
Home » Biosimilar drugs
Category Archives: Biosimilar drugs
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
FDA approves first biosimilar to Neulasta, Fulphila (pegfilgrastim) to help reduce the risk of infection during cancer treatment
The U.S. Food and Drug Administration today approved Fulphila (pegfilgrastim-jmdb) as the first biosimilar to Neulasta (pegfilgrastim) to decrease the chance of infection as suggested by febrile neutropenia (fever, often with other signs of infection, associated with an abnormally low number of infection-fighting white blood cells), in patients with non-myeloid (non-bone marrow) cancer who are receiving myelosuppressive chemotherapy that has a clinically significant incidence of febrile neutropenia.
June 4, 2018
Release
The U.S. Food and Drug Administration today approved Fulphila (pegfilgrastim-jmdb) as the first biosimilar to Neulasta (pegfilgrastim) to decrease the chance of infection as suggested by febrile neutropenia (fever, often with other signs of infection, associated with an abnormally low number of infection-fighting white blood cells), in patients with non-myeloid (non-bone marrow) cancer who are receiving myelosuppressive chemotherapy that has a clinically significant incidence of febrile neutropenia.
“Bringing new biosimilars to patients is a top priority for the FDA, and a key part of our efforts to help promote competition that can reduce drug costs and promote access,” said FDA Commissioner Scott Gottlieb, M.D. “We’ll continue to prioritize reviews of these products to help ensure that biosimilar medications are brought to the market efficiently and through a process that makes certain that these new medicines meet the FDA’s rigorous standard for approval. This summer, we’ll release a comprehensive new plan to advance new policy efforts that promote biosimilar product development. Biologics represent some of the most clinically important, but also costliest products that patients use to promote their health. We want to make sure that the pathway for developing biosimilar versions of approved biologics is efficient and effective, so that patients benefit from competition to existing biologics once lawful intellectual property has lapsed on these products.”
Biological products are generally derived from a living organism and can come from many sources, such as humans, animals, microorganisms or yeast. A biosimilar is a biological product that is approved based on data showing that it is highly similar to a biological product already approved by the FDA (reference product) and has no clinically meaningful differences in terms of safety, purity and potency (i.e., safety and effectiveness) from the reference product, in addition to meeting other criteria specified by law.
The FDA’s approval of Fulphila is based on review of evidence that included extensive structural and functional characterization, animal study data, human pharmacokinetic and pharmacodynamic data, clinical immunogenicity data, and other clinical safety and effectiveness data that demonstrates Fulphila is biosimilar to Neulasta. Fulphila has been approved as a biosimilar, not as an interchangeable product.
The most common side effects of Fulphila are bone pain and pain in extremities. Patients with a history of serious allergic reactions to human granulocyte colony-stimulating factors such as pegfilgrastim or filgrastim products should not take Fulphila.
Serious side effects from treatment with Fulphila include rupture of the spleen, acute respiratory distress syndrome, serious allergic reactions including anaphylaxis, acute inflammation of the kidney (glomerulonephritis), an abnormally high level of white blood cells (leukocytosis), capillary leak syndrome and the potential for tumor growth. Fatal sickle cell crises have occurred.
The FDA granted approval of Fulphila to Mylan GmbH.

//////////// pegfilgrastim, fda 2018, Fulphila, Neulasta, Mylan GmbH, biosimilars, MONOCLONAL ANTIBODY,
Sun Pharma and Merck & Co. Inc. Enter into Licensing Agreement for Tildrakizumab, MK 3222

Tildrakizumab (MK-3222)
| Company | Merck & Co. Inc. |
| Description | Anti-IL-23 antibody |
| Molecular Target | Interleukin-23 (IL-23) |
| Mechanism of Action | Antibody |
| Therapeutic Modality | Biologic: Antibody |
| Latest Stage of Development | Phase III |
| Standard Indication | Psoriasis |
| Indication Details | Treat moderate to severe chronic plaque psoriasis |
| Regulatory Designation | |
| Partner | Sun Pharmaceutical Industries Ltd. |
Tildrakizumab is a monoclonal antibody designed for the treatment of immunologically mediated inflammatory disorders.[1]
Tildrakizumab was designed to block interleukin-23, a cytokine that plays an important role in managing the immune system and autoimmune disease. Originally developed by Schering-Plough, this drug is now part of Merck‘s clinical program, following that company’s acquisition of Schering-Plough.
Sun Pharmaceutical acquired worldwide rights to tildrakizumab for use in all human indications from Merck in exchange for an upfront payment of U.S. $80 million. Upon product approval, Sun Pharmaceutical will be responsible for regulatory activities, including subsequent submissions, pharmacovigilance, post approval studies, manufacturing and commercialization of the approved product. [2]
As of March 2014, the drug was in phase III clinical trials for plaque psoriasis. The two trials will enroll a total of nearly 2000 patients, and preliminary results are expected in June, 2015. [3][4]

References
- 1 Statement On A Nonproprietary Name Adopted By The USAN Council – Tildrakizumab, American Medical Association.
- 2
- http://www.merck.com/licensing/our-partnership/sunpharma_partnership.html
- 3
- http://clinicaltrials.gov/ct2/show/NCT01729754?term=SCH-900222&phase=2&fund=2&rank=1
- 4
http://clinicaltrials.gov/ct2/show/NCT01722331?term=SCH-900222&phase=2&fund=2&rank=2
Sun Pharma and Merck & Co. Inc. Enter into Licensing Agreement for Tildrakizumab, MK 3222
WHITEHOUSE STATION, N.J., and MUMBAI, India, Wednesday, September 17, 2014 (BUSINESS WIRE) – Merck & Co., Inc., (NYSE:MRK), known as MSD outside the United States and Canada, and Sun Pharmaceutical Industries Ltd. (Reuters: SUN.BO, Bloomberg: SUNP IN, NSE: SUNPHARMA, BSE: 524715) through their respective subsidiaries, today announced an exclusive worldwide licensing agreement for Merck’s investigational therapeutic antibody candidate, tildrakizumab, (MK-3222), which is currently being evaluated in Phase 3 registration trials for the treatment of chronic plaque psoriasis, a skin ailment.
Under terms of the agreement, Sun Pharma will acquire worldwide rights to tildrakizumab for use in all human indications from Merck in exchange for an upfront payment of U.S. $80 million. Merck will continue all clinical development and regulatory activities, which will be funded by Sun Pharma. Upon product approval, Sun Pharma will be responsible for regulatory activities, including subsequent submissions, pharmacovigilance, post approval studies, manufacturing and commercialization of the approved product. Merck is eligible to receive undisclosed payments associated with regulatory (including product approval) and sales milestones, as well as tiered royalties ranging from mid-single digit through teen percentage rates on sales.
“Consistent with our previously announced global initiative to sharpen our commercial and R&D focus, including prioritizing our late stage pipeline candidates, we are pleased to enter into this agreement with Sun Pharma to help realize the potential of tildrakizumab for patients with chronic plaque psoriasis,” said Iain D. Dukes, Ph.D., senior vice president, Business Development and Licensing, Merck Research Laboratories.
“Sun Pharma is very pleased to enter into this collaboration with Merck, a recognized leader in the field of inflammatory/immunology therapies, for this late-stage candidate for chronic plaque psoriasis,” said Kirti Ganorkar, senior vice president, Business Development, Sun Pharma. “This collaboration is a part of our strategy towards building our pipeline of innovative dermatology products in a market with strong growth potential.”
The transaction is subject to customary closing conditions, including the requirements under the Hart Scott-Rodino Antitrust Improvements Act.
About Tildrakizumab
Tildrakizumab is an investigational humanized, anti-IL-23p19 monoclonal antibody that binds specifically to IL-23p19 and is therefore designed to selectively block the cytokine IL-23. Human genetics suggest that inhibiting IL-23 is effective for treating inflammatory conditions. In clinical studies for the treatment of chronic plaque psoriasis, tildrakizumab demonstrates efficacy in blocking inflammation by blocking IL-23. Other potential indications, which may be evaluated in future, include psoriatic arthritis and Crohn’s Disease.
Further details of the Phase 3 clinical trials can be found at: http://clinicaltrials.gov
About Merck
Today’s Merck is a global healthcare leader working to help the world be well. Merck is known as MSD outside the United States and Canada. Through our prescription medicines, vaccines, biologic therapies, and consumer care and animal health products, we work with customers and operate in more than 140 countries to deliver innovative health solutions. We also demonstrate our commitment to increasing access to healthcare through far-reaching policies, programs and partnerships. For more information, visit www.merck.com and connect with us on Twitter, Facebook and YouTube.
About Sun Pharma
Established in 1983, listed since 1994 and headquartered in India, Sun Pharmaceutical Industries Ltd. (Reuters: SUN.BO, Bloomberg: SUNP IN, NSE: SUNPHARMA, BSE: 524715) is an international specialty pharmaceutical company with over 75% sales from global markets. It manufactures and markets a large basket of pharmaceutical formulations as branded generics as well as generics in US, India and several other markets across the world. For the year ending March 2014, overall revenues were at US$2.7 billion, of which US contributed US$1.6 billion. In India, the company is a leader in niche therapy areas of psychiatry, neurology, cardiology, nephrology, gastroenterology, orthopedics and ophthalmology. The company has strong skills in product development, process chemistry, and manufacturing of complex dosage forms. More information about the company can be found at www.sunpharma.com.
| Monoclonal antibody | |
|---|---|
| Type | ? |
| Source | Humanized (from mouse) |
| Target | IL23 |
| Identifiers | |
| CAS Number | 1326244-10-3 |
| ATC code | none |
| ChemSpider | none |
| Chemical data | |
| Formula | C6426H9918N1698O2000S46 |
| Molar mass | 144.4 kg/mol |
///////Sun Pharma, Merck & Co. Inc, Licensing Agreement, Tildrakizumab, mk 3222
Reslizumab


Reslizumab
(Cinqair®) Approved Active, FDA 2016-03-23
An interleukin-5 (IL-5) antagonist used to treat severe asthma.
CAS 241473-69-8
![]()
Research Code CDP-835; CEP-38072; CTx-55700; SCH-5570; SCH-55700; TRFK-5,

Anti-interleukin-5 monoclonal antibody – Celltech/Schering-Plough
Reslizumab was approved by the U.S. Food and Drug Administration (FDA) on March 23, 2016. It was developed and marketed as Cinqair® by Teva.
Reslizumab is an interleukin-5 antagonist, which binds to human IL-5 and prevents it from binding to the IL-5 receptor, thereby reducing eosinophilic inflammation. It is indicated for the maintenance treatment of patients with severe asthma in patients aged 18 years and older.
Cinqair® is available as injection for intravenous infusion, containing 100 mg of reslizumab in 10 mL solution in single-use vials. The recommended dose is 3 mg/kg once every four weeks.
- Originator Celltech R&D; Schering-Plough
- Developer Celltech R&D; Teva Pharmaceutical Industries
- Class Antiasthmatics; Monoclonal antibodies
- Mechanism of Action Interleukin 5 receptor antagonists
- Orphan Drug Status Yes – Oesophagitis
- 23 Mar 2016 Registered for Asthma in USA (IV) – First global approval
- 04 Mar 2016 Pooled efficacy data from two phase III trials in Asthma presented at the 2016 Annual Meeting of the American Academy of Allergy, Asthma and Immunology (AAAAI-2016)
- 10 Dec 2015 Preregistration for Asthma in Canada (IV)
Reslizumab (trade name Cinqair) is a humanized monoclonal antibody intended for the treatment of eosinophil-meditated inflammations of the airways, skin and gastrointestinal tract.[1] The FDA approved reslizumab for use with other asthma medicines for the maintenance treatment of severe asthma in patients aged 18 years and older on March 23, 2016. Cinqair is approved for patients who have a history of severe asthma attacks (exacerbations) despite receiving their current asthma medicines.[2]
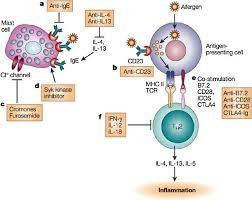

Teva Announces FDA Acceptance of the Biologics License Application for Reslizumab
Investigational Biologic for the Treatment of Inadequately Controlled Asthma in Patients with Elevated Blood Eosinophils Accepted for Review
JERUSALEM–(BUSINESS WIRE)–Jun. 15, 2015– Teva Pharmaceutical Industries Ltd., (NYSE: TEVA) announced today that the U.S. Food and Drug Administration (FDA) has accepted for review the Biologics License Application (BLA) for reslizumab, the company’s investigational humanized monoclonal antibody (mAb) which targets interleukin-5 (IL-5), for the treatment of inadequately controlled asthma in adult and adolescent patients with elevated blood eosinophils, despite an inhaled corticosteroid (ICS)-based regimen.
“Despite currently available medicines, uncontrolled asthma remains a serious problem for patients, physicians and healthcare systems, highlighting the need for targeted new treatment options,” said Dr. Michael Hayden, President of Global R&D and Chief Scientific Officer at Teva Pharmaceutical Industries Ltd. “The reslizumab BLA filing acceptance represents a significant milestone for Teva as we work toward serving a specific asthma patient population that is defined by elevated blood eosinophil levels and inadequately controlled symptoms despite standard of care therapy. In clinical trials, patients treated with reslizumab showed significant reductions in the rate of asthma exacerbations and significant improvement in lung function. If approved, we believe reslizumab will serve as an important new targeted treatment option to achieve better asthma control for patients with eosinophil-mediated disease.”
The BLA for reslizumab includes data from Teva’s Phase III BREATH clinical trial program. The program consisted of four separate placebo-controlled Phase III trials involving more than 1,700 adult and adolescent asthma patients with elevated blood eosinophils, whose symptoms were inadequately controlled with inhaled corticosteroid-based therapies. Results from these studies demonstrated that reslizumab, in comparison to placebo, reduced asthma exacerbation rates by at least half and provided significant improvement in lung function and other secondary measures of asthma control when added to an existing ICS-based therapy. Common adverse events in the reslizumab treatment group were comparable to placebo and included worsening of asthma, nasopharyngitis, upper respiratory infections, sinusitis, influenza and headache. Two anaphylactic reactions were reported and resolved following medical treatment at the study site.
Results from the reslizumab BREATH program were recently presented at the American Thoracic Society 2015 Annual Meeting and the American Academy of Allergy, Asthma and Immunology 2015 Annual Meeting, in addition to being published in The Lancet Respiratory Medicine. The BLA for reslizumab has been accepted for filing by the FDA for standard review, with FDA Regulatory Action expected in March 2016.
About Reslizumab
Reslizumab is an investigational humanized monoclonal antibody which targets interleukin-5 (IL-5). IL-5 is a key cytokine involved in the maturation, recruitment, and activation of eosinophils, which are inflammatory white blood cells implicated in a number of diseases, such as asthma. Elevated levels of blood eosinophils are a risk factor for future asthma exacerbations. Reslizumab binds circulating IL-5 thereby preventing IL-5 from binding to its receptor.
About Asthma
Asthma is a chronic (long term) disease usually characterized by airway inflammation and narrowing of the airways, which can vary over time. Asthma may cause recurring periods of wheezing (a whistling sound when you breathe), chest tightness, shortness of breath and coughing that often occurs at night or early in the morning. Without appropriate treatment, asthma symptoms may become more severe and result in an asthma attack, which can lead to hospitalization and even death.
About Eosinophils
Eosinophils are a type of white blood cell that are present at elevated levels in the lungs and blood of many asthmatics. Evidence shows that eosinophils play an active role in the pathogenesis of the disease. IL-5 has been shown to play a crucial role in maturation, growth and activation of eosinophils. Increased levels of eosinophils in the sputum and blood have been shown to correlate with severity and frequency of asthma exacerbations.
About Teva
Teva Pharmaceutical Industries Ltd. (NYSE and TASE: TEVA) is a leading global pharmaceutical company that delivers high-quality, patient-centric healthcare solutions to millions of patients every day. Headquartered in Israel, Teva is the world’s largest generic medicines producer, leveraging its portfolio of more than 1,000 molecules to produce a wide range of generic products in nearly every therapeutic area. In specialty medicines, Teva has a world-leading position in innovative treatments for disorders of the central nervous system, including pain, as well as a strong portfolio of respiratory products. Teva integrates its generics and specialty capabilities in its global research and development division to create new ways of addressing unmet patient needs by combining drug development capabilities with devices, services and technologies. Teva’s net revenues in 2014 amounted to $20.3 billion. For more information, visit www.tevapharm.com.
The U.S. Food and Drug Administration today approved Cinqair (reslizumab) for use with other asthma medicines for the maintenance treatment of severe asthma in patients aged 18 years and older. Cinqair is approved for patients who have a history of severe asthma attacks (exacerbations) despite receiving their current asthma medicines.
Asthma is a chronic disease that causes inflammation in the airways of the lungs. During an asthma attack, airways become narrow making it hard to breathe. Severe asthma attacks can lead to asthma-related hospitalizations because these attacks can be serious and even life-threatening. According to the Centers for Disease Control and Prevention, as of 2013, more than 22 million people in the U.S. have asthma, and there are more than 400,000 asthma-related hospitalizations each year.
“Health care providers and their patients with severe asthma now have another treatment option to consider when the disease is not well controlled by their current asthma therapies,” said Badrul Chowdhury, M.D., Ph.D., director of the Division of Pulmonary, Allergy, and Rheumatology Products in the FDA’s Center for Drug Evaluation and Research.
Cinqair is administered once every four weeks via intravenous infusion by a health care professional in a clinical setting prepared to manage anaphylaxis. Cinqair is a humanized interleukin-5 antagonist monoclonal antibody produced by recombinant DNA technology in murine myeloma non-secreting 0 (NS0) cells. Cinqair reduces severe asthma attacks by reducing the levels of blood eosinophils, a type of white blood cell that contributes to the development of asthma.
The safety and efficacy of Cinqair were established in four double-blind, randomized, placebo‑controlled trials in patients with severe asthma on currently available therapies. Cinqair or a placebo was administered to patients every four weeks as an add-on asthma treatment. Compared with placebo, patients with severe asthma receiving Cinqair had fewer asthma attacks, and a longer time to the first attack. In addition, treatment with Cinqair resulted in a significant improvement in lung function, as measured by the volume of air exhaled by patients in one second.
Cinqair can cause serious side effects including allergic (hypersensitivity) reactions. These reactions can be life-threatening. The most common side effects in clinical trials for Cinqair included anaphylaxis, cancer, and muscle pain.
Cinqair is made by Teva Pharmaceuticals in Frazer, Pennsylvania.


References
- 1Walsh, GM (2009). “Reslizumab, a humanized anti-IL-5 mAb for the treatment of eosinophil-mediated inflammatory conditions”. Current opinion in molecular therapeutics 11 (3): 329–36. PMID 19479666.
- 2http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm491980.htm
- http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm491980.htm
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Humanized (from rat) |
| Target | IL-5 |
| Clinical data | |
| Trade names | Cinquil |
| Identifiers | |
| ATC code | R03DX08 (WHO) |
| ChemSpider | none |
/////////CDP-835, CEP-38072, CTx-55700, SCH-5570, SCH-55700, TRFK-5, Reslizumab, Cinqair®, teva, interleukin-5 (IL-5) antagonist, severe asthma, FDA 2016, Orphan Drug StatuS
Kiran Mazumdar Shaw Conferred with ‘The Global Leadership in Engineering 2016’ Award by USC


http://societyofwomenengineers.swe.org/awards/individual-awards/4153-global-leadership
![]()
The Global Leadership Award honors a person with at least fifteen (15) years professional experience who has worked in and led an internationally based engineering, scientific or technology-based business or organization, and in doing so, serves as a role model to women engineers and technologists worldwide. A maximum of three (3) awards may be presented annually.

“This award is a recognition of Biocon’s significant role in harnessing the potential of Biotechnology to provide affordable access to highly complex bio-pharmaceuticals like Insulins and monoclonal antibodies for the benefit of patients the world over.” – Kiran Mazumdar-Shaw

//////Kiran Mazumdar Shaw, ‘The Global Leadership in Engineering 2016’ , Award by USC
Biocon’s Insulin Glargine gets approval in Japan

![]()
Avik Das | TNN | Mar 28, 2016, 02.52 PM IST
BENGALURU: Biopharmaceutical company Biocon said it got approval from Japan’s health ministry to sell its biosimilar Insulin Glargine in the country.
The product, which is a ready-to-use, prefilled disposable pen with 3 ml of 100IU Insulin Glargine, is expected to be launched in Japan in the first quarter of 2017 with its commercial partner FUJIFILM Pharma Co. Ltd, Biocon said on Monday.
The move will help Biocon capture a significant share of the Japanese Glargine market, which is about $144 million and second largest market outside of North America & Europe.
“The Insulin Glargine approval in the highly regulated market like Japan, marks a huge credibility milestone for Biocon. We see this as a significant achievement in our journey of making global impact in diabetes management through our affordable biosimilar insulins,” chairperson and managing director Kiran Mazumdar-Shaw said.

Kiran Mazumdar–Shaw

Biosimilars are biologic products, made inside living cells and has no clinical differences in terms of safety and effectiveness from the main product. They are however not considered duplicates, like generics, by regulators as it is impossible to manufacture exact copies of biotech drugs.



| Public company | |
| Traded as | BSE: 532523 NSE: BIOCON |
| Industry | Biotechnology |
| Founded | 1978 |
| Founder | Kiran Mazumdar-Shaw |
| Headquarters | Bangalore, Karnataka, India |
|
Key people
|
Kiran Mazumdar-Shaw, (Chairman & MD) |
| Products | Pharmaceuticals Enzymes |
| Revenue | ₹22.41 billion (US$330 million) (2014–15)[1] |
|
Number of employees
|
5,585 (Mar 2011)[1] |
| Subsidiaries | Syngene Clinigene |
| Website | www.biocon.com |
//////Biocon, Insulin Glargine, approval, Japan
Fresolimumab

Fresolimumab
GC 1008, GC1008
UNII-375142VBIA
cas 948564-73-6
Structure
- immunoglobulin G4, anti-(human transforming growth factors beta-1, beta-2 (G-TSF or cetermin) and beta-3), human monoclonal GC-1008 γ4 heavy chain (134-215′)-disulfide with human monoclonal GC-1008 κ light chain, dimer (226-226”:229-229”)-bisdisulfide
- immunoglobulin G4, anti-(transforming growth factor β) (human monoclonal GC-1008 heavy chain), disulfide with human monoclonal GC-1008 light chain, dimer
For Idiopathic Pulmonary Fibrosis, Focal Segmental Glomerulosclerosis,and Cancer
An anti-TGF-beta antibody in phase I clinical trials (2011) for treatment-resistant primary focal segmental glomerulosclerosis.
A pan-specific, recombinant, fully human monoclonal antibody directed against human transforming growth factor (TGF) -beta 1, 2 and 3 with potential antineoplastic activity. Fresolimumab binds to and inhibits the activity of all isoforms of TGF-beta, which may result in the inhibition of tumor cell growth, angiogenesis, and migration. TGF-beta, a cytokine often over-expressed in various malignancies, may play an important role in promoting the growth, progression, and migration of tumor cells.

Fresolimumab (GC1008) is a human monoclonal antibody[1] and an immunomodulator. It is intended for the treatment of idiopathic pulmonary fibrosis (IPF), focal segmental glomerulosclerosis, and cancer[2][3] (kidney cancer and melanoma).
It binds to and inhibits all isoforms of the protein transforming growth factor beta (TGF-β).[2]
History
Fresolimumab was discovered by Cambridge Antibody Technology (CAT) scientists[4] and was one of a pair of candidate drugs that were identified for the treatment of the fatal condition scleroderma. CAT chose to co-develop the two drugs metelimumab (CAT-192) and fresolimumab with Genzyme. During early development, around 2004, CAT decided to drop development of metelimumab in favour of fresolimumab.[5]
In February 2011 Sanofi-Aventis agreed to buy Genzyme for US$ 20.1 billion.[6]
As of June 2011 the drug was being tested in humans (clinical trials) against IPF, renal disease, and cancer.[7][8] On 13 August 2012, Genzyme applied to begin a Phase 2 clinical trial in primary focal segmental glomerulosclerosis[9] comparing fresolimumab versus placebo.
As of July 2014, Sanofi-Aventis continue to list fresolimumab in their research and development portfolio under Phase II development.[10]

References
2 National Cancer Institute: Fresolimumab
- 3 Statement On A Nonproprietary Name Adopted By The USAN Council – Fresolimumab
- 4 Grütter, Christian; Wilkinson, Trevor; Turner, Richard; Podichetty, Sadhana; Finch, Donna; McCourt, Matthew; Loning, Scott; Jermutus, Lutz; Grütter, Markus G. (2008-12-23). “A cytokine-neutralizing antibody as a structural mimetic of 2 receptor interactions”. Proceedings of the National Academy of Sciences 105 (51): 20251–20256. doi:10.1073/pnas.0807200106. ISSN 0027-8424. PMC 2600578. PMID 19073914.
- 5 http://www.independent.co.uk/news/business/news/cat-may-abandon-skin-drug-after-trial-results-disappoint-569445.html
- 6 http://www.bbc.co.uk/news/business-12477750
- 7 http://www.genengnews.com/gen-news-highlights/scientists-trigger-white-fat-to-become-brown-fat-like-to-treat-obesty-and-type-2-diabetes/81245389/
- 8 Clinicaltrials.gov for Fresolimumab
- 9 http://clinicaltrials.gov/show/NCT01665391
- 10 http://en.sanofi.com/rd/rd_portfolio/rd_portfolio.aspx
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | TGF beta 1, 2 and 3 |
| Clinical data | |
| Legal status |
|
| Identifiers | |
| CAS Number | 948564-73-6 |
| ATC code | None |
| ChemSpider | none |
| KEGG | D09620 |
| Chemical data | |
| Formula | C6392H9926N1698O2026S44 |
| Molar mass | 144.4 kDa |
////////////
Elotuzumab
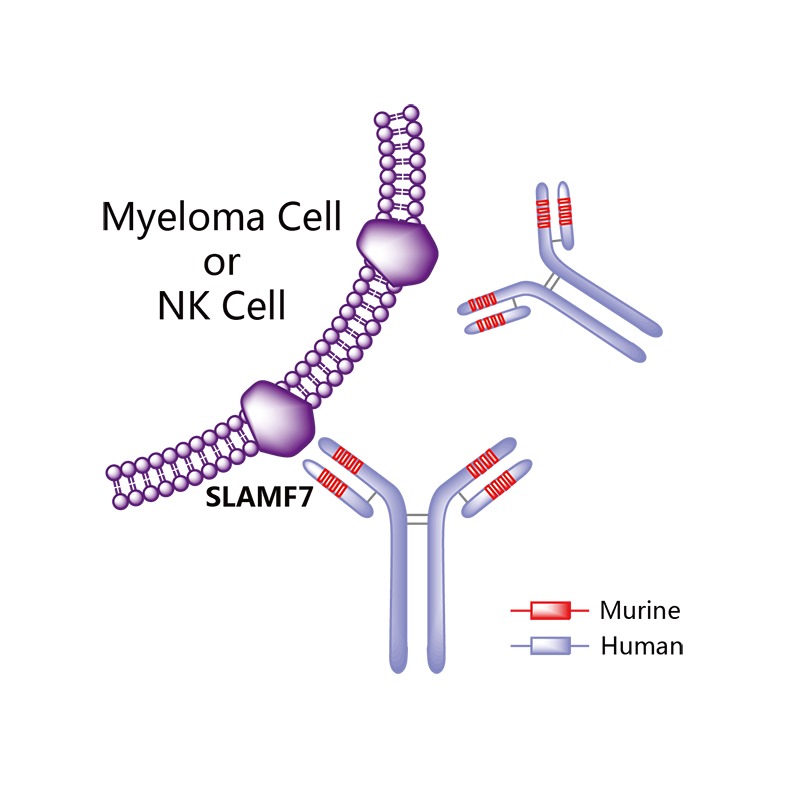
Elotuzumab
Approved nov 30 2012
A SLAMF7-directed immunostimulatory antibody used to treat multiple myeloma.
(Empliciti®)
HuLuc-63;BMS-901608
cas 915296-00-3
![]()


Elotuzumab (brand name Empliciti, previously known as HuLuc63) is a humanized monoclonal antibody used in relapsed multiple myeloma.[1] The package insert denotes its mechanism as a SLAMF7-directed (also known as CD 319) immunostimulatory antibody.[2]
Approvals and indications
In May 2014, it was granted “Breakthrough Therapy” designation by the FDA. [3] On November 30, 2015, FDA approved elotuzumab as a treatment for patients with multiple myeloma who have received one to three prior medications.[1] Elotuzumab was labeled for use with lenalidomide and dexamethasone. Each intravenous injection of elotuzumab should be premedicated with dexamethasone, diphenhydramine, ranitidine and acetaminophen.[2]
Elotuzumab is APPROVED for safety and efficacy in combination with lenalidomide and dexamethasone.
Monoclonal antibody therapy for multiple myeloma, a malignancy of plasma cells, was not very clinically efficacious until the development of cell surface glycoprotein CS1 targeting humanized immunoglobulin G1 monoclonal antibody – Elotuzumab. Elotuzumab is currently APPROVED in relapsed multiple myeloma.
Elotuzumab (HuLuc63) binds to CS1 antigens, highly expressed by multiple myeloma cells but minimally present on normal cells. The binding of elotuzumab to CS1 triggers antibody dependent cellular cytotoxicity in tumor cells expressing CS1. CS1 is a cell surface glycoprotein that belongs to the CD2 subset of immunoglobulin superfamily (IgSF). Preclinical studies showed that elotuzumab initiates cell lysis at high rates. The action of elotuzumab was found to be enhanced when multiple myeloma cells were pretreated with sub-therapeutic doses of lenalidomide and bortezomib. The impressive preclinical findings prompted investigation and analysis of elotuzumab in phase I and phase II studies in combination with lenalidomide and bortezomib.
Elotuzumab As Part of Combination Therapy: Clinical Trial Results
Elotuzumab showed manageable side effect profile and was well tolerated in a population of relapsed/refractory multiple myeloma patients, when treated with intravenous elotuzumab as single agent therapy. Lets’ take a look at how elotuzumab fared in combination therapy trials,
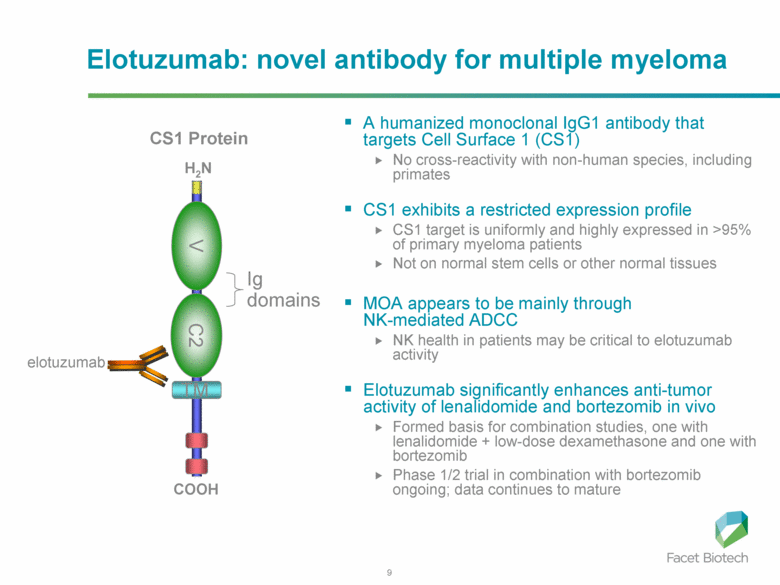
In phase I trial of elotuzumab in combination with Velcade/bortezomib in patients with relapsed/refractory myeloma, the overall response rate was 48% and activity was observed in patients whose disease had stopped responding to Velcade previously. The trial results found that elotuzumab enhanced Velcade activity.
A phase I/II trial in combination with lenalidomide and dexamethasone in refractory/relapsed multiple myeloma patients showed that 82% of patients responded to treatment with a partial response or better and 12% of patients showed complete response. Patients who had received only one prior therapy showed 91% response rate with elotuzumab in combination with lenalidomide and dexamethasone.
Phase I/II trials of the antibody drug has been very impressive and the drug is currently into Phase III trials. Two phase III trials are investigating whether addition of elotuzumab with Revlimid and low dose dexamethasone would increase the time to disease progression. Another phase III trial (ELOQUENT 2) is investigating and comparing safety and efficacy of lenalidomide plus low dose dexamethasone with or without 10mg/kg of elotuzumab in patients with relapsed/refractory multiple myeloma.
Elotuzumab is being investigated in many other trials too. It is being evaluated in combination with Revlimid and low-dose dexamethasone in multiple myeloma patients with various levels of kidney functions, while another phase II study is investigating elotuzumab’s efficacy in patients with high-risk smoldering myeloma.
The main target of multiple myeloma drug development is to satisfy the unmet need for drugs that would improve survival rates. Elotuzumab is an example that mandates much interest in this area and should be followed with diligence.

Empliciti’s Cost
Empliciti will be sold in the U.S. in two vials sizes: A smaller vial that contains 300 mg of the drug, and a larger vial that contains 400 mg.
Bristol-Myers Squibb has informed The Beacon that the wholesale price per vial of Empliciti will be $1,776 for the 300 mg vial and $2,368 for the 400 mg vial.
Using these prices and an assumed patient weight of between 154 and 176 pounds, Empliciti will cost $18,944 per four-week cycle for each of the first two cycles of treatment, and $9,472 per cycle thereafter. This means, in turn, that Empliciti’s cost per year will be $142,080 in the first year and $123,136 in subsequent years.
In comparison, Velcade costs between $4,800 and $8,500 per four-week cycle, depending on how often it is dosed. Ninlaro costs $8,670 per four-week cycle. And Kyprolis costs $10,500 per four-week cycle at the standard (20 – 27 mg/m2) dose.
Additional details about the FDA approval of Empliciti can be found in this press release from the FDA, a related press release from Bristol-Myers Squibb and AbbVie, and the full Empliciti prescribing information.
The results of the ELOQUENT-2 trial were published in Lonial, S. et al., “Elotuzumab Therapy for Relapsed or Refractory Multiple Myeloma,” The New England Journal of Medicine, June 2, 2015 (abstract). Slides from the ASCO presentation summarizing the ELOQUENT-2 results can be viewed here (PDF, courtesy of Dr. Lonial). This Beacon news article provides an in-depth look at the trial results.
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Humanized |
| Target | SLAMF7 (CD319) |
| Clinical data | |
| Trade names | Empliciti |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
IV |
| Pharmacokinetic data | |
| Bioavailability | 100% (IV) |
| Identifiers | |
| CAS Number | 915296-00-3 |
| ATC code | None |
| IUPHAR/BPS | 8361 |
| UNII | 1351PE5UGS |
| Chemical data | |
| Formula | C6476H9982N1714O2016S42 |
| Molecular mass | 145.5 kDa |
References
1 “Press Announcement—FDA approves Empliciti, a new immune-stimulating therapy to treat multiple myeloma”. U.S. Food and Drug Administration. Retrieved 3 December 2015.
2“Empliciti (elotuzumab) for Injection, for Intravenous Use. Full Prescribing Information” (PDF). Empliciti (elotuzumab) for US Healthcare Professionals. Bristol-Myers Squibb Company, Princeton, NJ 08543 USA.
3 “Bristol-Myers Squibb and AbbVie Receive U.S. FDA Breakthrough Therapy Designation for Elotuzumab, an Investigational Humanized Monoclonal Antibody for Multiple Myeloma” (Press release). Princeton, NJ & North Chicago, IL: Bristol-Myers Squibb. 2014-05-19. Retrieved 2015-02-05.
///////
Novartis launches first US ‘biosimilar’ drug at 15 percent discount

Zarxio, filgrastim-sndz
Novartis launches first US ‘biosimilar’ drug at 15 percent discount
LONDON/ZURICH: Novartis kicked off a new era in U.S. medicine on Thursday with the launch of the first “biosimilar” copy of a biotechnology drug approved in the United States, at a discount of 15 percent to the original.
The Swiss drugmaker’s generics unit Sandoz said Zarxio, its form of Amgen’s white blood cell-boosting product Neupogen, would increase access to an important treatment by offering a “high-quality, more affordable version”.
U.S. biotech group Amgen had tried to stop the sale of Zarxio, also known as filgrastim-sndz, but the Washington-based appeals court rejected its attempt to block the launch…..http://www.channelnewsasia.com/news/health/novartis-makes-history-wi/2097550.html

On March 6, 2015, FDA approved the first biosimilar under the Biologics Price Competition and Innovation Act (BPCIA), Sandoz’s Zarxio®. Sandoz submitted Zarxio®as a highly similar, not interchangeable biosimilar, for the same indications as the referenced product. The BPCIA was signed into law in March 2010.
FDA designated “filgrastim-sndz” as the placeholder nonproprietary name rather than the innovator’s name, filgrastim. FDA said that this nonproprietary name “should not be viewed as reflective of the agency’s decision on a comprehensive naming policy for biosimilar and other biological products. While the FDA has not yet issued draft guidance on how current and future biological [biosimilar?] products marketed in the United States should be named, the agency intends to do so in the near future.”
Accompanying the news release was a document “Biosimilars: More Treatment Options Are on the Way”. The document includes various quotes and paraphrased statements by Leah Christl, Ph.D., Associate Director for Therapeutic Biologics, to help describe to consumers what biosimilar medications are. Below are some quotes and information from that document:
Biologics are medicines that generally come from living organisms, which can include humans, animals and microorganisms such as yeast and bacteria.
. . .
“Biologics are different from conventional medications. Conventional medications—drugs—are generally made from chemicals, or chemically synthesized, and therefore their structure can be relatively easily defined,” explains Christl.
Unlike conventional medications, biologics can’t be made by following a chemical “recipe.” “Biologics come from living organisms which are variable in nature. In addition, they are generally more complex and not as easy to define and characterize,” Christl explains. For that reason, manufacturing biologics is a far more complex process than manufacturing drugs.
Just as it does for drugs, FDA rigorously and thoroughly evaluates a biologic’s safety and effectiveness before granting it licensure (approval). Currently, biologics are among the fastest growing segments of the prescription product market.
. . .
Christl explains that a biosimilar is a type of biologic that is highly similar to another, already FDA-approved biologic (known as the reference product).
“It is important to note that a biosimilar is not just like a generic drug,” she adds. “Because of the differences in complexity of the structure of the biologic and the process used to make a biologic, biosimilars are not as easy to produce as generics, which are copies of brand name drugs.” A biosimilar is not an exact duplicate of another biologic; rather, a biosimilar is highly similar to the reference product.
Before approving a biosimilar, FDA experts must also first verify that there are no clinically meaningful differences between the biosimilar and its reference product. In other words, it will work the same way as the reference product for its approved indications.
Also, the biosimilar must have the same strength and dosage form (injectable, for example) and route of administration as the reference product. The biosimilar must be manufactured followingCurrent Good Manufacturing Practices.
“Patients can rest assured that they’ll be able to rely upon the safety and effectiveness of an FDA-approved biosimilar, just as they can rely on the reference product that the biosimilar was compared to,” Christl says. Like other biologics, biosimilars generally must be prescribed by a physician.
. . .
“Biosimilars are likely to create greater competition in the medical marketplace,” saysChristl. This could not only increase treatment options for patients, but also lead to less expensive alternatives to comparable products. With an increasing number of biosimilars on the market, consumers may expect to get equally safe and effective treatment, but at lower costs, she says.
Despite the significant achievement for FDA to approve the first biosimilar under the BPCIA, significant questions other than nonproprietary naming remain. First, Sandoz chose not to take advantage of the pre-approval patent exchange mechanism of the BPCIA, which could have addressed possible patent challenges that may prevent Sandoz from marketing Zarxio®until certain patents are invalidated, are found unenforceable, or have expired. Second, because this and other non-interchangeable versions of biosimilars are not expected to have automatic substitution based on the BPCIA, it remains unclear how ready physicians or patients will be to try a biosimilar version over its referenced product. Third, company representatives from Sandoz and other biosimilar manufacturers have not indicated at what price their biosimilar products will be sold, at times suggesting “at parity,” which may cause reimbursement issues. Fourth, many states have enacted rules that include special physician notification provisions, even when interchangeable biosimilars are dispensed to patients. And there are still issues surrounding pharmacovigilance and risk management when there are innovator and corresponding biosimilar versions marketed. Nevertheless, FDA proclaims that more biosimilars are on the way, as additional companies have indicated that they have submitted or FDA has filed their biosimilar applications. Sandoz’s Zarxio® then is just the tip of the iceberg of what is coming with more issues to be resolved along the way.
ZARXIO (filgrastim-sndz) is a 175 amino acid human granulocyte colony-stimulating factor (G-CSF) manufactured by recombinant DNA technology.
ZARXIO is produced by Escherichia coli (E coli) bacteria into which has been inserted the human granulocyte colony-stimulating factor gene. ZARXIO has a molecular weight of 18,800 daltons. The protein has an amino acid sequence that is identical to the natural sequence predicted from humanDNA sequence analysis, except for the addition of an N-terminal methioninenecessary for expression in E coli. Because ZARXIO is produced in E coli, the product is non-glycosylated and thus differs from G-CSF isolated from a human cell.
ZARXIO injection is a sterile, clear, colorless to slightly yellowish , preservative-free liquid containing filgrastimsndz at a specific activity of 1.0 x 108 U/mg (as measured by a cell mitogenesis assay). The product is available in single-use prefilled syringes. The single-use prefilled syringes contain either 300 mcg/0.5 mL or 480 mcg/0.8 mL of filgrastim-sndz. See table below for product composition of each single-use prefilled syringe.
| 300 MCG/0.5 ML SYRINGE | 480 MCG/0.8 ML SYRINGE | |
| Filgrastim-sndz | 300 mcg | 480 mcg |
| Glutamic Acid | 0.736 mg | 1.178 mg |
| Polysorbate 80 | 0.02 mg | 0.032 mg |
| Sorbitol | 25 mg | 40 mg |
| Sodium hydroxide | q.s. | qs. |
| Water for Injection | ||
| USP q.s. ad* | ad 0.5 mL | ad 0.8 mL |
FDA approves first biosimilar product Zarxio

![]()
March 6, 2015
The U.S. Food and Drug Administration today approved Zarxio (filgrastim-sndz), the first biosimilar product approved in the United States.
Biological products are generally derived from a living organism. They can come from many sources, including humans, animals, microorganisms or yeast.
A biosimilar product is a biological product that is approved based on a showing that it is highly similar to an already-approved biological product, known as a reference product. The biosimilar also must show it has no clinically meaningful differences in terms of safety and effectiveness from the reference product. Only minor differences in clinically inactive components are allowable in biosimilar products.
Sandoz, Inc.’s Zarxio is biosimilar to Amgen Inc.’s Neupogen (filgrastim), which was originally licensed in 1991. Zarxio is approved for the same indications as Neupogen, and can be prescribed by a health care professional for:
- patients with cancer receiving myelosuppressive chemotherapy;
- patients with acute myeloid leukemia receiving induction or consolidation chemotherapy;
- patients with cancer undergoing bone marrow transplantation;
- patients undergoing autologous peripheral blood progenitor cell collection and therapy; and
- patients with severe chronic neutropenia.
“Biosimilars will provide access to important therapies for patients who need them,” said FDA Commissioner Margaret A. Hamburg, M.D. “Patients and the health care community can be confident that biosimilar products approved by the FDA meet the agency’s rigorous safety, efficacy and quality standards.”
The Biologics Price Competition and Innovation Act of 2009 (BPCI Act) was passed as part of the Affordable Care Act that President Obama signed into law in March 2010. The BPCI Act created an abbreviated licensure pathway for biological products shown to be “biosimilar” to or “interchangeable” with an FDA-licensed biological product, called the “reference product.” This abbreviated licensure pathway under section 351(k) of the Public Health Service Act permits reliance on certain existing scientific knowledge about the safety and effectiveness of the reference product, and enables a biosimilar biological product to be licensed based on less than a full complement of product-specific preclinical and clinical data.
A biosimilar product can only be approved by the FDA if it has the same mechanism(s) of action, route(s) of administration, dosage form(s) and strength(s) as the reference product, and only for the indication(s) and condition(s) of use that have been approved for the reference product. The facilities where biosimilars are manufactured must also meet the FDA’s standards.
The FDA’s approval of Zarxio is based on review of evidence that included structural and functional characterization, animal study data, human pharmacokinetic and pharmacodynamics data, clinical immunogenicity data and other clinical safety and effectiveness data that demonstrates Zarxio is biosimilar to Neupogen. Zarxio has been approved as biosimilar, not as an interchangeable product. Under the BPCI Act, a biological product that that has been approved as an “interchangeable” may be substituted for the reference product without the intervention of the health care provider who prescribed the reference product.
The most common expected side effects of Zarxio are aching in the bones or muscles and redness, swelling or itching at injection site. Serious side effects may include spleen rupture; serious allergic reactions that may cause rash, shortness of breath, wheezing and/or swelling around the mouth and eyes; fast pulse and sweating; and acute respiratory distress syndrome, a lung disease that can cause shortness of breath, difficulty breathing or increase the rate of breathing.
For this approval, the FDA has designated a placeholder nonproprietary name for this product as “filgrastim-sndz.” The provision of a placeholder nonproprietary name for this product should not be viewed as reflective of the agency’s decision on a comprehensive naming policy for biosimilar and other biological products. While the FDA has not yet issued draft guidance on how current and future biological products marketed in the United States should be named, the agency intends to do so in the near future.
Sandoz, a Novartis company, is based in Princeton, New Jersey. Neupogen is marketed by Amgen, based in Thousand Oaks, California.
Sandoz’s Zarzio (filgrastim) would be the first ‘biosimilar’ drug available in the US

A key advisory committee of the US Food and Drug Administration (FDA) has voted in favour of licencing a copycat version of a biological drug. If approved, Sandoz’s Zarxio (filgrastim) would be the first ‘biosimilar’ drug available in the US.
read at……..http://www.rsc.org/chemistryworld/2015/01/us-poised-approve-first-biosimilar-drug
On 7 January, the FDA’s Oncological Drugs Advisory Committee unanimously cleared Sandoz’ version of filgrastim – marketed as Neupogen by Amgen – for all five indications approved for the Amgen drug. The medication is used to prevent infection and low white blood cell counts caused by chemotherapy.


| Systematic (IUPAC) name | |
|---|---|
| Human granulocyte colony stimulating factor | |
| Clinical data | |
| Trade names | Neupogen |
| AHFS/Drugs.com | monograph |
| Legal status |
?
|
| Identifiers | |
| CAS number | 143011-72-7 |
| ATC code | L03AA02 |
| DrugBank | DB00099 |
| UNII | PVI5M0M1GW |
| ChEMBL | CHEMBL1201567 |
| Chemical data | |
| Formula | C845H1343N223O243S9 |
| Molecular mass | 18802.8 g/mol |
Filgrastim is a granulocyte colony-stimulating factor (G-CSF) analog used to stimulate the proliferation and differentiation ofgranulocytes;[1] it is a pharmaceutical analog of naturally occurring G-CSF. It is produced by recombinant DNA technology. The gene for human granulocyte colony-stimulating factor is inserted into the genetic material of Escherichia coli. The G-CSF then produced byE. coli is different from G-CSF naturally made in humans.

Commercialization
Filgrastim is marketed under several brand names, including:
| Company | Brand |
|---|---|
| Cadila Pharmaceuticals | Filcad |
| Abbott Laboratories | Imumax |
| Dr. Reddy’s Laboratories | Grafeel |
| Intas Biopharmaceuticals | Neukine |
| Amgen | Neupogen[2] |
| Emcure Pharmaceuticals | Emgrast |
| Reliance Life Sciences | Religrast |
| Sandoz | Zarzio |
| Biocon | Nufil |

Apricus Biosciences is currently developing and testing a product under the brand name Nupen which can deliver filgrastim through the skin to improve post-chemotherapy recovery of neutrophil counts.

Therapeutic uses
Filgrastim is used to treat neutropenia,[3] stimulating the bone marrow to increase production of neutrophils. Causes of neutropenia include chemotherapy and bone marrow transplantation.
Filgrastim is also used to increase the number of hematopoietic stem cells in the blood before collection by leukapheresis for use in hematopoietic stem cell transplantation.
Mechanism of Action: Filgrastim is a human granulocyte colony stimulating factor (G-CSF) produced by recombinant DNA technology. G-CSF regulates the production of neutrophils within the bone marrow; endogenous G-CSF is a glycoprotein produced by monocytes, fibroblasts, and endothelial cells.
G-CSF is a colony stimulating factor which has been shown to have minimal direct in vivo or in vitro effects on the production of other haematopoietic cell types.NEUPOGEN (filgrastim) is the name for recombinant methionyl human granulocyte colony stimulating factor (r-metHuG-CSF). ref: [1]
Contraindications
Filgrastim should not be used in patients with known hypersensitivity to E. coli-derived proteins.
Adverse effects
The most commonly observed adverse effect is mild-to-moderate bone pain after repeated administration and local skin reactions at the site of injection.[4] Other observed adverse effects include serious allergic reactions (including a rash over the whole body, shortness of breath, wheezing, dizziness, swelling around the mouth or eyes, fast pulse, and sweating), ruptured spleen (sometimes resulting in death), alveolar hemorrhage, acute respiratory distress syndrome, and hemoptysis.[4] Severe sickle cell crises, in some cases resulting in death, have been associated with the use of filgrastim in patients with sickle cell disorders.[5]
Interactions
Drug interactions between filgrastim and other drugs have not been fully evaluated. Drugs which may potentiate the release of neutrophils‚ such as lithium‚ should be used with caution.
Increased hematopoietic activity of the bone marrow in response to growth factor therapy has been associated with transient positive bone imaging changes; this should be considered when interpreting bone-imaging results.[6]
Filgrastim has not been studied in pregnant women and its effects on the foetus is unknown. If taking filgrastim while pregnant, it is possible that traces of the drug could be found in the baby’s blood. It is not known if the drug can get into human breast milk.
References
- Beveridge, R. A.; Miller, J. A.; Kales, A. N.; Binder, R. A.; Robert, N. J.; Harvey, J. H.; Windsor, K.; Gore, I.; Cantrell, J.; Thompson, K. A.; Taylor, W. R.; Barnes, H. M.; Schiff, S. A.; Shields, J. A.; Cambareri, R. J.; Butler, T. P.; Meister, R. J.; Feigert, J. M.; Norgard, M. J.; Moraes, M. A.; Helvie, W. W.; Patton, G. A.; Mundy, L. J.; Henry, D.; Sheridan, B.; Staddon, A.; Ford, P.; Katcher, D.; Houck, W.; Major, W. B. (1998). “A Comparison of Efficacy of Sargramostim (Yeast-Derived RhuGM-CSF) and Filgrastim (Bacteria-Derived RhuG-CSF) in the Therapeutic Setting of Chemotherapy-Induced Myelosuppression”. Cancer Investigation 16 (6): 366–373. doi:10.3109/07357909809115775. PMID 9679526.
- “FDA Reviews What Could Be First Biosimilar”. Discov. Dev. Mag. (Rockaway, New Jersey, United States). Associated Press. 25 July 2014.
- Crawford, J.; Glaspy, J. A.; Stoller, R. G.; Tomita, D. K.; Vincent, M. E.; McGuire, B. W.; Ozer, H. (2005). “Final Results of a Placebo-Controlled Study of Filgrastim in Small-Cell Lung Cancer: Exploration of Risk Factors for Febrile Neutropenia”. Supportive Cancer Therapy 3 (1): 36–46. doi:10.3816/SCT.2005.n.023. PMID 18632435.
- Neupogen “Neupogen: Patient Information Leaflet”. Amgen. Retrieved 24 June 2013.
- “NEUPOGEN® Patient Guide”. Amgen. Retrieved 24 June 2013.
- “Neupogen”. RxList. 4 June 2012. Retrieved 23 June 2013.
Further reading
- Budiono Santoso; Chris J. van Boxtel; Boxtel, Christoffel Jos van (2001). Drug benefits and risks: international textbook of clinical pharmacology. New York: Wiley. ISBN 0-471-89927-5.
- “Neupogen information”. Retrieved 20 October 2005.

{kind=link}