
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

Laporolimus
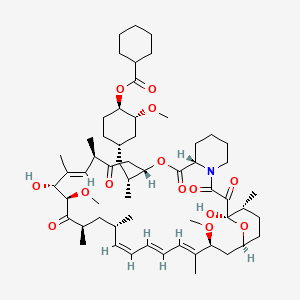
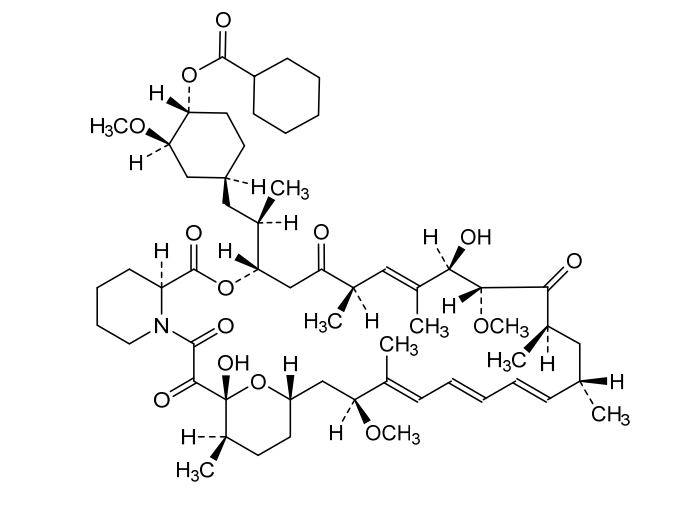
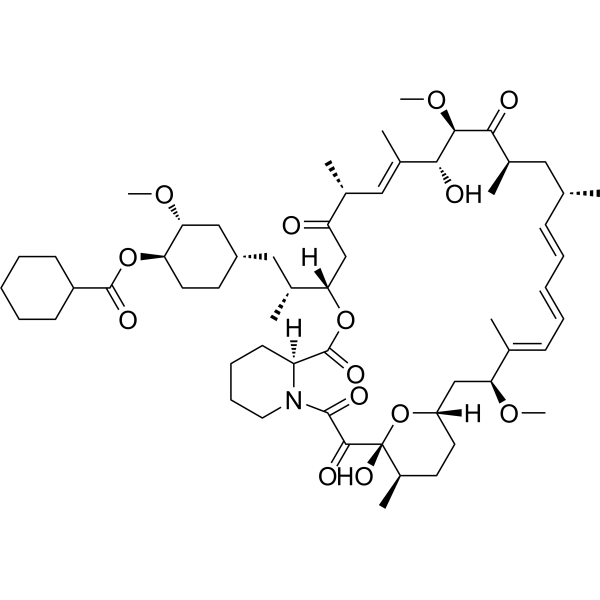
Laporolimus
Rapamycin, 42-cyclohexanecarboxylate
CAS 1504576-27-5
MF C58H89NO14 MW 1024.3 g/mol
[(1R,2R,4S)-4-[(2R)-2-[(1R,9S,12S,15R,16E,18R,19R,21R,23S,24E,26E,28E,30S,32S,35R)-1,18-dihydroxy-19,30-dimethoxy-15,17,21,23,29,35-hexamethyl-2,3,10,14,20-pentaoxo-11,36-dioxa-4-azatricyclo[30.3.1.04,9]hexatriaconta-16,24,26,28-tetraen-12-yl]propyl]-2-methoxycyclohexyl] cyclohexanecarboxylate
(1R,2R,4S)-4-{(2R)-2-[(3S,6R,7E,9R,10R,12R,14S,15E,17E,19E,21S,23S,26R,27R,34aS)-9,27-dihydroxy-10,21-dimethoxy6,8,12,14,20,26-hexamethyl-1,5,11,28,29-pentaoxo1,4,5,6,9,10,11,12,13,14,21,22,23,24,25,26,27,28,29,3
1,32,33,34,34a-tetracosahydro-3H-23,27-epoxypyrido[2,1-c][1,4]oxazacyclohentriacontin-3-yl]propyl}-2-methoxycyclohexyl
cyclohexanecarboxylate
immunosuppressant, CRC-015, CRC 015, F5041W3RVA, Rapamycin, 42-cyclohexanecarboxylate
Laporolimus is an experimental immunosuppressant compound that acts as an mTOR (mechanistic target of rapamycin) pathway inhibitor. It is chemically classified as a macrolide derivative and is also known by its chemical synonym, rapamycin 42-cyclohexanecarboxylate.
Currently, Laporolimus is designated for research use only and has not been approved for clinical medical applications in humans or animals.
Key Technical Details
- Mechanism of Action: It blocks the mTOR signaling pathway, which is responsible for regulating cell growth, proliferation, and immune cell activation.
Distinguishing Laporolimus from Clinical Alternatives
Because it ends with the suffix -limus, it shares structural and nomenclature similarities with widely used clinical immunosuppressants. However, its legal status and development stage differ significantly:
| Drug Name | Clinical Availability | Primary Mechanism | Primary Uses |
|---|---|---|---|
| Laporolimus | None (Research Only) | mTOR Inhibitor | Laboratory research |
| Sirolimus (Rapamycin) | Approved | mTOR Inhibitor | Transplant rejection, coating coronary stents |
| Tacrolimus | Approved | Calcineurin Inhibitor | Organ transplant prophylaxis, severe eczema |
If you are researching this compound for a laboratory study, you can review its structural data and biochemical properties via the PubChem Laporolimus Compound Page
Laporolimus (CAS 1504576-27-5) is an immunosuppressive agent and mTOR inhibitor structurally derived from rapamycin as a cyclohexanecarboxylate derivative. Its total chemical synthesis is highly complex, typically achieved via semisynthesis starting from natural macrolides produced by Streptomyces fermentation.
Semisynthetic Pathway
Because the core macrocyclic lactone (a 36-membered polyketide ring) is incredibly challenging to build from scratch, researchers and pharmaceutical manufacturers rely on a derivatization approach:
- Fermentation: The baseline macrolide is produced via large-scale fermentation of Streptomyces hygroscopicus (similar to the base rapamycin process).
- Purification: The naturally produced macrocyclic core is isolated and purified from the fermentation broth using column chromatography.
- Esterification: The C-42 hydroxyl group of the macrolide core is selectively protected and subjected to acylation with a cyclohexanecarboxylic acid derivative (or reactive cyclohexanecarbonyl chloride).
- Deprotection & Purification: The C-42 cyclohexanecarboxylate is then deprotected and purified via preparative chromatography to yield pure laporolimus.
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma



AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

PAT
/////////laporolimus, immunosuppressant, CRC-015, CRC 015, F5041W3RVA, Rapamycin, 42-cyclohexanecarboxylate
Lanoracopan
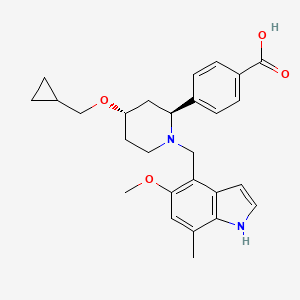
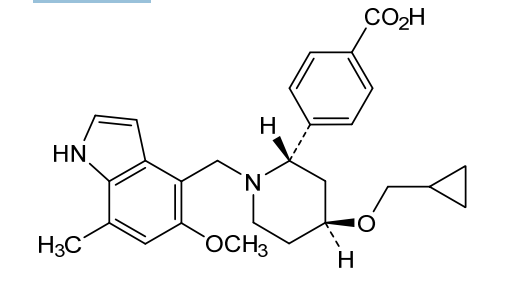
Lanoracopan
CAS 2797066-85-2
MFC27H32N2O4 MW448.6 g/mol
4-[(2S,4S)-4-(cyclopropylmethoxy)-1-[(5-methoxy-7-methyl-1H-indol-4-yl)methyl]piperidin-2-yl]benzoic acid
4-{(2S,4S)-4-(cyclopropylmethoxy)-1-[(5-methoxy-7-methyl-1H-indol-4-yl)methyl]piperidin-2-yl}benzoic acid
complement factor B inhibitor, MY 008211A, Factor B-IN-5, Y5UN7AE8SF
Lanoracopan (also known by its developmental code MY008211A or Factor B-IN-5) is an investigational small-molecule drug that acts as a potent complement factor B (CFB) inhibitor. It is designed to target and regulate the alternative pathway of the complement system, which is a crucial part of the body’s innate immune defense
Clinical Development & Indications
Originally developed by Shanghai Meiyue Biotech Development Co. Ltd., the drug has transitioned from early discovery into active clinical trials. It is primarily being evaluated for:
- Paroxysmal Nocturnal Hemoglobinuria (PNH): Lanoracopan (as MY008211A tablets) is currently undergoing Phase 2 and Phase 2/3 clinical trials. These studies assess its long-term safety, tolerability, and efficacy in patients suffering from PNH who experience active hemolysis (the premature destruction of red blood cells).
- Renal Impairment Studies: Clinical research is also actively evaluating the drug’s safety profile and pharmacokinetics in individuals with varying degrees of kidney function.
Current Status
Lanoracopan is recognized under the World Health Organization’s proposed International Nonproprietary Names (INN) registry. It is not yet approved for public use or commercial medical prescriptions by global regulatory bodies. Currently, it is primarily available to the scientific community as a reference standard for laboratory research use only
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023020566&_cid=P21-MQD5YH-25997-1
[0727]4-((2S,4S)-4-(cyclopropylmethoxy)-1-((5-methoxy-7-methyl-1H-indol-4-yl)methyl)piperidin-2-yl)benzoic acid (compound 4)
[0728]
4-((2S,4S)-4-(cyclopropylmethoxy)-1-((5-methoxy-7-methyl-1H-indol-4-yl)methyl)piperidin-2-yl)benzoic acid
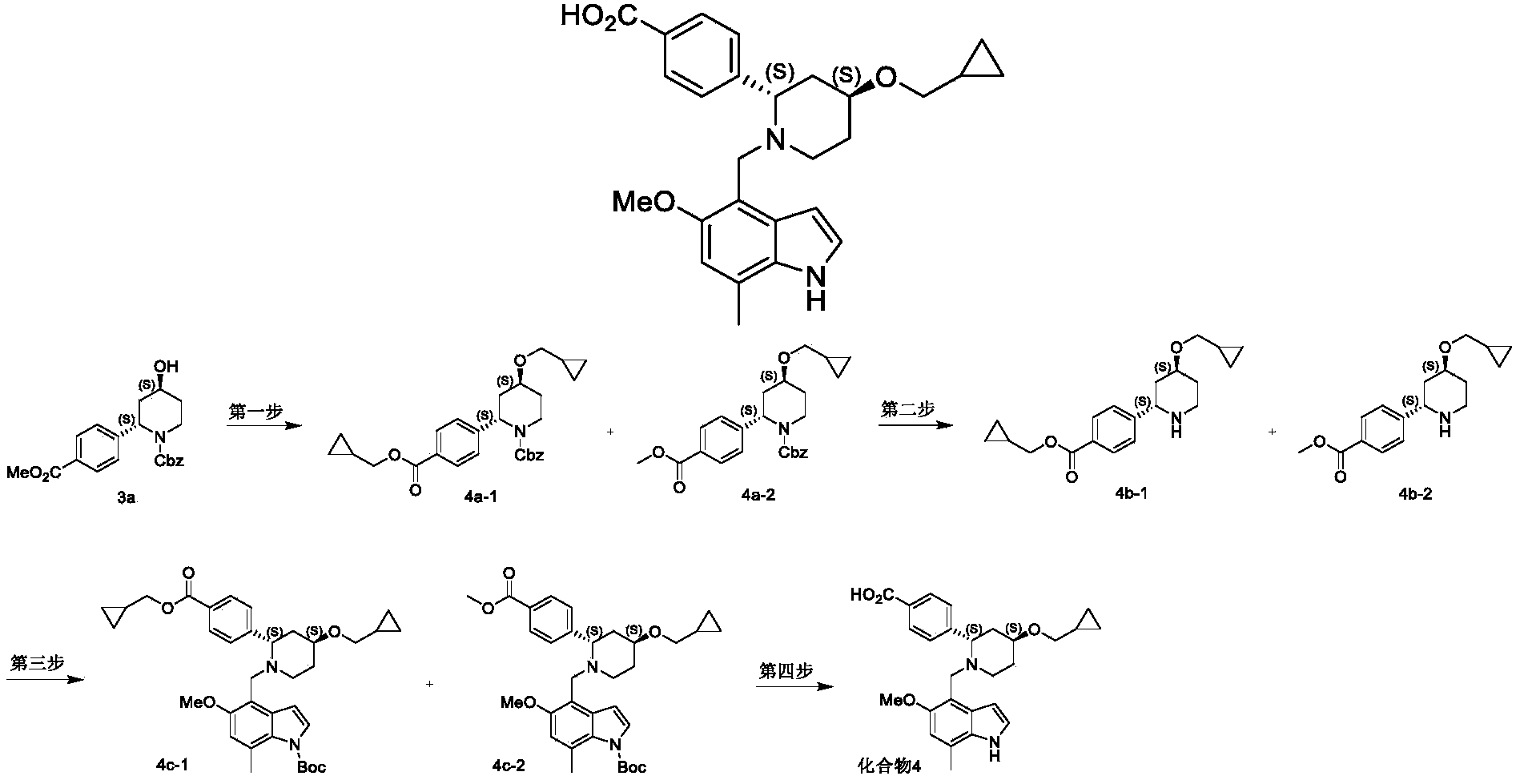
[0755]The mixture (130 mg) of the above-mentioned tert-butyl 4-(((2S,4S)-4-(cyclopropylmethoxy)-2-(4-(((cyclopropylmethoxy)carbonyl)phenyl)piperidin-1-yl)methyl)-5-methoxy-7-methyl-1H-indole-1-carboxylic acid (tert-butyl ester) and 4-(((2S,4S)-4-(cyclopropylmethoxy)-2-(4-(methoxycarbonyl)phenyl)piperidin-1-yl)methyl)-5-methoxy-7-methyl-1H-indole-1-carboxylic acid (tert-butyl ester) (4c-2) was dissolved in 10 mL of methanol, and solid potassium carbonate (149 mg, 1.08 mmol) was added. The mixture was heated to 85 °C and refluxed for 3 h. The reaction solution was cooled to room temperature and concentrated under reduced pressure to obtain the crude product. The crude product was dissolved in a mixed solvent of 10 mL THF, 5 mL methanol, and 2 mL water. Lithium hydroxide monohydrate (181 mg, 4.3 mmol) was added, and the mixture was stirred at room temperature for 16 h. The reaction system was concentrated under reduced pressure, and the crude product was subjected to Pre-HPLC (instrument and preparative column: Glison GX-281 preparative HPLC system; Sunfire C18 column, 5 μm, inner diameter × length = 30 mm × 150 mm). Preparation method: The crude product was dissolved in methanol and dimethyl sulfoxide, and filtered through a 0.45 μm filter membrane to prepare the sample solution. Mobile phase system: acetonitrile/aqueous solution containing 5 mmol/L ammonium acetate. Gradient elution method: Acetonitrile was used to elute 60% of the solution with a 5% gradient (elution time 15 min), and the solution was lyophilized to obtain 4-((2S,4S)-4-(cyclopropylmethoxy)-1-((5-methoxy-7-methyl-1H-indol-4-yl)methyl)piperidin-2-yl)benzoic acid (compound 4) (5 mg).
[0756]
1H NMR(400MHz,CD 3OD)δ8.10(d,2H),7.60(d,2H),7.28(d,1H),6.73(s,1H),6.32(s,1H),4.70–4.40(m,1H),4.32–4.14(m,1H),4.09–3.90(m,1H),3.88–3.79(m,1H),3.75(s, 3H),3.42–3.34(m,2H),3.30–3.14(m,2H),2.49(s,3H),2.26–2.10(m,2H),2.06–1.90(m,2H),1.19–1.04(m,1H),0.64–0.50(m,2H),0.31–0.22(m,2H).
[0757]
LCMS m/z=449.2[M+1] +
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
PAT
- Benzo nitrogen-containing heteroaromatic ring derivative and use thereof in medicinePublication Number: WO-2023020566-A1Priority Date: 2021-08-18
- Benzo nitrogen-containing heteroaromatic ring derivative and use thereof in medicinePublication Number: US-2024383917-A1Priority Date: 2021-08-18
- A benzazaaromatic ring derivative and its application in medicinePublication Number: CN-118043314-APriority Date: 2021-08-18
- Benzo nitrogen-containing heteroaromatic ring derivative and use thereof in medicinePublication Number: EP-4389742-A1Priority Date: 2021-08-18
- Complement factor b inhibitor, and pharmaceutical composition thereof, preparation method therefor and use thereofPublication Number: EP-4194451-A1Priority Date: 2020-08-07
- Complement factor b inhibitor, and pharmaceutical composition thereof, preparation method therefor and use thereofPublication Number: EP-4282486-A2Priority Date: 2020-08-07
- Complement factor b inhibitor, and pharmaceutical composition, preparation method and use thereofPublication Number: US-2023286947-A1Priority Date: 2020-08-07
/////////lanoracopan, anax labs, complement factor B inhibitor, MY 008211A, Factor B-IN-5, Y5UN7AE8SF
Lanisidenib
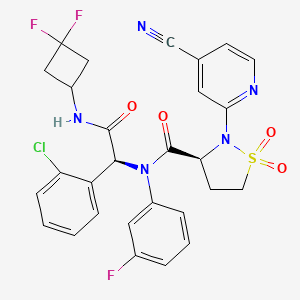
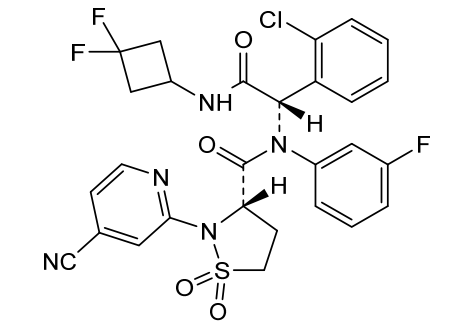
Lanisidenib
Cas 2135537-20-9
MF C28H23ClF3N5O4S MW618.03 g/mol
(3S)-N-[(1S)-1-(2-chlorophenyl)-2-[(3,3-difluorocyclobutyl)amino]-2-oxoethyl]-2-(4-cyano-2-pyridinyl)-N-(3-fluorophenyl)-1,1-dioxo-1,2-thiazolidine-3-carboxamide
IUPAC Name: (3S)-N-{(1S)-1-(2-chlorophenyl)-2-[(3, 3-difluorocyclobutyl)amino]-2-oxoethyl}-2-(4-cyanopyridin-2-yl)-N-(3-fluorophenyl)-1,1-dioxo-1λ⁶,2-thiazolidine-3-carboxamide
(3S)-N-{(1S)-1-(2-chlorophenyl)-2-[(3,3-difluorocyclobutyl)amino]-2-oxoethyl}-2-(4-cyanopyridin-2-yl)-N-(3-fluorophenyl)-1,1-dioxo1λ6,2-thiazolidine-3-carboxamide
isocitrate dehydrogenase inhibitor, antineoplastic, G5J396CG5J
Lanisidenib is a potent, selective isocitrate dehydrogenase (IDH) inhibitor that exhibits antineoplastic (anti-cancer) activity. It works by targeting abnormal IDH enzymes, which are frequently mutated in various malignancies, such as certain myeloid leukemias and solid tumours. By blocking these mutant enzymes, it halts the production of oncometabolites that drive cancer progression
Research and Availability
The compound is primarily utilized in biochemical research and preclinical drug screening platforms. Specialty chemical suppliers, such as MedChemExpress and AdooQ BioScience, distribute it exclusively for laboratory research
SYN
Inhibitors of Mutant Isocitrate Dehydrogenases 1 and 2 (mIDH1/2): An Update and Perspective
Publication Name: Journal of Medicinal Chemistry
Publication Date: 2018-05-31
PMID: 29847930
DOI: 10.1021/acs.jmedchem.8b00159
PAT
| Step F: (S)—N—((S)-1-(2-chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-2-(4-cyanopyridin-2-yl-N-(3-fluorophenyl)-isothiazolidine-3-carboxamide 1,1-dioxide |
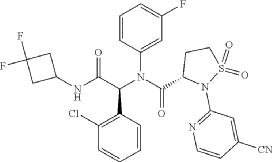
| At room temperature, 3-amino-5-Fluorouridine (57 mg, 0.508 mmol) and o-chlorobenzaldehyde (72 mg, 0.512 mmol) were dissolved in methanol, and stirred for 30 min. (S)-2-(4-cyanopyridin-2-yl)isothiazolidine-3-carboxylic acid 1,1-dioxide (136 mg, 0.508 mmol) was then added into the mixed solution, stirred for 10 min, then added with 1,1-difluoro-3-isocyanocyclobutane (prepared according to the method described in patent CN103097340, 60 mg, 0.508 mmol), and stirred overnight. The solvent was removed and the residue was separated by thin layer chromatography, to give the title compound (S)—N—((S)-1-(2-chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-2-(4-cyanopyridin-2-yl-N-(3-fluorophenyl)-isothiazolidine-3-carboxamide 1,1-dioxide (the compound of formula I). |
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019057142&_cid=P20-MQBQHW-03190-1
A sulfonamide compound with the structure shown in Formula I has the chemical name: (S)-N-((S)-1-(2-chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-2-(4-cyanopyridin-2-yl)-N-(3-fluorophenyl)-isothiazolidin-3-carboxamide 1,1-dioxide.
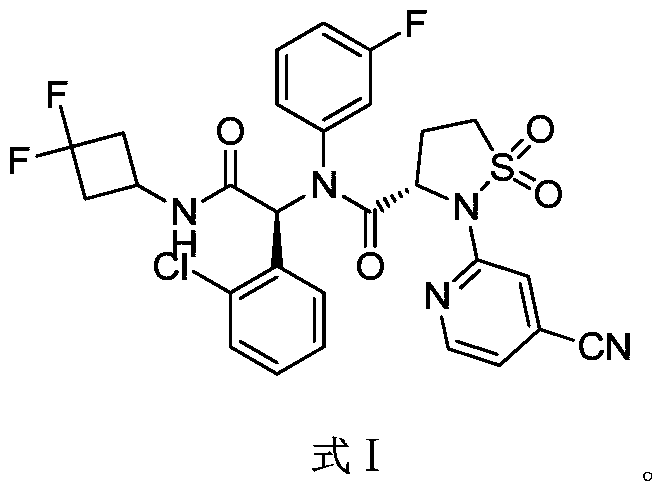
Step F: (S)-N-((S)-1-(2-chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-2-(4-cyanopyridin-2-yl)-N-(3-fluorophenyl)-isothiazolidin-3-carboxamide 1,1-dioxide
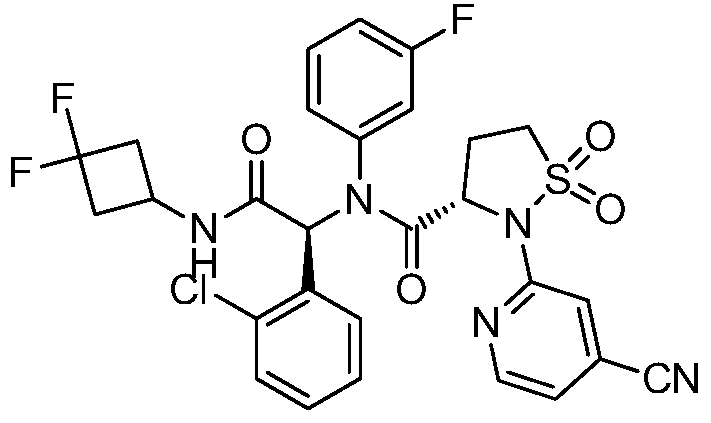
At room temperature, 3-amino-5-fluoropyridine (57 mg, 0.508 mmol) and o-chlorobenzaldehyde (72 mg, 0.512 mmol) were dissolved in methanol and stirred for 30 minutes. Then, (S)-2-(4-cyanopyridin-2-yl)isothiazolidin-3-carboxylic acid 1,1-dioxide (136 mg, 0.508 mmol) was added to the mixture and stirred for 10 minutes. Finally, 1,1-difluoro-3-isocyanocyclobutane (refer to the patent) was added. Prepared by the method described in CN103097340, 60 mg (0.508 mmol), stirred overnight, solvent removed, and separated by thin-layer chromatography to obtain the title compound (S)-N-((S)-1-(2-chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-2-(4-cyanopyridin-2-yl)-N-(3-fluorophenyl)-isothiazolidin-3-carboxamide 1,1-dioxide (compound of formula I).
[0134]
1H-NMR(400MHz,CDCl 3):δ=8.46(m,1H),7.67(d,J=8.8Hz,1H),7.63(s,1H),7.22-6.84(m,8H),6.47(d,J=3.6,1H),6.08(s,1H),4.82(d,J=6.1Hz,1H),4.33(m,1H),3.68-3.60(m,1H),3.40-3.28(m,1H),3.10-2.98(m,2H),2.68-2.38(m,4H)。
[0135]
m/z=618[M+H] +。
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
PAT
- Sultam compound and application method thereofPublication Number: US-11111240-B2Priority Date: 2016-03-22Grant Date: 2021-09-07
- Sultam Compound And Application Method ThereofPublication Number: US-2021047314-A1Priority Date: 2016-03-22
- Lactam compounds and methods of using the samePublication Number: CN-113666922-APriority Date: 2016-03-22
- Endosulfonamide compound and method of use thereofPublication Number: TW-201736354-APriority Date: 2016-03-22
- Sultam compound and application method thereofPublication Number: EP-3434671-B1Priority Date: 2016-03-22Grant Date: 2020-10-21
- Internal sulfonamide compounds and methods of use thereofPublication Number: CN-109071471-BPriority Date: 2016-03-22Grant Date: 2021-05-07
- Sultam compounds and methods of use thereofPublication Number: KR-102389985-B1Priority Date: 2016-03-22Grant Date: 2022-04-22
- Endosulfonamide compounds and methods of usePublication Number: TW-I729094-BPriority Date: 2016-03-22Grant Date: 2021-06-01
- Crystalline sulfamide compoundsPublication Number: KR-102707847-B1Priority Date: 2017-09-22Grant Date: 2024-09-23
- Crystalline sulfamide compoundPublication Number: KR-20200057049-APriority Date: 2017-09-22
- Crystalline sulfamide compoundPublication Number: CA-3076405-A1Priority Date: 2017-09-22
- Crystalline sulfamide compoundPublication Number: US-2020291012-A1Priority Date: 2017-09-22
- Crystalline sulfamide compoundPublication Number: US-11254665-B2Priority Date: 2017-09-22Grant Date: 2022-02-22
- Preparation method of lactam compoundPublication Number: CN-118580235-APriority Date: 2023-03-03
- A kind of internal sulfonamide compound crystalPublication Number: CN-111065630-APriority Date: 2017-09-22
- Crystalline sulfamide compoundPublication Number: EP-3686191-A1Priority Date: 2017-09-22
- A kind of internal sulfonamide compound crystallizationPublication Number: CN-111065630-BPriority Date: 2017-09-22Grant Date: 2022-12-30
- Crystalline sulfamide compoundPublication Number: EP-3686191-B1Priority Date: 2017-09-22Grant Date: 2022-12-14
///////////lanisidenib, anax labs, isocitrate dehydrogenase inhibitor, antineoplastic, G5J396CG5J
Itareparib
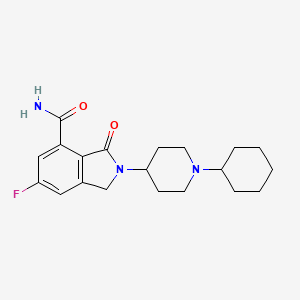
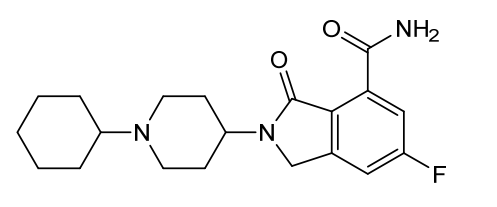
Itareparib
CAS 1606995-47-4
MF C20H26FN3O2 MW359.4 g/mol
2-(1-Cyclohexyl-4-piperidinyl)-6-fluoro-2,3-dihydro-3-oxo-1H-isoindole-4-carboxamide
1H-ISOINDOLE-4-CARBOXAMIDE, 2-(1-CYCLOHEXYL-4-PIPERIDINYL)-6-FLUORO-2,3-DIHYDRO-3-OXO-
2-(1-cyclohexylpiperidin-4-yl)-6-fluoro-3-oxo-2,3-dihydro-1H-isoindole4-carboxamide
poly (ADP-ribose) polymerase (PARP) inhibitor, antineoplastic, NMS-03305293, NMS-293, NMS 03305293, NMS 293, KFI1190L8L, NV 578,
Itareparib is the inhibitor for PARP and exhibits antineoplastic activity.
Itareparib (development code NMS-03305293 or NMS-293) is an experimental, next-generation PARP1-selective oral inhibitor being developed by the biopharmaceutical company Nerviano Medical Sciences for the treatment of various advanced solid tumors and brain cancers
Key Characteristics & Mechanism
Unlike first-generation poly(ADP-ribose) polymerase (PARP) inhibitors, itareparib features a highly specialized mechanism designed to improve clinical safety and versatility:
- Non-Trapping Profile: Traditional PARP inhibitors trap the PARP enzyme onto DNA, forming PARP-DNA complexes. This trapping causes significant bone marrow toxicity (myelosuppression), leading to severe side effects like anemia, neutropenia, and thrombocytopenia. Itareparib is engineered to be “non-trapping,” avoiding these complexes to protect healthy blood cells.
- High Brain Penetrance: It crosses the blood-brain barrier effectively, making it uniquely suitable for treating primary and secondary central nervous system (CNS) malignancies.
- Ideal Combinability: Because it does not cause overlapping bone marrow toxicity, it can be safely paired with other DNA-damaging therapies like traditional chemotherapies and antibody-drug conjugates (ADCs).
Clinical Development & Target Indications
Itareparib is currently advancing through Phase I and Phase II clinical trials. It is being investigated across several oncology settings:
- Glioblastoma (GBM): Evaluated in Phase II clinical studies for relapsed, IDH wild-type glioblastoma in combination with the chemotherapy drug temozolomide (TMZ).
- Ovarian Cancer: Evaluated in Phase Ia/Ib trials (such as trial NCT06930755) in combination with topotecan for patients with recurrent, platinum-resistant ovarian, fallopian tube, or peritoneal cancers. [1]
- Small Cell Lung Cancer (SCLC) & Astrocytoma: Explored in ongoing combination trials targeting highly aggressive tumors where conventional PARP inhibitors are limited by overlapping toxicity.
- Study of NMS-03305293 in Adult Patients With Relapsed Ovarian CancerCTID: NCT06930755Phase: Phase 1Status: RecruitingDate: 2026-05-28
- Study of NMS-03305293 in Adult Patient With Relapsed Small Cell Lung CancerCTID: NCT06931626Phase: Phase 1Status: RecruitingDate: 2025-11-12
- Ph I/II Study of NMS-03305293+TMZ in Adult Patients With Recurrent GlioblastomaCTID: NCT04910022Phase: Phase 1/Phase 2Status: Active, not recruitingDate: 2025-08-19
- Study of NMS-03305293 in Pts with Selected Advanced/Metastatic Solid TumorsCTID: NCT04182516Phase: Phase 1Status: TerminatedDate: 2024-09-19
A Phase I/II Combination Study of NMS-03305293 and Temozolomide in Adult Patients with Recurrent Glioblastoma
EudraCT: 2020-003417-35
Phase: Phase 1, Phase 2
Status: Trial now transitioned
Date: 2021-11-10
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US275481284&_cid=P10-MQA9O8-42416-1
2-(1-Cyclohexyl-piperidin-4-yl)-6-fluoro-3-oz-2,3-dihydro-1H-isoindole-4-carboxylic Acid Amide (I), cpd 29 [R═F; n=m=0; R1=piperidin-4-yl; R2=1-cyclohexyl]
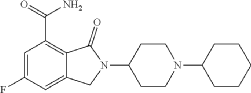
| To a stirred solution of 2-(1-cyclohexyl-piperidin-4-yl)-6-fluoro-3-oxo-2,3-dihydro-1H-isoindole-4-carbonitrile (IV) (100 mg, 0.3 mmol) in acetic acid (5 mL), concentrated sulfuric acid (2.7 mL) was added dropwise during 30 min. The reaction was then warmed at 80° C. for 9 h, cooled at room temperature and poured into cold water (10 mL). The aqueous phase was then made basic by adding concentrated aqueous ammonia and extracted with dichloromethane (3×10 mL). The combined organic phases were washed with 2N aqueous sodium hydroxide (2×12 mL) and brine, dried over Na 2SO 4 and evaporated to dryness in vacuo. The title compound was obtained as a white solid (43 mg, 40%) after purification through column chromatography ((dichloromethane/methanol/ammonia solution, 7N in methanol:97/2/1). |
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014064149&_cid=P10-MQA9K8-39764-1
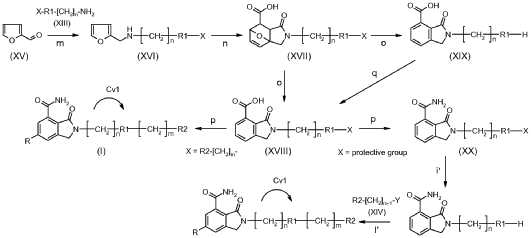
2-(1-Cyclohexyl-piperidin-4-yl)-6-fluoro-3-oxo-2,3-dihydro-1 H-isoindole-4-carboxylic acid amide (I), cpd 29
[R = F; n = m = 0; R1 = piperidin-4-yl; R2 = 1-cyclohexyl]
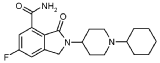
To a stirred solution of 2-(1-cyclohexyl-piperidin-4-yl)-6-fluoro-3-oxo-2,3-dihydro-1 H-isoindole-4-carbonitrile (IV) (100 mg, 0.3 mmol) in acetic acid (5 mL), concentrated sulfuric acid (2.7 mL) was added dropwise during 30 min. The reaction was then warmed at 80 °C for 9 h, cooled at room temperature and poured into cold water (10 mL). The aqueous phase was then made basic by adding concentrated aqueous ammonia and extracted with dichloromethane (3 x 10 mL). The combined organic phases were washed with 2N aqueous sodium hydroxide (2 X 12 mL) and brine, dried over Na2S04 and evaporated to dryness in vacuo. The title compound was obtained as a white solid (43 mg, 40%) after purification through column chromatography ((dichloromethane/methanol/ammonia solution, 7N in methanol: 97/2/1).
1H NMR (400.5 MHz, DMSO- cfe) δ ppm 1.00 – 1.14 (m, 1 H), 1.14 – 1.28 (m, 4 H), 1.53 – 1.61 (m, 1 H), 1.67 – 1.80 (m, 6 H), 2.25 – 2.36 (m, 3 H), 2.88 – 2.95 (m, 2 H), 3.94 – 4.03 (m, 1 H), 4.55 (s, 2 H), 7.66 (dd, JHF = 7.7, JHH = 2.6 Hz, 1 H), 7.85 (br. s., 1 H), 7.89 (dd, JHF = 10.9, JHH = 2.6 Hz, 1 H), 10.78 (br. s., 1 H).
HRMS (ESI+): calcd. for C20H27FN3O2 [M + H]+ 360.2082; found 360.2098
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
- 4-carboxamido-isoindolinone derivatives as selective parp-1 inhibitorsPublication Number: US-2020407314-A1Priority Date: 2012-10-26
- 4-carboxamido-isoindolinone derivatives as selective PARP-1 inhibitorsPublication Number: US-11773064-B2Priority Date: 2012-10-26Grant Date: 2023-10-03
- 4-Carboxamide-isoindolinone derivatives as selective PARP-1 inhibitorsPublication Number: JP-6314147-B2Priority Date: 2012-10-26Grant Date: 2018-04-18
- 4-carboxamido-isoindolinone derivatives as selective PARP-1 inhibitorsPublication Number: ES-2813530-T3Priority Date: 2012-10-26Grant Date: 2021-03-24
- 4-carboxamido-isoindolinone derivatives as selective PARP-1 inhibitorsPublication Number: US-10385018-B2Priority Date: 2012-10-26Grant Date: 2019-08-20
- 4-carboxamido-isoindolinone derivatives as selective parp-1 inhibitorsPublication Number: US-2015274662-A1Priority Date: 2012-10-26
- 4-carboxamido-isoindolinone derivatives as selective parp-1 inhibitorsPublication Number: WO-2014064149-A1Priority Date: 2012-10-26
- 4-carboxamido-isoindolinone derivatives as selective parp-1 inhibitorsPublication Number: US-11420940-B2Priority Date: 2012-10-26Grant Date: 2022-08-23
- DERIVATIVES OF 4-CARBOXAMIDO-ISOINDOLINONA AS SELECTIVE INHIBITORS OF PARP-1.Publication Number: MX-2015005245-APriority Date: 2012-10-26
- 4-carboxamido-isoindolinone derivatives as selective parp-1 inhibitorsPublication Number: US-2019330151-A1Priority Date: 2012-10-26
- COMPOUNDS DERIVED FROM 4-CARBOXAMIDO-ISOINDOLINONE, PROCESS OF PREPARATION OF THESE, IN VITRO METHOD TO SELECTIVELY INHIBIT PARP-1 PROTEIN ACTIVITY, PHARMACEUTICAL COMPOSITION AND USE OF THE REFERRED COMPOUNDSPublication Number: BR-112015009130-B1Priority Date: 2012-10-26
- 4-carboxamido-isoindolinone derivatives as selective parp-1 inhibitorsPublication Number: US-2022363636-A1Priority Date: 2012-10-26
- 4-carboxamido-isoindolinone derivatives as selective parp-1 inhibitorsPublication Number: CA-2889581-A1Priority Date: 2012-10-26
- 4-carboxamido-isoindolinone derivatives as selective PARP-1 inhibitorsPublication Number: US-10800739-B2Priority Date: 2012-10-26Grant Date: 2020-10-13
- 4-carboxamido-isoindolinone derivatives as selective parp-1 inhibitorsPublication Number: EP-2912032-A1Priority Date: 2012-10-26
- 4-carboxamido-isoindolinone derivatives as selective parp-1 inhibitorsPublication Number: EP-2912032-B1Priority Date: 2012-10-26Grant Date: 2020-05-27
- DERIVATIVES 4-CARBOXAMIDO-ISOINDOLINONE AS PARP-1 SELECTIVE INHIBITORS, METHOD FOR THEIR PRODUCTION AND APPLICATIONPublication Number: EA-028506-B1Priority Date: 2012-10-26
- 4-Formylamino-isoindolinone derivatives as selective PARP-1 inhibitorsPublication Number: CN-104768948-APriority Date: 2012-10-26
- 4-carboxamido-isoindolinone derivatives as selective parp-1 inhibitorsPublication Number: CA-2889581-CPriority Date: 2012-10-26Grant Date: 2021-06-29
////////itareparib, ANAX LABS, poly (ADP-ribose) polymerase (PARP) inhibitor, antineoplastic, NMS-03305293, NMS-293, NMS 03305293, NMS 293, KFI1190L8L, NV 578,
Irodanoprost
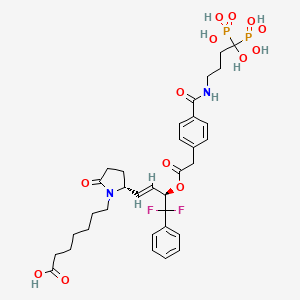
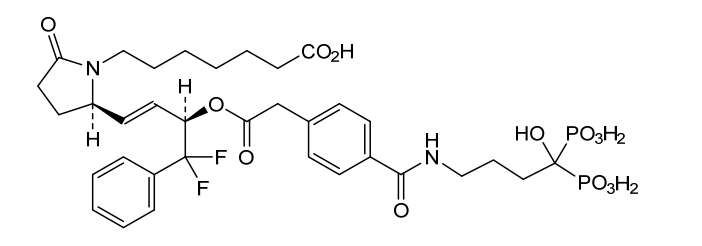
Irodanoprost
CAS 2055490-48-5
MF C34H44F2N2O13P2 MW788.7 g/mol
7-[(2R)-2-[(E,3R)-4,4-difluoro-3-[2-[4-[(4-hydroxy-4,4-diphosphonobutyl)carbamoyl]phenyl]acetyl]oxy-4-phenylbut-1-enyl]-5-oxopyrrolidin-1-yl]heptanoic acid
- 1-Pyrrolidineheptanoic acid, 2-[(1E,3R)-4,4-difluoro-3-[[2-[4-[[(4-hydroxy-4,4-diphosphonobutyl)amino]carbonyl]phenyl]acetyl]oxy]-4-phenyl-1-buten-1-yl]-5-oxo-, (2R)-
- 7-[(2R)-2-{(1E,3R)-4,4-difluoro-3-[({4-[(4-hydroxy-4,4-diphosphonobutyl)carbamoyl]phenyl}acetyl)oxy]-4-phenylbut-1-en-1-yl}-5-oxopyrrolidin-1-yl]heptanoic acid
7-[(2R)-2-{(1E,3R)-4,4-difluoro-3-[({4-[(4-hydroxy-4,4-diphosphonobutyl)carbamoyl]phenyl}acetyl)oxy]-4-phenylbut-1-en-1-yl}-5-oxopyrrolidin-1-yl]heptanoic acid
prostaglandin receptor agonist, osteogenesis-related diseases, KP8BK46Z6R, MES 1022
Irodanoprost (also known as MES-1022) is a clinical-stage, bone- and pathology-targeted small molecule prodrug that acts as a potent and selective agonist for the prostaglandin E2 receptor subtype 4 (EP4). Developed by the pharmaceutical company Mesentech Inc., the compound is designed to treat severe musculoskeletal and rare muscle-wasting diseases
Therapeutic Mechanism
Systemic activation of the EP4 receptor has long been known to stimulate bone and muscle regeneration. However, its clinical use was previously restricted due to toxic off-target side effects like severe hypotension and gastrointestinal issues.
Irodanoprost overcomes this barrier through a unique “pathology-targeted” conjugate design:
- Targeting Mechanism: The EP4 agonist is chemically linked to a moiety that binds selectively to calcium-rich tissues—such as bone matrices or damaged, dystrophic muscle fibers.
- Local Activation: Once it accumulates at the site of damage, it delivers localized therapeutic signaling while avoiding systemic tissues.
Primary Target Indications
The drug candidate is primarily under investigation for several rare or progressive degenerative diseases:
- Duchenne Muscular Dystrophy (DMD): Preclinical trials on advanced DMD rat models published in bioRxiv demonstrate that irodanoprost actively blocks the differentiation of fibro-adipogenic progenitors, effectively reversing established muscle fibrosis and restoring muscle mass to wild-type levels.
- Osteogenesis Imperfecta (Brittle Bone Disease): Utilizing its bone-anabolic pathway to promote bone growth and strength.
- Facioscapulohumeral Muscular Dystrophy (FSHD)
- Osteoporosis
PAT
EP-3307747-A1 US-10400000-B2 US-11312737-B2 US-20180170951-A1 US-20190345179-A1 WO-2016199111-A1
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016199111&_cid=P11-MQ7G6Y-06859-1
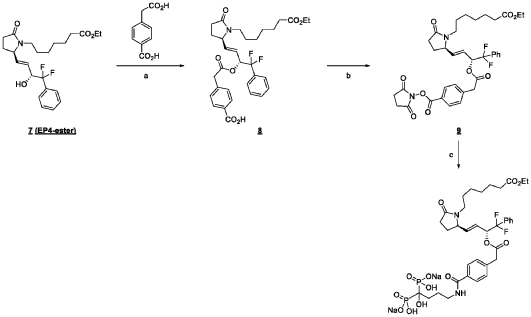
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
PAT
- Amide-linked EP4 agonist-bisphosphonate compounds and uses thereofPublication Number: US-10400000-B2Priority Date: 2015-06-12Grant Date: 2019-09-03
- Amide-linked ep4 agonist-bisphosphonate compounds and uses thereofPublication Number: US-2019345179-A1Priority Date: 2015-06-12
- Amide-linked ep4 agonist-bisphosphonate compounds and uses thereofPublication Number: US-2018170951-A1Priority Date: 2015-06-12
- Amide-linked ep4 agonist-bisphosphonate compounds and uses thereofPublication Number: WO-2016199111-A1Priority Date: 2015-06-12
- Amide-linked ep4 agonist-bisphosphonate compounds and uses thereofPublication Number: EP-3307747-A1Priority Date: 2015-06-12
- Amide-linked EP4 agonist-bisphosphonate compounds and uses thereof
- Publication Number: US-11312737-B2
- Priority Date: 2015-06-12
- Grant Date: 2022-04-26
////////////irodanoprost, ANAX LABS, prostaglandin receptor agonist, osteogenesis-related diseases, KP8BK46Z6R, MES 1022
Inidascamine
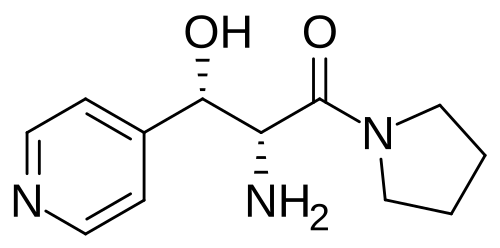
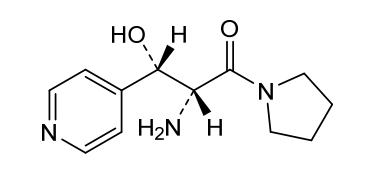
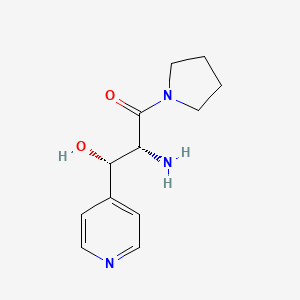
Inidascamine
CAS 903884-71-9
MF C12H17N3O2 MW235.28 g/mol
(-)-(2R,3S)-2-amino-3-hydroxy-3-(pyridin-4-yl)-1-(pyrrolidin-1-yl)propan-1-one
(2R,3S)-2-amino-3-hydroxy-3-pyridin-4-yl-1-pyrrolidin-1-ylpropan-1-one
(2R,3S)-2-amino-3-hydroxy-3-(pyridin-4-yl)-1-(pyrrolidin-1-yl)propan-1-one
schizophrenia, 3LW01V88B7, RL 007
Inidascamine (developmental code name RL-007 or FSV7-007) is an experimental, orally administered drug primarily studied for treating Cognitive Impairment Associated with Schizophrenia (CIAS). Developed jointly by Recognify Life Sciences and atai Life Sciences, the molecule targets the underlying neural mechanisms that restrict verbal learning, memory retention, and mental processing speed in schizophrenia patients
Mechanism of Action
The compound is designed to alter the brain’s complex excitatory and inhibitory balance to produce pro-cognitive effects. It accomplishes this by interacting with three major neurotransmitter systems simultaneously:
- Cholinergic system: Modulates acetylcholine pathways vital for attention and memory.
- Glutamatergic system: Interacts with NMDA/glutamate receptors to influence synaptic plasticity.
- GABAergic system: Targets \(GABA_{B}\) receptors to stabilize neural transmission.
Clinical Status & Current Data
- Phase 2b Trial Results: In July 2025, data from a Phase 2b clinical trial showed that while inidascamine produced numerical improvements in memory and processing speed compared to a placebo, it failed to achieve statistical significance on its primary efficacy endpoint.
- Safety Profile: The drug demonstrated excellent tolerability. It lacked common antipsychotic side effects like heavy sedation, rapid weight gain, or involuntary body movements.
- Commercial Backing: Following the trial shortfall, atai Life Sciences officially deprioritized the asset to shift its primary funding toward its wholly owned pipeline of psychedelic therapies.
Inidascamine (INNTooltip International Nonproprietary Name; developmental code names RL-007, FSV7-007) is an experimental drug which is under development for the treatment of cognitive impairment associated with schizophrenia (CIAS).[1][3][4][5][6][2] It is taken orally.[1][2] The drug is said to act on the cholinergic, NMDA, and GABAB receptor systems.[1][5][2] Inidascamine is being developed by Recognify Life Sciences and atai Life Sciences.[1][3] It was discovered via screening of compounds for effects on synaptic plasticity and cognition.[2] The drug shows structural similarities to phenethylamines and amphetamines.[7]
- A Study to Evaluate RL-007 in the Treatment of Cognitive Impairment Associated With Schizophrenia (CIAS)CTID: NCT05686239Phase: Phase 2Status: CompletedDate: 2025-07-30
- Safety, Biomarker Study of RL-007 in Subjects With SchizophreniaCTID: NCT04822883Phase: Phase 2Status: CompletedDate: 2022-04-27
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2006081273&_cid=P11-MQ616P-01336-1
OL-threo-2- Amino-3-hvdroxy-3 -(pyridin-4-yl)- 1 -(pyrrolidin- 1 -yDpropan- 1 -one dihydrochloride Compound 22.
Compound 22 was prepared following method E with trans-(4,5-άihydτo-5-(pyridin-4-yl)oxazol-4-yl)(pyrrolidin-l-yl)methanone Compound 19 (0.750 g, 3.07 mmol), hydrochloric acid 37 % (1.0 mL) and methanol (10 mL). After 3.0 h at 50 °C and work-up DL-tAreø-2-amino-3-hydroxy-3-(pyridin-4-yl)-l-(pyrrolidin-l-yl)propan-l-one dihydrochloride Compound 22 was obtained as a white solid (0.935 g, 99 % yield).
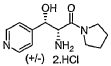
Compound 22
MW: 308.28; Yield: 99 %; White Solid; Mp (°C): 117.0.
1H-NMR (CD3OD3 δ): 1.75-2.03 (m, 4H, 2xCH2), 2.93-3.08 (m, 1H, CHN), 3.32-3.75 (m, 3H, 2xCH2), 4.54 (d, 1H, J= 5.9 Hz, CH1N), 5.40 (d, 1H, J = 5.9 Hz, CH-O), 8.21 (d, 2H, J= 5.8 Hz, ArH), 8.94 (d, 2H, J= 5.8 Hz, ArH).
MS-ESI m/z (% rel. int.): 236.1 ([MH]+, 17), 219 (25), 148 (100).
HPLC: Method A, detection UV 254 nm, Compound 22 RT = 0.8 min, peak area 96.3 %.
PAT
- 3-aryl-3-hydroxy-2-amino-propionic acid amides, 3-heteroaryl-3-hydroxy-2-amino-propionic acid amides and related compounds having analgesic and / or immunostimulatory activityPublication Number: ES-2565236-T3Priority Date: 2005-01-26Grant Date: 2016-04-01
- 3-ARYL-3-HYDROXY-2-AMINO-PROPIONIC ACID, 3-HETEROARYL-3-HYDROXY-2-AMINOPROPIONIC ACID AMIDES AND RELATED COMPOUNDS HAVING ANALGESIC AND/OR IMMUNOSTIMULATING ACTIVITYPublication Number: BR-122018068138-B1Priority Date: 2005-01-26
- 1-aryl-1-hydroxy-2,3-diamino-propyl amines, 1-heteroaryl-1-hydroxy-2,3-diamino-propyl amines and related compounds having analgesic and/or immuno stimulant activityPublication Number: US-2011288094-A1Priority Date: 2005-01-26
- 1-aryl-1-hydroxy-2,3-diamino-propyl amines, 1-heteroaryl-1-hydroxy-2,3-diamino-propyl amines and related compounds having analgesic and/or immuno stimulant activityPublication Number: US-9828349-B2Priority Date: 2005-01-26Grant Date: 2017-11-28
- Compounds having analgesic and/or immunostimulant activityPublication Number: US-2012157497-A1Priority Date: 2005-01-26
- 3-aryl-3-hydroxy-2-amino-propionic acid amides, 3-heteroaryl-3-hydroxy-2-amino-propionic acid amides and related compounds having analgesic and/or immunostimulant activityPublication Number: AU-2012241156-A1Priority Date: 2005-01-26
- 3-Aryl-3-hydroxy-2-amino-propionic acid amide, 3-heteroaryl-3-hydroxy-2-amino-propionic acid amide and related compounds having analgesic activity and / or immunostimulatory activityPublication Number: JP-2012162547-APriority Date: 2005-01-26
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
 | |
| Clinical data | |
|---|---|
| Other names | RL-007; RL007; FSV7-007 |
| Routes of administration | Oral[1][2] |
| Identifiers | |
| IUPAC name | |
| CAS Number | 903884-71-9 |
| PubChem CID | 11535990 |
| ChemSpider | 9710771 |
| UNII | 3LW01V88B7 |
| ChEMBL | ChEMBL5095258 |
| CompTox Dashboard (EPA) | DTXSID90238113 |
| Chemical and physical data | |
| Formula | C12H17N3O2 |
| Molar mass | 235.287 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- “RL 007”. AdisInsight. 10 June 2024. Retrieved 26 February 2025.
- Donello JE, Walker GA, Schweighoffer F, and Pando MP. Abstract Number: 149. RL-007, a novel oral neuromodulator, enhances synaptic plasticity and cognition in non-clinical models. American College of Neuropsychopharmacology (ACNP) annual meeting. December 5, 2023. https://ir.atai.life/static-files/06c60339-e93c-42fc-9323-c0e38e83f86e
- “Delving into the Latest Updates on RL-007 with Synapse”. Synapse. 23 January 2025. Retrieved 26 February 2025.
- Brady LS, Lisanby SH, Gordon JA (2023). “New directions in psychiatric drug development: promising therapeutics in the pipeline”. Expert Opinion on Drug Discovery. 18 (8): 835–850. doi:10.1080/17460441.2023.2224555. PMID 37352473.
- Vita A, Barlati S, Cavallaro R, Mucci A, Riva MA, Rocca P, et al. (2024). “Definition, assessment and treatment of cognitive impairment associated with schizophrenia: expert opinion and practical recommendations”. Frontiers in Psychiatry. 15 1451832. doi:10.3389/fpsyt.2024.1451832. PMC 11450451. PMID 39371908.
- Ye N, Wang Q, Li Y, Zhen X (March 2025). “Current emerging therapeutic targets and clinical investigational agents for schizophrenia: Challenges and opportunities”. Medicinal Research Reviews. 45 (2): 755–787. doi:10.1002/med.22086. PMID 39300769.
- “2-Amino-3-hydroxy-3-(pyridin-4-yl)-1-(pyrrolidin-1-yl)propan-1-one, (2R,3S)-“. PubChem. Retrieved 26 February 2025.
//////////inidascamine, ANAX LABS, schizophrenia, 3LW01V88B7, RL 007
Imofinostat
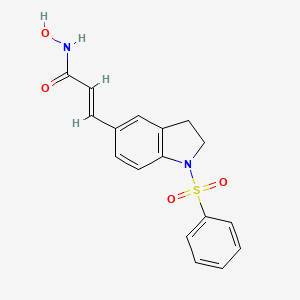
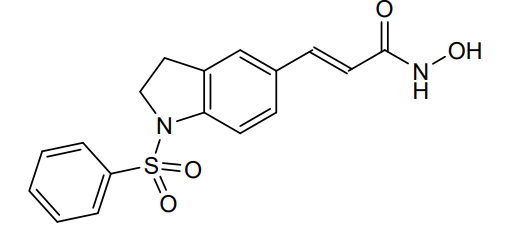
Imofinostat
CAS 1338320-94-7
MF C17H16N2O4S MW 344.4 g/mol
- 3-(1-(Benzenesulfonyl)-2,3-dihydro-1H-indol-5-yl)-N-hydroxyacrylamide
- (E)-3-[1-(benzenesulfonyl)-2,3-dihydroindol-5-yl]-N-hydroxyprop-2-enamide
(2E)-3-[1-(benzenesulfonyl)-2,3-dihydro-1H-indol-5-yl]-N-hydroxyprop2-enamide
histone deacetylase inhibitor, antineoplastic, ABT-301, MPT0E028, ABT 301, MPT0E 028, T65L58FI65
Imofinostat (also known as ABT-301 or MPT0E028) is an orally bioavailable, small-molecule histone deacetylase (HDAC) inhibitor primarily being developed as an innovative precision oncology treatment. Developed by companies like AnBogen Therapeutics and Formosa Pharmaceuticals, it is designed to reactivate tumor suppressor genes that cancer cells have silenced, thereby triggering cancer cell death (apoptosis) and stopping tumor growth.
Mechanism of Action
Imofinostat works through a distinct multi-modality approach to fight cancer cells:
- HDAC Inhibition: It acts as a potent inhibitor of human pan-histone deacetylase enzymes, showing preferential selectivity for Class I HDACs (especially HDAC3). This blocks the deacetylation of histone proteins, causing chromatin to remodel and forcing cancer cells to express tumor-suppressor genes.
- Akt Pathway Targeting: Independent of its epigenetic effects, it can directly target and reduce the activation (phosphorylation) of the Akt protein kinase, a major pathway that cancer cells use to survive and multiply.
- Microenvironment Modulation: Preclinical data shows it alters the tumor microenvironment by converting “cold tumors” (invisible to the immune system) into “hot tumors” by promoting the infiltration of CD8+ cytotoxic T cells.
Current Clinical Status & Indications
Imofinostat is actively moving through clinical trial pipelines, focusing heavily on combination therapies to overcome treatment resistance:
- Colorectal Cancer (CRC): It is currently being evaluated in a global Phase 1/2 clinical trial (NCT07244705). It is combined with the immune checkpoint inhibitor tislelizumab (Tevimbra®) and the anti-angiogenic drug bevacizumab to treat advanced, metastatic colorectal cancer.
- Pancreatic Cancer: Recent data presented at the 2026 American Association for Cancer Research (AACR) Annual Meeting demonstrates that imofinostat disrupts the HDAC3-NRF2 pathway. This action breaks down chemotherapy resistance in highly aggressive KRAS-mutant pancreatic ductal adenocarcinoma, making tumors much more sensitive to treatments like gemcitabine.
- Other Solid Tumors: Phase 1 monotherapy trials have confirmed that the drug possesses a highly competitive safety profile across a broad variety of advanced solid tumors.
Imofinostat is an orally bioavailable N-hydroxyacrylamide-derived inhibitor of both human pan-histone deacetylase (HDAC) enzymes and the serine/threonine protein kinase Akt (protein kinase B), with potential antineoplastic activity. Upon administration, imofinostat selectively binds to and inhibits HDACs, which inhibits deacetylation of histone proteins and leads to the accumulation of highly acetylated histones. This may result in both an induction of chromatin remodeling, and the selective transcription of tumor suppressor genes. This prevents cell division and induces both cell cycle arrest and apoptosis, which may inhibit the proliferation of susceptible tumor cells. In addition, imofinostat inhibits the phosphorylation and activation of Akt, which prevents the activation of downstream signaling pathways, independent of its HDAC inhibitory activity. HDACs, upregulated in many tumor cell types, are a family of enzymes that deacetylate histone proteins. Akt, overexpressed in many tumor cell types, plays a key role in tumor cell proliferation and survival.
Dose-Seeking Study of MPT0E028 in Subjects With Advanced Solid Malignancies Without Standard Treatment
CTID: NCT02350868
Phase: Phase 1
Status: Completed
Date: 2019-04-11
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2011126821&_cid=P11-MQ4LAI-84972-1
COMD 12
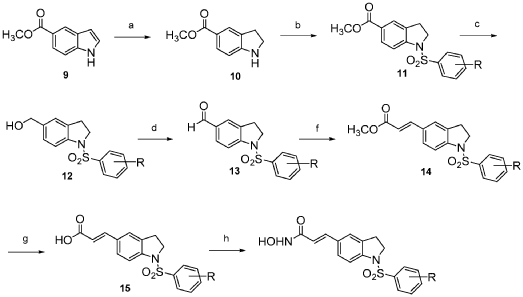
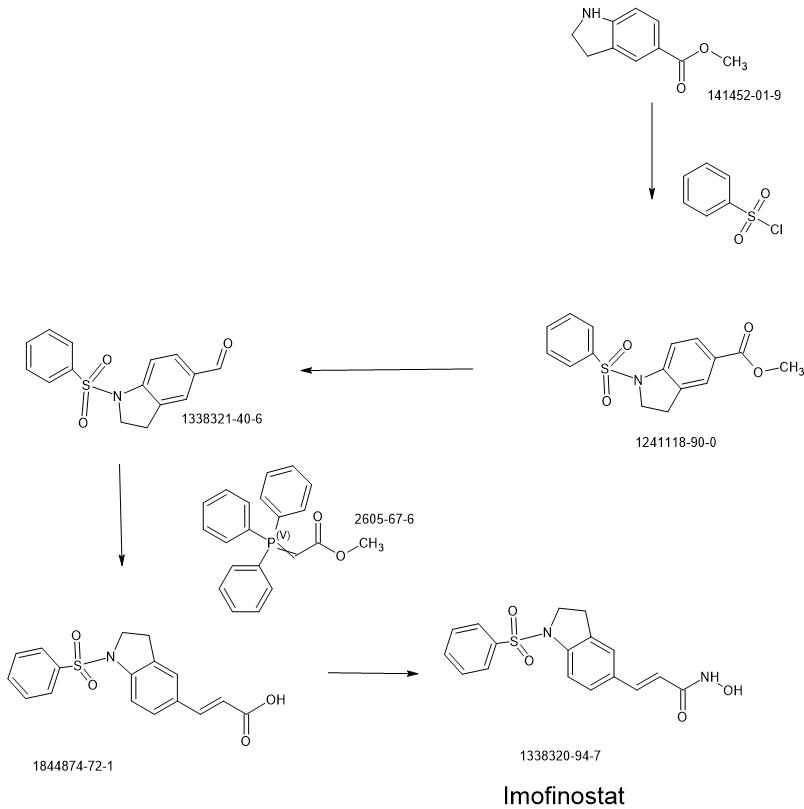
Compound 12 was synthesized via the route as shown in Scheme 3 above (reagents and conditions: (a) NaBH3CN, AcOH; (b) Benzenesulfonyl chloride, 4-methoxybenzenesulfonyl chloride, 3,4-dimethoxybenzenesulfonyl chloride, 4-fluorobenzenesulfonyl chloride, or 4-nitrobenzenesulfonyl chloride, pyridine; (c) L1AIH4, THF; (d) PDC, MS, CH2C12; f) Ph3P = CH-COOCH3, CH2C12; (g) 1M LiOH(aq), dioxane; (h) (i) NH2OTHP, PyBOP, NEt3, DMF; (ii) TFA, MeOH; (i) Fe, NH4C1, Isopropanol, H20).
2,3-Dihydro-lH-indole-5-carboxylic acid methyl ester (10): sodium cyanoborohydride (0.16 g, 2.57 mmol) was added to a solution of methyl indole-5-carboxylate (9) (0.30 g, 1.71 mmol) in AcOH (2 mL) at 0 °C. The reaction mixture was warmed to room temperature and stirred for 2 h before it was quenched with water at 0 °C. Concentrated NaOH was added to reach pH=10. The aqueous layer was extracted with CH2CI2 (15 mL x 3). The combined organic layer was dried over anhydrous MgS04 and concentrated under reduced pressure to give a yellow residue, which was purified by silica gel chromatography (EtOAc: n-hexane = 1 : 2) to afford 10 (0.28 g). 1H NMR (500MHz, CDC13): δ 3.06 (t, J= 8.5 Hz, 2H), 3.65 (t, J= 8.5 Hz, 2H), 3.84 (s, 3H), 6.53-6.55 (m, 1H), 7.75-7.76 (m, 2H).
l-Benzenesulfonyl-2,3-dihydro-lH-indole-5-carboxylic acid methyl ester (11): To a solution of 10 (0.28 g, 1.58 mmol) in pyridine (2 mL), benzenesulfonyl chloride (0.40 ml, 3.16 mmol) was added. The reaction mixture was refluxed overnight. The mixture was then purified by silica gel chromatography (EtOAc: n-hexane = 1 : 3) to afford 11 (0.40 g). 1H NMR (500MHz, CDCI3): δ 2.99 (t, J= 8.6 Hz, 2H), 3.87 (s, 3H), 3.97 (t, J= 8.6 Hz, 2H), 7.45-7.48 (m, 2H), 7.56-7.59 (m, 1H), 7.66 (d, J= 8.5 Hz, 1H), 7.75 (s, 1H), 7.82 (d, J= 7.7 Hz, 2H), 7.90 (d, J= 7.9 Hz, 1H).
(l-Benzenesulfonyl-2,3-dihydro-lH-indol-5-yl)-methanol (12): LAH (0.10 g, 2.52 mmol) was added to a solution of 11 (0.40 g, 1.26 mmol) in THF (10 mL) at 0 °C. The reaction mixture was warmed to room temperature and stirred for 2 h before it was quenched with water and then extracted with CH2CI2 (15 mL x 3). The combined organic layer was dried over anhydrous MgS04 and concentrated under reduced pressure. The reaction mixture was purified by silica gel chromatography (EtOAc: n-hexane = 1 : 1) to afford 12 (0.24 g). 1H NMR (500MHz, CDC13): δ 2.83 (t, J= 8.4 Hz, 2H), 3.92 (t, J= 8.5 Hz, 2H), 4.49 (s, 2H), 7.09 (s, 1H), 7.16 (d, J= 8.2 Hz, 1H), 7.46-7.49 (m, 2H), 7.53 (d, J= 8.2 Hz, 1H), 7.60 (t, J= 7.5 Hz, 1H), 7.76 (d, J= 7.7 Hz, 2H).
l-Benzenesulfonyl-2,3-dihydro-lH-indole-5-carbaldehyde (13): molecular sieves (0.63g) were added to a solution of 12 (0.24 g, 0.83 mmol) in CH2C12 (10 mL), PDC (0.63 g, 1.66 mmol). The mixture was stirred at room temperature overnight before it was filtered through celite. The organic layer was concentrated under reduced pressure then purified by silica gel chromatography (EtOAc: n-hexane = 1 : 2) to afford 13 (0.19 g). 1H NMR (500MHz, CDC13): δ 3.05 (t, J= 8.6 Hz, 2H), 4.01 (t, J= 8.7 Hz, 2H), 7.46-7,49 (m, 2H), 7.58-7.62 (m, 2H), 7.71 (d, J= 8.3 Hz, 1H), 7.75 (d, J= 8.3 Hz, 1H), 7.84 (d, J= 7.8 Hz, 2H), 9.85 (s, 1H).
3-(l-Benzenesulfonyl-2,3-dihydro-lH-indol-5-yl)-acrylic acid methyl ester (14): Methyl (triphenylphosphoranylidene) acetate (0.27 g, 0.79 mmol) was added to a solution of 13 (0.19g,
0.66 mmol) in CH2CI2 (10 mL). The mixture was stirred at room temperature for 3h before it was
quenched with water and then extracted with CH2CI2 (15 mL x 3). The combined organic layer was dried over anhydrous MgS04 and concentrated under reduced pressure to give a yellow residue, which was then purified by silica gel chromatography (EtOAc: n-hexane = 1 : 3) to afford 14
(0.20 g).
3-(l-Benzenesulfonyl-2,3-dihydro-lH-indol-5-yl)-acrylic acid (15): 1M LiOH aqueous solution (1.16 ml, 1.16 mmol) was added to a solution of 14 (0.20g, 0.58 mmol) in dioxane
(15 mL). The reaction mixture was stirred at 40 °C overnight before it was concentrated under reduced pressure. The residue was dissolved in water and concentrated HCl was added up to acidic pH to give the precipitation, which was dried by vacuum to afford 15 (0.16 g). 1H NMR (500MHz, CD3OD): δ 2.92 (t, J= 8.5 Hz, 2H), 3.96 (t, J= 8.5 Hz, 2H), 6.33 (d, J= 15.9 Hz, 1H), 7.38 (s, 1H), 7.41 (d, J= 8.5 Hz, 1H), 7.50-7.53 (m, 2H), 7.55 (d, J= 16.1 Hz, 1H), 7.58-7.64 (m, 2H), 7.82 (d, J = 7.6 Hz, 2H).
3-(l-Benzenesulfonyl-2,3-dihydro-lH-indol-5-yl)-N-hydroxy-acrylamide
(Compound 12): NH2OTHP (0.05 g, 0.44 mmol) was added to a solution of 15 (0.12 g, 0.37 mmol), PyBOP (0.20 g, 0.39 mmol), triethylamine (0.12 ml, 0.88 mmol) in DMF (1.5 mL). The reaction mixture was stirred at room temperature for 1 h before it was quenched with water, followed by extraction with EtOAc (15 mL x 3). The combined organic layer was dried over anhydrous MgS04 and concentrated under reduced pressure. The residue was purified by silica gel chromatography (CH2C12: CH3OH = 30 : 1 : l%NH3(aq)) to give a white solid, which was treated with TFA (1.13 ml, 15.21 mmol) in the presence of CH3OH (25 mL) and stirred overnight at room temperature. The reaction mixture was concentrated under reduced pressure to give a white residue, which was recrystallized by CH3OH to afford Compound 12 (0.12 g). 1H NMR (500MHz,
CD3OD): δ 2.91 (t, J= 8.5 Hz, 2H), 3.96 (t, J= 8.4 Hz, 2H), 6.32 (d, J= 15.8 Hz, 1H), 7.32 (s, 1H), 7.37-7.39 (m, 1H), 7.46 (d, J= 15.7 Hz, 1H), 7.50-7.53 (m, 2H), 7.58-7.64 (m, 2H), 7.82 (d, J= 7.8 Hz, 2H). MS (EI) mlz: 170 (100%), 344 (M+, 3.21%). HRMS (EI) for Ci7Hi6N204S (M+): calcd, 344.0831; found, 344.0829.
PAT
US20150368195
https://patentscope.wipo.int/search/en/detail.jsf?docId=US154007904&_cid=P11-MQ4M0P-01888-1
PAT
- Indolyl or indolinyl hydroxamate compoundsPublication Number: US-8846748-B2Priority Date: 2010-03-29Grant Date: 2014-09-30
- Indolyl or indolinyl hydroxamate compoundsPublication Number: US-9598364-B2Priority Date: 2010-03-29Grant Date: 2017-03-21
- Indolyl or indolinyl hydroxamate compoundsPublication Number: WO-2011126821-A2Priority Date: 2010-03-29
- Indolyl or indolinyl hydroxamate compoundsPublication Number: EP-2552887-A2Priority Date: 2010-03-29
- Indolyl or indolinyl hydroxamate compoundsPublication Number: US-2011245315-A1Priority Date: 2010-03-29
- Indolyl or indolinyl hydroxamate compoundsPublication Number: US-2014364477-A1Priority Date: 2010-03-29
- Indolyl or indolinyl hydroxamate compoundsPublication Number: EP-2552887-B1Priority Date: 2010-03-29Grant Date: 2018-10-24
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
//////////imofinostat, anax labs, histone deacetylase inhibitor, antineoplastic, ABT-301, MPT0E028, ABT 301, MPT0E 028, T65L58FI65
Ifupinostat
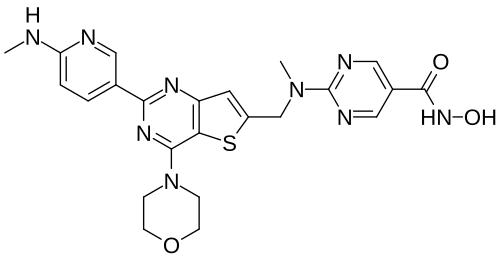
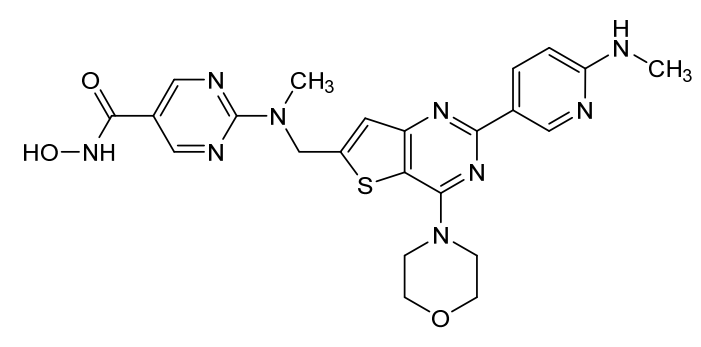
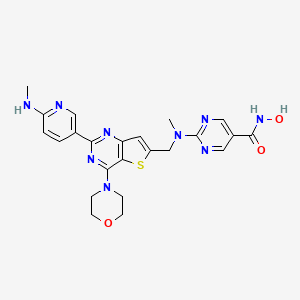
Ifupinostat
CAS 1235449-52-1
MF C23H25N9O3S MW507.6 g/mol
N-hydroxy-2-[methyl-[[2-[6-(methylamino)-3-pyridinyl]-4-morpholin-4-ylthieno[3,2-d]pyrimidin-6-yl]methyl]amino]pyrimidine-5-carboxamide
5-Pyrimidinecarboxamide, N-hydroxy-2-(methyl((2-(6-(methylamino)-3-pyridinyl)-4-(4-morpholinyl)thieno(3,2-d)pyrimidin-6-yl)methyl)amino)-
DQ7TD3X4ZJ, BEBT908 FREE BASE,
Ifupinostat (brand name Betlin; formerly known as BEBT-908) is a first-in-class, dual-action cancer medication used to treat specific types of blood cancer. It is developed by the biopharmaceutical company BeBetter Med.
Approved Clinical Use
The drug is conditionally approved in China as a monotherapy for adults with relapsed or refractory diffuse large B-cell lymphoma (r/r DLBCL). It is specifically indicated for patients who have already undergone at least two prior lines of systemic therapy.
Mechanism of Action
Unlike traditional cancer drugs that target a single pathway, ifupinostat is designed to simultaneously disrupt two major cellular mechanisms that drive tumor growth:
- PI3Kα Inhibition: It blocks phosphoinositide 3-kinase alpha (PI3Kα), shutting down the downstream PI3K/AKT/mTOR survival pathway within cancer cells.
- HDAC Inhibition: It blocks histone deacetylase (HDAC) enzymes, leading to epigenetic modifications (such as increased histone-3 acetylation) that trigger cancer cell death.
By hitting both targets at once, the drug suppresses tumor cell proliferation, downregulates the cancer-driving c-Myc protein, and induces cell death via ferroptosis (an iron-dependent form of programmed cell death).
Clinical Research and Future Outlook
- Combinations: Beyond its use as a single agent, ifupinostat is being evaluated in combination with the monoclonal antibody rituximab as a potential second-line treatment for r/r DLBCL. Early phase 1b clinical data presented at ASCO showed a promising 76.2% objective response rate (ORR).
- Brain Penetration: Lab studies indicate that the molecule successfully crosses the blood-brain barrier (BBB), showing therapeutic potential for central nervous system lymphomas.
- Ongoing Verification: Because its initial regulatory green light was given on a conditional basis, a confirmatory randomized phase 3 trial is currently underway to achieve full approval
Ifupinostat is an inhibitor of both phosphoinositide 3-kinase (PI3K) and histone deacetylase (HDAC) enzymes, with potential antineoplastic activity. Upon administration, ifupinostat binds to and inhibits the activity and mediated signaling of both PI3K and HDAC. In addition, ifupinostat may also inhibit other signaling pathways. This may prevent growth of PI3K and/or HDAC-expressing tumor cells.
Ifupinostat (trade name Betlin) is a drug used for the treatment of cancer. It is approved in China for adults with relapsed or refractory diffuse large B-cell lymphoma who have received at least two lines of systemic therapy.[1] It is being developed by BeBetter Med.[2]
Ifupinostat acts as both a phosphoinositide 3-kinase α (PI3Kα) inhibitor and a histone deacetylase (HDAC) inhibitor.[1][3][4]
SYN
- Hydroxamic acid hybrids: Histone deacetylase inhibitors with anticancer therapeutic potencyPublication Name: European Journal of Medicinal ChemistryPublication Date: 2023-12-15PMID: 37875056DOI: 10.1016/j.ejmech.2023.115879
- A Dual PI3K/HDAC Inhibitor Induces Immunogenic Ferroptosis to Potentiate Cancer Immune Checkpoint TherapyPublication Name: Cancer ResearchPublication Date: 2021-12-15PMID: 34711611DOI: 10.1158/0008-5472.can-21-1547
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2024086894&_cid=P21-MQ35ZX-16907-1
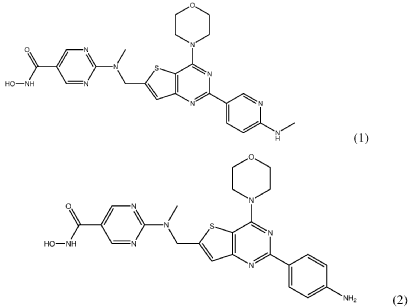
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2024086894&_cid=P21-MQ35ZX-16907-1
PAT
- Phosphoinositide 3-Kinase Inhibitors with a Zinc Binding MoietyPublication Number: US-2025230169-A1Priority Date: 2009-01-08
- Phosphoinositide 3-kinase inhibitors with a zinc binding moietyPublication Number: US-2023227467-A1Priority Date: 2009-01-08
- Phosphoinositide 3-kinase inhibitors with a zinc binding moietyPublication Number: US-11261195-B2Priority Date: 2009-01-08Grant Date: 2022-03-01
- PRMT5 inhibitors and uses thereofPublication Number: US-12448388-B2Grant Date: 2025-10-21
- KRAS G12D modulating compoundsPublication Number: US-12448400-B2Grant Date: 2025-10-21
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
 | |
| Clinical data | |
|---|---|
| Trade names | Betlin; 贝特琳 |
| Other names | BEBT-908 |
| Legal status | |
| Legal status | Rx in China |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1235449-52-1 |
| PubChem CID | 59474330 |
| ChemSpider | 45743497 |
| UNII | DQ7TD3X4ZJ |
| ChEMBL | ChEMBL5618885 |
| Chemical and physical data | |
| Formula | C23H25N9O3S |
| Molar mass | 507.57 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- Fung S (December 2025). “Ifupinostat: First Approval”. Drugs. 85 (12): 1629–1633. doi:10.1007/s40265-025-02248-z. PMID 41028651.
- “Ifupinostat – BeBetter Med”. AdisInsight. Springer Nature Switzerland AG.
- Wang N, Mo Z, Pan L, Zhou M, Ye X, Liu X, et al. (November 2023). “Dual PI3K/HDAC Inhibitor BEBT-908 Exhibits Potent Efficacy as Monotherapy for Primary Central Nervous System Lymphoma”. Targeted Oncology. 18 (6): 941–952. doi:10.1007/s11523-023-01006-z. PMID 37855991.
- Luzietti L, Pires GS, Ryan A, Regidor C, Hiller M, Sarti D, et al. (2025). “Design, synthesis, and biological evaluation of novel triazine-based dual HDAC/PI3K inhibitors for breast cancer therapy”. ChemRxiv. doi:10.26434/chemrxiv-2025-tzwbz.
/////////////ifupinostat, anax labs, Q7TD3X4ZJ, BEBT908 FREE BASE, BEBT 908
Gozanertinib
Gozanertinib
CAS 1226549-49-0
MF C32H31N5O3 MW533.6 g/mol
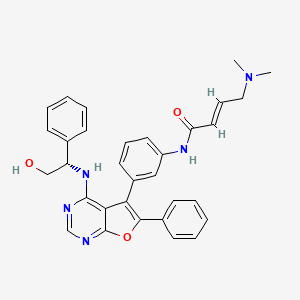
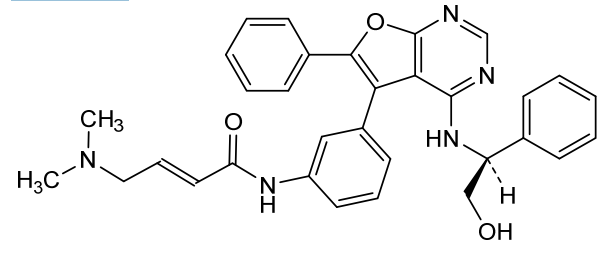
(E)-4-(dimethylamino)-N-[3-[4-[[(1S)-2-hydroxy-1-phenylethyl]amino]-6-phenylfuro[2,3-d]pyrimidin-5-yl]phenyl]but-2-enamide
(2E)-4-(dimethylamino)-N-[3-(4-{[(1S)-2-hydroxy-1-phenylethyl]amino}-6-phenylfuro[2,3-d]pyrimidin-5-yl)phenyl]but-2-
enamide
epidermal growth factor receptor tyrosine kinase inhibitor, antineoplastic, DBPR 112, ABT 101, 6G0COS33K4
Gozanertinib (also known as DBPR112 or ABT-101) is an orally bioavailable, advanced small-molecule dual kinase inhibitor designed to treat advanced non-small cell lung cancer (NSCLC). It targets alterations in the epidermal growth factor receptor (EGFR) and human epidermal growth factor receptor 2 (HER2) families.
Mechanism of Action
Gozanertinib is a furanopyrimidine-based tyrosine kinase inhibitor. It functions by entering the ATP-binding pocket of the receptor and forming an irreversible covalent bond with a specific cysteine residue (Cys797). By permanently blocking these receptors, it halts downstream oncogenic signaling pathways—specifically the RAS/RAF/MEK/ERK and PI3K/AKT cascades—thereby inducing cancer cell death and suppressing tumor expansion.
Target Profile and Key Mutations
Unlike earlier generations of tyrosine kinase inhibitors that only target standard configurations, gozanertinib is optimized to combat specific treatment-resistant mutations:
- EGFR Mutations: It effectively targets wild-type EGFR as well as the dual L858R/T790M resistance mutations.
- Exon 20 Insertions: A standout feature of gozanertinib is its preclinical potency against EGFR and HER2 exon 20 insertion (Ex20ins) mutations. According to chemical development findings published in the Journal of Medicinal Chemistry, it demonstrated ten times better potency against these specific insertions than the widely used third-generation inhibitor, osimertinib.
Development and Status
The drug was initially discovered through scaffold optimization by the National Health Research Institutes (NHRI) and is being co-developed with Anbogen Therapeutics. The International Nonproprietary Name (INN) “gozanertinib” was formally proposed for the compound in early 2025. Preclinical evaluations indicated favorable oral bioavailability and strong anti-tumor efficacy compared to older inhibitors like afatinib, advancing the compound into early-phase clinical trials
Gozanertinib is an orally bioavailable dual kinase inhibitor of epidermal growth factor receptor (EGFR; ErbB1) and human epidermal growth factor receptor 2 (HER2; EGFR2; ErbB2), including EGFR L858R, EGFR T790M and HER2 exon 20 insertion (Ex20ins) mutations, with potential antineoplastic activity. Upon oral administration, gozanertinib targets, binds to and inhibits the activity of EGFR or HER2 insertions or mutations. This prevents EGFR/HER2-mediated signaling, which may induce cell death and inhibit tumor growth in EGFR/HER2-overexpressing tumor cells. The ErbB receptor tyrosine kinase family is involved in key cellular functions, including cell growth and survival. EGFR and HER2 alterations constitutively upregulate kinase activity.
- Phase 1b/2 Study to Evaluate ABT-101 in Solid Tumor and NSCLC PatientsCTID: NCT05532696Phase: Phase 1/Phase 2Status: RecruitingDate: 2024-06-24
- A Study of DBPR112 in Patients With Head and Neck Cancer and EGFR Mutated Lung CancerCTID: NCT03246854Phase: Phase 1Status: TerminatedDate: 2020-12-17
PAT
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US43249513&_cid=P11-MQ1QG3-86325-1
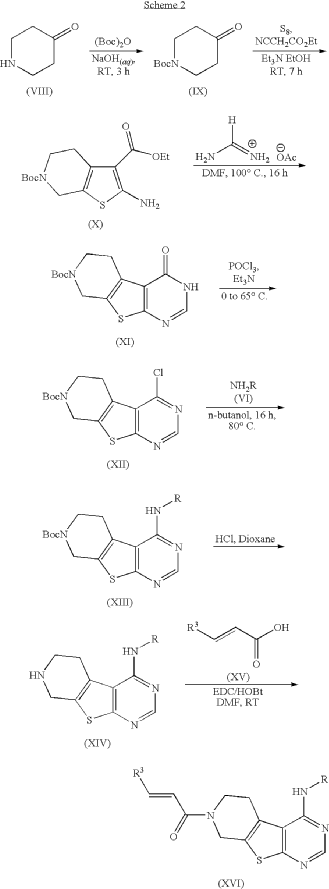
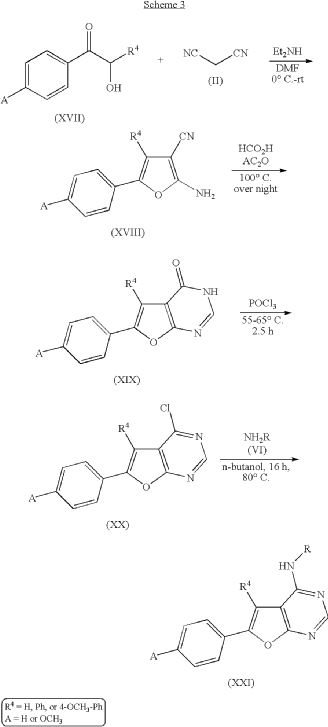
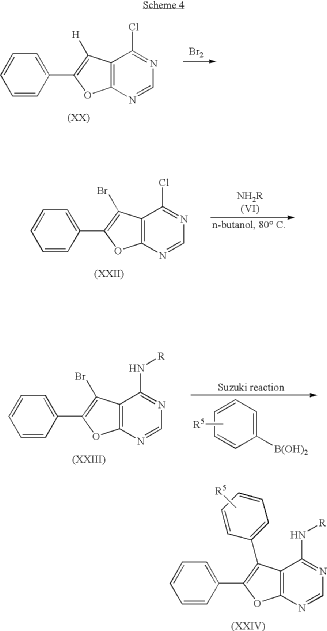
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- Development of Furanopyrimidine-Based Orally Active Third-Generation EGFR Inhibitors for the Treatment of Non-Small Cell Lung CancerPublication Name: Journal of Medicinal ChemistryPublication Date: 2023-02-07PMCID: PMC9969398PMID: 36749735DOI: 10.1021/acs.jmedchem.2c01434
- Discovery of a Furanopyrimidine-Based Epidermal Growth Factor Receptor Inhibitor (DBPR112) as a Clinical Candidate for the Treatment of Non-Small Cell Lung CancerPublication Name: Journal of Medicinal ChemistryPublication Date: 2019-09-27PMID: 31560541DOI: 10.1021/acs.jmedchem.9b00722
PAT
- Active cancer immunotherapy by immune modulation via globo series antigensPublication Number: EP-4248214-A1Priority Date: 2020-11-19
- Fused Bicyclic and Tricyclic Pyrimidine Compounds as Tyrosine Kinase InhibitorsPublication Number: US-2010120805-A1Priority Date: 2008-11-10
- Fused bicyclic and tricyclic pyrimidine compounds as tyrosine kinase inhibitorsPublication Number: US-8507502-B2Priority Date: 2008-11-10Grant Date: 2013-08-13
- Fused bicyclic and tricyclic pyrimidine compounds as tyrosine kinase inhibitorsPublication Number: WO-2010054285-A2Priority Date: 2008-11-10
- Fused bicyclic and polycyclic pyrimidine compounds as tyrosine kinase inhibitorsPublication Number: CN-102264745-APriority Date: 2008-11-10
- Active cancer immunotherapy through immune modulation via GLOBO series antigensPublication Number: CN-116847875-APriority Date: 2020-11-19
- Active cancer immunotherapy by immune modulation via globo series antigensPublication Number: CA-3200572-A1Priority Date: 2020-11-19
- Active cancer immunotherapy by immune modulation via globo series antigensPublication Number: WO-2022109601-A1Priority Date: 2020-11-19
- Active cancer immunotherapy by immune modulation via globo series antigensPublication Number: IL-302947-APriority Date: 2020-11-19
- Active cancer immunotherapy by immunomodulation through GLOBO family antigensPublication Number: KR-20230110529-APriority Date: 2020-11-19
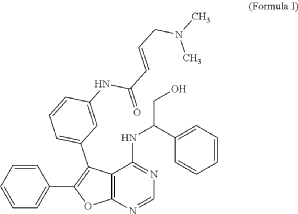
- Crystalline forms of (s, e)-4-(dimethylamino)-n-(3-(4-(2-hydroxy-1-phenylethylamino)-6-phenylfuro[2,3-d]pyrimidin-5-yl)phenyl)but-2-enamide free basePublication Number: TW-I809967-BPriority Date: 2021-07-06Grant Date: 2023-07-21
- Crystalline forms of (S, E)-4-(dimethylamino)-N-(3-(4-(2-hydroxy-1-phenylethylamino)-6-phenylfuro[2,3-d]pyrimidin-5-yl)phenyl)but-2- enamide free basePublication Number: US-12240858-B2Priority Date: 2021-07-06Grant Date: 2025-03-04
- Active cancer immunotherapy by immune modulation via globo series antigensPublication Number: TW-202237177-APriority Date: 2020-11-19
- Active cancer immunotherapy by immune modulation via globo series antigensPublication Number: US-2024139301-A1Priority Date: 2020-11-19
- Active cancer immunotherapy by immune modulation via globo series antigensPublication Number: AU-2021382807-A1Priority Date: 2020-11-19
- Crystalline forms of (s, e)-4-(dimethylamino)-n-(3-(4-(2-hydroxy-1-phenylethylamino)-6-phenylfuro[2,3-d]pyrimidin-5-yl)phenyl)but-2-enamide free basePublication Number: US-2023021909-A1Priority Date: 2021-07-06
- Crystalline forms of (s, e)-4-(dimethylamino)-n-(3-(4-(2-hydroxy-1-phenylethylamino)-6-phenylfuro[2,3-d]pyrimidin-5-yl)phenyl)but-2-enamide free basePublication Number: WO-2023283269-A1Priority Date: 2021-07-06
- Crystalline forms of (s, e)-4-(dimethylamino)-n-(3-(4-(2-hydroxy-1-phenylethylamino)-6-phenylfuro[2,3-d]pyrimidin-5-yl)phenyl)but-2-enamide free basePublication Number: US-2024368175-A1Priority Date: 2021-07-06
- Crystalline forms of (s, e)-4-(dimethylamino)-n-(3-(4-(2-hydroxy-1-phenylethylamino)-6-phenylfuro[2,3-d]pyrimidin-5-yl)phenyl)but-2-enamide free basePublication Number: TW-202309041-APriority Date: 2021-07-06
- Crystalline forms of (s, e)-4-(dimethylamino)-n- (3-(4-(2-hydroxy-1-phenylethylamino)-6-phenylfuro[2,3-d]pyrimidin-5-yl)phenyl)but-2-enamide free basePublication Number: EP-4330259-A1Priority Date: 2021-07-06
//////gozanertinib, ANAX LABS, epidermal growth factor receptor tyrosine kinase inhibitor, antineoplastic, DBPR 112, ABT 101, 6G0COS33K4
Gintemetostat
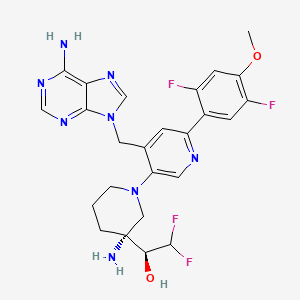
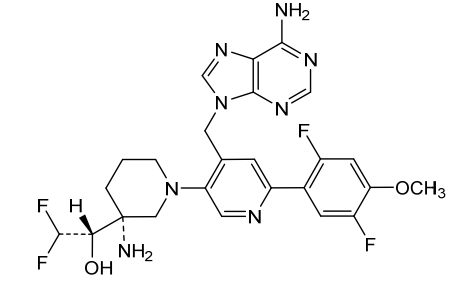
Gintemetostat
(1S)-1-[(3R)-3-amino-4′-[(6-amino-9H-purin-9-yl)methyl]-6′-(2,5-difluoro-4-methoxyphenyl)-3,4,5,6-tetrahydro-2H-[1,3′-bipyridin]-3-yl]-2,2-difluoroethan1-ol
antineoplastic, KTX 1001, NSD2 inhibitor 161, A48CGJ5UQM
CAS 2604513-16-6
MF C25H26F4N8O2 MW 546.5 g/mol
(S)-1-((R)-3-Amino-1-(4-((6-amino-9H-purin-9-yl)methyl)-6-(2,5-difluoro-4-methoxyphenyl)pyridin-3-yl)piperidin-3-yl)-2,2-difluoroethan-1-ol
Gintemetostat (also known as KTX-1001) is a first-in-class, orally administered small molecule being developed to treat relapsed and refractory multiple myeloma. It works as a selective inhibitor of NSD2 (also known as MMSET), targeting the epigenetic drivers of high-risk cancers.
How it Works
- Mechanism: Gintemetostat selectively binds to the catalytic SET domain of the NSD2 enzyme.
- Effect: By blocking this enzyme, it downregulates oncogenic signaling, decreases cancer cell growth, and can enhance T-cell activation against the tumor.
Target Patient Population
- High-Risk Myeloma: The drug focuses heavily on patients harboring the t(4;14) translocation, a genetic alteration found in 10-15% of patients that often causes aggressive relapses.
- Refractory Cases: It has shown notable single-agent activity in heavily pretreated patients who have exhausted standard-of-care, triple-class refractory treatment options.
Current Clinical Status
- Phase 1 Trial: Early data from phase 1 trials (such as NCT05651932) showed the drug has manageable safety profiles and offers clinical benefit (ranging from stable disease to very good partial response) in patients with aggressive, hard-to-treat multiple myeloma.
- Future Developments: Researchers are expanding studies to pair gintemetostat with other standard myeloma treatments, such as proteasome inhibitors and CELMoDs, to create stronger synergistic anti-cancer effects.
Gintemetostat is an orally available small molecule inhibitor of the histone-lysine N-methyltransferase nuclear receptor-binding SET domain protein 2 (NSD2; MMSET; WHSC1), with potential antineoplastic activity. Upon oral administration, gintemetostat selectively targets and binds to NSD2, and inhibits its catalytic activity and the mono- and di-methylation of histone H3 lysine 36 (H3K36). This modulates the expression of genes involved in cellular processes including cellular proliferation, which may lead to decreased growth of cancer cells. NSD2, a member of the NSD family of histone lysine methyltransferase enzymes that catalyzes the mono- and di-methylation of H3K36, is overexpressed and dysregulated in many types of cancers.
SYN
Discovery of a Highly Potent and Selective Inhibitor Targeting Protein Lysine Methyltransferase NSD2
Publication Name: Journal of Medicinal Chemistry
Publication Date: 2024-09-04
PMID: 39230932
DOI: 10.1021/acs.jmedchem.4c00639
SYN
PAT
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021028854&_cid=P12-MQ0AZT-13511-1
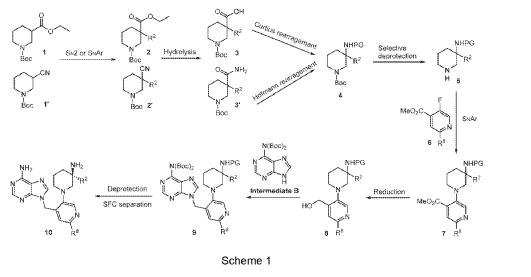
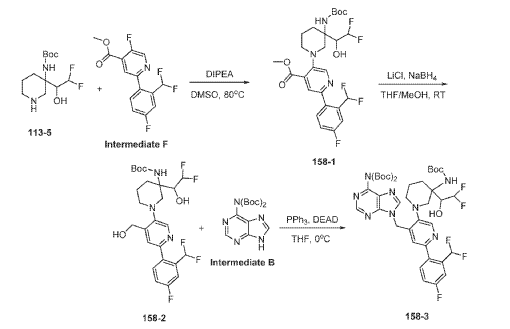
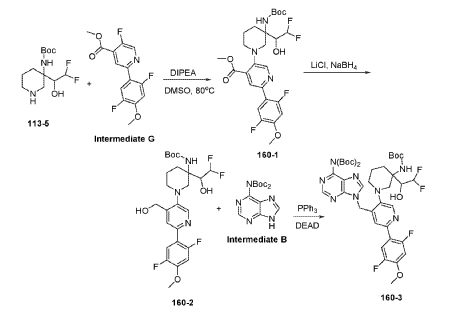
Example 160 and Example 161: (R)-1-((R)-3-amino-1-(4-((6-amino-9H-purin-9-yl)methyl)-6- (2,5-difluoro-4-methoxyphenyl)pyridin-3-yl)piperidin-3-yl)-2,2-difluoroethan-1-ol and (S)-1-((R)-3- amino-1-(4-((6-amino-9H-purin-9-yl)methyl)-6-(2,5-difluoro-4-methoxyphenyl)pyridin-3- yl)piperidin-3-yl)-2,2-difluoroethan-1-ol
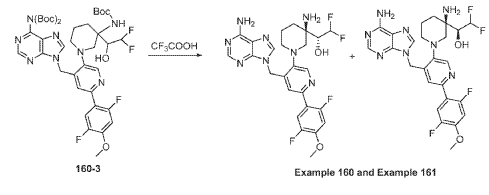
To a solution of tert-butyl (tert-butoxycarbonyl)(9-((5-(3-((tert-butoxycarbonyl)amino)-3-(2,2- difluoro-1-hydroxyethyl)piperidin-1-yl)-2-(2,5-difluoro-4-methoxyphenyl)pyridin-4-yl)methyl)-9H- purin-6-yl)carbamate (Intermediate 160-3) (200 mg, 0.237 mmol) in DCM (18 mL), was added TFA (36 mL), and the reaction mixrture was stirred at rt for 30 min under N2 atmosphere. The reaction mixture was concentrated in vacuo to give the crude product. The crude product was purifed by Pre-HPLC and SFC to afford (R)-1-((R)-3-amino-1-(4-((6-amino-9H-purin-9- yl)methyl)-6-(2,5-difluoro-4-methoxyphenyl)pyridin-3-yl)piperidin-3-yl)-2,2-difluoroethan-1-ol (Example 160) and (S)-1-((R)-3-amino-1-(4-((6-amino-9H-purin-9-yl)methyl)-6-(2,5-difluoro-4- methoxyphenyl)pyridin-3-yl)piperidin-3-yl)-2,2-difluoroethan-1-ol (Example 161).
Example 160: 1H NMR (400 MHz, CD3OD) d ppm 8.48 (s, 1H), 8.20 (d, J = 1.6 Hz, 2H), 7.58 (dd, J = 12.2, 7.3 Hz, 1H), 7.11 (d, J = 1.3 Hz, 1H), 6.90 (dd, J = 12.6, 7.1 Hz, 1H), 6.06 (td, J = 55.1, 3.9 Hz, 1H), 5.67 (s, 2H), 3.87 (s, 3H), 3.75 – 3.58 (m, 1H), 3.25 – 2.75 (m, 4H), 2.26 – 1.60 (m, 4H). LC-MS: [M+H]+ = 547.2, 548.2.
Example 161: 1H NMR (400MHz, CD3OD) d = 8.51 – 8.44 (m, 1H), 8.24 – 8.16 (m, 2H), 7.62 – 7.48 (m, 1H), 7.03 (s, 1H), 6.93 – 6.79 (m, 1H), 6.25 – 5.86 (m, 1H), 5.71 – 5.59 (m, 2H), 4.00 (m, 1H), 3.88 – 3.80 (m, 3H), 3.28 – 2.87 (m, 4H), 1.99 – 1.56 (m, 4H). LC-MS: [M+H]+ =547.4.
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- Piperidinyl-methyl-purineamines as nsd2 inhibitors and anti-cancer agentsPublication Number: EP-4559915-A1Priority Date: 2019-08-14
- Piperidinyl-methyl-purineamines as nsd2 inhibitors and anti-cancer agentsPublication Number: EP-4013755-B1Priority Date: 2019-08-14Grant Date: 2025-01-08
- Piperidinyl-methyl-purinamines as NSD2 inhibitors and anticancer agentsPublication Number: CN-114585622-APriority Date: 2019-08-14
- Piperidinyl-methyl-purineamines as NSD2 inhibitors and anti-cancer agentsPublication Number: US-12312353-B2Priority Date: 2019-08-14Grant Date: 2025-05-27
- Piperidinyl-methyl-purineamines as nsd2 inhibitors and anti-cancer agentsPublication Number: WO-2021026803-A1Priority Date: 2019-08-14
- Piperidinyl-methyl-purineamines as nsd2 inhibitors and anti-cancer agentsPublication Number: EP-4013755-A1Priority Date: 2019-08-14
- Piperidinyl-methyl-purineamines as nsd2 inhibitors and anti-cancer agentsPublication Number: WO-2021028854-A1Priority Date: 2019-08-14
- Piperidinyl-methyl-purineamine D-tartrate, crystalline forms, and use thereof in the treatment of medical diseases and conditionsPublication Number: CN-119744262-APriority Date: 2022-05-18
- Piperidinyl-methyl-purine amine d-tartaric acid salts, crystalline forms, and their use in treating medical diseases and conditionsPublication Number: EP-4526305-A1Priority Date: 2022-05-18
- Piperidinyl-methyl-purineamines as nsd2 inhibitors and anti-cancer agentsPublication Number: US-2023002388-A1Priority Date: 2019-08-14
- Piperidinyl-methyl-purinamines as NSD2 inhibitors and anticancer agentsPublication Number: CN-114585622-BPriority Date: 2019-08-14Grant Date: 2024-08-09
- Piperidinyl-methyl-purineamines as NSD2 inhibitors and anti-cancer agentsPublication Number: US-11420970-B1
- Piperidinyl-methyl-purine amine d-tartaric acid salts, crystalline forms, and their use in treating medical diseases and conditionsPublication Number: AU-2023273656-A1Priority Date: 2022-05-18
- Piperidinyl-methyl-purine amine salts, crystalline forms, and their use in treating medical diseases and conditionsPublication Number: WO-2023225144-A1Priority Date: 2022-05-18
- Piperidinyl-methyl-purine amine fumaric acid salts, crystalline forms, and their use in treating medical diseases and conditionsPublication Number: WO-2023225150-A1Priority Date: 2022-05-18
- Piperidinyl-methyl-purine amine salts, crystalline forms, and their use in treating medical diseases and conditionsPublication Number: US-2025326752-A1Priority Date: 2022-05-18
- Piperidinyl-methyl-purinic amine D-tartrate salt, crystalline form and their use in the treatment of medical diseases and conditionsPublication Number: KR-20250012083-APriority Date: 2022-05-18
- Pharmaceutical compositions containing a piperidinyl-methyl-purine amine and their use in treating diseases and conditionsPublication Number: US-2024207192-A1Priority Date: 2022-12-12
- Pharmaceutical compositions containing a piperidinyl-methyl-purine amine and their use in treating diseases and conditionsPublication Number: WO-2024129670-A1Priority Date: 2022-12-12
- Piperidinyl-methyl-purine amine d-tartaric acid salts, crystalline forms, and their use in treating medical diseases and conditionsPublication Number: US-2024002385-A1Priority Date: 2022-05-18
- Piperidinyl-methyl-purine amine salts, crystalline forms, and their use in treating medical diseases and conditionsPublication Number: WO-2023225154-A1Priority Date: 2022-05-18
- Piperidinyl-methyl-purine amine d-tartaric acid salts, crystalline forms, and their use in treating medical diseases and conditionsPublication Number: WO-2023225141-A1Priority Date: 2022-05-18
////////////gintemetostat, ANAX LABS, antineoplastic, KTX 1001, NSD2 inhibitor 161, A48CGJ5UQM
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY


{kind=link}