
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
RIDINILAZOLE

RIDINILAZOLE
SMT19969
- Molecular FormulaC24H16N6
- Average mass388.424 Da
- ридинилазол [Russian] [INN]ريدينيلازول [Arabic] [INN]利地利唑 [Chinese] [INN]
- リジニラゾール;
10075
2,2′-Di(4-pyridinyl)-3H,3’H-5,5′-bibenzimidazole
308362-25-6[RN]6,6′-Bi-1H-benzimidazole, 2,2′-di-4-pyridinyl-
Summit Therapeutics (formerly Summit Corp ) is developing ridinilazole the lead compound from oral narrow-spectrum, GI-restricted antibiotics, which also include SMT-21829, for the treatment of Clostridium difficile infection and prevention of recurrent disease.
Ridinilazole (previously known as SMT19969) is an investigational small molecule antibiotic being evaluated for oral administration to treat Clostridioides difficile infection (CDI). In vitro, it is bactericidal against C. difficile and suppresses bacterial toxin production; the mechanism of action is thought to involve inhibition of cell division.[1] It has properties which are desirable for the treatment of CDI, namely that it is a narrow-spectrum antibiotic which exhibits activity against C. difficile while having little impact on other normal intestinal flora and that it is only minimally absorbed systemically after oral administration.[2] At the time ridinilazole was developed, there were only three antibiotics in use for treating CDI: vancomycin, fidaxomicin, and metronidazole.[1][2] The recurrence rate of CDI is high, which has spurred research into other treatment options with the aim to reduce the rate of recurrence.[3][4]
As of 2019, two phase II trials have been completed and two phase III trials comparing ridinilazole to vancomycin for CDI are expected to be completed in September 2021.[2][5][6] Ridinilazole was designated as a Qualified Infectious Disease Product (QIDP) and was granted Fast Track status by the U.S. FDA.[2] Fast Track status is reserved for drugs designed to treat diseases where there is currently a gap in the treatment, or a complete lack thereof.[7] The QIDP designation adds five more years of exclusivity for ridinazole upon approval.[8]


PATENT
WO-2021009514
Process for preparing ridinilazole useful for treating Clostridium difficile infection. Also claimed is the crystalline form of a compound.
The present invention relates to processes for the preparation of 2,2′-di(pyridin-4-yl)-1/-/,T/-/-5,5′-bibenzo[d]imidazole (which may also be known as 5,5’-bis[2-(4-pyridinyl)-1/-/-benzimidazole], 2,2′-bis(4-pyridyl)-3/-/,3’/-/-5,5′-bibenzimidazole or 2-pyridin-4-yl-6-(2-pyridin-4-yl-3/-/-benzimidazol-5-yl)-1/-/-benzimidazole), referenced herein by the INN name ridinilazole, and pharmaceutically acceptable derivatives, salts, hydrates, solvates, complexes, bioisosteres, metabolites or prodrugs thereof. The invention also relates to various crystalline forms of ridinilazole, to processes for their preparation and to related pharmaceutical preparations and uses thereof (including their medical use and their use in the efficient large-scale synthesis of ridinilazole).
WO2010/063996 describes various benzimidazoles, including ridinilazole, and their use as antibacterials (including in the treatment of CDAD).
WO 2011/151621 describes various benzimidazoles and their use as antibacterials
(including in the treatment of CDAD).
W02007056330, W02003105846 and W02002060879 disclose various 2-amino benzimidazoles as antibacterial agents.
W02007148093 discloses various 2-amino benzothiazoles as antibacterial agents.
W02006076009, W02004041209 and Bowser et at. (Bioorg. Med. Chem. Lett., 2007, 17, 5652-5655) disclose various substituted benzimidazole compounds useful as anti-infectives that decrease resistance, virulence, or growth of microbes. The compounds are said not to exhibit intrinsic antimicrobial activity in vitro.
US 5,824,698 discloses various dibenzimidazoles as broad-spectrum antibiotics, disclosing activity against both Gram-negative and Gram-positive bacteria, including Staphylococcus spp.and Enterococcus spp. However, this document does not disclose activity against anaerobic spore-forming bacteria and in particular does not disclose activity against any Clostridioides spp. (including C. difficile).
US 2007/0112048 A1 discloses various bi- and triarylimidazolidines and bi- and
triarylamidines as broad-spectrum antibiotics, disclosing activity against both Gram negative and Gram-positive bacteria, including Staphylococcus spp., Enterococcus spp. and Clostridioides spp. However, this document does not disclose compounds of formula (I) as described herein.
Chaudhuri et al. (2007) J.Org. Chem. 72, 1912-1923 describe various bis-2-(pyridyl)-1 H-benzimidazoles (including compounds of formula I as described herein) as DNA binding agents. This document is silent as to potential antibacterial activity.
Singh et al. (2000) Synthesis 10: 1380-1390 describe a condensation reaction for producing 2,2′-di(pyridin-4-yl)-1/-/,T/-/-5,5′-bibenzo[d]imidazole using 4-pyridine
carboxaldehyde, FeCI3, 02, in DMF at 120°C.
Bhattacharya and Chaudhuri (2007) Chemistry – An Asian Journal 2: 648-655 describe a condensation reaction for producing 2,2′-di(pyridin-4-yl)-1/-/,T/-/-5,5′-bibenzo[d]imidazole using 4-pyridine carboxaldehyde and nitrobenzene at 120°C.
WO2019/068383 describes the synthesis of ridinilazole by metal-ion catalyzed coupling of 3,4,3’,4’-tetraaminobiphenyl with 4-pyridinecarboxaldehyde in the presence of oxygen, followed by the addition of a complexing agent.
PATENT
WO2010063996
claiming antibacterial compounds. Bicyclic heteroaromatic compounds, particularly bi-benzimidazole derivatives.
WO2007056330, WO2003105846 and WO2002060879 disclose various 2-amino benzimidazoles as antibacterial agents.
WO2007148093 discloses various 2-amino benzothiazoles as antibacterial agents.
WO2006076009, WO2004041209 and Bowser et al. (Bioorg. Med. Chem. Lett., 2007, 17, 5652-5655) disclose various substituted benzimidazole compounds useful as anti-infectives that decrease resistance, virulence, or growth of microbes. The compounds are said not to exhibit intrinsic antimicrobial activity in vitro.
US 5,824,698 discloses various dibenzimidazoles as broad-spectrum antibiotics, disclosing activity against both Gram-negative and Gram-positive bacteria, including Staphylococcus spp.and Enterococcus spp. However, this document does not disclose activity against anaerobic spore-forming bacteria and in particular does not disclose activity against any Clostridium spp. (including C. difficile).
US 2007/0112048 A1 discloses various bi- and triarylimidazolidines and bi- and triarylamidines as broad-spectrum antibiotics, disclosing activity against both Gram-negative and Gram-positive bacteria, including Staphylococcus spp., Enterococcus spp.
and Clostridium spp. However, this document does not disclose compounds of general formula (I) as described herein.
Chaudhuri et al. (J.Org. Chem., 2007, 72, 1912-1923) describe various bis-2-(pyridyl)-1 H-benzimidazoles (including compounds of formula I as described herein) as DNA binding agents. This document is silent as to potential antibacterial activity.
PATENT
Product PATENT, WO2010063996 ,
protection in the EP until 2029 and expire in the US in December 2029.
PAPER
https://www.frontiersin.org/articles/10.3389/fmicb.2018.01206/full
PAPER
Synthesis (2000), (10), 1380-1390.
https://www.thieme-connect.de/products/ejournals/abstract/10.1055/s-2000-7111
PAPERT
Chemistry – An Asian Journal (2007), 2(5), 648-655.
https://onlinelibrary.wiley.com/doi/abs/10.1002/asia.200700014
Studies of double‐stranded‐DNA binding have been performed with three isomeric bis(2‐(n‐pyridyl)‐1H‐benzimidazole)s (n=2, 3, 4). Like the well‐known Hoechst 33258, which is a bisbenzimidazole compound, these three isomers bind to the minor groove of duplex DNA. DNA binding by the three isomers was investigated in the presence of the divalent metal ions Mg2+, Co2+, Ni2+, Cu2+, and Zn2+. Ligand–DNA interactions were probed with fluorescence and circular dichroism spectroscopy. These studies revealed that the binding of the 2‐pyridyl derivative to DNA is dramatically reduced in the presence of Co2+, Ni2+, and Cu2+ ions and is abolished completely at a ligand/metal‐cation ratio of 1:1. Control experiments done with the isomeric 3‐ and 4‐pyridyl derivatives showed that their binding to DNA is unaffected by the aforementioned transition‐metal ions. The ability of 2‐(2‐pyridyl)benzimidazole to chelate metal ions and the conformational changes of the ligand associated with ion chelation probably led to such unusual binding results for the ortho isomer. The addition of ethylenediaminetetraacetic acid (EDTA) reversed the effects completely.
PAPER
Journal of Organic Chemistry (2007), 72(6), 1912-1923.
https://pubs.acs.org/doi/10.1021/jo0619433

Three symmetrical positional isomers of bis-2-(n-pyridyl)-1H-benzimidazoles (n = 2, 3, 4) were synthesized and DNA binding studies were performed with these isomeric derivatives. Like bisbenzimidazole compound Hoechst 33258, these molecules also demonstrate AT-specific DNA binding. The binding affinities of 3-pyridine (m-pyben) and 4-pyridine (p-pyben) derivatized bisbenzimidazoles to double-stranded DNA were significantly higher compared to 2–pyridine derivatized benzimidazole o-pyben. This has been established by combined experimental results of isothermal fluorescence titration, circular dichroism, and thermal denaturation of DNA. To rationalize the origin of their differential binding characteristics with double-stranded DNA, computational structural analyses of the uncomplexed ligands were performed using ab initio/Density Functional Theory. The molecular conformations of the symmetric head-to-head bisbenzimidazoles have been computed. The existence of intramolecular hydrogen bonding was established in o-pyben, which confers a conformational rigidity to the molecule about the bond connecting the pyridine and benzimidazole units. This might cause reduction in its binding affinity to double-stranded DNA compared to its para and meta counterparts. Additionally, the predicted stable conformations for p-, m-, and o-pyben at the B3LYP/6-31G* and RHF/6-31G* levels were further supported by experimental pKa determination. The results provide important information on the molecular recognition process of such symmetric head to head bisbenzimidazoles toward duplex DNA.
Patent
US 8975416
PATENT
WO 2019068383
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019068383
Clostridium difficile infection (CDI) is the leading cause of infectious healthcare-associated diarrhoea. CDI remains a challenge to treat clinically, because of a limited number of antibiotics available and unacceptably high recurrence rates. Because of this, there has been significant demand for creating innovative therapeutics, which has resulted in the development of several novel antibiotics.
Ridinilazole (SMT19969) is the INN name of 5,5’bis[2-(4-pyridinyl)-lH-benzimidazole], which is a promising non-absorbable small molecule antibiotic intended for oral use in the treatment of CDI. It has been shown to exhibit a prolonged post-antibiotic effect and treatment with ridinilazole has resulted in decreased toxin production. A phase 1 trial demonstrated that oral ridinilazole is well tolerated and specifically targets Clostridia whilst sparing other faecal bacteria.
Ridinilazole has the following chemical structure:
Bhattacharya & Chaudhuri (Chem. Asian J., 2007, No. 2, 648-655) report performing double-stranded DNA binding with three benzimidazole derivatives, including ridinilazole. The compounds have been prepared by dissolving the reactants in nitrobenzene, heating at 120°C for 8- 1 Oh and purifying the products by column chromatography over silica gel. The compounds were obtained in 65-70% yield. Singh et al., (Synthesis, 2000, No. 10, 1380-1390) describe a catalytic redox cycling approach based on Fe(III) and molecular oxygen as co-oxidant for providing access to benzimidazole and
imidazopyridine derivatives, such as ridinilazole. The reaction is performed at high temperatures of 120°C and the product is isolated in 91% yield by using silica flash chromatography.
Both processes are not optimal, for example in terms of yield, ease of handling and scalability. Thus, there is a need in the art for an efficient and scalable preparation of ridinilazole, which overcomes the problems of the prior art processes.
Example 1 : Preparation of crude ridinilazole free base
A solution of 3,4,3′,4′-tetraaminobiphenyl (3.28 g, 15.3 mmol) and isonicotinaldehyde (3.21 g, 30.0 mmol) in DMF (40 mL) was stirred at 23 °C for one hour. Then anhydrous ferric chloride (146 mg, 0.90 mmol), water (0.10 mL, 5.4 mmol) and additional DMF (2 mL) were added and fresh air was bubbled into the solution during vigorous stirring for 5 hours at room temperature. Next, water (80 mL) and EDTA (0.29 g) were added resulting in a brownish suspension, which was stirred overnight. The product was isolated by filtration, washed with water, and dried in a desiccator in vacuo as a brown powder (5.56 g; 95%). The addition of EDTA had held iron in solution and the crude ridinilazole contained significantly lower amounts of iron than comparative example 1.
Example 12: Formation of essentially pure ridinilazole free base
To a suspension von ridinilazole tritosylate (1 10 mg, 0.12 mmol) in water (35 mL) featuring a pH value of about 4.5 stirring at 70 °C sodium bicarbonate (580 mg, 6.9 mmol) were added and caused a change of color from orange to slightly tan. The mixture, now at a pH of about 8.5, was cooled down to room temperature and the solids were separated by filtration, washed with water (1 ML) and dried in vacuo providing 40 mg (85%) essentially pure ridinilazole as a brownish powder.
Spectroscopic analysis:
¾ NMR (DMSO-de, 300 MHz): δ 7.55 (d, J = 8.4 Hz, 2H), 7.70 (d, J = 8.4 Hz, 2H), 7.88 (s, 2H), 8.13 (d, J = 5.8 Hz, 4H), 8.72 (d, J = 5.8 Hz, 4H) ppm.
13C NMR (DMSO-d6, 75 MHz): δ 1 13.4 (2C), 1 16.4 (2C), 120.4 (4C), 121.8 (2C), 135.7 (2C), 138.7 (2C), 140.7 (2C), 141.4 (2C), 150.3 (4C), 151.1 (2C) ppm.
IR (neat): v 3033 (w), 1604 (s), 1429 (m), 1309 (m), 1217 (m), 1 1 15 (w), 998 (m), 964 (m), 824 (m), 791 (s), 690 (s), 502 (s) cm .
UV-Vis (MeOH): 257, 341 nm.
The sharp peaks in the ¾ NMR indicated that iron had been efficiently removed.
Comparative example 1 : Preparation of ridinilazole
A solution of 3,4,3′,4′-tetraaminobiphenyl (0.69 g, 3.2 mmol) and isonicotinaldehyde (0.64 g, 6.0 mmol) in DMF (20 mL) was stirred at 80°C for one hour. Then ferric chloride hexahydrate (49 mg, 0.18 mmol), water (0.10 mL, 5.4 mmol) and additional DMF (2 mL) were added and fresh air was bubbled into the solution during vigorous stirring for 10 hours at 120 °C. After cooling to room temperature water (50 mL) and the mixture was stirred for one hour. A black crude product was isolated by filtration and comprised ridinilazole and iron.
References
- ^ Jump up to:a b Cho JC, Crotty MP, Pardo J (March 2019). “Clostridium difficile infection”. Annals of Gastroenterology. 32 (2): 134–140. doi:10.20524/aog.2018.0336. PMC 6394264. PMID 30837785.
- ^ Jump up to:a b c d Carlson TJ, Endres BT, Bassères E, Gonzales-Luna AJ, Garey KW (April 2019). “Ridinilazole for the treatment of Clostridioides difficile infection”. Expert Opinion on Investigational Drugs. 28 (4): 303–310. doi:10.1080/13543784.2019.1582640. PMID 30767587.
- ^ Bassères E, Endres BT, Dotson KM, Alam MJ, Garey KW (January 2017). “Novel antibiotics in development to treat Clostridium difficile infection”. Current Opinion in Gastroenterology. 33 (1): 1–7. doi:10.1097/MOG.0000000000000332. PMID 28134686.
These tables highlight the increased drug development directed towards CDI due to the rise in prevalence of infections and to attempt to reduce the number of recurrent infections.
- ^ Vickers RJ, Tillotson G, Goldstein EJ, Citron DM, Garey KW, Wilcox MH (August 2016). “Ridinilazole: a novel therapy for Clostridium difficile infection”. International Journal of Antimicrobial Agents. 48 (2): 137–43. doi:10.1016/j.ijantimicag.2016.04.026. PMID 27283730.
there exists a significant unmet and increasing medical need for new therapies to treat CDI, specifically those that can reduce the rate of disease recurrence.
- ^ Clinical trial number NCT03595553 for “Ri-CoDIFy 1: Comparison of Ridinilazole Versus Vancomycin Treatment for Clostridium Difficile Infection” at ClinicalTrials.gov
- ^ Clinical trial number NCT03595566 for “Ri-CoDIFy 2: To Compare Ridinilazole Versus Vancomycin Treatment for Clostridium Difficile Infection” at ClinicalTrials.gov
- ^ “Fast Track”. U.S. Food and Drug Administration. 2018-11-03.
- ^ “”HHS spurs new antibiotic development for biodefense and common infections””. Public Health Emergency. U.S. Department of Health and Human Services. Retrieved 2020-12-04.
| Clinical data | |
|---|---|
| Other names | SMT19969 |
| ATC code | None |
| Identifiers | |
| IUPAC name[show] | |
| CAS Number | 308362-25-6 |
| PubChem CID | 16659285 |
| ChemSpider | 17592423 |
| UNII | 06DX01190R |
| KEGG | D11958 |
| Chemical and physical data | |
| Formula | C24H16N6 |
| Molar mass | 388.42 g/mol |
| 3D model (JSmol) | Interactive image |
| SMILES[hide]c6cc(c5nc4ccc(c3ccc2nc(c1ccncc1)[nH]c2c3)cc4[nH]5)ccn6 |
/////////RIDINILAZOLE, SMT19969, SMT 19969, ридинилазол , ريدينيلازول , 利地利唑 , リジニラゾール , Qualified Infectious Disease Product, QIDP, Fast Track , PHASE 3, Clostridioides difficile infection ,
OI 338
OI 338
OI338GT (NN1953)
NNC0123-0000-0338
Insulin oral (NN 1953); Insulin-338-GIPET-I; LAI 338; NN 1438; NN-1953; NNC-0123-0000-0338; NNC0123-0338; OI-338GT; Oral insulin 338 C10
- OriginatorNovo Nordisk
- ClassAntihyperglycaemics; Insulins; Pancreatic hormones
- Mechanism of ActionOrnithine decarboxylase stimulants; Phosphokinase stimulants; Protein tyrosine kinase stimulants
- Phase IIType 1 diabetes mellitus; Type 2 diabetes mellitus
- 28 Jul 2018No recent reports of development identified for phase-I development in Type-1 diabetes mellitus in Germany (SC, Injection)
- 28 Jul 2018No recent reports of development identified for phase-I development in Type-2-diabetes-mellitus in Denmark (SC, Injection)
- 11 Sep 2017Efficacy and adverse events data from a phase II trial in Type-2 diabetes mellitus presented at the 53rd Annual Meeting of the European Association for the Study of Diabetes (EASD-2017)
OI-338GT is a long-acting oral basal insulin analogue which had been in phase II clinical trials at Novo Nordisk for the treatment of patients with type 2 and type 1 diabetes. In 2016, the company discontinued the development of the product as the emergent product profile and required overall investments were not commercially viable in the increasingly challenging payer environment.
PAPERJ. Med. Chem. 2021, 64, 1, 616–628
Publication Date:December 28, 2020
https://doi.org/10.1021/acs.jmedchem.0c01576https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c01576

Recently, the first basal oral insulin (OI338) was shown to provide similar treatment outcomes to insulin glargine in a phase 2a clinical trial. Here, we report the engineering of a novel class of basal oral insulin analogues of which OI338, 10, in this publication, was successfully tested in the phase 2a clinical trial. We found that the introduction of two insulin substitutions, A14E and B25H, was needed to provide increased stability toward proteolysis. Ultralong pharmacokinetic profiles were obtained by attaching an albumin-binding side chain derived from octadecanedioic (C18) or icosanedioic acid (C20) to the lysine in position B29. Crucial for obtaining the ultralong PK profile was also a significant reduction of insulin receptor affinity. Oral bioavailability in dogs indicated that C18-based analogues were superior to C20-based analogues. These studies led to the identification of the two clinical candidates OI338 and OI320 (10 and 24, respectively).
Oral insulin 338 (I338) is a long-acting, basal insulin analogue formulated in a tablet with the absorption-enhancer sodium caprate. We investigated the efficacy and safety of I338 versus subcutaneous insulin glargine (IGlar) in patients with type 2 diabetes. METHODS: This was a phase 2, 8-week, randomised, double-blind, double-dummy, active-controlled, parallel trial completed at two research institutes in Germany. Insulin-naive adult patients with type 2 diabetes, inadequately controlled on metformin monotherapy or combined with other oral antidiabetic drugs (HbA1c 7·0-10·0%; BMI 25·0-40·0 kg/m(2)), were randomly assigned (1:1) to receive once-daily I338 plus subcutaneous placebo (I338 group) or once-daily IGlar plus oral placebo (IGlar group). Randomisation occurred by interactive web response system stratified by baseline treatment with oral antidiabetic drugs. Patients and investigators were masked to treatment assignment. Weekly insulin dose titration aimed to achieve a self-measured fasting plasma glucose (FPG) concentration of 4·4-7·0 mmol/L. The recommended daily starting doses were 2700 nmol I338 or 10 U IGlar, and maximum allowed doses throughout the trial were 16 200 nmol I338 or 60 U IGlar. The primary endpoint was treatment difference in FPG concentration at 8 weeks for all randomly assigned patients receiving at least one dose of trial product (ie, the full analysis set). The trial has been completed and is registered at ClinicalTrials.gov, number NCT02470039. FINDINGS: Between June 1, 2015, and Oct 19, 2015, 82 patients were screened for eligibility and 50 patients were randomly assigned to the I338 group (n=25) or the IGlar group (n=25). Mean FPG concentration at baseline was 9·7 (SD 2·8) in the I338 group and 9·1 (1·7) in the IGlar group. Least square mean FPG concentration at 8 weeks was 7·1 mmol/L (95% CI 6·4-7·8) in the I338 group and 6·8 mmol/L (6·5-7·1) in the IGlar group, with no significant treatment difference (0·3 mmol/L [-0·5 to 1·1]; p=0·46). I338 and IGlar were well tolerated by patients. Adverse events were reported in 15 (60%) patients in the I338 group and 17 (68%) patients in the IGlar group. The most common adverse events were diarrhoea (three [12%] patients in each group) and nasopharyngitis (five [20%] in the I338 group and two [8%] in the IGlar group). Most adverse events were graded mild (47 of 68 events), and no severe adverse events were reported. One patient in the IGlar group had a treatment-emergent serious adverse event (urogenital haemorrhage of moderate intensity, assessed by the investigator as unlikely to be related to treatment; the patient recovered). Incidence of hypoglycaemia was low in both groups (n=7 events in the I338 group; n=11 in the IGlar group), with no severe episodes. INTERPRETATION: I338 can safely improve glycaemic control in insulin-naive patients with type 2 diabetes with no evidence of a difference compared with insulin glargine, a widely used subcutaneously administered basal insulin. Further development of this particular oral insulin project was discontinued because I338 doses were high and, therefore, production of the required quantities of I338 for wide public use was deemed not commercially viable. Improvement of technologies involved in the product’s development is the focus of ongoing research. FUNDING: Novo Nordisk…..Halberg, I. B.; Lyby, K.; Wassermann, K.; Heise, T.; Zijlstra, E.; Plum-Mörschel, L. Efficacy and safety of oral basal insulin versus subcutaneous insulin glargine in type 2 diabetes: a randomised, double-blind, phase 2 trial. Lancet Diabetes Endocrinol. 2019, 7, 179– 188, DOI: 10.1016/s2213-8587(18)30372-3
ral insulin 338 is a novel tablet formulation of a long-acting basal insulin. This randomised, open-label, four-period crossover trial investigated the effect of timing of food intake on the single-dose pharmacokinetic properties of oral insulin 338. Methods: After an overnight fast, 44 healthy males received single fixed doses of oral insulin 338 administered 0, 30, 60 or 360 min before consuming a standardised meal (500 kcal, 57 energy percent [E%] carbohydrate, 13 E% fat, 30 E% protein). Blood samples for pharmacokinetic assessment were taken up to 288 h post-dose. Results: Total exposure (area under the concn.-time curve from time zero to infinity [AUCIns338,0-∞]) and max. concn. (Cmax,Ins338) of insulin 338 were both significantly lower for 0 vs. 360 min post-dose fasting (ratio [95% confidence interval (CI)]: 0.36 [0.26-0.49], p < 0.001, and 0.35 [0.25-0.49], p < 0.001, resp.). There were no significant differences in AUCIns338,0-∞ and Cmax,Ins338 for 30 or 60 vs. 360 min post-dose fasting (ratio [95% CI] 30 vs. 360 min: 0.85 [0.61-1.21], p = 0.36, and 0.86 [0.59-1.26], p = 0.42; ratio [95% CI] 60 vs. 360 min: 0.96 [0.72-1.28], p = 0.77, and 0.99 [0.75-1.31], p = 0.95). The mean half-life was ∼ 55 h independent of the post-dose fasting period. Oral insulin 338 was well-tolerated with no safety issues identified during the trial. Conclusions: Oral insulin 338 pharmacokinetics are not affected by food intake from 30 min after dosing, implying that patients with diabetes mellitus do not need to wait more than 30 min after a morning dose of oral insulin 338 before having their breakfast. This is considered important for convenience and treatment compliance. ClinicalTrials.gov identifier: NCT02304627./……Halberg, I. B.; Lyby, K.; Wassermann, K.; Heise, T.; Plum-Mörschel, L.; Zijlstra, E. The effect of food intake on the pharmacokinetics of oral basal insulin: A randomised crossover trial in healthy male subjects. Clin. Pharmacokinet. 2019, 58, 1497– 1504, DOI: 10.1007/s40262-019-00772-2
///////////////OI 338, OI338GT, NN1953, NNC0123-0000-0338, Insulin oral (NN 1953), Insulin-338-GIPET-I, LAI 338, NN 1438, NN-1953, NNC-0123-0000-0338, NNC0123-0338, OI-338GT, Oral insulin 338 C10
Devimistat

Devimistat
CPI-613
| Molecular Weight | 388.59 |
|---|---|
| Formula | C₂₂H₂₈O₂S₂ |
| CAS No. | 95809-78-2 |
| SMILES | O=C(O)CCCCC(SCC1=CC=CC=C1)CCSCC2=CC=CC=C2 |
phase III, hematological cancer
6,8-Bis(benzylsulfanyl)octanoic acid
Octanoic acid, 6,8-bis[(phenylMethyl)thio]-
Octanoic acid, 6,8-bis((phenylmethyl)thio)-
Rafael Pharmaceuticals (formerly Cornerstone Pharmaceuticals), a subsidiary of Rafael Holdings, is developing devimistat, the lead candidate from a program of thioctans and their derivatives that act as pyruvate dehydrogenase and alpha-ketoglutarate inhibitors and stimulators of pyruvate dehydrogenase kinase (PDK), using the company’s proprietary Altered Energy Metabolism Directed (AEMD) platform, for the iv treatment of hematological cancer [phase III, January 2021].
Devimistat (INN; development code CPI-613) is an experimental anti-mitochondrial drug being developed by Rafael Pharmaceuticals.[1] It is being studied for the treatment of patients with metastatic pancreatic cancer and relapsed or refractory acute myeloid leukemia (AML).
Devimistat’s mechanism of action differs from other drugs, operating on the tricarboxylic acid cycle and inhibiting enzymes involved with cancer cell energy metabolism. A lipoic acid derivative different from standard cytotoxic chemotherapy, devimistat is currently being studied in combination with modified FOLFIRINOX to treat various solid tumors and heme malignancies.
Regulation
The U.S. Food and Drug Administration (FDA) has designated devimistat as an orphan drug for the treatment of pancreatic cancer, AML, myelodysplastic syndromes (MDS), peripheral T-cell lymphoma, and Burkitt’s lymphoma, and given approval to initiate clinical trials in pancreatic cancer and AML.
Clinical trials
Clinical trials of the drug are underway including a Phase III open-label clinical trial[2] to evaluate efficacy and safety of devimistat plus modified FOLFIRINOX (mFFX) versus FOLFIRINOX (FFX) in patients with metastatic adenocarcinoma of the pancreas.
Developed as part of Rafael’s proprietary Altered Metabolism Directed (AMD) drug platform, CPI-613® was discovered at Stony Brook University. CPI-613® is designed to target the mitochondrial tricarboxylic acid (TCA) cycle, an indispensable process essential to tumor cell multiplication and survival, selectively in cancer cells.
The attacks of CPI-613® on the TCA cycle also substantially increases the sensitivity of cancer cells to a diverse range of chemotherapeutic agents. This synergy allows for combinations of CPI-613® with lower doses of these generally toxic drugs to be highly effective with lower patient side effects. Combinations with CPI-613® represent a diverse range of potential opportunities to substantially improve patient benefit in many different cancers.
The U.S. Food and Drug Administration (FDA) has given Rafael approval to initiate pivotal clinical trials in pancreatic cancer and acute myeloid leukemia (AML), and has designated CPI-613® as an orphan drug for the treatment of pancreatic cancer, AML, Myelodysplastic syndromes (MDS), peripheral T-cell lymphoma and Burkitt’s lymphoma. The EMA has granted orphan drug designation to CPI-613® for pancreatic cancer and AML.
Learn more about recent developments involving CPI-613®: CPI-613® (devimistat) Fact Sheet
he FDA granted a Fast Track designation to devimistat for the treatment of patients with acute myeloid leukemia.
The FDA has granted a Fast Track designation to devimistat (CPI-613) for the treatment of patients with acute myeloid leukemia (AML), Rafael Pharmaceuticals, announced in a press release.1
“This designation underscores the pressing need to find new ways to combat this aggressive disease,” said Jorge Cortes, MD, director of the Georgia Cancer Center at Augusta University, and principal investigator on the phase 3 clinical trial, in a statement. “It brings hope not only to clinicians, but to patients who hear that they have been diagnosed.”
The first-in-class agent devimistat targets enzymes that are involved in cancer cell energy metabolism. This therapy substantially increases the sensitivity of cancer cells to a diverse range of chemotherapies, and this synergy allows for potential combinations that could be more effective with devimistat and lower doses of drugs that are generally toxic.
“Receiving Fast Track designation, especially during a pandemic that has created significant challenges for many trials across the globe, is a testament to the dedicated work of the Rafael team,” stated Sanjeev Luther, president and CEO of Rafael Pharmaceuticals, Inc.
Devimistat combinations appear promising with a diverse range of potential opportunities to improve benefit in patients with various cancer types. Two pivotal phase 3 clinical trials, including the AVENGER 500 study in pancreatic cancer (NCT03504423) and ARMADA 2000 for AML (NCT03504410), have been approved for initiation by the FDA.
The primary end point of the multicenter, open-label, randomized ARMADA 2000 study is complete response (CR), and secondary end points include overall survival and CR plus CR with partial hematologic recovery rate. To be eligible to enroll to the study, patients must be aged ≥50 years with a documented AML diagnosis that has relapsed from or became refractory to previous standard therapy. Patients must have an ECOG performance status of 0 to 2 and an expected survival longer than 3 months.
Five hundred patients are expected to be enrolled and randomized in the study. To enroll, patients could not have received prior radiotherapy or cytotoxic chemotherapy for their current AML. Those with active central nervous system involvement, active uncontrolled bleeding, history of other malignancy, or known hypersensitivity to study drugs are ineligible to enroll to the trial as well.
This study aims to determine the safety and efficacy of devimistat in combination with high-dose cytarabine and mitoxantrone in older patients with relapsed/refractory AML compared with high-dose cytarabine and mitoxantrone therapy alone. Other control groups include patients treated with mitoxantrone, etoposide, and cytarabine and the combination of fludarabine, cytarabine, and filgrastim. The addition of devimistat is expected to improve the CR rate in patients who are aged 50 years or older with relapsed/refractory AML.
In a prior phase 1 study of devimistat plus high-dose cytarabine and mitoxantrone in patients with relapsed/refractory AML, the addition of devimistat sensitized AML cells to chemotherapy treatment.2
The objective response rate was 50% including CRs in 26 of 62 evaluable patients. Median overall survival was 6.7 months. In patients above age 60, the CR or CR with incomplete hematologic recovery rate was 47% and the median survival was 6.9 months.
This designation for this experimental anti-mitochondrial agent follows news of another Fast Track designation granted to devimistat for the treatment of patients with metastatic pancreatic cancer in November 2020, as well as an Orphan Drug designation granted in October 2020 for the treatment of patients with soft tissue sarcoma.
References
1. Rafael Pharmaceuticals Receives FDA Fast Track Designation for CPI-613® (devimistat) for the treatment of acute myeloid leukemia (AML). News Release. Rafael Pharmaceuticals, Inc. December 15, 2020. Accessed December 15, 2020. https://bit.ly/34g6YsR
2. Pardee TS, Anderson RG, Pladna KM, et al. A Phase I Study of CPI-613 in Combination with High-Dose Cytarabine and Mitoxantrone for Relapsed or Refractory Acute Myeloid Leukemia. Clin Cancer Res. 2018;24(9):2060-2073. doi:10.1158/1078-0432.CCR-17-2282 P[APERJournal of the American Chemical Society (1954), 76, 4109-12.https://pubs.acs.org/doi/abs/10.1021/ja01645a016
PAPERJournal of the American Chemical Society (1955), 77, 416-19.https://pubs.acs.org/doi/abs/10.1021/ja01607a057PAPERJustus Liebigs Annalen der Chemie (1958), 614, 66-83.https://chemistry-europe.onlinelibrary.wiley.com/doi/abs/10.1002/jlac.19586140108PATENTWO 2009123597WO 2009110859WO 2010110771PATENTCN 111362848
PATENT
WO-2021011334
Deuterated derivatives of 6,8-bis(benzylsulfanyl)octanoic acid (CPI-613 or devimistat ) or its salts for treating cancer.
CPI-613 (6,8-bis(benzylsulfanyl)octanoic acid) is a first-in-class investigational small-molecule (lipoate analog), which targets the altered energy metabolism unique to many cancer cells. CPI-613 is currently being evaluated in two phase III clinical trials, and has been granted orphan drug designation for the treatment of pancreatic cancer, acute myeloid leukemia (AML), peripheral T-cell lymphoma (PTCL), Burkitt lymphoma and myelodysplastic syndromes (MDS).
[0004] One limitation to the clinical utility of CPI-613 is its very rapid metabolism. After IV dosing the half-life of 6,8-bis(benzylsulfanyl)octanoic acid is only about 1-2 hours (Pardee,
T.S. et al, Clin Cancer Res. 2014, 20, 5255-64). The short half-life limits the patient’s overall exposure to the drug and necessitates administration of relatively high doses. For safety reasons, CPI-613 is administered via a central venous catheter as an IV infusion over 30-120 minutes, with higher doses requiring longer infusion times.
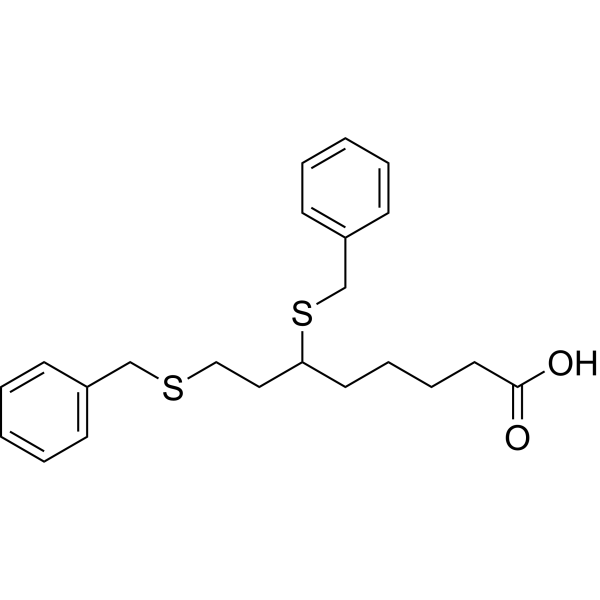
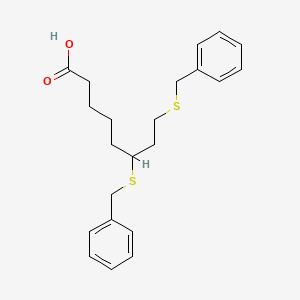
The terms“6,8-bis(benzylsulfanyl)octanoic acid” and“ 6,8-bis-benzylthio-octanoic acid” refer to the compound known as CPI-613 or devimistat, having the chemical structure
PATENT
WO2020132397
claiming the use of CPI-613 in combination with an autophagy inhibitor eg chloroquine for treating eg cancers.
CPI-613 (6,8-bis-benzylthio-octanoic acid) is a first-in-class investigational small-molecule (lipoate analog), which targets the altered energy metabolism that is common to many cancer cells. CPI-613 has been evaluated in multiple phase I, I/II, and II clinical studies, and has been granted orphan drug designation for the treatment of pancreatic cancer, acute myeloid leukemia (AML), peripheral T-cell lymphoma (PTCL), Burkitt lymphoma and myelodysplastic syndromes (MDS).
PAPER
https://pubs.acs.org/doi/10.1021/op200091t
An Efficient, Economical Synthesis of the Novel Anti-tumor Agent CPI-613
Cite this: Org. Process Res. Dev. 2011, 15, 4, 855–857
Publication Date:May 2, 2011
https://doi.org/10.1021/op200091t

An efficient and practical synthesis of the novel anti-tumor compound 6,8-dithiobenzyl octanoic acid, CPI-613 (2), was developed and executed on a practical scale. CPI-613 can be made in a single vessel from (±)-lipoic acid (1) via reductive opening of the disulfide ring followed by benzylation of the sulfhydryls with benzyl bromide. CPI-613 was isolated by simple crystallization in high yield and purity. The process is scaleable and has been demonstrated at up to 100 kg.CPI-613 (2) was isolated [4.7 kg (90%)] with an HPLC purity of 99.8 area %. Mp 66–67 °C. IR: 3050, 1710, 1400, 668 cm–1. 1H NMR (400 MHz, CDCl3) δ 7.40–7.20 (m, 10 H), 3.80–3.60 (m, 4 H), 2.60–2.50 (m, 2 H), 2.44 (t, J = 8.7, 2 H), 2.23 (t, J = 8.1, 2 H) 2.03–1.30 (m, 8 H). Anal. Calc for C22H28O2S2: C, 68.00; H, 7.26; S, 16.50. Found: C, 67.99; H, 7.31; S, 16.37.

References
- ^ “CPI-613”. Rafael Pharmaceuticals.
- ^ Philip PA, Buyse ME, Alistar AT, Rocha Lima CM, Luther S, Pardee TS, Van Cutsem E (October 2019). “A Phase III open-label trial to evaluate efficacy and safety of CPI-613 plus modified FOLFIRINOX (mFFX) versus FOLFIRINOX (FFX) in patients with metastatic adenocarcinoma of the pancreas”. Future Oncology. 15 (28): 3189–3196. doi:10.2217/fon-2019-0209. PMC 6854438. PMID 31512497.
| Clinical data | |
|---|---|
| Other names | CPI-613 |
| Legal status | |
| Legal status | Investigational |
| Identifiers | |
| IUPAC name[show] | |
| CAS Number | 95809-78-2 |
| PubChem CID | 24770514 |
| DrugBank | 12109 |
| ChemSpider | 28189062 |
| UNII | E76113IR49 |
| ChEMBL | ChEMBL3186849 |
| CompTox Dashboard(EPA) | DTXSID70914807 |
| ECHA InfoCard | 100.231.125 |
| Chemical and physical data | |
| Formula | C22H28O2S2 |
| Molar mass | 388.58 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES[hide]C1=CC=C(C=C1)CSCCC(CCCCC(=O)O)SCC2=CC=CC=C2 |
//////////devimistat, CPI-613, CPI 613, phase 3, hematological cancer , Fast Track designation, ORPHAN DRUG,
Fosnetupitant
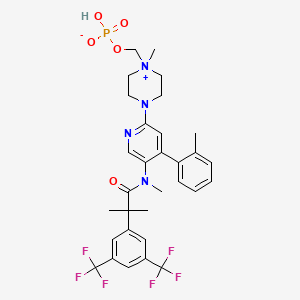

Fosnetupitant
- Molecular FormulaC31H35F6N4O5P
- Average mass688.598 Da
[4-[5-[[2-[3,5-bis(trifluoromethyl)phenyl]-2-methylpropanoyl]-methylamino]-4-(2-methylphenyl)pyridin-2-yl]-1-methylpiperazin-1-ium-1-yl]methyl hydrogen phosphate(4-{5-[{2-[3,5-Bis(trifluoromethyl)phenyl]-2-methylpropanoyl}(methyl)amino]-4-(2-methylphenyl)-2-pyridinyl}-1-methylpiperazin-1-ium-1-yl)methyl hydrogen phosphate07-PNET10146
CAS 1703748-89-3
HCL 1643757-72-5
FDA 2014 AND EMA 2015Фоснетупитант [Russian] [INN]فوسنيتوبيتانت [Arabic] [INN]磷奈匹坦 [Chinese] [INN]
- 07-PNET
In April 2018, the U.S. Food and Drug Administration (FDA) and the Swiss company Helsinn approved the intravenous formulation of AKYNZEO® (NEPA, a fixed antiemetic combination of fosnetupitant, 235mg, and palonosetron, 0.25mg) as an alternative treatment option for patients experiencing chemotherapy-induced nausea and vomiting. Fosnetupitant is the pro-drug form of netupitant. Generally, 25% to 30% of patients with a diagnosis of cancer receive chemotherapy as a treatment modality and 70% to 80% of these patients undergoing chemotherapy treatment may experience nausea and vomiting as major side effects. Considered one of the most distressing side effects of chemotherapy, nausea and vomiting has an immense impact on the quality of life of patients receiving certain antineoplastic therapies.
In April 2018, the U.S. Food and Drug Administration (FDA) and the Swiss company Helsinn approved the intravenous formulation of AKYNZEO® (NEPA, a fixed antiemetic combination of fosnetupitant, 235mg, and palonosetron, 0.25mg) as an alternative treatment option for patients experiencing chemotherapy-induced nausea and vomiting 3. Fosnetupitant is the pro-drug form of netupitant Label.
Generally, 25% to 30% of patients with a diagnosis of cancer receive chemotherapy as a treatment modality and 70% to 80% of these patients undergoing chemotherapy treatment may experience nausea and vomiting as major side effects. Considered one of the most distressing side effects of chemotherapy, nausea and vomiting has an immense impact on the quality of life of patients receiving certain antineoplastic therapies 1.
Fosnetupitant: Fosnetupitant is a selective antagonist of human substance P/neurokinin 1 (NK-1) receptors. Upon intravenous administration, Fosnetupitant is converted by phosphatases to its active form. It competitively binds to and blocks the activity of NK-1 receptors in the central nervous system, by inhibiting binding of substance P (SP) to NK-1 receptors. This prevents delayed emesis, which is associated with SP secretion. AKYNZEO is a combination of palonosetron, a serotonin-3 receptor antagonist, and Fosnetupitant (capsules for oral use) or Fosnetupitant (injections for intravenous use). AKYNZEO for injection is indicated in combination with dexamethasone in adults for the prevention of acute and delayed nausea and vomiting associated with initial and repeat courses of highly emetogenic cancer chemotherapy.
EMA
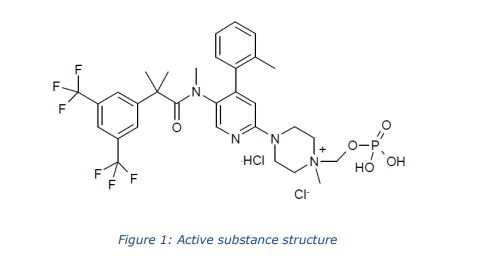
The chemical name of fosnetupitant chloride hydrochloride is 4-(5-(2-(3,5-bis(trifluoromethyl)phenyl)- N,2-dimethylpropanamido)-4-(o-tolyl)pyridin-2-yl)-1-methyl-1-((phosphonooxy)methyl)piperazin-1- ium chloride hydrochloride is corresponding to the molecular formula C31H37Cl2N4O5P. It has a relative molecular mass of 761.53 g/mol and the following structure:
The chemical structure of fosnetupitant chloride hydrochloride was elucidated by a combination of 1H and 13C NMR spectroscopy, infrared spectroscopy, mass spectrometry and elemental analysis. The active substance is achiral. The solid state properties of the active substance were measured by gravimetric vapour sorption and x-ray powder diffraction (XRPD). It is a white to off-white to yellowish, crystalline, hygroscopic solid. Three polymorphic forms have been identified following extensive screening, requiring isolation from different solvent mixtures. Fosnetupitant chloride hydrochloride is always isolated as form I following the commercial manufacturing process. Since it is dissolved and lyophilised during finished product manufacture, particle size and polymorphic form are not considered critical quality attributes (CQAs) of the active substance and are not included in the specification.
RX
AKYNZEO (300 mg netupitant/0.5 mg palonosetron) capsules are an oral combination product of netupitant, a substance P/neurokinin 1 (NK-1) receptor antagonist, and palonosetron hydrochloride, a serotonin-3 (5-HT3) receptor antagonist. Both netupitant and palonosetron hydrochloride are anti-nausea and anti-emetic agents.
Netupitant is chemically described: 2-[3,5-bis(trifluoromethyl)phenyl]-N, 2 dimethyl-N-[4-(2methylphenyl)-6-(4-methylpiperazin-1-yl)pyridin-3-yl] propanamide. The empirical formula is C30H32F6N4O, with a molecular weight of 578.61. Netupitant exists as a single isomer and has the following structural formula:
 |
Palonosetron hydrochloride is chemically described: (3aS)-2-[(S)-1-Azabicyclo [2.2.2]oct-3-yl]2,3,3a,4,5,6-hexahydro-1-oxo-1H-benz[de]isoquinoline hydrochloride. The empirical formula is C19H24N2O.HCl, with a molecular weight of 332.87. Palonosetron hydrochloride exists as a single isomer and has the following structural formula:
 |
Netupitant is white to off-white crystalline powder. It is freely soluble in toluene and acetone, soluble in isopropanol and ethanol, and very slightly soluble in water.
Palonosetron hydrochloride is a white to off-white crystalline powder. It is freely soluble in water, soluble in propylene glycol, and slightly soluble in ethanol and 2-propanol.
Each AKYNZEO capsule is composed of one white-caramel hard gelatin capsule which contains three tablets each containing 100 mg netupitant and one gelatin capsule containing 0.5 mg palonosetron (equivalent to 0.56 mg palonosetron hydrochloride). The inactive ingredients are butylated hydroxyanisole (BHA), croscarmellose sodium, gelatin, glycerin, magnesium stearate, microcrystalline cellulose, mono-and di-glycerides of capryl/capric acid, polyglyceryl dioleate, povidone K-30, purified water, red iron oxide, silicon dioxide, sodium stearyl fumarate, sorbitol, sucrose fatty acid esters, titanium dioxide and yellow iron oxide. It may contain traces of medium-chain triglycerides, lecithin, and denatured ethanol.
AKYNZEO (235 mg fosnetupitant/0.25 mg palonosetron) for injection is a combination product of fosnetupitant, a prodrug of netupitant, which is a substance P/neurokinin 1 (NK-1) receptor antagonist, and palonosetron hydrochloride, a serotonin-3 (5-HT3) receptor antagonist.
Fosnetupitant chloride hydrochloride is chemically described as 2-(3,5-bistrifluoromethylphenyl)-N-methyl-N-[6-(4-methyl-4-O-methylene-phosphatepiperazinium-1-yl)4-o-tolyl-pyridin-3-yl]-isobutyramide chloride hydrochloride. The empirical formula is C31H36F6N4O5P•Cl•HCl, with a molecular weight of 761.53. Fosnetupitant chloride hydrochloride exists as a single isomer and has the following structural formula:
 |
Fosnetupitant chloride hydrochloride is white to off-white to yellowish solid or powder. Its solubility is pH dependent: at acidic pH (pH 2), its solubility is 1.4 mg/mL; at basic pH (pH 10), its solubility is 11.5 mg/mL.
Palonosetron hydrochloride is described above in this section.
AKYNZEO for injection is available for intravenous infusion, and is supplied as a sterile lyophilized powder in a single-dose vial. Each vial contains 235 mg of fosnetupitant (equivalent to 260 mg fosnetupitant chloride hydrochloride) and 0.25 mg of palonosetron (equivalent to 0.28 mg of palonosetron hydrochloride). The inactive ingredients are edetate disodium (6.4 mg), mannitol (760 mg), sodium hydroxide and/or hydrochloric acid (for pH adjustment).
PATENT
WO 2013082102
https://patents.google.com/patent/WO2013082102A1/un



PATENT
US 20150011510
https://patents.google.com/patent/US20150011510A1/en
Step 1:
- [0160]
13.0 g (82.5 mMol) 6-Chloro-nicotinic acid in 65 ml THF were cooled to 0° C. and 206.3 ml (206.3 mMol) o-tolylmagnesium chloride solution (1M in THF) were added over 45 minutes. The solution obtained was further stirred 3 hours at 0° C. and overnight at room temperature. It was cooled to −60° C. and 103.8 ml (1.8 Mol) acetic acid were added, followed by 35 ml THF and 44.24 g (165 mMol) manganese(III) acetate dihydrate. After 30 minutes at −60° C. and one hour at room temperature, the reaction mixture was filtered and THF removed under reduced pressure. The residue was partitioned between water and dichloromethane and extracted. The crude product was filtered on silica gel (eluent: ethyl acetate/toluene/formic acid 20:75:5) then partitioned between 200 ml aqueous half-saturated sodium carbonate solution and 100 ml dichloromethane. The organic phase was washed with 50 ml aqueous half-saturated sodium carbonate solution. The combined aqueous phases were acidified with 25 ml aqueous HCI 25% and extracted with dichloromethane. The organic extracts were dried (Na2SO4) and concentrated under reduced pressure to yield 10.4 g (51%) of 6-chloro-4-o-tolyl-nicotinic acid as a yellow foam. MS (ISN): 246 (M−H, 100), 202 (M-CO2H, 85), 166 (36).
Step 2:
- [0161]
To a solution of 8.0 g (32.3 mMol) 6-chloro-4-o-tolyl-nicotinic acid in 48.0 ml THF were added 3.1 ml (42.0 mMol) thionylchloride and 143 .mu.l (1.8 mMol) DMF. After 2 hours at 50° C., the reaction mixture was cooled to room temperature and added to a solution of 72.5 ml aqueous ammonium hydroxide 25% and 96 ml water cooled to 0° C. After 30 minutes at 0° C., THF was removed under reduced pressure and the aqueous layer was extracted with ethyl acetate. Removal of the solvent yielded 7.8 g (98%) 6-chloro-4-o-tolyl-nicotinamide as a beige crystalline foam. MS (ISP): 247 (M+H+, 100).
Step 3:
- [0162]
1.0 g (4.05 mMol) 6-Chloro-4-o-tolyl-nicotinamide in 9.0 ml 1-methyl-piperazine was heated to 100° C. for 2 hours. The excess N-methyl-piperazine was removed under high vacuum and the residue was filtered on silica gel (eluent: dichloromethane) to yield 1.2 g (95%) 6-(4-methyl-piperazin-1-yl)-4-o-tolyl-nicotinamide as a light yellow crystalline foam. - [0163]
MS (ISP): 311 (M+H+, 100), 254 (62).
Step 4:
- [0164]
A solution of 0.2 g (0.6 mMol) 6-(4-methyl-piperazin-1-yl)-4-o-tolyl-nicotinamide in 1.0 ml methanol was added to a solution of 103 mg (2.6 mMol) sodium hydroxide in 1.47 ml (3.2 mMol) NaOCl (13%) and heated for 2 hours at 70° C. After removal of methanol, the aqueous layer was extracted with ethyl acetate. The combined organic extracts were dried (Na2SO4), concentrated under reduced pressure and the residue filtered on silica gel (eluent: dichloromethane/methanol 4:1) to yield 100 mg (70%) 6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-ylamine as a brown resin. MS (ISP): 283 (M+H+, 100), 226 (42).
Step 5:
- [0165]
2.15 mil (11.6 mMol) Sodium methoxide in methanol were added over 30 minutes to a suspension of 0.85 g (4.6 mMol) N-bromosuccinimide in 5.0 ml dichloromethane cooled to −5° C. The reaction mixture was stirred 16 hours at −5° C. Still at this temperature, a solution of 1.0 g (3.1 mMol) 6-(4-methyl-piperazin-1-yl)-4-o-tolyl-nicotinamide in 5.0 ml methanol was added over 20 minutes and stirred for 5 hours. 7.1 ml (7.1 mMol) Aqueous HCl 1N and 20 ml dichloromethane were added. The phases were separated and the organic phase was washed with deionized water. The aqueous phases were extracted with dichloromethane, brought to pH=8 with aqueous NaOH 1N and further extracted with dichloromethane. The latter organic extracts were combined, dried (Na2SO4) and concentrated to yield 1.08 g (quant.) [6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-carbamic acid methyl ester as a grey foam. - [0166]
MS (ISP): 341 (M+H+, 100), 284 (35).
Step 6:
- [0167]
A solution of 0.5 g (1.4 mMol) [6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-carbamic acid methyl ester in 3.0 ml dichloromethane was added over 10 minutes to a solution of 1.98 ml (6.9 mMol) Red-Al®. (70% in toluene) and 2.5 ml toluene (exothermic, cool with a water bath to avoid temperature to go >50° C.). The reaction mixture was stirred 2 hours at 50° C. in CH2Cl2, extracted with ethyl acetate and cooled to 0° C. 4 ml Aqueous NaOH 1N were carefully (exothermic) added over 15 minutes, followed by 20 ml ethyl acetate. The phases were separated and the aqueous phase was extracted with ethyl acetate. The combined organic extracts were washed with deionized water and brine, dried (Na2SO4) and concentrated under reduced pressure to yield 0.37 g (89%) methyl-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-amine as an orange resin. MS (ISP): 297 (M+H+, 100).
Synthesis of 2-(3,5-bis-Trifluoromethyl-phenyl)-2-methyl-propionyl Chloride
- [0168]
- [0169]
15.0 g (50 mmol) 2-(3,5-bis-trifluoromethyl-phenyl)-2-methyl-propionic acid were dissolved in 127.5 ml dichloromethane in the presence of 0.75 ml DMF. 8.76 ml (2 eq.) Oxalyl chloride were added and after 4.5 hours, the solution was rotary evaporated to dryness. 9 ml Toluene were added and the resulting solution was again rotary evaporated, then dried under high vacuum yielding 16.25 g (quant.) of 2-(3,5-bis-trifluoromethyl-phenyl)-2-methyl-propionyl chloride as a yellow oil of 86% purity according to HPLC analysis. NMR (250 MHz, CDCl3): 7.86 (br s, 1H); 7.77, (br s, 2H, 3Harom); 1.77 (s, 6H, 2 CH3).
Synthesis of 2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethyl-N-(6-(4-methylpiperazin-1-yl)-4-(o-tolyl)pyridin-3-yl)propanamide (Netupitant)
- [0170]
- [0171]
A solution of 20 g (67.5 mmol) methyl-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-amine and 17.5 ml (101 mmol) N-ethyldiisopropylamine in 200 ml dichloromethane was cooled in an ice bath and a solution of 24 g (75 mmol)2-(3,5-bis-trifluoromethyl-phenyl)-2-methyl-propionyl chloride in 50 ml dichloromethane was added dropwise. The reaction mixture was warmed to 35-40° C. for 3 h, cooled to room temperature again and was stirred with 250 ml saturated sodium bicarbonate solution. The organic layer was separated and the aqueous phase was extracted with dichloromethane. The combined organic layers were dried (magnesium sulfate) and evaporated. The residue was purified by flash chromatography to give 31.6 g (81%) of 2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethyl-N-(6-(4-methylpiperazin-1-yl)-4-(o-tolyl)pyridin-3-yl)propanamide as white crystals. - [0172]
M.P. 155-157° C.; MS m/e (%): 579 (M+H+, 100).
Synthesis of 5-(2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-2-(4-methylpiperazin-1-yl)-4-(o-tolyl)pyridine 1-oxide
Step 1:
- [0174]
The solution of 6-chloropyridin-3-amine (115 g, 0.898 mol) and (Boc)2O (215.4 g, 0.988 mol) in 900 mL of dioxane was refluxed overnight. The resulting solution was poured into 1500 mL of water. The resulting solid was collected, washed with water and re-crystallized from EtOAc to afford 160 g tert-butyl (6-chloropyridin-3-yl)carbamate as a white solid (Yield: 78.2%).
Step 2:
- [0175]
To the solution of tert-butyl (6-chloropyridin-3-yl)carbamate (160 g, 0.7 mol) in 1 L of anhydrous THF was added n-BuLi (600 mL, 1.5 mol) at −78° C. under N2 atmosphere. After the addition was finished, the solution was stirred at −78° C. for 30 min, and the solution of I2 (177.68 g, 0.7 mol) in 800 mL of anhydrous THF was added. Then the solution was stirred at −78° C. for 4 hrs. TLC indicated the reaction was over. Water was added for quench, and EtOAc was added to extract twice. The combined organic phases were washed with brine, dried over Na2SO4, filtered and purified by flash chromatography to afford 80 g of tert-butyl (6-chloro-4-iodopyridin-3-yl)carbamate as a yellow solid (32.3%).
Step 3:
- [0176]
To the solution of tert-butyl (6-chloro-4-iodopyridin-3-yl)carbamate (61 g, 0.172 mol) in 300 mL of anhydrous THF was added 60% NaH (7.6 g, 0.189 mol) at 0° C. under N2 atmosphere. After the addition was finished, the solution was stirred for 30 min, and then the solution of MeI (26.92 g, 0.189 mol) in 100 mL of dry THF was added. Then the solution was stirred at 0° C. for 3 hrs. TLC indicated the reaction was over. Water was added for quench, and EtOAc was added to extract twice. The combined organic phases were washed with brine, dried over Na2SO4, filtered and concentrated to afford 63 g of crude tert-butyl (6-chloro-4-iodopyridin-3-yl)(methyl)carbamate used into the following de-protection without the further purification.
Step 4:
- [0177]
To the solution of tert-butyl (6-chloro-4-iodopyridin-3-yl)(methyl)carbamate (62.5 g, 0.172 mol) in 500 mL of anhydrous DCM was added 180 mL of TFA. Then the solution was stirred at room temperature for 4 hrs. Concentrated to remove the solvent, and purified by flash chromatography to afford 45.1 g 6-chloro-4-iodo-N-methylpyridin-3-amine as a yellow solid (Yield: 97.3%).
Step 5:
- [0178]
To the solution of 6-chloro-4-iodo-N-methylpyridin-3-amine (40.3 g, 0.15 mol) and 2-methylbenzene boric acid (24.5 g, 0.18 mol) in 600 mL of anhydrous toluene was added 400 mL of 2 N aq. Na2CO3 solution, Pd(OAc)2 (3.36 g, 15 mmol) and PPh3 (7.87 g, 0.03 mmol). The solution was stirred at 100° C. for 2 hrs. Cooled to room temperature, and diluted with water. EtOAc was added to extract twice. The combined organic phases were washed with water and brine consecutively, dried over Na2SO4, concentrated and purified by flash chromatography to afford 19 g 6-chloro-N-methyl-4-(o-tolyl)pyridin-3-amine as a white solid (Yield: 54.6%).
Step 6:
- [0179]
To the solution of 6-chloro-N-methyl-4-(o-tolyl)pyridin-3-amine (18.87 g, 81.3 mmol) and DMAP (29.8 g, 243.9 mmol) in 200 mL of anhydrous toluene was added the solution of 2-(3,5-bis-trifluoromethyl-phenyl)-2-methyl-propionyl chloride (28.5 g, 89.4 mmol) in toluene under N2 atmosphere. The solution was heated at 120° C. for 23 hrs. Cooled to room temperature, poured into 1 L of 5% aq. NaHCO3 solution, and extracted with EtOAc twice. The combined organic phases were washed by water and brine consecutively, dried over Na2SO4, filtered and purified by flash chromatography to afford 35 g 2-(3,5-bis(trifluoromethyl)phenyl)-N-(6-chloro-4-(o-tolyl)pyridin-3-yl)-N,2-dimethylpropanamide as a white solid (Yield: 83.9%).
Step 7:
- [0180]
To the solution of 2-(3,5-bis(trifluoromethyl)phenyl)-N-(6-chloro-4-(o-tolyl)pyridin-3-yl)-N,2-dimethylpropanamide (5.14 g, 10 mmol) in 60 mL of DCM was added m-CPBA (6.92 g, 40 mmol) at 0° C. under N2 atmosphere. Then the solution was stirred overnight at room temperature. 1 N aq. NaOH solution was added to wash twice for removing the excess m-CPBA and a side product. The organic phase was washed by brine, dried over Na2SO4, filtered and concentrated to afford 5.11 g of crude 5-(2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-2-chloro-4-(o-tolyl)pyridine 1-oxide as a white solid (Yield: 96.4%).
Step 8:
- [0181]
To the solution of crude 5-(2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-2-chloro-4-(o-tolyl)pyridine 1-oxide (5.1 g, 9.62 mmol) in 80 mL of n-BuOH was added N-methylpiperazine (7.41 g, 74.1 mmol) under N2 atmosphere. Then the solution was stirred at 80° C. overnight. Concentrated and purified by flash chromatography to afford 4.98 g 5-(2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-2-(4-methylpiperazin-1-yl)-4-(o-tolyl)pyridine 1-oxide as a white solid (Yield: 87.2%). 1HNMR (CDCl3, 400 MHz) δ 8.15 (s, 1H), 7.93 (s, 1H), 7.78 (s, 2H), 7.38 (m, 2H), 7.28 (m, 1H), 7.17 (m, 1H), 7.07 (s, 1H), 5.50 (s, 3H), 2.72 (d, J=4.4 Hz, 4H), 2.57 (m, 3H), 2.40 (s, 3H), 2.23 (s, 3H), 1.45˜1.20 (m, 6H).
Synthesis of 4-(5-(2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-1-oxido-4-(o-tolyl)pyridin-2-yl)-1-methylpiperazine 1-oxide
- [0182]
- [0183]
To a solution of 5-(2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-2-(4-methylpiperazin-1-yl)-4-(o-tolyl)pyridine 1-oxide (3 g, 5.05 mmol) and NaHCO3 (0.354 g, 12.66 mmol) in 60 mL of MeOH and 15 mL of H2O were added potassium monopersulfate triple salt (1.62 g, 26.25 mmol) at room temperature during 15 min. After stirring for 4 hrs at room temperature under N2 atmosphere, the reaction mixture was concentrated in vacuo and purified by flash chromatography (eluent: MeOH). The product was dissolved into DCM, the formed solid was filtered off, and the solution was concentrated under reduced pressure to afford 1.77 g 4-(5-(2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-1-oxido-4-(o-tolyl)pyridin-2-yl)-1-methylpiperazine 1-oxide as a white solid (Yield: 57.4%). 1HNMR (CDCl3, 400 MHz) δ 8.06 (s, 1H), 7.78 (s, 1H), 7.60 (s, 2H), 7.37˜7.20 (m, 4H), 6.81 (s, 1H), 3.89 (s, 21H), 3.74 (m, 4H), 3.31 (m, 5H), 2.48 (s, 3H), 2.18 (s, 3H), 1.36 (s, 6H).
Synthesis of 1-(5-(2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-4-(o-tolyl)pyridin-2-yl)-4-methylpiperazine 1,4-dioxide
- [0184]
- [0185]
To the solution of 2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethyl-N-(6-(4-methylpiperazin-1-yl)-4-(o-tolyl)pyridin-3-yl)propanamide (11.1 g, 19.2 mmol) in 75 ml of Methanol was added sodium bicarbonate (3.38 g, 40.3 mmol) dissolved in 20 ml of water. Then Oxone (14.75 g, 48.0 mmol) was added to the stirred solution at room temperature in 3-4 portions. The suspension was heated for 4 h at 50° C. After filtration of the salts (washed with 3×8 ml of methanol), the solvent has been evaporated under reduced pressure and substituted by DCM (30 ml). The organic phase was washed with water (5×30 ml), dried over Na2SO4, filtered, concentrated and purified by precipitation in toluene to afford 9.3 g 1-(5-(2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-4-(o-tolyl)pyridin-2-yl)-4-methylpiperazine 1,4-dioxide as a white solid (Yield: 80%). 1H-NMR (CDCl3, 400 MHz, at 333K) δ 8.27 (s, 2H), 7.75 (s, 1H), 7.63 (s, 2H), 7.26˜7.19 (m, 2H), 7.14 (t, 1H, J=7.4 Hz), 7.09 (d, 1H, J=7.4 Hz), 4.93 (t, 2H, J=11.6 Hz), 4.70 (t, 2H, J=11.6 Hz), 4.12 (d, 2H, J=10.7 Hz), 3.84 (s, 3H), 3.50 (d, 2H, J=10.3 Hz), 2.47 (s, 3H), 2.12 (s, 3H), 1.40 (s, 6H).
Synthesis (A) of di-tert-butyl (chloromethyl)phosphate
- [0186]
- [0187]
Di-tert-butyl phosphohite (40.36 mmole) was combined with potassium bicarbonate (24.22 mmole) in 35 ml of water. The solution was stirred in an ice bath and potassium permanganate (28.25 mmole) was added in three equal portions over one hour’s time. The reaction as then allowed to continue at room temperature for an additional half hour. - [0188]
Decolorizing carbon (600 mg) was then incorporated as the reaction was heated to 60° C. for 15 minutes. The reaction was then vacuum filtered to remove solid magnesium dioxide. The solid was washed several times with water. The filtrate was then combined with one gram of decolorizing carbon and heated at 60° C. for an additional twenty minutes. The solution was again filtered to yield a colorless solution, which was then evaporated under vacuum to afford crude Di-tert-butyl phosphate potassium salt. Di-tert-butyl phosphate potassium salt (5 g, 20.14 mmole) was dissolved in methanol (15 g): to this solution at 0° C. a slight excess of concentrated HCl is slowly added with efficient stirring at 0° C. The addition of acid causes the precipitation of potassium chloride. The solid is then filtered and washed with methanol. The compound in the mother liquor is then converted to the ammonium form by adding an equal molar amount of tetramethylammonium hydroxide (3.65 g, 20.14 mmole) while keeping the reaction cooled by a salt/ice bath with efficient stirring. The resulting clear solution is placed under reduced pressure to give the crude product. To the tetramethylammonium di-tert-butyl-phosphate dissolved in refluxing dimethoxyethane is then added 4.3 grams of chloroiodomethane (24.16 mmole) and stirred for 1-2 hours. The reaction is then filtered and the filtrate is placed under reduced pressure to concentrate the solution in DME. The chloromethyl di-tert-butyl phosphate 12-16% in DME is used in the synthesis of 4-(5-(2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-4-(o-tolyl)pyridin-2-yl)-1-methyl-1-((phosphonooxy)methyl)piperazin-1-ium without further purifications (60% yield): 1HNMR (CD3OD, 300 MHz) δ 1.51 (s, 12H), 5.63 (d, 2H, J=14.8). 31P-NMR (CD3OD, 300 MHz) δ −11.3 (s, 1P).
Synthesis (B) of di-tert-butyl (chloromethyl)phosphate
- [0189]
- [0190]
Di-tert-butyl phosphate potassium salt (5 g, 20.14 mmole) is dissolved in methanol (15 g): to this solution at 0° C. a slight excess of concentrated HCl is slowly added with efficient stirring at 0° C. The addition of acid causes the precipitation of potassium chloride. The solid is then filtered and washed with methanol. The compound in the mother liquor is then converted to the ammonium form by adding an equal molar amount of tetrabuthylammonium hydroxide 1 M in methanol (20.14 mmole) while keeping the reaction cooled at 0° C. with efficient stirring. The resulting clear solution is placed under reduced pressure to give the intermediate product. The tetrabuthylammonium di-tert-butyl-phosphate dissolved in acetone is then added dropwise to 53.3 grams of chloroiodomethane (302.1 mmole) and stirred at 40° C. for 1-2 hours. The solvent and excess of chloroiodomethane are distilled off, the reaction mass suspended in TBME and then filtered. The filtrate is washed by a saturated solution of sodium bicarbonate and water and then placed under reduced pressure to substitute the solvent by acetone, i.e., to remove the solvent after which it is replaced with acetone. The chloromethyl di-tert-butyl phosphate 7-20% in acetone is used in the next step without further purifications (70-80% yield): 1H-NMR (CD3OD, 300 MHz) δ 1.51 (s, 12H), 5.63 (d, 2H, J=14.8). 31P-NMR (CD3OD, 300 MHz) δ −11.3 (s, 1P).
Stability studies of 4-(5-(2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-4-(o-tolyl)pyridin-2-yl)-1-methyl-1-((phosphonooxy)methyl)piperazin-1-ium salts
- [0191]
In order to further improve the stability and solubility of 4-(5-(2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-4-(o-tolyl)pyridin-2-yl)-1-methyl-1-((phosphonooxy)methyl)piperazin-1-ium, a variety of its derivative salts were synthesized and tested. Their synthesis employed either a) neutralization of the dried diacid phosphate species and its corresponding base salts or b) a direct acid deprotection starting from the dried di(tert-butyl)-protected phosphate species. Neutralization was performed with L-histidine, magnesium salt, N-methyl-D-glucamine (dimeglumine), and L-lysine. Both procedures were tried in the synthesis of citric derivatives whereas with other acids the direct deprotection reaction was used. The figures below show the most relevant structures. - [0192]
When the parent acid species was not stored in dry condition it was found to undergo over 8% degradation in the first week and over 65% degradation in the first six months. When the dried parent acid species was held at 30° C. in air it underwent 0.05% degradation in the first 7 days and at total of 7.03% degradation in six months. When the dried parent acid species was held under argon at room temperature it underwent up to 0.13% degradation in the first 7 days but then was essentially stable for six months. Results for various derivative salts are shown in Table 1 below. - TABLE 1 Representative Degradation Results for Salts Purity A % Solvents Additives Yield % HPLC Comments MeOH L-Histidine, 2 eq. 26.6% 95.94% Degradation: +0.70% in 6 days (in air) +0.46% in 6 days (in argon) MeOH Mg(OH)2, 2 eq. 48.6% 94.11% Degradation: +0.81% in 6 days (in air) +0.29% in 6 days (in argon) MeOH + Citric acid, 2 eq. N.A. 94.40% From protected species. DCM, 1:1 MeOH 1. HCl dioxane, 4 eq. >90% 94.50% From protected species. 2. Ca(OH)2 MeOH H3PO4, 85%, 2 eq. >90% 98.81% From protected species and retains 0.39% of that species. MeOH HBr, 48%, 4 eq. 84.6% 96.11% From protected species. Product degrades rapidly, MeOH + CH3SO3H N.A. 61.54% From protected species. DCM, Product NOT stable: contains 1:4 32.45% decomposition species. MeOH NaH2PO4, 4 eq. N.A. n.d. Only 1.27 of parent species formed. Poor reaction. MeOH N-methyl-D- N.A. 96.88% Degradation: glucamine +0.87% in 6 days (in air) (Meglumine), 2 eq. +1.52% in 11 days (in argon) MeOH N-methyl-D- >99% 97.42% Degradation: glucamine +0.77% in 6 days (in air) (Meglumine), 1 eq. +0.83% in 7 days (in argon) MeOH+ 1. NaOH, 3 eq 96.5% 97.49% Degradation: DCM, 2. Citric acid, 1 eq. +0.09% in 2 days (in argon) 5:2 +0.59% in 89 days (in argon) MeOH+ 1. NaOH, 3 eq. 93.8% 97.46% Degradation: DCM, 2. Fumaric acid, 1 eq. +1.95% in 14 days (in air) 5:2 +1.80% in 12 days (in argon) MeOH L-lysine, 1 eq. >99% 97.62% Degradation: +0.69% in 14 days (in air) +0.48% in 12 days (in argon)
- [0194]
- [0195]
The solution of chloromethyl di-tert-butyl phosphate in DME (250 g from a 10% solution, 96.64 mmole) was evaporated under reduced pressure until the formation of pale yellow oil, dissolved then at 50° C. with 318 ml of Acetonitrile. 17.2 g (80.54 mmole) of 1,8-bis(dimethylamino)naphtalene and 46.6 g (80.54 mmole) of 2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethyl-N-(6-(4-methylpiperazin-1-yl)-4-(o-tolyl)pyridin-3-yl)propanamide were added and the solution heated at 90° C. for at least 12 h. After the addition of 75 g of isopropylether, the precipitated crude product was cooled at room temperature, filtered and washed with acetonitrile, isopropylether/acetone, 3:1 and isopropylether, and dried under reduced pressure to afford 20-33 g of the 4-(5-{2-[3,5-bis(trifluoromethyl)phenyl]-N,2-dimethylpropanamido}-4-(o-tolyl)pyridin-2-yl)-1-methyl-1-{[(tert-butoxy)phosphoryl]oxymethyl}piperazin-1-ium as white solid (Yield: 30-50%). 1H-NMR (CD3OD, 400 MHz) δ 7.98 (s, 1H), 7.86 (s, 1H), 7.76 (s, 2H), 7.33-7.10 (m, 4H), 6.80 (s, 1H), 5.03 (d, 2H, JPH=8.5 Hz), 4.52 (s, 2H), 4.13 (m, 2H), 3.83 (m, 2H), 3.69 (m, 2H), 3.52 (m. 2H), 3.23 (s, 3H), 2.53 (s, 3H), 2.18 (s, 3H), 1.46 (s, 18H), 1.39 (s, 6H). 31P-NMR (CD3OD, 161 MHz) δ −5.01 (s, 1P). To 20 g (23.89 mmole) of the 4-(5-{2-[3,5-bis(trifluoromethyl)phenyl]-N,2-dimethylpropanamido}-4-(o-tolyl)pyridin-2-yl)-1-methyl-1-{[(tert-butoxy)phosphoryl]oxymethyl}piperazin-1-ium dissolved in 180 g of methanol and 400 g of dichloromethane was added HCl 4M in dioxane (18.8 g, 71.66 mmole) and the solution was heated for 3 h at reflux. After the addition of 200 g of dioxane, DCM and methanol were distilled under reduced pressure until precipitation of the product, which was filtered and washed with isopropylether (100 g), acetone (30 g) and pentane (2×60 g). The product was finally dried under reduced pressure at 55° C. to afford 15-17 g of 4-(5-(2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-4-(o-tolyl)pyridin-2-yl)-1-methyl-1-((phosphonooxy)methyl)piperazin-1-ium chloride hydrochloride as white solid (Yield: 88-93%). 1H-NMR (CD3OD, 400 MHz) δ 7.02 (s, 1H), 7.87 (s, 1H), 7.74 (s, 2H), 7.33-7.40 (m, 2H), 7.27 (m, 1H), 7.21 (s, 1H), 7.16 (d, 1H, J=8.2 Hz), 5.27 (d, 2H, JPH=7.9 Hz), 4.29 (m, 2H), 4.05 (m, 2H), 3.85 (m, 2H), 3.74 (m, 2H), 3.35 (s, 3H), 2.62 (s, 3H), 2.23 (s, 3H), 1.38 (s, 6H). 31P-NMR (CD3OD, 161 MHz) δ −2.81 (t, 1P, JPH=7.9 Hz).
Synthesis (B) of 4-(5-(2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-4-(o-tolyl)pyridin-2-yl)-1-methyl-1-((phosphonooxy)methyl)piperazin-1-ium chloride hydrochloride
- [0196]
- [0197]
To the solution of chloromethyl di-tert-butyl phosphate in Acetone (22.1 g from a 10% solution, 85.58 mmole), 15.5 g (103.24 mmole) of sodium iodide and 33.0 g (57.00 mmole) of netupitant were added and the solution heated at 50° C. for at 6-16 h. The precipitated salts were filtered off, the acetone distilled under reduced pressure and the crude product dissolved in 43.0 g of methanol and 43.0 g 1,4-dioxane. 12.6 g of HCl 4M in dioxane (113.85 mmole) were added, and then methanol is distilled off at 40° C. under reduced pressure. The solution is cooled at 5° C. and stirred at 5° C. for at least 2 h at 5° C. The product was isolated by filtration, purified by additional slurry in acetone (238 g), and filtered and washed with acetone (47 g) and pentane (2×72 g). - [0198]
The product was finally dried under reduced pressure at 60° C. to afford 22-30 g of white-yellowish solid (Yield: 50-70%) - [0199]
1H-NMR (CD3OD, 400 MHz) δ 7.02 (s, 1H), 7.87 (s, 1H), 7.74 (s, 2H), 7.33-7.40 (m, 2H), 7.27 (m, 1H), 7.21 (s, 1H), 7.16 (d, 1H, J=8.2 Hz), 5.27 (d, 2H, JPH=7.9 Hz), 4.29 (m, 2H), 4.05 (m, 2H), 3.85 (m, 2H), 3.74 (m, 2H), 3.35 (s, 3H), 2.62 (s, 3H), 2.23 (s, 3H), 1.38 (s, 6H). 31P-NMR (CD3OD, 161 MHz) δ −2.81 (t, IP, JPH=7.9 Hz).
PATENT
US 8,426,450
PATENT
US 9,403,772
SYN
https://chemistry-europe.onlinelibrary.wiley.com/doi/10.1002/chem.201901840
The synthesis of fosnetupitant (195) was developed by the Swiss company Helsinn (Scheme 34).[58] The synthesis started with the reaction of 6-chloronicotinic acid (196) with o-tolylmagnesium chloride followed by manganese(III) acetate to give acid derivative 197. This was converted to amide 198 after reaction with thionyl chloride and ammonium hydroxide. Next, reaction with N-methylpiperazine furnished intermediate 199, which was then transformed into carbamate 200 after reaction with NBS in methanol. Reduction with Red-Al followed by acylation with acyl chloride 202 afforded netupitant (203).
Finally, reaction with di-tert-butyl chloromethyl phosphate followed by the removal of the tert-butyl groups by treatment with HCl in dioxane afforded fosnetupitant (195).
L. Fadini, P. Manini, C. Pietra, C. Giuliano, E. Lovati, R. Cannella, S. Venturini, V. J. Stella, WO 082102 A1, 2013.


SYN
Fosnetupitant chloride HCl

PATENT
Fosnetupitant is a neurokynin-1 (“NK-1”) antagonist under development by Helsinn Healthcare SA, Lugano/Pazzallo Switzerland, for the treatment of chemotherapy induced nausea and vomiting. The compound is known chemically as 4-(5-(2-(3,5- bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-4-(o-tolyl)pyridin-2-yl)-l-methyl- 1 -((phosphonooxy)methyl)piperazin- 1 -ium, and has the following chemical structure in its acidic/free base form:

[004] The chloride monohydrochloride salt, and a method for its preparation, is described in WO 2013/082102. The chemical structure for this salt is reported as follows:

[005] The molecule can be challenging to manufacture, particularly in a highly pure crystalline form in a commercially acceptable yield. Solvents used in the manufacture of the product pose special challenges. Prior art processes have removed these solvents via evaporative techniques, which can degrade the fosnetupitant due to the excessive heat.
EXAMPLES
[089] In all the examples reported, unless otherwise reported, the starting compound was Form I of the chloride hydrochloride salt of 4-(5-(2-(3,5-bis(trifluoromethyl)phenyl)- N,2-dimethylpropanamido)-4-(o-tolyl)pyridin-2-yl)-l-methyl-l – ((phosphonooxy)methyl)piperazin-l-ium, produced substantially according to the methods described in WO 2013/082102.
EXAMPLE 1 : CHARACTERIZATION OF FOSNETUPITANT
1 . Experimental Methods
1.1 Solubility
[090] The solubility of the starting compound was determined in 25 pharmaceutically acceptable solvents (class II and III) of differing polarity. The procedure was as follows:
[091] Approximately 20 mg of material was weighed out into each glass vial.
[092] 5 volume aliquots of each solvent were added separately with stirring (i.e. 1 volume = 20 μΐ; hence, 5 volume = 100 μΐ (5 x 20 μΐ)).
[093] The mixture was stirred at RT for 5- 10 minutes. Visual checks were then made for solubility.
[094] If no solubility was achieved then steps (ii) and (iii) were repeated until either the solubility was achieved or the 50 volume aliquots of that solvent were added.
[095] Solubility was then approximated.
[096] Solubility was finally checked at the elevated temperature (40°C).
1.2 Polymorph Screen (including slurry studies)
[097] Using the information from the solubility study, the compound was slurried in the solvents outlined in Table I and two more mixtures of water/ MeOH (10:90) and water/ Acetone (1 :20) respectively with temperature cycling between 40°C and RT (4 hour periods at each temperature) over 48 hours. After the slurries the resulting solids were isolated and analyzed by Raman and XRPD (where enough material was available) for any change in physical form.
[098] The compound was also dissolved in the listed solvents and two more mixtures of water/organic solvent to yield saturated solutions, and crystallization was induced by: crash cooling (at ca. -1 8°C); evaporation (at RT); and addition of an anti-solvent. Solid materials generated were then isolated and examined by Raman and XRPD (where enough material was available).
1.3 Scale-up of any new polymorphic forms
[099] Any new potential polymorphic forms of the Form I fosnetupitant were then scaled-up to ~500mg level for further characterizations by PLM, SEM, DSC, TGA, GVS (XRPD post GVS) and NMR. Further studies of conversion between each polymorphic form were also performed. From this information, an understanding of the polymorphic space was achieved.
Synthetic Reference
Fadini, Luca; Manini, Peter; Pietra, Claudio; Giuliano, Claudio; Lovati, Emanuela; Cannella, Roberta; Venturini, Alessio; Stella, Valentino. (Assignee: Helsinn Healthcare SA, Switz). Substituted 4 – phenyl – pyridines for the treatment of nk-1 receptor related diseases. WO2013082102 (2013).
//////////Fosnetupitant, 07-PNET, Фоснетупитант , فوسنيتوبيتانت , 磷奈匹坦 , FDA 2014, EMA 2015
Centhaquine
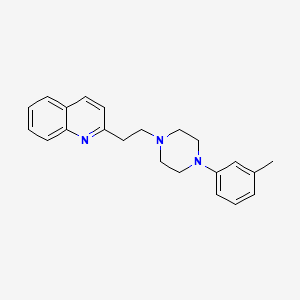
Centhaquine
PMZ-2010
CAS 57961-90-7
2-[2-[4-(3-methylphenyl)piperazin-1-yl]ethyl]quinoline
INDIA 2020, 14.05.2020, Centhaquine citrate bulk and Centhaquine citrate injection 1.0mg/vial, Add on resuscitative agent for hypovolemic shock
- OriginatorMidwestern University; Pharmazz
- DeveloperPharmazz
- ClassAnalgesics; Antihaemorrhagics; Antihypertensives; Cardiovascular therapies; Piperazines; Quinolines; Small molecules
- Mechanism of ActionAlpha 1 adrenergic receptor antagonists; Alpha 2 adrenergic receptor agonists
- RegisteredHaemorrhagic shock
- Phase IHeart arrest; Postoperative pain
- 20 Jul 2020Pharmazz plans to launch centhaquin for Haemorrhagic shock (Adjuvant therapy) in India by the middle of September 2020
- 20 Jul 2020Efficacy data from a phase III trial in Haemorrhagic shock released by Pharmazz
- 02 Jun 2020Centhaquine is still in phase I trials for Postoperative pain in USA (Pharmazz pipeline, June 2020)
SYNCenthaquin is a compound that produces hypotension and bradycardia in higher doses and resuscitation in lower doses. It is water insoluble, and is unsuitable for intravenous use. We prepared the citrate salt of centhaquin and evaluated its cardiovascular efficacy vs. centhaquin. Centhaquin citrate was prepared and characterized; its purity was determined by HPLC. Mean arterial pressure (MAP), heart rate (HR), pulse pressure (PP), cardiac output (CO), stroke volume (SV) and stroke work (SW) following intravenous administration of centhaquin and the citrate (0.05, 0.15 and 0.45 mg.kg(-1)) were determined in anaesthetized male Sprague-Dawley rats. Centhaquin citrate was 99.8% pure and water soluble. Centhaquin (0.05, 0.15 and 0.45 mg.kg(-1)) produced a maximal decrease in MAP of 15.6, 25.2 and 28.1%, respectively; while centhaquin citrate produced a greater (p<0.001) decrease of 35.7, 47.1 and 54.3%, respectively. The decrease in PP and HR produced by the citrate was greater than centhaquin (p<0.001). At 0.45 mg.kg(-1) centhaquin produced a maximal decrease of 20.9% (p<0.01) in CO, while centhaquin citrate produced a decrease of 42.1% (p<0.001). Reduction in SV (p<0.01) and SW (p<0.001) produced by centhaquin citrate were greater than centhaquin. Centhaquin citrate has greater cardiovascular activity compared to centhaquin.https://www.semanticscholar.org/paper/Synthesis-and-characterization-of-centhaquin-and-a-Reniguntala-Lavhale/6ca3975b114b0f23753e7a47710eff2467bc2dae

PATENT
https://patents.google.com/patent/WO2014035446A1/en
Shock due to severe hemorrhage accounts for a large proportion of posttraumatic deaths, particularly during early stages of injury (Wu, Dai et al. 2009). A majority of deaths due to hemorrhage occur within the first six hours after trauma (Shackford, Mackersie et al. 1993), but many of these deaths can be prevented (Acosta, Yang et al. 1998).
[0003] Shock is accompanied by circulatory failure which is the primary cause of mortality and morbidity. Presently, the recommended fluid therapy uses large volumes of Lactated Ringer’s solution (LR), which is effective in restoring hemodynamic parameters, but presents logistic and physiologic limitations (Vincenzi, Cepeda et al. 2009). For example, resuscitation using a large volume of crystalloids, like LR, has been associated with secondary abdominal compartment syndrome, pulmonary edema, cardiac dysfunction, and paralytic ileus (Balogh, McKinley et al. 2003). Therefore, a need exists in the art for a resuscitation agent that improves survival time, and can be used with a small volume of resuscitation fluid, for resuscitation in hypovolemic shock.
[0004] Centhaquin (2-[2-(4-(3-methyphenyl)-l-piperazinyl) ethyl-quinoline) is a centrally acting antihypertensive drug. The structure of centhaquin was determined (Bajpai et al., 2000) and the conformation of centhaquin was confirmed by X-ray diffraction (Carpy and Saxena, 1991).

Structure of centhaquin (2-[2-(4-(3-methyphenyl)- 1 -piperazinyl) ethyl] -quinoline) (as free base)
[0005] Centhaquin is an active cardiovascular agent that produces a positive inotropic effect and increases ventricular contractions of isolated perfused rabbit heart (Bhatnagar, Pande et al. 1985). Centhaquin does not affect spontaneous contractions of the guinea pig right auricle, but significantly potentiates positive inotropic effect of norepinephrine (NE) (Srimal, Mason et al. 1990). The direct or indirect positive inotropic effect of centhaquin can lead to an increase in cardiac output (CO). Centhaquin produces a decrease in mean arterial pressure (MAP) and heart rate (HR) in anesthetized rats and conscious freely moving cats and rats (Srimal, Gulati et al. 1990) due to its central sympatholytic activity (Murti, Bhandari et al. 1989; Srimal, Gulati et al. 1990; Gulati, Hussain et al. 1991). When administered locally into a dog femoral artery, centhaquin (10 and 20 μg) increased blood flow, which was similar to that observed with acetylcholine and papaverine. However, the vasodilator effect of centhaquin could not be blocked by atropine or dibenamine (Srimal, Mason et al. 1990). The direct vasodilator or central sympatholytic effect of centhaquin is likely to decrease systemic vascular resistance (SVR).
[0006] It was found that centhaquin enhances the resuscitative effect of hypertonic saline (HS) (Gulati, Lavhale et al. 2012). Centhaquin significantly decreased blood lactate and increases MAP, stroke volume, and CO compared to hypertonic saline alone. It is theorized, but not relied upon, that the cardiovascular actions of hypertonic saline and centhaquin are mediated through the ventrolateral medulla in the brain (Gulati, Hussain et al. 1991 ; Cavun and Millington 2001) and centhaquin may be augmenting the effect of hypertonic saline.
[0007] A large volume of LR (i.e., about three times the volume of blood loss) is the most commonly used resuscitation fluid therapy (Chappell, Jacob et al. 2008), in part because LR does not exhibit the centrally mediated cardiovascular effects of hypertonic saline. Large volume resuscitation has been used by emergency medical personnel and surgeons to reverse hemorrhagic shock and to restore end-organ perfusion and tissue oxygenation. However, there has been a vigorous debate with respect to the optimal methods of resuscitation (Santry

ased on the molecular weight of centhaquin (free base) (MW-332) and centhaquin citrate (MW-523), for identical doses of centhaquin (as free base) and centhaquin citrate, centhaquin citrate provides only 63.5% of centhaquin free base compared to the dose of centhaquin free base, e.g., a 0.05 mg dose of centhaquin citrate contains a 0.0318 mg of centhaquin (as free base). Similarly, a dose of centhaquin citrate dihydrate (MW-559) provides 59.4% centhaquin (free base) of the same dose as centhaquin (as free base), i.e., a 0.0005 mg dose of centhaquin citrate dihydrate contains 0.030 mg of centhaquin (as free base). Surprisingly, and as demonstrated below, at the same mg/kg dose centhaquin citrate and centhaquin citrate dihydrate provides greater cardiovascular effects than centhaquin free base.

Synthesis of Centhaquin

[0061] The synthesis of centhaquin was reported by Murthi and coworkers (Murthi et al U.S. Patent No. 3,983,121 ; Murti, Bhandari et al. 1989). In one procedure, reactants 1 and 2 were stirred at reflux for 15 hours. The resulting product was purified by evaporating the solvents to obtain an oil, which was heated in vacuo (100°C, 1 mm Hg). The remaining residue was recrystallized from ether-petroleum ether to obtain the final centhaquin product 3. The melting point reported for centhaquin was 76-77°C. In a subsequent publication (Murti, Bhandari et al. 1989), the reaction mixture was concentrated following 24 hours of reflux, diluted with water, and basified with aqueous NaOH. The basic mixture was extracted with ethyl acetate, and the ethyl acetate extracts were dried over anhydrous sodium sulfate and evaporated in vacuo to give centhaquin which was crystallized from hexane. The melting point of centhaquin (free base) obtained in this procedure was 82°C. The product obtained using either purification method is light tan in color, which is indicative of small amounts of impurities that were not completely removed using previously reported purification methods.
[0062] In accordance with the present invention, an improved purification method was found. According to the improved method, reactants 1 and 2 were stirred at reflux for 24 hours. The solvents were evaporated in vacuo and the resulting mixture was diluted with water and basified (10% NaOH). The basic mixture was extracted with ethyl acetate and the combined ethyl acetate extracts are dried over anhydrous sodium sulfate and evaporated in vacuo to obtain a residue, which was further purified with column chromatography (Si02, ethyl acetate). The resulting product can be decolorized using activated charcoal or directly crystallized from hot hexane to yield pure centhaquin. The resulting product is an off-white crystalline solid having a melting point of 94-95°C (free base). The product was
characterized using proton NMR, mass spectral, and elemental analysis and indicated high purity and superior quality.
[0063] Synthesis and characterization of centhaquin (free base): A mixture of 2- vinylquinoline (1) (5.0 g, 32.2 mmol, 98.5%) and 1 -(3-methylphenyl)piperazine (2) (5.68 g, 32.2 mmol, 99.0%) in absolute ethyl alcohol (150 ml) and glacial acetic acid (3.5 ml) was stirred at reflux for 24 hours in a round bottom flask. The reaction mixture was concentrated in vacuo, diluted with water (150 ml) and treated with 10% aqueous NaOH (150 ml). The residue was extracted with ethyl acetate (4 x 125 ml), dried with anhydrous Na2S04, and concentrated under reduced pressure to yield a crude product which was purified by column chromatography using silica gel (100-200 mesh) with ethyl acetate as an eluent. The resulting compound was recrystallized from hot hexane and filtered, to yield centhaquin as an off- white crystalline solid (7.75 g, 23.4 mmol, 73% yield); mp. 94-95°C; i? 0.30 (100% ethyl acetate); 1H NMR (300 MHz, CDC13): δ 8.07 (t, J= 7.5 Hz, 2 H), 7.78 (d, J= 7.8 Hz, 1 H), 7.70 (t, J= 7.8 Hz, 1 H), 7.50 (t, J= 7.5 Hz, 1 H), 7.36 (d, J= 8.4 Hz, 1 H), 7.16 (t, J = 7.5 Hz, 1 H), 6.77 – 6.74 (m, 2 H), 6.69 (d, J= 7.2 Hz, 1 H), 3.26- 3.21 (m, 6 H), 2.97 – 2.92 (m, 2 H), 2.76 – 2.73 (m, 4 H), 2.32 (s, 3 H); HRMS (ESI) m/z 332.2121 [M+l]+ (calcd for C22H26N3 332.2122); Anal. (C22H25N3) C, H, N.
[0064] Preparation of centhaquin citrate: Centhaquin (free base) (5.62 g, 16.98 mmol) was treated with citric acid (3.26 g, 16.98 mmol) in a suitable solvent and converted to the citrate salt obtained as an off-white solid (7.96 g, 15.2 mmol, 90%); m.p. 94-96°C ; Anal.
10065] Figs. 1(a) and 1(b) are high resolution mass spectral analyses of centhaquin free base (Fig 1(a)) and centhaquin citrate (Fig. 1(b)). Compound samples were analyzed following ionization using electrospray ionization (ESI).
[0066J For centhaquin free base in Fig 1(a), a base peak [M+l]+ was observed at m z 332.2141 (theory: 332.2121) consistent with the elemental composition of protonated centhaquin (C22H26N3).
[0067] For centhaquin citrate in Fig 1(b), the mass spectrum was identical to the mass spectrum obtained for the free base. An [M+l]+base peak was observed at m z 332.2141 (theory: 332.2121), which corresponds to the elemental composition of protonated centhaquin (C22H26N3). This result is typical of salts of organic bases to yield the [M+l]+ of the free base as observed here with centhaquin citrate.
[0068] Mass spectrometry is one of the most sensitive analytical methods, and examination of the mass spectra of Fig. 1 indicate that the samples are devoid of any extraneous peaks and are of homogeneous purity (>99.5).
PATENT
https://patents.google.com/patent/WO2014035446A1/en
////////////Centhaquine, PMZ-2010, PMZ 2010, INDIA 2020, 2020 APPROVALS
Esketamine

Esketamine
- Molecular FormulaC13H16ClNO
- Average mass237.725 Da
(+)-Ketamine(2S)-2-(2-Chlorophenyl)-2-(methylamino)cyclohexanone
(S)-Ketamine33643-46-8[RN]7884Cyclohexanone, 2-(2-chlorophenyl)-2-(methylamino)-, (2S)-Cyclohexanone, 2-(2-chlorophenyl)-2-(methylamino)-, (S)-
KetamineCAS Registry Number: 6740-88-1CAS Name: 2-(2-Chlorophenyl)-2-(methylamino)cyclohexanoneMolecular Formula: C13H16ClNOMolecular Weight: 237.73Percent Composition: C 65.68%, H 6.78%, Cl 14.91%, N 5.89%, O 6.73%Literature References: Prepn: C. L. Stevens, BE634208; idem,US3254124 (1963, 1966 both to Parke, Davis). Isoln of optical isomers: T. W. Hudyma et al.,DE2062620 (1971 to Bristol-Myers), C.A.75, 118119x (1971). Clinical pharmacology of racemate and enantiomers: P. F. White et al.,Anesthesiology52, 231 (1980). Toxicity: E. J. Goldenthal, Toxicol. Appl. Pharmacol.18, 185 (1971). Enantioselective HPLC determn in plasma: G. Geisslinger et al.,J. Chromatogr.568, 165 (1991). Comprehensive description: W. C. Sass, S. A. Fusari, Anal. Profiles Drug Subs.6, 297-322 (1977). Review of pharmacology and use in veterinary medicine: M. Wright, J. Am. Vet. Med. Assoc.180, 1462-1471 (1982). Review of pharmacology and clinical experience: D. L. Reich, G. Silvay, Can. J. Anaesth.36, 186-197 (1989); in pediatric procedures: S. M. Green, N. E. Johnson, Ann. Emerg. Med.19, 1033-1046 (1990).Properties: Crystals from pentane-ether, mp 92-93°. uv max (0.01N NaOH in 95% methanol): 301, 276, 268, 261 nm (A1%1cm 5.0, 7.0, 9.8, 10.5). pKa 7.5. pH of 10% aq soln 3.5.Melting point: mp 92-93°pKa: pKa 7.5Absorption maximum: uv max (0.01N NaOH in 95% methanol): 301, 276, 268, 261 nm (A1%1cm 5.0, 7.0, 9.8, 10.5)
Derivative Type: HydrochlorideCAS Registry Number: 1867-66-9Manufacturers’ Codes: CI-581Trademarks: Ketalar (Pfizer); Ketanest (Pfizer); Ketaset (Fort Dodge); Ketavet (Gellini); Vetalar (Bioniche)Molecular Formula: C13H16ClNO.HClMolecular Weight: 274.19Percent Composition: C 56.95%, H 6.25%, Cl 25.86%, N 5.11%, O 5.84%Properties: White crystals, mp 262-263°. Soly in water: 20 g/100 ml. LD50 in adult mice, rats (mg/kg): 224 ±4, 229 ±5 i.p. (Goldenthal).Melting point: mp 262-263°Toxicity data: LD50 in adult mice, rats (mg/kg): 224 ±4, 229 ±5 i.p. (Goldenthal)
NOTE: This is a controlled substance (depressant): 21 CFR, 1308.13.Therap-Cat: Anesthetic (intravenous).Therap-Cat-Vet: Anesthetic (intravenous).Keywords: Anesthetic (Intravenous).Esketamine hydrochloride, S enantiomer of ketamine, is in phase III clinical trials by Johnson & Johnson for the treatment of depression.Drug Name:Esketamine HydrochlorideResearchCode:JNJ-54135419MOA:Dopamine reuptake inhibitor; NMDA receptor antagonistIndication:DepressionStatus:Phase III (Active)Company:Johnson & Johnson (Originator)
| Molecular Weight | 274.19 |
| Formula | C13H16ClNO•HCl |
| CAS No. | 33643-46-8 (Esketamine); 33643-47-9 (Esketamine Hydrochloride); |
Reference:1. US6040479.
https://patents.google.com/patent/US6040479A/en
EXAMPLE 1
50 g (0.21 mol) R,S-ketamine are dissolved in 613 ml of acetone at the boiling point and subsequently mixed with 31.5 g (0.21 mol) L-(+)-tartaric acid. In order to obtain a clear solution, 40 ml of water are added thereto at the boiling point and subsequently the clear solution is filtered off while still hot. After the addition of seed crystals obtained in a small preliminary experiment, the whole is allowed to cool to ambient temperature while stirring. After standing overnight, the crystals formed are filtered off with suction and dried in a circulating air drying cabinet (first at ambient temperature and then at 50-60° C.).
Yield (tartrate): 64.8 g
m.p.: 161° C.
[α]D : +26.1° (c=2/H2 O)
Thereafter, the crystallisate is recrystallised in a mixture of 1226 ml acetone and 90 ml water. After cooling to ambient temperature and subsequently stirring for 4 hours, the crystals are filtered off with suction and dried in a circulating air drying cabinet (first at ambient temperature and then at 50-60° C). There are obtained 38.8 g of tartrate (95.29% of theory).
m.p.: 175.3° C.
[α]D : +68.9° (c=2/H2 O)
The base is liberated by taking up 38.8 g of tartrate in 420 ml of aqueous sodium hydroxide solution and stirring with 540 ml of diethyl ether. The ethereal phase is first washed with water and subsequently with a saturated solution of sodium chloride. The organic phase is dried over anhydrous sodium sulphate. After filtering, the solution is evaporated to dryness on a rotary evaporator, a crystalline, colourless product remaining behind.
Yield (crude base): 21.5 g=86.0% of theory
m.p.: 118.9° C. (literature: 120-122° C.)
[α]D : -55.8° (c=2/EtOH) (literature: [α]D : -56.35° ).
In order possibly to achieve a further purification, the base can be recrystallised from cyclohexane. For this purpose, 10.75 g of the crude base are dissolved in 43 ml cyclohexane at the boiling point. While stirring, the clear solution is slowly cooled to about 10° C. and then stirred at this temperature for about 1 hour. The crystallisate which precipitates out is filtered off with suction and dried to constant weight.
Yield (base): 10.3 g=82.4% of theory
m.p.: 120° C. (literature: 120-122° C.)
[α]D : -56.8° (c=2/EtOH) (literature: [α]D : -56.35° )
EXAMPLE 2
125 ml of water are taken and subsequently 31.5 g (0.21 mol) L-(+)-tartaric acid and 50 g (0.21 mol) R,S-ketamine added thereto. While stirring, this mixture is warmed to 50-60° C. until a clear solution results. After cooling to ambient temperature while stirring and subsequently stirring overnight, the crystals formed are filtered off with suction. Subsequently, the crystallisate is first washed with water (1-6° C.) and subsequently washed twice with, in each case, 20 ml of acetone. Drying in a circulating air drying cabinet (first at ambient temperature and then at 50-60° C.) gives 31.79 g of tartrate (78.23%) of theory).
EXAMPLE 3
150 ml of water are taken and subsequently mixed with 39.8 g (0.27 mol) L-(+)-tartaric acid and 50 g (0.21 mol) R,S-ketamine. While stirring, this mixture is warmed to 50-60° C. until a clear solution results.
After cooling to ambient temperature while stirring and subsequently stirring overnight, the crystals formed are filtered off with suction. Subsequently, the crystallisate is successively washed with 8 ml of water (1-6° C.) and thereafter twice with, in each case, 20 ml acetone.
Drying in a circulating air drying cabinet (first at ambient temperature and then at 50-60° C.) gives 32.58 g of tartrate (80.02% of theory).
EXAMPLE 4
150 ml of water and 50 ml isopropanol are taken. After the addition of 39.8 g (0.21 mol) L-(+)-tartaric acid and 50 g (0.21 mol) R,S-ketamine, the mixture is heated to reflux temperature while stirring until a solution results (possibly add water until all is dissolved).
Subsequently, while stirring, the solution is allowed to cool to ambient temperature and stirred overnight. The crystals are filtered off with suction and subsequently washed with a 1:2 mixture of 20 ml of water/isopropanol and dried in a circulating air drying cabinet (first at ambient temperature and then at 50-60° C.). There are obtained 24.45 g of tartrate (62.63% of theory).
EXAMPLE 5
50 g (0.21 mol) R,S-ketamine are dissolved at the boiling point in 300 ml acetone and subsequently mixed with 31.5 g (0.21 mol) L-(+)-tartaric acid and 100 ml of water. The whole is allowed to cool while stirring and possibly seeded.
After standing overnight, the crystals formed are filtered off with suction, then washed twice with, in each case, 20 ml acetone and dried in a circulating air drying cabinet (first at ambient temperature and then at 50-60° C.). There are obtained 30.30 g of tartrate (74.57% of theory).
EXAMPLE 6
75 ml of water and 50 ml isopropanol are taken and subsequently 39.8 g (0.27 mol) L-(+)-tartaric acid added thereto. While stirring, the mixture is heated to reflux temperature until a clear solution results. After cooling to ambient temperature while stirring and subsequently stirring overnight, the crystals formed are filtered off with suction. Subsequently, the crystallisate is washed with a 1:2 mixture of 20 ml water/isopropanol. After drying in a circulating air drying cabinet (first at ambient temperature and then at 50-60° C.), there are obtained 34.84 g of tartrate (85.74% of theory).
EXAMPLE 7
20 g of the S-(+)-tartrate obtained in Example 4 are dissolved in 100 ml of water at 30-40° C. With about 7 ml of 50% sodium hydroxide solution, an S-(-)-ketamine base is precipitated out up to about pH 13. It is filtered off with suction and washed neutral with water to pH 7-8. Subsequently, it is dried for about 24 hours at 50° C. in a circulating air drying cabinet. There are obtained 11.93 g S-(-)-ketamine (97.79% of theory).
EXAMPLE 8
5 g of the S-(-)-ketamine obtained in Example 7 are dissolved in 50 ml isopropanol at about 50° C. and possibly filtered off with suction over kieselguhr. Subsequently, gaseous hydrogen chloride is passed in at 50-60° C. until a pH value of 0-1 is reached. The reaction mixture is allowed to cool to ambient temperature, filtered off with suction and washed with about 5 ml isopropanol. The moist product is dried overnight at about 50° C. in a circulating air drying cabinet. There are obtained 5.09 g S-(+)-ketamine hydrochloride (88.06% of theory).
Route 2
Reference:1. J. Am. Chem. Soc. 2015, 137, 3205-3208.
https://pubs.acs.org/doi/10.1021/jacs.5b00229
Here we report the direct asymmetric amination of α-substituted cyclic ketones catalyzed by a chiral phosphoric acid, yielding products with a N-containing quaternary stereocenter in high yields and excellent enantioselectivities. Kinetic resolution of the starting ketone was also found to occur on some of the substrates under milder conditions, providing enantioenriched α-branched ketones, another important building block in organic synthesis. The utility of this methodology was demonstrated in the short synthesis of (S)-ketamine, the more active enantiomer of this versatile pharmaceutical.



CLIP

Initial reagent: cyclopentyl Grignard Step 0: Producing cyclopentyl Grignard Reacting cyclopentyl bromide with magnesium in solvent (ether or THF) Best results: distill solvent from Grignard under vacuum and replace with hydrocarbon solvent (e.g. benzene) Step 1: processing to (o-chlorophenyl)-cyclopentyl ketone Adding o-chlorobenzonitrile to cyclopentyl Grignard in solvent, stirring for long period of time (typically three days) Hydrolyzing reaction with mixture containing crushed ice, ammonium chloride and some ammonium hydroxide Extraction with organic solvent gives (o-chlorophenyl)-cyclopentyl ketone
Step 2: processing to alpha-bromo (o-chlorophenyl)-cyclopentyl ketone ketone processed with bromine in carbon tetrachloride at low temperature (typical T = 0°C), addition of bromine dropwise forming orange suspension Suspension washed in dilute aquerous solution of sodium bisufide and evaporated giving 1-bromocyclopentyl-(o-chlorophenyl)-ketone Note: bromoketone is unstable, immeadiate usage. Bromination carried out with NBromosuccinimide result higher yield (roughly 77%) Step 3: processing to 1-hydroxycyclopentyl-(o-chlorophenyl)-ketone-N-methylimine Dissolving bromoketone in liquid methylamine freebase (or benzene as possible solvent) After time lapse (1h): excess methylamine evaporated, residue dissolved in pentane and filtered evaporation of solvent yields 1-hydroxy-cyclopentyl-(o-chlorophenyl)-ketone N-methylimine Note: longer time span (4-5d) for evaporation of methylaminemay increase yield Step 4: processing to 2-Methylamino-2-(o-chlorophenyl)-cyclohexanone (Ketamine) Method: Thermal rearragement (qualitative yield after 30min in 180°C) N-methylimine dissolved in 15ml decalin, refluxed for 2.5h Evaporation of solvent under reduced temperature followed by extraction of residue with dilute hydrochloric acid Treatment with decolorizing charcoal (solution: acidic => basic) Recrystallization from pentane-ether Note – alternative to use of decalin: pressure bomb
racemic compound, in pharmaceutical preparation racemic more active enantiomere esketamine (S-Ketamine) available as Ketanest S, but Arketamine (R-Ketamine) never marketed for clinical use, Optical rotation: varies between salt and free base form free base form: (S)-Ketamine dextrorotation (S)-(+)-ketamine hydrochloridesalt: levorotation(S)-(-)-ketamine Reason found in molecular level: different orientation of substituents: freebase: o-chlorophenyl equatorial, methylamino axia
Sources: http://creationwiki.org/Ketamine#Synthesis http://www.lycaeum.org/rhodium/chemistry/pcp/ketamine.html https://pubchem.ncbi.nlm.nih.gov/compound/ketamine https://pubchem.ncbi.nlm.nih.gov/compound/ketamine#section=Drug-Warning http://www.rsc.org/chemistryworld/2014/02/ketamine-special-k-drugs-podcast http://drugabuse.com/library/the-effects-of-ketamine-use/ http://www.drugfreeworld.org/drugfacts/prescription/ketamine.html http://onlinelibrary.wiley.com/doi/10.1002/1615-9314(20021101)25:15/17%3C1155::AID-JSSC1155%3E3.0.CO;2-M/pdf
CLIP
Process Research and Impurity Control Strategy of Esketamine Organic Process Research & Development ( IF 3.023
) Pub Date: 2020-03-18 , DOI: 10.1021/acs.oprd.9b00553
Shenghua Gao; Xuezhi Gao; Zhezhou Yang; Fuli Zhang
An improved synthesis of ( S )-ketamine (esketamine) has been developed, which was cost-effective, and the undesired isomer could be recovered by racemization. Critical process parameters of each step were identified as well as the process-related impurities. The formation mechanisms and control strategies of most impurities were first discussed. Moreover, the ( S )-ketamine tartrate is a dihydrate, which was disclosed for the first time. The practicable racemization catalyzed by aluminum chloride was carried out in quantitative yield with 99% purity . The ICH-grade quality ( S)-ketamine hydrochloride was obtained in 51.1% overall yield (14.0% without racemization) by chiral resolution with three times recycling of the mother liquors. The robust process of esketamine could be industrially scalable.
Process Research and ketamine impurity control strategy
has been developed an improved ( S ) – ketamine (esketamine) synthesis, the high cost-effective way, the undesired isomer may be recycled by racemization. Determine the key process parameters and process-related impurities for each step. First, the formation mechanism and control strategy of most impurities are discussed. In addition, ( S )-ketamine tartrate is a dihydrate, which is the first time it has been published. The feasible racemization catalyzed by aluminum chloride proceeds in a quantitative yield with a purity of 99%. ICH grade quality ( S) 5-ketamine hydrochloride can be obtained through chiral resolution and three times the mother liquor recovery rate. The total yield is 51.1% (14.0% without racemization). The robust process of ketamine can be used in Industrial promotion.

CLIP

CLIP
https://link.springer.com/article/10.1007/s13738-018-1404-1#citeas
Taghizadeh, M.J., Gohari, S.J.A., Javidan, A. et al. A novel strategy for the asymmetric synthesis of (S)-ketamine using (S)-tert-butanesulfinamide and 1,2-cyclohexanedione. J IRAN CHEM SOC 15, 2175–2181 (2018). https://doi.org/10.1007/s13738-018-1404-1
Abstract
We present a novel asymmetric synthesis route for synthesis of (S)-ketamine using a chiral reagent according to the strategy (Scheme 1), with good enantioselectivity (85% ee) and yield. In this procedure, the (S)-tert-butanesulfinamide (TBSA) acts as a chiral auxiliary reagent to generate (S)-ketamine. A series of new intermediates were synthesized and identified for the first time in this work (2–4). The monoketal intermediate (1) easily obtained after partial conversion of one ketone functional group of 1,2-cyclohexanedione into a ketal using ethylene glycol. The sulfinylimine (2) was obtained by condensation of (S)-tert-butanesulfinamide (TBSA) with (1), 4-dioxaspiro[4.5]decan-6-one in 90% yield. The (S)-N–tert-butanesulfinyl ketamine (3) was prepared on further reaction of sulfinylimine (2) with appropriate Grignard reagent (ArMgBr) in which generated chiral center in 85% yield and with 85% diastereoselectivity. Methylation of amine afforded the product (4). Finally, the sulfinyl- and ketal-protecting groups were removed from the compound (4) by brief treatment with stoichiometric quantities of HCl in a protic solvent gave the (S)-ketamine in near quantitative yield.

Esketamine, sold under the brand name Spravato[4] among others,[6][7] is a medication used as a general anesthetic and for treatment-resistant depression.[4][1] Esketamine is used as a nasal spray or by injection into a vein.[4][1]
Esketamine acts primarily as a non-competitive N-methyl-D-aspartate (NMDA) receptor antagonist.[1][8] It also acts to some extent as a dopamine reuptake inhibitor but, unlike ketamine, does not interact with the sigma receptors.[1] The compound is the S(+) enantiomer of ketamine, which is an anesthetic and dissociative similarly.[1] It is unknown whether its antidepressant action is superior, inferior or equal to racemic ketamine and its opposite enantiomer, arketamine, which are both being investigated for the treatment of depression.
Esketamine was introduced for medical use in 1997.[1] In 2019, it was approved for use with other antidepressants, for the treatment of depression in adults in the United States.[9]
In August 2020, it was approved by the U.S. Food and Drug Administration (FDA) with the added indication for the short-term treatment of suicidal thoughts.[10]
Medical uses
Anesthesia
Esketamine is a general anesthetic and is used for similar indications as ketamine.[1] Such uses include induction of anesthesia in high-risk patients such as those with hemorrhagic shock, anaphylactic shock, septic shock, severe bronchospasm, severe hepatic insufficiency, cardiac tamponade, and constrictive pericarditis; anesthesia in caesarian section; use of multiple anesthetics in burns; and as a supplement to regional anesthesia with incomplete nerve blocks.[1]
Depression
See also: List of investigational antidepressants
Similarly to ketamine, esketamine appears to be a rapid-acting antidepressant.[8][11] It received a breakthrough designation from the FDA for treatment-resistant depression (TRD) in 2013 and major depressive disorder (MDD) with accompanying suicidal ideation in 2016.[12][11] The medication was studied for use in combination with an antidepressant in people with TRD who had been unresponsive to treatment;[12][8][11] six phase III clinical trials for this indication were conducted in 2017.[12][8][11] It is available as a nasal spray.[12][8][11]
In February 2019, an outside panel of experts recommended that the FDA approve the nasal spray version of esketamine,[13] provided that it be given in a clinical setting, with people remaining on site for at least two hours after. The reasoning for this requirement is that trial participants temporarily experienced sedation, visual disturbances, trouble speaking, confusion, numbness, and feelings of dizziness during immediately after.[14]
In January 2020, esketamine was rejected by the National Health Service of Great Britain. NHS questioned the benefits and claimed that it was too expensive. People who have been already using the medication were allowed to complete treatment if their doctors consider this necessary.[15]
Side effects
Most common side effects when used in those with treatment resistant depression include dissociation, dizziness, nausea, sleepiness, anxiety, and increased blood pressure.[16]
Pharmacology
Esketamine is approximately twice as potent as an anesthetic as racemic ketamine.[17] It is eliminated from the human body more quickly than arketamine (R(–)-ketamine) or racemic ketamine, although arketamine slows its elimination.[18]
A number of studies have suggested that esketamine has a more medically useful pharmacological action than arketamine or racemic ketamine[citation needed] but, in mice, that the rapid antidepressant effect of arketamine was greater and lasted longer than that of esketamine.[19] The usefulness of arketamine over eskatamine has been supported by other researchers.[20][21][22]
Esketamine inhibits dopamine transporters eight times more than arketamine.[23] This increases dopamine activity in the brain. At doses causing the same intensity of effects, esketamine is generally considered to be more pleasant by patients.[24][25] Patients also generally recover mental function more quickly after being treated with pure esketamine, which may be a result of the fact that it is cleared from their system more quickly.[17][26] This is however in contradiction with R-ketamine being devoid of psychotomimetic side effects.[27]
Unlike arketamine, esketamine does not bind significantly to sigma receptors. Esketamine increases glucose metabolism in frontal cortex, while arketamine decreases glucose metabolism in the brain. This difference may be responsible for the fact that esketamine generally has a more dissociative or hallucinogenic effect while arketamine is reportedly more relaxing.[26] However, another study found no difference between racemic and (S)-ketamine on the patient’s level of vigilance.[24] Interpretation of this finding is complicated by the fact that racemic ketamine is 50% (S)-ketamine.
History
Esketamine was introduced for medical use as an anesthetic in Germany in 1997, and was subsequently marketed in other countries.[1][28] In addition to its anesthetic effects, the medication showed properties of being a rapid-acting antidepressant, and was subsequently investigated for use as such.[8][12] In November 2017, it completed phase III clinical trials for treatment-resistant depression in the United States.[8][12] Johnson & Johnson filed a Food and Drug Administration (FDA) New Drug Application (NDA) for approval on September 4, 2018;[29] the application was endorsed by an FDA advisory panel on February 12, 2019, and on March 5, 2019, the FDA approved esketamine, in conjunction with an oral antidepressant, for the treatment of depression in adults.[9]
In the 1980s and ’90s, closely associated ketamine was used as a club drug known as “Special K” for its trip-inducing side effects.[30][31]
Society and culture
Names
Esketamine is the generic name of the drug and its INN and BAN, while esketamine hydrochloride is its BANM.[28] It is also known as S(+)-ketamine, (S)-ketamine, or (–)-ketamine, as well as by its developmental code name JNJ-54135419.[28][12]
Esketamine is marketed under the brand name Spravato for use as an antidepressant and the brand names Ketanest, Ketanest S, Ketanest-S, Keta-S for use as an anesthetic (veterinary), among others.[28]
Availability
Esketamine is marketed as an antidepressant in the United States;[9] and as an anesthetic in the European Union.[28]
Legal status
Esketamine is a Schedule III controlled substance in the United States.[4]
References
- ^ Jump up to:a b c d e f g h i j Himmelseher S, Pfenninger E (December 1998). “[The clinical use of S-(+)-ketamine–a determination of its place]”. Anasthesiologie, Intensivmedizin, Notfallmedizin, Schmerztherapie. 33 (12): 764–70. doi:10.1055/s-2007-994851. PMID 9893910.
- ^ “Spravato 28 mg nasal spray, solution – Summary of Product Characteristics (SmPC)”. (emc). Retrieved 24 November 2020.
- ^ “Vesierra 25 mg/ml solution for injection/infusion – Summary of Product Characteristics (SmPC)”. (emc). 21 February 2020. Retrieved 24 November2020.
- ^ Jump up to:a b c d e “Spravato- esketamine hydrochloride solution”. DailyMed. 6 August 2020. Retrieved 26 September 2020.
- ^ “Spravato EPAR”. European Medicines Agency (EMA). 16 October 2019. Retrieved 24 November 2020.
- ^ “Text search results for esketamine: Martindale: The Complete Drug Reference”. MedicinesComplete. London, UK: Pharmaceutical Press. Retrieved 20 August 2017.[dead link]
- ^ Brayfield A, ed. (9 January 2017). “Ketamine Hydrochloride”. MedicinesComplete. London, UK: Pharmaceutical Press. Retrieved 20 August2017.[dead link]
- ^ Jump up to:a b c d e f g Rakesh G, Pae CU, Masand PS (August 2017). “Beyond serotonin: newer antidepressants in the future”. Expert Review of Neurotherapeutics. 17 (8): 777–790. doi:10.1080/14737175.2017.1341310. PMID 28598698. S2CID 205823807.
- ^ Jump up to:a b c “FDA approves new nasal spray medication for treatment-resistant depression; available only at a certified doctor’s office or clinic”. U.S. Food and Drug Administration (FDA) (Press release). Retrieved 2019-03-06.
- ^ “FDA Approves A Nasal Spray To Treat Patients Who Are Suicidal”. NPR. 4 August 2020. Retrieved 27 September 2020.
- ^ Jump up to:a b c d e Lener MS, Kadriu B, Zarate CA (March 2017). “Ketamine and Beyond: Investigations into the Potential of Glutamatergic Agents to Treat Depression”. Drugs. 77 (4): 381–401. doi:10.1007/s40265-017-0702-8. PMC 5342919. PMID 28194724.
- ^ Jump up to:a b c d e f g “Esketamine – Johnson & Johnson – AdisInsight”. Retrieved 7 November 2017.
- ^ Koons C, Edney A (February 12, 2019). “First Big Depression Advance Since Prozac Nears FDA Approval”. Bloomberg News. Retrieved February 12, 2019.
- ^ Psychopharmacologic Drugs Advisory Committee (PDAC) and Drug Safety and Risk Management (DSaRM) Advisory Committee (February 12, 2019). “FDA Briefing Document” (PDF). Food and Drug Administration. Retrieved February 12, 2019.
Meeting, February 12, 2019. Agenda Topic: The committees will discuss the efficacy, safety, and risk-benefit profile of New Drug Application (NDA) 211243, esketamine 28 mg single-use nasal spray device, submitted by Janssen Pharmaceutica, for the treatment of treatment-resistant depression.
- ^ “Anti-depressant spray not recommended on NHS”. BBC News. 28 January 2020.
- ^ “Esketamine nasal spray” (PDF). U.S. Food and Drug Administration (FDA). Retrieved 21 October 2019.
- ^ Jump up to:a b Himmelseher S, Pfenninger E (December 1998). “[The clinical use of S-(+)-ketamine–a determination of its place]”. Anasthesiologie, Intensivmedizin, Notfallmedizin, Schmerztherapie (in German). 33 (12): 764–70. doi:10.1055/s-2007-994851. PMID 9893910.
- ^ Ihmsen H, Geisslinger G, Schüttler J (November 2001). “Stereoselective pharmacokinetics of ketamine: R(–)-ketamine inhibits the elimination of S(+)-ketamine”. Clinical Pharmacology and Therapeutics. 70 (5): 431–8. doi:10.1067/mcp.2001.119722. PMID 11719729.
- ^ Zhang JC, Li SX, Hashimoto K (January 2014). “R (-)-ketamine shows greater potency and longer lasting antidepressant effects than S (+)-ketamine”. Pharmacology, Biochemistry, and Behavior. 116: 137–41. doi:10.1016/j.pbb.2013.11.033. PMID 24316345. S2CID 140205448.
- ^ Muller J, Pentyala S, Dilger J, Pentyala S (June 2016). “Ketamine enantiomers in the rapid and sustained antidepressant effects”. Therapeutic Advances in Psychopharmacology. 6 (3): 185–92. doi:10.1177/2045125316631267. PMC 4910398. PMID 27354907.
- ^ Hashimoto K (November 2016). “Ketamine’s antidepressant action: beyond NMDA receptor inhibition”. Expert Opinion on Therapeutic Targets. 20 (11): 1389–1392. doi:10.1080/14728222.2016.1238899. PMID 27646666. S2CID 1244143.
- ^ Yang B, Zhang JC, Han M, Yao W, Yang C, Ren Q, Ma M, Chen QX, Hashimoto K (October 2016). “Comparison of R-ketamine and rapastinel antidepressant effects in the social defeat stress model of depression”. Psychopharmacology. 233 (19–20): 3647–57. doi:10.1007/s00213-016-4399-2. PMC 5021744. PMID 27488193.
- ^ Nishimura M, Sato K (October 1999). “Ketamine stereoselectively inhibits rat dopamine transporter”. Neuroscience Letters. 274 (2): 131–4. doi:10.1016/s0304-3940(99)00688-6. PMID 10553955. S2CID 10307361.
- ^ Jump up to:a b Doenicke A, Kugler J, Mayer M, Angster R, Hoffmann P (October 1992). “[Ketamine racemate or S-(+)-ketamine and midazolam. The effect on vigilance, efficacy and subjective findings]”. Der Anaesthesist (in German). 41 (10): 610–8. PMID 1443509.
- ^ Pfenninger E, Baier C, Claus S, Hege G (November 1994). “[Psychometric changes as well as analgesic action and cardiovascular adverse effects of ketamine racemate versus s-(+)-ketamine in subanesthetic doses]”. Der Anaesthesist (in German). 43 Suppl 2: S68-75. PMID 7840417.
- ^ Jump up to:a b Vollenweider FX, Leenders KL, Oye I, Hell D, Angst J (February 1997). “Differential psychopathology and patterns of cerebral glucose utilisation produced by (S)- and (R)-ketamine in healthy volunteers using positron emission tomography (PET)”. European Neuropsychopharmacology. 7 (1): 25–38. doi:10.1016/s0924-977x(96)00042-9. PMID 9088882. S2CID 26861697.
- ^ Yang C, Shirayama Y, Zhang JC, Ren Q, Yao W, Ma M, Dong C, Hashimoto K (September 2015). “R-ketamine: a rapid-onset and sustained antidepressant without psychotomimetic side effects”. Translational Psychiatry. 5 (9): e632. doi:10.1038/tp.2015.136. PMC 5068814. PMID 26327690.
- ^ Jump up to:a b c d e “Esketamine”. Drugs.com.
- ^ “Janssen Submits Esketamine Nasal Spray New Drug Application to U.S. FDA for Treatment-Resistant Depression”. Janssen Pharmaceuticals, Inc.
- ^ Marsa, Linda (January 2020). “A Paradigm Shift for Depression Treatment”. Discover. Kalmbach Media.
- ^ Hoffer, Lee (7 March 2019). “The FDA Approved a Ketamine-Like Nasal Spray for Hard-to-Treat Depression”. Vice. Retrieved 11 February 2020.
External links
- “Esketamine”. Drug Information Portal. U.S. National Library of Medicine.
- “Esketamine hydrochloride”. Drug Information Portal. U.S. National Library of Medicine.
/////////////Esketamine, JNJ 54135419, phase 3
Inclisiran
Inclisiran
CAS 1639324-58-5
- ALN-60212
- ALN-PCSsc
US FDA APPROVED
12/22/2021 |
To treat heterozygous familial hypercholesterolemia or clinical atherosclerotic cardiovascular disease as an add-on therapy, Leqvio
Inclisiran was first developed by Alnylam Pharmaceuticals, Inc. (Cambridge, Massachusetts, US). Development has now been assumed by The Medicines Company (Parsippany, New Jersey, US). One phase I and two phase II trials have been completed. Topline results of two phase III trials were also recently presented while other phase III trials are still ongoing as part of the ORION clinical development program. …..https://www.ncbi.nlm.nih.gov/books/NBK555477/
Inclisiran is a long-acting, synthetic small interfering RNA (siRNA) directed against proprotein convertase subtilisin-kexin type 9 (PCSK9), which is a serine protease that regulates plasma low-density lipoprotein cholesterol (LDL-C) levels. By binding to PCSK9 messenger RNA, inclisiran prevents protein translation of PCSK9, leading to decreased concentrations of PCSK9 and plasma concentrations of LDL cholesterol.1,2 Lowering circulating plasma LDL-C levels offers an additional benefit of reducing the risk of cardiovascular disease (CVD) and improving cardiovascular outcomes, as hypercholesterolemia is a major known risk factor for CVD.1,2
On December 11, 2020, the European Commission (EC) granted authorization for marketing inclisiran as the first and only approved siRNA for the treatment of adults with primary hypercholesterolemia (heterozygous familial and non-familial) or mixed dyslipidemia, alone or in combination with other lipid-lowering therapies. It is marketed under the trade name Leqvio 8 and is also currently under review by the FDA.
Inclisiran, sold under the brand name Leqvio, is a medication for the treatment of people with atherosclerotic cardiovascular disease (ASCVD), ASCVD risk equivalents and heterozygous familial hypercholesterolemia (HeFH). It is a small interfering RNA that inhibits translation of the protein PCSK9.[2][3][4] It is being developed by The Medicines Company which licensed the rights to inclisiran from Alnylam Pharmaceuticals.[5]
On 15 October 2020, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Leqvio, intended for the treatment for primary hypercholesterolaemia or mixed dyslipidaemia.[6] Inclisiran was approved for use in the European Union in December 2020.[1]
History
In 2019 The Medicines Company announced positive results from pivotal phase III study (all primary and secondary endpoints were met with efficacy consistent with Phase I and II studies). The company anticipates regulatory submissions in the U.S. in the fourth quarter of 2019, and in Europe in the first quarter of 2020.[7] The Medicines Company is being acquired by Novartis.[8]

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
References
- ^ Jump up to:a b “Leqvio EPAR”. European Medicines Agency. 13 October 2020. Retrieved 6 January 2021.
- ^ Fitzgerald K, White S, Borodovsky A, Bettencourt BR, Strahs A, Clausen V, et al. (January 2017). “A Highly Durable RNAi Therapeutic Inhibitor of PCSK9”. The New England Journal of Medicine. 376 (1): 41–51. doi:10.1056/NEJMoa1609243. PMC 5778873. PMID 27959715.
- ^ Spreitzer H (11 September 2017). “Neue Wirkstoffe: Inclisiran”. Österreichische Apotheker-Zeitung (in German) (19/2017).
- ^ “Proposed INN: List 114” (PDF). WHO Drug Information. WHO. 29 (4): 531f. 2015.
- ^ Taylor NP (26 August 2019). “Medicines Company’s PCSK9 drug hits phase 3 efficacy goals”. FierceBiotech.
- ^ “Leqvio: Pending EC decision”. European Medicines Agency (EMA). 16 October 2020. Retrieved 16 October 2020. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “The Medicines Company Announces Positive Topline Results from First Pivotal Phase 3 Trial of Inclisiran”. The Medicines Company. Retrieved 29 August 2019.
- ^ “Novartis acquires medicines company”. Novartis. Retrieved 15 January 2020.
Further reading
- Ray KK, Landmesser U, Leiter LA, et al. (April 2017). “Inclisiran in Patients at High Cardiovascular Risk with Elevated LDL Cholesterol” (PDF). N. Engl. J. Med. 376 (15): 1430–1440. doi:10.1056/NEJMoa1615758. hdl:10044/1/45416. PMID 28306389. S2CID 205101529.
- Ray KK, Wright RS, Kallend D, et al. (March 2020). “Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol”. N. Engl. J. Med. 382 (16): 1507–1519. doi:10.1056/NEJMoa1912387. PMID 32187462.
External links
- “Inclisiran”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03399370 for “Inclisiran for Participants With Atherosclerotic Cardiovascular Disease and Elevated Low-density Lipoprotein Cholesterol (ORION-10)” at ClinicalTrials.gov
- Clinical trial number NCT03400800 for “Inclisiran for Subjects With ACSVD or ACSVD-Risk Equivalents and Elevated Low-density Lipoprotein Cholesterol (ORION-11)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Leqvio |
| Other names | ALN-PCSsc, ALN-60212 |
| Routes of administration | Subcutaneous injection |
| ATC code | C10AX16 (WHO) |
| Legal status | |
| Legal status | EU: Rx-only [1] |
| Identifiers | |
| CAS Number | 1639324-58-5 |
| DrugBank | DB14901 |
| UNII | UOW2C71PG5 |
| KEGG | D11931 |
| Chemical and physical data | |
| Formula | C520H679F21N175O309P43S6 |
| Molar mass | 16248.27 g·mol−1 |
General References
- Kosmas CE, Munoz Estrella A, Sourlas A, Silverio D, Hilario E, Montan PD, Guzman E: Inclisiran: A New Promising Agent in the Management of Hypercholesterolemia. Diseases. 2018 Jul 13;6(3). pii: diseases6030063. doi: 10.3390/diseases6030063. [PubMed:30011788]
- German CA, Shapiro MD: Small Interfering RNA Therapeutic Inclisiran: A New Approach to Targeting PCSK9. BioDrugs. 2020 Feb;34(1):1-9. doi: 10.1007/s40259-019-00399-6. [PubMed:31782112]
- Doggrell SA: Inclisiran, the billion-dollar drug, to lower LDL cholesterol – is it worth it? Expert Opin Pharmacother. 2020 Nov;21(16):1971-1974. doi: 10.1080/14656566.2020.1799978. Epub 2020 Aug 4. [PubMed:32749892]
- Goldstein JL, Brown MS: Regulation of low-density lipoprotein receptors: implications for pathogenesis and therapy of hypercholesterolemia and atherosclerosis. Circulation. 1987 Sep;76(3):504-7. doi: 10.1161/01.cir.76.3.504. [PubMed:3621516]
- Pratt AJ, MacRae IJ: The RNA-induced silencing complex: a versatile gene-silencing machine. J Biol Chem. 2009 Jul 3;284(27):17897-901. doi: 10.1074/jbc.R900012200. Epub 2009 Apr 1. [PubMed:19342379]
- Leiter LA, Teoh H, Kallend D, Wright RS, Landmesser U, Wijngaard PLJ, Kastelein JJP, Ray KK: Inclisiran Lowers LDL-C and PCSK9 Irrespective of Diabetes Status: The ORION-1 Randomized Clinical Trial. Diabetes Care. 2019 Jan;42(1):173-176. doi: 10.2337/dc18-1491. Epub 2018 Nov 28. [PubMed:30487231]
- Cupido AJ, Kastelein JJP: Inclisiran for the treatment of hypercholesterolaemia: implications and unanswered questions from the ORION trials. Cardiovasc Res. 2020 Sep 1;116(11):e136-e139. doi: 10.1093/cvr/cvaa212. [PubMed:32766688]
- Novartis: Novartis receives EU approval for Leqvio (inclisiran), a first-in-class siRNA to lower cholesterol with two doses a year [Link]
- Summary of Product Characteristics: Leqvio (inclisiran), solution for subcutaneous injection [Link]
Summary
- Atherosclerotic cardiovascular disease (ASCVD) remains one of the leading causes of death in Canada. Cholesterol, specifically low-density lipoprotein cholesterol (LDL-C), is a major risk factor for cardiovascular disease (CVD) and is thereby targeted to reduce the likelihood of a cardiovascular event, such as a myocardial infarction (MI) and stroke.
- Inclisiran, first developed by Alnylam Pharmaceuticals, Inc. (Cambridge, Massachusetts, US) then by The Medicines Company (Parsippany, New Jersey, US), is a small interfering ribonucleic acid (siRNA) molecule being investigated for the treatment of hypercholesterolemia.
- ORION-1 was a phase II, double-blind, placebo-controlled, multi-centre, randomized controlled trial of 501 patients. Patients were included in the trial if they had a history of ASCVD or were at high risk of ASCVD. The treatment arms were administered 200 mg, 300 mg, or 500 mg of inclisiran on day 1, or 100 mg, 200 mg, or 300 mg of inclisiran on days 1 and 90. The comparator was either placebo on day 1 or placebo on days 1 and 90. The primary end point was percentage change in LDL-C at day 180 from baseline.
- The ORION-1 study demonstrated that inclisiran, administered at various doses and intervals, compared with placebo, resulted in a statistically significant reduction in LDL-C levels (P < 0.001 for all comparisons versus placebo). The greatest reduction in LDL-C levels was obtained with the 300 mg dose of inclisiran given at days 1 and 90 with a 52.6% (95% confidence interval [CI]: −57.1 to −48.1) reduction at day 180 compared with baseline, and a mean absolute reduction in LDL-C levels of 1.66 (standard deviation 0.54) mmol/L. Results from the ORION-1 trial provided the necessary data to make a decision regarding the dosing regimen to be used in subsequent phase III trials, in particular the ORION-11 phase III trial.
- The ORION-11 study was a phase III international, multi-centre, and double-blind trial which randomized 1,617 participants (87% with established ASCVD) to inclisiran 300 mg (n = 810) or placebo (n = 807). An initial inclisiran dose of 300 mg given subcutaneously was administered at day 1, day 90, and then every six months for two doses, that is at days 270 and 450. The mean baseline LDL-C level was 2.8 mmol/L (inclisiran) and 2.7 mmol/L (placebo); 96% of participants were on high-dose statin therapy. There was a 50% time-averaged reduction in LDL-C levels from day 90 to day 540 (P < 0.00001). Pre-specified exploratory cardiovascular composite end point (cardiac death, cardiac arrest, MI, or stroke) occurred in 7.8% of inclisiran treated patients versus 10.3% of patients on placebo; this lower rate was mainly driven by a reduction in MI and stroke. With respect to adverse effects, 4.69% of patients on inclisiran reported an injection site reaction, compared with 0.5% of patients on placebo. All reactions were transient. There was no evidence of liver, kidney, muscle, or platelet toxicity.
- Inclisiran may be an option in the future as a cholesterol-lowering medication, where it would likely be used in patients who are unable to achieve their LDL-C targets despite maximally tolerated statin therapy or who are intolerant to statin therapy. However, results from the inclisiran cardiovascular outcome trial (ORION-4), are needed to confirm its efficacy in reducing CVD and its long-term safety.
- Inclisiran is not yet approved by any regulatory authority, but its ORION clinical development program identifies the year 2021 as the goal to reach worldwide markets.
///////////Inclisiran, LEQVIO, ALN 60212, ALN PCSsc , NOVARTIS, Leqvio, APPROVALS 2021, FDA 2021

NEW DRUG APPROVALS
ONE TIME
$10.00
CIGLITAZONE
Ciglitazone
U-63287, ADD-3878
- Molecular FormulaC18H23NO3S
- Average mass333.445 Da
- 74772-77-3 [RN]
(±)-5-[4-(1-Methylcyclohexylmethoxy)benzyl]thiazolidine-2,4-dione2,4-Thiazolidinedione, 5-[[4-[(1-methylcyclohexyl)methoxy]phenyl]methyl]-5-[4-(1-methylcyclohexylmethoxy) benzyl]-thiazolidine-2,4-dione
Ciglitazone (INN) is a thiazolidinedione. Developed by Takeda Pharmaceuticals in the early 1980s, it is considered the prototypical compound for the thiazolidinedione class.[1][2][3][4]
Ciglitazone was never used as a medication, but it sparked interest in the effects of thiazolidinediones. Several analogues were later developed, some of which—such as pioglitazone and troglitazone—made it to the market.[2]
Ciglitazone significantly decreases VEGF production by human granulosa cells in an in vitro study, and may potentially be used in ovarian hyperstimulation syndrome.[5] Ciglitazone is a potent and selective PPARγ ligand. It binds to the PPARγ ligand-binding domain with an EC50 of 3.0 μM. Ciglitazone is active in vivo as an anti-hyperglycemic agent in the ob/ob murine model.[6] Inhibits HUVEC differentiation and angiogenesis and also stimulates adipogenesis and decreases osteoblastogenesis in human mesenchymal stem cells.[7]
SYN
T. Sohda, K. Mizuno, E. Imamiya, Y. Sugiyama, T. Fujita, and Y. Kawamatsu, Chem. Pharm. Bull., 30, 3580 (1982).

SYN
Ciglitazone (CAS NO.: ), with other name of , 5-((4-((1-methylcyclohexyl)methoxy)phenyl)methyl)-, (+-)-, could be produced through many synthetic methods.
Following is one of the reaction routes:

The reaction of 1-methylcyclohexylmethanol (II) with 4-chloronitrobenzene (III) by means of NaH in hot DMSO gives 4-(1-methylcyclohexylmethoxylnitrobenzene (III), which is reduced with H2 over Pd/C in methanol yielding 4-(1-methylcyclohexylmethoxylaniline (IV). Diazotation of (IV) with NaNO2 and HCl in water affords a solution of the corresponding diazonium chloride (V), which is condensed with methyl acrylate (VI) by means of Cu2O affording methyl 2-chloro-3-[4-(1-methylcyclohexylmethoxyl)phenyl]propionate (VII). The cyclization of (VII) with thiourea (VIII) by means of sodium acetate in hot 2-methoxyethanol gives 2-imino-5-[4-(1-methylcyclohexylmethoxy)benzyl]thiazolidin-4-one (IX), which is finally hydrolyzed with HCl in refluxing 2-methoxyethanol – water.
Syn
Chem Pharm Bull 1982,30(10),3580
The reaction of 1-methylcyclohexylmethanol (II) with 4-chloronitrobenzene (III) by means of NaH in hot DMSO gives 4-(1-methylcyclohexylmethoxylnitrobenzene (III), which is reduced with H2 over Pd/C in methanol yielding 4-(1-methylcyclohexylmethoxylaniline (IV). Diazotation of (IV) with NaNO2 and HCl in water affords a solution of the corresponding diazonium chloride (V), which is condensed with methyl acrylate (VI) by means of Cu2O affording methyl 2-chloro-3-[4-(1-methylcyclohexylmethoxyl)phenyl]propionate (VII). The cyclization of (VII) with thiourea (VIII) by means of sodium acetate in hot 2-methoxyethanol gives 2-imino-5-[4-(1-methylcyclohexylmethoxy)benzyl]thiazolidin-4-one (IX), which is finally hydrolyzed with HCl in refluxing 2-methoxyethanol – water.
By cyclization of (VIII) with methyl 2-(methanesulfonyloxy)-3-[4-(1-methylcyclohexylmethoxy)phenyl]propionate (X) by means of sodium acetate in hot 2-methoxyethanol, followed by hydrolysis with HCl in ethanol water.

paper
Vijay Kumar Sharma , Anup Barde & Sunita Rattan (2020): A short review on synthetic strategies toward glitazone drugs, Synthetic Communications, DOI: 10.1080/00397911.2020.1821223
Experimental process for synthesis of ciglitazone is fairly robust, albeit pyrophorophic NaH as base is utilized for synthesis of 15.


Scheme 4. Reagents and conditions for the preparation of (R)-ciglitazone 24 (a) (S)-1-phenylethan-1- amine 19 (0.9 mol. equiv.), EtOH, RT, 4 h; (b) 1 N HCl (2 vol.), diethyl ether, RT, 10 min; (c) CH2N2 in diethyl ether (ca. 3% w/w), diethyl ether, 0 C-RT, 30 min; (d) KSCN (1.5 mol. equiv.), DMSO, 90 C, 2 h; (e) 2 N HCl (10 vol.), and EtOH, reflux 4 h.
Chiral synthesis Racemic-ciglitazone 17 was resolved with optically active a-methylbenzylamine (PEA) 19 through asymmetric transformation of optical lability at the C-5 position of TZD ring. 2-chloro-3-(4-((1-methylcyclohexyl)methoxy)phenyl)propanoic acid 18 was resolved using (S)-()-1-Phenylethylamine 19 to isolate (S)-2-chloro-3-(4-((1- methylcyclohexyl) methoxy)phenyl)propanoicacid 21. Esterification followed by substitution with KSCN provided methyl (R)-3-(4-((1-methylcyclohexyl)methoxy)phenyl)-2- thiocyanatopropan-oate 23 which was then hydrolyzed to isolate (R)-ciglitazone 24. Similarly, S-isomer was also isolated with (R)-(þ)-1-phenylethylamine (Scheme 4).
[30]Sohda, T.; Mizuno, K.; Kawamatsu, Y. Studies on Antidiabetic Agents. VI. Asymmetric Transformation of (þ/-)-5-[4-(1-Methylcyclohexylmethoxy)Benzyl]-2,4- Thiazolidinedione (Ciglitazone) with Optically Active 1-Phenylethylamines. Chem. Pharm. Bull. 1984, 32, 4460–4465. DOI: 10.1248/cpb.32.4460.
References
- ^ Pershadsingh HA, Szollosi J, Benson S, Hyun WC, Feuerstein BG, Kurtz TW (June 1993). “Effects of ciglitazone on blood pressure and intracellular calcium metabolism”. Hypertension. 21 (6 Pt 2): 1020–3. doi:10.1161/01.hyp.21.6.1020. PMID 8505086.
- ^ Jump up to:a b Hulin B, McCarthy PA, Gibbs EM (1996). “The glitazone family of antidiabetic agents”. Current Pharmaceutical Design. 2: 85–102.
- ^ Imoto H, Imamiya E, Momose Y, Sugiyama Y, Kimura H, Sohda T (October 2002). “Studies on non-thiazolidinedione antidiabetic agents. 1. Discovery of novel oxyiminoacetic acid derivatives”. Chem. Pharm. Bull. 50 (10): 1349–57. doi:10.1248/cpb.50.1349. PMID 12372861.
- ^ Sohda T, Kawamatsu Y, Fujita T, Meguro K, Ikeda H (November 2002). “[Discovery and development of a new insulin sensitizing agent, pioglitazone]”. Yakugaku Zasshi (in Japanese). 122 (11): 909–18. doi:10.1248/yakushi.122.909. PMID 12440149.
- ^ Shah DK, Menon KM, Cabrera LM, Vahratian A, Kavoussi SK, Lebovic DI (April 2010). “Thiazolidinediones decrease vascular endothelial growth factor (VEGF) production by human luteinized granulosa cells in vitro”. Fertil. Steril. 93 (6): 2042–7. doi:10.1016/j.fertnstert.2009.02.059. PMC 2847675. PMID 19342033.
- ^ Willson, T.M.; Cobb, J.E.; Cowan, D.J.; et al. (1996). “The structure-activity relationship between peroxisome proliferator-activated receptor γ agonism and the antihyperglycemic activity of thiazolidinediones”. J Med Chem. 39 (3): 665–668. doi:10.1021/jm950395a. PMID 8576907.
- ^ Xin, X.; et al. (1999). “Peroxisome proliferator-activated receptor gamma ligands are potent inhibitors of angiogenesis in vitro and in vivo;”. J. Biol. Chem. 274 (13): 9116–21. doi:10.1074/jbc.274.13.9116. PMID 10085162.
| Clinical data | |
|---|---|
| ATC code | none |
| Identifiers | |
| IUPAC name[show] | |
| CAS Number | 74772-77-3 |
| PubChem CID | 2750 |
| IUPHAR/BPS | 2711 |
| DrugBank | DB09201 |
| ChemSpider | 2648 |
| UNII | U8QXS1WU8G |
| KEGG | D03493 |
| ChEMBL | ChEMBL7002 |
| CompTox Dashboard (EPA) | DTXSID0040757 |
| ECHA InfoCard | 100.220.474 |
| Chemical and physical data | |
| Formula | C18H23NO3S |
| Molar mass | 333.45 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES[hide]O=C1NC(=O)SC1Cc3ccc(OCC2(C)CCCCC2)cc3 | |
| InChI[hide]InChI=1S/C18H23NO3S/c1-18(9-3-2-4-10-18)12-22-14-7-5-13(6-8-14)11-15-16(20)19-17(21)23-15/h5-8,15H,2-4,9-12H2,1H3,(H,19,20,21) Key:YZFWTZACSRHJQD-UHFFFAOYSA-N |
/////////ciglitazone, U 63287, ADD 3878, DIABETES
BMS 262084
BMS-262084
CAS 253174-92-4
- Molecular FormulaC18H31N7O5
- Average mass425.483 Da
NII-I0IR71971G
I0IR71971G
(2S,3R)-1-[4-(tert-butylcarbamoyl)piperazine-1-carbonyl]-3-[3-(diaminomethylideneamino)propyl]-4-oxoazetidine-2-carboxylic acid(2S,3R)-1-{[4-(tert-butylcarbamoyl)piperazin-1-yl]carbonyl}-3-{3-[(diaminomethylidene)amino]propyl}-4-oxoazetidine-2-carboxylic acid
(2S,3R)-3-{3-[(Diaminomethylene)amino]propyl}-1-({4-[(2-methyl-2-propanyl)carbamoyl]-1-piperazinyl}carbonyl)-4-oxo-2-azetidinecarboxylic acid253174-92-4[RN]2-Azetidinecarboxylic acid, 3-[3-[(diaminomethylene)amino]propyl]-1-[[4-[[(1,1-dimethylethyl)amino]carbonyl]-1-piperazinyl]carbonyl]-4-oxo-, (2S,3R)-
Factor XIa inhibitors (thrombosis), BMS; Factor XIa inhibitors (thrombosis), Bristol-Myers Squibb; BMS-654457; Factor XIa inhibitors (cardiovascular diseases), BMS; BMS-724296
Novel crystalline forms of BMS-262084 as Factor XIa antagonist useful for treating cardiovascular diseases.
PHASE 2

PAPER
Bioorganic & Medicinal Chemistry Letters (2002), 12(21), 3229-3233.
https://www.sciencedirect.com/science/article/pii/S0960894X02006881
Abstract
A series of N1-activated C4-carboxy azetidinones was prepared and tested as inhibitors of human tryptase. The key stereochemical and functional features required for potency, serine protease specificity and aqueous stability were determined. From these studies compound 2, BMS-262084, was identified as a potent and selective tryptase inhibitor which, when dosed intratracheally in ovalbumin-sensitized guinea pigs, reduced allergen-induced bronchoconstriction and inflammatory cell infiltration into the lung.
BMS-262084 was identified as a potent and selective tryptase inhibitor that, when dosed intratracheally in ovalbumin-sensitized guinea pigs, reduced allergen-induced bronchoconstriction and inflammatory cell infiltration into the lung.

PAPER
https://pubs.acs.org/doi/10.1021/jo010757o
Journal of Organic Chemistry (2002), 67(11), 3595-3600.

A highly stereoselective synthesis of the novel tryptase inhibitor BMS-262084 was developed. Key to this synthesis was the discovery and development of a highly diastereoselective demethoxycarbonylation of diester 12 to form the trans-azetidinone 13. BMS-262084 was prepared in 10 steps from d-ornithine in 30% overall yield.
1 as a white powder (3.18 g, 99% yield). Mp: 213-215 °C dec. [α]25D = −65.9 (c 0.99, MeOH). 1H NMR (CD3OD): δ 4.17 (d, J = 3.29 Hz, 1H), 3.61−3.11 (m, 11H), 1.94−1.75 (m, 4H), 1.32 (s, 9H). 13C NMR (CD3OD): δ 176.6, 168.7, 159.4, 158.7, 152.3, 58.7, 53.2, 51.8, 46.5, 45.0, 41.8, 29.6, 27.4, 26.3. HRMS: calcd for C18H32N7O5(M+ + H) 426.2465, found 426.2470. IR (KBr): 3385, 3184, 1775, 1657, 1535, 1395, 1259, 1207, 996, 763 cm–1. Anal. Calcd for C18H31N7O5: C, 50.81, H, 7.34, N, 23.04. Found: C, 50.65, H, 7.42, N, 22.72. Chiral HPLC: ee 99.6%; Chiralpak OD column, 250 × 4.6 mm, 10 μm; mobile phase hexane/EtOH (85:15, v/v); isocratic at ambient temperature, 1.0 mL/min, 220 nm; concentration 0.25 mg/mL, 10 μL injection; RT = 18.6 min (enantiomer, RT = 15.7 min).


PATENT
WO2018133793
claiming macrocyclic compounds.
PATENT
WO-2020259366
Novel crystalline and solid forms of BMS-262084 (designates as monohydrate or 1.5 hydrate), processes for their preparation and compositions comprising them are claimed. BMS-262084 is disclosed to be Factor XIa antagonist, useful for treating cardiovascular diseases.MS-262084 (CAS number: 253174-92-4), the chemical name is (2S,3R)-1-[4-(tert-butylcarbamoyl)piperazine-1-carbonoyl]-3-[3- (Diaminomethylamino)propyl]-4-cyclopropanamide-2-carboxylic acid, also called compound (1) in the present invention, is developed by BMS (Bristol-Myers-Squibb) to treat cardiovascular diseases The drug, as an oral coagulation factor XIa inhibitor for thrombus, has the advantage of significantly reducing the risk of bleeding, and its structure is shown in formula (1):
Patent application WO 9967215A1 discloses the BMS-262084 compound, but the specific molecular formula of the solid substance obtained by the disclosed preparation process is C 18 H 31 N 7 O 5 ·1.56H 2 O, which is similar to the crystal of BMS-262084 described in this application. Type and amorphous water have different molecular weights.
“A stereoselective synthesis of BMS-262084 an azetidinone-based tryptase inhibitor” (Source: Journal of Organic Chemistry, 2002,67(11):3595-3600; Journal of Organic Chemistry,2002,67(11):3595-3600) It is mentioned that the preparation method of BMS-262084 is that hydrogenolysis under neutral conditions eliminates the benzene and Cbz protection groups, and obtains BMS-262084 (melting point 213-215℃). The inventors conducted experiments based on part of the contents disclosed in the document, and the test results obtained crystal form A and crystal form B. The X-ray powder diffraction patterns are shown in Figure 1 and Figure 2 respectively.Example 1
“A stereoselective synthesis of BMS-262084 an azetidinone-based tryptase inhibitor” (Source: Journal of Organic Chemistry, 2002,67(11):3595-3600; Journal of Organic Chemistry,2002,67(11):3595-3600) Only ethanol solvents are mentioned in the literature. Since no specific crystal refining process was provided, only part of the experiment was performed using ethanol solvent.
1) Ethanol solvent volatilization at room temperature: 50mg of BMS-262084 (amorphous) was added to 1.0 mL of ethanol solvent and completely dissolved at room temperature (about 25°C). After volatilizing at room temperature for two days, the solid product was obtained and its crystal form was tested. It is crystal form A, as shown in Figure 1. It is considered that it contains a small amount of amorphous form; but it is unstable and will undergo crystal transformation at room temperature. After standing for one day, the XRPD was tested, and it was found that it was converted to a mixture containing crystal form A, other crystal forms and amorphous forms.
2) Ethanol solvent high-temperature volatilization: 50mg BMS-262084 is added to 1.0mL ethanol solvent, completely dissolved at high temperature (about 60℃), and high-temperature volatilization is carried out in the open to obtain a solid product. The crystal form of the solid product is detected, and the crystal form is B (contains a lot of amorphous), see Figure 2.
SYN1
WO 9967215
The condensation of N-(tert-butyldimethylsilyl)-4-oxoazetidine-2(S)-carboxylic acid (I) with 1-chloro-3-iodopropane (II) by means of BuLi and triisopropylamine (TIA) in THF, followed by treatment with HCl, gives the 3(R)-(3-chloropropyl) derivative (III), which is treated with tetrabutylammonium azide and tetrabutylammonium iodide in DMF to yield the 3-azidopropyl derivative (IV). The reduction of (IV) with H2 over Pd/C in DMF affords the 3-aminopropyl compound (V), which is treated with 1-[N,N’-bis(benzyloxycarbonyl)-1H-pyrazole] (VI) in the same solvent to provide the protected 3-guanidinopropyl compound (VII). The esterification of (VII) with NaHCO3, tetrabutylammonium iodide and Bn-Br in DMF gives the benzyl ester (VIII), which is condensed with N-tert-butylpiperazine-1-carboxamide (IX) and phosgene by means of TEA in toluene to yield the protected precursor (X). Finally, this compound is debenzylated by hydrogenation with H2 over Pd/C in dioxane to give the target azetidine-carboxylic acid.
SYN 2
Ethyl nipecotate (I) was protected as the N-Boc derivative (II) and subsequently reduced to alcohol (III) by means of LiAlH4. Conversion of alcohol (III) into iodide (IV) was achieved by treatment with iodine and triphenylphosphine. The dianion of the chiral azetidinecarboxylic acid (V) was alkylated with iodide (IV) to furnish adduct (VI) as a diastereomeric mixture that was desilylated to (VII) using tetrabutylammonium fluoride. Benzyl ester (VIII) was then obtained by reaction of carboxylic acid (VII) with benzyl bromide and NaHCO3.
SYN 3
Coupling of 6-phenylhexanoic acid (X) with N-Boc-piperazine (IX) to give (XI), followed by acid deprotection of the Boc group of (XI), provided (6-phenylhexanoyl)piperazine (XII). This was converted to the carbamoyl chloride (XIII) upon treatment with phosgene. The condensation of carbamoyl chloride (XIII) with azetidinone (VIII) gave rise to the urea derivative (XIV). After acid cleavage of the Boc protecting group of (XIV), the resulting piperidine (XV) was condensed with N,N’-dicarbobenzoxy-S-methylisothiourea (XVI) in the presence of HgCl2, yielding the protected guanidine (XVII). This was finally deprotected by catalytic hydrogenolysis over Pd/C.
////////////////////////BMS-262084, BMS 262084, BMS 724296, Factor XIa inhibitors, thrombosis, Bristol-Myers Squibb, BMS 654457, PHASE 2
CC(C)(C)NC(=O)N1CCN(CC1)C(=O)N2C(C(C2=O)CCCN=C(N)N)C(=O)O
IDEBENONE
IDEBENONE
2-(10-Hydroxydecyl)-5,6-dimethoxy-3-methyl-1,4-benzoquinone
- Molecular FormulaC19H30O5
- Average mass338.439 Da
- 58186-27-9
- Idebenona, Idebenonum, CV 2619
IdesolKS-5193NemocebralSNT-MC17идебенонإيديبينون艾地苯醌
Puldysa (idebenone), for the treatment of Duchenne muscular dystrophyTitle: Idebenone
CAS Registry Number: 58186-27-9
CAS Name: 2-(10-Hydroxydecyl)-5,6-dimethoxy-3-methyl-2,5-cyclohexadiene-1,4-dione
Additional Names: 6-(10-hydroxydecyl)-2,3-dimethoxy-5-methyl-1,4-benzoquinone; 2,3-dimethoxy-5-methyl-6-(10¢-hydroxydecyl)-1,4-benzoquinone; 6-(10-hydroxydecyl)ubiquinone
Manufacturers’ Codes: CV-2619
Trademarks: Avan (Takeda); Daruma (Takeda); Lucebanol (Hormona); Mnesis (Takeda)
Molecular Formula: C19H30O5Molecular Weight: 338.44
Percent Composition: C 67.43%, H 8.93%, O 23.64%
Literature References: Ubiquinone derivative with protective effects against cerebral ischemia. Prepn: H. Morimoto et al.,DE2519730; eidem,US4271083 (1975, 1981 both to Takeda); K. Okamoto et al.,Chem. Pharm. Bull.30, 2797 (1982); C.-A. Yu, L. Yu, Biochemistry21, 4096 (1982). Effect on ischemia-induced amnesia in rats: N. Yamazaki et al.,Jpn. J. Pharmacol.36, 349 (1984). Metabolism in animals: T. Kobayashi et al.,J. Pharmacobio-Dyn.8, 448 (1985). Disposition: H. Torii et al.,ibid. 457. Pharmacokinetics and tolerance in humans: M. F. Barkworth et al.,Arzneim.-Forsch.35, 1704 (1985). Series of articles on pharmacology and clinical studies: Arch. Gerontol. Geriatr.8, 193-366 (1989). Review of chemistry, toxicology and pharmacology: I. Zs-Nagy, Arch. Gerontol. Geriatr.11, 177-186 (1990).Properties: Orange needles from ligroin, mp 46-50° (Morimoto); also reported as crystals from hexane + ethyl acetate, mp 52-53° (Okamoto). Sol in organic solvents. Practically insol in water.Melting point: mp 46-50° (Morimoto); mp 52-53° (Okamoto)Therap-Cat: Nootropic.Keywords: Nootropic.
Idebenone is a member of the class of 1,4-benzoquinones which is substituted by methoxy groups at positions 2 and 3, by a methyl group at positions 5, and by a 10-hydroxydecyl group at positions 6. Initially developed for the treatment of Alzheimer’s disease, benefits were modest; it was subsequently found to be of benefit for the symptomatic treatment of Friedreich’s ataxia. It has a role as an antioxidant. It is a primary alcohol and a member of 1,4-benzoquinones.
Idebenone (pronounced eye-deb-eh-known, trade names Catena, Raxone, Sovrima, among others) is a drug that was initially developed by Takeda Pharmaceutical Company for the treatment of Alzheimer’s disease and other cognitive defects.[1] This has been met with limited success. The Swiss company Santhera Pharmaceuticals has started to investigate it for the treatment of neuromuscular diseases. In 2010, early clinical trials for the treatment of Friedreich’s ataxia[2] and Duchenne muscular dystrophy[3] have been completed. As of December 2013 the drug is not approved for these indications in North America or Europe. It is approved by the European Medicines Agency (EMA) for use in Leber’s hereditary optic neuropathy (LHON) and was designated an orphan drug in 2007.[4]
Chemically, idebenone is an organic compound of the quinone family. It is also promoted commercially as a synthetic analog of coenzyme Q10 (CoQ10).
Uses
Indications that are or were approved in some territories
Nootropic effects and Alzheimer’s disease
Idebenone improved learning and memory in experiments with mice.[5] In humans, evaluation of Surrogate endpoints like electroretinography, auditory evoked potentials and visual analogue scales also suggested positive nootropic effects,[6] but larger studies with hard endpoints are missing.
Research on idebenone as a potential therapy of Alzheimer’s disease have been inconsistent, but there may be a trend for a slight benefit.[7][8] In May 1998, the approval for this indication was cancelled in Japan due to the lack of proven effects. In some European countries, the drug is available for the treatment of individual patients in special cases.[1]
Friedreich’s ataxia (Sovrima)
Preliminary testing has been done in humans and found idebenone to be a safe treatment for Friedreich’s ataxia (FA), exhibiting a positive effect on cardiac hypertrophy and neurological function.[9] The latter was only significantly improved in young patients.[10] In a different experiment, a one-year test on eight patients, idebenone reduced the rate of deterioration of cardiac function, but without halting the progression of ataxia.[11]
The drug was approved for FA in Canada in 2008 under conditions including proof of efficacy in further clinical trials.[12] However, on February 27, 2013, Health Canada announced that idebenone would be voluntarily recalled as of April 30, 2013 by its Canadian manufacturer, Santhera Pharmaceuticals, due to the failure of the drug to show efficacy in the further clinical trials that were conducted.[13] In 2008, the European Medicines Agency (EMA) refused a marketing authorisation for this indication.[1] As of 2013 the drug was not approved for FA in Europe[14] nor in the US, where there is no approved treatment.[15]
Leber’s hereditary optic neuropathy (Raxone)
Leber’s hereditary optic neuropathy (LHON) is a mitochondrially inherited (mother to all offspring) degeneration of retinal ganglion cells (RGCs) and their axons that leads to an acute or subacute loss of central vision; this affects predominantly young adult males. Santhera completed a Phase III clinical trial in this indication in Europe with positive results,[16] and submitted an application to market the drug to European regulators in July 2011.[17] It is approved by EMA for this indication and was designated an orphan drug in 2007.[4]
Indications being explored
Duchenne muscular dystrophy (Catena)
After experiments in mice[18] and preliminary studies in humans, idebenone has entered Phase II clinical trials in 2005[3] and Phase III trials in 2009.[19]
Other neuromuscular diseases
Phase I and II clinical trials for the treatment of MELAS (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes)[20] and primary progressive multiple sclerosis[21] are ongoing as of December 2013.
Life style
Idebenone is claimed to have properties similar to CoQ10 in its antioxidant properties, and has therefore been used in anti-aging on the basis of free-radical theory. Clinical evidence for this use is missing. It has been used in topical applications to treat wrinkles.[22]
Pharmacology
In cellular and tissue models, idebenone acts as a transporter in the electron transport chain of mitochondria and thus increases the production of adenosine triphosphate (ATP) which is the main energy source for cells, and also inhibits lipoperoxide formation. Positive effects on the energy household of mitochondria has also been observed in animal models.[1][23] Clinical relevance of these findings has not been established.
Pharmacokinetics
Idebenone is well absorbed from the gut but undergoes excessive first pass metabolism in the liver, so that less than 1% reach the circulation. This rate can be improved with special formulations (suspensions) of idebenone and by administering it together with fat food; but even taking these measures bioavailability still seems to be considerably less than 14% in humans. More than 99% of the circulating drug are bound to plasma proteins. Idebenone metabolites include glucuronides and sulfates, which are mainly (~80%) excreted via the urine.[1]
SYN
https://www.sciencedirect.com/science/article/abs/pii/S0040402014014306


SYN
The palladium-catalyzed olefination of a sp2 or benzylic carbon attached to a (pseudo)halogen is known as the Heck reaction.2,63 It is a powerful tool, mainly used for the synthesis of vinylarenes, and it has also been employed for the construction of conjugated double bonds. The widespread application of this reaction can be illustrated by numerous examples in both academia small-scale64 and industrial syntheses.5 As an example, in 2011, a idebenone (124) total synthesis based on a Heck reaction was described (Scheme 35).65 This compound, initially designed for the treatment of Alzheimer’s and Parkinson’s diseases, presented a plethora of other interesting activities, such as free radical scavenging and action against some muscular illnesses. The key step in the synthesis was the coupling of 2-bromo-3,4,5-trimethoxy-1-methylbenzene (125) with dec-9-en-1-ol affording products 126. Under non-optimized conditions (Pd(OAc)2, PPh3, Et3N, 120 ºC), a mixture composed of 60% linear olefins 126 and 15% of the undesired branched product 127 was obtained after three days of reaction. Therefore, the conditions were optimized, allowing the preparation of 126 in 67% yield with no detection of 127 after only 30 min of reaction employing DMF, Pd(PPh3)4, iPr2NEt under microwave heating. To conclude the synthesis, the Heck adducts were submitted to hydroxyl protection/deprotection, hydrogenation, and ring oxidation. After these reactions, idebenone was obtained with 20% overall yield over 6 steps.

Scheme 35 Synthesis of idebenone (124) based on Heck reaction of 2-bromo-3,4,5-trimethoxy-1-methylbenzene with dec-9-en-1-ol under microwave irradiation.
Syn
- Duveau, Damien Y.; Bioorganic & Medicinal Chemistry 2010, V18(17), P6429-6441
- Okada, Taiiti; EP 289223 A1 1988
- Watanabe, Masazumi; EP 58057 A1 1982
- Okamoto, Kayoko; Chemical & Pharmaceutical Bulletin 1982, V30(8), P2797-819
- “Drugs – Synonyms and Properties” data were obtained from Ashgate Publishing Co. (US)
Paper
Tsoukala, Anna; Organic Process Research & Development 2011, V15(3), P673-680
https://pubs.acs.org/doi/10.1021/op200051v
An environmentally benign, convenient, high yielding, and cost-effective synthesis leading to idebenone is disclosed. The synthesis includes a bromination process for the preparation of 2-bromo-3,4,5-trimethoxy-1-methylbenzene, a protocol for the Heck cross-coupling reaction using either thermal or microwave heating, olefin reduction by palladium catalyzed hydrogenation, and a green oxidation protocol with hydrogen peroxide as oxidant to achieve the benzoquinone framework. The total synthesis is composed of six steps that provide an overall yield of 20% that corresponds to a step yield of 76%.





PAPER
Bioorganic & Medicinal Chemistry 2010, V18(17), P6429-6441
https://www.sciencedirect.com/science/article/abs/pii/S0968089610006322

Analogues of mitoQ and idebenone were synthesized to define the structural elements that support oxygen consumption in the mitochondrial respiratory chain. Eight analogues were prepared and fully characterized, then evaluated for their ability to support oxygen consumption in the mitochondrial respiratory chain. While oxygen consumption was strongly inhibited by mitoQ analogues 2–4 in a chain length-dependent manner, modification of idebenone by replacement of the quinone methoxy groups by methyl groups (analogues 6–8) reduced, but did not eliminate, oxygen consumption. Idebenone analogues 6–8 also displayed significant cytoprotective properties toward cultured mammalian cells in which glutathione had been depleted by treatment with diethyl maleate.
Idebenone (5)18 To a stirred solution containing 200 mg (0.467 mmol) of 2,3- dimethoxy-6-methyl-5-benzyloxydecyl-p-benzoquinone (38) in 5 mL of anhydrous methanol at 23 C was added 15 mg of 10 % Pd/C in one portion. The reaction mixture was stirred at 23 C under an atmosphere of hydrogen for 24 h. Air was then bubbled through the reaction mixture at 23 C for 24 h. The suspension was filtered through Celite and the filtrate was concentrated under diminished pressure to afford idebenone (5) as an orange solid: yield 130 mg (82%); mp: 46–47 C; 1 H NMR (400 MHz, CDCl3) d 1.34 (m, 14H), 1.60 (quint, 2H, J = 7.6 Hz), 2.04 (s, 3H), 2.44 (t, 2H, J = 8.0 Hz), 3.63 (t, 2H, J = 6.8 Hz), and 3.99 (s, 6H); 13C NMR (100 MHz, CDCl3) d 11.9, 25.7, 26.4, 28.7, 29.3, 29.3, 29.4, 29.5, 29.8, 32.7
References
- ^ Jump up to:a b c d e “CHMP Assessment Report for Sovrima” (PDF). European Medicines Agency. 20 November 2008: 6, 9–11, 67f.
- ^ Clinical trial number NCT00229632 for “Idebenone to Treat Friedreich’s Ataxia” at ClinicalTrials.gov
- ^ Jump up to:a b Clinical trial number NCT00654784 for “Efficacy and Tolerability of Idebenone in Boys With Cardiac Dysfunction Associated With Duchenne Muscular Dystrophy (DELPHI)” at ClinicalTrials.gov
- ^ Jump up to:a b “Raxone”. http://www.ema.europa.eu. Retrieved 12 July 2019.
- ^ Liu, XJ; Wu, WT (1999). “Effects of ligustrazine, tanshinone II A, ubiquinone, and idebenone on mouse water maze performance”. Zhongguo Yao Li Xue Bao. 20 (11): 987–90. PMID 11270979.
- ^ Schaffler, K; Hadler, D; Stark, M (1998). “Dose-effect relationship of idebenone in an experimental cerebral deficit model. Pilot study in healthy young volunteers with piracetam as reference drug”. Arzneimittel-Forschung. 48 (7): 720–6. PMID 9706371.
- ^ Gutzmann, H; Kühl, KP; Hadler, D; Rapp, MA (2002). “Safety and efficacy of idebenone versus tacrine in patients with Alzheimer’s disease: results of a randomized, double-blind, parallel-group multicenter study”. Pharmacopsychiatry. 35 (1): 12–8. doi:10.1055/s-2002-19833. PMID 11819153.
- ^ Parnetti, L; Senin, U; Mecocci, P (1997). “Cognitive enhancement therapy for Alzheimer’s disease. The way forward”. Drugs. 53 (5): 752–68. doi:10.2165/00003495-199753050-00003. PMID 9129864. S2CID 46987059.
- ^ Di Prospero NA, Baker A, Jeffries N, Fischbeck KH (October 2007). “Neurological effects of high-dose idebenone in patients with Friedreich’s ataxia: a randomised, placebo-controlled trial”. Lancet Neurol. 6 (10): 878–86. doi:10.1016/S1474-4422(07)70220-X. PMID 17826341. S2CID 24749816.
- ^ Tonon C, Lodi R (September 2008). “Idebenone in Friedreich’s ataxia”. Expert Opin Pharmacother. 9 (13): 2327–37. doi:10.1517/14656566.9.13.2327. PMID 18710357. S2CID 73285881.
- ^ Buyse G, Mertens L, Di Salvo G, et al. (May 2003). “Idebenone treatment in Friedreich’s ataxia: neurological, cardiac, and biochemical monitoring”. Neurology. 60 (10): 1679–81. doi:10.1212/01.wnl.0000068549.52812.0f. PMID 12771265. S2CID 36556782.
- ^ “Heath Canada Fact Sheet – Catena”. Archived from the original on 19 June 2014.
- ^ Voluntary Withdrawal of Catena from the Canadian Market
- ^ Margaret Wahl for Quest Magazine, MAY 28, 2010. FA Research: Idebenone Strikes Out Again
- ^ NINDS Fact Sheet
- ^ Klopstock, T; et al. (2011). “A randomized placebo-controlled trial of idebenone in Leber’s hereditary optic neuropathy”. Brain. 134 (9): 2677–86. doi:10.1093/brain/awr170. PMC 3170530. PMID 21788663.
- ^ Staff (26 July 2011). “Santhera publishes pivotal trial results of idebenone and goes for EU approval”. European Biotechnology News. Archived from the original on 2013-02-17.
- ^ Buyse, GM; Van Der Mieren, G; Erb, M; D’hooge, J; Herijgers, P; Verbeken, E; Jara, A; Van Den Bergh, A; et al. (2009). “Long-term blinded placebo-controlled study of SNT-MC17/idebenone in the dystrophin deficient mdx mouse: cardiac protection and improved exercise performance”. European Heart Journal. 30 (1): 116–24. doi:10.1093/eurheartj/ehn406. PMC 2639086. PMID 18784063.
- ^ Clinical trial number NCT01027884 for “Phase III Study of Idebenone in Duchenne Muscular Dystrophy (DMD) (DELOS)” at ClinicalTrials.gov
- ^ Clinical trial number NCT00887562 for “Study of Idebenone in the Treatment of Mitochondrial Encephalopathy Lactic Acidosis & Stroke-like Episodes (MELAS)” at ClinicalTrials.gov
- ^ Clinical trial number NCT00950248 for “Double Blind Placebo-Controlled Phase I/II Clinical Trial of Idebenone in Patients With Primary Progressive Multiple Sclerosis (IPPoMS)” at ClinicalTrials.gov
- ^ McDaniel D, Neudecker B, Dinardo J, Lewis J, Maibach H (September 2005). “Clinical efficacy assessment in photodamaged skin of 0.5% and 1.0% idebenone”. J Cosmet Dermatol. 4 (3): 167–73. doi:10.1111/j.1473-2165.2005.00305.x. PMID 17129261. S2CID 2394666.
- ^ Suno M, Nagaoka A (May 1988). “[Effect of idebenone and various nootropic drugs on lipid peroxidation in rat brain homogenate in the presence of succinate]”. Nippon Yakurigaku Zasshi (in Japanese). 91 (5): 295–9. doi:10.1254/fpj.91.295. PMID 3410376.
| Clinical data | |
|---|---|
| Trade names | Catena, Raxone, Sovrima |
| AHFS/Drugs.com | International Drug Names |
| License data | EU EMA: by INN |
| ATC code | N06BX13 (WHO) |
| Legal status | |
| Legal status | In general: ℞ (Prescription only) |
| Pharmacokinetic data | |
| Bioavailability | <1% (high first pass effect) |
| Protein binding | >99% |
| Elimination half-life | 18 hours |
| Excretion | Urine (80%) and feces |
| Identifiers | |
| IUPAC name[show] | |
| CAS Number | 58186-27-9 |
| PubChem CID | 3686 |
| ChemSpider | 3558 |
| UNII | HB6PN45W4J |
| KEGG | D01750 |
| ChEMBL | ChEMBL252556 |
| CompTox Dashboard (EPA) | DTXSID0040678 |
| Chemical and physical data | |
| Formula | C19H30O5 |
| Molar mass | 338.444 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES[hide]O=C1/C(=C(\C(=O)C(\OC)=C1\OC)C)CCCCCCCCCCO | |
| InChI[hide]InChI=1S/C19H30O5/c1-14-15(12-10-8-6-4-5-7-9-11-13-20)17(22)19(24-3)18(23-2)16(14)21/h20H,4-13H2,1-3H3 Key:JGPMMRGNQUBGND-UHFFFAOYSA-N |
////////////IDEBENONE, Puldysa, Duchenne muscular dystrophy, Idesol, KS 5193, Nemocebral, SNT MC17, идебенон, إيديبينون , 艾地苯醌 , CV 2619
CC1=C(C(=O)C(=C(C1=O)OC)OC)CCCCCCCCCCO

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}