

| Patent | Submitted | Granted |
|---|---|---|
| Compound useful for the treatment of degenerative and inflammatory diseases [US8088764] | 2010-12-30 | 2012-01-03 |
| NOVEL COMPOUNDS USEFUL FOR THE TREATMENT OF DEGENERATIVE AND INFLAMMATORY DISEASES [US2011190260] | 2011-08-04 |
Home » EU 2020
Category Archives: EU 2020
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Isatuximab
(A chain)
QVQLVQSGAE VAKPGTSVKL SCKASGYTFT DYWMQWVKQR PGQGLEWIGT IYPGDGDTGY
AQKFQGKATL TADKSSKTVY MHLSSLASED SAVYYCARGD YYGSNSLDYW GQGTSVTVSS
ASTKGPSVFP LAPSSKSTSG GTAALGCLVK DYFPEPVTVS WNSGALTSGV HTFPAVLQSS
GLYSLSSVVT VPSSSLGTQT YICNVNHKPS NTKVDKKVEP KSCDKTHTCP PCPAPELLGG
PSVFLFPPKP KDTLMISRTP EVTCVVVDVS HEDPEVKFNW YVDGVEVHNA KTKPREEQYN
STYRVVSVLT VLHQDWLNGK EYKCKVSNKA LPAPIEKTIS KAKGQPREPQ VYTLPPSRDE
LTKNQVSLTC LVKGFYPSDI AVEWESNGQP ENNYKTTPPV LDSDGSFFLY SKLTVDKSRW
QQGNVFSCSV MHEALHNHYT QKSLSLSPGK
(B chain)
QVQLVQSGAE VAKPGTSVKL SCKASGYTFT DYWMQWVKQR PGQGLEWIGT IYPGDGDTGY
AQKFQGKATL TADKSSKTVY MHLSSLASED SAVYYCARGD YYGSNSLDYW GQGTSVTVSS
ASTKGPSVFP LAPSSKSTSG GTAALGCLVK DYFPEPVTVS WNSGALTSGV HTFPAVLQSS
GLYSLSSVVT VPSSSLGTQT YICNVNHKPS NTKVDKKVEP KSCDKTHTCP PCPAPELLGG
PSVFLFPPKP KDTLMISRTP EVTCVVVDVS HEDPEVKFNW YVDGVEVHNA KTKPREEQYN
STYRVVSVLT VLHQDWLNGK EYKCKVSNKA LPAPIEKTIS KAKGQPREPQ VYTLPPSRDE
LTKNQVSLTC LVKGFYPSDI AVEWESNGQP ENNYKTTPPV LDSDGSFFLY SKLTVDKSRW
QQGNVFSCSV MHEALHNHYT QKSLSLSPGK
(C chain)
DIVMTQSHLS MSTSLGDPVS ITCKASQDVS TVVAWYQQKP GQSPRRLIYS ASYRYIGVPD
RFTGSGAGTD FTFTISSVQA EDLAVYYCQQ HYSPPYTFGG GTKLEIKRTV AAPSVFIFPP
SDEQLKSGTA SVVCLLNNFY PREAKVQWKV DNALQSGNSQ ESVTEQDSKD STYSLSSTLT
LSKADYEKHK VYACEVTHQG LSSPVTKSFN RGEC
(D chain)
DIVMTQSHLS MSTSLGDPVS ITCKASQDVS TVVAWYQQKP GQSPRRLIYS ASYRYIGVPD
RFTGSGAGTD FTFTISSVQA EDLAVYYCQQ HYSPPYTFGG GTKLEIKRTV AAPSVFIFPP
SDEQLKSGTA SVVCLLNNFY PREAKVQWKV DNALQSGNSQ ESVTEQDSKD STYSLSSTLT
LSKADYEKHK VYACEVTHQG LSSPVTKSFN RGEC
(Disulfide bridge: A22-A96, A147-A203, A223-C214, A229-B229, A232-B232, A264-A324, A370-A428, B22-B96, B147-B203, B223-D214, B264-B324, B370-B428, C23-C88, C134-C194, D23-D88, D134-D194)
Isatuximab
イサツキシマブ (遺伝子組換え)
APPROVED USFDA 2020/3/2, Sarclisa
EU APPROVED 2020/5/30
JAPAN APPROVED 2020/6/29
CAS 1461640-62-9
| Antineoplastic, Anti-CD38 antibody | |
| Disease | Multiple myeloma |
|---|
Isatuximab, sold under the brand name Sarclisa, is a monoclonal antibody (mAb) medication for the treatment of multiple myeloma.[4][3]
The most common side effects include neutropenia (low levels of neutrophils, a type of white blood cell), infusion reactions, pneumonia (infection of the lungs), upper respiratory tract infection (such as nose and throat infections), diarrhoea and bronchitis (inflammation of the airways in the lungs).[3]
Isatuximab is an anti-CD38 mAb intended to treat relapsed or refractory multiple myeloma.[5] It entered in Phase II trials for multiple myeloma[6] and T-cell leukemia in 2015.[7]
Medical uses
In the United States it is indicated, in combination with pomalidomide and dexamethasone, for the treatment of adults with multiple myeloma who have received at least two prior therapies including lenalidomide and a proteasome inhibitor.[8][9][10]
In the European Union it is indicated, in combination with pomalidomide and dexamethasone, for the treatment of adults with relapsed and refractory multiple myeloma (MM) who have received at least two prior therapies including lenalidomide and a proteasome inhibitor (PI) and have demonstrated disease progression on the last therapy.[3]
History
It was granted orphan drug designation for multiple myeloma by the European Medicines Agency (EMA) in April 2014, and by the U.S. Food and Drug Administration (FDA) in December 2016.[3][11]
Researchers started a Phase I study with isatuximab in combination with pomalidomide and dexamethasone for the treatment of patients with multiple myeloma (MM). The results during the Phase I trial showed that 26 out of the 45 patients discontinued the treatment due to progression of the disease. The patients had already been heavily pretreated. The latter lead to a manageable safety profile where the dose of isatuximab in combination with pomalidomide and dexamethasone would be capped to the maximum of 10 mg/kg weekly every two weeks for future studies.[12]
Based on the remarkable findings during the Phase I trial, a Phase II trial was launched where researchers investigated isatuximab as a single agent in patients with MM. The heavily pretreated patients reacted well to the single administration of isatuximab during Phase II of the trial.[13]
A Phase III combination trial for plasma cell myeloma is comparing pomalidomide and dexamethasone with and without isatuximab is in progress with an estimated completion date of 2021.[medical citation needed]
Additionally, two Phase III trials were added in 2017. The first trial highlights whether there is an added value in the combination of isatuximab with bortezomib, lenalidomide and dexamethasone. The latter will be tested in patients with newly diagnosed MM who are not qualified for a transplant (IMROZ trial). The second trial evaluates the combinations of isatuximab with carfilzomib and dexamethasone compared to carfilzomib with dexamethasone. The second trial was designed for patients who were previously treated with one to three prior lines (IKEMA). There is currently[when?] no treatment for MM, however promising improvements have been made and the study is still ongoing.[14][15]
In March 2020, it was approved for medical use in the United States.[8][9][10]
The U.S. Food and Drug Administration (FDA) approved isatuximab-irfc in March 2020, based on evidence from a clinical trial (NCT02990338) of 307 subjects with previously treated multiple myeloma.[10] The trial was conducted at 102 sites in Europe, North America, Asia, Australia and New Zealand.[10]
The trial evaluated the efficacy and side effects of isatuximab-irfc in subjects with previously treated multiple myeloma.[10] Subjects were randomly assigned to receive either isatuximab-irfc (in combination with pomalidomide and low-dose dexamethasone) or active comparator (pomalidomide and low-dose dexamethasone).[10] Treatment was administered in both groups in 28-day cycles until disease progression or unacceptable toxicity.[10] Both subjects and health care providers knew which treatment was given.[10] The trial measured the time patients lived without the cancer growing (progression-free survival or PFS).[10]
It was approved for medical use in the European Union in May 2020.[3]
Structure and reactivity
The structure of isatuximab consists of two identical immunoglobulin kappa light chains and also two equal immunoglobulin gamma heavy chains. Chemically, isatuximab is similar to the structure and reactivity of daratumumab, hence both drugs show the same CD38 targeting. However, isatuximab shows a more potent inhibition of its ectozyme function. The latter gives potential for some non-cross reactivity. Isatuximab shows action of an allosteric antagonist with the inhibition of the CD38 enzymatic activity. Additionally, isatuximab shows potential where it can induce apoptosis without cross linking.[16] Lastly, Isatuximab reveals direct killing activity when a larger increase in apoptosis is detected in CD38 expressing cancer cells. Furthermore, isatuximab demonstrated a dose dependent inhibition of CD38 enzymatic activity. However, daratumumab with the same experimental conditions shows a more limited inhibition without a dose response.[17]
Reactions
Isatuximab binds uniquely to an epitope on the CD38 receptor and is the only CD38 antibody which can start apoptosis directly.[18] Isatuximab binds to a different CD38 epitope amino-acid sequence than does the anti-CD38 monoclonal antibody daratumumab.[19] The binding with the CD38 receptor is mainly via the gamma heavy chains and are more potent than other CD38 antibodies such as daratumumab which can inhibit the enzymatic activity of CD38. Moreover, isatuximab inhibits the hydrolase activity of CD38.[medical citation needed]
The antibodies show signs of improving antitumor immunity by eliminating regulatory T cells, B cells and myeloid-derived suppressor cells. The difference in binding between isatuximab and daratumumab is in the recognition of the different amino acid groups. Isatuximab identifies 23 amino acids of CD38 to the contrary with daratumumab who has 27. The residue of Glu233 has a flexible sidechain and faces the N-terminal of Asp1 residue in the isatuximab light chain. The latter light chain of isatuximab is also flexible which makes the interaction between CD38/Glu233 and the Asp1 weaker than the other interactions between CD38 and isatuximab. The caspase-dependent apoptotic pathway and the lysosomal mediated cell death pathway in MM cells is induced by isatuximab. The MM cell death follows the downstream reactions of the lysosomal activation. The latter also activates the production of reactive oxygen species.[20]
Available forms
Isatuximab or isatuximab-irfc is available as a drug in an intravenous infusion form. Injection doses are 100 mg/5 mL (20 mg/mL) solution in single-dose vial or 500 mg/25 mL (20 mg/mL) solution in single-dose vial.[4]
Mechanism of action
Cancer of the blood that is distinguished by an overproduction of malignant plasma cells in the bone marrow is called multiple myeloma. The myeloma cells are marked with uniformed overexpression of CD38 surface glycoproteins. Although these proteins are also expressed on other myeloid and lymphoid cells, the extent is relatively minor compared to myeloma cells. The fact that CD38 glycoproteins carry out various important cellular functions, and that they are plentiful on the surface of myeloma cells, has made them an appealing target for multiple myeloma treatment.[21] CD38 was first described as an activation marker, but later the molecule displayed functions in adhesion to endothelial CD31 proteins, e.g. as an aiding component of the synapse complex, as well as an ectoenzyme implicated in the metabolism of extracellular NAD+ and cytoplasmic NADP. The tumour cells can evade the immune system, possibly due to adenosine, an immunosuppressive molecule that arises as a product of the ectoenzymatic activity of CD38.[22]
Isatuximab-irfc is an IgG1-derived monoclonal antibody that selectively binds to the CD38 that exists on the exterior of hematopoietic and multiple myeloma cells (as well as other tumor cells). This drug induces apoptosis of tumor cells and activates immune effector mechanisms such as complement dependent cytotoxicity (CDC), antibody-dependent cellular phagocytosis (ADCP), and antibody-dependent cell-mediated cytotoxicity (ADCC). Isatuximab-irfc is able to stimulate natural killer (NK) cells in the absence of CD38-positive target tumor cells and blocks CD38-positive T-regulatory cells.[4] Furthermore, the NADase activity of CD38 is adjusted by isatuximab, similarly to other CD38 antibodies. Contrarily to daratumumab however, isatuximab can incite apoptosis directly without cross-linking, and in its binding epitope.[23] According to the FDA, isatuximab-irfc alone has reduced ADCC and direct tumor cell killing activity in vitro in comparison to when it is combined with pomalidomide. As well as increased anti-tumor activity as opposed to isatuximab-irfc or pomalidomide only in a human multiple myeloma xenograft model.[4]
Metabolism and toxicity
Metabolism
Isatuximab-irfc is likely to be metabolized through catabolic pathways into smaller peptides. When isatuximab is at a constant state it is expected that the ≥99% elimination will occur approximately two months after the last dose was administered. The clearance percentage diminished when the dosages were increased over time, as well as when multiple doses were administered. However, the elimination of isatuximab-irfc did not differ when applied as a single agent or as a combination therapy.[4]
Toxicity
A dose-limiting toxicity (DLT) has characterized been characterized as the development of any of the following: grade ≥ 3 non-hematologic toxicity; grade 4 neutropenia or grade 4 thrombocytopenia lasting more than 5 days; grade ≥ 2 allergic reactions or hypersensitivity (i.e., infusion reactions); or any other toxicity considered to be dose-limiting by the investigators or sponsor. Grade ≤ 2 infusion reactions were excluded from the DLT definition, because, with suitable care, patients that suffered a grade 2 infusion reaction prior to completion of the infusion were able to finalize isatuximab administration.[23]
There is no recommended reduced dose of isatuximab-irfc. In the eventuality of hematological toxicity it may be necessary to delay administration so that the blood count may be recovered.[4] Although there is no counteracting agent for isatuximab, clinical experience with overdoses is seemingly nonexistent as well. Overdose symptoms will probably be in line with the side effects attached to isatuximab. Therefore, infusion reactions, gastrointestinal disturbances and an elevated risk of infections may occur. It is necessary to carefully monitor the patient in case of an overdose and to employ clinically indicated symptomatic and supportive procedures.[21]
No studies have been conducted with isatuximab concerning carcinogenicity, genotoxicity or fertility.[4]
Pregnancy
When given to pregnant women isatuximab-irfc can cause fetal injury, due to the mechanism of action. It can precipitate depletion of immune cells as well as decreased bone density in the fetus. Pregnant women are therefore notified of the potential risks to a fetus, and women that are able to reproduce are advised to use effective contraceptives during treatment and at least five months subsequent to the last dose of isatuximab-irfc.
Furthermore, it is not recommended to combine isatuximab-irfc with pomalidomide in women that are carrying a child, because pomalidomide may cause birth defects and death of the unborn child.[4]
Indications
Isatuximab is indicated as a CD38-directed cytolytic antibody. By inhibiting the enzymatic activity of CD38.
The binding of isatuximab to CD38 on multiple myeloma (MM) cells leads to a trigger to several mechanisms leading to direct apoptosis of target cancer cells. The triggered pathways are the caspase-dependent apoptotic and the lysosome-mediated cell death pathway in MM cells.[24]
It is used in a combination with dexamethasone and pomalidomide. The drug is thus to treat patients with multiple myeloma. Restrictions for the use of isatuximab is that the patients have to be adults who have at least received two previous treatments with lenalidomide and a proteasome inhibitor.[4]
Isatuximab is currently[when?] also being tested in a Phase II trial as a monotherapy against refractory/recurrent systemic light-chain amyloidosis.[24]
Efficacy and side effects
Efficacy
A Phase III study of patients with refractory and relapsed MM, who were resistant to lenalidomide and a proteasome inhibitor, and could not have received daratumumab, another anti-CD38 monoclonal antibody was published in 2019 (ICARIA-MM). The addition of isatuximab to pomalidomide and dexamethasone improved progression free survival to 11.5 months compared to 6.5 months, with an overall response rate of 63%.[25]
Side effects
Adverse reactions to isatuximab-irfc may include neutropenia, infusion-related reactions and/or secondary primary malignancies.[4] Of these three the most commonly occurring ones are the infusion-related reactions.[24] Examples of the most frequent symptoms of infusion-related reactions are dyspnea, cough, chills, and nausea, while the severest signs and symptoms included hypertension and dyspnea.[4]
Effects on animals
The activity of isatuximab has been researched in mouse tumor models. It has been proven that isatuximab leads to antitumor activity in MM cells. Furthermore, the combination of isatuximab and pomalidomide will lead to an extra enhanced antitumor activity in MM cells. Thus, pomalidomide in vivo and in vitro leads to an increase of the activity of isatuximab.[24]
Animal studies in reproduction toxicity have not yet been carried out. So, the risks of birth defects and miscarriage risks are unknown.[4]
Names
Isatuximab is the United States Adopted Name (USAN).[26]
It was developed by ImmunoGen and Sanofi-Aventis with the development name SAR-650984.
References
- ^ Jump up to:a b “Sarclisa Australian prescription medicine decision summary”. Therapeutic Goods Administration (TGA). 14 May 2020. Retrieved 16 August 2020.
- ^ “Isatuximab (Sarclisa) Use During Pregnancy”. Drugs.com. 25 March 2020. Retrieved 25 June 2020.
- ^ Jump up to:a b c d e f “Sarclisa EPAR”. European Medicines Agency (EMA). 24 March 2020. Retrieved 25 June 2020. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b c d e f g h i j k l “Sarclisa- isatuximab injection, solution, concentrate”. DailyMed. 2 March 2020. Retrieved 26 March 2020.
- ^ ImmunoGen, Inc. Announces Data Presentations at Upcoming 57th ASH Annual Meeting and Exposition
- ^ Martin T (2015). “A Dose Finding Phase II Trial of Isatuximab (SAR650984, Anti-CD38 mAb) As a Single Agent in Relapsed/Refractory Multiple Myeloma”. Blood. 126 (23): 509. doi:10.1182/blood.V126.23.509.509.
- ^ “Safety and Efficacy of Isatuximab in Lymphoblastic Leukemia”. ClinicalTrials.gov. Retrieved 4 March 2020.
- ^ Jump up to:a b “FDA approves isatuximab-irfc for multiple myeloma”. U.S. Food and Drug Administration (FDA). 2 March 2020. Retrieved 2 March 2020.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b “FDA Approves New Therapy for Patients with Previously Treated Multiple Myeloma”. U.S. Food and Drug Administration (FDA) (Press release). 2 March 2020. Retrieved 4 March 2020. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c d e f g h i “Drug Trials Snapshots: Sarclisa”. U.S. Food and Drug Administration(FDA). 2 March 2020. Retrieved 25 March 2020. This article incorporates text from this source, which is in the public domain.
- ^ “Isatuximab Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 24 December 1999. Retrieved 4 March 2020.
- ^ Mikhael J, Richardson P, Usmani SZ, Raje N, Bensinger W, Karanes C, et al. (July 2019). “A phase 1b study of isatuximab plus pomalidomide/dexamethasone in relapsed/refractory multiple myeloma”. Blood. 134 (2): 123–133. doi:10.1182/blood-2019-02-895193. PMC 6659612. PMID 30862646.
- ^ Martin T (7 December 2015). “A Dose Finding Phase II Trial of Isatuximab (SAR650984, Anti-CD38 mAb) As a Single Agent in Relapsed/Refractory Multiple Myeloma”. ASH.
- ^ Orlowski RZ, Goldschmidt H, Cavo M, Martin TG, Paux G, Oprea C, Facon T (20 May 2018). “Phase III (IMROZ) study design: Isatuximab plus bortezomib (V), lenalidomide (R), and dexamethasone (d) vs VRd in transplant-ineligible patients (pts) with newly diagnosed multiple myeloma (NDMM)”. Journal of Clinical Oncology. 36 (15_suppl): TPS8055. doi:10.1200/JCO.2018.36.15_suppl.TPS8055.
- ^ Moreau P, Dimopoulos MA, Yong K, Mikhael J, Risse ML, Asset G, Martin T (January 2020). “Isatuximab plus carfilzomib/dexamethasone versus carfilzomib/dexamethasone in patients with relapsed/refractory multiple myeloma: IKEMA Phase III study design”. Future Oncology. 16 (2): 4347–4358. doi:10.2217/fon-2019-0431. PMID 31833394.
- ^ Rajan AM, Kumar S (July 2016). “New investigational drugs with single-agent activity in multiple myeloma”. Blood Cancer Journal. 6 (7): e451. doi:10.1038/bcj.2016.53. PMC 5030378. PMID 27471867.
- ^ Martin T, Baz R, Benson DM, Lendvai N, Wolf J, Munster P, et al. (June 2017). “A phase 1b study of isatuximab plus lenalidomide and dexamethasone for relapsed/refractory multiple myeloma”. Blood. 129 (25): 3294–3303. doi:10.1182/blood-2016-09-740787. PMC 5482100. PMID 28483761.
- ^ Martin TG, Corzo K, Chiron M (2019). “Therapeutic Opportunities with Pharmacological Inhibition of CD38 with Isatuximab”. Cells. 8 (12): 1522. doi:10.3390/cells8121522. PMC 6953105. PMID 31779273.
- ^ Dhillon S (2020). “Isatuximab: First Approval”. Drugs. 80 (9): 905–912. doi:10.1007/s40265-020-01311-1. PMID 32347476. S2CID 216597315.
- ^ Martin TG, Corzo K, Chiron M, Velde HV, Abbadessa G, Campana F, et al. (November 2019). “Therapeutic Opportunities with Pharmacological Inhibition of CD38 with Isatuximab”. Cells. 8 (12): 1522. doi:10.3390/cells8121522. PMC 6953105. PMID 31779273.
- ^ Jump up to:a b “Isatuximab”. Drugbank. 20 May 2019.
- ^ Morandi F, Horenstein AL, Costa F, Giuliani N, Pistoia V, Malavasi F (28 November 2018). “CD38: A Target for Immunotherapeutic Approaches in Multiple Myeloma”. Frontiers in Immunology. 9: 2722. doi:10.3389/fimmu.2018.02722. PMC 6279879. PMID 30546360.
- ^ Jump up to:a b Martin T, Strickland S, Glenn M, Charpentier E, Guillemin H, Hsu K, Mikhael J (March 2019). “Phase I trial of isatuximab monotherapy in the treatment of refractory multiple myeloma”. Blood Cancer Journal. 9 (4): 41. doi:10.1038/s41408-019-0198-4. PMC 6440961. PMID 30926770.
- ^ Jump up to:a b c d Martin TG, Corzo K, Chiron M, Velde HV, Abbadessa G, Campana F, et al. (November 2019). “Therapeutic Opportunities with Pharmacological Inhibition of CD38 with Isatuximab”. Cells. 8 (12): 1522. doi:10.3390/cells8121522. PMC 6953105. PMID 31779273.
- ^ Attal, Michel; Richardson, Paul G; Rajkumar, S Vincent; San-Miguel, Jesus; Beksac, Meral; Spicka, Ivan; Leleu, Xavier; Schjesvold, Fredrik; Moreau, Philippe; Dimopoulos, Meletios A; Huang, Jeffrey Shang-Yi (2019). “Isatuximab plus pomalidomide and low-dose dexamethasone versus pomalidomide and low-dose dexamethasone in patients with relapsed and refractory multiple myeloma (ICARIA-MM): a randomised, multicentre, open-label, phase 3 study”. The Lancet. 394 (10214): 2096–2107. doi:10.1016/s0140-6736(19)32556-5. ISSN 0140-6736. PMID 31735560. S2CID 208049235.
- ^ Statement On A Nonproprietary Name Adopted By The USAN Council – Isatuximab, American Medical Association
External links
- “Isatuximab”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT02990338 for “Multinational Clinical Study Comparing Isatuximab, Pomalidomide, and Dexamethasone to Pomalidomide and Dexamethasone in Refractory or Relapsed and Refractory Multiple Myeloma Patients (ICARIA-MM)” at ClinicalTrials.gov
| Isatuximab (pale blue) binding CD38 (purple). PDB: 4CMH | |
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Chimeric (mouse/human) |
| Target | CD38 |
| Clinical data | |
| Trade names | Sarclisa |
| Other names | SAR-650984, isatuximab-irfc |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a620023 |
| License data | US DailyMed: Sarclisa |
| Pregnancy category | AU: C[1]US: N (Not classified yet)[2] |
| Routes of administration | Intravenous |
| Drug class | Antineoplastic |
| ATC code | None |
| Legal status | |
| Legal status | AU: S4 (Prescription only) [1]US: ℞-onlyEU: Rx-only [3] |
| Identifiers | |
| CAS Number | 1461640-62-9 |
| DrugBank | DB14811 |
| ChemSpider | none |
| UNII | R30772KCU0 |
| KEGG | D11050 |
| Chemical and physical data | |
| Formula | C6456H9932N1700O2026S44 |
| Molar mass | 145190.99 g·mol−1 |
////////Isatuximab, Sarclisa, 2020APPROVALS, JAPAN 2020, US 2020, EU 2020, PEPTIDE, SANOFI , イサツキシマブ (遺伝子組換え) ,
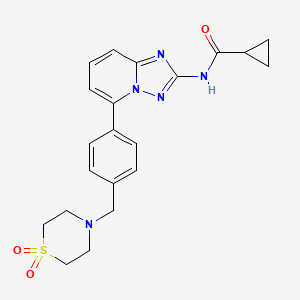
Viltolarsen
Viltolarsen
维托拉生
ビルトラルセン
| Formula | C244H381N113O88P20 |
|---|---|
| CAS | 2055732-84-6 |
| Mol weight | 6924.8155 |
APPROVED FDA 2020/8/12, Viltepso
APPROVED JAPAN PMDA 2020/3/25, VILTEPSO
- NCNP-01
- NS-065
- NS-065/NCNP-01
- WHO 10771
- WHO-10771
| NAME | DOSAGE | STRENGTH | ROUTE | LABELLER | MARKETING START | MARKETING END | ||
|---|---|---|---|---|---|---|---|---|
| Viltepso | Injection, solution | 250 mg/1 | Intravenous | Ns Pharma, Inc. | 2020-08-13 | Not applicable |  |
SYNWatanabe N, Nagata T, Satou Y, Masuda S, Saito T, Kitagawa H, Komaki H, Takagaki K, Takeda S: NS-065/NCNP-01: An Antisense Oligonucleotide for Potential Treatment of Exon 53 Skipping in Duchenne Muscular Dystrophy. Mol Ther Nucleic Acids. 2018 Dec 7;13:442-449. doi: 10.1016/j.omtn.2018.09.017.
| PATENT NUMBER | PEDIATRIC EXTENSION | APPROVED | EXPIRES (ESTIMATED) | |
|---|---|---|---|---|
| US9079934 | No | 2011-08-31 | 2031-08-31 | |
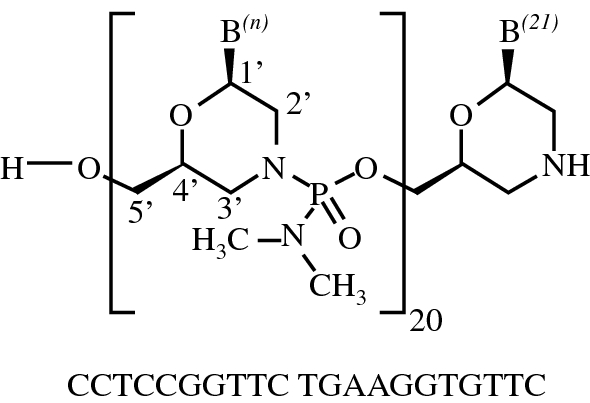
Viltolarsen
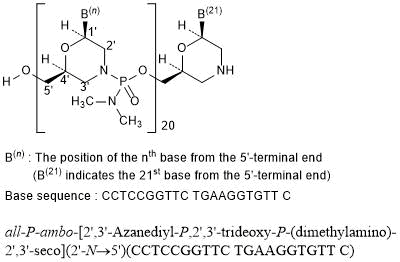
all-P-ambo-[2′,3′-Azanediyl-P,2′,3′-trideoxy-P-(dimethylamino)-2′,3′-seco](2′-N→5′)(CCTCCGGTTC TGAAGGTGTT C)
C244H381N113O88P20 : 6924.82
[2055732-84-6]
Viltolarsen, sold under the brand name Viltepso, is a medication used for the treatment of Duchenne muscular dystrophy (DMD).[3][4][2] Viltolarsen is an antisense oligonucleotide.[3][2]
The most common side effects include upper respiratory tract infection, injection site reaction, cough, and pyrexia (fever).[3][4][2]
Viltolarsen was approved for medical use in the United States in August 2020.[3][4] After golodirsen was approved in December 2019, viltolarsen is the second approved targeted treatment for people with this type of mutation in the United States.[3][5] Approximately 8% of people with DMD have a mutation that is amenable to exon 53 skipping.[3]
Medical uses
Viltolarsen is indicated for the treatment of Duchenne muscular dystrophy (DMD) in people who have a confirmed mutation of the DMD gene that is amenable to exon 53 skipping.[3][2]
DMD is a rare genetic disorder characterized by progressive muscle deterioration and weakness.[3] It is the most common type of muscular dystrophy.[3] DMD is caused by mutations in the DMD gene that results in an absence of dystrophin, a protein that helps keep muscle cells intact.[3] The first symptoms are usually seen between three and five years of age and worsen over time.[3] DMD occurs in approximately one out of every 3,600 male infants worldwide; in rare cases, it can affect females.[3]
Adverse effects
The most common side effects include upper respiratory tract infection, injection site reaction, cough, and pyrexia (fever).[3][4][2]
Although kidney toxicity was not observed in the clinical studies, the clinical experience is limited, and kidney toxicity, including potentially fatal glomerulonephritis, has been observed after administration of some antisense oligonucleotides.[3]
History
Viltolarsen was evaluated in two clinical studies with a total of 32 participants, all of whom were male and had genetically confirmed DMD.[3] The increase in dystrophin production was established in one of those two studies, a study that included sixteen DMD participants, with eight participants receiving viltolarsen at the recommended dose.[3] In the study, dystrophin levels increased, on average, from 0.6% of normal at baseline to 5.9% of normal at week 25.[3] Trial 1 provided data for evaluation of the benefits of viltolarsen.[4] The combined populations from both trials provided data for evaluation of the side effects of viltolarsen.[4] Trial 1 was conducted at six sites in the United States and Canada and Trial 2 was conducted at five sites in Japan.[4] All participants in both trials were on a stable dose of corticosteroids for at least three months before entering the trials.[4]
The U.S. Food and Drug Administration (FDA) concluded that the applicant’s data demonstrated an increase in dystrophin production that is reasonably likely to predict clinical benefit in people with DMD who have a confirmed mutation of the dystrophin gene amenable to exon 53 skipping.[3] A clinical benefit of the drug has not been established.[3] In making this decision, the FDA considered the potential risks associated with the drug, the life-threatening and debilitating nature of the disease, and the lack of available therapies.[3]
The application for viltolarsen was granted priority review designation and the FDA granted the approval to NS Pharma, Inc.[3]
References
- ^ https://www.drugs.com/pregnancy/viltolarsen.html
- ^ Jump up to:a b c d e f “Viltepso- viltolarsen injection, solution”. DailyMed. 12 August 2020. Retrieved 18 August 2020.
- ^ Jump up to:a b c d e f g h i j k l m n o p q r s t u “FDA Approves Targeted Treatment for Rare Duchenne Muscular Dystrophy Mutation”. U.S. Food and Drug Administration (FDA) (Press release). 12 August 2020. Retrieved 12 August 2020. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c d e f g h “Drug Trials Snapshots: Viltepso”. U.S. Food and Drug Administration. 12 August 2020. Retrieved 18 August 2020. This article incorporates text from this source, which is in the public domain.
- ^ Anwar S, Yokota T (August 2020). “Golodirsen for Duchenne muscular dystrophy”. Drugs of Today. 56 (8): 491–504. doi:10.1358/dot.2020.56.8.3159186. PMID 33025945.
Further reading
- Dhillon S (July 2020). “Viltolarsen: First Approval”. Drugs. 80 (10): 1027–1031. doi:10.1007/s40265-020-01339-3. PMID 32519222. S2CID 219542850.
- Dzierlega K, Yokota T (June 2020). “Optimization of antisense-mediated exon skipping for Duchenne muscular dystrophy”. Gene Ther. 27 (9): 407–416. doi:10.1038/s41434-020-0156-6. PMID 32483212. S2CID 219157034.
- Hwang J, Yokota T (October 2019). “Recent advancements in exon-skipping therapies using antisense oligonucleotides and genome editing for the treatment of various muscular dystrophies”. Expert Rev Mol Med. 21: e5. doi:10.1017/erm.2019.5. PMID 31576784.
- Roshmi RR, Yokota T (October 2019). “Viltolarsen for the treatment of Duchenne muscular dystrophy”. Drugs Today. 55 (10): 627–639. doi:10.1358/dot.2019.55.10.3045038. PMID 31720560.
External links
- “Viltolarsen”. Drug Information Portal. U.S. National Library of Medicine (NLM).
- Clinical trial number NCT02740972 for “Safety and Dose Finding Study of NS-065/NCNP-01 in Boys With Duchenne Muscular Dystrophy (DMD)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Viltepso |
| Other names | NS-065/NCNP-01 |
| AHFS/Drugs.com | Monograph |
| License data | US DailyMed: Viltolarsen |
| Pregnancy category | US: N (Not classified yet)[1] |
| Routes of administration | Intravenous |
| Drug class | Antisense oligonucleotide |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [2]In general: ℞ (Prescription only) |
| Identifiers | |
| CAS Number | 2055732-84-6 |
| DrugBank | DB15005 |
| ChemSpider | 71115970 |
| UNII | SXA7YP6EKX |
| KEGG | D11528 |
| ChEMBL | ChEMBL4298062 |
| Chemical and physical data | |
| Formula | C244H381N113O88P20 |
| Molar mass | 6924.910 g·mol−1 |
//////////Viltolarsen, Viltepso, 维托拉生 , FDA 2020, EU 2020, APPROVALS 2020, NCNP-01, NS-065, NS-065/NCNP-01, WHO 10771, WHO-10771, ビルトラルセン
Bulevirtide acetate

Bulevirtide acetate
(N-Myristoyl-glycyl-L-threonyl-L-asparaginyl-L-leucyl-L-seryl-L-valyl-Lprolyl-L-asparaginyl-L-prolyl-L-leucyl-glycyl-L-phenylalanyl-L-phenylalanyl-L-prolyl-L-aspartyl-L-histidyl-Lglutaminyl-L-leucyl-L-aspartyl-L-prolyl-L-alanyl-L-phenylalanyl-glycyl-L-alanyl-L-asparaginyl-L-seryl-Lasparaginyl-L-asparaginyl-Lprolyl-L-aspartyl-L-tryptophanyl-L-aspartyl-L-phenylalanyl-L-asparaginyl-L-prolylL-asparaginyl-L-lysyl-L-aspartyl-L-histidyl-L-tryptophanyl-L-prolyl-L-glutamyl-L-alanyl-L-asparaginyl-L-lysylL-valylglycinamide, acetate salt.
molecular formula C248H355N65O72,
molecular mass is 5398.9 g/mol
ブレビルチド酢酸塩;
APROVED 2020/7/31, EU, Hepcludex
MYR GmbH
|
Antiviral, Entry inhibitor
|
|
| Disease |
Hepatitis delta virus infection
|
|---|
Bulevirtide is a 47-amino acid peptide with a fatty acid, a myristoyl residue, at the N-terminus and an amidated C-terminus. The active substance is available as acetate salt. The counter ion acetate is bound in ionic form to basic groups of the peptide molecule and is present in a non-stoichiometric ratio. The chemical name of bulevirtide is (N-Myristoyl-glycyl-L-threonyl-L-asparaginyl-L-leucyl-L-seryl-L-valyl-Lprolyl-L-asparaginyl-L-prolyl-L-leucyl-glycyl-L-phenylalanyl-L-phenylalanyl-L-prolyl-L-aspartyl-L-histidyl-Lglutaminyl-L-leucyl-L-aspartyl-L-prolyl-L-alanyl-L-phenylalanyl-glycyl-L-alanyl-L-asparaginyl-L-seryl-Lasparaginyl-L-asparaginyl-Lprolyl-L-aspartyl-L-tryptophanyl-L-aspartyl-L-phenylalanyl-L-asparaginyl-L-prolylL-asparaginyl-L-lysyl-L-aspartyl-L-histidyl-L-tryptophanyl-L-prolyl-L-glutamyl-L-alanyl-L-asparaginyl-L-lysylL-valylglycinamide, acetate salt. It corresponds to the molecular formula C248H355N65O72, its relative molecular mass is 5398.9 g/mol
Bulevirtide appears as a white or off-white hygroscopic powder. It is practically insoluble in water and soluble at concentrations of 1 mg/ml in 50% acetic acid and about 7 mg/ml in carbonate buffer solution at pH 8.8, respectively. The structure of the active substance (AS) was elucidated by a combination of infrared spectroscopy (IR), mass spectrometry (MS), amino acid analysis and sequence analysis Other characteristics studied included ultraviolet (UV) spectrum, higher order structure (1D- and 2D- nuclear magnetic resonance spectroscopy (NMR)) and aggregation (Dynamic Light Scattering). Neither tertiary structure nor aggregation states of bulevirtide have been identified. With regard to enantiomeric purity, all amino acids are used in L-configuration except glycine, which is achiral by nature. Two batches of bulevirtide acetate were evaluated for enanatiomeric purity and no relevant change in configuration during synthesis was detected.
Bulevirtide is manufactured by a single manufacturer. It is a chemically synthesised linear peptide containing only naturally occurring amino acids. The manufacturing of this peptide is achieved using standard solidphase peptide synthesis (SPPS) on a 4-methylbenzhydrylamine resin (MBHA resin) derivatised with Rink amide linker in order to obtain a crude peptide mixture. This crude mixture is purified through a series of washing and preparative chromatography steps. Finally, the purified peptide is freeze-dried prior to final packaging and storage. The process involves further four main steps: synthesis of the protected peptide on the resin while side-chain functional groups are protected as applicable; cleavage of the peptide from the resin, together with the removal of the side chain protecting groups to obtain the crude peptide; purification; and lyophilisation. Two chromatographic systems are used for purification. No design space is claimed. Resin, Linker Fmoc protected amino acids and myristic acid are starting materials in line with ICH Q11. Sufficient information is provided on the source and the synthetic route of the starting materials. The active substance is obtained as a nonsterile, lyophilised powder. All critical steps and parameters were presented and clearly indicated in the description of the manufacturing process. The process description includes also sufficient information on the type of equipment for the SPPS, in-process controls (IPCs). The circumstances under which reprocessing might be performed were clearly presented. No holding times are proposed. Overall the process is sufficiently described.
The finished product is a white to off white lyophilised powder for solution for injection supplied in single-use vials. Each vial contains bulevirtide acetate equivalent to 2 mg bulevirtide. The composition of the finished product was presented. The powder is intended to be dissolved in 1 ml of water for injection per vial. After reconstitution the concentration of bulevirtide net peptide solution in the vial is 2 mg/ml. The components of the formulation were selected by literature review and knowledge of compositions of similar products available on the market at that time, containing HCl, water, mannitol, sodium carbonate, sodium hydrogen carbonate and sodium hydroxide. All excipients are normally used in the manufacture of lyophilisates. The quality of the excipients complies with their respective Ph. Eur monographs. The intrinsic properties of the active substance and the compounding formulation do not support microbiological growth as demonstrated by the stability data. No additional preservatives are therefore needed.
Hepcludex is an antiviral medicine used to treat chronic (long-term) hepatitis delta virus (HDV) infection in adults with compensated liver disease (when the liver is damaged but is still able to work), when the presence of viral RNA (genetic material) has been confirmed by blood tests.
HDV is an ‘incomplete’ virus, because it cannot replicate in cells without the help of another virus, the hepatitis B virus. Because of this, patients infected with the virus always also have hepatitis B.
HDV infection is rare, and Hepcludex was designated an ‘orphan medicine’ (a medicine used in rare diseases) on 19 June 2015. For further information on the orphan designation, see EU/3/15/1500.
Hepcludex contains the active substance bulevirtide.
Bulevirtide, sold under the brand name Hepcludex, is an antiviral medication for the treatment of chronic hepatitis D (in the presence of hepatitis B).[2]
The most common side effects include raised levels of bile salts in the blood and reactions at the site of injection.[2]
Bulevirtide works by attaching to and blocking a receptor (target) through which the hepatitis delta and hepatitis B viruses enter liver cells.[2] By blocking the entry of the virus into the cells, it limits the ability of HDV to replicate and its effects in the body, reducing symptoms of the disease.[2]
Bulevirtide was approved for medical use in the European Union in July 2020.[2]
Medical uses
Bulevirtide is indicated for the treatment of chronic hepatitis delta virus (HDV) infection in plasma (or serum) HDV-RNA positive adult patients with compensated liver disease.[2][3]
Pharmacology
Mechanism of action
Bulevirtide binds and inactivates the sodium/bile acid cotransporter, blocking both viruses from entering hepatocytes.[4]
The hepatitis B virus uses its surface lipopeptide pre-S1 for docking to mature liver cells via their sodium/bile acid cotransporter (NTCP) and subsequently entering the cells. Myrcludex B is a synthetic N-acylated pre-S1[5][6] that can also dock to NTCP, blocking the virus’s entry mechanism.[7]
The drug is also effective against hepatitis D because the hepatitis D virus is only infective in the presence of a hepatitis B virus infection.[7]
References
- ^ Deterding, K.; Wedemeyer, H. (2019). “Beyond Pegylated Interferon-Alpha: New Treatments for Hepatitis Delta”. Aids Reviews. 21 (3): 126–134. doi:10.24875/AIDSRev.19000080. PMID 31532397.
- ^ Jump up to:a b c d e f g “Hepcludex EPAR”. European Medicines Agency (EMA). 26 May 2020. Retrieved 12 August 2020. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “Summary of opinion: Hepcludex” (PDF). European Medicines Agency. 28 May 2020.
- ^ Francisco, Estela Miranda (29 May 2020). “Hepcludex”. European Medicines Agency. Retrieved 6 August 2020.
- ^ Volz T, Allweiss L, Ben MBarek M, Warlich M, Lohse AW, Pollok JM, et al. (May 2013). “The entry inhibitor Myrcludex-B efficiently blocks intrahepatic virus spreading in humanized mice previously infected with hepatitis B virus”. Journal of Hepatology. 58 (5): 861–7. doi:10.1016/j.jhep.2012.12.008. PMID 23246506.
- ^ Abbas Z, Abbas M (August 2015). “Management of hepatitis delta: Need for novel therapeutic options”. World Journal of Gastroenterology. 21 (32): 9461–5. doi:10.3748/wjg.v21.i32.9461. PMC 4548107. PMID 26327754.
- ^ Jump up to:a b Spreitzer H (14 September 2015). “Neue Wirkstoffe – Myrcludex B”. Österreichische Apothekerzeitung (in German) (19/2015): 12.
External links
- “Bulevirtide”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Hepcludex |
| Other names | MyrB, Myrcludex-B[1] |
| License data | |
| Routes of administration |
Subcutaneous injection |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| DrugBank | |
| UNII | |
| KEGG | |
| ChEMBL | |
/////////Bulevirtide acetate, ブレビルチド酢酸塩 , orphan designation, MYR GmbH, PEPTIDE, EU 2020, 2020 APPROVALS
Imlifidase
MDSFSANQEI RYSEVTPYHV TSVWTKGVTP PANFTQGEDV FHAPYVANQG WYDITKTFNG
KDDLLCGAAT AGNMLHWWFD QNKDQIKRYL EEHPEKQKIN FNGEQMFDVK EAIDTKNHQL
DSKLFEYFKE KAFPYLSTKH LGVFPDHVID MFINGYRLSL TNHGPTPVKE GSKDPRGGIF
DAVFTRGDQS KLLTSRHDFK EKNLKEISDL IKKELTEGKA LGLSHTYANV RINHVINLWG
ADFDSNGNLK AIYVTDSDSN ASIGMKKYFV GVNSAGKVAI SAKEIKEDNI GAQVLGLFTL
STGQDSWNQT N
Imlifidase
イムリフィダーゼ;
| Formula |
C1575H2400N422O477S6
|
|---|---|
| CAS |
1947415-68-0
|
| Mol weight |
35070.8397
|
EMA APPROVED, 2020/8/25, Idefirix
Pre-transplant treatment to make patients with donor specific IgG eligible for kidney transplantation
Immunosuppressant, Immunoglobulin modulator (enzyme)
Imlifidase is under investigation in clinical trial NCT02854059 (IdeS in Asymptomatic Asymptomatic Antibody-Mediated Thrombotic Thrombocytopenic Purpura (TTP) Patients).
Imlifidase, brand name Idefirix, is a medication for the desensitization of highly sensitized adults needing kidney transplantation, but unlikely to receive a compatible transplant.[1]
Imlifidase is a cysteine protease derived from the immunoglobulin G (IgG)‑degrading enzyme of Streptococcus pyogenes.[1] It cleaves the heavy chains of all human IgG subclasses (but no other immunoglobulins), eliminating Fc-dependent effector functions, including CDC and antibody-dependent cell-mediated cytotoxicity (ADCC).[1] Thus, imlifidase reduces the level of donor specific antibodies, enabling transplantation.[1]
The benefits with imlifidase are its ability to convert a positive crossmatch to a negative one in highly sensitized people to allow renal transplantation.[1] The most common side effects are infections and infusion related reactions.[1]
In June 2020, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) recommended the approval of Imlifidase.[1][2]
Medical uses
Per the CHMP recommendation, imlifidase will be indicated for desensitization treatment of highly sensitized adult kidney transplant people with positive crossmatch against an available deceased donor.[1] The use of imlifidase should be reserved for people unlikely to be transplanted under the available kidney allocation system including prioritization programmes for highly sensitized people.[1]
History
Imlifidase was granted orphan drug designations by the European Commission in January 2017, and November 2018,[3][4] and by the U.S. Food and Drug Administration (FDA) in both February and July 2018.[5][6]
In February 2019, Hansa Medical AB changed its name to Hansa Biopharma AB.[4]
| PHASE | STATUS | PURPOSE | CONDITIONS | COUNT |
|---|---|---|---|---|
| 2 | Recruiting | Treatment | Anti-Glomerular Basement Membrane Disease | 1 |
| 2 | Recruiting | Treatment | Guillain-Barré Syndrome (GBS) | 1 |
| 2 | Recruiting | Treatment | Kidney Transplant Rejection | 1 |
| 2 | Terminated | Treatment | Thrombotic Thrombocytopenic Purpura (TTP) | 1 |
| Not Available | Recruiting | Not Available | Kidney Transplant Failure and Rejection | 1 |
References
- ^ Jump up to:a b c d e f g h i “Imlifidase: Pending EC decision”. European Medicines Agency (EMA). 25 June 2020. Retrieved 26 June 2020. This article incorporates text from this source, which is in the public domain.
- ^ “New treatment to enable kidney transplant in highly sensitised patients”. European Medicines Agency (Press release). 26 June 2020. Retrieved 26 June 2020. This article incorporates text from this source, which is in the public domain.
- ^ “EU/3/16/1826”. European Medicines Agency (EMA). 12 January 2017. Retrieved 27 June 2020. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b “EU/3/18/2096”. European Medicines Agency (EMA). 13 February 2019. Retrieved 27 June 2020. This article incorporates text from this source, which is in the public domain.
- ^ “Imlifidase Orphan Drug Designation and Approval”. U.S. Food and Drug Administration (FDA). 3 July 2018. Retrieved 27 June 2020.
- ^ “Imlifidase Orphan Drug Designation and Approval”. U.S. Food and Drug Administration (FDA). 14 February 2018. Retrieved 27 June 2020.
Further reading
- Ge S, Chu M, Choi J, Louie S, Vo A, Jordan SC, et al. (October 2019). “Imlifidase Inhibits HLA Antibody-Mediated NK Cell Activation and Antibody-Dependent Cell-Mediated Cytotoxicity (ADCC) In Vitro”. Transplantation. doi:10.1097/TP.0000000000003023. PMID 31644495.
- Lin J, Boon L, Bockermann R, Robertson AK, Kjellman C, Anderson CC (March 2020). “Desensitization using imlifidase and EndoS enables chimerism induction in allosensitized recipient mice”. Am. J. Transplant. doi:10.1111/ajt.15851. PMID 32185855.
- Lonze BE, Tatapudi VS, Weldon EP, Min ES, Ali NM, Deterville CL, et al. (September 2018). “IdeS (Imlifidase): A Novel Agent That Cleaves Human IgG and Permits Successful Kidney Transplantation Across High-strength Donor-specific Antibody”. Ann. Surg. 268 (3): 488–496. doi:10.1097/SLA.0000000000002924. PMID 30004918.
- Lorant T, Bengtsson M, Eich T, Eriksson BM, Winstedt L, Järnum S, et al. (November 2018). “Safety, immunogenicity, pharmacokinetics, and efficacy of degradation of anti-HLA antibodies by IdeS (imlifidase) in chronic kidney disease patients”. Am. J. Transplant. 18 (11): 2752–2762. doi:10.1111/ajt.14733. PMC 6221156. PMID 29561066.
External links
- “Imlifidase”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Pronunciation | im lif’ i dase |
| Trade names | Idefirix |
| Other names | HMED-IdeS |
| Routes of administration |
Intravenous |
| ATC code | |
| Identifiers | |
| CAS Number | |
| DrugBank | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C1575H2400N422O477S6 |
| Molar mass | 35071.36 g·mol−1 |
//////////Imlifidase, Idefirix, PEPTIDE, イムリフィダーゼ , 2020 APPROVALS, EMA 2020, EU 2020
Filgotinib

Filgotinib
EU APPROVED 2020/9/24, JYSELECA
JAPAN APPROVED2020/9/25
- C21H23N5O3S
- MW425.504
- Elemental Analysis: C, 59.28; H, 5.45; N, 16.46; O, 11.28; S, 7.54
1206161-97-8
Cyclopropanecarboxamide, N-[5-[4-[(1,1-dioxido-4-thiomorpholinyl)methyl]phenyl][1,2,4]triazolo[1,5-a]pyridin-2-yl]-
G146034
GLPG0634
N-(5-(4-((1,1-dioxidothiomorpholino)methyl)phenyl)-[1,2,4]triazolo[1,5-a]pyridin-2-yl)cyclopropanecarboxamide
Galapagos Nv INNOVATOR
PHASE 3, Crohn’s disease, Rheumatoid arthritis, Ulcerative colitis
Filgotinib is an orally available inhibitor of JAK1/JAK2 and TYK2 in phase III clinical development at Galapagos and Gilead for the treatment of rheumatoid arthritis, moderate or severe Crohn’s disease and ulcerative colitis
IL-6 antagonist; Jak1 tyrosine kinase inhibitor; Tyk2 tyrosine kinase inhibitor; Jak3 tyrosine kinase inhibitor; Jak2 tyrosine kinase inhibitor
Autoimmune disease; Cancer; Colitis; Crohns disease; Inflammatory disease; Neoplasm; Rheumatoid arthritis; Transplant rejection
In 2017, orphan drug designation was assigned to the compound in the U.S. for the treatment of pediatric Crohn’s disease and pediatric ulcerative colitis.
GlaxoSmithKline had been developing filgotinib preclinically for the treatment of rheumatoid arthritis pursuant to a license; however, in 2010, the compound was re-acquired by Galapagos. In 2012, the product was licensed to Abbott for development and marketing. In January 2013, Abbott spun-off its research-based pharmaceutical business into a newly-formed company AbbVie. The license agreement between Galapagos and Abbott was terminated in September 2015, Galapagos regaining all rights to the product. The same year, Galapagos and Gilead entered into a global partnership and Gilead obtained the global rights of codevelopment and commercialization for the treatment of inflammatory diseases
Filgotinib (GLPG0634), by the Belgian biotech company Galápagos NV, is a drug which is currently under investigation for the treatment of rheumatoid arthritis and Crohn’s disease.
Filgotinib (GLPG0634) is an orally-available, selective inhibitor of JAK1 (Janus kinase 1) for the treatment of rheumatoid arthritis and potentially other inflammatory diseases. Filgotinib (GLPG0634) dose-dependently inhibited Th1 and Th2 differentiation and to a lesser extent the differentiation of Th17 cells in vitro. GLPG0634 was well exposed in rodents upon oral dosing, and exposure levels correlated with repression of Mx2 expression in leukocytes. The JAK1 selective inhibitor GLPG0634 (Filgotinib) is a promising novel therapeutic with potential for oral treatment of rheumatoid arthritis and possibly other immune-inflammatory diseases. Filgotinib (GLPG0634) is currently in a Phase 2 study in Crohn’s disease.

Mechanism of action
Filgotinib is a Janus kinase inhibitor with selectivity for subtype JAK1 of this enzyme. It is considered a promising agent as it inhibits JAK1 selectively. Less selective JAK inhibitors (e.g. tofacitinib) are already being marketed. They show long-term efficacy in the treatment of various inflammatory diseases. However, their lack of selectivity leads to dose-limiting side effects.[1] It is thought that inhibition of all JAK isoenzymes is beneficial in rheumatoid arthritis. However, pan-JAK inhibition might also lead to unwanted side effects that might not outweigh its benefits. This is the rationale for the development of newer and more selective inhibitors like filgotinib.
The signal transmission of large numbers of proinflammatory cytokines is dependent on JAK1. Inhibition of JAK2 may also contribute to the efficacy against RA. Nonetheless it is thought that JAK2 inhibition might lead to anemia and thrombopenia by interference witherythropoietin and thrombopoietin and granulocyte-macrophage colony-stimulating factor. Therefore one might prefer to choose a more selective JAK1 inhibitor as a primary therapeutic option. Filgotinib exerts a 30-fold selectivity for JAK1 compared to JAK2.[2] It is however still to be seen to what extent JAK2 inhibition should be avoided.
Novel crystalline forms of filgotinib salts, particularly hydrochloride salt, useful for treating JAK-mediated diseases eg inflammatory diseases, autoimmune diseases, proliferative diseases, allergy and transplant rejection. Galapagos and licensee AbbVie are developing filgotinib, a selective JAK-1 inhibitor, for treating rheumatoid arthritis (RA) and Crohn’s disease (CD). In August 2015, the drug was reported to be in phase 2 clinical development for treating RA and CD. The drug is also being investigated for the treatment of colitis and was discovered as part of the company’s arthritis alliance with GSK; however in August 2010 Galapagos reacquired the full rights. See WO2013189771, claiming use of filgotinib analog for treating inflammatory diseases. Also see WO2010010190 (co-assigned with GSK and Abbott) and WO2010149769 (assigned to Galapagos) claiming filgotinib, generically and specifically, respectively.
Clinical trials and approval
The efficacy of filgotinib is currently studied in a phase2b program (DARWIN trial 1, 2) with involvement of 886 rheumatoid arthritis patients and 180 Crohn’s disease patients.
Phase 1 study
It was shown in phase 1 studies that the pharmacokinetics of filgotinib metabolism is independent of hepatic CYP450 enzymatic degradation. The drug metabolism is however mediated by carboxylesterases. There is no interference reported with the metabolism of methotrexate nor with any of the investigated transport proteins.[3]
Phase 2 study: Proof of concept (2011)
In november 2011 Galápagos released the results of their phase 2 study (identification: NCT01384422, Eudract: 2010-022953-40) in which 36 patients were treated who showed a suboptimal clinical response to methotrexate treatment. Three groups of twelve patients were treated either with 200 mg filgotinib in a single dose, 200 mg divided in two doses or placebo. The primary end-point was the ACR20 score, which monitors improvements in the symptomatology of the patient. After the scheduled 4 weeks of treatment, 83% of the respondents showed an improved ACR20-score. Half of the treated patients showed a complete (or near complete) remission of the disease. There were no reports ofanemia nor changes in lipidemia. The company stated in their press release that filgotinib is the first selective JAK1 inhibitor that shows clinical efficacy. As a result of this study, the company stated that “GLPG0634 shows one of the highest initial response rates ever reported for rheumatoid arthritis treatments”.[4]
DARWIN 1 trial
The DARWIN 1 trial is a 24 week double blind placebo-controlled trial with 599 rheumatoid arthritis patients enrolled. All participants have moderate to severe RA and showed an insufficient response to standard methotrexate treatment. The trial compares three dosages of filgotinib as a once or twice per day regimen. During the trial all participants remain on their methotrexate treatment. According to the company, the results of this trial are expected in July 2015.[5]
DARWIN 2 trial
The DARWIN 2 trial is a double blind placebo-controlled trial with 280 rheumatoid arthritis patients enrolled who show an insufficient response to standard methotrexate treatment. This trial, in contrast to the previous DARWIN 1 trial, methotrexate is discontinued. Therefore, this trial investigates filgotinib as a monotherapy.[6] The recruitment of DARWIN trial 2b ended in november 2014.[7] Preliminary results are expected in the second quarter of 2015 and a full completion of the study is expected in the third quarter of 2015.
DARWIN 3 trial
Patients who complete DARWIN 1 and 2 will be eligible for DARWIN 3.
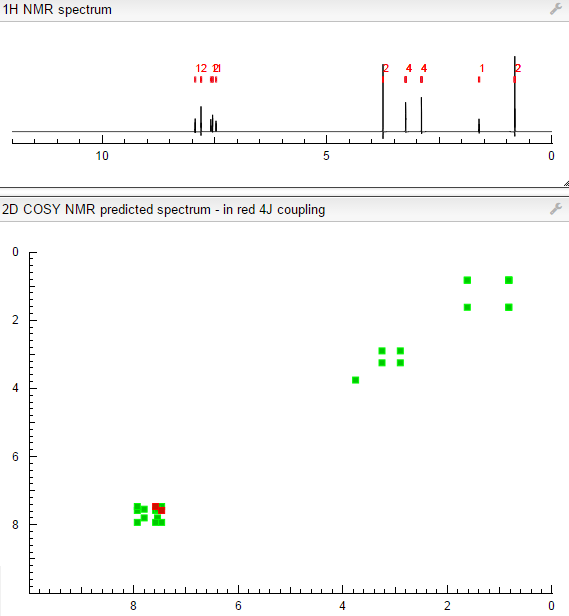
COSY PREDICT
Time line
- june 2011: results of first phase 2 trial
- november 2014: initiation of DARWIN 1 and 2 trials
- april 2015: expected date of DARWIN 1 trial results
- june 2015: expected date of DARWIN 2 trial results
NMR FROM NET….ABMOLE, DMSOD6
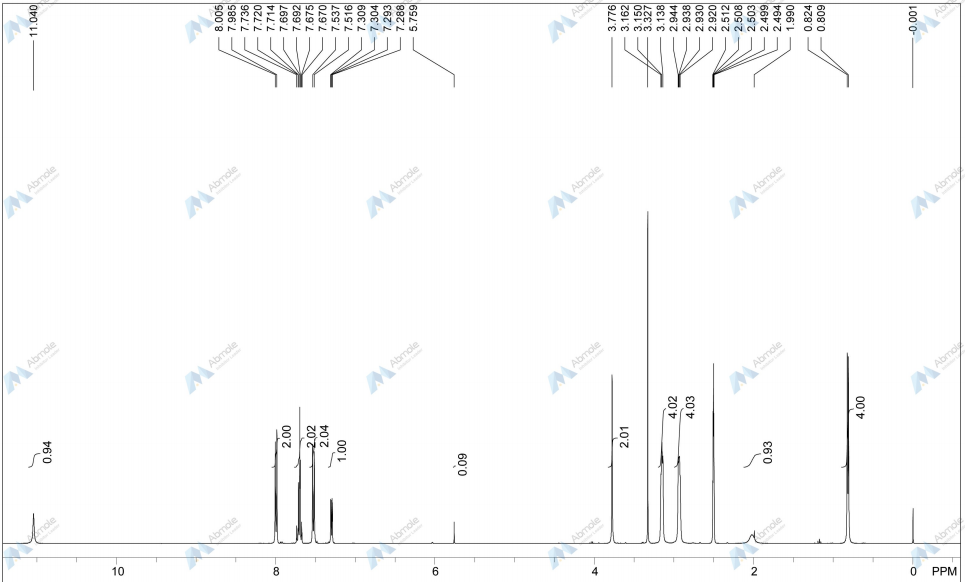
NMR MEDKOO DMSOD6

CHEMIETEK
1H NMR PREDICT


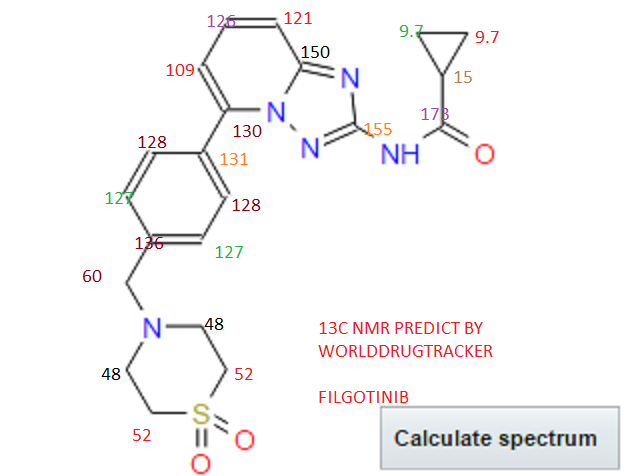
13C NMR PREDICT
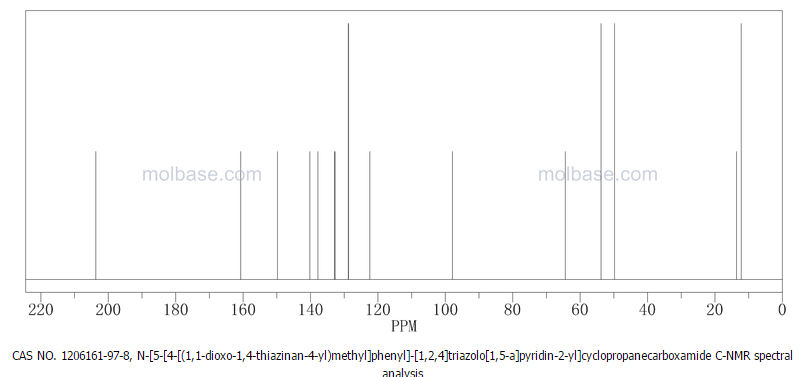

……………………
MORE PREDICTS
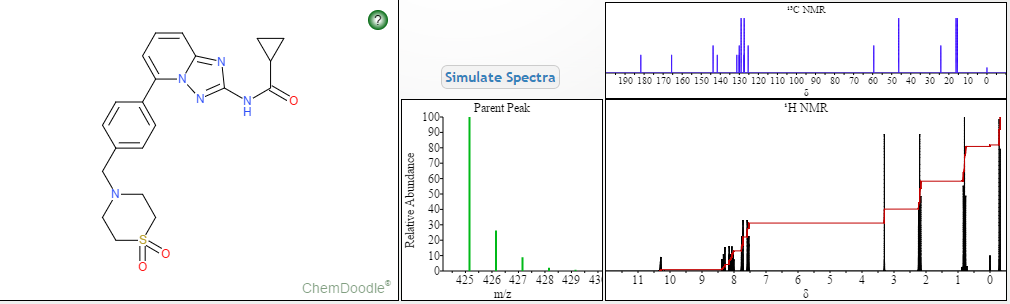
1H NMR PREDICT
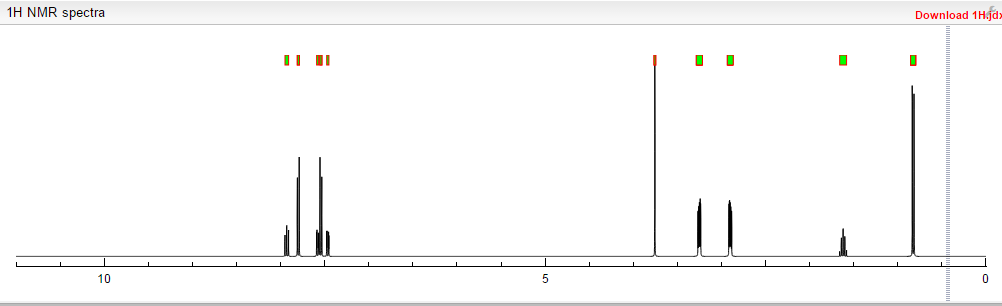
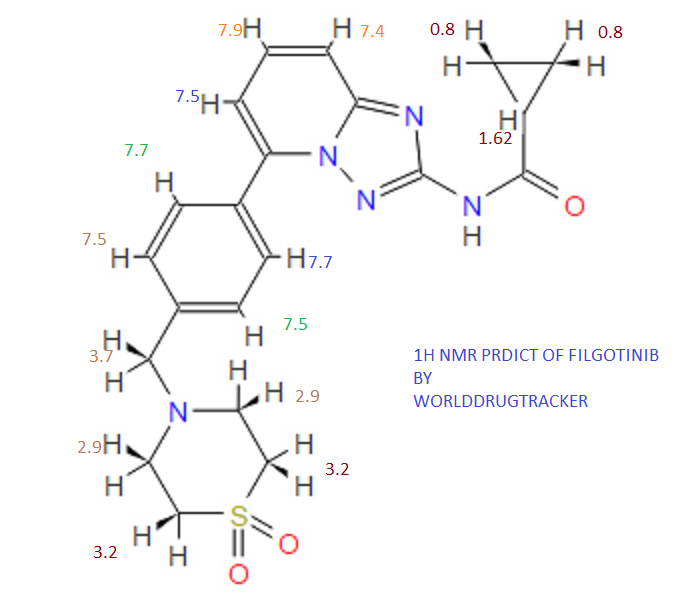
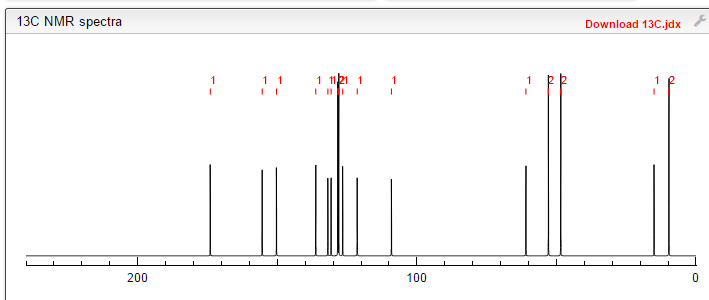
13C NMR PREDICT
PRODUCT PATENT
http://www.google.com/patents/WO2010149769A1?cl=en
| Applicants: | GALAPAGOS NV [BE/BE]; Generaal De Wittelaan L11/A3 B-2800 Mechelen (BE) (For All Designated States Except US). MENET, Christel Jeanne Marie [FR/BE]; (BE) (For US Only). SMITS, Koen Kurt [BE/BE]; (BE) (For US Only) |
| Inventors: | MENET, Christel Jeanne Marie; (BE). SMITS, Koen Kurt; (BE) |
| International Filing Date: | 25.06.2010 |
ESTIMATED EXP 2030
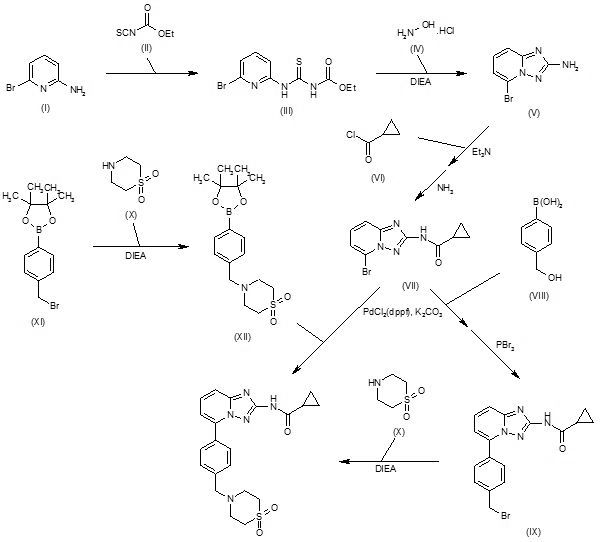
Condensation of 2-amino-6-bromopyridine (I) with ethoxycarbonyl isothiocyanate (II) in CH2Cl2 gives 1-(6-bromopyridin-2-yl)-3-carboethoxythiourea (III), which upon cyclization with hydroxylamine hydrochloride (IV) in the presence of DIEA in EtOH/MeOH yields 2-amino-5-bromo[1,2,4]triazolo[1,5-a]pyridine (V). N-Acylation of amine (V) with cyclopropanecarbonyl chloride (VI) using Et3N in acetonitrile, and subsequent treatment with methanolic ammonia furnishes the carboxamide (VII) (1-3), which upon Suzuki coupling with 4-(hydroxymethyl)phenylboronic acid (VIII) in the presence of PdCl2(dppf) and K2CO3 in dioxane/H2O at 90 °C, followed by bromination with PBr3 in CHCl3 affords intermediate (IX). Condensation of benzyl bromide derivative (IX) with thiomorpholine-1,1-dioxide (X) using DIEA in CH2Cl2/MeOH yields filgotinib (1,2). Alternatively, condensation of (4-bromomethylphenyl)-4,4,5,5-tetramethyl-[1,3,2]dioxaborolane (XI) with thiomorpholine 1,1-dioxide (X) in the presence of DIEA in CH2Cl2/MeOH gives intermediate (XII), which undergoes Suzuki coupling with aryl bromide (VII) in the presence of PdCl2(dppf) and K2CO3 in dioxane/H2O at 90 °C to afford the target filgotinib
The present invention is based on the discovery that the compound of the invention is able to act as an inhibitor of JAK and that it is useful for the treatment of inflammatory conditions, autoimmune diseases, proliferative diseases, transplantation rejection, diseases involving impairment of cartilage turnover, congenital cartilage malformations, and/or diseases associated with hypersecretion of IL6. In a specific aspect the compound is an inhibitor of JAKl and JAK2. The present invention also provides methods for the production of this compound, a pharmaceutical composition comprising this compound and methods for treating inflammatory conditions, autoimmune diseases, proliferative diseases, transplantation rejection, diseases involving impairment of cartilage turnover, congenital cartilage malformations, and/or diseases associated with hypersecretion of IL6 by administering the compound of the invention.
Accordingly, in a first aspect of the invention, a compound of the invention is provided having a formula (I):

[0017] The compound of the invention is a novel inhibitor of JAK that appears to exhibit a dramatically improved in vivo potency as compared to structurally similar compounds. In a particular embodiment the compound of the invention is an inhibitor of JAKl and JAK2. In particular it appears to exhibit this increase in potency at lower in vivo exposure levels compared to structurally similar compounds. The use of a compound with these improvements is expected to result in a lower dosage requirement (and therefore an improved dosing schedule).
General Synthetic Method Scheme 1

1. RCOCI, Et3N 2. NH3 / MeOH CH3CN, 20 0C 2O 0C

wherein Ar represents phenyl-Ll-heterocycloalkyl, where Ll is a bond, -CH2– or -CO- and the heterocycloalkyl group is optionally substituted.
General
1.1.1 l-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea (2)

(2)
[00117] To a solution of 2-amino-6-bromopyridine (1) (253.8 g, 1.467 mol) in DCM (2.5 L) cooled to 5 0C is added ethoxycarbonyl isothiocyanate (173.0 mL, 1.467 mol) dropwise over 15 min. The reaction mixture is then allowed to warm to room temp. (20 0C) and stirred for 16 h. Evaporation in vacuo gives a solid which may be collected by filtration, thoroughly washed with petrol (3×600 mL) and air-dried to afford (2). The thiourea may be used as such for the next step without any purification. 1H (400 MHz, CDCl3) δ 12.03 (IH, br s, NH), 8.81 (IH, d, J 7.8 Hz, H-3), 8.15 (IH, br s, NH), 7.60 (IH, t, J 8.0 Hz, H-4), 7.32 (IH, dd, J 7.7 and 0.6 Hz, H-5), 4.31 (2H, q, J 7.1 Hz, CH2), 1.35 (3H, t, J 7.1 Hz, CH3).
7.7.2 5-Bromo-[l, 2, 4]triazolo[l, 5-a]pyridin-2-ylamine (3)

[00118] To a suspension of hydroxylamine hydrochloride (101.8 g, 1.465 mol) in EtOH/MeOH
(1 :1, 900 mL) is added N,N-diisopropylethylamine (145.3 mL, 0.879 mol) and the mixture is stirred at room temp. (20 0C) for 1 h. l-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea (2) (89.0 g, 0.293 mol) is then added and the mixture slowly heated to reflux (Note: bleach scrubber is required to quench H2S evolved). After 3 h at reflux, the mixture is allowed to cool and filtered to collect the precipitated solid. Further product is collected by evaporation in vacuo of the filtrate, addition Of H2O (250 mL) and filtration. The combined solids are washed successively with H2O (250 mL), EtOH/MeOH (1 : 1, 250 mL) and Et2O (250 mL) then dried in vacuo to afford the triazolopyridine derivative (3) as a solid. The compound may be used as such for the next step without any purification. 1H (400 MHz, DMSO-t/β) δ 7.43-7.34 (2H, m, 2 x aromatic-H), 7.24 (IH, dd, J 6.8 and 1.8 Hz, aromatic-H), 6.30 (2H, br, NH2); m/z 213/215 (1 :1, M+H+, 100%).
7.7.3 General procedure for mono-acylation to afford intermediate (4):

[00119] To a solution of the 2-amino-triazolopyridine (3) (7.10 g, 33.3 mmol) in dry CH3CN
(150 mL) at 5 0C is added Et3N (11.6 mL, 83.3 mmol) followed by cyclopropanecarbonyl chloride (83.3 mmol). The reaction mixture is then allowed to warm to ambient temperature and stirred until all starting material (3) is consumed. If required, further Et3N (4.64 mL, 33.3 mmol) and cyclopropanecarbonyl chloride (33.3 mmol) is added to ensure complete reaction. Following solvent evaporation in vacuo the resultant residue is treated with 7 N methanolic ammonia solution (50 mL) and stirred at ambient temp, (for 1-16 h) to hydro lyse any bis-acylated product. Product isolation is made by removal of volatiles in vacuo followed by trituration with Et2O (50 mL). The solids are collected by filtration, washed with H2O (2x50mL), acetone (50 mL) and Et2O (50 mL), then dried in vacuo to give the required bromo intermediate (4).
Method A
Preparation of compounds of the invention via Suzuki coupling (5):
[00120] An appropriate boronic acid (2eq.) is added to a solution of bromo intermediate (4) in
1 ,4-dioxane/water (5:1). K2CO3 (2 eq.) and PdCl2dppf (5%) are added to the solution. The resulting mixture is then heated in a microwave at 140 0C for 30 min (this reaction can also be carried out by traditional heating in an oil bath at 900C for 16h under N2). Water is added and the solution is extracted with ethyl acetate. The organic layers are dried over anhyd. MgSθ4 and evaporated in vacuo. The final compound is obtained after purification by flash chromatography or preparative HPLC. HPLC: Waters
XBridge Prep Cl 8 5μm ODB 19mm ID x 100mm L (Part No.186002978). All the methods are using
MeCN/H2O gradients. H2O contains either 0.1% TFA or 0.1% NH3.
Method B

Bl. 4 4-[2-(Cyclopropanecarbonyl-amino)-[ 1 , 2, 4]triazolo[l, 5-a] pyridin-5-yl] -benzoyl chloride

[00121] 2 Drops of DMF are added to a solution of 4-[2-(cyclopropanecarbonyl-amino)- [l,2,4]triazolo[l,5-a]pyridin-5-yl]-benzoic acid (1 eq) obtained by Method A using 4-carboxyphenylboronic acid in DCM under N2 atmosphere. Then oxalyl chloride (2 eq) is added dropwise to this resulting solution (gas release). The mixture is stirred at room temperature for 2 hours. After completion of the reaction by LCMS, the solvent is removed. The crude acid chloride is used without further purification in next step.
B2. Amide formation (General Method)

[00122] An appropriate amine (1.1 eq) and Et3N (5 eq) are dissolved in DCM under N2 atmosphere and cooled at 00C. The acid chloride (Bl, 1 eq) dissolved in DCM is added dropwise to this solution. The reaction is stirred at room temperature for 16 h. After this time, reaction is complete. The compound is extracted with EtOAc and water, washed with brine and dried over anhyd. MgSO4. Organic layers are filtered and evaporated. The final compound is isolated by preparative HPLC. Preparative HPLC: Waters XBridge Prep C18 5μm ODB 19mm ID x 100mm L (Part No.186002978). All the methods are using MeCN/H2O gradients. H2O contains either 0.1% TFA or 0.1% NH3.
Method C

Wherein R3a or R3b together with the nitrogen atom to which they are attached, may form a heterocycloalkyl.
Reductive alkylation (general method)
[00123] An appropriate amine (2 eq.), cyclopropanecarboxylic acid (for example cyclopropanecarboxylic acid [5-(4-formyl-phenyl)-[l,2,4]triazolo[l,5-a]pyridine-2-yl]-amide) prepared by method A (1 eq.) and Ti(OPr)4 are mixed and stirred at room temperature for 3 hrs. The mixture is diluted in ethanol and Na(CN)BH3 (leq.) is added. The resulting solution is stirred at room temperature for 16 hrs. The mixture is diluted in water and filtered. The filtrate is washed with ethanol. The combined solvent phases are evaporated under vacuum. The final compound is isolated by preparative HPLC.
Method D 
wherein R1 and R2 together with the Nitrogen atom to which they are attached, may form a heterocycloalkyl.
Reaction ofalkylation

[00124] 2-(4-Bromomethyl-phenyl)-4,4,5,5-tetramethyl-[l,3,2]dioxaborolane (leq) and Et3N (2 eq) (or AgCO3) are dissolved in DCM/MeOH (4:1 v:v) under N2 and an amine (2 eq) is added dropwise. The resulting solution is stirred at room temperature for 16h. After this time, the reaction is complete. The solvent is evaporated. The compound is extracted with EtOAc and water, washed with brine and dried over anhyd. MgSθ4. Organic layers are filtered and evaporated. The final compound is isolated by flash chromatography.
Suzuki coupling

[00125] The obtained boronic acid (2eq.) is added to a solution of cyclopropanecarboxylic acid
(5-bromo-[l,2,4]triazolo[l,5-a]pyridin-2-yl)-amide (4) in 1 ,4-dioxane/water (5:1). K2CO3 (2 eq.) and PdCl2dppf (5%) are added to the solution. The resulting mixture is then heated in a microwave at 140 0C for 30 min (This reaction can also be carried out by traditional heating in an oil bath at 900C for 16h under N2). Water is added and the solution is extracted with ethyl acetate. The organic layers are dried over anhyd. MgSθ4 and evaporated in vacuo. The final compound is obtained after purification by flash chromatography or preparative HPLC. HPLC: Waters XBridge Prep C18 5μm ODB 19mm ID x 100mm L (Part No.186002978). All the methods are using MeCN/H2O gradients. H2O contains either 0.1% TFA or 0.1% NH3.
Synthesis of the compound of the invention and comparative examples
Compound l(the compound of the invention)
Step 1:

[00126] 2-(4-Bromomethyl-phenyl)-4,4,5,5-tetramethyl-[l,3,2]dioxaborolane (leq) and DIPEA
(2 eq) were dissolved in DCM/MeOH (5:1 v:v) under N2 and thiomorpholine 1,1 -dioxide (2 eq) was added portionwise. The resulting solution was stirred at room temperature for 16h. After this time, the reaction was complete. The solvent was evaporated. The compound was extracted with EtOAc and water, washed with brine and dried over anhyd. MgS O4. Organic layers were filtered and evaporated. The final compound was isolated without further purification.
Step 2: Suzuki coupling

[00127] 4-[4-(4,4,5,5-Tetramethyl-[l,3,2]dioxaborolan-2-yl)-benzyl]-thiomorpholine-l,l-dioxide
(l.leq.) was added to a solution of cyclopropanecarboxylic acid (5-bromo-[l,2,4]triazolo[l,5-a]pyridin-2-yl)-amide in 1 ,4-dioxane/water (4:1). K2CO3 (2 eq.) and PdCl2dppf (0.03 eq.) were added to the solution. The resulting mixture was then heated in an oil bath at 900C for 16h under N2. Water was added and the solution was extracted with ethyl acetate. The organic layers were dried over anhyd. MgSθ4 and evaporated in vacuo. The final compound was obtained after purification by flash chromatography.
[00128] Alternatively, after completion of the reaction, a palladium scavenger such as 1,2-bis(diphenylphosphino)ethane, is added, the reaction mixture is allowed to cooled down and a filtration is performed. The filter cake is reslurried in a suitable solvent (e.g. acetone), the solid is separated by filtration, washed with more acetone, and dried. The resulting solid is resuspended in water, aqueous HCl is added, and after stirring at RT, the resulting solution is filtered on celite (Celpure P300). Aqueous NaOH is then added to the filtrate, and the resulting suspension is stirred at RT, the solid is separated by filtration, washed with water and dried by suction. Finally the cake is re-solubilised in a mixture of THF/H2O, treated with a palladium scavenger (e.g. SMOPEX 234) at 500C, the suspension is filtered, the organic solvents are removed by evaporation, and the resulting slurry is washed with water and methanol, dried and sieved, to obtain the title compound as a free base.
Alternative route to Compound l(the compound of the invention):
Step 1:

[00129] 4-(Hydroxymethyl)phenylboronic acid (l.leq.) was added to a s o luti o n o f cyclopropanecarboxylic acid (5-bromo-[l,2,4]triazolo[l,5-a]pyridin-2-yl)-amide in 1 ,4-dioxane/water (4:1). K2CO3 (2 eq.) and PdCl2dppf (0.03 eq.) were added to the solution. The resulting mixture was then heated in an oil bath at 900C for 16h under N2. Water was added and the solution was extracted with ethyl acetate. The organic layers were dried over anhyd. MgSθ4 and evaporated in vacuo. The resulting mixture was used without further purification.
Step 2:

[00130] To a solution of cyclopropanecarboxylic acid [5-(4-hydroxymethyl-phenyl)- [l,2,4]triazolo[l,5-a]pyridin-2-yl]-amide (1.0 eq) in chloroform was slowly added phosphorus tribromide (1.0 equiv.). The reaction mixture was stirred at room temperature for 20 hours, quenched with ice and water (20 mL) and extracted with dichloromethane. The organic layer was dried over anhyd. MgSθ4, filtered and concentrated to dryness. The resulting white residue was triturated in dichloromethane/diethyl ether 2:1 to afford the expected product as a white solid.
Step 3:

[00131] Cyclopropanecarboxylic acid [5-(4-bromomethyl-phenyl)-[l,2,4]triazolo[l,5-a]pyridin- 2-yl]-amide (leq) and DIPEA (2 eq) were dissolved in DCM/MeOH (5:1 v:v) under N2 and thiomorpholine 1,1 -dioxide (1.1 eq) was added dropwise. The resulting solution was stirred at room temperature for 16h. After this time, the reaction was complete. The solvent was evaporated. The compound was dissolved in DCM, washed with water and dried over anhyd. MgSO^ Organic layers were filtered and evaporated. The final compound was isolated by column chromatography using EtOAc to afford the desired product.
PATENT
WO 2010010190
WO 2013173506
WO 2013189771
WO 2015117980
WO 2015117981
POLYMORPH
CN 105061420
https://encrypted.google.com/patents/CN105061420A?cl=en
JAK inhibitor N-(5-(4-(1,1-dioxothiomorpholinyl)methyl)phenyl)-[1,2,4]triazolo[1,5-a]pyridin-2-yl)cyclopropanecarboxamide, and methods for preparing the four crystal forms, wherein the four crystal forms respectively are a crystal form H1, a crystal form H2, a crystal form H3 and a crystal form H4,
POLYMORPH
E CRYSTAL
CN 105111206
D CRYSTAL
CN 105111207
H CRYSYAL
CN 105198876
G
CN 105198877
F CN 105198878
C CN 105198880
POLYMORPH
WO 2016105453
POLYMORPH
Preparation of solid forms of filgotinib free base
WO 2017012773
The present invention relates to cryst. filgotinib free base (I), a method of its prepn. and a pharmaceutical compn. comprising the same. Thus, reacting the compd. II with thiomorpholine dioxide afforded 92% I (purity of 98.6%) which was then converted into its hydrochloride salt. Pharmaceutical compn. comprising compd. I was disclosed.
POLYMORPH
CN 105669669
The present invention provides a crystal form A, B, D, G and M of N-[5-[4-[(1,1-dioxido-4-thiomorpholinyl)methyl]phenyl][1,2,4]triazolo[1,5-a]pyridin-2-yl]cyclopropanecarboxamide hydrochloride.
PAPER
Future Medicinal Chemistry (2015), 7(2), 203-235. | Language: English, Database: CAPLUSA review. The discovery of the JAK-STAT pathway was a landmark in cell biol. The identification of these pathways has changed the landscape of treatment of rheumatoid arthritis and other autoimmune diseases. The two first (unselective) JAK inhibitors have recently been approved by the US FDA for the treatment of myelofibrosis and rheumatoid arthritis and many other JAK inhibitors are currently in clin. development or at the discovery stage. Research groups have demonstrated the different roles of JAK member and the therapeutic potential of targeting them selectively. ………..
https://www.future-science.com/doi/10.4155/fmc.14.149
PAPER
Journal of Pharmaceutical Sciences (Philadelphia, PA, United States) (2018), 107(6), 1624-1632.
PATENT
US2010/331319 A1, ; Page/Page column 13-14
http://www.google.com/patents/US20100331319
Synthetic Preparation of the Compound of the Invention and Comparative Examples
The compound of the invention and the comparative examples can be produced according to the following scheme.

wherein Ar represents phenyl-L1-heterocycloalkyl, where L1 is a bond, —CH2— or —CO— and the heterocycloalkyl group is optionally substituted.
General 1.1.1 1-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea (2)

To a solution of 2-amino-6-bromopyridine (1) (253.8 g, 1.467 mol) in DCM (2.5 L) cooled to 5° C. is added ethoxycarbonyl isothiocyanate (173.0 mL, 1.467 mol) dropwise over 15 min. The reaction mixture is then allowed to warm to room temp. (20° C.) and stirred for 16 h. Evaporation in vacuo gives a solid which may be collected by filtration, thoroughly washed with petrol (3×600 mL) and air-dried to afford (2). The thiourea may be used as such for the next step without any purification. 1H (400 MHz, CDCl3) δ 12.03 (1H, br s, NH), 8.81 (1H, d, J=7.8 Hz, H-3), 8.15 (1H, br s, NH), 7.60 (1H, t, J=8.0 Hz, H-4), 7.32 (1H, dd, J 7.7 and 0.6 Hz, H-5), 4.31 (2H, q, J 7.1 Hz, CH2), 1.35 (3H, t, J 7.1 Hz, CH3).
1.1.2 5-Bromo-[1,2,4]triazolo[1,5-a]pyridin-2-ylamine (3)

To a suspension of hydroxylamine hydrochloride (101.8 g, 1.465 mol) in EtOH/MeOH (1:1, 900 mL) is added N,N-diisopropylethylamine (145.3 mL, 0.879 mol) and the mixture is stirred at room temp. (20° C.) for 1 h. 1-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea (2) (89.0 g, 0.293 mol) is then added and the mixture slowly heated to reflux (Note: bleach scrubber is required to quench H2S evolved). After 3 h at reflux, the mixture is allowed to cool and filtered to collect the precipitated solid. Further product is collected by evaporation in vacuo of the filtrate, addition of H2O (250 mL) and filtration. The combined solids are washed successively with H2O (250 mL), EtOH/MeOH (1:1, 250 mL) and Et2O (250 mL) then dried in vacuo to afford the triazolopyridine derivative (3) as a solid. The compound may be used as such for the next step without any purification. 1H (400 MHz, DMSO-d6) δ 7.43-7.34 (2H, m, 2×aromatic-H), 7.24 (1H, dd, J 6.8 and 1.8 Hz, aromatic-H), 6.30 (2H, br, NH2); m/z 213/215 (1:1, M+H+, 100%).
1.1.3 General Procedure for Mono-Acylation to Afford Intermediate (4)

To a solution of the 2-amino-triazolopyridine (3) (7.10 g, 33.3 mmol) in dry CH3CN (150 mL) at 5° C. is added Et3N (11.6 mL, 83.3 mmol) followed by cyclopropanecarbonyl chloride (83.3 mmol). The reaction mixture is then allowed to warm to ambient temperature and stirred until all starting material (3) is consumed. If required, further Et3N (4.64 mL, 33.3 mmol) and cyclopropanecarbonyl chloride (33.3 mmol) is added to ensure complete reaction. Following solvent evaporation in vacuo the resultant residue is treated with 7 N methanolic ammonia solution (50 mL) and stirred at ambient temp. (for 1-16 h) to hydrolyse any bis-acylated product. Product isolation is made by removal of volatiles in vacuo followed by trituration with Et2O (50 mL). The solids are collected by filtration, washed with H2O (2×50 mL), acetone (50 mL) and Et2O (50 mL), then dried in vacuo to give the required bromo intermediate (4).
Method A Preparation of Compounds of the Invention Via Suzuki Coupling (5):
An appropriate boronic acid (2 eq.) is added to a solution of bromo intermediate (4) in 1,4-dioxane/water (5:1). K2CO3 (2 eq.) and PdCl2dppf (5%) are added to the solution. The resulting mixture is then heated in a microwave at 140° C. for 30 min (this reaction can also be carried out by traditional heating in an oil bath at 90° C. for 16 h under N2). Water is added and the solution is extracted with ethyl acetate. The organic layers are dried over anhyd. MgSO4 and evaporated in vacuo. The final compound is obtained after purification by flash chromatography or preparative HPLC. HPLC: Waters XBridge Prep C18 5 μm ODB 19 mm ID×100 mm L (Part No. 186002978). All the methods are using MeCN/H2O gradients. H2O contains either 0.1% TFA or 0.1% NH3.
Method B

B1. 4 4-[2-(Cyclopropanecarbonyl-amino)-[1,2,4]triazolo[1,5-a]pyridin-5-yl]-benzoyl chloride

2 Drops of DMF are added to a solution of 4-[2-(cyclopropanecarbonyl-amino)-[1,2,4]triazolo[1,5-a]pyridin-5-yl]-benzoic acid (1 eq) obtained by Method A using 4-carboxyphenylboronic acid in DCM under N2 atmosphere. Then oxalyl chloride (2 eq) is added dropwise to this resulting solution (gas release). The mixture is stirred at room temperature for 2 hours. After completion of the reaction by LCMS, the solvent is removed. The crude acid chloride is used without further purification in next step.
B2. Amide Formation (General Method)

An appropriate amine (1.1 eq) and Et3N (5 eq) are dissolved in DCM under N2 atmosphere and cooled at 0° C. The acid chloride (B1, 1 eq) dissolved in DCM is added dropwise to this solution. The reaction is stirred at room temperature for 16 h. After this time, reaction is complete. The compound is extracted with EtOAc and water, washed with brine and dried over anhyd. MgSO4. Organic layers are filtered and evaporated. The final compound is isolated by preparative HPLC. Preparative HPLC: Waters XBridge Prep C18 5 μm ODB 19 mm ID×100 mm L (Part No. 186002978). All the methods are using MeCN/H2O gradients. H2O contains either 0.1% TFA or 0.1% NH3.
…
Synthesis of the Compound of the Invention and Comparative Examples Compound 1 (the Compound of the Invention) Step 1:

2-(4-Bromomethyl-phenyl)-4,4,5,5-tetramethyl-[1,3,2]dioxaborolane (1 eq) and DIPEA (2 eq) were dissolved in DCM/MeOH (5:1 v:v) under N2 and thiomorpholine 1,1-dioxide (2 eq) was added portionwise. The resulting solution was stirred at room temperature for 16 h. After this time, the reaction was complete. The solvent was evaporated. The compound was extracted with EtOAc and water, washed with brine and dried over anhyd. MgSO4. Organic layers were filtered and evaporated. The final compound was isolated without further purification.
STEP 2: Suzuki coupling

4-[4-(4,4,5,5-Tetramethyl-[1,3,2]dioxaborolan-2-yl)-benzyl]-thiomorpholine-1,1-dioxide (1.1 eq.) was added to a solution of cyclopropanecarboxylic acid (5-bromo-[1,2,4]triazolo[1,5-a]pyridin-2-yl)-amide in 1,4-dioxane/water (4:1). K2CO3 (2 eq.) and PdCl2dppf (0.03 eq.) were added to the solution. The resulting mixture was then heated in an oil bath at 90° C. for 16 h under N2. Water was added and the solution was extracted with ethyl acetate. The organic layers were dried over anhyd. MgSO4 and evaporated in vacuo. The final compound was obtained after purification by flash chromatography.
Alternatively, after completion of the reaction, a palladium scavenger such as 1,2-bis(diphenylphosphino)ethane, is added, the reaction mixture is allowed to cooled down and a filtration is performed. The filter cake is reslurried in a suitable solvent (e.g. acetone), the solid is separated by filtration, washed with more acetone, and dried. The resulting solid is resuspended in water, aqueous HCl is added, and after stirring at RT, the resulting solution is filtered on celite (Celpure P300). Aqueous NaOH is then added to the filtrate, and the resulting suspension is stirred at RT, the solid is separated by filtration, washed with water and dried by suction. Finally the cake is re-solubilised in a mixture of THF/H2O, treated with a palladium scavenger (e.g. SMOPEX 234) at 50° C., the suspension is filtered, the organic solvents are removed by evaporation, and the resulting slurry is washed with water and methanol, dried and sieved, to obtain the title compound as a free base.
Alternative Route to Compound 1 (the Compound of the Invention): Step 1:

4-(Hydroxymethyl)phenylboronic acid (1.1 eq.) was added to a solution of cyclopropanecarboxylic acid (5-bromo-[1,2,4]triazolo[1,5-a]pyridin-2-yl)-amide in 1,4-dioxane/water (4:1). K2CO3 (2 eq.) and PdCl2dppf (0.03 eq.) were added to the solution. The resulting mixture was then heated in an oil bath at 90° C. for 16 h under N2. Water was added and the solution was extracted with ethyl acetate. The organic layers were dried over anhyd. MgSO4 and evaporated in vacuo. The resulting mixture was used without further purification.
Step 2:

To a solution of cyclopropanecarboxylic acid [5-(4-hydroxymethyl-phenyl)-[1,2,4]triazolo[1,5-a]pyridin-2-yl]-amide (1.0 eq) in chloroform was slowly added phosphorus tribromide (1.0 equiv.). The reaction mixture was stirred at room temperature for 20 hours, quenched with ice and water (20 mL) and extracted with dichloromethane. The organic layer was dried over anhyd. MgSO4, filtered and concentrated to dryness. The resulting white residue was triturated in dichloromethane/diethyl ether 2:1 to afford the expected product as a white solid.
Step 3:

Cyclopropanecarboxylic acid [5-(4-bromomethyl-phenyl)-[1,2,4]triazolo[1,5-a]pyridin-2-yl]-amide (1 eq) and DIPEA (2 eq) were dissolved in DCM/MeOH (5:1 v:v) under N2 and thiomorpholine 1,1-dioxide (1.1 eq) was added dropwise. The resulting solution was stirred at room temperature for 16 h. After this time, the reaction was complete. The solvent was evaporated. The compound was dissolved in DCM, washed with water and dried over anhyd. MgSO4. Organic layers were filtered and evaporated. The final compound was isolated by column chromatography using EtOAc to afford the desired product.
…………………….
PATENT
Novel salts and pharmaceutical compositions thereof for the treatment of inflammatory disorders
Also claims a method for preparing filgotinib hydrochloride trihydrate. The present filing forms a pair with this week’s filing, WO2015117980, claiming a tablet composition comprising filgotinib hydrochloride.
The compound cyclopropanecarboxylic acid {5-[4-(l,l-dioxo-thiomorpholin-4-ylmethyl)-phenyl]-[l,2,4]triazolo[l,5-a]pyridin-2-yl -amide (Compound 1), which has the chemical structure:

is disclosed in our earlier application WO 2010/149769 (Menet C. J., 2010) as being an inhibitor of JAK and as being useful in the treatment of inflammatory conditions, autoimmune diseases, proliferative diseases, allergy, transplant rejection, diseases involving impairment of cartilage turnover, congenital cartilage malformations, and/or diseases associated with hypersecretion of IL6 or interferons. Hereafter this compound is named Compound 1. The data presented in WO 2010/149769 demonstrate that despite similar in vitro activities, Compound 1 has unexpectedly high in vivo potency compared with structurally similar compounds.
Example 1. Preparation of Compound 1
1.1. Route 1
1.1.1. 4-[4-(4,4,5,5-Tetramethyl-[l,3,2]dioxaborolan-2-yl)-benzyl]-thiomorpholine-l,l-dioxide

[00205] 2-(4-Bromomethyl-phenyl)-4,4,5,5-tetramethyl-[l,3,2]dioxaborolane (1 eq) and DIPEA (2 eq) are dissolved in DCM/MeOH (5:1 v:v) under N2 and thiomorpholine 1,1 -dioxide (2 eq) is added portionwise. The resulting solution is stirred at room temperature for 16h. After this time, the reaction is complete. The solvent is evaporated. The compound is extracted with EtOAc and water, washed with brine and dried over anhydrous MgSO i. Organic layers are filtered and evaporated. The final compound is isolated without further purification.
1.1.2. Cyclopropanecarboxylic acid (5-bromo-[l,2,4]triazolo[l,5-a]pyridin-2-yl)-amide

1.1.2.1. Step i): l-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea
[00206] To a solution of 2-amino-6-bromopyridine (1) (253.8 g, 1.467 mol) in DCM (2.5 L) cooled to 5°C is added ethoxycarbonyl isothiocyanate (173.0 mL, 1.467 mol) dropwise over 15 min. The reaction
mixture is then allowed to warm to room temp. (20 °C) and stirred for 16 h. Evaporation in vacuo gives a solid which may be collected by filtration, thoroughly washed with petrol (3 x 600 niL) and air-dried to afford the desired product. The thiourea may be used as such for the next step without any purification. lH (400 MHz, CDC13) δ 12.03 (1H, br s), 8.81 (1H, d), 8.15 (1H, br s), 7.60 (1H, t), 7.32 (1H, dd), 4.31 (2H, q), 1.35 (3H, t).
1.1.2.2. Step ii): 5-Bromo-[l,2,4]triazolo[l,5-a]pyridin-2-ylamine
[00207] To a suspension of hydroxylamine hydrochloride (101.8 g, 1.465 mol) in EtOH/MeOH (1 : 1, 900 mL) is added NN-diisopropylethylamine (145.3 mL, 0.879 mol) and the mixture is stirred at room temp. (20 °C) for 1 h. l-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea (2) (89.0 g, 0.293 mol) is then added and the mixture slowly heated to reflux (Note: bleach scrubber is required to quench H2S evolved). After 3h at reflux, the mixture is allowed to cool and filtered to collect the precipitated solid. Further product is collected by evaporation in vacuo of the filtrate, addition of H20 (250 mL) and filtration. The combined solids are washed successively with H20 (250 mL), EtOH/MeOH (1 : 1, 250 mL) and Et20 (250 mL) then dried in vacuo to afford the triazolopyridine derivative (3) as a solid. The compound may be used as such for the next step without any purification. lH (400 MHz, DMSO-i¼) δ 7.43-7.34 (2H, m, 2 x aromatic-H), 7.24 (1H, dd, J 6.8 and 1.8 Hz, aromatic-H), 6.30 (2H, br, NH2); m/z 213/215 (1 : 1, M+H+, 100%).
1.1.2.3. Step Hi): Cyclopropanecarboxylic acid (5-bromo-[l ,2,4]triazolo[l ,5-a]pyridin-2-yl)-amide
[00208] To a solution of the 2-amino-triazolopyridine obtained in the previous step (7.10 g, 33.3 mmol) in dry MeCN (150 mL) at 5°C is added Et3N (11.6 mL, 83.3 mmol) followed by cyclopropanecarbonyl chloride (83.3 mmol). The reaction mixture is then allowed to warm to ambient temperature and stirred until all starting material is consumed. If required, further Et3N (4.64 mL, 33.3 mmol) and cyclopropanecarbonyl chloride (33.3 mmol) is added to ensure complete reaction. Following solvent evaporation in vacuo the resultant residue is treated with 7 N methanolic ammonia solution (50 mL) and stirred at ambient temp, (for 1-16 h) to hydro lyse any bis-acylated product. Product isolation is made by removal of volatiles in vacuo followed by trituration with Et20 (50 mL). The solids are collected by filtration, washed with H20 (2x50mL), acetone (50 mL) and Et20 (50 mL), then dried in vacuo to give the desired compound.
1.1.3. Compound 1

[00209] 4-[4-(4,4,5,5-Tetramethyl-[l ,3,2]dioxaborolan-2-yl)-benzyl] hiomoφholine , l -dioxide (l . l eq.) is added to a solution of cyclopropanecarboxylic acid (5-bromo-[l ,2,4]triazolo[l ,5-a]pyridin-2-yl)-amide in 1 ,4-dioxane/water (4: 1). K2CO3 (2 eq.) and PdC^dppf (0.03 eq.) are added to the solution. The resulting mixture is then heated in an oil bath at 90°C for 16h under N2. Water is added and the solution is extracted with ethyl acetate. The organic layers are dried over anhydrous MgS04 and evaporated in vacuo.
[00210] The final compound is obtained after purification by flash chromatography.
[00211] Alternatively, after completion of the reaction, a palladium scavenger such as 1 ,2-bis(diphenylphosphino)ethane, is added, the reaction mixture is allowed to cool down and a filtration is performed. The filter cake is reslurried in a suitable solvent (e.g. acetone), the solid is separated by filtration, washed with more acetone, and dried. The resulting solid is resuspended in water, aqueous HC1 is added, and after stirring at room temperature, the resulting solution is filtered on celite (Celpure P300). Aqueous NaOH is then added to the filtrate, and the resulting suspension is stirred at room temperature, the solid is separated by filtration, washed with water and dried by suction. Finally the cake is re-solubilised in a mixture of THF/H20, treated with a palladium scavenger (e.g. SMOPEX 234) at 50°C, the suspension is filtered, the organic solvents are removed by evaporation, and the resulting slurry is washed with water and methanol, dried and sieved, to obtain the desired compound as a free base.
1.2. Route 2
1.2.1. Step 1: cyclopropanecarboxylic acid [5-(4-hydroxymethyl-phenyl)-[l,2, 4]triazolo[l, 5- a] pyridin-2-yl] -amide

[00212] 4-(Hydroxymethyl)phenylboronic acid (l . l eq.) is added to a solution of cyclopropanecarboxylic acid (5-bromo-[l ,2,4]triazolo[l ,5-a]pyridin-2-yl)-amide in 1 ,4-dioxane/water
(4:1). K2CO3 (2 eq.) and PdC^dppf (0.03 eq.) are added to the solution. The resulting mixture is then heated in an oil bath at 90°C for 16h under N2. Water is added and the solution is extracted with ethyl acetate. The organic layers are dried over anhydrous MgS04 and evaporated in vacuo. The resulting mixture is used without further purification.
1.2.2. Step 2: Cyclopropanecarboxylic acid [5-(4-bromomethyl-phenyl)-[l,2,4]triazolo[l,5- a Jpyridin-2-ylJ -amide

[00213] To a solution of cyclopropanecarboxylic acid [5-(4-hydroxymethyl-phenyl)-[l,2,4]triazolo[l,5-a]pyridin-2-yl] -amide (1.0 eq) in chloroform is slowly added phosphorus tribromide (1.0 eq.). The reaction mixture is stirred at room temperature for 20 h, quenched with ice and water (20 mL) and extracted with dichloromethane. The organic layer is dried over anhydrous MgSO i, filtered and concentrated to dryness. The resulting white residue is triturated in dichloromethane/diethyl ether 2:1 to afford the desired product.
1.2.3. Step 3:

[00214] Cyclopropanecarboxylic acid [5-(4-bromomethyl-phenyl)-[l,2,4]triazolo[l,5-a]pyridin-2-yl]-amide (l eq) and DIPEA (2 eq) are dissolved in DCM/MeOH (5: 1 v:v) under N2 and thiomorpho line 1,1-dioxide (1.1 eq) is added dropwise. The resulting solution is stirred at room temperature for 16h. After this time, the reaction is complete. The solvent is evaporated. The compound is dissolved in DCM, washed with water and dried over anhydrous MgSO i. Organic layers are filtered and evaporated. The final compound is isolated by column chromatography using EtOAc to afford the desired product.
…………………
PATENT
http://www.google.co.in/patents/WO2013189771A1?cl=en
Example 1. Synthesis of the compounds
1.1. Route 1
1.1.1. Synthesis of 5-Bromo-[l,2,4]triazolo[l,5-a]pyridin-2-ylamine (Intermediate 3)

led to 5 °C was added ethoxycarbonyl isothiocyanate (173.0 mL, 1.467 mol) dropwise over 15 min. The reaction mixture was then allowed to warm to room temp. (20 °C) and stirred for 16 h. Evaporation in vacuo gave a solid which was collected by filtration, thoroughly washed with petrol (3×600 mL) and air-dried to afford (2). The thiourea was used as such in the next step without any purification.
[00157] lH (400 MHz, CDC13) δ 12.03 (IH, br s, NH), 8.81 (IH, d, J 7.8 Hz, H-3), 8.15 (IH, br s, NH), 7.60 (IH, t, J 8.0 Hz, H-4), 7.32 (IH, dd, J 7.7 and 0.6 Hz, H-5), 4.31 (2H, q, J 7.1 Hz, CH2), 1.35 (3H, t, J 7.1 Hz, CH3).
1.1.1.2. 5-Bromo-f 1,2, 4]triazolo[ 1 ,5-a] pyridin-2-ylamine (3)
[00158] To a suspension of hydroxylamine hydrochloride (101.8 g, 1.465 mol) in EtOH/MeOH (1 : 1, 900 mL) was added NN-diisopropylethylamine (145.3 mL, 0.879 mol) and the mixture was stirred at room temp. (20 °C) for 1 h. l-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea (2) (89.0 g, 0.293 mol) was then added and the mixture slowly heated to reflux (Note: bleach scrubber was required to quench H2S evolved). After 3 h at reflux, the mixture was allowed to cool and filtered to collect the precipitated solid. Further product was collected by evaporation in vacuo of the filtrate, addition of H20 (250 mL) and filtration. The combined solids were washed successively with H20 (250 mL), EtOH/MeOH (1 : 1, 250 mL) and Et20 (250 mL) then dried in vacuo to afford the triazolopyridine derivative (3) as a solid. The compound was used as such in the next step without any purification.
[00159] lH (400 MHz, DMSO-i¼) δ 7.43-7.34 (2H, m, 2 x aromatic-H), 7.24 (1H, dd, J 6.8 and 1.8 Hz, aromatic-H), 6.30 (2H, br, NH2); m/z 213/215 (1 : 1, M+H+, 100%).
1.1.2. Synthesis of 4-[ 4-(4, 4, 5, 5-Tetramethyl-f 1, 3,2] ‘ dioxaborolan-2-yl) -benzyl] ‘- thiomor holine- 1, 1 -dioxide (Intermediate 4)

[00160] 2-(4-Bromomethyl-phenyl)-4,4,5,5-tetramethyl-[l,3,2]dioxaborolane (1 eq) and DIPEA (2 eq) were dissolved in DCM/MeOH (5:1 v:v) under N2 and thiomorpholine 1,1 -dioxide (2 eq) was added portion wise. The resulting solution was stirred at room temperature for 16h. After this time, the reaction was complete. The solvent was evaporated. The compound was extracted with EtOAc and water, washed with brine and dried over anhydrous MgSO i. Organic layers were filtered and evaporated. The final compound was isolated without further purification.
1.1.3. Synthesis of 5-[4-(l, l-Dioxothiomorpholin-4-ylmethyl)-phenyl]-[l,2,4]triazolo[l,5- a ridin-2-ylamine (Formula I)

[00161] 4-[4-(4,4,5,5-Tetramethyl-[l,3,2]dioxaborolan-2-yl)-benzyl]-thiomorpholine-l,l-dioxide (l .leq.) was added to a solution of 5-bromo-[l,2,4]triazolo[l,5-a]pyrid in-2-ylamine (4: 1). K2CO3 (2 eq.) and PdC^dppf (0.03 eq.) were added to the solution. The resulting mixture was then heated in an oil bath at 90°C for 16h under N2. Water was added and the solution was extracted with ethyl acetate. The organic layers were dried over anhydrous MgSC>4 and evaporated in vacuo. The final compound was obtained after purification by flash chromatography.
[00162] lH (400 MHz, CDC13) δ 7.94-7.92 (d, 2H), 7.52-7.48 (m, 3H), 7.37-7.34 (m, 1H), 7.02-7.00 (m, 1H), 6.00 (d, 2H), 3.76 (d, 2H), 3.15-3.13 (m, 4H), 2.93-2.91 (m, 4H).
[00163] m/z 358.2 (M+H+, 100%). 1.2. Route 2
1.2.1. Cyclopropanecarboxylic acid {5-[4-(l, l-dioxo-thiomorpholin-4-ylmethyl)-phenylJ- [l,2,4]triazolo[l,5-a]pyridin-2-yl}-amide (Formula II)
[00164] The compound according to Formula II may be synthesized according to the procedure described in WO 2010/149769.
1.2.2. Synthesis of 5-[4-(l, l-Dioxothiomorpholin-4-ylmethyl)-phenyl]-[l,2,4]triazolo[l,5- aJpyridin-2-ylamine (Formula I)
[00165] The compound according to Formula I can also be produced by hydrolysis of the compound accor ing to Formula II:

[00166] Hydrochloric acid 30% aq (12.06 kg; 3.9 rel. volumes) was added to a slurry of the compound according to Formula II (3.45 kg; 1.0 equiv.) in demineralized water (10.0 kg; 3.0 rel. volumes). Subsequently, a line rinse was performed with demineralized water (3.4 kg; 1.0 rel. volumes). The reaction mixture was heated to 80±5°C for 14.5 h. After completion of the reaction (conversion > 99%>), the reaction mixture was cooled to 20±5°C. The reaction mixture was diluted with demineralized water (6.8 kg; 2.0 rel. volumes) and sodium hydroxide 33%> aq (9.52 kg; 3.7 rel volumes) was dosed at such a rate that the temperature of the reactor contents remained below 35°C. An additional amount of sodium hydroxide 33%> aq (2.55 kg; 1.0 rel. volumes) was needed to get the pH > 10. The product was filtered off, washed twice with demineralized water (1.5 rel. volumes) and dried under vacuum for 1 h, thus yielding the crude compound according to Formula I.
[00167] The crude compound according to Formula I (5.70 kg) was re-slurried in demineralized water (23.0 kg; 8.5 rel. volumes). Hydrochloric acid 30%> aq (1.65 kg; 0.7 rel. volumes) and demineralized water (4.3 kg; 1.6 rel. volumes) were added and the reaction mixture was stirred at 20±5°C for 45 min. As the compound according to Formula I was not dissolved completely, the reaction mixture was stirred at 45±5°C for 1 h. The reaction mixture was filtered and the residue was washed with demineralized water (2.0 kg 0.75 rel. volumes). Sodium hydroxide 33%> aq (1.12 kg; 0.6 rel volumes) was added to the filtrate. An additional amount of sodium hydroxide 33%> aq (1.01 kg) was needed to get the pH > 10. The resulting reaction mixture was stirred at 20±5°C for about 3 h. The product was filtered off, washed twice with demineralized water (4.1 kg; 1.5 rel. volumes), and twice with methyl tert-butyl ether (MTBE; 3.0 kg; 1.5 rel. volumes) and dried under vacuum for 15.5 h on the filter. The product was further dried in a vacuum oven at 40±5°C for 202 h, thus affording the desired compound according to Formula I.
Update
Scheme 1: General S nthesis of Compounds of Formula I or A

Formula A
Scheme 7.

(16) (17) (18)
(18a): R3a=R3b=R2a=R (18b): R3a=R3b=D; R2a 18c): R3a=R3b=H; R2a

References
- Namour, Florence; Diderichsen, Paul Matthias; Cox, Eugène; Vayssière, Béatrice; Van der Aa, Annegret; Tasset, Chantal; Van’t Klooster, Gerben (2015-02-14). “Pharmacokinetics and Pharmacokinetic/Pharmacodynamic Modeling of Filgotinib (GLPG0634), a Selective JAK1 Inhibitor, in Support of Phase IIB Dose Selection”. Clin Pharmacokinet. Epub ahead of print.doi:10.1007/s40262-015-0240-z.
- Van Rompaey, L; Galien, R; Van der Aar, E; Clement-Lacroix, P; Van der Aar, E; Nelles, L; Smets, B; Lepescheux, L; Cristophe, T; Conrath, K; Vandeghinste, N; Vayssiere, B; De Vos, S; Fletcher, S; Brys, R; Van’t Klooster, G; Feyen, J; Menet, C (2013-10-01). “Preclinical characterization of GLPG0634, a selective inhibitor of JAK1 for the treatment of inflammatory diseases”. J Immunol. 191(7). doi:10.4049/jimmunol.1201348.
- http://acrabstracts.org/abstracts/phase-1-and-phase-2-data-confirm-that-glpg0634-a-selective-jak1-inhibitor-has-a-low-potential-for-drug-drug-interactions/
- “Galapagos’ GLPG0634 shows excellent efficacy and safety in rheumatoid arthritis Phase II study” (PDF) (Press release). Retrieved 2015-02-26.
- “Galapagos reports that the last patient in DARWIN 1 has completed 12 weeks of treatment” (PDF) (Press release). Retrieved 2015-02-26.
- “Galapagos completes recruitment for Darwin 1 study with GLPG0634 (filgotinib) in RA”. EuroInvestor. Retrieved 2015-02-26.
- NASDAQ OMX Corporate Solutions. “Galapagos completes recruitment for Darwin 2 monotherapy study with GLPG0634 (filgotinib) in RA”. Yahoo Finance. Retrieved 2015-02-26.
| US8551980 | Nov 17, 2010 | Oct 8, 2013 | Bayer Intellectual Property Gmbh | Substituted triazolopyridines |
| US8796457 | Jun 25, 2010 | Aug 5, 2014 | Galapagos Nv | Compound useful for the treatment of degenerative and inflammatory diseases |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
N-[5-[4-[(1,1-dioxo-1,4-thiazinan-4-yl)methyl]phenyl]-[1,2,4]triazolo[1,5-a]pyridin-2-yl]cyclopropanecarboxamide
|
|
| Clinical data | |
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Biological half-life | 6 hours[1] |
| Identifiers | |
| CAS Registry Number | 1206161-97-8 |
| ATC code | L01XE18 |
| IUPHAR/BPS | 7913 |
| ChemSpider | 28189566 |
| UNII | 3XVL385Q0M |
| ChEMBL | CHEMBL3301607 |
| Chemical data | |
| Formula | C21H23N5O3S |
| Molecular mass | 425.50402 g/mol |
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये। औकात बस इतनी देना, कि औरों का भला हो जाये।
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE


Join me on twitter

 LIONEL MY SON
LIONEL MY SON
He was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, Now I keep him as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son happy

सुकून उतना ही देना प्रभू, जितने से
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
Read all about Organic Spectroscopy on ORGANIC SPECTROSCOPY INTERNATIONAL
/////////Galapagos, GLPG0634, Filgotinib, PHASE 2, orphan drug designation, PHASE 3, Crohn’s disease, Rheumatoid arthritis, Ulceraticolitis
ve SMILES code: O=C(C1CC1)NC2=NN3C(C4=CC=C(CN5CCS(CC5)(=O)=O)C=C4)=CC=CC3=N2

{kind=link}
{kind=link}