2hrs. After reaction completion, the mixture is cooled to room temperature and adjusted with 32% HCl solution to pH value of approximately 2.5. The mixture is stirred for
2hrs to allow the solids to precipitate. The mixture is then adjusted pH value of approximately 9 using 30% NaOH solution while maintaining an internal temperature below 30°C and stirred at room temperature for about 30 min. The solids are removed by filtration. The filtrate is concentrated at 50°C. The concentrated residual is suspended with isopropanol (4160g, 8 vol)
/water (416g, 0.8 vol) and heated to 70°C for about lhr. The solution is then cooled to room
temperature and stirred for 2hr before cooling to 5-10°C for 30min. The un-dissolved solids are
removed by filtration. The filtrate is concentrated at 50°C. The concentrated residue is charged
with dichloromefhane (2700g, 5vol) and heated to 40 °C for 30min. The suspension is cooled to 5-
10°C and stirred for 30mins. The solid is collected by filtration and dried under vacuum at 40°C to obtain pyridine-2,6-diyldimethanol; 540.77g, purity 100%, yield 85.86%.
[0323] Preparation of 2,6-6 s(chloromethyl)pyridine:
2,6-bis(chloromethyl)pyridine (400g, leq) is suspended in DCM (2000g) and then cooled to 10- 15°C. Thionyl chloride (SOCb; 775g, 3eq) is charged with CH2CI2 (775g) and then added drop- wised into the reaction vessel while maintaining the internal temperature below 20 °C. The reaction is then warmed to room temperature and held for approximately 2hrs. After reaction completion, the 15% aqueous solution of a2C03 (9038g) is pre-cooled to 10-15°C before charging the reaction mixture into the carbonate solution while maintaining internal temperature below 20 °C. The mixture is stirred until gas-evolution is no longer observed. The organic layer is extracted with water (2 x 3200g) and then concentrated at 50°C to a crude product. The concentrated crude is purified by recrystallization using heptane (946g). The mixture is cooled to 5-10°C for 30min. The solid is collected by filtration and wet-cake is washed with heptane and dried at 40°C under vacuum to obtain 2,6-6zs(chloromethyl)pyridine; 442.6g, purity 100%, yield 87.0%.
[0324] Preparation of (3r,5r,7r)-l-((6-(chloromethyl)pyridin-2-yl)methyl)-l,3,5,7-tetraazaadamantan-l-ium:
2,6-to(chloromethyl)pyridine (420g, leq) is dissolved in CH2CI2 (8400g), HMTA (336g, leq) is added into the reaction vessel. The reaction is heated to approximately 40 °C for about 3hrs. Additional HMTA (168g, 0.5eq) is added into the reaction mixture and stirred overnight at room
temperature. The product is collected by filtration. The wet-cake is washed with CthCkand dried under vacuumat 50°C to obtain (3r,5r,7r)-l -((6-(chloromethyl)pyridin-2-yl)methyl)- 1 ,3, 5,7-tetraazaadamantan- 1 -ium; 730g, purity 97.01%, yield 96.58%.
[0325] Preparation of (6-(chloromethyl)pyridin-2-yl)methanamine dihydrochloride:
(3r,5r,7r)- 1 -((6-(chloromethyl)pyridin-2-yl)methyl)- 1 ,3 ,5 ,7-tetraazaadamantan- 1 -ium (730g, leq) is suspended in EtOH (4380g) before charging 37% HC1 (159g). The mixture is heated to approximately 60 °C for about lhr. After reaction completion, it is cooled to 25°C. MTBE
(1200g) is charged into the suspension. The suspension is then stirred for about 30 min and cooled to 5-10°C for about lhr. The solids are collected by filtration and washed with MTBE and dried at 50°C under vacuum to obtain (6-(chloromethyl)pyridin-2-yl)methanamine dihydrochloride; 449.56g (after assay correction), purity 98.15%, yield85.23%.
[0326] Preparation of tert-butyl ((6-(chloromethyl)pyridin-2-yl)methyl)carbamate:
(6-(chloromethyl)pyridin-2-yl)methanamine dihydrochloride [422.56g (after assay correction), leq] is dissolved in CH2CI2 (5600g) and pre-cooled to 10-15°C. K2CO3 (1632g) pre-dissolved in water (4000g) is charged into the reaction solution solution. The mixture is stirred for about lOmin and then cooled to 10-15°C. Boc-anhydride (603g) is pre-dissolved in CH2CI2 (1808g) before charging into the reactor. The mixture is warmed to room temperature and held for about an hour. After reaction completion, the organic layer is extracted with water (4000g), The organic layer is concentrated to dryness at 50 °C to obtain tert-butyl ((6-(chloromethyl)pyridin-2-yl)methyl)carbamate; 382.93g [after assay correction); purity 99.01%; yield 81%].
[0327] Preparation of tert-butyl ((6-(iodomethyl)pyridin-2-yl)methyl)carbamate:
tert-butyl ((6-(chloromethyl)pyridin-2-yl)methyl)carbamat [ 382.93g (after assay correction) , leq] is dissolved in THF (1 150) and Nal (720g) is added, the reaction is at room temperature for approximately 4hr. After reaction completion, excess Nal and NaCl are filtered off and the filtrate is concentrated at 40°C. The concentrated residue is re-dissolved in ethyl acetate (2300g) and extracted with water (2900g), the organic layer is washed with 10% aqueous solution of Na2S203 (2600g) followed by 5% brine solution (2900g). The organic layer is concentrated to a residue. The residue is re-dissolved in ethyl acetate (4200g), and then filtered. The filtrate is oncentrated and taken up in ethyl acetate (765g) and stirred at room temperature for about 2hr before slowly adding heptane (380g). The solids are filtered and dried at 50°C under vacuum to
obtain tert-butyl ((6-(iodomethyl)pyridin-2-yl)methyl)carbamate; 440g; purity 100%, Yield 85%.
[0328] Preparation of tert-butyl (S)-((6-(((tetrahydrofuran-3-yl)oxy)methyl)pyridin-2-yl)methyl)carbamate:
A solution of t-BuOK (113g in THF (1.1 kg) is pre-cooled to 5- 10°C, before charging asolutionof (S)-tetrahydrofuran-3-ol (166g) in THF (220g). The mixture is stirred at room temperature for about lhr. A solution of tert-butyl ((6-(iodomethyl)pyridin-2-yl)methyl)carbamate (440g, leq) in THF (880g) is pre-cooled to 10-15°C before. The tetrahydrofuranyl solution is slowly charged into reaction solution while maintaining an internal temperature below 1 °C. After about 1 hour another solution of pre-cooled solution of t-BuOK (50g) and (S)-tetrahydrofuran-3-ol (66g) in THF (405g) kg) is slowly added into reaction mixture while maintaining internal temperature below 10 °C. The mixture is stirred at about 10 °C for approximately 1 hour. After reaction completion, the mixture is quenched with water (2200g) and extracted with toluene (4400g). The organic layer is washed with 5% brine (2x 2200g). The organic layer is concentrated to dryness at 50°C under vacuum to obtain tert-butyl (S)-((6-(((tetrahydrofuran-3-yl)oxy)methyl)pyridin-2-yl)methyl)carbamate; 389g, purity 89.63%, yield 105%.
[0329] Preparation of CP-56 free base:
tert-butyl (S)-((6-(((tetrahydrofuran-3-yl)oxy)methyl)pyridin-2-yl)methyl)carbamate (389g, leq) is dissolved in CH2CI2 (1556g) and pre-cooled to 0-5°C before charging drop-wise methanesulfonic acid ( MSA; 600g) into the reaction solution while maintaining internal temperature below 20°C. The mixture is warmed to room temperature and hold for about lhr. After reaction completion, water (389g) is added and cooled to 5-10°C. 30% NaOH is charged to adjust the reactor pH to approximately 12.5. The mixture is stirred for about 30 min before extracting with CH2CI2 (1556g). The organic layer is collected and extracted with an aqueous saturated solution of brine (584g). The organic layer is concentrated under vacuum. The residue is re-dissolved in toluene (1560g andthenconcentrated. The concentrated residue is re-dissolved in toluene (1560g) and then filtered. The filtrate is concentrated to dryness at 50°C under vacuum to obtain CP-56 free base; 221g (after assay correction), purity 91%, yield 84.23%.
[0330] Preparation of CP-56:
CP-56 free base (22 lg (after assay correction), leq) is dissolved in MeOH (260g) and EtOH (1300g) and then cooled about 15°C. Oxalic acid (47), pre-dissolved in MeOH (1 lOg is charged into reaction mixture. The reaction is at 15-20°C for 3hr. The mixture is cooled to 0-5°C and
stirred for about an Ihr. The solid is collected by filtration and the wet-cake is washed with EtOH (390g). The solid is dried under vacuum at 50°C to obtain CP-56 crude. Crude CP-56 is re-crystallized from isopropanol (865g) and H20 (lOOg). The mixture is heated to about 70°C to obtain a solution. The solution is slowly cooled to 50°C for Ihr. The mixture is cooled to 0-5°C for about another Ihr. The solid is filtered and washed with isopropanol. The wet-cake is dried at 50°C under vacuum to obtain CP-56; 164g, purity 99%, yield 95%.
[0331] Alternatively, CP-56 can be formed using the following process:
Reaction Scheme 2
7 8 9
[0332] Preparation of Dimethyl pyridine-2,6-dicarboxylate (compound 2):
Charge diacid (1; 628g) into reactor containing methanol (2Kg) and heat to reflux. After reaction completion the reaction is cooled to 30 C and stirred. The wet-cake is filtered and washed with methanol (500g). The wet-cake is dried under vacuum at about 55 °C to obtain diester (680 g, purity >99%; yield 85%).
[0333] Preparation of 6-(hydroxymethyl)picolinamide (compound 4):
Charge diester (2; 600 g) into reactor containing methanol (1.8 kg) and tetrahydrofuran (1.2 kg). Charge slowly sodium borohydride ( aBH4; about 130 g) into the reaction solution while maintaining an internal temperature below 30 °C. After reaction completion aqueous hydrochloric acid (about 350 g of 32% HC1) is charged into the reaction solution. The mixture is concentrated and then charged with dichloromethane (1.8 kg). The organic solution is extracted with water (600 g) and then concentrated to obtain the crude product (3). Crude 3 was dissolved in methanol (1.3 kg) and then charge ammonium hydroxide (20%; 1.3 kg). The solution was stirred until reaction completion before concentrating solution. The residue was taken up in water (600g) and heated to about 60 °C before cooling to 0 °C. The wet-cake was filtered, washed with water and dried in vacuum oven to obtain 6-(hydroxymethyl)picolinamide (about 220 g, >99% purity).
[0334] Preparation of 6-(chloromethyl)picolinonitrile (compound 5):
Charge 6-(hydroxymethyl)picolinamide (about 220 g) into a rector containing acetonitrile (450 g). Charge POCb (519 g and agitate at about 70 °C. After reaction completion the solution is
cooled to about 30 °C before slowly charging into a pre-cool (about 10 °C) reactor with water
(305 g). Charge toluene (1.4 kg) to extract the solution mixture. The toluene phase is washed in sequence with 20 % NaOH (600 g), saturated NaHC03 (300 g) and water (300 g). Toluene is concentrated to obtain crude Cl-nitrile, 5. Isopropyl alcohol (400 g) is charged to dissolve the wet-cake at about 45 °C before cooling to about 0 °C. The wet-cake was filtrated and washed with heptane (150 g) and dried in vacuum oven to obtain 6-(chloromethyl)picolinonitrile (180 g; > 99%.
[0335] Preparation of (S)-6-(((tetrahydrofuran-3-yl)oxy)methyl)picolinonitrile (compound 7):
Charge Cl-nitrile (180 g) into a rector containing THF (540 g). Charge Nal (185.7 g) to the reactor and stirred at 50 °C. After reaction completion, the reactor is cooled to 0 °C. In another
reactor, charge t-BuOK (145.6 g) and THF (320 g). Add (S)-tetrahydrofuran-3-ol (31 1.9 g) into the reactor while maintaining internal temperature below 50 °Cto deprotonate the alcohol. Stir
until t-BuOK dissolves. Add THF-OK / THF solution into 6-(iodomethyl)picolinonitrile solution (compound 6) while maintaining internal temperature below 10 °C. Stir at room
temperature until reaction completion. Concentrate the solution to remove THF solvent. Add
ethyl acetate (630 g) and wash by water (420 g). Extract water phase by ethyl acetate (630 g). Combine organic layer and concentrate to obtain oil crude 374 g. The residue was distilled under vaccum (P=3~4 torr, internal temperature 174 °C to 188 °C) to obtain (S)-6-
(((tetrahydrofuran-3-yl)oxy)methyl)picolinonitrile (compound 7) as an oily product (204g, >96% purity; 74% yield).
[0336] Preparation of (S)-(6-(((tetrahydrofuran-3-yl)oxy)methyl)pyridin-2-yl)methanamine (compound 9):
Charge (S)-6-(((tetrahydrofuran-3-yl)oxy)methyl)picolinonitrile (180 g) into a rector containing MeOH (1620 g). Charge NaOMe (95.3 g) to the reactor and stirred for 30 min at 30 °C until
reaction completion. The methyl (S)-6-(((tetrahydrofuran-3-yl)oxy)methyl)picolinimidate solution (compound 8) was transferred to hydrogenation apparatus containing 50% Ni (60 g). Purge with N2 and then increase the H2 pressure. Under H2 pressure of 5 kg / cm2 and temperature of 30 °C until reaction completion. The reaction is filtered through celite. The filtrate is concentrated. Toluene is charged (1kg) and then concentrated. Then add toluene (1000 g) and filter to remove salt by-products. The filtrate was concentrated to obtain the oil residue of (S)-(6-(((tetrahydrofuran-3-yl)oxy)methyl)pyridin-2-yl)methanamine (136 g; 85% yield, assay 80%, >91% purity).
[0337] Preparation of CP-56:
Charge (S)-(6-(((tetrahydrofuran-3-yl)oxy)methyl)pyridin-2-yl)methanamine (170 g) into a rector containing isopropyl alcohol (600 g). Set internal temperature of 75 °C. In another reactor,
charge oxalic acid (41.1 g) and water (60 g) and heat solution. Add oxalic acid solution into
CP-56 free-base solution. Cool to 30 °C for about 4 hours and agitate. The wet-cake was filtered
and washed with isopropyl alcohol (175 g) and dried under vacuum drying with heat to obtain crude CP-56 (136.2 g). Charge CP-56 crude (123 g) into a rector containing methanol (1295 g). Stir until CP-56 was dissolved completely. Filter through celite to remove insoluble salt. The filtrate is concentrated. Charge isopropyl alcohol (500 g) and water (50 g) to dissolve CP-56 using heat. Cool to about 30 °C for about 3 hours and stir. The wet-cake was filtrated and
washed by isopropyl alcohol (165 g) and dried under vacuum drying with heat to obtain CP-56 (1 13.4 g. purity = >99 %, > 99% ee).
EXAMPLE 4. Preparation of CPI-444
CP-58 CP-60
C15H16CIN702 CPI-444
1H-17BO3
W: 361 .79 MW: 208.06 C20H21N O3
MW: 407.43
[0349] It is to be noted that other Pd coupling reagents can also be used such as Pd(PPh3)4 or Pd(PPh3)2Cl2.
[0350] A solution of CP-58 (30.0 g, 1 equiv.), CP-60 (approximately 20.8 g, 1.2 equiv.), in THF (approximately 180 mL), K2C03 (approximately 17.5 g), Pd(dtbpf)Cl2(approximately 337 mg), and water (approximately 100 mL) were stirred and heated to about 60 °C until reaction completion. The reaction was cooled to about 50 °C and the layers were allowed to separate. The aqueous layer was removed and back extracted with THF (approximately 30 mL). The THF layers were combined and water (approximately 450 ml) was added to precipitate out crude CPI-444. The slurry was cooled to about 20 °C and stirred for approximately 60 min and the slurry was filtered. The cake was washed in sequence with water (approximately 120 ml) and 2-propanol (approximately 30 ml). The wet-cake was dried in the vacuum oven to provide an off- white solid (29.74 g, 88% yield) with a purity of 98.5 %. Crude CPI-444 conforms to reference.
-444 can be prepared by the following process:
EDA and DAP are used to remove Palladium during CPI-444 formation.
[0352] The solution of CP-58 (10 g), CP-60 (6.9 g) , Pd(dtbpf)C12 (approx. 0.0015 mol eq) and K2C03 (5.8 g) in THF (6V) and H20 (3V) is heated to approximately 60 °C. The reaction is complete after approximately 30 minutes. The solution is cooled to 50 °C and aqueous layer is separated. The aqueous layer is extracted with THF (9 mL); the THF layer is added to organic solution. The organics are cooled to 40 °C, 1 ,3-diaminopropane (DAP; approximately 50 g) or ethylene diamine (EDA; approximately 45 g) is added and the mixture stirred for 1 hour. H20 (15V) is added to the organic layer over 10 min. The slurry is cooled to 20 °C for 2 hours, and stirred for an additional 1 hour. The slurry is filtered and washed with H20 (2V x 2) and z‘PrOH (IV). CPI-444 wet-cake is dried at 50 °C under full vacuum. (Yield = 90 %; purity > 99.0%).
[0353] Alternatively, CPI-444 can be prepared by the following process:
using cysteine in TNF to remove Palladium during CPI-444 formation
[0354] CP-58 (1 kg), K2C03 (0.58 kg), water (3 kg), CP-60 (0.69 kg), and THF (5.3 kg),
Pd(dtbpf)Cb (3 g). The solution is heated to 60 °C. The reaction is complete after approximately 30 minutes. Charge THF (4.5 kg) and cool to 50 °C. The aqueous layer is separated. The organic layer is charged with cysteine (0.32 kg) and water (5 kg). The mixture is agitated. NH4OH (1.1 kg) is charged to the reaction mixture and agitate for approximately 15 minutes. The layers are allowed to separate and the lower aqueous layer is separated. The organic layer is charged with cysteine (0.32 kg) and water (5 kg). The mixture is agitated. NH4OH (1.1 kg) is charged to the reaction mixture and agitate for approximately 15 minutes. The layers are allowed to separate and the lower aqueous layer is separated. THF is distilled to approximately 7 volumes under atmospheric pressure. The solution is cooled to 50 °C before charging NH4OH (0.5 kg) and agitate for 30 min. Water (14.5 kg) is charged while maintaining the internal temperature >40 °C. The reactor is cooled to 20 °C for 2 hours and hold for an additional 1 hour. CPI-444 is filtered and washed with water followed by isopropanol. CPI-444 wet-cake is dried under vacuum at 50 °C. Purity > 99%, yield 85%.
EXAMPLE 5. Removal of Residual Palladium With Biocap Filter Cartridge
[0355] A mixture of CPI-444 crude (16.00 g), THF (approximately 190 ml), L-cysteine
(approximately 8 g), and H20 (approximately 90 ml) were mixed and heated to a solution at about 60 °C for 1 hour. A solution of 28% NH OH (approximately 20 ml) was added and heated for an additional 15 minutes. The agitation was turned off to allow the layers allowed to settle. The aqueous layer was removed; the THF layer was washed with brine solution (approximately 15 ml). The combined aqueous solutions were back extracted with THF (approximately 15 ml). A 3M Biocap filter (BC0025LR55SP; available from 3M) was pretreated with THF (approximately 150 ml) at about 50 °C. The combined organic layers were recirculated through the Biocap at about 10 ml/min for approximately 3 hours and then filtered forward. The Biocap filter was rinsed with THF (approximately 130 ml) at about 50 °C. The combined filtrates were concentrated. Water
(approximately 80 ml) was added, and distilled to remove residual THF. 2-Propanol (approximately 1 10 ml) was added to the slurry, and the mixture was heated to a solution. The solution was cooled to 20 °C and water (approximately 240 ml) was added. The slurry was performed in series by heating to about 55 °C and held that that temperature for approximately 30 minutes, cooled to 20 °C over 30 minutes, and held at 20 °C for 30 minutes. This heating cycle was repeated two more. The slurry was then held at 20 °C for approximately 12 hours. The slurry was filtered, and the product was washed with water (approximately 300 ml). The wet cake (about 23 g) was dried in the vacuum oven to obtain an off white solid (13.6 g; 85% yield;99.9% purity; Pd = 25 ppm).
[0356] Reprocess of step 4. AFC-825-106
[0357] CPI-444 (16.02 g, AFC-825-48) and THF (approximately 280 ml) were charged to a flask and heated to about 50 °C for about 30 minutes to obtain a solution. A 3M Biocap filter
(BC0025LR55SP) was pretreated with THF (approximately 150 ml) at about 50 °C . The CPI-444 solution was passed through the Biocap at aboutl O ml/min. The Biocap filter was rinsed with THF (approximately 130 ml) at about 50 °C. The combined filtrates were transferred to a reactor and concentrated. Water (approximately 80 ml) was added, and distilled to remove residual THF solvent. 2-Propanol (approximately 1 10 ml) was added to the slurry and heated to about 65 °C to obtain a solution. The solution was cooled to about 20 °C before adding water (approximately 240 ml). The slurry was heated to 55 °C over 30 minutes, held at 55 °C for 30 minutes, cooled to 20 °C over 30 minutes, and held at 20 °C for 30 minutes. This heating cycle was two more times. The slurry was then held at 20 °C for 12 hours. The slurry was filtered, and the product was washed with water (approximately 300 ml). The wet cake (26.6 g) was dried in the vacuum oven overnight to obtain 15 as a white solid (95% yield; 99% purity; Pd = 5 ppm).
EXAMPLE 6. Removal of Residual Palladium With Darco KB-G
Crude CPI-444
CPI-444 Drug Substance
[0358] Crude CPI-444 (475 g, 1.17 mol, 1.00 eq), 2-MeTHF (1 1.9 L, 25.0 vol) and WFI water (2.6 L, 5.5 vol) were charged to a 19 L jacketed reactor. The mixture was mechanically agitated under a nitrogen blanket. Nitrogen was bubbled through the solution for 20 minutes. L-Cysteine (242 g, 1.99 mol, 1.71 eq) was then charged. The solution in the reactor was heated to 55±5 °C. Upon reaching 50 °C, the reaction mixture was stirred for 1 hour. 28-30% NH4OH (594 mL, 1.25 vol) was charged via addition funnel, and then the reaction mixture was stirred for 15 min. Agitation was stopped and the reaction was allowed to separate for 1 hour. The aqueous layer was removed. The organic layer was allowed to cool to ambient. The organic layer was filtered and the frit was washed with 2-MeTHF (618 mL, 1.3 vol). The organics were concentrated off by rotary evaporation. WFI water (2.42 L, 5.1 vol) and IPA (2.38 L, 5.0 vol) were used to charge the concentrated slurry to a clean 19 L jacketed reactor under N2. The mixture was heated to 65±5 °C, and then was stirred for 1 hour to obtain solution. Darco KB-G activated carbon (71.3 g, 15 wt%) was charged. The reactor was heated to 75±5 °C and stirred for 15 hours. A I L pocket filter was prepared with filter cloth and a heating jacket and heated to 70±5 °C. Reactor contents were filtered through the pocket filter using N2 pressure. The pocket filter was rinsed with a mixture of IPA/WFI water (1 : 1, 950 mL, 2 vol) followed by a mixture of IPA/WFI water (1 : 1, 1.90 L, 4 vol) and IPA/WFI water ( 1 : 1 , 1.90 L, 4 vol). Inside a 22 L three neck round bottom flask the filtrates were mechanically agitated under a N2 blanket. WFI water (7.13 L, 15 vol) was slowly added via addition funnel over 1 h at ambient temperature, and aged for 1 h. The slurry was heated to 55±5 °C and maintained the temperature for 30 min. This heating and subsequent cooling were repeated twice more. After reaching ambient
temperature the final time, the mixture was stirred for at least 2 hours. The reaction mixture was filtered and the reactor rinsed with WFI water (2.38 L, 5.0 vol, 3x). The cake was dried under N2 for 30 minutes and then transferred to a glass dish. The material was dried under full vacuum at 55±5 °C. The desired product was obtained 368.1 g (77%) as light yellow solids. This material was 99.6% pure by HPLC and had a Pd content of 3.6 ppm.
EXAMPLE 7. Removal of Residual Palladium With Polymer-Bound Thiol (SiST)
[0359] Crude CPI-444 (24.48 g, pd = 1267 ppm) and THF (244.8 mL, 10 vol) were charged to a 500 mL 4-necked flask fitted with mechanical agitation, a condenser with nitrogen balloon and a thermometer. The slurry was heated to 60 °C for 20 minutes and then slowly cooled to 45 °C. SiST (36.72 g) was added to the solution and the mixture was stirred at 42 °C for 14 h. The mixture was filtered and washed by THF (24 mL, 1 vol, twice; Pd= 13.12 ppm). H20 (120 mL, 5 vol) and IPA (120 mL, 5vol) were charged to the flask. The slurry was heated to 70 °C and maintained for 1 h (the slurry became solution). The solution was slowly cooled to room temperature and the slurry was added H20 (360 mL, 15 vol) and heated to 55 °C for 1 h. The slurry was cooled to room temperature and then heated to 55 °C for 1 h. The slurry was cooled to rt. and stirred at rt. for 2 h. The slurry was filtered and washed by H20 (100 mL, 4 vol, three times). The wet cake (28.36 g) was dried by 10 mmHg and 50 °C for overnight (14h) and the weight of CPI-444 was 19.31 g (79% recovery).
EXAMPLE 8. Removal of Residual Palladium By Recrystallization
[0360] CUNO Filter Cartridge 55 S
[0361] CPI-444 (5.0 g, Pd 14.06 ppm) and THF (50 mL, 10 vol) were charged to a 100 mL 3-necked flask fitted with stirring bar, a condenser with nitrogen balloon and a thermometer. The slurry was heated to 60 °C for 20 minutes and added CUNO 55S filter (0.75 g, 15w%). The mixture was stirred at 60 °C for 1 h. The mixture was filtered and washed by THF (5 mL, 1 vol, twice). The filtrate was concentrated. The solid, H20 (25 mL, 5 vol) and IPA (25 mL, 5vol) were charged to 250 mL 3 -necked flask fitted with stirring bar, a condenser with nitrogen balloon and a thermometer. The slurry was heated to 70 °C and maintained for 1 h (the slurry became solution). The solution was slowly cooled to rt.(40 minutes) The slurry was added H20 (75 mL, 15 vol) and then heated to 55 °C for 1 h. The slurry was cooled to rt. (30 minutes) and stirred at rt. for 2 h. The slurry was filtered and washed by H20 (20 mL, 4 vol, three times). The cake (6.355 g) was dried by 10 mmHg and 50 °C
for overnight (16 h) and the weight of CPI-444 was 4.281 g (85% recovery). Pd content(ppm) = 2.02 ppm.
[0362] Polymer-bound Thiol: SiST
[0363] CPI-444(5 g; Pd 14.06ppm) was dissolved in THF (50 mL) at 60 °C. The solution was cooled to 55 °C and SiST (7.5 g) was added to the solution. The solution was stirred at 50-55 °C for 16 h. The solution was filtered through celite and a 0.2 micron filter. The filtrate was tested for Pd content. Result: 2.43 ppm.
Catalyst
Molecular Weight: 291.6990
Molecular Weight: 337.3430
[0364] 1. A solution of S.M., CP-60, Pd(PPh3)2Cl2 and K2C03 in THF – H20 (7.9 mL, 1 : 1) was put in oil-bath at 70-75 °C.
[0365] 2. After 2 h, 0.047 g CP-60 was added to the reaction at 70-75 °C.
[0366] 3. After 1 hr, the reaction was cooled to rt. and 10 mL H20 was added to the reaction.
[0367] 4. The reaction was filtered to provide wet cake (0.812 g).
[0368] 5. The solid wet cake was dried at 45 °C and 20 mmHg for 2h to provide weight 0.499 g. (86%).
[0369] 6. The solid wet cake was stirred in 2 mL DMF for 30 mins (slurry) and then filtered. The solid was dried by 45 °C and 10 mmHg for 12h to provide weight 0.40 g; 69% yield; 98.1% purity.
//////////CIFORADENANT, CPI-444, CPI 444, CPI444, V81444, V-81444, V 81444, UNII 8KFO2187CP, Corvus Pharmaceuticals, Inc., PHASE 1,
NC1=NC2=C(N=NN2CC3=NC(CO[C@H]4CCOC4)=CC=C3)C(C5=CC=C(O5)C)=N1
TRILACICLIB, G1T28
Trilaciclib
update 2021/2/12 US FDA APPROVED COSELA
- Molecular FormulaC24H30N8O
- Average mass446.548 Da
- G1T 28
CAS 1374743-00-6
2′-{[5-(4-Methyl-1-piperazinyl)-2-pyridinyl]amino}-7′,8′-dihydro-6’H-spiro[cyclohexane-1,9′-pyrazino[1′,2′:1,5]pyrrolo[2,3-d]pyrimidin]-6′-one
G1T28, SHR 6390
Spiro[cyclohexane-1,9′(6’H)-pyrazino[1′,2′:1,5]pyrrolo[2,3-d]pyrimidin]-6′-one, 7′,8′-dihydro-2′-[[5-(4-methyl-1-piperazinyl)-2-pyridinyl]amino]-
- 7′,8′-Dihydro-2′-[[5-(4-methyl-1-piperazinyl)-2-pyridinyl]amino]spiro[cyclohexane-1,9′(6’H)-pyrazino[1′,2′:1,5]pyrrolo[2,3-d]pyrimidin]-6′-one
- 2′-[[5-(4-Methylpiperazin-1-yl)pyridin-2-yl]amino}-7′,8′-dihydro-6’H-spiro[cyclohexane-1,9′-pyrazino[1′,2′:1,5]pyrrolo[2,3-d]pyrimidin]-6′-one
UNII:U6072DO9XG
Reduction of Chemotherapy-Induced Myelosuppression
Trilaciclib dihydrochloride
1977495-97-8
In phase II clinical development as a chemoprotectant at G1 Therapeutics for first- or second-line treatment in patients with metastatic triple negative breast cancer, in combination with gemcitabine and carboplatin
![]()
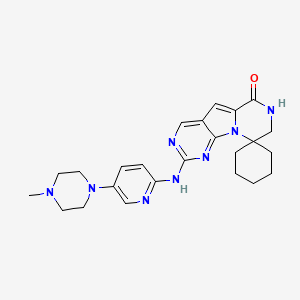
PATENT, WO 2014144326, Compound 89 (also referred to as Compound T)
| WO2014144847A3 | |
| Inventors | Norman E. Sharpless, Jay Copeland Strum, John Emerson Bisi, Patrick Joseph Roberts, Francis Xavier Tavares |
| Applicant | G1 Therapeutics, Inc. |
| Norman Sharpless | |
|---|---|
 |
|
| Born | Norman Edward Sharpless September 20, 1966 Greensboro, North Carolina |
| Nationality | American |
| Other names | Ned Sharpless |
| Occupation | Director, Lineberger Comprehensive Cancer Center Founder, G1 Therapeutics ($GTHX) |
| Notable work | Wellcome Distinguished Professor, American Society of Clinical Investigation Member, Association of American Cancer Institute board of directors, |
NCI Director Dr. Norman E. Sharpless

NCI Director Dr. Norman E. Sharpless, Credit: National Institutes of Health
Norman E. “Ned” Sharpless, M.D., was officially sworn in as the 15th director of the National Cancer Institute (NCI) on October 17, 2017. Prior to his appointment, Dr. Sharpless served as the director of the University of North Carolina (UNC) Lineberger Comprehensive Cancer Center, a position he held since January 2014.
Dr. Sharpless was a Morehead Scholar at UNC–Chapel Hill and received his undergraduate degree in mathematics. He went on to pursue his medical degree from the UNC School of Medicine, graduating with honors and distinction in 1993. He then completed his internal medicine residency at the Massachusetts General Hospital and a hematology/oncology fellowship at Dana-Farber/Partners Cancer Care, both of Harvard Medical School in Boston.
After 2 years on the faculty at Harvard Medical School, he joined the faculty of the UNC School of Medicine in the Departments of Medicine and Genetics in 2002. He became the Wellcome Professor of Cancer Research at UNC in 2012.
Dr. Sharpless is a member of the Association of American Physicians as well as the American Society for Clinical Investigation (ASCI), the nation’s oldest honor society for physician–scientists, and served on the ASCI council from 2011 to 2014. Dr. Sharpless was an associate editor of Aging Cell and deputy editor of the Journal of Clinical Investigation. He has authored more than 150 original scientific papers, reviews, and book chapters, and is an inventor on 10 patents. He cofounded two clinical-stage biotechnology companies: G1 Therapeutics and HealthSpan Diagnostics.
In addition to serving as director of NCI, Dr. Sharpless continues his research in understanding the biology of the aging process that promotes the conversion of normal self-renewing cells into dysfunctional cancer cells. Dr. Sharpless has made seminal contributions to the understanding of the relationship between aging and cancer, and in the preclinical development of novel therapeutics for melanoma, lung cancer, and breast cancer.
Synthesis
WO 2016040858


Trilaciclib (G1T28)
Trilaciclib is a potential first-in-class short-acting CDK4/6 inhibitor in development to preserve hematopoietic stem cells and enhance immune system function during chemotherapy. Trilaciclib is administered intravenously prior to chemotherapy and has the potential to significantly improve treatment outcomes.
G1 is currently evaluating trilaciclib in four Phase 2 clinical trials: three studies in patients with small-cell lung cancer (SCLC), and one study in patients with triple-negative breast cancer (TNBC). Preliminary data from the SCLC trials were presented at the American Society of Clinical Oncology 2017 Annual Meeting and at the 2016 World Conference on Lung Cancer.
Data from a Phase 1 trial in healthy volunteers were presented at the American Society of Clinical Oncology 2015 Annual Meeting and published in Science Translational Medicine. Trilacicilib has been extensively studied in animals; these preclinical data have been presented at several scientific meetings and published in Molecular Cancer Therapeutics, Science Translational Medicine, and Cancer Discovery.
Trilaciclib is a small molecule, competitive inhibitor of cyclin dependent kinases 4 and 6 (CDK4/6), with potential antineoplastic and chemoprotective activities. Upon intravenous administration, trilaciclib binds to and inhibits the activity of CDK4/6, thereby blocking the phosphorylation of the retinoblastoma protein (Rb) in early G1. This prevents G1/S phase transition, causes cell cycle arrest in the G1 phase, induces apoptosis, and inhibits the proliferation of CDK4/6-overexpressing tumor cells. In patients with CDK4/6-independent tumor cells, G1T28 may protect against multi-lineage chemotherapy-induced myelosuppression (CIM) by transiently and reversibly inducing G1 cell cycle arrest in hematopoietic stem and progenitor cells (HSPCs) and preventing transition to the S phase. This protects all hematopoietic lineages, including red blood cells, platelets, neutrophils and lymphocytes, from the DNA-damaging effects of certain chemotherapeutics and preserves the function of the bone marrow and the immune system. CDKs are serine/threonine kinases involved in the regulation of the cell cycle and may be overexpressed in certain cancer cell types. HSPCs are dependent upon CDK4/6 for proliferation.
Trilaciclib (G1T28) is a CDK4/6 inhibitor in phase II clinical development as a chemoprotectant at G1 Therapeutics for first- or second-line treatment in patients with metastatic triple negative breast cancer, in combination with gemcitabine and carboplatin. Also, phase II trials are ongoing in newly diagnosed, treatment-naive small-cell lung cancer patients, in combination with carboplatin, etoposide, and atezolizumab and phase I trials in previously treated small-cell lung cancer patients, in combination with topotecan.
U.S. Patent Nos. 8,822,683; 8,598,197; 8,598,186, 8,691,830, 8,829,102, 8,822,683, 9, 102,682, 9,499,564, 9,481,591, and 9,260,442, filed by Tavares and Strum and assigned to Gl Therapeutics describe a class of N-(heteroaryl)-pyrrolo[3,2-d]pyrimidin-2-amine cyclin dependent kinase inhibitors including those of the formula with variables as defined therein):

U.S. Patent Nos. 9,464,092, 9,487,530, and 9,527,857 which are also assigned to Gl Therapeutics describe the use of the above pyrimidine-based agents in the treatment of cancer.
These patents provide a general synthesis of the compounds that is based on a coupling reaction of a fused chloropyrimidine with a heteroaryl amine to form the central disubstituted amine. Such coupling reactions are sometimes referred to as Buchwald coupling (see WO Ί56 paragraph 127; reference WO 2010/020675). The lactam of the fused chloropyrimidine, for example, a 2-chloro-spirocyclo-pyrrolo[2,3-d]pyrimidine-one such as Intermediate K as shown below can be prepared by dehydration of the corresponding carboxylic acid. The reported process to prepare intermediate IK requires seven steps.

(Intermediate IK; page 60, paragraph 215 of WO Ί56)
WO 2013/148748 (U.S. S.N. 61/617,657) entitled “Lactam Kinase Inhibitors” filed by Tavares, and also assigned to Gl Therapeutics likewise describes the synthesis of N-(heteroaryl)-pyrrolo[3,2-d]pyrimidin-2-amines via the coupling reaction of a fused chloropyrimidine with a heteroaryl amine to form the central disubstituted amine.
WO 2013/163239 (U.S. S.N. 61/638,491) “Synthesis of Lactams” describes a method for the synthesis of this class of compounds with the variation that in the lactam preparation step, a carboxylic acid can be cyclized with a protected amine in the presence of a strong acid and a dehydrating agent, which can be together in one moiety as a strong acid anhydride. The purported improvement is that cyclization can occur without losing the protecting group on the amine before cyclization. The typical leaving group is “tBOC” (t-butoxycarbonyl). The application teaches (page 2 of WO 2013/163239) that the strong acid is, for example, trifluoroacetic acid anhydride, tribromoacetic acid anhydride, trichloroacetic acid anhydride or mixed anhydrides. An additional step may be necessary to take off the N-protecting group. The dehydrating agent can be a carbodiimide-based compound such as DCC (Ν,Ν-dicyclohexylcarbodiimide), EDC (l-ethyl-3-(3-dimethylaminopropyl)carbodiimide, or DIC (Ν,Ν-diisopropylcarbodiimide). DCC and DIC are in the same class of reagents-carbodiimides. DIC is sometimes considered better because it is a liquid at room temperature, which facilitates reactions.
WO 2015/061407 filed by Tavares and licensed to Gl Therapeutics also describes the synthesis of these compounds via the coupling of a fused chloropyrimidine with a heteroaryl amine to form the central disubstituted amine. WO ‘407 focuses on the lactam production step and in particular describes that the fused lactams of these compounds can be prepared by treating the carboxylic acid with an acid and a dehydrating agent in a manner that a leaving group on the amine is not removed during the amide-forming ring closing step.
Other publications that describe compounds of this general class include the following. WO 2014/144326 filed by Strum et al. and assigned to Gl Therapeutics describes compounds and methods for protection of normal cells during chemotherapy using pyrimidine based CDK4/6 inhibitors. WO 2014/144596 filed by Strum et al. and assigned to Gl Therapeutics describes compounds and methods for protection of hematopoietic stem and progenitor cells against ionizing radiation using pyrimidine based CDK4/6 inhibitors. WO 2014/144847 filed by Strum et al. and assigned to Gl Therapeutics describes HSPC-sparing treatments of abnormal cellular proliferation using pyrimidine based CDK4/6 inhibitors. WO2014/144740 filed by Strum et al. and assigned to Gl Therapeutics describes highly active anti -neoplastic and anti-proliferative pyrimidine based CDK 4/6 inhibitors. WO 2015/161285 filed by Strum et al. and assigned to Gl Therapeutics describes tricyclic pyrimidine based CDK inhibitors for use in radioprotection. WO 2015/161287 filed by Strum et al. and assigned to Gl Therapeutics describes analogous tricyclic pyrimidine based CDK inhibitors for the protection of cells during chemotherapy. WO 2015/161283 filed by Strum et al. and assigned to Gl Therapeutics describes analogous tricyclic pyrimidine based CDK inhibitors for use in HSPC-sparing treatments of RB-positive abnormal cellular proliferation. WO 2015/161288 filed by Strum et al. and assigned to Gl Therapeutics describes analogous tricyclic pyrimidine based CDK inhibitors for use as anti -neoplastic and anti-proliferative agents. WO 2016/040858 filed by Strum et al. and assigned to Gl Therapeutics describes the use of combinations of pyrimidine based CDK4/6 inhibitors with other anti-neoplastic agents. WO 2016/040848 filed by Strum et al. and assigned to Gl Therapeutics describes compounds and methods for treating certain Rb-negative cancers with CDK4/6 inhibitors and topoisomerase inhibitors.
Other biologically active fused spirolactams and their syntheses are described, for example, in the following publications. Griffith, D. A., et al. (2013). “Spirolactam-Based Acetyl-CoA Carboxylase Inhibitors: Toward Improved Metabolic Stability of a Chromanone Lead Structure.” Journal of Medicinal Chemistry 56(17): 7110-7119, describes metabolically stable spirolactams wherein the lactam resides on the fused ring for the inhibition of acetyl-CoA carboxylase. WO 2013/169574 filed by Bell et al. describes aliphatic spirolactams as CGRP receptor antagonists wherein the lactam resides on the spiro ring. WO 2007/061677 filed by Bell et al. describes aryl spirolactams as CGRP receptor antagonists wherein the lactam resides on the spiro ring. WO 2008/073251 filed by Bell et al. describes constrained spirolactam compounds wherein the lactam resides on the spiro ring as CGRP receptor antagonists. WO 2006/031606 filed by Bell et al. describes carboxamide spirolactam compounds wherein the spirolactam resides on the spiro ring as CGRP receptor antagonists. WO 2006/031610, WO 2006/031491, and WO 2006/029153 filed by Bell et al. describe anilide spirolactam compounds wherein the spirolactam resides on the spiro ring; WO 2008/109464 filed by Bhunai et al. describes spirolactam compounds wherein the lactam resides on the spiro ring which is optionally further fused.
Given the therapeutic activity of selected N-(heteroaryl)-pyrrolo[3,2-d]pyrimidin-2-amines, it would be useful to have additional methods for their preparation. It would also be useful to have new intermediates that can be used to prepare this class of compounds.
PATENT
WO 2014144596
PATENT
Compound 89 (also referred to as Compound T)
| WO2014144847A3 | |
| Inventors | Norman E. Sharpless, Jay Copeland Strum, John Emerson Bisi, Patrick Joseph Roberts, Francis Xavier Tavares |
| Applicant | G1 Therapeutics, Inc. |
EXAMPLES
Intermediates B, E, K, L, 1A, IF and 1CA were synthesized according to US 8,598,186 entitled CDK Inhibitors to Tavares, F.X. and Strum, J.C..
The patents WO 2013/148748 entitled Lactam Kinase Inhibitors to Tavares, F.X., WO 2013/163239 entitled Synthesis of Lactams to Tavares, F.X., and US 8,598,186 entitled CDK Inhibitors to Tavares, F.X. and Strum, J.C. are incorporated by reference herein in their entirety. Example 1
Synthesis of tert-butyl N- [2- [(5-bromo-2-chloro-pyrimidin-4yl)amino] ethyl] carbamate, Compound 1

To a solution of 5-bromo-2,4-dichloropyrimidine (3.2 g, 0.0135 mol) in ethanol (80 mL) was added Hunig’s base (3.0 mL) followed by the addition of a solution of N-(tert- butoxycarbonyl)-l,2-diaminoethane (2.5 g, 0.0156 mole) in ethanol (20 mL). The contents were stirred overnight for 20 hrs. The solvent was evaporated under vacuum. Ethyl acetate (200 mL) and water (100 mL) were added and the layers separated. The organic layer was dried with magnesium sulfate and then concentrated under vacuum. Column chromatography on silica gel using hexane/ethyl acetate (0- 60%) afforded tert-butyl N-[2-[(5-bromo-2-chloro-pyrimidin-4- yl)amino]ethyl]carbamate. 1HNMR (d6-DMSO) δ ppm 8.21 (s, 1H), 7.62 (brs, 1H), 7.27 (brs, 1H), 3.39 (m, 2H), 3.12 (m, 2H), 1.34 (s, 9H). LCMS (ESI) 351 (M + H).
Example 2
Synthesis of tert-butyl N-[2-[[2-chloro-5-(3,3-diethoxyprop-l-ynyl)pyrimidin-4- yl] amino] ethyl] carbamate, Compound 2

To tert-butyl N-[2-[(5-bromo-2-chloro-pyrimidin-4-yl)amino]ethyl]carbamate (1.265 g, 6 mmol) in THF (10 mL) was added the acetal (0.778 mL, 5.43 mmol), Pd(dppf)CH2Cl2 (148 g), and triethylamine (0.757 mL, 5.43 mmol). The contents were degassed and then purged with nitrogen. To this was then added Cul (29 mg). The reaction mixture was heated at reflux for 48 hrs. After cooling, the contents were filtered over CELITE™ and concentrated. Column chromatography of the resulting residue using hexane/ethyl acetate (0- 30%) afforded tert-butyl N- [2- [ [2-chloro-5 -(3 ,3 -diethoxyprop- 1 -ynyl)pyrimidin-4-yl]amino] ethyl] carbamate. 1HNMR (d6-DMSO) δ ppm 8.18 (s, 1H), 7.63 (brs, 1H), 7.40 (brs, 1H), 5.55 (s, 1H), 3.70 (m, 2H), 3.60 (m, 2H), 3.42 (m, 2H), 3.15 (m, 2H), 1.19 – 1.16 (m, 15H). LCMS (ESI) 399 (M + H).
Example 3
Synthesis of tert-butyl N-[2-[2-chloro-6-(diethoxymethyl)pyrrolo[2,3-d]pyrimidin-7- yl] ethyl] carbamate, Compound 3

To a solution of the coupled product (2.1 g, 0.00526 mole) in THF (30 mL) was added TBAF solid (7.0 g). The contents were heated to and maintained at 65 degrees for 2 hrs. Concentration followed by column chromatography using ethyl acetate/hexane (0-50%) afforded tert-butyl N-[2-[2-chloro-6-(diethoxymethyl)pyrrolo[2,3-d]pyrimidin-7-yl]ethyl]carbamate as a pale brown liquid (1.1 g). 1FiNMR (d6-DMSO) δ ppm 8.88 (s, 1H), 6.95 (brs, 1H), 6.69 (s, 1H), 5.79 (s, 1H), 4.29 (m, 2H), 3.59 (m, 4H), 3.34 (m, 1H), 3.18 (m, 1H), 1.19 (m, 9H), 1.17 (m, 6H). LCMS (ESI) 399 (M + H).
Example 4
Synthesis of tert-buty\ N-[2-(2-chloro-6-formyl-pyrrolo [2,3-d] pyrimidin-7- yl)ethyl] carbamate, Compound 4

To the acetal (900 mg) from the preceeding step was added AcOH (8.0 mL) and water
(1.0 mL). The reaction was stirred at room temperature for 16 hrs. Cone, and column chromatography over silica gel using ethyl acetate/hexanes (0- 60%) afforded tert-butyl N-[2-(2- chloro-6-formyl-pyrrolo[2,3-d]pyrimidin-7-yl)ethyl]carbamate as a foam (0.510 g). 1HNMR (d6-DMSO) δ ppm 9.98 (s, 1H), 9.18 (s, 1H), 7.66 (s, 1H), 6.80 (brs, 1H), 4.52 (m, 2H), 4.36 (m, 2H), 1.14 (s, 9H). LCMS (ESI) 325 (M + H).
Example 5
Synthesis of 7- [2-(teri-butoxycarbonylamino)ethyl] -2-chloro-pyrrolo [2,3-d] pyrimidine-6- carboxylic acid, Compound 5

To the aldehyde (0.940 g) from the preceeding step in DMF (4 mL) was added oxone (1.95 g, 1.1 eq). The contents were stirred at room temp for 7 hrs. Silica gel column chromatography using hexane/ethyl acetate (0- 100%) afforded l-\2-(tert- butoxycarbonylamino)ethyl]-2-chloro-pyrrolo[2,3-d]pyrimidine-6-carboxylic acid (0.545 g). 1HNMR (d6-DMSO) δ ppm 9.11 (s, 1H), 7.39 (s, 1H), 4.38 (m, 2H), 4.15 (m, 2H), 1.48 (m, 9H). LCMS (ESI) 341(M + H).
Example 6
Synthesis of methyl 7-[2-(teri-butoxycarbonylamino)ethyl]-2-chloro-pyrrolo[2,3- d]pyrimidine-6-carboxylate, Compound 6

To a solution of 2-chloro-7-propyl-pyrrolo[2,3-d]pyrimidine-6-carboxylic acid (0.545 g, 0.00156 mole) from the preceeding step in toluene (3.5 mL) and MeOH (1 mL) was added TMS- diazomethane (1.2 mL). After stirring overnight at room temperature, the excess of TMS- diazomethane was quenched with acetic acid (3 mL) and the reaction was concentrated under vacuum. The residue was purified by silica gel column chromatography with hexane/ethyl acetate (0- 70%) to afford methyl 7-[2-(tert-butoxycarbonylamino)ethyl]-2-chloro-pyrrolo[2,3- d]pyrimidine-6-carboxylate as an off white solid (0.52 g). 1HNMR (d6-DMSO) δ ppm 9.10 (s, 1H), 7.45 (s, 1H), 6.81 (brs, 1H) 4.60 (m, 2H), 3.91 (s, 3H), 3.29 (m, 2H), 1.18 (m, 9H) LCMS (ESI) 355 (M + H).
Example 7
Synthesis of Chloro tricyclic amide, Compound 7

To methyl 7- [2-(tert-butoxycarbonylamino)ethyl] -2-chloro-pyrrolo [2,3 -d]pyrimidine-6- carboxylate (0.50 g, 0.0014 mole) from the preceeding step in dichloromethane (2.0 mL) was added TFA (0.830 mL). The contents were stirred at room temperature for 1 hr. Concentration under vacuum afforded the crude amino ester which was suspended in toluene (5 mL) and Hunig’s base (0.5 mL). The contents were heated at reflux for 2 hrs. Concentration followed by silica gel column chromatography using hexane/ethyl acetate (0- 50%) afforded the desired chloro tricyclic amide (0.260 g). 1HNMR (d6-DMSO) δ ppm 9.08 (s, 1H), 8.48 (brs, 1H), 7.21 (s, 1H) 4.33 (m, 2H), 3.64 (m, 2H). LCMS (ESI) 223 (M + H).
Example 8
Synthesis of chloro-N-methyltricyclic amide, Compound 8

To a solution of the chloro tricycliclactam, Compound 7, (185 mg, 0.00083 mole) in DMF (2.0 mL) was added sodium hydride (55% dispersion in oil, 52 mg). After stirring for 15 mins, methyl iodide (62 μί, 1.2 eq). The contents were stirred at room temperature for 30 mins. After the addition of methanol (5 mL), sat NaHCOs was added followed by the addition of ethyl acetate. Separation of the organic layer followed by drying with magnesium sulfate and concentration under vacuum afforded the N-methylated amide in quantitative yield. 1FiNMR (d6-DMSO) δ ppm 9.05 (s, 1H), 7.17 (s, 1H) 4.38 (m, 2H), 3.80 (m, 2H), 3.05 (s, 3H). LCMS (ESI) 237 (M + H). Example 9
Synthesis of l-methyl-4-(6-nitro-3-pyridyl)piperazine, Compound 9

To 5-bromo-2-nitropyridine (4.93 g, 24.3 mmole) in DMF (20 mL) was added N- methylpiperazine (2.96 g, 1.1 eq) followed by the addition of DIPEA (4.65 mL, 26.7 mmole). The contents were heated at 90 degrees for 24 hrs. After addition of ethyl acetate (200 mL), water (100 mL) was added and the layers separated. Drying followed by concentration afforded the crude product which was purified by silica gel column chromatography using (0-10%) DCM/Methanol. 1HNMR (d6-DMSO) δ ppm 8.26 (s, 1H), 8.15 (1H, d, J = 9.3 Hz), 7.49 (1H, d, J = 9.4 Hz), 3.50 (m, 4H), 2.49 (m, 4H), 2.22 (s, 3H).
Example 10
Synthesis of 5-(4-methylpiperazin-l-yl)pyridin-2-amine, Compound 10

To l-methyl-4-(6-nitro-3-pyridyl)piperazine (3.4 g) in ethyl acetate (100 mL) and ethanol (100 mL) was added 10%> Pd/C (400 mg) and then the reaction was stirred under hydrogen (10 psi) overnight. After filtration through CELITE™, the solvents were evaporated and the crude product was purified by silica gel column chromatography using DCM/ 7N ammonia in MeOH (0- 5%) to afford 5-(4-methylpiperazin-l-yl)pyridin-2-amine (2.2 g). 1HNMR (d6-DMSO) δ ppm 7.56 (1H, d, J = 3 Hz), 7.13 (1H, m), 6.36 (1H, d, J = 8.8 Hz), 5.33 (brs, 2H), 2.88 (m, 4H), 2.47 (m, 4H), 2.16 (s, 3H).
Example 11
Synthesis of tert-butyl 4-(6-amino-3-pyridyl)piperazine-l-carboxylate, Compound 11

This compound was prepared as described in WO 2010/020675 Al .
Synthesis of Compound 89 (also referred to as Compound T)

Compound 89 was synthesized in a similar manner to that described for compound 78 and was converted to an HCl salt. 1HNMR (600 MHz, DMSO-d6) δ ppm 1.47 (br. s., 6 H) 1.72 (br. s., 2 H) 1.92 (br. s., 2 H) 2.77 (br. s., 3 H) 3.18 (br. s., 2 H) 3.46 (br. s., 2 H) 3.63 (br. s., 2 H) 3.66 (d, J=6.15 Hz, 2 H) 3.80 (br. s., 2 H) 7.25 (s, 1 H) 7.63 (br. s., 2 H) 7.94 (br. s., 1 H) 8.10 (br. s., 1 H) 8.39 (br. s., 1 H) 9.08 (br. s., 1 H) 11.59 (br. s., 1 H). LCMS (ESI) 447 (M + H)
PATENT
WO 2014144740
PATENT
Preparation of Active Compounds
Syntheses
The disclosed compounds can be made by the following general schemes:

Scheme 1
In Scheme 1, Ref-1 is WO 2010/020675 Al; Ref-2 is White, J. D.; et al. J. Org. Chem. 1995, 60, 3600; and Ref-3 Presser, A. and Hufher, A. Monatshefte fir Chemie 2004, 135, 1015.

Scheme 2
In Scheme 2, Ref-1 is WO 2010/020675 Al; Ref-4 is WO 2005/040166 Al; and Ref-5 is Schoenauer, K and Zbiral, E. Tetrahedron Letters 1983, 24, 573.

92


93 
3) Pd/C/H2 ![]()
Scheme 6

Scheme 7
NHfOH

Scheme 8
In Scheme 8, Ref-1 is WO 2010/020675 Al; Ref-2 is WO 2005/040166 Al; and Ref-3 is Schoenauer, K and Zbiral, E. Tetrahedron Letters 1983, 24, 573.
Alternatively, the lactam can be generated by reacting the carboxylic acid with a protected amine in the presence of a strong acid and a dehydrating agent, which can be together in one moiety as a strong acid anhydride. Examples of strong acid anhydrides include, but are not limited to, trifluoroacetic acid anhydride, tribromoacetic acid anhydride, trichloroacetic acid anhydride, or mixed anhydrides. The dehydrating agent can be a carbodiimide based compound such as but not limited to DCC (Ν,Ν-dicyclohexylcarbodiimide), EDC (l-ethyl-3-(3-
dimethylaminopropyl)carbodiimide or DIC (Ν,Ν-diisopropylcarbodiimide). An additional step may be necessary to take off the N-protecting group and the methodologies are known to those skilled in the art.
Alternatively, the halogen moiety bonded to the pyrimidine ring can be substituted with any leaving group that can be displaced by a primary amine, for example to create an intermediate for a final product such as Br, I, F, SMe, SO2Me, SOalkyl, SO2alkyl. See, for Exmaple PCT /US2013/037878 to Tavares.
Other amine intermediates and final amine compounds can be synthesized by those skilled in the art. It will be appreciated that the chemistry can employ reagents that comprise reactive functionalities that can be protected and de-protected and will be known to those skilled in the art at the time of the invention. See for example, Greene, T.W. and Wuts, P.G.M., Greene’s Protective Groups in Organic Synthesis, 4th edition, John Wiley and Sons.

Scheme 9
CDK4/6 Inhibitors of the present invention can be synthesized according to the generalized Scheme 9. Specific synthesis and characterization of the Substituted 2-aminopyrmidines can be found in, for instance, WO2012/061156.
Compounds T, Q, GG, and U were prepared as above and were characterized by mass spectrometry and NMR as shown below:
Compound T
1H NMR (600 MHz, DMSO- d6) ppm 1.47 (br. s., 6 H) 1.72 (br. s., 2 H) 1.92 (br. s., 2 H) 2.77 (br. s., 3 H) 3.18 (br. s., 2 H) 3.46 (br. s., 2 H) 3.63 (br. s., 2 H) 3.66 (d, J=6.15 Hz, 2 H) 3.80 (br. s., 2 H) 7.25 (s, 1 H) 7.63 (br. s., 2 H) 7.94 (br. s., 1 H) 8.10 (br. s., 1 H) 8.39 (br. s., 1 H) 9.08 (br. s., 1 H) 11.59 (br. s., 1 H). LCMS ESI (M + H) 447.
PATENT
Synthesis of N-(heteroaryl)-pyrrolo[3,2-d]pyrimidin-2-amines. The application appears to be particularly focused on methods for the preparation of trilaciclib and an analog of it. Trilaciclib is the company’s lead CDK4/6 inhibitor presently in phase II trials against small-cell lung cancer and triple negative breast cancer. Interestingly, the company is working on a second CDK4/6 inhibitor, G1T38 , which is in a phase II trial against breast cancer.
GENERAL METHODS
The structure of starting materials, intermediates, and final products was confirmed by standard analytical techniques, including NMR spectroscopy and mass spectrometry. Unless otherwise noted, reagents and solvents were used as received from commercial suppliers. Proton nuclear magnetic resonance spectra were obtained on a Bruker AVANCE 500 at 500 MHz in DMSO-dis. HPLC analyses were performed on a Waters HPLC using the below HPLC method.
HPLC Method
Column: Atlantis T3 (150 χ 4.6, 3 μιη)
Column Temperature: 40°C
Flow Rate: 1 mL/min
Detection: UV @ 275 nm
Analysis Time: 36 min
Mobile Phase A: Water (with 0.1% Trifluoroacetic Acid)
Mobile Phase B : Acetonitrile (with 0.1% Trifluoroacetic Acid)
Sample preparation: dissolve PC sample, wet or dry solid (~1 mg of active compound) in acetonitrile/water (1/1) to achieve complete dissolution.
HPLC Method Gradient

Example 1. General Routes of Synthesis

Scheme 1-1 : Starting from an appropriately substituted halo pyrimidine, compounds of the present invention can be prepared. In Step 1 the appropriately substituted halo pyrimidine is subjected to l,4-diazaspiro[5.5]undecan-3-one in the presence of base and heat to afford a substituted spirolactam. In Step 2 the appropriately substituted spirolactam is protected with a group selected from R2. In Step 3 the protected spirolactam is cyclized in the presence of base to afford a fused spirolactam. The fused spirolactam can be optionally oxidized to a sulfoxide or sulfone after Step 3, Step 4, Step 5, or Step 6. Oxidation prior to Step 3 results in undesired byproducts. In Step 4 the hydroxyl group of the fused spirolactam is converted to a leaving group.
In Step 5 the leaving group is dehydrated to afford a compound of Formula IV. In Step 6 the compound of Formula IV is optionally deprotected.

Scheme 1-2: Starting from an appropriately substituted halo pyrimidine compounds of the present invention can be prepared. In Step 1 the appropriately substituted halo pyrimidine is subjected to l,4-diazaspiro[5.5]undecan-3-one in the presence of base and heat to afford a substituted spirolactam. In Step 2 the appropriately substituted spirolactam is protected with a group selected from R2. In Step 3 the protected spirolactam is cyclized in the presence of base to afford a fused spirolactam of Formula IV. The fused spirolactam can be optionally oxidized to a sulfoxide or sulfone after Step 3 or Step 4. Oxidation prior to Step 3 results in undesired byproducts. In Step 4 the compound of Formula IV is optionally deprotected.

Scheme 1-3 : Starting from an appropriately substituted alkyl glycinate, compounds of the present invention can be prepared. In Step 1 the appropriately substituted alkyl glycinate is subjected to cyclohexanone and TMSCN in the presence of base to afford a cyano species. In Step 2 the appropriately substituted cyanospecies is reduced and subsequently cyclized to afford a compound of Formula I.
Scheme 1-4

Scheme 1-4: Starting from an appropriately substituted l-(aminomethyl)cyclohexan-l-amine, compounds of the present invention can be prepared. In Step 1 the appropriately substituted l-(aminomethyl)cyclohexan-l -amine is reductively aminated with an aldehyde. In Step 2 the appropriately substituted cyclohexane amine is optionally deprotected (i.e.: the group selected from R2 if not H is optionally replaced by H). In Step 3 the cyclohexane amine is cyclized to afford a compound of Formula I. In Step 4 the compound of Formula I is optionally protected.
1-5

Conversion

Scheme 1-5: Starting from an appropriately substituted halo pyrimidine, compounds of the present invention can be prepared. In Step 1 the appropriately substituted halo pyrimidine is subjected to l,4-diazaspiro[5.5]undecan-3-one in the presence of base and heat to afford a
substituted spirolactam. In Step 2 the protected spirolactam is cyclized in the presence of base to afford a fused spirolactam. The fused spirolactam can be optionally oxidized to a sulfoxide or sulfone after Step 2, Step 3, Step 4, or Step 5. Oxidation prior to Step 2 results in undesired byproducts. In Step 3 the hydroxyl group of the fused spirolactam is converted to a leaving group. In Step 4 the leaving group is dehydrated to afford a compound of Formula IV. In Step 5 the compound of Formula IV is optionally deprotected.
S

Scheme 1-6: Starting from an appropriately substituted halo pyrimidine compounds of the present invention can be prepared. In Step 1 the appropriately substituted halo pyrimidine is subjected to l,4-diazaspiro[5.5]undecan-3-one in the presence of base and heat to afford a substituted spirolactam. In Step 2 the protected spirolactam is cyclized in the presence of base to afford a fused spirolactam of Formula IV. The fused spirolactam can be optionally oxidized to a sulfoxide or sulfone after Step 2 or Step 3. Oxidation prior to Step 2 results in undesired byproducts. In Step 3 the compound of Formula IV is optionally deprotected.

Scheme 1-7: Starting from compound of Formula IV a CDK4/6 inhibitor can be prepared. In Step 1 a heteroaryl amine is subjected to a base and a compound of Formula IV is added slowly under chilled conditions to afford a nucleophilic substitution reaction. The compound of Formula IV can previously be prepared as described in the schemes herein.
Example 2. Representative Routes of Synthesis
Scheme 2-1

quant, yield 2 steps
isolated

70% yield 2 steps 75% yield 95% yield
isolated isolated isolated
Scheme 2-1 : An ester route is one embodiment, of the present invention. Ideally, the best synthesis scheme would afford crystalline intermediates to provide material of consistent purity without column chromatography, and high yielding steps while using safe and cost effective reagents when possible.
The first step in the ester route is a SNAr nucleophilic substitution of CI group in commercially available ester 3 using spirolactam 4. Due to low reactivity of 4, a reaction temperature of 85-95 °C was required. Because of the temperature requirements, DIPEA and dimethylacetamide were selected as the base and solvent, respectively. The reaction follows second-order kinetics and usually stalls after -85% conversion. Therefore, the reaction was typically stopped after 60 hours by first cooling it to room temperature at which point solid formation was observed. The mixture was then partitioned between MTBE and water and product was filtered with excellent purity with -53% yield of the desired product 5. The obtained
compound 5 was protected with a Boc group using Boc anhydride and DMAP as the catalyst and dichloromethane as the solvent. The intermediate 6 was obtained in a quantitative yield. Due to the semi-solid nature of compound 6, the material was taken to the next step without further purification. The Dieckmann condensation was initially performed with strong bases such as LiHMDS and tBuOK. A similar result to the aldehyde route (Scheme 2-2) was obtained: a partial deprotection of Boc group was observed that required column chromatography. However, the best results were obtained when DBU was used as base and THF as solvent. The reaction outcome was complete, clean conversion of 6 to 7. Moreover, the product crystallized from the reaction mixture upon seeding, and a quantitative yield was obtained for the two steps.
The hydroxyl group of 7 was removed via a two-step procedure. First, compound 7 was converted completely into triflate 8 using triflic anhydride and triethylamine in dichloromethane. The reaction was found to proceed well at 0°C. Due to the potential instability of the triflate intermediate, it was not isolated. It was immediately taken to the next step and reduced with triethylsilane and palladium tetrakis to afford the product 9 after ethyl acetate crystallization in -70% yield. The Boc group of 9 was removed using trifluoroacetic acid in dichloromethane to afford 10. Intermediate 10 was converted into the final sulfone 11 using Oxone™ in acetonitrile/water solvent system.
The obtained sulfone 11 was use-tested in the coupling step and was found to perform well. In conclusion, the route to sulfone 11 was developed which eliminated the use of column chromatography with good to excellent yields on all steps.
Scheme 2-2

Molecular Weight: 421 
Scheme 2-2: The first step of Scheme 2-2 consistently afforded product 13 contaminated with one major impurity found in substantial amount. Thorough evaluation of the reaction impurity profile by LC-MS and 2D MR was performed, which showed the impurity was structurally the condensation of two aldehyde 12 molecules and one molecule of lactam 4. Therefore, column chromatography was required to purify compound 13, which consistently resulted in a modest 30% yield. A solvent screen revealed that sec-butanol, amyl alcohol, dioxane, and tert-butanol can all be used in the reaction but a similar conversion was observed in each case. However, tert-butanol provided the cleanest reaction profile, so it was selected as a solvent for the reaction. Assessing the impact of varying the stoichiometric ratio of 4 and 12 on the reaction outcome was also investigated. The reaction was performed with 4 equivalents of amine 4 in an attempt to disrupt the 2: 1 aldehyde/amine composition of the impurity. The result was only a marginal increase in product 13 formation. The temperature impact on the reaction outcome was evaluated next. The coupling of aldehyde 12 and 4 was investigated at two different temperatures: 50 °C and 40 °C with 1 : 1 ratio of aldehyde/amine. Reactions were checked at 2 and 4 hours and then every 12 hours. The reaction progress was slow at 50°C and was accompanied by growth of other impurities. The reaction at 40°C was much cleaner; however the conversion was lower in the same time period. The mode of addition of the reagents was investigated as well at 80°C with a slow addition (over 6 hours) of either aldehyde 12 or amine 4 to the reaction mixture. The product distribution did not change and an about 1 to 1 ratio was observed between product and impurity when amine 4 was added slowly to the reaction mixture containing aldehyde 12 and
DIPEA at reflux. The product distribution did change when aldehyde 12 was added slowly to the mixture of amine 4 and DIPEA. However, the major product of the reaction was the undesired impurity. Other organic bases were tried as well as different ratios of DIPEA. No product was observed when potassium carbonate was used as a base. The results of the experiments are presented in Table 1 below.
Table 1

Compound 13 was successfully formed in three cases: triethylamine, 2,6-lutidine and DIPEA, with the DIPEA result being the best. The use of Boc protected spirolactam 4 had no effect on the impurity formation as well. Its utilization was speculated to be beneficial in performing the coupling step together with the following step, preparation of compound 14.
The major impurity formed during Step 1 of Scheme 2-2 is:

Chemical Formula:€2)Η;Μ(¾ 6( 2ί>2
Molecular Weight: 527.4903
The second step (Boc protection of the free lactam) proceeded well using DMAP as a catalyst in dichloromethane at room temperature. The product 14 is a thick oil, and, therefore, cannot be purified by crystallization. The Boc protected intermediate 14 was cyclized successfully into the desired pentacyclic structure 10 upon treatment with a strong base such as LiHMDS or tBuOK. Surprisingly, the Boc group was partially removed during the reaction. The level of deprotection was independent from the internal reaction temperature and was positively correlated with excess of base used. Therefore the mixture of the desired product 10 and 10-Boc compound was treated with acid to completely deprotect Boc group. The conversion of methyl sulfide into the final sulfone 11 was carried out with Oxone™. Initially a mixture of methanol and water was used for the reaction. As the result, a partial displacement of sulfone by methoxy group was detected. The methanol was replaced with acetonitrile and the sulfone displacement was eliminated.
In summary, the ester route (Scheme 2-1) is preferred because:
1. Formation of the impurity during the first step of Scheme 2-2 was unavoidable and resulted in yields of < 35%.
2. Column purification was required to isolate intermediate 14.
3. The aldehyde starting material was not commercially available and required two synthetic steps from the corresponding ester.

Scheme 2-3 : Starting with cyclohexanone, compounds of the present invention can be prepared. In Step 1 the methyl glycinate is subjected to cyclohexanone and TMSCN in the presence of tri ethyl amine in DCM to afford 15. In Step 2 15 hydrogenated with hydrogen gas in the presence of catalytic platinum oxide and subsequently undergoes an intramolecular cyclization to afford compound 16 which is used in the schemes above.

Scheme 2-4: Starting with compound 17, compounds of the present invention can be prepared. In Step 1 compound 17 is subjected to ethyl 2-oxoacetate in the presence platinum on carbon and hydrogen gas to afford compound 18. In Step 2 compound 18 is Boc-deprotected with hydrochloric acid. In Step 3 compound 18 is cyclized to afford compound 16 which is used in the schemes above.
Scheme 2-5

11 19
Scheme 2-5: Starting from compound 11 the CDK 4/6 inhibitor 19 can be prepared. In Step 1 5-(4-methylpiperazin-l-yl)pyridin-2-amine is subjected to LiHMDS and compound 11 is added slowly under chilled conditions to afford a nucleophilic substitution reaction and compound 19. Compound 11 can be prepared as described in the schemes herein.

Scheme 2-6: Starting from compound 11 the CDK 4/6 inhibitor 20 can be prepared. In Step 1 5-(4-isopropylpiperazin-l-yl)pyridin-2-amine is subjected to LiHMDS and compound 11 is added slowly under chilled conditions to afford a nucleophilic substitution reaction and compound 20. Compound 11 can be prepared as described in the schemes herein.
Preparation of Compound 5:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet, and reflux condenser was charged with ethyl 4-chloro-2-(methylthio)pyrimidine-5-carboxylate 3 (49.2 g, 0.21 mol, 1.00 equiv.), spirolactam 4 (39.2 g, 0.23 mol, 1.10 equiv.), DIPEA (54.7 g, 0.42 mol, 2.00 equiv.), and DMAc (147.6 mL, 3 vol). The batch was heated to 90-95 °C, and after 60 h, IPC confirmed -14% (AUC) of ethyl 4-chloro-2-(methylthio)pyrimidine-5-carboxylate remained. The batch was cooled to RT, and precipitate formation was observed. The suspension was diluted with MTBE (100 mL, 2 vol) and water (442 mL, 9 vol) and stirred for 2 h at RT. The product was isolated by vacuum filtration and washed with MTBE (49 mL, 1 vol). The solid cake was conditioned for 1 h and dried under vacuum at 40 °C for 16 h to afford compound 5 [41.0 g, 53% yield] as an off-white solid with a purity of >99% AUC. ¾ MR (CDCh): δ 8.76 (d, J = 2.0 Hz, 1H), 6.51-6.29 (br, 1H), 4.33 (q, J = 7.0 Hz, 2H), 3.78 (s, 2H), 3.58 (s, 2H), 2.92 (s, 2H), 2.53 (s, 3H), 1.63-1.37 (m, 12H). LCMS (ESI, m/z = 365.3 [M+H]).
Preparation of Compound 6:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged with 5 [41.0 g, 0.11 mol, 1.00 equiv.], Boc-anhydride (36.8 g, 0.17 mol, 1.50 equiv.), DMAP (1.37 g, 0.01 mol, 0.10 equiv.), and dichloromethane (287 mL, 7 vol). The batch was stirred for 3 h at RT. IPC confirmed no starting material remained (AUC). The batch was concentrated into a residue under reduced pressure and taken to the next step (a quantitative yield is assumed for this step). An aliquot (200 mg) was purified by column chromatography (heptanes/ethyl acetate 0 to 100%) to afford compound 6. 1H MR (CDCh): δ 8.64 (s, 1H), 4.31 (q, J = 7.0 Hz, 2H), 4.07 (s, 2H), 3.83 (S, 2H), 3.15 (m, 2H), 2.56 (s, 3H), 172 (m, 3H), 1.59 (m, 15H), 1.42 (t, J= 7.0 Hz, 3H). LCMS (ESI, m/z = 465.2 [M+H]).
Preparation of Compound 7:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged with compound 6 [residue from a previous step, quantitative yield assumed, 52.2 g, 0.11 mol, 1.00 equiv.], and THF (261 mL, 5 vol). The batch was cooled to 0°C and 1,8-diazabicyclo[5.4.0]un-dec-7-ene (17.1 g, 0.11 mmol, 1.00 equiv.) was added keeping the internal temperature in 0-10°C range. After the addition was complete, the cooling bath was removed and the reaction mixture was allowed to warm up to RT and after 2 h, IPC confirmed no starting material remained. The batch was seeded with the product (1.0 g) and was cooled to 0°C. The slurry was stirred at 0°C for 2 h. The product was isolated by vacuum filtration and washed with cold (0°C) THF (50 mL, 1 vol). The solid cake was conditioned for 1 h and dried under vacuum at 40°C for 16 h to afford 7 [47 g, quantitative yield] as a light orange solid with a purity of >99% AUC. The color of the product changed into yellow once the batch was exposed to air for an extended period of time (~ 1 day). Material was isolated with substantial amount DBU, according to proton NMR. However, it did not interfere with the next step. 1H MR (CDCh): δ 8.71 (s, 1H), 4.03 (s, 2H), 2.57 (s, 3H), 1.85 (m, 10H), 1.51 (s, 9H). LCMS (ESI, m/z = 419.2 [M+H]).
Preparation of Compound 8:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged with 7 [40.8 g, 0.10 mol, 1.00 equiv.], triethylamine (31.5 g, 0.31 mol, 3.20 equiv.), and dichloromethane (408 mL, 10 vol). The batch was purged with N2 for 15 min and was cooled to 0°C. Triflic anhydride (44.0 g, 0.16 mol, 1.60 equiv.) was added keeping the
internal temperature in 0-10°C range. The batch was stirred at 0°C and after 3 h, IPC confirmed -7.0% (AUC) of 7 remained. [It was speculated that the product was hydrolyzing back into starting material during the analysis.] Once the reaction was deemed complete, the batch was transferred to a 1 L, separatory funnel and was washed with 50% saturated sodium bicarbonate (200 mL, 5 vol). [It was prepared by mixing saturated sodium bicarbonate (100 mL) with water (100 mL)).] The aqueous layer was separated and was extracted with DCM (2×40 mL, 1 vol). The organic layers were combined and concentrated into a residue under reduced pressure and taken to the next step. LCMS (ESI, m/z = 551.6 [M+H]).
Preparation of Compound 9:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged with compound 8 [residue from a previous step, quantitative yield assumed, 53.7 g, 0.10 mol, 1.00 equiv.], and THF (110 mL, 2 vol). The solvent was removed under vacuum distillation and the procedure was repeated two times. The flask was charged with triethylsilane (22.7 g, 0.20 mol, 2.00 equiv.), and DMF (268 mL, 5 vol). The batch was degassed by five cycles of evacuation, followed by backfilling with nitrogen. The flask was charged with tetrakis(triphenylphosphine)palladium(0) (11.3 g, 0.01 mol, 0.1 equiv.). The batch was heated to 45-50°C, and after 14 h, IPC confirmed no starting material remained. The batch was transferred to a 500 mL, separatory funnel while still warm. The reaction was partitioned between water (5 vol) and ethyl acetate (5 vol). The aqueous layer was extracted with ethyl acetate (3 x3 vol). The organic layers were combined and concentrated down to 2 volumes. The precipitate was filtered and washed with ethyl acetate (2x 1 vol). The solid cake was conditioned for 1 h and dried under vacuum at 40°C for 16 h to afford 9 [27.5 g, 70% yield] as a yellow solid with a purity of -98% AUC. Proton NMR showed some triphenylphosphine oxide present. ¾ NMR (DMSO-i¾):5 9.01 (s, 1H), 7.40 (s, 1H), 4.30 (s, 2H), 2.58 (m, 2H), 2.58 (s, 3H), 1.81 (m, 5H), 1.51 (s, 9H). LCMS (ESI, m/z = 403.4 [M+H]).
Preparation of Compound 10 from the Scheme 2-1 route:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged 9 (12.8 g, 31.8 mmol, 1.00 equiv.) and dichloromethane (64 mL, 5 vol). Trifluoroacetic acid (18.2 g, 159 mmol, 5.00 equiv.) was added over 20 min and the solution was stirred for 2 h at RT. IPC confirmed reaction was complete. The batch was transferred to a 500 mL, separatory funnel and washed with saturated sodium bicarbonate (200 mL). The aqueous layer was extracted with dichlorom ethane (3 x3 vol). The organic layers were combined and concentrated down to 1 volume. The precipitate was filtered and conditioned for 1 h and dried under vacuum at 40 °C for 16 h to afford 9 [6.72 g, 70% yield] as an off-white solid with a purity of 99.1% AUC. ¾ NMR (DMSO-dis): δ 8.95 (s, 1H), 8.32 (s, 1H), 7.15 (s, 1H), 3.68 (d, J = 1.0 Hz, 2H), 2.86 (m, 2H), 2.57 (s, 3H), 1.92 (m, 2H), 1.73 (m, 3H), 1.39 (m, 3H). LCMS, ESI, m/z = 303.2 [M+H]).
Preparation of Compound 10 from Scheme 2-2 route:
A 50 mL, three-neck flask equipped with a magnetic stirring bar, thermocouple, N2 inlet was charged 14 (680 mg, 1.62 mmol, 1.00 equiv.) and THF (6.8 mL, 10 vol). A I M solution of potassium tert-butoxide (3.2 mL, 3.24 mmol, 2.00 equiv.) in THF was added over 10 min and the solution was stirred for 2 h at RT. IPC confirmed reaction was complete. The batch was acidified with 4 N hydrogen chloride solution in dioxane (2.4 mL, 9.72 mmol, 6.00 equiv.) and stirred for additional 1 h. The batch was transferred to a 500 mL, separatory funnel and washed with saturated sodium bicarbonate (100 mL). The aqueous layer was extracted with ethyl acetate (3 x20 vol). The organic layers were combined and concentrated down to 3volumes and product precipitated. The precipitate was filtered and conditioned for 1 h and dried under vacuum at 40 °C for 16 h to afford 9 [489 mg, quantitative yield] as an off-white solid.
Preparation of Compound 11 :
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged with 10 (9.00 g, 29.8 mmol, 1.00 equiv.), and acetonitrile (180 mL, 20 vol). A solution of Oxone™ (45.9 g, 0.15 mol, 5.00 equiv.) in water (180 mL, 20 vol) was added to the batch over 20 min. The batch was stirred for 2 h and IPC confirmed the reaction was complete. The batch was concentrated down to ½ of the original volume and was extracted with dichloromethane DCM (4x 10 vol). The organic layers were combined; polish filtered and concentrated down to -10 vol of DCM. The product was slowly crystallized out by addition of heptanes (-30 vol). The mixture was cooled to 0°C and the product was filtered and dried under vacuum at 40 °C for 16 h to afford 11 [9.45 g, 95% yield] as an off-white solid with a purity of >99% AUC. ¾ NMR (CDCb): 5 9.24 (s, 1H), 7.78 (s, 1H), 7.46 (s, 1H), 3.89 (d, J= 2.0 Hz, 2H), 3.43 (s, 3H), 2.98 (m, 2H), 2.10 (m, 2H), 1.86 (m, 3H), 1.50 (m, 3H). LCMS (ESI, m/z = 335.2 [M+H]).
Preparation of Compound 13:
A 250 mL, single-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet, and reflux condenser was charged with 4-chloro-2-(methylthio)pyrimidine-5-carbaldehyde (2.00 g, 10.6 mmol, 1.00 equiv.), spirolactam 4 (1.96 g, 11.7 mmol, 1.10 equiv.), DIPEA (2.74 g, 21.2 mmol, 2.00 equiv.), and fert-butanol (20 mL, 10 vol). The batch was heated to 80-85 °C, and after 24 h, IPC confirmed no aldehyde 12 remained. The batch was cool to RT and concentrated into a residue, which was loaded on silica gel column. The product was eluted with ethyl acetate/heptanes (0% to 100%). The product containing fractions were pulled out and concentrated to afford 13 [0.98 g, 29% yield] as an off-white solid.
Preparation of Compound 14:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged with 13 [0.98 g, 3.00 mmol, 1.00 equiv.], Boc-anhydride (4.90 g, 21.5 mmol, 7.00 equiv.), DMAP (36 mg, 0.30 mmol, 0.10 equiv.), and dichloromethane (7 mL, 7 vol). The batch was stirred for 3 h at RT. IPC confirmed no starting material remained. The batch was cool to RT and concentrated into a residue, which was loaded on silica gel column. The product was eluted with ethyl acetate/heptanes (0% to 100%). The product containing fractions were pulled out and concentrated to afford 14 [0.98 g, 29% yield] as an off-white solid.
Preparation of Compound 15:
To a suspension of methyl glycinate (500 g, 3.98 mol, 1 eq) in DCM (10 L) was added
TEA dropwise at rt under nitrogen atmosphere, followed by the addition of cyclohexanone (781 g, 7.96 mol, 2 eq) dropwise over 15 min. To the resulting mixture was added TMSCN (591 g, 5.97 mol, 1.5 eq) dropwise over 1 hour while maintaining the internal reaction temperature below 35
°C. After stirred at rt for 2 hrs, the suspension became a clear solution. The progress of the reaction was monitored by H- MR.
When the methyl glycinate was consumed completely as indicated by H-NMR analysis, the reaction was quenched by water (5 L). The layers were separated. The aqueous layer was extracted with DCM (1 L). The combined organic phase was washed with water (5 L X 2) and
dried over Na2S04 (1.5 Kg). After filtration and concentration, 1.24 Kg of crude 15 was obtained as oil.
The crude 15 was dissolved in IPA (4 L). The solution was treated with HC1/IPA solution (4.4 mol/L, 1.1L) at RT. A large amount of solid was precipitated during the addition. The resulting suspension was stirred for 2 hrs. The solid product was collected by vacuum filtration and rinsed with MTBE (800 mL). 819 g of pure 15 was obtained as a white solid. The yield was 88.4%. ¾- MR (300 MHz, CD3OD) 4.20 (s, 2H), 3.88 (s, 3H), 2.30-2.40 (d, J = 12 Hz, 2H), 1.95-2.02 (d, J = 12 Hz, 2H), 1.55-1.85 (m, 5H), 1.20-1.40 (m, 1H).
Preparation of Compound 16:
To a solution of 15 (10 g, 43 mmol) in MeOH (100 mL) was added methanolic hydrochloride solution (2 .44 mol/L, 35.3 mL, 2 eq) and Pt02 (0.5 g, 5 wt %). The reaction suspension was stirred with hydrogen bubble at 40 °C for 6 hours. H- MR analysis showed consumption of 15. To the reaction mixture was added K2CO3 (15 g, 108 mmol, 2.5 eq) and the mixture was stirred for 3 hrs. The suspension was filtered and the filtrate was concentrated to dryness. The residual oil was diluted with DCM (100 mL) and resulting suspension was stirred for 3 hrs. After filtration, the filtrate was concentrated to provide crude 16 (6.6 g) as an oil. The crude 16 was diluted with EtOAc/hexane (1 : 1, 18 mL) at rt for 2 hrs. After filtration, 16 (4 g) was isolated. The obtained 16 was dissolved in DCM (16.7 mL) and hexane (100 mL) was added dropwise to precipitate the product. After further stirred for 1 h, 2.8 g of the pure 16 was isolated as a white solid. The yield was 39%. HPLC purity was 98.3%; MS (ESI): 169.2 (MH+); 1 H-NMR (300 MHz, D2O) 3.23 (s, 3H), 3.07 (s, 3H), 1.37-1.49 (m, 10H).
Preparation of compound 19:
5-(4-methylpiperazin-l-yl)pyridin-2-amine (803.1 g; 3.0 equivalents based on sulfone 11) was charged to a 22 L flask. The flask was blanketed with N2 and anhydrous THF added (12.4 kg). The resulting black-purple solution was cooled in an ice bath to < 5°C. 1M LiHMDS (4.7 L; 1.2 equivalents based on sulfone 11) was added via an addition funnel in three equal additions to keep the temperature below 10°C. Upon the completion of the addition, the reaction mixture was warmed to 16°C. The sulfone 11 (455.1 g; 1.00 equivalents) was added in five additions. Reaction proceeded until HPLC analysis of an IPC sample indicated less than 3% of sulfone 11 remained.
To quench the reaction, the contents of the 22L flask were transferred to a 100 L flask containing water. After stirring for 30 minutes at <30°C, the crude product was collected by filtration, washed with water and dried to afford 19 (387 g, 99.1% purity, 63.7% yield).
Preparation of compound 20:
5-(4-isopropylpiperazin-l-yl)pyridin-2-amine (1976.2 g; 3.0 equivalents based on sulfone 11) was charged to a 50 L flask. The flask was blanketed with N2 and anhydrous THF added (10.7 kg). The resulting black-purple solution was cooled in an ice bath to < 5°C. 1M LiHMDS (9.6 kg; 3.6 equivalents based on sulfone) was added via an addition funnel at a rate to keep the temperature below 10°C. Upon the completion of the addition, the reaction mixture was warmed to 16°C over 120 minutes by removing the ice bath. The sulfone (1000 g; 1.00 mol) was added in five additions. The reaction proceeded until HPLC analysis of an IPC sample indicated less than 1% of sulfone 11 remained. After completion of the reaction, ammonium chloride was added to the reaction mixture. The mixture stirred at < 32°C for at least 30 minutes and the solids collected by filtration to afford 20 (900 g, 99.1% purity, 64.2% yield).
Alternate Route to Spirolactam via cyclohexanone:
Scheme 2-7

26
In one embodiment the spirolactam is made via the synthetic scheme above. By reducing the nitrile group before addition of the glycinate group the reaction sequence proceeds in higher yield. The chemistry used in Step 1 is described in the literature (J. Org. Chem. 2005, 70,8027-8034), and was performed on a kilogram scale. The chemistry to convert Compound 24 into the
spirolactam was also demonstrated on kilogram scale. The Boc protection of Compound 23, is carried out at -70°C in order to limit formation of the di-Boc protected product. Experimental details of a 200 g pilot run are described below.
Step 1

200 g of cyclohexanone 21 was converted to 22 using Ti(Oi-Pr)4 /TMSCN/NH3. After work-up, 213 g of 22 was obtained. The H- MR was clean. The yield was 84%. The titanium salts were removed by vacuum filtration. In one embodiment, the titanium salts are removed by centrifugation or Celite filtration.
Step 2

190 g of 22 was mixed with LAH (2 eq) in MTBE for 30 minutes at 45°C. After work-up, 148 g of crude 23 was obtained.
Step 3

136 g of the crude 23 from step 2 was converted to 24 with 0.9 eq of B0C2O at -70°C. The reaction was completed and worked up. After concentration, 188 g of 24 was obtained. The yield was 86%. The H-NMR and C-NMR spectra confirmed that the compound was pure.
Step 4

188 g of 24 was subjected to methyl 2-bromoacetate and K2CO3 in acetonitrile to afford 25. 247 g of crude 25 was obtained.
Step 5

247 g of 25 was subjected to TFA in DCE heated to reflux to afford 26. After work-up, 112 g of 6 as TFA salt was obtained. H- MR was clean.
Step 6

26 27
Compound 26 was stirred in EtOH in the presence at room temperature overnight to afford 27. In one embodiment DCM is used as the solvent instead of EtOH.
Example 3. Purge of residual palladium from Step 5 Scheme 2-1:
Since palladium was used in Step 5 of Scheme 2-1, the levels of residual Pd present in the subsequent synthetic steps was determined. Table 2 below and Figure 3 show the palladium levels in the isolated solids.
Table 2

The material after Step 5 was isolated containing 1.47% (14700 ppm) of residual palladium. This data represents the highest level of palladium in the worst case scenario. The workup conditions of the latter steps purged nearly all of the palladium and the final product, 19 bis HC1 salt, contained 14 ppm of Pd, which is below the standard 20 ppm guidline. The Pd levels will likely be even lower once the catal st loading is optimized in Step 5.

19
The process developed in this route was a significant improvement over the one used for the first generation synthesis. Overall, the scheme consists of seven steps with five isolations, all by crystallization. No silica column chromatography is employed in the synthesis, which makes the process highly scalable. The process workup conditions can successfully purge the 1.47% of residual palladium after step 5 of Scheme 2-1.
///////////////TRILACICLIB, G1T28, G1T 28, SHR 6390, PHASE 2, G1 Therapeutics, Inc.
CN1CCN(CC1)C2=CN=C(C=C2)NC3=NC=C4C=C5C(=O)NCC6(N5C4=N3)CCCCC6
ALCAFTADINE, WO 2017211246, NEW PATENT, SHENZHEN TARGETRX, INC.

Alcaftadine
NEW PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017211246&redirectedID=true
WO-2017211246, SHENZHEN TARGETRX, INC.
SUBSTITUTED FUSED IMIDAZOLE CYCLIC COMPOUND AND PHARMACEUTICAL COMPOSITION THEREOF
WANG, Yihan; (CN).
XING, Qingfeng; (CN)
Novel deuterated analogs of substituted fused imidazole cyclic compounds, particularly alcaftadine are histamine H1-receptor antagonists and mast cell stabilizers, useful for treating allergy and nasal congestion.
The present invention relates to a substituted fused imidazole cyclic compound and a composition containing said compound and application thereof. Specifically disclosed is the fused imidazole cyclic compound represented by formula (I), or a pharmaceutical composition of its crystalline form, pharmaceutically acceptable salt, prodrug, stereoisomer, hydrate, or solvate. The compound of the present invention may be used as a histamine H1-receptor antagonist and mast-cell stabilizer, and is capable of inhibiting mast-cell release of histamine and preventing histamine function, thereby reducing allergic reaction

Example 1 Preparation of 6,11-dihydro -11- (1- (d3- methyl) piperidin-4-ylidene) -5H- imidazo [2,1-b] [3] benzazepine – 3- aldehyde (compound 8)
Step 1. Synthesis of compound 3.
N-benzyloxycarbonylpiperidine-4-carboxylic acid (2.63 g, 10 mmol) was dissolved in 20 mL of dichloromethane, 6 mL of oxalyl chloride and 1 drop of DMF were added and the mixture was reacted at room temperature for 2 hours under nitrogen. The reaction mixture was concentrated to dryness under reduced pressure, dissolved in 20 mL of acetonitrile, and added with triethylamine (4.1 mL, 30 mmol) in an ice bath and stirred for 3 minutes. A solution of 1-phenethyl-1H-imidazole (2.06 g, 12 mmol) in 5 mL of acetonitrile was slowly added dropwise and the reaction was allowed to warm to room temperature overnight after the addition was completed. The reaction was completed, concentrated to dryness, 30 mL of ethyl acetate and 20 mL of water were added and the mixture was stirred for 5 minutes. The layers were separated and the aqueous phase was extracted with ethyl acetate. The combined organic phases were dried over anhydrous sodium sulfate and concentrated to give 3.34 g of a colorless oil, benzyl-4- (1-phenethyl-1H-imidazole-2-formyl) piperidine-1-carboxylate (Compound 3) was obtained in a yield of 80%. ESI-MS: 418 [M ++ 1].
Step 2. Synthesis of compound 4.
Benzyl-4- (1-phenylethyl-1H-imidazole-2-formyl) piperidine-1-carboxylate (3.34 g, 8 mmol) was dissolved in 30 mL of absolute ethanol and 300 mg of 10% palladium on carbon , Hydrogen was substituted three times and stirred overnight at room temperature under a hydrogen atmosphere of 1 atmosphere. After completion of the reaction, the palladium carbon was filtered off and the filtrate was concentrated. 2.04 g of (1-phenethyl-1H-imidazol-2-yl) (piperidin-4-yl) methanone (Compound 4) 90%. ESI-MS: 284 [M ++ 1].
Step 3. Synthesis of compound 5.
(Piperidin-4-yl) methanone (2.04 g, 7.2 mmol) was dissolved in 10 mL of DMF and potassium carbonate (1.98 g, 14.4 mmol) The solution was cooled to -15 ° C and deuterated methyl iodide (1.02 g, 7.2 mmol) was slowly added dropwise under the protection of nitrogen. After the addition was completed, the mixture was stirred at room temperature for 0.5 hour. The mixture was extracted with ethyl acetate and extracted with ethyl acetate. The organic phase was washed once with 20 mL of water and 20 mL of saturated brine, dried over anhydrous sodium sulfate, concentrated and separated on a silica gel column (1- (methyl-d3) piperidine (1-phenethyl-1H-imidazol-2-yl) methanone (Compound 5) was obtained in an amount of 70%. 1 H NMR (300 MHz, CDCl 3 ) δ 7.23 (d, J = 2.0Hz, 1H), 7.06 (td, J = 4.2,3.8,1.7Hz, 3H), 6.86 (d, J = 1.0Hz, 1H) (Dd, J = 10.2, 5.8 Hz, 2H), 3.09 (t, J = 7.2 Hz, 2H) J = 7.2 Hz, 2H), 2.85-2.65 (m, 2H), 2.15 (td, J = 7.5, 3.9 Hz, 4H); ESI-MS: 301 [M ++ l ].
Step 4. Synthesis of Compound 6.
(1-phenethyl-1H-imidazol-2-yl) methanone (1.5 g, 5.1 mmol) was placed in a reaction flask and the mixture was purged with nitrogen three times , 7mL trifluoromethanesulfonic acid was added dropwise, the reaction was warmed to 110 ° C overnight. Cooled to room temperature, the reaction solution was poured into 30mL ice water, 50% sodium hydroxide solution was added dropwise to adjust the pH = 10-11, extracted with dichloromethane, the organic phase was washed once with 20mL of water and 20mL of saturated brine, Dried over sodium sulfate, concentrated and separated by silica gel column to obtain 0.85 g of compound 6, yield 60%. 1 H NMR (300 MHz, CDCl 3 ) δ 7.28 (d, J = 4.4 Hz, 2H), 7.23 (d, J = 5.0 Hz, 1H), 7.13 (d, J = 7.0 Hz, 1H), 7.02 (D, J = 1.3Hz, 1H), 4.38 (dt, J = 12.7, 3.9Hz, 1H), 4.02 (td, J = 13.3,3.1Hz, 1H), 3.59 -3.34 (m, 3H), 3.21 (s, 2H), 3.04-2.87 (m, 3H), 2.78-2.63 (m, 2H). ESI-MS: 283 [M ++ l ].
Step 5. Synthesis of compound 7.
Compound 6 (850 mg, 3 mmol) was placed in a reaction flask, followed by the addition of 0.5 mL of acetic acid, 5 mL of 37% formaldehyde and sodium acetate (87 mg, 1.1 mmol) and warming to 100 ° C overnight. After the reaction was cooled to room temperature completely, 30 mL of methylene chloride was added to the reaction solution, 50% sodium hydroxide solution was added dropwise to adjust pH = 11-12, stirred for 0.5 hour, and the layers were separated and the organic phase was washed with 10 mL of saturated saline , Dried over anhydrous sodium sulfate, concentrated and separated on a silica gel column to give the compound 7 340 mg, yield 36%. ESI-MS: 313 [M ++ 1].
Step 6. Synthesis of Compound 8.
Compound 7 (340 mg, 1.1 mmol) was dissolved in 20 mL of dichloromethane and 4-dimethylaminopyridine (DMAP, 13 mg, 0.11 mmol) and Dess-Martin Periodinane 1.3 mmol) and reacted at room temperature for 3 hours. Join 20mL saturated sodium bicarbonate solution and 20mL dichloromethane, stirred for 5 minutes, filtered and the filtrate was separated. The organic phase was washed with saturated brine, dried over anhydrous sodium sulfate and concentrated. The compound 8 270mg was obtained by silica gel column, and the yield was 80%. 1 H NMR (300 MHz, CDCl 3 ) δ 9.64 (s, 1H), 7.76 (s, 1H), 7.34-7.26 (m, 3H), 7.16 (d, J = 6.7 Hz, 1H), 4.74 J = 14.5, 3.9 Hz, 1H), 4.31 (td, J = 14.1, 3.2 Hz, 1H), 3.53 (td, 3.03-2.89 (m, 4H), 2.64-2.81 (m, 4H); ESI-MS: 311 [M ++ l ].
//////////////
FDA approves first drug Actemra (tocilizumab) to specifically treat giant cell arteritis
05/22/2017
The U.S. Food and Drug Administration today expanded the approved use of subcutaneous Actemra (tocilizumab) to treat adults with giant cell arteritis. This new indication provides the first FDA-approved therapy, specific to this type of vasculitis.
May 22, 2017
Release
The U.S. Food and Drug Administration today expanded the approved use of subcutaneous Actemra (tocilizumab) to treat adults with giant cell arteritis. This new indication provides the first FDA-approved therapy, specific to this type of vasculitis.
“We expedited the development and review of this application because this drug fulfills a critical need for patients with this serious disease who had limited treatment options,” said Badrul Chowdhury, M.D., Ph.D., director of the Division of Pulmonary, Allergy, and Rheumatology Products in the FDA’s Center for Drug Evaluation and Research.
Giant cell arteritis is a form of vasculitis, a group of disorders that results in inflammation of blood vessels. This inflammation causes the arteries to narrow or become irregular, impeding adequate blood flow. In giant cell arteritis, the vessels most involved are those of the head, especially the temporal arteries (located on each side of the head). For this reason, the disorder is sometimes called temporal arteritis. However, other blood vessels, including large ones like the aorta, can become inflamed in giant cell arteritis. Standard treatment involves high doses of corticosteroids that are tapered over time.
The efficacy and safety of subcutaneous (injected under the skin) Actemra for giant cell arteritis were established in a double-blind, placebo-controlled study with 251 patients with giant cell arteritis. The primary efficacy endpoint was the proportion of patients achieving sustained remission from Week 12 through Week 52. Sustained remission was defined as the absence of symptoms of giant cell arteritis, normalization of inflammatory laboratory tests, and tapering the use of prednisone (a steroid drug). A greater proportion of patients receiving subcutaneous Actemra with standardized prednisone regimens achieved sustained remission from Week 12 through Week 52 as compared to patients receiving placebo with standardized prednisone regimens. The cumulative prednisone dose was lower in treated patients with Actemra relative to placebo.
The overall safety profile observed in the Actemra treatment groups was generally consistent with the known safety profile of Actemra. Actemra carries a Boxed Warning for serious infections. Patients treated with Actemra who develop a serious infection should stop that treatment until the infection is controlled. Live vaccines should be avoided during treatment with Actemra. Actemra should be used with caution in patients at increased risk of gastrointestinal perforation. Hypersensitivity reactions, including anaphylaxis and death, have occurred. Laboratory monitoring is recommended due to potential consequences of treatment-related changes in neutrophils (type of white blood cell), platelets, lipids and liver function tests.
Subcutaneous Actemra was previously approved for the treatment of moderate to severely active rheumatoid arthritis. Intravenous Actemra was also previously approved for the treatment of moderate to severely active rheumatoid arthritis, systemic juvenile idiopathic arthritis and polyarticular juvenile idiopathic arthritis. Intravenous administration is not approved for giant cell arteritis.
The FDA granted this application a Breakthrough Therapy designation and a Priority Review.
The FDA granted the supplemental approval of Actemra to Hoffman La Roche, Inc.
//////////Actemra, tocilizumab, fda 2017, Breakthrough Therapy designation, Priority Review, supplemental approval, Hoffman La Roche, Inc.
FDA approves Xermelo (telotristat ethyl) for carcinoid syndrome diarrhea

Telotristat ethyl
Molecular Formula, C27-H26-Cl-F3-N6-O3,
Molecular Weight, 574.9884,
RN: 1033805-22-9
UNII: 8G388563M
LX 1032
(2S)-2-Amino-3-[4-[2-amino-6-[[(1R)-1-[4-chloro-2-(3-methylpyrazol-1-yl)phenyl]-2,2,2-trifluoroethyl]oxy]pyrimidin-4-yl]phenyl]propionic acid ethyl ester
Ethyl-4-(2-amino-6-{(1R)-1-[4-chlor-2-(3-methyl-1H-pyrazol-1-yl)phenyl]-2,2,2-trifluorethoxy}-4-pyrimidinyl)-L-phenylalaninat
L-Phenylalanine, 4-[2-amino-6-[(1R)-1-[4-chloro-2-(3-methyl-1H-pyrazol-1-yl)phenyl]-2,2,2-trifluoroethoxy]-4-pyrimidinyl]-, ethyl ester
SEE……………

Telotristat etiprate,
(S)-ethyl 2-amino-3-(4-(2-amino-6-((R)-1-(4-chloro-2-(3-methyl-1H-pyrazol-1-yl)phenyl)-2,2,2-trifluoroethoxy)pyrimidin-4-yl)phenyl)propanoate 2-benzamidoacetate .
CAS: 1137608-69-5 (etiprate), LX 1606
Chemical Formula: C36H35ClF3N7O6
Molecular Weight: 754.16
CAS: 1137608-69-5 (etiprate), LX 1606
Chemical Formula: C36H35ClF3N7O6
Molecular Weight: 754.16
L-Phenylalanine, 4-[2-amino-6-[(1R)-1-[4-chloro-2-(3-methyl-1H-pyrazol-1-yl)phenyl]-2,2,2-trifluoroethoxy]-4-pyrimidinyl]-, ethyl ester, compd. with N-benzoylglycine (1:1)
- LX 1032 hippurate
- LX 1606
SEE ALSO………….

Telotristat, also known as LX1033, 1033805-28-5 CAS OF ACID FORM
WO 2008073933, PRODUCT PATENT


02/28/2017
The U.S. Food and Drug Administration today approved Xermelo (telotristat ethyl) tablets in combination with somatostatin analog (SSA) therapy for the treatment of adults with carcinoid syndrome diarrhea that SSA therapy alone has inadequately controlled.
February 28, 2017
The U.S. Food and Drug Administration today approved Xermelo (telotristat ethyl) tablets in combination with somatostatin analog (SSA) therapy for the treatment of adults with carcinoid syndrome diarrhea that SSA therapy alone has inadequately controlled.
Carcinoid syndrome is a cluster of symptoms sometimes seen in people with carcinoid tumors. These tumors are rare, and often slow-growing. Most carcinoid tumors are found in the gastrointestinal tract. Carcinoid syndrome occurs in less than 10 percent of patients with carcinoid tumors, usually after the tumor has spread to the liver. The tumors in these patients release excess amounts of the hormone serotonin, resulting in diarrhea. Complications of uncontrolled diarrhea include weight loss, malnutrition, dehydration, and electrolyte imbalance.
“Today’s approval will provide patients whose carcinoid syndrome diarrhea is not adequately controlled with another treatment option,” said Julie Beitz, M.D., director of the Office of Drug Evaluation III in the FDA’s Center for Drug Evaluation and Research.
Xermelo, in a regimen with SSA therapy, is approved in tablet form to be taken orally three times daily with food. Xermelo inhibits the production of serotonin by carcinoid tumors and reduces the frequency of carcinoid syndrome diarrhea.
The safety and efficacy of Xermelo were established in a 12-week, double-blind, placebo-controlled trial in 90 adult participants with well-differentiated metastatic neuroendocrine tumors and carcinoid syndrome diarrhea. These patients were having between four to 12 daily bowel movements despite the use of SSA at a stable dose for at least three months. Participants remained on their SSA treatment, and were randomized to add placebo or treatment with Xermelo three times daily. Those receiving Xermelo added on to their SSA treatment experienced a greater reduction in average bowel movement frequency than those on SSA and placebo. Specifically, 33 percent of participants randomized to add Xermelo on to SSA experienced an average reduction of two bowel movements per day compared to 4 percent of patients randomized to add placebo on to SSA.
The most common side effects of Xermelo include nausea, headache, increased levels of the liver enzyme gamma-glutamyl transferase, depression, accumulation of fluid causing swelling (peripheral edema), flatulence, decreased appetite and fever. Xermelo may cause constipation, and the risk of developing constipation may be increased in patients whose bowel movement frequency is less than four bowel movements per day. Patients treated with a higher than recommended dosage of Xermelo developed severe constipation in clinical trials. One patient required hospitalization and two other patients developed complications of either intestinal perforation or intestinal obstruction. Patients should be monitored for severe constipation. If a patient experiences severe constipation or severe, persistent or worsening abdominal pain, they should discontinue Xermelo and contact their healthcare provider.
The FDA granted this application fast track designation and priority review. The drug also received orphan drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
Xermelo is manufactured by Woodlands, Texas-based Lexicon Pharmaceuticals, Inc.
SYNTHESIS…….WO 2011100285

5.67. Synthesis of (S)-2-Amino-3-[4-(2-amino-6-{R-l-[4-chloro-2-(3-methyl-pyrazol-l-yll- phenyll-2,2,2-trifluoro-ethoxy)-pyrimidin-4-yl)-phenyll-propionic acid ethyl ester

The title compound was prepared stepwise, as described below:
Step 1: Synthesis of l-(2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-ethanone. To a 500 ml 2 necked RB flask containing anhydrous methanol (300 ml) was added thionyl chloride (29.2 ml, 400 mmol) dropwise at 0-5°C (ice water bath) over 10 minutes. The ice water bath was removed, and 2-bromo-4-chloro-benzoic acid (25 g, 106 mmol) was added. The mixture was heated to mild reflux for 12h. Progress of the reaction was monitored by TLC and LCMS. After completion of the reaction, the reaction mixture was concentrated. Crude product was dissolved in dichloromethane (DCM, 250 ml), washed with water (50 ml), sat. aq. NaHC03 (50 ml), brine (50 ml), dried over sodium sulfate, and concentrated to give the 2- bromo-4-chloro-benzoic acid methyl ester (26 g, 99 %), which was directly used in the following step.
2-Bromo-4-chloro-benzoic acid methyl ester (12.4 g, 50 mmol) in toluene (200 ml) was cooled to -70°C, and trifluoromethyl trimethyl silane (13 ml, 70 mmol) was added.
Tetrabutylamonium fluoride (1M, 2.5 ml) was added dropwise, and the mixture was allowed to warm to room temperature over 4h, after which it was stirred for 10 hours at room temperature. The reaction mixture was concentrated to give the crude [l-(2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-l-methoxy-ethoxy]-trimethyl-silane. The crude intermediate was dissolved in methanol (100 ml) and 6N HCI (100 ml) was added. The mixture was kept at 45-50°C for 12h. Methanol was removed, and the crude was extracted with dichloromethane (200 ml). The combined DCM layer was washed with water (50 ml), NaHC03 (50 ml), brine (50 ml), and dried over sodium sulfate. Removal of solvent gave a crude product, which was purified by ISCO column chromatography, using 1-2% ethyl acetate in hexane as solvent, to afford l-(2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-ethanone (10 g, 70%). !H-NMR (300 MHz, CDC ): δ (ppm) 7.50 (d,lH), 7.65(d,lH), 7.80(s,lH).
Step 2: Synthesis of R-l-(2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-ethanol. To catechol borane (1M in THF 280 ml, 280 mmol) in a 2L 3-necked RB flask was added S-2-methyl-CBS oxazaborolidine (7.76 g, 28 mmol) under nitrogen, and the resulting mixture was stirred at room temperature for 20 min. The reaction mixture was cooled to -78°C (dry ice/acetone bath), and 1-(2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-ethanone (40 g, 139 mmol) in THF (400 ml) was added dropwise over 2 hours. The reaction mixture was allowed to warm to -36°C, and was stirred at that temperature for 24 hours, and further stirred at -32 °C for another 24h. 3N NaOH (250 ml) was added, and the cooling bath was replaced by ice-water bath. Then 30 % hydrogen peroxide in water (250 ml) was added dropwise over 30 minutes. The ice water bath was removed, and the mixture was stirred at room temperature for 4 hours. The organic layer was separated, concentrated and re-dissolved in ether (200 ml). The aqueous layer was extracted with ether (2 x 200 ml). The combined organic layers were washed with IN aq. NaOH (4 x 100 ml), brine, and dried over sodium sulfate. Removal of solvent gave crude product which was purified by column chromatography using 2 to 5% ethyl acetate in hexane as solvent to give desired alcohol 36.2 g (90 %, e.e. >95%). The alcohol (36.2 g) was crystallized from hexane (80 ml) to obtain R-l-(2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-ethanol 28.2 g (70 %; 99-100 % e.e.). !H-NMR (400 MHz, CDCIs) δ (ppm) 5.48 (m, 1H), 7.40 (d, 1H), 7.61 (d, 2H).
Step 3: Synthesis of R-l-[4-chloro-2-(3-methyl-pyrazol-l-yl)-phenyll-2.2.2-trifluoro-ethanol. R-l-(2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-ethanol (15.65 g, 54.06 mmol), 3-methylpyrazole (5.33 g, 65 mmol), Cul (2.06 g, 10.8 mmol), 2CO3 (15.7 g, 113.5 mmol), (lR,2R)-N,N’-dimethyl-cyclohexane-l,2-diamine (1.54 g, 10.8 mmol) and toluene (80 ml) were combined in a 250 ml pressure tube and heated to 130°C (oil bath temperature) for 12 hours. The reaction mixture was diluted with ethyl acetate and washed with H2O (4 x 100 ml), brine, and dried over sodium sulfate. Removal of solvent gave a crude product, which was purified by ISCO column chromatography using 5-10 % ethyl acetate in hexane as solvent to get R-l-[4-chloro-2-(3-methyl-pyrazol-l-yl)-phenyl]-2,2,2-trifluoro-ethanol (13.5 g; 86 %). i-H-NMR (400 MHz, CDC ): δ (ppm) 2.30(s, 3H), 4.90(m, 1H), 6.20(s, 1H), 6.84(d, 1H), 7.20(s, 1H), 7.30(d, 1H), 7.50(d, 1H).
Step 4: Synthesis of (S)-2-Amino-3- 4-(2-amino-6-fR-l-[4-chloro-2-(3-methyl-pyrazol-l-yl)-phenyll^^^-trifluoro-ethoxyl-pyrimidin^-yll-phenvD-propionic acid ethyl ester. R-l-[4-chloro-2-(3-methyl-pyrazol-l-yl)-phenyl]-2,2,2-trifluoro-ethanol (17.78 g, 61.17 mmol), (S)-3-[4-(2-amino-6-chloro-pyrimidine-4-yl)-phenyl]-2-tert-butoxycarbonylamino-propionic acid (20.03 g, 51 mmol), 1,4-dioxane (250 ml), and CS2CO3 (79.5 g, 244 mmol) were combined in a 3-necked 500 ml RB flask and heated to 100°C (oil bath temperature) for 12-24 hours. The progress of reaction was monitored by LCMS. After the completion of the reaction, the mixture was cooled to 60°C, and water (250 ml) and THF (400 ml) were added. The organic layer was separated and washed with brine (150 ml). The solvent was removed to give crude BOC protected product, which was taken in THF (400 ml), 3N HCI (200 ml). The mixture was heated at 35-40 °C for 12 hours. THF was removed in vacuo. The remaining aqueous layer was extracted with isopropyl acetate (2x 100 ml) and concentrated separately to recover the unreacted alcohol (3.5 g). Traces of remaining organic solvent were removed from the aqueous fraction under vacuum.
To a 1L beaker equipped with a temperature controller and pH meter, was added H3PO4 (40 ml, 85 % in water) and water (300 ml) then 50 % NaOH in water to adjust pH to 6.15. The temperature was raised to 58 °C and the above acidic aqueous solution was added dropwise into the buffer with simultaneous addition of 50 % NaOH solution in water so that the pH was maintained between 6.1 to 6.3. Upon completion of addition, precipitated solid was filtered and washed with hot water (50-60°C) (2 x 200 ml) and dried to give crude (S)-2-amino-3-[4-(2-amino-6-[R-l-[4-chloro-2-(3-methyl-pyrazol-l-yl)-phenyl]-2,2,2-trifluoro-ethoxy}-pyrimidin-4-yl)-phenyl}^ propionic acid (26.8 g; 95 %). LCMS and HPLC analysis indicated the compound purity was about 96-97 %.
To anhydrous ethanol (400 ml) was added SOC (22 ml, 306 mmol) dropwise at 0-5°C.
Crude acid (26.8 ) from the above reaction was added. The ice water bath was removed, and the reaction mixture was heated at 40-45°C for 6-12 hours. After the reaction was completed, ethanol was removed in vacuo. To the residue was added ice water (300 ml), and extracted with isopropyl acetate (2 x 100 ml). The aqueous solution was neutralized with saturated Na2C03 to adjust the pH to 6.5. The solution was extracted with ethyl acetate (2 x 300 ml). The combined ethyl acetate layer was washed with brine and concentrated to give 24 g of crude ester (HPLC purity of 96-97 %). The crude ester was then purified by ISCO column chromatography using 5 % ethanol in DCM as solvent to give (S)-2-amino-3-[4-(2-amino-6-{R-l-[4-chloro-2-(3-methyl-pyrazol-l-yl)-phenyl]-2,2,2-trifluoro-ethoxy}-pyrimidin-4-yl)-phenyl}-propionic acid ethyl ester (20.5g; 70 %; HPLC purity of 98 %). LCMS M+l = 575. !H-NMR (400 MHz, CDsOD): δ (ppm) 1.10 (t, 3H), 2.25 (s, 3H), 2.85 (m, 2H), 3.65 (m, IH), 4.00 (q, 2H), 6.35 (s, IH), 6.60 (s, IH), 6.90 (m, IH), 7.18 (d, 2H), 7.45 (m, 2H), 7.70 (d, IH), 7.85 (m, 3H).
SYNTHESIS OF INTERMEDIATE
WO 2009048864

https://google.com/patents/WO2009048864A1?cl=en
6.15. Preparation of 6SV3-(4-(2-Amino-6-chloropyrimidin-4-yl)phenyl)-2- (fert-butoxycarbonylamino)propanoic Acid Using the Lithium Salt of (S)-2-(te^-butoxycarbonylamino)-3-(4-(4,4,5,5-tetramethyl-l,3,2- dioxaborolan-2-yl)phenyl)propanoic Acid

During preparation of compound 7, the isolation of the free acid can be optionally omitted. Thus, an aqueous solution of the lithium salt of compound 7 in 100 ml water, prepared from 5.0 g of Boc-Tyr-OMe (4, 17 mmol), was mixed 2-amino-4,6- dichloropyrimidine (3.3 g, 1.2 eq), potassium bicarbonate (5.0 g, 3 eq), bis(triphenylphosphine)palladium(II) dichloride (60 mg, 0.5 mol%), and 100 ml ethanol. The resulting mixture was heated at 700C for 5 hours. Additional 2-amino-4,6- dichloropyrimidine (1.1 g, 0.4 eq) was added and heating was continued at 7O0C for an additional 2 hours. HPLC analysis showed about 94% conversion. Upon cooling and filtration, the filtrate was analyzed by HPLC against a standard solution of compound 8. The assay indicated 3.9 g compound 8 was contained in the solution (59% yield from compound 4).
6.16. Alternative Procedure for Preparation of (S)-3-(4-f2-Amino-6- chloropyrimidin-4-yl)phenyl)-2-(fe^-butoxycarbonylamino)propanoic Acid Using Potassium Carbonate as Base

The boronic acid compound 11 (Ryscor Science, Inc., North Carolina, 1.0 g, 4.8 mmol) and potassium carbonate (1.32 g, 2 eq) were mixed in aqueous ethanol (15 ml ethanol and 8 ml water). Di-ter£-butyldicarbonate (1.25 g, 1.2 eq) was added in one portion. After 30 minutes agitation at room temperature, HPLC analysis showed complete consumption of the starting compound 11. The 2-amino-4,6- dichloropyrimidine (1.18 g, 1.5 eq) and the catalyst bis(triphenylphosphine)palladium(II) dichloride (34 mg, 1 mol%) were added and the resulting mixture was heated at 65-700C for 3 hours. HPLC analysis showed complete consumption of compound 12. After concentration and filtration, HPLC analysis of the resulting aqueous solution against a standard solution of compound 8 showed 1.26 g compound 8 (67% yield).
6.17. Alternative procedure for preparation of (5)-3-(4-(2-Amino-6-

The boronic acid compound 11 (10 g, 48 mmol) and potassium bicarbonate (14.4 g, 3 eq) were mixed in aqueous ethanol (250 ml ethanol and 50 ml water). Oi-tert- butyldicarbonate (12.5 g, 1.2 eq) was added in one portion. HPLC analysis indicated that the reaction was not complete after overnight stirring at room temperature. Potassium carbonate (6.6 g, 1.0 eq) and additional di-te/t-butyldicarbonate (3.1 g, 0.3 eq) were added. After 2.5 hours agitation at room temperature, HPLC analysis showed complete consumption of the starting compound 11. The 2-amino-4,6-dichloropyrimidine (11.8 g, 1.5 eq) and the catalyst bis(triphenylphosphine)-palladium(II) dichloride (0.34 g, 1 mol%” were added and the resulting mixture was heated at 75-8O0C for 2 hours. HPLC analysis showed complete consumption of compound 12. The mixture was concentrated under reduced pressure and filtered. The filtrate was washed with ethyl acetate (200 ml) and diluted with 3 : 1 THF/MTBE (120 ml). This mixture was acidified to pH about 2.4 by 6 N hydrochloric acid. The organic layer was washed with brine and concentrated under reduced pressure. The residue was precipitated in isopropanol, filtered, and dried at 500C under vacuum to give compound 8 as an off-white solid (9.0 g, 48% yield). Purity: 92.9% by HPLC analysis. Concentration of the mother liquor yielded and additional 2.2 g off-white powder (12% yield). Purity: 93.6% by HPLC analysis
PATENT
https://www.google.com/patents/WO2013059146A1?cl=en
This invention is directed to solid pharmaceutical dosage forms in which an active pharmaceutical ingredient (API) is (S)-ethyl 2-amino-3-(4-(2-amino-6-((R)-l-(4-chloro-2-(3- methyl-lH-pyrazol-l-yl)phenyl)-2,2,2-trifluoroethoxy)pyrimidin-4-yl)phenyl)propanoate
(telotristat):

or a pharmaceutically acceptable salt thereof. The compound, its salts and crystalline forms can be obtained by methods known in the art. See, e.g., U.S. patent no. 7,709,493.
PATENT
http://www.google.co.in/patents/WO2008073933A2?cl=en
6.19. Synthesis of (S)-2-Amino-3-r4-q-amino-6-{R-l-r4-chloro-2-(3-methyl- Pyrazol-l-yl)-phenyll-2,2,2-trifluoro-ethoxy}-pyrimidin-4-yl)-phenyll- propionic acid ethyl ester

The title compound was prepared stepwise, as described below: Step 1 : Synthesis of l-(2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-ethanone. To a 500 ml 2 necked RB flask containing anhydrous methanol (300 ml) was added thionyl chloride (29.2 ml, 400 mmol) dropwise at 0-50C (ice water bath) over 10 min. The ice water bath was removed, and 2-bromo-4-chloro-benzoic acid (25 g, 106 mmol) was added. The mixture was heated to mild reflux for 12h. Progress of the reaction was monitored by TLC and LCMS. After completion of the reaction, the reaction mixture was concentrated. Crude product was dissolved in dichloromethane (DCM, 250 ml), washed with water (50 ml), sat. aq. NaHCO3 (50 ml), brine (50 ml), dried over sodium sulfate, and concentrated to give the 2- bromo-4-chloro-benzoic acid methyl ester (26 g, 99 %), which was directly used in the following step.
2-Bromo-4-chloro-benzoic acid methyl ester (12.4 g, 50 mmol) in toluene (200 ml) was cooled to -700C, and trifluoromethyl trimethyl silane (13 ml, 70 mmol) was added. Tetrabutylamonium fluoride (IM, 2.5 ml) was added dropwise, and the mixture was allowed to warm to room temperature over 4h, after which it was stirred for 1Oh at room temperature. The reaction mixture was concentrated to give the crude [l-(2-bromo-4-chloro-phenyl)-2,2,2- trifluoro-l-methoxy-ethoxy]-trimethyl-silane. The crude intermediate was dissolved in methanol (100 ml) and 6N HCl (100 ml) was added. The mixture was kept at 45-500C for 12h. Methanol was removed, and the crude was extracted with dichloromethane (200 ml). The combined DCM layer was washed with water (50 ml), NaHCO3 (50 ml), brine (50 ml), and dried over sodium sulfate. Removal of solvent gave a crude product, which was purified by ISCO column chromatography, using 1-2% ethyl acetate in hexane as solvent, to afford 1- (2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-ethanone (10 g, 70%). 1H-NMR (300 MHz, CDCl3): δ (ppm) 7.50 (d,lH), 7.65(d,lH), 7.80(s,lH).
Step 2: Synthesis of R-l-(2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-ethanol. To catechol borane (IM in THF 280 ml, 280 mmol) in a 2L 3-necked RB flask was added S-2- methyl-CBS oxazaborolidine (7.76 g, 28 mmol) under nitrogen, and the resulting mixture was stirred at room temperature for 20 min. The reaction mixture was cooled to -78°C (dry ice/acetone bath), and l-(2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-ethanone (40 g, 139 mmol) in THF (400 ml) was added dropwise over 2h. The reaction mixture was allowed to warm to -36°C, and was stirred at that temperature for 24 h, and further stirred at -32°C for another 24h. 3N NaOH (250 ml) was added, and the cooling bath was replaced by ice-water bath. Then 30 % hydrogen peroxide in water (250 ml) was added dropwise over 30 minutes. The ice water bath was removed, and the mixture was stirred at room temperature for 4h. The organic layer was separated, concentrated and re-dissolved in ether (200 ml). The aqueous layer was extracted with ether (2 x 200 ml). The combined organic layers were washed with IN aq. NaOH (4 x 100 ml), brine, and dried over sodium sulfate. Removal of solvent gave crude product which was purified by column chromatography using 2 to 5% ethyl acetate in hexane as solvent to give desired alcohol 36.2 g (90 %, e.e. >95%). The alcohol (36.2 g) was crystallized from hexane (80 ml) to obtain R-l-(2-bromo-4-chloro- phenyl)-2,2,2-trifiuoro-ethanol 28.2 g (70 %; 99-100 % e.e.). 1H-NMR (400 MHz, CDCl3) δ (ppm) 5.48 (m, IH), 7.40 (d, IH), 7.61 (d, 2H). Step 3: Synthesis of R-l-r4-chloro-2-(3-methyl-pyrazol-l-vπ-phenyl1-2.2.2-trifluoro- ethanol. R-l-(2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-ethanol (15.65g, 54.06 mmol), 3- methylpyrazole (5.33 g, 65 mmol), CuI (2.06 g, 10.8 mmol), K2CO3 (15.7 g, 113.5 mmol), (lR,2R)-N,N’-dimethyl-cyclohexane-l,2-diamine (1.54 g, 10.8 mmol) and toluene (80 ml) were combined in a 250 ml pressure tube and heated to 1300C (oil bath temperature) for 12 h. The reaction mixture was diluted with ethyl acetate and washed with H2O (4 x 100 ml), brine, and dried over sodium sulfate. Removal of solvent gave a crude product, which was purified by ISCO column chromatography using 5-10 % ethyl acetate in hexane as solvent to get R-I- [4-chloro-2-(3-methyl-pyrazol-l-yl)-phenyl]-2,2,2-trifluoro-ethanol (13.5 g; 86 %). 1H-NMR (400 MHz, CDCl3): δ (ppm) 2.30(s, 3H), 4.90(m, IH), 6.20(s, IH), 6.84(d, IH), 7.20(s, IH), 7.30(d, IH), 7.50(d, IH).
Step 4: Synthesis of (S)-2-Amino-3- r4-(2-amino-6- (R-I- r4-chloro-2-(3-methyl- pyrazol- 1 -ylVphenyl~|-2,2.,2-trifluoro-ethoxy| -pyrimidin-4-yl)-phenyU -propionic acid ethyl ester. R-l-[4-chloro-2-(3-methyl-pyrazol-l-yl)-phenyl]-2,2,2-trifluoro-ethanol (17.78 g, 61.17 mmol), (S)-3-[4-(2-amino-6-chloro-pyrimidine-4-yl)-phenyl]-2-tert- butoxycarbonylamino-propionic acid (20.03 g, 51 mmol), 1,4-dioxane (250 ml), and Cs2CO3 (79.5 g, 244 mmol) were combined in a 3-necked 500 ml RB flask and heated to 1000C (oil bath temperature) for 12-24 h. The progress of reaction was monitored by LCMS. After the completion of the reaction, the mixture was cooled to 600C, and water (250 ml) and THF (400 ml) were added. The organic layer was separated and washed with brine (150 ml). The solvent was removed to give crude BOC protected product, which was taken in THF (400 ml), 3N HCl (200 ml). The mixture was heated at 35-400C for 12h. THF was removed in vacuo. The remaining aqueous layer was extracted with isopropyl acetate (2x 100 ml) and concentrated separately to recover the unreacted alcohol (3.5 g). Traces of remaining organic solvent were removed from the aqueous fraction under vacuum.
To a IL beaker equipped with a temperature controller and pH meter, was added H3PO4 (40 ml, 85 % in water) and water (300 ml) then 50 % NaOH in water to adjust pH to 6.15. The temperature was raised to 58°C and the above acidic aqueous solution was added dropwise into the buffer with simultaneous addition of 50 % NaOH solution in water so that the pH was maintained between 6.1 to 6.3. Upon completion of addition, precipitated solid was filtered and washed with hot water (50-600C) (2 x 200 ml) and dried to give crude (S)-2- amino-3-[4-(2-amino-6-{R-l-[4-chloro-2-(3-methyl-pyrazol-l-yl)-phenyl]-2,2,2-trifluoro- ethoxy}-pyrimidin-4-yl)-phenyl} -propionic acid (26.8 g; 95 %). LCMS and HPLC analysis indicated the compound purity was about 96-97 %. To anhydrous ethanol (400 ml) was added SOCl2 (22 ml, 306 mmol) dropwise at 0-
5°C. Crude acid (26.8 g ) from the above reaction was added. The ice water bath was removed, and the reaction mixture was heated at 40-450C for 6-12h. After the reaction was completed, ethanol was removed in vacuo. To the residue was added ice water (300 ml), and extracted with isopropyl acetate (2 x 100 ml). The aqueous solution was neutralized with saturated Na2CO3 to adjust the pH to 6.5. The solution was extracted with ethyl acetate (2 x 300 ml). The combined ethyl acetate layer was washed with brine and concentrated to give 24 g of crude ester (HPLC purity of 96-97 %). The crude ester was then purified by ISCO column chromatography using 5 % ethanol in DCM as solvent to give (S)-2-amino-3-[4-(2- amino-6- (R- 1 -[4-chloro-2-(3-methyl-pyrazol- 1 -yl)-phenyl]-2,2,2-trifluoro-ethoxy} – pyrimidin-4-yl)-phenyl} -propionic acid ethyl ester (20.5g; 70 %; HPLC purity of 98 %). LCMS M+l = 575. 1H-NMR (400 MHz, CD3OD): δ (ppm) 1.10 (t, 3H), 2.25 (s, 3H), 2.85 (m, 2H), 3.65 (m, IH), 4.00 (q, 2H), 6.35 (s, IH), 6.60 (s, IH), 6.90 (m, IH), 7.18 (d, 2H), 7.45 (m, 2H), 7.70 (d, IH), 7.85 (m, 3H).
PATENT
WO 2011056916
https://www.google.com/patents/WO2011056916A1?cl=en
PATENT
WO 2010065333
CLIP,……..PL CHECK ERROR

CONFUSION ON CODES, CLEAR PIC BELOW……LINK
Description of Telotristat Etiprate
Telotristat etiprate is the hippurate salt of telotristat ethyl.
Telotristat ethyl, also known as LX1032, has the chemical name, CAS identifier, and chemical structure shown below:
Chemical name: (S)-ethyl 2-amino-3-(4-(2-amino-6-((R)-1-(4-chloro-2-(3-methyl-1H-pyrazol-1-yl)phenyl)-2,2,2-trifluoroethoxy)pyrimidin-4-yl)phenyl)propanoate
CAS Registry number: 1033805-22-9
Chemical structure:

Telotristat etiprate, also known as LX1606, is the hippurate salt of telotristat ethyl, and has the chemical name, CAS identifier, and chemical structure shown below:
Chemical Name: (S)-ethyl 2-amino-3-(4-(2-amino-6-((R)-1-(4-chloro-2-(3-methyl-1H-pyrazol-1-yl)phenyl)-2,2,2-trifluoroethoxy)pyrimidin-4-yl)phenyl)propanoate 2-benzamidoacetate
CAS Registry number: 1137608-69-5
Chemical Structure:

Description of LX1033
Telotristat, also known as LX1033, has the chemical name, CAS identifier and chemical structure shown below:
Chemical Name: (S)-2-amino-3-(4-(2-amino-6-((R)-1-(4-chloro-2-(3-methyl-1H-pyrazol-1-yl)phenyl)-2,2,2-trifluoroethoxy)pyrimidin-4-yl)phenyl)propanoic acid
CAS Registry number: 1033805-28-5
Chemical Structure:
REFERENCES
Kulke, M.H.; Hoersch, D.; Caplin, M.E.; et al.
Telotristat ethyl, a tryptophan hydroxylase inhibitor for the treatment of carcinoid syndrome
J Clin Oncol 2017, 35(1): 14
| WO2010056992A1 * | Nov 13, 2009 | May 20, 2010 | The Trustees Of Columbia University In The City Of New York | Methods of preventing and treating low bone mass diseases |
| US7709493 | May 20, 2009 | May 4, 2010 | Lexicon Pharmaceuticals, Inc. | 4-phenyl-6-(2,2,2-trifluoro-1-phenylethoxy)pyrimidine-based compounds and methods of their use |
| US20090088447 * | Sep 25, 2008 | Apr 2, 2009 | Bednarz Mark S | Solid forms of (s)-ethyl 2-amino-3-(4-(2-amino-6-((r)-1-(4-chloro-2-(3-methyl-1h-pyrazol-1-yl)phenyl)-2,2,2-trifluoroethoxy)-pyrimidin-4-yl)phenyl)propanoate and methods of their use |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US9199994 | Sep 5, 2014 | Dec 1, 2015 | Karos Pharmaceuticals, Inc. | Spirocyclic compounds as tryptophan hydroxylase inhibitors |
| US9512122 | Sep 1, 2015 | Dec 6, 2016 | Karos Pharmaceuticals, Inc. | Spirocyclic compounds as tryptophan hydroxylase inhibitors |
///////////telotristat ethyl, fast track designation,priority review,orphan drug designation, Xermelo , Woodlands, Texas-based, Lexicon Pharmaceuticals, Inc, fda 2017, LX 1606, LX 1032
O=C(OCC)[C@@H](N)Cc1ccc(cc1)c2cc(nc(N)n2)O[C@H](c3ccc(Cl)cc3n4ccc(C)n4)C(F)(F)F
O=C(OCC)[C@@H](N)CC1=CC=C(C2=NC(N)=NC(O[C@H](C3=CC=C(Cl)C=C3N4N=C(C)C=C4)C(F)(F)F)=C2)C=C1.O=C(O)CNC(C5=CC=CC=C5)=O
Belinostat (PXD101), a novel HDAC inhibitor

Belinostat (PXD101)
FAST TRACK FDA , ORPHAN STATUS
PXD101;PX105684;PXD-101;PXD 101;PX-105684
UNII:F4H96P17NZ
N-Hydroxy-3-(3-phenylsulphamoylphenyl)acrylamide
N-HYDROXY-3-[3-[(PHENYLAMINO)SULFONYL]PHENYL]-2-PROPENAMIDE
NSC726630

July 3, 2014 — The U.S. Food and Drug Administration today approved Beleodaq (belinostat) for the treatment of patients with peripheral T-cell lymphoma (PTCL), a rare and fast-growing type of non-Hodgkin lymphoma (NHL). The action was taken under the agency’s accelerated approval program.
Belinostat (PXD101) is a novel HDAC inhibitor with IC50 of 27 nM, with activity demonstrated in cisplatin-resistant tumors.
CLINICAL TRIALS…http://clinicaltrials.gov/search/intervention=Belinostat+OR+PXD101
MP 172–174 °C, (lit.(@) 172 °C). 1H NMR (400 MHz, DMSO-d6) δ = 10.75–10.42 (m, 2H), 9.15 (s, 1H), 7.92 (s, 1H), 7.78 (d, J = 7.8 Hz, 1H), 7.71 (d, J = 7.8 Hz, 1H), 7.56 (d, J = 7.8 Hz, 1H),7.47 (d, J = 15.8 Hz, 1H), 7.24 (m, 2H), 7.10–7.01 (m, 3H), 6.51 (d, J = 15.8 Hz, 1H). MS (ESI): m/z = 318.6 [M+H] +.
@ Finn, P. W.; Bandara, M.; Butcher, C.; Finn, A.; Hollinshead, R.; Khan, N.; Law, N.; Murthy, S.; Romero,R.; Watkins, C.; Andrianov, V.; Bokaldere, R. M.; Dikovska, K.; Gailite, V.; Loza, E.; Piskunova, I.;Starchenkov, I.; Vorona, M.; Kalvinsh, I. Helv. Chim. Acta 2005, 88, 1630, DOI: 10.1002/hlca.200590129
Beleodaq and Folotyn are marketed by Spectrum Pharmaceuticals, Inc., based in Henderson, Nevada. Istodax is marketed by Celgene Corporation based in Summit, New Jersey.
Belinostat was granted orphan drug status for the treatment of Peripheral T-cell lymphoma (PTCL) in the US in September 2009 and the EU in October 2012. In July 2015, an orphan drug designation has also been granted for malignant thymoma in the EU.
Belinostat received its first global approval in the US-FDA on 3 July 2014 for the intravenous (IV) treatment of relapsed or refractory PTCL in adults.
Belinostat was approved by the U.S. Food and Drug Administration (FDA) on July 3, 2014. It was originally developed by CuraGen Pharma,then developed by Spectrum Pharmaceuticals cooperating with Onxeo, then marketed as Beleodaq® by Spectrum.
Beleodaq is a pan-histone deacetylase (HDAC) inhibitor selectively causing the accumulation of acetylated histones and other proteinsin tumor cells. It is indicated for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma (PTCL).
Beleodaq® is available as lyophilized powder for intravenous infusion, containing 500 mg of free Belinostat. The recommended dose is 1,000 mg/m2 once daily on days 1-5 of a 21-day cycle.

Index:
| MW 318.07 | |
| MF | C15H14N2O4S |
414864-00-9 cas no
866323-14-0
(2E)-N-hydroxy-3-[3-(phenylsulfamoyl)phenyl]acrylamide
A novel HDAC inhibitor
PTCL comprises a diverse group of rare diseases in which lymph nodes become cancerous. In 2014, the National Cancer Institute estimates that 70,800 Americans will be diagnosed with NHL and 18,990 will die. PTCL represents about 10 to 15 percent of NHLs in North America.Belinostat inhibits the growth of tumor cells (A2780, HCT116, HT29, WIL, CALU-3, MCF7, PC3 and HS852) with IC50 from 0.2-0.66 μM. PD101 shows low activity in A2780/cp70 and 2780AD cells. Belinostat inhibits bladder cancer cell growth, especially in 5637 cells, which shows accumulation of G0-G1 phase, decrease in S phase, and increase in G2-M phase. Belinostat also shows enhanced tubulin acetylation in ovarian cancer cell lines. A recent study shows that Belinostat activates protein kinase A in a TGF-β signaling-dependent mechanism and decreases survivin mRNA.


Beleodaq works by stopping enzymes that contribute to T-cells, a type of immune cell, becoming cancerous. It is intended for patients whose disease returned after treatment (relapsed) or did not respond to previous treatment (refractory).
“This is the third drug that has been approved since 2009 for the treatment of peripheral T-cell lymphoma,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Today’s approval expands the number of treatment options available to patients with serious and life-threatening diseases.”
The FDA granted accelerated approval to Folotyn (pralatrexate) in 2009 for use in patients with relapsed or refractory PTCL and Istodax (romidepsin) in 2011 for the treatment of PTCL in patients who received at least one prior therapy.
The safety and effectiveness of Beleodaq was evaluated in a clinical study involving 129 participants with relapsed or refractory PTCL. All participants were treated with Beleodaq until their disease progressed or side effects became unacceptable. Results showed 25.8 percent of participants had their cancer disappear (complete response) or shrink (partial response) after treatment.
The most common side effects seen in Beleodaq-treated participants were nausea, fatigue, fever (pyrexia), low red blood cells (anemia), and vomiting.
The FDA’s accelerated approval program allows for approval of a drug based on surrogate or intermediate endpoints reasonably likely to predict clinical benefit for patients with serious conditions with unmet medical needs. Drugs receiving accelerated approval are subject to confirmatory trials verifying clinical benefit. Beleodaq also received orphan product designation by the FDA because it is intended to treat a rare disease or condition.
Belinostat (trade name Beleodaq, previously known as PXD101) is a histone deacetylase inhibitor drug developed by TopoTargetfor the treatment of hematological malignancies and solid tumors.[2]
It was approved in July 2014 by the US FDA to treat peripheral T-cell lymphoma.[3]
In 2007 preliminary results were released from the Phase II clinical trial of intravenous belinostat in combination with carboplatin andpaclitaxel for relapsed ovarian cancer.[4] Final results in late 2009 of a phase II trial for T-cell lymphoma were encouraging.[5]Belinostat has been granted orphan drug and fast track designation by the FDA,[6] and was approved in the US for the use againstperipheral T-cell lymphoma on 3 July 2014.[3] It is not approved in Europe as of August 2014.[7]
The approved pharmaceutical formulation is given intravenously.[8]:180 Belinostat is primarily metabolized by UGT1A1; the initial dose should be reduced if the recipient is known to be homozygous for the UGT1A1*28 allele.[8]:179 and 181
NCI: A novel hydroxamic acid-type histone deacetylase (HDAC) inhibitor with antineoplastic activity. Belinostat targets HDAC enzymes, thereby inhibiting tumor cell proliferation, inducing apoptosis, promoting cellular differentiation, and inhibiting angiogenesis. This agent may sensitize drug-resistant tumor cells to other antineoplastic agents, possibly through a mechanism involving the down-regulation of thymidylate synthase


The study of inhibitors of histone deacetylases indicates that these enzymes play an important role in cell proliferation and differentiation. The inhibitor Trichostatin A (TSA) (Yoshida et al., 1990a) causes cell cycle arrest at both G1 and G2 phases (Yoshida and Beppu, 1988), reverts the transformed phenotype of different cell lines, and induces differentiation of Friend leukaemia cells and others (Yoshida et al., 1990b). TSA (and SAHA) have been reported to inhibit cell growth, induce terminal differentiation, and prevent the formation of tumours in mice (Finnin et al., 1999).
Trichostatin A (TSA)

Suberoylanilide Hydroxamic Acid (SAHA)

Cell cycle arrest by TSA correlates with an increased expression of gelsolin (Hoshikawa et al., 1994), an actin regulatory protein that is down regulated in malignant breast cancer (Mielnicki et al., 1999). Similar effects on cell cycle and differentiation have been observed with a number of deacetylase inhibitors (Kim et al., 1999). Trichostatin A has also been reported to be useful in the treatment of fibrosis, e.g., liver fibrosis and liver cirrhosis. See, e.g., Geerts et al., 1998.
Recently, certain compounds that induce differentiation have been reported to inhibit histone deacetylases. Several experimental antitumour compounds, such as trichostatin A (TSA), trapoxin, suberoylanilide hydroxamic acid (SAHA), and phenylbutyrate have been reported to act, at least in part, by inhibiting histone deacetylase (see, e.g., Yoshida et al., 1990; Richon et al., 1998; Kijima et al., 1993). Additionally, diallyl sulfide and related molecules (see, e.g., Lea et al., 1999), oxamflatin (see, e.g., Kim et al., 1999), MS-27-275, a synthetic benzamide derivative (see, e.g., Saito et al., 1999; Suzuki et al., 1999; note that MS-27-275 was later re-named as MS-275), butyrate derivatives (see, e.g., Lea and Tulsyan, 1995), FR901228 (see, e.g., Nokajima et al., 1998), depudecin (see, e.g., Kwon et al., 1998), and m-carboxycinnamic acid bishydroxamide (see, e.g., Richon et al., 1998) have been reported to inhibit histone deacetylases. In vitro, some of these compounds are reported to inhibit the growth of fibroblast cells by causing cell cycle arrest in the G1 and G2 phases, and can lead to the terminal differentiation and loss of transforming potential of a variety of transformed cell lines (see, e.g., Richon et al, 1996; Kim et al., 1999; Yoshida et al., 1995; Yoshida & Beppu, 1988). In vivo, phenybutyrate is reported to be effective in the treatment of acute promyelocytic leukemia in conjunction with retinoic acid (see, e.g., Warrell et al., 1998). SAHA is reported to be effective in preventing the formation of mammary tumours in rats, and lung tumours in mice (see, e.g., Desai et al., 1999).
The clear involvement of HDACs in the control of cell proliferation and differentiation suggest that aberrant HDAC activity may play a role in cancer. The most direct demonstration that deacetylases contribute to cancer development comes from the analysis of different acute promyelocytic leukaemias (APL). In most APL patients, a translocation of chromosomes 15 and 17 (t(15;17)) results in the expression of a fusion protein containing the N-terminal portion of PML gene product linked to most of RARσ (retinoic acid receptor). In some cases, a different translocation (t(11 ;17)) causes the fusion between the zinc finger protein PLZF and RARα. In the absence of ligand, the wild type RARα represses target genes by tethering HDAC repressor complexes to the promoter DNA. During normal hematopoiesis, retinoic acid (RA) binds RARα and displaces the repressor complex, allowing expression of genes implicated in myeloid differentiation. The RARα fusion proteins occurring in APL patients are no longer responsive to physiological levels of RA and they interfere with the expression of the RA- inducible genes that promote myeloid differentiation. This results in a clonal expansion of promyelocytic cells and development of leukaemia. In vitro experiments have shown that TSA is capable of restoring RA-responsiveness to the fusion RARα proteins and of allowing myeloid differentiation. These results establish a link between HDACs and oncogenesis and suggest that HDACs are potential targets for pharmaceutical intervention in APL patients. (See, for example, Kitamura et al., 2000; David et al., 1998; Lin et al., 1998).
BELINOSTAT
Furthermore, different lines of evidence suggest that HDACs may be important therapeutic targets in other types of cancer. Cell lines derived from many different cancers (prostate, coloreetal, breast, neuronal, hepatic) are induced to differentiate by HDAC inhibitors (Yoshida and Horinouchi, 1999). A number of HDAC inhibitors have been studied in animal models of cancer. They reduce tumour growth and prolong the lifespan of mice bearing different types of transplanted tumours, including melanoma, leukaemia, colon, lung and gastric carcinomas, etc. (Ueda et al., 1994; Kim et al., 1999).
Psoriasis is a common chronic disfiguring skin disease which is characterised by well-demarcated, red, hardened scaly plaques: these may be limited or widespread. The prevalence rate of psoriasis is approximately 2%, i.e., 12.5 million sufferers in the triad countries (US/Europe/Japan). While the disease is rarely fatal, it clearly has serious detrimental effects upon the quality of life of the patient: this is further compounded by the lack of effective therapies. Present treatments are either ineffective, cosmetically unacceptable, or possess undesired side effects. There is therefore a large unmet clinical need for effective and safe drugs for this condition. Psoriasis is a disease of complex etiology. Whilst there is clearly a genetic component, with a number of gene loci being involved, there are also undefined environmental triggers. Whatever the ultimate cause of psoriasis, at the cellular level, it is characterised by local T-cell mediated inflammation, by keratinocyte hyperproliferation, and by localised angiogenesis. These are all processes in which histone deacetylases have been implicated (see, e.g., Saunders et al., 1999; Bernhard et al, 1999; Takahashi et al, 1996; Kim et al , 2001 ). Therefore HDAC inhibitors may be of use in therapy for psoriasis. Candidate drugs may be screened, for example, using proliferation assays with T-cells and/or keratinocytes.
CLIP
PXD101/Belinostat®
(E)-N-hydroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide, also known as PXD101 and Belinostat®, shown below, is a well known histone deacetylate (HDAC) inhibitor. It is being developed for treatment of a range of disorders mediated by HDAC, including proliferative conditions (such as cancer and psoriasis), malaria, etc.

PXD101 was first described in WO 02/30879 A2. That document describes a multi-step method of synthesis which may conveniently be illustrated by the following scheme.


PATENT
GENERAL SYNTHESIS
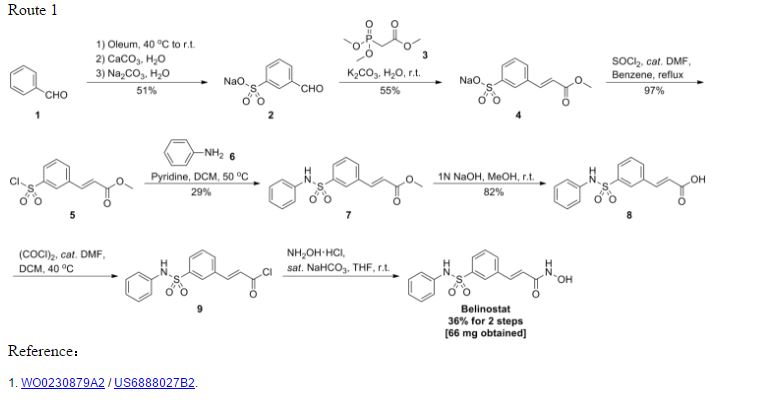
IGNORE 10

ENTRY 45 IS BELINOSTAT
Scheme 1

By using amines instead of aniline, the corresponding products may be obtained. The use of aniline, 4-methoxyaniline, 4-methylaniline, 4-bromoaniline, 4-chloroaniline, 4-benzylamine, and 4-phenethyamine, among others, is described in the Examples below.
In another method, a suitable amino acid (e.g., ω-amino acid) having a protected carboxylic acid (e.g., as an ester) and an unprotected amino group is reacted with a sulfonyl chloride compound (e.g., RSO2CI) to give the corresponding sulfonamide having a protected carboxylic acid. The protected carboxylic acid is then deprotected using base to give the free carboxylic acid, which is then reacted with, for example, hydroxylamine 2-chlorotrityl resin followed by acid (e.g., trifluoroacetic acid), to give the desired carbamic acid.
One example of this approach is illustrated below, in Scheme 2, wherein the reaction conditions are as follows: (i) RSO2CI, pyridine, DCM, room temperature, 12 hours; (ii) 1 M LiOH or 1 M NaOH, dioxane, room temperature, 3-48 hours; (iii) hydroxylamine 2-chlorotrityl resin, HOAt, HATU, DIPEA, DCM, room temperature, 16 hours; and (iv) TFA/DCM (5:95, v/v), room temperature, 1.5 hours.
Scheme 2

Additional methods for the synthesis of compounds of the present invention are illustrated below and are exemplified in the examples below.
Scheme 3A

Scheme 3B

Scheme 4


Scheme 8

Scheme 9

PATENT
SYNTHESIS
Example 1
3-Formylbenzenesulfonic acid, sodium salt (1)

Oleum (5 ml) was placed in a reaction vessel and benzaldehyde (2.00 g, 18.84 mmol) was slowly added not exceeding the temperature of the reaction mixture more than 30°C. The obtained solution was stirred at 40°C for ten hours and at ambient temperature overnight. The reaction mixture was poured into ice and extracted with ethyl acetate. The aqueous phase was treated with CaC03 until the evolution of C02 ceased (pH~6-7), then the precipitated CaSO4was filtered off and washed with water. The filtrate was treated with Na2CO3 until the pH of the reaction medium increased to pH 8, obtained CaCO3 was filtered off and water solution was evaporated in vacuum. The residue was washed with methanol, the washings were evaporated and the residue was dried in desiccator over P2Oβ affording the title compound (2.00 g, 51%). 1H NMR (D20), δ: 7.56-8.40 (4H, m); 10.04 ppm (1 H, s).
Example 2 3-(3-Sulfophenyl)acrylic acid methyl ester, sodium salt (2)

Sodium salt of 3-formylbenzenesulfonic acid (1) (1.00 g, 4.80 mmol), potassium carbonate (1.32 g, 9.56 mmol), trimethyl phosphonoacetate (1.05 g, 5.77 mmol) and water (2 ml) were stirred at ambient temperature for 30 min., precipitated solid was filtered and washed with methanol. The filtrate was evaporated and the title compound (2) was obtained as a white solid (0.70 g, 55%). 1H NMR (DMSO- dβl HMDSO), δ: 3.68 (3H, s); 6.51 (1 H, d, J=16.0 Hz); 7.30-7.88 (5H, m).
Example 3 3-(3-Chlorosulfonylphenyl)acrylic acid methyl ester (3)

To the sodium salt of 3-(3-sulfophenyl)acrylic acid methyl ester (2) (0.670 g, 2.53 mmol) benzene (2 ml), thionyl chloride (1.508 g, 0.9 ml, 12.67 mmol) and 3 drops of dimethylformamide were added and the resultant suspension was stirred at reflux for one hour. The reaction mixture was evaporated, the residue was dissolved in benzene (3 ml), filtered and the filtrate was evaporated to give the title compound (0.6’40 g, 97%).
Example 4 3-(3-Phenylsulfamoylphenyl)acrylic acid methyl ester (4a)

A solution of 3-(3-chlorosulfonylphenyl)acrylic acid methyl ester (3) (0.640 g, 2.45 mmol) in dichloromethane (2 ml) was added to a mixture of aniline (0.465 g, 4.99 mmol) and pyridine (1 ml), and the resultant solution was stirred at 50°C for one hour. The reaction mixture was evaporated and the residue was partitioned between ethyl acetate and 10% HCI. The organic layer was washed successively with water, saturated NaCl, and dried (Na2S0 ). The solvent was removed and the residue was chromatographed on silica gel with chloroform-ethyl acetate (7:1 , v/v) as eluent. The obtained product was washed with diethyl ether to give the title compound (0.226 g, 29%). 1H NMR (CDCI3, HMDSO), δ: 3.72 (3H, s); 6.34 (1H, d, J=16.0 Hz); 6.68 (1 H, br s); 6.92-7.89 (10H, m).
Example 5 3-(3-Phenylsulfamoylphenyl)acrylic acid (5a)

3-(3-Phenylsulfamoylphenyl)acrylic acid methyl ester (4a) (0.220 g, 0.69 mmol) was dissolved in methanol (3 ml), 1N NaOH (2.08 ml, 2.08 mmol) was added and the resultant solution was stirred at ambient temperature overnight. The reaction mixture was partitioned between ethyl acetate and water. The aqueous layer was acidified with 10% HCI and stirred for 30 min. The precipitated solid was filtered, washed with water and dried in desiccator over P2Os to give the title compound as a white solid (0.173 g, 82%). Example 6 3-(3-Phenylsulfamoylphenyl)acryloyl chloride (6a)

To a suspension of 3-(3-phenylsulfamoylphenyl)acrylic acid (5a) (0.173 g, 0.57 mmol) in dichloromethane (2.3 ml) oxalyl chloride (0.17 ml, 1.95 mmol) and one drop of dimethylformamide were added. The reaction mixture was stirred at 40°C for one hour and concentrated under reduced pressure to give crude title compound (0.185 g).
Example 7
N-Hydroxy-3-(3-phenylsulfamoylphenyl)acrylamide (7a) (PX105684) BELINOSTAT

To a suspension of hydroxylamine hydrochloride (0.200 g, 2.87 mmol) in tetrahydrofuran (3.5 ml) a saturated NaHCOβ solution (2.5 ml) was added and the resultant mixture was stirred at ambient temperature for 10 min. To the reaction mixture a 3-(3-phenylsulfamoylphenyl)acryloyl chloride (6a) (0.185 g) solution in tetrahydrofuran (2.3 ml) was added and stirred at ambient temperature for one hour. The reaction mixture was partitioned between ethyl acetate and 2N HCI. The organic layer was washed successively with water and saturated NaCl, the solvent was removed and the residue was washed with acetonitrile and diethyl ether.
The title compound was obtained as a white solid (0.066 g, 36%), m.p. 172°C. BELINOSTAT

1H NMR (DMSO-d6, HMDSO), δ: 6.49 (1 H, d, J=16.0 Hz); 7.18-8.05 (10H, m); 9.16 (1 H, br s); 10.34 (1 H, s); 10.85 ppm (1 H, br s).
HPLC analysis on Symmetry C18column: impurities 4% (column size 3.9×150 mm; mobile phase acetonitrile – 0.1 M phosphate buffer (pH 2.5), 40:60; sample concentration 1 mg/ml; flow rate 0.8 ml/ min; detector UV 220 nm).
Anal. Calcd for C15Hι4N204S, %: C 56.59, H 4.43, N 8.80. Found, %: C 56.28, H 4.44, N 8.56.
PATENT
https://www.google.com/patents/CN102786448A?cl=en
Example: belinostat (compound of formula I) Preparation of

Methods of operation:
The compound of formula II (4. Og) added to the reactor, was added methanol 30ml, and stirred to dissolve, was added IM aqueous sodium hydroxide solution (38mL), stirred at room temperature overnight, the reaction was completed, ethyl acetate was added (IOmL) ^ K (20mL), stirred for 5 minutes, phase separation, the ethyl acetate phase was discarded, the aqueous phase was acidified with 10% hydrochloric acid to pH2, stirred at room temperature for 30 minutes, filtered, washed with water, and dried to give hydrolyzate 3. lg, yield rate of 81.6%.
The hydrolyzate (3. Og) added to the reactor, was added methylene chloride (53. 2g), dissolved with stirring, was added oxalyl chloride (2.8mL, 0.0032mol) at room temperature was added I drop DMF, reflux I hours, concentrated and the residue was dissolved in THF (30mL) alternate, the other to take a reaction flask was added hydroxylamine hydrochloride (3. 5g, 0.05mol), THF (50mL), saturated aqueous sodium bicarbonate (40mL), the mixture at room temperature under stirring for 10 minutes, then was added to spare, stirred at room temperature for I hour, the reaction was complete, at – at room temperature was added ethyl acetate (50mL), 2M hydrochloric acid (50mL), stirred for 5 minutes the phases were separated, the aqueous phase was discarded, the organic layer was washed with water, saturated brine, dried, filtered and concentrated to give crude product belinostat, recrystallized from ethyl acetate, 50 ° C and dried for 8 hours to give white crystals 2. 6g, yield 83.8%. . 1H-NMR (DMS0-d6, 400MHz) δ: 6 50 (1H, d, J = 16. OHz); 7 07 (d, J = 7. 8Hz, 2H); 7 16 (t.. , J = 7. 3Hz, 1H);. 7 25 (m, 2H);. 7 45 (t, J = 7. 8Hz, 1H);. 7 60 (d, J = 15. 9Hz, 1H); 7 . 62 (d, J = 7. 7Hz, 1H);. 7 75 (d, J = 7. 8Hz, 1H);. 7 88 (br s. ‘1H);. 9 17 (br s’ 1H); 10. 35 (s, 1H);. 10 82ppm (br s, 1H). ·
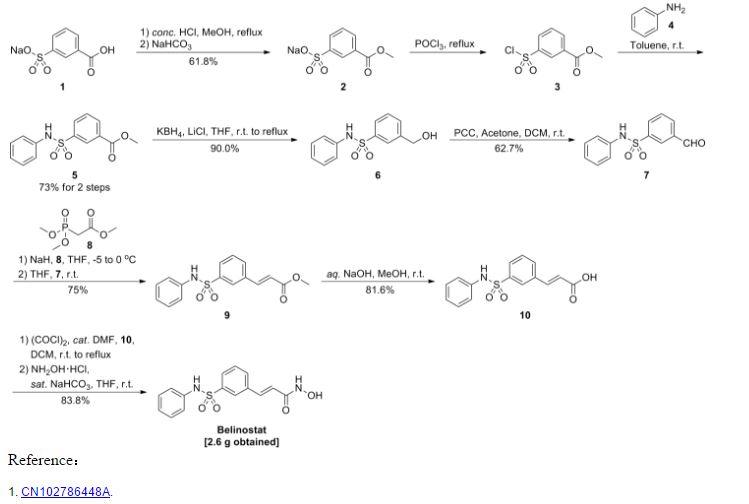
Step a): Preparation of Compound III

The carboxy benzene sulfonate (224g, Imol), anhydrous methanol (2300g), concentrated hydrochloric acid (188. 6g) refluxing
3-5 hours, filtered and the filtrate was added anhydrous sodium bicarbonate powder (200g), stirred for I hour, filtered, the filter residue was discarded, the filtrate was concentrated. The concentrate was added methanol (2000g), stirred at room temperature for 30 minutes, filtered and the filtrate was concentrated to dryness, 80 ° C and dried for 4 hours to give a white solid compound III147g, yield 61.8%.
Step b): Preparation of Compound IV

Compound III (50g, 0. 21mol), phosphorus oxychloride (250mL) was refluxed for 2_6 hours, completion of the reaction, cooled to
0-5 ° C, was slowly added to ice water, stirred for 2 hours and filtered to give a brown solid compound IV40 g, due to the instability of Compound IV, directly into the next reaction without drying.
Preparation of Compound V: [0040] Step c)

The aniline (5. 58g, 0. 06mol) and 30mL of toluene added to the reactor, stirred to dissolve, in step b) the resulting compound IV (7. 05g, O. 03mol) was dissolved in 60 ml of toluene, at room temperature dropwise added to the reactor, the addition was completed, stirring at room temperature for 1-2 hours, the reaction was completed, the filtered solid washed with water, and then recrystallized from toluene, 50 ° C and dried for 4 hours to obtain a white crystalline compound V6. Og, yield 73%. mp:.. 144 4-145 2. . .
1H- bandit R (CDCl3, 400MHz) δ:…. 3 92 (s, 3H); 6 80 (. Br s, 1H); 7 06-7 09 (m, 2H); 7 11. . -7 15 (m, 1H);.. 7 22-7 26 (m, 2H);. 7 51 (t, J = 7. 8Hz, 1H);.. 7 90-7 93 (dt, J = . 1.2,7 8Hz, 1H); 8 18-8 21 (dt, J = I. 4, 7. 8Hz, 1H);… 8 48 (t, J = L 6Hz, 1H).
IR v ™ r: 3243,3198,3081,2953,1705,1438,1345,766,702,681cm-1.
Step d): Preparation of Compound VI

The anhydrous lithium chloride 2. 32g, potassium borohydride 2. 96g, THF50mL added to the reactor, stirring evenly, Compound V (8g, 0. 027mol) was dissolved in 7mL of tetrahydrofuran, was slowly dropped into the reactor was heated under reflux for 5 hours, the reaction was completed, the force mouth 40mL water and ethyl acetate 40mL, stirred for half an hour, allowed to stand for separation, the organic layer was washed with 40mL water, concentrated under reduced pressure to give the crude product, the crude product was recrystallized from toluene, solid 50 V dried for 4 hours to give a white crystalline compound VI6. 82g, yield 90. O%. mp:.. 98 2-98 6. . .
1H-NMR (DMS0-d6, 400ΜΗζ) δ:….. 4 53 (s, 2H); 5 39 (s, 1H); 6 99-7 03 (m, 1H); 7 08- 7. ll (m, 2H);.. 7 19-7 24 (m, 2H);.. 7 45-7 52 (m, 2H);.. 7 61-7 63 (dt, J = I. 8 , 7 4Hz, 1H);.. 7 79 (br s, 1H);. 10. 26 (s, 1H).
IRv =: 3453,3130,2964,1488,1151,1031, 757,688cm_10
Step e): Preparation of Compound VII

After Compound VI (7.5g, 0.028mol) dissolved in acetone was added 7ml, dichloromethane was added 60mL, supported on silica gel was added PCC at room temperature 20g, stirred at room temperature for 12-24 hours, the reaction was complete, filtered and the filtrate was purified The layers were separated and the aqueous layer was discarded after the organic phase is washed 30mL5% aqueous sodium bicarbonate, evaporated to dryness under reduced pressure to give the crude product, the crude product was recrystallized from toluene, 50 ° C and dried for 8 hours to give white crystalline compound VII4. 7g, yield 62.7%. mp:.. 128 1-128 5 ° C.
1H- bandit R (CDCl3,400MHz) δ:…. 7 08-7 15 (m, 4Η); 7 · 23-7 27 (m, 2H); 7 · 60-7 64 (t, J = 7 7Hz, 1Η);.. 8 00 (d, J = 7. 6Hz, 1Η);. 8 04 (d, J = 7. 6Hz, 1Η);. 8 30 (br s’ 1Η).; 10. 00 (S, 1Η).
IR ν ™ Γ: 3213,3059,2964,2829,1687,1480,1348,1159,1082,758,679cm_10
Preparation of compounds of formula II: [0055] Step f)

phosphoryl trimethylorthoacetate (2. 93g, 0. 0161mol) added to the reaction vessel, THF30mL, stirring to dissolve, cooled to -5-0 ° C, was added sodium hydride (O. 8g, content 80%) , the addition was completed, stirring for 10-20 minutes, was added dropwise the compound VII (4g, O. 0156mol) and THF (20mL) solution, stirred for 1_4 hours at room temperature, the reaction was complete, 10% aqueous ammonium chloride solution was added dropwise 50mL, and then After addition of 50mL of ethyl acetate, stirred 30min rested stratification, the aqueous layer was discarded, the organic phase was concentrated under reduced pressure to give the crude product, the crude product was recrystallized from methanol 60mL, 50 ° C and dried for 8 hours to give white crystalline compound 113. 6g, yield 75%. mp:.. 152 0-152 5 ° C.
1H-Nmr (Cdci3JOOmHz) δ:…. 3 81 (s, 3H); 6 40 (d, J = 16. 0Hz, 1H); 6 79 (. Br s, 1H); 7 08 ( d, J = 7. 8Hz, 2H);. 7 14 (t, J = 7. 3Hz, 1H);. 7 24 (m, 2H);. 7 46 (t, J = 7. 8Hz, 1H); 7. 61 (d, J = 16. ΟΗζ, ΙΗ);. 7 64 (d, J = 7. 6Hz, 1H);. 7 75 (d, J = 7. 8Hz, 1H);. 7 89 (br . s, 1H).
IR v ^ :: 3172,3081,2954,2849,1698,1475,1345,1157,773,714,677cm-1.
PATENT
SYNTHESIS
US20100286279

CLIP
SYNTHESIS AND SPECTRAL DATA
Journal of Medicinal Chemistry, 2011 , vol. 54, 13 pg. 4694 – 4720
(E)-N-Hydroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide (28, belinostat, PXD101).
http://pubs.acs.org/doi/full/10.1021/jm2003552
http://pubs.acs.org/doi/suppl/10.1021/jm2003552/suppl_file/jm2003552_si_001.pdf
The methyl ester (27) (8.0 g) was prepared according to reported synthetic route,
(Watkins, C. J.; Romero-Martin, M.-R.; Moore, K. G.; Ritchie, J.; Finn, P. W.; Kalvinsh, I.;
Loza, E.; Dikvoska, K.; Gailite, V.; Vorona, M.; Piskunova, I.; Starchenkov, I.; Harris, C. J.;
Duffy, J. E. S. Carbamic acid compounds comprising a sulfonamide linkage as HDAC
inhibitors. PCT Int. Appl. WO200230879A2, April 18, 2002.)
but using procedure D (Experimental Section) or method described for 26 to convert the methyl ester to crude
hydroxamic acid which was further purified by chromatography (silica, MeOH/DCM = 1:10) to
afford 28 (PXD101) as off-white or pale yellow powder (2.5 g, 31%).
LC–MS m/z 319.0 ([M +H]+).
1H NMR (DMSO-d6) 12–9 (very broad, 2H), 7.90 (s, 1H), 7.76 (d, J = 7.7 Hz, 1H), 7.70 (d, J
= 7.8 Hz, 1H), 7.56 (t, J = 7.8 Hz, 1H), 7.44 (d, J = 15.8 Hz, 1H), 7.22 (t, J = 7.8 Hz, 2H), 7.08 (d,J = 7.8 Hz, 2H), 7.01 (t, J = 7.3 Hz, 1H), 6.50 (d, J = 15.8 Hz, 1H);
13C NMR (DMSO-d6) 162.1, 140.6, 138.0, 136.5, 135.9, 131.8, 130.0, 129.2, 127.1, 124.8, 124.1, 121.3, 120.4.
Anal.
(C15H14N2O4S) C, H, N
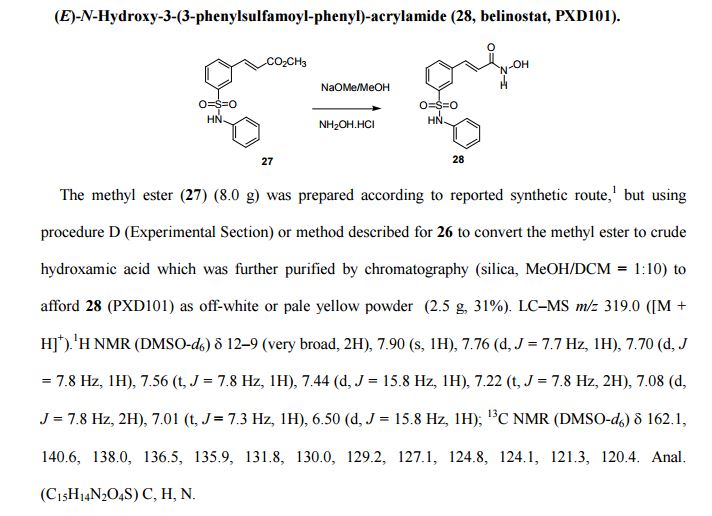
PATENT
SYNTHESIS
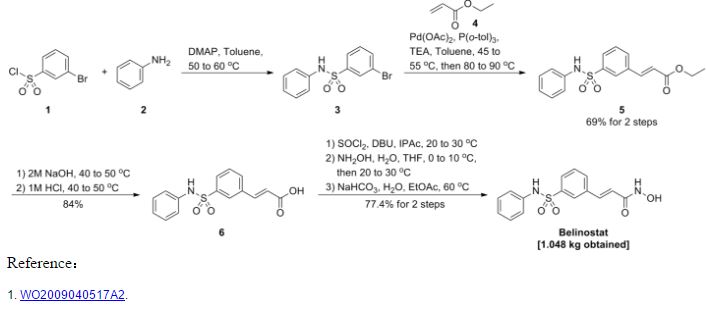
PXDIOI / Belinostat®
(E)-N-hydroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide, also known as PXD101 and Belinostat®, shown below, is a well known histone deacetylate (HDAC) inhibitor. It is being developed for treatment of a range of disorders mediated by HDAC, including proliferative conditions (such as cancer and psoriasis), malaria, etc.

PXD101 was first described in WO 02/30879 A2. That document describes a multi-step method of synthesis which may conveniently be illustrated by the following scheme.
Scheme 1
Not isolated

ed on (A)
on (D)

d on (H)

There is a need for alternative methods for the synthesis of PXD101 and related compounds for example, methods which are simpler and/or employ fewer steps and/or permit higher yields and/or higher purity product.
Scheme 5

DMAP, toluene



Synthesis 1 3-Bromo-N-phenyl-benzenesulfonamide (3)

To a 30 gallon (-136 L) reactor was charged aniline (2) (4.01 kg; 93.13 g/mol; 43 mol), toluene (25 L), and 4-(dimethylamino)pyridine (DMAP) (12 g), and the mixture was heated to 50-600C. 3-Bromobenzenesulfonyl chloride (1) (5 kg; 255.52 g/mol; 19.6 mol) was charged into the reactor over 30 minutes at 50-600C and progress of the reaction was monitored by HPLC. After 19 hours, toluene (5 L) was added due to losses overnight through the vent line and the reaction was deemed to be complete with no compound (1) being detected by HPLC. The reaction mixture was diluted with toluene (10 L) and then quenched with 2 M aqueous hydrochloric acid (20 L). The organic and aqueous layers were separated, the aqueous layer was discarded, and the organic layer was washed with water (20 L), and then 5% (w/w) sodium bicarbonate solution (20 L), while maintaining the batch temperature at 45-55°C. The batch was then used in the next synthesis.
Synthesis 2 (E)-3-(3-Phenylsulfamoyl-phenyl)-acrylic acid ethyl ester (5)

To the batch containing 3-bromo-N-phenyl-benzenesulfonamide (3) (the treated organic layer obtained in the previous synthesis) was added triethylamine (2.97 kg; 101.19 g/mol; 29.4 mol), tri(o-tolyl)phosphine (119 g; 304.37 g/mol; 0.4 mol), and palladium (II) acetate (44 g; 224.51 g/mol; 0.2 mol), and the resulting mixture was degassed four times with a vacuum/nitrogen purge at 45-55°C. Catalytic palladium (0) was formed in situ. The batch was then heated to 80-900C and ethyl acrylate (4) (2.16 kg; 100.12 g/mol; 21.6 mol) was slowly added over 2.75 hours. The batch was sampled after a further 2 hours and was deemed to be complete with no compound (3) being detected by HPLC. The batch was cooled to 45-55°C and for convenience was left at this temperature overnight.
The batch was then reduced in volume under vacuum to 20-25 L, at a batch temperature of 45-55°C, and ethyl acetate (20 L) was added. The batch was filtered and the residue washed with ethyl acetate (3.5 L). The residue was discarded and the filtrates were sent to a 100 gallon (-454 L) reactor, which had been pre-heated to 600C. The 30 gallon (-136 L) reactor was then cleaned to remove any residual Pd, while the batch in the 100 gallon (-454 L) reactor was washed with 2 M aqueous hydrochloric acid and water at 45-55°C. Once the washes were complete and the 30 gallon (-136 L) reactor was clean, the batch was transferred from the 100 gallon (-454 L) reactor back to the 30 gallon (-136 L) reactor and the solvent was swapped under vacuum from ethyl acetate/toluene to toluene while maintaining a batch temperature of 45-55°C (the volume was reduced to 20-25 L). At this point, the batch had precipitated and heptanes (10 L) were added to re-dissolve it. The batch was then cooled to 0-100C and held at this temperature over the weekend in order to precipitate the product. The batch was filtered and the residue was washed with heptanes (5 L). A sample of the wet-cake was taken for Pd analysis. The Pd content of the crude product (5) was determined to be 12.9 ppm.
The wet-cake was then charged back into the 30 gallon (-136 L) reactor along with ethyl acetate (50 L) and heated to 40-500C in order to obtain a solution. A sparkler filter loaded with 12 impregnated Darco G60® carbon pads was then connected to the reactor and the solution was pumped around in a loop through the sparkler filter. After 1 hour, a sample was taken and evaporated to dryness and analysed for Pd content. The amount of Pd was found to be 1.4 ppm. A second sample was taken after 2 hours and evaporated to dryness and analysed for Pd content. The amount of Pd had been reduced to 0.6 ppm. The batch was blown back into the reactor and held at 40-500C overnight before the solvent was swapped under vacuum from ethyl acetate to toluene while maintaining a batch temperature of 45-55°C (the volume was reduced to 20-25 L). At this point, the batch had precipitated and heptanes (10 L) were added to re-dissolve it and the batch was cooled to 0-100C and held at this temperature overnight in order to precipitate the product. The batch was filtered and the residue was washed with heptanes (5 L). The filtrate was discarded and the residue was dried at 45-55°C under vacuum for 25 hours. A first lot of the title compound (5) was obtained as an off-white solid (4.48 kg, 69% overall yield from 3-bromobenzenesulfonyl chloride (1)) with a Pd content of 0.4 ppm and a purity of 99.22% (AUC) by HPLC.
Synthesis 3 (E)-3-(3-Phenylsulfamoyl-phenyl)-acrvlic acid (6)

To the 30 gallon (-136 L) reactor was charged the (E)-3-(3-phenylsulfamoyl-phenyl)- acrylic acid ethyl ester (5) (4.48 kg; 331.39 g/mol; 13.5 mol) along with 2 M aqueous sodium hydroxide (17.76 L; -35 mol). The mixture was heated to 40-50°C and held at this temperature for 2 hours before sampling, at which point the reaction was deemed to be complete with no compound (5) being detected by HPLC. The batch was adjusted to pH 2.2 using 1 M aqueous hydrochloric acid while maintaining the batch temperature between 40-500C. The product had precipitated and the batch was cooled to 20-300C and held at this temperature for 1 hour before filtering and washing the cake with water (8.9 L). The filtrate was discarded. The batch was allowed to condition on the filter overnight before being charged back into the reactor and slurried in water (44.4 L) at 40-500C for 2 hours. The batch was cooled to 15-20°C, held for 1 hour, and then filtered and the residue washed with water (8.9 L). The filtrate was discarded. The crude title compound (6) was transferred to an oven for drying at 45-55°C under vacuum with a slight nitrogen bleed for 5 days (this was done for convenience) to give a white solid (3.93 kg, 97% yield). The moisture content of the crude material was measured using Karl Fischer (KF) titration and found to be <0.1% (w/w). To the 30 gallon (-136 L) reactor was charged the crude compound (6) along with acetonitrile (47.2 L). The batch was heated to reflux (about 80°C) and held at reflux for 2 hours before cooling to 0-10°C and holding at this temperature overnight in order to precipitate the product. The batch was filtered and the residue was washed with cold acetonitrile (7.9 L). The filtrate was discarded and the residue was dried under vacuum at 45-55°C for 21.5 hours. The title compound (6) was obtained as a fluffy white solid (3.37 kg, 84% yield with respect to compound (5)) with a purity of 99.89% (AUC) by HPLC.
Synthesis 4 (E)-N-Hvdroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide (PXD101) BELINOSTAT

To the 30 gallon (-136 L) reactor was charged (E)-3-(3-phenylsulfamoyl-phenyl)-acrylic acid (6) (3.37 kg; 303.34 g/mol; 11.1 mol) and a pre-mixed solution of 1 ,8-diazabicyclo[5.4.0]undec-7-ene (DBU) in isopropyl acetate (IPAc) (27 g in 30 L; 152.24 g/mol; 0.18 mol). The slurry was stirred and thionyl chloride (SOCI2) (960 mL; density ~1.631 g/mL; 118.97 g/mol; -13 mol) was added to the reaction mixture and the batch was stirred at 20-300C overnight. After 18.5 hours, the batch was sampled and deemed to be complete with no compound (6) being detected by HPLC. The resulting solution was transferred to a 100 L Schott reactor for temporary storage while the
30 gallon (-136 L) reactor was rinsed with isopropyl acetate (IPAc) and water. Deionized water (28.9 L) was then added to the 30 gallon (-136 L) reactor followed by 50% (w/w) hydroxylamine (6.57 L; -1.078 g/mL; 33.03 g/mol; -214 mol) and another charge of deionized water (1.66 L) to rinse the lines free of hydroxylamine to make a 10% (w/w) hydroxylamine solution. Tetrahydrofuran (THF) (6.64 L) was then charged to the
30 gallon (-136 L) reactor and the mixture was stirred and cooled to 0-100C. The acid chloride solution (from the 100 L Schott reactor) was then slowly charged into the hydroxylamine solution over 1 hour maintaining a batch temperature of 0-10°C during the addition. The batch was then allowed to warm to 20-300C. The aqueous layer was separated and discarded. The organic layer was then reduced in volume under vacuum while maintaining a batch temperature of less than 300C. The intention was to distill out 10-13 L of solvent, but this level was overshot. A larger volume of isopropyl acetate (IPAc) (16.6 L) was added and about 6 L of solvent was distilled out. The batch had precipitated and heptanes (24.9 L) were added and the batch was held at 20-30°C overnight. The batch was filtered and the residue was washed with heptanes (6.64 L). The filtrate was discarded and the residue was dried at 45-55°C under vacuum with a slight nitrogen bleed over the weekend. The title compound (PXD101) was obtained as a light orange solid (3.11 kg, 89% yield with respect to compound (6)) with a purity of 99.25% (AUC) by HPLC.
The title compound (PXD101) (1.2 kg, 3.77 mol) was dissolved in 8 volumes of 1:1 (EtOH/water) at 600C. Sodium bicarbonate (15.8 g, 5 mol%) was added to the solution. Water (HPLC grade) was then added at a rate of 65 mL/min while keeping the internal temperature >57°C. After water (6.6 L) had been added, crystals started to form and the water addition was stopped. The reaction mixture was then cooled at a rate of 10°C/90 min to a temperature of 0-10cC and then stirred at ambient temperature overnight. The crystals were then filtered and collected. The filter cake was washed by slurrying in water (2 x 1.2 L) and then dried in an oven at 45°C for 60 hours with a slight nitrogen bleed. 1.048 kg (87% recovery) of a light orange solid was recovered. Microscopy and XRPD data showed a conglomerate of irregularly shaped birefringant crystalline particles. The compound was found to contain 0.02% water.
As discussed above: the yield of compound (5) with respect to compound (1) was 69%. the yield of compound (6) with respect to compound (5) was 84%. the yield of PXD101 with respect to compound (6) was 89%.
PAPER
Synthetic Commun. 2010, 40, 2520-2524.
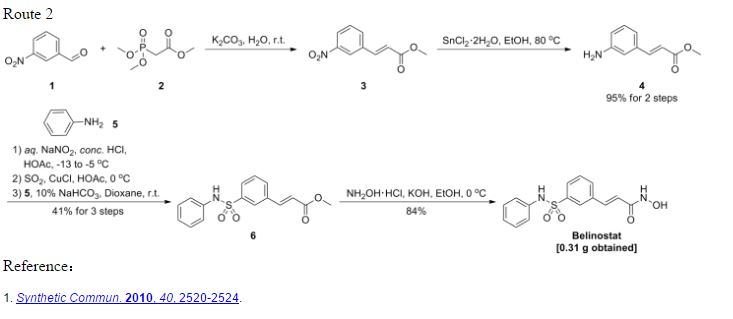
PATENT
FORMULATION
Formulation Studies
These studies demonstrate a substantial enhancement of HDACi solubility (on the order of a 500-fold increase for PXD-101) using one or more of: cyclodextrin, arginine, and meglumine. The resulting compositions are stable and can be diluted to the desired target concentration without the risk of precipitation. Furthermore, the compositions have a pH that, while higher than ideal, is acceptable for use.

UV Absorbance
The ultraviolet (UV absorbance E\ value for PXD-101 was determined by plotting a calibration curve of PXD-101 concentration in 50:50 methanol/water at the λmax for the material, 269 nm. Using this method, the E1i value was determined as 715.7.
Methanol/water was selected as the subsequent diluting medium for solubility studies rather than neat methanol (or other organic solvent) to reduce the risk of precipitation of the cyclodextrin.
Solubility in Demineralised Water
The solubility of PXD-101 was determined to be 0.14 mg/mL for demineralised water. Solubility Enhancement with Cvclodextrins
Saturated samples of PXD-101 were prepared in aqueous solutions of two natural cyclodextrins (α-CD and γ-CD) and hydroxypropyl derivatives of the α, β and Y cyclodextrins (HP-α-CD, HP-β-CD and HP-γ-CD). All experiments were completed with cyclodextrin concentrations of 250 mg/mL, except for α-CD, where the solubility of the cyclodextrin was not sufficient to achieve this concentration. The data are summarised in the following table. HP-β-CD offers the best solubility enhancement for PXD-101.

Phase Solubility Determination of HP-β-CD
The phase solubility diagram for HP-β-CD was prepared for concentrations of cyclodextrin between 50 and 500 mg/mL (5-50% w/v). The calculated saturated solubilities of the complexed HDACi were plotted against the concentration of cyclodextrin. See Figure 1.

CLIP
SPECTRUM
Tiny Biotech With Three Cancer Drugs Is More Alluring Takeover Bet Now
Forbes
The drug is one of Spectrum’s two drugs undergoing phase 3 clinical trials. Allergan paid Spectrum $41.5 million and will make additional payments of up to $304 million based on achieving certain milestones. So far, Raj Shrotriya, Spectrum’s chairman, …
CLIP
Copenhagen, December 10, 2013
Topotarget announces the submission of a New Drug Application (NDA) for belinostat for the treatment of relapsed or refractory (R/R) peripheral T-cell lymphoma (PTCL) to the US Food and Drug Administration (FDA). The NDA has been filed for Accelerated Approval with a request for Priority Review. Response from the FDA regarding acceptance to file is expected within 60 days from the FDA receipt date.
read all this here
PAPER
The Development of an Effective Synthetic Route of Belinostat
Key Laboratory of Structure-Based Drug Design & Discovery, Ministry of Education, Shenyang Pharmaceutical University, Shenyang 110016, China
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.6b00170
Publication Date (Web): July 12, 2016
Copyright © 2016 American Chemical Society
*E-mail: guoliang222@gmail.com.

A practical synthetic route of belinostat is reported. Belinostat was obtained via a five-step process starting from benzaldehyde and including addition reaction with sodium bisulfite, sulfochlorination with chlorosulfonic acid, sulfonamidation with aniline, Knoevenagel condensation, and the final amidation with hydroxylamine. Key to the strategy is the preparation of 3-formylbenzenesulfonyl chloride using an economical and practical protocol. The main advantages of the route include inexpensive starting materials and acceptable overall yield. The scale-up experiment was carried out to provide 169 g of belinostat with 99.6% purity in 33% total yield.
(E)-N-Hydroxy-3-((phenylamino)sulfonyl)phenyl)acrylamide (Belinostat, 1)
1
mp 172–174 °C, (lit.(@) 172 °C). 1H NMR (400 MHz, DMSO-d6) δ = 10.75–10.42 (m, 2H), 9.15 (s, 1H), 7.92 (s, 1H), 7.78 (d, J = 7.8 Hz, 1H), 7.71 (d, J = 7.8 Hz, 1H), 7.56 (d, J = 7.8 Hz, 1H),7.47 (d, J = 15.8 Hz, 1H), 7.24 (m, 2H), 7.10–7.01 (m, 3H), 6.51 (d, J = 15.8 Hz, 1H). MS (ESI): m/z = 318.6 [M+H] +.
@ Finn, P. W.; Bandara, M.; Butcher, C.; Finn, A.; Hollinshead, R.; Khan, N.; Law, N.; Murthy, S.; Romero,R.; Watkins, C.; Andrianov, V.; Bokaldere, R. M.; Dikovska, K.; Gailite, V.; Loza, E.; Piskunova, I.;Starchenkov, I.; Vorona, M.; Kalvinsh, I. Helv. Chim. Acta 2005, 88, 1630, DOI: 10.1002/hlca.200590129
Clip
Belinostat (Beleodaq),
Belinostat is a drug which was developed by Spectrum Pharmaceuticals and is currently marketed by Onxeo as Beleodaq. The
drug, which received fast track designation by the United States Food and Drug Administration (US FDA) and was approved for
the treatment of hematological malignancies and solid tumors associated with peripheral T-cell lymphoma (PTCL) in 2014,58 is a histone deacetylase (HDAC) inhibitor and is the third such treatment to receive accelerated approval for PTCL, the others being
vorinostat (Zolinza) and pralatrexate (Folotyn).58 Although belinostat was not yet approved in Europe as of August 2014,58 the
compound exhibits a safety profile considered to be acceptable for HDAC inhibitors–less than 25% of patients reported adverse
effects and these most frequently were nausea, fatigue, pyrexia,anemia, and emesis.58 While several different synthetic approaches
have been reported for the preparation of belinostat and related HDAC inhibitors,59–62 the most likely process-scale approach has
been described in a patent application filed by Reisch and co-workers at Topotarget UK, which exemplifies the synthesis described in
Scheme 8 on kilogram scale.63
Commercially available 3-bromobenzenesulfonyl chloride (41) was reacted with aniline in the presence of aqueous sodium carbonate
to deliver sulfonamide 42 in 94% yield. Next, this aryl bromide was subjected to a Heck reaction involving ethyl acrylate to
give rise to cinnamate ester 43, which was immediately saponified under basic conditions and acidic workup to furnish the corresponding acid 44. This acid was activated as the corresponding acid chloride prior to subjection to hydroxylamine under basic conditions to form the hydroxamic acid, which was then recrystallized from an 8:1 ethanol/water mixture in the presence of a catalytic
amount of sodium bicarbonate to furnish crystalline belinostat (VI) in 87% overall yield from acid 44.61
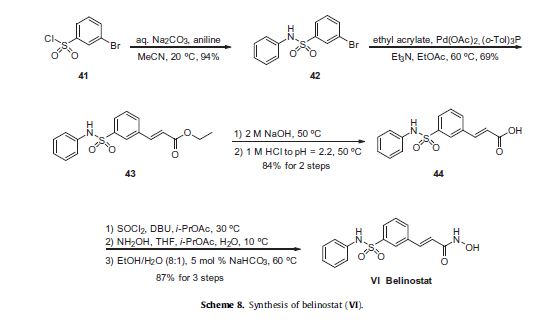
Lee, H. Z.; Kwitkowski, V. E.; Del Valle, P. L.; Ricci, M. S.; Saber, H.;Habtemariam, B. A.; Bullock, J.; Bloomquist, E.; Li Shen, Y.; Chen, X. H.;Brown, J.; Mehrotra, N.; Dorff, S.; Charlab, R.; Kane, R. C.; Kaminskas, E.;Justice, R.; Farrell, A. T.; Pazdur, R. Clin. Cancer Res. 2015, 21, 2666.
59. Qian, J.; Zhang, G.; Qin, H.; Zhu, Y.; Xiao, Y. CN Patent 102786448A, 2012.
60. Wang, H.; Yu, N.; Chen, D.; Lee, K. C.; Lye, P. L.; Chang, J. W.; Deng, W.; Ng, M.C.; Lu, T.; Khoo, M. L.; Poulsen, A.; ngthongpitag, K.; Wu, X.; Hu, C.; Goh, K.C.; Wang, X.; Fang, L.; Goh, K. L.; Khng, H. H.; Goh, S. K.; Yeo, P.; Liu, X.; Bonday, Z.; Wood, J. M.; Dymock, B. W.; Kantharaj, E.; Sun, E. T. J. Med. Chem.2011, 54, 4694.
61. Yang, L.; Xue, X.; Zhang, Y. Synth. Comm. 2010, 40, 2520.
CLIP
Let’s Research !!!!!

Helv Chim Acta 2005, 88(7), 1630-1657: It is first reported synthesis for Belinostat and many other derivatives. The procedure uses oleum, thionyl chloride (SOCl2) as well as oxalyl chloride (COCl)2, no wonder better procedures were derived from it. ABOVE
Synth Comm 2010, 40(17), 2520–2524: The synthesis avoids the use of the extremely corrosive oleum and thionyl chloride (SOCl2) and therefore is possibly better for scaled-up production. Second, synthetic steps do not involve tedious separations and give a better overall yield. BELOW Identifications:
Identifications:
Synth Comm 2010, 40(17), 2520–2524: The synthesis avoids the use of the extremely corrosive oleum and thionyl chloride (SOCl2) and therefore is possibly better for scaled-up production. Second, synthetic steps do not involve tedious separations and give a better overall yield. BELOW
Identifications:| 1H NMR (Estimated) for Belinostat |

Experimental: 1H NMR (300 MHz, DMSO-d6): δ 6.52 (d, J=15.9 Hz, 1H), 6.81–7.12 (m, 6H), 7.33 (d, J=15.9 Hz, 1H), 7.47–7.67 (m, 3 H), 7.87 (s, 1H), 9.00–11.20 (br, 3H).


SEE COMPILATION ON SIMILAR COMPOUNDS AT …………..http://drugsynthesisint.blogspot.in/p/nostat-series.html
HPLC
ANALYTICAL HPLC TEST METHOD
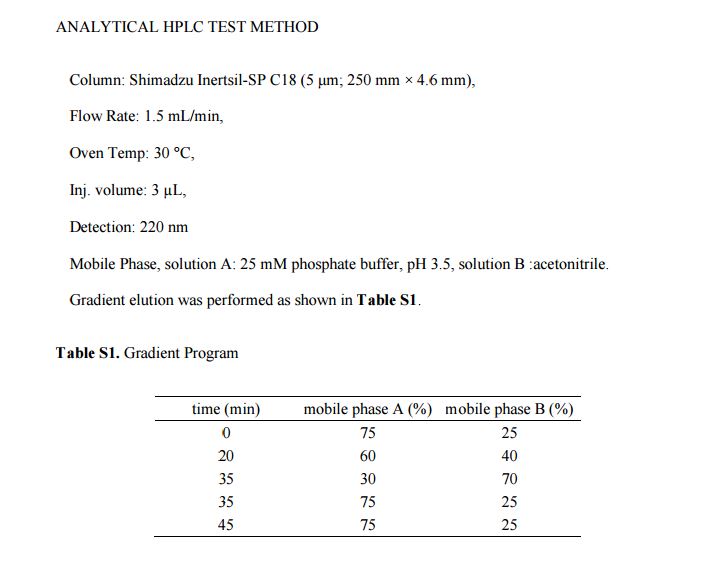
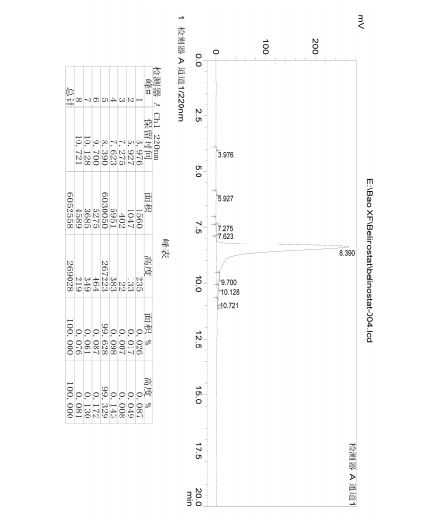
HPLC spectrum of Belinostat.
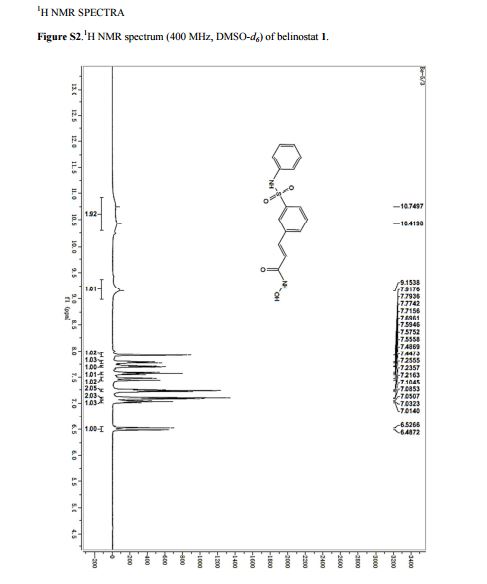
PATENT
http://www.google.si/patents/CN102531972A?cl=en
Belinostat synthesis process related to the first report of the literature of W002 / 30879 A2, including preparation for Belinostat described as follows:


Example 3:
3- (3-sulfonate-yl) phenyl – acrylate preparation:
First, 3-bromophenyl sulfonate 37. Ig (257. 90g / mol, 0. 1439mol) was dissolved with stirring in 260mL toluene IL reactor was then added triethylamine 36. 5g (101. 19g / mol, 0. 3604mol), tri (o-methylphenyl) phosphine 0. 875g (304. 37g / mol, 0. 002874mol), palladium acetate 0. 324g (224. 51g, 0. 001441mol), the reaction mixture was heated to 45- 55 ° C with nitrogen pumping ventilation four, this time in the reaction system to generate the catalytically active 1 ^ (0). The temperature of the reaction system was raised to 80-90 ° C, within 2. 75h dropwise methacrylate 13. 6g (86. 04g / mol, 0. 1586mol), the reaction was continued after the cell by HPLC 3- bromophenyl sulfonyl chloride was completion of the reaction. The temperature of the reaction system was reduced to 45-55 ° C.
[0021] In at 45-55 ° C, the reaction mixture was concentrated under reduced pressure, ethyl acetate and n-heptane and recrystallized to give the product 29. 4g, 83% yield.
[0022] The spectral data:
1HNMR (DMS0-d6, HMDS0), δ (ppm): 3. 65 (3H, S, H-1); 6. 47 (1H, d, J = 16 0 Hz, H-2.); 7. 30 -8 00 (5H, m, H-3, H_4, H_5, H_6, H_7) m / e:. 264. 23

References
-
- “Beleodaq (belinostat) For Injection, For Intravenous Administration. Full Prescribing Information” (PDF). Spectrum Pharmaceuticals, Inc. Irvine, CA 92618. Retrieved 21 November2015.
- Plumb JA; Finn PW; Williams RJ; et al. (2003). “Pharmacodynamic Response and Inhibition of Growth of Human Tumor Xenografts by the Novel Histone Deacetylase Inhibitor PXD101”. Molecular Cancer Therapeutics 2 (8): 721–728.PMID 12939461.
- “FDA approves Beleodaq to treat rare, aggressive form of non-Hodgkin lymphoma”. FDA. 3 July 2014.
- “CuraGen Corporation (CRGN) and TopoTarget A/S Announce Presentation of Belinostat Clinical Trial Results at AACR-NCI-EORTC International Conference”. October 2007.
- Final Results of a Phase II Trial of Belinostat (PXD101) in Patients with Recurrent or Refractory Peripheral or Cutaneous T-Cell Lymphoma, December 2009
- “Spectrum adds to cancer pipeline with $350M deal.”. February 2010.
- H. Spreitzer (4 August 2014). “Neue Wirkstoffe – Belinostat”.Österreichische Apothekerzeitung (in German) (16/2014): 27.
- Lexicomp, (corporate author) (2016). Bragalone, DL, ed.Drug Information Handbook for Oncology (14th ed.). Wolters Kluwer. ISBN 9781591953517.
- Helvetica Chimica Acta, 2005 , vol. 88, 7 PG. 1630 – 1657, MP 172
- WO2009/40517 A2, ….
- WO2006/120456 A1, …..
- Synthetic Communications, 2010 , vol. 40, 17 PG. 2520 – 2524, MP 172
- Journal of Medicinal Chemistry, 2011 , vol. 54, 13 PG. 4694 – 4720, NMR IN SUP INFO
Drug@FDA, NDA206256 Pharmacology Review(s).
Biochem. J. 2008, 409, 581-589.
J. Transl. Med. 2007, 5, 1-12.
Mol. Cancer Ther. 2006, 5, 2086-2095.
Int. J. Cancer 2008, 122, 1400-1410.
Synthetic Commun. 2010, 40, 2520-2524.
| CN101868446A * | Sep 23, 2008 | Oct 20, 2010 | 托波塔吉特英国有限公司 | Methods of synthesis of certain hydroxamic acid compounds |
| CN102531972A * | Dec 31, 2010 | Jul 4, 2012 | 北京万全阳光医药科技有限公司 | Preparation method of intermediate of antitumor medicament Belinostat |
| EP2093292A2 * | Mar 26, 2001 | Aug 26, 2009 | Methylgene, Inc. | Inhibition of specific histone deacetylase isoforms |
| GB2378179A * | Title not available | |||
| WO2002030879A2 * | Sep 27, 2001 | Apr 18, 2002 | Prolifix Limited | Carbamic acid compounds comprising a sulfonamide linkage as hdac inhibitors |
| WO2008068170A1 * | Nov 27, 2007 | Jun 12, 2008 | William Paul Jackson | Hdac inhibitors |
| WO2009146871A1 * | Jun 1, 2009 | Dec 10, 2009 | William Paul Jackson | 5-lipoxygenase inhibitors |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| CN104478769A * | Dec 22, 2014 | Apr 1, 2015 | 深圳万乐药业有限公司 | Belinostatsynthesis method suitable for industrial production |
| CN104478769B * | Dec 22, 2014 | Jan 6, 2016 | 深圳万乐药业有限公司 | 一种适合工业化生产的贝利司他合成方法 |
| CN104610100A * | Jan 9, 2015 | May 13, 2015 | 华东理工大学 | Nitrogen-chlorine type chlorination agent |
| US2008274120 | 11-7-2008 | Histone Deacetylase (Hdac) Inhibitors (Pxd101) for the Treatment of Cancer Alone or in Combination With Chemotherapeutic Agent |
| US2008227845 | 9-19-2008 | CYCLOOXYGENASE-2 INHIBITOR/HISTONE DEACETYLASE INHIBITOR COMBINATION |
| US2008213399 | 9-5-2008 | Combination Therapies Using Hdac Inhibitors |
| US2008194690 | 8-15-2008 | Pharmaceutical Formulations Of Hdac Inhibitors |
| US7407988 | 8-6-2008 | Carbamic acid compounds comprising a sulfonamide linkage as HDAC inhibitors |
| US7402603 | 7-23-2008 | Cyclooxygenase-2 inhibitor/histone deacetylase inhibitor combination |
| US7183298 | 2-28-2007 | Carbamic acid compounds comprising a sulfonamide linkage as HDAC inhibitors |
| US2005107445 | 5-20-2005 | Carbamic acid compounds comprising a sulfonamide linkage as HDAC inhibitors |
| US6888027 | 5-4-2005 | Carbamic acid compounds comprising a sulfonamide linkage as hdac inhibitors |
| WO2002030879A2 | Sep 27, 2001 | Apr 18, 2002 | Prolifix Ltd | Carbamic acid compounds comprising asulfonamide linkage as hdac inhibitors |
| US7973181 | 7-6-2011 | HYDROXAMIC ACID DERIVATIVES AS INHIBITORS OF HDAC ENZYMATIC ACTIVITY |
| US7928081 | 4-20-2011 | Combined Use of Prame Inhibitors and Hdac Inhibitors |
| US2011077305 | 3-32-2011 | 5-LIPOXYGENASE INHIBITORS |
| US2011003777 | 1-7-2011 | Methods of Treatment Employing Prolonged Continuous Infusion of Belinostat |
| US2010286279 | 11-12-2010 | Methods of Synthesis of Certain Hydroxamic Acid Compounds |
| US2010190694 | 7-30-2010 | Methods for identifying patients who will respond well to cancer treatment |
| US2010010010 | 1-15-2010 | HDAC INHIBITORS |
| US2009312311 | 12-18-2009 | COMBINATION OF ORGANIC COMPOUNDS |
| US2009192211 | 7-31-2009 | CYCLOOXYGENASE-2 INHIBITOR/HISTONE DEACETYLASE INHIBITOR COMBINATION |
| US7557140 | 7-8-2009 | CARBAMIC ACID COMPOUNDS COMPRISING A SULFONAMIDE LINKAGE AS HDAC INHIBITORS |
| WO1998038859A1 * | Mar 4, 1998 | Sep 11, 1998 | Thomas E Barta | Sulfonyl divalent aryl or heteroaryl hydroxamic acid compounds |
| WO1999024399A1 * | Nov 12, 1998 | May 20, 1999 | Darwin Discovery Ltd | Hydroxamic and carboxylic acid derivatives having mmp and tnf inhibitory activity |
| WO2000056704A1 * | Mar 22, 2000 | Sep 28, 2000 | Duncan Batty | Hydroxamic and carboxylic acid derivatives |
| WO2000069819A1 * | May 12, 2000 | Nov 23, 2000 | Thomas E Barta | Hydroxamic acid derivatives as matrix metalloprotease inhibitors |
| WO2001038322A1 * | Nov 22, 2000 | May 31, 2001 | Methylgene Inc | Inhibitors of histone deacetylase |
| EP0570594A1 * | Dec 7, 1992 | Nov 24, 1993 | SHIONOGI & CO., LTD. | Hydroxamic acid derivative based on aromatic sulfonamide |
| EP0931788A2 * | Dec 16, 1998 | Jul 28, 1999 | Pfizer Inc. | Metalloprotease inhibitors |
| GB2312674A * | Title not available |
| WO2002030879A2 | Sep 27, 2001 | Apr 18, 2002 | Prolifix Ltd | Carbamic acid compounds comprising a sulfonamide linkage as hdac inhibitors |
| WO2005063806A1 | Dec 30, 2003 | Jul 14, 2005 | Council Scient Ind Res | Arginine hydrochloride enhances chaperone-like activity of alpha crystallin |
| US4642316 | May 20, 1985 | Feb 10, 1987 | Warner-Lambert Company | Parenteral phenytoin preparations |
| WO2008090585A2 * | Jan 25, 2008 | Jul 31, 2008 | Univ Roma | Soluble forms of inclusion complexes of histone deacetylase inhibitors and cyclodextrins, their preparation processes and uses in the pharmaceutical field |
| WO2009109861A1 * | Mar 6, 2009 | Sep 11, 2009 | Topotarget A/S | Methods of treatment employing prolonged continuous infusion of belinostat |
| WO2010048332A2 * | Oct 21, 2009 | Apr 29, 2010 | Acucela, Inc. | Compounds for treating ophthalmic diseases and disorders |
| WO2011064663A1 | Nov 24, 2010 | Jun 3, 2011 | Festuccia, Claudio | Combination treatment employing belinostat and bicalutamide |
| US20110003777 * | Mar 6, 2009 | Jan 6, 2011 | Topotarget A/S | Methods of Treatment Employing Prolonged Continuous Infusion of Belinostat |
| CN102786448A * | 9 avg 2012 | 21 nov 2012 | 深圳万乐药业有限公司 | Method of synthesizing belinostat |
| CN102786448B | 9 avg 2012 | 12 mar 2014 | 深圳万乐药业有限公司 | Method of synthesizing belinostat |
CLIP
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(2E)-N-Hydroxy-3-[3-(phenylsulfamoyl)phenyl]prop-2-enamide
|
|
| Clinical data | |
| Trade names | Beleodaq |
| AHFS/Drugs.com | beleodaq |
| Pregnancy category |
|
| Routes of administration |
Intravenous (IV) |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | 100% (IV) |
| Protein binding | 92.9–95.8%[1] |
| Metabolism | UGT1A1 |
| Excretion | Urine |
| Identifiers | |
| CAS Number | 866323-14-0 |
| ATC code | L01XX49 (WHO) |
| PubChem | CID 6918638 |
| ChemSpider | 5293831 |
| UNII | F4H96P17NZ |
| ChEBI | CHEBI:61076 |
| ChEMBL | CHEMBL408513 |
| Synonyms | PXD101 |
| Chemical data | |
| Formula | C15H14N2O4S |
| Molar mass | 318.348 g/mol |
////////////Belinostat, PXD101, novel HDAC inhibitor, Beleodaq, Folotyn, Spectrum Pharmaceuticals, Inc., Henderson, Nevada, Istodax, Celgene Corporation, Summit, New Jersey, CuraGen Pharma, FDA 2014
O=S(=O)(Nc1ccccc1)c2cc(\C=C\C(=O)NO)ccc2
SEE COMPILATION ON SIMILAR COMPOUNDS AT …………..http://drugsynthesisint.blogspot.in/p/nostat-series.html
TAK-243, AOB 87172, MLN-7243

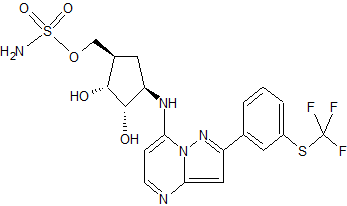
TAK-243, AOB 87172, MLN-7243
CAS 1450833-55-2
Chemical Formula: C19H20F3N5O5S2
Molecular Weight: 519.5142
Sulfamic acid, [(1R,2R,3S,4R)-2,3-dihydroxy-4-[[2-[3-[(trifluoromethyl)thio]phenyl]pyrazolo[1,5-a]pyrimidin-7-yl]amino]cyclopentyl]methyl ester
((lR,2R,3S,4R)-2,3-dihydroxy-4-(2-(3-(trifluoromethylthio)phenyl)pyrazolo[l ,5-a]pyrimidin-7-ylamino)cyclopentyl)methyl sulfamate
methyl ((1S,2R,3S,4R)-2,3-dihydroxy-4-((2-(3-((trifluoromethyl)thio)phenyl)pyrazolo[1,5-a]pyrimidin-7-yl)amino)cyclopentyl)sulfamate
Phase I
Millennium Pharmaceuticals, Inc. INNOVATOR
Roushan AFROZE, Indu T. Bharathan,Jeffrey P. CIAVARRI, Paul E. Fleming,Jeffrey L. Gaulin, Mario Girard, Steven P. Langston, Francois R. SOUCY, Tzu-Tshin WONG, Yingchun Ye,
A UAE inhibitor potentially for the treatment of solid tumors.
TAK-243, also known as MLN7243 and AOB87172, is a small molecule inhibitor of ubiquitin-activating enzyme (UAE), with potential antineoplastic activity. UAE inhibitor MLN7243 binds to and inhibits UAE, which prevents both protein ubiquitination and subsequent protein degradation by the proteasome. This results in an excess of proteins in the cells and may lead to endoplasmic reticulum (ER) stress-mediated apoptosis. This inhibits tumor cell proliferation and survival. UAE, also called ubiquitin E1 enzyme (UBA1; E1), is more active in cancer cells than in normal, healthy cells.
![]()
Research Code TAK-243; MLN-7243, TAK-243; TAK 243; TAK243; MLN7243; MLN-7243; MLN 7243; AOB87172; AOB-87172; AOB 87172.
CAS No. 1450833-55-2(MLN 7243)
- Originator Millennium
- Developer Takeda Oncology
- Class Antineoplastics
- Mechanism of Action Ubiquitin-protein ligase inhibitors
- Phase I Solid tumours

Most Recent Events
- 01 Feb 2014 Phase-I clinical trials in Solid tumours (late-stage disease, second-line therapy or greater) in USA (IV)
- 18 Dec 2013 Preclinical trials in Solid tumours in USA (IV)
- 18 Dec 2013 Millennium plans a phase I trial for Solid tumours (late-stage disease, second-line therapy or greater) in USA (NCT02045095)
Cancer is the second most common cause of death in the U.S. and accounts for one of every eight deaths globally (American Cancer Society, Cancer Facts and Figures, 2014). The American Cancer Society expects that in 2014 at least 1,665,540 new cancer cases will be diagnosed in the US and 585,720 Americans are expected to die of cancer, almost 1 ,600 people per day. Currently available paradigms for treating solid tumors may include systemic treatment such as chemotherapy, hormonal therapy, use of targeted agents and biological agents, either as single agents or in combination. These treatments can be delivered in combination with localized treatments such as surgery or radiotherapy. These anti-cancer paradigms can be use in the curative setting as adjuvant or neo-adjuvant treatments or in the metastatic setting as palliative case for prolonged survival and to help manage symptoms and side-effects. In hematological cancers, stem cell transplants may also be an option in certain cancers as well as chemotherapy and/or radiation. Although medical advances have improved cancer survival rates, there remains a continuing need for new and more effective treatments.
Ubiquitin is a small 76-amino acid protein that is the founding member of a family of posttranslational modifiers known as the ubiquitin-like proteins (Ubls). Ubls play key roles in controlling many biological processes including cell division, cell signaling and the immune response. There are 8 known human Ubl activating enzymes (known as Els) (Schulman, B.A., and J.W. Harper, 2009, Ubiquitin-like protein activation by El enzymes: the apex for downstream signalling pathways, Nat Rev Mol Cell Biol 10:319-331). Ubiquitin and other Ubls are activated by a specific El enzyme which catalyzes the formation of an acyl-adenylate intermediate with the C-terminal glycine of the Ubl. The activated Ubl molecule is then transferred to the catalytic cysteine residue within the El enzyme through formation of a thioester bond intermediate. The El -Ubl intermediate and an E2 enzyme interact, resulting in a thioester exchange wherein the Ubl is transferred from the El to active site cysteine on the E2. The Ubl is then conjugated, i.e. transferred, to the target protein, either directly or in conjunction with an E3 ligase enzyme, through isopeptide bond formation with the amino group of a lysine side chain in the target protein. Eukaryotic cells possess ~35 ubiquitin E2 enzymes and >500 ubiquitin E3 enzymes. The E3 enzymes are the specificity factors of the ubiquitin pathway which mediate the selective targeting of specific cellular substrate proteins (Deshaies, R.J., and C.A. Joazeiro, 2009, RING domain E3 ubiquitin ligases, Annu Rev Biochem 78:399-434; Lipkowitz, S., and A.M. Weissman, 2011, RTNGs of good and evil: RING finger ubiquitin ligases at the crossroads of tumour suppression and oncogenesis, Nat Rev Cancer 11 :629-643; Rotin, D., and S. Kumar, 2009, Physiological functions of the HECT family of ubiquitin ligases, Nat Rev Mol Cell Biol 10:398-409).
Two El enzymes have been identified for ubiquitin, UAE (ubiquitin-activating enzyme) and UBA6 (Jin, J., et al., 2007, Dual El activation systems for ubiquitin differentially regulate E2 enzyme charging, Nature 447: 1135-1138). UAE is the El responsible for the majority (>99%) of ubiquitin flux within the cell. UAE is capable of charging each of the approximately -35 E2 enzymes with the exception of Usel, which is the only E2 known to exclusively work with UBA6 (Jin et al., 2007). Inhibition of UAE is sufficient to dramatically impair the great majority of ubiquitin-dependent cellular processes (Ciechanover, A., et al., 1984, Ubiquitin dependence of selective protein degradation demonstrated in the mammalian cell cycle mutant ts85, Cell 37:57-66; Finley, D., A. et al., 1984, Thermolability of ubiquitin-activating enzyme from the mammalian cell cycle mutant ts85, Cell 37:43-55).
The cellular signals generated by ubiquitin are diverse. Ubiquitin can be attached to substrates as a single entity or as polyubiquitin polymers generated through isopeptide linkages between the C-terminus of one ubiquitin and one of the many lysines on a second ubiquitin. These varied modifications are translated into a variety of cellular signals. For example, conjugation of a lysine 48 -linked polyubiquitin chain to a substrate protein is predominantly associated with targeting the protein for removal by the 26S proteasome. A single ubiquitin modification, or monoubiquination, typically affects protein localization and/or function. For example, monoubiquitination modulates the following: 1) the function of Histones 2a and 2b (Chandrasekharan, M.B., et al., 2010, Histone H2B ubiquitination and beyond: Regulation of nucleosome stability, chromatin dynamics and the trans-histone H3 methylation, Epigenetics 5:460-468), 2) controls the nucleo-cytoplasmic shuttling of PTEN (Trotman, L,C, et al., 2007, 3) ubiquitination regulates PTEN nuclear import and tumor suppression, Cell 128: 141-156), 4) drives localization of the FANCD2 protein to sites of DNA damage (Gregory, R.C., et al., 2003, Regulation of the Fanconi anemia pathway by monoubiquitination, Semin Cancer Biol 13:77-82) and 5) promotes the internalization and endosomal/lysosomal turnover of some cell surface receptors, like EGFR (Mosesson, Y., and Y. Yarden, 2006, Monoubiquitylation: a recurrent theme in membrane proteintransport. Isr Med Assoc J 8:233-237). Other forms of polyubiquitination chains occur on lysine positions 11, 29 and 63, impacting various cellular roles including cell cycle, DNA repair and autophagy (Behrends, C, and J.W. Harper, 2011, Constructing and decoding unconventional ubiquitin chains, Nat Struct Mol Biol 18:520-528; Bennett, E.J., and J.W. Harper, 2008, DNA damage: ubiquitin marks the spot, Nat Struct Mol Biol 15:20-22; Komander, D., 2009, The emerging complexity of protein ubiquitination, Biochem Soc Trans 37:937-953).
UAE-initiated ubiquitin conjugation plays an important role in protein homeostasis, cell surface receptor trafficking, transcription factor turnover and cell cycle progression. Many of these processes are important for cancer cell survival and it is believed that tumor cells may have increased sensitivity to UAE inhibition as a result of their rapid growth rate, increased metabolic demands and oncogene fueled protein stress. Preclinical studies with PYZD-4409, a UAE inhibitor, demonstrated this compound induced cell death in both leukemia and myeloma cell lines and induced anti-tumor activity in a mouse acute myeloid leukemia (AML model). (Xu, W.G., et al., 2010, The ubiquitin-activating enzyme El as a therapeutic target for the treatment of leukemia and multipie myeloma, Blood, 115:2251-59). Thus, UAE represents a protein homeostasis target opportunity for the treatment of cancer.
Abstracts: AACR-NCI-EORTC International Conference: Molecular Targets and Cancer Therapeutics; November 5-9, 2015; Boston, MA
Abstract
Clinical results of VELCADE® (bortezomib) For Injection have prompted evaluation of other enzymes within the ubiquitin proteasome system (UPS) as druggable targets for human cancer. We have identified a first in class investigational drug, TAK-243 (MLN7243), which targets the ubiquitin activating enzyme, UAE (UBA1), an essential cellular enzyme responsible for activating > 99% of all cellular ubiquitin. Ubiquitin is involved in multiple cellular processes including ubiquitin-dependent protein turnover, cell cycle progression, regulation of apoptosis, protein localization and response to DNA damage. Experiments combining targeted siRNA knockdown with TAK-243 identified DNA damage repair genes necessary for UAE inhibitor-induced cell death. A more focused approach revealed TAK-243 treatment blocked essential monoubiquitination events within the Translesion synthesis (TLS), Fanconi Anemia (FA) and Homologous recombination (HR) pathways. Inhibition of UAE prevented mono-ubiquitin signaling of key mediators within these pathways, including PCNA and FANCD2, by blocking formation of their specific E2-ubiquitin thioesters. In vitro cell-based assays combining TAK-243 with ultraviolet (UV) and radiation, both known to induce DNA damage, yielded inhibition of cell growth and enhanced DNA damage as observed through colony formation assays and Comet assay detection, respectively. Xenograft tumor bearing mice were treated with carboplatin or docetaxel, combined with TAK-243, to evaluate combination benefits in vivo. Synergistic and additive anti-tumor combination benefits were observed in animals treated with TAK-243 + carboplatin and TAK-243 + docetaxel. These important mechanistic in vitro and in vivo studies indicate the dependency of ubiquitination signaling in DNA damage repair and provide a mechanistic rationale for combining radiation, carboplatin or docetaxel with TAK-243 in the clinical setting. Currently, TAK-243 is being evaluated in a solid tumor phase I clinical trial evaluating safety, tolerability, pharmacokinetics, pharmacodynamics and anti-tumor activity (ClinicalTrials.gov identifier: NCT02045095).
Citation Format: Michael A. Milhollen, Judi Shi, Tary Traore, Jessica Huck, Darshan Sappal, Jennifer Duffy, Eric Lightcap, Yuko Ishii, Jeff Ciavarri, Paul Fleming, Neil Bence, Marc L. Hyer. The small molecule UAE inhibitor TAK-243 (MLN7243) prevents DNA damage repair and reduces cell viability/tumor growth when combined with radiation, carboplatin and docetaxel. [abstract]. In: Proceedings of the AACR-NCI-EORTC International Conference: Molecular Targets and Cancer Therapeutics; 2015 Nov 5-9; Boston, MA. Philadelphia (PA): AACR; Mol Cancer Ther 2015;14(12 Suppl 2):Abstract nr A164.
PATENT
WO 2013123169
https://www.google.com/patents/WO2013123169A1?cl=en
Scheme 1 : General route for 2-substituted ((1R,2R,3S,4R)-2,3-dihydroxy-4- (pyrazolo[1,5-a]pyrimidin-7-ylamino)cyclopentyl)methyl sulfamates

A genera! route for the synthesis of compounds represented by structure iv wherein Z is an optionally substituted fused or non-fused aryl or heteroaryl ring is outlined above in Scheme 1. Compound i (obtained by coupling an appropriately protected cyclopentylamine or salt thereof with 2-bromo-7-chloropyrazolo[1 ,5-a]pyrimidine in the presence of a suitable base as described below in the procedure of Examples 1a and 1b) is transformed to a compound of formula iii by coupling with a metal substituted compound Z-M via a palladium catalyzed reaction. A compound of formula iii can also be obtained by first transforming i to a metal substituted compound of formula ii using suitable boron or tin containing reagents, and then coupling with a halogen substituted compound Z-X via a palladium catalyzed reaction. Compounds of formula iv are then obtained by reaction with an appropriate sulfamating reagent (for example chlorosulfonamide or see Armitage, I. et. al. U.S. Patent Application US2009/0036678, and Armitage, I. et. al. Org. Lett., 2012, 14 (10), 2626-2629) followed by appropriate deprotection conditions.
Scheme 2: General route for 5-halogen substituted, 2 -substituted ((1R,2R,3S,4R)- 2,3-dihydroxy-4-(pyrazolo[1,5-a]pyrimidin-7-ylamino)cyclopentyl)methyl sulfamates


A general route for the synthesis of compounds represented by structure ix wherein Z is an optionally substituted fused or non-fused aryl or heteroaryl ring and X is a halogen is outlined above in Scheme 2. Cyclization of amino-pyrazole v with a suitable diester and an appropriate base at an elevated temperature is followed by reaction with an appropriate halogenating reagent such as POCI3 at an elevated temperature to give compounds of formula vii. Compounds of formula viii are then obtained by reaction with an appropriately protected cyc!opentylamine or a salt thereof in the presence of a suitable base. Sulfamation and deprotection following Method 1 as described previously provides compounds of formula ix.
Scheme 3: General route for 5-alkyl substituted, 2-substituted ((1R,2R,3S,4R)-2,3- dihydroxy-4-(pyrazolo[1 ,5-a]pyrimidin-7-ylamino)cyclopentyl)methyl sulfamates

SIMILAR COMPD
Example 17. Synthesis of (s.e.)-{(1 ,2R,3S,4R)-4-[(3,6-dichloro-2-{3- [(trifluoromethyl)sulfanyl]phenyl}pyrazolo[1,5-a]pyrimidin-7-yl)amino]-2,3- dihydroxycyclopentyl}methyl sulfamate (1-124) and (s.e.)-{(1 ,2R,3S,4R)-4-[(6-chloro-2-{3- [(trifluoromethyl)sulfanyl]phenyl}pyrazolo[1,5^]pyrimidin-7-y[)arnino]-2,3- dihydroxycyclopentyl}methyl sulfamate 0-125).

SIMILAR NOT SAME
Step 1. To a vial containing s.e {(1 ,2 ,3S,4 )-2,3-dihydroxy-4-t(2-{3- [(t rif I u orometh y l)sulf a nyl] phen l}p^
sulfamate (0.82 g, 0.0015 mol) and cooled to 0 °C is added N-chlorosuccinimide (126 mg, 0.000943 mol) as a solution in 12 mL of N,N-dimethy)formamide. The reaction mixture is stirred overnight with warming to rt. Saturated sodium bicarbonate solution is added and the reaction mixture is extracted with ethyl acetate, washed with brine, dried over sodium sulfate and concentrated in vacuo. The crude material is first purified by column chromatography (eluent: methanol/methylene chloride) and then purified by HPLC to afford both the dichloro (LCMS: (FA) +1 588) and mono chloro (LCMS: (FA) M+1 554) titlecompounds.
PATENT
UAE inhibitors are disclosed in patent application publications WO2013/123169 and US 2014/0088096. In one embodiment, the UAE inhibitor is a compound having the following structure (Compound 1):

(Compound 1);
or a pharmaceutically acceptable salt thereof. The Compound 1 is named ((lR,2R,3S,4R)-2,3-dihydroxy-4-(2-(3-(trifluoromethylthio)phenyl)pyrazolo[l ,5-a]pyrimidin-7-ylamino)cyclopentyl)methyl sulfamate.
process for making Compound 1 :

Compound 1;
or pharmaceutically acceptable salt thereof, comprising the steps of:
a) contacting Compound 9 or a salt, solvate or hydrate thereof with 2,2-dimethyl-l,3-dioxane-4,6-dione (Meldrum’s acid):

Compound 9
under coupling conditions to provide compound 8 or a salt, solvate or hydrate thereof:

Compound 8
b) subjecting compound 8 or a salt, solvate or hydrate thereof to cyclization conditions to provide compound 7 or a salt, solvate or hydrate thereof

Compound 7
c) contacting Compound 7 or a salt, solvate or hydrate thereof with benzotriazole under chlorination displacement conditions to provide Compound 5 or a salt, complex, solvate or hydratei thereof

; Compound 5
d) contacting Compound 5 or a salt, complex, solvate or hydrate thereof with Compound 6 or a solvate or hydrate thereof:

; Compound 6
under displacement reaction conditions to provide Compound 3 or a salt, solvate or hydrate thereof
solvate or hydrate thereof with Compound

Cl ; Compound 4
under sulfamoylating reaction conditions to provide Compound 2 or a salt, solvate or hydrate thereof

; Compound 2
f) contacting Compound 2 or a salt, solvate or hydrate thereof with an acid under sulfamoylation conditions to provide Compound 1 or a pharmaceutically acceptable salt thereof
 COMPD1
COMPD1
Example 1: Synthesis of S-iB-Ktrifluoromethyltsulfanyllphenyll-lH-pyrazol-S-amine
Step A: 3-((trifluoromethyl)thio)benzoate
[0148] To dimethylcarbonate (68 mL) was added 3-((trifluoromethyl)thio)benzoic acid (100 g, Beta Pharma Scientific) and a catalytic amount of sulfuric acid (2.4 mL). The mixture was then heated to 90°C for 5h. The reaction was then cooled to room temperature and quenched with sodium bicarbonate (1.0 L). To the aqueous layer was with ethyl acetate (1.0 L). The phases were separated and this process was repeated with ethyl acetate (1.0 L). The organic layers were combined and concentrated with a rotovap to give a light orange oil. The methyl 3-((trifluoromethyl)thio)benzoate (105g, 99%) was taken on crude to the next reaction. Ή NMR (300 MHz, CHLOROFORM-^ δ ppm 3.99 (s, 3 H) 7.49 – 7.58 (m, 1 H) 7.85 (d, J=l.62 Hz, 1 H) 8.17 (dt, J=7.69, 1.43 Hz, 1 H) 8.32 – 8.44 (m, 1 H).
[0149] Step B: 3-oxo-3-(3-((trifluoromcthvnthio)phcnyl>proDaneiiitrilc
[0150] Methyl 3-((trifluoromethyl)thio)benzoate (100.0 g) in tetrahydrofuran (1.0 L) was added acetonitrile (44.2 mL, 847 rnmol) and 1M (in THF) potassium tert-butoxide (95.01 g). The reaction was complete in 10 min by HPLC analysis. The reaction was quenched with 1M HC1 (1.0 L) and then extracted with three times with (1.0 L) of ethyl acetate. The organic layers with 3-oxo-3-(3-((trifluoromethyl)thio)phenyl)propanenitrile were then concentrated to dryness. This material (lOO.Og, 96.3%) was taken on crude with further purification. Ή NMR (300 MHz, CHLOROFORM-rf) δ ppm 4.12 (s, 2 H) 7.51 – 7.75 (m, 1 H) 7.89 – 8.01 (m, 1 H) 8.01 – 8.10 (m, 1 H) 8.20 (s, 1 H)
[0151] Step C: 3-}3-htrifliioromethv sulfan llphenyl}-lH-pyrazol-5-amine
[0152] To 3-oxo-3-{3-[(trifluoromethyl)sulfanyl]phenyl}propanenitrile (100.0 g,) in ethanol (1000.0 mL) was added hydrazine hydrate (59.52 mL). The reaction was heated to 100°C for lh at which point HPLC analysis showed the reaction was complete. The reaction was concentrated to dryness on a rotovap to give a brown oil. The oil was taken up in ethyl acetate (1.0 L) and extracted with water (1.0 L). The phases were separated and the organic phase was concentrated. Upon concentration 3-{3-[(trifluoromethyl)sulfanyl]phenyl}-lH-pyrazol-5-amine was obtained (80.8 g; Yield = 76.4%) . !H NMR (300 MHz, CHLOROFORM-^ δ ppm 5.95 (s, 1 H) 6.73 (br s, 1 H) 7.13 – 7.34 (m, 2 H) 7.42 – 7.74 (m, 3 H) 7.85 (s, 1 H).
[0153] Example 2: f R.2R.3St4RV2.3-dihvdroxy-4-ff2-r3- ((trifluoromethylHhio)phenvnpyrazolo[l,5-alpyrimidin-7-yl¼mino)cvclopentyl)metliyl sulfamate
[0154] Step 1: f2.2-dimethyl-5-ffl3-(3-((triiluoromethvnthio phenvn-lH-pyrazol-5- amino methyleBC>-1.3-dioxane-4,6-dione)
[0155] To trimethoxy orthoformate (2.0 L), at 20°C and under a blanket of nitrogen, was added 2,2-dimethyl-l,3-dioxane-4,6-dione (361.35 g). The resulting white suspension went clear within minutes and was heated to 85°C over 15 minutes. The reaction was held at 85°C for 120 minutes. While the reaction was heated and stirred another solution of 3-(3-((trifluoromethyl)thio)pheny])-lH-pyrazol-5-amine (500.0 g) was made. To a 4L RBF was added 3-(3-((trifluoromethyl)thio)phenyl)-lH-pyrazol-5-amine (500.0 g) and then trimethoxy orthoformate (1.4 L) added into this solid. This solution was mixed to dissolve the solids and resulted a dark brown solution. The solution of 3-(3-((trifluoromethyl)thio)phenyl)-lH-pyrazol-5-amine (-1.8L in trimethoxy orthoformate) was added to the reactor over 30 minutes while maintaining the reaction temperature at 85°C. The reaction was then stirred for 20 minutes with white solids forming in the solution. After 20 minutes the reaction was sampled and the UPLC showed the complete conversion of 3-(3-((trifluoromethyl)thio)phenyl)-lH-pyrazol-5 -amine to 2,2-dimethyl-5-(((3-(3-((trifluoromethyl)thio)phenyl)-lH-pyrazol-5-yl)amino)methylene)-l ,3-dioxane-4,6-dione. The reaction was cooled to 20 °C over 20 minutes and maintained at that temperature for 20 additional minutes. At this point, a thick white slurry had formed and the reaction was filtered using a Nutche Filter over 15 minutes. The reactor was washed with 1L of ethyl acetate and this solution was then mixed with the filter cake and removed by filtration. The cake was dried for -40 minutes on the filter and then transferred to a vacuum oven and heated at 40°C under full vacuum overnight (16 hours). The reaction was then analyzed by FfPLC and NMR to give 2,2-dimethyl-5-(((3-(3 -((trifluoromethyl)thio)phenyi lH-pyrazol-5-yl)amino)methylene)- 1 ,3-dioxane-4,6-dione (635.3 g, 79%) XH NMR (300 MHz, DMSO-cfe) δ ppm 1.68 (s, 6 H) 7.05 (d, J=2.05 Hz, 1 H) 7.64 -7.77 (m, 2 H) 7.77 – 8.03 (m, 1 H) 8.12 (s, 1 H) 8.72 (d, J=14.36 Hz, 1 H) 1 1.35 (d, J=14.66 Hz, 1 H) 13.47 (s, 1 H).
[0156] Step 2: 2-( 3-f(trifluoromethyl)thio phenyl)pyrazoIo [1,5-al pyrimidin-7-ol
[0157] A solution of 2,2-dimethyl-5-(((3-(3-((trifluoromethyl)thio)phenyl)-lH-pyrazol-5-yl)amino)methylene)-l,3-dioxane-4,6-dione (615.00 g) in 1,2-dichIorobenzene (6.3 L) was stirred at ambient temperature for 10 minutes. The solution was then heated to 150°C over 75 minutes. The reaction was maintained at this temperature for 16 hours. An sample was taken after 16 hours and the UPLC analysis showed the complete conversion of 2,2-dimethyl-5-(((3-(3-((trifluoromethyl)thio)phenyl)-lH-pyrazol-5-yI)amino)methylene)-l,3-dioxane-4,6-dione to 2-(3- ((trifluoromethyl)tmo)phenyl)pyrazolo[l,5-a]pyrimidin-7-ol. The reaction was cooled to 20°C over 130 minutes. At this point, a thick white slurry had formed and the reaction was filtered using a Nutche Filter over 15 minutes. The reactor was washed with 1.8 L of acetonitrile and this solution was then mixed with the filter cake and then the solvent was removed by filtration. The cake was dried for ~40 minutes on the filter and then transferred to a vacuum oven and heated at 40°C under full vacuum overnight (16 hours). The reaction was then analyzed by HPLC and NMR to give 2-(3-((trifluoromethyl)thio)phenyl)pyrazolo[l,5-a]pyrimidin-7-ol (331.2 g, 72%) Ή NMR (300 MHz, METHANOL-^) δ ppm 6.55 (d, J=7.33 Hz, 1 H) 7.59 (s, 1 H) 8.40 – 8.52 (m, 1 H) 8.53 – 8.64 (m, 1 H) 8.69 (d, J=7.62 Hz, 1 H) 9.01 (dt, J=7.77, 1.39 Hz, 1 H) 9.12 (s, 1 H) 13.34 (s, 1 H).
[0158] Step 3: l-(2-(3-(f trffluoromethvmhiotohenvnpyrazolo n.5-al pyrimidin-7-vn-lH-benzofdiri.2.31triazole: triethylamine: hydrochloride complex (1:1.25:1.25 molesimolestmolest
[0159] To a solution of 2-(3-((trifluoromethyl)thio)phenyl)pyrazolo[l,5-a]pyrimidin-7-ol (30.00 g), benzotriazole (287.02 g) in acetonitrile (3000 mL) and triethylamine (403.00 mL) at 0°C, was added phosphoryl chloride (108 mL) slowly under a blanket of nitrogen, maintaining < 10°C. The reaction was then warmed to 80°C over 45 minutes and stirred for 240 minutes. HPLC indicated complete
consumption of starting material. To the reaction mixture was added acetonitrile (3000 mL) while maintaining the temperature at 80°C. The reaction was then cooled to 20°C over 80 minutes. The reaction was then stirred at ambient temperature for 14 hours. At this point, a thick slurry had formed and the reaction was filtered using a Nutche filter over 15 minutes. The reactor was washed twice with 900 mL of acetonitrile and this solution was then mixed with the filter cake and then the solvent was removed by filtration. The cake was dried for -40 minutes on the filter and then transferred to a vacuum oven and heated at 40°C under full vacuum overnight (16h). The reaction was then analyzed by HPLC and NMR to give l-(2-(3-((trifluorometJiyl)thio)phe
triethylamine: hydrochloride complex (1:1.25:1.25 moles:moles:moles) (438.1 g, 83%). ¾ NMR (300 MHz, DMSO-</6) δ ppm 1.19 (t, J=7.33 Hz, 12 H) 3.07 (qd, J=7.28, 4.84 Hz, 8 H) 7.60 – 7.78 (m, 6 H) 7.80 – 7.87 (m, 1 H) 8.15 (dt, J=7.99, 1.28 Hz, 1 H) 8.24 (s, 1 H) 8.33 (dt, J=8.14, 0.92 Hz, 1 H) 8.85 (d, J=4.69 Hz, 1 H).
[0160] Step 4: ff3aR4R.6R.6aS 2.2-dimethyl-6-ff2-f3~mrifluoromethyl)thio)phenvnpyrazoloil.5-alD\timidin-7-yl¼mino)tctralivdro-3aH-cvcLoDentaldlll,31dioxol-4-vnincthanol
[0161] To the reactor was added l-(2 3-((trifluoromethyl)thio)phenyl)pyrazolo[l,5-a]pyrimidin-7-yl)-lH-benzo[d][l,2,3]triazole: triethylamine: hydrochloride complex (1 :1.25: 1.25 moles :moles:moles) (430.0 g) and ((3aR,4R,6R,6aS)-6-amino-2,2-dimethyltetrahydro-3aH-cyclopenta[d][l,3]dioxol-4-yl)methanol hydrochloride (209.0 g) and then triethylamine (2103 mL) was added. The reaction was then heated to 80°C, under a blanket of nitrogen. After 360 minutes, HPLC analysis indicated that the reaction mixture contained <1% starting material and the reaction was cooled to 20°C over 60 minutes. To the reaction was added ethyl acetate (3.5 L) and water (3.5 L). After stirring for 10 minutes the phases were separated and the aqueous layer was back extracted with ethyl acetate (3.5 L). The organic layers were combined and concentrated to form a dark, brown oil. Acetonitrile (4.5 L) was added and the solution was concentrated to dryness to give an orange solid. The solids was transferred back to the reaction with water (4.3 L), heated to 50°C, and stirred for 20 minutes. White solids formed in this hot solution and were isolated by filtration using a Nutche Filter over 15 minutes. The solids were dried under vacuum for 15 minutes on the filter and then dissolved in acetonitrile (4.0 L) at 0°C. The solution was stirred for 1 minutes. The solution was then filtered through a fritted funnel to remove the hydrolysis solid by product and the solution was concentrated to dryness. The solids were dried in a vacuum oven at full vacuum overnight (40°C, 16 hours). The reaction was then analyzed by HPLC and NMR to give ((3aR,4R,6R,6aS)-2,2-dimethyl-6-((2-(3 -((trifluoromethyl)thio)phenyl)pyrazolo[ 1 ,5 -a]pyrimidin-7-yl)amino)tetrahydro-3aH-cyclopenta[d][l,3]dioxol-4-yl)methanoI (349.2 g, 88%). Ή NMR (300 MHz, DMSO-<¾) δ ppm 1.25 (s, 3 H) 1.47 (s, 3 H) 1.76 – 1.90 (m, 1 H) 2.25 (br d, J-3.22 Hz, 1 H) 2.33 – 2.47 (m, 1 H) 3.46 – 3.67 (m, 2 H) 4.08 (br d, J=5.57 Hz, 1 H) 4.48 – 4.64 (m, 2 H) 5.19 (t, J=4.40 Hz, 1 H) 6.28 (d, J=5.28 Hz, 1 H) 7.06 (s, 1 H) 7.58 – 7.71 (m, 1 H) 7.72 – 7.80 (m, 1 H) 8.12 – 8.24 (m, 2 H) 8.31 (d, J=7.62 Hz, 1 H) 8.42 (s, 1 H).
[0162] Step 5: ((3aR.4R.6R.6aS 2.2-dimethyl-6-ff2-f3-fftrifluoroinethYmhio)phenvnpyrazolo[1.5-al Dyrimidin-7-vnan] iiio>tetrahvdro-3aH-cvclonen ta [dl [1,31 dioTOl-4-yl )meth yl tert-bntoxycarbonylsulfamate
[0163] ((3aR,4R,6R,6aS)-2,2-dime l-6-((2-(3-((trifluorome
7-yl)amino)tetrahydro-3aH-cyclopenta[d][l,3]dioxol-4-yl)methanol (6.0 g) was dissolved in 2-methyltetrahedrafuran (60.0 mL) and to this solution was added pyridinium p-toluenesulfonate (5.9 g). This formed a precipitated and to this white slurry was added (4-aza-l-azoniabicyclo[2.2.2]oct-l-ylsulfonyl)(tert-butoxycarbonyl)azanide-l,4-diazabicyclo[2.2.2]octane (1 :1) hydrochloride1 (17.0 g). The mixture was stirred at ambient temperature until the HPLC showed <1% ((3aR,4R,6R,6aS)-2,2-dimethyl-6-((2-(3-((trifluoromethyl)thio)phenyl)pyrazolo[l,5-a]pyrimidin-7-yl)amino)tetrahydro-3aH-cyclopenta[d][l,3]dioxol-4-yl)methanol remaining starting material (-300 minutes). The reaction was quenched with water (60 mL) and the phases were separated. To the organic layer was added acetonitrile (60 mL) and the mixture was concentrated using a rotovap at 50°C to ~60 mL. The mixture was allowed to cool to room temperature and stirred overnight. During this time a white slurry formed. White solids were filtered using a medium fritted filter. The solid was dried in a vacuum oven at full vacuum overnight (40 °C). The reaction was then analyzed by HPLC and NMR to give ((3aR,4R,6R,6aS)-2,2-dimethyl-6-((2-(3-((trifluoromethyI)tM^
cyclopenta[d][l,3]dioxol-4-yl)methyl tert-butoxycarbonylsulfamate (5.03 g, 68%). [H NMR (300 MHz, DMSO- 6) δ ppm 1.26 (s, 3 H) 1.42 (s, 9 H) 1.51 (s, 3 H) 2.33 – 2.48 (m, 2 H) 3.30 (br s, 1 H) 4.06 – 4.21 (m, 1 H) 4.29 (d, J=5.28 Hz, 2 H) 4.52 (dd, J=7.18, 5.13 Hz, 1 H) 4.76 (dd, J=7.18, 4.54 Hz, 1 H) 6.35 (d, J=5.57 Hz, 1 H) 7.08 (s, 1 H) 7.63 – 7.72 (m, 1 H) 7.74 – 7.82 (m, 1 H) 8.01 (d, ^=7. 2 Hz, 1 H) 8.21 (d, J=5.28 Hz, 1 H) 8.31 (dt, J=7.84, 1.36 Hz, 1 H) 8.48 (s, 1 H) 1 1.92 (br s, 1 H)
[0164] Step 6: f R,2R3S.4R)-2J-dihvdroxy-4-((2-(3-fftrifluoromethvDthio^phenvnpyrazolori.5-a]pyrimidin-7-yl)aminokvcl nent\l)methyl sulfamate
[0165] To a solution of ((3aR,4R,6R!6aS)-2,2-dimethyl-6-((2-(3- ((trifluoromethyl)thio)phenyl)pyrazolo[l,5-a]pyrimidin-7-yl)amino)tetrahydro-3aH-cyclopenta[d][l,3]dioxol-4-yl)methyl tert-butoxycarbonylsulfamate (2.0 g) in acetonitrile (11 mL) at 0°C was added phosphoric acid (1 1 mL) while maintaining the temperature below 10°C. This mixture was warmed to ambient temperature and stirred for 4 hours. At this time HPLC analysis showed that <1% ((3aR,4R,6R,6aS)-2,2-dimethyl-6-((2-(3 -((trifluoromethyl)thio)phenyl)pyrazolo[ 1 ,5-a]pyrimidin-7-yl)amino)tetrahydro-3aH-cyclopenta[d][l,3]dioxol-4-yl)methyl tert-butoxycarbonylsulfamate starting material or reaction intermediates remained. To the reaction was added ethyl acetate (1 1 mL) and water (11 mL) and saturated Na2C03 (10 mL) dropwise. After this addition was complete saturated Na2C03 was added until the pH was between 6-7. The phases were separated and to the organic layer was added acetonitrile (30 mL) and the mixture was concentrated on a rotovap to ~16 mL. The mixture was stirred overnight. During this time a white slurry formed. The white solids were filtered using a medium filtted filter. The solid was dried in a vacuum oven at full vacuum overnight (40°C). The reaction was then analyzed by HPLC and NMR to give ((lR,2R,3S,4R)-2,3-dihydroxy-4-((2-(3-((trifluoromethyl)thio)phenyl)pyrazolo[ 1 ,5-a]pyrimidin-7-yl)amino)cyclopentyl)methyl sulfamate (1.5g ,84%). lH NMR (300 MHz, DMSO-c¾) δ ppm 1.44 – 1.61 (m, 1 H) 2.20 – 2.42 (m, 2 H) 3.78 (q, J-4.50 Hz, 1 H) 3.90 – 4.09 (m, 3 H) 4.09 – 4.22 (m, 1 H) 4.80 (d, ^5.28 Hz, 1 H) 5.03 (d, J=5.28 Hz, 1 H) 6.31 (d, J=5.57 Hz, 1 H) 7.05 (s, 1 H) 7.48 (s, 2 H) 7.62 – 7.72 (m, 1 H) 7.77 (d, J=7.92 Hz, 2 H) 8.17 (d, J=5.28 Hz, 1 H) 8.31 (dt, ^7.70, 1.43 Hz, 1 H) 8.47 (s, 1 H).
[0166] Example 3: fflR.2R.3S.4RV2.3-dihvdrosy-4-ff2-f3- ( ( trifluoroniethyl )thio)ph en vDpyrazolo 11,5-a I pyi Lmidin-7-Yl)amino)cvclopcntyl>m ethyl sulfama te
[0167] Step 1: .2-dimethyl-5-ff -(3-frtrifluoromethvnthio)phenvn-lH-pyrazol-5-yl)ainino)methylene -l,3-dioxane-4,6-dione)
[0168] Under a blanket of nitrogen at 20°C, Meldrum’s acid (18.6 Kg) and isopropanol (33 L) were placed in a 100 L glass-lined reactor. Trimethyl orthoformate (15.5 Kg (16.0L)) and isopropanol (11 L) were added and the mixture was heated to 80 °C for 40 min, whereby a small amount of methanol distilled off (<0.5 L). The mixture was stirred for 2 h at 80 °C. in a separate 160 L glass-lined reactor under nitrogen at 20 °C, 3-(3-((trifluoromethyl)thio)phenyl)-lH-pyrazol-5-amine (prepared in the manner described above) was mixed with isopropanol ( 10.9 kg, 42.0 mmol) and heated up to 80 °C within 60 min. The content of the 100 L reactor was transferred into the reaction mixture in the 160 L reactor at 80 °C, which was completed after 3 min. The reaction mixture was stirred for 30 min at 78 °C, the reaction was then cooled to 60 °C. HPLC analysis showed the reaction was 99.56% complete (product%/(product%+starting material0/.). The reaction mixture was cooled to 20 °C within 100 min, then the mixture was stirred for further 100 min at 20 °C. The suspension was then transferred onto a pressure filter. At 1.2 bar nitrogen, the solids were collected on the filter. The filter cake was washed 4 x with ethyl acetate (18 L each time). The wet cake was dried on the filter for 17 h at 20°C using a slight stream of nitrogen/vacuum (200-100 mbar). The wet product (14.7 kg) was further dried at the rotavap for approx. 24 h at 40-50 °C. 11,75 kg of the crude title compound was obtained (68% yield). NMRspectrum was consistent with that described above in Example 2.
[0169] Step 2: 2-(3-fftrifluoromethvnthio)phenYnpyrazolori.S-a1pyrimidin-7-ol
[0170] Under nitrogen at 20 °C, (2,2-dimethyl-5-(((3-(3-((trifluoromethyl)thio)phenyl)-lH-pyrazol-5-yl)amino)methylene)-l ,3-dioxane-4,6-dione) was placed in the reactor. 1 ,2-Dichlorobenzene (117 L) was added. The suspension was heated to 147°C for 90 min to give a solution, then it was stirred at 147°C for 18 h. Before sampling, the reaction was cooled to 60°C. HPLC analysis showed the reaction was 92.28% completion (product%/(product%+starting material%). The mixture was heated up again to 147°C and stirred for further 5 h at this temperature. HPLC analysis showed the reaction was 96.51% complete (product%/(product%+starting material%). The mixture was then stirred for 48 hours at 20°C, then it was heated again to 147°C und stirred at this temperature for 5 h. Before sampling, the reaction was cooled to 60°C. HPLC analysis showed the reaction was 98.47% completion (product%/(product%+starting material%). The mixture was heated up again to 146°C and stirred for further 5 h at this temperature.
Before sampling, the reaction was cooled to 60°C. HPLC analysis showed the reaction was 99.35% complete (product%/(product%+starting material%). The reaction was cooled to 20°C and the suspension was transferred in a pressure filter. The solids were collected on the filter at 1.8-3 bar N2 over a greater than 10 hour period. The filter cake was washed 4 x with acetonitrile (17 L), then it was dried on the filter for 18 h at 20°C/200-100 mbar, using a slight stream of N2. The material was transferred to a 50 L flask and dried on a rotavap at 50-60°C / 24-14 mbar for 2 d. 6.118 kg of the crude title compound was obtained (70% yield). NMR spectrum was consistent with that described above in Example 2.
[0171] Step 3: l-f2-f3- trifluoromethYnthio^phenvnpyrazoIo[1.5-alpyriinidiii-7-vn-lH-benzofdl [1.2.31 triazolc: triethylamine: hydrochloride complex ( 1 :0.21:0.21 moles:moles:moles)
[0172] Under N2 at 20°C, acetonitrile (30 L) was placed in the reactor, 2-(3-((trifluoromethyl)thio)phenyl)pyrazolo[l,5-a]pyrimidin-7-ol (6.00 kg) and lH-benzotriazol (5.83 kg) was added. A further portion of acetonitrile (30 L) was added, then the mixture was stirred at 20°C. Stirring proceeded over night. Triethylamine (8.16 L) was added at 20°C over 6 min. The yellow suspension was heated up to 45°C for 40 min. While stirring at 150 rpm, phosphoryl chloride (4.562 kg) was slowly added for 45 min. By controlling the addition, the reagent was dropped directly into the mixture to avoid the formation of lumps. The addition was exothermic, a maximum temperature of 53°C was observed. The brown suspension was heated up to 80°C over 1 h, then the reaction mixture was stirred for 5 h at this temperature. Acetonitrile (30 L) was added over 20 min keeping the internal temperature between 75-80°C. HPLC analysis showed the reaction was 98.31% completion (product%/(product%+starting material%).The mixture (brown suspension) was further stirred at 80°C for 70 min. HPLC analysis showed the reaction was 99.48% completion (product%/(product%+starting material%). Acetonitrile (61 L) was added over 30 min maintaining the temperature between 75-80°C. The pale brown suspension was stirred at 80°C for 90 min, then it was cooled to 20°C over 2.5 h. The mixture was stirred for 12 h at 20°C. The mixture was transferred in a pressure filter. The filter cake was washed twice with acetonitrile ( 18 L). Both wash steps were done at 3.5-4 bar N2. Each of these filtrations took overnight to go to completion. The filter cake was dried on the filter for 7.5 h. The material was transferred in a 50 L flask and dried at the rotavap at Ta 40-50°C / 50-11 mbar for 3 d to get a dry mass of 99.88% . The yield of l -(2-(3-((trifluoromethyl)t]iio)phenyl)pyrazolo[l ,5-a]pyrimidin-7-yl)-lH-benzo[d][l,2,3]triazole: triethylamine: hydrochloride complex (1 :0.21 :0.21 moles:moles:moles) was 7.948 kg (75%). NMR spectrum was consistent with that described above in Example 2.
[0173] Step 4: 3aR4R.6R,6aS)-2,2-dimethYl-6-f(2-f3-ffMfluoromethvnthio phenvnpyrazolori.5-alDyrimidin-7-yl)amino)tetrahvdro-3aH- vclopenta Idl [1.31 dioxol-4-vDmethanol
[0174] Under N2 in a 160 L glasslined reactor, triethylamine (21%) compound with l -(2-(3-((trifluoromethyl)thio)phenyl)pyrazolo[ 1 ,5 -a] pyrimidin-7-yI) – 1 H-benzo [d] [ 1 ,2,3 Jtriazole (21 %) hydrochloride (7.86 kg) was dissolved in triethylamine (23.3 L) at 20°C. ((3aR,4R,6R,6aS)-6-amino-2,2-dimethyltetrahydro-3aH-cyclopenta[d][l,3]dioxol-4-yl)methanol hydrochloride (4.49 kg) was added, followed by triethylamine (23 L). The reaction mixture was heated up to 80°C over 1 h, and then the mixture was stirred for 8 h at 80°C. The mixture was then cooled to 20°C. HPLC analysis showed the reaction was 99.97% complete (product%/(product%+starting material%). Water (66 L) was then added over 30 min at 20-25°C (exotherm), whereby a brown suspension was obtained. The mixture was concentrated at 60°C, 150-95 mbar, until 42 L solvent was distilled off. The suspension was heated to 50°C, and the solids were collected on a 90 L pressure filter (1.2 bar N2), which took 40 min. During this process, the material on the filter was not actively heated. The remaining solids in the reactor were rinsed with 15 L of the mother liquor. The wet filter cake was transferred back in the reactor. Water (64 L) was added. The mixture was heated up to 50°C over 30 min. The washed solids were collected on the 90 L pressure filter. Remaining mother liquor in the filter cake was pressed off at 1.2 bar N2 for 50 min (50 L mother liquor was used to rinse the reactor). The filter cake was dried on the pressure filter for 13.5 h, applying a slight stream of N2 / vac at 20°C to afford 10.247 kg of crude ((3aR,4R,6R,6aS)-2,2-dimethyl-6-((2-(3-((trifluoromethyl)tWo)phenyl)pyrazolo[l ,5-a]pyrimidin-7-yl)ammo)tetrahydro-3aH-cyclopenta[d][l,3]dioxol-4-yl)methanol. The wet filter cake was isolated. The wet filter cake was loaded into the reactor. Acetonitrile (65 L) was added, followed by activated charcoal (6.59 kg). The mixture was heated to 50°C for 30 min and stirred for 2 h at 50°C. Meanwhile a bed of celite (4.25 kg) had been prepared in the 90 L pressure filter, using acetonitrile (20 L) for conditioning. The bed was heated at 50°C. The black suspension was transferred on the filter and pushed through the Celite plug at 2 bar. The filtrate was transferred to a 200 L stirring tank via a heat resistant tube and a 0.45 μιη inline filter. The operation needed 18 min for completion. For washing, acetonitrile (50 L) which had been warmed up in the reactor to 50°C and transferred over the warmed filter cake and pushed through at 2 bar. Again, the filtrate was transferred in the 200 L stirring tank via a heat resistant tube and a 0.45 μιη inline filter. The operation needed 10 min for completion. The reactor was cleaned to remove attached charcoal (abrasive cleaning, using NaCl /acetone). The filtrate in the stirring tank was transferred in the reactor and concentrated at 50°C / 120 mbar until 63 L were distilled off. While well stirring (300 rpm) and 50°C, Water (1 10 L) was slowly added over 2 h. A pale yellow suspension was formed. The concentrate was cooled to 20°C for 3 h, then stirred at this temperature for 13 h. The solids were collected on a 50 L filter, using 1.2 bar N2 to push the filtrate through. The filter cake was washed twice with water (18 L), then dried on the filter for 24 h at 200-100 mbar, using a slight stream of N2. 4.563 kg of the title compound was obtained 55% yield. NMR spectrum was consistent with that described above in Example 2.
[0175] Step 5: (f3aR,4R,6R,6aS)-2^-dimethyl-6-(f2-f3-fftrifluorQmethvnthio phenvnpyrazolo[1.5- |pyrimidm-7-vnamino)teti ahYclro-3aH-cvclopenta|d||1.3ldioxol-4-yl mcthyl tert-butoxycarbonylsutfamatc
[0176] Under N2 at 20°C, ((3aR,4R,6R,6aS)-2,2-dimethyl-6-((2-(3- ((trifluoromethyl)thio)phenyl)pyrazolo[ 1 , 5 -a]pyrimidin-7-yl)amino)tetrahydro-3 aH-cyclopenta[d][l,3]dioxol-4-yl)methanol (4.019 kg) was placed in a 160 L glasslined reactor, then 2-methyl-tetrahydrofuran (40 L) was added. The mixture was stirred at 150 rpm for 30 min at 20°C, whereby a clear solution was formed. A KF measurement was taken and showed the water content to be 0.036% H20. The solution was stirred over night at 20 °C. The next morning, PPTS (2.2 kg) was loaded into the reactor. At 20°C, (4-aza-l-azoniabicyclo[2.2.2]oct-l-yIsulfonyl)(tert-butoxyc£u-bonyl)azanide-l,4-diazabicyclo[2.2.2]octane (1:1) hydrochloride (10.2 kg) was added. Stirring of the heterogeneous mixture was started at 130 rpm. The reaction was stirred with 200 rpm for 1 h at 20°C, then with increased speed of 250 rpm for an additional hour. HPLC analysis showed the conversion to be 87.3%. The reaction mass was stirred with 300 rpm for 2 h at 20°C. HPLC analysis showed the conversion to be 95.6%. The reaction mass was stirred with 300 rpm for 2 h at 20°C. HPLC analysis showed the conversion to be 97.7%. NaHC03 3.7% (40 L) was added to the mixture at 20°C and the reaction was stirred at 300 rpm for 10 min. Most of the solids from the reaction mixture went into solution. To dissolve remaining material which was attached at the top of the reactor, the bilayered mixture was stir up shortly by a N2 stream from the bottom. The layers were separated, which was completed after 13 min. The aqueous layer was discharged, the organic layer remained in the reactor. The org. layer was a brown solution, the aqueous layer was colorless and turbid. The pH of aqueous layer was approx. 8 (pH stick). NaHC03 3.7% (40 L) was added to the mixture at 20°C and it was stirred at 300 rpm for 10 min. The layers were separated, which was completed after 27 min. The aqueous layer was discharged, the organic layer remained in the reactor. The organic layer was a brown solution, the aqueous layer was colorless and turbid. The pH of aqueous layer was approx. 8-9 (pH stick) and the pH of organic layer was approx. 8 (pH stick, wet). The product in organic layer was transferred in the feeding tank and stored temporarily (approx. 30 min) at 20°C. The reactor was optically cleaned using a mixture of 2-methyltetrahydrofuran (30 L) and H20 (20 L). The org. layer was placed in the reactor and stored at -20°C for 14.5 h . While stirring at 150 rpm, the org. layer (suspension) was diluted with acetonitrile (16 L) and water (15 L) and warmed up to 5°C. At 5°C, acetic acid (0.172 kg) was added over 5 min. to a pH of 6; resulting in a mixture that was a pale brown solution. ((3aR,4R,6R,6aS)-2,2-dimethyl-6-((2-(3-((trifluoromethyl)thio)phenyl)pyrazolo[l,5-a]pyrimidin-7-yl)amino)tetrahydro-3aH-cyclopenta[d][l,3]dioxol-4-yl)methyl tert-butoxycarbonylsulfamate (2.0 g; prepared in a similar manner to that described above Example 2, Step 5) was added as seed. At 5°C, acetic acid (0.515 kg) was added over 15 min. to pH 4-5; a suspension formed. The feeding tank was rinsed with water (1.6 L). The mixture was stirred at 5°C with 90 rpm for 1.5 h, then it was transferred in a 50 L filter and filtered at 1.2 bar N2, in only 4 min. The filter cake was washed 4 x with cold acetonitrile (8 L, 0-5°C), then it was dried on the filter at 20°C for 8 h at 200 mbar, using a slight stream of N2. The yield of the title compound was 3.594 kg (62%). MR spectrum was consistent with that described above in Example 2.
[0177] Step 6: friR.2R.3S.4R 2.3-dihvdroxY-4-ff2-f3-fftrifluoromethvntliio phenvnDyrazolori.5-alpyrimidin-7-yl)aminokvciopent>T)mcthyl sulfamate Compound 1
[0178] 3.538 kg of ((3aR,4R,6R,6aS)-2,2-dimethyl-6-((2-(3-((trifluoromethyl)thio)phenyl)pyrazolo[l,5-a]pyrimidin-7-yl)amino)tetrahydro-3aH-cyclopenta[d][l,3]dioxol-4-yl)methyl tert-butoxycarbonylsulfamate was suspended in 13.5 kg of acetonitrile and cooled to 5°C. To this mixture was added 27.3 kg of H3PO4 over 1 hour and 50 minutes. The reaction was warmed to 20°C over 50 minutes and then stirred for 8h at 22°C. HPLC analysis showed the reaction was 99.69% complete. To the first portion (50% of the reaction mixture) was added 8.9 kg of water and 7.95 kg of ethyl acetate. The pH was then adjusted to 6.5 with 48 L of saturated sodium carbonate. 7.7 kg of ethyl acetate was added and the phases were separated. To the second portion (50% of the reaction mixture) was added 8.9 kg of water and 7.95 kg of ethyl acetate. The pH was then adjusted to 6.15 with 48 L of saturated sodium carbonate. 7.7 kg of ethyl acetate was added and the phases were separated. The organic phases were combined in a vessel (rinsed with 1.8 kg of ethyl acetate) and washed with 17.8 kg of water. The phases were separated and 17.8 kg of water and 0.237 kg of NaCl were added and the phases were separated. A repeat of wash with 17.8 kg of water and 0.237 kg of NaCl was added and the phases were separated. The organic layers were then combined and the temperature of the mixture was raised to 40°C and the pressure was reduced to 300-142 mbar. 27 L of liquid was distilled off over 4h. 31.7 kg of acetonitrile were then added to the solution and the temperature of the mixture was raised to 38°C and the pressure was reduced to 320-153 mbar. 26 L of liquid was distilled over 3h. 31.7 kg of acetonitrile were then added to the solution and the temperature of the mixture was raised to 37°C and the pressure was reduced to 320-153 mbar. 34 L of liquid was distilled over 2h. The suspension was stirred for lh at 50°C and then cooled to 20-25°C over 3h. The reaction was stirred overnight and the product was filtered and washed with 8.9 kg of acetonitrile twice. The cake was dried for 2h at 20°C (33 mbar) then at 40-45°C (1 mbar) to afford 2.08 kg (75.8%) of the title compound. 2.066 kg of ((lR,2R,3S,4R)-2,3-dihydroxy-4-((2-(3 -((trifluoromethyl)thio)phenyl)pyrazolo[ 1 , 5 -a]pyrimidin-7-yl)amino)cyclopenty l)methy 1 sulfamate was loaded into a reactor with 9.76 kg of acetronitrile and 4.12 kg of water and heated at a temperature of 56 °C for 1 hour and 10 minutes until dissolved. The solution was polished filtered and the filter was
rinsed with 3.16 kg acetonitrile and 1.37 kg of water. To the resulting solution was added with 11.0 kg of water over 45 minutes while maintaining the reaction temperature between 52-55°C. 0.009 kg of (( 1 R,2R,3S,4R)-2,3 -dihydroxy-4-((2-(3 -((trifluoromethyl)thio)phenyl)pyrazolo[ 1 ,5-a]pyrimidin-7-yl)amino)cyclopentyl)methyl sulfamate was added as seed (prepared in a similar manner to that described above Example 2, Step 5). A suspension was visible after 10 minutes of stirring. To the solution was added 9.62 kg of water over 3h while maintaining the reaction temperature between 50-55°C. The suspension was then cooled over 3h to 20°C and stirred for 12h at 22-23°C. The suspension was then filtered and washed twice with 13.7 kg of water. The product was dried at 40°C. 1.605 kg of the title compound was obtained in 78% yield. NMR spectrum was consistent with that described above in Example 2.
PATENT
WO2016069392
SYNTHESIS
![]()
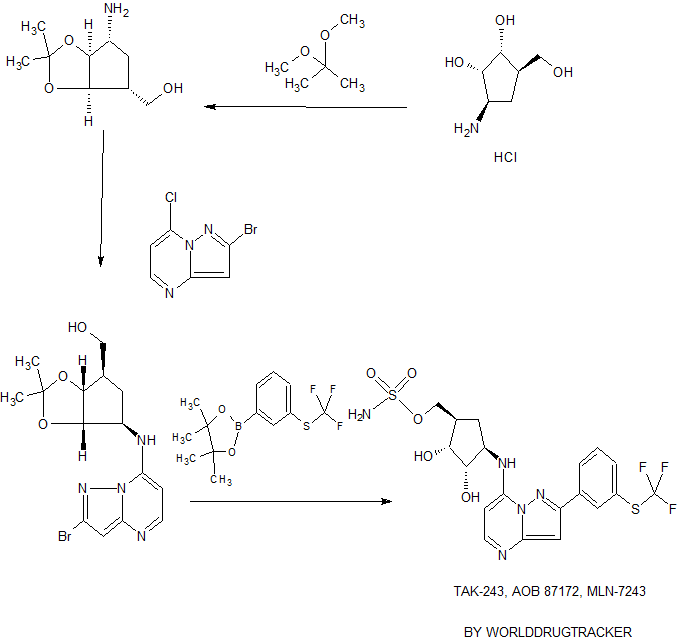
![]()
///////////////1450833-55-2, MLN 7243, TAK-243, TAK 243, TAK243, MLN7243; MLN-7243, MLN 7243, AOB87172, AOB-87172, AOB 87172, Millennium Pharmaceuticals, Inc., PHASE 1, TAKEDA ONCOLOGY
COS(=O)(=O)N[C@H]1C[C@H]([C@@H]([C@@H]1O)O)NC2=CC=NC3=CC(=NN23)C4=CC(=CC=C4)SC(F)(F)F
TAK-058 (ENV-8058)

TAK-058 , ENV-8058
5-HT 3 receptor antagonist
1-(1-methyl-1H-pyrazol-4-yl)-N-((1R,5S,7S)-9-methyl-3-oxa-9-azabicyclo[3.3.1]nonan-7-yl)-1H-indole-3-carboxamide
l-(l-methyl-lH-pyrazol-4-yl)-N-((lR,5 .7S)-9-methyl-3-oxa-9-azabicyclo[3.3.11nonan-7-yl)-lH-indole-3-carboxamide
1-(1-methyl-1H- pyrazol-4-yl)-N- ((1R,5S,7S)- 9-methyl-3- oxa-9-azabicyclo [3.3.1]nonan-7- yl)-1H-indole-3- carboxamide, 2,2,2- trifluoroacetic acid salt
N-(9-methyl-3-oxa-9-azabicyclo[3.3.1]nonan-7-yl)-1-(1-methylpyrazol-4-yl)indole-3-carboxamide
| Molecular Formula: | C21H25N5O2 |
|---|---|
| Molecular Weight: | 379.4555 g/mol |
https://clinicaltrials.gov/ct2/show/NCT02153099
Phase I Schizophrenia
| Latest Stage of Development | Phase I |
| Standard Indication | Schizophrenia |
| Indication Details | Treat schizophrenia |
- 01 Dec 2015 Phase-I clinical trials in Schizophrenia (Combination therapy) in USA (PO)
- 01 Dec 2015 Takeda completes a phase I trial in Healthy volunteers in USA (NCT02389881)
- 28 Nov 2015 Takeda plans a phase I trial in Schizophrenia (Combination therapy) in USA (NCT02614586)


1 -( 1 -methyl- 1 H-pyrazol-4-yl)-N-((lR,5S,7S)-9-methyl-3-oxa-9-azabicyclo[3.3.1]nonan-7-yl)-lH-indole-3-carboxamide, free base, which is an antagonist of the 5-HT3 receptor. 1 -(1 -Methyl- 1 H-pyrazol-4-yl)-N-((lR,5S,7S)-9-methyl-3-oxa-9-azabicyclo[3.3.1]nonan-7-yl)-lH-indole-3-carboxamide, 2,2,2-trifluoroacetic acid salt, is disclosed in PCT Publication No. WO
2014/014951, published January 23, 2014.
1-(1-methyl-1H-pyrazol-4-yl)-N-((1R,5S,7S)-9-methyl-3-oxa-9-azabicyclo[3.3.1]nonan-7-yl)-1H-indole-3-carboxamide a 5-HT3 receptor antagonist, useful for treating anxiety, depression, eating disorder, schizophrenia, cognitive dysfunction, Parkinson’s disease, Huntington’s Chorea, presenile dementia, Alzheimer’s disease and atherosclerosis.
This compound was originally claimed in WO2014014951, Takeda, following its acquisition of Envoy Therapeutics, is developing TAK-058 (ENV-8058), a 5-HT3 receptor antagonist, as an oral solution for treating schizophrenia, especially cognitive impairment associated with schizophrenia.
In July 2015, the drug was listed as being in phase I development. TAK-058 may have emerged from a schizophrenia therapy program which used Envoy’s bacTRAP translational profiling technology to identify a protein target in the brain.
PATENT
Example 5
Synthesis of l-(l-methyl-lH-pyrazol-4-yl)-N-((lR,5 .7S)-9-methyl-3-oxa-9-azabicyclo[3.3.11nonan-7-yl)-lH-indole-3-carboxamide. 2.2.2-trifluoroacetic acid salt
Step 1 : methyl 1-(1 -methyl- lH-pyrazol-4-yl)-lH-indole-3-carboxylate. TFA
To a sealed tube was added copper(I) iodide (65.2 mg, 0.342 mmol), methyl 1H-indole-3-carboxylate (200 mg, 1.142 mmol) and potassium phosphate (509 mg, 2.397 mmol), then the reaction vessel was evacuated and purged with nitrogen (3x). Next, 4-bromo-l-methyl-lH-pyrazole (184 mg, 1.142 mmol) and (lR,2R)- ,N2-dimethylcyclohexane-l,2-diamine (109 μΐ, 0.685 mmol) were added, followed by toluene (1 142 μΐ). The reaction tube was evacuated and purged with nitrogen, then sealed and heated at 1 10 °C for 24 h. HPLC purification provided the title compound as a colorless oil.
Step 2: 1-(1 -methyl- lH-pyrazol-4-yl)-lH-indole-3-carboxylic acid hydrochloride
To a solution of methyl 1-(1 -methyl- lH-pyrazol-4-yl)-lH-indole-3-carboxylate, TFA
(3.5 mg, 9.48 μιηοΐ) in MeOH (95 μΐ) was added a solution of aq. KOH (33.2 μΐ, 0.066 mmol, 2 M). The reaction mixture was stirred at RT overnight, then acidified with IN HC1.
The solvent was evaporated under reduced pressure and the residue was dried under vacuum overnight. The title compound was used without further purification.
Step 3 : l-(l-methyl-lH-pyrazol-4-yl)-N-((lR,5 .7S)-9-methyl-3-oxa-9-azabicyclor3.3.11nonan-7-yl)-lH-indole-3-carboxamide, 2,2,2-trifluoroacetic acid salt
To a mixture of 1-(1 -methyl- lH-pyrazol-4-yl)-lH-indole-3-carboxylic acid hydrochloride (2.6 mg, 9.36 μιηοΐ) in DMF (187 μΐ) was added HATU (4.27 mg, 0.01 1 mmol) and DIPEA (8.18 μΐ, 0.047 mmol). After the reaction mixture was stirred at RT for 15 min, (lR,5S,7S)-9-methyl-3-oxa-9-azabicyclo[3.3.1]nonan-7-amine, TFA (3.04 mg, 0.01 1 mmol) was added and stirring was continued for 2 h. HPLC purification afforded the title compound as a white solid. MS (ESI, pos. ion) m/z: 380.30 (M+l).
PATENT
EXAMPLE 1 : l-(l-methyl-lH-pyrazol-4-yl)-N-((lR,5S,7S)-9-methyl-3-oxa-9-azabicyclo[3.3.1 ]nonan-7-yl)- lH-indole-3-carboxamide
l-(l-Methyl-lH-pyrazol-4-yl)-lH-indole-3-carboxylic acid (128.7 g, 0.53 mol,) and anhydrous THF (645 mL) was heated to about 43°C. Oxalyl chloride (137.7 g, 92 mL, 1.08 mol) was added dropwise between 40 and 50°C. Gas evolution ceased in approximately 30 minutes. The resulting suspension was stirred for 2 hours at 50°C, allowed to cool to room temperature, and then stirred overnight. The suspension was diluted with heptane (1.5 L), stirred for 10 minutes, and allowed to settle. The supernatant was removed. The addition of heptane (1.5 L), followed by stirring, settling, and decanting was repeated two more times.
The resulting suspension was diluted with anhydrous THF (645 mL) and the ratio between THF and heptane was determined by NMR to be 3:2. The reaction mixture was cooled to 5°C and to the mixture was added DIPEA base (138 g, 1.07 mol) at such a rate that the temperature did not exceed 20°C. Next (li?,55*,7S)-9-methyl-3-oxa-9-azabicyclo[3.3.1]nonan-7-amine (101.4 g, 0.63 mol) in 500 mL of anhydrous THF was added. The reaction mixture was warmed to ambient temperature and stirred at 20 to 23°C overnight to give a suspension.
The suspension was filtered and the cake was dissolved in IN HC1 (2.6 L). The aqueous layer was washed with EtOAc (3 x 2.6 L). The aqueous layer was cooled to 5°C and was basified to pH 12 with aqueous potassium hydroxide (230 g) solution in water (500 mL). The mixture was stirred at 5 to 10°C overnight to give a solid. The product was filtered, washed with water (2 x 1.2 L), followed by MTBE (2 x 1.2 L), and then dried to give 128 g (64%) of the (crude) title compound.
Patent
https://www.google.co.in/patents/US20140024644
1-(1-methyl-1H- pyrazol-4-yl)-N- ((1R,5S,7S)- 9-methyl-3- oxa-9-azabicyclo [3.3.1]nonan-7- yl)-1H-indole-3- carboxamide, 2,2,2- trifluoroacetic acid salt
Synthetic Procedures Reference 1 Synthesis of (1R,5S,7S)-tert-butyl 7-hydroxy-3-oxa-9-azabicyclo[3.3.1]nonane-9-carboxylate
-

-
Sodium borohydride (259 mg, 6.84 mmol) was added portion-wise to a solution of (1R,5S)-tert-butyl 7-oxo-3-oxa-9-azabicyclo[3.3.1]nonane-9-carboxylate (550 mg, 2.279 mmol) in MeOH (4559 μl) at 0° C. After 5 min, the reaction mixture was allowed to warm to RT then stirred for 30 min. The mixture was concentrated under reduced pressure, dissolved in EtOAc and washed with brine. The combined organic layers were dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure to afford the title compound as a white solid, which was used without further purification.
Example 4 Synthesis of N-((1R,5S,7S)-9-methyl-3-oxa-9-azabicyclo[3.3.1]nonan-7-yl)-1-(1H-pyrazol-4-yl)-1H-indole-3-carboxamide, 2,2,2-trifluoroacetic acid salt
-

-
A mixture of 1-((1-benzyl-1H-pyrazol-4-yl)-N-((1R,5S,7S)-9-methyl-3-oxa-9-azabicyclo[3.3.1]nonan-7-yl)-1H-indole-3-carboxamide 2,2,2-trifluoroacetate (85 mg, 0.149 mmol) and 10% Pd—C (120 mg) in MeOH (1.0 ml) was stirred at RT under H2 for 2 days. Filtration and concentration afforded the title compound as a white solid. MS (ESI, pos. ion) m/z: 366.20 (M+1).
Example 5 Synthesis of 1-(1-methyl-1H-pyrazol-4-yl)-N-((1R,5S,7S)-9-methyl-3-oxa-9-azabicyclo[3.3.1]nonan-7-yl)-1H-indole-3-carboxamide, 2,2,2-trifluoroacetic acid salt

Step 1: methyl 1-(1-methyl-1H-pyrazol-4-yl)-1H-indole-3-carboxylate, TFA
-
To a sealed tube was added copper(I) iodide (65.2 mg, 0.342 mmol), methyl 1H-indole-3-carboxylate (200 mg, 1.142 mmol) and potassium phosphate (509 mg, 2.397 mmol), then the reaction vessel was evacuated and purged with nitrogen (3×). Next, 4-bromo-1-methyl-1H-pyrazole (184 mg, 1.142 mmol) and (1R,2R)—N1,N2-dimethylcyclohexane-1,2-diamine (109 μl, 0.685 mmol) were added, followed by toluene (1142 μl). The reaction tube was evacuated and purged with nitrogen, then sealed and heated at 110° C. for 24 h. HPLC purification provided the title compound as a colorless oil.
Step 2: 1-(1-methyl-1H-pyrazol-4-yl)-1H-indole-3-carboxylic acid hydrochloride
-
To a solution of methyl 1-(1-methyl-1H-pyrazol-4-yl)-1H-indole-3-carboxylate, TFA (3.5 mg, 9.48 μmol) in MeOH (95 μl) was added a solution of aq. KOH (33.2 μl, 0.066 mmol, 2 M). The reaction mixture was stirred at RT overnight, then acidified with 1N HCl. The solvent was evaporated under reduced pressure and the residue was dried under vacuum overnight. The title compound was used without further purification.
Step 3: 1-(1-methyl-1H-pyrazol-4-yl)-N-((1R,5S,7S)-9-methyl-3-oxa-9-azabicyclo[3.3.1]nonan-7-yl)-1H-indole-3-carboxamide, 2,2,2-trifluoroacetic acid salt
-
To a mixture of 1-(1-methyl-1H-pyrazol-4-yl)-1H-indole-3-carboxylic acid hydrochloride (2.6 mg, 9.36 μmol) in DMF (187 μl) was added HATU (4.27 mg, 0.011 mmol) and DIPEA (8.18 μl, 0.047 mmol). After the reaction mixture was stirred at RT for 15 min, (1R,5S,7S)-9-methyl-3-oxa-9-azabicyclo[3.3.1]nonan-7-amine, TFA (3.04 mg, 0.011 mmol) was added and stirring was continued for 2 h. HPLC purification afforded the title compound as a white solid. MS (ESI, pos. ion) m/z: 380.30 (M+1).
| 15 |
 |
 |
TFA |
| 379.456 MW | 380.30 MS +1
|
/////////TAK-058 , ENV-8058, phase I, takeda, 5-HT 3 receptor antagonist, Envoy Therapeutics, Inc., Phase I, Schizophrenia
C12CC(CC(N1C)COC2)NC(c4c3ccccc3n(c4)c5cnn(c5)C)=O
CN1C=C(C=N1)N2C=C(C3=CC=CC=C32)C(=O)NC4CC5COCC(C4)N5C
FDA approves first recombinant von Willebrand factor to treat bleeding episodes

12/08/2015 02:44
The U.S. Food and Drug Administration today approved Vonvendi, von Willebrand factor (Recombinant), for use in adults 18 years of age and older who have von Willebrand disease (VWD). Vonvendi is the first FDA-approved recombinant von Willebrand factor, and is approved for the on-demand (as needed) treatment and control of bleeding episodes in adults diagnosed with VWD.
| Company | Baxalta Inc. |
| Description | Recombinant human von Willebrand factor (vWF) |
| Molecular Target | von Willebrand factor (vWF) |
| Mechanism of Action | |
| Therapeutic Modality | Biologic: Protein |
| Latest Stage of Development | Registration |
| Standard Indication | Bleeding |
| Indication Details | Treat and prevent bleeding episodes in von Willebrand disease (vWD) patients; Treat von Willebrand disease (vWD) |
| Regulatory Designation | U.S. – Orphan Drug (Treat and prevent bleeding episodes in von Willebrand disease (vWD) patients); EU – Orphan Drug (Treat and prevent bleeding episodes in von Willebrand disease (vWD) patients); Japan – Orphan Drug (Treat and prevent bleeding episodes in von Willebrand disease (vWD) patients) |
December 8, 2015
Release
The U.S. Food and Drug Administration today approved Vonvendi, von Willebrand factor (Recombinant), for use in adults 18 years of age and older who have von Willebrand disease (VWD). Vonvendi is the first FDA-approved recombinant von Willebrand factor, and is approved for the on-demand (as needed) treatment and control of bleeding episodes in adults diagnosed with VWD.
VWD is the most common inherited bleeding disorder, affecting approximately 1 percent of the U.S. population. Men and women are equally affected by VWD, which is caused by a deficiency or defect in von Willebrand factor, a protein that is critical for normal blood clotting. Patients with VWD can develop severe bleeding from the nose, gums, and intestines, as well as into muscles and joints. Women with VWD may have heavy menstrual periods lasting longer than average and may experience excessive bleeding after childbirth.
“Patients with heritable bleeding disorders should meet with their health care provider to discuss appropriate measures to reduce blood loss,” said Karen Midthun, M.D., director of the FDA’s Center for Biologics Evaluation and Research. “The approval of Vonvendi provides an additional therapeutic option for the treatment of bleeding episodes in patients with von Willebrand disease.”
The safety and efficacy of Vonvendi were evaluated in two clinical trials of 69 adult participants with VWD. These trials demonstrated that Vonvendi was safe and effective for the on-demand treatment and control of bleeding episodes from a variety of different sites in the body. No safety concerns were identified in the trials. The most common adverse reaction observed was generalized pruritus (itching).
The FDA granted Vonvendi orphan product designation for these uses. Orphan product designation is given to drugs intended to treat rare diseases in order to promote their development.
Vonvendi is manufactured by Baxalta U.S., Inc., based in Westlake Village, California.
//////////
New “mTOR” inhibitor from Exelixis, Inc., XL 388

XL 388
A Novel Class of Highly Potent, Selective, ATP-Competitive, and Orally Bioavailable Inhibitors of the Mammalian Target of Rapamycin (mTOR)
Benzoxazepine-Containing Kinase Inhibitor

[7-(6-Aminopyridin-3-yl)-2,3-dihydro-1,4-benzoxazepin-4(5H)-yl][3-fluoro-2-methyl-4-(methylsulfonyl)phenyl]methanone
[7-(6-amino-3-pyridinyl)-2,3-dihydro-1,4-benzoxazepin-4(5H)-yl][3-fluoro-2-methyl-4-(methylsulfonyl)phenyl]-Methanone,
(7-(6-Aminopyridin-3-yl)-2,3-dihydrobenz[f][1,4]oxazepin-4(5H)-yl)(3-fluoro-2-methyl-4-(methylsulfonyl)phenyl)methanone
MW 455.50, CAS 1251156-08-7, MF C23 H22 F N3 O4 S
Exelixis, Inc. INNOVATOR, IND Filed
½H2O
C23H22FN3O4S.½H2O , Molecular Weight: 464.51
MONO HYDROCHLORIDE…..CAS 1777807-51-8, [7-(6-Aminopyridin-3-yl)-2,3-dihydro-1,4-benzoxazepin-4(5H)-yl][3-fluoro-2-methyl-4-(methylsulfonyl)phenyl]methanone Hydrochloride (1·HCl)
TLC Rf = 0.33 (Dichloromethane:Methanol [95:5])
Potent and selective mTOR inhibitor (IC50 = 9.9 nM). Inhibits mTOR activity in an ATP-competitive manner. Exhibits >300-fold selectivity for mTOR over PI 3-K and a range of other kinases. Displays antitumor activity in athymic nude mice implanted with tumor xenografts.
SYNTHESIS
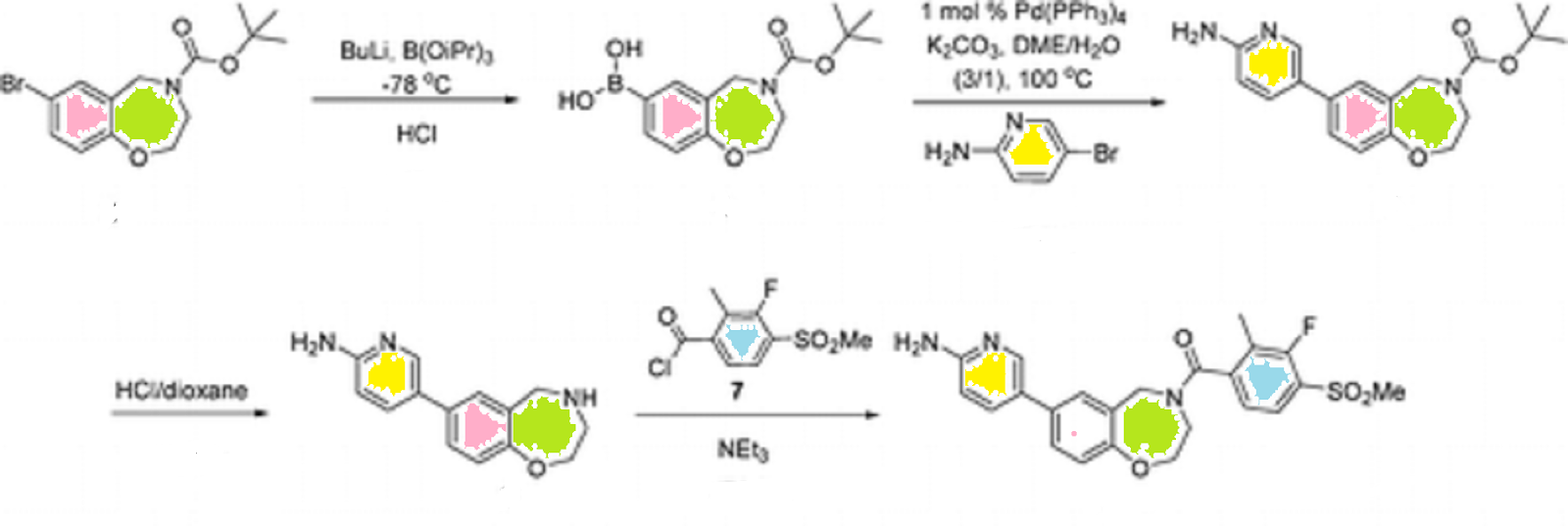
CLICK ON IMAGE FOR CLEAR VIEW……………..

Tyrosine kinases are important enzymes for signal transduction in cells. Therefore, they are often targets for the treatment of diseases that are caused by dysregulation of cellular processes, such as cancers. Mammalian target of rapamycin (mTOR) is a kinase in the phosphatidylinositol-3-kinase (PI3K) family of enzymes and is implicated in the regulation of cell growth and proliferation. Various inhibitors of mTOR have been explored as possible agents for treatment of various cancers
The mammalian target of rapamycin (mTOR) is a large protein kinase that integrates both extracellular and intracellular signals of cellular growth, proliferation, and survival. Both extracellular mitogenic growth factor signaling from cell surface receptors and intracellular signals that convey hypoxic stress, energy, and nutrient status converge at mTOR. mTOR exists in two distinct multiprotein complexes: mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2).
mTORC1 is a key mediator of translation and cell growth, via its substrates p70S6 kinase (p70S6K) and eIF4E binding protein 1 (4E-BP1), and promotes cell survival via the serum and glucocorticoid-activated kinase (SGK), whereas mTORC2 promotes activation of prosurvival kinase AKT. mTORC1, but not mTORC2, can be inhibited by an intracellular complex between rapamycin and FK506 binding protein (FKBP). However, rapamycin–FKBP may indirectly inhibit mTORC2 in some cells by sequestering mTOR protein and thereby inhibiting assembly of mTORC2.
Given the role of mTOR signaling in cellular growth, proliferation, and survival as well as its frequent deregulation in cancers, several rapamycin analogues (rapalogues) that are selective allosteric mTORC1 inhibitors have been extensively evaluated in a number of cancer clinical trials.

Demonstrated clinical efficacy for rapalogues is currently limited to patients with advanced, metastatic renal cell carcinoma (RCC) despite extensive development efforts.
This result is likely attributed not only to a lack of inhibition of mTORC2 by rapalogues that leads to upregulation of Akt through a negative feedback loop, but also to only partial inhibition of mTORC1.Therefore, ATP-competitive mTOR inhibitors that should simultaneously inhibit both mTORC1 and mTORC2 may offer a clinical advantage over rapalogues.
As a key component of the phosphoinositide 3-kinase-related kinase (PIKK) family, which is comprised of phosphoinositide 3-kinases (PI3Ks), DNA-PK, ATM, and ATR, mTOR shares the highly conserved ATP binding pockets of the PI3K family with sequence similarity of 25% in the kinase catalytic domain.
In light of this fact, it is not surprising that many of the first reported ATP-competitive mTOR inhibitors such as BEZ235 and GDC-0980 also inhibited PI3Ks. PI3Ks are responsible for the production of 3-phosphoinositide lipid second messengers such as phosphatidylinositol 3,4,5-triphosphate (PIP3), which are involved in a number of critical cellular processes, including cell proliferation, cell survival, angiogenesis, cell adhesion, and insulin signaling.
Therefore, the development of ATP-competitive mTOR inhibitors that are selective over PI3Ks may offer an improved therapeutic potential relative to rapalogues as well as dual PI3K/mTOR inhibitors. Recently, several selective ATP-competitive mTOR inhibitors such as Torin 2 and AZD8055 have been reported with sufficient promise to warrant clinical trials.
PATENT
WO 2010118208
Example 2:
[7-(6-Aminopyridin-3-yl)-2,3-dihydro-l,4-benzoxazepin-4(5H)-yl] [3-fluoro- 2-methyl-4-(methylsulfonyl)phenyl]methanone

tørt-Butyl 7-(6-aminopyridin-3-yl)-2,3-dihydrobenzo[/] [l,4]oxazepine-4(5H)- carboxylate. To a mixture of 4-(te/t-butoxycarbonyl)-2,3,4,5- tetrahydrobenzo[/][l,4]oxazepin-7-ylboronic acid (1.52 g, 5.2 mmol), prepared as described in Reference Example 5, 2-amino-5-bromopyridine (900 mg, 5.2 mmol), and potassium carbonate (1.73 g, 12.5 mmol) in 1 ,2-dimethoxyethane/water (30 mL/10 mL) was added tetrakis(triphenylphosphine)palladium(0) (90 mg, 1.5 mol%) and the reaction mixture was purged with nitrogen and stirred at reflux for 3 h. The reaction was cooled to rt, diluted with water/ethyl acetate (50 mL/50 mL), and the separated aqueous layer was extracted with ethyl acetate. The resulting emulsion was removed by filtration. The combined organic layer was washed with brine, dried with sodium sulfate, filtered and concentrated under reduced pressure, and the residue was triturated with toluene for 1 h. The resulting off-white solid was isolated by filtration to give the desired product (1.37 g, 77 %) as an off-white solid. MS (EI) for Ci9H23N3O3: 342 (MH+).
5-(2,3,4,5-Tetrahydrobenzo[/] [l,4]oxazepin-7-yl)pyridine-2-amine. To a stirred solution of tert-butyl 7-(6-aminopyridin-3-yl)-2,3-dihydrobenzo[/][l,4]oxazepine- 4(5H)-carboxylate (1.36 g, 3.98 mmol) in 1,4-dioxane (5 mL) was added 4 N hydrogen chloride in 1 ,4-dioxane (5 mL) and the reaction mixture was stirred at rt overnight. The reaction was concentrated on a rotary evaporator and the residue was triturated with ether. The solid was isolated by filtration. This solid was dissolved in water (5 mL) and made basic with 5 N sodium hydroxide to pH 11-12. The brownish sticky oil that aggregated at the bottom was isolated and the aqueous layer was extracted with 5 % methanol in ethyl acetate. The extracts were dried with sodium sulfate and concentrated on a rotary evaporator. The brownish sticky oil was dissolved with a mixture of methanol/ethyl acetate, combined with the isolated organic residue and concentrated under reduced pressure to give a yellow solid. This solid was triturated with dichloromethane (10 mL) for 1 h and a yellow solid was isolated by filtration and dried under high vacuum to give amine the desired product (920 mg, 96 %). MS (EI) for Ci4Hi5N3O: 242 (MH+).
[7-(6-Aminopyridin-3-yl)-2,3-dihydro-l,4-benzoxazepin-4(5H)-yl][3-fluoro-2- methyl-4-(methylsulfonyl)phenyl]methanone.
To a stirred suspension of 5-(2, 3,4,5- tetrahydrobenzo[/][l,4]oxazepin-7-yl)pyridine-2-amine (85 mg, 352 μmol) and triethylamine (54 μL, 387 μmol) in dichloromethane (10 mL) was added 3-fluoro-2-methyl-4- (methylsulfonyl)benzoyl chloride (91 mg, in 3 mL of dichloromethane), prepared as described in Reference Example 1, at 0 0C for 2 h. After stirring for an additional 1 h at rt, the reaction mixture was diluted with water (5 mL) and the separated aqueous layer was extracted with dichloromethane. The combined extracts were dried with sodium sulfate, filtered and concentrated under reduced pressure to give a light-yellow solid that was purified via silica gel chromatography to give the desired product (113 mg, 70%) as a white solid.
1H NMR (400 MHz, DMSO-d6): δ 8.24-8.03 (dd, IH), 7.79-7.71 (m, IH), 7.71-7.69 (dd, 0.5H), 7.57-7.57 (d, 0.5H), 7.44-7.40 (m, 1.5H), 7.29-7.19 (dd, IH), 7.05-7.01 (dd, IH), 6.64-6.63 (d, 0.5H), 6.54-6.45 (dd, IH), 6.06 (s, 2H), 4.93-4.31 (m, 2H), 4.31-3.54 (m, 4H), 3.37-3.36(d, 3H), 2.12-1.77 (d, 3H).
MS (EI) C23H22FN3O4S: 456 (MH+).
PAPER
Journal of Medicinal Chemistry (2013), 56(6), 2218-2234.
J. Med. Chem., 2013, 56 (6), pp 2218–2234
DOI: 10.1021/jm3007933

A series of novel, highly potent, selective, and ATP-competitive mammalian target of rapamycin (mTOR) inhibitors based on a benzoxazepine scaffold have been identified. Lead optimization resulted in the discovery of inhibitors with low nanomolar activity and greater than 1000-fold selectivity over the closely related PI3K kinases. Compound 28 (XL388) inhibited cellular phosphorylation of mTOR complex 1 (p-p70S6K, pS6, and p-4E-BP1) and mTOR complex 2 (pAKT (S473)) substrates. Furthermore, this compound displayed good pharmacokinetics and oral exposure in multiple species with moderate bioavailability. Oral administration of compound 28 to athymic nude mice implanted with human tumor xenografts afforded significant and dose-dependent antitumor activity.
(7-(6-Aminopyridin-3-yl)-2,3-dihydrobenz[f][1,4]oxazepin-4(5H)-yl)(3-fluoro-2-methyl-4-(methylsulfonyl)phenyl)methanone (28)
1H NMR (400 MHz, DMSO-d6): δ (rotamers are observed) 8.24 and 8.03 (d, J = 2.4 Hz, 1H), 7.77 and 7.72 (t, J = 7.6 Hz, 1H), 7.71–7.39 (m, 2H), 7.57 and 6.63 (d, J = 2.4 Hz, 1H), 7.28 and 7.19 (d, J = 7.6 Hz, 1H), 7.04 and 7.02 (d, J = 8.0 Hz, 1H), 6.52 and 6.46 (d, J = 8.8 Hz, 1H), 6.05 (br s, 2H), 4.93–4.31 (m, 2H), 4.28–3.56 (m, 4H), 3.37 and 3.34 (s, 3H), 2.12 and 1.77 (d,J = 1.6 Hz, 3H). 13C NMR (100 MHz, DMSO-d6): δ 167.3, 167.2, 166.6, 166.6, 158.9, 158.9, 158.4, 158.4, 157.4, 157.2, 155.9, 155.8, 145.4, 145.1, 145.1, 144.0, 143.9, 135.0, 134.7, 132.9, 132.8, 129.4, 129.2, 128.2, 128.2, 128.1, 128.0, 127.0, 126.9, 126.8, 125.9, 125.6, 125.4, 123.6, 123.5, 123.3, 123.1, 122.8, 122.0, 122.0, 121.9, 121.9, 121.2, 120.7, 107.8, 107.8, 70.9, 70.8, 51.1, 51.1, 47.4, 46.5, 43.5, 43.5, 43.5, 43.4, 11.0, 10.9, 10.7, 10.6. IR (KBr pellet): 1623, 1487, 1457, 1423, 1385, 1314, 1269, 1226, 1193, 1144, 1133, 1054, 1031, 962, 821, 768 cm–1. Mp: 204–205 °C. MS (EI): m/z for C23H22FN3O4S, 456.0 (MH+). High-resolution MS (FAB MS using glycerol as the matrix): m/z calcd for C23H22FN3O4S 456.13878, found 456.13943.
PATENT
- SYNTHETIC EXAMPLES
-

-
1-Bromo-3,4-difluoro-2-methylbenzene. To a stirred mixture of 2,3-difluorotoluene (1.9 g, 14.8 mmol) and iron (82.7 mg, 1.48 mmol) in chloroform (10 mL) at rt was added bromine (76 μL, 14.8 mmol) over 2 h. The resulting mixture was stirred at rt overnight. Excess water (10 mL) was added and the reaction mixture was diluted with ether (20 mL). The separated organic layer was washed with aqueous sodium thiosulfate, brine, dried over sodium sulfate and concentrated on a rotary evaporator. The residue was distilled to give the desired product (2.49 g, 81%) as a colorless oil.
-
3,4-Difluoro-2-methylbenzoic acid. To a stirred solution of 1-bromo-3,4-difluoro-2-methylbenzene (940 mg, 4.54 mmol) in tetrahydrofuran (5 mL) was added isopropylmagnesium bromide (3.0 mL, 6.0 mmol) over 1 h at 0° C. The resulting mixture was stirred at rt for 24 h. Carbon dioxide (CO2), generated from dry ice, was introduced to the reaction mixture over 2 h and the resulting mixture was stirred for an additional 30 min. The reaction mixture was quenched with addition of an excess amount of water (5 mL) and the tetrahydrofuran was removed on a rotary evaporator. The resulting aqueous layer was diluted with water (5 mL) and acidified with concentrated hydrochloric acid to pH 1-2. The white precipitate was filtered and washed with water and cold hexanes and dried under high vacuum to give the desired product (630 mg, 81%) as a white powder. MS (EI) for C8H6F2O2: 171 (MH−).
-
3-Fluoro-2-methyl-4-(thiomethyl)benzoic acid. To a stirred solution of acid 3,4-difluoro-2-methylbenzoic acid (700 mg, 4.1 mmol) in dimethylsulfoxide (5 mL) was added powdered potassium hydroxide (274 mg, 4.9 mmol) and the mixture was stirred at rt for 30 min. Sodium thiomethoxide (342 mg, 4.9 mmol) was added to the mixture and the resulting mixture was stirred at 55-60° C. for 4 h. Additional powdered potassium hydroxide (70 mg, 1.2 mmol), sodium thiomethoxide (60 mg, 0.8 mmol), and dimethylsulfoxide (2 mL) were added to the reaction mixture. After stirring for 4 h, the mixture was cooled to 0° C. and quenched with excess water (10 mL). The resulting suspension was acidified at 0° C. with concentrated hydrochloric acid to pH 1-2. The white precipitate was collected by suction filtration, washed with water and dried under vacuum overnight to give the desired product (870 mg, 100%). The intermediate sulfide was used in the next step without further purification. MS (EI) for C9H9FO2S: 199.1 (MH−).
-
3-Fluoro-2-methyl-4-(methylsulfonyl)benzoic acid. To a stirred suspension of 3-fluoro-2-methyl-4-(thiomethyl)benzoic acid in an acetone/water (1 mL/10 mL) mixture was added sodium hydroxide (330 mg, 8.25 mmol) and sodium bicarbonate (680 mg, 8.1 mmol). Oxone (˜4 g) was added portionwise to the reaction mixture at 0° C. over 2 h. The reaction was monitored by LC/MS. Concentrated hydrochloric acid was added to adjust the pH 2-3 and the white precipitate was collected by suction filtration, washed with water, and dried under vacuum. Dried precipitate was suspended in water (10 mL), stirred vigorously at rt for 1 h, filtered, washed with water, and hexanes and dried under vacuum to give the desired product (886 mg, 94%) as a white powder. MS (EI) for C9H9FO4S: 231 (MH−).
-
3-Fluoro-2-methyl-4-(methylsulfonyl)benzoyl chloride. A mixture of 3-fluoro-2-methyl-4-(methylsulfonyl)benzoic acid (860 mg, 3.7 mmol) in thionyl chloride (10 mL) was heated to reflux for 3 h. (the reaction mixture became homogenous). The reaction mixture was concentrated on a rotary evaporator to give the crude acid chloride. This acid chloride was triturated with dichloromethane (2 mL) and concentrated under reduced pressure. The trituration process was repeated 3 times until the product (900 mg, 98%) was obtained as a white powder.
- Reference Example 13-Fluoro-2-methyl-4-(methylsulfonyl)benzoyl chloride

Reference Example 2Ethyl 4-(2,3,4,5-tetrahydro-1,4-benzoxazepin-7-yl)benzoate hydrochloride salt
-

-
4-(ethoxycarbonyl)phenylboronic acid (22.16 g, 114 mmol), tert-butyl 7-bromo-2,3-dihydrobenzo[f][1,4]oxazepin-4(5H)-carboxylate (34.08 g, 104 mmol), prepared as described in Reference Example 4, Pd(dppf)Cl2 and TEA (21 g, 208 mmol) were combined in a mixture of dioxane (200 mL) and water (20 mL). The reaction mixture was heated to 90° C. for 2 h, then cooled and the solvent removed. Purification of the residue by silica chromatography gave the desired product ester (31.3 g, 69% yield).
-
To the solution of tert-butyl 7-(4-(ethoxycarbonyl)phenyl)-2,3-dihydrobenzo[f][1,4]oxazepine-4(5H)-carboxylate (10.3 g, 25.93 mmol) in MeOH (120 mL) was added a solution of 4 N HCl in dioxane (50 mL). The reaction mixture was heated to 50° C. for 3 h (monitored by LC/MS). The reaction mixture was allowed to cool to rt. Ethyl 4-(2,3,4,5-tetrahydro-1,4-benzoxazepin-7-yl)benzoate as the hydrochloride salt (8.8 g, 99% yield) was collected by suction filtration.

-

-
tert-Butyl-5-bromo-2-hydroxybenzyl(2-hydroxyethyl)carbamate. Commercially-available 5-bromo-2-hydroxybenzaldehyde (4.0 g, 10 mmol) and 2-aminoethanol were combined in THF/MeOH (100 mL, 10:1) and sodium borohydride (0.76 g, 2.0 mmol) was added with stirring. The resulting reaction mixture was stirred at 40° C. for 4 h, concentrated on a rotary evaporator then diluted with EtOAc (50 mL) and saturated NaHCO3 (30 mL). To this suspension was added di-tert-butyl dicarbonate (2.83 g, 13 mmol). The mixture was stirred at rt overnight. The organic layer was washed with water, dried over anhydrous magnesium sulfate, filtered, and concentrated on a rotary evaporator. Hexane was subsequently added to the crude reaction product which resulted in the formation of a white solid. This slurry was filtered to obtain the desired product (6.8 g, 98%) as a white solid. MS (EI) for C14H20BrNO4, found 346 (MH+).
-
tert-Butyl-7-bromo-2,3-dihydrobenzo[f][1,4]oxazepine-4(5H)-carboxylate. tert-Butyl-5-bromo-2-hydroxybenzyl(2-hydroxyethyl)carbamate (3.46 g, 10 mmol) and triphenylphosphine (3.96 g, 15 mmol) were combined in DCM (100 mL) and diisopropyl azodicarboxylate (3.03 g, 15 mmol) was added. The resulting reaction mixture was stirred at rt for 12 h. The reaction mixture was washed with water, dried, filtered, and concentrated on a rotary evaporator. The resulting crude product was purified via silica gel chromatography eluting with 8:2 hexane/ethyl acetate to give the desired product (1.74 g, 53%) as a white solid. MS (EI) for C14H18BrNO3, found 328 (MH+).
- Reference Example 4tert-Butyl-7-bromo-2,3-dihydrobenzo[f][1,4]oxazepine-4(5H)-carboxylate

Reference Example 54-(tert-Butoxycarbonyl)-2,3,4,5-tetrahydrobenzo[f][1,4]oxazepin-7-ylboronic acid
-

-
To a stirred solution of tert-butyl-7-bromo-2,3-dihydrobenzo[f][1,4]oxazepine-4(5H)-carboxylate (10 g, 30.5 mmol), prepared as described in Reference Example 4, and triisopropylborate (9.1 mL, 40 mmol) in dry tetrahydrofuran (100 mL) was added dropwise n-butyllithium in tetrahydrofuran (1.6 M, 25 mL, 40 mmol) while maintaining the temperature below −60° C. Upon completion of addition, the reaction mixture was stirred for 30 min, then quenched with 1 N aqueous hydrochloric acid (35 mL) and allowed to warm to rt. The reaction mixture was extracted with ethyl acetate, dried over anhydrous magnesium sulfate, filtered and concentrated on a rotary evaporator. Hexane was subsequently added to the crude reaction product which resulted in the formation of a white solid. This slurry was stirred for 1 h and filtered to obtain 4-(tert-butoxycarbonyl)-2,3,4,5-tetrahydrobenzo[f][1,4]oxazepin-7-ylboronic acid (8.6 g, 95%) as a white solid. MS (EI) for C14H20BNO5: 194 (M-Boc).

- Example 2[7-(6-Aminopyridin-3-yl)-2,3-dihydro-1,4-benzoxazepin-4(5H)-yl][3-fluoro-2-methyl-4-(methylsulfonyl)phenyl]methanone
-

-
tert-Butyl 7-(6-aminopyridin-3-yl)-2,3-dihydrobenzo[f][1,4]oxazepine-4(5H)-carboxylate. To a mixture of 4-(tert-butoxycarbonyl)-2,3,4,5-tetrahydrobenzo[f][1,4]oxazepin-7-ylboronic acid (1.52 g, 5.2 mmol), prepared as described in Reference Example 5, 2-amino-5-bromopyridine (900 mg, 5.2 mmol), and potassium carbonate (1.73 g, 12.5 mmol) in 1,2-dimethoxyethane/water (30 mL/10 mL) was added tetrakis(triphenylphosphine)palladium(0) (90 mg, 1.5 mol %) and the reaction mixture was purged with nitrogen and stirred at reflux for 3 h. The reaction was cooled to rt, diluted with water/ethyl acetate (50 mL/50 mL), and the separated aqueous layer was extracted with ethyl acetate. The resulting emulsion was removed by filtration. The combined organic layer was washed with brine, dried with sodium sulfate, filtered and concentrated under reduced pressure, and the residue was triturated with toluene for 1 h. The resulting off-white solid was isolated by filtration to give the desired product (1.37 g, 77%) as an off-white solid. MS (EI) for C19H23N3O3: 342 (MH+).
-
5-(2,3,4,5-Tetrahydrobenzo[f][1,4]oxazepin-7-yl)pyridine-2-amine. To a stirred solution of tert-butyl 7-(6-aminopyridin-3-yl)-2,3-dihydrobenzo[f][1,4]oxazepine-4(5H)-carboxylate (1.36 g, 3.98 mmol) in 1,4-dioxane (5 mL) was added 4 N hydrogen chloride in 1,4-dioxane (5 mL) and the reaction mixture was stirred at rt overnight. The reaction was concentrated on a rotary evaporator and the residue was triturated with ether. The solid was isolated by filtration. This solid was dissolved in water (5 mL) and made basic with 5 N sodium hydroxide to pH 11-12. The brownish sticky oil that aggregated at the bottom was isolated and the aqueous layer was extracted with 5% methanol in ethyl acetate. The extracts were dried with sodium sulfate and concentrated on a rotary evaporator. The brownish sticky oil was dissolved with a mixture of methanol/ethyl acetate, combined with the isolated organic residue and concentrated under reduced pressure to give a yellow solid. This solid was triturated with dichloromethane (10 mL) for 1 h and a yellow solid was isolated by filtration and dried under high vacuum to give amine the desired product (920 mg, 96%). MS (EI) for C14H15N3O: 242 (MH+).
-
[7-(6-Aminopyridin-3-yl)-2,3-dihydro-1,4-benzoxazepin-4(5H)-yl][3-fluoro-2-methyl-4-(methylsulfonyl)phenyl]methanone. To a stirred suspension of 5-(2,3,4,5-tetrahydrobenzo[f][1,4]oxazepin-7-yl)pyridine-2-amine (85 mg, 352 μmol) and triethylamine (54 μL, 387 μmol) in dichloromethane (10 mL) was added 3-fluoro-2-methyl-4-(methylsulfonyl)benzoyl chloride (91 mg, in 3 mL of dichloromethane), prepared as described in Reference Example 1, at 0° C. for 2 h. After stirring for an additional 1 h at rt, the reaction mixture was diluted with water (5 mL) and the separated aqueous layer was extracted with dichloromethane. The combined extracts were dried with sodium sulfate, filtered and concentrated under reduced pressure to give a light-yellow solid that was purified via silica gel chromatography to give the desired product (113 mg, 70%) as a white solid. 1H NMR (400 MHz, DMSO-d6): δ 8.24-8.03 (dd, 1H), 7.79-7.71 (m, 1H), 7.71-7.69 (dd, 0.5H), 7.57-7.57 (d, 0.5H), 7.44-7.40 (m, 1.5H), 7.29-7.19 (dd, 1H), 7.05-7.01 (dd, 1H), 6.64-6.63 (d, 0.5H), 6.54-6.45 (dd, 1H), 6.06 (s, 2H), 4.93-4.31 (m, 2H), 4.31-3.54 (m, 4H), 3.37-3.36 (d, 3H), 2.12-1.77 (d, 3H). MS (EI) C23H22FN3O4S: 456 (MH+).

PAPER
Org. Process Res. Dev., 2015, 19 (7), pp 721–734
DOI: 10.1021/acs.oprd.5b00037
http://pubs.acs.org/doi/abs/10.1021/acs.oprd.5b00037

The benzoxazepine core is present in several kinase inhibitors, including the mTOR inhibitor 1. The process development for a scalable synthesis of 7-bromobenzoxazepine and the telescoped synthesis of 1 are reported. Compound 1 consists of three chemically rich, distinct fragments: the tetrahydrobenzo[f][1,4]oxazepine core, the aminopyridyl fragment, and the substituted (methylsulfonyl)benzoyl fragment. Routes were developed for the preparation of 3-fluoro-2-methyl-4-(methylsulfonyl)benzoic acid (17) and tert-butyl 7-bromo-2,3-dihydrobenzo[f][1,4]oxazepine-4(5H)-carboxylate (2). The processes for the two compounds were scaled up, and over 15 kg of each starting material was prepared in overall yields of 42% and 58%, respectively.
A telescoped sequence beginning with compound 2 afforded 7.5 kg of the elaborated intermediate 5-(2,3,4,5-tetrahydrobenzo[f][1,4]oxazepin-2-amine dihydrochloride (6) in 63% yield. Subsequent coupling with benzoic acid 17 gave 7.6 kg of the target compound 1 in 84% yield. The preferred hydrochloride salt was eventually prepared. The overall yield for the synthesis of inhibitor 1 was 21% over eight isolated synthetic steps, and the final salt was obtained with 99.7% HPLC purity.
[7-(6-Aminopyridin-3-yl)-2,3-dihydro-1,4-benzoxazepin-4(5H)-yl][3-fluoro-2-methyl-4-(methylsulfonyl)phenyl]methanone (1)
Compound 1 was observed as a mixture of two rotational isomers in the 1H and 13C NMR spectra.
1H NMR (400 MHz, DMSO-d6): δ 8.24–8.03 (dd, 1H), 7.79–7.71 (m, 1H), 7.71–7.69 (dd, 0.5H), 7.57–7.57 (d, 0.5H), 7.44–7.40 (m, 1.5H), 7.29–7.19 (dd, 1H), 7.05–7.01 (dd, 1H), 6.64–6.63 (d, 0.5H), 6.54–6.45 (dd, 1H), 6.06 (s, 2H), 4.93–4.31 (m, 2H), 4.31–3.54 (m, 4H), 3.37–3.36 (d, 3H), 2.12–1.77 (d, 3H). 13C NMR (100 MHz, DMSO-d6): δ 167.3, 167.2, 166.6, 166.6, 158.9, 158.9, 158.4, 158.4, 157.4, 157.2, 155.9. 155.8, 145.4, 145.1, 145.1, 144.0, 143.9, 135.0, 134.7, 132.9, 132.8, 129.4, 129.2, 128.2, 128.2, 128.1, 128.0, 127.0, 126.9, 126.8, 125.9, 125.6, 125.4, 123.6, 123.5, 123.3, 123.1, 122.8, 122.0, 122.0, 121.9, 121.9, 121.2, 120.7, 107.8, 107.8, 70.9, 70.8, 51.1, 51.1, 47.4, 46.5, 43.5, 43.5, 43.5, 43.4, 11.0, 10.9, 10.7, 10.6. IR (KBr pellet): 1623, 1487, 1457, 1423, 1385, 1314, 1269, 1226, 1193, 1144, 1133, 1054, 1031, 962, 821, 768 cm–1. MS (EI) C23H22FN3O4S: found 456.2 ([M + H]+). High-resolution MS (FAB-MS using glycerol as a matrix) for C23H22FN3O4S: found 456.13943 ([M + H]+), calcd 456.13878.
[7-(6-Aminopyridin-3-yl)-2,3-dihydro-1,4-benzoxazepin-4(5H)-yl][3-fluoro-2-methyl-4-(methylsulfonyl)phenyl]methanone Hydrochloride (1·HCl)
1·HCl as a white solid (7.81 kg, 95%, 99.7% purity by AN-HPLC).
Analyses: OVI: DMF < 100 ppm, DMC < 100 ppm, acetone = 3081 ppm, MTBE < 100 ppm, iPAc < 100 ppm, THF < 100 ppm. Heavy metals: Pd ≤ 0.2 ppm, others < 20 ppm (USP ⟨231⟩). 1H NMR (400 MHz, DMSO-d6), equimolar amounts of two rotamers: δ 8.20–8.40 (br s, 2H), 8.33 (s, 0.5H), 8.31 (d, J = 2.8 Hz, 0.5H), 8.15 (d, J = 2.0 Hz, 0.5H), 7.96 (dd, J = 9.7, 2.0 Hz, 0.5H), 7.70–7.78 (m, 1.5H), 7.55–7.57 (m, 0.5H), 7.51–7.55 (m, 0.5H), 7.28 (d, J = 8.6 Hz, 0.5H), 7.17 (d, J = 3.1 Hz, 0.5H), 7.15 (d, J = 5.1 Hz, 0.5H), 7.05–7.11 (m, 1.5H), 6.83 (d, J = 2.7 Hz, 0.5H), 4.86–4.99 (m, 1H), 4.29–4.56 (m, 1H), 4.10–4.27 (m, 2H), 3.93–4.04 (m, 0.5H), 3.45–3.65 (m, 1.5H), 3.37 (s, 1.5 H), 3.35 (s, 1.5H), 2.12 (d, J = 2.0 Hz, 1.5H), 1.76 (d, J = 2.0 Hz, 1.5H). 13C NMR (100 MHz, DMSO-d6), equimolar amounts of two rotamers: δ 168.1, 167.5, 159.4, 159.2, 159.1, 156.6, 153.9, 153.8, 144.6, 142.9, 142.3, 133.0, 132.7, 130.0, 129.9, 129.7, 129.5, 129.1, 129.0, 128.9, 128.8, 128.5, 127.7, 127.6, 127.5, 127.1, 126.9, 124.4, 124.3, 124.1, 122.7, 122.1, 121.6, 114.4, 71.2, 51.7, 51.3, 47.9, 46.9, 44.3, 44.2, 11.7, 11.4.
REFERENCES
Anand, N.; Benzoxazepines as Inhibitors of PI3K/mTOR and Methods of their Use and Manufacture. U.S. Patent 8,648,066, Feb 11, 2014.
Aay, N.; Benzoxazepines as Inhibitors of PI3K/mTOR and Methods of their Use and Manufacture. U.S. Patent 8,637,499, Jan 28,2014.
| US8637499 * | May 25, 2010 | Jan 28, 2014 | Exelixis, Inc. | Benzoxazepines as inhibitors of PI3K/mTOR and methods of their use and manufacture |
| US20120258953 * | May 25, 2010 | Oct 11, 2012 | Exelixis, Inc. | Benzoxazepines as Inhibitors of PI3K/mTOR and Methods of Their Use and Manufacture |
PROFILE

Senior Director
Chemical Development at Dermira, Inc.
Lives San jose caifornia
E-mail: sriram.naganathan@dermira.com.

LINKS
https://www.linkedin.com/pub/sriram-naganathan/3/50a/5b6
https://www.facebook.com/sriram.naganathan.5
snaganat@exelixis.com, sriramrevathi@yahoo.com
Summary
Chemical process-development and CMC professional offering 20 years of experience from preclinical development through commercialization of small molecules and peptides.
Hands-on experience in multi-step synthesis, route-scouting, process development, scale-up, tech transfer to CRO/CMO, including manufacture under cGMP and process validation.
Extensive knowledge of CMC regulatory landscape (FDA, EMEA) including preparation of CMC sections of IND, IMPD, NDA and MAA
Experience
Senior Director, Chemical Development
Dermira, Inc.
January 2015 – Present (10 months)Menlo Park, CA

Consultant
Intarcia Therapeutics
December 2014 – January 2015 (2 months)

Senior Director
Exelixis
March 2013 – November 2014 (1 year 9 months)South San Francisco, CA
210 E. Grand Ave
South San Francisco , California 94080
United States
United States
Company Description: Exelixis, Inc. (Exelixis) is developing therapies for cancer and other serious diseases. Through its drug discovery and development activities, the Company is… more
Senior Scientist II
Exelixis
August 2004 – January 2008 (3 years 6 months)
Associate Director
CellGate, Inc.
2000 – 2004 (4 years)
Research Scientist
Roche Bioscience
1997 – 2000 (3 years)
Research Scientist
Cultor
1995 – 1997 (2 years)

Research Scientist
Pfizer
1994 – 1997 (3 years)

Research Assistant Professor
University of Pittsburgh
April 1992 – October 1994 (2 years 7 months)
Worked on Vitamin K mechanism in the labs of (Late) Prof Paul Dowd
Education


Vivekananda College (University of Madras), India
Bachelor of Science (B.Sc.), Chemistry
1980 – 1983
![]()

(Above) Former Group members join Professor Block at the National ACS Meeting in San Francisco, March 2010: from left, Dr. Shuhai Zhao, Dr. Sherida Johnson, Professor Block, Dr. Sriram Naganathan.
Sriram Naganathan, Ph.D. 1992, snaganat@exelixis.com, sriramrevathi@yahoo.com

As many things change, many things remain constant. One such constant is the frequent reminder that “You can take the boy out of sulfur chemistry but you cannot take sulfur chemistry out of the boy”. At every stage of my professional career organic chemistry of sulfur and sulfur-containing compounds have followed me (or is it the other way around?). Not many can point to the cover of an Angewandte Chemie issue as a synopsis of his/her thesis work – I will be forever grateful for that opportunity received in the Block Group.
As a post-doc in the late Prof. Paul Dowd’s lab at the University of Pittsburgh we used sulfur-containing analogs of vitamin K to probe the mechanism of action. I was then hired at Pfizer Central Research in Groton, CT in the Specialty Chemicals Division to investigate possible decomposition pathways of sulfur-containing high-intensity artificial sweeteners.
At Roche Bioscience (Palo Alto, CA) and Exelixis (South San Francisco, CA – my current job………CHANGED……Dermira) I was involved in process development for the preparation of therapeutic agents, several of them sulfur-containing molecules. Between those two positions I was a Senior Scientist at CellGate (Sunnyvale, CA).
We attempted to exploit the chemistry of sulfur-containing linkers to target the delivery active pharmaceutical agents, using the transport properties of polyarginines. Although I thought I was only training to become a synthetic organic chemist, I did not realize that my passion was really organic reaction mechanisms until I arrived in the Block lab – the two arms of the science are truly inseparable.
I realize after many years that the seed was really sown and nurtured during the many friendly and sometimes-fiery discussions in the lab, and further solidified in my post-doc years. I learned that every “blip-in-the-baseline” cannot to be ignored, and is part of the whole story.
As a process chemist in the pharma industry, I can attribute much of my success to lessons about careful and critical evaluation of primary data and thorough knowledge of reaction mechanisms. I am currently Director, Chemical Development, at Exelixis.NOW DERMIRA.
My primary responsibility involves the manufacture and potential commercialization of our primary product, cabozantinib. It was only natural that I developed a strong interest in the science of cooking and food. I have been pursuing this avenue since moving to Northern California.
I am also an avid gardener, experimenting with growing interesting varieties of chilies, tomatoes and then combining those with all sorts of alliums. It does help that I live close enough to Gilroy, CA, that I can often smell what they are famous for as I walk out of the front door!! I have shared my knowledge in several lectures at the Tech Museum (San Jose, CA) where I was a volunteer exhibit explainer.
My family (my wife Revathi and our two high-school-age daughters Swetha and Sandhya) like to travel and also enjoy the outdoor recreation so abundant in Northern California. We try to take in a new country each year and accomplish personal challenges. After many interesting years in the tech-industry, Revathi is a full-time mom. She is also a fitness instructor at the Y. Swetha and Sandhya are part of the water polo and swim teams at their school.
Swetha is very active in a leadership role for the robotics team, and Sandhya belongs to the quiz team. Revathi and I climbed Half Dome (Yosemite) a few years ago and I just completed a 100-mile bicycle ride around Lake Tahoe.
I remain a highly-opinionated baseball and college basketball fan (favorite teams: in order, Kansas, North Carolina and whoever happens to be playing Missouri and Duke). I am still an avid photographer, although I spend no money on film (I thought I was going to be the last guy on the planet still shooting film!!). I greatly value the many friendships developed during my stay in Albany and keep in touch with many.
In fact, one of my roommates from the SUNY days was instrumental in me getting my present position. Of course, this also means that I have lost touch with several friends during the past decades. If you are reading this and haven’t contacted me in a few years, please do, via e-mail.
We enjoy entertaining guests who drop by – so now you have no excuse not to contact us, especially when you visit the SF Bay Area.

OLD PROFLE……Dr Sriram Naganathan received his Ph.D. from SUNY-Albany where he studied organosulfur chemistry. He is currently an Associate Director at CellGate, Inc. located in Sunnyvale, California. CellGate is involved in the commercialization of novel medicines by utilizing proprietary transporter technology, based on oligomers of arginine, to enhance the therapeutic potential of existing drugs. His responsibilities include process development, scale-up and GMP production of clinical candidates, as well some basic research. He previously held positions at Pfizer Central Research and Roche Bioscience.
Dermira


Thomas G. Wiggans | Founder & Chief Executive Officer……..http://dermira.com/about-us/management-team/

CEO TOM WIGGANS, LEFT AND CMO GENE GAUER, RIGHT


Exelixis, Inc.
![]()

210 East Grand Avenue
So. San Francisco, CA 94080
(650) 837-7000 phone
(650) 837-8300 fax
Directions to Exelixis, Inc.
101 Northbound from San Francisco Airport:
- Take 101 North toward San Francisco.
- Take the Grand Avenue exit, exit 425A, toward So San Francisco.
- Turn right onto East Grand Ave.
- 210 East Grand Ave is on your right-hand side.
101 Southbound from San Francisco:
- Take 101 South.
- Take the Grand Avenue exit. Turn left at the first light.
- Immediately turn left at the first light onto Grand Avenue (which will become East Grand Avenue)
- 210 East Grand Ave is on your right-hand side.

////////////mTOR inhibitor, Exelixis, Inc., PI3K, phosphatidylinositol-3-kinase, XL 388, XL388, IND Filed
