
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

Bulevirtide-gmod

Bulevirtide-gmod
CAS 2012558-47-1.
MF C248H355N65O72 MW 5399 g/mol
FDA 2026, APPROVALS 2026, 5/22/2026, Hepcludex, WKM56H3TLB
To treat chronic hepatitis delta virus infection in adults without cirrhosis or with compensated cirrhosis
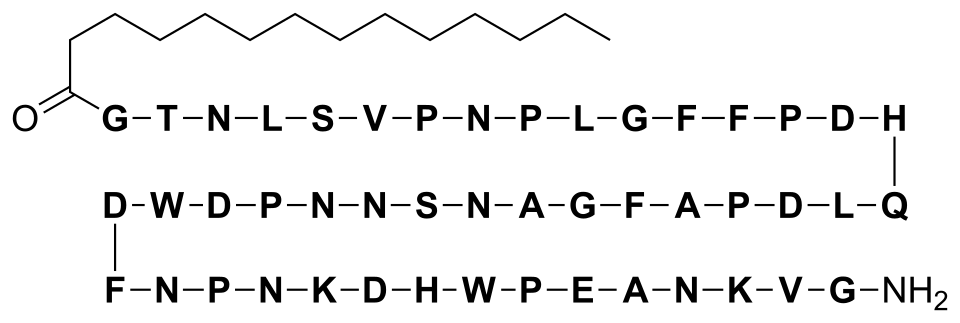
N-myristoyl-glycyl-L-threonyl-L-asparagyl-L-leucyl-L-seryl-L-valyl-L-prolyl-L-asparagyl-L-prolyl-L-leucyl-glycyl-L-phenylalanyl-L-phenylalanyl-L-prolyl-L-alpha-aspartyl-L-histidyl-L-glutaminyl-L-leucyl-L-alpha-aspartyl-L-prolyl-L-alanyl-L-phenylalanyl-glycyl-L-alanyl-L-asparagyl-L-seryl-L-asparagyl-L-asparagyl-L-prolyl-L-alpha-aspartyl-L-tryptophyl-L-alpha-aspartyl-L-phenylalanyl-L-asparagyl-L-prolyl-L-asparagyl-L-lysyl-L-alpha-aspartyl-L-histidyl-L-tryptophyl-L-prolyl-L-alpha-glutamyl-L-alanyl-L-asparagyl-L-lysyl-L-valyl-glycinamide
(4S)-4-[[(2S)-1-[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-6-amino-2-[[(2S)-4-amino-2-[[(2S)-1-[(2S)-4-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-1-[(2S)-4-amino-2-[[(2S)-4-amino-2-[[(2S)-2-[[(2S)-4-amino-2-[[(2S)-2-[[2-[[(2S)-2-[[(2S)-2-[[(2S)-1-[(2S)-2-[[(2S)-2-[[(2S)-5-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-1-[(2S)-2-[[(2S)-2-[[2-[[(2S)-2-[[(2S)-1-[(2S)-4-amino-2-[[(2S)-1-[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-4-amino-2-[[(2S,3R)-3-hydroxy-2-[[2-(tetradecanoylamino)acetyl]amino]butanoyl]amino]-4-oxobutanoyl]amino]-4-methylpentanoyl]amino]-3-hydroxypropanoyl]amino]-3-methylbutanoyl]pyrrolidine-2-carbonyl]amino]-4-oxobutanoyl]pyrrolidine-2-carbonyl]amino]-4-methylpentanoyl]amino]acetyl]amino]-3-phenylpropanoyl]amino]-3-phenylpropanoyl]pyrrolidine-2-carbonyl]amino]-3-carboxypropanoyl]amino]-3-(1H-imidazol-4-yl)propanoyl]amino]-5-oxopentanoyl]amino]-4-methylpentanoyl]amino]-3-carboxypropanoyl]pyrrolidine-2-carbonyl]amino]propanoyl]amino]-3-phenylpropanoyl]amino]acetyl]amino]propanoyl]amino]-4-oxobutanoyl]amino]-3-hydroxypropanoyl]amino]-4-oxobutanoyl]amino]-4-oxobutanoyl]pyrrolidine-2-carbonyl]amino]-3-carboxypropanoyl]amino]-3-(1H-indol-3-yl)propanoyl]amino]-3-carboxypropanoyl]amino]-3-phenylpropanoyl]amino]-4-oxobutanoyl]pyrrolidine-2-carbonyl]amino]-4-oxobutanoyl]amino]hexanoyl]amino]-3-carboxypropanoyl]amino]-3-(1H-imidazol-4-yl)propanoyl]amino]-3-(1H-indol-3-yl)propanoyl]pyrrolidine-2-carbonyl]amino]-5-[[(2S)-1-[[(2S)-4-amino-1-[[(2S)-6-amino-1-[[(2S)-1-[(2-amino-2-oxoethyl)amino]-3-methyl-1-oxobutan-2-yl]amino]-1-oxohexan-2-yl]amino]-1,4-dioxobutan-2-yl]amino]-1-oxopropan-2-yl]amino]-5-oxopentanoic acid
Bulevirtide-gmod, sold under the brand name Hepcludex, is the first and only FDA-approved medication for treating chronic hepatitis delta virus (HDV) infection in adults. Developed by Gilead Sciences, it received accelerated approval from the U.S. Food and Drug Administration (FDA) on May 22, 2026, filling a critical gap for patients with this severe viral liver disease.
Indication and Clinical Use
- Target Patient Profile: Approved for adults with chronic HDV who have compensated cirrhosis or no cirrhosis.
- The Clinical Need: HDV only occurs as a co-infection in individuals who already have Hepatitis B (HBV). It is considered the most aggressive form of viral hepatitis, often accelerating liver scarring (fibrosis), liver failure, and liver cancer.
- Basis of Approval: The FDA granted accelerated approval based on Phase 3 MYR301 study data, which demonstrated a significant reduction in viral HDV RNA and the normalization of alanine aminotransferase (ALT) liver enzymes.
Mechanism of Action
Bulevirtide-gmod is a first-in-class entry inhibitor. It works by binding to and blocking the sodium taurocholate co-transporting polypeptide (NTCP) receptor on liver cells. Because HDV and HBV rely on this specific receptor to enter hepatocytes, the drug successfully disrupts the viral life cycle and prevents the virus from spreading to healthy liver cells.
Dosage and Administration
- Form: Supplied as a lyophilized powder for injection.
- Dose: The recommended dose is 8.5 mg once daily.
- Administration: Delivered via subcutaneous injection (under the skin).
Safety and Side Effects
- Boxed Warning: The drug carries a prominent warning regarding the risk of severe acute exacerbations of hepatitis D and B if treatment is discontinued. Stopping the medication can cause severe, life-threatening viral flares, requiring close medical monitoring for at least 6 months post-treatment.
- Common Side Effects: The most frequent adverse reactions of patients) include:
- Injection site reactions
- Headache
- Abdominal pain
- Fatigue
- Pruritus (itching)
Bulevirtide, sold under the brand name Hepcludex, is an antiviral medication used for the treatment of chronic hepatitis D (in the presence of hepatitis B).[8]
The most common side effects include raised levels of bile salts in the blood and reactions at the site of injection.[8]
Bulevirtide works by attaching to and blocking a receptor (target) through which the hepatitis delta and hepatitis B viruses enter liver cells.[8] By blocking the entry of the virus into the cells, it limits the ability of HDV to replicate and its effects in the body, reducing symptoms of the disease.[8]
Bulevirtide was approved for medical use in the European Union in July 2020,[8] and in Canada in August 2025.[5]
Medical uses
Bulevirtide is indicated for the treatment of chronic hepatitis delta virus (HDV) infection in plasma (or serum) HDV-RNA positive adult patients with compensated liver disease.[8][10]
Pharmacology
Mechanism of action
Bulevirtide binds and inactivates the sodium/bile acid cotransporter, blocking both hepatitis B and hepatitis D viruses from entering hepatocytes.[11]
The hepatitis B virus uses its surface lipopeptide pre-S1 for docking to mature liver cells via their sodium/bile acid cotransporter (NTCP) and subsequently entering the cells. Myrcludex B is a synthetic N-acylated pre-S1[12][13] that can also dock to NTCP, blocking the virus’s entry mechanism.[14]
Bulevirtide is also effective against hepatitis D because the hepatitis D virus uses the same entry receptor as the hepatitis B virus and is only effective in the presence of a hepatitis B virus infection.[14]
Pre-clinical data in mice suggests that pharmacological inhibition of NTCP-mediated bile salt uptake may also be effective to lower hepatic bile salt accumulation in cholestatic conditions. This reduces hepatocellular damage.[15] An increased ratio of phospholipid to bile salts seen in bile upon NTCP inhibition may further contribute to the protective effect as bile salts are less toxic in presence of phospholipids.[16]
Structural formula
Bulevirtide is a 47-amino acid peptide with the following sequence:[17]
CH3(CH2)12CO–Gly–Thr–Asn–Leu–Ser–Val–Pro-Asn-Pro-Leu-Gly-Phe-Phe-Pro-Asp–His–Gln-Leu-Asp-Pro-Ala-Phe-Gly-Ala-Asn-Ser-Asn-Asn-Pro-Asp-Trp-Asp-Phe-Asn-Pro-Asn-Lys-Asp-His-Trp-Pro-Glu-Ala-Asn-Lys-Val-Gly-NH2 (C13H27CO-GTNLSVPNPLGFFPDHQLDPAFGANSNNPDWDFNPNKDHWPEANKVG-NH2)
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2024073572&_cid=P11-MPNG4J-82875-1
PATENTS
- Therapy of atherosclerosis, primary biliary cirrhosis and nrlp3 inflammasome-associated disease by htcp inhibitorsPublication Number: EP-3392267-A1Priority Date: 2017-04-18
- Therapy of atherosclerosis, primary biliary cirrhosis and nrlp3 inflammasome-associated disease by htcp inhibitorsPublication Number: US-2018296634-A1Priority Date: 2017-04-18
- Therapy of atherosclerosis, primary biliary cirrhosis and nrlp3 inflammasome-associated disease by htcp inhibitorsPublication Number: US-2021196786-A1Priority Date: 2017-04-18
- Therapy of atherosclerosis, primary biliary cirrhosis and NRLP3 inflammasome-associated disease by HTCP inhibitorsPublication Number: US-10925925-B2Priority Date: 2017-04-18Grant Date: 2021-02-23
- Combination therapy of hbv and hdv infectionPublication Number: EP-3204030-B1Priority Date: 2014-10-07Grant Date: 2022-04-27
- Combination therapy of hbv and hdv infectionPublication Number: EP-4098273-A1Priority Date: 2014-10-07
- Combination therapy of hbv and hdv infectionPublication Number: US-2022040178-A1Priority Date: 2014-10-07
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma



AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

References
References
- Deterding K, Wedemeyer H (2019). “Beyond Pegylated Interferon-Alpha: New Treatments for Hepatitis Delta”. AIDS Reviews. 21 (3): 126–134. doi:10.24875/AIDSRev.19000080. PMID 31532397. S2CID 202674681.
- “Hepcludex (bulevirtide acetate)”. Therapeutic Goods Administration (TGA). 12 August 2024. Retrieved 12 October 2024.
- “Therapeutic Goods (Poisons Standard—June 2024) Instrument 2024”. Federal Register of Legislation. 30 May 2024. Retrieved 10 June 2024.
- “Hepcludex (Gilead Sciences Pty Ltd)”. Therapeutic Goods Administration (TGA). 13 September 2024. Retrieved 15 September 2024.
- “Hepcludex Product information”. Health Canada. 8 August 2025. Retrieved 20 August 2025.
- “Summary Basis of Decision for Hepcludex”. Drug and Health Products Portal. 29 September 2025. Retrieved 12 October 2025.
- “Hepcludex 2 mg powder for solution for injection – Summary of Product Characteristics (SmPC)”. (emc). 30 March 2022. Retrieved 1 July 2022.
- “Hepcludex EPAR”. European Medicines Agency (EMA). 26 May 2020. Retrieved 12 August 2020. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- “Hepcludex Product information”. Union Register of medicinal products. Retrieved 3 March 2023.
- “Summary of opinion: Hepcludex” (PDF). European Medicines Agency (EMA). 28 May 2020.
- Francisco EM (29 May 2020). “Hepcludex”. European Medicines Agency (EMA). Archived from the original on 15 June 2020. Retrieved 6 August 2020.
- Volz T, Allweiss L, Ben MBarek M, Warlich M, Lohse AW, Pollok JM, et al. (May 2013). “The entry inhibitor Myrcludex-B efficiently blocks intrahepatic virus spreading in humanized mice previously infected with hepatitis B virus”. Journal of Hepatology. 58 (5): 861–867. doi:10.1016/j.jhep.2012.12.008. PMID 23246506.
- Abbas Z, Abbas M (August 2015). “Management of hepatitis delta: Need for novel therapeutic options”. World Journal of Gastroenterology. 21 (32): 9461–9465. doi:10.3748/wjg.v21.i32.9461. PMC 4548107. PMID 26327754.
- Spreitzer H (14 September 2015). “Neue Wirkstoffe – Myrcludex B”. Österreichische Apothekerzeitung (in German) (19/2015): 12.
- Na+ -taurocholate cotransporting polypeptide inhibition has hepatoprotective effects in cholestasis in mice. Slijepcevic D, Roscam Abbing RLP, Fuchs CD, Haazen LCM, Beuers U, Trauner M, Oude Elferink RPJ, van de Graaf SFJ. Hepatology. 2018 Sep;68(3):1057-1069. doi: 10.1002/hep.29888
- Roscam Abbing RL, Slijepcevic D, Donkers JM, Havinga R, Duijst S, Paulusma CC, et al. (January 2020). “Blocking Sodium-Taurocholate Cotransporting Polypeptide Stimulates Biliary Cholesterol and Phospholipid Secretion in Mice”. Hepatology. 71 (1): 247–258. doi:10.1002/hep.30792. PMC 7003915. PMID 31136002.
- Sauter M, Blank A, Stoll F, Lutz N, Haefeli WE, Burhenne J (September 2021). “Intact plasma quantification of the large therapeutic lipopeptide bulevirtide”. Analytical and Bioanalytical Chemistry. 413 (22): 5645–5654. doi:10.1007/s00216-021-03384-7. PMC 8410713. PMID 34018034.
| Clinical data | |
|---|---|
| Pronunciation | /bjuːˈlɛvɪrtaɪd/ byoo-LEH-vir-tyde |
| Trade names | Hepcludex |
| Other names | MyrB, Myrcludex-B[1] |
| License data | US DailyMed: Bulevirtide |
| Pregnancy category | AU: B1[2] |
| Routes of administration | Subcutaneous |
| ATC code | J05AX28 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only)[3][4][2]CA: ℞-only[5][6]UK: POM (Prescription only)[7]EU: Rx-only[8][9] |
| Identifiers | |
| CAS Number | 2012558-47-1 |
| DrugBank | DB15248 |
| ChemSpider | 129157549 |
| UNII | WKM56H3TLB |
| KEGG | D11877as salt: D11878 |
| ChEMBL | ChEMBL4297711 |
| Chemical and physical data | |
| Formula | C248H355N65O72 |
| Molar mass | 5398.951 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
/////////Bulevirtide-gmod, ANAX LABS, FDA 2026, APPROVALS 2026, Hepcludex, WKM56H3TLB, ANTIVIRALS
Flormotridazum (18F)
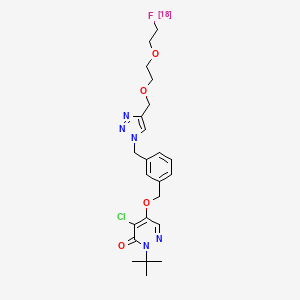
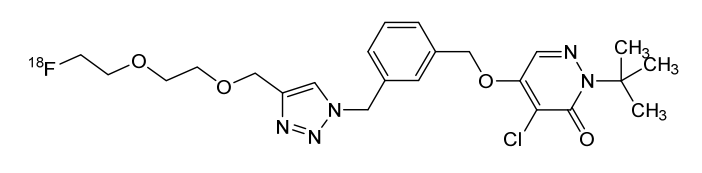
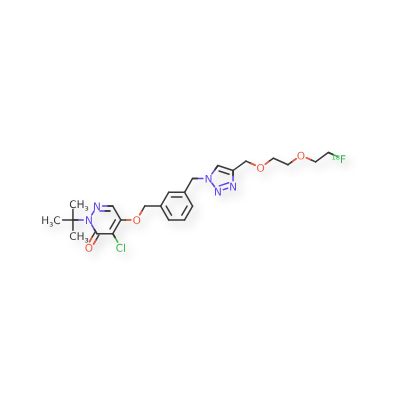
Flormotridazum (18F)
CAS 2798832-03-6
MF C23H29Cl18FN5O4 MW492.961
2-tert-butyl-4-chloro-5-[(3-{[4-({2-[2-(18F)fluoroethoxy]ethoxy}methyl)-1H-1,2,3-triazol-1-yl]methyl}phenyl)methoxy]pyridazin-3(2H)-one
| 3(2H)-Pyridazinone, 4-chloro-2-(1,1-dimethylethyl)-5-[[3-[[4-[[2-[2-(fluoro-18F)ethoxy]ethoxy]methyl]-1H-1,2,3-triazol-1-yl]methyl]phenyl]methoxy]- |
2-tert-butyl-4-chloro-5-[(3-{[4-({2-[2-(18F)fluoroethoxy]ethoxy}methyl)-1H-1,2,3-triazol-1-yl]methyl}phenyl)methoxy]pyridazin-3(2H)-one
imaging agent, 7AR6ZH8YUU
Flormotridaz (18F) (also referred to by its International Nonproprietary Name, flormotridazum) is an advanced radiopharmaceutical compound utilized in nuclear medicine. It is specifically engineered as a radioactive diagnostic tracer containing the fluorine-18 positron-emitting isotope.
Core Characteristics & Chemical Profile
- Substance Classification: Radioactive Diagnostic Agent / Small Molecule.
- Mechanism Basis: It shares core structural similarities and structural lineage with pyridazinone-based mitochondrial complex 1 (MC-1) inhibitors, heavily linking its functionality to target-specific tissues with high metabolic or mitochondrial activity.
Mechanism and Clinical Application
Like related fluorine-18 labeled pyridazinone analogues, this agent is designed for Positron Emission Tomography (PET) imaging workflows. [1]
- Administration: The agent is administered intravenously as a sterile unit dose before scanning.
- Cellular Targeting: It binds selectively to specific intracellular molecular targets (such as mitochondrial pathways) within highly active tissues.
- PET Imaging: As the Fluorine-18 radioisotope decays, it emits positrons. These positrons encounter electrons to produce gamma rays, which the PET scanner captures to map high-resolution, three-dimensional metabolic layouts of internal organ systems.
Contextual Comparison
In clinical nuclear medicine, molecular tracers tagged with Fluorine-18 offer significant clinical benefits over older Single-Photon Emission Computed Tomography (SPECT) agents. Their 110-minute half-life allows them to be manufactured at centralized cyclotron facilities and distributed directly to regional medical centres as ready-to-use unit doses, eliminating the need for an on-site cyclotron
Flormotridaz (\(^{18}\text{F}\)):
- CN112807276B: “Preparation method and application of a pyridazinone myocardial perfusion PET radiopharmaceutical” (Covers the definitive radiosynthesis scheme).
- CN115947775A: “Method for preparing compound (I), compound (I), and uses thereof”.
- WO2024008073A1 / CN114832118B: “Compound I liquid composition, preparation method and use thereof” (Covers final formulation stabilization utilizing vitamin C and gentisic acid)
PAT
https://patents.google.com/patent/WO2024008073A1/zh
Compound I, chemically named 2-tert-butyl-4-chloro-5-((3-((4-((2-(2-fluoro[ 18F ]ethoxy)ethoxy)methyl)-1H-1,2,3-triazol-1-yl)methyl)benzyl)oxy)pyridazine-3(2H)-one. Chemical structural formula:Molecular formula : C₂₃H₂₉Cl₁₈FN₅O₄
Molecular weight: 492.97The mechanism of action of compound I as a myocardial perfusion PET imaging agent: Once compound I enters cardiomyocytes, it can rapidly interact with respiratory chain complex I (MC-I) in mitochondria and remain in the myocardium for a long time. Preliminary animal studies showed that it has high cardiac uptake and low hepatic uptake 15 minutes after injection, and maintains a good heart-liver ratio 60 minutes after injection, showing good potential for myocardial perfusion imaging.In this application, Compound I liquid composition or Compound I is used as a myocardial perfusion PET imaging agent.Precursor of Compound I: Chemical name is methyl 2-(2-((1-(3-(((1-(tert-butyl)-5-chloro-6-oxo-1,6-dihydropyridazin-4-yl)oxy)methyl)benzyl)-1H-1,2,3-triazol-4-yl)methoxy)ethoxy)ethyl-4-methylbenzenesulfonate, chemical structural formula is:Molecular formula : C30H36ClN5O7S
Molecular weight: 646.16Amino polyethers (K222 ) are tribridged crown ether molecules with cavitary structures, and are typical nitrogen-containing cavitary ethers, belonging to the category of cavitary ethers. Due to their unique coordination properties, nitrogen-containing cavitary ethers can effectively and selectively complex transition metal and heavy metal cations, forming more stable complexes. Furthermore, they possess both lipophilic and hydrophilic properties, thus showing promising research potential.In existing technologies, the classic synthetic method for amino polyether (K
222 ) is the highly diluted method proposed by Lehn et al., which is a typical non-template ion synthesis method. The specific steps involve dissolving the starting materials 1,8-diamino-3,6-dioxane and 1,8-diacyl chloride-3,6-dioxane in a large amount of benzene solvent and heating the reaction for 8 hours. Then, a reduction reaction with lithium aluminum hydride is performed for 24 hours, followed by column chromatography separation and recrystallization to obtain amino polyether (K
222 ). This method requires a large amount of solvent, such as benzene, has a long synthetic route, is complex, has a low yield, and is not economically efficient. Besides the highly diluted method, another classic synthetic method for amino polyether (K222 ) is proposed by Kulstad and Malmsten, which uses Na 2CO 3
as a template to obtain a sodium iodide complex of amino polyether (K 222 ) in acetonitrile , and then decomplexes it using a resin to obtain amino polyether (K 222 ). The specific steps are as follows: 1,2-bis(2-iodoethoxy)ethane and benzylamine are refluxed in acetonitrile solution for 3 days. An intermediate is then obtained through post-processing. This intermediate is recrystallized from acetone and filtered to obtain a NaI complex. This complex is then decomplexed under acidic conditions using cation exchange resins and anion exchange resins to prepare amino polyether (K222 ) . This method uses simple equipment, requires little solvent, and has relatively mild reaction conditions. However, the applicant has found that the decomplexing method using ion exchange resins fails to proceed when the sodium ion content decreases to a certain level, resulting in a low yield.
PAT
https://patents.google.com/patent/CN114773179B/en
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
//////////flormotridazum (18F), anax labs, imaging agent, 7AR6ZH8YUU
Florensocatib
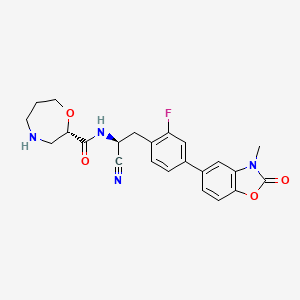
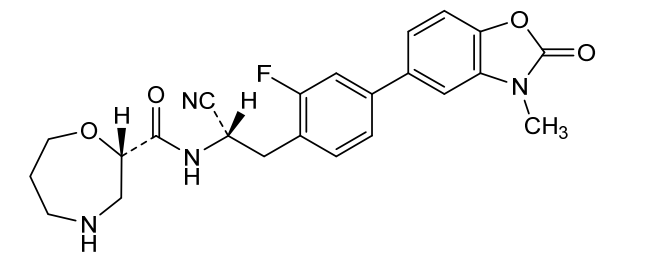
Florensocatib
CAS 2762114-61-2
MF C23H23FN4O4 MW438.5 g/mol
(2S)-N-[(1S)-1-cyano-2-[2-fluoro-4-(3-methyl-2-oxo-1,3-benzoxazol-5-yl)phenyl]ethyl]-1,4-oxazepane-2-carboxamide
(2S)-N-{(1S)-1-cyano-2-[2-fluoro-4-(3-methyl-2-oxo-2,3-dihydro1,3-benzoxazol-5-yl)phenyl]ethyl}-1,4-oxazepane-2-
carboxamide
cathepsin inhibitor, HSK 31858, CHF 10196, DPP1-IN-1, RWC743JRK7
Florensocatib (originally designated as HSK31858 or CHF10196) is an investigative, highly potent, oral reversible inhibitor of dipeptidyl peptidase 1 (DPP1). It is being actively researched for its ability to reduce the frequency of pulmonary exacerbations in adults suffering from inflammatory respiratory diseases like bronchiectasis
Mechanism of Action
DPP1 (also known as cathepsin C) is a lysosomal protease enzyme responsible for activating neutrophil serine proteases (NSPs). In conditions like non-cystic fibrosis bronchiectasis, hyperactive neutrophils accumulate in the airways, causing severe tissue damage, chronic inflammation, and airway widening.
By inhibiting DPP1, florensocatib prevents the activation of these damaging enzymes, effectively targeting the primary driver of neutrophilic inflammation in the lungs.
Clinical Development & Trial Progress
Florensocatib is undergoing global evaluation across multiple advanced clinical trials:
- The SAVE-BE Trial: An earlier clinical phase where the drug demonstrated high potency and favorable efficacy profiles in treating inflammatory lung conditions.
- The HOPE-BE Trial: A definitive Phase III protocol launched to evaluate the long-term safety and overall reduction of pulmonary exacerbation frequencies specifically among Chinese adults.
- Global Phase III Status: According to records on ClinicalTrials.gov, randomised, double-blind trials are evaluating the drug against a placebo in participants aged 12 to 85 for treatment windows stretching up to 78 weeks.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US395653715&_cid=P21-MPKKTA-33002-1
Example 1: (S)—N—((S)-1-cyano-2-(2-fluoro-4-(3-methyl-2-oxo-2,3-dihydrobenzo[d]oxazol-5-yl)phenyl)ethyl)-1,4-oxazepane-2-carboxamide (Compound 1)
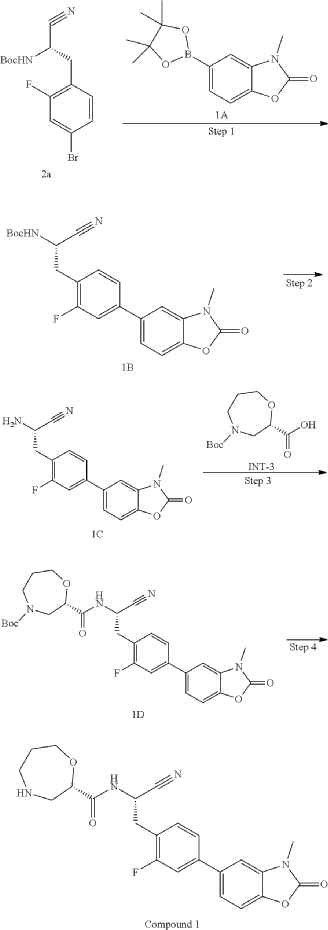
Step 4: (S)—N—((S)-1-cyano-2-(2-fluoro-4-(3-methyl-2-oxo-2,3-dihydrobenzo[d]oxazol-5-yl)phenyl)ethyl)-1,4-oxazepane-2-carboxamide (Compound 1)
| 1H NMR (400 MHz, CDCl 3) δ 7.43-7.22 (m, 5H), 7.12 (d, 1H), 5.19 (dd, 1H), 4.18-4.04 (m, 1H), 4.05-3.95 (m, 1H), 3.78 (m, 1H), 3.46 (s, 3H), 3.41-3.17 (m, 3H), 3.03-2.87 (m, 3H), 1.88 (m, 2H). |
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US395653715&_cid=P21-MPKKYT-35548-1
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2022042591&_cid=P21-MPKKS9-32509-1
(S)-N-((S)-1-cyano-2-(2-fluoro-4-(3-methyl-2-oxo-2,3-dihydrobenzo[d]oxazol-5-yl)phenyl)ethyl)-1,4-oxazepane-2-carboxamide(compound 1)
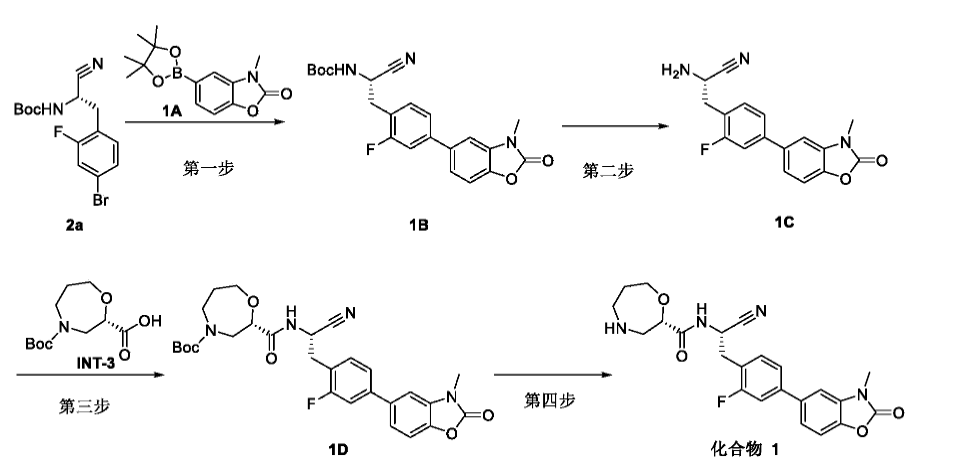
Step 4: (S)-N-((S)-1-cyano-2-(2-fluoro-4-(3-methyl-2-oxo-2,3-dihydrobenzo[d]oxazol-5-yl)phenyl)ethyl)-1,4-oxazacycloheptane-2-carboxamide (Compound 1)
[0360]
(S)-N-((S)-1-cyano-2-(2-fluoro-4-(3-methyl-2-oxo-2,3-dihydrobenzo[d]oxazol-5-yl)phenyl)ethyl)-1,4-oxazepane-2-carboxamide(compound 1)
[0361]1D (0.32 g, 0.59 mmol) was dissolved in formic acid (2.5 mL), and the mixture was reacted at 50 °C for 10 min after the addition was complete. The solution was concentrated to dryness, and ethyl acetate (20 mL) was added. The pH was adjusted to approximately 8 by dropwise addition of saturated sodium bicarbonate solution. The organic layer was separated and extracted with ethyl acetate (25 mL × 3). The organic phases were combined, dried over anhydrous sodium sulfate, filtered, concentrated, and the residue was purified by silica gel column chromatography (dichloromethane:methanol (v/v) = 20:1) to give title compound 1 (0.15 g, 58.0%). LC-MS (ESI): m/z = 439.1 [M+H] + .
[0362]
1H NMR(400MHz,CDCl 3)δ7.43–7.22(m,5H),7.12(d,1H),5.19(dd,1H),4.18–4.04(m,1H),4.05–3.95(m,1H),3.78(m,1H),3.46(s,3H),3.41–3.17(m,3H),3.03–2.87(m,3H),1.88(m,2H).
ADVT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- Nitrile derivative that acts as inhibitor of dipeptidyl peptidase 1 and use thereofPublication Number: US-2023121807-A1Priority Date: 2020-08-26
- Nitrile derivative that acts as inhibitor of dipeptidyl peptidase 1 and use thereofPublication Number: WO-2022042591-A1Priority Date: 2020-08-26
- Nitrile derivative as dipeptidyl peptidase 1 inhibitor and application thereofPublication Number: CN-115996923-APriority Date: 2020-08-26
- Nitrile derivative that acts as inhibitor of dipeptidyl peptidase 1 and use thereofPublication Number: EP-4129989-A1Priority Date: 2020-08-26
- Dipeptidyl peptidase 1 inhibitor polymorph and preparation method and application thereofPublication Number: CN-118696040-APriority Date: 2022-02-22
- Salt and crystal form of dipeptidyl peptidase inhibitor compoundPublication Number: EP-4484414-A1Priority Date: 2022-02-22
- Nitrile derivative that acts as inhibitor of dipeptidyl peptidase 1 and use thereofPublication Number: US-11807635-B2Priority Date: 2020-08-26Grant Date: 2023-11-07
- A kind of nitrile derivative as dipeptidyl peptidase 1 inhibitor and use thereofPublication Number: TW-202214616-APriority Date: 2020-08-26
- Nitrile derivatives acting as inhibitors of dipeptidyl peptidase 1 and uses thereofPublication Number: KR-20230084134-APriority Date: 2020-08-26
- Salt and crystal form of dipeptidyl peptidase inhibitor compoundPublication Number: WO-2023160542-A1Priority Date: 2022-02-22
- Dipeptidyl peptidase 1 inhibitor polymorph, preparation method and use thereforPublication Number: WO-2023160579-A1Priority Date: 2022-02-22
- Salts and crystalline forms of dipeptidyl peptidase inhibitor compoundsPublication Number: CN-118660873-APriority Date: 2022-02-22
- Preparation method of nitrogen-containing heterocyclic compoundPublication Number: WO-2023160541-A1Priority Date: 2022-02-22
- Preparation method of nitrogen-containing heterocyclic compoundPublication Number: CN-118742547-APriority Date: 2022-02-22
- Dipeptidyl peptidase 1 inhibitors and uses thereofPublication Number: WO-2025059526-A1Priority Date: 2023-09-15
- Pharmaceutical composition containing dipeptidyl peptidase small molecule inhibitorPublication Number: WO-2024193695-A1Priority Date: 2023-03-23
- Uses of certain 1,4-oxazepane-2-carboxamides as dpp1 inhibitorsPublication Number: WO-2024173556-A2Priority Date: 2023-02-15
- Novel dpp1 inhibitors and uses thereofPublication Number: WO-2024026433-A2Priority Date: 2022-07-28
- Novel dpp1 inhibitors and uses thereofPublication Number: EP-4561565-A2Priority Date: 2022-07-28
/////////florensocatib, anax labs, cathepsin inhibitor, HSK 31858, CHF 10196, DPP1-IN-1, RWC743JRK7
Florcicaper (18F)
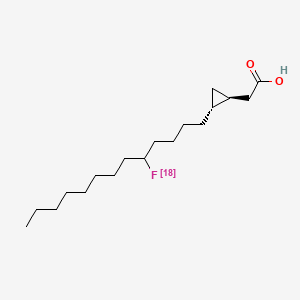
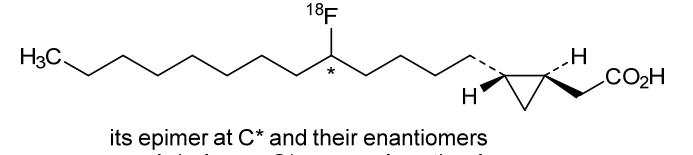
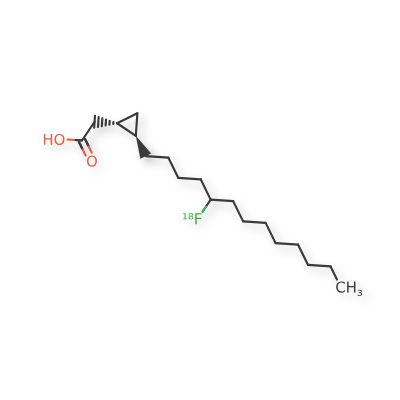
Florcicaper (18F)
CAS 855927-17-2
MF C18H3318FO2, MW 299.4544
2-[(1S,2R)-2-(5-(18F)fluorotridecyl)cyclopropyl]acetic acid
| 2-((1S,2R)-2-(5-(FLUORO-18F)TRIDECYL)CYCLOPROPYL)ACETIC ACID |
| TRANS-9(RS)-18F-FLUORO-3,4(RS,RS)-METHYLENEHEPTADECANOIC ACID |
rac-{(1R,2S)-2-[(5RS)-5-(18F)fluorotridecyl]cyclopropyl}aceticacid
imaging agent, CARDIOPET, (18F FCPHA), FDG79C95XB
CardioPET is: An F-18 labeled, modified fatty acid that provides insight into regions with decreased blood flow or metabolic insufficiency in the myocardium.; CardioPET may be used to: Identify patients that will benefit from PCI or revascularization and guide intervention, Assess myocardial viability, Evaluate CAD in patients that cannot exercise.; Agent: Muscle State Imaging Agent, Type: Fatty Acid (Labeled with Fluorine 18), Condition: Coronary Artery Disease, Status: completed enrollment.;This imaging agent exploits the dietary needs of the heart as it relates to glucose and fatty acids. By introducing a radio-labeled analog to the natural fatty acids utilized as an energy source by the heart we can visualize the anatomic location and state of the muscle within the areas defined by the specific coronary artery blood flow distribution and detect problems in advance of symptoms that would lead to a stress test.
Cardiopet is under investigation in clinical trial NCT01826773 (Cardiopet as PET Imaging Agent to Assess Myocardial Perfusion and Fatty Acid Uptake in Known or Suspected CAD Subjects).
A Phase I Study in Healthy Volunteers to Evaluate the Safety of CardioPET™ in Detection of Coronary Artery Disease
CTID: NCT00413647
Phase: Phase 1
Status: Completed
Date: 2013-06-12
PATENTS
CA-2876139-A1 CN-104684546-A CN-114736112-A CN-115141087-A CN-115141087-B CN-115141125-A CN-115141125-B CN-115181013-A CN-115181013-B CN-115850224-A CN-115850224-B CN-115959978-A CN-115959978-B CN-116041169-A CN-116199658-A CN-116217356-A EP-2858630-A1 EP-4133284-A1 US-10533059-B2 US-11701429-B2 US-2015361110-A1 US-2017014528-A1 US-2020199249-A1 US-2020297854-A1 US-20230314449-A1 US-20230381092-A1 US-9409927-B2 WO-2013185032-A1 WO-2022082327-A1 WO-2023085674-A1 WO-2023236978-A1 WO-2023237092-A1
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2013185032&_cid=P11-MPJ5UV-89230-1
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2022082327&_cid=P11-MPJ5WJ-90048-1
ADVT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- Nanotherapeutics for drug targetingPublication Number: WO-2013185032-A1Priority Date: 2012-06-07
- Nanotherapeutics for drug targetingPublication Number: EP-2858630-A1Priority Date: 2012-06-07
- Super lewis acidic borate esters as 18F-labeled PET probesPublication Number: US-9409927-B2Priority Date: 2013-01-24Grant Date: 2016-08-09
- Super lewis acidic borate esters as 18f-labeled pet probesPublication Number: US-2015361110-A1Priority Date: 2013-01-24
- Nanotherapeutics for drug targetingPublication Number: CA-2876139-A1Priority Date: 2012-06-07
- Nanotherapeutics for drug targetingPublication Number: US-2020297854-A1Priority Date: 2012-06-07
- Nanotherapeutics for drug targetingPublication Number: CN-104684546-APriority Date: 2012-06-07
- Diagnosis of stage b2 dmvdPublication Number: US-2023314449-A1Priority Date: 2020-04-07
- Targeted drug delivery through affinity based linkersPublication Number: US-2017014528-A1Priority Date: 2014-03-12
- Targeted drug delivery through affinity based linkersPublication Number: US-2020199249-A1Priority Date: 2014-03-12
- Targeted drug delivery through affinity based linkersPublication Number: US-10533059-B2Priority Date: 2014-03-12Grant Date: 2020-01-14
- Targeted drug delivery through affinity based linkersPublication Number: US-11701429-B2Priority Date: 2014-03-12Grant Date: 2023-07-18
//////////florcicaper (18F), anax labs, imaging agent, CARDIOPET, (18F FCPHA), FDG79C95XB
Fexlamose
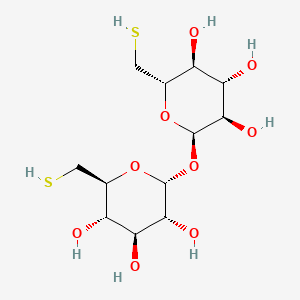
Fexlamose
CAS 1285607-08-0
MFC12H22O9S2 MW374.4 g/mol
(2S,3S,4S,5R,6R)-2-(sulfanylmethyl)-6-[(2R,3R,4S,5S,6S)-3,4,5-trihydroxy-6-(sulfanylmethyl)oxan-2-yl]oxyoxane-3,4,5-triol
- 6-Thio-alpha-D-glucopyranosyl 6-thio-alpha-D-glucopyranoside
- alpha-D-Glucopyranoside, 6-thio-alpha-D-glucopyranosyl 6-thio-
- 6-thio-alpha-D-glucopyranosyl 6-thio-alpha-D-glucopyranoside; 6,6′-dithiotrehalose
6-thio-α-D-glucopyranosyl 6-thio-α-D-glucopyranoside; 6,6′-dithiotrehalose
mucolytic, AER-01, AER 01, VY9GAK6EVR, MUC-031, MUC031
Fexlamose (formerly known as AER-01) is an experimental inhaled small-molecule drug developed by Aer Therapeutics to treat muco-obstructive lung diseases like COPD (Chronic Obstructive Pulmonary Disease) and asthma. It is a thiol-modified carbohydrate designed to break down mucus plugs in the airways
How it Works
- Mechanism: Fexlamose acts as a mucolytic by cleaving the disulfide bonds in mucus, thinning the thick secretions that block airways.
- The Problem It Targets: While many respiratory drugs manage inflammation or relax airway muscles, currently no approved treatments directly tackle mucus plugs, which are a major cause of breathing difficulties in COPD.
- Delivery: It is administered as an inhalation solution (or potentially a dry powder) directly into the lungs.
- Current Clinical Status
- Fexlamose is an investigational drug and is not yet approved for public use or commercial prescription.
- Clinical Trials: It is undergoing Phase 2a clinical studies (such as the AER-01-002 trial) to evaluate its safety, tolerability, and efficacy in adults with moderate to severe COPD.
- Study Design: These trials utilize specialized CT mucus plug scoring to identify patients who are most likely to benefit from the therapy.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017197360&_cid=P10-MPHQBA-11926-1
Syn
US-20230172885-A1
https://patentscope.wipo.int/search/en/detail.jsf?docId=US399239351&_cid=P10-MPHQQF-20898-1
Syn
US-20230181607-A1
https://patentscope.wipo.int/search/en/detail.jsf?docId=US399582635&_cid=P10-MPHQRV-21687-1
ADVT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
//////////fexlamose, ANAX LABS, mucolytic, AER-01, AER 01, VY9GAK6EVR, MUC-031, MUC031
Famlasertib
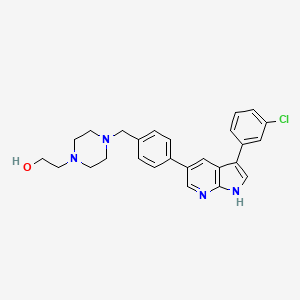
Famlasertib
CAS 2375591-69-6
MFC26H27ClN4O MW 447.0 g/mol
4-[[4-[3-(3-Chlorophenyl)-1H-pyrrolo[2,3-b]pyridin-5-yl]phenyl]methyl]-1-piperazineethanol
2-[4-({4-[3-(3-chlorophenyl)-1H-pyrrolo[2,3-b]pyridin-5-yl]phenyl}methyl)piperazin-1-yl]ethan-1-ol
serine/threonine kinase inhibitor, amyotrophic lateral sclerosis, Prosetin, WJP32276AY
Prosetin is an orally administered blocker of MAP4K under investigation for the treatment of amyotrophic lateral sclerosis.
Famlasertib (also known as Prosetin or Prostetin/12k) is a highly potent, small-molecule inhibitor targeting the mitogen-activated protein kinase kinase kinase kinase (MAP4K) family. It is an experimental drug primarily under investigation for its neuroprotective capabilities in treating neurodegenerative disorders like amyotrophic lateral sclerosis (ALS) and as an anti-invasive agent in certain cancers
- Target Pathways: MAP4K4 (HGK), MLK1, and MLK3
- Key Properties: Orally active, blood-brain barrier penetrant (CNS-penetrant)
Mechanism of Action
Famlasertib functions by blocking the activation of the MAP4K protein family, specifically demonstrating powerful inhibitory values (\(\text{IC}_{50}\)) against subfamilies like HGK (MAP4K4), MLK3, and MLK1. By inhibiting these kinases, the compound: [1]
- Reduces Endoplasmic Reticulum (ER) Stress: It helps mitigate the unfolded protein response that triggers programmed cell death in neurons affected by misfolded protein accumulation.
- Suppresses Inflammation: It blocks inflammatory pathways associated with neurodegeneration and cell damage.
- Restrains Cell Motility: In oncology contexts, it disrupts kinase signaling linked to actin cytoskeleton remodeling, preventing malignant cells from migrating.
Primary Areas of Research
1. Amyotrophic Lateral Sclerosis (ALS)
In motor neuron models of ALS, cellular stress frequently triggers neurodegeneration. Because famlasertib easily passes through the blood-brain barrier, it is capable of directly shielding motor neurons from ER-stress-mediated cell death, extending cell viability in laboratory models.
2. Oncology (Medulloblastoma)
Recent findings published on bioRxiv indicate that famlasertib acts as a “migrastatic” agent in medulloblastoma (a type of pediatric brain tumor). It suppresses the highly invasive behavior and single-cell motility of tumor cells without exhibiting developmental toxicity.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020163594&_cid=P21-MPGALG-31359-1
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US317630245&_cid=P21-MPGALG-31359-1
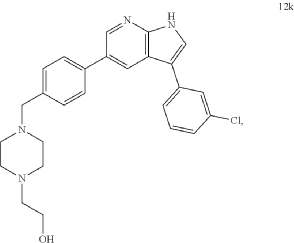
Preparation of 2-(4-(4-(3-(3-Chlorophenyl)-1H-pyrrolo[2,3-b]pyridin-5-yl)benzyl)piperazin-1-yl)ethan-1-ol (Compound 12k)
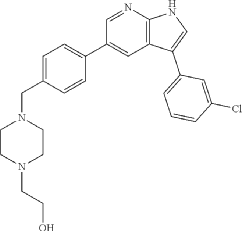
Following the general procedure described above, with 4-(3-(3-chlorophenyl)-1-tosyl-1H-pyrrolo[2,3-b]pyridin-5-yl)benzaldehyde (10c, 418 mg, 0.86 mmol) and 1-(2-hydroxyethyl)piperazine (224 mg, 211 μL, 1.72 mmol, 2.0 eq) as the starting materials, 2-(4-(4-(3-(3-chlorophenyl)-1H-pyrrolo[2,3-b]pyridin-5-yl)benzyl)piperazin-1-yl)ethan-1-ol (12k) was isolated as an off-white solid (139.9 mg, 36% yield over two steps). 1H NMR (400 MHz, Methanol-d 4) δ 8.57 (d, J=2.0 Hz, 1H), 8.54 (d, J=2.0 Hz, 1H), 7.80 (s, 1H), 7.76 (d, J=8.3 Hz, 2H), 7.65 (t, J=1.9 Hz, 1H), 7.64-7.57 (m, 3H), 7.42 (t, J=7.9 Hz, 1H), 7.29 (ddd, J=8.0, 2.1, 1.0 Hz, 1H), 4.27 (s, 2H), 3.93-3.86 (m, 2H), 3.62 (s, 4H), 3.41 (s, 4H), 3.35-3.31 (m, 2H) ppm. HRMS (APCI +, m/z): calcd. for C 26H 28N 40Cl [M+H +]: 447.1952, found: 447.1954.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US469942811&_cid=P21-MPGAUU-39605-1
ADVT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- Design, synthesis of new 3H-imidazo[4,5-b]pyridine derivatives and evaluation of their inhibitory properties as mixed lineage kinase 3 inhibitorsPublication Name: Bioorganic & Medicinal Chemistry LettersPublication Date: 2024-03-15PMID: 38346577DOI: 10.1016/j.bmcl.2024.129652
- The emerging role of mixed lineage kinase 3 (MLK3) and its potential as a target for neurodegenerative diseases therapiesPublication Name: European Journal of Medicinal ChemistryPublication Date: 2023-09-05PMID: 37247505DOI: 10.1016/j.ejmech.2023.115511
- Development of MAP4 Kinase Inhibitors as Motor Neuron-Protecting AgentsPublication Name: Cell Chemical BiologyPublication Date: 2019-12-19PMCID: PMC7253076PMID: 31676236DOI: 10.1016/j.chembiol.2019.10.005
PAT
- Immunophilin binding agents and uses thereofPublication Number: US-2023063768-A1Priority Date: 2019-02-07
- Immunophilin binding agents and uses thereofPublication Number: WO-2020163594-A1Priority Date: 2019-02-07
- Immunophilin-dependent inhibitors and uses thereofPublication Number: US-2022193242-A1Priority Date: 2019-02-07
- Immunophilin-dependent inhibitors and uses thereofPublication Number: WO-2020163598-A1Priority Date: 2019-02-07
- Compounds, compositions and methods for inhibiting toxic endoplasmic reticulum stress – Patents.comPublication Number: JP-7487170-B2Priority Date: 2018-03-01Grant Date: 2024-05-20
- Compounds, compositions, and methods for suppressing toxic endoplasmic reticulum stress
- Publication Number: US-2021040091-A1
- Priority Date: 2018-03-01G
///////famlasertib, serine/threonine kinase inhibitor, amyotrophic lateral sclerosis, Prosetin, WJP32276AY
Epaldeudomide
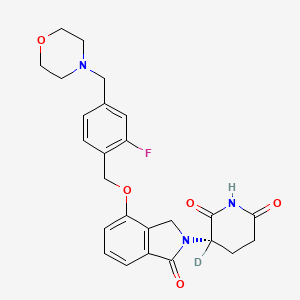
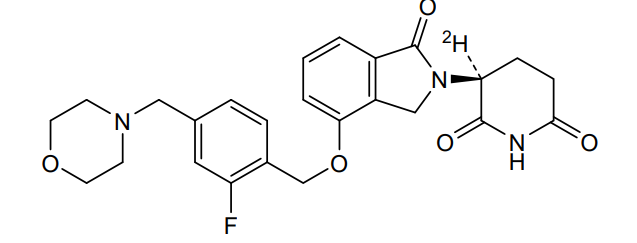
Epaldeudomide
CAS 1918159-31-5
MF C25H252HFN3O5, MW 468.5 g/mol
(3S)-3-deuterio-3-[7-[[2-fluoro-4-(morpholin-4-ylmethyl)phenyl]methoxy]-3-oxo-1H-isoindol-2-yl]piperidine-2,6-dione

KPG-818, KPG 818, ANTINEOPLASTIC, KV0TBL8MUS
Epaldeudomide (also known as KPG-818) is an investigational, next-generation immunomodulatory drug and “molecular glue” developed by Kangpu Biopharmaceuticals. Designed as a targeted therapy, it works by binding to the CRL4-CRBN E3 ubiquitin ligase complex to degrade specific proteins, showing promise in treating blood cancers, solid tumors, and autoimmune diseases.
Mechanism of Action
- Molecular Glue: It is a small molecule that acts as a modulator of the cereblon (CRBN) E3 ligase.
- Protein Degradation: It targets and induces the rapid degradation of two Ikaros zinc-finger transcription factors: IKZF3 (Aiolos) and IKZF1 (Ikaros).
- Immunomodulation: By degrading these targets, epaldeudomide triggers broad-spectrum immune responses, reduces tumor proliferation, and suppresses inflammation (such as the production of TNF-\(\alpha \)).
Therapeutic Pipeline and Research
Epaldeudomide is currently undergoing clinical evaluation to assess its safety, tolerability, and efficacy.
- Hematology/Oncology: It is being studied for the treatment of hematologic malignancies (such as multiple myeloma and lymphomas). It demonstrates potent anti-tumor and anti-angiogenic activity without several severe side effects typically associated with earlier immunomodulatory drugs.
- Autoimmune and Inflammatory Disorders: Because of its broad anti-inflammatory effects and ability to inhibit TNF-\(\alpha \), it is being explored for use against autoimmune conditions and inflammatory arthritis.
- OriginatorKangpu Biopharmaceuticals
- ClassAnti-inflammatories; Antineoplastics; Small molecules
- Mechanism of ActionCRBN protein modulators; Ubiquitin protein ligase complex modulators
- Phase IIInflammatory bowel diseases
- Phase I/IISystemic lupus erythematosus
- Phase IHaematological malignancies
- PreclinicalBehcet’s syndrome; Crohn’s disease; Multiple myeloma
- 06 Dec 2025Efficacy, pharmacokinetics and adverse events data from a phase I trial in Haematological malignancies presented at 67th American Society of Hematology Annual Meeting and Exposition (ASH-Hem-2025)
- 26 Nov 2025Epaldeudomide is still in phase I trials for Haematological malignancies (Second-line therapy or greater) in USA (PO, Capsule) (NCT04283097)
- 18 Nov 2025Efficacy and adverse events data from a phase I trial in Haematological malignancies released by Kangpu Biopharmaceuticals
SYN
US-10017492-B2
US-20170313676-A1
SYN
EP-3643709-A1
EP-3643709-B1
https://patentscope.wipo.int/search/en/detail.jsf?docId=EP293972088&_cid=P21-MPEVL7-37300-1
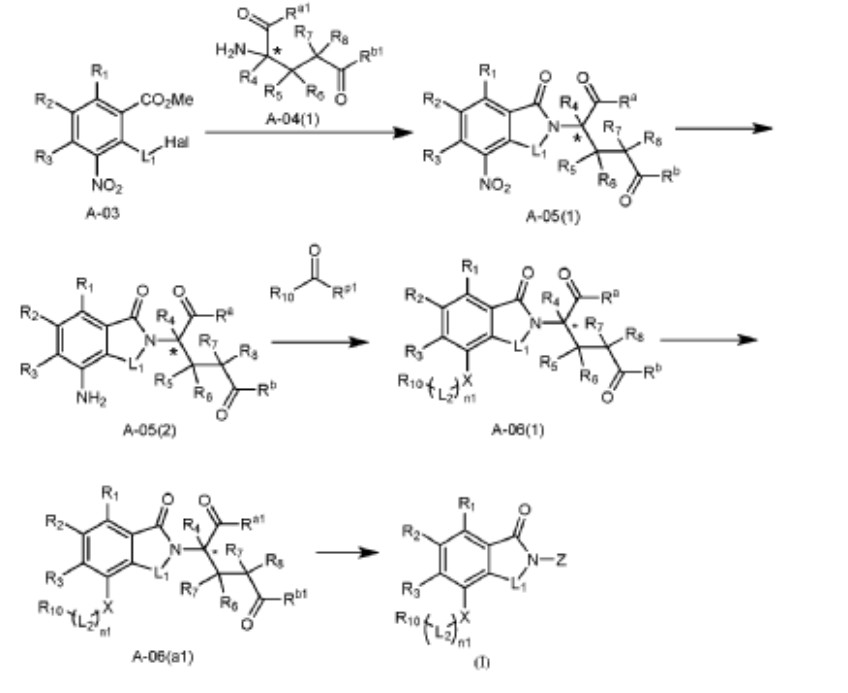
Example 37: Compound A382
[0196] 3-(4-((2-fluoro-5-(3-morpholinopropoxy)benzyl)amino)-1-oxoisoindolin-2-yl)piperidine-2,6-dione, A382.
[0197] 1H NMR (DMSO- d 6, 300 MHz): δ 11.00 (s, 1H), 7.32 (t, J = 7.8 Hz, 1H), 7.22 (t, J = 7.8 Hz, 1H), 7.05-7.13 (m, 2H), 6.93 (d, J = 7.5 Hz, 1H), 6.64 (d, J = 7.8 Hz, 1H), 6.28 (t, J = 6.3 Hz, 1H), 5.07-5.13 (m, 1H), 4.38 (d, J= 5.7 Hz, 2H), 4.28 (d, J= 17.4 Hz, 1H), 4.16 (d, J= 17.4 Hz, 1H), 3.54 (t, J= 4.5 Hz, 4H), 3.42 (s, 2H), 2.85-2.97 (m, 1H), 2.57-2.63 (m, 1H), 2.26-2.38 (m, 5H), 2.00-2.09 (m, 1H). LCMS: 467.2 ([M+1] +).
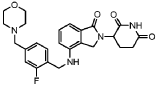
Example 69: Compound A406
[0296] ( S)-3-deuterium-3-(4-((2-fluoro-4-(morpholinomethyl)benzyl)oxy)-1-oxoisoindolin-2-yl)piperidine-2,6-dione, A406.
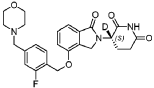
[0297] 1H NMR (DMSO- d 6, 300 MHz): δ 10.98 (s, 1H),7.47-7.55 (m, 2H), 7.31-7.38 (m, 2H), 7.16-7.20 (m, 2H), 5.24 (s, 2H), 5.06-5.12 (m, 0.04H), 4.35 (d, J = 18.0 Hz, 1H), 4.19 (d, J = 18.0 Hz, 1H), 3.55 (br, 4H), 3.47 (s, 2H), 2.82-2.94 (m, 1H), 2.48-2.57 (m, 1H), 2.33-2.42 (m, 5H), 1.91-1.96 (m, 1H). LCMS: 469.2 ([M+1] +).
ADVT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- Isoindoline derivative, intermediate, preparation method, pharmaceutical composition and use thereofPublication Number: US-10017492-B2Priority Date: 2014-10-30Grant Date: 2018-07-10
- Isoindoline derivative, intermediate, preparation method, pharmaceutical composition and use thereofPublication Number: EP-3643709-A1Priority Date: 2014-10-30
- Isoindoline derivative, intermediate, preparation method, pharmaceutical composition and use thereofPublication Number: US-2017313676-A1Priority Date: 2014-10-30
- Isoindoline derivative, intermediate, preparation method, pharmaceutical composition and use thereofPublication Number: EP-3643709-B1Priority Date: 2014-10-30Grant Date: 2021-10-20
/////////epaldeudomide, ANAX LABS, KPG-818, KPG 818, ANTINEOPLASTIC, KV0TBL8MUS
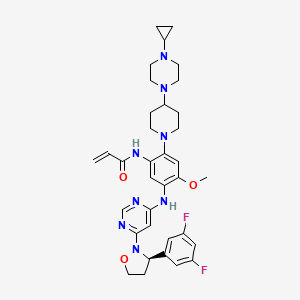
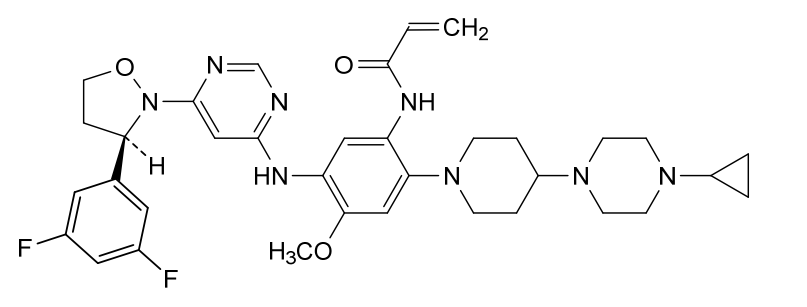
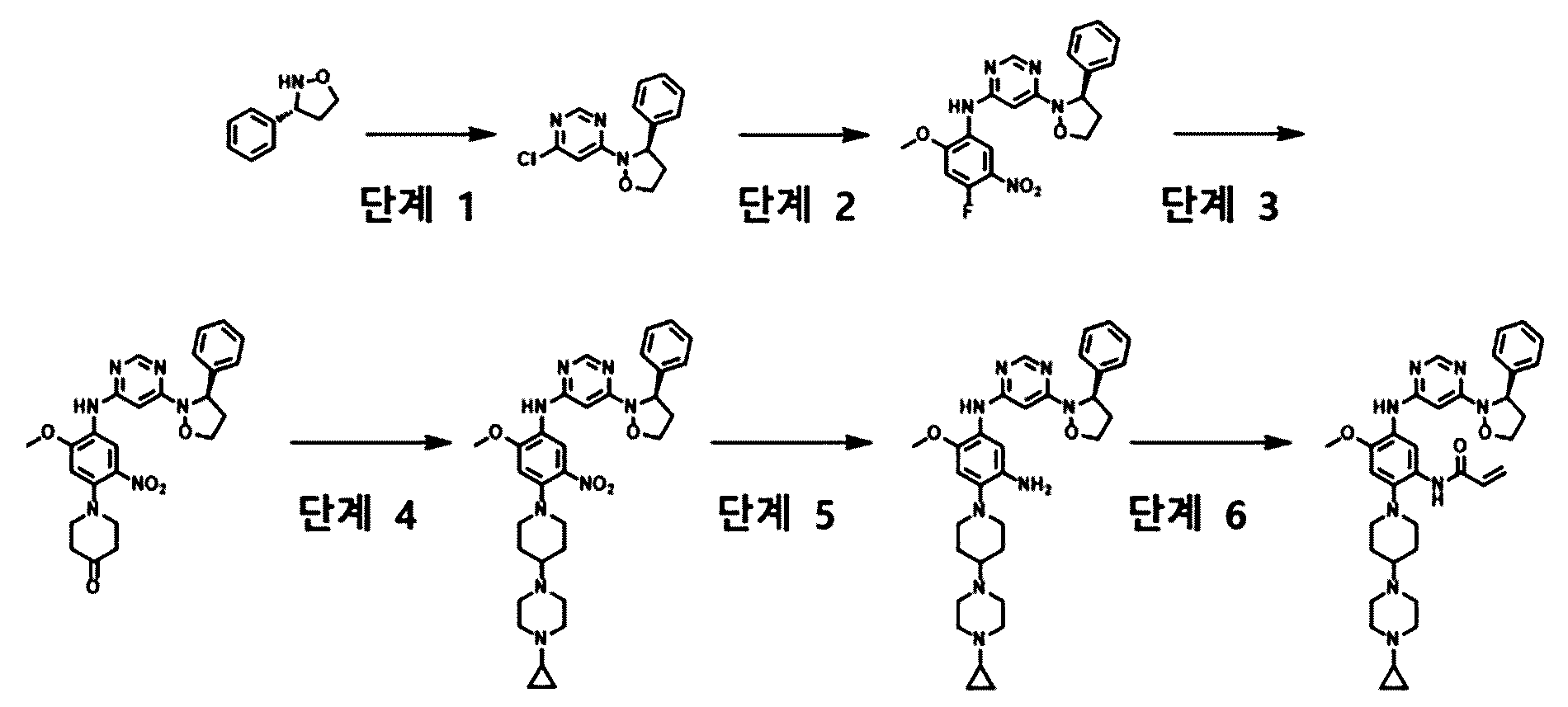
Enozertinib
Enozertinib
CAS 2489185-38-6
MF C35H42F2N8O3 MW660.8
N-[2-[4-(4-cyclopropylpiperazin-1-yl)piperidin-1-yl]-5-[[6-[(3R)-3-(3,5-difluorophenyl)-1,2-oxazolidin-2-yl]pyrimidin-4-yl]amino]-4-methoxyphenyl]prop-2-enamide
- N-(2-(4-(4-cyclopropylpiperazine-1-yl)piperidine-1-yl)-5-((6-((R)-3-(3,5-difluorophenyl)isoxazolidine-2-yl)pyrimidine-4-yl)amino)-4-methoxyphenyl)acrylamide
- N-[2-[4-(4-Cyclopropyl-1-piperazinyl)-1-piperidinyl]-5-[[6-[(3R)-3-(3,5-difluorophenyl)-2-isoxazolidinyl]-4-pyrimidinyl]amino]-4-methoxyphenyl]-2-propenamide
- N-[2-[4-(4-cyclopropylpiperazin-1-yl)piperidin-1-yl]-5-[[6-[(3R)-3-(3,5-difluorophenyl)-1,2-oxazolidin-2-yl]pyrimidin-4-yl]amino]-4-methoxyphenyl]prop-2-enamide
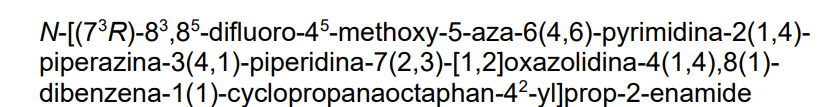
epidermal growth factor receptor tyrosine kinase inhibitor, antineoplastic, ORIC-114, ORIC 114, DU24UP8R94
Enozertinib (formerly ORIC-114) is an investigational, orally bioavailable, and brain-penetrant dual EGFR/HER2 inhibitor developed by ORIC Pharmaceuticals. It targets cancers with exon 20 insertion and atypical EGFR mutations. Its core profile highlights its ability to cross the blood-brain barrier.
How it Works
Enozertinib acts as an irreversible, mutant-selective covalent inhibitor. By blocking overactive EGFR and HER2 signaling, it induces cell death and inhibits tumor growth. Because it penetrates the central nervous system (CNS), it is uniquely suited to treat both primary brain tumors and brain metastases—a common complication in non-small cell lung cancer (NSCLC).
Enozertinib is an orally bioavailable, central nervous system (CNS) penetrating, mutant-selective covalent inhibitor of epidermal growth factor receptor (EGFR; ErbB1) and human epidermal growth factor receptor 2 (HER2; EGFR2; ErbB2) alterations, including exon 20 insertion (Ex20ins) mutations, with potential antineoplastic activity. Upon oral administration, enozertinib selectively targets, irreversibly binds to and inhibits the activity of EGFR or HER2 insertions or mutations. This prevents EGFR/HER2-mediated signaling. This may induce cell death and inhibit tumor growth in EGFR/HER2-overexpressing tumor cells. Enozertinib is able to penetrate the blood-brain-barrier (BBB) and may therefore exert its activity against EGFR Ex20ins-driven CNS primary tumors and CNS metastases. The ErbB receptor tyrosine kinase family is involved in key cellular functions, including cell growth and survival. EGFR and HER2 alterations constitutively upregulate kinase activity.
SYN
https://drughunter.com/molecule/enozertinib-oric-114
SYN

PAT

SIMILAR SYNTHESIS
ADVT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- Egfr inhibitor for treating cancers comprising atypical egfr mutationsPublication Number: WO-2024233313-A1Priority Date: 2023-05-05
- Fumarate, tartrate, malate, and citrate salts of an egfr inhibitorPublication Number: WO-2024096624-A1Priority Date: 2022-11-03
- Malonate and glycolate salts of an egfr inhibitorPublication Number: WO-2024097848-A1Priority Date: 2022-11-03
- Malonate and glycolate salts of an egfr inhibitorPublication Number: EP-4611902-A1Priority Date: 2022-11-03
- Fumarate, tartrate, malate, and citrate salts of an egfr inhibitorPublication Number: EP-4612147-A1Priority Date: 2022-11-03
- Fumarate, tartrate, malate and citrate salts of EGFR inhibitorsPublication Number: CN-120035590-APriority Date: 2022-11-03
- Heteroaryl derivative, method for producing same, and pharmaceutical composition comprising same as effective componentPublication Number: US-11466000-B2Priority Date: 2019-03-19Grant Date: 2022-10-11
- Heteroaryl derivatives, their preparation methods, and pharmaceutical compositions containing them as active ingredientsPublication Number: CN-115838369-APriority Date: 2019-03-19
- Heteroaryl derivatives, methods for their preparation, and pharmaceutical compositions containing them as active ingredientsPublication Number: CN-114605400-APriority Date: 2019-03-19
- HETEROARYL DERIVATIVE, METHOD FOR PRODUCING THE SAME, AND PHARMACEUTICAL COMPOSITION INCLUDING THE SAME AS AN EFFECTIVE COMPONENTPublication Number: BR-112021018704-B1Priority Date: 2019-03-19
- Heteroaryl derivative, method for producing same, and pharmaceutical composition comprising same as effective componentPublication Number: US-2022289733-A1Priority Date: 2019-03-19
- Heteroaryl derivatives, methods for producing heteroaryl derivatives, and pharmaceutical compositions containing heteroaryl derivatives as active ingredientsPublication Number: JP-7394298-B2Priority Date: 2019-03-19Grant Date: 2023-12-08
- Heteroaryl derivatives, methods for their preparation, and pharmaceutical compositions containing them as active ingredientsPublication Number: CN-113993866-APriority Date: 2019-03-19
//////////enozertinib, anax labs, epidermal growth factor receptor tyrosine kinase inhibitor, antineoplastic, ORIC-114, ORIC 114, DU24UP8R94
Engasertib
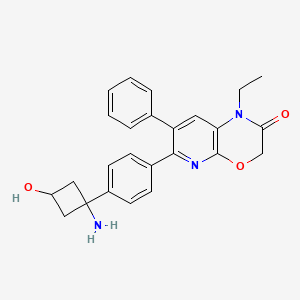
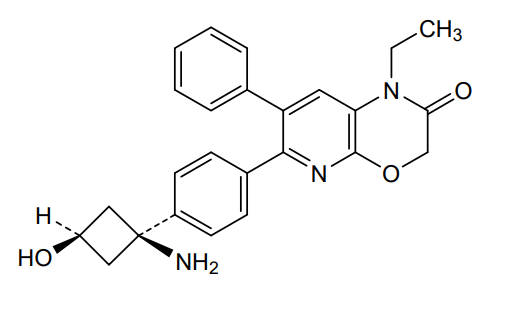
Engasertib
CAS 1313439-71-2
MF C25H25N3O3 MW415.5 g/mol
6-[4-(1-amino-3-hydroxycyclobutyl)phenyl]-1-ethyl-7-phenylpyrido[2,3-b][1,4]oxazin-2-one
6-{4-[(1S,3S)-1-amino-3-hydroxycyclobutyl]phenyl}-1-ethyl-7-phenyl-1H-pyrido[2,3-b][1,4]oxazin-2(3H)-one
serine/threonine kinase inhibitor, ALM-301, VAD-044, ALM 301, VAD 044, Orphan Drug, K2US8HW4TQ
Engasertib is an oral, once-daily AKT inhibitor developed by Vaderis Therapeutics, primarily investigated as a targeted therapy for Hereditary Hemorrhagic Telangiectasia (HHT). Clinical trials show it safely reduces the frequency and duration of bleeding episodes without an FDA-approved equivalent currently available
Core Information
- Mechanism of Action: Engasertib is a highly selective inhibitor of AKT1 and AKT2. In HHT, mutations in the ALK1 pathway lead to abnormal blood vessel growth driven by an excess of the AKT protein. By inhibiting AKT, the drug promotes vascular stability and reduces vessel fragility.
- Target Indication: Hereditary Hemorrhagic Telangiectasia (HHT) — a rare, severe genetic disorder causing vascular abnormalities and frequent, heavy bleeding, particularly nosebleeds (epistaxis)
Clinical Efficacy & Safety
- Proof-of-Concept Trial: A 12-week, placebo-controlled study with 75 HHT patients evaluated daily doses of 30 mg and 40 mg.
- The 40 mg cohort demonstrated a 41% reduction in mean bleeding duration and a 28% reduction in bleeding frequency, compared to 24% and 18% in the placebo group.
- 61% of patients in the 40 mg group rated their clinical condition as “Much Better”.
- Extended Efficacy: In long-term open-label extensions, benefits were sustained and amplified over 12 months, resulting in a 66% reduction in mean bleeding duration and a 55% reduction in bleeding frequency.
- Side Effects: Generally well-tolerated. The most common side effects (reversible and manageable with supportive care) were mild-to-moderate rash and hyperglycemia
- OriginatorAlmac Discovery
- DeveloperVaderis Therapeutics
- ClassAntineoplastics; Small molecules; Vascular disorder therapies
- Mechanism of ActionProto-oncogene protein c-akt inhibitors
- Orphan Drug StatusYes – Hereditary haemorrhagic telangiectasia
- Phase IVascular disorders
- PreclinicalBreast cancer; Prostate cancer
- No development reportedHereditary haemorrhagic telangiectasia
- 28 Dec 2025No recent reports of development identified for phase-I development in Hereditary haemorrhagic telangiectasia in Belgium (PO, Capsule)
- 28 Dec 2025No recent reports of development identified for phase-I development in Hereditary haemorrhagic telangiectasia in France (PO, Capsule)
- 28 Dec 2025No recent reports of development identified for phase-I development in Hereditary haemorrhagic telangiectasia in Italy (PO, Capsule)
SYN
Example 139: 6-(4-((1s.3s)-1-amino-3-hydroxycyclobutyl)phenyl)-1-ethyl-7-phenyl-1H-pyrido[2,3-b][1,4]oxazin-2(3H)-one
Step 1: tert-butyl ((1s.3s)-1-(4-(1-ethyl-2-oxo-7-phenyl-2.3-dihydro-1H-pyrido[2,3-b][1,4]oxazin-6-yl)phenyl)-3-hydroxycyclobutyl)carbamate
In a 15 mL reaction tube was added 6-bromo-1-ethyl-7-phenyl-1H-pyrido[2,3-b][1,4]oxazin-2(3H)-one (50 mg, 0.150 mmol), tert-butyl ((1s,3s)-3-hydroxy-1-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)cyclobutyl)carbamate (49 mg, 0.125 mmol) and cesium carbonate (204 mg, 0.625 mmol) in a mixture of 1,4-dioxane (2.3 ml) and water (0.8 ml) to give a colourless solution. This was degassed by bubbling nitrogen for 15 minutes, followed by the addition of [1,1′-bis(diphenylphosphino)ferrocene]dichloropalladium(II) dichloromethane adduct (20 mg, 0.025 mmol) and degassing for a further 5 minutes. The reaction mixture was heated to 50°C under a nitrogen atmosphere for one hour then allowed to cool to room temperature, diluted with water (5 ml) and extracted into ethyl acetate (3 x 5 ml). The combined organic phases were dried over Na2SO4, filtered and concentrated to dryness under reduced pressure. The residue was purified by Biotage chromatography (cyclohexane:ethyl acetate, gradient elution from 90:10 to 0:100) to give the desired product as an off-white solid (45 mg, 70% yleld). Ή-NMR (500 MHz, CDCl3) δ 7.29-7.35 (5H, m), 7.28 (1H, s), 7.18-7.24 (4H, m), 4.96 (1H, br s), 4.88 (2H, s), 4.05 (1H, br s), 4.01 (2H, q), 2.98 (2H, br s), 2.75 (2H, br s), 1.20-1.51 (9H, br m), 1.32 (3H, t). LCMS (Method D) RT = 1.25 min, M+H+ = 516.20.
Step 2: 6-(4-((1s,3s)-1-amino-3-hydroxycyclobutyl)phenyl)-1-ethyl-7-phenyl-1H-pyrido[2,3-b][1,4]oxazin-2(3H)-one
tert-butyl ((1s,3s)-1-(4-(1-ethyl-2-oxo-7-phenyl-2,3-dihydro-1H-pyrido[2,3-b][1,4]oxazin-6-yl)phenyl)-3-hydroxycyclobutyl)carbamate (45 mg, 0.087 mmol) was dissolved in TFA (1 mL) and stirred for 30 seconds. The solution was immediately concentrated to dryness under reduced pressure. The residue was dissolved in diethyl ether (~3 mL) and concentrated to dryness under reduced pressure three times. The residue was then slurried in diethyl ether (3 mL) and after settling the supernatant solvent removed by pipette. This was repeated three times. The remaining solvent was removed by freeze drylng overnight to give the desired compound as an off-white solid (33 mg, 71% yleld).
1H-NMR (500 MHz, MeOD) δ 7.55 (1H, s), 7.39-7.42 (4H, m), 7.27-7.31 (3H, m), 7.20-7.24 (2H, m), 4.93 (2H, s), 4.01-4.11 (3H, m), 3.03-3.11 (2H, m), 2.42-2.50 (2H, m), 1.28 (3H, t). LCMS (Method D) RT = 0.74 min, M+H+ = 416.20.
SYN
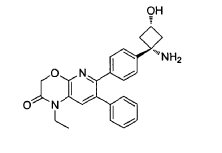
EXAMPLES
Example 1: Synthesis of 6-(4-(l-amino-3-hvdroxycvclobutyl)phenyl)-l-ethyl-7-phenyl-lH-pyrido[2,3-b][l,4]oxazin-2(3H)-one
6-(4-(l-amino-3-hydroxycyclobutyl)phenyl)-l-ethyl-7-phenyl-lH-pyrido[2,3-b][l,4]oxazin-2(3H)-one (referred to herein as “VAD044 free base”) was synthesized in accordance with the protocol as set out in W02011077098 – see in particular Examples 97, 113 and 139 (reproduced below):
Synthesis of 6-(4-((ls,3s)-l-amino-3-hvdroxycvclobutyl)phenyl)-l-ethyl-7-phenyl-lH-pyrido[2,3-bHl,41oxazin-2(3H)-one: from WO2Q11077098 Example 139:
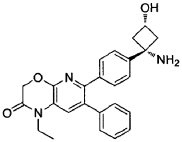
Step 1: tert-butyl((ls,3s)-l-(4-(l-ethyl-2-oxo-7-phenyl-2,3-dihydro-lH-pyrido[2,3-b][l,4]oxazin-6-yl)phenyl)-3-hvdroxycvclobutyl)carbamate
In a 15 mL reaction tube was added 6-bromo-l-ethyl-7-phenyl-lH-pyrido[2,3-b][l,4]oxazin-2(3H)-one* (50 mg, 0.150 mmol), tert-butyl((ls,3s)-3-hydroxy-l-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl)cyclobutyl)carbamate** (49 mg, 0.125 mmol) and cesium carbonate (204 mg, 0.625 mmol) in a mixture of 1,4-dioxane (2.3 ml) and water (0.8 ml) to give a colourless solution. This was degassed by bubbling nitrogen for 15 minutes, followed by the addition of [1,1′-bis(diphenylphosphino)ferrocene]dichloropalladium(ll) dichloromethane adduct (20 mg, 0.025 mmol) and degassing for a further 5 minutes. The reaction mixture was heated to 50°C under a nitrogen atmosphere for one hour then allowed to cool to room temperature, diluted with water (5 ml) and extracted into ethyl acetate (3 x 5 ml). The combined organic phases were dried over Na2SO4, filtered and concentrated to dryness under reduced pressure. The residue was purified by Biotage chromatography (cyclohexane:ethyl acetate, gradient elution from 90:10 to 0:100) to give the desired product as an off- white solid (45 mg, 70% yield). 1H-NMR (500 MHz, CDCI3) 6 7.29-7.35 (5H, m), 7.28 (1H, s), 7.18-7.24 (4H, m), 4.96 (1H, br s), 4.88 (2H, s), 4.05 (1H, br s), 4.01 (2H, q), 2.98 (2H, br s), 2.75 (2H, br s), 1.20-1.51 (9H, br m), 1.32 (3H, t). LCMS (Method D) RT = 1.25 min, M+H+ = 516.20.

tert-butyl((ls,3s)-l-(4-(l-ethyl-2-oxo-7-phenyl-2,3-dihydro-lH-pyrido[2,3-b][l,4]oxazin-6-yl)phenyl)-3- hydroxycyclobutyl)carbamate (45 mg, 0.087 mmol) was dissolved in TFA (1 mL) and stirred for 30 seconds. The solution was immediately concentrated to dryness under reduced pressure. The residue was dissolved in diethyl ether (~3 mL) and concentrated to dryness under reduced pressure three times. The residue was then slurried in diethyl ether (3 mL) and after settling the supernatant solvent removed by pipette. This was repeated three times. The remaining solvent was removed by freeze drying overnight to give the desired compound as an off-white solid (33 mg, 71% yield). 1H-NMR (500 MHz, MeOD) 6 7.55 (1H, s), 7.39- 7.42 (4H, m), 7.27-7.31 (3H, m), 7.20- 7.24 (2H, m), 4.93 (2H, s), 4.01-4.11 (3H, m), 3.03-3.1 1 (2H, m), 2.42-2.50 (2H, m), 1.28 (3H, t). LCMS (Method D) RT = 0.74 min, M+H+ = 416.20.

To a suspension of sodium hydride (5.31 g, 133 mmol) in 1,4-dioxane (250 ml), ethyl glycolate (12.56 ml,
133 mmol) was added drop wise over a period of 30 minutes ensuring that the temperature was maintained below 30°C. The resulting thick suspension was stirred at room temperature for 15 minutes.
In a separate II round- bottomed flask was added 5-bromo-2-chloro-3-nitropyridine (21 g, 88 mmol) in
1,4-dioxane (150 ml) to give a brown solution. The suspension of sodium hydride and ethyl glycolate was added drop wise over a period of 30 minutes at 0°C. The resulting reaction mixture was heated to 80°C overnight.
The reaction mixture was concentrated under reduced pressure and the crude residue was purified by
Biotage silica chromatography (gradient 0% to 10% ethyl acetate in n-hexanes) to give the title compound
(1 ,8g, 44%).1H NMR (500 MHz, CDCI3) 6 8.48 (1H, s), 8.42 (1H, s), 5.07 (2H, s), 4.28-4.24 (2H, q), 1.31-1.28
(3H, t).
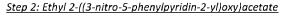
In a II round-bottomed flask was added ethyl 2-(5-bromo-3-nitropyridin-2-yloxy)acetate (18.33 g, 60.1 mmol), phenylboronic acid (10.99 g, 90 mmol), triphenylphosphine (4.73 g, 18.02 mmol) and cesium fluoride (45.6 g, 300 mmol) in 1,2-dimethoxyethane (300 ml) to give a yellow solution. The reaction mixture was degassed by bubbling nitrogen for 30 minutes. Pallad ium (II ) acetate (2.023 g, 9.01 mmol) was added and the mixture was heated to 75°C under a nitrogen atmosphere overnight. The reaction mixture was allowed to cool to room temperature and concentrated to dryness under reduced pressure to give a brown solid. This was re-dissolved in dichloromethane, filtered and concentrated to dryness under reduced pressure to give a brown solid The crude residue was purified via Biotage chromatography (gradient 5% to 60% ethyl acetate in n-hexanes) to give the title compound (6.9g, 38%). 1H NMR (500 MHz, CDCI3) 6 8.58 (1H, s), 8.56 (1H, s), 7.59-7.52 (2H, m), 7.48-7.46 (2H, m), 7.45-7.43 (1H, m), 5.13 (2H, s), 4.30-4.26 (2H, q), 1.33-1.30 (3H, t).
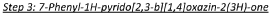
In a 500 ml round-bottomed flask was added ethyl 2-(3-nitro-5-phenylpyridin-2-yloxy)acetate (4.6 g, 15.22 mmol) in hydrochloric acid, 37% (40 ml) to give a yellow suspension. The mixture was cooled to 0-5°C followed by the portion wise addition of tin powder (9.94 g, 84 mmol). The addition proved to be exothermic. Caution should be taken while adding. The mixture was then stirred at room temperature for further 30 minutes until all foaming ceased. The reaction mixture was heated to 80°C under a nitrogen atmosphere for 3 hours. The reaction mixture cooled to room temperature and diluted with water (800ml). The white precipitate was isolated by filtration, washed with water (100 ml) and sucked dry to give a white solid. The solid was azeotroped with toluene (3 x 30 ml) to give a white solid as the title compound (2.6g, 77%). XH NMR (500 MHz, (CD3)2SO) 6 10.41 (1H, s), 8.10 (1H, s), 7.59 (2H, d), 7.49-7.42 (2H, t), 7.39-7-38 (1H, d), 4.83 (2H, s).
Step 4: 6-bromo-7-phenyl-lH-pyrido[2,3-b][l,4]oxazin-2(3H)-one
In a 10ml microwave vial was 7-phenyl-l H-pyrido[2,3-b][l ,4]oxazin-2(3H)-one (50 mg, 0.221 mmol) and N-bromosuccinimide (78.6 mg, 0.441 mmol) in dimethylformamide (1 ml). The reaction mixture was heated to 80°C under microwave irradiation for 30 minutes. The reaction mixture was cooled to room
temperature and diluted with ethyl acetate (10ml). The organic solution was washed with water (2x10ml) and brine (2x10ml). The organic phase was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude residue was purified via Biotage chromatography (gradient 0% to 5% methanol in dichloromethane) to give the title compound as a yellow solid (61 mg, 90%). 1H NMR (500 MHz, CD3OD) 6 7.48-7.32 (5H, m), 7.12 (1 H, s), 4.82 (2H, s).
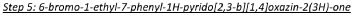
In a 15 mL reaction tube was added 6-bromo-7-phenyl-lH-pyrido[2,3-b][l,4]oxazin-2(3H)-one (300 mg, 0.983 mmol), iodoethane (0.095 mL, 1.180 mmol) and potassium carbonate (408 mg, 2.95 mmol) in anhydrous N,N-dimethylformamide (1 mL) to give a brown suspension. This was stirred at 50 °C under a nitrogen atmosphere for 60 minutes. The reaction mixture was diluted with saturated sodium bicarbonate solution (5 mL) and extracted into ethyl acetate (3 x 5 mL). The combined organic phases were washed with 50:50 water.brine (3 x 5 mL), dried over Na2SO4, filtered and concentrated to dryness under reduced pressure to give a brown solid. This was purified by Biotage chromatography (25g silica cartridge, cyclohexane:ethyl acetate, gradient elution from 90:10 to 20:80) to give the title compound as a beige solid (160 mg, 48.8 % yield). XH NMR (500 MHz, CDCI3) 6 7.58-7.37 (5H, m), 7.21 (1H, s), 4.86 (2H, s), 3.96 (2H, q), 1.27 (3H, t). LCMS (Method D) RT 1.293 min, M+l= 334.

n a 40 mL reaction tube was added tert-butyl(ls,3s)-l-(4-bromophenyl)-3- hydroxycyclobutylcarbamate*** (0.25 g, 0.731 mmol) in anhydrous tetrahydrofuran (14 ml) to give a colourless solution. This was degassed by bubbling nitrogen for 20 minutes, followed by the addition of [l,l’-bis(diphenylphosphino)ferrocene]dichloropalladium(ll) dichloromethane adduct (60 mg, 0.073 mmol). After bubbling nitrogen for a further 15 minutes, potassium acetate (143 mg, 1.461 mmol) and bis(pinacolato)diboron (223 mg, 0.877 mmol) were added. The reaction mixture was heated to reflux overnight then concentrated to dryness under reduced pressure and purified by Biotage chromatography (cyclohexane:ethyl acetate, gradient elution from 88:12 to 0:100) to give the desired product as a colourless oil that solidified upon standing (240 mg, 84% yield). 1H-NMR (500 MHz, CDCI3) 6 7.71 (2H, d), 7.44 (2H, d), 4.15 (1H, br s), 2.87-2.98 (2H, m), 2.27-2.44 (2H, m), 1.22-1.49 (21H, br m).
(*** synthesis described in WO2009148887 and WO2009148916)
ADVT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- Inhibitors of akt activityPublication Number: EP-2516435-B1Priority Date: 2009-12-23Grant Date: 2014-08-06
- Inhibitors of akt activityPublication Number: WO-2011077098-A1Priority Date: 2009-12-23
- Inhibitors of akt activityPublication Number: EP-2516435-B8Priority Date: 2009-12-23Grant Date: 2014-10-15
- Inhibitors of akt activityPublication Number: EP-2516435-A1Priority Date: 2009-12-23
- Inhibitors of AKT activityPublication Number: US-9221838-B2Priority Date: 2009-12-23Grant Date: 2015-12-29
- Allosteric akt inhibitors for use in the treatment of hereditary hemorrhagic telangiectasiaPublication Number: US-2024092801-A1Priority Date: 2020-09-30
- Allosteric akt inhibitors for use in the treatment of hereditary hemorrhagic telangiectasiaPublication Number: WO-2022069552-A1Priority Date: 2020-09-30
- Allosteric akt inhibitors for use in the treatment of hereditary hemorrhagic telangiectasiaPublication Number: EP-4221713-A1Priority Date: 2020-09-30
- Inhibitors of akt activityPublication Number: US-2013116243-A1Priority Date: 2009-12-23
- Inhibitors of akt activityPublication Number: WO-2011077098-A9Priority Date: 2009-12-23
////////engasertib, anax labs, serine/threonine kinase inhibitor, ALM-301, VAD-044, ALM 301, VAD 044, Orphan Drug, K2US8HW4TQ
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY


{kind=link}
{kind=link}