
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
Filgotinib
EU APPROVED 2020/9/24, JYSELECA
JAPAN APPROVED2020/9/25
- C21H23N5O3S
- MW425.504
- Elemental Analysis: C, 59.28; H, 5.45; N, 16.46; O, 11.28; S, 7.54
IL-6 antagonist; Jak1 tyrosine kinase inhibitor; Tyk2 tyrosine kinase inhibitor; Jak3 tyrosine kinase inhibitor; Jak2 tyrosine kinase inhibitor
Autoimmune disease; Cancer; Colitis; Crohns disease; Inflammatory disease; Neoplasm; Rheumatoid arthritis; Transplant rejection
In 2017, orphan drug designation was assigned to the compound in the U.S. for the treatment of pediatric Crohn’s disease and pediatric ulcerative colitis.
GlaxoSmithKline had been developing filgotinib preclinically for the treatment of rheumatoid arthritis pursuant to a license; however, in 2010, the compound was re-acquired by Galapagos. In 2012, the product was licensed to Abbott for development and marketing. In January 2013, Abbott spun-off its research-based pharmaceutical business into a newly-formed company AbbVie. The license agreement between Galapagos and Abbott was terminated in September 2015, Galapagos regaining all rights to the product. The same year, Galapagos and Gilead entered into a global partnership and Gilead obtained the global rights of codevelopment and commercialization for the treatment of inflammatory diseases
Filgotinib (GLPG0634), by the Belgian biotech company Galápagos NV, is a drug which is currently under investigation for the treatment of rheumatoid arthritis and Crohn’s disease.
Filgotinib (GLPG0634) is an orally-available, selective inhibitor of JAK1 (Janus kinase 1) for the treatment of rheumatoid arthritis and potentially other inflammatory diseases. Filgotinib (GLPG0634) dose-dependently inhibited Th1 and Th2 differentiation and to a lesser extent the differentiation of Th17 cells in vitro. GLPG0634 was well exposed in rodents upon oral dosing, and exposure levels correlated with repression of Mx2 expression in leukocytes. The JAK1 selective inhibitor GLPG0634 (Filgotinib) is a promising novel therapeutic with potential for oral treatment of rheumatoid arthritis and possibly other immune-inflammatory diseases. Filgotinib (GLPG0634) is currently in a Phase 2 study in Crohn’s disease.

Mechanism of action
Filgotinib is a Janus kinase inhibitor with selectivity for subtype JAK1 of this enzyme. It is considered a promising agent as it inhibits JAK1 selectively. Less selective JAK inhibitors (e.g. tofacitinib) are already being marketed. They show long-term efficacy in the treatment of various inflammatory diseases. However, their lack of selectivity leads to dose-limiting side effects.[1] It is thought that inhibition of all JAK isoenzymes is beneficial in rheumatoid arthritis. However, pan-JAK inhibition might also lead to unwanted side effects that might not outweigh its benefits. This is the rationale for the development of newer and more selective inhibitors like filgotinib.
The signal transmission of large numbers of proinflammatory cytokines is dependent on JAK1. Inhibition of JAK2 may also contribute to the efficacy against RA. Nonetheless it is thought that JAK2 inhibition might lead to anemia and thrombopenia by interference witherythropoietin and thrombopoietin and granulocyte-macrophage colony-stimulating factor. Therefore one might prefer to choose a more selective JAK1 inhibitor as a primary therapeutic option. Filgotinib exerts a 30-fold selectivity for JAK1 compared to JAK2.[2] It is however still to be seen to what extent JAK2 inhibition should be avoided.
Novel crystalline forms of filgotinib salts, particularly hydrochloride salt, useful for treating JAK-mediated diseases eg inflammatory diseases, autoimmune diseases, proliferative diseases, allergy and transplant rejection. Galapagos and licensee AbbVie are developing filgotinib, a selective JAK-1 inhibitor, for treating rheumatoid arthritis (RA) and Crohn’s disease (CD). In August 2015, the drug was reported to be in phase 2 clinical development for treating RA and CD. The drug is also being investigated for the treatment of colitis and was discovered as part of the company’s arthritis alliance with GSK; however in August 2010 Galapagos reacquired the full rights. See WO2013189771, claiming use of filgotinib analog for treating inflammatory diseases. Also see WO2010010190 (co-assigned with GSK and Abbott) and WO2010149769 (assigned to Galapagos) claiming filgotinib, generically and specifically, respectively.
Clinical trials and approval
The efficacy of filgotinib is currently studied in a phase2b program (DARWIN trial 1, 2) with involvement of 886 rheumatoid arthritis patients and 180 Crohn’s disease patients.
Phase 1 study
It was shown in phase 1 studies that the pharmacokinetics of filgotinib metabolism is independent of hepatic CYP450 enzymatic degradation. The drug metabolism is however mediated by carboxylesterases. There is no interference reported with the metabolism of methotrexate nor with any of the investigated transport proteins.[3]
Phase 2 study: Proof of concept (2011)
In november 2011 Galápagos released the results of their phase 2 study (identification: NCT01384422, Eudract: 2010-022953-40) in which 36 patients were treated who showed a suboptimal clinical response to methotrexate treatment. Three groups of twelve patients were treated either with 200 mg filgotinib in a single dose, 200 mg divided in two doses or placebo. The primary end-point was the ACR20 score, which monitors improvements in the symptomatology of the patient. After the scheduled 4 weeks of treatment, 83% of the respondents showed an improved ACR20-score. Half of the treated patients showed a complete (or near complete) remission of the disease. There were no reports ofanemia nor changes in lipidemia. The company stated in their press release that filgotinib is the first selective JAK1 inhibitor that shows clinical efficacy. As a result of this study, the company stated that “GLPG0634 shows one of the highest initial response rates ever reported for rheumatoid arthritis treatments”.[4]
DARWIN 1 trial
The DARWIN 1 trial is a 24 week double blind placebo-controlled trial with 599 rheumatoid arthritis patients enrolled. All participants have moderate to severe RA and showed an insufficient response to standard methotrexate treatment. The trial compares three dosages of filgotinib as a once or twice per day regimen. During the trial all participants remain on their methotrexate treatment. According to the company, the results of this trial are expected in July 2015.[5]
DARWIN 2 trial
The DARWIN 2 trial is a double blind placebo-controlled trial with 280 rheumatoid arthritis patients enrolled who show an insufficient response to standard methotrexate treatment. This trial, in contrast to the previous DARWIN 1 trial, methotrexate is discontinued. Therefore, this trial investigates filgotinib as a monotherapy.[6] The recruitment of DARWIN trial 2b ended in november 2014.[7] Preliminary results are expected in the second quarter of 2015 and a full completion of the study is expected in the third quarter of 2015.
DARWIN 3 trial
Patients who complete DARWIN 1 and 2 will be eligible for DARWIN 3.
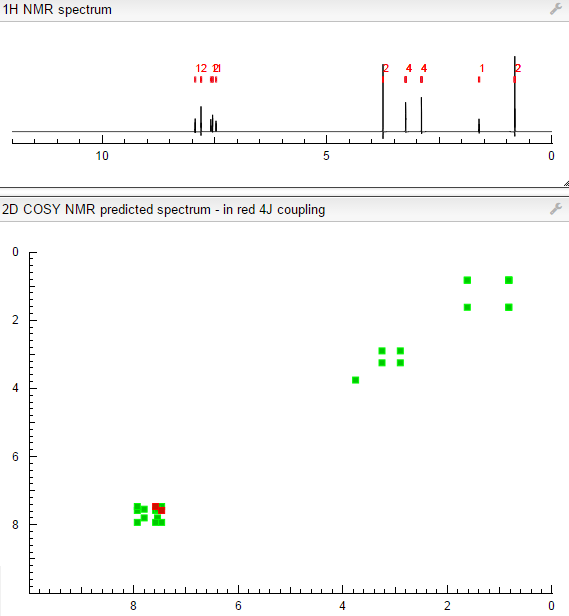
COSY PREDICT
Time line
- june 2011: results of first phase 2 trial
- november 2014: initiation of DARWIN 1 and 2 trials
- april 2015: expected date of DARWIN 1 trial results
- june 2015: expected date of DARWIN 2 trial results
NMR FROM NET….ABMOLE, DMSOD6
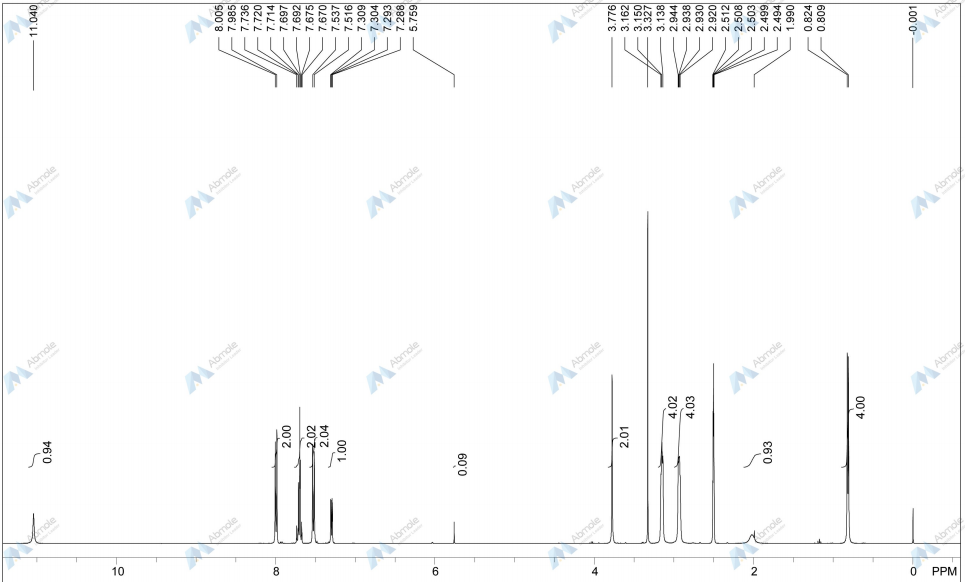
NMR MEDKOO DMSOD6

CHEMIETEK


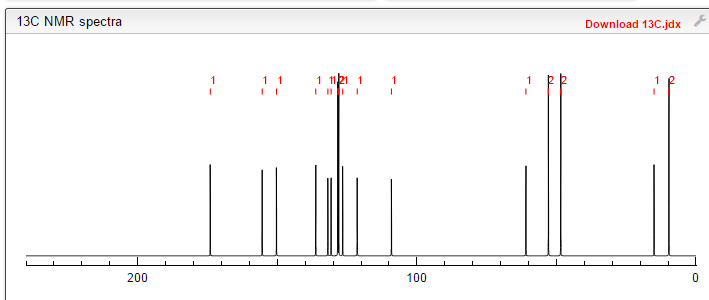
13C NMR PREDICT
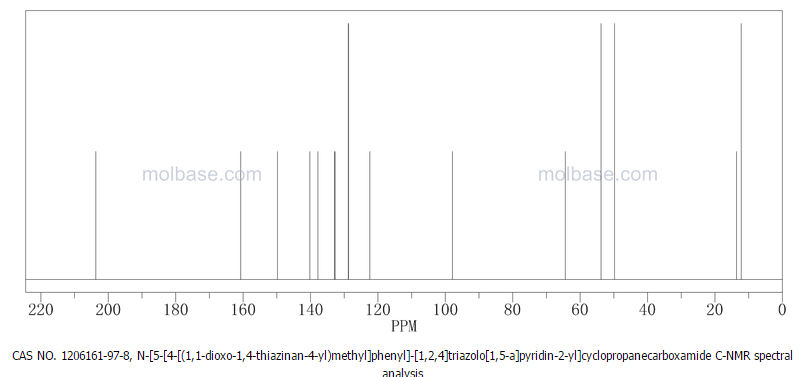

……………………
MORE PREDICTS
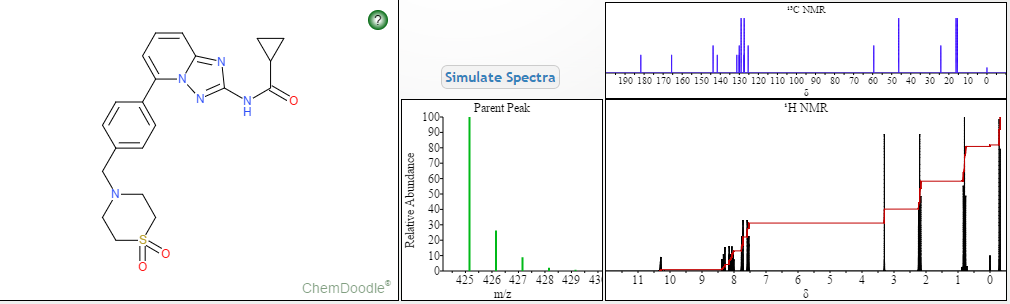
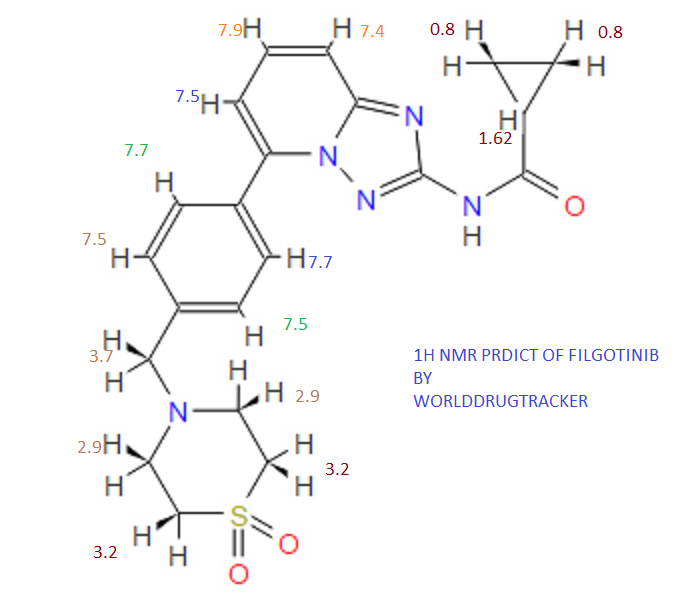
1H NMR PREDICT
13C NMR PREDICT
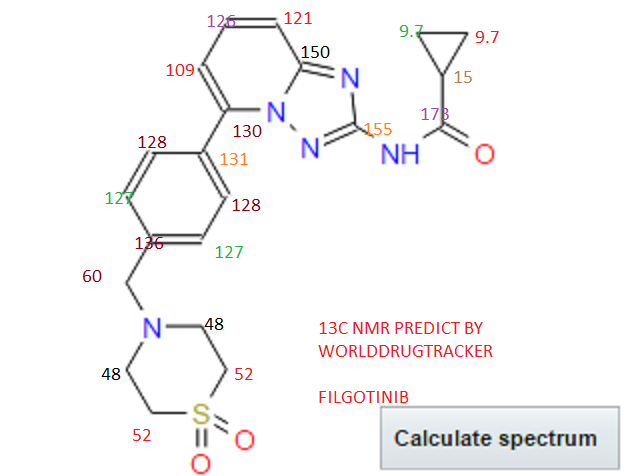
PRODUCT PATENT
http://www.google.com/patents/WO2010149769A1?cl=en
| Applicants: | GALAPAGOS NV [BE/BE]; Generaal De Wittelaan L11/A3 B-2800 Mechelen (BE) (For All Designated States Except US). MENET, Christel Jeanne Marie [FR/BE]; (BE) (For US Only). SMITS, Koen Kurt [BE/BE]; (BE) (For US Only) |
| Inventors: | MENET, Christel Jeanne Marie; (BE). SMITS, Koen Kurt; (BE) |
| International Filing Date: | 25.06.2010 |
ESTIMATED EXP 2030
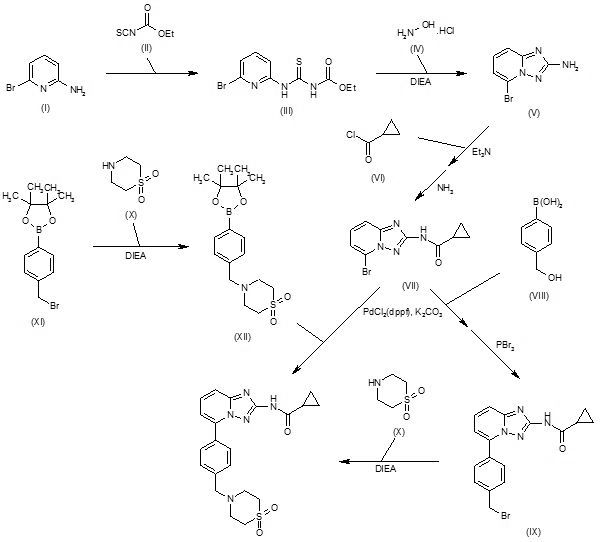
Condensation of 2-amino-6-bromopyridine (I) with ethoxycarbonyl isothiocyanate (II) in CH2Cl2 gives 1-(6-bromopyridin-2-yl)-3-carboethoxythiourea (III), which upon cyclization with hydroxylamine hydrochloride (IV) in the presence of DIEA in EtOH/MeOH yields 2-amino-5-bromo[1,2,4]triazolo[1,5-a]pyridine (V). N-Acylation of amine (V) with cyclopropanecarbonyl chloride (VI) using Et3N in acetonitrile, and subsequent treatment with methanolic ammonia furnishes the carboxamide (VII) (1-3), which upon Suzuki coupling with 4-(hydroxymethyl)phenylboronic acid (VIII) in the presence of PdCl2(dppf) and K2CO3 in dioxane/H2O at 90 °C, followed by bromination with PBr3 in CHCl3 affords intermediate (IX). Condensation of benzyl bromide derivative (IX) with thiomorpholine-1,1-dioxide (X) using DIEA in CH2Cl2/MeOH yields filgotinib (1,2). Alternatively, condensation of (4-bromomethylphenyl)-4,4,5,5-tetramethyl-[1,3,2]dioxaborolane (XI) with thiomorpholine 1,1-dioxide (X) in the presence of DIEA in CH2Cl2/MeOH gives intermediate (XII), which undergoes Suzuki coupling with aryl bromide (VII) in the presence of PdCl2(dppf) and K2CO3 in dioxane/H2O at 90 °C to afford the target filgotinib
The present invention is based on the discovery that the compound of the invention is able to act as an inhibitor of JAK and that it is useful for the treatment of inflammatory conditions, autoimmune diseases, proliferative diseases, transplantation rejection, diseases involving impairment of cartilage turnover, congenital cartilage malformations, and/or diseases associated with hypersecretion of IL6. In a specific aspect the compound is an inhibitor of JAKl and JAK2. The present invention also provides methods for the production of this compound, a pharmaceutical composition comprising this compound and methods for treating inflammatory conditions, autoimmune diseases, proliferative diseases, transplantation rejection, diseases involving impairment of cartilage turnover, congenital cartilage malformations, and/or diseases associated with hypersecretion of IL6 by administering the compound of the invention.
Accordingly, in a first aspect of the invention, a compound of the invention is provided having a formula (I):

[0017] The compound of the invention is a novel inhibitor of JAK that appears to exhibit a dramatically improved in vivo potency as compared to structurally similar compounds. In a particular embodiment the compound of the invention is an inhibitor of JAKl and JAK2. In particular it appears to exhibit this increase in potency at lower in vivo exposure levels compared to structurally similar compounds. The use of a compound with these improvements is expected to result in a lower dosage requirement (and therefore an improved dosing schedule).
General Synthetic Method Scheme 1

1. RCOCI, Et3N 2. NH3 / MeOH CH3CN, 20 0C 2O 0C

wherein Ar represents phenyl-Ll-heterocycloalkyl, where Ll is a bond, -CH2– or -CO- and the heterocycloalkyl group is optionally substituted.
General
1.1.1 l-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea (2)

(2)
[00117] To a solution of 2-amino-6-bromopyridine (1) (253.8 g, 1.467 mol) in DCM (2.5 L) cooled to 5 0C is added ethoxycarbonyl isothiocyanate (173.0 mL, 1.467 mol) dropwise over 15 min. The reaction mixture is then allowed to warm to room temp. (20 0C) and stirred for 16 h. Evaporation in vacuo gives a solid which may be collected by filtration, thoroughly washed with petrol (3×600 mL) and air-dried to afford (2). The thiourea may be used as such for the next step without any purification. 1H (400 MHz, CDCl3) δ 12.03 (IH, br s, NH), 8.81 (IH, d, J 7.8 Hz, H-3), 8.15 (IH, br s, NH), 7.60 (IH, t, J 8.0 Hz, H-4), 7.32 (IH, dd, J 7.7 and 0.6 Hz, H-5), 4.31 (2H, q, J 7.1 Hz, CH2), 1.35 (3H, t, J 7.1 Hz, CH3).
7.7.2 5-Bromo-[l, 2, 4]triazolo[l, 5-a]pyridin-2-ylamine (3)

[00118] To a suspension of hydroxylamine hydrochloride (101.8 g, 1.465 mol) in EtOH/MeOH
(1 :1, 900 mL) is added N,N-diisopropylethylamine (145.3 mL, 0.879 mol) and the mixture is stirred at room temp. (20 0C) for 1 h. l-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea (2) (89.0 g, 0.293 mol) is then added and the mixture slowly heated to reflux (Note: bleach scrubber is required to quench H2S evolved). After 3 h at reflux, the mixture is allowed to cool and filtered to collect the precipitated solid. Further product is collected by evaporation in vacuo of the filtrate, addition Of H2O (250 mL) and filtration. The combined solids are washed successively with H2O (250 mL), EtOH/MeOH (1 : 1, 250 mL) and Et2O (250 mL) then dried in vacuo to afford the triazolopyridine derivative (3) as a solid. The compound may be used as such for the next step without any purification. 1H (400 MHz, DMSO-t/β) δ 7.43-7.34 (2H, m, 2 x aromatic-H), 7.24 (IH, dd, J 6.8 and 1.8 Hz, aromatic-H), 6.30 (2H, br, NH2); m/z 213/215 (1 :1, M+H+, 100%).
7.7.3 General procedure for mono-acylation to afford intermediate (4):

[00119] To a solution of the 2-amino-triazolopyridine (3) (7.10 g, 33.3 mmol) in dry CH3CN
(150 mL) at 5 0C is added Et3N (11.6 mL, 83.3 mmol) followed by cyclopropanecarbonyl chloride (83.3 mmol). The reaction mixture is then allowed to warm to ambient temperature and stirred until all starting material (3) is consumed. If required, further Et3N (4.64 mL, 33.3 mmol) and cyclopropanecarbonyl chloride (33.3 mmol) is added to ensure complete reaction. Following solvent evaporation in vacuo the resultant residue is treated with 7 N methanolic ammonia solution (50 mL) and stirred at ambient temp, (for 1-16 h) to hydro lyse any bis-acylated product. Product isolation is made by removal of volatiles in vacuo followed by trituration with Et2O (50 mL). The solids are collected by filtration, washed with H2O (2x50mL), acetone (50 mL) and Et2O (50 mL), then dried in vacuo to give the required bromo intermediate (4).
Method A
Preparation of compounds of the invention via Suzuki coupling (5):
[00120] An appropriate boronic acid (2eq.) is added to a solution of bromo intermediate (4) in
1 ,4-dioxane/water (5:1). K2CO3 (2 eq.) and PdCl2dppf (5%) are added to the solution. The resulting mixture is then heated in a microwave at 140 0C for 30 min (this reaction can also be carried out by traditional heating in an oil bath at 900C for 16h under N2). Water is added and the solution is extracted with ethyl acetate. The organic layers are dried over anhyd. MgSθ4 and evaporated in vacuo. The final compound is obtained after purification by flash chromatography or preparative HPLC. HPLC: Waters
XBridge Prep Cl 8 5μm ODB 19mm ID x 100mm L (Part No.186002978). All the methods are using
MeCN/H2O gradients. H2O contains either 0.1% TFA or 0.1% NH3.
Method B

Bl. 4 4-[2-(Cyclopropanecarbonyl-amino)-[ 1 , 2, 4]triazolo[l, 5-a] pyridin-5-yl] -benzoyl chloride

[00121] 2 Drops of DMF are added to a solution of 4-[2-(cyclopropanecarbonyl-amino)- [l,2,4]triazolo[l,5-a]pyridin-5-yl]-benzoic acid (1 eq) obtained by Method A using 4-carboxyphenylboronic acid in DCM under N2 atmosphere. Then oxalyl chloride (2 eq) is added dropwise to this resulting solution (gas release). The mixture is stirred at room temperature for 2 hours. After completion of the reaction by LCMS, the solvent is removed. The crude acid chloride is used without further purification in next step.
B2. Amide formation (General Method)

[00122] An appropriate amine (1.1 eq) and Et3N (5 eq) are dissolved in DCM under N2 atmosphere and cooled at 00C. The acid chloride (Bl, 1 eq) dissolved in DCM is added dropwise to this solution. The reaction is stirred at room temperature for 16 h. After this time, reaction is complete. The compound is extracted with EtOAc and water, washed with brine and dried over anhyd. MgSO4. Organic layers are filtered and evaporated. The final compound is isolated by preparative HPLC. Preparative HPLC: Waters XBridge Prep C18 5μm ODB 19mm ID x 100mm L (Part No.186002978). All the methods are using MeCN/H2O gradients. H2O contains either 0.1% TFA or 0.1% NH3.
Method C

Wherein R3a or R3b together with the nitrogen atom to which they are attached, may form a heterocycloalkyl.
Reductive alkylation (general method)
[00123] An appropriate amine (2 eq.), cyclopropanecarboxylic acid (for example cyclopropanecarboxylic acid [5-(4-formyl-phenyl)-[l,2,4]triazolo[l,5-a]pyridine-2-yl]-amide) prepared by method A (1 eq.) and Ti(OPr)4 are mixed and stirred at room temperature for 3 hrs. The mixture is diluted in ethanol and Na(CN)BH3 (leq.) is added. The resulting solution is stirred at room temperature for 16 hrs. The mixture is diluted in water and filtered. The filtrate is washed with ethanol. The combined solvent phases are evaporated under vacuum. The final compound is isolated by preparative HPLC.
Method D 
wherein R1 and R2 together with the Nitrogen atom to which they are attached, may form a heterocycloalkyl.
Reaction ofalkylation

[00124] 2-(4-Bromomethyl-phenyl)-4,4,5,5-tetramethyl-[l,3,2]dioxaborolane (leq) and Et3N (2 eq) (or AgCO3) are dissolved in DCM/MeOH (4:1 v:v) under N2 and an amine (2 eq) is added dropwise. The resulting solution is stirred at room temperature for 16h. After this time, the reaction is complete. The solvent is evaporated. The compound is extracted with EtOAc and water, washed with brine and dried over anhyd. MgSθ4. Organic layers are filtered and evaporated. The final compound is isolated by flash chromatography.
Suzuki coupling

[00125] The obtained boronic acid (2eq.) is added to a solution of cyclopropanecarboxylic acid
(5-bromo-[l,2,4]triazolo[l,5-a]pyridin-2-yl)-amide (4) in 1 ,4-dioxane/water (5:1). K2CO3 (2 eq.) and PdCl2dppf (5%) are added to the solution. The resulting mixture is then heated in a microwave at 140 0C for 30 min (This reaction can also be carried out by traditional heating in an oil bath at 900C for 16h under N2). Water is added and the solution is extracted with ethyl acetate. The organic layers are dried over anhyd. MgSθ4 and evaporated in vacuo. The final compound is obtained after purification by flash chromatography or preparative HPLC. HPLC: Waters XBridge Prep C18 5μm ODB 19mm ID x 100mm L (Part No.186002978). All the methods are using MeCN/H2O gradients. H2O contains either 0.1% TFA or 0.1% NH3.
Synthesis of the compound of the invention and comparative examples
Compound l(the compound of the invention)
Step 1:

[00126] 2-(4-Bromomethyl-phenyl)-4,4,5,5-tetramethyl-[l,3,2]dioxaborolane (leq) and DIPEA
(2 eq) were dissolved in DCM/MeOH (5:1 v:v) under N2 and thiomorpholine 1,1 -dioxide (2 eq) was added portionwise. The resulting solution was stirred at room temperature for 16h. After this time, the reaction was complete. The solvent was evaporated. The compound was extracted with EtOAc and water, washed with brine and dried over anhyd. MgS O4. Organic layers were filtered and evaporated. The final compound was isolated without further purification.
Step 2: Suzuki coupling

[00127] 4-[4-(4,4,5,5-Tetramethyl-[l,3,2]dioxaborolan-2-yl)-benzyl]-thiomorpholine-l,l-dioxide
(l.leq.) was added to a solution of cyclopropanecarboxylic acid (5-bromo-[l,2,4]triazolo[l,5-a]pyridin-2-yl)-amide in 1 ,4-dioxane/water (4:1). K2CO3 (2 eq.) and PdCl2dppf (0.03 eq.) were added to the solution. The resulting mixture was then heated in an oil bath at 900C for 16h under N2. Water was added and the solution was extracted with ethyl acetate. The organic layers were dried over anhyd. MgSθ4 and evaporated in vacuo. The final compound was obtained after purification by flash chromatography.
[00128] Alternatively, after completion of the reaction, a palladium scavenger such as 1,2-bis(diphenylphosphino)ethane, is added, the reaction mixture is allowed to cooled down and a filtration is performed. The filter cake is reslurried in a suitable solvent (e.g. acetone), the solid is separated by filtration, washed with more acetone, and dried. The resulting solid is resuspended in water, aqueous HCl is added, and after stirring at RT, the resulting solution is filtered on celite (Celpure P300). Aqueous NaOH is then added to the filtrate, and the resulting suspension is stirred at RT, the solid is separated by filtration, washed with water and dried by suction. Finally the cake is re-solubilised in a mixture of THF/H2O, treated with a palladium scavenger (e.g. SMOPEX 234) at 500C, the suspension is filtered, the organic solvents are removed by evaporation, and the resulting slurry is washed with water and methanol, dried and sieved, to obtain the title compound as a free base.
Alternative route to Compound l(the compound of the invention):
Step 1:

[00129] 4-(Hydroxymethyl)phenylboronic acid (l.leq.) was added to a s o luti o n o f cyclopropanecarboxylic acid (5-bromo-[l,2,4]triazolo[l,5-a]pyridin-2-yl)-amide in 1 ,4-dioxane/water (4:1). K2CO3 (2 eq.) and PdCl2dppf (0.03 eq.) were added to the solution. The resulting mixture was then heated in an oil bath at 900C for 16h under N2. Water was added and the solution was extracted with ethyl acetate. The organic layers were dried over anhyd. MgSθ4 and evaporated in vacuo. The resulting mixture was used without further purification.
Step 2:

[00130] To a solution of cyclopropanecarboxylic acid [5-(4-hydroxymethyl-phenyl)- [l,2,4]triazolo[l,5-a]pyridin-2-yl]-amide (1.0 eq) in chloroform was slowly added phosphorus tribromide (1.0 equiv.). The reaction mixture was stirred at room temperature for 20 hours, quenched with ice and water (20 mL) and extracted with dichloromethane. The organic layer was dried over anhyd. MgSθ4, filtered and concentrated to dryness. The resulting white residue was triturated in dichloromethane/diethyl ether 2:1 to afford the expected product as a white solid.
Step 3:

[00131] Cyclopropanecarboxylic acid [5-(4-bromomethyl-phenyl)-[l,2,4]triazolo[l,5-a]pyridin- 2-yl]-amide (leq) and DIPEA (2 eq) were dissolved in DCM/MeOH (5:1 v:v) under N2 and thiomorpholine 1,1 -dioxide (1.1 eq) was added dropwise. The resulting solution was stirred at room temperature for 16h. After this time, the reaction was complete. The solvent was evaporated. The compound was dissolved in DCM, washed with water and dried over anhyd. MgSO^ Organic layers were filtered and evaporated. The final compound was isolated by column chromatography using EtOAc to afford the desired product.
PATENT
WO 2010010190
WO 2013173506
WO 2013189771
WO 2015117980
WO 2015117981
POLYMORPH
CN 105061420
https://encrypted.google.com/patents/CN105061420A?cl=en
JAK inhibitor N-(5-(4-(1,1-dioxothiomorpholinyl)methyl)phenyl)-[1,2,4]triazolo[1,5-a]pyridin-2-yl)cyclopropanecarboxamide, and methods for preparing the four crystal forms, wherein the four crystal forms respectively are a crystal form H1, a crystal form H2, a crystal form H3 and a crystal form H4,
POLYMORPH
E CRYSTAL
CN 105111206
D CRYSTAL
CN 105111207
H CRYSYAL
CN 105198876
G
CN 105198877
F CN 105198878
C CN 105198880
POLYMORPH
WO 2016105453
POLYMORPH
Preparation of solid forms of filgotinib free base
WO 2017012773
The present invention relates to cryst. filgotinib free base (I), a method of its prepn. and a pharmaceutical compn. comprising the same. Thus, reacting the compd. II with thiomorpholine dioxide afforded 92% I (purity of 98.6%) which was then converted into its hydrochloride salt. Pharmaceutical compn. comprising compd. I was disclosed.
POLYMORPH
CN 105669669
The present invention provides a crystal form A, B, D, G and M of N-[5-[4-[(1,1-dioxido-4-thiomorpholinyl)methyl]phenyl][1,2,4]triazolo[1,5-a]pyridin-2-yl]cyclopropanecarboxamide hydrochloride.
PAPER
Future Medicinal Chemistry (2015), 7(2), 203-235. | Language: English, Database: CAPLUSA review. The discovery of the JAK-STAT pathway was a landmark in cell biol. The identification of these pathways has changed the landscape of treatment of rheumatoid arthritis and other autoimmune diseases. The two first (unselective) JAK inhibitors have recently been approved by the US FDA for the treatment of myelofibrosis and rheumatoid arthritis and many other JAK inhibitors are currently in clin. development or at the discovery stage. Research groups have demonstrated the different roles of JAK member and the therapeutic potential of targeting them selectively. ………..
https://www.future-science.com/doi/10.4155/fmc.14.149
PAPER
Journal of Pharmaceutical Sciences (Philadelphia, PA, United States) (2018), 107(6), 1624-1632.
PATENT
US2010/331319 A1, ; Page/Page column 13-14
http://www.google.com/patents/US20100331319
Synthetic Preparation of the Compound of the Invention and Comparative Examples
The compound of the invention and the comparative examples can be produced according to the following scheme.

wherein Ar represents phenyl-L1-heterocycloalkyl, where L1 is a bond, —CH2— or —CO— and the heterocycloalkyl group is optionally substituted.
General 1.1.1 1-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea (2)

To a solution of 2-amino-6-bromopyridine (1) (253.8 g, 1.467 mol) in DCM (2.5 L) cooled to 5° C. is added ethoxycarbonyl isothiocyanate (173.0 mL, 1.467 mol) dropwise over 15 min. The reaction mixture is then allowed to warm to room temp. (20° C.) and stirred for 16 h. Evaporation in vacuo gives a solid which may be collected by filtration, thoroughly washed with petrol (3×600 mL) and air-dried to afford (2). The thiourea may be used as such for the next step without any purification. 1H (400 MHz, CDCl3) δ 12.03 (1H, br s, NH), 8.81 (1H, d, J=7.8 Hz, H-3), 8.15 (1H, br s, NH), 7.60 (1H, t, J=8.0 Hz, H-4), 7.32 (1H, dd, J 7.7 and 0.6 Hz, H-5), 4.31 (2H, q, J 7.1 Hz, CH2), 1.35 (3H, t, J 7.1 Hz, CH3).
1.1.2 5-Bromo-[1,2,4]triazolo[1,5-a]pyridin-2-ylamine (3)

To a suspension of hydroxylamine hydrochloride (101.8 g, 1.465 mol) in EtOH/MeOH (1:1, 900 mL) is added N,N-diisopropylethylamine (145.3 mL, 0.879 mol) and the mixture is stirred at room temp. (20° C.) for 1 h. 1-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea (2) (89.0 g, 0.293 mol) is then added and the mixture slowly heated to reflux (Note: bleach scrubber is required to quench H2S evolved). After 3 h at reflux, the mixture is allowed to cool and filtered to collect the precipitated solid. Further product is collected by evaporation in vacuo of the filtrate, addition of H2O (250 mL) and filtration. The combined solids are washed successively with H2O (250 mL), EtOH/MeOH (1:1, 250 mL) and Et2O (250 mL) then dried in vacuo to afford the triazolopyridine derivative (3) as a solid. The compound may be used as such for the next step without any purification. 1H (400 MHz, DMSO-d6) δ 7.43-7.34 (2H, m, 2×aromatic-H), 7.24 (1H, dd, J 6.8 and 1.8 Hz, aromatic-H), 6.30 (2H, br, NH2); m/z 213/215 (1:1, M+H+, 100%).
1.1.3 General Procedure for Mono-Acylation to Afford Intermediate (4)

To a solution of the 2-amino-triazolopyridine (3) (7.10 g, 33.3 mmol) in dry CH3CN (150 mL) at 5° C. is added Et3N (11.6 mL, 83.3 mmol) followed by cyclopropanecarbonyl chloride (83.3 mmol). The reaction mixture is then allowed to warm to ambient temperature and stirred until all starting material (3) is consumed. If required, further Et3N (4.64 mL, 33.3 mmol) and cyclopropanecarbonyl chloride (33.3 mmol) is added to ensure complete reaction. Following solvent evaporation in vacuo the resultant residue is treated with 7 N methanolic ammonia solution (50 mL) and stirred at ambient temp. (for 1-16 h) to hydrolyse any bis-acylated product. Product isolation is made by removal of volatiles in vacuo followed by trituration with Et2O (50 mL). The solids are collected by filtration, washed with H2O (2×50 mL), acetone (50 mL) and Et2O (50 mL), then dried in vacuo to give the required bromo intermediate (4).
Method A Preparation of Compounds of the Invention Via Suzuki Coupling (5):
An appropriate boronic acid (2 eq.) is added to a solution of bromo intermediate (4) in 1,4-dioxane/water (5:1). K2CO3 (2 eq.) and PdCl2dppf (5%) are added to the solution. The resulting mixture is then heated in a microwave at 140° C. for 30 min (this reaction can also be carried out by traditional heating in an oil bath at 90° C. for 16 h under N2). Water is added and the solution is extracted with ethyl acetate. The organic layers are dried over anhyd. MgSO4 and evaporated in vacuo. The final compound is obtained after purification by flash chromatography or preparative HPLC. HPLC: Waters XBridge Prep C18 5 μm ODB 19 mm ID×100 mm L (Part No. 186002978). All the methods are using MeCN/H2O gradients. H2O contains either 0.1% TFA or 0.1% NH3.
Method B

B1. 4 4-[2-(Cyclopropanecarbonyl-amino)-[1,2,4]triazolo[1,5-a]pyridin-5-yl]-benzoyl chloride

2 Drops of DMF are added to a solution of 4-[2-(cyclopropanecarbonyl-amino)-[1,2,4]triazolo[1,5-a]pyridin-5-yl]-benzoic acid (1 eq) obtained by Method A using 4-carboxyphenylboronic acid in DCM under N2 atmosphere. Then oxalyl chloride (2 eq) is added dropwise to this resulting solution (gas release). The mixture is stirred at room temperature for 2 hours. After completion of the reaction by LCMS, the solvent is removed. The crude acid chloride is used without further purification in next step.
B2. Amide Formation (General Method)

An appropriate amine (1.1 eq) and Et3N (5 eq) are dissolved in DCM under N2 atmosphere and cooled at 0° C. The acid chloride (B1, 1 eq) dissolved in DCM is added dropwise to this solution. The reaction is stirred at room temperature for 16 h. After this time, reaction is complete. The compound is extracted with EtOAc and water, washed with brine and dried over anhyd. MgSO4. Organic layers are filtered and evaporated. The final compound is isolated by preparative HPLC. Preparative HPLC: Waters XBridge Prep C18 5 μm ODB 19 mm ID×100 mm L (Part No. 186002978). All the methods are using MeCN/H2O gradients. H2O contains either 0.1% TFA or 0.1% NH3.
…
Synthesis of the Compound of the Invention and Comparative Examples Compound 1 (the Compound of the Invention) Step 1:

2-(4-Bromomethyl-phenyl)-4,4,5,5-tetramethyl-[1,3,2]dioxaborolane (1 eq) and DIPEA (2 eq) were dissolved in DCM/MeOH (5:1 v:v) under N2 and thiomorpholine 1,1-dioxide (2 eq) was added portionwise. The resulting solution was stirred at room temperature for 16 h. After this time, the reaction was complete. The solvent was evaporated. The compound was extracted with EtOAc and water, washed with brine and dried over anhyd. MgSO4. Organic layers were filtered and evaporated. The final compound was isolated without further purification.
STEP 2: Suzuki coupling

4-[4-(4,4,5,5-Tetramethyl-[1,3,2]dioxaborolan-2-yl)-benzyl]-thiomorpholine-1,1-dioxide (1.1 eq.) was added to a solution of cyclopropanecarboxylic acid (5-bromo-[1,2,4]triazolo[1,5-a]pyridin-2-yl)-amide in 1,4-dioxane/water (4:1). K2CO3 (2 eq.) and PdCl2dppf (0.03 eq.) were added to the solution. The resulting mixture was then heated in an oil bath at 90° C. for 16 h under N2. Water was added and the solution was extracted with ethyl acetate. The organic layers were dried over anhyd. MgSO4 and evaporated in vacuo. The final compound was obtained after purification by flash chromatography.
Alternatively, after completion of the reaction, a palladium scavenger such as 1,2-bis(diphenylphosphino)ethane, is added, the reaction mixture is allowed to cooled down and a filtration is performed. The filter cake is reslurried in a suitable solvent (e.g. acetone), the solid is separated by filtration, washed with more acetone, and dried. The resulting solid is resuspended in water, aqueous HCl is added, and after stirring at RT, the resulting solution is filtered on celite (Celpure P300). Aqueous NaOH is then added to the filtrate, and the resulting suspension is stirred at RT, the solid is separated by filtration, washed with water and dried by suction. Finally the cake is re-solubilised in a mixture of THF/H2O, treated with a palladium scavenger (e.g. SMOPEX 234) at 50° C., the suspension is filtered, the organic solvents are removed by evaporation, and the resulting slurry is washed with water and methanol, dried and sieved, to obtain the title compound as a free base.
Alternative Route to Compound 1 (the Compound of the Invention): Step 1:

4-(Hydroxymethyl)phenylboronic acid (1.1 eq.) was added to a solution of cyclopropanecarboxylic acid (5-bromo-[1,2,4]triazolo[1,5-a]pyridin-2-yl)-amide in 1,4-dioxane/water (4:1). K2CO3 (2 eq.) and PdCl2dppf (0.03 eq.) were added to the solution. The resulting mixture was then heated in an oil bath at 90° C. for 16 h under N2. Water was added and the solution was extracted with ethyl acetate. The organic layers were dried over anhyd. MgSO4 and evaporated in vacuo. The resulting mixture was used without further purification.
Step 2:

To a solution of cyclopropanecarboxylic acid [5-(4-hydroxymethyl-phenyl)-[1,2,4]triazolo[1,5-a]pyridin-2-yl]-amide (1.0 eq) in chloroform was slowly added phosphorus tribromide (1.0 equiv.). The reaction mixture was stirred at room temperature for 20 hours, quenched with ice and water (20 mL) and extracted with dichloromethane. The organic layer was dried over anhyd. MgSO4, filtered and concentrated to dryness. The resulting white residue was triturated in dichloromethane/diethyl ether 2:1 to afford the expected product as a white solid.
Step 3:

Cyclopropanecarboxylic acid [5-(4-bromomethyl-phenyl)-[1,2,4]triazolo[1,5-a]pyridin-2-yl]-amide (1 eq) and DIPEA (2 eq) were dissolved in DCM/MeOH (5:1 v:v) under N2 and thiomorpholine 1,1-dioxide (1.1 eq) was added dropwise. The resulting solution was stirred at room temperature for 16 h. After this time, the reaction was complete. The solvent was evaporated. The compound was dissolved in DCM, washed with water and dried over anhyd. MgSO4. Organic layers were filtered and evaporated. The final compound was isolated by column chromatography using EtOAc to afford the desired product.
…………………….
PATENT
Novel salts and pharmaceutical compositions thereof for the treatment of inflammatory disorders
Also claims a method for preparing filgotinib hydrochloride trihydrate. The present filing forms a pair with this week’s filing, WO2015117980, claiming a tablet composition comprising filgotinib hydrochloride.
The compound cyclopropanecarboxylic acid {5-[4-(l,l-dioxo-thiomorpholin-4-ylmethyl)-phenyl]-[l,2,4]triazolo[l,5-a]pyridin-2-yl -amide (Compound 1), which has the chemical structure:

is disclosed in our earlier application WO 2010/149769 (Menet C. J., 2010) as being an inhibitor of JAK and as being useful in the treatment of inflammatory conditions, autoimmune diseases, proliferative diseases, allergy, transplant rejection, diseases involving impairment of cartilage turnover, congenital cartilage malformations, and/or diseases associated with hypersecretion of IL6 or interferons. Hereafter this compound is named Compound 1. The data presented in WO 2010/149769 demonstrate that despite similar in vitro activities, Compound 1 has unexpectedly high in vivo potency compared with structurally similar compounds.
Example 1. Preparation of Compound 1
1.1. Route 1
1.1.1. 4-[4-(4,4,5,5-Tetramethyl-[l,3,2]dioxaborolan-2-yl)-benzyl]-thiomorpholine-l,l-dioxide

[00205] 2-(4-Bromomethyl-phenyl)-4,4,5,5-tetramethyl-[l,3,2]dioxaborolane (1 eq) and DIPEA (2 eq) are dissolved in DCM/MeOH (5:1 v:v) under N2 and thiomorpholine 1,1 -dioxide (2 eq) is added portionwise. The resulting solution is stirred at room temperature for 16h. After this time, the reaction is complete. The solvent is evaporated. The compound is extracted with EtOAc and water, washed with brine and dried over anhydrous MgSO i. Organic layers are filtered and evaporated. The final compound is isolated without further purification.
1.1.2. Cyclopropanecarboxylic acid (5-bromo-[l,2,4]triazolo[l,5-a]pyridin-2-yl)-amide

1.1.2.1. Step i): l-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea
[00206] To a solution of 2-amino-6-bromopyridine (1) (253.8 g, 1.467 mol) in DCM (2.5 L) cooled to 5°C is added ethoxycarbonyl isothiocyanate (173.0 mL, 1.467 mol) dropwise over 15 min. The reaction
mixture is then allowed to warm to room temp. (20 °C) and stirred for 16 h. Evaporation in vacuo gives a solid which may be collected by filtration, thoroughly washed with petrol (3 x 600 niL) and air-dried to afford the desired product. The thiourea may be used as such for the next step without any purification. lH (400 MHz, CDC13) δ 12.03 (1H, br s), 8.81 (1H, d), 8.15 (1H, br s), 7.60 (1H, t), 7.32 (1H, dd), 4.31 (2H, q), 1.35 (3H, t).
1.1.2.2. Step ii): 5-Bromo-[l,2,4]triazolo[l,5-a]pyridin-2-ylamine
[00207] To a suspension of hydroxylamine hydrochloride (101.8 g, 1.465 mol) in EtOH/MeOH (1 : 1, 900 mL) is added NN-diisopropylethylamine (145.3 mL, 0.879 mol) and the mixture is stirred at room temp. (20 °C) for 1 h. l-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea (2) (89.0 g, 0.293 mol) is then added and the mixture slowly heated to reflux (Note: bleach scrubber is required to quench H2S evolved). After 3h at reflux, the mixture is allowed to cool and filtered to collect the precipitated solid. Further product is collected by evaporation in vacuo of the filtrate, addition of H20 (250 mL) and filtration. The combined solids are washed successively with H20 (250 mL), EtOH/MeOH (1 : 1, 250 mL) and Et20 (250 mL) then dried in vacuo to afford the triazolopyridine derivative (3) as a solid. The compound may be used as such for the next step without any purification. lH (400 MHz, DMSO-i¼) δ 7.43-7.34 (2H, m, 2 x aromatic-H), 7.24 (1H, dd, J 6.8 and 1.8 Hz, aromatic-H), 6.30 (2H, br, NH2); m/z 213/215 (1 : 1, M+H+, 100%).
1.1.2.3. Step Hi): Cyclopropanecarboxylic acid (5-bromo-[l ,2,4]triazolo[l ,5-a]pyridin-2-yl)-amide
[00208] To a solution of the 2-amino-triazolopyridine obtained in the previous step (7.10 g, 33.3 mmol) in dry MeCN (150 mL) at 5°C is added Et3N (11.6 mL, 83.3 mmol) followed by cyclopropanecarbonyl chloride (83.3 mmol). The reaction mixture is then allowed to warm to ambient temperature and stirred until all starting material is consumed. If required, further Et3N (4.64 mL, 33.3 mmol) and cyclopropanecarbonyl chloride (33.3 mmol) is added to ensure complete reaction. Following solvent evaporation in vacuo the resultant residue is treated with 7 N methanolic ammonia solution (50 mL) and stirred at ambient temp, (for 1-16 h) to hydro lyse any bis-acylated product. Product isolation is made by removal of volatiles in vacuo followed by trituration with Et20 (50 mL). The solids are collected by filtration, washed with H20 (2x50mL), acetone (50 mL) and Et20 (50 mL), then dried in vacuo to give the desired compound.
1.1.3. Compound 1

[00209] 4-[4-(4,4,5,5-Tetramethyl-[l ,3,2]dioxaborolan-2-yl)-benzyl] hiomoφholine , l -dioxide (l . l eq.) is added to a solution of cyclopropanecarboxylic acid (5-bromo-[l ,2,4]triazolo[l ,5-a]pyridin-2-yl)-amide in 1 ,4-dioxane/water (4: 1). K2CO3 (2 eq.) and PdC^dppf (0.03 eq.) are added to the solution. The resulting mixture is then heated in an oil bath at 90°C for 16h under N2. Water is added and the solution is extracted with ethyl acetate. The organic layers are dried over anhydrous MgS04 and evaporated in vacuo.
[00210] The final compound is obtained after purification by flash chromatography.
[00211] Alternatively, after completion of the reaction, a palladium scavenger such as 1 ,2-bis(diphenylphosphino)ethane, is added, the reaction mixture is allowed to cool down and a filtration is performed. The filter cake is reslurried in a suitable solvent (e.g. acetone), the solid is separated by filtration, washed with more acetone, and dried. The resulting solid is resuspended in water, aqueous HC1 is added, and after stirring at room temperature, the resulting solution is filtered on celite (Celpure P300). Aqueous NaOH is then added to the filtrate, and the resulting suspension is stirred at room temperature, the solid is separated by filtration, washed with water and dried by suction. Finally the cake is re-solubilised in a mixture of THF/H20, treated with a palladium scavenger (e.g. SMOPEX 234) at 50°C, the suspension is filtered, the organic solvents are removed by evaporation, and the resulting slurry is washed with water and methanol, dried and sieved, to obtain the desired compound as a free base.
1.2. Route 2
1.2.1. Step 1: cyclopropanecarboxylic acid [5-(4-hydroxymethyl-phenyl)-[l,2, 4]triazolo[l, 5- a] pyridin-2-yl] -amide

[00212] 4-(Hydroxymethyl)phenylboronic acid (l . l eq.) is added to a solution of cyclopropanecarboxylic acid (5-bromo-[l ,2,4]triazolo[l ,5-a]pyridin-2-yl)-amide in 1 ,4-dioxane/water
(4:1). K2CO3 (2 eq.) and PdC^dppf (0.03 eq.) are added to the solution. The resulting mixture is then heated in an oil bath at 90°C for 16h under N2. Water is added and the solution is extracted with ethyl acetate. The organic layers are dried over anhydrous MgS04 and evaporated in vacuo. The resulting mixture is used without further purification.
1.2.2. Step 2: Cyclopropanecarboxylic acid [5-(4-bromomethyl-phenyl)-[l,2,4]triazolo[l,5- a Jpyridin-2-ylJ -amide

[00213] To a solution of cyclopropanecarboxylic acid [5-(4-hydroxymethyl-phenyl)-[l,2,4]triazolo[l,5-a]pyridin-2-yl] -amide (1.0 eq) in chloroform is slowly added phosphorus tribromide (1.0 eq.). The reaction mixture is stirred at room temperature for 20 h, quenched with ice and water (20 mL) and extracted with dichloromethane. The organic layer is dried over anhydrous MgSO i, filtered and concentrated to dryness. The resulting white residue is triturated in dichloromethane/diethyl ether 2:1 to afford the desired product.
1.2.3. Step 3:

[00214] Cyclopropanecarboxylic acid [5-(4-bromomethyl-phenyl)-[l,2,4]triazolo[l,5-a]pyridin-2-yl]-amide (l eq) and DIPEA (2 eq) are dissolved in DCM/MeOH (5: 1 v:v) under N2 and thiomorpho line 1,1-dioxide (1.1 eq) is added dropwise. The resulting solution is stirred at room temperature for 16h. After this time, the reaction is complete. The solvent is evaporated. The compound is dissolved in DCM, washed with water and dried over anhydrous MgSO i. Organic layers are filtered and evaporated. The final compound is isolated by column chromatography using EtOAc to afford the desired product.
…………………
PATENT
http://www.google.co.in/patents/WO2013189771A1?cl=en
Example 1. Synthesis of the compounds
1.1. Route 1
1.1.1. Synthesis of 5-Bromo-[l,2,4]triazolo[l,5-a]pyridin-2-ylamine (Intermediate 3)

led to 5 °C was added ethoxycarbonyl isothiocyanate (173.0 mL, 1.467 mol) dropwise over 15 min. The reaction mixture was then allowed to warm to room temp. (20 °C) and stirred for 16 h. Evaporation in vacuo gave a solid which was collected by filtration, thoroughly washed with petrol (3×600 mL) and air-dried to afford (2). The thiourea was used as such in the next step without any purification.
[00157] lH (400 MHz, CDC13) δ 12.03 (IH, br s, NH), 8.81 (IH, d, J 7.8 Hz, H-3), 8.15 (IH, br s, NH), 7.60 (IH, t, J 8.0 Hz, H-4), 7.32 (IH, dd, J 7.7 and 0.6 Hz, H-5), 4.31 (2H, q, J 7.1 Hz, CH2), 1.35 (3H, t, J 7.1 Hz, CH3).
1.1.1.2. 5-Bromo-f 1,2, 4]triazolo[ 1 ,5-a] pyridin-2-ylamine (3)
[00158] To a suspension of hydroxylamine hydrochloride (101.8 g, 1.465 mol) in EtOH/MeOH (1 : 1, 900 mL) was added NN-diisopropylethylamine (145.3 mL, 0.879 mol) and the mixture was stirred at room temp. (20 °C) for 1 h. l-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea (2) (89.0 g, 0.293 mol) was then added and the mixture slowly heated to reflux (Note: bleach scrubber was required to quench H2S evolved). After 3 h at reflux, the mixture was allowed to cool and filtered to collect the precipitated solid. Further product was collected by evaporation in vacuo of the filtrate, addition of H20 (250 mL) and filtration. The combined solids were washed successively with H20 (250 mL), EtOH/MeOH (1 : 1, 250 mL) and Et20 (250 mL) then dried in vacuo to afford the triazolopyridine derivative (3) as a solid. The compound was used as such in the next step without any purification.
[00159] lH (400 MHz, DMSO-i¼) δ 7.43-7.34 (2H, m, 2 x aromatic-H), 7.24 (1H, dd, J 6.8 and 1.8 Hz, aromatic-H), 6.30 (2H, br, NH2); m/z 213/215 (1 : 1, M+H+, 100%).
1.1.2. Synthesis of 4-[ 4-(4, 4, 5, 5-Tetramethyl-f 1, 3,2] ‘ dioxaborolan-2-yl) -benzyl] ‘- thiomor holine- 1, 1 -dioxide (Intermediate 4)

[00160] 2-(4-Bromomethyl-phenyl)-4,4,5,5-tetramethyl-[l,3,2]dioxaborolane (1 eq) and DIPEA (2 eq) were dissolved in DCM/MeOH (5:1 v:v) under N2 and thiomorpholine 1,1 -dioxide (2 eq) was added portion wise. The resulting solution was stirred at room temperature for 16h. After this time, the reaction was complete. The solvent was evaporated. The compound was extracted with EtOAc and water, washed with brine and dried over anhydrous MgSO i. Organic layers were filtered and evaporated. The final compound was isolated without further purification.
1.1.3. Synthesis of 5-[4-(l, l-Dioxothiomorpholin-4-ylmethyl)-phenyl]-[l,2,4]triazolo[l,5- a ridin-2-ylamine (Formula I)

[00161] 4-[4-(4,4,5,5-Tetramethyl-[l,3,2]dioxaborolan-2-yl)-benzyl]-thiomorpholine-l,l-dioxide (l .leq.) was added to a solution of 5-bromo-[l,2,4]triazolo[l,5-a]pyrid in-2-ylamine (4: 1). K2CO3 (2 eq.) and PdC^dppf (0.03 eq.) were added to the solution. The resulting mixture was then heated in an oil bath at 90°C for 16h under N2. Water was added and the solution was extracted with ethyl acetate. The organic layers were dried over anhydrous MgSC>4 and evaporated in vacuo. The final compound was obtained after purification by flash chromatography.
[00162] lH (400 MHz, CDC13) δ 7.94-7.92 (d, 2H), 7.52-7.48 (m, 3H), 7.37-7.34 (m, 1H), 7.02-7.00 (m, 1H), 6.00 (d, 2H), 3.76 (d, 2H), 3.15-3.13 (m, 4H), 2.93-2.91 (m, 4H).
[00163] m/z 358.2 (M+H+, 100%). 1.2. Route 2
1.2.1. Cyclopropanecarboxylic acid {5-[4-(l, l-dioxo-thiomorpholin-4-ylmethyl)-phenylJ- [l,2,4]triazolo[l,5-a]pyridin-2-yl}-amide (Formula II)
[00164] The compound according to Formula II may be synthesized according to the procedure described in WO 2010/149769.
1.2.2. Synthesis of 5-[4-(l, l-Dioxothiomorpholin-4-ylmethyl)-phenyl]-[l,2,4]triazolo[l,5- aJpyridin-2-ylamine (Formula I)
[00165] The compound according to Formula I can also be produced by hydrolysis of the compound accor ing to Formula II:

[00166] Hydrochloric acid 30% aq (12.06 kg; 3.9 rel. volumes) was added to a slurry of the compound according to Formula II (3.45 kg; 1.0 equiv.) in demineralized water (10.0 kg; 3.0 rel. volumes). Subsequently, a line rinse was performed with demineralized water (3.4 kg; 1.0 rel. volumes). The reaction mixture was heated to 80±5°C for 14.5 h. After completion of the reaction (conversion > 99%>), the reaction mixture was cooled to 20±5°C. The reaction mixture was diluted with demineralized water (6.8 kg; 2.0 rel. volumes) and sodium hydroxide 33%> aq (9.52 kg; 3.7 rel volumes) was dosed at such a rate that the temperature of the reactor contents remained below 35°C. An additional amount of sodium hydroxide 33%> aq (2.55 kg; 1.0 rel. volumes) was needed to get the pH > 10. The product was filtered off, washed twice with demineralized water (1.5 rel. volumes) and dried under vacuum for 1 h, thus yielding the crude compound according to Formula I.
[00167] The crude compound according to Formula I (5.70 kg) was re-slurried in demineralized water (23.0 kg; 8.5 rel. volumes). Hydrochloric acid 30%> aq (1.65 kg; 0.7 rel. volumes) and demineralized water (4.3 kg; 1.6 rel. volumes) were added and the reaction mixture was stirred at 20±5°C for 45 min. As the compound according to Formula I was not dissolved completely, the reaction mixture was stirred at 45±5°C for 1 h. The reaction mixture was filtered and the residue was washed with demineralized water (2.0 kg 0.75 rel. volumes). Sodium hydroxide 33%> aq (1.12 kg; 0.6 rel volumes) was added to the filtrate. An additional amount of sodium hydroxide 33%> aq (1.01 kg) was needed to get the pH > 10. The resulting reaction mixture was stirred at 20±5°C for about 3 h. The product was filtered off, washed twice with demineralized water (4.1 kg; 1.5 rel. volumes), and twice with methyl tert-butyl ether (MTBE; 3.0 kg; 1.5 rel. volumes) and dried under vacuum for 15.5 h on the filter. The product was further dried in a vacuum oven at 40±5°C for 202 h, thus affording the desired compound according to Formula I.
Update
Scheme 1: General S nthesis of Compounds of Formula I or A

Formula A
Scheme 7.

(16) (17) (18)
(18a): R3a=R3b=R2a=R (18b): R3a=R3b=D; R2a 18c): R3a=R3b=H; R2a

References
- Namour, Florence; Diderichsen, Paul Matthias; Cox, Eugène; Vayssière, Béatrice; Van der Aa, Annegret; Tasset, Chantal; Van’t Klooster, Gerben (2015-02-14). “Pharmacokinetics and Pharmacokinetic/Pharmacodynamic Modeling of Filgotinib (GLPG0634), a Selective JAK1 Inhibitor, in Support of Phase IIB Dose Selection”. Clin Pharmacokinet. Epub ahead of print.doi:10.1007/s40262-015-0240-z.
- Van Rompaey, L; Galien, R; Van der Aar, E; Clement-Lacroix, P; Van der Aar, E; Nelles, L; Smets, B; Lepescheux, L; Cristophe, T; Conrath, K; Vandeghinste, N; Vayssiere, B; De Vos, S; Fletcher, S; Brys, R; Van’t Klooster, G; Feyen, J; Menet, C (2013-10-01). “Preclinical characterization of GLPG0634, a selective inhibitor of JAK1 for the treatment of inflammatory diseases”. J Immunol. 191(7). doi:10.4049/jimmunol.1201348.
- http://acrabstracts.org/abstracts/phase-1-and-phase-2-data-confirm-that-glpg0634-a-selective-jak1-inhibitor-has-a-low-potential-for-drug-drug-interactions/
- “Galapagos’ GLPG0634 shows excellent efficacy and safety in rheumatoid arthritis Phase II study” (PDF) (Press release). Retrieved 2015-02-26.
- “Galapagos reports that the last patient in DARWIN 1 has completed 12 weeks of treatment” (PDF) (Press release). Retrieved 2015-02-26.
- “Galapagos completes recruitment for Darwin 1 study with GLPG0634 (filgotinib) in RA”. EuroInvestor. Retrieved 2015-02-26.
- NASDAQ OMX Corporate Solutions. “Galapagos completes recruitment for Darwin 2 monotherapy study with GLPG0634 (filgotinib) in RA”. Yahoo Finance. Retrieved 2015-02-26.
| US8551980 | Nov 17, 2010 | Oct 8, 2013 | Bayer Intellectual Property Gmbh | Substituted triazolopyridines |
| US8796457 | Jun 25, 2010 | Aug 5, 2014 | Galapagos Nv | Compound useful for the treatment of degenerative and inflammatory diseases |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
N-[5-[4-[(1,1-dioxo-1,4-thiazinan-4-yl)methyl]phenyl]-[1,2,4]triazolo[1,5-a]pyridin-2-yl]cyclopropanecarboxamide
|
|
| Clinical data | |
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Biological half-life | 6 hours[1] |
| Identifiers | |
| CAS Registry Number | 1206161-97-8 |
| ATC code | L01XE18 |
| IUPHAR/BPS | 7913 |
| ChemSpider | 28189566 |
| UNII | 3XVL385Q0M |
| ChEMBL | CHEMBL3301607 |
| Chemical data | |
| Formula | C21H23N5O3S |
| Molecular mass | 425.50402 g/mol |
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये। औकात बस इतनी देना, कि औरों का भला हो जाये।
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE


Join me on twitter

 LIONEL MY SON
LIONEL MY SON
He was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, Now I keep him as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son happy

सुकून उतना ही देना प्रभू, जितने से
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
Read all about Organic Spectroscopy on ORGANIC SPECTROSCOPY INTERNATIONAL 
/////////Galapagos, GLPG0634, Filgotinib, PHASE 2, orphan drug designation, PHASE 3, Crohn’s disease, Rheumatoid arthritis, Ulceraticolitis
ve SMILES code: O=C(C1CC1)NC2=NN3C(C4=CC=C(CN5CCS(CC5)(=O)=O)C=C4)=CC=CC3=N2

