
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

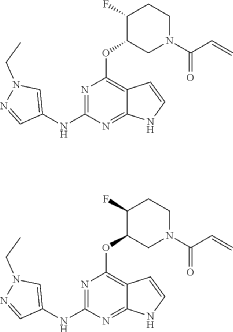
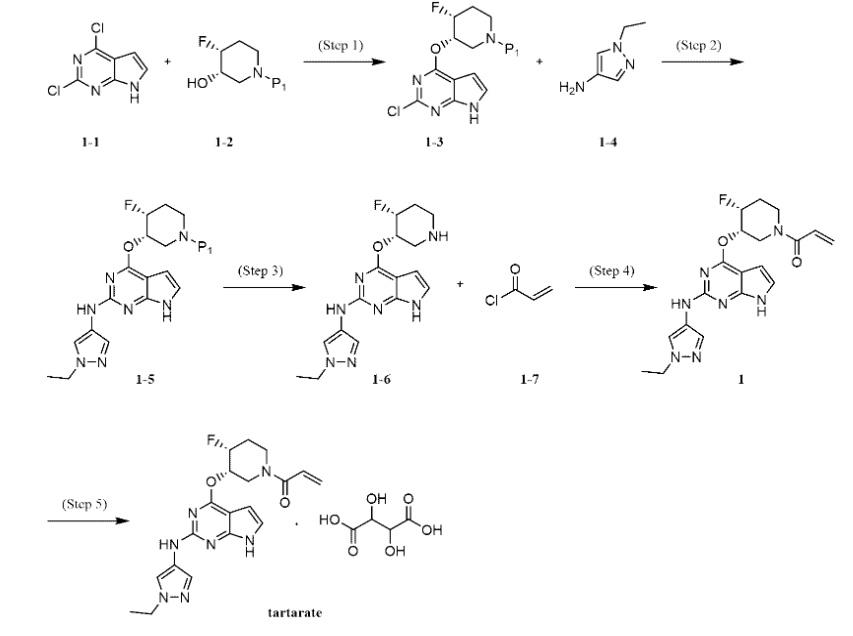
Plodicitinib
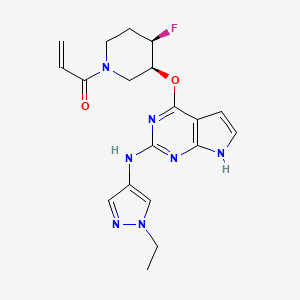
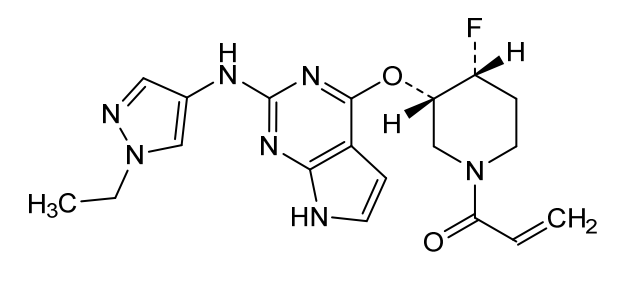
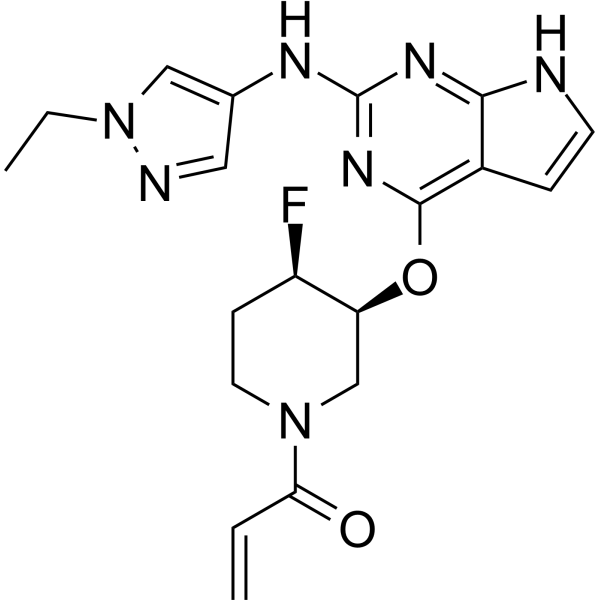
Plodicitinib
CAS 2360992-48-7
MF C19H22FN7O2 MW399.42
1-[(3S,4R)-3-[[2-[(1-ethylpyrazol-4-yl)amino]-7H-pyrrolo[2,3-d]pyrimidin-4-yl]oxy]-4-fluoropiperidin-1-yl]prop-2-en-1-one
1-[(3S,4R)-3-({2-[(1-ethyl-1H-pyrazol-4-yl)amino]-7Hpyrrolo[2,3-d]pyrimidin-4-yl}oxy)-4-fluoropiperidin-1-yl]prop2-en-1-one
Janus tyrosine kinase 3/TEC family kinase inhibitor, antiinflammatory, veterinary, SX5UEP3JXA
Plodicitinib is a Janus tyrosine kinase 3/TEC family kinase inhibitor with anti-inflammatory activity.
Plodicitinib is a small-molecule, dual Janus tyrosine kinase 3 (JAK3) and TEC family kinase (specifically BTK) inhibitor that exhibits strong anti-inflammatory properties.
Mechanism of Action
The compound blocks specific enzymatic pathways involved in cellular signaling:
- JAK3 Inhibition: It targets Janus kinase 3, which plays an essential role in transmitting signals for cytokines that regulate immune cell development and activation.
- TEC/BTK Inhibition: It blocks Bruton’s tyrosine kinase (BTK), a component vital for B-cell development and activation. [1, 2]
- Combined Effect: By blocking these pathways, it suppresses the overactive immune and inflammatory responses that drive autoimmune and allergic conditions.
Applications and Development Status
- Veterinary Medicine: In early 2026, Daewoong Pharmaceutical submitted an application to the Animal and Plant Quarantine Agency in South Korea for commercial approval of plodicitinib. It is being positioned as a specialized, companion animal-only treatment to manage atopic dermatitis in dogs.
- Research Use: In the scientific community, it is actively utilized as a laboratory tool compound to study kinase signaling pathways and inflammatory disease models.
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US306969271&_cid=P22-MSL6QJ-67927-1
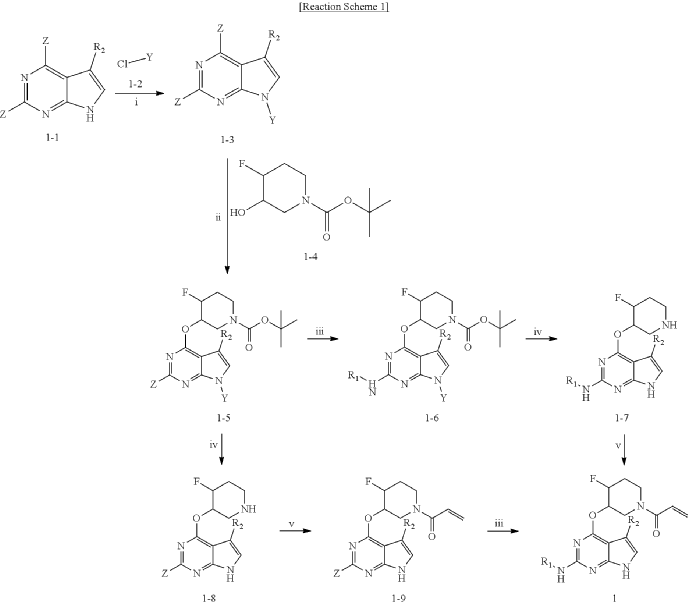
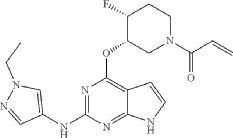
Example 4: Preparation of 1-(cis-3-((2-((1-ethyl-1H-pyrazol-4-yl)amino)-7H-pyrrolo[2,3-d]pyrimidin-4-yl)oxy)-4-fluoropiperidin-1-yl)prop-2-en-1-one
19.3 mg (yield: 27.8%) of the title compound was obtained in the same manner as in Example 1, except that cis-tert-butyl-4-fluoro-3-hydroxypiperidine-1-carboxylate was used instead of trans-tert-butyl-4-fluoro-3-hydroxypiperidine-1-carboxylate in Example 1.
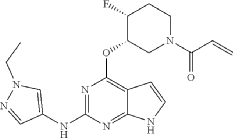
Example 5: Preparation of 1-((3S,4R)-3-((2-((1-ethyl-1H-pyrazol-4-yl)amino)-7H-pyrrolo[2,3-d]pyrimidin-4-yl)oxy)-4-fluoropiperidin-1-yl)prop-2-en-1-one
| 16.2 mg (yield: 57.4%) of the title compound was obtained in the same manner as in Example 1, except that tert-butyl(3S,4R)-4-fluoro-3-hydroxypiperidine-1-carboxylate was used instead of trans-tert-butyl-4-fluoro-3-hydroxypiperidine-1-carboxylate in Example 1. |
Example 25: Preparation of 1-((3S,4S)-3-((2-((1-ethyl-1H-pyrazol-4-yl)amino)-7H-pyrrolo[2,3-d]pyrimidin-4-yl)oxy)-4-fluoropiperidin-1-yl)prop-2-en-1-one
The compound of Example 1 was separated by CHIRALCEL OZ-H column to obtain the title compound with an analysis time of 10.1 minutes.
1H NMR (500 MHz, CD 3OD) δ 7.98-7.95 (m, 1H), 7.57-7.55 (m, 1H), 6.84-6.53 (m, 2H), 6.26-6.08 (m, 2H), 5.78-5.52 (m, 1H), 5.41-5.40 (m, 1H), 5.10-5.04 (m, 1H), 4.50-4.06 (m, 4H), 3.89-3.86 (m, 1H), 3.55-3.50 (m, 1H), 2.19-2.16 (m, 1H), 1.95-1.94 (m, 1H), 1.45-1.41 (m, 3H)
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2025121903&_cid=P22-MSL6W2-73218-1
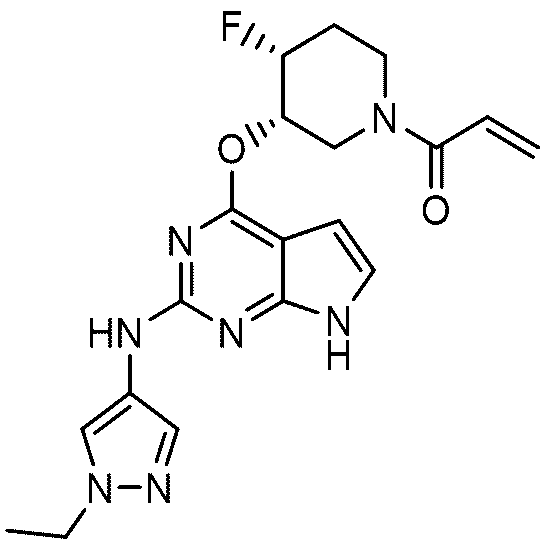
1-((3S,4R)-3-((2-((1-ethyl-1H-pyrazole-4-yl)amino)-7H-pyrrolo[2,3-d]pyrimidine-4-yl)oxy)-4-fluoropiperidin-1-yl)prop-2-en-1-one is represented by the following chemical formula 1:
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma



AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

References
- Tartrate salt of 1-((3s,4r)-3-((2-((1-ethyl-1h-pyrazol-4-yl)amino)-7h-pyrrolo[2,3-d]pyrimidin-4-yl)oxy)-4-fluoropiperidin-1-yl)prop-2-en-1-one, crystalline form thereof, and method for preparing samePublication Number:WO-2025121903-A1Priority Date:2023-12-05
- Method for preparation of 1-((3s,4r)-3-((2-((1-ethyl-1h-pyrazol-4-yl)amino)-7h-pyrrolo[2,3-d]pyrimidin-4-yl)oxy)-4-fluoropiperidin-1-yl)prop-2-en-1-one, and intermediate compounds thereofPublication Number:WO-2025121899-A1Priority Date:2023-12-05
- Phosphate salt of 1-((3s,4r)-3-((2-((1-ethyl-1h-pyrazol-4-yl)amino)-7h-pyrrolo[2,3-d]pyrimidin-4-yl)oxy)-4-fluoropiperidin-1-yl)prop-2-en-1-one, crystalline form thereof, and method for preparation thereofPublication Number:WO-2025121900-A1Priority Date:2023-12-05
- Oxy-fluoropiperidine derivative as kinase inhibitorPublication Number:EP-3733673-A1Priority Date:2017-12-28
- Oxy-fluoropiperidine Derivative as Kinase InhibitorPublication Number:US-2020308177-A1Priority Date:2017-12-28
- Oxy-fluoropiperidine compounds as kinase inhibitors, pharmaceutical composition comprising the same and their use in the prevention or treatment of inflammatory diseases, autoimmune diseases, proliferative diseases or hyperproliferative diseases and immune-mediated diseases, cancers, tumorsPublication Number:BR-112020013141-B1Priority Date:2017-12-28
- Substituted piperidines as kinase inhibitorsPublication Number:US-11339167-B2Priority Date:2017-12-28Grant Date:2022-05-24
- Oxy-haloperidine derivatives as kinase inhibitorsPublication Number:CN-111527091-APriority Date:2017-12-28
- Oxy-fluoropiperidine derivatives as kinase inhibitorPublication Number:KR-102318929-B1Priority Date:2017-12-28Grant Date:2021-10-28
- Oxy-fluoropiperidine derivative as a kinase inhibitorPublication Number:JP-6995428-B2Priority Date:2017-12-28Grant Date:2022-01-14
- Oxy-fluoropiperidine derivatives as kinase inhibitorPublication Number:CA-3084962-A1Priority Date:2017-12-28
- Oxy-fluoropiperidine derivative as a kinase inhibitorPublication Number:ES-2922633-T3Priority Date:2017-12-28Grant Date:2022-09-19
- Oxy-fluoropiperidine derivatives as kinase inhibitorPublication Number:KR-20190080803-APriority Date:2017-12-28
- Oxy-fluoropiperidine derivatives as kinase inhibitorPublication Number:CA-3084962-CPriority Date:2017-12-28Grant Date:2022-08-09
- Oxy-fluoropiperidine derivative as kinase inhibitorPublication Number:EP-3733673-B1Priority Date:2017-12-28Grant Date:2022-06-29
- Oxy-haloperidine derivatives as kinase inhibitorsPublication Number:CN-111527091-BPriority Date:2017-12-28Grant Date:2023-03-28
- Oxy-fluoropiperidine derivatives as kinase inhibitorPublication Number:KR-20210062618-APriority Date:2017-12-28
- Oxy-fluoropiperidine derivatives as kinase inhibitorPublication Number:KR-102592083-B1Priority Date:2017-12-28Grant Date:2023-10-20
- OXY-FLUOROPIPERIDINE DERIVATIVE AS A KINASE INHIBITORPublication Number:HR-P20221043-T1Priority Date:2017-12-28
- Oxy-fluoropiperidine derivative as kinase inhibitorPublication Number:IL-275207-BPriority Date:2017-12-28
/////////anax labs, plodicitinib, Janus tyrosine kinase 3/TEC family kinase inhibitor, antiinflammatory, veterinary, SX5UEP3JXA
Peturadol
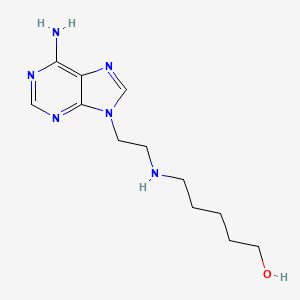
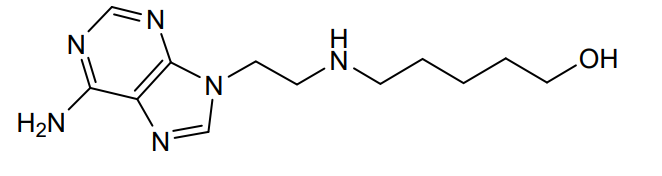
Peturadol
CAS 686301-48-4
MFC12H20N6O MW264.33 g/mol
5-{[2-(6-amino-9H-purin-9-yl)ethyl]amino}pentan-1-ol
central analgesic, NB001, NB 001, HTS 09836, J89QT81NBQ
NB-001 has been investigated for the treatment of Recurrent Herpes Labialis.
NB001 (HTS 09836) is an adenylcyclase 1 (AC1) inhibitor which has effect on neural and non-neural pain by modulating AC1 activity
Peturadol (also known by its developmental code NB001) is a potent, selective, and orally active adenylyl cyclase 1 (AC1) inhibitor. It is primarily recognized as a specialized chemical compound used in advanced medical and pharmacological laboratory research.
Because drug names can sometimes look or sound very similar, please check the spelling carefully. If you are looking for a medication prescribed to you by a doctor, it is highly likely you mean Patradol (a combination painkiller containing tramadol and paracetamol) or Pentadol / Tapentadol (an opioid analgesic).
Clinical Trial
| NCT Number | Sponsor | Condition | Start Date | Phase |
|---|---|---|---|---|
| NCT01324466 | NanoBio Corporation | Recurrent Herpes Labialis | 2011-04 | PHASE3 |
| NCT05290493 | Nobias Therapeutics, Inc. | 22q11 Deletion Syndrome | 2022-02-10 | PHASE2 |
| NCT01695187 | NanoBio Corporation | Herpes Labialis | 2012-10 | PHASE3 |
| NCT01321359 | NanoBio Corporation | Recurrent Herpes Simplex Labialis | 2011-04 | PHASE3 |
| NCT00453401 | NanoBio Corporation | Herpes Labialis | 2007-02 | PHASE2 |
- NB-001 in Children and Adolescents With 22q11 Deletion SyndromeCTID:NCT05290493Phase:Phase 2Status:CompletedDate:2025-02-10
- NB-001 Treatment of Recurrent Herpes LabialisCTID:NCT01695187Phase:Phase 3Status:Unknown statusDate:2013-06-14
- A Multicenter Study of NB-001 in the Treatment of Recurrent Herpes Labialis (SHaRCS)CTID:NCT01324466Phase:Phase 3Status:CompletedDate:2013-05-23
- Safety, Pharmacokinetics, and Efficacy Study of NB-001 to Treat Recurrent Herpes LabialisCTID:NCT00453401Phase:Phase 2Status:CompletedDate:2008-05-30
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2007041863&_cid=P22-MSFGZW-54602-1
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US210409779&_cid=P22-MSFGZW-54602-1
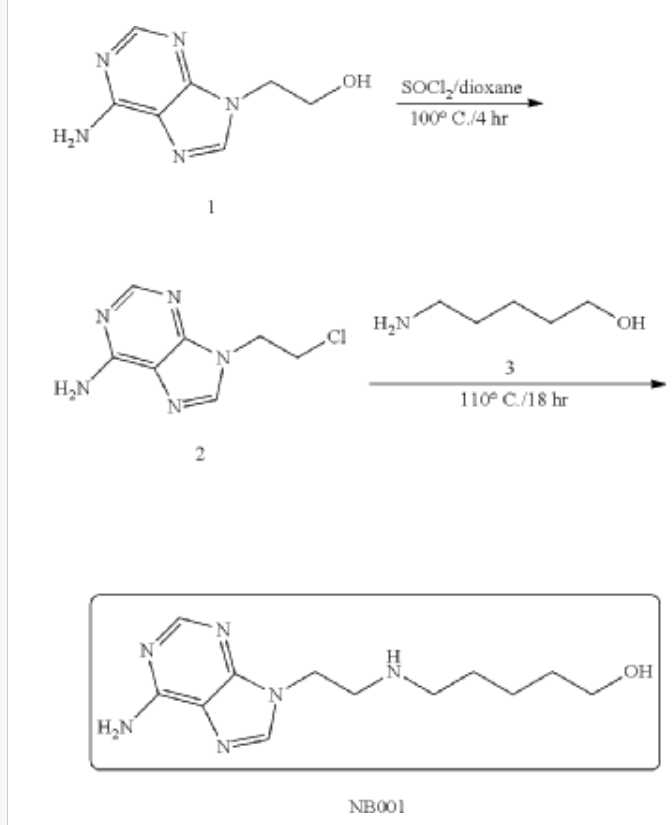
Synthesis and Purification of Intermediate 2
(2) Synthesis and Purification of NB001
PAT
- Method for treating neuronal and non-neuronal painPublication Number:CA-2625553-CPriority Date:2005-10-14Grant Date:2014-03-11
- Method for treating neuronal and non-neuronal painPublication Number:WO-2007041863-A1Priority Date:2005-10-14
- Methods for treating herpes virus infectionsPublication Number:CA-2721510-A1Priority Date:2008-04-18
- Method for treating neuronal and non-neuronal painPublication Number:EP-1948182-B1Priority Date:2005-10-14Grant Date:2012-12-12
- Method for Treating Neuronal and Non-Neuronal PainPublication Number:US-2009233922-A1Priority Date:2005-10-14
- Method for treating neuronal and non-neuronal painPublication Number:CA-2625553-A1Priority Date:2005-10-14
- Methods for treating neural and non-neuralgiaPublication Number:JP-5404045-B2Priority Date:2005-10-14Grant Date:2014-01-29
- Neuronal stem cell differentiationPublication Number:WO-2015055987-A1Priority Date:2013-10-14
- Phosphodiesterase Inhibitor TreatmentPublication Number:US-2024108627-A1Priority Date:2013-03-15
- Phosphodiesterase Inhibitor TreatmentPublication Number:US-2022226332-A1Priority Date:2013-03-15
- Process for preparing an enantiomerically enriched, deuterated secondary alcohol from a corresponding ketone without reducing deuterium incorporationPublication Number:US-9074233-B2Priority Date:2010-09-01Grant Date:2015-07-07
- Methods for treating herpes virus infectionsPublication Number:US-2010075914-A1Priority Date:2008-04-18
- Anti-hsv pre-exposure prophylaxisPublication Number:EP-3166680-B1Priority Date:2014-07-07Grant Date:2023-11-15
- Viral prophylaxis treatment methods and pre-exposure prophylaxis kitsPublication Number:EP-4342545-A2Priority Date:2014-07-07
- Neuronal stem cell differentiationPublication Number:EP-3058064-A1Priority Date:2013-10-14
- neuronal stem cell differentiationPublication Number:CN-105658787-APriority Date:2013-10-14
- Neuronal stem cell differentiationPublication Number:US-2016257930-A1Priority Date:2013-10-14
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
[1]. Min Zhuo. Method for treating neuronal and non-neuronal pain. US8124599B2.
[2]. Wang H, et al., Identification of an adenylyl cyclase inhibitor for treating neuropathic and inflammatory pain. Sci Transl Med. 2011 Jan 12;3(65):65ra3. [Content Brief]
[3]. Zhou Z, et al., Inhibition of calcium-stimulated adenylyl cyclase subtype 1 (AC1) for the treatment of pain and anxiety symptoms in Parkinson’s disease mice model. Mol Pain. 2024 Jan-Dec;20:17448069241266683. [Content Brief]
////////////peturadol, anax labs, central analgesic, NB001, NB 001, HTS 09836, J89QT81NBQ
Petadeferitrin
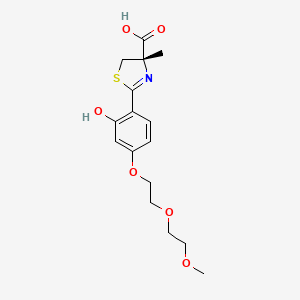
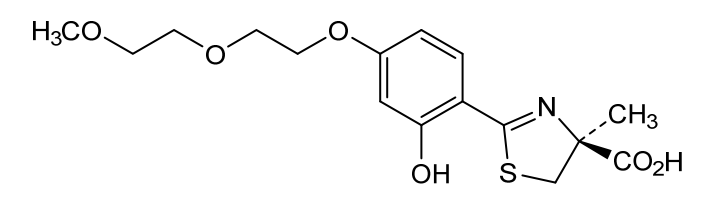
Petadeferitrin
CAS911714-45-9
MFC16H21NO6S MW355.4 g/mol
(4S)-2-[2-hydroxy-4-[2-(2-methoxyethoxy)ethoxy]phenyl]-4-methyl-5H-1,3-thiazole-4-carboxylic acid
- (4S)-4,5-Dihydro-2-(2-hydroxy-4-(2-(2-methoxyethoxy)ethoxy)phenyl)-4-methyl-4-thiazolecarboxylic acid
- 4-Thiazolecarboxylic acid, 4,5-dihydro-2-(2-hydroxy-4-(2-(2-methoxyethoxy)ethoxy)phenyl)-4-methyl-, (4S)-
- (4S)-4,5-dihydro-2-[2-hydroxy-4-[2-(2-methoxyethoxy)ethoxy]phenyl]-4-methyl-4-thiazolecarboxylic acid
(4S)-2-{2-hydroxy-4-[2-(2-methoxyethoxy)ethoxy]phenyl}-4-methyl4,5-dihydro-1,3-thiazole-4-carboxylic acid
iron chelating agent, SP 420, WBX54NZ436
Petadeferitrin is an orally bioavailable iron-chelating agent and derivative of desferrithiocin, with iron chelating and protective activities in diseases of iron overload. Upon oral administration, petadeferitrin targets, binds to and chelates free iron. This induces the excretion of iron, prevents iron accumulation and prevents cellular and/or tissue damage associated with iron overload.
Petadeferitrin (formerly known as SP-420) is an investigational, orally bioavailable, small-molecule iron chelator being developed by Pharmacosmos (and its subsidiary Abfero Pharmaceuticals) to treat patients with transfusion-dependent iron overload. The drug works by binding to excess free iron in the body and forming complexes that are primarily excreted through bile and feces.
Key Characteristics & Mechanisms
- Drug Class: It is a tridentate iron chelator and a derivative of desferrithiocin.
- Enhanced Efficiency: In preclinical studies, it demonstrated a higher iron clearance efficiency (ICE value of 26.7) compared to desferrithiocin.
- Brain-Penetrant: It is uniquely characterized as a brain-penetrant agent, which could expand its potential protective use in specific diseases associated with iron accumulation.
Clinical Development Status
- Investigational Status: The drug remains investigational and has not yet been approved for commercial use anywhere in the world.
- Target Diseases: Clinical evaluation focuses on individuals who suffer from iron overload due to frequent blood transfusions, such as patients with β-thalassemia and sickle cell disease.
- Ongoing Studies: Pharmacosmos is actively evaluating the drug in Phase II clinical trials (such as ClinicalTrials.gov ID NCT05693909) to assess its safety, tolerability, and dosing advantages over existing options. Early human data suggests it may offer effective clearance with less frequent dosing compared to some currently approved alternatives.
- A Trial Testing SP-420 in Subjects With Transfusion-dependent β-thalassemia or Low-risk Myelodysplastic SyndromesCTID:NCT05693909Phase:Phase 2Status:RecruitingDate:2025-09-24
- Safety of SP-420 in the Treatment of Transfusional Iron OverloadCTID:NCT04741542Phase:Phase 1Status:TerminatedDate:2024-12-27
- SP-420 in Subjects With Transfusion-dependent Beta-Thalassemia or Other Rare AnemiasCTID:NCT03801889Phase:Phase 2Status:WithdrawnDate:2020-10-05
- Safety and Pharmacokinetic Study of Escalating Doses of SP-420, an Iron Chelator, in Patients With β-ThalassemiaCTID:NCT02274233Phase:Phase 1Status:TerminatedDate:2015-09-29
An open-label, dose-escalation, dose-finding, and proof-of-concept trial of SP-420 in subjects with transfusion-dependent β-thalassemiaEudraCT:2022-002395-36
Phase:Phase 2, Status:Trial now transitioned, Date:2022-12-16
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US42268401&_cid=P21-MSCM3W-69668-1
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2006107626&_cid=P21-MSCM3W-69668-1
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US43268075&_cid=P21-MSCM3W-69668-1
PAT
Desferrithiocin polyether analoguesPublication Number:
US-2017217912-A1Priority Date:
2005-04-04
- Deferithiocin polyether analoguePublication Number:JP-6178816-B2Priority Date:2005-04-04Grant Date:2017-08-09
- Deferithiocin polyether analoguePublication Number:JP-2008536833-APriority Date:2005-04-04
- Desferrithiocin polyether analoguesPublication Number:EP-3190106-A1Priority Date:2005-04-04
- Desferrithiocin polyether analoguesPublication Number:US-9567309-B2Priority Date:2005-04-04Grant Date:2017-02-14
- Desferrithiocin polyether analoguesPublication Number:US-2013030028-A1Priority Date:2005-04-04
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- Target Class Profiling of Small-Molecule MethyltransferasesPublication Name:ACS Chemical BiologyPublication Date:2023-03-28PMCID:PMC10983791PMID:36976909DOI:10.1021/acschembio.3c00124
- Therapeutic candidates for the Zika virus identified by a high-throughput screen for Zika protease inhibitorsPublication Name:Proceedings of the National Academy of Sciences of the United States of AmericaPublication Date:2020-11-23PMCID:PMC7733812PMID:33229545DOI:10.1073/pnas.2005463117
- Cytotoxic Profiling of Annotated and Diverse Chemical Libraries Using Quantitative High-Throughput ScreeningPublication Name:SLAS discovery : advancing life sciences R & DPublication Date:2020-01PMCID:PMC10791069PMID:31498718DOI:10.1177/2472555219873068
- A High-Throughput Screen of a Library of Therapeutics Identifies Cytotoxic Substrates of P-glycoproteinPublication Name:Molecular PharmacologyPublication Date:2019-11PMCID:PMC6790066PMID:31515284DOI:10.1124/mol.119.115964
- Safety and pharmacokinetics of the oral iron chelator SP‐420 in β‐thalassemiaPublication Name:American Journal of HematologyPublication Date:2017-10-31PMID:28940308DOI:10.1002/ajh.24914
- Metabolically programmed iron chelatorsPublication Name:Bioorganic & Medicinal ChemistryPublication Date:2015-09-01PMCID:PMC4608554PMID:26231739DOI:10.1016/j.bmc.2015.06.059
- Substituent Effects on Desferrithiocin and Desferrithiocin Analogue Iron-Clearing and Toxicity ProfilesPublication Name:Journal of Medicinal ChemistryPublication Date:2012-08-13PMCID:PMC3583384PMID:22889170DOI:10.1021/jm300509y
- CCDC 757291: Experimental Crystal Structure DeterminationPublication Date:2011DOI:10.5517/cctf0rz
- The Impact of Polyether Chain Length on the Iron Clearing Efficiency and Physiochemical Properties of Desferrithiocin AnaloguesPublication Name:Journal of Medicinal ChemistryPublication Date:2010-04-08PMCID:PMC2951135PMID:20232803DOI:10.1021/jm9018146
/////////petadeferitrin, ANAX LABS, iron chelating agent, SP 420, WBX54NZ436
Perzebertinib, Bizrolertinib
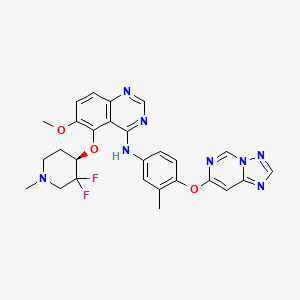
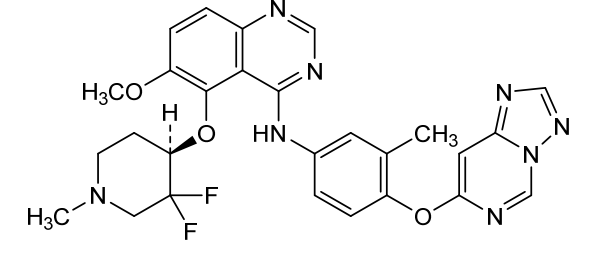
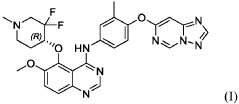
Perzebertinib, Bizrolertinib
CAS 2414056-31-6
MFC27H26F2N8O3 MW548.5 g/mol
5-[(4R)-3,3-difluoro-1-methylpiperidin-4-yl]oxy-6-methoxy-N-[3-methyl-4-([1,2,4]triazolo[1,5-c]pyrimidin-7-yloxy)phenyl]quinazolin-4-amine
- 4-Quinazolinamine, 5-[[(4R)-3,3-difluoro-1-methyl-4-piperidinyl]oxy]-6-methoxy-N-[3-methyl-4-([1,2,4]triazolo[1,5-c]pyrimidin-7-yloxy)phenyl]-
- 5-[[(4R)-3,3-Difluoro-1-methyl-4-piperidinyl]oxy]-6-methoxy-N-[3-methyl-4-([1,2,4]triazolo[1,5-c]pyrimidin-7-yloxy)phenyl]-4-quinazolinamine
5-{[(4R)-3,3-difluoro-1-methylpiperidin-4-yl]oxy}-6-methoxy-N-{3-methyl-4-[([1,2,4]triazolo[1,5-c]pyrimidin-7-yl)oxy]phenyl}quinazolin4-amine
epidermal growth factor receptor tyrosine kinase inhibitor, antineoplastic, ZN-A-1041, ZN 1041, RG 6596, Bizrolertinib, UN8TM5120C
Perzebertinib (also known as bizrolertinib or by developmental codes ZN-A-1041, ZN-1041, and RG6596) is an orally active, potent, and highly selective HER2 (ERBB2) tyrosine kinase inhibitor (TKI) designed to treat advanced solid tumors, primarily HER2-positive breast cancer.
Mechanism of Action
Perzebertinib functions as a selective, irreversible inhibitor of the HER2 tyrosine kinase. It blocks the ATP-binding site of the receptor to stop autophosphorylation. This action shuts down downstream signaling via the PI3K/AKT and MAPK pathways, successfully suppressing the growth, survival, and migration of tumor cells overexpressing HER2.
Key Clinical Advantages
- Blood-Brain Barrier (BBB) Penetration: The drug is designed to cross the blood-brain barrier effectively. This makes it highly valuable for treating brain metastases, a common and aggressive complication in advanced HER2-positive breast cancers.
- EGFR Sparing: Unlike older pan-EGFR/HER2 inhibitors, perzebertinib is engineered to spare wild-type EGFR. Sparing EGFR helps minimise common on-target side effects like severe skin rash and diarrhea.
- Efflux Resistance: It is not a substrate for P-gp or BCRP efflux pumps, allowing it to maintain high concentrations within central nervous system (CNS) tissues.
Development and Clinical Status
Initially discovered and developed by Suzhou Zanrong Pharmaceutical Technology (Zion Pharma), the asset is being co-developed in partnership with Roche and Genentech.
The drug has progressed through Phase 1 clinical studies evaluating its safety and pharmacokinetics in advanced solid tumors, moving forward into Phase 2/3 evaluations for HER2-positive advanced or locally advanced metastatic breast cancer. It is frequently evaluated as a monotherapy or in combination regimens alongside established therapies like capecitabine, trastuzumab, or pertuzumab
PAT
US11723908, Example 37, EG 77
https://patentscope.wipo.int/search/en/detail.jsf?docId=US344952565&_cid=P10-MS9QYW-06569-1
PAT
International Patent Publication No. WO 2020/057511 A1, which is incorporated herein by reference in its entirety, discloses quinazoline compounds that inhibit type I receptor tyrosine kinases, demonstrate good brain penetration in animals, and possess favorable toxicity profiles (for example a decreased activity against hERG), and thus particularly useful in the treatment of type I receptor tyrosine kinases mediated diseases or conditions, in particular ErbB2-associated disease or conditions, including cancer (e.g., metastatic cancer, such as brain metastases). A specific compound, which is identified as (R)-N-(4-([1,2,4]triazolo[1,5-c]pyrimidin-7-yloxy)-3-methylphenyl)-5-((3,3-difluoro-1-methylpiperidin-4-yl)oxy)-6-methoxyquinazolin-4-amine (also referred to as compound (I) herein),
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020057511&_cid=P10-MS9R3W-09545-1
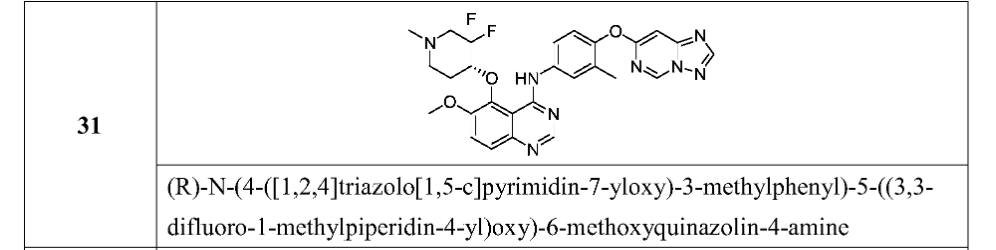

[0681]
(S) -N- (4- ( [1, 2, 4] triazolo [1, 5-c] pyrimidin-7-yloxy) -3-methylphenyl) -5- ( (3, 3-difluoro-1-methylpiperidin-4-yl) oxy) -6-methoxyquinazolin-4-amine

Step 5: (R) -N- (4- ( [1, 2, 4] triazolo [1, 5-c] pyrimidin-7-yloxy) -3-methylphenyl) -5- ( (3, 3-difluoro-1-methylpiperidin-4-yl) oxy) -6-methoxyquinazolin-4-amine and
[0696]
(S) -N- (4- ( [1, 2, 4] triazolo [1, 5-c] pyrimidin-7-yloxy) -3-methylphenyl) -5- ( (3, 3-difluoro-1-methylpiperidin-4-yl) oxy) -6-methoxyquinazolin-4-amine
[0697]
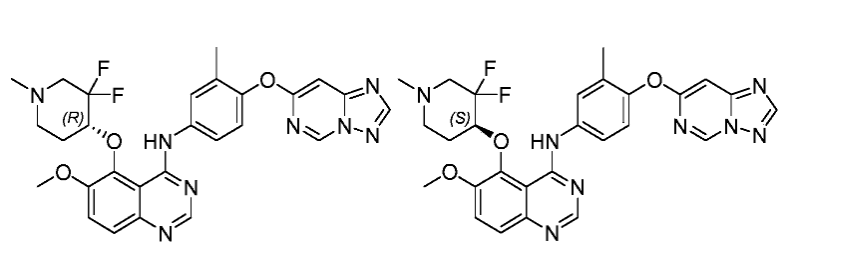
[0698]
To a solution of 4-chloro-5- ( (3, 3-difluoro-1-methylpiperidin-4-yl) oxy) -6-methoxyquinazoline (410 mg, 1.19 mmol) in Propan-2-ol (60 mL) was added TsOH. H 2O (68 mg, 0.36 mmol) and 4- ( [1, 2, 4] triazolo [1, 5-c] pyrimidin-7-yloxy) -3-methylaniline (259 mg, 1.07 mmol) . The resulting mixture was stirred at 100℃ under Ar 2protection and concentrated. The residue was dissolved in H 2O (100 mL) , basified with aq. NaHCO 3to pH =7-8, extracted with DCM: MeOH = 20: 1 (100 mLx3) . The combined organic layers were dried over anhydrous Na 2SO 4, filtered and concentrated. The residue was purified by column chromatography (DCM/MeOH=30/1) to give product (300 mg, 46%yield) as white solid. The racemic material was subsequently separated by chiral SFC to give two isomers:
[0699]
(R) -N- (4- ( [1, 2, 4] triazolo [1, 5-c] pyrimidin-7-yloxy) -3-methylphenyl) -5- ( (3, 3-difluoro-1-methylpiperidin-4-yl) oxy) -6-methoxyquinazolin-4-amine (Peak 1, retention time 6.241 min, ee: >99%) (100 mg, 67%) as a white solid. MS (ESI) m/z: 549.2 (M+H) +. 1H NMR (400 MHz, CDCl 3) δ 10.04 (s, 1H) , 9.20 (s, 1H) , 8.61 (s, 1H) , 8.33 (s, 1H) , 7.88 (d, J = 2.0 Hz, 1H) , 7.79-7.76 (m, 1H) 7.69 (d, J = 9.2 Hz, 1H) , 7.53 (d, J = 9.2 Hz, 1H) , 7.11 (d, J = 8.8 Hz, 1H) , 6.90 (s, 1H) , 4.84-4.79 (m, 1H) , 4.03 (s, 3H) , 3.22-3.21 (m, 1H) , 2.93 (d, J = 7.2 Hz, 1H) , 2.38 (s, 3H) , 2.41-2.34 (m, 1H) , 2.34-2.27 (m, 1H) , 2.19 (s, 3H) , 2.16-2.10 (m, 2H) .
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- Quinazoline derivatives as antitumor agentsPublication Number:US-2023293533-A1Priority Date:2018-09-18
- Quinazoline derivatives as antitumor agentsPublication Number:ES-2971927-T3Priority Date:2018-09-18Grant Date:2024-06-10
- Quinazoline derivatives as antitumor agentsPublication Number:EP-4360713-B1Priority Date:2018-09-18Grant Date:2024-10-30
- Quinazoline derivatives as antitumor agentsPublication Number:ES-3007082-T3Priority Date:2018-09-18Grant Date:2025-03-19
- Quinazoline derivatives as antitumor agentsPublication Number:EP-4360713-A2Priority Date:2018-09-18
- Quinazoline derivatives as antitumor agentsPublication Number:EP-3853220-B1Priority Date:2018-09-18Grant Date:2024-01-03
- Quinazoline derivatives as antitumor agentsPublication Number:EP-3853220-A1Priority Date:2018-09-18
- Quinazoline derivatives as antitumor agentsPublication Number:US-2021386742-A1Priority Date:2018-09-18
- Quinazoline derivatives as antitumor agentsPublication Number:US-11723908-B2Priority Date:2018-09-18Grant Date:2023-08-15
- Crystalline forms of quinazoline derivatives, preparation, compositions and uses thereofPublication Number:CN-120623183-APriority Date:2021-10-20
- Crystalline forms of quinazoline derivatives, preparation, compositions and uses thereofPublication Number:CN-120309621-APriority Date:2021-10-20
- Quinazoline derivatives as antitumor agentsPublication Number:WO-2020057511-A1Priority Date:2018-09-18
- Quinazoline Derivatives as Antitumor AgentsPublication Number:JP-7546550-B2Priority Date:2018-09-18Grant Date:2024-09-06
- Quinazoline derivatives as antitumor agentsPublication Number:CA-3099776-A1Priority Date:2018-09-18
/////////perzebertinib, anax labs, epidermal growth factor receptor tyrosine kinase inhibitor, antineoplastic, ZN-A-1041, ZN 1041, RG 6596, Bizrolertinib, UN8TM5120C
Pasodacigib
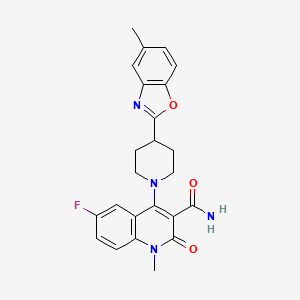
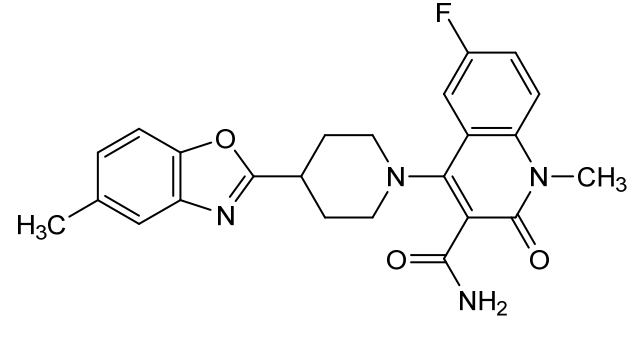
Pasodacigib
Cas 2648721-77-9
MFC24H23FN4O3 mw 434.5 g/mol
6-fluoro-1-methyl-4-[4-(5-methyl-1,3-benzoxazol-2-yl)piperidin-1-yl]-2-oxo-1,2-dihydroquinoline-3-carboxamide
6-fluoro-1-methyl-4-[4-(5-methyl-1,3-benzoxazol-2-yl)piperidin-1-yl]-2-oxo-1,2-dihydroquinoline-3-carboxamide
diacylglycerol kinase inhibitor, antineoplastic, BAY 2862789, BAY-2862789, XM6U88YE6H
Pasodacigib (also known by its developmental code BAY 2862789 or BAY-2862789) is an investigational small-molecule drug developed by Bayer AG. It functions as a potent and selective diacylglycerol kinase alpha (DGKα) inhibitor designed for cancer immunotherapy.
🧪 Mechanism of Action
- Targeting DGKα: Diacylglycerol kinase alpha (DGKα) is an enzyme that converts diacylglycerol (DAG) into phosphatidic acid (PA) within cells.
- T-Cell Reactivation: In the tumor microenvironment, overactive DGKα metabolises DAG, which depletes the signaling required for T-cell activation. By blocking this enzyme, pasodacigib aims to restore DAG levels, reactivating the patient’s own T-cells to mount a clinically beneficial anti-tumor immune response.
📋 Key Details & Chemical Properties
- Developer: Bayer AG
- CAS Registry Number: 2648721-77-9
- Molecular Formula: C₂₄H₂₃FN₄O₃
- Molecular Weight: 434.46 g/mol
- Research Status: It is an investigational drug that has entered clinical trial assessment (such as the Bayer-led study NCT05858164) to evaluate its safety and efficacy in treating advanced malignancies.
⚠️ Important Medical Disclaimer
Pasodacigib is strictly an investigational compound undergoing clinical development and laboratory research. It is not approved by the FDA or any other global regulatory authority for prescription, public medical use, or patient purchase.
If you are looking into this molecule for scientific research or a clinical study, please let me know if you need specific details regarding its chemical structure, information on related DGK inhibitors, or updates on active oncology clinical trials
Pat
https://patentscope.wipo.int/search/en/detail.jsf?docId=US400268899&_cid=P22-MS6W4B-66581-1
Example 298
6-fluoro-1-methyl-4-[4-(5-methyl-1,3-benzoxazol-2-yl)piperidin-1-yl]-2-oxo-1,2-dihydroquinoline-3-carboxamide
| 1H NMR (400 MHz, DMSO-d 6) δ ppm 2.02-2.15 (m, 2H) 2.17-2.26 (m, 2H) 2.44 (s, 3H) 3.13-3.25 (m, 3H) 3.35-3.43 (m, 2H) 3.59 (s, 3H) 7.18 (d, 1H) 7.47-7.63 (m, 6H) 7.73 (br s, 1H). |
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021105117&_cid=P22-MS6W2Y-65422-1
Intermediate 101
6-fluoro-1-methyl-2H-3,1-benzoxazine-2,4(1 H)-dione
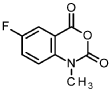
To a solution of 5.00 g 6-fluoro-2H-3,1-benzoxazine-2,4(1 H)-dione (26.8 mmol, CAS 321-69-7) and 9.3 ml. N,N-diisopropylethylamine (54 mmol) in 40 ml. dimethylformamide was added 5.1 ml. iodomethane (80 mmol) at rt and the mixture was stirred overnight. The reaction mixture was diluted with 1000 ml. water and the resulting solid was collected by filtration, the filter cake was washed with water and dried in vacuum to give 4.93 g of the title compound (99 % purity, 93 % yield).
1H NMR (400 MHz, DMSO-cfe) d ppm 3.47 (s, 3H), 7.40-7.61 (m, 1 H), 7.69-7.93 (m, 2H).Intermediate 102
6-fluoro-4-hydroxy-1-methyl-2-oxo-1 ,2-dihydroquinoline-3-carbonitrile
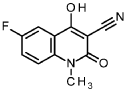
4.85 g 6-fluoro-1 -methyl-2H-3,1 -benzoxazine-2,4(1 H)-dione (intermediate 101 , 24.6 mmol,) was solubilised in 50 ml. tetrahydrofurane, 34 ml. triethylamine (250 mmol) and then 15.1 ml. ethyl cyanoacetate (133 mmol) were added carefully and the suspension was stirred 72 h at 900. The reaction mixture was cooled down to rt, concentrated under reduced pressure, the residue was diluted with water and ethyl acetate (1 :1) and the mixture was adjusted to pH = 1 with hydrogen chloride solution (2 M in water). The resulting solid was filtered and the filter cake was washed with less water and ethyl acetate to give 4.40 g of the title compound (100 % purity, 82 % yield).
1H NMR (400 MHz, DMSO-cfe) d ppm 3.34 (br s, 3H), 7.01 -7.44 (m, 2H), 7.51 -7.70 (m, 1 H).
Intermediate 103
4-chloro-6-fluoro-1-methyl-2-oxo-1,2-dihydroquinoline-3-carbonitrile
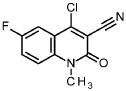
A mixture of 4.40 g 6-fluoro-4-hydroxy-1-methyl-2-oxo-1 ,2-dihydroquinoline-3-carbonitrile (intermediate 102, 20.0 mmol) and 19 ml. phosphoric trichloride (200 mmol) was stirred overnight at 900. The reaction mixture was cooled down to rt, diluted with hexane and the resulting solid was filtered. The filter cake was carefully added to a half saturated solution of sodium bicarbonate, the resulting suspension was filtered, the solid was washed with water, ethyl acetate and then with ethanol and dried in vacuum to give 4.17 g of the title compound (100 % purity, 88 % yield).
1H NMR (400 MHz, DMSO-cfe) d ppm 3.67 (s, 3H); 7.58 – 8.09 (m, 3H).
Example 217
6-fluoro-1-methyl-4-[4-(5-methyl-1,3-benzoxazol-2-yl)pipendin-1-yl]-2-oxo-1,2-dihydroquinoline-3-carbonitrile
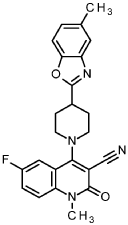
80 mg 4-chloro-6-fluoro-1-methyl-2-oxo-1 ,2-dihydroquinoline-3-carbonitrile (338 pmol, intermediate 103) was suspended in 2.5 ml. 2-propanol, 180 mI_ N,N-diisopropylethylamine (1.0 mmol) and 87.7 mg 5-methyl-2-(piperidin-4-yl)-1 ,3-benzoxazole (406 pmol, CAS 199292-77-8) were added and the mixture was stirred for 2 h at 900. The reaction mixture was cooled down to rt and the suspension was diluted with water and stirred for 15 min. The solid was filtered off and washed with water and ethanol to give 127 mg of the title compound (98 % purity, 88 % yield).
1H NMR (400 MHz, DMSO-cfe) d ppm 2.07 – 2.19 (m, 2 H) 2.27 – 2.35 (m, 2 H) 2.44 (s, 3 H) 3.43 (tt, 1 H) 3.55 – 3.64 (m, 5 H) 3.81 (br d, 2 H) 7.18 (dd, 1 H) 7.53 (d, 1 H) 7.54 – 7.61 (m, 2 H) 7.61 – 7.70 (m, 2 H).
LC-MS (Method 2): R, = 1.32 min; MS (ESIpos): m/z = 417.4 [M+H]+
Example 298
6-fluoro-1-methyl-4-[4-(5-methyl-1,3-benzoxazol-2-yl)pipendin-1-yl]-2-oxo-1,2-dihydroquinoline-3-carboxamide
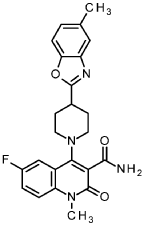
68 mg 6-fluoro-1 -methyl-4-[4-(5-methyl-1 ,3-benzoxazol-2-yl)piperidin-1-yl]-2-oxo-1 ,2-dihydroquinoline-3-carbonitrile (160 pmol, example 217), 9 mg palladium(ll)acetate (40 pmol) and 142 mg acetaldoxime (2.4 mmol) were stirred in 1 .5 ml. ethanol for 5 h at 80TT The reaction mixture was diluted with water, extracted with ethyl acetate two times, the combined organic layers were filtered through a waterresistant filter and the filtrate was concentrated under reduced pressure. The residue was purified by RP-HPLC (column: X-Bridge C18 5pm 100x30mm, mobile phase: acetonitrile / water (0.2 vol. % ammonia 32 %)-gradient) to give 41 .4 mg of the title compound (100 % purity, 60 % yield).
1H NMR (400 MHz, DMSO-cfe) d ppm 2.02 – 2.15 (m, 2 H) 2.17 – 2.26 (m, 2 H) 2.44 (s, 3 H) 3.13 – 3.25 (m, 3 H) 3.35 – 3.43 (m, 2 H) 3.59 (s, 3 H) 7.18 (d, 1 H) 7.47 – 7.63 (m, 6 H) 7.73 (br s, 1 H).
LC-MS (Method 2): R, = 1 .17 min; MS (ESIpos): m/z = 435.4 [M+H]
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- DGK alpha inhibitors as immune activation substituted aminoquinolones of (2)Publication Number:CN-119523983-APriority Date:2019-11-28
- Cyclopropyl modulators of p2y12 receptorPublication Number:US-2017002015-A1Priority Date:2009-07-27
- Substituted aminoquinolones as dgkalpha inhibitors for immune activationPublication Number:EP-4065574-A1Priority Date:2019-11-28
- Substituted aminoquinolones as dgkalpha inhibitors for immune activationPublication Number:WO-2021105117-A1Priority Date:2019-11-28
- Substituted aminoquinolones as immunoactive DGK alpha inhibitorsPublication Number:CN-115003665-APriority Date:2019-11-28
- Substituted aminoquinolones as inhibitors of immune-activated DGKαPublication Number:CN-119564686-APriority Date:2019-11-28
- DGK alpha inhibitors as immune activation substituted aminoquinolones of (2)Publication Number:CN-119424429-APriority Date:2019-11-28
- Combinations of dgk (diacylglycerol kinase) inhibitors and immune checkpoint inhibitors and modulatorsPublication Number:TW-202448461-APriority Date:2023-02-06
- Substituted aminoquinolones as DGKalpha inhibitors for immune activationPublication Number:US-11998539-B2Priority Date:2019-11-28Grant Date:2024-06-04
- Substituted aminoquinolones as dgkalpha inhibitors for immune activationPublication Number:US-2023148194-A1Priority Date:2019-11-28
- Substituted aminoquinolones as dgkalpha inhibitors for immune activationPublication Number:US-2023201186-A1Priority Date:2019-11-28
- DGK alpha inhibitors as immune activation substituted aminoquinolones of (2)Publication Number:CN-115003665-BPriority Date:2019-11-28Grant Date:2024-11-08
- Combination of ccr8 antibodies with dgk inhibitors in the treatment of cancerPublication Number:WO-2024165468-A1Priority Date:2023-02-06
- Combination of ccr8 antibodies with dgk inhibitorsPublication Number:TW-202436351-APriority Date:2023-02-06
- Combinations of dgk (diacylglycerol kinase) inhibitorsPublication Number:WO-2024165470-A1Priority Date:2023-02-06
- Combinations of dgk (diacylglycerol kinase) inhibitors and immune checkpoint inhibitors and modulatorsPublication Number:WO-2024165469-A1Priority Date:2023-02-06
- Combinations of dgk (diacylglycerol kinase) inhibitorsPublication Number:TW-202448460-APriority Date:2023-02-06
/////////pasodacigib, anax labs, diacylglycerol kinase inhibitor, antineoplastic, BAY 2862789, BAY-2862789, XM6U88YE6H
Pariceract
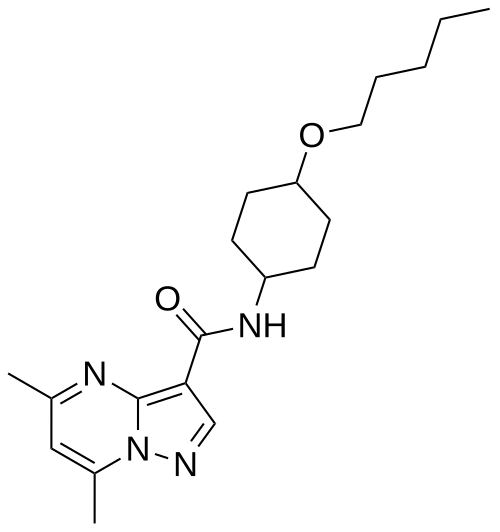
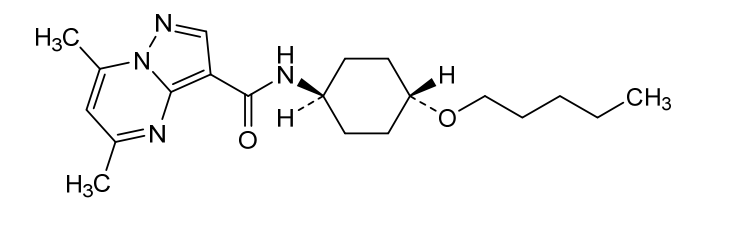
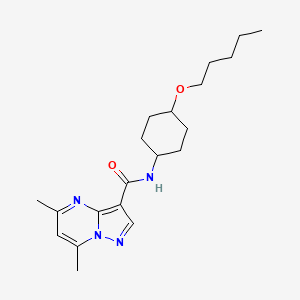
Pariceract
CAS 1919820-28-2
MFC20H30N4O2 MW358.5 g/mol
- 5,7-dimethyl-N-((1r,4r)-4-(pentyloxy)cyclohexyl)pyrazolo[1,5-a]pyrimidine-3-carboxamide
- 5,7-Dimethyl-N-(4-pentoxycyclohexyl)pyrazolo[1,5-a]pyrimidine-3-carboxamide
- Pyrazolo(1,5-a)pyrimidine-3-carboxamide, 5,7-dimethyl-N-(trans-4-(pentyloxy)cyclohexyl)-
5,7-dimethyl-N-[trans-4-(pentyloxy)cyclohexyl]pyrazolo[1,5-a]pyrimidine-3-carboxamide
beta-glucocerebrosidase activator, antiparkinsonian, BIA 28-6156, LTI-291, LIT291, LTI-00291, BIA28-6156, LTI- 91, LIT 291, LTI 00291, Gcase activator 1, V9WUN9UUU8
Pariceract (INNTooltip International Nonproprietary Name; developmental code name BIA 28-6156 or LTI-291) is a β-glucocerebrosidase (GCase) activator or positive allosteric modulator which is under development for the treatment of Parkinson’s disease.[1][2][3][4][5] It is taken orally.[1] GCase is a lysosomal enzyme encoded by the gene GBA1.[4][5] Loss-of-function mutations in this gene are thought to promote α-synuclein accumulation and are among the leading genetic risk factors for Parkinson’s disease.[4][5][6] As such, activation of GCase might provide a disease-modifying therapy for treatment of Parkinson’s disease.[4][5] Pariceract was first described in the scientific literature by 2017.[6] It was originated by Lysosomal Therapeutics and is under development by Lysosomal Therapeutics and Bial.[1][2][3] As of October 2025, it is in phase 2 clinical trials.[1][2][3]
PAT
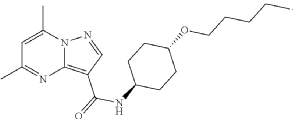
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2025198486&_cid=P12-MS41BL-88178-1
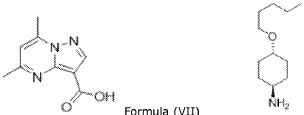
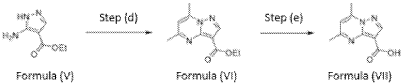
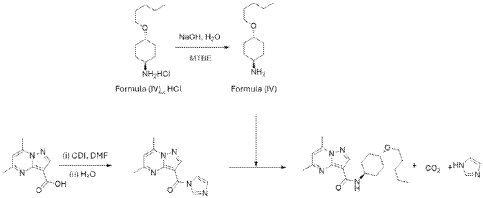
Example 3 – Preparation of 5,7-dimethyl-/V-((l/?,4/?)-4- (pentyloxy)cyclohexyl)pyrazolo[1,5-a]pyrimidine-3-carboxamide (Compound of Formula (A))
[00168] Part 1 : Under nitrogen atmosphere, purified water (41.32 L, 2.0 V) and NaOH (5.99 kg, 1.4 eq.) were charged to a reactor at 20 °C; after this, purified water (10.33 L, 0.5 V) was added to wash the walls and the solution was left stirring for 0.5-3 hours until the solid was dissolved; the solution was transferred from the reactor to a clean drum.
[00169] Part 2: Under nitrogen atmosphere, purified water (30.99 L, 1.5 V) and NaCI
(2.27 kg, 0.11 wt.%) were charged to the reactor at 20 °C; after this, purified water (10.33 L, 0.5 V) was charged to wash the walls and the solution was left stirring for 0.5-3 hours until the solid was dissolved; the solution was transferred from the reactor to a clean drum.
[00170] Part 3: Under nitrogen atmosphere, purified water (62 L, 3.0 V) and the compound of Formula (IV) in HCI salt form (27.84 kg, 1.15 eq.), from step 3 of Example 1, were charged to the reactor at 25 °C; after this, purified water (16.5 L, 0.8 V) was charged to wash the walls and then MTBE (165.28 L, 8.0 V) was charged to the reactor and the solution was left stirring for 0.5-1 hour; after this, the solution was cooled down to 5 °C and the NaOH (5.99 kg, 1.4 eq.) aqueous solution (prepared in advance in part 1) was charged in 0.5-2 hours; then, the temperature of the reaction mixture was increased to 25 °C and it was left stirring for 1-3 hours. The agitation was stopped, and the phases were separated. The organic phase was washed two times: the first with 5% NaCI aqueous (41.32 L, 2.0 V) prepared in advance in part 2 and the second with purified water (41.32 L, 2.0 V). Then, the organic phase was concentrated under vacuum until the total volume was 51.65 L – 72.31 L (2.5-3.5 V). MTBE (62 L, 3.0 V) was charged to the reactor and the solution was concentrated under vacuum until the total volume was 51.65 L – 72.31 L (2.5-3.5 V).
[00171] Part 4: Under nitrogen atmosphere, DMF (92.97 L, 4.5 V), the compound of Formula (VII) (20.66 kg, 1.0 eq.), from step 2 of Example 2, and l,l’-carbonyldiimidazole (21.07 kg, 1.2 eq.) were charged to the reactor at 25 °C; DMF (10.33 L, 0.5 V) was added to wash the walls. The temperature of the reaction mixture was increased to 28 °C, and it was left stirring for 2-5 hours. If necessary, charge additional l,l’-carbonyldiimidazole (1.76 kg, 0.1 eq.) to the reactor at 28 °C and leave it stirring for 2-5 hours. After this, purified water (1.178 kg, 0.6 eq.) was charged to the reactor and it was left stirring for 1-4 hours. Under nitrogen atmosphere, the compound of Formula (IV) Free Base in MTBE solution (27.84 kg ,1.15 eq.), from Part 3, was charged to the reactor in 1-2 hours; MTBE (4.132 L, 0.2 V) was charged to rinse the drum and then it was transferred to the reactor. The solution was left stirring for 16-24 hours and then DMF (20.66 L, 1.0 V) was charged to the reactor. The solution was concentrated under vacuum until the total volume was 123.96 L – 165.28 L (6.0-8.0 V).
[00172] Part 5: The reaction mixture temperature was increased to 40 °C and purified water (41.32 L, 2.0 V) was added dropwise to the reactor in 1-2 hours; the reaction mixture was left stirring for 2-5 hours before cooling down to 20 °C; the slurry was left stirring for 10-15 hours and then purified water (82.64 L, 4.0 V) was added dropwise in 1-3 hours to the reactor; the slurry was left stirring for 3-6 hours. After this, the solid was centrifuged and the cake was washed with DMF:H2O (1: 1, 20.66 L, 1.0 V x 2) two times and then washed with purified water (41.32 L, 2.0 V) one time*. The cake was dried under vacuum at 55 °C for 15-24 hours and 32.35 kg were obtained (83.3% yield and 100% purity by HPLC).
* In one campaign, at this stage the wet cake was dissolved in acetonitrile (25.5 V), the temperature adjusted to 27°C and stirred for 2-6 hours. The solution was filtered via a polish filter (0.22 pirn). The solution was concentrated under vacuum to 5.0 – 7.0 V at a temperature below 35°C. The temperature was adjusted to 20°C and purified water was added (14.0 V) in 2-4 hours. The slurry was stirred for 4-8 hours at 20°C, and afterwards filtered and washed with purified water (4.0 V).
Example 4 – Recrystallisation of 5,7-dimethyl-/V-((l/?,4R)-4- (pentyloxy)cyclohexyl)pyrazolo[1,5-a]pyrimidine-3-carboxamide (Compound of Formula (A))
[00173] Under nitrogen atmosphere, MEK (192 L, 6.0 V) and the compound of Formula (A) from Example 3 in crude (32 kg, 1.0 eq.) were charged to a reactor at 25 °C; MEK (160 L, 5.0 V) was added to wash the walls. The solution was left stirring for 1-3 hours until the solid was dissolved. After this, the solution from the reactor was transferred to a clean drum.
[00174] Under nitrogen atmosphere, the crude compound of Formula (A) in MEK solution was transferred to another reactor via a polish filter. The temperature was set at 17 °C and n-heptane (115.2 kg, 3.6 V) was charged in 2-4 hours. The solution was concentrated under vacuum until the total volume was 320 L – 352 L (10-11 V). In the event that there is no solid precipitation, proceed to the 2nd concentration directly; if there is solid precipitation, take a solid sample for XRPD test, and ensure that the crystal form is polymorph form B; if it is not form B, proceed to the 2nd concentration (and repeat if required). The reactor temperature was kept at 17 °C before cooling down to 0 °C and it was left stirring for 6-12 hours. A solid sample was taken for XRPD test to make sure that the crystal form is form B; if not form B, keep at 0 °C and stir for 6-12 hours; sample solid every 6-10 hours for XRPD until the crystal form is form B. The solid was centrifuged and washed with MEK:n-heptane (1 : 10, 57.6 L, 1.8 V ) and then with n-heptane (57.6 L, 1.8
V). The cake was dried under vacuum at 30 °C for 16-24 hours and 30.67 kg were obtained (95% yield and purity of 100% by HPLC).
PAT
- Substituted pyrazolo[1,5-a]pyrimidines and their use in the treatment of medical disordersPublication Number:US-2017334916-A1Priority Date:2014-11-06
- Substituted pyrazolo[1,5-a]pyrimidines and their use in the treatment of medical disordersPublication Number:US-2025066364-A1Priority Date:2014-11-06
- SUBSTITUTED PYRAZOLO[1,5-a]PYRIMIDINES AND THEIR USE IN THE TREATMENT OF MEDICAL DISORDERSPublication Number:US-2017183354-A1Priority Date:2014-11-06
- Substituted pyrazolo[1,5-a]pyrimidines and their use in the treatment of medical disordersPublication Number:US-9732089-B2Priority Date:2014-11-06Grant Date:2017-08-15
- Substituted pyrazolo[1,5-a]pyrimidines and their use in the treatment of medical disordersPublication Number:US-11091492-B2Priority Date:2014-11-06Grant Date:2021-08-17
- Substituted pyrazolo[1,5-a]pyrimidines and their use in treating medical disorders – Patents.comPublication Number:JP-7519221-B2Priority Date:2014-11-06Grant Date:2024-07-19
- Substituted pyrazolo[1,5-a]pyrimidines and their use in the treatment of medical disordersPublication Number:US-2022169652-A1Priority Date:2014-11-06
- Substituted pyrazolo[1,5-a]pyrimidines and their use in the treatment of medical disordersPublication Number:US-11932645-B2Priority Date:2014-11-06Grant Date:2024-03-19
- Substituted pyrazolo(1,5-a)pyrimidines and their use in the treatment of medical disordersPublication Number:WO-2016073895-A1Priority Date:2014-11-06
- Substituted pyrazolo[1,5-A]pyrimidines and their use in the treatment of medical disordersPublication Number:US-10570135-B2Priority Date:2014-11-06Grant Date:2020-02-25
- Substituted pyrazolo(1,5-a)pyrimidines and their use in the treatment of medical disordersPublication Number:EP-4406616-A2Priority Date:2014-11-06
- Substituted pyrazolo[1,5-a]pyrimidines and their use in the treatment of medical disordersPublication Number:US-2020385390-A1Priority Date:2014-11-06
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
References
- “LTI 291”. AdisInsight. Springer Nature Switzerland AG. 15 October 2025. Retrieved 19 February 2026.
- “Delving into the Latest Updates on Pariceract with Synapse”. Synapse. 8 May 2025. Retrieved 19 February 2026.
- “Pariceract Drug Profile”. Ozmosi. 1 January 1900. Retrieved 19 February 2026.
- den Heijer JM, Kruithof AC, Moerland M, Walker M, Dudgeon L, Justman C, et al. (July 2023). “A Phase 1B Trial in GBA1-Associated Parkinson’s Disease of BIA-28-6156, a Glucocerebrosidase Activator”. Movement Disorders. 38 (7): 1197–1208. doi:10.1002/mds.29346. hdl:1887/3722043. PMID 37195859.
- den Heijer JM, Kruithof AC, van Amerongen G, de Kam ML, Thijssen E, Grievink HW, et al. (September 2021). “A randomized single and multiple ascending dose study in healthy volunteers of LTI-291, a centrally penetrant glucocerebrosidase activator”. British Journal of Clinical Pharmacology. 87 (9): 3561–3573. doi:10.1111/bcp.14772. PMC 8451761. PMID 33576113.
- Ellis JM, Fell MJ (September 2017). “Current approaches to the treatment of Parkinson’s Disease”. Bioorganic & Medicinal Chemistry Letters. 27 (18): 4247–4255. doi:10.1016/j.bmcl.2017.07.075. PMID 28869077.
Increased degradation of a-synuclein is another potential mechanism explored in the clinic. Currently, the leading candidates for this approach center on modulating the activity of the Glucocerebrosidase (GBA) pathway. Mutations in GBA are the leading genetic risk factor for sporadic PD and reductions in GBA activity are thought to lead to an accumulation of a-synuclein. Lysosomal Therapeutics is currently developing a brain penetrant, small molecule enhancer of GBA activity called LTI-291 (structure not reported) which is expected to enter Phase 1 testing in 2017.
External links
 | |
| Clinical data | |
|---|---|
| Other names | BIA 28-6156; LTI-291; LIT291; LTI-00291 |
| Routes of administration | Oral[1] |
| Drug class | β-Glucocerebrosidase (GCase) activator or positive allosteric modulator |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1919820-28-2 |
| PubChem CID | 121327414 |
| ChemSpider | 68028144 |
| UNII | V9WUN9UUU8 |
| Chemical and physical data | |
| Formula | C20H30N4O2 |
| Molar mass | 358.486 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
////////pariceract, ANAX LABS, beta-glucocerebrosidase activator, antiparkinsonian, BIA 28-6156, LTI-291, LIT291, LTI-00291, BIA28-6156, LTI- 91, LIT 291, LTI 00291, Gcase activator 1, V9WUN9UUU8
Palacaparib
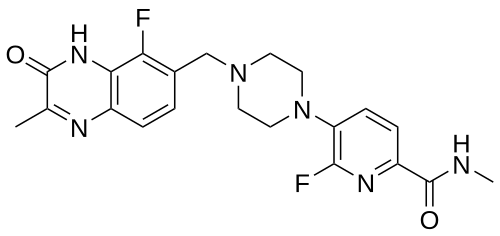
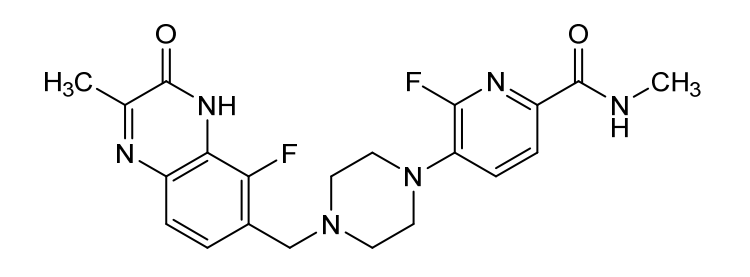
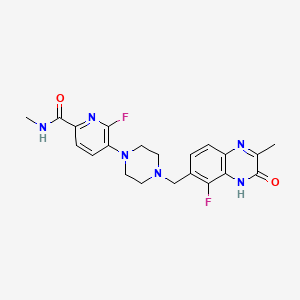
Palacaparib
CAS 2756333-39-6
MFC21H22F2N6O2 MW428.4 g/mol
6-fluoro-5-[4-[(5-fluoro-2-methyl-3-oxo-4H-quinoxalin-6-yl)methyl]piperazin-1-yl]-N-methylpyridine-2-carboxamide
2-Pyridinecarboxamide, 6-fluoro-5-(4-((5-fluoro-3,4-dihydro-2-methyl-3-oxo-6-quinoxalinyl)methyl)-1-piperazinyl)-N-methyl-
6-fluoro-5-{4-[(5-fluoro-2-methyl-3-oxo-3,4-dihydroquinoxalin-6-yl)methyl]piperazin-1-yl}-N-methylpyridine-2-carboxamide
poly (ADP-ribose) polymerase (PARP) inhibitor, antineoplastic, AZD 9574, 9UG32UQW48
Palacaparib is an investigational new drug that is being evaluated by AstraZeneca for the treatment of prostate cancer.[1] It is a selective PARP1 inhibitor.[2][3]
Palacaparib (also known as AZD9574) is an investigational, orally bioavailable cancer drug developed by AstraZeneca. It belongs to a class of medications called PARP inhibitors.
Key Characteristics
- High Selectivity: It selectively targets the PARP1 enzyme. It features an 8,000-fold greater selectivity for PARP1 over other PARP forms like PARP2. This targeted approach aims to lower typical bone marrow toxicities linked with older, non-selective PARP inhibitors.
- Brain Penetrance: Unlike many earlier options, it successfully crosses the blood-brain barrier. This trait makes it a prime candidate for managing primary brain tumors and central nervous system (CNS) metastases.
Mechanism of Action
- Enzyme Binding: Palacaparib tightly binds to the PARP1 enzyme at single-strand DNA break locations.
- DNA Trapping: It traps the enzyme on the damaged DNA, blocking the base excision repair pathway.
- Synthetic Lethality: This stalling stalls replication forks and forces single-strand breaks to progress into double-strand breaks.
- Cell Death: In tumors with homologous recombination deficiency (HRD)—such as BRCA1/2 mutations—cells cannot fix these severe breaks, triggering apoptosis (programmed cell death).
Clinical Research and Targets
According to active registries from the National Cancer Institute (NCI), Palacaparib is undergoing early-phase human trials both as a single agent and alongside other treatments. Researchers are testing its efficacy across several oncological areas:
- Advanced Solid Malignancies: Studies focus heavily on HRD-positive tumors, including specific types of breast, ovarian, and pancreatic cancers.
- Prostate Cancer: Active monotherapy and combination trials target metastatic prostate cancer.
- CNS Malignancies: Preclinical designs show promise in treating gliomas when paired with radiation or alkylating agents like temozolomide.
- OriginatorAstraZeneca
- ClassAntineoplastics; Carbamates; Fluorinated hydrocarbons; Ketones; Piperazines; Pyridines; Quinoxalines; Small molecules
- Mechanism of ActionPoly(ADP-ribose) polymerase-1 inhibitors
- Phase I/IIProstate cancer; Solid tumours
- 03 Jun 2026Phase-I/II clinical trials in Prostate cancer (Combination therapy, Hormone refractory, Second-line therapy or greater, Metastatic disease) in USA (PO) (NCT07590934)
- 14 May 2026AstraZeneca plans a phase I/II trial for Prostate cancer (Metastatic disease, Combination therapy, Second-line therapy or greater, Hormone-refractory) in USA, Australia, Germany, Italy, South Korea, Spain, United Kingdom (PO) in May 2026 (NCT07590934) (EudraCT2025-524920-23)
- 19 Feb 2026Chemical structure information added.
Palacaparib is an orally bioavailable central nervous system (CNS) penetrant and inhibitor of nuclear enzyme poly(ADP-ribose) polymerase (PARP) 1, with potential antineoplastic activity. Upon oral administration, palacaparib selectively binds to PARP1, thereby preventing repair of damaged DNA via the base excision repair (BER) pathway. This agent enhances the accumulation of DNA strand breaks and promotes genomic instability eventually leading to apoptosis. Palacaparib may enhance the cytotoxicity of DNA-damaging agents and reverse tumor cell chemo- and radioresistance. PARP1 catalyzes post-translational ADP-ribosylation of nuclear proteins that signal and recruit other proteins to repair damaged DNA and plays a key role in the repair of single strand DNA (ssDNA) breaks and double-strand break (DSBs). Palacaparib is able to penetrate the blood-brain barrier (BBB).
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021260092&_cid=P22-MS178J-58931-1
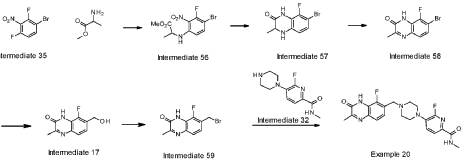
Example 20: 6-fluoro-5-[4-[(5-fluoro-2-methyl-3-oxo-4H-ciuinoxalin-6-yl)methvnDiDerazin-1-vn-N-methyl-pyridine-2-carboxamide
Triethylphosphane (20.90 ml, 145.06 mmol) was added dropwise with an addition funnel to a stirred suspension of 8-fluoro-7-(hydroxymethyl)-3-methyl-1 H-quinoxalin-2-one (intermediate 17) (15.1 g, 72.53 mmol) and 1 ,2-dibromo-1 ,1 ,2,2-tetrachloroethane (52.0 g, 159.56 mmol) in DCM (400 mL) at 0°C under nitrogen. The mixture was stirred at r.t for 3 h gave a light-yellow suspension. Crude LCMS indicated full conversion. DCM was removed under vacuum; the residue was slurry in 300 mL diethyl ether at rt and the light yellow ppt was filtered and washed with 200 ml ether. The solid was taken into 300 ml of water, stirred at rt for 10 min, the solid was collected by filtration, thorough wash (200 ml) with water to remove the salts. The solid was dried under vacuum for overnight (no heat). The solid was washed with hexanes and dried in vacuum in a bushel funnel to give 7-(bromomethyl)-8-fluoro-3-methylquinoxalin-2(1 H)-one (intermediate 59) (22.76 g, 116 %, likely contains some inorganic salts) as an off white solid. Used as such for next reaction. 1 H NMR (500 MHz, DMSO-c/6) 2.42 (3H, s), 4.65 – 4.93 (2H, m), 7.28 – 7.42 (1 H, m), 7.51 (1 H, d), 12.53 (1 H, br s); m/z (ES+) [M+H]+ = 271 , 273.
To a flask charged with 7-(bromomethyl)-8-fluoro-3-methylquinoxalin-2(1 H)-one (intermediate 59) (22.76 g) and 6-fluoro-N-methyl-5-(piperazin-1-yl)picolinamide, 2HCI ( intermediate 32) (24.24 g, 77.9 mmol) in acetonitrile (350 ml), was added DIPEA (38.0 ml, 217.59 mmol) at rt and the resulting mixture was stirred at 70°C for 4 h. Reaction was not complete. To the mixture was added 5 g of Kl and 2 g of Nal and the mixture was stirred at 50°C for 20 h. More 540 mgs (~0.03eq) of 6-fluoro-N-methyl-5-(piperazin-l-yl)picolinamide, 2HCI (intermediate 32) was added to the mixture and the stirring continued at 50°C for 2 h. The solid from the reaction suspension was collected by filtration, washed with acetonitrile and dried. The resulting material was then suspended in water (~400 ml), slurred at rt for 20 min, filtered and dried (97% purity by LCMS). The solid was then dissolved into a mixture of DCM/MeOH (3/1) (about 1.5 L) at reflux, filtered through a pad of silica gel, removed most of the DCM until solid precipitate out and the mixture was kept at rt for 20 min. The solid was collected by filtration and repeated the procedure for the filtrate, and the solids were combined to yield the product 6-fluoro-5-[4-[(5-fluoro-2-methyl-3-oxo-4H-quinoxalin-6-yl)methyl]piperazin-1-yl]-N-methyl-pyridine-2-carboxamide (example 20) (26 g, 84%) as a light yellow solid. 1 H NMR (500 MHz, DMSO-c/6) 2.41 (3H, s), 2.57 – 2.69 (4H, m), 2.76 (3H, d), 3.16 (4H, br s), 3.70 (2H, s), 7.29 (1 H, br t), 7.40 – 7.60 (2H, m), 7.83 (1 H, d), 8.38 (1 H, br d), 12.44 (1 H, br s); m/z (ES+) [M+H]+ = 429.
PAT
- Chemical compoundsPublication Number:US-12421208-B2Priority Date:2020-06-25Grant Date:2025-09-23
- Chemical compoundsPublication Number:US-11795158-B2Priority Date:2020-06-25Grant Date:2023-10-24
- Chemical compoundsPublication Number:US-2022009901-A1Priority Date:2020-06-25
- Combining ATR inhibitors and PARP inhibitors for cancer treatmentPublication Number:IL-309388-APriority Date:2021-06-16
- Use of ATR inhibitors in combination with PARP inhibitors to treat cancerPublication Number:KR-20240021884-APriority Date:2021-06-16
- Quinoxaline derivatives as anticancer drugsPublication Number:CN-115768760-APriority Date:2020-06-25
- Quinoxaline derivatives as anti-cancer drugsPublication Number:WO-2021260092-A1Priority Date:2020-06-25
- Quinoxaline derivatives as anti-cancer drugsPublication Number:EP-4172152-A1Priority Date:2020-06-25
SYN
- Discovery of 6-Fluoro-5-{4-[(5-fluoro-2-methyl-3-oxo-3,4-dihydroquinoxalin-6-yl)methyl]piperazin-1-yl}-N-methylpyridine-2-carboxamide (AZD9574): A CNS-Penetrant, PARP1-Selective InhibitorPublication Name:Journal of Medicinal ChemistryPublication Date:2024-12-10PMID:39655996DOI:10.1021/acs.jmedchem.4c01725
- New Horizons of Synthetic Lethality in Cancer: Current Development and Future PerspectivesPublication Name:Journal of Medicinal ChemistryPublication Date:2024-07-02PMCID:PMC11284803PMID:38955347DOI:10.1021/acs.jmedchem.4c00113
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
 | |
| Clinical data | |
|---|---|
| Other names | AZD-9574 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2756333-39-6 |
| PubChem CID | 162524593 |
| IUPHAR/BPS | 11946 |
| ChemSpider | 115008044 |
| UNII | 9UG32UQW48 |
| ChEMBL | ChEMBL5095223 |
| PDB ligand | A1H64 (PDBe, RCSB PDB) |
| Chemical and physical data | |
| Formula | C21H22F2N6O2 |
| Molar mass | 428.444 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- “Palacaparib”. AdisInsight. Springer Nature Switzerland AG. Retrieved 5 July 2026.
- Johannes JW, Balazs AY, Barratt D, Bista M, Chuba MD, Cosulich S, et al. (December 2024). “Discovery of 6-Fluoro-5-{4-[(5-fluoro-2-methyl-3-oxo-3,4-dihydroquinoxalin-6-yl)methyl]piperazin-1-yl}-N-methylpyridine-2-carboxamide (AZD9574): A CNS-Penetrant, PARP1-Selective Inhibitor”. Journal of Medicinal Chemistry. 67 (24): 21717–21728. doi:10.1021/acs.jmedchem.4c01725. PMID 39655996.
- Shkil DO, Chesnokova NA, Ivashchenko AA, Petersen EV, Maximov PY (May 2026). “Structural Determinants of PARP1 Selectivity from Molecular Dynamics Analysis of PARP1 and PARP2 Complexes”. Molecules. 31 (10). Basel, Switzerland: 1592. doi:10.3390/molecules31101592. PMC 13210057. PMID 42197145.
///////////palacaparib, ANAX LABS, poly (ADP-ribose) polymerase (PARP) inhibitor, antineoplastic, AZD 9574, 9UG32UQW48
Zidesamtinib
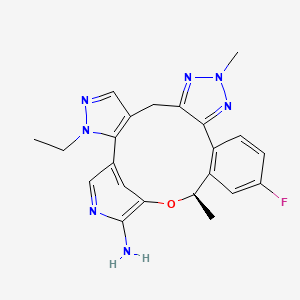
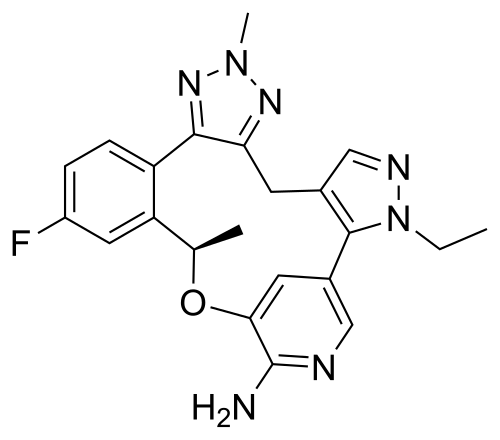
Zidesamtinib
CAS 2739829-00-4
MF C22H22FN7O MW419.5 g/mol
(19R)-3-ethyl-16-fluoro-10,19-dimethyl-20-oxa-3,4,9,10,11,23-hexazapentacyclo[19.3.1.02,6.08,12.013,18]pentacosa-1(25),2(6),4,8,11,13(18),14,16,21,23-decaen-22-amine
- (14R)-7-ethyl-16-fluoro-2,14-dihydro-2,14-dimethyl-7H-8,12-Metheno-4H-pyrazolo[3,4-h]-1,2,3-triazolo[4,5-k][2,5]benzoxaazacyclotetradecin-11-a mine
- (14R)-7-Ethyl-16-fluoro-2,14-dihydro-2,14-dimethyl-7H-8,12-metheno-4H-pyrazolo[3,4-h]-1,2,3-triazolo[4,5-k][2,5]benzoxaazacyclotetradecin-11-amine
- (19R)-3-ethyl-16-fluoro-10,19-dimethyl-20-oxa-3,4,9,10,11,23-hexazapentacyclo[19.3.1.02,6.08,12.013,18]pentacosa-1(25),2(6),4,8,11,13(18),14,16,21,23-decaen-22-amine
- 7H-8,12-Metheno-4H-pyrazolo[3,4-h]-1,2,3-triazolo[4,5-k][2,5]benzoxaazacyclotetradecin-11-amine, 7-ethyl-16-fluoro-2,14-dihydro-2,14-dimethyl-, (14R)-
To treat adults with locally advanced or metastatic ROS1-positive non-small cell lung cancer after receiving a ROS1 kinase inhibitor
FDA 2026, APPROVALS 2026, Jideytro, NVL-520, NUV-520, NU-520, NVL 520, NUV 520, NU 520, MX5KQV5XHC
Zidesamtinib (sold under the brand name Jideytro) is an oral, highly selective, next-generation kinase inhibitor approved by the U.S. Food and Drug Administration (FDA) on July 22, 2026, to treat adults with locally advanced or metastatic ROS1-positive non-small cell lung cancer (NSCLC) who have previously been treated with at least one ROS1 kinase inhibitor. Developed originally by Nuvalent and subsequently acquired by GSK, it represents a major milestone as GSK’s first approved therapeutic targeting lung cancer.
Mechanism of Action
Zidesamtinib functions by targeting and inhibiting the receptor tyrosine kinase c-ros oncogene 1 (ROS1). It is custom-engineered to solve the primary clinical challenges that limit previous therapies:
- Overcoming Resistance Mutations: It binds tightly to wild-type ROS1 and remains robustly active against a broad array of treatment-emergent point mutants. This includes G2032R (the most common solvent-front resistance mutation), as well as S1986Y/F, L2026M, and D2033N mutations.
- Blood-Brain Barrier Penetration: It features high central nervous system (CNS) penetrance to effectively treat and control brain metastases, which are frequent in aggressive ROS1-positive cancers.
- TRK-Sparing Design: Unlike older dual-acting inhibitors, it deliberately avoids inhibiting the structurally similar tropomyosin receptor kinase (TRK) family. This minimizes off-target TRK-related neurological toxicities like severe dizziness and ataxia.
Clinical Trial Outcomes
The FDA approval was heavily supported by data from the ongoing global, single-arm, Phase 1/2 ARROS-1 clinical trial (N=117 heavily pretreated patients):
- Overall Response: Delivered an Objective Response Rate (ORR) of 44% in patients who had exhausted alternative TKI options.
- Subgroup Efficacy: Achieved a 51% ORR in patients who had received only one prior ROS1 inhibitor, a 54% ORR in those harboring the G2032R mutation, and an intracranial ORR of 48% for patients with active brain metastases.
- Durability: Showed prolonged disease control, with a 12-month duration of response (DOR) rate standing at 69%.
Administration and Side Effects
Jideytro is formulated as an oral tablet taken once daily, with or without food. It demonstrates a highly tolerable safety profile, with only a 10% dose reduction rate and a 2% treatment discontinuation rate due to adverse events.
- Common Adverse Reactions (≥ 15%): Edema (swelling), peripheral neuropathy, constipation, fatigue, and dyspnea (shortness of breath).
- Warnings & Precautions: Includes risks of mild CNS reactions (dizziness, cognitive alterations), QTc interval prolongation, skeletal fractures, pancreatic toxicity, and interstitial lung disease (ILD)/pneumonitis.
Zidesamtinib, sold under the brand name Jideytro, is an anti-cancer medication used for the treatment of previously treated locally advanced or metastatic ROS1+ non-small cell lung cancer.[1][2][3] It is taken by mouth once daily.[1][2][3]
Medical uses
Indication
Zidesamtinib is a prescription medicine used to treat adults with non-small cell lung cancer that has spread within the chest or other parts of the body and is caused by an abnormal ROS1 gene, and who have received a ROS1 kinase inhibitor.[1][2][3]
Mechanism of action
Zidesamtinib is a kinase inhibitor that works by blocking ROS1, an abnormal protein that drives some lung cancers to grow, including forms that have become resistant to earlier ROS1 treatments.[4] Jideytro also works on the related proteins ALK and TRK. In laboratory and animal studies, Zidesamtinib stopped cancer cells with ROS1 changes from growing and slowed tumor growth, including tumors in the brain.[2]
PAT
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023056405&_cid=P10-MRYBTX-59205-1
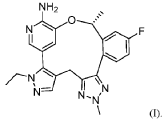
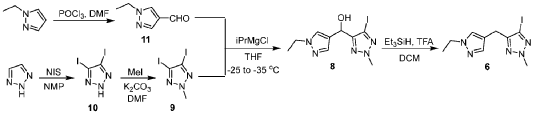
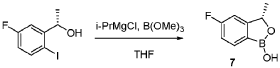
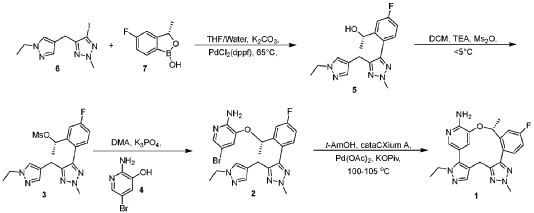
Scheme 3. Synthesis of Compound 1.
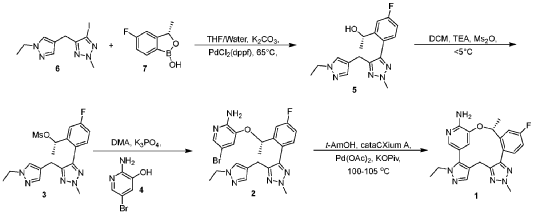
[00548] Synthesis of Compound 5. To a reactor was charged THF (10 vol ), water (1 vol ), followed by Compound 6 (850.0 g, 2.68 mol, 1 equiv.) and Compound 7 (534.0 g, 3.22 mol, 1.2 equiv ) at 20~30°C. The solids were completely dissolved at 20~30°C while stirring for 15 min and K2CO3 (1.11 kg, 3 equiv.) was added in portions over 10-15 min at 20~30°C. The reaction mixture was fully refdled with nitrogen, and was added Pd(dppf)C12 (78.5 g, 0.04 equiv.) in one portion under nitrogen. The reaction mixture was fully refdled with nitrogen again, then heated to 60-65 °C and stirred at 60~65°C for 16 h under nitrogen. The reaction mixture was cooled to 20~30°C, fdtered through a 10 cm celite pad (2X, 2.4 kg celite). The combined fdtrates were washed with EtOAc (10 vol., 21 L) and separated. The organic phase was washed with water (5 vol., 10.5 L) and separated. The organic phase was stirred for 1 h at 40-45°C in 5 w% aqueous L-cysteine (2.0 eq., 1.61 kg in 30.6 kg water) and separated. The organic phase was washed with water (5 vol., 10.5 L) and separated. The resulting organic phase was concentrated at 45-50°C in vacuum to afford crude product as a light brown oil (2.28 kg). To the crude product was charged MTBE (228 mL, 0.1 vol. relative to crude product), heated to 50°C over 15 min, followed by isopropyl ether (2.28 L, 1 vol.) dropwise over 1 h at 45~50°C, then cooled to 10°C over 2 h. A large amount of solids came out and the resulting slurry was stirred for 2 h at 10-15°C. The solids were collected by fdtration, dried in oven at 45°C for 16 h to get crude Compound 5 as a pale-yellow solid (1.67 kg, 96.3% /220 nm, >99.9%/220 nm chiral purity). 1.67 kg of crude Compound 5 was purified by silica gel chromatography (EtOAc/ n-heptane=l: 1, 2.5X silica gel, 100-200 meshes) to get Compound 5 as an off-white solid (1.58 kg, 99.6%/220 nm, >99.9%/220 nm chiral purity, 97.9 w%, 72% yield). H NMR (400 MHz, DMSO) 5 7.44 (dd, J = 10.5, 2.5
Hz, 1H), 7.36 (s, 1H), 7.22 (dd, J = 8.3, 6.0 Hz, 1H), 7.16 – 7.08 (m, 2H), 5.25 (d, J = 4.2 Hz, 1H), 4.86 – 4.68 (m, 1H), 4.14 (s, 3H), 4.00 (q, J = 7.2 Hz, 2H), 3.72 (s, 2H), 1.27 (t, J = 7.3 Hz, 3H), 1.11 (d, J = 6.3 Hz, 3H). MS (ESI, m/z): 330.20 (M + H)+.
[00549] In another example, a similar procedure was run in a 0.5:2 biphasic mixture of toluene and water (2.5 vol.) with a catalystic amount (e.g. 0.002 mol equiv.) Pd(Amphos)C12 (instead of 0.04 mol equiv. of PdidppfhCE) used as the catalyst. Potassium phosphate (K3PO4 3 H2O) substituted potassium carbonate (K2CO3) 3.0 mol equiv. as the base, and the amount of Compound 7 employed was 1.02 mol equiv. The improved process was conducted at 50 °C. At the end of the reaction, the organic layer was fdtered and treated with activated carbon and concentrated, and the final material was crystallized from toluene/heptane/water to give Compound 5 in 92% yield and 99.9% purity.
[00550] Synthesis of Compound 3. To a 50 L reactor was charged dichloromethane (11.25 L), Compound 5 (750 g, >99.9%/220 nm chiral purity) and triethylamine (920.0 g) at r t. (20~30°C). The resulting mixture was refilled with nitrogen and cooled to 0°C. To it was added a solution of MS2O (793.0 g) in dichloromethane (3.75 L) drop-wise over 45 min while keeping the temperature at 0~5°C. The reaction mixture was stirred at 0~5°C for 1 h under nitrogen. The reaction mixture was quenched with cooled water (7.5 L) at 5~15°C and separated. The organic phase was washed with cooled water (3.75 L) and separated. The organic phase was dried over anhydrous Na2SC>4, filtered and concentrated at 25~30°C in vacuum to around 2 vol., then switched to n-heptane (2.25 L) and concentrated at 25~30°C in vacuum to around 2 vol. of Compound 3 in n-heptane. n-heptane /EtOAc (3.0 L, lOv/lv) was added to the above mixture and the mixture was slurried for 1 h at 0~10°C under nitrogen and filtered. The filter cake was washed with n-heptane (1.5 L), dried in vacuum at 25~30°C for 5 h to afford Compound 3 as an off-white solid (845 g, 98.9 w%, 99.98%/220 nm chiral purity, 91% yield). H NMR (400 MHz, CDC13) 5 7.35 (dd, J = 9.6, 2.5 Hz, 1H), 7.24 – 7.18 (m, 2H), 7.12 (s, 1H), 7.08 (td, J = 8.3, 2.6 Hz, 1H), 5.78 (d, J = 6.4 Hz, 1H), 4.21 (s, 3H), 4.05 (q, J = 7.3 Hz, 2H), 3.90 – 3.76 (m, 2H), 2.78 (s, 3H), 1.58 (d, J = 6.5 Hz, 3H), 1.40 (t, J = 7.3 Hz, 3H). MS (ESI, m/z): 408.20 (M + H)+.
[00551] In another example, triethylamine base (1.3 mol equiv.), MS2O (1.2 mol equiv.), and dichloromethane solvent (10 vol) were used. The reaction mixture was quenched with aqueous sodium bicarbonate to remove excess MS2O, and crystallization from dichloromenthane/hexane results in 98% yield with 100% purity of Compound 3.
[00552] Synthesis of Compound 2. A 20 L reactor was refilled with nitrogen, then charged with DMA (12.6 L) at r.t. (20~25°C) To the reactor was charged Compound 4 (390.0 g) and Compound 3 (840.0 g, 99.98%/220 nm chiral purity) in one portion at 20~25°C through a dry nitrogen flow. The reaction mixture was heated to 35°C over 15 min and stirred for 5-10 min at 35~40°C to get a clear solution. To the reaction mixture was charged powder K3PO4 (875.0 g) in one portion at 35~45°C. After complete addition, the resulting mixture was heated to 60°C over 20 min and stirred at 58~63°C for 1.5 h through a dry nitrogen flow. The reaction mixture was cooled to 25~30°C, filtered through a celite pad (5 cm, 1.5 kg) and rinsed the filter cake with EtOAc (2 L, 2-3 vol.). The filtrate was poured into water (16.8 L, 20 vol.) at 0-10°C, extracted with EtOAc (10 L, 12 vol.) and separated. The aqueous phase was extracted with EtOAc (5 L, 6 vol.). The combined organic phases were washed with water (5 L*3, 6 vol. *3), concentrated at 50°C in vacuum to afford crude product as a gray solid (956 g). The crude product was dissolved in EtOAc (950 mL, 1 vol. relative to crude product) at 35~40°C, then was added dropwise n-heptane (950 mL, 1 vol. relative to crude product) at 30~40°C over 20 min. The resulting mixture was cooled to 20~25°C over 30 min and stirred for 1 h at 30-40°C. Some solids came out slowly and n-heptane (1.9 L, 2 vol. relative to crude product) was added dropwise to the slurry mixture at 20~25°C over 30 min. The precipitates were stirred at 15~20°C for 3 h and filtered. The filter cake was washed with n-heptane (1.5 L) and dried in oven at 45-50°C for 16 h to afford Compound 2 as a pale-yellow solid (743 g, 98.6%/220 nm, 96.9 w%, 99.98%/220 nm chiral purity, 0.48%KF, 72% yield). H NMR (400 MHz, DMSO) 5 7.54 (dd, J = 10.2, 2.7 Hz, 1H), 7.51 (d, J = 1.9 Hz, 1H), 7.42 (s, 1H), 7.31 (dd, J = 8.5, 5.8 Hz, 1H), 7.22 (td, J = 8.4, 2.7 Hz, 1H), 7.17 (s, 1H), 6.92 (d, J = 1.8 Hz, 1H), 6.14 (s, 2H), 5.47 (q, J = 6.0 Hz, 1H), 4.22 (s, 3H), 4.02 (q, J = 7.3 Hz, 2H), 3.78 (q, J = 16.1 Hz, 2H), 1.40 (d, J = 6.3 Hz, 3H), 1.29 (t, J = 7.3 Hz, 3H). MS (ESI, m/z): 500.30 (M + H)+.
[00553] In another example, a process was developed where Compound 4 (1.1 mol equiv. to Compound 3) was used. Potassium phosphate base (K2PO4, 4. 1 mol equiv.) and DMA (16 vol.) were substituted with cesium carbonate (CS2CO3, 2.2 mol equiv.) and NMP (5.6 vol.). The reaction was carried out at 20~30°C. Following completion of the reaction, the crude product was precipitated with water. The material was then dissolved in ethyl acetate, washed with water, and treated with activated carbon. The product is subsequently crystallized from toluene/ethyl acetate/heptane to give Compound 2 in 80% yield and 99.9% purity.
[00554] Synthesis of Compound 1. To a reactor was charged t-AmOH (20 vol.), Compound 2 (700.0 g, 99.99% chiral purity) and potassium pivalate (588.0 g). The reaction mixture was fully refilled with nitrogen. To the reaction mixture was added cataCXium A (120.4 g) and Pd(OAc)2 (37.8 g) at r.t. under nitrogen. The resulting mixture was heated to 100°C and stirred for 18 h under nitrogen. The reaction mixture was cooled to 30°C , filtered through a celite pad and washed the filter cake with EtOAc (3 vol.). The filtrate was washed with water (5 vol. *2) and separated. The upper organic phase was concentrated in vacuum to afford a brown oil. The oil was dissolved in EtOAc (27 L) then added 5w% aqueous L-cysteine (0.98 kg in 18.6 kg water), stirred for 1 h at 40~45°C and separated. The organic phase was washed with water (6.75 L) and separated. 5w% aqueous L-cysteine (0.98 kg in 18.6 kg water) was charged to the above organic phase, stirred for 1 h at 40~45°C and separated. The organic phase was washed with water (6.75 L.) and separated. The organic phase was concentrated in vacuum at 45~50°C to afford a brown solid (1.12 kg). The crude solid (1.12 kg) was further purified by silica gel chromatography eluted with EtOAc/DCM (dry loading, 3X, 100-200 meshes, EtOAc:DCM=l : 1) to afford a pale-yellow solid ( 1.02 kg). The solid was dissolved in EtOAc (600 mL, 2 vol.) at 50~60°C, then was added n-heptane (1.8 L, 6 vol.) dropwise over 50 min at 50~60°C. A large of solids came out during addition. The resulting slurry was cooled to 15~20°C over 50 min and stirred for 30 min at 15~20°C. The slurry was concentrated in vacuum at 45~50°C to 2-3 vol. mixture, n-heptane (1.2 L, 4 vol.) was added to the
above mixture (2-3 vol.), concentrated in vacuum at 45~50°C to 2-3 vol. mixture. The mixture was cooled to 10~15°C over 2 h, stirred at 10~15°C for 1 h and filtered. The filtered cake was rinsed with n-heptane (600 mb) and dried in vacuum at 50°C for 20 h to afford Form 1 of Compound 1 as an off-white solid (280 g, 99.0%). H NMR (400 MHz, DMSO) 57.79 (dd, J = 10.3, 2.2 Hz, 1H), 7.58 (s, 1H), 7.43 (d, J = 1.8 Hz, 1H), 7.24 – 7.16 (m, 2H), 6.13 (s, 2H), 6.08 (d, J = 1.7 Hz, 1H), 5.31 – 5.23 (m, 1H), 4.16 (s, 3H), 4.05 – 3.94 (m, 2H), 3.78 (d, J = 15.6 Hz, 1H), 2.98 (d, J = 15.5 Hz, 1H), 1.71 (d, J = 6.2 Hz, 3H), 1.26 (t, J = 7.2 Hz, 3H). MS (ESI, m/z): 420.30 (M + H)+. XRPD (FIG. 1), TG/DTA (FIG. 2), DSC (FIG. 3), DVS (FIG. 4), and FT-IR (FIG. 5) results for a sample of Form 1 were obtained.
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
References
- “JIDEYTRO™ (zidesamtinib) for Patients”. Jideytro. 2026-02-05. Retrieved 2026-07-23.
- “Jideytro: Uses, Dosage, Side Effects & Warnings”. Drugs.com. Retrieved 2026-07-23.
- “Nuvalent Announces FDA Acceptance of New Drug Application for Zidesamtinib for the Treatment of TKI Pre-treated Patients with Advanced ROS1-positive NSCLC”. Nuvalent Investors. Retrieved 2026-07-23.
- Wespiser M, Gille R, Pérol M (2026). “ROS1-positive non-small cell lung cancer: from genomics to treatment decisions”. Frontiers in Oncology. 16 1739598. doi:10.3389/fonc.2026.1739598. PMC 12907153. PMID 41704605.
- “New FDA Drug Approvals for 2026”. Drugs.com. Retrieved 2026-07-23.
- Center for Drug Evaluation and Research (2026-07-22). “Novel Drug Approvals for 2026”. FDA.
- Zidesamtinib Selective Targeting of Diverse ROS1 Drug-Resistant MutationsPublication Name:Molecular Cancer TherapeuticsPublication Date:2025-05-09PMCID:PMC12214885PMID:40299789DOI:10.1158/1535-7163.mct-25-0025
- Targeting Solvent-Front Mutations for Kinase Drug Discovery: From Structural Basis to Design StrategiesPublication Name:Journal of Medicinal ChemistryPublication Date:2024-08-15PMID:39143914DOI:10.1021/acs.jmedchem.4c00361
- NVL-520 Is a Selective, TRK-Sparing, and Brain-Penetrant Inhibitor of ROS1 Fusions and Secondary Resistance MutationsPublication Name:Cancer DiscoveryPublication Date:2022-12-13PMCID:PMC9975673PMID:36511802DOI:10.1158/2159-8290.cd-22-0968
- Electron transport by C-type cytochromes. I. The reaction of horse heart cytochrome c with anionic reductantsPublication Name:Biophysics of structure and mechanismPublication Date:1975-02-19PMID:10021DOI:10.1007/bf00539772
PAT
- Solid forms, pharmaceutical compositions and preparation of heteroaromatic macrocyclic ether compoundsPublication Number:WO-2023056405-A1Priority Date:2021-10-01
- Solid forms, pharmaceutical compositions and preparation of heteroaromatic macrocyclic ether compoundsPublication Number:US-12043626-B2Priority Date:2021-10-01Grant Date:2024-07-23
- Methods of treating solid tumor using heteroaromatic macrocyclic ether compoundsPublication Number:US-2024398768-A1Priority Date:2021-10-01
- Heteroaromatic macrocyclic ether chemotherapeutic agentsPublication Number:US-12054498-B2Priority Date:2020-05-05Grant Date:2024-08-06
- Heteroaromatic macrocyclic ether chemotherapeutic agentsPublication Number:US-2023107663-A1Priority Date:2020-05-05
- Heteroaromatic macrocyclic ether chemotherapeutic agentsPublication Number:US-11542278-B1Priority Date:2020-05-05Grant Date:2023-01-03
- Heteroaromatic macrocyclic ether chemotherapeutic agentsPublication Number:WO-2021226208-A2Priority Date:2020-05-05
- Heteroaromatic macrocyclic ether chemotherapeutic agentsPublication Number:US-2024352037-A1Priority Date:2020-05-05
 | |
| Clinical data | |
|---|---|
| Pronunciation | jih-DAY-troh[1][2] |
| Trade names | Jideytro |
| Other names | NUV-520; NVL 520 |
| AHFS/Drugs.com | jideytro |
| Routes of administration | By mouth |
| Drug class | Tyrosine kinase inhibitor |
| Legal status | |
| Legal status | US: ℞-only |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2739829-00-4 |
| PubChem CID | 166560233 |
| IUPHAR/BPS | 12392 |
| DrugBank | DB21623 |
| ChemSpider | 128922073 |
| UNII | MX5KQV5XHC |
| KEGG | D12899 |
| ChEBI | CHEBI:747901 |
| ChEMBL | ChEMBL5314497 |
| Chemical and physical data | |
| Formula | C22H22FN7O |
| Molar mass | 419.464 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
////////zidesamtinib, anax labs, CANCER, FDA 2026, APPROVALS 2026, Jideytro, NVL-520, NUV-520, NU-520, NVL 520, NUV 520, NU 520, MX5KQV5XHC
Gedatolisib
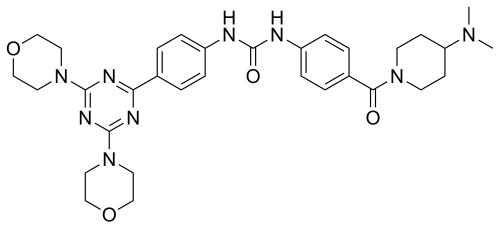
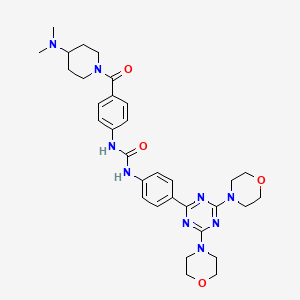
Gedatolisib
Approvals 3026, FDA 2026, 7/14/2026, Revtorpyk
| PF-05212384; PF-5212384; PKI-587 CAS 1197160-78-3 Chemical Formula: C32H41N9O4 Molecular Weight: 615.72 1-(4-{[4-(Dimethylamino)-1-piperidinyl]carbonyl}phenyl)-3-{4-[4,6-di(4-morpholinyl)-1,3,5-triazin-2-yl]phenyl}urea 3-{4-[bis(morpholin-4-yl)-1,3,5-triazin-2-yl]phenyl}-1-{4-[4-(dimethylamino)piperidine-1-carbonyl]phenyl}urea N-[4-[[4-(Dimethylamino)-1-piperidinyl]carbonyl]phenyl]-N’-[4-[4,6-di(4-morpholinyl)-1,3,5-triazin-2-yl]phenyl]urea гедатолисиб [Russian] [INN] غيداتوليسيب [Arabic] [INN] 吉达利塞 [Chinese] [INN] |
1-(4-(4-(Dimethylamino)piperidine-1-carbonyl)phenyl)-3-(4-(4,6-dimorpholino-1,3,5-triazin-2-yl)phenyl)urea
In combination with fulvestran, to treat hormone receptor-positive, human epidermal growth factor receptor 2-negative, locally advanced or metastatic breast cancer without a PIK3CA mutation detected following progression on or after treatment with at least one line of endocrine therapy in the metastatic setting
Gedatolisib, sold under the brand name Revtorpyk, is an anti-cancer drug used for the treatment of breast cancer.[1] It is under development by Celcuity, Inc. Gedatolisib is a kinase inhibitor.[1] The mechanism of action is accomplished by binding the different p110 catalytic subunit isoforms of PI3K and the kinase site of mTOR.[2] Gedatolisib is administered by intravenous infusion.[1]
Gedatolisib was approved for medical use in the United States in July 2026.[1][3]
Medical uses
Gedatolisib is indicated in combination with fulvestrant, with or without palbociclib, for the treatment of adults with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative locally advanced or metastatic breast cancer without a PIK3CA mutation detected following progression on or after treatment with at least one line of endocrine therapy in the metastatic setting.[1]
- Synthesis of New Dialkyl 2,2′-[Carbonylbis(azanediyl)]dibenzoates via Curtius RearrangementDOI: 10.1055/s-0040-1706643Publication Date: 2021Publication Name: Synthesis
- A New One-Pot Three-Component Synthesis of 4-Aryl-6-cycloamino-1,3,5-triazin-2-amines under Microwave IrradiationDOI: 10.1055/a-1401-2795Publication Date: 2021Publication Name: Synthesis
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US42900900&_cid=P22-MRVGV9-51327-1
Example 76
Preparation of 1-(4-(4-(dimethylamino)piperidine-1-carbonyl)phenyl)-3-(4-(4,6-dimorpholino-1,3,5-triazin-2-yl)phenyl)urea
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2010096619&_cid=P22-MRVGV9-51327-1
Scheme 1
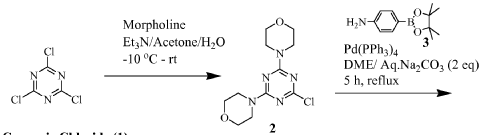
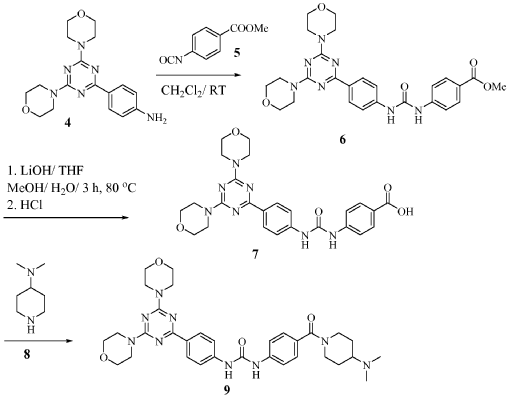
Preparation of 1-(4-(4-(dimethylamino) piperidine-1-carbonyl)phenyl-3-(4-(4,6- dimorpholino-1 ,3,5-triazine-2-yl)phenyl) urea (9)
To a slurry of 4-(3-(4-(4,6-dimorpholino-1 ,3,5-triazine-2- yl)phenyl)ureido)benzoic acid (7, 45.5 g, 0.09 mol) in dry THF (1.6 L) heated to 50 0C was added N,N’-carbonyl diimidazole (28 g, 0.17 mol). The reaction mixture was heated for 2 hours and followed by dimethylaminopiperidine (8, 23.5 g, 0.18 mol) and stirred at 53 0C for 16 hours. The reaction mixture was cooled to the room temperature and filtered. The cake was washed with 2-propanol and air-dried to give 97 % pure white powder in 88% yield (49.2 g, 0.08 mol). To the solids stirred in dimethyl acetamide (DMAC, 165 ml) at 70° C for 1 hour was added 2-propanol (640 ml) and the mixture was stirred at 65 0C for additional 1 hour. The solids were filtered, washed with 2-propanol and dried in a vacuum oven at 700C for 16 hour to give crystalline white powder (45 g) with >99% purity. The above-mentioned work up process and crystallization procedure gave a Pd residue of 20 ppm. Alternate procedures for the formation of 1-(4-(4-(dimethylamino) piperidine-1 – carbonyl)phenyl-3-(4-(4,6-dimorpholino-1 ,3,5-triazine-2-yl)phenyl) urea (9)
To the solution of 4-(4,6-dimorpholin-4-yl-1 ,3,5-triazin-2-yl) aniline (4, 18 g, 0.052 mol) in dichloromethane (300 ml) was added methyl 4-isocyanato benzoate (5, 10.5 g, 0.061 mol) and the reaction mixture was stirred for 5 hours. The separated solids were filtered, washed with ether and air dried to give beige solids (21 g, 0.04 mol). Yield 77%. 90 % pure by HPLC; Mass: 520.1 (M+H). Preparation of 4-(3-(4-(4,6-dimorpholino-1 ,3,5-triazine-2-yl)phenyl)ureido) benzoic acid (7)
The mixture of methyl 4-(3-(4-(4,6-dimorpholino-1 ,3,5-triazine-2-yl)ureido)benzoate (6, 21 g, 0.04mol) and lithium hydroxide monohydrate (3.8 g, 0.09 mol) in THF (120 ml), MeOH (60 ml), and water (60 ml) was heated at 80 0C for 3 hours. The dark brown solution was cooled to room temperature and made acidic with concentrated HCI. The solids were filtered, washed with water, washed with acetone , washed with ether, and dried in a vacuum oven at 60 0C for 48 hours to give off white solids of 4-(3-(4-(4,6-dimorpholino-1 ,3,5-triazine-2-yl)phenyl)ureido) benzoic acid (19.2 g, 0.038 mol). Mass: 506.3 (M+H)+; Yield.94%. 1 -(4-(4-(dimethylamino) piperidine-1 -carbonyl)phenyl-3-(4-(4,6-dimorpholino- 1 ,3,5-triazine-2-yl)phenyl) urea (9)
The suspension of 4-(3-(4-(4,6-dimorpholino-1 ,3,5-triazine-2-yl)phenyl)ureido) benzoic acid (7, 17 g, 33.66 mmol) and N-(3-dimethylaminopropyl)ethyl carbodiimide hydrochloride (9.5 g, 49.5 mmol) in THF (200 ml) and acetonitrile (50 ml) was stirred for 10 min and followed by addition of 1-hydroxybenzotriazole hydrate (6.4 g, 47.88 mmol). The reaction mixture was stirred for 30 min and 4-dimethylaminopiperidine (8, 8.86 g, 69.2 mmol) was added by drops. After being stirred for additional 16 hours, the reaction mixture was concentrated to min. The solids were filtered and washed thoroughly with water (very fine suspension). The cake was slurred in hot ethanol, filtered and dried in a vacuum oven at 68 0C for 16 hours to give off white solids (10.3 g, 16.77 mmol). M. p. 238-240 0C. 99 % pure. Mass: 616.3 (M+H)+; Yield 50 %.
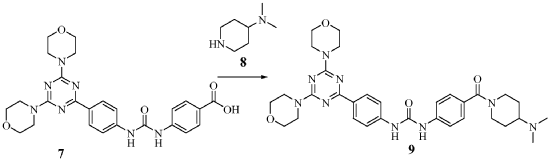
PATENT
WO 2009143317
WO 2010096619
WO 2012148540
WO 2014151147
PATENT
US 20170119778
PAPER
Journal of Medicinal Chemistry (2010), 53(6), 2636-2645
http://pubs.acs.org/doi/abs/10.1021/jm901830p
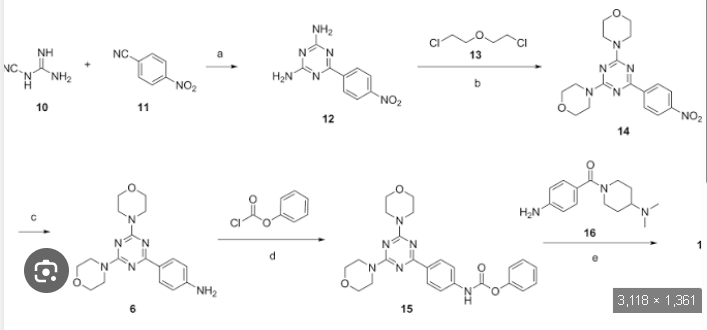
J. Med. Chem., 2010, 53 (6), pp 2636–2645
DOI: 10.1021/jm901830p
Abstract
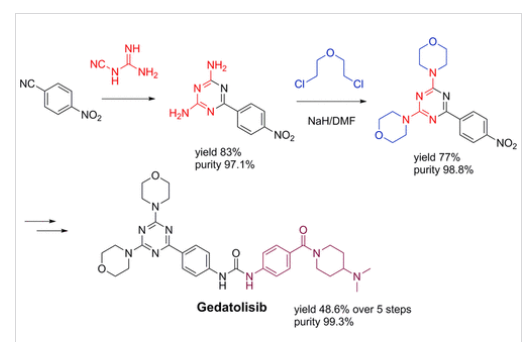
The PI3K/Akt signaling pathway is a key pathway in cell proliferation, growth, survival, protein synthesis, and glucose metabolism. It has been recognized recently that inhibiting this pathway might provide a viable therapy for cancer. A series of bis(morpholino-1,3,5-triazine) derivatives were prepared and optimized to provide the highly efficacious PI3K/mTOR inhibitor 1-(4-{[4-(dimethylamino)piperidin-1-yl]carbonyl}phenyl)-3-[4-(4,6-dimorpholin-4-yl-1,3,5-triazin-2-yl)phenyl]urea 26 (PKI-587). Compound 26 has shown excellent activity in vitro and in vivo, with antitumor efficacy in both subcutaneous and orthotopic xenograft tumor models when administered intravenously. The structure−activity relationships and the in vitro and in vivo activity of analogues in this series are described.
Preparation of 1-(4-{[4-(Dimethylamino)piperidin-1-yl]carbonyl}phenyl)-3-[4-(4,6-dimorpholin-4- yl-1,3,5-triazin-2-yl)phenyl]urea (26)
MS (ESI) m/z = 616.7. HRMS: calcd for C32H41N9O4 + H+, 616.335 43; found (ESI-FTMS, [M + H]+), 616.334 24. Purity by analytical HPLC 99.3%. (Prodigy ODS3, 0.46 cm × 15 cm, 20 min gradient acetonitrile in water, trifluoroacetic acid, detector wavelengths, 215 and 254 nm.) 1H NMR (DMSO-d6) δ 1.29−1.36 (m, 6H), 2.6 (m, 4H), 2.9 (m,1H), 3.3 (m, 4H), 3.6 (m, 8H), 3.7 (m, 8H), 7.3 (d, J = 8.3 Hz, 2H), 7.51−7.57 (m, 4H), 8.3 (d, J = 8.3 Hz 2H), 8.9 (s, 1H), 9.0 (s, 1H) ppm. Anal. Calcd for C32H41N9O4: C 62.42%, H 6.71%, N 20.47%. Found: C 62.34%, H 6.67%, N 20.39%.
PAPER
Bioorganic & Medicinal Chemistry Letters (2011), 21(16), 4773-4778.
http://www.sciencedirect.com/science/article/pii/S0960894X11008468
PAPER
New and Practical Synthesis of Gedatolisib
http://pubs.acs.org/doi/10.1021/acs.oprd.7b00298
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.7b00298
Abstract
A new, practical, and convergent synthetic route of gedatolisib, an antitumor agent, is developed on a hectogram scale which avoids the Pd coupling method. The key step is adopting 6-(4-nitrophenyl)-1,3,5-triazine-2,4-diamine and 2,2′-dichlorodiethyl ether to prepare the key 4,4′-(6-(4-nitrophenyl)-1,3,5-triazine-2,4-diyl)dimorpholine in 77% yield and 98.8% purity. Gedatolisib is obtained in 48.6% yield over five simple steps and 99.3% purity (HPLC). Purification methods of the intermediates and the final product involved in the route are given.
off-white solid. 1H NMR (400 MHz, DMSO-d6): δ 1.46 (brs, 2H), 1.89 (brs, 2H), 2.29 (s, 6H), 2.94 (brs, 2H), 3.76 (m, 8H), 3.89 (m, 8H), 7.09 (d, J = 8.4 Hz, 2H), 7.20 (d, J = 8.4 Hz, 2H), 7.50 (d, J = 8.7 Hz, 2H), 8.28 (s, 1H), 8.31 (d, J = 8.6 Hz, 2H), 8.48 (s, 1H). ESI-MS (m/z) 615.9 (M + H). HPLC conditions: Column: Agilent Eclipse XDB-C18 (250 mm × 4.6 mm × 5 μm); Detection: 254 nm; Flow rate: 0.8 mL/min; Temperature: 30 °C; Injection load: 1 μL; Solvent: MeOH; Concentration: 0.5 mg/mL; Run time: 20 min; Mobile phase A: water; Mobile phase B: MeOH/TEA = 100:0.1; Gradient program: time (min): 20; % of mobile phase A: 10; % of mobile phase B: 90; tR = 2.598 min, purity: 99.34%
- Zhao, X.; Tan, Q.; Zhang, Z.; Zhao, Y. Med. Chem. Res. 2014, 23, 5188– 5196 DOI: 10.1007/s00044-014-1084-z
- Khafizova, G.; Potoski, J. R. PCT Int. Appl. WO 2010096619, 2010.
- Venkatesan, A. M.; Chen, Z.; Dehnhardt, C. M.; Dos Santos, O.; Delos Santos, E. G.; Zask, A.; Verheijen, J. C.; Kaplan, J. A.; Richard, D. J.; Ayral-Kaloustian, S.; Mansour, T. S.; Gopalsamy, A.; Curran, K. J.; Shi, M. PCT Int. Appl. WO 2009143317, 2009.
REFERENCES
1: Gedaly R, Galuppo R, Musgrave Y, Angulo P, Hundley J, Shah M, Daily MF, Chen C, Cohen DA, Spear BT, Evers BM. PKI-587 and sorafenib alone and in combination on inhibition of liver cancer stem cell proliferation. J Surg Res. 2013 Nov;185(1):225-30. doi: 10.1016/j.jss.2013.05.016. Epub 2013 May 25. PubMed PMID: 23769634.
2: Gedaly R, Angulo P, Hundley J, Daily MF, Chen C, Evers BM. PKI-587 and sorafenib targeting PI3K/AKT/mTOR and Ras/Raf/MAPK pathways synergistically inhibit HCC cell proliferation. J Surg Res. 2012 Aug;176(2):542-8. doi: 10.1016/j.jss.2011.10.045. Epub 2011 Nov 21. PubMed PMID: 22261591.
3: Dehnhardt CM, Venkatesan AM, Chen Z, Delos-Santos E, Ayral-Kaloustian S, Brooijmans N, Yu K, Hollander I, Feldberg L, Lucas J, Mallon R. Identification of 2-oxatriazines as highly potent pan-PI3K/mTOR dual inhibitors. Bioorg Med Chem Lett. 2011 Aug 15;21(16):4773-8. doi: 10.1016/j.bmcl.2011.06.063. Epub 2011 Jun 21. PubMed PMID: 21763134.
4: Mallon R, Feldberg LR, Lucas J, Chaudhary I, Dehnhardt C, Santos ED, Chen Z, dos Santos O, Ayral-Kaloustian S, Venkatesan A, Hollander I. Antitumor efficacy of PKI-587, a highly potent dual PI3K/mTOR kinase inhibitor. Clin Cancer Res. 2011 May 15;17(10):3193-203. doi: 10.1158/1078-0432.CCR-10-1694. Epub 2011 Feb 15. PubMed PMID: 21325073.
5: Venkatesan AM, Chen Z, dos Santos O, Dehnhardt C, Santos ED, Ayral-Kaloustian S, Mallon R, Hollander I, Feldberg L, Lucas J, Yu K, Chaudhary I, Mansour TS. PKI-179: an orally efficacious dual phosphatidylinositol-3-kinase (PI3K)/mammalian target of rapamycin (mTOR) inhibitor. Bioorg Med Chem Lett. 2010 Oct 1;20(19):5869-73. doi: 10.1016/j.bmcl.2010.07.104. Epub 2010 Jul 30. PubMed PMID: 20797855.
6: Venkatesan AM, Dehnhardt CM, Delos Santos E, Chen Z, Dos Santos O, Ayral-Kaloustian S, Khafizova G, Brooijmans N, Mallon R, Hollander I, Feldberg L, Lucas J, Yu K, Gibbons J, Abraham RT, Chaudhary I, Mansour TS. Bis(morpholino-1,3,5-triazine) derivatives: potent adenosine 5′-triphosphate competitive phosphatidylinositol-3-kinase/mammalian target of rapamycin inhibitors: discovery of compound 26 (PKI-587), a highly efficacious dual inhibitor. J Med Chem. 2010 Mar 25;53(6):2636-45. doi: 10.1021/jm901830p. PubMed PMID: 20166697.
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- PRMT5 inhibitors and uses thereofPublication Number: US-12448388-B2Grant Date: 2025-10-21
- KRAS G12D modulating compoundsPublication Number: US-12448400-B2Grant Date: 2025-10-21
- TRIAZINE COMPOUNDS AS P13 KINASE AND MTOR INHIBITORSPublication Number: PT-2294072-TPriority Date: 2008-05-23
- TRIASINE UNITS AS P13 KINASE INHIBITORS AND MOTORPublication Number: ME-01111-BPriority Date: 2008-05-23
- HER2 mutation inhibitorsPublication Number: US-12447153-B2Grant Date: 2025-10-21
- Anti-hiv compoundsPublication Number: US-2025326779-A1
- Aryl aminopyrimidines as dual MerTK and TYRO3 inhibitors and methods thereofPublication Number: US-12448365-B2Grant Date: 2025-10-21
References
- https://celcuity.com/revtorpyk/REVTORPYK_PI_2026.pdf
- Dehnhardt CM, Venkatesan AM, Chen Z, Delos-Santos E, Ayral-Kaloustian S, Brooijmans N, et al. (August 2011). “Identification of 2-oxatriazines as highly potent pan-PI3K/mTOR dual inhibitors”. Bioorganic & Medicinal Chemistry Letters. 21 (16): 4773–8. doi:10.1016/j.bmcl.2011.06.063. PMID 21763134.
- “FDA approves gedatolisib with fulvestrant, with or without palbociclib, for HR-positive, HER2-negative locally advanced or metastatic breast cancer”. U.S. Food and Drug Administration (FDA). 14 July 2026. Retrieved 20 July 2026.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - Sabatini DM (November 2017). “Twenty-five years of mTOR: Uncovering the link from nutrients to growth”. Proceedings of the National Academy of Sciences of the United States of America. 114 (45): 11818–11825. Bibcode:2017PNAS..11411818S. doi:10.1073/pnas.1716173114. PMC 5692607. PMID 29078414.
- Tian T, Li X, Zhang J (February 2019). “mTOR Signaling in Cancer and mTOR Inhibitors in Solid Tumor Targeting Therapy”. International Journal of Molecular Sciences. 20 (3): 755. doi:10.3390/ijms20030755. PMC 6387042. PMID 30754640.
- Hua H, Kong Q, Zhang H, Wang J, Luo T, Jiang Y (July 2019). “Targeting mTOR for cancer therapy”. Journal of Hematology & Oncology. 12 (1) 71. doi:10.1186/s13045-019-0754-1. PMC 6612215. PMID 31277692.
- Vanhaesebroeck B, Perry MW, Brown JR, André F, Okkenhaug K (October 2021). “PI3K inhibitors are finally coming of age”. Nature Reviews. Drug Discovery. 20 (10): 741–769. doi:10.1038/s41573-021-00209-1. PMC 9297732. PMID 34127844. S2CID 235437841.
- Millis SZ, Ikeda S, Reddy S, Gatalica Z, Kurzrock R (December 2016). “Landscape of Phosphatidylinositol-3-Kinase Pathway Alterations Across 19 784 Diverse Solid Tumors”. JAMA Oncology. 2 (12): 1565–1573. doi:10.1001/jamaoncol.2016.0891. PMID 27388585.
- Anderson EJ, Mollon LE, Dean JL, Warholak TL, Aizer A, Platt EA, et al. (2020). “A Systematic Review of the Prevalence and Diagnostic Workup of PIK3CA Mutations in HR+/HER2- Metastatic Breast Cancer”. International Journal of Breast Cancer. 2020 3759179. doi:10.1155/2020/3759179. PMC 7322582. PMID 32637176.
- Clinical trial number NCT01420081 for “A Study Of Two Dual PI3K/mTOR Inhibitors, PF-04691502 And PF-05212384 In Patients With Recurrent Endometrial Cancer” at ClinicalTrials.gov
- Clinical trial number NCT01925274 for “A Study Of PF-05212384 Plus Irinotecan Vs Cetuximab Plus Irinotecan In Patients With KRAS And NRAS Wild Type Metastatic Colorectal Cancer” at ClinicalTrials.gov
- Clinical trial number NCT02438761 for “PF-05212384 (PKI-587) for t-AML/MDS or de Novo Relapsed or Refractory Acute Myeloid Leukemia (AML)” at ClinicalTrials.gov
- Clinical trial number NCT03698383 for “Phase II Study of Herzuma® Plus Gedatolisib in Patients With HER-2 Positive Metastatic Breast Cancer” at ClinicalTrials.gov
- Clinical trial number NCT03911973 for “Gedatolisib Plus Talazoparib in Advanced Triple Negative or BRCA1/2 Positive, HER2 Negative Breast Cancers” at ClinicalTrials.gov
- Clinical trial number NCT03065062 for “Study of the CDK4/6 Inhibitor Palbociclib (PD-0332991) in Combination With the PI3K/mTOR Inhibitor Gedatolisib (PF-05212384) for Patients With Advanced Squamous Cell Lung, Pancreatic, Head & Neck and Other Solid Tumors” at ClinicalTrials.gov
- Clinical trial number NCT02626507 for “Phase I Study of Combination of Gedatolisib With Palbociclib and Faslodex in Patients With ER+/HER2- Breast Cancer” at ClinicalTrials.gov
- “Celcuity Announces FDA Approval of Revtorpyk (gedatolisib) for the Treatment of HR+/HER2-, PIK3CA Wild-Type Locally Advanced or Metastatic Breast Cancer” (Press release). Celcuity. 14 July 2026. Retrieved 20 July 2026 – via GlobeNewswire.
- World Health Organization (2015). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 73”. WHO Drug Information. 29 (1). hdl:10665/331088.
External links
- Clinical trial number NCT05501886 for “Gedatolisib Plus Fulvestrant With or Without Palbociclib vs Standard-of-Care for the Treatment of Patients With Advanced or Metastatic HR+/HER2- Breast Cancer (VIKTORIA-1) (VIKTORIA-1)” at ClinicalTrials.gov
 | |
| Clinical data | |
|---|---|
| Trade names | Revtorpyk |
| Other names | PF-05212384; PKI-587 |
| AHFS/Drugs.com | revtorpyk |
| License data | US DailyMed: Gedatolisib |
| Routes of administration | Intravenous infusion |
| Drug class | Antineoplastic |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1197160-78-3 |
| PubChem CID | 44516953 |
| IUPHAR/BPS | 7940 |
| DrugBank | DB11896 |
| ChemSpider | 24644946 |
| UNII | 96265TNH2R |
| KEGG | D10635 |
| ChEMBL | ChEMBL592445 |
| CompTox Dashboard (EPA) | DTXSID40152557 |
| Chemical and physical data | |
| Formula | C32H41N9O4 |
| Molar mass | 615.739 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
/////////Gedatolisib, anax labs, approvals 3026, FDA 2026, PF 05212384, PF 5212384, PKI-587, PF-05212384; PF-5212384; PKI 587, gedatolisib, antitumor agent, PHASE 3, PFIZER, гедатолисиб , غيداتوليسيب , 吉达利塞 , 96265TNH2R
O=C(NC1=CC=C(C2=NC(N3CCOCC3)=NC(N4CCOCC4)=N2)C=C1)NC5=CC=C(C(N6CCC(N(C)C)CC6)=O)C=C5
Journal of Medicinal Chemistry (2017), 60(17), 7524-7538 PQR 309
Opakalim
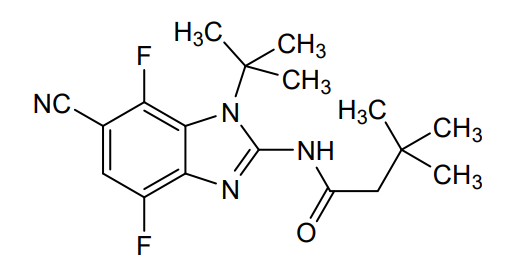
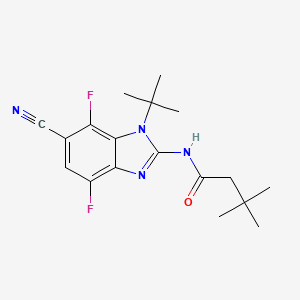
Opakalim
CAS 2376397-93-0
MF C18H22F2N4O MW 348.4 g/mol
N-(1-tert-butyl-6-cyano-4,7-difluorobenzimidazol-2-yl)-3,3-dimethylbutanamide
- N-(1-tert-butyl-6-cyano-4,7-difluorobenzimidazol-2-yl)-3,3-dimethylbutanamide
- Butanamide, N-[6-cyano-1-(1,1-dimethylethyl)-4,7-difluoro-1H-benzimidazol-2-yl]-3,3-dimethyl-
- N-[6-Cyano-1-(1,1-dimethylethyl)-4,7-difluoro-1H-benzimidazol-2-yl]-3,3-dimethylbutanamide
N-(1-tert-butyl-6-cyano-4,7-difluoro-1H-1,3-benzimidazol-2-yl)-3,3-dimethylbutanamide
potassium channel activator, antiepileptic, BHV-7000; BHV7 000, BPN-25203, BPN 25203, KB-3061, KB 3061, 73H9J7RBA2
| BHV-7000 is under investigation in clinical trial NCT06419608 (Efficacy and Safety Study of BHV-7000 Monotherapy in Major Depression). |
Opakalim (also known as BHV-7000) is an investigational, small-molecule medication that acts as a selective activator of Kv7.2 and Kv7.3 potassium channels. Developed by Biohaven Pharmaceuticals, it targets a clinically validated pathway to regulate neuronal hyperexcitability, primarily for the treatment of epilepsy and focal seizures.
Key Characteristics
- Mechanism of Action: It opens Kv7.2/7.3 potassium channels. This stabilizes electrical activity in the brain. Unlike older class drugs, it has minimal to no (GABA_A) receptor activity.
- Safety Benefit: Its precision prevents typical central nervous system side effects. Patients experience much lower rates of somnolence, fatigue, and severe dizziness compared to other anti-seizure medications.
- Administration: It is taken orally once a day. It requires no titration period before reaching therapeutic dosing.
Current Clinical Status
- Refractory Focal Epilepsy: The drug is undergoing phase 2/3 clinical evaluation. The pivotal RISE 3 study completed patient enrollment, with high-profile top-line efficacy data anticipated in the second half of 2026.
- Idiopathic Generalized Epilepsy (IGE): Recent clinical data highlights a three-fold prolongation in the time to a second generalized tonic-clonic seizure compared to a placebo.
- Other Explored Indications: While it is actively tested for conditions like bipolar disorder and erythromelalgia (pain), an exploratory phase 2 trial for major depressive disorder failed to meet its primary goals.
Opakalim (INNTooltip International Nonproprietary Name, USANTooltip United States Adopted Name; developmental code names BHV-7000, BPN-25203, and KB-3061) is a highly selective Kv7.2 and Kv7.3 potassium channel opener which is under development for the treatment of bipolar disorders, epilepsy, partial epilepsies, major depressive disorder, erythromelalgia, pain, infantile spasms, and mood disorders.[1][2][3][4] It is taken orally.[1] The drug was originated by Channel Biosciences and was under development by Biohaven Pharmaceuticals or Biohaven Therapeutics.[1][2] As of April 2026, it is in phase 2/3 clinical trials for bipolar disorders, epilepsy, and partial epilepsies, phase 2 trials for major depressive disorder, and phase 1 trials for erythromelalgia and pain, whereas no recent development has been reported for infantile spasms and mood disorders.[1][2] A phase 2 trial for major depressive disorder failed to meet its primary efficacy endpoint, resulting in focus more on epilepsy instead.[3]
- Study to Determine if BHV-7000 is Effective and Safe in Adults With Refractory Focal Onset EpilepsyCTID: NCT06309966Phase: Phase 2/Phase 3Status: Active, not recruitingDate: 2026-07-01
- A Study to Determine if BHV-7000 is Effective and Safe in Adults With Refractory Focal Onset EpilepsyCTID: NCT06132893Phase: Phase 2/Phase 3Status: RecruitingDate: 2026-05-01
- Long-term Safety and Tolerability of BHV-7000CTID: NCT06443463Phase: Phase 2Status: Enrolling by invitationDate: 2026-05-01
- A Phase 1b Study of BHV-7000 in Participants With Inherited ErythromelalgiaCTID: NCT07262268Phase: Phase 1Status: Enrolling by invitationDate: 2026-04-08
- Long-term Safety Study of BHV-7000 in Participants With Major Depressive Disorder (MDD)CTID: NCT06423781Phase: Phase 2Status: CompletedDate: 2026-04-01
- A Study to Determine if BHV-7000 is Effective and Safe in Adults With Idiopathic Generalized Epilepsy With Generalized Tonic-clonic SeizuresCTID: NCT06425159Phase: Phase 2/Phase 3Status: TerminatedDate: 2026-03-24
- Efficacy and Safety Study of BHV-7000 Monotherapy in Major DepressionCTID: NCT06419608Phase: Phase 2Status: CompletedDate: 2026-01-07
- BHV-7000 Acute Treatment of Bipolar ManiaCTID: NCT06419582Phase: Phase 2/Phase 3Status: CompletedDate: 2025-12-16
- BHV-7000 Open-Label Extension Bipolar Mania StudyCTID: NCT06423794Phase: Phase 2Status: TerminatedDate: 2025-12-16
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US383826737&_cid=P20-MRSLRP-32698-1
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2025035066&_cid=P20-MRSLVM-39325-2
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- Kv7 channel activators compositions and methods of usePublication Number: US-11724990-B2Priority Date: 2018-03-19Grant Date: 2023-08-15
- KV7 channel activators compositions and methods of usePublication Number: US-12286408-B2Priority Date: 2018-03-19Grant Date: 2025-04-29
- KV7 channel activators compositions and methods of usePublication Number: US-11261162-B2Priority Date: 2018-03-19Grant Date: 2022-03-01
- Kv7 channel activators compositions and methods of usePublication Number: US-2019284142-A1Priority Date: 2018-03-19
- Kv7 channel activators compositions and methods of usePublication Number: US-10851067-B2Priority Date: 2018-03-19Grant Date: 2020-12-01
- KV7 channel activator composition and method of use thereofPublication Number: KR-20200133259-APriority Date: 2018-03-19
- Kv7 channel activators compositions and methods of usePublication Number: US-2021130299-A1Priority Date: 2018-03-19
- KV7 channel activator composition and method of use thereofPublication Number: KR-102731128-B1Priority Date: 2018-03-19Grant Date: 2024-11-18
- Methods of use for kv7 channel activatorsPublication Number: US-2025312316-A1Priority Date: 2019-09-17
- Methods of use for KV7 channel activatorsPublication Number: US-12370176-B2Priority Date: 2019-09-17Grant Date: 2025-07-29
- How to use KV7 channel activatorPublication Number: CN-118986972-APriority Date: 2019-09-17
- Kv7 channel activators compositions and methods of usePublication Number: US-2024002349-A1Priority Date: 2018-03-19
- Kv7 channel activators compositions and methods of usePublication Number: US-2023000831-A1Priority Date: 2018-03-19
References
- “Biohaven Pharmaceuticals”. AdisInsight. 15 April 2026. Retrieved 31 May 2026.
- “Delving into the Latest Updates on Opakalim with Synapse”. Synapse. 2 April 2026. Retrieved 31 May 2026.
- Pelorosso C, Balestrini S, Guerrini R (May 2026). “Potassium channel agonists emerging as treatment options for focal epilepsy: are we breaking new ground?”. Expert Opinion on Emerging Drugs: 1–8. doi:10.1080/14728214.2026.2675274. hdl:2158/1473832. PMID 42153277.
- Pong AW (December 2025). “Expanding the toolkit: An update on the evolution of new therapies for Lennox-Gastaut Syndrome”. Seminars in Pediatric Neurology. 56 101242. doi:10.1016/j.spen.2025.101242. PMID 41371876.
 | |
| Clinical data | |
|---|---|
| Other names | BHV-7000; BHV7000; BPN-25203; BPN25203; KB-3061; KB3061 |
| Routes of administration | Oral[1] |
| Drug class | Kv7.2 and Kv7.3 potassium channel opener |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2376397-93-0 |
| PubChem CID | 139487180 |
| DrugBank | DB22041 |
| ChemSpider | 129910012 |
| UNII | 73H9J7RBA2 |
| KEGG | D13061 |
| Chemical and physical data | |
| Formula | C18H22F2N4O |
| Molar mass | 348.398 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
////////opakalim, anax labs, potassium channel activator, antiepileptic, BHV-7000; BHV7 000, BPN-25203, BPN 25203, KB-3061, KB 3061, 73H9J7RBA2
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY


{kind=link}