
Home » osteoarthritis
Category Archives: osteoarthritis
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Rutoside, Rutin

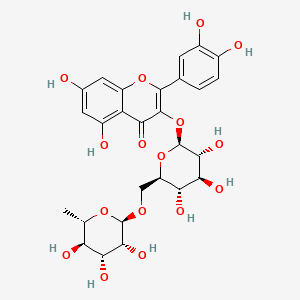

Rutoside
RUTIN
- Molecular FormulaC27H30O16
- Average mass610.518
- рутозид [Russian] [INN]ルチン [Japanese]روتوسيد [Arabic] [INN]芦丁 [Chinese] [INN]
CAS 153-18-4
- C.I. 75730
- NSC-9220
2-(3,4-dihydroxyphenyl)-5,7-dihydroxy-3-[(2S,3R,4S,5S,6R)-3,4,5-trihydroxy-6-[[(2R,3R,4R,5R,6S)-3,4,5-trihydroxy-6-methyloxan-2-yl]oxymethyl]oxan-2-yl]oxychromen-4-one


Rutin trihydrate | CAS 250249-75-3 RutinCAS Registry Number: 153-18-4
CAS Name: 3-[[6-O-(6-Deoxy-a-L-mannopyranosyl)-b-D-glucopyranosyl]oxy]-2-(3,4-dihydroxyphenyl)-5,7-dihydroxy-4H-1-benzopyran-4-one
Additional Names: rutoside; quercetin-3-rutinoside; 3,3¢,4¢,5,7-pentahydroxyflavone-3-rutinoside; melin; phytomelin; eldrin; ilixathin; sophorin; globularicitrin; paliuroside; osyritrin; osyritin; myrticolorin; violaquercitrin
Trademarks: Birutan (Merck KGaA)
Molecular Formula: C27H30O16Molecular Weight: 610.52Percent Composition: C 53.12%, H 4.95%, O 41.93%
Literature References: Identity with ilixanthin: Schindler, Herb, Arch. Pharm.288, 372 (1955). Found in many plants, especially the buckwheat plant (Fagopyrum esculentum Moench., Polygonaceae) which contains about 3% (dry basis): Couch et al.,Science103, 197 (1946). From tobacco (Nicotiana tabacum L., Solanaceae) Couch, Krewson, U.S. Dept. Agr., Eastern Regional Res. Lab.AIC-52 (1944).
In forsythia [Forsythia suspensa (Thunb.) Vahl. var. fortunei (Lindl.) Rehd., Oleaceae], in hydrangea (Hydrangea paniculata Sieb., Saxifragaceae), in pansies (Viola sp., Violaceae).
General extraction procedure: BeilsteinXXXI, 376. From leaves of Eucalyptus macroryncha F. v. Muell., Myrtaceae: Attree, Perkin, J. Chem. Soc.1927, 234.
Industrial production from Eucalyptus spp.: Humphreys, Econ. Bot.18, 195 (1964).
Structure: Zemplén, Gerecs, Ber.68B, 1318 (1935).
Synthesis: Shakhova et al.,Zh. Obshch. Khim.32, 390 (1962), C.A.58, 1426e (1963). Rutin is hydrolyzed by rhamnodiastase from the seed of Rhamnus utilis Decne, Rhamnaceae (Chinese buckthorn); emulsin is not effective: Bridel, Charaux, Compt. Rend.181, 925 (1925). Toxicity data: Harrison et al.,J. Am. Pharm. Assoc.39, 557 (1950). Book: J. Q. Griffith, Jr., Rutin and Related Flavonoids (Mack, Easton, Pa., 1955).
Comprehensive description: T. I. Khalifa et al.,Anal. Profiles Drug Subs.12, 623-681 (1983).UV






Properties: Pale yellow needles from water, gradual darkening on exposure to light. The crystals contain 3 H2O and become anhydr after 12 hrs at 110° and 10 mm Hg. Anhydr rutin browns at 125°, becomes plastic at 195-197°, and dec 214-215° (with effervescence). [a]D23 +13.82° (ethanol); [a]D23 -39.43° (pyridine). Anhydr rutin is hygroscopic. One gram dissolves in about 8 liters water, about 200 ml boiling water, 7 ml boiling methanol. Sol in pyridine, formamide and alkaline solns; slightly sol in alcohol, acetone, ethyl acetate. Practically insol in chloroform, carbon bisulfide, ether, benzene, petr solvents. Dil solns give green color with ferric chloride. Rutin is colored brown by tobacco enzyme under experimental conditions: Neuberg, Kobel, Naturwissenschaften23, 800 (1935). LD50 i.v. in mice: 950 mg/kg (propylene glycol soln) (Harrison).
Optical Rotation: [a]D23 +13.82° (ethanol); [a]D23 -39.43° (pyridine)
Toxicity data: LD50 i.v. in mice: 950 mg/kg (propylene glycol soln) (Harrison)Therap-Cat: Capillary protectant.Keywords: Vasoprotectant.
C13




MASS

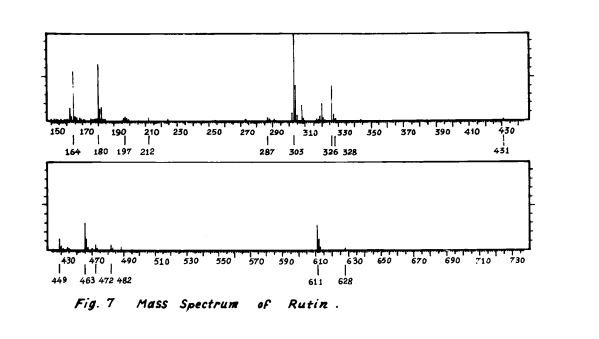
Rutin, also called rutoside, quercetin-3-O-rutinoside and sophorin, is the glycoside combining the flavonol quercetin and the disaccharide rutinose (α-L-rhamnopyranosyl-(1→6)-β-D-glucopyranose). It is a citrus flavonoid found in a wide variety of plants including citrus.
Rutin, also called rutoside, is the glycoside flavonoid found in a certain fruits and vegetables. Most rutine-rich foods are capers, olives, buckwheat (whole grain flour), asparagus, raspberry.In a clinical trial, rutin was found to aid control of intraocular pressure in patients with primary open angle glaucoma. As a component of dietary supplement Phlogenzym, rutin is used for treatment of osteoarthritis. Rutin is also used for treatment of post-surgical swelling of the arm after breast cancer surgery. Traditionally, rutin is used to prevent mucositis due to cancer treatment, to treat blood vessel disease such as varicose veins, bleeding, hemorrhoids.
Occurrences
Rutin is one of the phenolic compounds found in the invasive plant species Carpobrotus edulis and contributes to the antibacterial[3] properties of the plant.
Its name comes from the name of Ruta graveolens, a plant that also contains rutin.
Various citrus fruit peels contain 32 to 49 mg/g of flavonoids expressed as rutin equivalents.[4]
Citrus leaves contain rutin at concentrations of 11 and 7 g/kg in orange and lime trees respectively.[5]
Metabolism
The enzyme quercitrinase can be found in Aspergillus flavus.[6] It is an enzyme in the rutin catabolic pathway.[7]
In food
Rutin is a citrus flavonoid glycoside found in many plants including buckwheat,[8] the leaves and petioles of Rheum species, and asparagus. Tartary buckwheat seeds have been found to contain more rutin (about 0.8–1.7% dry weight) than common buckwheat seeds (0.01% dry weight).[8] Rutin is one of the primary flavonols found in ‘clingstone’ peaches.[9] It is also found in green tea infusions.[10]
Approximate rutin content per 100g of selected foods, in milligrams per 100 milliliters:[11]
| Numeric | Alphabetic |
|---|---|
| 332 | Capers, spice |
| 45 | Olive [Black], raw |
| 36 | Buckwheat, whole grain flour |
| 23 | Asparagus, raw |
| 19 | Black raspberry, raw |
| 11 | Red raspberry, raw |
| 9 | Buckwheat, groats, thermally treated |
| 6 | Buckwheat, refined flour |
| 6 | Greencurrant |
| 6 | Plum, fresh |
| 5 | Blackcurrant, raw |
| 4 | Blackberry, raw |
| 3 | Tomato (Cherry), whole, raw |
| 2 | Prune |
| 2 | Fenugreek |
| 2 | Marjoram, dried |
| 2 | Tea (Black), infusion |
| 1 | Grape, raisin |
| 1 | Zucchini, raw |
| 1 | Apricot, raw |
| 1 | Tea (Green), infusion |
| 0 | Apple |
| 0 | Redcurrant |
| 0 | Grape (green) |
| 0 | Tomato, whole, raw |
Research
Rutin (rutoside or rutinoside)[12] and other dietary flavonols are under preliminary clinical research for their potential biological effects, such as in reducing post-thrombotic syndrome, venous insufficiency, or endothelial dysfunction, but there was no high-quality evidence for their safe and effective uses as of 2018.[12][13][14][needs update] As a flavonol among similar flavonoids, rutin has low bioavailability due to poor absorption, high metabolism, and rapid excretion that collectively make its potential for use as a therapeutic agent limited.[12]
Biosynthesis
The biosynthesis pathway of rutin in mulberry (Morus alba L.) leaves begins with phenylalanine, which produces cinnamic acid under the action of phenylalanine ammonia lyase (PAL). Cinnamic acid is catalyzed by cinnamic acid-4-hydroxylase (C4H) and 4-coumarate-CoA ligase (4CL) to form p–coumaroyl-CoA. Subsequently, chalcone synthase (CHS) catalyzes the condensation of p-coumaroyl-CoA and three molecules of malonyl-CoA to produce naringenin chalcone, which is eventually converted into naringenin flavanone with the participation of chalcone isomerase (CHI). With the action of flavanone 3-hydroxylas (F3H), dihydrokaempferol (DHK) is generated. DHK can be further hydroxylated by flavonoid 3´-hydroxylase (F3’H) to produce dihydroquercetin (DHQ), which is then catalyzed by flavonol synthase (FLS) to form quercetin. After quercetin is catalyzed by UDP-glucose flavonoid 3-O-glucosyltransferase (UFGT) to form isoquercitrin, finally, the formation of rutin from isoquercitrin is catalyzed by flavonoid 3-O-glucoside L-rhamnosyltransferase.[15]

SYN
https://www.sciencedirect.com/science/article/abs/pii/S100184171300017X
The compound 2 was synthesized for the first time by highly selective esterification reaction and fully characterized. The by-products of the reaction were complex, which brought out many considerable difficulties in separation and purification of the target product. Our work was the first in using the improved pyrogallol autoxidation method to test the antioxidant activities of these two flavonoids compounds in vitro and discovered that the compound 2 was much more effective as a free radical scavenger than the compound 1.

SYN
Synthesis of Rutin
The synthesis of rutin can be achieved according to the following three schemes. These schemes differ in the synthesis of ouercetin (the aglycone moiety of rutin).
Scheme 1: Kostanecki — et al. 1904 (33 ). Based upon the Claisen reaction between 2-hydroxy4, 6-dimethoxyacetophenone [l] and 3, 4-dimethoxybenzaldehyde [2] to give the intermediate [3] which upon treatment with HC1, cyclization occurs to give 5, 7, 3 , which upon treatment with F2SO4 enolisation occurs to give 5, 7, 1’3 ,’4 -tetramethoxyflavonol [6]. tion with HI affords quercetin [7].
Scheme 2: Robinson et al. 1926 (34 ). , ‘4 -tetramethoxyflavonone [4]. Oximination affords [5] Demethyla- — Condensation ofw-methoxypholoroacetophenone [I] with veratric acid anhydride [2] in the presence of the potassium salt of veratric acid to give the diarylester [3]. On hydrolysis with alcoholic KOH affords 5, 7-dihydroxy-3, /3 , ‘4 -trimethoxyf lavone [ 41 , which on demethylation with HI gives quercetin [5].
Scheme 3: Shakhova et al. 1962 (35), complete synthesis of rutin. W-methoxyphloroacetophenone [2] was condensed with 0-benzylvanillinic acid, anhydride [ 13 in triethylamine to give 5 , 7-dihydroxy-4 -benzyloxy-3, /3 -dimethoxyf lavone [3]. On treatment with AcOH-HC1 mixture gave 5, 7, ‘4 -trihydroxy-3,’3 -dimethoxyflavone [4]. Demethylation of the latter with HI yielded (about 802) quercetin [5]. Ouercetin potassium salt [6] was produced upon treating [5] with AcOK in ethanol. Levoglucosan [7] was acetylated with Ac20 in the presence of AcONa to give 2, 3, 4-triacetyllevoglucosan [8] which with TIC14 gave 1-chloro-2, 3, 4-triacetyl Dglucose [9]. L-rhamnose tetraacetate [lo] treated with TiBr4 in CHC13 gave 1-bromo-2, 3, I-triacetyl-L-rhamnose [ll]. [lo] + [11] heated with Hg (OAC)~ in C6H6 gave (53x) CC – acetochloro-f3-l-L-rhamnosido-6-D-glucose [12]. [12] was treated with AgOAc and acetylated with Ac20 to prodilce (68.703 B-heptaacet yl-f3-1-L-rhamnos ido-6-D-glucose [13]. This with 33% HBr in AcOH gave (61%) d – acetobromo-~-l-L-rhamnosido-6-D-glucose [14]. [14] and quercetin potassium salt [6] were dissolved in NH40H which was evaporated and treated with methanol andpurified over a chromatographic column packed with polycaprolactum resin to give rutin [151.









References
- ^ Merck Index, 12th Edition, 8456
- ^ Krewson CF, Naghski J (Nov 1952). “Some physical properties of rutin”. Journal of the American Pharmaceutical Association. 41 (11): 582–7. doi:10.1002/jps.3030411106. PMID 12999623.
- ^ van der Watt E, Pretorius JC (2001). “Purification and identification of active antibacterial components in Carpobrotusedulis L.”. Journal of Ethnopharmacology. 76 (1): 87–91. doi:10.1016/S0378-8741(01)00197-0. PMID 11378287.
- ^ [1] p. 280 Table 1
- ^ [2] p.8 fig. 7
- ^ quercitrinase on www.brenda-enzymes.org
- ^ Tranchimand S, Brouant P, Iacazio G (Nov 2010). “The rutin catabolic pathway with special emphasis on quercetinase”. Biodegradation. 21 (6): 833–59. doi:10.1007/s10532-010-9359-7. PMID 20419500. S2CID 30101803.
- ^ Jump up to:a b Kreft S, Knapp M, Kreft I (Nov 1999). “Extraction of rutin from buckwheat (Fagopyrum esculentumMoench) seeds and determination by capillary electrophoresis”. Journal of Agricultural and Food Chemistry. 47 (11): 4649–52. doi:10.1021/jf990186p. PMID 10552865.
- ^ Chang S, Tan C, Frankel EN, Barrett DM (Feb 2000). “Low-density lipoprotein antioxidant activity of phenolic compounds and polyphenol oxidase activity in selected clingstone peach cultivars”. Journal of Agricultural and Food Chemistry. 48 (2): 147–51. doi:10.1021/jf9904564. PMID 10691607.
- ^ Malagutti AR, Zuin V, Cavalheiro ÉT, Henrique Mazo L (2006). “Determination of Rutin in Green Tea Infusions Using Square-Wave Voltammetry with a Rigid Carbon-Polyurethane Composite Electrode”. Electroanalysis. 18 (10): 1028–1034. doi:10.1002/elan.200603496.
- ^ “foods in which the polyphenol Quercetin 3-O-rutinoside is found”. Phenol-Explorer v 3.6. June 2015.
- ^ Jump up to:a b c “Flavonoids”. Micronutrient Information Center, Linus Pauling Institute, Oregon State University, Corvallis, Oregon. November 2015. Retrieved 25 February 2018.
- ^ Morling, J. R; Yeoh, S. E; Kolbach, D. N (November 2018). “Rutosides for treatment of post-thrombotic syndrome”. Cochrane Database of Systematic Reviews. 11 (11): CD005625. doi:10.1002/14651858.CD005625.pub4. PMC 6517027. PMID 30406640.
- ^ Martinez-Zapata, M. J; Vernooij, R. W; Uriona Tuma, S. M; Stein, A. T; Moreno, R. M; Vargas, E; Capellà, D; Bonfill Cosp, X (2016). “Phlebotonics for venous insufficiency”. Cochrane Database of Systematic Reviews. 4: CD003229. doi:10.1002/14651858.CD003229.pub3. PMC 7173720. PMID 27048768.
- ^ Yu X, Liu J, Wan J, Zhao L, Liu Y, Wei Y, Ouyang Z. Cloning, prokaryotic expression, and enzyme activity of a UDP-glucose flavonoid 3-o-glycosyltransferase from mulberry (Morus alba L.) leaves. Phcog Mag 2020;16:441-7
| Names | |
|---|---|
| IUPAC name3′,4′,5,7-Tetrahydroxy-3-[α-L-rhamnopyranosyl-(1→6)-β-D-glucopyranosyloxy]flavone | |
| Preferred IUPAC name(42S,43R,44S,45S,46R,72R,73R,74R,75R,76S)-13,14,25,27,43,44,45,73,74,75-Decahydroxy-76-methyl-24H-3,6-dioxa-2(2,3)-[1]benzopyrana-4(2,6),7(2)-bis(oxana)-1(1)-benzenaheptaphane-24-one | |
| Other namesRutoside (INN) Phytomelin Sophorin Birutan Eldrin Birutan Forte Rutin trihydrate Globularicitrin Violaquercitrin Quercetin rutinoside | |
| Identifiers | |
| CAS Number | 153-18-4 |
| 3D model (JSmol) | Interactive image |
| ChemSpider | 4444362 |
| DrugBank | DB01698 |
| ECHA InfoCard | 100.005.287 |
| KEGG | C05625 |
| PubChem CID | 5280805 |
| RTECS number | VM2975000 |
| UNII | 5G06TVY3R7 |
| CompTox Dashboard (EPA) | DTXSID3022326 |
| showInChI | |
| showSMILES | |
| Properties | |
| Chemical formula | C27H30O16 |
| Molar mass | 610.521 g·mol−1 |
| Appearance | Solid |
| Melting point | 242 °C (468 °F; 515 K) |
| Solubility in water | 12.5 mg/100 mL[1] 13 mg/100mL[2] |
| Pharmacology | |
| ATC code | C05CA01 (WHO) |
| Hazards | |
| NFPA 704 (fire diamond) |  200 200 |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
| Infobox references |
/////////Rutoside, RUTIN, рутозид , ルチン , روتوسيد , 芦丁 , C.I. 75730, NSC 9220,
CC1C(C(C(C(O1)OCC2C(C(C(C(O2)OC3=C(OC4=CC(=CC(=C4C3=O)O)O)C5=CC(=C(C=C5)O)O)O)O)O)O)O)O

NEW DRUG APPROVALS
ONE TIME
$10.00
IMRECOXIB
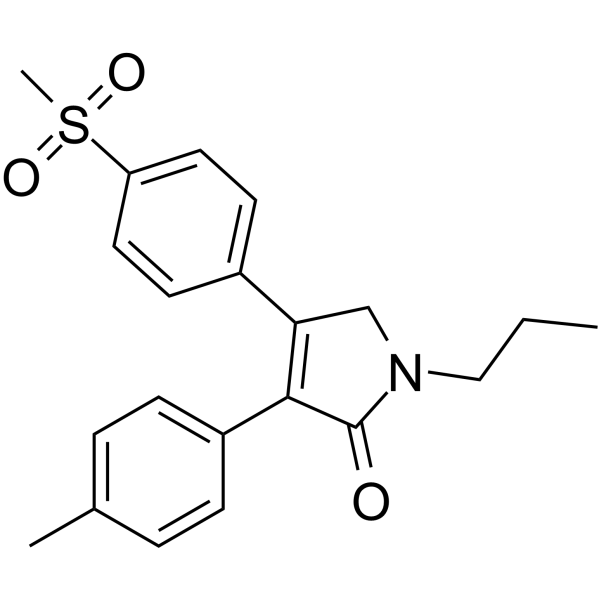
Imrecoxib (Hengyang)
CHINA 2012 osteoarthritis2H-Pyrrol-2-one, 1,5-dihydro-3-(4-methylphenyl)-4-[4-(methylsulfonyl)phenyl]-1-propyl-
3-(4-Methylphenyl)-4-[4-(methylsulfonyl)phenyl]-1-propyl-1,5-dihydro-2H-pyrrol-2-one395683-14-4[RN]
Imrecoxib was approved by China Food and Drug Administration (CFDA) on May 20, 2011. It was developed and marketed as 恒扬® by HengRui Pharmaceuticals.
Imrecoxib is a selective COX-2 inhibitor indicated for treatment of osteoarthritis.
恒扬® is available as tablet for oral use, containing 100 mg of free Imrecoxib, and the recommend dose is 100 mg twice daily.
Common name: Imrecoxib; BAP-909; BAP 909; BAP909
Trademarks: Hengyang
Molecular Formula: C21H23NO3S
CAS Registry Number: 395683-14-4
IUPAC Name: 4-(4-methane-sulfonyl-phenyl)-1-propyl-3-p-tolyl-1,5-dihydropyrrol-2-one
Molecular Weight: 369.48
SMILES: O=C1N(CCC)CC(C2=CC=C(S(=O)(C)=O)C=C2)=C1C3=CC=C(C)C=C3
Mechanism: COX-2 Inhibitor; Cyclooxygenase-2 Inhibitor
Activity: Treatment of Osteoarthritis; Analgesic; Antipyritic; Antiinflammatory Drug
Status: Launched 2011 (China)
Originator: HengRuiDrug Name:ImrecoxibResearch Code:BAP-909Trade Name:恒扬®MOA:Selective cyclooxygenase-2 (COX-2) inhibitorIndication:Osteoarthritis (OA)Status:ApprovedCompany:HengRui (Originator)Sales:ATC Code:Approved Countries or Area
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2011-05-20 | Marketing approval | 恒扬 | Osteoarthritis (OA) | Tablet, Film coated | 100 mg | HengRui |
Reference:1. US7112605B2.Route 2
Reference:1. CN102206178A.
2. Chinese Chem. Lett. 2001, 12, 775-778.Route 3
Reference:1. CN104193664A.
Imrecoxib | NSAID | Treatment of Osteoarthritis | COX-2 Inhibitor
Imrecoxib [4-(4-methane-sulfonyl-phenyl)-1-propyl-3-p-tolyl-1,5-dihydropyrrol-2-one] is a novel and moderately selective cyclooxygenase-2 (COX-2) inhibitor that possesses anti-inflammatory effect by inhibition of COX-2 mRNA expression. It belongs to the family of non-steroid anti-inflammtory drugs (NSAIDs). Imrecoxib was found to inhibit COX-1 and COX-2 with IC50 value of 115 ± 28 nM and 18 ± 4 nM, respectively [1].
| Imrecoxib: 2D and 3D Structure |
Imrecoxib effectively inhibited carrageenan-induced acute inflammation at the doses of 5, 10, and 20 mg-kg-1 ig and adjuvant-induced chronic inflammation at the doses of 10 and 20 mg-kg -1·d-1 ig.
NSAIDs and Imrecoxib:
Non-steroidal anti-inflammatory drugs (NSAIDs) are used extensively for the treatment of inflammatory conditions, including pain-releasing, anti-pyretic and rheumatoid arthritis. These functions are believed to inhibit the enzyme cyclooxygenase (COX) that is involved in the biosynthesis of prostaglandins G and H from arachidonic acid. So far two isozymes of COX are known: COX-1 and COX-2. COX-1 is constitutively produced in a variety of tissues and appears to be important to the maintenance of normal physiological functions, including gastric and renal cytoprotection. The COX-2 is an inducible isozyme, which is produced in cells under the stimulation of endotoxins, cytokines, and hormones and catalyzes the production of prostaglandins which cause inflammation.
The currently therapeutic use of NSAIDs has been associated with the inhibition of both COX-1 and COX-2 and causes well-known side effects at the gastrointestinal and renal level. Therefore, the selective COX-2 inhibitors could provide anti-inflammatory agents devoid of the undesirable effects associated with classical, nonselective NSAIDs. In addition, COX-2 is over-expressed in colon cancer tissue. COX-2 inhibitors possess potential prophylactic and therapeutic application to colon cancer.
Imrecoxib is designed in a manner such that it has “moderate selectivity” for COX-2 over COX-1. This balanced inhibition to both COX-1 and COX-2 was pursued to maintain the homeostasis of the two enzymes in the body,which is presumably critical to normal functions of the cardiovascular system.
Imrecoxib was launched in China with the trade name of Hengyang for the treatment of osteoarthritis in May 2011. Hengyang is available as tablet for oral use, containing 100 mg of free Imrecoxib, and the recommend dose is 100 mg twice daily.
SYN
Imrecoxib Synthesis
Chin Chem Lett 2001, 12, 775-778 (also Ref 2. This route is quoted as industrial method in various texts)
CN104193664A (an improvement here as Br is replaced with Cl)

US7112605B2 (primary reference for synthesis routes)

Identification:

| 1H NMR (Estimated) for Imrecoxib |
Experimental: 1H-NMR (CDCl3, TMS, 400MHz) 1.008 (3H, t, J = 7.2Hz), 1.701-1.756 (2H, m), 2.376 (3H, s), 3.078 (3H, s), 3.575 (2H, t, J = 7.2Hz), 4.317 (2H, s), 7. 175 (2H, d, J = 8.0Hz), 7.294 (2H, d, J = 8.0Hz), 7.505 (2H, t, J = 6.8Hz), 7.870 (2H, t, J = 6.8Hz)
Sideeffects:
Being a mild COX-2 inhibitor, it is expected not to cause any serious cardiovascular risks. Similarly, it should not have any serious gastrointestinal problems too, as it not a good inhibitor of COX-1. None of the reports though have listed any serious adverse event reported by patients in the clinical trials.
References:
- Cheng, G. F.;et. al. Imrecoxib: A novel and selective cyclooxygenase 2 inhibitor with anti-inflammatory effect. Acta Pharmacol Sin 2004, 25(7), 927-931.
- Zhang, F.;et. al.Method for preparing imrecoxib. CN102206178A
- Chao, W.;et. al. Synthesis method of imrecoxib. CN104193664A
- Bai, A. P.;et. al. Synthesis and in vitro Evaluation of a New Class of Novel Cyclooxygenase-2 Inhibitors: 3, 4-diaryl-3-pyrrolin-2 ones.Chin Chem Lett 2001, 12, 775-7785.
- Guo, Z. Discovery of imrecoxib. Chin J New Drugs2012, 21, 223.
- Guo, Z.;et. al. Sulfonyl-containing 3,4-diaryl-3-pyrrolin-2-ones, preparation method, and medical use thereof. US7112605B2
![MS 2 spectrum of the [M þ H] þ ion (m/z 370) of imrecoxib (inset, full-scan mass spectrum).](https://www.researchgate.net/profile/Dafang_Zhong/publication/6930659/figure/fig4/AS:394580018122760@1471086616006/MS-2-spectrum-of-the-M-th-H-th-ion-m-z-370-of-imrecoxib-inset-full-scan-mass.png)
SYN
Imrecoxib (Hengyang)
Imrecoxib, a new non-steroid anti-inflammtory drug (NSAID), was launched in China with the trade name of Hengyang for the treatment of osteoarthritis in 2012. It was originally designed and synthesized by Guo and co-workers at the Institute of Materia Medica (IMM) of the Chinese Academy of Medical Sciences in collaboration with Hengrui Pharmaceuticals.88 Imrecoxib, which is a moderately selective COX-2 inhibitor (with IC50 values against COX-1 and COX-2 being 115 ± 28 and 18 ± 4 nM, respectively),89 is the subject of twwo synthetic routes reported across several publications.90–93
The most likely process-scale route to this drug is described in Scheme 15, 93 which began with 2-bromo-40 -(methylsulfonyl)-acetophenone (84) and p-tolylacetic acid (85) as starting materials. In the presence of base, a-bromoketone 84 was treated with acid 85 which resulted in lactone 86 in 72% yield across the two-step sequence. Exposure of lactone 86 with propylamine triggered a ring-opening-ring closing reaction, which resulted in imrecoxib (XIII) directly in 85% yield.93

88. Guo, Z. R. Chin. J. New Drugs 2012, 21, 223.
89. Chen, X. H.; Bai, J. Y.; Shen, F.; Bai, A. P.; Guo, Z. R.; Cheng, G. F. Acta Pharmacol. Sin. 2004, 25, 927.
90. Bai, A. P.; Guo, Z. R.; Hu, W. H.; Shen, F.; Cheng, G. F. Chin. Chem. Lett. 2001, 12, 775.
91. Guo, Z.; Cheng, G.; Chu, F.; Yang, G.; Xu, B. CN Patent 1134413 C, 2001.
92. Guo, Z.; Cheng, G.; Chu, F. US Patent 2004/0029951 A1, 2004.
93. Zhang, F. Y.; Shen, X. M.; Sun, P. Y. CN Patent 102206178 A, 2011
Patent
CN 111747879
PATENT
CN 111747874
CN 111747873
CN 110386891
CN 109553564
CN 109553563
CN 108997188
CN 108947884
CN 108912030
CN 108864003
CN 108707100
CN 107586268
CN 104193664
CN 102206178
CN 101774958
US 20040029951
PATENT
CN 109678775
https://patents.google.com/patent/CN102206178A/en
Ai Rui former times cloth (N-n-propyl-3-p-methylphenyl-4-is to methylsulfonyl phenyl-3-pyrrolidin-2-one) is the nonsteroidal anti-inflammatory drug that a kind of appropriateness suppresses COX-2; put down in writing the synthetic method of Ai Rui former times cloth in the prior art (US20040029951), may further comprise the steps:
1) is raw material with the methylsulfonyl methyl phenyl ketone, makes alpha-brominated methylsulfonyl methyl phenyl ketone through bromo;
2) sodium borohydride reduction of alpha-brominated methylsulfonyl methyl phenyl ketone obtains the Styrene oxide 98min. derivative;
3) reaction of Styrene oxide 98min. derivative and Tri N-Propyl Amine generates N-n-propyl-capable biology of beta-hydroxyphenyl ethamine;
4) tolyl-acetic acid and the reaction of excessive thionyl chloride are generated the methylbenzene Acetyl Chloride 98Min.;
5) methylbenzene Acetyl Chloride 98Min. and the capable biological respinse of N-n-propyl-beta-hydroxyphenyl ethamine are generated N-n-propyl-N-[2-hydroxyl-2-to the methylsulfonyl styroyl] phenylacetamide;
6) Jone ‘ s reagent or pyridine chromium trioxide oxidation N-n-propyl-N-[2-hydroxyl-2-are to the methylsulfonyl styroyl] phenylacetamide obtains the capable biology of oxo phenylacetamide;
7) the above-mentioned oxo phenylacetamide of condensation makes end product Ai Rui former times cloth under the alkaline medium effect.
Because existing preparation method’s route is longer, and relate to reduction, oxidation, steps such as acid amides coupling, solvent load is big, the cost height, particularly to use oxygenants such as Jone ‘ s reagent or pyridine chromium trioxide in the oxidation step, low and the product of this oxidation step productive rate is difficult for separation and purification, and is difficult to control because chromium metal residual quantity control criterion in bulk drug is extremely strict, thereby makes this preparation method be difficult to be applicable to scale operation.
Synthetic route 1
Step 1), preparation alpha-brominated methylsulfonyl methyl phenyl ketone (III)
51.0g 4-methylsulfonyl methyl phenyl ketone and 760mL acetic acid is added to has magnetic agitation, in three mouthfuls of glass flask of the 1000mL of thermometer and constant pressure funnel.Be heated to 40 ℃, beginning slowly drips 41.1g liquid bromine, after dripping, continues to stir 30 minutes at 40 ℃.Reaction solution 50 ℃ concentrate after, add entry, stir, filter, washing, oven dry obtains the thick product of 70.5g, adds ethyl acetate/normal hexane mixed solvent, reflux 1 hour, slowly be cooled to 25 ℃, filtering drying gets the alpha-brominated methylsulfonyl methyl phenyl ketone of 56.5g off-white color solid (III), yield 80.0%.
1H-NMR(CDCl 3,TMS,400MHz):3.120(3H,s),4.485(2H,s),8.101(2H,dd,J=2.0Hz),8.191(2H,dd,J=2.0Hz)
MS(M+1):279.05
Step 2), prepare 4-(4-methylsulfonyl phenyl)-3-(4-aminomethyl phenyl)-2,5-dihydrofuran-2-ketone (II)
Experiment condition A
With the alpha-brominated methylsulfonyl methyl phenyl ketone of 44.3g (III), 24.0g 4-methylphenyl acetic acid and 600mL acetonitrile are added to and have magnetic agitation, in the 500mL there-necked flask of thermometer and constant pressure funnel.Be added dropwise to the 24.0mL triethylamine by constant pressure funnel, temperature is controlled at 25 ℃, after adding, continues to stir 1 hour.Add the 36.0mL triethylamine again, reaction solution is heated to 75 ℃, stirring reaction 18 hours.Cool to 25 ℃, concentrate, add ethyl acetate, washing, organic phase concentrates the back and adds ethyl acetate and ethanol, stirs, and filters and obtains 28.0g light yellow solid compound (II), yield 53.4%.
1H-NMR(CDCl 3,TMS)2.398(3H,s),3.091(3H,s),5.192(2H,s),7.216(2H,d,J=8.0Hz),7.292(2H,d,J=8.0Hz),7.543(2H,d,J=8.0Hz),7.933(2H,d,J=8.0Hz)
MS(M+1):329.02
Similarly, compound (II) can prepare under experiment condition B, C, D.
Experiment condition B
With the alpha-brominated methylsulfonyl methyl phenyl ketone of 5.0g (III), 2.7g 4-methylphenyl acetic acid and 70mL acetonitrile are added to and have magnetic agitation, in the 100mL there-necked flask of thermometer and constant pressure funnel.Be added dropwise to the 2.3mL tetramethyl guanidine by constant pressure funnel, temperature is controlled at 20 ℃, after adding, continues to stir 1.5 hours.Add the 4.6mL tetramethyl guanidine again, 20 ℃ of stirring reactions 2 hours.Concentrate, add ethyl acetate, washing, organic phase concentrates the back and adds ethyl acetate and ethanol, stirs, and filters and obtains 2.5g light yellow solid compound (II), yield 42.0%.
Experiment condition C
With the alpha-brominated methylsulfonyl methyl phenyl ketone of 1.85g (III), 1.0g 4-methylphenyl acetic acid and 20mL ethanol are added to and have magnetic agitation, in the 50mL there-necked flask of thermometer and constant pressure funnel.Be added dropwise to the 1.0mL triethylamine by constant pressure funnel, temperature is controlled at 25 ℃, after adding, continues to stir 3 hours.Add the 2.0mL triethylamine again, 80 ℃ of stirring reactions 18 hours.Concentrate, add ethyl acetate, washing, organic phase concentrates the back and adds ethyl acetate and ethanol, stirs, and filters and obtains 0.83g light yellow solid compound (II), yield 38.1%.
Experiment condition D
With the alpha-brominated methylsulfonyl methyl phenyl ketone of 1.0g (III), 0.54g 4-methylphenyl acetic acid and 12mL acetonitrile are added to and have magnetic agitation, in the 50mL there-necked flask of thermometer and constant pressure funnel.Add 1.0g salt of wormwood, 25 ℃ were reacted 2 hours.50 ℃ of stirring reactions are 5 hours then.Concentrate, add ethyl acetate, washing, organic phase concentrates the back and adds ethyl acetate and ethanol, stirs, and filters and obtains 0.13g light yellow solid compound (II), yield 11%.
Step 3), preparation N-n-propyl-3-p-methylphenyl-4-are to methylsulfonyl phenyl-3-pyrrolidin-2-one (Ai Rui former times cloth (I))
Experiment condition A
With the 25.0mL Tri N-Propyl Amine, be added drop-wise in the 17.5mL acetic acid at 10 ℃, add the back and stir, in the Tri N-Propyl Amine acetate that generates, add 10.0g compound (II).Under the nitrogen protection, be heated to 160 ℃, stirring reaction 8 hours.Cool to 40 ℃, add methylene dichloride and water, standing demix.Organic phase concentrates in the residue of back and adds ethanol, and reflux cools to 25 ℃, filters, and oven dry obtains 8.2g white solid product compound (I), yield 72.8%.
1H-NMR(CDCl 3,TMS,400MHz)1.008(3H,t,J=7.2Hz),1.701-1.756(2H,m),2.376(3H,s),3.078(3H,s),3.575(2H,t,J=7.2Hz),4.317(2H,s),7.175(2H,d,J=8.0Hz),7.294(2H,d,J=8.0Hz),7.505(2H,t,J=6.8Hz),7.870(2H,t,J=6.8Hz)
MS(M+1):370.17
Similarly, compound (I) can prepare under experiment condition B, C, D.
Experiment condition B
2.9g Tri N-Propyl Amine hydrochloride and 1.0g compound (II) are mixed, under the nitrogen protection, be heated to 170 ℃, stirring reaction 2 hours.Cool to 40 ℃, add methylene dichloride and water, standing demix.Organic phase concentrates in the residue of back and adds ethanol, and reflux cools to 25 ℃, filters, and oven dry obtains 0.9g white solid product compound (I), yield 80.0%.
Experiment condition C
Digest compound (II) with 2.0,3 milliliters of Tri N-Propyl Amines, 1.75 gram Tri N-Propyl Amine hydrochlorides add in the tube sealing of nitrogen protections, are heated to 140 ℃, react 20 hours.Be cooled to room temperature, add methylene dichloride and water, standing demix.Organic phase concentrates in the residue of back and adds ethanol, and reflux cools to 25 ℃, filters, and oven dry obtains 1.8g white solid product compound (I), yield 80.0%.
Experiment condition D
With the 0.5mL Tri N-Propyl Amine, be added drop-wise in the 0.35mL acetic acid at 10 ℃, add the back and stir, in the Tri N-Propyl Amine acetate that generates, add 0.5g compound (II).Under the nitrogen protection, be heated to 120 ℃, stirring reaction 4 hours.Cool to 40 ℃, add methylene dichloride and water, standing demix.Obtain 0.14g compound (I) after organic phase is concentrated and purified, yield 24.2%.Publication numberPriority datePublication dateAssigneeTitleCN104072467A *2014-07-072014-10-01太仓博亿化工有限公司Synthesis method of 5-chloro-2-benzofuranyl-p-chlorophenyl-oneCN104193664A *2014-08-222014-12-10山东铂源药业有限公司Synthesis method of imrecoxibCN107586268A *2016-07-072018-01-16江苏恒瑞医药股份有限公司A kind of preparation method of imrecoxib and its intermediateCN108864003A *2018-06-152018-11-23江苏美迪克化学品有限公司A kind of preparation method of imrecoxib intermediate and imrecoxibCN108947884A *2018-06-292018-12-07江苏美迪克化学品有限公司A kind of Preparation Method And Their Intermediate of imrecoxibCN109553564A *2017-09-252019-04-02江苏恒瑞医药股份有限公司A kind of purification process of imrecoxibCN109678775A *2017-10-182019-04-26江苏恒瑞医药股份有限公司A kind of crystal form and preparation method thereof of COX-2 selective depressantCN107586268B *2016-07-072021-01-19江苏恒瑞医药股份有限公司Preparation method of dapoxib and intermediate thereofPublication numberPriority datePublication dateAssigneeTitleUS5489693A *1992-04-281996-02-06Linz; GuenterCyclic imino derivatives, pharmaceutical compositions containing these compounds and processes preparing themCN101386590A *2007-09-132009-03-18中国医学科学院药物研究所Pyrrolidone containing hydroxymethyl and carboxyl, preparation method and medicament composition and use thereofCN101497580A *2009-01-092009-08-05华南理工大学HIV-1 inhibitor 2-pyrrolidinone derivative, as well as synthesizing method and use thereof
PAPER
Chinese Chemical Letters (2001), 12(9), 775-778.
PATENT
CN 110386891,

CLIP
For Chinese drugmaker Hengrui, R&D plans pan out
Ambitious program to launch innovative drugs starts to pay off for generics producerby Jean-François TremblayJULY 17, 2017 | APPEARED IN VOLUME 95, ISSUE 29

Credit: Jean-François Tremblay/C&ENHengrui recently invested in a custom-made phage-display library screening system for its Shanghai lab.
Launching their own innovative pharmaceuticals is a common goal for managers of generic drug firms. But it remains a dream for many. Jiangsu Hengrui Medicine, one of China’s largest generic drug makers, has advanced further than most. It has already launched two of its own drugs in China and licensed rights to another to a U.S. firm.
JIANGSU HENGRUI MEDICINE AT A GLANCE
▸ Headquarters: Lianyungang, Jiangsu, China
▸ 2016 sales: $1.6 billion
▸ 2016 profits: $390 million
▸ Employees: More than 13,000, 2,000 of whom work for a Shanghai-based unit developing and commercializing innovative drugs
▸ Innovative drug R&D staff: 800
Obtaining these results required substantial resources, though. Back in 2004, Hengrui built a large R&D lab in Shanghai, hired world-class researchers to lead it, equipped the facility with the latest instruments, and staffed it with hundreds of scientists.
Initially, the project looked like a money pit. In Chinese industry circles, many doubted that it would amount to anything. But revenues from the company’s innovative drugs are starting to pour in, and R&D at Hengrui is well on its way to financial sustainability.
Over the past 10 years, China has made great strides in growing an innovative drug industry. For all the talk, cynics say, China has yet to foster a blockbuster with $1 billion or more in annual sales. But as Hengrui and other Chinese firms launch their own drugs at home and license the foreign rights to others, it is becoming clear that an innovative drug industry is taking root.
“Producing generic drugs funds our R&D,” says Weikang Tao, a Hengrui vice president who doubles as chief executive officer of Shanghai Hengrui, the company’s innovative drug subsidiary.
Overall, Hengrui invests more than 10% of its sales in R&D, “which is big by Chinese standards,” Tao says. The drug giant Pfizer by comparison spent about 15% of its sales on R&D in 2016. With sales of $1.6 billion last year, Hengrui does most of its business in China. But it also exports finished drugs to the U.S., making it one of the few Chinese firms to have the U.S. Food & Drug Administration’s okay to do so.
Hengrui was formed in 1970 as a state-owned company. It began investing in its own R&D in 2004 and has since cultivated an innovative drug subsidiary that employs 2,000 people, including more than 800 at a Shanghai lab and about 20 at a subsidiary in Princeton, N.J. Other staffers work in the usual functions found in an innovative drug firm: clinical trial management, regulatory affairs, marketing and sales, and so on.
The Shanghai subsidiary recruits in China and internationally. Tao, who joined Hengrui in 2014, is a Chinese-trained physician who earned a Ph.D. in molecular and cell biology at the University of Medicine & Dentistry of New Jersey. He focused on tumor cell biology during a postdoc at Princeton University and worked in research at Merck & Co. for 10 years. Hengrui is constantly hiring, he notes.
Hengrui’s research facilities appear to be well equipped. Earlier this year the firm opened a biologics drug lab and a pilot plant for process development in Shanghai. “We spent nearly $7 million just on equipment for the biologics lab,” Tao says.

The lab is equipped with a custom-made automated phage-display library screening system that speeds up the process of discovering antibody drugs. “The machine can do automatically in a few hours what would otherwise take days for several scientists,” says Jiakang Sun, group leader of in vivo pharmacology at Shanghai Hengrui Pharmaceutical. With the phage-display system, Sun adds, a library displaying millions of human antibodies can be screened in vitro to find antibodies that bind to a specific antigen.
However well-staffed and well-equipped, Hengrui’s labs are still smaller than those of Merck or other major drug firms. But Hengrui has made notable strides recently. In 2015, it became the first Chinese firm to license a drug candidate to a U.S. firm. Incyte agreed to pay $25 million up front, and several hundred million dollars more once certain milestones are met, for the rights outside China and Taiwan to camrelizumab, a cancer treatment in Phase III human clinical trials in China.

In China, Hengrui’s priority market, the firm launched the osteoarthritis treatment imrecoxib in 2011 and the gastric cancer drug apatinib in 2014. The two will eventually achieve combined annual sales of $160 million, Hengrui expects.
Together with the licensing deal with Incyte, this will allow the firm to nearly recoup its R&D investment. Launching a few more compounds, particularly in the U.S., would make innovative R&D at Hengrui solidly profitable. The company is making good progress in that direction. A neutropenia treatment awaits final market approval in China, and five others have reached Phase III trials. Hengrui also has drugs in Phase I clinical trials in the U.S.
“I wouldn’t say that our lab is more productive than a lab operated by a multinational drug firm,” Tao says. Merck and other major players operate excellent facilities staffed by top people, he says. “But I would say that our researchers work very hard, and our decision-making at the top is very quick.”
Unlike biotech start-ups that tend to be built around groundbreaking technology, promising drug leads, or star researchers, Hengrui at first approached innovative drug development with a conservative strategy designed to reduce the risk of failure.
Relying on developmental compounds licensed from other organizations, the company initially aimed to develop drugs with the same mechanisms of action as others already on the market. Imrecoxib, for example, is part of the well-known family of COX-2 inhibitor anti-inflammatory drugs.
Later, it sought to invent compounds offering slight improvements over existing ones. Today, Hengrui is aiming to launch pharmaceuticals that are clearly superior to the competition. The company’s ultimate goal, Tao says, is to develop groundbreaking pharmaceuticals.
“We went from me-too to me-better to now best in class, and then we will do first in class,” he says.
And as Hengrui’s research strategy has become more ambitious, its scientists have broadened the range of diseases and drugs that they work on. Six or seven years ago, Tao says, Hengrui limited itself to the development of small-molecule drugs that treat cancer. Today the company is looking at small molecules, peptides, antibodies, antibody-drug conjugates, and other drug types to treat diseases as diverse as psoriasis and diabetes. “We have expanded our focus,” Tao says.
Most research is conducted in-house, Tao says. This includes medicinal chemistry, process chemistry, biology, drug metabolism, and pharmacokinetics. But the company leans on contract research firms for certain specific tasks, such as developing animal models. “They help accelerate our R&D,” Tao says.
Although Hengrui is a pioneer in launching new drugs in China, several other Chinese firms have made progress in advancing their own drug development. For instance, in the southern city of Dongguan, the generic drug producer HEC Pharma is conducting Phase II trials of the hepatitis C drug yimitasvir.
In Beijing, the biotech firm BeiGene just sold the U.S. company Celgene rights in much of the world to one of its immuno-oncology compounds for $263 million. Celgene also agreed to inject $150 million into BeiGene.
Chinese companies increasingly have the resources required to sustain innovative drug discovery and development, China watchers say. “Hengrui has the financial resources and the commitment to become a world-class innovative drugmaker,” says George Baeder, a former pharmaceutical industry executive who is now a director of China Global Insight, a California-based think tank.
But developing drugs and selling them are two different things, Baeder warns. “It is easy to underestimate the complexity a firm faces when moving into the arena of innovative medicines,” he says. “Chinese companies typically lack the capabilities in medical affairs, marketing, and sales needed to build a successful franchise.”
For the time being, Hengrui’s innovative drug subsidiary will stay focused on developing new drugs and not worry about the fine points of marketing them. Tao expects the former will keep his firm busy. “Don’t be surprised if several of our drugs begin clinical trials in the U.S., Europe, and Australia in the next year or two,” he says.
Rich pipeline
Hengrui boasts a diverse portfolio of drugs in late-stage development.
| China approval stage | Name | Application | Mechanism or target |
|---|---|---|---|
| Phase II clinical trials | Hetrombopaga | Idiopathic thrombocytopenia | Thrombopoietin receptor agonist |
| HR7056 | Anesthesia | na | |
| Pyrotiniba | Non-small cell lung cancer | EGFR/HER2 | |
| SHR3680b | Prostate cancer | Androgen receptor | |
| Phase III | Apatinib | Liver and non-small cell lung cancer | VEGFR-2 |
| Camrelizumabc | Cancer | PD-1 blocker | |
| Pyrotinib | HER2-positive breast cancer | EGFR/HER2 | |
| Retagliptin | Type 2 diabetes | Dipeptidyl peptidase 4 | |
| SHR3824 | Type 2 diabetes | SGLT2 inhibitor | |
| New drug application (China) | Mecapegfilgrastim | Neutropenia | PEG G-CSF |
| Launched | Apatinib | Gastric cancer | VEGFR-2 |
| Imrecoxib | Osteoarthritis | COX-2 inhibitor |
a In Phase I in U.S. b In Phase I in Australia. c The U.S. firm Incyte has acquired the rights to this drug outside China. na = not available. Source: Hengrui
////////////////Imrecoxib, Hengyang, CHINA 2012, osteoarthritis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}