
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

XL 388
A Novel Class of Highly Potent, Selective, ATP-Competitive, and Orally Bioavailable Inhibitors of the Mammalian Target of Rapamycin (mTOR)
Benzoxazepine-Containing Kinase Inhibitor

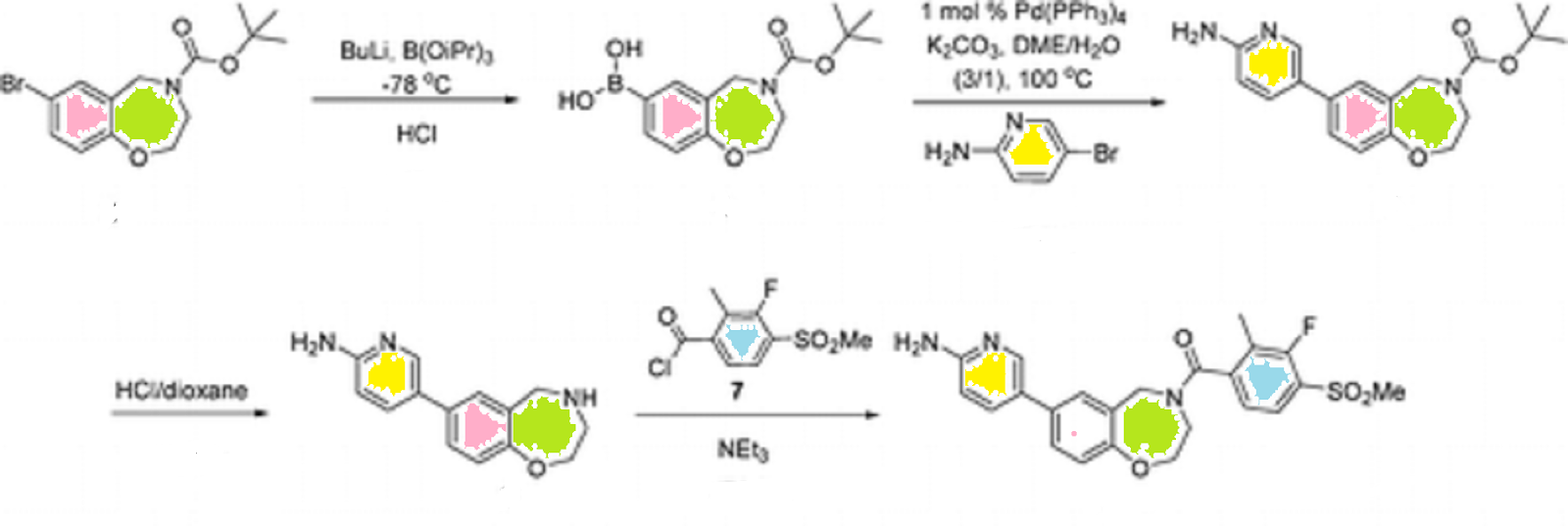


PATENT
Example 2:
[7-(6-Aminopyridin-3-yl)-2,3-dihydro-l,4-benzoxazepin-4(5H)-yl] [3-fluoro- 2-methyl-4-(methylsulfonyl)phenyl]methanone

tørt-Butyl 7-(6-aminopyridin-3-yl)-2,3-dihydrobenzo[/] [l,4]oxazepine-4(5H)- carboxylate. To a mixture of 4-(te/t-butoxycarbonyl)-2,3,4,5- tetrahydrobenzo[/][l,4]oxazepin-7-ylboronic acid (1.52 g, 5.2 mmol), prepared as described in Reference Example 5, 2-amino-5-bromopyridine (900 mg, 5.2 mmol), and potassium carbonate (1.73 g, 12.5 mmol) in 1 ,2-dimethoxyethane/water (30 mL/10 mL) was added tetrakis(triphenylphosphine)palladium(0) (90 mg, 1.5 mol%) and the reaction mixture was purged with nitrogen and stirred at reflux for 3 h. The reaction was cooled to rt, diluted with water/ethyl acetate (50 mL/50 mL), and the separated aqueous layer was extracted with ethyl acetate. The resulting emulsion was removed by filtration. The combined organic layer was washed with brine, dried with sodium sulfate, filtered and concentrated under reduced pressure, and the residue was triturated with toluene for 1 h. The resulting off-white solid was isolated by filtration to give the desired product (1.37 g, 77 %) as an off-white solid. MS (EI) for Ci9H23N3O3: 342 (MH+).
5-(2,3,4,5-Tetrahydrobenzo[/] [l,4]oxazepin-7-yl)pyridine-2-amine. To a stirred solution of tert-butyl 7-(6-aminopyridin-3-yl)-2,3-dihydrobenzo[/][l,4]oxazepine- 4(5H)-carboxylate (1.36 g, 3.98 mmol) in 1,4-dioxane (5 mL) was added 4 N hydrogen chloride in 1 ,4-dioxane (5 mL) and the reaction mixture was stirred at rt overnight. The reaction was concentrated on a rotary evaporator and the residue was triturated with ether. The solid was isolated by filtration. This solid was dissolved in water (5 mL) and made basic with 5 N sodium hydroxide to pH 11-12. The brownish sticky oil that aggregated at the bottom was isolated and the aqueous layer was extracted with 5 % methanol in ethyl acetate. The extracts were dried with sodium sulfate and concentrated on a rotary evaporator. The brownish sticky oil was dissolved with a mixture of methanol/ethyl acetate, combined with the isolated organic residue and concentrated under reduced pressure to give a yellow solid. This solid was triturated with dichloromethane (10 mL) for 1 h and a yellow solid was isolated by filtration and dried under high vacuum to give amine the desired product (920 mg, 96 %). MS (EI) for Ci4Hi5N3O: 242 (MH+).
[7-(6-Aminopyridin-3-yl)-2,3-dihydro-l,4-benzoxazepin-4(5H)-yl][3-fluoro-2- methyl-4-(methylsulfonyl)phenyl]methanone.
To a stirred suspension of 5-(2, 3,4,5- tetrahydrobenzo[/][l,4]oxazepin-7-yl)pyridine-2-amine (85 mg, 352 μmol) and triethylamine (54 μL, 387 μmol) in dichloromethane (10 mL) was added 3-fluoro-2-methyl-4- (methylsulfonyl)benzoyl chloride (91 mg, in 3 mL of dichloromethane), prepared as described in Reference Example 1, at 0 0C for 2 h. After stirring for an additional 1 h at rt, the reaction mixture was diluted with water (5 mL) and the separated aqueous layer was extracted with dichloromethane. The combined extracts were dried with sodium sulfate, filtered and concentrated under reduced pressure to give a light-yellow solid that was purified via silica gel chromatography to give the desired product (113 mg, 70%) as a white solid.
1H NMR (400 MHz, DMSO-d6): δ 8.24-8.03 (dd, IH), 7.79-7.71 (m, IH), 7.71-7.69 (dd, 0.5H), 7.57-7.57 (d, 0.5H), 7.44-7.40 (m, 1.5H), 7.29-7.19 (dd, IH), 7.05-7.01 (dd, IH), 6.64-6.63 (d, 0.5H), 6.54-6.45 (dd, IH), 6.06 (s, 2H), 4.93-4.31 (m, 2H), 4.31-3.54 (m, 4H), 3.37-3.36(d, 3H), 2.12-1.77 (d, 3H).
MS (EI) C23H22FN3O4S: 456 (MH+).
PAPER

A series of novel, highly potent, selective, and ATP-competitive mammalian target of rapamycin (mTOR) inhibitors based on a benzoxazepine scaffold have been identified. Lead optimization resulted in the discovery of inhibitors with low nanomolar activity and greater than 1000-fold selectivity over the closely related PI3K kinases. Compound 28 (XL388) inhibited cellular phosphorylation of mTOR complex 1 (p-p70S6K, pS6, and p-4E-BP1) and mTOR complex 2 (pAKT (S473)) substrates. Furthermore, this compound displayed good pharmacokinetics and oral exposure in multiple species with moderate bioavailability. Oral administration of compound 28 to athymic nude mice implanted with human tumor xenografts afforded significant and dose-dependent antitumor activity.
(7-(6-Aminopyridin-3-yl)-2,3-dihydrobenz[f][1,4]oxazepin-4(5H)-yl)(3-fluoro-2-methyl-4-(methylsulfonyl)phenyl)methanone (28)
PATENT
- SYNTHETIC EXAMPLES
-

-
1-Bromo-3,4-difluoro-2-methylbenzene. To a stirred mixture of 2,3-difluorotoluene (1.9 g, 14.8 mmol) and iron (82.7 mg, 1.48 mmol) in chloroform (10 mL) at rt was added bromine (76 μL, 14.8 mmol) over 2 h. The resulting mixture was stirred at rt overnight. Excess water (10 mL) was added and the reaction mixture was diluted with ether (20 mL). The separated organic layer was washed with aqueous sodium thiosulfate, brine, dried over sodium sulfate and concentrated on a rotary evaporator. The residue was distilled to give the desired product (2.49 g, 81%) as a colorless oil.
-
3,4-Difluoro-2-methylbenzoic acid. To a stirred solution of 1-bromo-3,4-difluoro-2-methylbenzene (940 mg, 4.54 mmol) in tetrahydrofuran (5 mL) was added isopropylmagnesium bromide (3.0 mL, 6.0 mmol) over 1 h at 0° C. The resulting mixture was stirred at rt for 24 h. Carbon dioxide (CO2), generated from dry ice, was introduced to the reaction mixture over 2 h and the resulting mixture was stirred for an additional 30 min. The reaction mixture was quenched with addition of an excess amount of water (5 mL) and the tetrahydrofuran was removed on a rotary evaporator. The resulting aqueous layer was diluted with water (5 mL) and acidified with concentrated hydrochloric acid to pH 1-2. The white precipitate was filtered and washed with water and cold hexanes and dried under high vacuum to give the desired product (630 mg, 81%) as a white powder. MS (EI) for C8H6F2O2: 171 (MH−).
-
3-Fluoro-2-methyl-4-(thiomethyl)benzoic acid. To a stirred solution of acid 3,4-difluoro-2-methylbenzoic acid (700 mg, 4.1 mmol) in dimethylsulfoxide (5 mL) was added powdered potassium hydroxide (274 mg, 4.9 mmol) and the mixture was stirred at rt for 30 min. Sodium thiomethoxide (342 mg, 4.9 mmol) was added to the mixture and the resulting mixture was stirred at 55-60° C. for 4 h. Additional powdered potassium hydroxide (70 mg, 1.2 mmol), sodium thiomethoxide (60 mg, 0.8 mmol), and dimethylsulfoxide (2 mL) were added to the reaction mixture. After stirring for 4 h, the mixture was cooled to 0° C. and quenched with excess water (10 mL). The resulting suspension was acidified at 0° C. with concentrated hydrochloric acid to pH 1-2. The white precipitate was collected by suction filtration, washed with water and dried under vacuum overnight to give the desired product (870 mg, 100%). The intermediate sulfide was used in the next step without further purification. MS (EI) for C9H9FO2S: 199.1 (MH−).
-
3-Fluoro-2-methyl-4-(methylsulfonyl)benzoic acid. To a stirred suspension of 3-fluoro-2-methyl-4-(thiomethyl)benzoic acid in an acetone/water (1 mL/10 mL) mixture was added sodium hydroxide (330 mg, 8.25 mmol) and sodium bicarbonate (680 mg, 8.1 mmol). Oxone (˜4 g) was added portionwise to the reaction mixture at 0° C. over 2 h. The reaction was monitored by LC/MS. Concentrated hydrochloric acid was added to adjust the pH 2-3 and the white precipitate was collected by suction filtration, washed with water, and dried under vacuum. Dried precipitate was suspended in water (10 mL), stirred vigorously at rt for 1 h, filtered, washed with water, and hexanes and dried under vacuum to give the desired product (886 mg, 94%) as a white powder. MS (EI) for C9H9FO4S: 231 (MH−).
-
3-Fluoro-2-methyl-4-(methylsulfonyl)benzoyl chloride. A mixture of 3-fluoro-2-methyl-4-(methylsulfonyl)benzoic acid (860 mg, 3.7 mmol) in thionyl chloride (10 mL) was heated to reflux for 3 h. (the reaction mixture became homogenous). The reaction mixture was concentrated on a rotary evaporator to give the crude acid chloride. This acid chloride was triturated with dichloromethane (2 mL) and concentrated under reduced pressure. The trituration process was repeated 3 times until the product (900 mg, 98%) was obtained as a white powder.
- Reference Example 13-Fluoro-2-methyl-4-(methylsulfonyl)benzoyl chloride

Reference Example 2Ethyl 4-(2,3,4,5-tetrahydro-1,4-benzoxazepin-7-yl)benzoate hydrochloride salt
-

-
4-(ethoxycarbonyl)phenylboronic acid (22.16 g, 114 mmol), tert-butyl 7-bromo-2,3-dihydrobenzo[f][1,4]oxazepin-4(5H)-carboxylate (34.08 g, 104 mmol), prepared as described in Reference Example 4, Pd(dppf)Cl2 and TEA (21 g, 208 mmol) were combined in a mixture of dioxane (200 mL) and water (20 mL). The reaction mixture was heated to 90° C. for 2 h, then cooled and the solvent removed. Purification of the residue by silica chromatography gave the desired product ester (31.3 g, 69% yield).
-
To the solution of tert-butyl 7-(4-(ethoxycarbonyl)phenyl)-2,3-dihydrobenzo[f][1,4]oxazepine-4(5H)-carboxylate (10.3 g, 25.93 mmol) in MeOH (120 mL) was added a solution of 4 N HCl in dioxane (50 mL). The reaction mixture was heated to 50° C. for 3 h (monitored by LC/MS). The reaction mixture was allowed to cool to rt. Ethyl 4-(2,3,4,5-tetrahydro-1,4-benzoxazepin-7-yl)benzoate as the hydrochloride salt (8.8 g, 99% yield) was collected by suction filtration.

-

-
tert-Butyl-5-bromo-2-hydroxybenzyl(2-hydroxyethyl)carbamate. Commercially-available 5-bromo-2-hydroxybenzaldehyde (4.0 g, 10 mmol) and 2-aminoethanol were combined in THF/MeOH (100 mL, 10:1) and sodium borohydride (0.76 g, 2.0 mmol) was added with stirring. The resulting reaction mixture was stirred at 40° C. for 4 h, concentrated on a rotary evaporator then diluted with EtOAc (50 mL) and saturated NaHCO3 (30 mL). To this suspension was added di-tert-butyl dicarbonate (2.83 g, 13 mmol). The mixture was stirred at rt overnight. The organic layer was washed with water, dried over anhydrous magnesium sulfate, filtered, and concentrated on a rotary evaporator. Hexane was subsequently added to the crude reaction product which resulted in the formation of a white solid. This slurry was filtered to obtain the desired product (6.8 g, 98%) as a white solid. MS (EI) for C14H20BrNO4, found 346 (MH+).
-
tert-Butyl-7-bromo-2,3-dihydrobenzo[f][1,4]oxazepine-4(5H)-carboxylate. tert-Butyl-5-bromo-2-hydroxybenzyl(2-hydroxyethyl)carbamate (3.46 g, 10 mmol) and triphenylphosphine (3.96 g, 15 mmol) were combined in DCM (100 mL) and diisopropyl azodicarboxylate (3.03 g, 15 mmol) was added. The resulting reaction mixture was stirred at rt for 12 h. The reaction mixture was washed with water, dried, filtered, and concentrated on a rotary evaporator. The resulting crude product was purified via silica gel chromatography eluting with 8:2 hexane/ethyl acetate to give the desired product (1.74 g, 53%) as a white solid. MS (EI) for C14H18BrNO3, found 328 (MH+).
- Reference Example 4tert-Butyl-7-bromo-2,3-dihydrobenzo[f][1,4]oxazepine-4(5H)-carboxylate

Reference Example 54-(tert-Butoxycarbonyl)-2,3,4,5-tetrahydrobenzo[f][1,4]oxazepin-7-ylboronic acid
-

-
To a stirred solution of tert-butyl-7-bromo-2,3-dihydrobenzo[f][1,4]oxazepine-4(5H)-carboxylate (10 g, 30.5 mmol), prepared as described in Reference Example 4, and triisopropylborate (9.1 mL, 40 mmol) in dry tetrahydrofuran (100 mL) was added dropwise n-butyllithium in tetrahydrofuran (1.6 M, 25 mL, 40 mmol) while maintaining the temperature below −60° C. Upon completion of addition, the reaction mixture was stirred for 30 min, then quenched with 1 N aqueous hydrochloric acid (35 mL) and allowed to warm to rt. The reaction mixture was extracted with ethyl acetate, dried over anhydrous magnesium sulfate, filtered and concentrated on a rotary evaporator. Hexane was subsequently added to the crude reaction product which resulted in the formation of a white solid. This slurry was stirred for 1 h and filtered to obtain 4-(tert-butoxycarbonyl)-2,3,4,5-tetrahydrobenzo[f][1,4]oxazepin-7-ylboronic acid (8.6 g, 95%) as a white solid. MS (EI) for C14H20BNO5: 194 (M-Boc).

- Example 2[7-(6-Aminopyridin-3-yl)-2,3-dihydro-1,4-benzoxazepin-4(5H)-yl][3-fluoro-2-methyl-4-(methylsulfonyl)phenyl]methanone
-

-
tert-Butyl 7-(6-aminopyridin-3-yl)-2,3-dihydrobenzo[f][1,4]oxazepine-4(5H)-carboxylate. To a mixture of 4-(tert-butoxycarbonyl)-2,3,4,5-tetrahydrobenzo[f][1,4]oxazepin-7-ylboronic acid (1.52 g, 5.2 mmol), prepared as described in Reference Example 5, 2-amino-5-bromopyridine (900 mg, 5.2 mmol), and potassium carbonate (1.73 g, 12.5 mmol) in 1,2-dimethoxyethane/water (30 mL/10 mL) was added tetrakis(triphenylphosphine)palladium(0) (90 mg, 1.5 mol %) and the reaction mixture was purged with nitrogen and stirred at reflux for 3 h. The reaction was cooled to rt, diluted with water/ethyl acetate (50 mL/50 mL), and the separated aqueous layer was extracted with ethyl acetate. The resulting emulsion was removed by filtration. The combined organic layer was washed with brine, dried with sodium sulfate, filtered and concentrated under reduced pressure, and the residue was triturated with toluene for 1 h. The resulting off-white solid was isolated by filtration to give the desired product (1.37 g, 77%) as an off-white solid. MS (EI) for C19H23N3O3: 342 (MH+).
-
5-(2,3,4,5-Tetrahydrobenzo[f][1,4]oxazepin-7-yl)pyridine-2-amine. To a stirred solution of tert-butyl 7-(6-aminopyridin-3-yl)-2,3-dihydrobenzo[f][1,4]oxazepine-4(5H)-carboxylate (1.36 g, 3.98 mmol) in 1,4-dioxane (5 mL) was added 4 N hydrogen chloride in 1,4-dioxane (5 mL) and the reaction mixture was stirred at rt overnight. The reaction was concentrated on a rotary evaporator and the residue was triturated with ether. The solid was isolated by filtration. This solid was dissolved in water (5 mL) and made basic with 5 N sodium hydroxide to pH 11-12. The brownish sticky oil that aggregated at the bottom was isolated and the aqueous layer was extracted with 5% methanol in ethyl acetate. The extracts were dried with sodium sulfate and concentrated on a rotary evaporator. The brownish sticky oil was dissolved with a mixture of methanol/ethyl acetate, combined with the isolated organic residue and concentrated under reduced pressure to give a yellow solid. This solid was triturated with dichloromethane (10 mL) for 1 h and a yellow solid was isolated by filtration and dried under high vacuum to give amine the desired product (920 mg, 96%). MS (EI) for C14H15N3O: 242 (MH+).
-
[7-(6-Aminopyridin-3-yl)-2,3-dihydro-1,4-benzoxazepin-4(5H)-yl][3-fluoro-2-methyl-4-(methylsulfonyl)phenyl]methanone. To a stirred suspension of 5-(2,3,4,5-tetrahydrobenzo[f][1,4]oxazepin-7-yl)pyridine-2-amine (85 mg, 352 μmol) and triethylamine (54 μL, 387 μmol) in dichloromethane (10 mL) was added 3-fluoro-2-methyl-4-(methylsulfonyl)benzoyl chloride (91 mg, in 3 mL of dichloromethane), prepared as described in Reference Example 1, at 0° C. for 2 h. After stirring for an additional 1 h at rt, the reaction mixture was diluted with water (5 mL) and the separated aqueous layer was extracted with dichloromethane. The combined extracts were dried with sodium sulfate, filtered and concentrated under reduced pressure to give a light-yellow solid that was purified via silica gel chromatography to give the desired product (113 mg, 70%) as a white solid. 1H NMR (400 MHz, DMSO-d6): δ 8.24-8.03 (dd, 1H), 7.79-7.71 (m, 1H), 7.71-7.69 (dd, 0.5H), 7.57-7.57 (d, 0.5H), 7.44-7.40 (m, 1.5H), 7.29-7.19 (dd, 1H), 7.05-7.01 (dd, 1H), 6.64-6.63 (d, 0.5H), 6.54-6.45 (dd, 1H), 6.06 (s, 2H), 4.93-4.31 (m, 2H), 4.31-3.54 (m, 4H), 3.37-3.36 (d, 3H), 2.12-1.77 (d, 3H). MS (EI) C23H22FN3O4S: 456 (MH+).

PAPER
http://pubs.acs.org/doi/abs/10.1021/acs.oprd.5b00037

The benzoxazepine core is present in several kinase inhibitors, including the mTOR inhibitor 1. The process development for a scalable synthesis of 7-bromobenzoxazepine and the telescoped synthesis of 1 are reported. Compound 1 consists of three chemically rich, distinct fragments: the tetrahydrobenzo[f][1,4]oxazepine core, the aminopyridyl fragment, and the substituted (methylsulfonyl)benzoyl fragment. Routes were developed for the preparation of 3-fluoro-2-methyl-4-(methylsulfonyl)benzoic acid (17) and tert-butyl 7-bromo-2,3-dihydrobenzo[f][1,4]oxazepine-4(5H)-carboxylate (2). The processes for the two compounds were scaled up, and over 15 kg of each starting material was prepared in overall yields of 42% and 58%, respectively.
A telescoped sequence beginning with compound 2 afforded 7.5 kg of the elaborated intermediate 5-(2,3,4,5-tetrahydrobenzo[f][1,4]oxazepin-2-amine dihydrochloride (6) in 63% yield. Subsequent coupling with benzoic acid 17 gave 7.6 kg of the target compound 1 in 84% yield. The preferred hydrochloride salt was eventually prepared. The overall yield for the synthesis of inhibitor 1 was 21% over eight isolated synthetic steps, and the final salt was obtained with 99.7% HPLC purity.
[7-(6-Aminopyridin-3-yl)-2,3-dihydro-1,4-benzoxazepin-4(5H)-yl][3-fluoro-2-methyl-4-(methylsulfonyl)phenyl]methanone (1)
[7-(6-Aminopyridin-3-yl)-2,3-dihydro-1,4-benzoxazepin-4(5H)-yl][3-fluoro-2-methyl-4-(methylsulfonyl)phenyl]methanone Hydrochloride (1·HCl)
REFERENCES
Anand, N.; Benzoxazepines as Inhibitors of PI3K/mTOR and Methods of their Use and Manufacture. U.S. Patent 8,648,066, Feb 11, 2014.
| US8637499 * | May 25, 2010 | Jan 28, 2014 | Exelixis, Inc. | Benzoxazepines as inhibitors of PI3K/mTOR and methods of their use and manufacture |
| US20120258953 * | May 25, 2010 | Oct 11, 2012 | Exelixis, Inc. | Benzoxazepines as Inhibitors of PI3K/mTOR and Methods of Their Use and Manufacture |
PROFILE

Senior Director
Chemical Development at Dermira, Inc.
Lives San jose caifornia

https://www.linkedin.com/pub/sriram-naganathan/3/50a/5b6
https://www.facebook.com/sriram.naganathan.5
snaganat@exelixis.com, sriramrevathi@yahoo.com
Summary
Chemical process-development and CMC professional offering 20 years of experience from preclinical development through commercialization of small molecules and peptides.
Hands-on experience in multi-step synthesis, route-scouting, process development, scale-up, tech transfer to CRO/CMO, including manufacture under cGMP and process validation.
Extensive knowledge of CMC regulatory landscape (FDA, EMEA) including preparation of CMC sections of IND, IMPD, NDA and MAA
Experience
Senior Director, Chemical Development
Dermira, Inc.
January 2015 – Present (10 months)Menlo Park, CA

Consultant
Intarcia Therapeutics
December 2014 – January 2015 (2 months)

Senior Director
Exelixis
March 2013 – November 2014 (1 year 9 months)South San Francisco, CA
210 E. Grand Ave
United States
Senior Scientist II
Exelixis
August 2004 – January 2008 (3 years 6 months)
Associate Director
CellGate, Inc.
2000 – 2004 (4 years)
Research Scientist
Roche Bioscience
1997 – 2000 (3 years)
Research Scientist
Cultor
1995 – 1997 (2 years)

Research Scientist
Pfizer
1994 – 1997 (3 years)

Research Assistant Professor
University of Pittsburgh
April 1992 – October 1994 (2 years 7 months)
Worked on Vitamin K mechanism in the labs of (Late) Prof Paul Dowd
Education


Vivekananda College (University of Madras), India
Bachelor of Science (B.Sc.), Chemistry
1980 – 1983
![]()

Sriram Naganathan, Ph.D. 1992, snaganat@exelixis.com, sriramrevathi@yahoo.com

As many things change, many things remain constant. One such constant is the frequent reminder that “You can take the boy out of sulfur chemistry but you cannot take sulfur chemistry out of the boy”. At every stage of my professional career organic chemistry of sulfur and sulfur-containing compounds have followed me (or is it the other way around?). Not many can point to the cover of an Angewandte Chemie issue as a synopsis of his/her thesis work – I will be forever grateful for that opportunity received in the Block Group.
As a post-doc in the late Prof. Paul Dowd’s lab at the University of Pittsburgh we used sulfur-containing analogs of vitamin K to probe the mechanism of action. I was then hired at Pfizer Central Research in Groton, CT in the Specialty Chemicals Division to investigate possible decomposition pathways of sulfur-containing high-intensity artificial sweeteners.
At Roche Bioscience (Palo Alto, CA) and Exelixis (South San Francisco, CA – my current job………CHANGED……Dermira) I was involved in process development for the preparation of therapeutic agents, several of them sulfur-containing molecules. Between those two positions I was a Senior Scientist at CellGate (Sunnyvale, CA).
We attempted to exploit the chemistry of sulfur-containing linkers to target the delivery active pharmaceutical agents, using the transport properties of polyarginines. Although I thought I was only training to become a synthetic organic chemist, I did not realize that my passion was really organic reaction mechanisms until I arrived in the Block lab – the two arms of the science are truly inseparable.
I realize after many years that the seed was really sown and nurtured during the many friendly and sometimes-fiery discussions in the lab, and further solidified in my post-doc years. I learned that every “blip-in-the-baseline” cannot to be ignored, and is part of the whole story.
As a process chemist in the pharma industry, I can attribute much of my success to lessons about careful and critical evaluation of primary data and thorough knowledge of reaction mechanisms. I am currently Director, Chemical Development, at Exelixis.NOW DERMIRA.
My primary responsibility involves the manufacture and potential commercialization of our primary product, cabozantinib. It was only natural that I developed a strong interest in the science of cooking and food. I have been pursuing this avenue since moving to Northern California.
I am also an avid gardener, experimenting with growing interesting varieties of chilies, tomatoes and then combining those with all sorts of alliums. It does help that I live close enough to Gilroy, CA, that I can often smell what they are famous for as I walk out of the front door!! I have shared my knowledge in several lectures at the Tech Museum (San Jose, CA) where I was a volunteer exhibit explainer.
My family (my wife Revathi and our two high-school-age daughters Swetha and Sandhya) like to travel and also enjoy the outdoor recreation so abundant in Northern California. We try to take in a new country each year and accomplish personal challenges. After many interesting years in the tech-industry, Revathi is a full-time mom. She is also a fitness instructor at the Y. Swetha and Sandhya are part of the water polo and swim teams at their school.
Swetha is very active in a leadership role for the robotics team, and Sandhya belongs to the quiz team. Revathi and I climbed Half Dome (Yosemite) a few years ago and I just completed a 100-mile bicycle ride around Lake Tahoe.
I remain a highly-opinionated baseball and college basketball fan (favorite teams: in order, Kansas, North Carolina and whoever happens to be playing Missouri and Duke). I am still an avid photographer, although I spend no money on film (I thought I was going to be the last guy on the planet still shooting film!!). I greatly value the many friendships developed during my stay in Albany and keep in touch with many.
In fact, one of my roommates from the SUNY days was instrumental in me getting my present position. Of course, this also means that I have lost touch with several friends during the past decades. If you are reading this and haven’t contacted me in a few years, please do, via e-mail.
We enjoy entertaining guests who drop by – so now you have no excuse not to contact us, especially when you visit the SF Bay Area.

OLD PROFLE……Dr Sriram Naganathan received his Ph.D. from SUNY-Albany where he studied organosulfur chemistry. He is currently an Associate Director at CellGate, Inc. located in Sunnyvale, California. CellGate is involved in the commercialization of novel medicines by utilizing proprietary transporter technology, based on oligomers of arginine, to enhance the therapeutic potential of existing drugs. His responsibilities include process development, scale-up and GMP production of clinical candidates, as well some basic research. He previously held positions at Pfizer Central Research and Roche Bioscience.
Dermira


Thomas G. Wiggans | Founder & Chief Executive Officer……..http://dermira.com/about-us/management-team/

CEO TOM WIGGANS, LEFT AND CMO GENE GAUER, RIGHT


Exelixis, Inc.
![]()

210 East Grand Avenue
So. San Francisco, CA 94080
(650) 837-7000 phone
(650) 837-8300 fax
Directions to Exelixis, Inc.
101 Northbound from San Francisco Airport:
- Take 101 North toward San Francisco.
- Take the Grand Avenue exit, exit 425A, toward So San Francisco.
- Turn right onto East Grand Ave.
- 210 East Grand Ave is on your right-hand side.
101 Southbound from San Francisco:
- Take 101 South.
- Take the Grand Avenue exit. Turn left at the first light.
- Immediately turn left at the first light onto Grand Avenue (which will become East Grand Avenue)
- 210 East Grand Ave is on your right-hand side.

////////////mTOR inhibitor, Exelixis, Inc., PI3K, phosphatidylinositol-3-kinase, XL 388, XL388, IND Filed


important cancer drug
LikeLike
Reblogged this on Leaders in Pharmaceutical Business Intelligence and commented:
interesting article
LikeLike
Reblogged this on ORGANIC CHEMISTRY SELECT.
LikeLike