
Home » Articles posted by DR ANTHONY MELVIN CRASTO Ph.D
Author Archives: DR ANTHONY MELVIN CRASTO Ph.D
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Gedatolisib
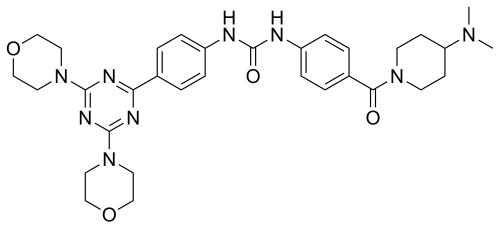
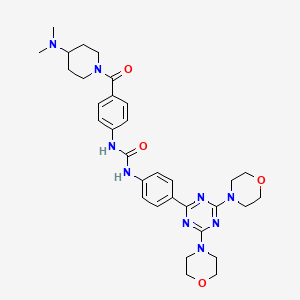
Gedatolisib
Approvals 3026, FDA 2026, 7/14/2026, Revtorpyk
| PF-05212384; PF-5212384; PKI-587 CAS 1197160-78-3 Chemical Formula: C32H41N9O4 Molecular Weight: 615.72 1-(4-{[4-(Dimethylamino)-1-piperidinyl]carbonyl}phenyl)-3-{4-[4,6-di(4-morpholinyl)-1,3,5-triazin-2-yl]phenyl}urea 3-{4-[bis(morpholin-4-yl)-1,3,5-triazin-2-yl]phenyl}-1-{4-[4-(dimethylamino)piperidine-1-carbonyl]phenyl}urea N-[4-[[4-(Dimethylamino)-1-piperidinyl]carbonyl]phenyl]-N’-[4-[4,6-di(4-morpholinyl)-1,3,5-triazin-2-yl]phenyl]urea гедатолисиб [Russian] [INN] غيداتوليسيب [Arabic] [INN] 吉达利塞 [Chinese] [INN] |
1-(4-(4-(Dimethylamino)piperidine-1-carbonyl)phenyl)-3-(4-(4,6-dimorpholino-1,3,5-triazin-2-yl)phenyl)urea
In combination with fulvestran, to treat hormone receptor-positive, human epidermal growth factor receptor 2-negative, locally advanced or metastatic breast cancer without a PIK3CA mutation detected following progression on or after treatment with at least one line of endocrine therapy in the metastatic setting
Gedatolisib, sold under the brand name Revtorpyk, is an anti-cancer drug used for the treatment of breast cancer.[1] It is under development by Celcuity, Inc. Gedatolisib is a kinase inhibitor.[1] The mechanism of action is accomplished by binding the different p110 catalytic subunit isoforms of PI3K and the kinase site of mTOR.[2] Gedatolisib is administered by intravenous infusion.[1]
Gedatolisib was approved for medical use in the United States in July 2026.[1][3]
Medical uses
Gedatolisib is indicated in combination with fulvestrant, with or without palbociclib, for the treatment of adults with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative locally advanced or metastatic breast cancer without a PIK3CA mutation detected following progression on or after treatment with at least one line of endocrine therapy in the metastatic setting.[1]
- Synthesis of New Dialkyl 2,2′-[Carbonylbis(azanediyl)]dibenzoates via Curtius RearrangementDOI: 10.1055/s-0040-1706643Publication Date: 2021Publication Name: Synthesis
- A New One-Pot Three-Component Synthesis of 4-Aryl-6-cycloamino-1,3,5-triazin-2-amines under Microwave IrradiationDOI: 10.1055/a-1401-2795Publication Date: 2021Publication Name: Synthesis
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US42900900&_cid=P22-MRVGV9-51327-1
Example 76
Preparation of 1-(4-(4-(dimethylamino)piperidine-1-carbonyl)phenyl)-3-(4-(4,6-dimorpholino-1,3,5-triazin-2-yl)phenyl)urea
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2010096619&_cid=P22-MRVGV9-51327-1
Scheme 1
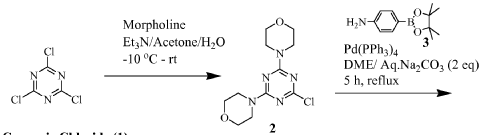
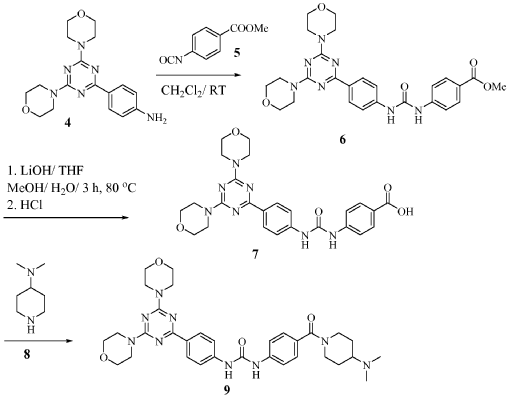
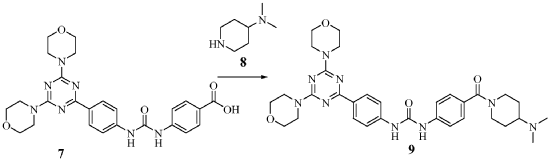
Preparation of 1-(4-(4-(dimethylamino) piperidine-1-carbonyl)phenyl-3-(4-(4,6- dimorpholino-1 ,3,5-triazine-2-yl)phenyl) urea (9)
To a slurry of 4-(3-(4-(4,6-dimorpholino-1 ,3,5-triazine-2- yl)phenyl)ureido)benzoic acid (7, 45.5 g, 0.09 mol) in dry THF (1.6 L) heated to 50 0C was added N,N’-carbonyl diimidazole (28 g, 0.17 mol). The reaction mixture was heated for 2 hours and followed by dimethylaminopiperidine (8, 23.5 g, 0.18 mol) and stirred at 53 0C for 16 hours. The reaction mixture was cooled to the room temperature and filtered. The cake was washed with 2-propanol and air-dried to give 97 % pure white powder in 88% yield (49.2 g, 0.08 mol). To the solids stirred in dimethyl acetamide (DMAC, 165 ml) at 70° C for 1 hour was added 2-propanol (640 ml) and the mixture was stirred at 65 0C for additional 1 hour. The solids were filtered, washed with 2-propanol and dried in a vacuum oven at 700C for 16 hour to give crystalline white powder (45 g) with >99% purity. The above-mentioned work up process and crystallization procedure gave a Pd residue of 20 ppm. Alternate procedures for the formation of 1-(4-(4-(dimethylamino) piperidine-1 – carbonyl)phenyl-3-(4-(4,6-dimorpholino-1 ,3,5-triazine-2-yl)phenyl) urea (9)
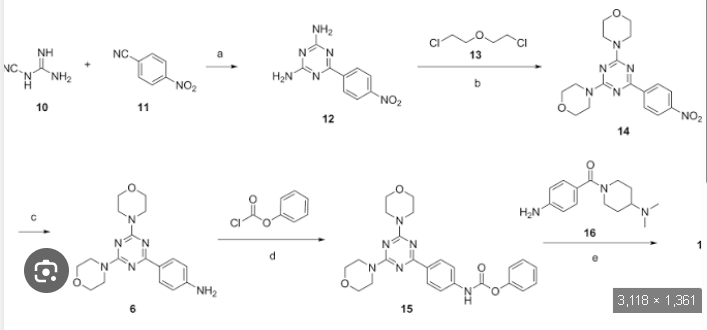
To the solution of 4-(4,6-dimorpholin-4-yl-1 ,3,5-triazin-2-yl) aniline (4, 18 g, 0.052 mol) in dichloromethane (300 ml) was added methyl 4-isocyanato benzoate (5, 10.5 g, 0.061 mol) and the reaction mixture was stirred for 5 hours. The separated solids were filtered, washed with ether and air dried to give beige solids (21 g, 0.04 mol). Yield 77%. 90 % pure by HPLC; Mass: 520.1 (M+H). Preparation of 4-(3-(4-(4,6-dimorpholino-1 ,3,5-triazine-2-yl)phenyl)ureido) benzoic acid (7)
The mixture of methyl 4-(3-(4-(4,6-dimorpholino-1 ,3,5-triazine-2-yl)ureido)benzoate (6, 21 g, 0.04mol) and lithium hydroxide monohydrate (3.8 g, 0.09 mol) in THF (120 ml), MeOH (60 ml), and water (60 ml) was heated at 80 0C for 3 hours. The dark brown solution was cooled to room temperature and made acidic with concentrated HCI. The solids were filtered, washed with water, washed with acetone , washed with ether, and dried in a vacuum oven at 60 0C for 48 hours to give off white solids of 4-(3-(4-(4,6-dimorpholino-1 ,3,5-triazine-2-yl)phenyl)ureido) benzoic acid (19.2 g, 0.038 mol). Mass: 506.3 (M+H)+; Yield.94%. 1 -(4-(4-(dimethylamino) piperidine-1 -carbonyl)phenyl-3-(4-(4,6-dimorpholino- 1 ,3,5-triazine-2-yl)phenyl) urea (9)
The suspension of 4-(3-(4-(4,6-dimorpholino-1 ,3,5-triazine-2-yl)phenyl)ureido) benzoic acid (7, 17 g, 33.66 mmol) and N-(3-dimethylaminopropyl)ethyl carbodiimide hydrochloride (9.5 g, 49.5 mmol) in THF (200 ml) and acetonitrile (50 ml) was stirred for 10 min and followed by addition of 1-hydroxybenzotriazole hydrate (6.4 g, 47.88 mmol). The reaction mixture was stirred for 30 min and 4-dimethylaminopiperidine (8, 8.86 g, 69.2 mmol) was added by drops. After being stirred for additional 16 hours, the reaction mixture was concentrated to min. The solids were filtered and washed thoroughly with water (very fine suspension). The cake was slurred in hot ethanol, filtered and dried in a vacuum oven at 68 0C for 16 hours to give off white solids (10.3 g, 16.77 mmol). M. p. 238-240 0C. 99 % pure. Mass: 616.3 (M+H)+; Yield 50 %.
PATENT
WO 2009143317
WO 2010096619
WO 2012148540
WO 2014151147
PATENT
US 20170119778
PAPER
Journal of Medicinal Chemistry (2010), 53(6), 2636-2645
http://pubs.acs.org/doi/abs/10.1021/jm901830p
J. Med. Chem., 2010, 53 (6), pp 2636–2645
DOI: 10.1021/jm901830p
Abstract
The PI3K/Akt signaling pathway is a key pathway in cell proliferation, growth, survival, protein synthesis, and glucose metabolism. It has been recognized recently that inhibiting this pathway might provide a viable therapy for cancer. A series of bis(morpholino-1,3,5-triazine) derivatives were prepared and optimized to provide the highly efficacious PI3K/mTOR inhibitor 1-(4-{[4-(dimethylamino)piperidin-1-yl]carbonyl}phenyl)-3-[4-(4,6-dimorpholin-4-yl-1,3,5-triazin-2-yl)phenyl]urea 26 (PKI-587). Compound 26 has shown excellent activity in vitro and in vivo, with antitumor efficacy in both subcutaneous and orthotopic xenograft tumor models when administered intravenously. The structure−activity relationships and the in vitro and in vivo activity of analogues in this series are described.
Preparation of 1-(4-{[4-(Dimethylamino)piperidin-1-yl]carbonyl}phenyl)-3-[4-(4,6-dimorpholin-4- yl-1,3,5-triazin-2-yl)phenyl]urea (26)
MS (ESI) m/z = 616.7. HRMS: calcd for C32H41N9O4 + H+, 616.335 43; found (ESI-FTMS, [M + H]+), 616.334 24. Purity by analytical HPLC 99.3%. (Prodigy ODS3, 0.46 cm × 15 cm, 20 min gradient acetonitrile in water, trifluoroacetic acid, detector wavelengths, 215 and 254 nm.) 1H NMR (DMSO-d6) δ 1.29−1.36 (m, 6H), 2.6 (m, 4H), 2.9 (m,1H), 3.3 (m, 4H), 3.6 (m, 8H), 3.7 (m, 8H), 7.3 (d, J = 8.3 Hz, 2H), 7.51−7.57 (m, 4H), 8.3 (d, J = 8.3 Hz 2H), 8.9 (s, 1H), 9.0 (s, 1H) ppm. Anal. Calcd for C32H41N9O4: C 62.42%, H 6.71%, N 20.47%. Found: C 62.34%, H 6.67%, N 20.39%.
PAPER
Bioorganic & Medicinal Chemistry Letters (2011), 21(16), 4773-4778.
http://www.sciencedirect.com/science/article/pii/S0960894X11008468
PAPER
New and Practical Synthesis of Gedatolisib
http://pubs.acs.org/doi/10.1021/acs.oprd.7b00298
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.7b00298
Abstract
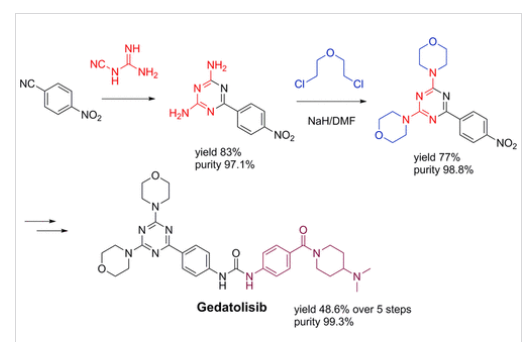
A new, practical, and convergent synthetic route of gedatolisib, an antitumor agent, is developed on a hectogram scale which avoids the Pd coupling method. The key step is adopting 6-(4-nitrophenyl)-1,3,5-triazine-2,4-diamine and 2,2′-dichlorodiethyl ether to prepare the key 4,4′-(6-(4-nitrophenyl)-1,3,5-triazine-2,4-diyl)dimorpholine in 77% yield and 98.8% purity. Gedatolisib is obtained in 48.6% yield over five simple steps and 99.3% purity (HPLC). Purification methods of the intermediates and the final product involved in the route are given.
off-white solid. 1H NMR (400 MHz, DMSO-d6): δ 1.46 (brs, 2H), 1.89 (brs, 2H), 2.29 (s, 6H), 2.94 (brs, 2H), 3.76 (m, 8H), 3.89 (m, 8H), 7.09 (d, J = 8.4 Hz, 2H), 7.20 (d, J = 8.4 Hz, 2H), 7.50 (d, J = 8.7 Hz, 2H), 8.28 (s, 1H), 8.31 (d, J = 8.6 Hz, 2H), 8.48 (s, 1H). ESI-MS (m/z) 615.9 (M + H). HPLC conditions: Column: Agilent Eclipse XDB-C18 (250 mm × 4.6 mm × 5 μm); Detection: 254 nm; Flow rate: 0.8 mL/min; Temperature: 30 °C; Injection load: 1 μL; Solvent: MeOH; Concentration: 0.5 mg/mL; Run time: 20 min; Mobile phase A: water; Mobile phase B: MeOH/TEA = 100:0.1; Gradient program: time (min): 20; % of mobile phase A: 10; % of mobile phase B: 90; tR = 2.598 min, purity: 99.34%
- Zhao, X.; Tan, Q.; Zhang, Z.; Zhao, Y. Med. Chem. Res. 2014, 23, 5188– 5196 DOI: 10.1007/s00044-014-1084-z
- Khafizova, G.; Potoski, J. R. PCT Int. Appl. WO 2010096619, 2010.
- Venkatesan, A. M.; Chen, Z.; Dehnhardt, C. M.; Dos Santos, O.; Delos Santos, E. G.; Zask, A.; Verheijen, J. C.; Kaplan, J. A.; Richard, D. J.; Ayral-Kaloustian, S.; Mansour, T. S.; Gopalsamy, A.; Curran, K. J.; Shi, M. PCT Int. Appl. WO 2009143317, 2009.
REFERENCES
1: Gedaly R, Galuppo R, Musgrave Y, Angulo P, Hundley J, Shah M, Daily MF, Chen C, Cohen DA, Spear BT, Evers BM. PKI-587 and sorafenib alone and in combination on inhibition of liver cancer stem cell proliferation. J Surg Res. 2013 Nov;185(1):225-30. doi: 10.1016/j.jss.2013.05.016. Epub 2013 May 25. PubMed PMID: 23769634.
2: Gedaly R, Angulo P, Hundley J, Daily MF, Chen C, Evers BM. PKI-587 and sorafenib targeting PI3K/AKT/mTOR and Ras/Raf/MAPK pathways synergistically inhibit HCC cell proliferation. J Surg Res. 2012 Aug;176(2):542-8. doi: 10.1016/j.jss.2011.10.045. Epub 2011 Nov 21. PubMed PMID: 22261591.
3: Dehnhardt CM, Venkatesan AM, Chen Z, Delos-Santos E, Ayral-Kaloustian S, Brooijmans N, Yu K, Hollander I, Feldberg L, Lucas J, Mallon R. Identification of 2-oxatriazines as highly potent pan-PI3K/mTOR dual inhibitors. Bioorg Med Chem Lett. 2011 Aug 15;21(16):4773-8. doi: 10.1016/j.bmcl.2011.06.063. Epub 2011 Jun 21. PubMed PMID: 21763134.
4: Mallon R, Feldberg LR, Lucas J, Chaudhary I, Dehnhardt C, Santos ED, Chen Z, dos Santos O, Ayral-Kaloustian S, Venkatesan A, Hollander I. Antitumor efficacy of PKI-587, a highly potent dual PI3K/mTOR kinase inhibitor. Clin Cancer Res. 2011 May 15;17(10):3193-203. doi: 10.1158/1078-0432.CCR-10-1694. Epub 2011 Feb 15. PubMed PMID: 21325073.
5: Venkatesan AM, Chen Z, dos Santos O, Dehnhardt C, Santos ED, Ayral-Kaloustian S, Mallon R, Hollander I, Feldberg L, Lucas J, Yu K, Chaudhary I, Mansour TS. PKI-179: an orally efficacious dual phosphatidylinositol-3-kinase (PI3K)/mammalian target of rapamycin (mTOR) inhibitor. Bioorg Med Chem Lett. 2010 Oct 1;20(19):5869-73. doi: 10.1016/j.bmcl.2010.07.104. Epub 2010 Jul 30. PubMed PMID: 20797855.
6: Venkatesan AM, Dehnhardt CM, Delos Santos E, Chen Z, Dos Santos O, Ayral-Kaloustian S, Khafizova G, Brooijmans N, Mallon R, Hollander I, Feldberg L, Lucas J, Yu K, Gibbons J, Abraham RT, Chaudhary I, Mansour TS. Bis(morpholino-1,3,5-triazine) derivatives: potent adenosine 5′-triphosphate competitive phosphatidylinositol-3-kinase/mammalian target of rapamycin inhibitors: discovery of compound 26 (PKI-587), a highly efficacious dual inhibitor. J Med Chem. 2010 Mar 25;53(6):2636-45. doi: 10.1021/jm901830p. PubMed PMID: 20166697.
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma



AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

References
- PRMT5 inhibitors and uses thereofPublication Number: US-12448388-B2Grant Date: 2025-10-21
- KRAS G12D modulating compoundsPublication Number: US-12448400-B2Grant Date: 2025-10-21
- TRIAZINE COMPOUNDS AS P13 KINASE AND MTOR INHIBITORSPublication Number: PT-2294072-TPriority Date: 2008-05-23
- TRIASINE UNITS AS P13 KINASE INHIBITORS AND MOTORPublication Number: ME-01111-BPriority Date: 2008-05-23
- HER2 mutation inhibitorsPublication Number: US-12447153-B2Grant Date: 2025-10-21
- Anti-hiv compoundsPublication Number: US-2025326779-A1
- Aryl aminopyrimidines as dual MerTK and TYRO3 inhibitors and methods thereofPublication Number: US-12448365-B2Grant Date: 2025-10-21
References
- https://celcuity.com/revtorpyk/REVTORPYK_PI_2026.pdf
- Dehnhardt CM, Venkatesan AM, Chen Z, Delos-Santos E, Ayral-Kaloustian S, Brooijmans N, et al. (August 2011). “Identification of 2-oxatriazines as highly potent pan-PI3K/mTOR dual inhibitors”. Bioorganic & Medicinal Chemistry Letters. 21 (16): 4773–8. doi:10.1016/j.bmcl.2011.06.063. PMID 21763134.
- “FDA approves gedatolisib with fulvestrant, with or without palbociclib, for HR-positive, HER2-negative locally advanced or metastatic breast cancer”. U.S. Food and Drug Administration (FDA). 14 July 2026. Retrieved 20 July 2026.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - Sabatini DM (November 2017). “Twenty-five years of mTOR: Uncovering the link from nutrients to growth”. Proceedings of the National Academy of Sciences of the United States of America. 114 (45): 11818–11825. Bibcode:2017PNAS..11411818S. doi:10.1073/pnas.1716173114. PMC 5692607. PMID 29078414.
- Tian T, Li X, Zhang J (February 2019). “mTOR Signaling in Cancer and mTOR Inhibitors in Solid Tumor Targeting Therapy”. International Journal of Molecular Sciences. 20 (3): 755. doi:10.3390/ijms20030755. PMC 6387042. PMID 30754640.
- Hua H, Kong Q, Zhang H, Wang J, Luo T, Jiang Y (July 2019). “Targeting mTOR for cancer therapy”. Journal of Hematology & Oncology. 12 (1) 71. doi:10.1186/s13045-019-0754-1. PMC 6612215. PMID 31277692.
- Vanhaesebroeck B, Perry MW, Brown JR, André F, Okkenhaug K (October 2021). “PI3K inhibitors are finally coming of age”. Nature Reviews. Drug Discovery. 20 (10): 741–769. doi:10.1038/s41573-021-00209-1. PMC 9297732. PMID 34127844. S2CID 235437841.
- Millis SZ, Ikeda S, Reddy S, Gatalica Z, Kurzrock R (December 2016). “Landscape of Phosphatidylinositol-3-Kinase Pathway Alterations Across 19 784 Diverse Solid Tumors”. JAMA Oncology. 2 (12): 1565–1573. doi:10.1001/jamaoncol.2016.0891. PMID 27388585.
- Anderson EJ, Mollon LE, Dean JL, Warholak TL, Aizer A, Platt EA, et al. (2020). “A Systematic Review of the Prevalence and Diagnostic Workup of PIK3CA Mutations in HR+/HER2- Metastatic Breast Cancer”. International Journal of Breast Cancer. 2020 3759179. doi:10.1155/2020/3759179. PMC 7322582. PMID 32637176.
- Clinical trial number NCT01420081 for “A Study Of Two Dual PI3K/mTOR Inhibitors, PF-04691502 And PF-05212384 In Patients With Recurrent Endometrial Cancer” at ClinicalTrials.gov
- Clinical trial number NCT01925274 for “A Study Of PF-05212384 Plus Irinotecan Vs Cetuximab Plus Irinotecan In Patients With KRAS And NRAS Wild Type Metastatic Colorectal Cancer” at ClinicalTrials.gov
- Clinical trial number NCT02438761 for “PF-05212384 (PKI-587) for t-AML/MDS or de Novo Relapsed or Refractory Acute Myeloid Leukemia (AML)” at ClinicalTrials.gov
- Clinical trial number NCT03698383 for “Phase II Study of Herzuma® Plus Gedatolisib in Patients With HER-2 Positive Metastatic Breast Cancer” at ClinicalTrials.gov
- Clinical trial number NCT03911973 for “Gedatolisib Plus Talazoparib in Advanced Triple Negative or BRCA1/2 Positive, HER2 Negative Breast Cancers” at ClinicalTrials.gov
- Clinical trial number NCT03065062 for “Study of the CDK4/6 Inhibitor Palbociclib (PD-0332991) in Combination With the PI3K/mTOR Inhibitor Gedatolisib (PF-05212384) for Patients With Advanced Squamous Cell Lung, Pancreatic, Head & Neck and Other Solid Tumors” at ClinicalTrials.gov
- Clinical trial number NCT02626507 for “Phase I Study of Combination of Gedatolisib With Palbociclib and Faslodex in Patients With ER+/HER2- Breast Cancer” at ClinicalTrials.gov
- “Celcuity Announces FDA Approval of Revtorpyk (gedatolisib) for the Treatment of HR+/HER2-, PIK3CA Wild-Type Locally Advanced or Metastatic Breast Cancer” (Press release). Celcuity. 14 July 2026. Retrieved 20 July 2026 – via GlobeNewswire.
- World Health Organization (2015). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 73”. WHO Drug Information. 29 (1). hdl:10665/331088.
External links
- Clinical trial number NCT05501886 for “Gedatolisib Plus Fulvestrant With or Without Palbociclib vs Standard-of-Care for the Treatment of Patients With Advanced or Metastatic HR+/HER2- Breast Cancer (VIKTORIA-1) (VIKTORIA-1)” at ClinicalTrials.gov
 | |
| Clinical data | |
|---|---|
| Trade names | Revtorpyk |
| Other names | PF-05212384; PKI-587 |
| AHFS/Drugs.com | revtorpyk |
| License data | US DailyMed: Gedatolisib |
| Routes of administration | Intravenous infusion |
| Drug class | Antineoplastic |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1197160-78-3 |
| PubChem CID | 44516953 |
| IUPHAR/BPS | 7940 |
| DrugBank | DB11896 |
| ChemSpider | 24644946 |
| UNII | 96265TNH2R |
| KEGG | D10635 |
| ChEMBL | ChEMBL592445 |
| CompTox Dashboard (EPA) | DTXSID40152557 |
| Chemical and physical data | |
| Formula | C32H41N9O4 |
| Molar mass | 615.739 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
/////////Gedatolisib, anax labs, approvals 3026, FDA 2026, PF 05212384, PF 5212384, PKI-587, PF-05212384; PF-5212384; PKI 587, gedatolisib, antitumor agent, PHASE 3, PFIZER, гедатолисиб , غيداتوليسيب , 吉达利塞 , 96265TNH2R
O=C(NC1=CC=C(C2=NC(N3CCOCC3)=NC(N4CCOCC4)=N2)C=C1)NC5=CC=C(C(N6CCC(N(C)C)CC6)=O)C=C5
Journal of Medicinal Chemistry (2017), 60(17), 7524-7538 PQR 309
Opakalim
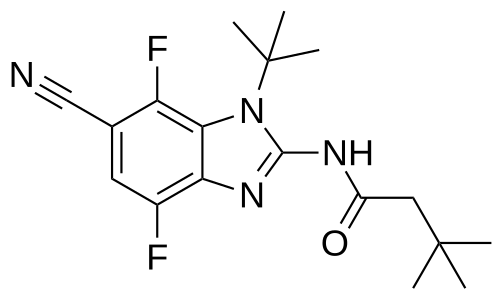
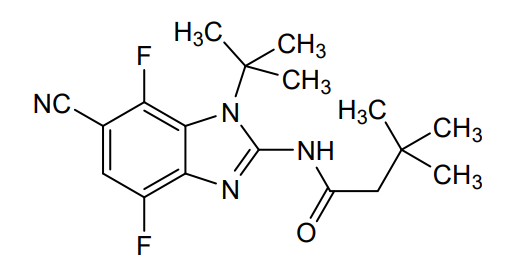
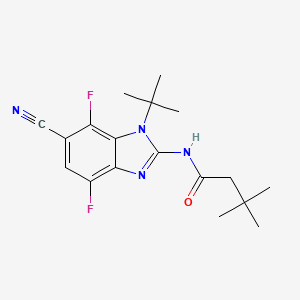
Opakalim
CAS 2376397-93-0
MF C18H22F2N4O MW 348.4 g/mol
N-(1-tert-butyl-6-cyano-4,7-difluorobenzimidazol-2-yl)-3,3-dimethylbutanamide
- N-(1-tert-butyl-6-cyano-4,7-difluorobenzimidazol-2-yl)-3,3-dimethylbutanamide
- Butanamide, N-[6-cyano-1-(1,1-dimethylethyl)-4,7-difluoro-1H-benzimidazol-2-yl]-3,3-dimethyl-
- N-[6-Cyano-1-(1,1-dimethylethyl)-4,7-difluoro-1H-benzimidazol-2-yl]-3,3-dimethylbutanamide
N-(1-tert-butyl-6-cyano-4,7-difluoro-1H-1,3-benzimidazol-2-yl)-3,3-dimethylbutanamide
potassium channel activator, antiepileptic, BHV-7000; BHV7 000, BPN-25203, BPN 25203, KB-3061, KB 3061, 73H9J7RBA2
| BHV-7000 is under investigation in clinical trial NCT06419608 (Efficacy and Safety Study of BHV-7000 Monotherapy in Major Depression). |
Opakalim (also known as BHV-7000) is an investigational, small-molecule medication that acts as a selective activator of Kv7.2 and Kv7.3 potassium channels. Developed by Biohaven Pharmaceuticals, it targets a clinically validated pathway to regulate neuronal hyperexcitability, primarily for the treatment of epilepsy and focal seizures.
Key Characteristics
- Mechanism of Action: It opens Kv7.2/7.3 potassium channels. This stabilizes electrical activity in the brain. Unlike older class drugs, it has minimal to no (GABA_A) receptor activity.
- Safety Benefit: Its precision prevents typical central nervous system side effects. Patients experience much lower rates of somnolence, fatigue, and severe dizziness compared to other anti-seizure medications.
- Administration: It is taken orally once a day. It requires no titration period before reaching therapeutic dosing.
Current Clinical Status
- Refractory Focal Epilepsy: The drug is undergoing phase 2/3 clinical evaluation. The pivotal RISE 3 study completed patient enrollment, with high-profile top-line efficacy data anticipated in the second half of 2026.
- Idiopathic Generalized Epilepsy (IGE): Recent clinical data highlights a three-fold prolongation in the time to a second generalized tonic-clonic seizure compared to a placebo.
- Other Explored Indications: While it is actively tested for conditions like bipolar disorder and erythromelalgia (pain), an exploratory phase 2 trial for major depressive disorder failed to meet its primary goals.
Opakalim (INNTooltip International Nonproprietary Name, USANTooltip United States Adopted Name; developmental code names BHV-7000, BPN-25203, and KB-3061) is a highly selective Kv7.2 and Kv7.3 potassium channel opener which is under development for the treatment of bipolar disorders, epilepsy, partial epilepsies, major depressive disorder, erythromelalgia, pain, infantile spasms, and mood disorders.[1][2][3][4] It is taken orally.[1] The drug was originated by Channel Biosciences and was under development by Biohaven Pharmaceuticals or Biohaven Therapeutics.[1][2] As of April 2026, it is in phase 2/3 clinical trials for bipolar disorders, epilepsy, and partial epilepsies, phase 2 trials for major depressive disorder, and phase 1 trials for erythromelalgia and pain, whereas no recent development has been reported for infantile spasms and mood disorders.[1][2] A phase 2 trial for major depressive disorder failed to meet its primary efficacy endpoint, resulting in focus more on epilepsy instead.[3]
- Study to Determine if BHV-7000 is Effective and Safe in Adults With Refractory Focal Onset EpilepsyCTID: NCT06309966Phase: Phase 2/Phase 3Status: Active, not recruitingDate: 2026-07-01
- A Study to Determine if BHV-7000 is Effective and Safe in Adults With Refractory Focal Onset EpilepsyCTID: NCT06132893Phase: Phase 2/Phase 3Status: RecruitingDate: 2026-05-01
- Long-term Safety and Tolerability of BHV-7000CTID: NCT06443463Phase: Phase 2Status: Enrolling by invitationDate: 2026-05-01
- A Phase 1b Study of BHV-7000 in Participants With Inherited ErythromelalgiaCTID: NCT07262268Phase: Phase 1Status: Enrolling by invitationDate: 2026-04-08
- Long-term Safety Study of BHV-7000 in Participants With Major Depressive Disorder (MDD)CTID: NCT06423781Phase: Phase 2Status: CompletedDate: 2026-04-01
- A Study to Determine if BHV-7000 is Effective and Safe in Adults With Idiopathic Generalized Epilepsy With Generalized Tonic-clonic SeizuresCTID: NCT06425159Phase: Phase 2/Phase 3Status: TerminatedDate: 2026-03-24
- Efficacy and Safety Study of BHV-7000 Monotherapy in Major DepressionCTID: NCT06419608Phase: Phase 2Status: CompletedDate: 2026-01-07
- BHV-7000 Acute Treatment of Bipolar ManiaCTID: NCT06419582Phase: Phase 2/Phase 3Status: CompletedDate: 2025-12-16
- BHV-7000 Open-Label Extension Bipolar Mania StudyCTID: NCT06423794Phase: Phase 2Status: TerminatedDate: 2025-12-16
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US383826737&_cid=P20-MRSLRP-32698-1
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2025035066&_cid=P20-MRSLVM-39325-2
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- Kv7 channel activators compositions and methods of usePublication Number: US-11724990-B2Priority Date: 2018-03-19Grant Date: 2023-08-15
- KV7 channel activators compositions and methods of usePublication Number: US-12286408-B2Priority Date: 2018-03-19Grant Date: 2025-04-29
- KV7 channel activators compositions and methods of usePublication Number: US-11261162-B2Priority Date: 2018-03-19Grant Date: 2022-03-01
- Kv7 channel activators compositions and methods of usePublication Number: US-2019284142-A1Priority Date: 2018-03-19
- Kv7 channel activators compositions and methods of usePublication Number: US-10851067-B2Priority Date: 2018-03-19Grant Date: 2020-12-01
- KV7 channel activator composition and method of use thereofPublication Number: KR-20200133259-APriority Date: 2018-03-19
- Kv7 channel activators compositions and methods of usePublication Number: US-2021130299-A1Priority Date: 2018-03-19
- KV7 channel activator composition and method of use thereofPublication Number: KR-102731128-B1Priority Date: 2018-03-19Grant Date: 2024-11-18
- Methods of use for kv7 channel activatorsPublication Number: US-2025312316-A1Priority Date: 2019-09-17
- Methods of use for KV7 channel activatorsPublication Number: US-12370176-B2Priority Date: 2019-09-17Grant Date: 2025-07-29
- How to use KV7 channel activatorPublication Number: CN-118986972-APriority Date: 2019-09-17
- Kv7 channel activators compositions and methods of usePublication Number: US-2024002349-A1Priority Date: 2018-03-19
- Kv7 channel activators compositions and methods of usePublication Number: US-2023000831-A1Priority Date: 2018-03-19
References
- “Biohaven Pharmaceuticals”. AdisInsight. 15 April 2026. Retrieved 31 May 2026.
- “Delving into the Latest Updates on Opakalim with Synapse”. Synapse. 2 April 2026. Retrieved 31 May 2026.
- Pelorosso C, Balestrini S, Guerrini R (May 2026). “Potassium channel agonists emerging as treatment options for focal epilepsy: are we breaking new ground?”. Expert Opinion on Emerging Drugs: 1–8. doi:10.1080/14728214.2026.2675274. hdl:2158/1473832. PMID 42153277.
- Pong AW (December 2025). “Expanding the toolkit: An update on the evolution of new therapies for Lennox-Gastaut Syndrome”. Seminars in Pediatric Neurology. 56 101242. doi:10.1016/j.spen.2025.101242. PMID 41371876.
 | |
| Clinical data | |
|---|---|
| Other names | BHV-7000; BHV7000; BPN-25203; BPN25203; KB-3061; KB3061 |
| Routes of administration | Oral[1] |
| Drug class | Kv7.2 and Kv7.3 potassium channel opener |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2376397-93-0 |
| PubChem CID | 139487180 |
| DrugBank | DB22041 |
| ChemSpider | 129910012 |
| UNII | 73H9J7RBA2 |
| KEGG | D13061 |
| Chemical and physical data | |
| Formula | C18H22F2N4O |
| Molar mass | 348.398 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
////////opakalim, anax labs, potassium channel activator, antiepileptic, BHV-7000; BHV7 000, BPN-25203, BPN 25203, KB-3061, KB 3061, 73H9J7RBA2
Ontunisertib

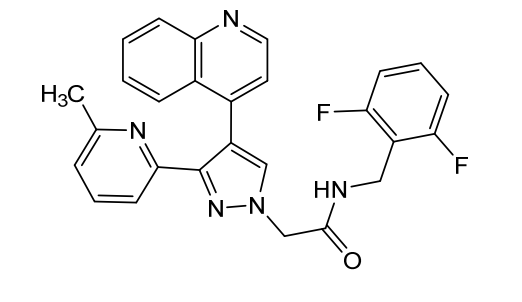
Ontunisertib
CAS 2647949-48-0
MFC27H21F2N5O MW469.5 g/mol
- 1H-Pyrazole-1-acetamide, N-[(2,6-difluorophenyl)methyl]-3-(6-methyl-2-pyridinyl)-4-(4-quinolinyl)-
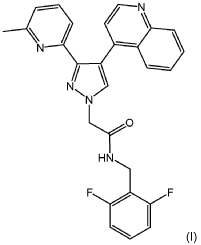
- N-(2,6-Difluorobenzyl)-2-(3-(6-methylpyridin-2-yl)-4-(quinolin-4-yl)-1H-pyrazol-1-yl)acetamide
N-[(2,6-difluorophenyl)methyl]-2-[3-(6-methyl-2-pyridinyl)-4-quinolin-4-ylpyrazol-1-yl]acetamide
N-(2,6-difluorobenzyl)-2-(3-(6-methylpyridin-2-yl)-4-(quinolin-4-yl)-1H-pyrazol-1-yl)acetamide
N-[(2,6-difluorophenyl)methyl]-2-[3-(6-methylpyridin-2-yl)-4-(quinolin-4-yl)-1H-pyrazol-1-yl]acetamide
serine/threonine kinase inhibitor, AGMB 129, SF6HGC94LK
Ontunisertib (AGMB-129) is an experimental, orally active small-molecule drug developed by Agomab Therapeutics to treat Fibrostenosing Crohn’s Disease (FSCD). It acts as a highly selective inhibitor of ALK5 (also known as Transforming Growth Factor-beta type I receptor or TGF-β RI).
Mechanism of Action
- Local Targeting: Designed to act specifically within the gastrointestinal (GI) tract.
- High Tissue Exposure: Provides high local exposure in inflamed and scarred intestinal tissues.
- Liver Inactivation: Undergoes rapid first-pass metabolism in the liver immediately after GI absorption.
- Safety Feature: Converts into an inactive metabolite to prevent systemic exposure and avoid cardiac toxicity.
Clinical Development Status
- FDA Status: Granted Fast Track Designation by the U.S. FDA.
- Phase 2a Results: Successfully completed the STENOVA clinical trial. Results demonstrated excellent safety, high local tissue penetration, and positive structural improvements in bowel strictures.
- Phase 2b Trial: Enrolling patients for the global, 52-week NOV-ERA trial to test multiple doses against a placebo. The primary goal is assessing the endoscopically confirmed widening of narrowed intestinal strictures
- NOV-ERA – A Clinical Trial to Assess the Efficacy and Safety of Ontunisertib Compared to Placebo in Patients With Fibrostenosing Crohn’s DiseaseCTID: NCT07683325Phase: Phase 2Status: Not yet recruitingDate: 2026-07-06
- Human Mass Balance Study of [14C] Ontunisertib in Healthy VolunteersCTID: NCT07672574Phase: Phase 1Status: Not yet recruitingDate: 2026-06-29
- A Multiple Ascending Dose Study With AGMB-129 in Healthy ParticipantsCTID: NCT07118878Phase: Phase 1Status: CompletedDate: 2025-11-21
- STENOVA – A Study to Evaluate Safety, Tolerability, PK and PD of AGMB-129 in Patients With Fibrostenotic Crohn’s DiseaseCTID: NCT05843578Phase: Phase 2Status: Active, not recruitingDate: 2025-11-21
- Drug-Drug Interaction Study With AGMB-129 and Midazolam in Healthy ParticipantsCTID: NCT05937386Phase: Phase 1Status: CompletedDate: 2024-06-18
SYN
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021105317&_cid=P20-MRPQUQ-14721-1

Example 24: N-(2,6-difluorobenzyl)-2-(3-(6-methylpyridin-2-yl)-4-(quinolin-4-yl)-1H-pyrazol-1 -yl)acetamide
1H-NMR (300 MHz, DMSO-d6): δ = 8.84 (d, J = 4.4 Hz, 1H), 8.76 (t, J = 5.3 Hz, 1H), 8.11-7.97 (m, 2H), 7.75-7.28 (m, 7H), 7.13 (t, J = 7.8 Hz, 2H), 6.97 (d, J = 7.5 Hz, 1 H), 4.99 (s, 2H), 4.43 (d, J = 5.3 Hz, 2H), 1.83 (s, 3H).
HPLC-MS: Rt 17.513 m/z 470.0 [M+H]+.
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2025176835&_cid=P20-MRPR39-19345-1
Potent inhibitors of TGFpRII-TGFpRI (ALK5) have been described in W02021/105317 including the compound (/V-(2,6-difluorobenzyl)-2-(3-(6-methylpyridin-2-yl)-4-(quinolin-4-yl)-1/7-pyrazol-1-yl)acetamide) which is the compound of formula (I) as shown below (see Example 24):
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
///////////ontunisertib, anax labs, serine/threonine kinase inhibitor, AGMB 129, SF6HGC94LK
Olisutrigine bromide
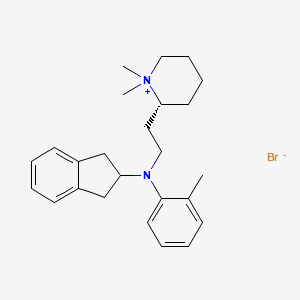
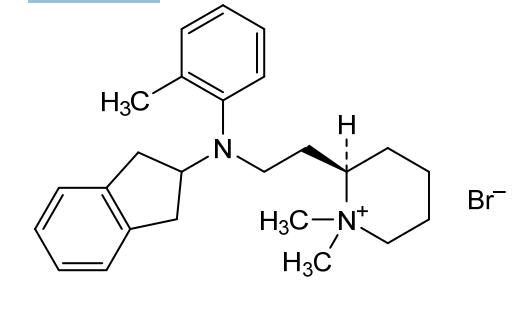
Olisutrigine bromide
Cas 1393836-45-7
MF C25H35BrN2 MW443.5 g/mol
N-[2-[(2R)-1,1-dimethylpiperidin-1-ium-2-yl]ethyl]-N-(2-methylphenyl)-2,3-dihydro-1H-inden-2-amine bromide
(2R)-2-{2-[N-(2,3-dihydro-1H-inden-2-yl)-2-methylanilino]ethyl}-1,1-dimethylpiperidin-1-ium bromide
sodium channel blocker, analgesic, ASN008, ASN 008, EN 3427, 0M9Q318030
Olisutrigine bromide (also known as ASN-008 or EN3427) is an investigational, permanently charged sodium channel blocker being studied for its potent, long-lasting analgesic (pain-relieving) properties.
Key Characteristics
- Mechanism of Action: It acts as a membrane-impermeant sodium channel blocker. Because it carries a permanent cationic charge, it cannot easily cross healthy cell membranes on its own.
- Targeted Delivery: It often requires a “vehicle” or a combination drug (like lidocaine) to activate specific channels (such as TRP channels), allowing olisutrigine entry into pain-sensing neurons where it becomes entrapped and blocks pain signaling.
- Efficacy: Rodent pain models demonstrate that its analgesic effects are significantly longer-lasting than lidocaine alone.
- Chemical Profile: Its molecular formula is C₂₅H₃₅BrN₂ with a molecular weight of 443.5 g/mol, and its CAS registry number is 1393836-45-7.
Current Status
- Investigational Drug: It is not approved for human or veterinary medical use and remains in the research and development phase.
- Availability: It is strictly available as a reference standard compound for laboratory and preclinical research through chemical suppliers like MedChemExpress and BenchChem.
A Study to Evaluate the Anti-pruritic Effectiveness of ASN008 in Adults With Mild to Moderate Atopic Dermatitis
CTID: NCT05870865
Phase: Phase 2
Status: Completed
Date: 2025-05-16
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2012112969&_cid=P11-MRILQZ-81481-1
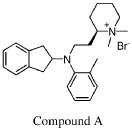
Example 43: General Procedure M – Preparation of (R)-1,1 -dimethyl-2-[2-((indan-2-yl)(2-methylphenyl)amino)ethyl]piperidinium bromide
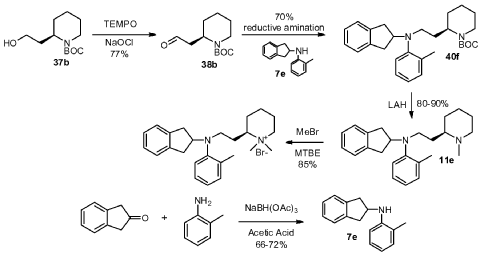
Alcohol 37b was synthesized as previouslydescribed (Tetrahedron 2007, 63, 3000-3005)
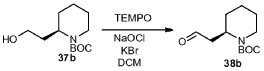
To a 250 mL round bottom flask was charged 2-(2-hydroxyethyl)piperidine-1-carboxylic acid tert-butyl ester 37b (5.0 g, 21.80 mmol), dichloromethane (7.50 mL), a solution of KBr (0.52 g, 4.36 mmol) in 2.0 mL of water and TEMPO (0.1 g, 0.64 mmol). The mixture was cooled to about -5 °C. A solution of NaOCl (31.1 mL, 5.25%, 24.1 mmol) was added slowly over 20 minutes while maintaining the temperature at 0 °C. The mixture was further stirred at 0°C for 20 minutes. The organic layer was separated, and the aqueous layer was extracted with
dichloromethane. The combined dichloromethane extract was washed with water (50 mL), followed by brine. After drying over MgSO4, the mixture was filtered and concentrated. The crude was purified with silica gel column chromatography to give product 38b (4.1 g, 83%) as colorless oil.
O
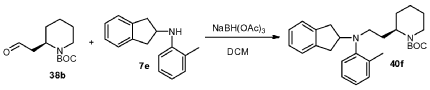
To a clean and dry 250 mL round bottom flask was charged sodium triacetoxyborohydride (5.59 g, 26.40 mmol), 4 A molecular sieves (10.0 g), amine 7e (7.37 g, 33.00 mmol) and dichloromethane (20.0 mL). The mixture was stirred and cooled to about 0 °C, and a solution of aldehyde 38b (5.0 g, 22.00 mmol) in 40 mL of dichloromethane was added. The mixture was then stirred further at 0 °C for about 1 hour and at ambient temperature for an additional 40 minutes. The reaction mixture was quenched with aqueous saturated NaHCO3 (100 mL). After separation of organic layer, the mixture was extracted with dichloromethane. After drying over MgSO4, the organic layer was concentrated. The crude product was purified by silica gel column chromatography to give product 40f (7.2 g, 75.3%) as colorless oil.

To a clean and dry 250 mL round bottom flask was charged lithium aluminum hydride (1.53 g, 40.27 mmol) and THF (30.0 mL). The mixture was heated to reflux. A solution of carbamate 40f (7.0 g, 16.11 mmol) in THF (40.0 mL) was added dropwise over 5 minutes. After refluxing for 15 h, the reaction mixture was cooled to 0 °C, and water (1.55 mL) was added slowly and carefully, followed by THF (100 mL) and 15% NaOH (1.55 mL). After stirring the mixture at room temperature for 1.0 h, MgSO4 was added, and the mixture was stirred further for 15 minutes. The mixture was filtered and concentrated to obtain the crude product, which was purified by silica gel column chromatography to afford product 11e (4.7 g, 84%) as pale yellow oil. Optical purity by chiral HPLC: 99.3% ee.
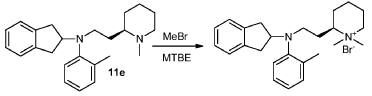
To a clean and dry 250 mL round bottom flask was charged diamine 11e (4.70 g, 13.49 mmol) and 1.07 M bromomethane in MTBE (126.0 mL, 134.8 mmol). After stirring at room temperature for 20 h, the reaction mixture was filtered. The solid cake was washed with MTBE to give the product (4.40 g, 73%) as white powder. Optical purity by chiral HPLC: 99.3% ee.
PAT
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2025250498&_cid=P11-MRILME-78328-1
Compound 1 is described in WO2012/112969, wherein Compound 1 is reported at Example 43, and certain formulations of Compound 1 are described in WO2020/113050,
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
AMINOINDANE COMPOUNDS AND THEIR APPLICATION FOR THE TREATMENT OF PAIN
Publication Number: RU-2013142433-A
Priority Date: 2011-02-18
- Aminoindane compounds and use thereof in treating painPublication Number: US-8865741-B2Priority Date: 2011-02-18Grant Date: 2014-10-21
- Aminoindan compound and its use in treating painPublication Number: CN-107540599-BPriority Date: 2011-02-18Grant Date: 2021-06-04
- Aminoindane compounds and their use in the treatment of painPublication Number: ES-2675510-T3Priority Date: 2011-02-18Grant Date: 2018-07-11
- Aminoindane compounds and use thereof in treating painPublication Number: EP-2675787-B1Priority Date: 2011-02-18Grant Date: 2018-03-28
- AMINOINDANE COMPOUNDS AND THEIR APPLICATION FOR THE TREATMENT OF PAINPublication Number: RU-2016151688-APriority Date: 2011-02-18
- Aminoindane compounds and use thereof in treating painPublication Number: RU-2612959-C2Priority Date: 2011-02-18Grant Date: 2017-03-14
- AMINOINDAN COMPOUNDS AND USE THEREOF IN THE TREATMENT OF PAINPublication Number: DK-2675787-T3Priority Date: 2011-02-18Grant Date: 2018-07-16
- Load Aminoinden Compounds and Therapeutic Compounds Containing Them and Uses in the Treatment of Itching and PainPublication Number: IL-227967-APriority Date: 2011-02-18
- Aminoindane compounds and use thereof in treating painPublication Number: EP-3363437-B1Priority Date: 2011-02-18Grant Date: 2021-07-28
- Aminoindane compounds and use thereof in treating painPublication Number: KR-20200125765-APriority Date: 2011-02-18
- Aminoindane compounds and use thereof in treating painPublication Number: WO-2012112969-A1Priority Date: 2011-02-18
- Aminoindane compounds and use thereof in treating painPublication Number: CA-2826648-CPriority Date: 2011-02-18Grant Date: 2019-06-04
- Aminoindane compounds for use in treating urological painPublication Number: EP-3970723-A1Priority Date: 2011-02-18
- Aminoindane compounds and use thereof in treating painPublication Number: CA-2826648-A1Priority Date: 2011-02-18
- Aminoindane Compounds and Use Thereof in Treating PainPublication Number: US-2012214809-A1Priority Date: 2011-02-18
- Use of aminoindane compounds in treating overactive bladder and interstitial cystitisPublication Number: US-9044482-B2Priority Date: 2012-08-15Grant Date: 2015-06-02
- Aminoindane compounds and their use in the treatment of painPublication Number: ES-2898910-T3Priority Date: 2011-02-18Grant Date: 2022-03-09
- Aminoindane compounds and use thereof in treating painPublication Number: AU-2012219254-A1Priority Date: 2011-02-18
- Aminoindane compounds and use thereof in treating painPublication Number: EP-2675787-A1Priority Date: 2011-02-18
- Aminoindane compounds and use thereof in treating painPublication Number: EP-3363437-A1Priority Date: 2011-02-18
- 6-(4-Nitro-phenoxy)-2H-pyridazin-3-one and 6-(4-amino-phenoxy)-2H-pyridazin-3-one as intermediates of thyroid hormone analogs The preparation method of ketone derivativePublication Number: CN-116406356-APriority Date: 2020-10-23
- Use of Aminoindane Compounds in Treating Overactive Bladder and Interstitial CystitisPublication Number: US-2014051702-A1Priority Date: 2012-08-15
- Use of aminoindane compounds in treating overactive bladder and interstitial cystitisPublication Number: US-2015290182-A1Priority Date: 2012-08-15
- Use of aminoindane compounds in treating overactive bladder and interstitial cystitisPublication Number: US-9375423-B2Priority Date: 2012-08-15Grant Date: 2016-06-28
- Use of aminoindane compounds in treating overactive bladder and interstitial cystitisPublication Number: WO-2014028675-A1Priority Date: 2012-08-15
///////////olisutrigine bromide, anax labs, sodium channel blocker, analgesic, ASN008, ASN 008, EN 3427, 0M9Q318030
Netanasvir
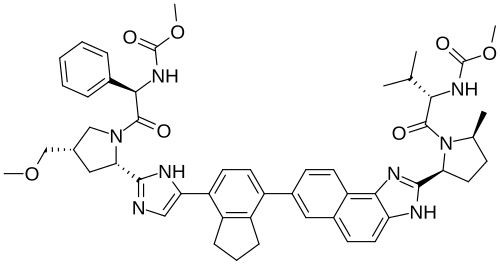
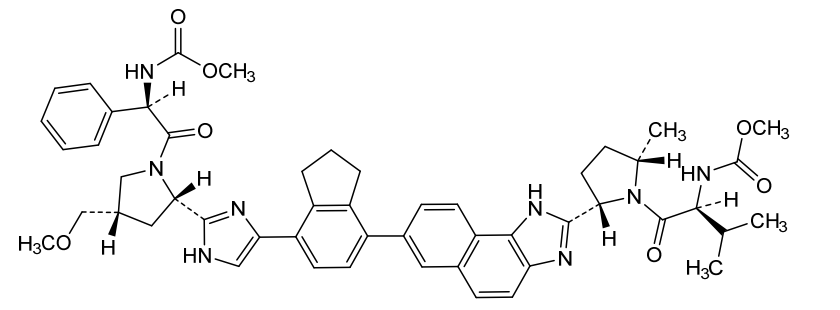

Netanasvir
CAS 2007900-70-9
MF C51H58N8O7 MW895.1 g/mol
methyl N-[(1R)-2-[(2S,4S)-2-[5-[7-[2-[(2S,5S)-1-[(2S)-2-(methoxycarbonylamino)-3-methylbutanoyl]-5-methylpyrrolidin-2-yl]-3H-benzo[e]benzimidazol-7-yl]-2,3-dihydro-1H-inden-4-yl]-1H-imidazol-2-yl]-4-(methoxymethyl)pyrrolidin-1-yl]-2-oxo-1-phenylethyl]carbamate
- Carbamic acid, N-[(1R)-2-[(2S,4S)-2-[5-[2,3-dihydro-7-[2-[(2S,5S)-1-[(2S)-2-[(methoxycarbonyl)amino]-3-methyl-1-oxobutyl]-5-methyl-2-pyrrolidinyl]-1H-naphth[1,2-d]imidazol-7-yl]-1H-inden-4-yl]-1H-imidazol-2-yl]-4-(methoxymethyl)-1-pyrrolidinyl]-2-oxo-1-phenylethyl]-, methyl ester
- Methyl ((S)-1-((2S,5S)-2-(7-(7-(2-((2S,4S)-1-((R)-2-((methoxycarbonyl)amino)-2-phenylacetyl)-4-(methoxymethyl)pyrrolidin-2-yl)-1H-imidazol-5-yl)-2,3-dihydro-1H-inden-4-yl)-1H-naphtho[1,2-d]imidazol-2-yl)-5-methylpyrrolidin-1-yl)-3-methyl-1-oxobutan-2-yl)carbamate
- methyl {(2S)-1-[(12S,15S,52S,54S,7R)-7-[(methoxycarbonyl)amino]-54-(methoxymethyl)-15-methyl-6-oxo-32,33-dihydro-21H,31H,41H-2(2,7)-naphtho[1,2-d]imidazola-4(4,2)-imidazola-1(2),5(2,1)-dipyrrolidina-3(4,7)-indena-8(1)-benzenaoctaphan-11-yl]-3-methyl-1-oxobutan-2-yl}carbamate
- Methyl N-[(1R)-2-[(2S,4S)-2-[5-[2,3-dihydro-7-[2-[(2S,5S)-1-[(2S)-2-[(methoxycarbonyl)amino]-3-methyl-1-oxobutyl]-5-methyl-2-pyrrolidinyl]-1H-naphth[1,2-d]imidazol-7-yl]-1H-inden-4-yl]-1H-imidazol-2-yl]-4-(methoxymethyl)-1-pyrrolidinyl]-2-oxo-1-phenylethyl]carbamate

antiviral, Dongweizhuo, HCV, Antaitavir Hasophate, HEC 74647 PA, 4Y7YD32BYY
Netanasvir (commonly prescribed as netanasvir phosphate) is a direct-acting antiviral medication primarily used to treat chronic hepatitis C virus (HCV) infection. It was approved by the China National Medical Products Administration (NMPA) under the trade name Dongweizhuo.
Key Clinical Information
- Mechanism of Action: It acts as a potent and selective NS5A inhibitor. By targeting the HCV NS5A protein, it successfully blocks viral RNA replication, virion assembly, and viral clearance mechanisms.
- Combination Therapy: It is exclusively indicated for use in combination with encofosbuvir (an NS5B polymerase inhibitor).
- Target Genotypes: The combination regimen effectively treats adult patients with chronic HCV genotypes 1, 2, 3, or 6.
- Patient Profiles: It is suitable for patients who are either treatment-naïve or have been previously treated with interferon, with or without compensated liver cirrhosis.
- Administration & Elimination: Taken orally, the drug demonstrates high metabolic stability, with feces serving as its primary elimination pathway
- OriginatorSunshine Lake Pharma
- ClassAntivirals
- Mechanism of ActionHepatitis C virus NS 5 protein inhibitors
- RegisteredHepatitis C
- 08 Feb 2025Registered for Hepatitis C (Combination therapy, Treatment-experienced) in China (PO)
- 08 Feb 2025Registered for Hepatitis C (Combination therapy, Treatment-naive) in China (PO)
- 31 Dec 2023NMPA, China accepts NDA for netanasvir for Hepatitis C for review
Netanasvir is an antiviral drug used to treat hepatitis C virus (HCV).[1] In China, netansavir is approved for use in combination with encofosbuvir for the treatment of adult patients with chronic HCV genotypes 1, 2, 3, or 6, who are either treatment-naive or have been previously treated with interferon.[2]
PAT
Compounds as hepatitis c virus inhibitors and pharmaceutical uses thereof
Publication Number: WO-2016141890-A1
Priority Date: 2015-03-12
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016141890&_cid=P22-MRH62L-18160-1
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- “Netanasvir phosphate – Sunshine Lake Pharma”. Adis Insight. Springer Nature Switzerland AG.
- “Netanasvir Phosphate Capsules Approved for Marketing by China NMPA”. National Medical Products Administration. 2025-06-11.
 | |
| Clinical data | |
|---|---|
| Trade names | 东卫卓; Dongweizhuo |
| Legal status | |
| Legal status | Rx in China |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2007900-70-9 |
| PubChem CID | 122535557 |
| UNII | 4Y7YD32BYY |
| Chemical and physical data | |
| Formula | C51H58N8O7 |
| Molar mass | 895.074 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
//////////netanasvir, anax labs, APPROVALS 2025, CHINA 2025, antiviral, Dongweizhuo, HCV, Antaitavir Hasophate, HEC 74647 PA, 4Y7YD32BYY
Nelremagpran
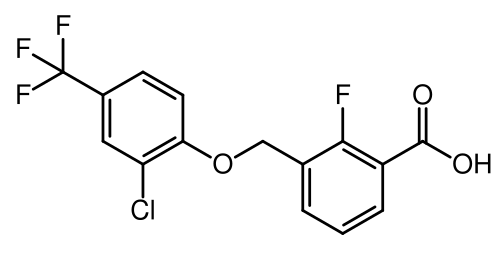
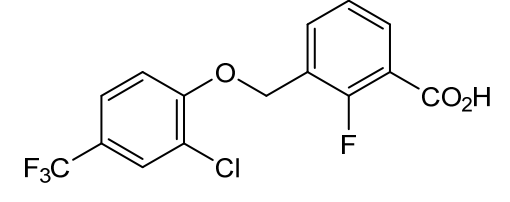

Nelremagpran
CAS 2492595-24-9
MFC15H9ClF4O3 MW 348.67 g/mol
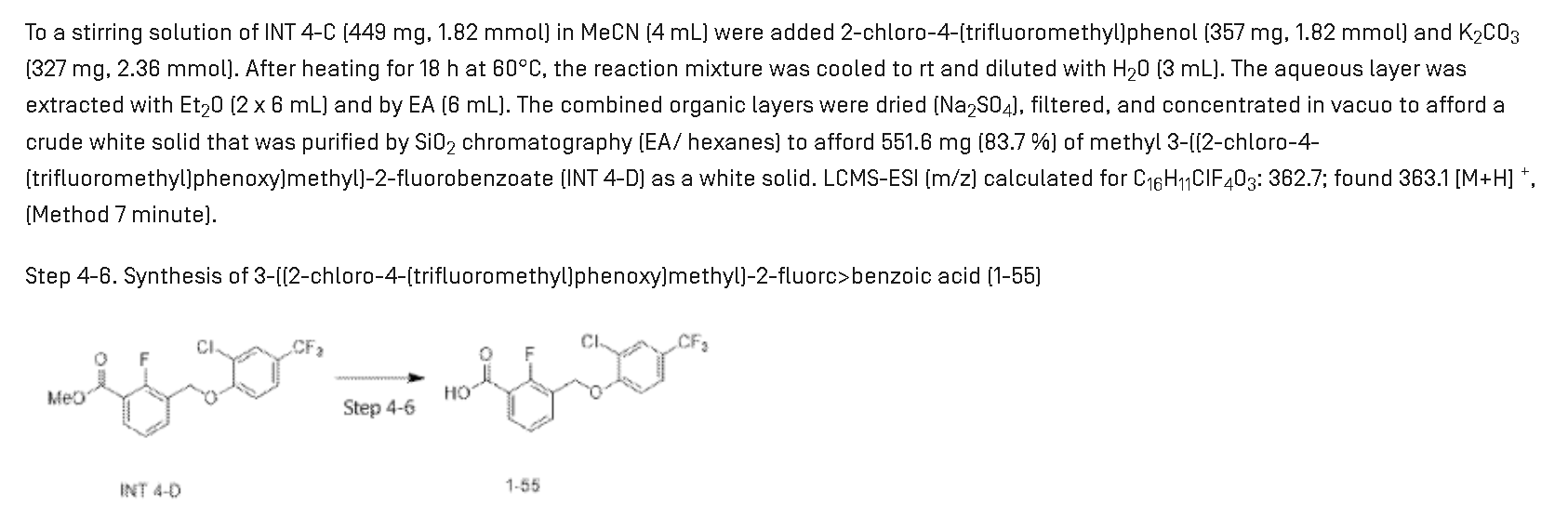
3-[[2-Chloro-4-(trifluoromethyl)phenoxy]methyl]-2-fluorobenzoic acid
3-{[2-chloro-4-(trifluoromethyl)phenoxy]methyl}-2-fluorobenzoic acid
Mas-related G protein-coupled receptor inverse agonist, anti-inflammatory, MRGPRX4 modulator-2, BL83FAK6DZ,
Nelremagpran is an experimental drug that acts as a potent and selective antagonist (or possibly inverse agonist) of the MAS-related Gq protein-coupled receptor X4 (MRGPRX4). It has antiinflammatory effects in animal studies. This receptor is poorly characterised but is thought to be involved in immune system function, and development of selective ligands is essential for researching its role in the body.[1][2]
Nelremagpran is an experimental drug that acts as a potent and selective antagonist or inverse agonist of the Mas-related G-protein-coupled receptor X4 (MRGPRX4). It was initially developed by Escient Pharmaceuticals, Inc. for its anti-inflammatory and anti-itch properties.
🧪 Core Mechanism
The drug targets MRGPRX4, a poorly characterized receptor found in the immune and nervous systems. It functions with high potency, exhibiting an half-maximal inhibitory concentration (IC50) of less than 100 nM.
🔬 Potential Research Applications
Because the receptor plays a significant role in mediating severe itch and pain pathways, Nelremagpran is actively used as a chemical probe to study:
- Chronic Pruritus: Conditions involving persistent, debilitating itch sensations, such as cholestatic pruritus.
- Autoimmune Diseases: Underlying mechanisms in conditions like psoriasis, multiple sclerosis, and Stevens-Johnson Syndrome.
- Inflammatory Sensation: How the body signals pain and immune hypersensitivity
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020198537&_cid=P12-MREAZN-03524-1

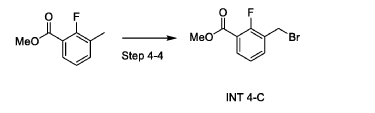
Synthesis of Compound 1-55

Step 4-4. Synthesis of methyl 3-(bromomethyl)-2-fluorc>benzoate (INT 4-C)



ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
 | |
| Identifiers | |
|---|---|
| IUPAC name | |
| CAS Number | 2492595-24-9 |
| PubChem CID | 155145968 |
| ChemSpider | 115008493 |
| UNII | BL83FAK6DZ |
| ChEMBL | ChEMBL4855031 |
| Chemical and physical data | |
| Formula | C15H9ClF4O3 |
| Molar mass | 348.68 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- WO 2020/198537, Yeager A, Selfridge B, Sainz M, Martinborough E, Boehm M, Huang, “Modulators of mas-related G-protein receptor x4 and related products and methods.”, published 1 October 2020, assigned to Escient Pharmaceuticals, Inc.
- “International Nonproprietary Names for Pharmaceutical Substances (INN)” (PDF). WHO Drug Information. 38 (4). 2024.
- Modulators of Mas-related G-protein receptor X4 and related products and methodsPublication Number: US-11840521-B2Priority Date: 2019-03-28Grant Date: 2023-12-12
- Modulators of mas-related g-protein receptor x4 and related products and methodsPublication Number: EP-3946304-A1Priority Date: 2019-03-28
- Modulators of mas-related g-protein receptor x4 and related products and methodsPublication Number: US-2023159474-A1Priority Date: 2019-03-28
- Modulators of mas-related g-protein receptor x4 and related products and methodsPublication Number: US-2023053860-A1Priority Date: 2019-03-28
- Modulators of mas-related g-protein receptor x4 and related products and methodsPublication Number: US-2021032213-A1Priority Date: 2019-03-28
- Modulators of mas-related G-protein receptor X4 and related products and methodsPublication Number: US-11643399-B2Priority Date: 2019-03-28Grant Date: 2023-05-09
- Modulators of mas-related g-protein receptor x4 and related products and methodsPublication Number: US-2024300907-A1Priority Date: 2019-03-28
- Modulators of mas-related g-protein receptor x4 and related products and methodsPublication Number: WO-2020198537-A1Priority Date: 2019-03-28
- Modulators of MAS-related G protein receptor X4 and related products and methodsPublication Number: CN-113939288-BPriority Date: 2019-03-28Grant Date: 2025-04-25
//////////nelremagpran, anax labs, Mas-related G protein-coupled receptor inverse agonist, anti-inflammatory, MRGPRX4 modulator-2, BL83FAK6DZ,
Nedizantrep
Nedizantrep
CAS 2376824-99-4
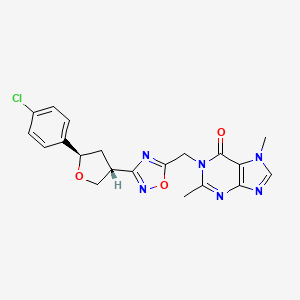
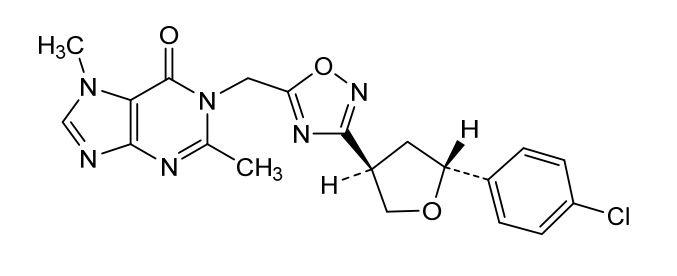
MF C20H19ClN6O3 MW 426.9 g/mol
- 1-[[3-[(3R,5R)-5-(4-chlorophenyl)oxolan-3-yl]-1,2,4-oxadiazol-5-yl]methyl]-2,7-dimethylpurin-6-one
- 1-[[3-[(3R,5R)-5-(4-Chlorophenyl)tetrahydro-3-furanyl]-1,2,4-oxadiazol-5-yl]methyl]-1,7-dihydro-2,7-dimethyl-6H-purin-6-one
- 6H-Purin-6-one, 1-[[3-[(3R,5R)-5-(4-chlorophenyl)tetrahydro-3-furanyl]-1,2,4-oxadiazol-5-yl]methyl]-1,7-dihydro-2,7-dimethyl-
1-[[3-[(3R,5R)-5-(4-chlorophenyl)oxolan-3-yl]-1,2,4-oxadiazol-5-yl]methyl]-2,7-dimethylpurin-6-one
1-({3-[(3R,5R)-5-(4-chlorophenyl)oxolan-3-yl]-1,2,4-oxadiazol-5-yl}methyl)-2,7-dimethyl-1,7-dihydro-6Hpurin-6-one
transient receptor potential (TRP) ion channel antagonist, GDC-6599, GDC 6599, RG 6341, RG-6341, ENQ95FVE4X
Nedizantrep (also known by its developmental code GDC-6599) is a potent, selective, and orally active chemical compound developed as a transient receptor potential ankyrin 1 (TRPA1) cation channel antagonist. It was originally designed by Roche Holding AG/Genentech to manage respiratory conditions, particularly chronic cough associated with asthma and Chronic Obstructive Pulmonary Disease (COPD).
Key Characteristics & Mechanism
- Target Engagement: It acts as a TRPA1 inhibitor with high potency, exhibiting an IC50 value of 5.3 nM in humans. [1, 2]
- Therapeutic Purpose: It blocks TRPA1 cation channels, which are thermoTRP channels known to function as sensor transducers for temperature, pain, and environmental irritants. Inhibiting this pathway prevents the sensory nerve hypersensitivity that triggers chronic coughing fits.
- Research Status: As per data recorded by platforms like the IUPHAR/BPS Guide to PHARMACOLOGY, its chemical structure was publically disclosed in late 2023, and it has been evaluated through Phase 2 clinical trials
- A Study To Evaluate The Efficacy, Safety, Pharmacokinetics, And Pharmacodynamic Effects Of GDC-6599 In Patients With Chronic Cough
- CTID: NCT05660850
- Phase: Phase 2
- Status: Completed
- Date: 2025-12-16
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US251637879&_cid=P12-MRBG82-53830-1
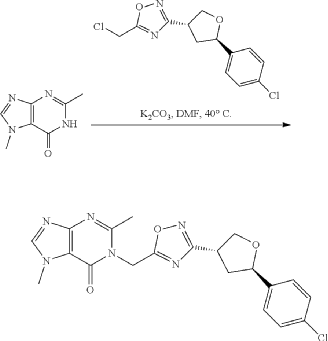
Example 8: 1-((3-((3R,5R)-5-(4-chlorophenyl)tetrahydrofuran-3-yl)-1,2,4-oxadiazol-5-yl)methyl)-2,7-dimethyl-1H-purin-6(7H)-one
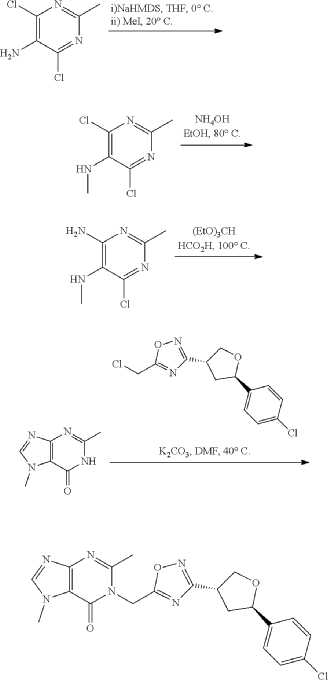
Step 4: Preparation of 1-((3-((3R,5R)-5-(4-chlorophenyl)tetrahydrofuran-3-yl)-1,2,4-oxadiazol-5-yl)methyl)-2,7-dimethyl-1H-purin-6(7H)-one
PAT
EG 8
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
Oxadiazole compounds as transient receptor potential channel inhibitors
Publication Number: EP-3768260-B1
Priority Date: 2018-03-19
Grant Date: 2025-10-08
- Oxadiazole transient receptor potential channel inhibitorsPublication Number: TW-202003510-APriority Date: 2018-03-19
- Oxadiazole transient receptor potential channel inhibitorsPublication Number: US-10710994-B2Priority Date: 2018-03-19Grant Date: 2020-07-14
- Oxadiazole transient receptor potential channel inhibitorsPublication Number: TW-I829676-BPriority Date: 2018-03-19Grant Date: 2024-01-21
- Oxadiazole transient receptor potential channel inhibitorsPublication Number: US-2025122184-A1Priority Date: 2018-03-19
- Oxadiazole transient receptor potential channel inhibitorsPublication Number: EP-3768260-A1Priority Date: 2018-03-19
- Oxadiazole transient receptor potential channel inhibitorsPublication Number: US-2019284179-A1Priority Date: 2018-03-19
- Oxadiazole transient receptor potential channel inhibitorsPublication Number: KR-20230070333-APriority Date: 2018-03-19
- Oxadiazole transient receptor potential channel inhibitorsPublication Number: US-12202824-B2Priority Date: 2018-03-19Grant Date: 2025-01-21
- Oxadiazole transient receptor potential channel inhibitorPublication Number: KR-102533094-B1Priority Date: 2018-03-19Grant Date: 2023-05-16
- Oxadiazole transient receptor potential channel inhibitorsPublication Number: TW-202438496-APriority Date: 2018-03-19
- Oxadiazole transient receptor potential channel inhibitorsPublication Number: US-2020308161-A1Priority Date: 2018-03-19
- Oxadiazole transient receptor potential channel inhibitorsPublication Number: US-2023121700-A1Priority Date: 2018-03-19
- Oxadiazole transient receptor potential channel inhibitorsPublication Number: WO-2019182925-A1Priority Date: 2018-03-19
- Oxadiazole transient receptor potential channel inhibitorsPublication Number: US-11655245-B2Priority Date: 2018-03-19Grant Date: 2023-05-23
- Oxadiazole transient receptor potential channel inhibitorsPublication Number: CN-111867585-APriority Date: 2018-03-19
////////////nedizantrep, anax labs, transient receptor potential (TRP) ion channel antagonist, GDC-6599, GDC 6599, RG 6341, RG-6341, ENQ95FVE4X
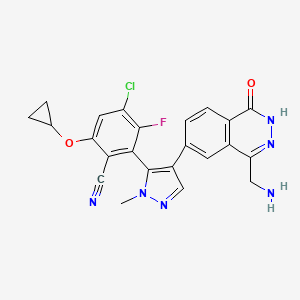
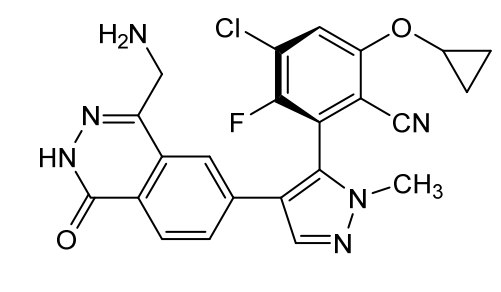
Navlimetostat
Navlimetostat
CAS 2630904-45-7
ALSO 2630904-44-6
MF C23H18ClFN6O2 MW464.9 g/mol
- Benzonitrile, 2-(4-(4-(aminomethyl)-1,2-dihydro-1-oxo-6-phthalazinyl)-1-methyl-1H-pyrazol-5-yl)-4-chloro-6-(cyclopropyloxy)-3-fluoro-, (2S)-
- (M)-27
- 2-[4-[4-(aminomethyl)-1-oxo-2H-phthalazin-6-yl]-2-methylpyrazol-3-yl]-4-chloro-6-cyclopropyloxy-3-fluorobenzonitrile
(2M)-2-{4-[4-(aminomethyl)-1-oxo-1,2-dihydrophthalazin-6-yl] -1-methyl-1H-pyrazol-5-yl}-4-chloro-6-(cyclopropyloxy)-3-fluorobenzonitrile
antineoplastic, MRTX-1719, BMS-986504, MRTX 1719, BMS 986504
Navlimetostat (also known as MRTX-1719 or BMS-986504) is an investigational, first-in-class oral targeted cancer therapy being developed by Bristol-Myers Squibb. It works by selectively binding to the PRMT5-MTA complex, exploiting synthetic lethality to kill cancer cells with MTAP gene deletions while sparing healthy cells.
Navlimetostat is currently in Phase 1/2 clinical trials for advanced solid tumors, including MTAP-deficient non-small cell lung cancer (NSCLC), pancreatic cancer, and glioblastoma.
Key highlights and ongoing research:
- Mechanism: In MTAP-deleted cancer cells, a metabolite called MTA accumulates and binds to PRMT5. Navlimetostat targets and inhibits this specific PRMT5-MTA complex, leading to tumor cell death.
- Clinical Trials: It is currently being investigated as a monotherapy (e.g., in MTAP-deleted advanced solid tumors) and in combination with other agents like pumitamig
- OriginatorMirati Therapeutics
- DeveloperBristol-Myers Squibb; Mirati Therapeutics
- ClassAntineoplastics; Small molecules
- Mechanism of ActionPRMT5 protein inhibitors
- Phase II/IIIAdenocarcinoma; Non-small cell lung cancer
- Phase I/IIMesothelioma; Neurilemmoma; Pancreatic cancer; Solid tumours
- 22 May 2026University of Southampton in collaboration with Bristol-Myers Squibb plans a phase II SELECTmeso1 trial for Malignant mesothelioma (Second-line therapy or greater) in United Kingdom in May 2026 (PO, Tablet) (NCT07602946)
- 13 May 2026Northwestern University plans a phase Ib/II trial for Solid tumours (Metastatic disease, Second-line therapy or greater, Combination therapy) in USA(PO) in December 2027 (NCT07594626)
- 12 May 2026M.D. Anderson Cancer Center plans a phase I/II trial for Non-small cell lung cancer (Combination therapy, Late-stage disease, Metastatic disease, Second-line therapy or greater) in USA (PO), in November 2026 (NCT07579221)
PRMT5 Inhibitor BMS-986504 is an orally bioavailable methylthioadenosine (MTA)-selective inhibitor of the protein arginine methyltransferase 5 (PRMT5), with potential antineoplastic activity. Upon oral administration, PRMT5 inhibitor BMS-986504 targets, binds to and inhibits PRMT5 that is bound to MTA, a complex that is elevated in methylthioadenosine phosphorylase (MTAP)-deleted cancer cells, thereby specifically inhibiting the function of PRMT5 solely within MTAP-deleted cancer cells and not in normal, healthy cells. By inhibiting the methyltransferase activity of PRMT5, levels of both monomethylated and dimethylated arginine residues in histones H2A, H3 and H4 are decreased. This modulates the expression of genes involved in several cellular processes, including cellular proliferation. This may increase the expression of antiproliferative genes and/or decrease the expression of genes that promote cell proliferation, which may lead to decreased growth of rapidly proliferating cancer cells. BMS-986504 also causes dysregulated RNA splicing and decreased retinoblastoma protein (pRb). Together, this decreases proliferation and increases apoptosis specifically in MTAP-deleted cancer cells. PRMT5, a type II methyltransferase that catalyzes the formation of both omega-N monomethylarginine (MMA) and symmetric dimethylarginine (sDMA) on histones and a variety of other protein substrates involved in signal transduction and cellular transcription, is essential for the viability of cancer and normal cells. It is overexpressed in several neoplasms. Elevated levels are associated with decreased patient survival. MTAP is deleted in certain cancer cells leading to an accumulation of the metabolite MTA; MTA binds to and partially inhibits the activity of PRMT5.
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021050915&_cid=P12-MR76CL-04796-1
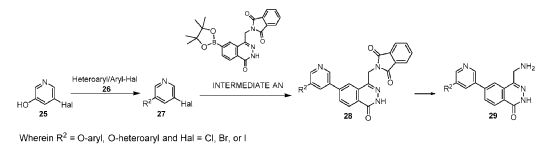
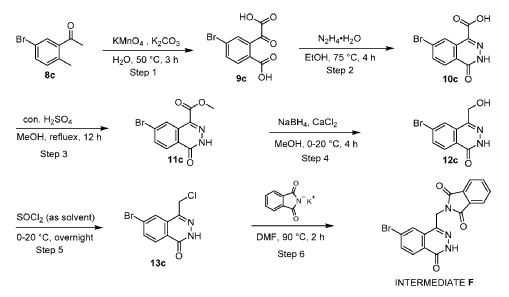
[0186] Step 6: To a solution of 6-bromo-4-(chloromethyl)-2H-phthalazin-1-one 13c (148 g, crude) in DMF (1.5 L) was added (1,3-dioxoisoindolin-2-yl)potassium (121 g, 653 mmol). The reaction mixture was stirred at 90 °C for 2 hours and then cooled to 25 °C. The formed precipitate was filtered and washed with DMF (200 mL x 2) and the filter cake triturated with water (1.00 L), filtered and dried to give Intermediate F, 2-[(7-bromo-4-oxo-3H-phthalazin-1-yl)methyl]isoindoline-1,3-dione (162 g, 413 mmol, 76% yield) as a white solid.1H NMR (400 MHz, DMSO-d6) d = 12.59 (s, 1H), 8.43 (d, J = 1.2 Hz, 1H), 8.18 (d, J = 8.4 Hz, 1H), 8.07 (dd, J = 1.6, 8.4 Hz, 1H), 7.97 – 7.93 (m, 2H), 7.92 – 7.86 (m, 2H), 5.19 (s, 2H). LCMS [M+1]: 383.9.
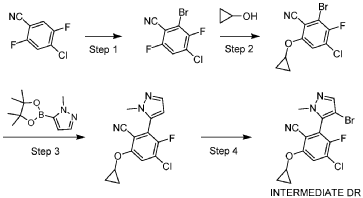
[0327] Step 4: A mixture of 4-chloro-6-(cyclopropoxy)-3-fluoro-2-(2-methylpyrazol-3-yl)benzonitrile (180 mg, 0.617 mmol, 1.00 eq) and N-bromosuccinimide (220 mg, 1.23 mmol, 2.00 eq.) in acetonitrile (10 mL) was stirred at 40 °C for 2 hours under a nitrogen atmosphere. After such time the mixture was concentrated and the residue was purified by prep-TLC (SiO2, petroleum ether: ethyl acetate 20%) to give 2-(4-bromo-2-methyl-pyrazol-3-yl)-4-chloro-6-(cyclopropoxy)-3-fluoro-benzonitrile (170 mg, 0.455 mmol, 74% yield) as a white solid. LCMS [M+1] + = 371.8; 1H NMR (400 MHz, CDCl3) d = 7.61 (s, 1H), 7.55 (d, J = 6.0 Hz, 1H), 3.93 – 3.85 (m, 1H), 3.80 (s, 4H), 0.97 – 0.94 (m, 4H).
EXAMPLE 16-7 and 16-8
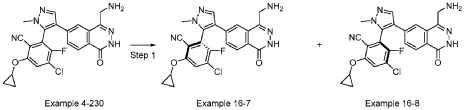
[0590] Example 4-230, 2-(4-(4-(aminomethyl)-1-oxo-1,2-dihydrophthalazin-6-yl)-1-methyl-1H-pyrazol-5-yl)-4-chloro-6-cyclopropoxy-3-fluorobenzonitrile (30 mg, 0.065 mmol) separated by SFC (DAICEL CHIRALPAK IC (250 mm × 30 mm x 10 mm); mobile phase:
[0.1% NH3H2O isopropanol]; B%: 40% isocratic, 4.1 min cycle; 120 min total ) to give example 16-7 (ee > 99%, 13 mg, 0.026 mmol, 25% yield) as a yellow solid and example 16-8 (8 mg, ee = 84% ). Example 16-8 was then then further separated by SFC (DAICEL CHIRALPAK IC (250 mm × 30 mm,10 mm); mobile phase: [0.1% NH3H2O EtOH]; B%: 60% isocratic, 3.1 min cycle; total 50 min) to give Example 16-8 (ee > 99%, 4 mg, 0.007 mmol, 7% yield) as a yellow gum. Spectra data for Example 16-7: LCMS [M+1] + = 465.1; 1H NMR (400 MHz, DMSO-d6) d = 12.59 – 12.44 (s, 1H), 8.29 (s, 1H), 8.15 (d, J = 8.4 Hz, 1H), 8.01 (d, J = 6.0 Hz, 1H), 7.75 (s, 1H), 7.67 (br d, J = 7.6 Hz, 1H), 4.23 – 4.17 (m, 1H), 3.86 (br s, 2H), 3.78 (s, 3H), 0.94 – 0.88 (m, 2H), 0.84 – 0.79 (m, 2H). Spectra data for Example 16-8: LCMS [M+1] + = 465.1; 1H NMR (400 MHz, DMSO-d6) d = 12.49 – 12.37 (s, 1H), 8.26 (s, 1H), 8.15 (d, J = 8.4 Hz, 1H), 8.00 (d, J = 6.0 Hz, 1H), 7.73 (d, J = 1.6 Hz, 1H), 7.72 – 7.68 (m, 1H), 4.19 (m, 1H), 3.80 (s, 2H), 3.77 (s, 3H), 0.93 – 0.87 (m, 2H), 0.83 – 0.78 (m, 2H).
PAT
- MTA-synergistic PRMT5 inhibitorsPublication Number: CN-114728912-APriority Date: 2019-09-12
- Mta-cooperative prmt5 inhibitorsPublication Number: WO-2021050915-A1Priority Date: 2019-09-12
- MTA-Cooperative PRMT5 InhibitorsPublication Number: US-2021078994-A1Priority Date: 2019-09-12
- MTA-Cooperative PRMT5 InhibitorsPublication Number: US-2021079003-A1Priority Date: 2019-09-12
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- Dynamic Kinetic Resolution of Axially Chiral MRTX1719Publication Name: SynfactsPublication Date: 2022-10-18DOI: 10.1055/s-0041-1738738
- Design and evaluation of achiral, non-atropisomeric 4-(aminomethyl)phthalazin-1(2H)-one derivatives as novel PRMT5/MTA inhibitorsPublication Name: Bioorganic & Medicinal ChemistryPublication Date: 2022-10-01PMID: 35926325DOI: 10.1016/j.bmc.2022.116947
- Synthesis of MRTX1719Publication Name: SynfactsPublication Date: 2022-03-18DOI: 10.1055/s-0041-1737934
- Fragment-Based Discovery of MRTX1719, a Synthetic Lethal Inhibitor of the PRMT5•MTA Complex for the Treatment of MTAP -Deleted CancersPublication Name: Journal of Medicinal ChemistryPublication Date: 2022-01-18PMID: 35041419DOI: 10.1021/acs.jmedchem.1c01900
- Disordered methionine metabolism in MTAP/CDKN2A-deleted cancers leads to dependence on PRMT5Publication Name: Science (New York, N.Y.)Publication Date: 2016-03-11PMID: 26912361DOI: 10.1126/science.aad5944
- Synthesis of MRTX1719DOI: 10.1055/s-0041-1737934Publication Date: 2022Publication Name: Synfacts
- Dynamic Kinetic Resolution of Axially Chiral MRTX1719DOI: 10.1055/s-0041-1738738Publication Date: 2022Publication Name: Synfacts
////////navlimetostat, anax labs, antineoplastic, MRTX-1719, BMS-986504, MRTX 1719, BMS 986504
Moxetomidate
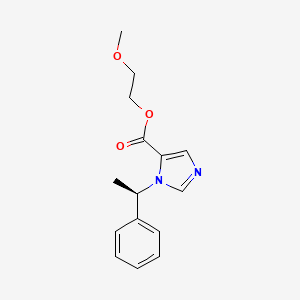
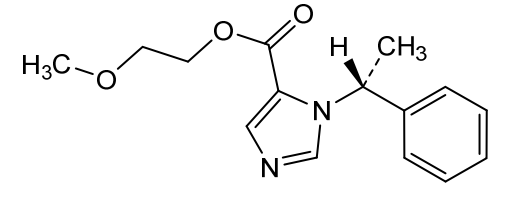
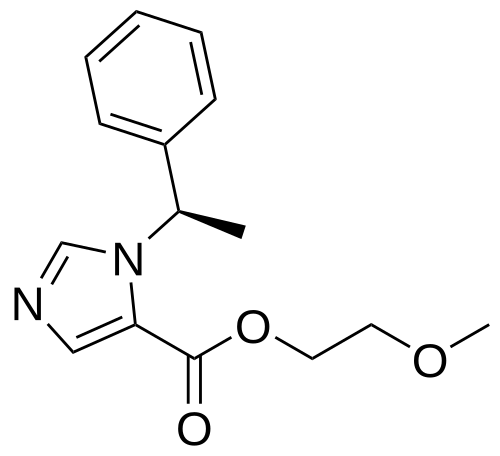
Moxetomidate
CAS 1567838-90-7
MF C15H18N2O3 MW 274.31 g/mol
- 2-Methoxyethyl 1-[(1R)-1-phenylethyl]-1H-imidazole-5-carboxylate
- 1H-Imidazole-5-carboxylic acid, 1-[(1R)-1-phenylethyl]-, 2-methoxyethyl ester
2-methoxyethyl 3-[(1R)-1-phenylethyl]imidazole-4-carboxylate
2-methoxyethyl 1-[(1R)-1-phenylethyl]-1H-imidazole-5-carboxylate
GABAA receptor agonist, hypnotic, ET-26, ET 26, LPQ2K767W2
Moxetomidate (also known as methoxyetomidate or ET-26) is a novel, investigational short-acting intravenous anesthetic and sedative-hypnotic agent. It functions as a{GABA}_A} receptor agonist and is designed as a “soft drug” analogue of the traditional anesthetic etomidate.
The Purpose of Its Development
Traditional etomidate is highly valued in clinical settings for its exceptional cardiovascular stability, making it the agent of choice for inducing anesthesia in patients with low blood pressure, trauma, or severe heart conditions. However, its primary drawback is that it causes prolonged adrenocortical suppression by inhibiting the enzyme 11β-hydroxylase. This limits its safety for continuous infusions or repeated uses.
Moxetomidate was synthesized to overcome this exact limitation. It retains the favorable, heart-safe properties of etomidate while undergoing rapid metabolic breakdown into an inactive compound. This prevents prolonged suppression of the adrenal gland.
Key Scientific Properties
- Mechanism of Action: It acts as a positive allosteric modulator and agonist at the \(GABA}_A}) receptor site, depressing the central nervous system to induce hypnosis and sedation.
- Chemical Profile: Its chemical formula is C₁₅H₁₈N₂O₃ with a molecular weight of 274.31 g/mol. Its IUPAC name is 2-methoxyethyl (R)-1-(1-phenylethyl)-1H-imidazole-5-carboxylate.
- Clinical Trial Status: Developed by entities including Jinzhou Ahon Pharmaceutical Co., Ltd., moxetomidate hydrochloride (ET-26 HCl) has progressed into Phase 3 clinical trials as an induction anesthetic.
Methoxyetomidate is an investigational anesthetic agent being developed by Jinzhou Ahon Pharmaceutical Co., Ltd. It is a short-acting intravenous anesthetic that acts as a positive allosteric modulator of GABAA receptors.[1][2] As of 2024, methoxyetomidate is undergoing Phase 3 clinical trials for use in anesthesia.[3][1]
PAT
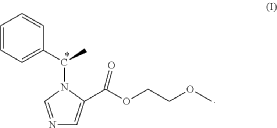
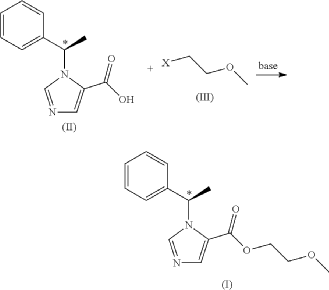
EG 1
Preparation of the Formula (I) Compound Guided by the Present Invention
| 1H-NMR (400 MHz CDCl 3) δ: 1.862 (3H, d, J=7.2 Hz), 3.393 (3H, s), 3.645 (3H, t, J=4.8 Hz), 4.31˜4.405 (2H, m), 6.348 (1H, q, J=7.2 Hz), 7.17˜7.359 (m, 5H), 7.742 (s, 1H), 7.827 (s, 1H). |
| 13C-NMR (100 MHz CDCl 3) δ: 22.30, 55.48, 59.15, 63.53, 70.48, 122.39, 126.36, 128.08, 128.93, 138.63, 140.02, 141.20, 160.26. |
PAT
- N-substituted imidazole carboxylic ester chiral compound containing an ether side chain, its preparation and applicationPublication Number: US-9969695-B2Priority Date: 2013-12-23Grant Date: 2018-05-15
- N-substituted imidazole carboxylic ester chiral compound containing ether side chain, preparation method and applicationPublication Number: EP-3088394-A1Priority Date: 2013-12-23
- N-Substituted Imidazole Carboxylic Ester Chiral Compound Containing an Ether Side Chain, Its Preparation and ApplicationPublication Number: US-2017001963-A1Priority Date: 2013-12-23
- N-substituted imidazole carboxylic ester chiral compound containing ether side chain, preparation method and applicationPublication Number: WO-2015096551-A1Priority Date: 2013-12-23
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
References
- “ET-26 Methoxyetomidate Hydrochloride Development Information”. Synapse by Patsnap. Retrieved 2024-11-23.
- Wang B, Chen S, Yang J, Yang L, Liu J, Zhang W (2017). “ET-26 hydrochloride (ET-26 HCl) has similar hemodynamic stability to that of etomidate in normal and uncontrolled hemorrhagic shock (UHS) rats”. PLOS ONE. 12 (8) e0183439. Bibcode:2017PLoSO..1283439W. doi:10.1371/journal.pone.0183439. PMC 5557577. PMID 28813523.
- “ET-26 Clinical Trials Overview”. Synapse by Patsnap. Retrieved 2024-11-23.
 | |
| Clinical data | |
|---|---|
| Other names | ET-26; Moxetomidate |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1567838-90-7 |
| PubChem CID | 74766803 |
| ChemSpider | 133326476 |
| UNII | LPQ2K767W2 |
| Chemical and physical data | |
| Formula | C15H18N2O3 |
| Molar mass | 274.320 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
//////////moxetomidate, anax labs, GABAA receptor agonist, hypnotic, ET-26, ET 26, LPQ2K767W2
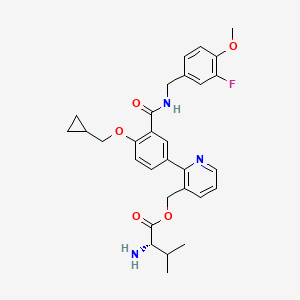
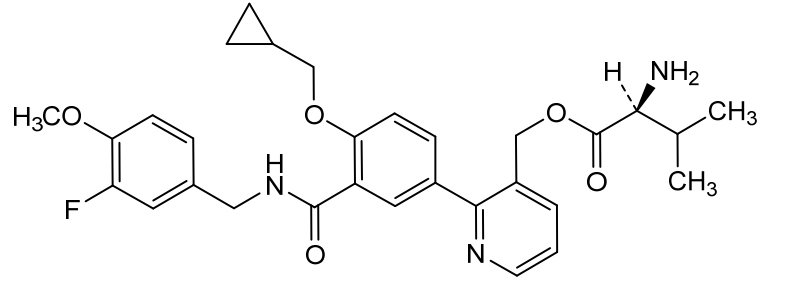
Mirivadelgat
Mirivadelgat
CAS 1804941-96-5
MF C30H34FN3O5 MW535.6 g/mol
[2-[4-(cyclopropylmethoxy)-3-[(3-fluoro-4-methoxyphenyl)methylcarbamoyl]phenyl]-3-pyridinyl]methyl (2S)-2-amino-3-methylbutanoate
L-Valine, [2-[4-(cyclopropylmethoxy)-3-[[[(3-fluoro-4-methoxyphenyl)methyl]amino]carbonyl]phenyl]-3-pyridinyl]methyl ester
{2-[4-(cyclopropylmethoxy)-3-{[(3-fluoro-4-methoxyphenyl)methyl]carbamoyl}phenyl]pyridin-3-yl}methyl
L-valinate
aldehyde dehydrogenase 2 activator, antianaemic, FP 045, 22TB7Q431D
Mirivadelgat (also known as FP-045) is a first-in-class, orally bioavailable small molecule that acts as a selective activator of mitochondrial aldehyde dehydrogenase 2 (ALDH2). Developed by Foresee Pharmaceuticals, this novel therapy helps cells clear toxic reactive aldehydes, reducing mitochondrial stress, cellular inflammation, and fibrosis. It is currently being evaluated in clinical trials for its disease-modifying potential across several serious conditions.
Current Clinical Development
The drug is undergoing clinical evaluation for multiple indications:
- Pulmonary Hypertension with Interstitial Lung Disease (PH-ILD): Mirivadelgat is currently in a multinational, Phase 2 trial known as the WINDWARD Study. The double-blind trial is assessing whether the drug can safely improve pulmonary vascular resistance and overall lung and cardiac function.
- Parkinson’s Disease: The drug was selected for the SLEIPNIR multi-drug Phase 2a platform trial funded by Cure Parkinson’s. It will evaluate mirivadelgat’s safety, brain penetration, and ability to clear the toxic waste products that drive Parkinson’s progression.
- Fanconi’s Anaemia: It has also reached Phase I/II testing for this rare genetic condition.
- Other Conditions: Early-stage Phase I research has explored its utility for metabolic and kidney disorders.
Mechanism of Action
When administered once daily, mirivadelgat targets deep cellular mechanics:
- ALDH2 Activation: It binds selectively to the ALDH2 enzyme in the mitochondria.
- Waste Clearance: It accelerates the metabolism of toxic reactive aldehydes produced by oxidative stress.
- Mitochondrial Protection: Lowering toxic aldehyde levels protects cellular structures and shields tissues from fibrosis and dysfunction.
Mirivadelgat is an orally bioavailable selective activator of the mitochondrial isoform of aldehyde dehydrogenase (ALDH2), with potential protective activity. Upon oral administration, mirivadelgat increases ALDH2 activity. This increases the metabolism of toxic reactive aldehydes and lowers the level of the toxic aldehydes. This may protect cells from the toxic aldehydes and prevent mitochondria diseases and disorders. ALDH2, a mitochondrial regulator of toxic aldehyde metabolism, plays an important role in the metabolism of toxic aldehydes. Toxic aldehydes are produced by oxidative stress.
Study to Evaluate Safety and Efficacy of Mirivadelgat in PH-ILD
CTID: NCT06475781, Phase: Phase 2, Status: Recruiting, Date: 2025-10-06
- OriginatorForesee Pharmaceuticals
- ClassAntianaemics; Antihypertensives; Cardiovascular therapies; Hepatoprotectants; Small molecules
- Mechanism of ActionAldehyde dehydrogenase 2 stimulants
- Phase IIPulmonary hypertension
- Phase I/IIFanconi’s anaemia
- Phase IKidney disorders; Metabolic disorders
- No development reportedCardiovascular disorders; Non-alcoholic fatty liver disease; Non-alcoholic steatohepatitis
- 08 Jun 2026Foresee Pharmaceuticals in collaboration with Haukeland University Hospital plans a phase IIa SLEIPNIR Platform trial for Parkinson’s disease (PO)
- 02 Jun 2026Helse Bergen HF, Cure Parkinson’s, Norwegian Parkinson’s Association plan the phase IIa SLEIPNIR-2 trial for Parkinson’s disease in Norway (PO, Capsule), in September 2026 , (CTIS2025-523570-17-00)
- 28 Oct 2025No recent reports of development identified for phase-I development in Cardiovascular-disorders in USA (PO)
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015127137&_cid=P20-MR2WCE-70172-1
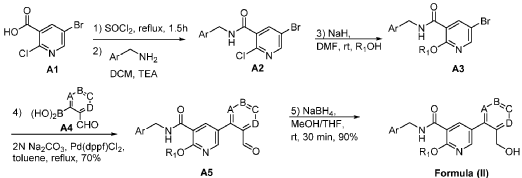
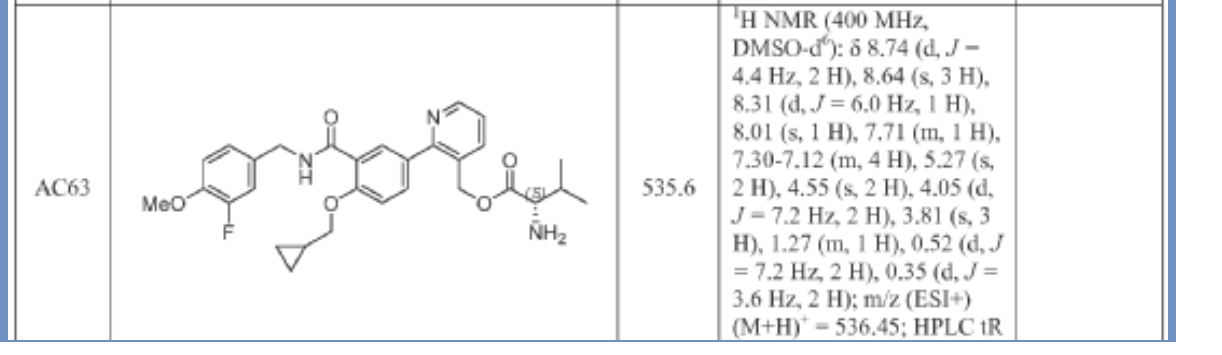
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- Mitochondrial aldehyde dehydrogenase-2 binding compounds and methods of use thereofPublication Number: US-9879036-B2Priority Date: 2014-02-19Grant Date: 2018-01-30
- Mitochondrial aldehyde dehydrogenase-2 binding compounds and methods of use thereofPublication Number: US-2017057982-A1Priority Date: 2014-02-19
- Mitochondrial aldehyde dehydrogenase 2 (aldh2) binding polycyclic amides and their use for the treatment of cancerPublication Number: EP-3107899-B1Priority Date: 2014-02-19Grant Date: 2020-08-12
//////////mirivadelgat, anax labs, aldehyde dehydrogenase 2 activator, antianaemic, FP 045, 22TB7Q431D

{kind=link}