
Home » Articles posted by DR ANTHONY MELVIN CRASTO Ph.D (Page 2)
Author Archives: DR ANTHONY MELVIN CRASTO Ph.D
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
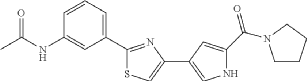
Milpecitinib
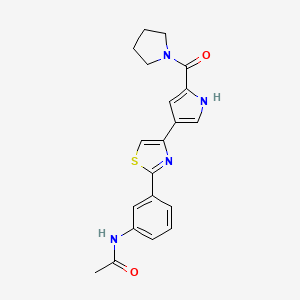
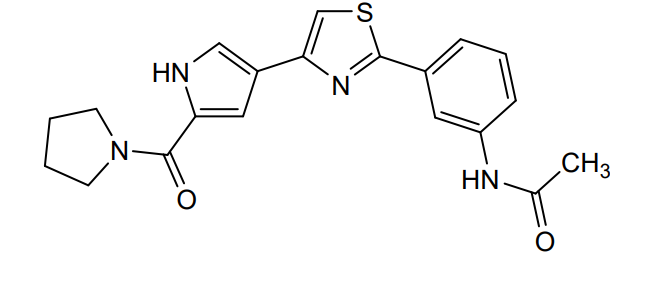
Milpecitinib
CAS 1415819-54-3
MF C20H20N4O2S MW380.5 g/mol
N-[3-[4-[5-(pyrrolidine-1-carbonyl)-1H-pyrrol-3-yl]-1,3-thiazol-2-yl]phenyl]acetamide
N-(3-{4-[5-(pyrrolidine-1-carbonyl)-1H-pyrrol-3-yl]-1,3-thiazol-2-yl}phenyl)acetamide
Janus tyrosine kinase inhibitor, anti-inflammatory, veterinary, PF-06263276, PF 06263276, Ph 1012, DNX 04013, CPh 1012, Ph-1012, CVXL 0074-02, 4Q8TT4B4GN
Milpecitinib is a small molecule drug. Milpecitinib has a monoisotopic molecular weight of 380.13 Da.
Milpecitinib (also known by its developmental codes PF-06263276, Ph-1012, and DNX-04013) is a potent, small-molecule Janus kinase (JAK) inhibitor used primarily in veterinary medicine and laboratory research. It functions as an ATP-competitive, broad-spectrum (pan-JAK) inhibitor that targets all four members of the JAK family: JAK1, JAK2, JAK3, and Tyrosine Kinase 2 (TYK2).
Primary Indication and Target
- Veterinary Use: Milpecitinib is designated for the control of pruritus (itching) associated with canine allergic dermatitis and the management of canine atopic dermatitis (CAD).
- Sponsorship: The United States Adopted Name (USAN) for this drug was officially adopted following sponsorship by Phibro Animal Health.
- Research Use: In laboratory settings, it is utilized to study complex inflammatory pathways, immune disorders, and certain cancers.
Mechanism of Action
Milpecitinib works by blocking the ATP-binding site of JAK enzymes. This inhibition halts the JAK-STAT signaling pathway, which plays a critical role in cellular responses to inflammatory cytokines. By blocking this cascade, the drug prevents the production and signaling of pro-inflammatory cytokines that cause severe itching and skin inflammation in dogs.
SYN
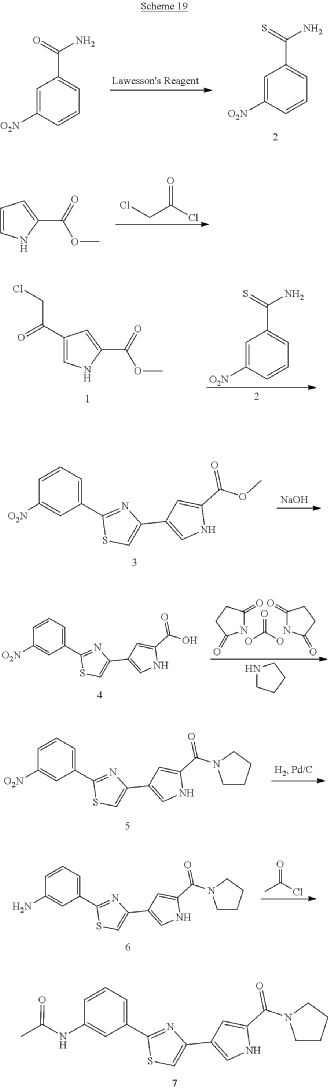
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma



AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

References
- Compositions and methods for modulating kinasesPublication Number: JP-6054379-B2Priority Date: 2011-06-07Grant Date: 2016-12-27
- Compositions and methods for modulating a kinasePublication Number: EP-2718290-B1Priority Date: 2011-06-07Grant Date: 2016-05-04
- Compositions and methods for modulating a kinasePublication Number: CA-2837268-CPriority Date: 2011-06-07Grant Date: 2020-05-12
- Compositions and Methods for Modulating a KinasePublication Number: US-2012316148-A1Priority Date: 2011-06-07
- Compositions and methods for modulating a kinasePublication Number: EP-2718290-A2Priority Date: 2011-06-07
- Compositions and methods for modulating a kinasePublication Number: WO-2012172438-A9Priority Date: 2011-06-07
- Compositions and methods for modulating a kinasePublication Number: US-8937065-B2Priority Date: 2011-06-07Grant Date: 2015-01-20
- Compositions and methods for modulating kinasesPublication Number: JP-2014520108-APriority Date: 2011-06-07
- Compositions and methods for modulating a kinasePublication Number: WO-2012172438-A2Priority Date: 2011-06-07
- Compositions and methods to modulate a kinasePublication Number: ES-2585244-T3Priority Date: 2011-06-07Grant Date: 2016-10-04
- Compound for modulating a kinase
- Publication Number: BR-112013031121-B1
- Priority Date: 2011-06-07
//////////milpecitinib, anax labs, Janus tyrosine kinase inhibitor, anti-inflammatory, veterinary, PF-06263276, PF 06263276, Ph 1012, DNX 04013, CPh 1012, Ph-1012, CVXL 0074-02, 4Q8TT4B4GN
Mazisotine



Mazisotine
CAS 1638588-92-7
MF C16H23N3O2 MW 289.37 g/mol
(1S,5R)-N-[2-methyl-1-[(3-methyl-2-pyridinyl)oxy]propan-2-yl]-3-azabicyclo[3.1.0]hexane-6-carboxamide
rac-(1R,5S,6R)-N-{2-methyl-1-[(3-methylpyridin-2-yl)oxy]propan-2-yl}-3-azabicyclo[3.1.0]hexane-6-carboxamide
(1R,5S,6r)-N-{2-methyl-1-[(3-methylpyridin-2-yl)oxy]propan-2-yl}-3-azabicyclo[3.1.0]hexane-6-carboxamide
somatostatin receptor receptor agonist, LY3556050, CNTX-0290, LY 3556050, CNTX 0290, D3M32WP3MH
Mazisotine (also known as LY3556050 or CNTX-0290) is an experimental, non-opioid chemical compound designed to treat chronic and neuropathic pain. It functions as a selective somatostatin receptor 4 (SSTR4) agonist, meaning it activates specific peripheral nerve pathways to block pain signals without activating the central nervous system’s opioid receptors.
While it showed early promise in animal models, Eli Lilly and Company removed mazisotine from its clinical development pipeline after disappointing results in Phase II clinical trials. It currently remains in use strictly as a chemical tool for laboratory pain research.
Key Facts and Clinical History
- Mechanism of Action: It binds to and activates SSTR4. This triggers a cellular response that suppresses pain and inflammation in peripheral sensory neurons.
- Intended Indications: It was being evaluated to treat diabetic peripheral neuropathic pain, osteoarthritis pain, and chronic low back pain.
- Development Partners: The compound was originally licensed by Eli Lilly from Centrexion Therapeutics in 2019 for an upfront fee of $47.5 million.
- Discontinuation: In mid-2025, Eli Lilly officially dropped the drug. Phase II clinical trials revealed that its efficacy did not meet the high success thresholds required to continue human testing.
- Side Effects: In clinical studies, reported treatment-emergent adverse effects were generally mild to moderate. They included constipation, nausea, dizziness, fatigue, and headaches.
Mazisotine (LY3556050, CNTX-0290) is a chemical compound which acts as an agonist at somatostatin receptor 4. It has analgesic effects and has been researched for the treatment of pain associated with arthritis and neuropathic pain. It was not pursued for human medical use following disappointing results in Phase II clinical trials, but continues to be used in research into the role of SST4 receptors in pain perception.[1][2]
- A Study of LY3556050 in Adult Participants With Diabetic Peripheral Neuropathic PainCTID: NCT06074562Phase: Phase 2Status: CompletedDate: 2025-07-03
- Chronic Pain Master Protocol (CPMP): A Study of LY3556050 in Participants With OsteoarthritisCTID: NCT04627038Phase: Phase 2Status: CompletedDate: 2023-11-02
- Chronic Pain Master Protocol (CPMP): A Study of LY3556050 in Participants With Diabetic Peripheral Neuropathic PainCTID: NCT04707157Phase: Phase 2Status: TerminatedDate: 2023-11-02
- Chronic Pain Master Protocol (CPMP): A Study of LY3556050 in Participants With Chronic Low Back PainCTID: NCT04874636Phase: Phase 2Status: CompletedDate: 2023-11-02
- A Study of Effect of LY3556050 on Metformin in Healthy ParticipantsCTID: NCT05615467Phase: Phase 1Status: CompletedDate: 2023-01-18
- A Study of Single and Repeated Doses of LY3556050 in Healthy ParticipantsCTID: NCT05341102Phase: Phase 1Status: CompletedDate: 2022-04-22
- A Study of LY3556050 in Healthy ParticipantsCTID: NCT04156750Phase: Phase 1Status: CompletedDate: 2020-08-19
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2025096300&_cid=P12-MQYLW5-94119-1

SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US214328185&_cid=P12-MQYLW3-94084-1
PAT

reparation 1
[0081] Methyl (E)-4-((4-methylphenyl)sulfonamido)but-2-enoate

[0082] Methyl 4-bromobut-2-enoate (36.29g, 202.7mmol) was dissolved in MeCN (500 mL) at 15-25 ºC. tert-Butyl tosylcarbamate (50.00 g, 184.3 mmol) was added at 15-25 ºC. K2CO3 (30.57 g, 221.2mmol) and KI (3.06 g, 202.7 mmol) were added to the solution at 15-25 ºC, and warmed under nitrogen at 30 ºC for 20 hrs. The solution was cooled to 20 ºC and the mixture filtered. The filtered residue was washed with MeCN (100 mL) to give methyl (E)-4-((N-(tert-butoxycarbonyl)-4-methylphenyl)sulfonamido)but-2-enoate. TFA (101.03 g, 886.06 mmol) was added to the methyl (E)-4-((N-(tert-butoxycarbonyl)-4-methylphenyl)sulfonamido)but-2-enoate in MeCN solution (483.72 g, 143 mmol) and heated to 55-60 ºC for 16 hrs. The reaction solution was concentrated in vacuo to ~50 mL and solvent exchanged with toluene (2 x 250 mL). Toluene (500 mL) was added followed by EtOAc (50 mL) at 15-25 °C, and heated to 60 ºC for 1 hr., then cooled to 0 ºC for 12 hrs. The solution was filtered, and the wet cake was rinsed with n-heptane (50 mL). The cake was dried in vacuum at 50 ºC to give the title compound (37.85 g, 74.4%) as a white solid.1H NMR (CDCl3) δ 7.68 (d, J = 8.0Hz, 2H) 7.25 (d, J = 8.0Hz, 2H) 6.71 (dt, J = 15.6, 5.2Hz, 1H) 5.88 (dt, J = 15.6, 1.6Hz, 1H) 4.55 (t, J = 6.4Hz, 1H) 3.71 – 3.67 (m, 2H) 3.65 (s, 3H) 2.37 (s, 3H); HRMS (ESI+) Calculated for [C12H15NO4S+H] +: 270.0795, Found: 270.0788 (M+H).
Preparation 2
[0083] (1R,5S,6r)-3-Tosyl-3-azabicyclo[3.1.0]hexane-6-carboxylic acid

0084] Methyl (E)-4-((4- 2-enoate (51.90 g) was dissolved in 2-MeTHF (600mL) at 0 ºC . Added (2-bromoethyl)diphenylsulfonium triflate (36.50 g), KF (6.47 g), KOH (18.75 g) to the solution at 0 ºC. Warmed the solution to 15 ºC for 22 hrs. then 30 ºC for 3 hrs. Added water (100 mL) and MeOH (100 mL) into the solution. Added LiOH.H2O (4.77 g) and stirred at 30 ºC for 16 hrs. Cooled to 15-25oC and added n-heptane (100 mL). Stirred at 15-25oC for 10 min. Separated and collected the aqueous phase and washed the aqueous phase with n-heptane/2-MeTHF (50 mL/200 mL × 2). Concentrated the aqueous phase in vacuo to ~50 mL and added 3M aq. HCl
dropwise to adjust pH to 1~2. Stirred the mixture at 20-30 ºC for 2 hrs. Filtered the solution and rinsed through with EtOH/H2O (15 mL1:4). Dried the wet cake at 45 ºC for 8-10 hrs. to give the title compound as white solid (20.37 g, 65%) 1H NMR (CDCl3) δ 7.67 (d, J = 8.2 Hz, 2 H) 7.34 (d, J = 8.2 Hz, 2 H) 3.63 (d, J = 9.4 Hz, 2 H) 3.12 (d, J = 9.4 Hz, 2 H) 2.46 – 2.40 (m, 4 H) 2.07 – 2.01 (m, 2 H); HRMS (ESI+) Calcd. for [C13H15NO4S+H] +: 282.0795, Found: 282.0795 (M+H).
Preparation 3
[0085] 2-Methyl-1-((3-methylpyridin-2-yl)oxy)propan-2-amine

0086] 2-Amino-2-methylpropan- mmol) was dissolved in toluene (250 mL) at 15-25 ºC. Added potassium tert-butoxide (60.59 g, 534.6 mmol) to the solution and warmed to 30-40 ºC. Added 2-fluoro-3-methylpyridine (50.00 g, 450.0 mmol) to the solution and warmed to 80 ºC. Stirred the mixture for 18 hrs. at 80 ºC. Cooled the mixture to 15-25 ºC, added water (100 mL) and stirred for 30 min. Separated the aqueous layer and washed the organics with 10% aq. NaCl (300 mL x 3). Added toluene (250 mL) to the organics and concentrated in vacuo to give the title compound as a liquid (107.50 g, 98%). 1H NMR (CDCl3) δ 7.92 – 7.82 (m, 1H) 7.32 – 7.22 (m, 1H) 6.74 – 6.66 (m, 1H) 3.97 (s, 3H) 2.14 (s, 3H) 1.15 (s, 6H).
Preparation 4
[0087] (1R,5S,6r)-N-(2-Methyl-1-((3-methylpyridin-2-yl)oxy)propan-2-yl)-3-tosyl-3-azabicyclo[3.1.0]hexane-6-carboxamide

[0088] Dissolved
6-carboxylic acid (32.33 g, 88.2% purity) in toluene (320 mL) at 15~25 ºC. Added DMF (741.0 mg) and (COCl)2 (19.20 g) to the solution and heated to 45-55 ºC for 3~4 hrs. Concentrated the mixture in vacuo and exchanged with THF (100 mL × 2). Added THF (320 mL) and cooled to 0-10 ºC. Added 2-methyl-1-((3-methylpyridin-2-yl)oxy)propan-2-amine (23.60 g), TEA (30.80 g), DMAP (620.0 mg) at 0-10 ºC then warmed to 15-25 ºC for 2~4 hrs.
Concentrated the mixture in vacuo and solvent exchanged with EtOH (140 mL × 2). Concentrated in vacuo to 140 mL and heated to 50 ºC until the solid was dissolved. Added water (210 mL) dropwise into the solution at 40 ºC. Cooled the solution to 10 ºC for 14 hrs. Filtered and rinsed with EtOH/H2O (75 mL, 1:1.5). Dried the wet cake at 45 ºC for 20 hrs. to give the title compound as a solid (41.87 g, 87.4%).1H NMR (CDCl3) δ 7.94 – 7.87 (m, 1 H) 7.60 (d, J=8.0 Hz, 2 H) 7.37 (d, J=6.6 Hz, 1 H) 7.26 (d, J=8.0 Hz, 2 H) 6.78 (dd, J=6.6, 5.0 Hz, 1 H) 6.48 (s, 1 H) 4.25 (s, 2 H) 3.52 (d, J=9.4 Hz, 2 H) 2.95 (d, J=9.4 Hz, 2 H) 2.39 – 2.33 (m, 3 H) 2.17 (s, 3 H) 1.84 (s, 2 H) 1.39 (s, 6 H); HRMS (ESI+) Calcd. for [C23H29N3O4S+H] +: 444.1952, Found: 444.2089 (M+H).
Preparation 5
[0089] (1R,5S,6r)-N-(2-Methyl-1-((3-methylpyridin-2-yl)oxy)propan-2-yl)-3-azabicyclo[3.1.0]hexane-6-carboxamide

[0090] Dissolved 2-yl)oxy)propan-2-yl)-3-tosyl-3-azabicyclo[3.1.0]hexane-6-carboxamide (5.00 g) in MTBE (50 mL) under nitrogen. Cooled to -70-60 ºC and added dropwise 1M Ph2PK in THF (56 mL, 56 mmol) into the solution. Stirred the mixture for 6-8 hrs. at -70-60 ºC. Added 2M aq. HCl (50 mL) to the solution allowing the temperature to rise to 15-25 ºC. Separated and collected the aqueous phase and washed with 2-MeTHF (50 mL × 3). Added 2-MeTHF (50 mL) and adjusted the pH to 8~9 with K2CO3 powder. Separated and extracted the aqueous phase with 2-MeTHF (50 mL). Combined the organics and concentrated in vacuo to give the title compound as a yellow-brown solid (3.05 g, 89.4%) 1H NMR (CDCl3) δ 8.01 – 7.92 (m, 1 H) 7.45 – 7.36 (m, 1 H) 6.81 (dd, J=7.0, 5.0 Hz, 1 H) 6.39 (s, 1 H) 4.31 (s, 2 H) 3.09 – 2.89 (m, 4 H) 2.21 (s, 3 H) 1.94 – 1.86 (m, 2 H) 1.69 (s, 1 H) 1.47 (s, 6 H) 1.12 (t, J=2.8 Hz, 1 H); HRMS (ESI+) Calcd. for [C16H23N3O2+H] +: 290.1863, Found:
290.1917 (M+H).
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
References
- Stevens R, Corradini L, Doods H (April 2019). “Preclinical Evaluation of Human Somatostatin Receptor 4 (hSSTR4) Agonist CNTX-0290 for Mixed Pain Conditions”. The Journal of Pain. 20 (4): S73. doi:10.1016/j.jpain.2019.02.092.
- Nguyen TH, Saito T, Chang W, Navarro A, Davies HM (March 2026). “Diastereoselective Cyclopropanation with Secondary Diazoacetamides to Access endo-Azabicyclo[3.1.0]hexane-6-carboxamides”. Organic Letters. 28 (9): 3063–3067. doi:10.1021/acs.orglett.6c00392. PMC 12973298. PMID 41729728.
 | |
| Clinical data | |
|---|---|
| Other names | LY3556050, CNTX-0290 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1638588-92-7 |
| PubChem CID | 86294067 |
| ChemSpider | 77005706 |
| UNII | D3M32WP3MH |
| ChEMBL | ChEMBL5874446 |
| Chemical and physical data | |
| Formula | C16H23N3O2 |
| Molar mass | 289.379 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
PAT
- Novel methods for the preparation of 3-azabicylco[3.1,0]hexane-6-carboxamide derivativesPublication Number: WO-2025096300-A1Priority Date: 2023-10-30
- Methods for the preparation and dose regimens for use of sstr4 agonists and salts thereofPublication Number: WO-2024191686-A1Priority Date: 2023-03-10
- Methods for the preparation and dose regimens for use of sstr4 agonists and salts thereofPublication Number: US-2024307349-A1Priority Date: 2023-03-10
- Methods for preparation and dose regimens for use of sstr4 agonists and salts thereofPublication Number: JP-2024170613-APriority Date: 2023-03-10
- Methods for the preparation of SSTR4 agonists and their salts and dosage regimens for usePublication Number: JP-7569953-B2Priority Date: 2023-03-10Grant Date: 2024-10-18
- Sstr4 agonist saltsPublication Number: US-2024067628-A1Priority Date: 2021-09-14
- SSTR4 agonist saltsPublication Number: US-11834435-B2Priority Date: 2021-09-14Grant Date: 2023-12-05
- New somatostatin receptor subtype 4 (sstr4) agonistsPublication Number: EP-2997021-B1Priority Date: 2013-05-17Grant Date: 2017-12-20
- Somatostatin receptor subtype 4 (SSTR4) agonistsPublication Number: US-9371282-B2Priority Date: 2013-05-17Grant Date: 2016-06-21
- Somatostatin receptor subtype 4 (SSTR4) agonistsPublication Number: US-10166214-B2Priority Date: 2013-05-17Grant Date: 2019-01-01
- New somatostatin receptor subtype 4 (sstr4) agonistsPublication Number: US-2019269650-A1Priority Date: 2013-05-17
- New somatostatin receptor subtype 4 (sstr4) agonistsPublication Number: WO-2014184275-A1Priority Date: 2013-05-17
- Somatostatin receptor subtype 4 (SSTR4) agonistsPublication Number: US-10675268-B2Priority Date: 2013-05-17Grant Date: 2020-06-09
- New Somatostatin receptor subtype 4 (SSTR4) agonistsPublication Number: US-2014343065-A1Priority Date: 2013-05-17
- Somatostatin receptor subtype 4 (SSTR4) agonistsPublication Number: US-9789082-B2Priority Date: 2013-05-17Grant Date: 2017-10-17
- New somatostatin receptor subtype 4 (sstr4) agonistsPublication Number: US-2017014381-A1Priority Date: 2013-05-17
- Somatostatin receptor subtype 4 (sstr4) agonistsPublication Number: US-2018092880-A1Priority Date: 2013-05-17
////////mazisotine, ANAX LABS, somatostatin receptor receptor agonist, LY3556050, CNTX-0290, LY 3556050, CNTX 0290, D3M32WP3MH, PAIN, NEUROPATHIC PAIN
Lonitoclax
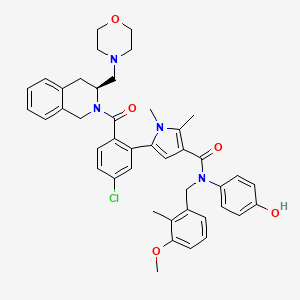
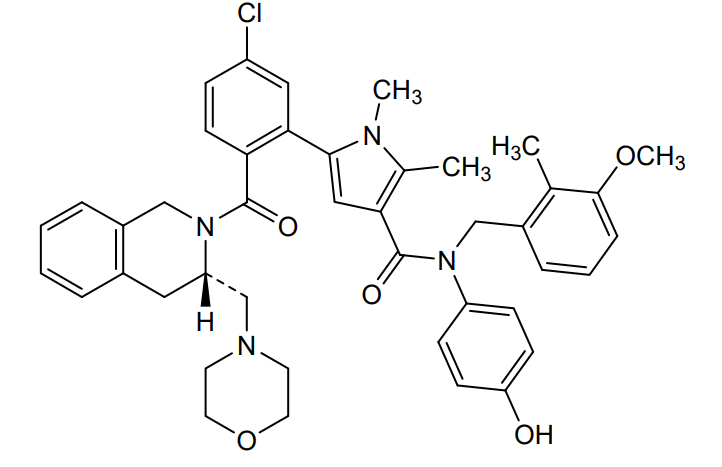
Lonitoclax
CAS 2952589-57-8
MF C43H45ClN4O5 MW733.3 g/mol
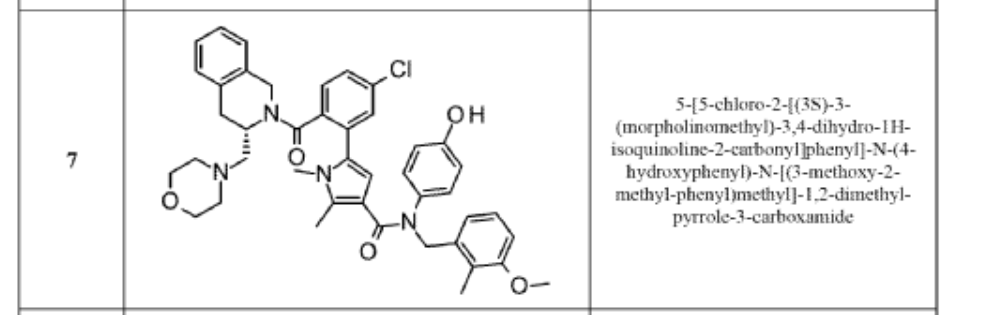
5-[5-chloro-2-[(3S)-3-(morpholin-4-ylmethyl)-3,4-dihydro-1H-isoquinoline-2-carbonyl]phenyl]-N-(4-hydroxyphenyl)-N-[(3-methoxy-2-methylphenyl)methyl]-1,2-dimethylpyrrole-3-carboxamide
- 1H-Pyrrole-3-carboxamide, 5-[5-chloro-2-[[(3S)-3,4-dihydro-3-(4-morpholinylmethyl)-2(1H)-isoquinolinyl]carbonyl]phenyl]-N-(4-hydroxyphenyl)-N-[(3-methoxy-2-methylphenyl)methyl]-1,2-dimethyl-
- 5-(5-Chloro-2-(((3S)-3-(morpholin-4-ylmethyl)-3,4-dihydroisoquinolin-2-(1-H)-yl)carbonyl)phenyl)-N-4-hydroxyphenyl)-N-(3-methoxy-2-methylbenzyl)-1,2-dimethyl-1H-pyrrole-3-carboxamide
- 5-[5-chloro-2-[(3S)-3-(morpholinomethyl)- 3,4-dihydro-1H-isoquinoline-2- carbonyl]phenyl]-N-(4-hydroxyphenyl)-N- [(3-methoxy-2-methyl-phenyl)methyl]-1,2- dimethyl-pyrrole-3-carboxamide
- 5-{5-Chloro-2-[(3S)-3-[(morpholin-4-yl)methyl]-3,4-dihydroisoquinoline-2(1H)-carbonyl]phenyl}-N-(4-hydroxyphenyl)-N-[(3-methoxy-2-methylphenyl)methyl]-1,2-dimethyl-1H-pyrrole-3-carboxamide
5-(5-chloro-2-{(3S)-3-[(morpholin-4-yl)methyl]-3,4-dihydroisoquinoline-2(1H)-carbonyl}phenyl)-N-(4-
hydroxyphenyl)-N-[(3-methoxy-2-methylphenyl)methyl]-1,2-dimethyl-1H-pyrrole-3-carboxamide
B-cell lymphoma 2 (Bcl-2) inhibitor, antineoplastic, ZE50-0134, ZE50 0134, Lomond Therapeutics, CANCER, 76NBC3X6A3
Lonitoclax (also known as ZE50-0134) is an investigational, next-generation, orally administered B-cell lymphoma 2 (Bcl-2) inhibitor being developed for the treatment of hematologic malignancies like Acute Myeloid Leukemia (AML) and Chronic Lymphocytic Leukemia (CLL). Developed by Lomond Therapeutics, the drug is engineered as a highly selective option to improve upon existing first-generation Bcl-2 inhibitors like venetoclax.
Mechanism and Advantages Over Venetoclax
Unlike earlier therapies, lonitoclax features a unique binding mode and a structurally distinct chemotype. Its design yields several pharmacology advantages:
- Higher Selectivity: It binds tightly to Bcl-2 while demonstrating exceptional selectivity over Bcl-xL, which helps lower hematologic toxicities.
- Limited Immune Suppression: In preclinical data, lonitoclax spared healthy non-malignant immune cells (B cells, CD8 T cells, and NK cells), a major shift from the immunosuppressive profile of venetoclax.
- Reduced Drug Interaction & Accumulation: It features a shorter half-life (~9–10 hours) and minimal CYP3A4 (P4503A4) inhibition. This prevents the drug from building up dangerously and mitigates the risk of Tumor Lysis Syndrome (TLS), potentially enabling safer outpatient treatments.
Clinical Development Status
Lonitoclax is currently advancing through early-phase clinical trials:
- IND Clearances: The U.S. FDA cleared Investigational New Drug (IND) applications evaluating lonitoclax for CLL/SLL and as a combination treatment for relapsed or refractory AML.
- Healthy Volunteer Studies: Phase 1 single ascending dose (SAD) studies in healthy adults confirmed that the drug is well tolerated with linear pharmacokinetics and no significant safety issues. Target engagement was confirmed through plasma apoptosis assays.
- Combination Trials: Active Phase 1b multicenter trials are underway evaluating the safety, efficacy, and synergy of lonitoclax when combined with hypomethylating agents like azacitidine in AML patients.
SYN
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023129553&_cid=P11-MQVQMH-93381-1
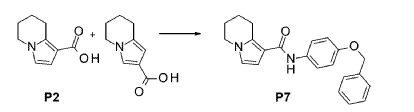
ADVERISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- Bcl-2 inhibitorsPublication Number: WO-2023129553-A1Priority Date: 2021-12-29
- BCL-2 InhibitorsPublication Number: US-2025115577-A1Priority Date: 2021-12-29
- Bcl-2 inhibitorsPublication Number: EP-4457223-A1Priority Date: 2021-12-29
///////Lonitoclax, ANAX LABS, B-cell lymphoma 2 (Bcl-2) inhibitor, antineoplastic, ZE50-0134, ZE50 0134, Lomond Therapeutics, CANCER, 76NBC3X6A3
Lomonitinib
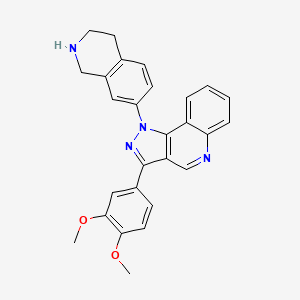
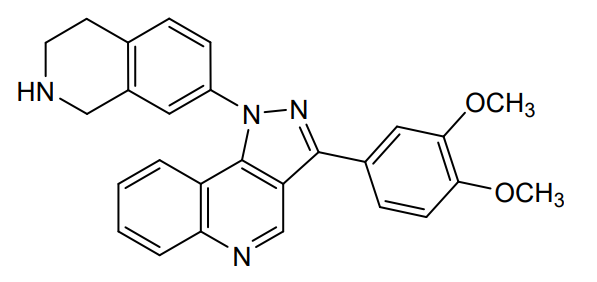
Lomonitinib
CAS 2923221-56-9
MF C27H24N4O2 MW436.5 g/mol
3-(3,4-dimethoxyphenyl)-1-(1,2,3,4-tetrahydroisoquinolin-7-yl)pyrazolo[4,5-c]quinoline
- 3-(3,4-dimethoxyphenyl)-1-(1,2,3,4-tetrahydroisoquinolin-7-yl)pyrazolo[4,3-c]quinoline
- 1H-Pyrazolo[4,3-c]quinoline, 3-(3,4-dimethoxyphenyl)-1-(1,2,3,4-tetrahydro-7-isoquinolinyl)-
- 3-(3,4-dimethoxyphenyl)-1-(1,2,3,4-tetrahydroisoquinolin-7-yl)-1H-pyrazolo[4,3-c]quinoline
- 7-[3-(3,4-dimethoxyphenyl)-1H- pyrazolo[4,3-c]quinolin-1-yl]-1,2,3,4- tetrahydroisoquinoline
- 7-[3-(3,4-Dimethoxyphenyl)-1H-pyrazolo[4,3-c]quinolin-1-yl]-1,2,3,4-tetrahydroisoquinoline
3-(3,4-dimethoxyphenyl)-1-(1,2,3,4-tetrahydroisoquinolin-7-yl)-1H-pyrazolo[4,3-c]quinoline
tyrosine kinase inhibitor, antineoplastic, ZE46-0134, Eilean Therapeutics, U4DPU7W7QU
Lomonitinib (also known as ZE46-0134) is a highly potent, selective, orally bioavailable pan-FLT3 and IRAK4 small molecule inhibitor being developed for the treatment of Acute Myeloid Leukemia (AML). Developed by Eilean Therapeutics in collaboration with Expert Systems, it uniquely targets both primary mutations and the major drug-resistance pathways that cause other AML therapies to fail.
Mechanism of Action
Lomonitinib utilizes a dual-targeting framework to bypass conventional drug resistance:
- Pan-FLT3 Inhibition: It binds to and blocks FMS-like tyrosine kinase 3 (FLT3) mutations. This includes the challenging FLT3-ITD-F691L “gatekeeper” mutation, which typically confers resistance to all currently approved standard FLT3 inhibitors like gilteritinib.
- IRAK4 Inhibition: It simultaneously targets interleukin-1 receptor-associated kinase 4 (IRAK4). IRAK4 activation acts as a key “escape pathway” that cancer cells use to survive and build adaptive resistance to standalone FLT3 therapy.
Key Clinical Advantages
According to preclinical models and clinical data presented at the American Society of Hematology (ASH), lomonitinib offers unique benefits:
- Superior Efficacy: In vivo models demonstrate stronger anti-tumor activity and deeper responses in gatekeeper mutation-dependent disease compared to gilteritinib.
- Favorable Loading Strategy: Because of its wide therapeutic index and low toxicity, clinicians can administer a high loading dose on Day 1 followed by a smaller maintenance dose. This achieves effective therapeutic drug levels by Day 4, a rapid target engagement not possible with older long-half-life FLT3 inhibitors.
- Low Drug Interactions: Clinical profiles show minimal pharmacokinetic interference from proton pump inhibitors (PPIs) or CYP3A4 inhibitors like itraconazole.
Development Status
Lomonitinib is currently classified as an investigational new drug:
- Clinical Trials: It is undergoing open-label, dose-escalation Phase 1/1b trials in both Australia and the United States (such as trial NCT06366789) evaluating adults with FLT3-mutated relapsed or refractory AML.
- Partnerships: The drug is being studied in the US in collaboration with The Leukemia & Lymphoma Society as part of their Beat AML master clinical trial portfolio
Lomonitinib is an orally bioavailable inhibitor of FMS-like tyrosine kinase 3 (FLT3; CD135; STK1; FLK2) mutations and interleukin-1 receptor-associated kinase 4 (IRAK4), with potential antineoplastic activity. Upon oral administration, lomonitinib targets, binds to and inhibits the activity of FLT3 mutations, including the FLT3-ITD-F691L gatekeeper mutation, while sparing the wild-type form of FLT3. This inhibits the proliferation of FLT3 mutant-expressing cancer cells. In addition, lomonitinib targets, binds to, and inhibits the kinase activity of IRAK4. This inhibits IRAK4-mediated signaling and may reduce adaptive resistance to FLT3 inhibition as toll-like receptor (TLR) activation plays an important role in resistance to FLT3 inhibition. FLT3, a class III receptor tyrosine kinase (RTK), is overexpressed or mutated in most B-lineage neoplasms and in acute myeloid leukemias. IRAK4, a serine/threonine-protein kinase, plays a key role in both the TLR and IL-1R signaling pathways.
- Dose Escalation and Expansion Study to Evaluate the Safety, PK, PD and Efficacy of ZE46-0134 in Adults With FLT3 Mutated or Spliceosome Mutated Relapsed or Refractory Acute Myeloid LeukemiaCTID: NCT06366789Phase: Phase 1Status: RecruitingDate: 2025-12-31
- Study of Biomarker-Based Treatment of Acute Myeloid LeukemiaCTID: NCT03013998Phase: Phase 1/Phase 2Status: RecruitingDate: 2025-12-17
- Study of Single and Multiple Ascending Doses of ZE46-0134 in Healthy VolunteersCTID: NCT06399315Phase: Phase 1Status: CompletedDate: 2025-12-09
SYN
PAT
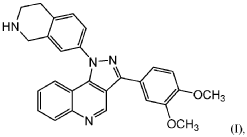
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US445571045&_cid=P12-MQUBPX-10324-1

Example 34: 3-(3,4-dimethoxyphenyl)-1-(1,2,3,4-tetrahydroisoquinolin-7-yl)-1H-pyrazolo[4,3-c]quinoline dihydrochloride (1.31)
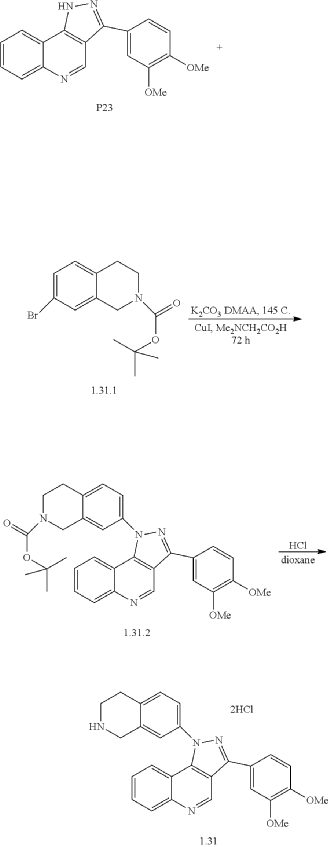
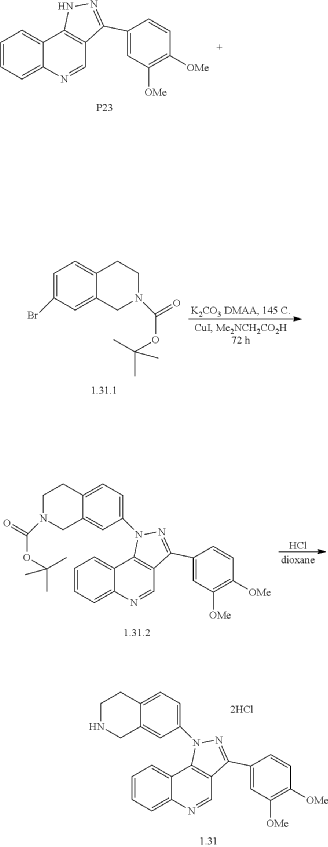
| A mixture of 3-(3,4-dimethoxyphenyl)-1H-pyrazolo[4,3-c]quinoline (P23) (153 mg, 0.5 mmol), tert-butyl 7-bromo-3,4-dihydroisoquinoline-2(1H)-carboxylate (1.31.1) (172 mg, 0.55 mmol), K 2CO 3 (83 mg, 0.6 mmol), CuI (10 mg, 0.05 mmol), N,N-dimethylglycine (11 mg, 0.1 mmol), and DMAA (2 mL) was stirred under Ar at 145° C. for 72 h, cooled to ambient temperature, diluted with CHCl 3, washed with 1% aq. solution of Na 2EDTA, and concentrated under reduced pressure. The residue was subjected to HPLC to afford 87 mg (33%) of tert-Butyl 7-(3-(3,4-dimethoxyphenyl)-1H-pyrazolo[4,3-c]quinolin-1-yl)-3,4-dihydroisoquinoline-2(1H)-carboxylate (1.31.2). 1H NMR (400 MHz, DMSO-d 6): δ 9.57 (s, 1H), 8.19 (d, J=8.4 Hz, 1H), 7.77 (m, 1H), 7.69 (dd, J 1=8.0 Hz, J 2=1.6 Hz, 1H), 7.63 (s, 1H), 7.57 (m, 3H), 7.51 (m, 2H), 7.17 (d, J=8.4 Hz, 1H), 4.63 (s, 2H), 3.88 (s, 3H), 3.86 (s, 3H), 3.68 (m, 2H), 2.98 (m, 2H), 1.45 (s, 9H). LCMS (ESI) m/z 538 [MH] +. |
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- Substituted 1H-pyrazolo [4,3-c ] quinolines, methods of preparation and uses thereofPublication Number: CN-118076605-APriority Date: 2021-10-15
- SUBSTITUTED 1H-PYRAZOLO [4,3-c] QUINOLINES, METHODS OF PREPARATION, AND USE THEREOFPublication Number: US-2025011319-A1Priority Date: 2021-10-15
////////lomonitinib, anax labs, tyrosine kinase inhibitor, antineoplastic, ZE46-0134, Eilean Therapeutics, U4DPU7W7QU
Lixosicone
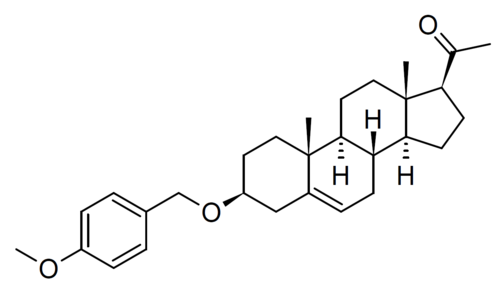
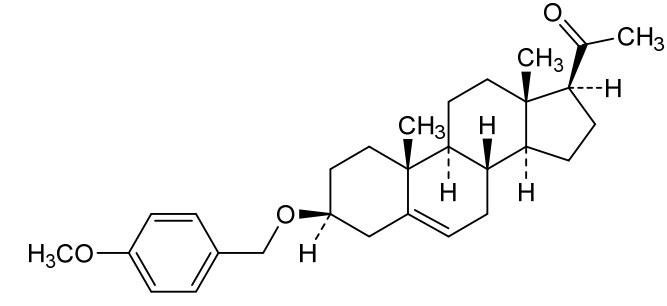
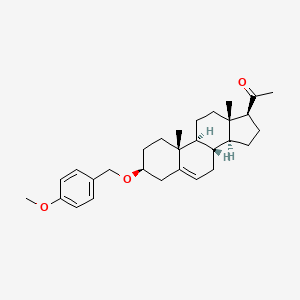
Lixosicone
CAS 1610878-71-1
MF C29H40O3 MW 436.6 g/mol
1-[(3S,8S,9S,10R,13S,14S,17S)-3-[(4-methoxyphenyl)methoxy]-10,13-dimethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-17-yl]ethanone
- 3beta-[(4-methoxyphenyl)methoxy]pregn-5-en-20-one
- (3beta)-3-[(4-Methoxyphenyl)methoxy]pregn-5-en-20-one
- Pregn-5-en-20-one, 3-[(4-methoxyphenyl)methoxy]-, (3beta)-
- 1-[(3S,8S,9S,10R,13S,14S,17S)-3-[(4-methoxyphenyl)methoxy]-10,13-dimethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-17-yl]ethanone
- 3?-(4-Methoxybenzyloxy)pregn-5-en-20-one; 1-((3S,8S,9S,10R,13S,14S,17S)-3-((4-Methoxybenzyl)oxy)-10,13-dimethyl-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-17-yl)ethan-1-one
3β-[(4-methoxyphenyl)methoxy]pregn-5-en-20-one
cannabinoid CB1 receptor signalling inhibitor, AEF0117, AEF 0117, 9LG9CT78SV
AEF0117 is a small molecule drug. AEF0117 is under investigation in clinical trial NCT05554926 (Study of [4-14C] AEF0117 Following a Single Oral Dose in Healthy Male Subjects).
Lixosicone (AEF0117, 3β-(4-methoxybenzyloxy)pregn-5-en-20-one) is a compound derived from pregnenolone by Aelis Farma, which acts as a biased negative allosteric modulator of the cannabinoid CB1 receptor, representing a new class of compounds referred to as CB1-selective signalling-specific inhibitors (CB1-SSi). It binds to an allosteric site on the CB1 receptor and modifies the downstream signalling produced as a result of CB1 activation, preventing CB1 mediated changes to mitogen-activated protein kinase (MAPK) phosphorylation but without affecting the signalling mediated by cyclic AMP. Unlike pregnenolone, AEF0117 is specific for the CB1-SSi activity and lacks the neurosteroid action typical of many structurally related compounds.[1]
In Phase II human clinical trials in patients diagnosed with cannabis use disorder, AEF0117 was found to partly but not completely block the effects of THC, and reduced cannabis self-administration but without producing an acute withdrawal syndrome and with relatively mild side effects. It is hoped that compounds of this type may be useful either as medications for the treatment of cannabinoid dependence, or could be used alongside medicinal cannabis to reduce unwanted side effects while retaining therapeutic efficacy.[2]
As of March 2026, lixosicone is in phase 2 clinical trials for treatment of substance-related disorders.[3] It is being developed by Aelis Farma.[3]
Clinical Development
The compound has shown promising results in clinical settings:
- Phase 2a results: In clinical trials evaluated by the National Institute on Drug Abuse, lixosicone significantly reduced the positive subjective effects (“the high”) of cannabis by 19% at lower doses and up to 38% at higher doses compared to a placebo.
- Reduced consumption: Testing demonstrated that the drug successfully reduced cannabis self-administration.
- Safety profile: Across early-stage evaluations, the drug was found to be safe, well-tolerated, and did not precipitate adverse behavioral withdrawal symptoms
SYN
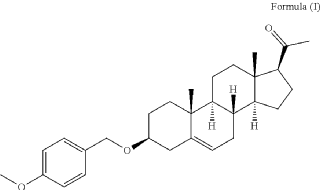
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN309682512&_cid=P10-MQRGHV-67541-1
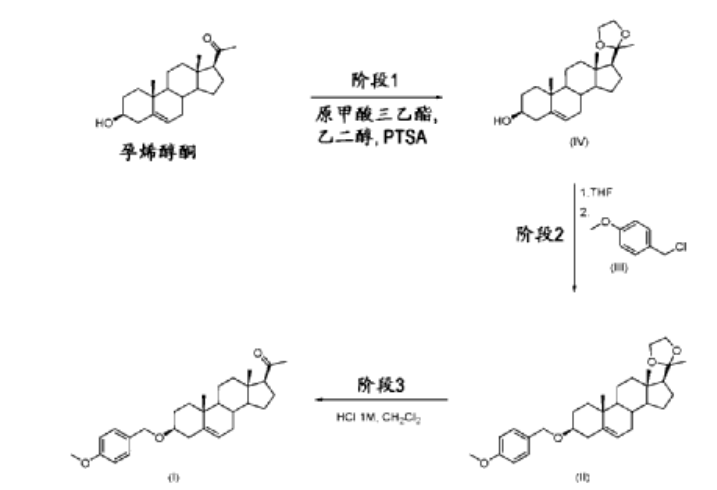
| Stage 1: Compound of formula (IV): (3S,8S,9S,10R,13S,14S,17S)-10,13-dimethyl-17-(2-methyl) (-1,3-dioxane-2-yl)-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecano-1H-cyclopentadiene Preparation of [a]phenanthrene-3-ol |
| Phase 1 was carried out in batches. Ethylene glycol (11.676 kg), pregnenolone (6.992 kg), and p-toluenesulfonic acid (0.840 kg, 4.42 mol, 0.2 equivalents) were charged into the reactor. The reaction mixture was stirred at 15°C–25°C for 25 minutes. Triethyl orthoformate (20.939 kg) was added in three portions, and the mixture was stirred at 15°C–25°C for at least 1 hour. Once complete, the reaction mixture was collected and slowly poured into a sodium bicarbonate solution (2.943 kg in 35.5 L of water) at 0°C–10°C. At the end of the addition, the reaction mixture was stirred at 0°C–10°C for 1 hour, then filtered and washed with water (12 L). The filtrate was also washed with 2-propanol (12 L) and dried under vacuum under a nitrogen stream. The dried solids were collected and charged into the reactor with 2-propanol (35 L). The slurry was heated under reflux for 2 hours. The reaction mixture was cooled to room temperature and stirred at room temperature for 12 hours. The reaction mixture was then cooled to between 0°C and 10°C and stirred for 2 hours. The solid was filtered and washed with 2-propanol (12 l), and then dried under vacuum under a nitrogen stream. Compound (IV) (8.031 kg) was obtained in a yield of 100.8% (uncorrected yield). |
| Phase 2: Compound of formula (II): 2-(3S,8S,9S,10R,13S,14S,17S)-3-((4-methoxybenzyl)oxy (10,13-dimethyl-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecano-1-H-cyclopentadien[a]) Preparation of phenanthrene-17-yl)-2-methyl-1,3-dioxolane |
| Compound (IV) (3.460 kg) and tetrahydrofuran (THF) (69 l) were charged into a reactor. The reaction mixture was stirred at 20°C–25°C for 80 minutes. The reaction mixture was filtered, and the solution of compound (IV) in THF was charged into the reactor. t-BuOK (2.835 kg) was added in portions to the THF solution of compound (IV) at 20°C–25°C. At the end of the addition, p-methoxybenzyl chloride (2.832 kg) and THF (4 l) of formula (III) were added to the reaction mixture via a feeding funnel. The reaction mixture was heated at 38°C–42°C. TBAI (1.555 kg) was added in portions to the reaction mixture at 38°C–42°C. The reaction mixture was heated at 55°C–60°C for 16 hours and 30 minutes. |
| Once complete, the reaction mixture is concentrated under vacuum to distill off 34-36 l of THF. The reaction mixture is then cooled to room temperature. Water (52 l) is added to the reactor, which is then cooled to 0-10 °C. The reaction mixture is carefully poured onto the water while maintaining the temperature at 0-10 °C. At the end of the addition, the reaction mixture is stirred at 0-10 °C for 1 hour and 50 minutes. The reaction mixture is filtered and washed with water (13 l). The filtrate is washed with acetonitrile (13.5 l), and the solids are dried under vacuum for 4 days under a nitrogen stream. |
| The solid was collected and acetonitrile (13 L) was added to the reactor. The mixture was heated under reflux for 4 hours. An additional acetonitrile (11 L) was added to the reactor and heated under reflux until a clear solution was obtained. The reaction mixture was cooled to room temperature and stirred at room temperature for 14 hours. The reaction mixture was cooled to 0 °C–10 °C and stirred at 0 °C–10 °C for 45 minutes, then filtered. Acetonitrile (10.5 L) was added to the reactor, cooled to 0 °C–10 °C, and then added to the filter to wash the filtrate. The solid was dried under vacuum under a nitrogen stream for 21 hours. Compound (II) (2.449 kg) was obtained in a yield of 59.2%. |
| • Stage 3: Preparation of compound (I): 3pMBP |
| Compound (II) (2.448 kg) and dichloromethane (10 L) were charged into a reactor. The solution was stirred for 20 minutes. 1 M hydrochloric acid (4.9 L) was added to the solution at 15 °C–25 °C. The reaction mixture was stirred until complete at 15 °C–25 °C. Dichloromethane (8 L) was added (to completely dissolve any precipitate) and phase separation was allowed. The organic layer was washed twice with water (5 L). The organic layer was collected and 2-propanol (24.5 L) was charged into a reactor at 15 °C–25 °C. The reaction mixture was concentrated under vacuum at a temperature below 40 °C. After completion, the reaction mixture was heated to reflux. 2-propanol (40 L) was added until a clear solution was observed. The reaction mixture was cooled to room temperature and stirred at room temperature for 12 hours. The reaction mixture was cooled to 0 °C–10 °C and stirred at 0 °C–-10 °C for 1 hour. The solid was filtered and washed with 2-propanol (5 l), then dried under vacuum with a nitrogen flow rate while the filter was heated at 35 °C–45 °C for 20 hours. Compound (I) was obtained in 85.8% yield (1.907 kg). |
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019162328&_cid=P10-MQRGHV-67541-1
PAT
- 3β-(4-Methoxybenzyloxy)pregnant-5-en-20-one for the treatment of cannabinoid-related disordersPublication Number: CN-111757740-BPriority Date: 2018-02-20Grant Date: 2023-12-15
- 3beta-(4-methoxybenzyloxy)paragan-5-en-20-one for use in the treatment of cannabinoid-related disordersPublication Number: IL-276697-B2Priority Date: 2018-02-20
- 3-BETA-(4-METHOXYBENZYLOXY)PREGN-5-EN-20-ONE FOR USE IN THE TREATMENT OF CANNABINOID-RELATED DISORDERSPublication Number: PT-3755339-TPriority Date: 2018-02-20
- 3beta-(4-methoxybenzyloxy)pregn-5-en-20-one for use in the treatment of Cannabinoids-Related DisordersPublication Number: US-2023226076-A1Priority Date: 2018-02-20
- 3beta-(4-methoxybenzyloxy)pregn-5-en-20-one for use in the treatment of cannabinoids-related disordersPublication Number: EP-3755339-A1Priority Date: 2018-02-20
- 3beta-(4-methoxybenzyloxy)pregn-5-en-20-one for use in the treatment of Cannabinoids-Related DisordersPublication Number: US-2021030768-A1Priority Date: 2018-02-20
- 3-beta-(4-methoxybenzyloxy)pregn-5-en-20-one for use in the treatment of cannabinoids-related disordersPublication Number: EP-3755339-B1Priority Date: 2018-02-20Grant Date: 2024-01-03
- 3BETA-(4-METHOXYBENZYLOXY)PREGN-5-EN-20-ONE FOR USE IN THE TREATMENT OF CANNABINOIDS RELATED DISORDERSPublication Number: MA-51891-B1Priority Date: 2018-02-20
- 3Beta-(4-Methoxybenzyloxy)pregn-5-en-20-one for use in the treatment of cannabinoid-related disordersPublication Number: MD-3755339-T2Priority Date: 2018-02-20
- 3-BETA-(4-METHOXYBENZYLLOXY)PREGN-5-EN-20-ONE FOR USE IN THE TREATMENT OF CANNABINOID-RELATED DISORDERSPublication Number: HR-P20240276-T1Priority Date: 2018-02-20
- 3beta-(4-methoxybenzyloxy)pregn-5-en-20-one for use in the treatment of cannabinoids-related disordersPublication Number: WO-2019162328-A1Priority Date: 2018-02-20
- 3BETA-(4-METHOXYBENCYLOXY)PREGN-5-EN-20-ONE FOR USE IN THE TREATMENT OF CANNABINOID-RELATED DISORDERS.Publication Number: MX-2020008687-APriority Date: 2018-02-20
- 3β-(4-methoxybenzyloxy)pregn-5-en-20-one for use in the treatment of cannabinoids-related disordersPublication Number: US-11484537-B2Priority Date: 2018-02-20Grant Date: 2022-11-01
- 3beta-(4-methoxybenzyloxy)pregn-5-en-20-one for use in the treatment of cannabinoids-related disordersPublication Number: AU-2019223049-A1Priority Date: 2018-02-20
- 3beta-(4-methoxybenzyloxy)pregn-5-en-20-one for use in the treatment of cannabinoids-related disordersPublication Number: CA-3090975-A1Priority Date: 2018-02-20
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
“AEF 0117”. AdisInsight. 13 March 2026. Retrieved 17 April 2026.
US 11484537, Piazza PV, Fabre S, Metna M, Monlezun S, Busquet-Garcia A, Cota D, Marsicano G, Revest JM, Vallée M, “3β-(4-methoxybenzyloxy)pregn-5-en-20-one for use in the treatment of cannabinoids-related disorders.”, issued 1 November 2022, assigned to Universite de Bordeaux.
Haney M, Vallée M, Fabre S, Collins Reed S, Zanese M, Campistron G, et al. (June 2023). “Signaling-specific inhibition of the CB1 receptor for cannabis use disorder: phase 1 and phase 2a randomized trials”. Nature Medicine. 29 (6): 1487–1499. doi:10.1038/s41591-023-02381-w. PMC 10287566. PMID 37291212.
 | |
| Clinical data | |
|---|---|
| Other names | AEF0117; AEF-0117; 3β-(4-Methoxybenzyloxy)pregn-5-en-20-one |
| Drug class | Cannabinoid CB1 receptor negative allosteric modulator |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1610878-71-1 |
| PubChem CID | 139433957 |
| ChemSpider | 129421614 |
| Chemical and physical data | |
| Formula | C29H40O3 |
| Molar mass | 436.636 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
//////////lixosicone, anax labs, cannabinoid CB1 receptor signalling inhibitor, AEF0117, AEF 0117, 9LG9CT78SV
Lirucitinib
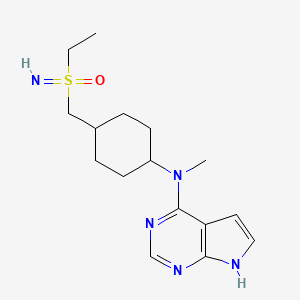
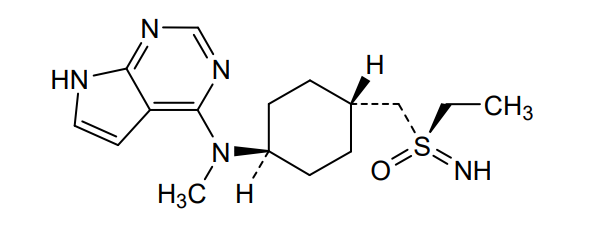
Lirucitinib
CAS 2458115-78-9
MF C16H25N5OS MW335.5 g/mol
N-[4-[(ethylsulfonimidoyl)methyl]cyclohexyl]-N-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine
(R)-ethyl(imino)({trans-4-[methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]cyclohexyl}methyl)-λ6
-sulfanone
Janus tyrosine kinase inhibitor, anti-inflammatory, GGW101, GGW 101
Lirucitinib is a Janus kinase (JAK) inhibitor primarily known as a novel, Class I veterinary drug. It specifically targets the JAK1 enzyme to block itch-inducing (pruritic) and inflammation-causing cytokines in the body.
Core Information
- Primary Use: The drug is developed for veterinary medicine to treat acute and chronic pruritic (severe itch) skin diseases in dogs, which are commonly caused by allergies, parasites, or infections.
- Approval Status: It received a Class I New Veterinary Drug Certificate from the Ministry of Agriculture and Rural Affairs (MARA) in China.
- Human Medicine: As of 2026, there are no clinical indications or indications that Lirucitinib is being tested for use in humans.
- Chemical Profile: It is an orally active small molecule with the chemical formula C₁₆H₂₅N₅OS and acts specifically as the (R)-enantiomer of the compound.
SYN
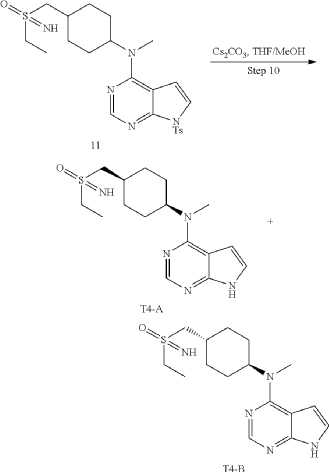
11 (1.0 g, 2.04 mmol) (prepared in step 9), tetrahydrofuran/methanol (10 mL), and cesium carbonate (1.33 g, 4.08 mmol) were added into a 25 mL single-necked flask, refluxed for 12 h, concentrated, and poured into dichloromethane and saturated salt solution, the organic phase was dried with anhydrous sodium sulfate, concentrated, and subjected to a conventional preparation method and a chiral preparation method to obtain product A as a white solid (20 mg, yield: 2.9%), LC-MS: 336 [M+H]+, H 1-NMR: 1H NMR (400 MHz, DMSO) δ 11.61 (s, 1H), 8.09 (s, 1H), 7.13 (s, 1H), 6.54 (s, 1H), 4.67 (s, 1H), 3.90-3.83 (m, 1H), 3.17 (s, 3H), 3.06-2.93 (m, 4H), 2.12-2.01 (m, 3H), 1.73-1.70 (m, 4H), 1.31-1.22 (m, 5H) and product B as a white solid (25 mg, yield: 3.7%), LC-MS: 336 [M+H]+, H 1—NMR: 1H NMR (400 MHz, DMSO) δ 11.59 (s, 1H), 8.09 (s, 1H), 7.12 (dd, J=3.3, 2.6 Hz, 1H), 6.54 (s, 1H), 4.67 (s, 1H), 3.58 (s, 1H), 3.17 (s, 3H), 3.06-2.89 (m, 4H), 2.16-1.93 (m, 3H), 1.74-1.69 (m, 4H), 1.25-1.23 (m, 5H).
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
PAT
- Jak inhibitor and preparation method thereforPublication Number: EP-3915989-B1Priority Date: 2019-01-30Grant Date: 2023-06-28
- Jak inhibitor and preparation method thereforPublication Number: US-2022106319-A1Priority Date: 2019-01-30
- Jak inhibitor and preparation method thereforPublication Number: WO-2020155931-A1Priority Date: 2019-01-30
- Jak inhibitor and preparation method thereforPublication Number: EP-3915989-A1Priority Date: 2019-01-30
/////////lirucitinib, anax labs, Janus tyrosine kinase inhibitor, anti-inflammatory, GGW101, GGW 101
Linvemastat
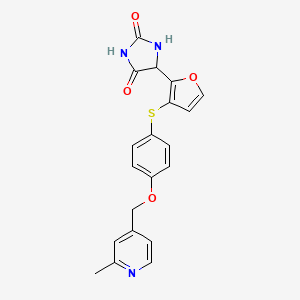
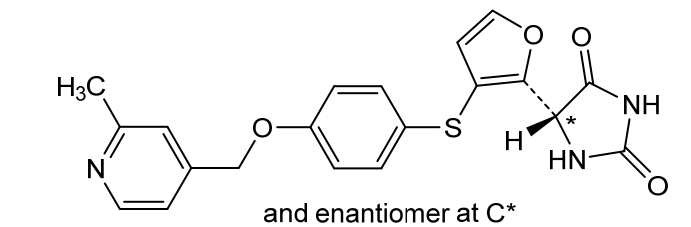
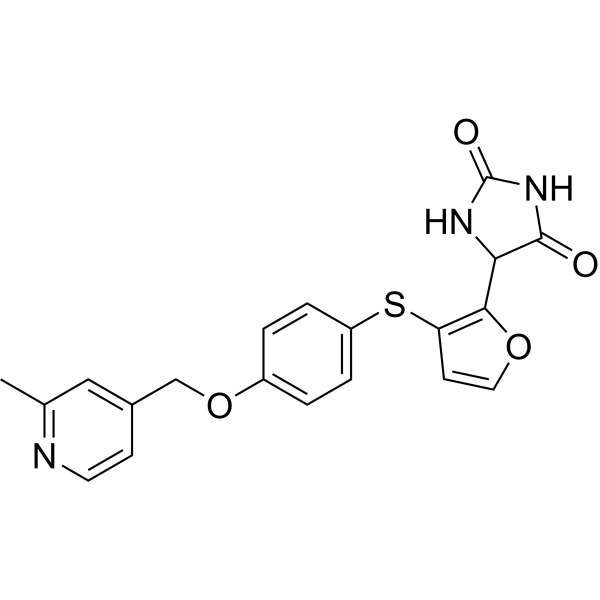
Linvemastat
CAS 2389060-50-6
MF C20H17N3O4S MW395.43
5-[3-[4-[(2-methyl-4-pyridinyl)methoxy]phenyl]sulfanylfuran-2-yl]imidazolidine-2,4-dione
- 2,4-Imidazolidinedione, 5-[3-[[4-[(2-methyl-4-pyridinyl)methoxy]phenyl]thio]-2-furanyl]-
- 5-[3-[[4-[(2-Methyl-4-pyridinyl)methoxy]phenyl]thio]-2-furanyl]-2,4-imidazolidinedione
- rac-(14R)-72-methyl-5-oxa-3-thia-7(4)-pyridina-1(4)-imidazolidina-2(2,3)-furana-4(1,4)-benzenaheptaphane-12,15-dione

matrix metalloproteinase-12 inhibitor, lung diseases, FP 020, FP2AMM4SVF
Linvemastat (also known by its developmental code FP-020) is an investigational, orally active small-molecule drug designed to selectively inhibit matrix metalloproteinase-12 (MMP-12). It is being developed as a potential disease-modifying therapy for chronic inflammatory and fibrotic diseases, primarily targeting respiratory conditions and gastrointestinal disorders.
How Linvemastat Works
- Targeting MMP-12: MMP-12 is an enzyme secreted by macrophages that breaks down extracellular matrix proteins like elastin.
- Controlling Damage: Overexpression of MMP-12 is strongly linked to tissue destruction, chronic inflammation, and fibrosis in the lungs and gut.
- High Selectivity: Unlike older matrix metalloproteinase inhibitors, linvemastat targets MMP-12 with high specificity, avoiding off-target interactions with other vital MMP enzymes.
Primary Therapeutic Indications under Research
- Severe, Uncontrolled Asthma: Evaluated to reduce airway inflammation and improve overall lung function.
- Chronic Obstructive Pulmonary Disease (COPD): Investigated for preventing structural lung damage and progressive emphysema.
- Inflammatory Bowel Disease (IBD): Researched to minimize continuous gut wall inflammation and intestinal fibrosis.
- Other Fibrotic Conditions: Explored in preclinical models for conditions like idiopathic pulmonary fibrosis (IPF) and kidney damage.
Current Clinical Status
Initially developed by Foresee Pharmaceuticals, global development rights for the program were transitioned to Primevera Therapeutics LLC.
- Phase 1 Trial: Completed trials in healthy volunteers demonstrated a favorable safety and pharmacokinetic profile, with only minor, recoverable side effects like mild nausea or headache.
- Phase 2 Trial: Undergoing mid-stage clinical evaluation, such as the global, randomized syMMPonia study. This trial is measuring changes in forced expiratory volume (FEV₁) to assess the drug’s impact on adults with partially controlled, moderate-to-severe asthma.
Effect of Linvemastat in Patients With Partially Controlled Asthma (syMMPonia)
CTID: NCT07191535
Phase: Phase 2
Status: Not yet recruiting
Date: 2025-10-20
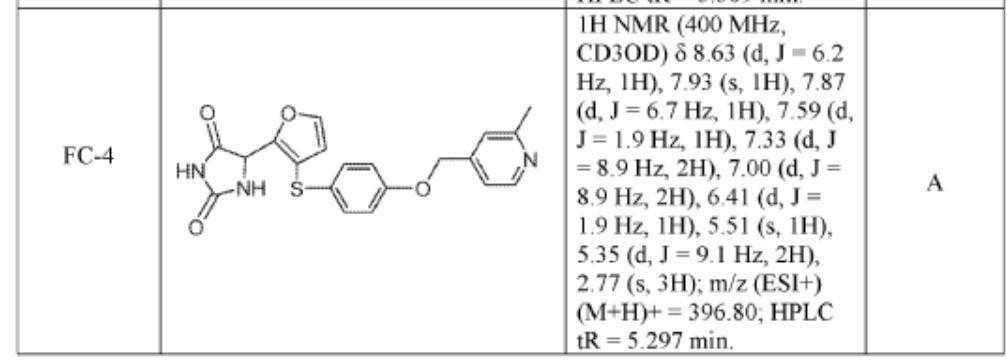
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019222157&_cid=P10-MQOLFP-71317-1
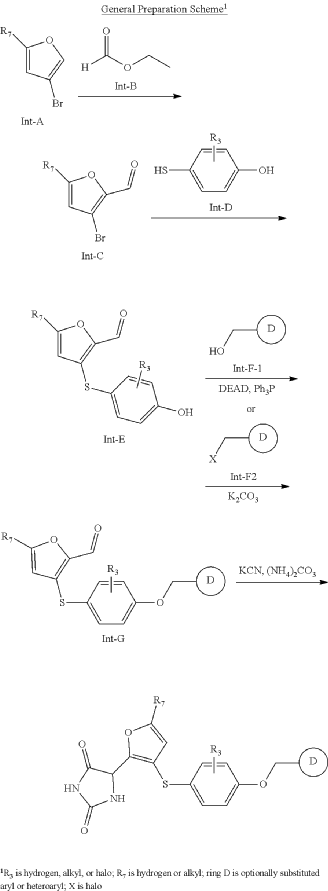
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US276884719&_cid=P10-MQOLBE-66887-1

SYN
PAT
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
PAT
- Matrix metalloproteinase (MMP) inhibitors and methods of use thereofPublication Number: US-10851089-B2Priority Date: 2018-05-15Grant Date: 2020-12-01
- Matrix metalloproteinase (mmp) inhibitors and methods of use thereofPublication Number: US-2019352287-A1Priority Date: 2018-05-15
//////////////linvemastat, anax labs, matrix metalloproteinase-12 inhibitor, lung diseases, FP 020, FP2AMM4SVF
Gadoquatrane
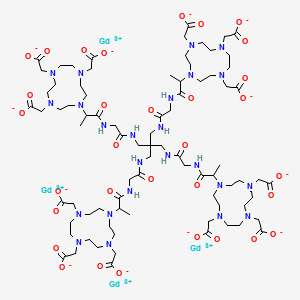
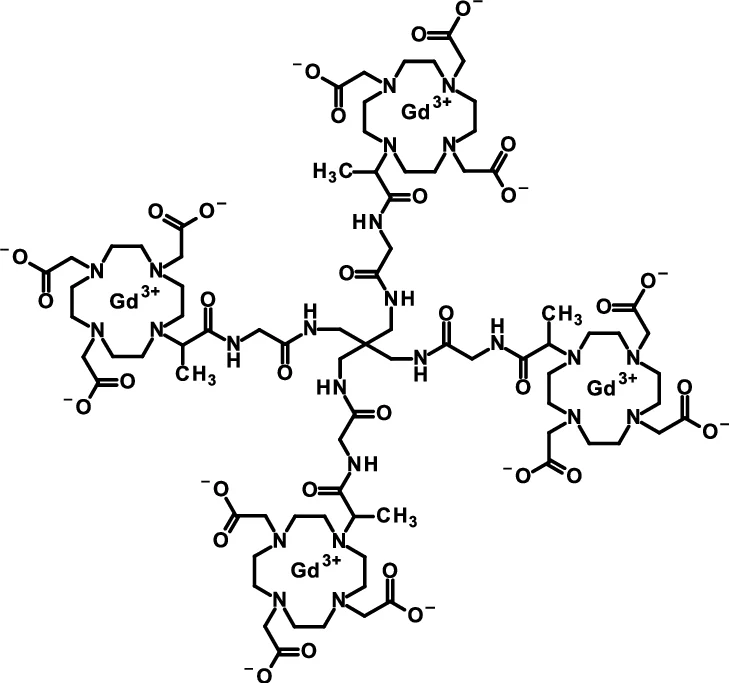
Gadoquatrane
CAS2048221-65-2MW2579.0 g/mol
FDA 2026, APPROVALS 2026, Ambelvist, OZG7J613HK, BAY-1747846, BAY 1747846
2-[4,10-bis(carboxylatomethyl)-7-[1-oxo-1-[[2-oxo-2-[[3-[[2-[2-[4,7,10-tris(carboxylatomethyl)-1,4,7,10-tetrazacyclododec-1-yl]propanoylamino]acetyl]amino]-2,2-bis[[[2-[2-[4,7,10-tris(carboxylatomethyl)-1,4,7,10-tetrazacyclododec-1-yl]propanoylamino]acetyl]amino]methyl]propyl]amino]ethyl]amino]propan-2-yl]-1,4,7,10-tetrazacyclododec-1-yl]acetate;tetrakis(gadolinium(3+))
To detect and visualize lesions with abnormal vascularity, in conjunction with MRI
Gadoquatrane (marketed as AMBELVIST®) is a low-dose, macrocyclic gadolinium-based contrast agent (GBCA) developed by Bayer for use in magnetic resonance imaging (MRI). It is designed to enhance the visualization of lesions in the central nervous system (CNS) and other body regions in adult and pediatric patients.
Core Highlights:
- Lower Gadolinium Exposure: It requires a dose of 0.04 mmol/kg, which results in 60% less gadolinium exposure compared to standard macrocyclic GBCAs.
- Regulatory Approval: The FDA approved it in June 2026 for use in adults and pediatric patients, including term neonates. It was also approved in Japan in March 2026.
- Efficacy & Safety: Phase III clinical trials (the QUANTI studies) showed it effectively detects lesions with abnormal vascularity while maintaining an efficacy and safety profile comparable to other standard macrocyclic agents.
- Structure: Gadoquatrane features a tetrameric, macrocyclic structure that gives it high relaxivity and stability in the body
SYN
https://patents.google.com/patent/US20250114485A1
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
PAT
- Gadolinium chelate compounds for use in magnetic resonance imagingPublication Number: US-11491245-B2Priority Date: 2015-06-04Grant Date: 2022-11-08
- Gadolinium chelate compounds for use in magnetic resonance imagingPublication Number: US-2025114485-A1Priority Date: 2015-06-04
- Formulation of contrast media and process of preparation thereofPublication Number: US-2024252690-A1Priority Date: 2018-11-23
- Formulation of contrast media and process of preparation thereofPublication Number: US-12303573-B2Priority Date: 2018-11-23Grant Date: 2025-05-20
- New gadolinium chelate compounds for use in magnetic resonance imagingPublication Number: EP-3611169-A1Priority Date: 2015-06-04
- New gadolinium chelate compounds for use in magnetic resonance imagingPublication Number: US-2023113481-A1Priority Date: 2015-06-04
- New gadolinium chelate compounds for use in magnetic resonance imagingPublication Number: EP-3303307-B1Priority Date: 2015-06-04Grant Date: 2019-09-04
- Generation of artificial contrast-enhanced computed tomography imagesPublication Number: US-2024346718-A1Priority Date: 2023-04-13
- Generation of artificial contrast-enhanced radiological imagesPublication Number: WO-2024100233-A1Priority Date: 2022-11-12
- Generation of artificial contrast-enhanced radiological imagesPublication Number: EP-4616420-A1Priority Date: 2022-11-12
- Automated analysis of radiological imagesPublication Number: WO-2024083466-A1Priority Date: 2022-10-17
- Formulation of contrast media and process of preparation thereofPublication Number: US-11944690-B2Priority Date: 2018-11-23Grant Date: 2024-04-02
/////////gadoquatrane, anax labs, FDA 2026, APPROVALS 2026, Ambelvist, OZG7J613HK, BAY-1747846, BAY 1747846
Lasmotinib
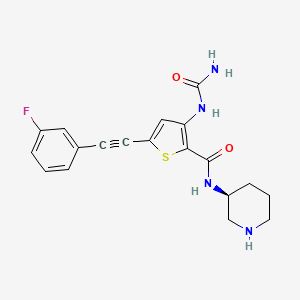
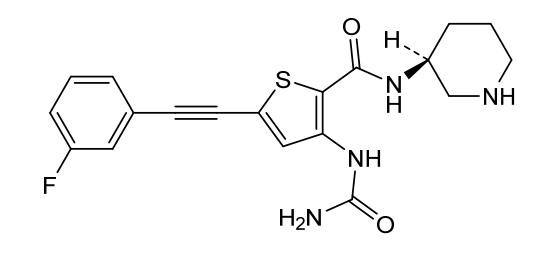
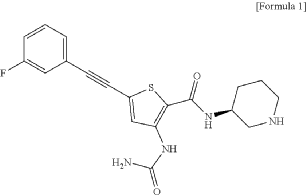
Lasmotinib
CAS 2127107-15-5
MF C19H19FN4O2S MW386.4 g/mol
3-(carbamoylamino)-5-[2-(3-fluorophenyl)ethynyl]-N-[(3S)-piperidin-3-yl]thiophene-2-carboxamide
- N-(2-(N-((3S)(3-Piperidyl))carbamoyl)-5-(2-(3-fluorophenyl)ethynyl)(3-thienyl))aminamide
- 2-Thiophenecarboxamide, 3-((aminocarbonyl)amino)-5-(2-(3-fluorophenyl)ethynyl)-N-(3S)-3-piperidinyl-
- 3-(carbamoylamino)-5-[2-(3-fluorophenyl)ethynyl]-N-[(3S)-piperidin-3-yl]thiophene-2-carboxamide
- 2-Thiophenecarboxamide, 3-[(aminocarbonyl)amino]-5-[2-(3-fluorophenyl)ethynyl]-N-(3S)-3-piperidinyl-
- 3-(carbamoylamino)-5-(2-(3-fluorophenyl)ethynyl)-N-((3S)-piperidin-3-yl)thiophene-2-carboxamide
3-(carbamoylamino)-5-[(3-fluorophenyl)ethynyl]-N-[(3S)-piperidin-3-yl]thiophene-2-carboxamide
tyrosine kinase inhibitor, antineoplastic, PHI-101, PHI 101, U2UY9TBQ8Z
Lasmotinib (also known by its research code PHI-101) is a next-generation, orally bioavailable targeted cancer therapy. It functions as a dual FLT3 and CHK2 inhibitor. It is primarily being investigated to treat Acute Myeloid Leukemia (AML) and ovarian cancer.
How It Works
- FLT3 Inhibition: It targets FMS-like tyrosine kinase 3 (FLT3), an enzyme that is often mutated in AML. Lasmotinib is designed to attack not just single activating mutations (ITD or TKD), but also difficult-to-treat double and triple-resistant mutations.
- CHK2 Inhibition: It also inhibits Checkpoint Kinase 2 (CHK2), preventing cancer cells from repairing DNA damage. This causes the cancer cells to undergo apoptosis (programmed cell death).
Key Clinical Advantages
- High Efficacy: In relapsed or refractory AML patients who have previously failed other FLT3 inhibitors, lasmotinib has demonstrated high rates of composite complete remission.
- Safety Profile: Preclinical and early-stage trials indicate a promising safety profile with a very low or 0% occurrence rate of cardiotoxicity (heart damage), which is a common hurdle for some other FLT3-targeting drugs.
Current Development & Combinations
- Developer: Discovered by Seoul National University Hospital and being developed by Pharos iBio.
- Synergistic Therapies: Lasmotinib is currently moving into global clinical trials as a powerful combination therapy. Research shows it synergizes strongly with existing treatments like Venetoclax or Azacytidine, as well as with emerging Menin inhibitors (such as bleximenib) to achieve deep tumor growth inhibition
Lasmotinib is an orally bioavailable inhibitor of checkpoint kinase 2 (chk2), with potential antineoplastic and chemopotentiating activities. Upon oral administration, lasmotinib binds to and inhibits the activity of chk2, which may prevent the repair of DNA damage caused by DNA-damaging agents. This may result in tumor cell apoptosis and potentiate the antitumor efficacies of various chemotherapeutic agents. Chk2, an ATP-dependent serine–threonine kinase, is a key component in the DNA replication-monitoring checkpoint system and is activated by double-stranded breaks (DSBs); activated chk2 is overexpressed by a variety of cancer cell types.
- Chk2 Inhibitor for Recurrent EpitheliAl periToneal, fallopIan or oVarian cancEr (CREATIVE Phase IA Trial)CTID: NCT04678102Phase: Phase 1Status: Unknown statusDate: 2023-06-26
- Evaluation of the Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of PHI 101 for the Treatment of AMLCTID: NCT04842370Phase: Phase 1Status: Unknown statusDate: 2021-04-20
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=JP405710409&_cid=P21-MQIVJB-43702-2
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US465154324&_cid=P21-MQIVJB-43702-2
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2024015484&_cid=P21-MQIVJB-43702-2
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2025210599&_cid=P21-MQIVJB-43702-2
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
PAT
- Inhibitors of brutons tyrosine kinasePublication Number: US-2021070748-A1Priority Date: 2015-06-02
- New, substituted quinoline compounds as inhibitors of S-nitrosoglutathion reductasePublication Number: HU-E025653-T2Priority Date: 2010-10-08
- New hybrid oligomers. Their preparation process and pharmaceutical compositions containing themPublication Number: AU-2006256439-A1Priority Date: 2005-03-18
- NEW THIOPHENE COMPOUND SUBSTITUTED IN POSITIONS 2,3,5, USED AS A PROTEIN KINASE INHIBITORPublication Number: BR-112018016729-B1Priority Date: 2016-02-16
- 2, 3, 5-substituted thiophene compounds as protein kinase inhibitorsPublication Number: CN-108884066-BPriority Date: 2016-02-16Grant Date: 2021-08-24
- 2,3,5-substituted thiophene compound as protein kinase inhibitorPublication Number: US-10442796-B2Priority Date: 2016-02-16Grant Date: 2019-10-15
- Novel compound of 2,3,5-substituted thiophene as a protein kinase inhibitorPublication Number: RU-2724957-C2Priority Date: 2016-02-16Grant Date: 2020-06-29
- Novel 2,3,5-substituted thiophene compounds as protein kinase inhibitorsPublication Number: KR-101965326-B1Priority Date: 2016-02-16Grant Date: 2019-04-03
- Novel 2,3,5-substituted thiophene compound as protein kinase inhibitorPublication Number: WO-2017142325-A1Priority Date: 2016-02-16
- Novel 2,3,5-substituted thiophene compound as protein kinase inhibitorPublication Number: US-2019047993-A1Priority Date: 2016-02-16
- Novel 2,3,5-substituted thiophene compounds as protein kinase inhibitorsPublication Number: KR-20180136425-APriority Date: 2016-02-16
- Novel 2,3,5-substituted thiophene compound as protein kinase inhibitorPublication Number: EP-3418275-B1Priority Date: 2016-02-16Grant Date: 2021-03-17
- Novel 2,3,5-substituted thiophene compounds that are protein kinase inhibitorsPublication Number: JP-2019504900-APriority Date: 2016-02-16
- Use of 2,3,5-substituted thiophene compound for enhancement of radiotherapyPublication Number: EP-3804719-A1Priority Date: 2018-05-30
- Use of 2,3,5-substituted thiophene compound to prevent, ameliorate, or treat breast cancersPublication Number: EP-3804718-A1Priority Date: 2018-05-30
- Novel 2,3,5-substituted thiophene compound as protein kinase inhibitorPublication Number: EP-3418275-A1Priority Date: 2016-02-16
- New 2,3,5-substituted thiophene compound as a protein kinase inhibitorPublication Number: RU-2018130703-APriority Date: 2016-02-16
- Novel 2,3,5-substituted thiophene compounds as protein kinase inhibitorsPublication Number: KR-20190035671-APriority Date: 2016-02-16
- Radiotherapy-enhancing applications of 2,3,5-substituted thiophene compoundsPublication Number: JP-2021525285-APriority Date: 2018-05-30
- Use of 2,3,5-substituted thiophene compound for enhancement of radiotherapyPublication Number: US-2021205289-A1Priority Date: 2018-05-30
- Use of 2,3,5-Substituted Thiophene Compound for Prevention, Improvement or Treatment of Breast CancerPublication Number: KR-102227117-B1Priority Date: 2018-05-30Grant Date: 2021-03-15
- Use of 2,3,5-Substituted Thiophene Compound for Prevention, Improvement or Treatment of Breast CancerPublication Number: KR-20190136976-APriority Date: 2018-05-30
- Use of 2,3,5-substituted thiophene compound to prevent, ameliorate, or treat breast cancersPublication Number: US-2021205290-A1Priority Date: 2018-05-30
////////lasmotinib, anax labs, tyrosine kinase inhibitor, antineoplastic, PHI-101, PHI 101, U2UY9TBQ8Z
Larubrilstat
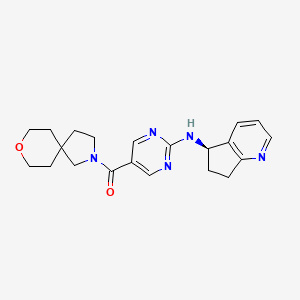
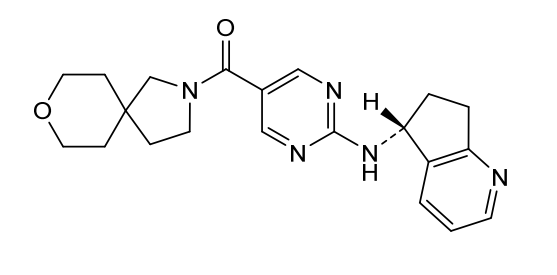
Larubrilstat
CAS 2765226-31-9
MF C21H25N5O2 MW379.5 g/mol
[2-[[(5R)-6,7-dihydro-5H-cyclopenta[b]pyridin-5-yl]amino]pyrimidin-5-yl]-(8-oxa-2-azaspiro[4.5]decan-2-yl)methanone
- [2-[[(5R)-6,7-dihydro-5H-cyclopenta[b]pyridin-5-yl]amino]pyrimidin-5-yl]-(8-oxa-2-azaspiro[4.5]decan-2-yl)methanone
- Methanone, [2-[[(5R)-6,7-dihydro-5H-cyclopenta[b]pyridin-5-yl]amino]-5-pyrimidinyl]-8-oxa-2-azaspiro[4.5]dec-2-yl-
(2-{[(5R)-6,7-dihydro-5H-cyclopenta[b]pyridin-5-yl]amino}pyrimidin-5-yl)(8-oxa-2-azaspiro[4.5]decan-2-
yl)methanone
vascular non-inflammatory molecule-1 (VNN1) inhibitor, AG6K4Y29B4
Larubrilstat is the International Nonproprietary Name (INN) for an experimental, small-molecule vascular non-inflammatory molecule-1 (VNN1) inhibitor. VNN1, also commonly known as Vanin-1 or pantetheinase, is an enzyme involved in tissue response to oxidative stress and inflammation.
Current Status
- Development Context: Larubrilstat is a designated compound linked to therapeutic exploration in inflammatory pathways. Research and patent filings, such as those cataloged by the IUPHAR/BPS Guide to PHARMACOLOGY, track its evaluation alongside similar Vanin-1 inhibitors
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US425298584&_cid=P20-MQHGA8-93141-1
COMP 2-1 IS PRODUCT
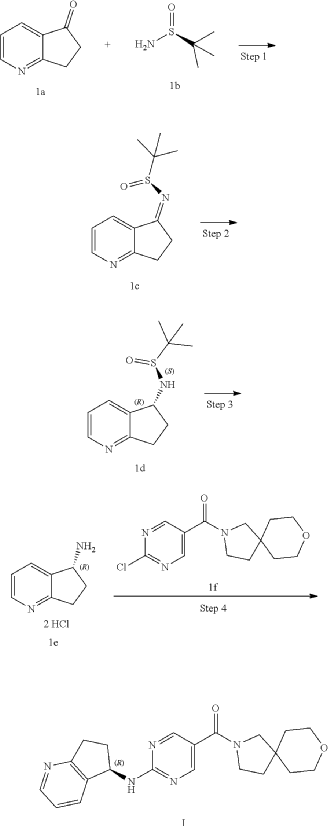
Example 2: Synthesis of Compound 2, Compound 2-1 and Compound 2-2
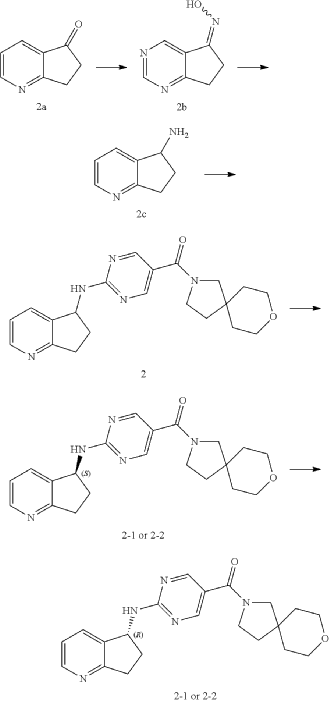
| Step 1 |
Step 2
Step 3
| Compound 2 (66 mg) was obtained from compound if (168 mg) and compound 2c (100 mg) according to the method of Example 1. |
| Two enantiomers 2-1 (retention time: 8.483 min) and 2-2 (retention time: 13.580 min) were obtained by chiral separation of compound 2. |
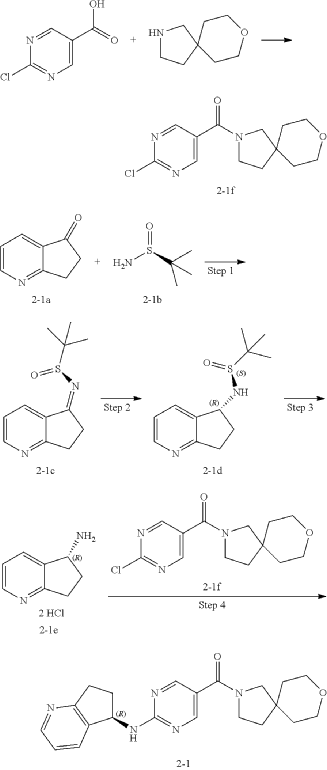
Step 4: Preparation of Compound 2-1
| The absolute stereochemical configuration of compound 2-1 was determined by comparative determination of the above preparation method of the chiral compounds. |
PAT
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
PAT
- Pyrimidine carboxamide compound and application thereofPublication Number: US-2024083873-A1Priority Date: 2020-09-25
- Pyrimidine carboxamide compound and application thereofPublication Number: WO-2022063197-A1Priority Date: 2020-09-25
- Pyrimidine carboxamide compound and application thereofPublication Number: WO-2022063333-A1Priority Date: 2020-09-25
////////larubrilstat, ANAX LABS, vascular non-inflammatory molecule-1 (VNN1) inhibitor, AG6K4Y29B4

{kind=link}