


A series of novel, potent orthopoxvirus egress inhibitors was identified during high-throughput screening of the ViroPharma small molecule collection. Using structure−activity relationship information inferred from early hits, several compounds were synthesized, and compound 14was identified as a potent, orally bioavailable first-in-class inhibitor of orthopoxvirus egress from infected cells. Compound 14 has shown comparable efficaciousness in three murine orthopoxvirus models and has entered Phase I clinical trials.
Home » MONKEY POX
Category Archives: MONKEY POX
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
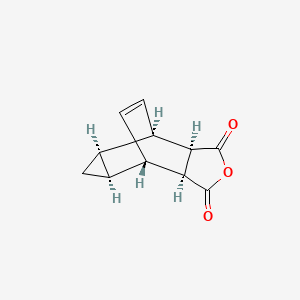
Tecovirimat

Tecovirimat
- Molecular FormulaC19H15F3N2O3
- Average mass376.329 Da
816458-31-8 [RN]
869572-92-9 [RN]
UNII-F925RR824R
тековиримат [Russian]
تيكوفيريمات [Arabic]
替韦立马 [Chinese]
Benzamide, N-[(3aR,4R,4aR,5aS,6S,6aS)-3,3a,4,4a,5,5a,6,6a-octahydro-1,3-dioxo-4,6-ethenocycloprop[f]isoindol-2(1H)-yl]-4-(trifluoromethyl)-
N-[(1R,2R,6S,7S,8S,10R)-3,5-Dioxo-4-azatetracyclo[5.3.2.02,6.08,10]dodec-11-en-4-yl]-4-(trifluoromethyl)benzamide
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Tecovirimat monohydrate | SB96YO2BR8 | 1162664-19-8 | QRHXYGPOQKLBJP-NPIFKJBVSA-N |
Tecovirimat, sold under the brand name Tpoxx among others,[6] is an antiviral medication with activity against orthopoxviruses such as smallpox and monkeypox.[4][7][8] It is the first antipoxviral drug approved in the United States.[9][10] It is an inhibitor of the orthopoxvirus VP37 envelope wrapping protein.[4]
The drug works by blocking cellular transmission of the virus, thus preventing the disease.[11] Tecovirimat has been effective in laboratory testing; it has been shown to protect animals from monkeypox and rabbitpox and causes no serious side effects in humans.[6] Tecovirimat was first used for treatment in December 2018, after a laboratory-acquired vaccinia virus infection.[12]
Two million doses of tecovirimat are stockpiled in the US Strategic National Stockpile should an orthopoxvirus-based bioterror attack occur.[13][14] The U.S. Food and Drug Administration (FDA) considers it to be a first-in-class medication.[15]
The World Health Organization declared smallpox, a contagious and sometimes fatal infectious disease, eradicated in 1980. However, there have been longstanding concerns that smallpox may be used as a bioweapon.2,5 Tecovirimat is an antiviral drug that was identified via a high-throughput screen in 2002.2 It is effective against all orthopoxviruses, including vaccinia, cowpox, ectromelia, rabbitpox, monkeypox, and Variola (smallpox) virus.1,4
Tecovirimat was approved by the FDA in July 2018 as the first drug ever approved to treat smallpox.6,5 Tecovirimat was later approved by Health Canada in December 2021,7 followed by the approval from the European Commission in January 2022.9 Other than smallpox, tecovirimat is also indicated to treat complications due to replication of the vaccinia virus following vaccination against smallpox, and to treat monkeypox and cowpox in adults and children.8 Tecovirimat is available as both oral and intravenous formulations.10
Medical uses
In the United States, tecovirimat is indicated for the treatment of human smallpox disease.[4] In the European Union it is indicated for the treatment of smallpox, monkeypox, and cowpox.[5]
Mechanism of action
Tecovirimat inhibits the function of a major envelope protein required for the production of extracellular virus. The drug prevents the virus from leaving an infected cell, hindering the spread of the virus within the body.[16]
Chemistry
The first synthesis of tecovirimat was published in a patent filed by scientists at Siga Technologies in 2004. It is made in two steps from cycloheptatriene.[17]
A Diels–Alder reaction with maleic anhydride forms the main ring system[18] and subsequent reaction with 4-trifluormethylbenzhydrazide gives the cyclic imide of the drug.[17][19]
Synthesis
US 9,546,137 [2017, to SIGA TECH INC]

SYNTHESIS FROM SMARTCHEM
The scheme has taken from SmartChem a knowledgebase by ROW2 Technologies, Inc. (www.row2technologies.com)
A perfect amalgamation of information on chemicals and global suppliers. A database where you can search for information on more than 150,000 chemicals and around 15,000 Global chemicals suppliers, including routes of synthesis, Applications, end uses, and validated contact details of global suppliers. For more information, please visit www.row2technologies.com or contact,
Anand Ramakrishnan (VP- Sales)
Tel : +1 973 795 1141
Mobile: +91 9821384045
Harshada Shivalkar (Manager-Business Development)
Mobile: +91 9323945301
hshivalkar@row2technologies.com
SYN 1

Synthetic Description
Reference: Dong, Ming-xin; Li, Hai-tao; Wang, Xiao-hua; Mao, Wen-xiang; Zhou, Shang-min; Dai, Qiu-yun. Preparation and structural determination of tecovirimat monohydrate crystal. Zhongguo Xinyao Zazhi. Volume 21. Issue 23. Pages 2736-2739. (2012).
SYN 2

Synthetic Description
Reference: Dai, Dongcheng. Process for the preparation of tecovirimat. Assignee Siga Technologies, Inc., USA. WO 2014028545. (2014).
SYN 3

Synthetic Description
Reference: Medical composition containing ST-246, its preparation and anti-poxvirus application. Assignee Institute of Microbiology and Epidemiology, Academy of Military Medical Sciences, PLA, Peop. Rep. China. CN 101912389. (2010).
EMA
Click to access tecovirimat-siga-epar-public-assessment-report_en.pdf
PATENT
https://patents.google.com/patent/US9546137B2/en
The present invention provides a process for making ST-246 outlined in Scheme 1

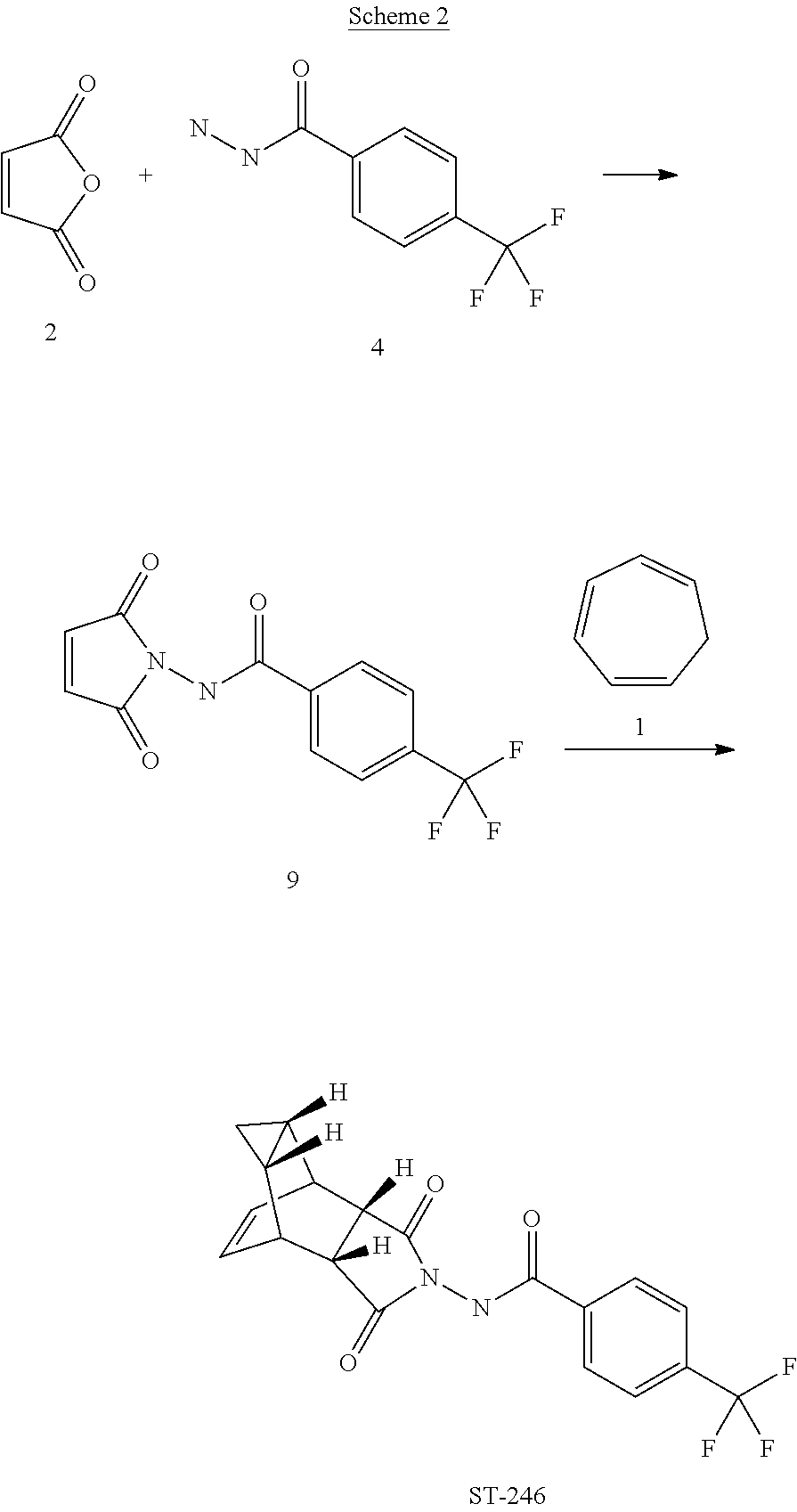
The present invention also provides a process for making ST-246 outlined in Scheme 2
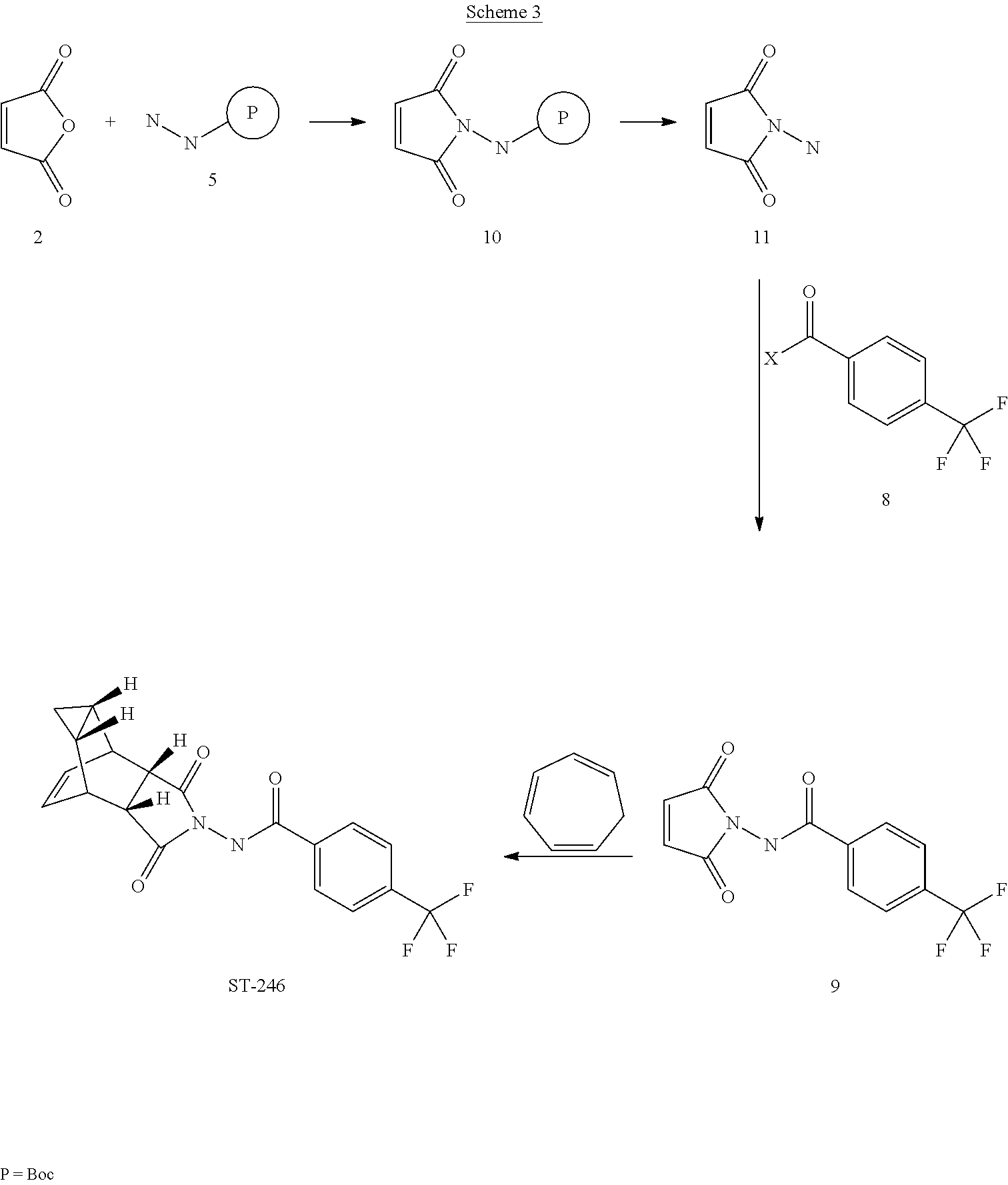
The present invention further provides a process for making ST-246 outlined in Scheme 3
The present invention also provides a process for making ST-246 outlined in Scheme 4

The present invention further provides a process for making ST-246 outlined in Scheme 5

The present invention also provides the following compounds useful in the synthesis of ST-246:
EXAMPLE 1Synthetic Route I

Step A. Synthesis of Compound 6 (P=Boc)

Step A. Synthesis of Compound 9


Step A. Synthesis of Compound 10

Step B. Synthesis of Compound 11 (HCl salt)

Step A. Synthesis of Compound 10

Step B. Synthesis of Compound 6

Step A. Synthesis of Compound 13
Step A. Synthesis of Compound 6 (P=Boc)
To a mixture of compound 3 (5.0 g, 26.3 mmol, synthesized according to WO04112718) in EtOH (80 mL, EMD, AX0441-3) was added tert-butyl carbazate 5 (3.65 g, 27.6 mmol, Aldrich, 98%). The reaction mixture was heated to reflux for 4 h under nitrogen atmosphere. LC-MS analysis of the reaction mixture showed less than 5% of compound 3 remained. The reaction mixture was evaporated under reduced pressure. The residue was recrystallized from EtOAc-hexanes, the solid was filtered, washed with hexanes (50 mL) and dried under vacuum to afford compound 6 (3.1 g, 39% yield) as a white solid. The filtrate was concentrated and purified by column chromatography eluting with 25% EtOAc in hexanes to give an additional 3.64 g (46% yield) of compound 6 as a white solid. Total yield: 6.74 g (84% yield). 1H NMR in CDCl3: δ 6.30 (br s, 1H), 5.79 (t, 2H), 3.43 (s, 2H), 3.04 (s, 2H), 1.46 (s, 9H), 1.06-1.16 (m, 2H), 0.18-0.36 (m, 2H); Mass Spec: 327.2 (M+Na)+
Step B. Synthesis of Compound 7 (HCl Salt)
Compound 6 (3.6 g, 11.83 mmol) was dissolved in i-PrOAc (65 mL, Aldrich, 99.6%). 4M HCl in dioxane (10.4 mL, 41.4 mmol, Aldrich) was added drop-wise to the above solution keeping the temperature below 20° C. The reaction mixture was stirred at room temperature overnight (18 h) under nitrogen atmosphere. The resulting solid was filtered, washed with i-PrOAc (15 mL) and dried under vacuum to yield HCl salt of compound 7 (1.9 g, 67% yield) as a white solid. The filtrate was concentrated to ⅓ its volume and stirred at 10-15° C. for 30 min. The solid was filtered, washed with minimal volume of i-PrOAc and dried to afford additional 0.6 g (21% yield) of compound 7. Total yield: 2.5 g (88% yield). 1H NMR in DMSO-d6: δ 6.72 (br s, 3H), 5.68 (m, 2H), 3.20 (s, 2H), 3.01 (s, 2H), 1.07-1.17 (m, 2H), 0.18-0.29 (m, 1H), −0.01-0.07 (m, 1H); Mass Spec: 205.1 (M+H)+
Step C. Synthesis of ST-246
To a mixture of compound 7 (0.96 g, 4 mmol) in dry dichloromethane (19 mL) was added triethylamine (1.17 mL, 8.4 mmol, Aldrich) keeping the temperature below 20° C. The resulting solution was stirred for 5 minutes at 15-20° C., to it was added drop-wise 4-(trifluoromethyl)benzoyl chloride 8 (0.63 mL, 4.2 mmol, Aldrich, 97%) and the reaction mixture was stirred at room temperature overnight (18 h). LC-MS and TLC analysis showed the correct molecular weight and Rf value of ST-246 but the reaction was not complete. Additional 0.3 mL (2 mmol, 0.5 eq) of 4-(trifluoromethyl)benzoyl chloride 8 was added to the reaction mixture at 15-20° C. The reaction was then stirred at room temperature overnight (19 h). LC-MS analysis indicated ca. 5% of starting material 7 still remained. The reaction was stopped and dichloromethane (30 mL) was added. The organic phase was washed with water (30 mL), saturated aqueous NH4Cl (30 mL), water (15 mL) and saturated aqueous NaHCO3 (30 mL). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 30-50% EtOAc in hexanes to afford ST-246 (0.34 g, 23% yield) as an off-white solid. Analytical data (1H NMR, LC-MS and HPLC by co-injection) were matched with those of ST-246 synthesized according to WO04112718 and were consistent.
EXAMPLE 2Synthetic Route II
Step A. Synthesis of Compound 9
A mixture of compound 4 (2.0 g, 9.8 mmol) and maleic anhydride 2 (0.96 g, 9.8 mmol, Aldrich powder, 95%) in o-xylene (100 mL, Aldrich anhydrous, 97%) was heated to reflux using a Dean-Stark trap apparatus overnight. After 18 h, LC-MS analysis at 215 nm showed the desired product 9 (86%), an uncyclized product (2.6%) and a dimer by-product (11.6%).
The reaction mixture was cooled to 45° C. and evaporated under reduced pressure. The residue was dissolved in EtOAc (50 mL) and the insoluble solid (mostly uncyclized product) was removed by filtration. The filtrate was concentrated and purified by column chromatography eluting with 50% EtOAc in hexanes to yield compound 9 (1.5 g, 54% yield) as an off-white solid. 1H NMR in CDCl3: δ 8.44 (s, 1H), 7.91 (d, 2H), 7.68 (d, 2H), 6.88 (s, 2H); Mass Spec: 285.1 (M+H)+
Step B. Synthesis of ST-246 (Route II)
A mixture of compound 9 (0.97 g, 3.4 mmol) and cycloheptatriene 1 (0.51 mL, 4.42 mmol, distilled before use, Aldrich tech 90%) in toluene (50 mL, Aldrich anhydrous) was heated at 95° C. under nitrogen atmosphere. After 1.5 h at 95° C., LC-MS analysis at 254 nm showed 29% conversion to the desired product (endo:exo=94:6). The resulting solution was continued to be heated at same temperature overnight. After 18 h at 95° C., LC-MS analysis indicated 75% conversion with an endo:exo ratio of 94:6. The reaction temperature was increased to 110° C. and the reaction was monitored. After heating at 110° C. for 7 h, LC-MS analysis at 254 nm showed 96.4% conversion to the desired product (endo:exo=94:6). The volatiles were removed by evaporation under reduced pressure and the reside was purified by column chromatography eluting with 30% EtOAc in hexanes to afford ST-246 (0.29 g, 22.6% yield, HPLC area 99.7% pure and 100% endo isomer) as a white solid. Analytical data (1H NMR, LC-MS and HPLC by co-injection) were matched with those of ST-246 synthesized according to WO04112718 and were consistent. An additional 0.5 g of ST-246 (38.9% yield, endo:exo=97:3) was recovered from column chromatography. Total Yield: 0.84 g (65.4% yield). 1H NMR of ST-246 exo isomer in CDCl3: δ 8.62 (s, 1H), 7.92 (d, 2H), 7.68 (d, 2H), 5.96 (m, 2H), 3.43 (s, 2H), 2.88 (s, 2H), 1.17 (s, 2H), 0.24 (q, 1H), 0.13 (m, 1H); Mass Spec: 377.1 (M+H)+
EXAMPLE 3Synthetic Route III
Step A. Synthesis of Compound 10
A mixture of maleic anhydride 2 (15.2 g, 155 mmol, Aldrich powder 95%) and tert-butyl carbazate 5 (20.5 g, 155 mmol, Aldrich, 98%) in anhydrous toluene (150 mL, Aldrich anhydrous) was heated to reflux using a Dean-Stark trap apparatus under nitrogen atmosphere. After refluxing for 2 h, no starting material 2 remained and LC-MS analysis at 254 nm showed the desired product 10 (20% by HPLC area), imine by-product (18%) and disubstituted by-product (56%). The reaction mixture was concentrated and purified by column chromatography eluting with 25% EtOAc in hexanes to afford compound 10 (5.98 g, 18% yield, HPLC area >99.5% pure) as a white solid. 1H NMR in DMSO-d6: δ 9.61 (s, 1H), 7.16 (s, 2H), 1.42 (s, 9H); Mass Spec: 235.1 (M+Na)+.
Step B. Synthesis of Compound 11 (HCl salt)
Compound 10 (3.82 g, 18 mmol) was dissolved in i-PrOAc (57 mL, Aldrich, 99.6%). 4M HCl in dioxane (15.8 mL, 63 mmol, Aldrich) was added drop-wise to the above solution keeping the temperature below 20° C. The solution was stirred overnight (24 h) at room temperature under nitrogen atmosphere. The resulting solid was filtered, washed with i-PrOAc (10 mL) and dried at 45° C. under vacuum for 1 h to afford HCl salt of compound 11 (2.39 g, 89% yield) as a white solid. 1H NMR in CD3OD: δ 6.98 (s, 2H); Mass Spec: 113.0 (M+H)+
Step C. Synthesis of Compound 9 (Route III)
To a mixture of compound 11 (1.19 g, 8 mmol) in dry dichloromethane (24 mL) was added diisopropylethylamine (2.93 mL, 16.8 mmol, Aldrich redistilled grade) keeping the temperature below 20° C. The resulting solution was stirred for 5 minute at 15-20° C. and to it was added 4-(trifluoromethyl)benzoyl chloride 8 (1.31 mL, 8.8 mmol, Aldrich, 97%) drop-wise. The reaction was stirred at room temperature for 5 h. LC-MS analysis showed the correct MW but the reaction was not complete. Additional 0.48 mL (0.4 equiv) of 4-(trifluoromethyl)benzoyl chloride 8 was added to the reaction mixture at 15-20° C. and the reaction mixture was stirred at room temperature overnight (21 h). The reaction was stopped and dichloromethane (50 mL) was added. The organic phase was washed with water (50 mL), saturated aqueous NH4Cl (50 mL), water (30 mL) and saturated aqueous NaHCO3 (30 mL). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 30-35% EtOAc in hexanes to afford compound 9 (0.8 g, 35% yield) as a light pink solid. Analytical data (1H NMR and LC-MS) were consistent with those of compound 9 obtained in Synthetic Route II.
Step D. Synthesis of ST-246 (Route III)
A mixture of compound 9 (0.5 g, 1.76 mmol) and cycloheptatriene 1 (0.33 mL, 3.17 mmol, distilled before to use, Aldrich tech 90%) in toluene (10 mL, Aldrich anhydrous) was heated at 110-115° C. under nitrogen atmosphere. After 6 h, LC-MS analysis at 254 nm showed 95% conversion to the desired product (endo:exo=94:6). The resulting solution was heated at same temperature overnight (22 h). LC-MS analysis at 254 nm showed no starting material 9 remained and the desired product (endo:exo=93:7). The reaction mixture was concentrated and purified by column chromatography eluting with 25-35% EtOAc in hexanes to afford ST-246 (0.39 g, HPLC area >99.5% pure with a ratio of endo:exo=99:1) as a white solid. Analytical data (1H NMR, LC-MS and HPLC by co-injection) were compared with those of ST-246 synthesized according to WO04112718 and were found to be consistent. An additional 0.18 g of ST-246 (HPLC area >99.5% pure, endo:exo=91:9) was recovered from column chromatography. Total Yield: 0.57 g (86% yield).
EXAMPLE 4Synthetic Route IV
Step A. Synthesis of Compound 10
A mixture of maleic anhydride 2 (3.4 g, 34.67 mmol, Aldrich powder, 95%) and tert-butyl carbazate 5 (4.6 g, 34.67 mmol, Aldrich, 98%) in anhydrous toluene (51 mL, Aldrich) was heated to reflux using a Dean-Stark trap apparatus under nitrogen atmosphere. After refluxing for 2.5 h, no starting material 2 remained and LC-MS analysis at 254 nm showed the desired product 10 (19% HPLC area), imine by-product (18%) and another by-product (56%). The reaction mixture was concentrated and purified by column chromatography eluting with 30% EtOAc in hexanes to afford compound 10 (1.0 g, 13.6% yield, HPLC area >99% pure) as a white solid. Analytical data (1H NMR and LC-MS) were consistent with those of compound 10 obtained in Synthetic Route III.
Step B. Synthesis of Compound 6
A mixture of compound 10 (4.4 g, 20.74 mmol) and cycloheptatriene 1 (3.22 mL, 31.1 mmol, distilled before to use, Aldrich tech 90%) in toluene (88 mL, 20 volume, Aldrich anhydrous) was heated at 95° C. under nitrogen atmosphere. After 15 h at 95° C., LC-MS analysis showed 83% conversion to the desired product. The reaction mixture was heated at 105° C. overnight. After total 40 h at 95-105° C., LC-MS analysis at 254 nm showed ˜99% conversion to the desired product (endo:exo=93:7). The reaction mixture was concentrated and the crude was purified by column chromatography eluting with 25-50% EtOAc in hexanes to afford compound 6 (2.06 g, 32.6% yield, HPLC area 99.9% pure and 100% endo isomer) as a white solid. 1H NMR and LC-MS were consistent with those of compound 6 obtained in Synthetic Route I. An additional 4.0 g of 6 (63.4% yield, HPLC area 93% pure with a ratio of endo:exo=91:9) was recovered from column chromatography. Total Yield: 6.06 g (96% yield).
Step C. Synthesis of Compound 7 (HCl salt)
Compound 6 (2.05 g, 6.74 mmol) was dissolved in i-PrOAc (26 mL, Aldrich, 99.6%). 4M HCl in dioxane (5.9 mL, 23.58 mmol, Aldrich) was added drop-wise to the above solution keeping the temperature below 20° C. The solution was stirred overnight (18 h) at room temperature under nitrogen atmosphere. The resulting solid was filtered, washed with i-PrOAc (5 mL) and dried under vacuum to yield HCl salt of compound 7 (1.57 g, 97% yield) as a white solid. Analytical data (1H NMR and LC-MS) were consistent with those of compound 7 in Synthetic Route I.
Step D. Synthesis of ST-246 (Route IV)
To a mixture of compound 7 (0.84 g, 3.5 mmol) in dichloromethane (13 mL) was added diisopropylethylamine (1.34 mL, 7.7 mmol) keeping the temperature below 20° C. and the resulting solution was stirred for 5-10 minutes. 4-(Trifluoromethyl)benzoyl chloride 8 (0.57 mL, 3.85 mmol, Aldrich, 97%) was added to above solution keeping the temperature below 20° C. The reaction mixture was stirred at room temperature for 2 h. Additional 0.2 mL (0.4 equiv) of 4-(trifluoromethyl)benzoyl chloride 8 was added to the reaction keeping the temperature below 20° C. The reaction was stirred at room temperature overnight (24 h). The reaction mixture was diluted with dichloromethane (20 mL). The organic phase was washed with water (20 mL), saturated aqueous NH4Cl (20 mL), water (20 mL) and saturated aqueous NaHCO3 (20 mL). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 30-35% EtOAc in hexanes to afford ST-246 (0.25 g, 19% yield, HPLC area >99.5% pure) as a white solid. Analytical data (1H NMR and LC-MS) were consistent with those of ST-246 synthesized according to WO04112718.
EXAMPLE 5Synthetic Route V
Step A. Synthesis of Compound 13
To a mixture of compound 7 (1.6 g, 6.65 mmol, synthesized according to Synthetic Route I) in dichloromethane (80 mL,) was added triethylamine (2.04 mL, 14.63 mmol) keeping the temperature below 20° C. and the resulting solution was stirred for 5-10 minute. 4-Iodobenzoyl chloride 12 (1.95 g, 7.31 mmol, 1.1 equiv, Aldrich) was added portion-wise under nitrogen atmosphere to the above solution keeping the temperature below 20° C. The reaction mixture was stirred at room temperature overnight. After 17 h and 19 h, additional 0.35 g (0.2 equiv) of acid chloride 12 was added to the reaction keeping the temperature below 20° C. After 24 h, additional 0.18 g (0.1 equiv, used total 1.6 equiv) of acid chloride 12 was added and the reaction was continued to stir at room temperature overnight (total 43 h). LC-MS analysis at 215 nm showed 43% of the desired product (13) and ˜5% of compound 7. The reaction was diluted with dichloromethane (100 mL). The organic phase was washed with saturated aqueous NH4Cl (100 mL), water (100 mL) and saturated aqueous NaHCO3 (100 mL). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 25-50% EtOAc in hexanes to afford compound 13 (1.63 g, 57% yield, HPLC area 93% pure) as a white solid. 1H NMR in DMSO-d6: δ 11.19 and 10.93 (two singlets with integration ratio of 1.73:1, total of 1H, same proton of two rotamers), 7.93 (d, 2H), 7.66 (d, 2H), 5.80 (s, 2H), 3.36 (s, 2H), 3.27 (s, 2H), 1.18 (s, 2H), 0.27 (q, 1H), 0.06 (s, 1H); Mass Spec: 435.0 (M+H)+
Step B. Synthesis of ST-246 (Route V)
Anhydrous DMF (6 mL) was added to a mixture of compound 13 (0.2 g, 0.46 mmol), methyl 2,2-difluoro-2-(fluorosulfonyl)acetate (0.44 mL, 3.45 mmol, Aldrich) and copper (I) iodide (90 mg, 0.47 mmol). The reaction mixture was stirred at −90° C. for 4 h. LC-MS analysis at 254 nm indicated no starting material 13 remained and showed 48% HPLC area of ST-246. The reaction mixture was cooled to 45° C. and DMF was removed under reduced pressure. The residue was slurried in EtOAc (30 mL) and insoluble solid was removed by filtration. The filtrate was concentrated and purified by column chromatography eluting with 25-35% EtOAc in hexanes to afford ST-246 (55 mg, 32% yield, 95% pure by HPLC at 254 nm) as off-white solid. Analytical data (1H NMR and LC-MS) were consistent with those of ST-246 synthesized according to WO04112718.
PATENT
Example 1 : Synthetic Route I:

P = Boc
Scheme 1
Step A. Synthesis of Compound 6 (P = Boc)
To a mixture of compound 3 (5.0 g, 26.3 mmol, synthesized according to WO041 12718) in EtOH (80 mL, EMD, AX0441 -3) was added terf-butyl carbazate 5 (3.65 g, 27.6 mmol, Aldrich, 98%). The reaction mixture was heated to reflux for 4 h under nitrogen atmosphere. LC-MS analysis of the reaction mixture showed less than 5% of compound 3 remained. The reaction mixture was evaporated under reduced pressure. The residue was recrystallized from EtOAc – hexanes, the solid was filtered, washed with hexanes (50 mL) and dried under vacuum to afford compound 6 (3.1 g, 39% yield) as a white solid. The filtrate was concentrated and purified by column chromatography eluting with 25% EtOAc in hexanes to give an additional 3.64 g (46% yield) of compound 6 as a white solid. Total yield: 6.74 g (84% yield). 1H NMR in CDCI3: δ 6.30 (br s, 1 H), 5.79 (t, 2H), 3.43 (s, 2H), 3.04 (s, 2H), 1 .46 (s, 9H), 1 .06-1 .16 (m, 2H), 0.18-0.36 (m, 2H); Mass Spec: 327.2 (M+Na)+
Step B. Synthesis of Compound 7 (HCI salt) Compound 6 (3.6 g, 1 1 .83 mmol) was dissolved in /‘-PrOAc (65 mL, Aldrich, 99.6%). 4M HCI in dioxane (10.4 mL, 41 .4 mmol, Aldrich) was added drop-wise to the above solution keeping the temperature below 20 °C. The reaction mixture was stirred at room temperature overnight (18 h) under nitrogen atmosphere. The resulting solid was filtered, washed with /‘-PrOAc (15 mL) and dried under vacuum to yield HCI salt of compound 7 (1 .9 g, 67% yield) as a white solid. The filtrate was concentrated to 1/3 its volume and stirred at 10 – 15 °C for 30 min. The solid was filtered, washed with minimal volume of /‘-PrOAc and dried to afford additional 0.6 g (21 % yield) of compound 7. Total yield: 2.5 g (88% yield). 1 H NMR in DMSO-d6: δ 6.72 (br s, 3H), 5.68 (m, 2H), 3.20 (s, 2H), 3.01 (s, 2H), 1 .07-1 .17 (m, 2H), 0.18-0.29 (m, 1 H), -0.01 -0.07 (m, 1 H); Mass Spec: 205.1 (M+H)+
Step C. Synthesis of ST-246
To a mixture of compound 7 (0.96 g, 4 mmol) in dry dichloromethane (19 mL) was added triethylamine (1 .17 mL, 8.4 mmol, Aldrich) keeping the temperature below 20 °C. The resulting solution was stirred for 5 minutes at 15 – 20 °C, to it was added drop-wise 4-(trifluoromethyl)benzoyl chloride 8 (0.63 mL, 4.2 mmol, Aldrich, 97%) and the reaction mixture was stirred at room temperature overnight (18 h). LC-MS and TLC analysis showed the correct molecular weight and Rf value of ST-246 but the reaction was not complete. Additional 0.3 mL (2 mmol, 0.5 eq) of 4-(trifluoromethyl)benzoyl chloride 8 was added to the reaction mixture at 15 – 20 °C. The reaction was then stirred at room temperature overnight (19 h). LC-MS analysis indicated ca. 5% of starting material 7 still remained. The reaction was stopped and dichloromethane (30 mL) was added. The organic phase was washed with water (30 mL), saturated aqueous NH CI (30 mL), water (15 mL) and saturated aqueous NaHCO3 (30 mL). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 30 – 50% EtOAc in hexanes to afford ST-246 (0.34 g, 23% yield) as an off-white solid. Analytical data (1H NMR, LC-MS and HPLC by co-injection) were matched with those of ST-246 synthesized according to WO041 12718 and were consistent. Example 2: Synthetic Route II

Scheme 2
Step A. Synthesis of Compound 9
A mixture of compound 4 (2.0 g, 9.8 mmol) and maleic anhydride 2 (0.96 g, 9.8 mmol, Aldrich powder, 95%) in o-xylene (100 mL, Aldrich anhydrous, 97%) was heated to reflux using a Dean-Stark trap apparatus overnight. After 18 h, LC-MS analysis at 215 nm showed the desired product 9 (86%), an uncyclized product (2.6%) and a dimer by-product (1 1 .6%).

Uncyclized product (MS = 303) Dimer by-product (MS = 489)
The reaction mixture was cooled to 45 °C and evaporated under reduced pressure. The residue was dissolved in EtOAc (50 mL) and the insoluble solid (mostly uncyclized product) was removed by filtration. The filtrate was concentrated and purified by column chromatography eluting with 50% EtOAc in hexanes to yield compound 9 (1 .5 g, 54% yield) as an off-white solid. 1 H NMR in CDCI3: δ 8.44 (s, 1 H), 7.91 (d, 2H), 7.68 (d, 2H), 6.88 (s, 2H); Mass Spec: 285.1 (M+H)+
Step B. Synthesis of ST-246 (Route II)
A mixture of compound 9 (0.97 g, 3.4 mmol) and cycloheptatriene 1 (0.51 mL, 4.42 mmol, distilled before use, Aldrich tech 90%) in toluene (50 mL, Aldrich anhydrous) was heated at 95 °C under nitrogen atmosphere. After 1 .5 h at 95 °C, LC-MS analysis at 254 nm showed 29% conversion to the desired product (endo:exo = 94:6). The resulting solution was continued to be heated at same temperature overnight. After 18 h at 95 °C, LC-MS analysis indicated 75% conversion with an endo:exo ratio of 94:6. The reaction temperature was increased to 1 10 °C and the reaction was monitored. After heating at 1 10 °C for 7 h, LC-MS analysis at 254 nm showed 96.4% conversion to the desired product (endo:exo = 94:6). The volatiles were removed by evaporation under reduced pressure and the reside was purified by column chromatography eluting with 30% EtOAc in hexanes to afford ST-246 (0.29 g, 22.6% yield, HPLC area 99.7% pure and 100% endo isomer) as a white solid. Analytical data (1H NMR, LC-MS and HPLC by co- injection) were matched with those of ST-246 synthesized according to WO041 12718 and were consistent. An additional 0.5 g of ST-246 (38.9% yield, endo:exo = 97: 3) was recovered from column chromatography. Total Yield: 0.84 g (65.4% yield). 1H NMR of ST-246 exo isomer in CDCI3: δ 8.62 (s, 1 H), 7.92 (d, 2H), 7.68 (d, 2H), 5.96 (m, 2H), 3.43 (s, 2H), 2.88 (s, 2H), 1 .17 (s, 2H), 0.24 (q, 1 H), 0.13 (m, 1 H); Mass Spec: 377.1 (M+H)+
Example 3: Synthetic Route III

ST-246 9
P = Boc Scheme 3
Step A. Synthesis of Compound 10
A mixture of maleic anhydride 2 (15.2 g, 155 mmol, Aldrich powder 95%) and terf-butyl carbazate 5 (20.5 g, 155 mmol, Aldrich, 98%) in anhydrous toluene (150 mL, Aldrich anhydrous) was heated to reflux using a Dean-Stark trap apparatus under nitrogen atmosphere. After refluxing for 2 h, no starting material 2 remained and LC-MS analysis at 254 nm showed the desired product 10 (20% by HPLC area), imine byproduct (18%) and disubstituted by-product (56%). The reaction mixture was concentrated and purified by column chromatography eluting with 25% EtOAc in hexanes to afford compound 10 (5.98 g, 18% yield, HPLC area >99.5% pure) as a white solid. 1 H NMR in DMSO-d6: δ 9.61 (s, 1 H), 7.16 (s, 2H), 1 .42 (s, 9H); Mass Spec: 235.1 (M+Na)+. duct

C9H12N204 C14H22N405
Mol. Wt.: 212.2 Mol. Wt.: 326.35
Step B. Synthesis of Compound 11 (HCI salt)
Compound 10 (3.82 g, 18 mmol) was dissolved in /‘-PrOAc (57 mL, Aldrich, 99.6%). 4M HCI in dioxane (15.8 mL, 63 mmol, Aldrich) was added drop-wise to the above solution keeping the temperature below 20 °C. The solution was stirred overnight (24 h) at room temperature under nitrogen atmosphere. The resulting solid was filtered, washed with /‘-PrOAc (10 mL) and dried at 45 °C under vacuum for 1 h to afford HCI salt of compound 11 (2.39 g, 89% yield) as a white solid. 1 H NMR in CD3OD: δ 6.98 (s, 2H); Mass Spec: 1 13.0 (M+H)+ Step C. Synthesis of Compound 9 (Route III)
To a mixture of compound 11 (1 .19 g, 8 mmol) in dry dichloromethane (24 mL) was added diisopropylethylannine (2.93 mL, 16.8 mmol, Aldrich redistilled grade) keeping the temperature below 20 °C. The resulting solution was stirred for 5 minute at 15 – 20 °C and to it was added 4-(trifluoromethyl)benzoyl chloride 8 (1 .31 mL, 8.8 mmol, Aldrich, 97%) drop-wise. The reaction was stirred at room temperature for 5 h. LC-MS analysis showed the correct MW but the reaction was not complete. Additional 0.48 mL (0.4 equiv) of 4-(trifluoromethyl)benzoyl chloride 8 was added to the reaction mixture at 15 – 20 °C and the reaction mixture was stirred at room temperature overnight (21 h). The reaction was stopped and dichloromethane (50 mL) was added. The organic phase was washed with water (50 mL), saturated aqueous NH4CI (50 mL), water (30 mL) and saturated aqueous NaHCO3 (30 mL). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 30 – 35% EtOAc in hexanes to afford compound 9 (0.8 g, 35% yield) as a light pink solid. Analytical data (1H NMR and LC-MS) were consistent with those of compound 9 obtained in Synthetic Route II.
Step D. Synthesis of ST-246 (Route III)
A mixture of compound 9 (0.5 g, 1 .76 mmol) and cycloheptatriene 1 (0.33 mL, 3.17 mmol, distilled before to use, Aldrich tech 90%) in toluene (10 mL, Aldrich anhydrous) was heated at 1 10 – 1 15 °C under nitrogen atmosphere. After 6 h, LC-MS analysis at 254 nm showed 95% conversion to the desired product (endo:exo = 94:6). The resulting solution was heated at same temperature overnight (22 h). LC-MS analysis at 254 nm showed no starting material 9 remained and the desired product (endo:exo = 93:7). The reaction mixture was concentrated and purified by column chromatography eluting with 25 – 35% EtOAc in hexanes to afford ST-246 (0.39 g, HPLC area >99.5% pure with a ratio of endo:exo = 99:1 ) as a white solid. Analytical data (1 H NMR, LC-MS and HPLC by co-injection) were compared with those of ST-246 synthesized according to WO041 12718 and were found to be consistent. An additional 0.18 g of ST-246 (HPLC area >99.5% pure, endo:exo = 91 : 9) was recovered from column chromatography. Total Yield: 0.57 g (86% yield).
Example 4 ; Synthetic Route IV:

P = Boc
Scheme 4
Step A. Synthesis of Compound 10
A mixture of maleic anhydride 2 (3.4 g, 34.67 mmol, Aldrich powder, 95%) and terf-butyl carbazate 5 (4.6 g, 34.67 mmol, Aldrich, 98%) in anhydrous toluene (51 ml_, Aldrich) was heated to reflux using a Dean-Stark trap apparatus under nitrogen atmosphere. After refluxing for 2.5 h, no starting material 2 remained and LC-MS analysis at 254 nm showed the desired product 10 (19% HPLC area), imine by-product (18%) and another by-product (56%). The reaction mixture was concentrated and purified by column chromatography eluting with 30% EtOAc in hexanes to afford compound 10 (1 .0 g, 13.6% yield, HPLC area >99% pure) as a white solid. Analytical data (1H NMR and LC-MS) were consistent with those of compound 10 obtained in Synthetic Route III. Im ine by-product

Mol. Wt.: 212.2
Step B. Synthesis of Compound 6
A mixture of compound 10 (4.4 g, 20.74 mmol) and cycloheptatriene 1 (3.22 mL, 31 .1 mmol, distilled before to use, Aldrich tech 90%) in toluene (88 mL, 20 volume, Aldrich anhydrous) was heated at 95 °C under nitrogen atmosphere. After 15 h at 95 °C, LC-MS analysis showed 83% conversion to the desired product. The reaction mixture was heated at 105 °C overnight. After total 40 h at 95 – 105 °C, LC-MS analysis at 254 nm showed -99% conversion to the desired product (endo:exo = 93:7). The reaction mixture was concentrated and the crude was purified by column chromatography eluting with 25 – 50 % EtOAc in hexanes to afford compound 6 (2.06 g, 32.6% yield, HPLC area 99.9% pure and 100% endo isomer) as a white solid. 1 H NMR and LC-MS were consistent with those of compound 6 obtained in Synthetic Route I. An additional 4.0 g of 6 (63.4% yield, HPLC area 93% pure with a ratio of endo:exo = 91 : 9) was recovered from column chromatography. Total Yield: 6.06 g (96% yield).
Step C. Synthesis of Compound 7 (HCI salt)
Compound 6 (2.05 g, 6.74 mmol) was dissolved in /‘-PrOAc (26 mL, Aldrich, 99.6%). 4M HCI in dioxane (5.9 mL, 23.58 mmol, Aldrich) was added drop-wise to the above solution keeping the temperature below 20 °C. The solution was stirred overnight (18 h) at room temperature under nitrogen atmosphere. The resulting solid was filtered, washed with /‘-PrOAc (5 mL) and dried under vacuum to yield HCI salt of compound 7 (1 .57 g, 97% yield) as a white solid. Analytical data (1 H NMR and LC-MS) were consistent with those of compound 7 in Synthetic Route I.
Step D. Synthesis of ST-246 (Route IV) To a mixture of compound 7 (0.84 g, 3.5 mmol) in dichloromethane (13 mL) was added diisopropylethylamine (1 .34 mL, 7.7 mmol) keeping the temperature below 20 °C and the resulting solution was stirred for 5 – 10 minutes. 4-(Trifluoromethyl)benzoyl chloride 8 (0.57 mL, 3.85 mmol, Aldrich, 97%) was added to above solution keeping the temperature below 20 °C. The reaction mixture was stirred at room temperature for 2 h. Additional 0.2 mL (0.4 equiv) of 4-(trifluoromethyl)benzoyl chloride 8 was added to the reaction keeping the temperature below 20 °C. The reaction was stirred at room temperature overnight (24 h). The reaction mixture was diluted with dichloromethane (20 mL). The organic phase was washed with water (20 mL), saturated aqueous NH4CI (20 mL), water (20 mL) and saturated aqueous NaHCO3 (20 mL). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 30 – 35% EtOAc in hexanes to afford ST-246 (0.25 g, 19% yield, HPLC area >99.5% pure) as a white solid. Analytical data (1H NMR and LC-MS) were consistent with those of ST-246 synthesized according to WO041 12718.
Example 5: Synthetic Route V:

Scheme 5 Step A. Synthesis of Compound 13
To a mixture of compound 7 (1 .6 g, 6.65 mmol, synthesized according to Synthetic Route I) in dichloromethane (80 ml_,) was added triethylamine (2.04 ml_, 14.63 mmol) keeping the temperature below 20 °C and the resulting solution was stirred for 5 – 10 minute. 4-lodobenzoyl chloride 12 (1 .95 g, 7.31 mmol, 1 .1 equiv, Aldrich) was added portion-wise under nitrogen atmosphere to the above solution keeping the temperature below 20 °C. The reaction mixture was stirred at room temperature overnight. After 17 h and 19 h, additional 0.35 g (0.2 equiv) of acid chloride 12 was added to the reaction keeping the temperature below 20 °C. After 24 h, additional 0.18 g (0.1 equiv, used total 1 .6 equiv) of acid chloride 12 was added and the reaction was continued to stir at room temperature overnight (total 43 h). LC-MS analysis at 215 nm showed 43% of the desired product (13) and -5% of compound 7. The reaction was diluted with dichloromethane (100 ml_). The organic phase was washed with saturated aqueous NH4CI (100 ml_), water (100 ml_) and saturated aqueous NaHCO3 (100 ml_). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 25 – 50% EtOAc in hexanes to afford compound 13 (1 .63 g, 57% yield, HPLC area 93% pure) as a white solid. 1 H NMR in DMSO-d6: δ 1 1 .19 and 10.93 (two singlets with integration ratio of 1 .73:1 , total of 1 H, same proton of two rotamers), 7.93 (d, 2H), 7.66 (d, 2H), 5.80 (s, 2H), 3.36 (s, 2H), 3.27 (s, 2H), 1 .18 (s, 2H), 0.27 (q, 1 H), 0.06 (s,1 H); Mass Spec: 435.0 (M+H)+
Step B. Synthesis of ST-246 (Route V)
Anhydrous DMF (6 ml_) was added to a mixture of compound 13 (0.2 g, 0.46 mmol), methyl 2, 2-difluoro-2-(fluorosulfonyl)acetate (0.44 ml_, 3.45 mmol, Aldrich) and copper (I) iodide (90 mg, 0.47 mmol). The reaction mixture was stirred at -90 °C for 4 h. LC-MS analysis at 254 nm indicated no starting material 13 remained and showed 48% HPLC area of ST-246. The reaction mixture was cooled to 45 °C and DMF was removed under reduced pressure. The residue was slurried in EtOAc (30 mL) and insoluble solid was removed by filtration. The filtrate was concentrated and purified by column chromatography eluting with 25 – 35% EtOAc in hexanes to afford ST-246 (55 mg, 32% yield, 95% pure by HPLC at 254 nm) as off-white solid. Analytical data (1H NMR and LC-MS) were consistent with those of ST-246 synthesized according to WO041 12718.
History
Originally researched by the National Institute of Allergy and Infectious Diseases, the drug was owned by Viropharma and discovered in collaboration with scientists at the United States Army Medical Research Institute of Infectious Diseases.[] It is owned and manufactured by Siga Technologies. Siga and Viropharma were issued a patent for tecovirimat in 2012.[20]
Clinical trials
As of 2009, the results of clinical trials support its use against smallpox and other related orthopoxviruses. It shows potential for a variety of uses including preventive healthcare, as a post-exposure therapeutic, as a therapeutic, and an adjunct to vaccination.[21][
Tecovirimat can be taken by mouth and as of 2008, was permitted for phase II trials by the U.S. Food and Drug Administration (FDA). In phase I trials, tecovirimat was generally well tolerated with no serious adverse events.[22] Due to its importance for biodefense, the FDA designated tecovirimat for fast-track status, creating a path for expedited FDA review and eventual regulatory approval. On 13 July 2018, the FDA announced approval of tecovirimat.[23]
Society and culture
Legal status
In November 2021, the Committee for Medicinal Products for Human Use of the European Medicines Agency adopted a positive opinion, recommending the granting of a marketing authorization under exceptional circumstances for the medicinal product tecovirimat siga, intended for the treatment of orthopoxvirus disease (smallpox, monkeypox, cowpox, and vaccinia complications) in adults and in children who weigh at least 13 kilograms (29 lb)[24] The applicant for this medicinal product is Siga Technologies Netherlands B.V.[24] Tecovirimat was approved for medical use in the European Union in January 2022.[5][25]
In December 2021, Health Canada approved oral tecovirimat for the treatment of smallpox in people weighing at least 13 kilograms (29 lb).[26][27]
As of August 2022, Tpoxx is available in the US only through the Strategic National Stockpile as a Centers for Disease Control and Prevention investigational new drug.[28][29] Intravenous Tpoxx has no lower weight cap and can be used in infants under the investigational new drug protocol.[30]
/////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
References
- ^ “Notice: Multiple Additions to the Prescription Drug List (PDL) [2022-01-24]”. Health Canada. 24 January 2022. Archived from the original on 29 May 2022. Retrieved 28 May 2022.
- ^ “New Medicines Approved in 2018”. Health Canada. 15 January 2020. Archived from the original on 29 May 2022. Retrieved 28 May 2022.
- ^ “Summary Basis of Decision (SBD) for Tpoxx”. Health Canada. 23 October 2014. Archived from the original on 29 May 2022. Retrieved 29 May 2022.
- ^ Jump up to:a b c d “Tpoxx- tecovirimat monohydrate capsule”. DailyMed. 2 December 2021. Archived from the original on 23 April 2022. Retrieved 23 April 2022.
- ^ Jump up to:a b c “Tecovirimat Siga EPAR”. European Medicines Agency. 10 November 2021. Archived from the original on 16 May 2022. Retrieved 23 April 2022. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b McNeil Jr DG (13 July 2018). “Drug to Treat Smallpox Approved by F.D.A., a Move Against Bioterrorism”. The New York Times. Archived from the original on 28 March 2019. Retrieved 16 July 2018.
- ^ Nakoune E, Olliaro P (May 2022). “Waking up to monkeypox”. BMJ. 377: o1321. doi:10.1136/bmj.o1321. PMID 35613732. S2CID 249047112.
- ^ Adler H, Gould S, Hine P, Snell LB, Wong W, Houlihan CF, et al. (May 2022). “Clinical features and management of human monkeypox: a retrospective observational study in the UK”. The Lancet. Infectious Diseases. 22 (8): 1153–1162. doi:10.1016/S1473-3099(22)00228-6. PMC 9300470. PMID 35623380. S2CID 249057804.
- ^ “FDA approves the first drug with an indication for treatment of smallpox”. U.S. Food and Drug Administration (FDA) (Press release). 13 July 2018. Archived from the original on 23 April 2019. Retrieved 1 August 2018.
- ^ “U.S. Food and Drug Administration Approves Siga Technologies’ Tpoxx (tecovirimat) for the Treatment of Smallpox”. Siga (Press release). Archived from the original on 21 September 2018. Retrieved 14 July 2018.
- ^ Grosenbach DW, Honeychurch K, Rose EA, Chinsangaram J, Frimm A, Maiti B, et al. (July 2018). “Oral Tecovirimat for the Treatment of Smallpox”. The New England Journal of Medicine. 379 (1): 44–53. doi:10.1056/NEJMoa1705688. PMC 6086581. PMID 29972742.
- ^ Whitehouse ER, Rao AK, Yu YC, Yu PA, Griffin M, Gorman S, et al. (October 2019). “Novel Treatment of a Vaccinia Virus Infection from an Occupational Needlestick – San Diego, California, 2019” (PDF). MMWR. Morbidity and Mortality Weekly Report. 68 (42): 943–946. doi:10.15585/mmwr.mm6842a2. PMC 6812835. PMID 31647789. Archived (PDF) from the original on 2 August 2022. Retrieved 2 August 2022.
- ^ Damon IK, Damaso CR, McFadden G (May 2014). “Are we there yet? The smallpox research agenda using variola virus”. PLOS Pathogens. 10 (5): e1004108. doi:10.1371/journal.ppat.1004108. PMC 4006926. PMID 24789223.
- ^ Cunningham A (13 July 2018). “FDA approves the first smallpox treatment”. Archived from the original on 12 July 2018. Retrieved 4 May 2018.
- ^ New Drug Therapy Approvals 2018 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2019. Archived from the original on 17 September 2020. Retrieved 16 September 2020.
- ^ Yang G, Pevear DC, Davies MH, Collett MS, Bailey T, Rippen S, et al. (October 2005). “An orally bioavailable antipoxvirus compound (ST-246) inhibits extracellular virus formation and protects mice from lethal orthopoxvirus Challenge”. Journal of Virology. 79 (20): 13139–13149. doi:10.1128/JVI.79.20.13139-13149.2005. PMC 1235851. PMID 16189015.
- ^ Jump up to:a b AU patent 2004249250, Bailey, Thomas R.; Jordan, Robert & Rippin, Susan R., “Compounds, compositions and methods for treatment and prevention of orthopoxvirus infections and associated diseases”, published 2004-12-29, assigned to Siga Pharmaceuticals Inc
- ^ Ishitobi, Hiroyuki; Tanida, Hiroshi; Tori, Kazuo; Tsuji, Teruji (1971). “Re-examination of the Cycloaddition of Cycloheptatriene with Maleic Anhydride”. Bulletin of the Chemical Society of Japan. 44 (11): 2993–3000. doi:10.1246/bcsj.44.2993.
- ^ Hughes, David L. (2019). “Review of the Patent Literature: Synthesis and Final Forms of Antiviral Drugs Tecovirimat and Baloxavir Marboxil”. Organic Process Research & Development. 23 (7): 1298–1307. doi:10.1021/acs.oprd.9b00144. S2CID 197172102.
- ^ U.S. Patent 8,124,643
- ^ “Siga Technologies”. Archived from the original on 20 February 2012. Retrieved 18 September 2009.
- ^ Jordan R, Tien D, Bolken TC, Jones KF, Tyavanagimatt SR, Strasser J, et al. (May 2008). “Single-dose safety and pharmacokinetics of ST-246, a novel orthopoxvirus egress inhibitor”. Antimicrobial Agents and Chemotherapy. 52 (5): 1721–1727. doi:10.1128/AAC.01303-07. PMC 2346641. PMID 18316519.
- ^ Commissioner, Office of the (24 March 2020). “Press Announcements – FDA approves the first drug with an indication for treatment of smallpox”. U.S. Food and Drug Administration (FDA). Archived from the original on 23 April 2019. Retrieved 14 July 2018.
- ^ Jump up to:a b “Tecovirimat Siga: Pending EC decision”. European Medicines Agency. 11 November 2021. Archived from the original on 13 November 2021. Retrieved 13 November 2021. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “Summary of Product Characteristics” (PDF). European Medicines Agency. Archived (PDF) from the original on 21 May 2022. Retrieved 24 May 2022.
- ^ “Notice: Multiple Additions to the Prescription Drug List (PDL) [2022-01-24]”. Health Canada. 24 January 2022. Archived from the original on 29 May 2022. Retrieved 28 May 2022.
- ^ “Siga Announces Health Canada Regulatory Approval of Oral Tpoxx” (Press release). Siga Technologies. 1 December 2021. Archived from the original on 24 May 2022. Retrieved 24 May 2022.
- ^ “Information for Healthcare Providers on Obtaining and Using TPOXX (Tecovirimat) for Treatment of Monkeypox”. U.S. Centers for Disease Control and Prevention (CDC). 22 July 2022. Archived from the original on 31 July 2022. Retrieved 1 August 2022.
- ^ “Steps for Clinicians to Order Medication to Treat Monkeypox”. Coca Now. 19 July 2022. Archived from the original on 2 August 2022. Retrieved 24 July 2022.
- ^ “Monkeypox Outbreak: Updates on the Epidemiology, Testing, Treatment, and Vaccination” (PDF). U.S. Centers for Disease Control and Prevention. Archived (PDF) from the original on 2 August 2022. Retrieved 27 July 2022.
External links
- “Tecovirimat”. Drug Information Portal. U.S. National Library of Medicine.
- “Tecovirimat monohydrate”. Drug Information Portal. U.S. National Library of Medicine.
 |
|
| Clinical data | |
|---|---|
| Trade names | Tpoxx |
| Other names | ST-246 |
| AHFS/Drugs.com | Monograph |
| License data |
|
| Routes of administration |
By mouth, intravenous |
| ATC code | |
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C19H15F3N2O3 |
| Molar mass | 376.335 g·mol−1 |
| 3D model (JSmol) | |
FDA approves the first drug with an indication for treatment of smallpox
The U.S. Food and Drug Administration today approved TPOXX (tecovirimat), the first drug with an indication for treatment of smallpox. Though the World Health Organization declared smallpox, a contagious and sometimes fatal infectious disease, eradicated in 1980, there have been longstanding concerns that smallpox could be used as a bioweapon.
“To address the risk of bioterrorism, Congress has taken steps to enable the development and approval of countermeasures to thwart pathogens that could be employed as weapons. Today’s approval provides an important milestone in these efforts. This new treatment affords us an additional option should smallpox ever be used as a bioweapon,” said FDA Commissioner Scott Gottlieb, M.D. “This is the first product to be awarded a Material Threat Medical Countermeasure priority review voucher. Today’s action reflects the FDA’s commitment to ensuring that the U.S. is prepared for any public health emergency with timely, safe and effective medical products.”
July 13, 2018
Release
The U.S. Food and Drug Administration today approved TPOXX (tecovirimat), the first drug with an indication for treatment of smallpox. Though the World Health Organization declared smallpox, a contagious and sometimes fatal infectious disease, eradicated in 1980, there have been longstanding concerns that smallpox could be used as a bioweapon.
“To address the risk of bioterrorism, Congress has taken steps to enable the development and approval of countermeasures to thwart pathogens that could be employed as weapons. Today’s approval provides an important milestone in these efforts. This new treatment affords us an additional option should smallpox ever be used as a bioweapon,” said FDA Commissioner Scott Gottlieb, M.D. “This is the first product to be awarded a Material Threat Medical Countermeasure priority review voucher. Today’s action reflects the FDA’s commitment to ensuring that the U.S. is prepared for any public health emergency with timely, safe and effective medical products.”
Prior to its eradication in 1980, variola virus, the virus that causes smallpox, was mainly spread by direct contact between people. Symptoms typically began 10 to 14 days after infection and included fever, exhaustion, headache and backache. A rash initially consisting of small, pink bumps progressed to pus-filled sores before finally crusting over and scarring. Complications of smallpox could include encephalitis (inflammation of the brain), corneal ulcerations (an open sore on the clear, front surface of the eye) and blindness.
TPOXX’s effectiveness against smallpox was established by studies conducted in animals infected with viruses that are closely related to the virus that causes smallpox, and was based on measuring survival at the end of the studies. More animals treated with TPOXX lived compared to the animals treated with placebo. TPOXX was approved under the FDA’s Animal Rule, which allows efficacy findings from adequate and well-controlled animal studies to support an FDA approval when it is not feasible or ethical to conduct efficacy trials in humans.
The safety of TPOXX was evaluated in 359 healthy human volunteers without a smallpox infection. The most frequently reported side effects were headache, nausea and abdominal pain.
The FDA granted this application Fast Track and Priority Review designations. TPOXX also received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases and a Material Threat Medical Countermeasure Priority Review Voucher, which provides additional incentives for certain medical products intended to treat or prevent harm from specific chemical, biological, radiological and nuclear threats.
The FDA granted approval of TPOXX to SIGA Technologies Inc.
TPOXX was developed in conjunction with the U.S. Department of Health and Human Services’ Biomedical Advanced Research and Development Authority (BARDA).
Tecovirimat


Tecovirimat
4-trifluoromethyl-N-(3,3a,4,4a,5,5a,6,6a-octahydro-1,3-dioxo-4,6-ethenocycloprop(f)isoindol-2(1H)-yl)-benzamide
N- [(3aR,4R,4aR,5aS,6S, 6aS)- 3,3a,4,4a,5,5a,6,6a- octahydro-1,3-dioxo- 4,6- ethenocycloprop[f]iso- indol-2(1H)-yl]-4- (trifluoromethyl)- benzamide
4 -trifluoromethyl -N- (3, 3a, 4, 4a, 5, 5a, 6, 6a- octahydro-1, 3 -dioxo-4, 6 -ethenocycloprop [f] isoindol -2 ( 1H) -yl ) – benzamide
Details
NDA FILED IN US
2006 ORPHAN DRUG DESIGNATION IN US FOR SMALL POX
2010 ORPHAN DRUG DESIGNATION IN US FOR ORTHOPOX VIRUS
A core protein cysteine protease inhibitor potentially for treatment of smallpox infection.
SIGA TECHNOLOGIES INNOVATOR
SIGA-246; ST-246
SYN
Tecovirimat (Tpoxx)
Tecovirimat is a drug used for the
treatment or prophylaxis of viral infections, particularly those caused by the
orthopoxvirus (Figure 12).
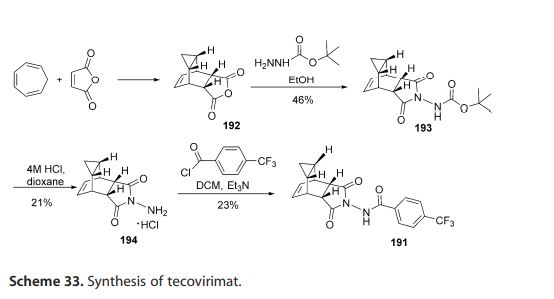
In 2015, Dai described a procedure
for the preparation of tecovirimat in a
US patent (Scheme 33).[57 ] The developed method started with a cycloaddition reaction of cycloheptatriene
with maleic anhydride in xylene to
yield adduct 192, which after reaction
with tert-butyl carbazate provided compound 193. Deprotection in acidic media gave rise to hydrazine derivative 194 and
subsequent reaction with p-trifluoromethylbenzoyl chloride afforded tecovirimat (191).
57 [57] D. Dai, US Patent 0322010, 2015.
Synthesis
RAW MATERIAL
Key RM is, 4,6-Etheno-1H-cycloprop[f]isobenzofuran-1,3(3aH)-dione, 3a,4,4a,5,5a,6-hexahydro-, (3aR,4R,4aR,5aS,6S,6aS)-rel–
cas 944-41-2, [US7655688]
| Molecular Formula: | C11H10O3 |
|---|---|
| Molecular Weight: | 190.1953 g/mol |
- 4,6-Etheno-1H-cycloprop[f]isobenzofuran-1,3(3aH)-dione, 4,4a,5,5a,6,6a-hexahydro-, (3aα,4β,4aα,5aα,6β,6aα)-
- Tricyclo[3.2.2.02,4]non-8-ene-6,7-dicarboxylic anhydride, stereoisomer (8CI)
- 3,6-Cyclopropylene-Δ4-tetrahydrophthalic anhydride
MP 94-96 °C
Ref, Dong, Ming-xin; European Journal of Medicinal Chemistry 2010, V45(9), Pg 4096-4103
SMILES……….
O=C1OC(=O)[C@H]4[C@@H]1[C@H]3C=C[C@@H]4[C@@H]2C[C@@H]23
SYNTHESIS CONTINUED…….
ST-246
Patent
WO2014028545
The present invention provides a process for making ST-246 outlined in Scheme 1
P = Boc
Scheme 1
The present invention also provides a process for making ST-246 outlined in, Scheme 2
Scheme 2
The present invention further provides a process for making ST-246 outlined in Scheme 3
ST-246
P = Boc
Scheme 3
P = Boc
Scheme 4
The present invention further provides a process for making ST-246 outlined in
Scheme 5
Scheme 5
Example 1 : Synthetic Route I:
P = Boc
Scheme 1
Step A. Synthesis of Compound 6 (P = Boc)
To a mixture of compound 3 (5.0 g, 26.3 mmol, synthesized according to WO041 12718) in EtOH (80 mL, EMD, AX0441 -3) was added terf-butyl carbazate 5 (3.65 g, 27.6 mmol, Aldrich, 98%). The reaction mixture was heated to reflux for 4 h under nitrogen atmosphere. LC-MS analysis of the reaction mixture showed less than 5% of compound 3 remained. The reaction mixture was evaporated under reduced pressure. The residue was recrystallized from EtOAc – hexanes, the solid was filtered, washed with hexanes (50 mL) and dried under vacuum to afford compound 6 (3.1 g, 39% yield) as a white solid. The filtrate was concentrated and purified by column chromatography eluting with 25% EtOAc in hexanes to give an additional 3.64 g (46% yield) of compound 6 as a white solid. Total yield: 6.74 g (84% yield). 1H NMR in CDCI3: δ 6.30 (br s, 1 H), 5.79 (t, 2H), 3.43 (s, 2H), 3.04 (s, 2H), 1 .46 (s, 9H), 1 .06-1 .16 (m, 2H), 0.18-0.36 (m, 2H); Mass Spec: 327.2 (M+Na)+
Step B. Synthesis of Compound 7 (HCI salt)
Compound 6 (3.6 g, 1 1 .83 mmol) was dissolved in /‘-PrOAc (65 mL, Aldrich, 99.6%). 4M HCI in dioxane (10.4 mL, 41 .4 mmol, Aldrich) was added drop-wise to the above solution keeping the temperature below 20 °C. The reaction mixture was stirred at room temperature overnight (18 h) under nitrogen atmosphere. The resulting solid was filtered, washed with /‘-PrOAc (15 mL) and dried under vacuum to yield HCI salt of compound 7 (1 .9 g, 67% yield) as a white solid. The filtrate was concentrated to 1/3 its volume and stirred at 10 – 15 °C for 30 min. The solid was filtered, washed with minimal volume of /‘-PrOAc and dried to afford additional 0.6 g (21 % yield) of compound 7. Total yield: 2.5 g (88% yield). 1 H NMR in DMSO-d6: δ 6.72 (br s, 3H), 5.68 (m, 2H), 3.20 (s, 2H), 3.01 (s, 2H), 1 .07-1 .17 (m, 2H), 0.18-0.29 (m, 1 H), -0.01 -0.07 (m, 1 H); Mass Spec: 205.1 (M+H)+
Step C. Synthesis of ST-246
To a mixture of compound 7 (0.96 g, 4 mmol) in dry dichloromethane (19 mL) was added triethylamine (1 .17 mL, 8.4 mmol, Aldrich) keeping the temperature below 20 °C. The resulting solution was stirred for 5 minutes at 15 – 20 °C, to it was added drop-wise 4-(trifluoromethyl)benzoyl chloride 8 (0.63 mL, 4.2 mmol, Aldrich, 97%) and the reaction mixture was stirred at room temperature overnight (18 h). LC-MS and TLC analysis showed the correct molecular weight and Rf value of ST-246 but the reaction was not complete. Additional 0.3 mL (2 mmol, 0.5 eq) of 4-(trifluoromethyl)benzoyl chloride 8 was added to the reaction mixture at 15 – 20 °C. The reaction was then stirred at room temperature overnight (19 h). LC-MS analysis indicated ca. 5% of starting material 7 still remained. The reaction was stopped and dichloromethane (30 mL) was added. The organic phase was washed with water (30 mL), saturated aqueous NH CI (30 mL), water (15 mL) and saturated aqueous NaHCO3 (30 mL). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 30 -50% EtOAc in hexanes to afford ST-246 (0.34 g, 23% yield) as an off-white solid. Analytical data (1H NMR, LC-MS and HPLC by co-injection) were matched with those of ST-246 synthesized according to WO041 12718 and were consistent.
Example 2: Synthetic Route II
Scheme 2
Step A. Synthesis of Compound 9
A mixture of compound 4 (2.0 g, 9.8 mmol) and maleic anhydride 2 (0.96 g, 9.8 mmol, Aldrich powder, 95%) in o-xylene (100 mL, Aldrich anhydrous, 97%) was heated to reflux using a Dean-Stark trap apparatus overnight. After 18 h, LC-MS analysis at 215 nm showed the desired product 9 (86%), an uncyclized product (2.6%) and a dimer by-product (1 1 .6%).
Uncyclized product (MS = 303) Dimer by-product (MS = 489)
The reaction mixture was cooled to 45 °C and evaporated under reduced pressure. The residue was dissolved in EtOAc (50 mL) and the insoluble solid (mostly uncyclized product) was removed by filtration. The filtrate was concentrated and purified by column chromatography eluting with 50% EtOAc in hexanes to yield compound 9 (1 .5 g, 54% yield) as an off-white solid. 1 H NMR in CDCI3: δ 8.44 (s, 1 H), 7.91 (d, 2H), 7.68 (d, 2H), 6.88 (s, 2H); Mass Spec: 285.1 (M+H)+
Step B. Synthesis of ST-246 (Route II)
A mixture of compound 9 (0.97 g, 3.4 mmol) and cycloheptatriene 1 (0.51 mL, 4.42 mmol, distilled before use, Aldrich tech 90%) in toluene (50 mL, Aldrich anhydrous) was heated at 95 °C under nitrogen atmosphere. After 1 .5 h at 95 °C, LC-MS analysis at 254 nm showed 29% conversion to the desired product (endo:exo = 94:6). The resulting solution was continued to be heated at same temperature overnight. After 18 h at 95 °C, LC-MS analysis indicated 75% conversion with an endo:exo ratio of 94:6. The reaction temperature was increased to 1 10 °C and the reaction was monitored. After heating at 1 10 °C for 7 h, LC-MS analysis at 254 nm showed 96.4% conversion to the desired product (endo:exo = 94:6). The volatiles were removed by evaporation under reduced pressure and the reside was purified by column chromatography eluting with 30% EtOAc in hexanes to afford ST-246 (0.29 g, 22.6% yield, HPLC area 99.7% pure and 100% endo isomer) as a white solid. Analytical data (1H NMR, LC-MS and HPLC by co-injection) were matched with those of ST-246 synthesized according to WO041 12718 and were consistent. An additional 0.5 g of ST-246 (38.9% yield, endo:exo = 97: 3) was recovered from column chromatography. Total Yield: 0.84 g (65.4% yield). 1H NMR of ST-246 exo isomer in CDCI3: δ 8.62 (s, 1 H), 7.92 (d, 2H), 7.68 (d, 2H), 5.96 (m, 2H), 3.43 (s, 2H), 2.88 (s, 2H), 1 .17 (s, 2H), 0.24 (q, 1 H), 0.13 (m, 1 H); Mass Spec: 377.1 (M+H)+
Example 3: Synthetic Route III
ST-246 9
P = Boc
Scheme 3
Step A. Synthesis of Compound 10
A mixture of maleic anhydride 2 (15.2 g, 155 mmol, Aldrich powder 95%) and terf-butyl carbazate 5 (20.5 g, 155 mmol, Aldrich, 98%) in anhydrous toluene (150 mL, Aldrich anhydrous) was heated to reflux using a Dean-Stark trap apparatus under nitrogen atmosphere. After refluxing for 2 h, no starting material 2 remained and LC-MS analysis at 254 nm showed the desired product 10 (20% by HPLC area), imine byproduct (18%) and disubstituted by-product (56%). The reaction mixture was concentrated and purified by column chromatography eluting with 25% EtOAc in hexanes to afford compound 10 (5.98 g, 18% yield, HPLC area >99.5% pure) as a white solid. 1 H NMR in DMSO-d6: δ 9.61 (s, 1 H), 7.16 (s, 2H), 1 .42 (s, 9H); Mass Spec: 235.1 (M+Na)+.
duct
C9H12N204 C14H22N405
Mol. Wt.: 212.2 Mol. Wt.: 326.35
C9H12N204 C14H22N405
Mol. Wt.: 212.2 Mol. Wt.: 326.35
Step B. Synthesis of Compound 11 (HCI salt)
Compound 10 (3.82 g, 18 mmol) was dissolved in /‘-PrOAc (57 mL, Aldrich, 99.6%). 4M HCI in dioxane (15.8 mL, 63 mmol, Aldrich) was added drop-wise to the above solution keeping the temperature below 20 °C. The solution was stirred overnight (24 h) at room temperature under nitrogen atmosphere. The resulting solid was filtered, washed with /‘-PrOAc (10 mL) and dried at 45 °C under vacuum for 1 h to afford HCI salt of compound 11 (2.39 g, 89% yield) as a white solid. 1 H NMR in CD3OD: δ 6.98 (s, 2H); Mass Spec: 1 13.0 (M+H)+
Step C. Synthesis of Compound 9 (Route III)
To a mixture of compound 11 (1 .19 g, 8 mmol) in dry dichloromethane (24 mL) was added diisopropylethylannine (2.93 mL, 16.8 mmol, Aldrich redistilled grade) keeping the temperature below 20 °C. The resulting solution was stirred for 5 minute at 15 – 20 °C and to it was added 4-(trifluoromethyl)benzoyl chloride 8 (1 .31 mL, 8.8 mmol, Aldrich, 97%) drop-wise. The reaction was stirred at room temperature for 5 h. LC-MS analysis showed the correct MW but the reaction was not complete. Additional 0.48 mL (0.4 equiv) of 4-(trifluoromethyl)benzoyl chloride 8 was added to the reaction mixture at 15 – 20 °C and the reaction mixture was stirred at room temperature overnight (21 h). The reaction was stopped and dichloromethane (50 mL) was added. The organic phase was washed with water (50 mL), saturated aqueous NH4CI (50 mL), water (30 mL) and saturated aqueous NaHCO3 (30 mL). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 30 – 35% EtOAc in hexanes to afford compound 9 (0.8 g, 35% yield) as a light pink solid. Analytical data (1H NMR and LC-MS) were consistent with those of compound 9 obtained in Synthetic Route II.
Step D. Synthesis of ST-246 (Route III)
A mixture of compound 9 (0.5 g, 1 .76 mmol) and cycloheptatriene 1 (0.33 mL, 3.17 mmol, distilled before to use, Aldrich tech 90%) in toluene (10 mL, Aldrich anhydrous) was heated at 1 10 – 1 15 °C under nitrogen atmosphere. After 6 h, LC-MS analysis at 254 nm showed 95% conversion to the desired product (endo:exo = 94:6). The resulting solution was heated at same temperature overnight (22 h). LC-MS analysis at 254 nm showed no starting material 9 remained and the desired product (endo:exo = 93:7). The reaction mixture was concentrated and purified by column chromatography eluting with 25 – 35% EtOAc in hexanes to afford ST-246 (0.39 g, HPLC area >99.5% pure with a ratio of endo:exo = 99:1 ) as a white solid. Analytical data (1 H NMR, LC-MS and HPLC by co-injection) were compared with those of ST-246 synthesized according to WO041 12718 and were found to be consistent. An additional 0.18 g of ST-246 (HPLC area >99.5% pure, endo:exo = 91 : 9) was recovered from column chromatography. Total Yield: 0.57 g (86% yield).
Example 4 ; Synthetic Route IV:
P = Boc
Scheme 4
Step A. Synthesis of Compound 10
A mixture of maleic anhydride 2 (3.4 g, 34.67 mmol, Aldrich powder, 95%) and terf-butyl carbazate 5 (4.6 g, 34.67 mmol, Aldrich, 98%) in anhydrous toluene (51 ml_, Aldrich) was heated to reflux using a Dean-Stark trap apparatus under nitrogen atmosphere. After refluxing for 2.5 h, no starting material 2 remained and LC-MS analysis at 254 nm showed the desired product 10 (19% HPLC area), imine by-product (18%) and another by-product (56%). The reaction mixture was concentrated and purified by column chromatography eluting with 30% EtOAc in hexanes to afford compound 10 (1 .0 g, 13.6% yield, HPLC area >99% pure) as a white solid. Analytical data (1H NMR and LC-MS) were consistent with those of compound 10 obtained in Synthetic Route III.
Im ine by-product
Mol. Wt.: 212.2
Step B. Synthesis of Compound 6
A mixture of compound 10 (4.4 g, 20.74 mmol) and cycloheptatriene 1 (3.22 mL, 31 .1 mmol, distilled before to use, Aldrich tech 90%) in toluene (88 mL, 20 volume, Aldrich anhydrous) was heated at 95 °C under nitrogen atmosphere. After 15 h at 95 °C, LC-MS analysis showed 83% conversion to the desired product. The reaction mixture was heated at 105 °C overnight. After total 40 h at 95 – 105 °C, LC-MS analysis at 254 nm showed -99% conversion to the desired product (endo:exo = 93:7). The reaction mixture was concentrated and the crude was purified by column chromatography eluting with 25 – 50 % EtOAc in hexanes to afford compound 6 (2.06 g, 32.6% yield, HPLC area 99.9% pure and 100% endo isomer) as a white solid. 1 H NMR and LC-MS were consistent with those of compound 6 obtained in Synthetic Route I. An additional 4.0 g of 6 (63.4% yield, HPLC area 93% pure with a ratio of endo:exo = 91 : 9) was recovered from column chromatography. Total Yield: 6.06 g (96% yield).
Step C. Synthesis of Compound 7 (HCI salt)
Compound 6 (2.05 g, 6.74 mmol) was dissolved in /‘-PrOAc (26 mL, Aldrich, 99.6%). 4M HCI in dioxane (5.9 mL, 23.58 mmol, Aldrich) was added drop-wise to the above solution keeping the temperature below 20 °C. The solution was stirred overnight (18 h) at room temperature under nitrogen atmosphere. The resulting solid was filtered, washed with /‘-PrOAc (5 mL) and dried under vacuum to yield HCI salt of compound 7 (1 .57 g, 97% yield) as a white solid. Analytical data (1 H NMR and LC-MS) were consistent with those of compound 7 in Synthetic Route I.
Step D. Synthesis of ST-246 (Route IV)
To a mixture of compound 7 (0.84 g, 3.5 mmol) in dichloromethane (13 mL) was added diisopropylethylamine (1 .34 mL, 7.7 mmol) keeping the temperature below 20 °C and the resulting solution was stirred for 5 – 10 minutes. 4-(Trifluoromethyl)benzoyl chloride 8 (0.57 mL, 3.85 mmol, Aldrich, 97%) was added to above solution keeping the temperature below 20 °C. The reaction mixture was stirred at room temperature for 2 h. Additional 0.2 mL (0.4 equiv) of 4-(trifluoromethyl)benzoyl chloride 8 was added to the reaction keeping the temperature below 20 °C. The reaction was stirred at room temperature overnight (24 h). The reaction mixture was diluted with dichloromethane (20 mL). The organic phase was washed with water (20 mL), saturated aqueous NH4CI (20 mL), water (20 mL) and saturated aqueous NaHCO3 (20 mL). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 30 – 35% EtOAc in hexanes to afford ST-246 (0.25 g, 19% yield, HPLC area >99.5% pure) as a white solid. Analytical data (1H NMR and LC-MS) were consistent with those of ST-246 synthesized according to WO041 12718.
Example 5: Synthetic Route V:
Scheme 5
Step A. Synthesis of Compound 13
To a mixture of compound 7 (1 .6 g, 6.65 mmol, synthesized according to Synthetic Route I) in dichloromethane (80 ml_,) was added triethylamine (2.04 ml_, 14.63 mmol) keeping the temperature below 20 °C and the resulting solution was stirred for 5 – 10 minute. 4-lodobenzoyl chloride 12 (1 .95 g, 7.31 mmol, 1 .1 equiv, Aldrich) was added portion-wise under nitrogen atmosphere to the above solution keeping the temperature below 20 °C. The reaction mixture was stirred at room temperature overnight. After 17 h and 19 h, additional 0.35 g (0.2 equiv) of acid chloride 12 was added to the reaction keeping the temperature below 20 °C. After 24 h, additional 0.18 g (0.1 equiv, used total 1 .6 equiv) of acid chloride 12 was added and the reaction was continued to stir at room temperature overnight (total 43 h). LC-MS analysis at 215 nm showed 43% of the desired product (13) and -5% of compound 7. The reaction was diluted with dichloromethane (100 ml_). The organic phase was washed with saturated aqueous NH4CI (100 ml_), water (100 ml_) and saturated aqueous NaHCO3 (100 ml_). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 25 – 50% EtOAc in hexanes to afford compound 13 (1 .63 g, 57% yield, HPLC area 93% pure) as a white solid. 1 H NMR in DMSO-d6: δ 1 1 .19 and 10.93 (two singlets with integration ratio of 1 .73:1 , total of 1 H, same proton of two rotamers), 7.93 (d, 2H), 7.66 (d, 2H), 5.80 (s, 2H), 3.36 (s, 2H), 3.27 (s, 2H), 1 .18 (s, 2H), 0.27 (q, 1 H), 0.06 (s,1 H); Mass Spec: 435.0 (M+H)+
Step B. Synthesis of ST-246 (Route V)
Anhydrous DMF (6 ml_) was added to a mixture of compound 13 (0.2 g, 0.46 mmol), methyl 2, 2-difluoro-2-(fluorosulfonyl)acetate (0.44 ml_, 3.45 mmol, Aldrich) and copper (I) iodide (90 mg, 0.47 mmol). The reaction mixture was stirred at -90 °C for 4 h. LC-MS analysis at 254 nm indicated no starting material 13 remained and showed 48% HPLC area of ST-246. The reaction mixture was cooled to 45 °C and DMF was removed under reduced pressure. The residue was slurried in EtOAc (30 mL) and insoluble solid was removed by filtration. The filtrate was concentrated and purified by column chromatography eluting with 25 – 35% EtOAc in hexanes to afford ST-246 (55
mg, 32% yield, 95% pure by HPLC at 254 nm) as off-white solid. Analytical data (1H NMR and LC-MS) were consistent with those of ST-246 synthesized according to WO041 12718.
PAPER
N-(3,3a,4,4a,5,5a,6,6a-Octahydro-1,3-dioxo-4,6- ethenocycloprop[f]isoindol-2-(1H)-yl)carboxamides: Identification of Novel Orthopoxvirus Egress Inhibitors
ViroPharma Incorporated, 397 Eagleview Boulevard, Exton, Pennsylvania 19341, United States Army Medical Research Institute of Infectious Diseases, 1425 Porter Street, Frederick, Maryland 21702, University of Alabama, Birmingham, Alabama 35294, and SIGA Technologies, Inc., 4575 SW Research Way, Corvallis, Oregon 97333
J. Med. Chem., 2007, 50 (7), pp 1442–1444
DOI: 10.1021/jm061484y
http://pubs.acs.org/doi/suppl/10.1021/jm061484y/suppl_file/jm061484ysi20070204_060607.pdf
General Procedure for synthesis of compounds 2-14, 16-18.
N-(3,3a,4,4a,5,5a,6,6aoctahydro-1,3-dioxo-4,6-ethenocycloprop[f]isoindol-2(1H)-yl)-4- (trifluoromethyl)benzamide (14).
A mixture of 2.00 g (9.8 mmol) of 4-(trifluoromethyl) benzoic acid hydrazide, 1.86 g (9.8 mmol) of 4,4a,5,5a,6,6a-hexahydro-4,6-etheno-1Hcycloprop[f]isobenzofuran-1,3(3aH)-dione, and one drop of diisopropylethylamine in 40 mL of absolute ethanol was refluxed for 4.5 h. Upon cooling to rt, 4 mL of water was added, and the product began to crystallize. The suspension was cooled in an ice bath, and the precipitate collected by filtration. The crystalline solid was air-dried affording 3.20 g (87%) of the product as a white solid;
Mp 194-195 ºC. 1 H NMR, (300 MHz, d6 -DMSO) δ 11.20, 11.09 (2 brs from rotamers, 1H), 8.06 (d, J= 7.8 Hz, 2H), 7.90 (d, J= 7.8 Hz, 2H), 5.78 (m, 2H), 3.26 (m, 4H), 1.15 (m, 2H), 0.24 (dd, J= 7.2, 12.9 Hz, 1H), 0.04 (m, 1H).
Anal. calcd. for C19H15F3N2O3● 0.25H2O: %C, 59.92; %H, 4.10; %F, 14.97; %N, 7.36; %O, 13.65. Found: %C, 59.97; %H, 4.02; %F, 14.94; %N, 7.36; %O, 13.71.
CLICK ON IMAGE
PATENT
US20140316145

CLICK ON IMAGE
http://www.google.com/patents/US8802714
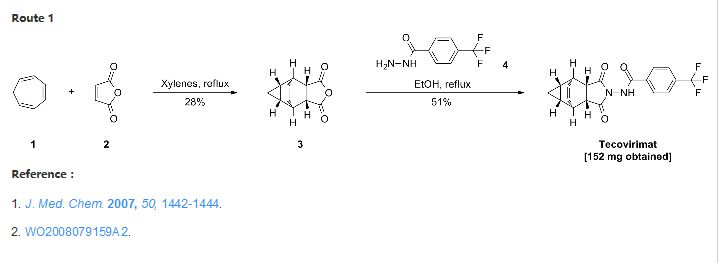
Example 1
Preparation of 4-trifluoromethyl-N-(3,3a,4,4a,5,5a,6,6a-octahydro-1,3-dioxo-4,6-ethenocycloprop[f]isoindol-2(1H)-yl)-benzamide
a. Preparation of Compounds 1(a) and 1(b).

A mixture of cycloheptatriene (5 g, 54.26 mmol) and maleic anhydride (6.13 g, 62.40 mmol) in xylenes (35 mL) was heated at reflux under argon overnight. The reaction was cooled to room temperature and a tan precipitate was collected by filtration and dried to give 2.94 grams (28%) of the desired product, which is a mixture of compounds 1(a) and 1(b). Compound 1(a) is normally predominant in this mixture and is at least 80% by weight. The purity of Compound 1(a) may be further enhanced by recrystallization if necessary. Compound 1(b), an isomer of compound 1(a) is normally less than 20% by weight and varies depending on the conditions of the reaction. Pure Compound 1(b) was obtained by concentrating the mother liquid to dryness and then subjecting the residue to column chromatography. Further purification can be carried out by recrystallization if necessary. 1H NMR (500 MHz) in CDCl3: δ 5.95 (m, 2H), 3.42 (m, 2H), 3.09 (m, 2H), 1.12 (m, 2H), 0.22 (m, 1H), 0.14 (m, 1H).
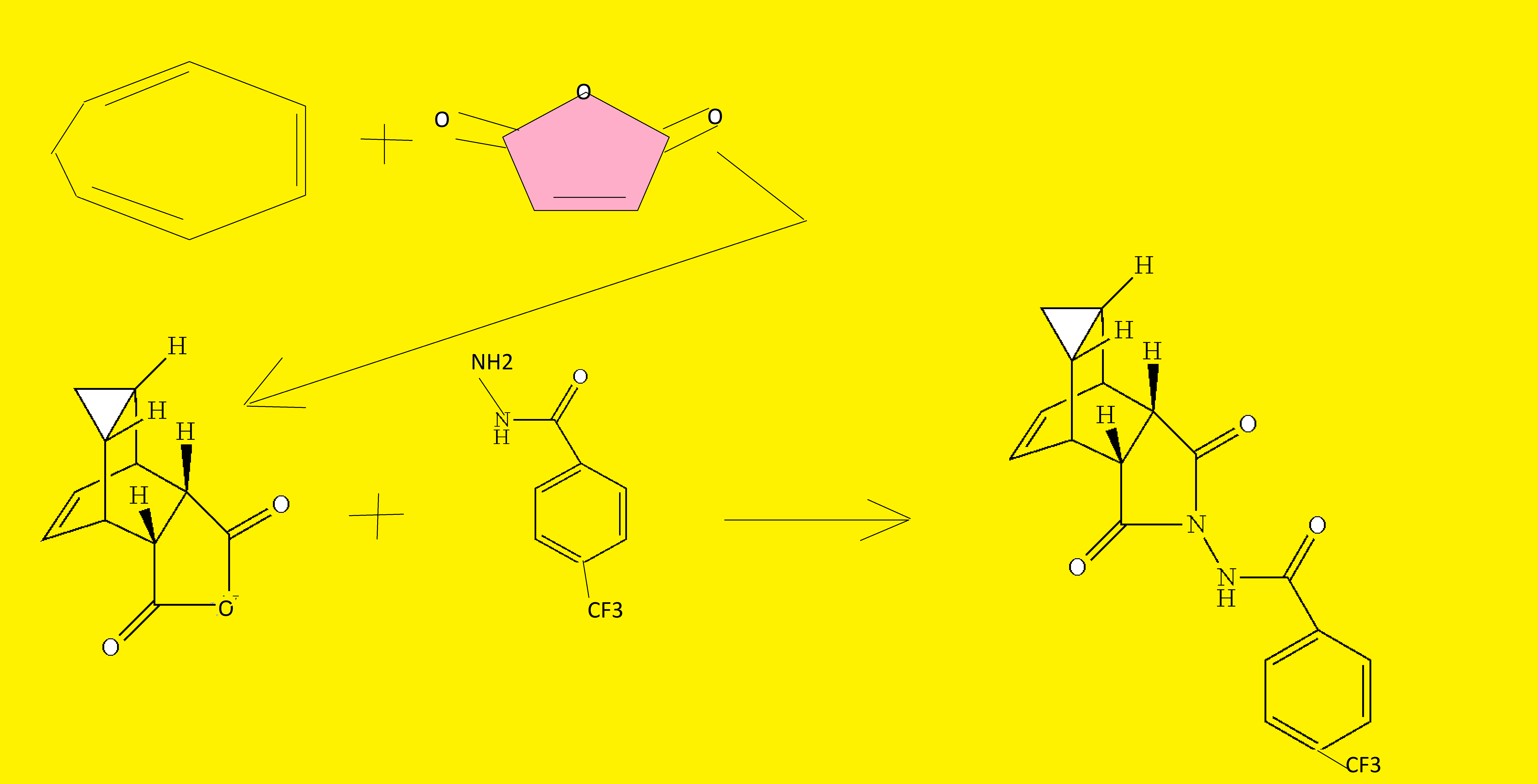
b. Preparation of N-[(3aR,4R,4aR,5aS,6S,6aS)-3,3a,4,4a,5,5a,6,6a-octahydro-1,3-dioxo-4,6-ethenocycloprop[f]isoindol-2(1H)-yl]-4-(trifluoromethyl)-benzamide. desired
A mixture of compound 1(a) (150 mg, 0.788 mmol) and 4-trifluoromethylbenzhydrazide (169 mg, 0.827 mmol) in ethanol (10 mL) was heated under argon overnight. The solvent was removed by rotary evaporation. Purification by column chromatography on silica gel using 1/1 hexane/ethyl acetate provided 152 mg (51%) of the product as a white solid.
c. Preparation of N-[(3aR,4S,4aS,5aR,6R,6aS)-3,3a,4,4a,5,5a,6,6a-octahydro-1,3-dioxo-4,6-ethenocycloprop[f]isoindol-2(1H)-yl]-4-(trifluoromethyl)-benzamide. UNWANTED
N-[(3aR,4S,4aS,5aR,6R,6aS)-3,3a,4,4a,5,5a,6,6a-octahydro-1,3-dioxo-4,6-ethenocycloprop[f]isoindol-2(1H)-yl]4-(trifluoromethyl)-benzamide was prepared and purified in the same fashion as for N-[(3aR,4R,4aR,5aS,6S,6aS)-3,3a,4,4a,5,5a,6,6a-octahydro-1,3-dioxo-4,6-ethenocycloprop[f]isoindol-2(1H)-yl]-4-(trifluoromethyl)-benzamide by replacing 1(a) with 1(b) and was obtained as a white solid. 1H NMR (300 MHz) in CDCl3: δ 8.62 (s, 1H), 7.92 (d, 2H), 7.68 (d, 2H), 5.96 (m, 2H), 3.43 (s, 2H), 2.88 (s, 2H), 1.17 (s, 2H), 0.24 (q, 1H), 0.13 (m, 1H); Mass Spec: 377.1 (M+H)+.
FINAL COMPD SYNTHESIS
| TABLE 1 | ||||
| Example | **Mass | |||
| Number | R6 | *NMR | Spec | Name |
| 1 |
|
1H NMR in DMSO-d6: δ 11.35 (d, 1H); 11.09 (d, 1H); 8.08 (d, 2H); 7.92 (d, 2H); 5.799 (s, 2H); 3.29 (brs, 4H); 1.17 (m, 2H); 0.26 (m, 1H); 0.078 (s, 1H) | 375 (M − H)− | N-[(3aR,4R,4aR,5aS,6S, 6aS)-3,3a,4,4a,5,5a,6,6a- octahydro-1,3-dioxo- 4,6-ethenocycloprop[f] isoindol-2(1H)-yl]-4- (trifluoromethyl)- benzamide |
TABLE 1 EXAMPLE 1
N- [(3aR,4R,4aR,5aS,6S, 6aS)- 3,3a,4,4a,5,5a,6,6a- octahydro-1,3-dioxo- 4,6- ethenocycloprop[f]iso- indol-2(1H)-yl]-4- (trifluoromethyl)- benzamide
1H NMR in DMSO-d6: δ 11.35 (d, 1H); 11.09 (d, 1H); 8.08 (d, 2H); 7.92 (d, 2H); 5.799 (s, 2H); 3.29 (brs, 4H); 1.17 (m, 2H); 0.26 (m, 1H); 0.078 (s, 1H), 375 (M − H)−
EXAMPLE 42 Characterization of 4-trifluoromethyl-N-(3,3a,4,4a,5,5a,6,6a-octahydro-1,3-dioxo-4,6-ethenocycloprop[f]isoindol-2(1H)-yl)-benzamide (“ ”)
In the present application, ST-246 refers to: N-[(3aR,4R,4aR,5aS,65,6aS)-3,3a,4,4a,5,5a,6,6a-octahydro-1,3-dioxo-4,6-ethenocycloprop[f]isoindol-2(1H)-yl]-4-(trifluoromethyl)-benzamide.
Physico-Chemical Properties
Appearance: ST-246 is a white to off-white powder.
Melting Point: Approximately 196° C. by DSC.
Permeability: The calculated log P is 2.94. Based on the partition coefficient, ST-246 is expected to have good permeability.
Particle Size: The drug substance is micronized to improve its dissolution in the gastrointestinal fluids. The typical particle size of the micronized material is 50% less than 5 microns.
Solubility: The solubility of ST-246 is low in water (0.026 mg/mL) and buffers of the gastric pH range. Surfactant increases its solubility slightly. ST-246 is very soluble in organic solvents. The solubility data are given in Table 5.


CLICK ON IMAGE
PATENT
http://www.google.com/patents/CN101445478A?cl=en


References
- Damon, Inger K.; Damaso, Clarissa R.; McFadden, Grant (2014). “Are We There Yet? The Smallpox Research Agenda Using Variola Virus”. PLoS Pathogens 10 (5): e1004108.doi:10.1371/journal.ppat.1004108. PMID 24789223.
- Siga Technologies
- Jordan, R; Tien, D; Bolken, T. C.; Jones, K. F.; Tyavanagimatt, S. R.; Strasser, J; Frimm, A; Corrado, M. L.; Strome, P. G.; Hruby, D. E. (2008). “Single-Dose Safety and Pharmacokinetics of ST-246, a Novel Orthopoxvirus Egress Inhibitor”. Antimicrobial Agents and Chemotherapy 52 (5): 1721–1727. doi:10.1128/AAC.01303-07. PMC 2346641. PMID 18316519.
- Yang, G; Pevear, D. C.; Davies, M. H.; Collett, M. S.; Bailey, T; Rippen, S; Barone, L; Burns, C; Rhodes, G; Tohan, S; Huggins, J. W.; Baker, R. O.; Buller, R. L.; Touchette, E; Waller, K; Schriewer, J; Neyts, J; Declercq, E; Jones, K; Hruby, D; Jordan, R (2005). “An Orally Bioavailable Antipoxvirus Compound (ST-246) Inhibits Extracellular Virus Formation and Protects Mice from Lethal Orthopoxvirus Challenge”. Journal of Virology 79 (20): 13139–13149. doi:10.1128/JVI.79.20.13139-13149.2005. PMC 1235851. PMID 16189015.
Referenced by
Citing Patent Filing date Publication date Applicant Title
CN101912389A * Aug 9, 2010 Dec 15, 2010 中国人民解放军军事医学科学院微生物流行病研究所 Pharmaceutical composition containing ST-246 and preparation method and application thereof
CN102406617A * Nov 30, 2011 Apr 11, 2012 中国人民解放军军事医学科学院生物工程研究所 Tecovirimat dry suspension and preparation method thereof
CN102406617B Nov 30, 2011 Aug 28, 2013 中国人民解放军军事医学科学院生物工程研究所 Tecovirimat dry suspension and preparation method thereof
CN103068232B * Mar 23, 2011 Aug 26, 2015 西佳科技股份有限公司 多晶型物形式st-246和制备方法
US8530509 Jul 29, 2011 Sep 10, 2013 Siga Technologies, Inc. Compounds, compositions and methods for treatment and prevention of orthopoxvirus infections and associated diseases
US8802714 Aug 14, 2013 Aug 12, 2014 Siga Technologies, Inc. Compounds, compositions and methods for treatment and prevention of orthopoxvirus infections and associated diseases
US9045418 Jul 3, 2014 Jun 2, 2015 Siga Technologies, Inc. Compounds, compositions and methods for treatment and prevention of Orthopoxvirus infections and associated diseases
Patent Citations
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US20070287735 * | Apr 23, 2007 | Dec 13, 2007 | Siga Technologies, Inc. | Chemicals, compositions, and methods for treatment and prevention of orthopoxvirus infections and associated diseases |
| US20090011037 * | Apr 23, 2008 | Jan 8, 2009 | Cydex Pharmaceuticals, Inc. | Sulfoalkyl Ether Cyclodextrin Compositions and Methods of Preparation Thereof |
| Citing Patent | Filing date | Publication date | Applicant | Title |
| US8530509 | Jul 29, 2011 | Sep 10, 2013 | Siga Technologies, Inc. | Compounds, compositions and methods for treatment and prevention of orthopoxvirus infections and associated diseases |
| US8802714 | Aug 14, 2013 | Aug 12, 2014 | Siga Technologies, Inc. | Compounds, compositions and methods for treatment and prevention of orthopoxvirus infections and associated diseases |
| US9045418 | Jul 3, 2014 | Jun 2, 2015 | Siga Technologies, Inc. | Compounds, compositions and methods for treatment and prevention of Orthopoxvirus infections and associated diseases |
//////////////////Tecovirimat, FDA 2018, ORPHAN DRUG DESIGNATION, TPOXX, SIGA Technologies Inc, Fast Track, Priority Review,
UNII-F925RR824R, тековиримат , تيكوفيريمات , 替韦立马 ,
FC(F)(F)c1ccc(cc1)C(=O)NN1C(=O)C2C(C3C=CC2C2CC32)C1=O

