
Home » Posts tagged 'phase 2'
Tag Archives: phase 2
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
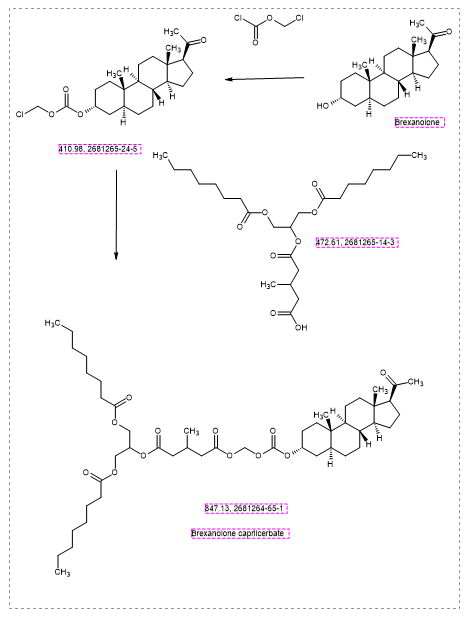
Brexanolone caprilcerbate



Brexanolone caprilcerbate
CAS 2681264-65-1
MFC48H78O12 MW 847.1 g/mol
1-O-[[(3R,5S,8R,9S,10S,13S,14S,17S)-17-acetyl-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-3-yl]oxycarbonyloxymethyl] 5-O-[1,3-di(octanoyloxy)propan-2-yl] 3-methylpentanedioate
1-[1,3-bis(octanoyloxy)propan-2-yl] 5-[({[(20-oxo-5α-pregnan3α-yl)oxy]carbonyl}oxy)methyl] 3-methylpentanedioate
GABAA receptor positive allosteric modulator, K3KLQ9T6WM, PHASE 2,
Brexanolone caprilcerbate (INNTooltip International Nonproprietary Name; developmental code names LYT-300, SPT-300) is an orally active prodrug of brexanolone (allopregnanolone) which is under development for the treatment of anxiety disorders.[1][2][3][4] It is a absorbed via the lymphatic system with oral administration.[5] The drug is being developed by Seaport Therapeutics and PureTech Health.[1][2] As of January 2025, it is in phase 2 clinical trials.[1]

SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US335021515&_cid=P22-MKAJEO-33027-1
PAT
- Lipid prodrugs of neurosteroidsPublication Number: WO-2021159021-A1Priority Date: 2020-02-05
- Lipid prodrugs of neurosteroidsPublication Number: US-2023338552-A1Priority Date: 2020-02-05
- Lipid prodrugs of neurosteroidsPublication Number: US-2022395513-A1Priority Date: 2020-02-05
- Lipid prodrugs of neurosteroidsPublication Number: US-2021268115-A1Priority Date: 2020-02-05



AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
References
- “Allopregnanolone prodrug”. AdisInsight. 28 January 2025. Retrieved 26 February 2025.
- “Delving into the Latest Updates on Brexanolone caprilcerbate with Synapse”. Synapse. 15 February 2025. Retrieved 26 February 2025.
- “Proposed INN: List 131 International Nonproprietary Names for Pharmaceutical Substances (INN)” (PDF). WHO Drug Information. 38 (2): 270. 2024.
brexanolonum caprilcerbas brexanolone caprilcerbate 1-[1,3-bis(octanoyloxy)propan-2-yl] 5-[({[(20-oxo-5α-pregnan3α-yl)oxy]carbonyl}oxy)methyl] 3-methylpentanedioate GABAA receptor positive allosteric modulator C48H78O12 2681264-65-1
- Carlini SV, Osborne LM, Deligiannidis KM (December 2023). “Current pharmacotherapy approaches and novel GABAergic antidepressant development in postpartum depression”. Dialogues in Clinical Neuroscience. 25 (1): 92–100. doi:10.1080/19585969.2023.2262464. PMC 10557560. PMID 37796239.
- Alashal N, Hussain N (2025). “Approach to the use of rescue medications in children for prolonged epileptic seizures in the community”. Paediatrics and Child Health. 35 (4): 113–117. doi:10.1016/j.paed.2025.01.004.
| Clinical data | |
|---|---|
| Other names | LYT-300; LYT300; SPT-300; SPT300; Allopregnanolone 3-O-caprilcerbate |
| Routes of administration | Oral[1] |
| Drug class | GABAA receptor positive allosteric modulator; Neurosteroid |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2681264-65-1 |
| PubChem CID | 158098654 |
| UNII | K3KLQ9T6WM |
| Chemical and physical data | |
| Formula | C48H76O12 |
| Molar mass | 845.124 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
/////////////Brexanolone caprilcerbate, GABAA receptor positive allosteric modulator, K3KLQ9T6WM, PHASE 2,
Privosegtor
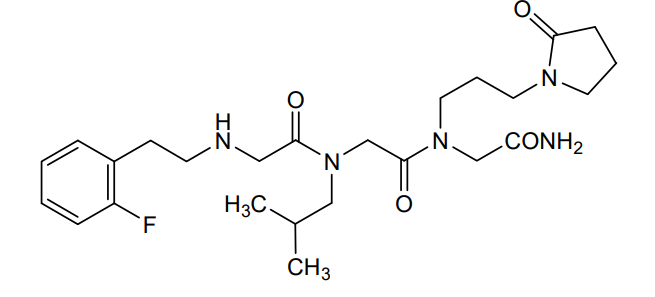
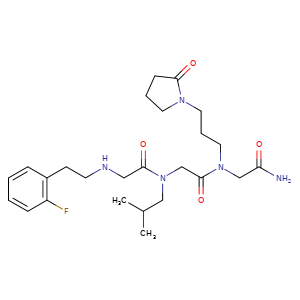
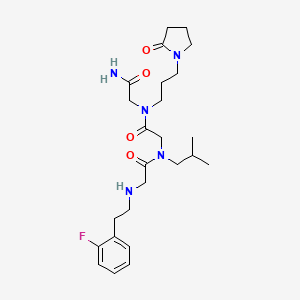
Privosegtor
CAS 1361200-34-1
MF C25H38FN5O4, MW 491.6 g/mol
GLYCINAMIDE, N-(2-(2-FLUOROPHENYL)ETHYL)GLYCYL-N-(2-METHYLPROPYL)GLYCYL-N2-(3-(2-OXO-1-PYRROLIDINYL)PROPYL)-
N-(2-(2-FLUOROPHENYL)ETHYL)GLYCYL-N-(2-METHYLPROPYL)GLYCYL-N2-(3-(2-OXO-1-PYRROLIDINYL)PROPYL)GLYCINAMIDE
N-(2-(2-FLUOROPHENYL)ETHYL)GLYCYL-N-(2-METHYLPROPYL)GLYCYL-N2-(3-(2-OXOPYRROLIDIN-1-YL)PROPYL)GLYCINAMIDE
N-[2-(2-fluorophenyl)ethyl]glycyl-N-(2-methylpropyl)glycyl-N2[3-(2-oxopyrrolidin-1-yl)propyl]glycinamide
serum/ glucocorticoid-regulated kinase 2 (Sgk2) activator, Phase 2, Optic neuritis, orphan drug, BN-201, BN 201, G-79, G 79, KCN37L7EIH
- OriginatorBionure
- DeveloperBionure; Oculis Pharma
- ClassAnti-inflammatories; Antiglaucomas; Eye disorder therapies; Neuroprotectants; Peptides; Small molecules
- Mechanism of ActionBrain derived neurotrophic factor agonists; Insulin-like growth factor I stimulants; Neuron modulators; Serum-glucocorticoid regulated kinase stimulants
- Orphan Drug StatusYes – Optic neuritis
- Phase IIOptic neuritis
- PreclinicalMultiple sclerosis; Neurotrophic keratopathy
- No development reportedGlaucoma; Neuromyelitis optica
- 06 Oct 2025Oculis Holding plans the PIONEER-2 trial in Optic neuritis in first half of 2026
- 06 Oct 2025Oculis Holding plans the PIONEER-3 trial in Optic nerve disorders in mid-2026
- 06 Oct 2025Oculis Holding completes End-of-phase II meeting with US FDA and receives positive feedback for registrational PIONEER program in Optic neuritis and Optic nerve disorders
OCS-05 in Patients With Optic Neuritis
CTID: NCT04762017
Phase: Phase 2
Status: Completed
Date: 2025-09-22
N-[2-[(2-amino-2-oxoethyl)-[3-(2-oxopyrrolidin-1-yl)propyl]amino]-2-oxoethyl]-2-[2-(2-fluorophenyl)ethylamino]-N-(2-methylpropyl)acetamide (BN201) is a small peptide molecule, a first-in-class neuroprotective compound. BN201 promotes the survival of cultured neural cells when subjected to oxidative stress or when deprived of trophic factors. BN201 promotes neuronal differentiation, the differentiation of precursor cells to mature oligodendrocytes in vitro, and the myelination of new axons. BN201 modulates several kinases participating in the insulin growth factor 1 pathway including serum-glucocorticoid kinase and midkine, inducing the phosphorylation of NDRG1 and the translocation of the transcription factor Foxo3 to the cytoplasm. In vivo, BN201 prevents axonal and neuronal loss, and it promotes remyelination in models of multiple sclerosis, chemically induced demyelination, and glaucoma. Bionure, a spin-off from Hospital Clínic de Barcelona that is based in California, is developing BN201 for multiple sclerosis, acute optic neuritis (AON) and glaucoma. BN201 was granted with orphan designation status for optic neuritis by the FDA. Optic neuritis is often an early sign of multiple sclerosis. The efficacy, safety, and capacity of the drug to cross the blood-brain barrier have been demonstrated in animal models, but the drug has not yet entered clinical testing.
PAT
Agonists of neurotrophin receptors and their use as medicaments
Publication Number: WO-2012028959-A1
Priority Date: 2010-08-31
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2012028959&_cid=P10-MIDYQ0-58943-1
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021084013&_cid=P10-MIDYSN-60542-1
In another embodiment, optionally in combination with one or more features of the various embodiments described above or below throughout all the description, the compound of formula (I) is selected from the group consisting of G79 ([N-(2-(2′-fluorophenyl)ethyl)- glycyl]-[N-(2-methylpropyl)-glycyl]-N-[3-(2′-oxopyrrolidinyl)-propyl]glycinamide, BN201 , Chemical Formula: C25H38FN5O4; MW 491.5987), G-80 ([N-(2-(2′-fluorophenyl)ethyl)- glycyl]-[N-(2-methyl-propyl)glycyl]-N-[2-(4′-sulfamoyl-phenyl)ethyl]glycinamide, BN 119, Chemical Formula: C26H36FN5O5S; MW 549.658) and G81 ([N-(2-(1 -pyrrolidinyl)ethyl)- glycyl]-[N-(2-methyl-propyl)glycyl]-N-[2-(4′-sulfamoyl-phenyl)ethyl]glycinamide, BN 120, Chemical Formula: C24H4oN6OS; MW 524.6766):
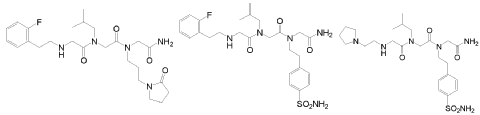
G79 (BN201) G80 (BN119) G81 (BN120)
Compounds of formula (I) can be prepared as disclosed in WO2012028959.
PAT
- Agonists of Neurotrophin Receptors and Their Use as MedicamentsPublication Number: US-2012052094-A1Priority Date: 2010-08-31
- Agonists of Neurotrophin Receptors and Their Use as MedicamentsPublication Number: US-2015005239-A1Priority Date: 2010-08-31
- Agonists of neurotrophin receptors and their use as medicamentsPublication Number: US-2017121367-A1Priority Date: 2010-08-31
- Agonists of neurotrophin receptors and their use as medicamentsPublication Number: US-8791076-B2Priority Date: 2010-08-31Grant Date: 2014-07-29
- Agonists of neurotrophin receptors and their use as medicamentsPublication Number: US-9453047-B2Priority Date: 2010-08-31Grant Date: 2016-09-27
- Combination Therapy Methods, Compositions and KitsPublication Number: KR-20220109378-APriority Date: 2019-07-03
- Combination therapy methods, compositions and kitsPublication Number: US-2022378866-A1Priority Date: 2019-07-03
- Agonists of neurotrophin receptors and their use as medicamentsPublication Number: EP-2611775-A1Priority Date: 2010-08-31
- Agonists of neurotrophin receptors and their use as medicamentsPublication Number: EP-2611775-B1Priority Date: 2010-08-31Grant Date: 2016-03-16
- Agonists of neurotrophin receptors and their use as medicamentsPublication Number: US-10106577-B2Priority Date: 2010-08-31Grant Date: 2018-10-23
- Combination therapy methods, compositions and kitsPublication Number: WO-2021001464-A1Priority Date: 2019-07-03
- Combination therapy methods, compositions and kitsPublication Number: AU-2020298782-A1Priority Date: 2019-07-03
- Combination therapy methods, compositions and kitsPublication Number: CN-114206329-APriority Date: 2019-07-03
- Combination therapy methods, compositions and kitsPublication Number: EP-3993784-A1Priority Date: 2019-07-03
- Combination therapy methods, compositions and kitsPublication Number: JP-2022539999-APriority Date: 2019-07-03
- Boron-nitrogen compound, organic electroluminescence composition, and organic electroluminescence device containing samePublication Number: WO-2022121951-A1Priority Date: 2020-12-10
- New treatment regimen for the treatment of neurological diseases or conditionsPublication Number: WO-2021084013-A1Priority Date: 2019-10-30
- Novel Therapeutic Approaches for the Treatment of Neurological Diseases or ConditionsPublication Number: CN-115052595-APriority Date: 2019-10-30
- New treatment regimen for the treatment of neurological diseases or conditionsPublication Number: EP-4051263-A1Priority Date: 2019-10-30
- New treatment regiment for the treatment of neurological diseases or conditionsPublication Number: US-2022387385-A1Priority Date: 2019-10-30
- A plant zinc-increasing compound inoculant and its preparation method and applicationPublication Number: CN-117286034-APriority Date: 2023-09-11
- A plant zinc-enhancing composite bacterial agent and its preparation method and applicationPublication Number: CN-117286034-BPriority Date: 2023-09-11Grant Date: 2024-11-15
- Compound, pharmaceutical composition comprising the same, and process for synthesizing the samePublication Number: TW-202432095-APriority Date: 2022-12-22
- Synthesis of small molecule agonists of neuroptrophinPublication Number: WO-2024133860-A1Priority Date: 2022-12-22
- Boron-nitrogen compound, organic electroluminescent composition and organic electroluminescent device containing samePublication Number: WO-2022121920-A1Priority Date: 2020-12-10
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
- Development and validation of PAMPA-BBB QSAR model to predict brain penetration potential of novel drug candidatesPublication Name: Frontiers in PharmacologyPublication Date: 2023-12-01PMCID: PMC10722238PMID: 38108064DOI: 10.3389/fphar.2023.1291246
- A Phase 1 randomized study on the safety and pharmacokinetics of OCS-05, a neuroprotective disease modifying treatment for Acute Optic Neuritis and Multiple SclerosisPublication Name: Scientific ReportsPublication Date: 2023-03-29PMCID: PMC10060579PMID: 36991169DOI: 10.1038/s41598-023-32278-0
- Retrospective assessment of rat liver microsomal stability at NCATS: data and QSAR modelsPublication Name: Scientific ReportsPublication Date: 2020-11-26PMCID: PMC7693334PMID: 33244000DOI: 10.1038/s41598-020-77327-0
- A High-Throughput Screen of a Library of Therapeutics Identifies Cytotoxic Substrates of P-glycoproteinPublication Name: Molecular PharmacologyPublication Date: 2019-11PMCID: PMC6790066PMID: 31515284DOI: 10.1124/mol.119.115964
- Predictive models of aqueous solubility of organic compounds built on A large dataset of high integrityPublication Name: Bioorganic & Medicinal ChemistryPublication Date: 2019-07-15PMCID: PMC8274818PMID: 31176566DOI: 10.1016/j.bmc.2019.05.037
/////////Privosegtor, Phase 2, Optic neuritis, orphan drug, BN-201, BN 201, G-79, G 79, KCN37L7EIH
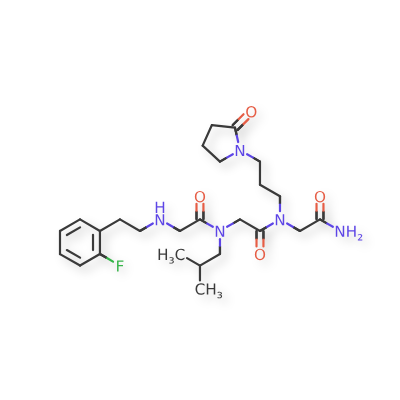
Pilavapadin
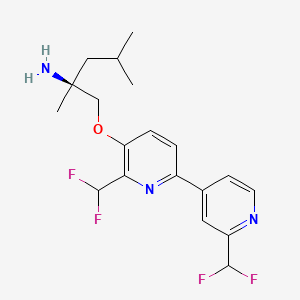

Pilavapadin
CAS1815613-42-3
MFC19H23F4N3O MW 385.4 g/mol
(2S)-1-{[2′,6-bis(difluoromethyl)[2,4′-bipyridin]-5-yl]oxy}-2,4-dimethylpentan-2-amine
(2S)-1-[[2-(difluoromethyl)-6-[2-(difluoromethyl)-4-pyridinyl]-3-pyridinyl]oxy]-2,4-dimethylpentan-2-amine
adaptor protein 2-associated kinase 1 (AAK1) inhibitor, LX9211, BMS-986176, LX 9211, BMS 986176, Phase 2, Neuropathic pain, Postherpetic neuralgia, AAK1-IN-1, 9G4RLM5X6Z
Pilavapadin (also known as LX9211 or BMS-986176) is an investigational, orally available small molecule developed by Lexicon Pharmaceuticals for the treatment of neuropathic pain, primarily diabetic peripheral neuropathic pain (DPNP).
Key Information
- Mechanism of Action: Pilavapadin is a selective inhibitor of AAK1 (AP2 associated kinase 1), a novel target identified through Lexicon’s gene science research. It is designed to inhibit the reuptake and recycling of neurotransmitters involved in pain signaling in the central nervous system without affecting opiate pathways.
- Indication: It is being investigated for the management of chronic and debilitating conditions such as diabetic peripheral neuropathic pain (DPNP), chemotherapy-induced peripheral neuropathy (CIPN), and multiple sclerosis (MS) pain.
- Development Stage: Pilavapadin has completed Phase 2 clinical trials for DPNP and is expected to advance to a Phase 3 trial.
- Status/Designation: The U.S. Food and Drug Administration (FDA) has granted Fast Track designation for the development of pilavapadin in DPNP.
Clinical Trial Results
Phase 2 studies (RELIEF-DPN-1 and PROGRESS) demonstrated that pilavapadin can provide meaningful pain reduction in adults with DPNP.
- In a post-hoc analysis of the PROGRESS study, the 10 mg dose was found to be effective, achieving a clinically meaningful, two-point reduction in average daily pain scores from baseline, with an acceptable safety and tolerability profile.
- The data has been presented at several medical meetings, including the European Association for the Study of Diabetes (EASD).
- OriginatorBristol-Myers Squibb; Lexicon Pharmaceuticals
- DeveloperLexicon Pharmaceuticals
- ClassAnalgesics; Small molecules
- Mechanism of ActionAdaptor-associated kinase 1 inhibitors
- Phase IINeuropathic pain; Postherpetic neuralgia
- 20 Jun 2025Updated efficacy data from the phase II PROGRESS trial in Neuropathic pain presented at 85th Annual Scientific Sessions of the American Diabetes Association (ADA-2025)
- 13 May 2025Lexicon Pharmaceuticals plans an End of Phase 2 meeting with FDA for Pilavapadin
- 13 May 2025Updated efficacy data from the phase II PROGRESS trial in Neuropathic pain released by Lexicon Pharmaceuticals
- A Dose-ranging Study in Patients With Diabetic Peripheral Neuropathic Pain (DPNP)CTID: NCT06203002Phase: Phase 2Status: CompletedDate: 2025-08-29
- Efficacy, Safety, and PK of LX9211 in Participants With Diabetic Peripheral Neuropathic PainCTID: NCT04455633Phase: Phase 2Status: CompletedDate: 2025-06-25
- Efficacy and Safety of LX9211 in Participants With Postherpetic NeuralgiaCTID: NCT04662281Phase: Phase 2Status: CompletedDate: 2023-11-18
Molecular FormulaC19H23F4N3O.H3O4P
Molecular Weight483.4
CAS 2977251-24-2
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US215884039&_cid=P11-MI4ESM-19570-1
Example 123
(S)-1-((2′,6-bis(difluoromethyl)-[2,4′-bipyridin]-5-yl)oxy)-2,4-dimethylpentan-2-amine

Part A: (2-(difluoromethyl)pyridin-4-yl)boronic acid

Part B: (S)-1-((2′,6-bis(difluoromethyl)-[2,4′-bipyridin]-5-yl)oxy)-2,4-dimethylpentan-2-amine
SYN
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021216441&_cid=P11-MI4EP8-16561-2
REF
- Discovery and Optimization of Biaryl Alkyl Ethers as a Novel Class of Highly Selective, CNS-Penetrable, and Orally Active Adaptor Protein-2-Associated Kinase 1 (AAK1) Inhibitors for the Potential Treatment of Neuropathic PainPublication Name: Journal of Medicinal ChemistryPublication Date: 2022-03-09PMID: 35261239DOI: 10.1021/acs.jmedchem.1c02132
- Discovery of (S)-1-((2′,6-Bis(difluoromethyl)-[2,4′-bipyridin]-5-yl)oxy)-2,4-dimethylpentan-2-amine (BMS-986176/LX-9211): A Highly Selective, CNS Penetrable, and Orally Active Adaptor Protein-2 Associated Kinase 1 Inhibitor in Clinical Trials for the Treatment of Neuropathic PainPublication Name: Journal of Medicinal ChemistryPublication Date: 2022-03-08PMID: 35257579DOI: 10.1021/acs.jmedchem.1c02131
- Discovery, Structure–Activity Relationships, and In Vivo Evaluation of Novel Aryl Amides as Brain Penetrant Adaptor Protein 2-Associated Kinase 1 (AAK1) Inhibitors for the Treatment of Neuropathic PainPublication Name: Journal of Medicinal ChemistryPublication Date: 2021-07-16PMID: 34270254DOI: 10.1021/acs.jmedchem.1c00472
PAT
- Biaryl kinase inhibitorsPublication Number: US-2021277001-A1Priority Date: 2014-04-02
- Biaryl kinase inhibitorsPublication Number: KR-102379518-B1Priority Date: 2014-04-02Grant Date: 2022-03-25
- Biaryl kinase inhibitorsPublication Number: US-12065437-B2Priority Date: 2014-04-02Grant Date: 2024-08-20
- Biaryl kinase inhibitorsPublication Number: US-2024360131-A1Priority Date: 2014-04-02
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
////////////Pilavapadin, LX9211, BMS-986176, LX 9211, BMS 986176, Phase 2, Neuropathic pain, Postherpetic neuralgia, AAK1-IN-1, 9G4RLM5X6Z
Matsupexole
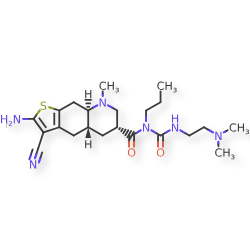
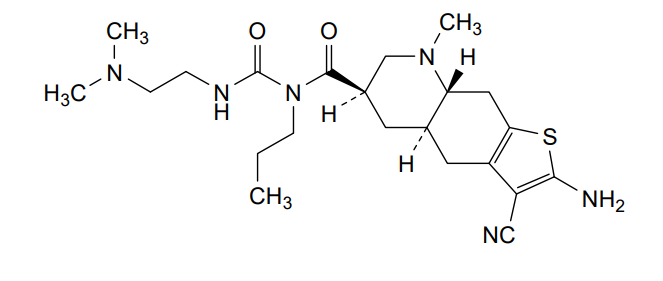
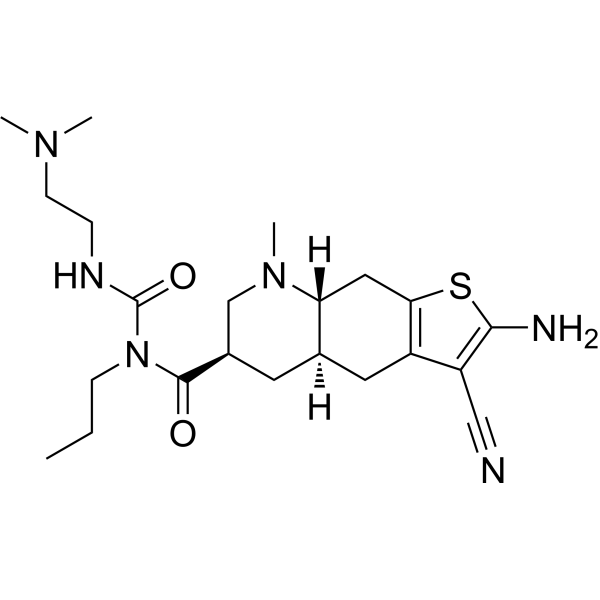

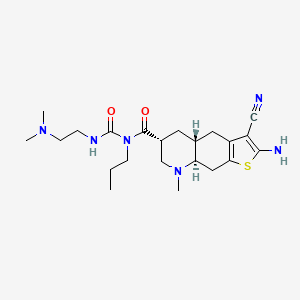
Matsupexole
CAS 1399442-97-7
MF C22H34N6O2S, Molecular Weight, 446.61
(4aR,6R,8aR)-2-amino-3-cyano-N-{[2-(dimethylamino)ethyl]carbamoyl}-8-methyl-N-propyl 4,4a,5,6,7,8,8a,9-octahydrothieno[3,2-g]quinoline-6-carboxamide
(4aR,6R,8aR)-2-amino-3-cyano-N-[2-(dimethylamino)ethylcarbamoyl]-8-methyl-N-propyl-4a,5,6,7,8a,9-hexahydro-4H-thieno[3,2-g]quinoline-6-carboxamide
dopamine receptor agonist, Phase 2, Parkinson’s disease, K4UEG65HTX
- OriginatorKissei Pharmaceutical
- DeveloperAffaMed Therapeutics; Kissei Pharmaceutical
- ClassAmides; Amines; Antiparkinsonians; Dimethylamines; Ethylenediamines; Nitriles; Quinolines; Small molecules; Thiophenes; Urea compounds
- Mechanism of ActionDopamine receptor agonists
- Phase IIParkinson’s disease
- 28 Aug 2025Chemical structure information added.
- 06 Sep 2021Kissei Pharmaceutical completes a phase II trial in Parkinson’s disease (In adults, In elderly) in Japan (PO) (NCT04867551)
- 04 Aug 2021Phase-II clinical trials in Parkinson’s disease in China (PO) (Kissei Pharmaceutical pipeline, August 2021)
PAT
- Novel octahydrothienoquinoline derivative, pharmaceutical composition comprising derivative, and use of thesePublication Number: WO-2012124649-A1Priority Date: 2011-03-14
- COMPOUNDS DERIVED FROM OCTAIDROTHIENOQUINOLINE, PHARMACEUTICAL COMPOSITION AND PHARMACEUTICAL AGENT COMPRISING SUCH COMPOUNDSPublication Number: BR-112013023575-B1Priority Date: 2011-03-14
- New octahydrothienoquinoline derivative, pharmaceutical composition containing derivative, and using themPublication Number: RU-2573399-C2Priority Date: 2011-03-14Grant Date: 2016-01-20
- Novel octahydrothienoquinoline derivative, pharmaceutical composition comprising derivative, and use of thesePublication Number: SG-193400-A1Priority Date: 2011-03-14
- Novel octahydrothienoquinoline derivatives, pharmaceutical compositions containing the same, and their usesPublication Number: TW-I537274-BPriority Date: 2011-03-14Grant Date: 2016-06-11
- Novel octahydrothienoquinoline derivative, pharmaceutical composition comprising derivative, and use of thesePublication Number: US-2014243311-A1Priority Date: 2011-03-14
- Octahydrothienoquinoline derivative, pharmaceutical composition comprising derivative, and use of thesePublication Number: US-9138434-B2Priority Date: 2011-03-14Grant Date: 2015-09-22
- Novel octahydrothienoquinoline derivative, pharmaceutical composition comprising derivative, and use of thesePublication Number: HU-E033449-T2Priority Date: 2011-03-14
- Novel octahydrothienoquinoline derivatives, pharmaceutical compositions containing them and their usePublication Number: JP-5563716-B2Priority Date: 2011-03-14Grant Date: 2014-07-30
- Novel octahydrothienoquinoline derivatives, pharmaceutical compositions containing them and their usePublication Number: JP-WO2012124649-A1Priority Date: 2011-03-14
- Novel octahydrothienoquinoline derivatives, pharmaceutical compositions containing them and uses thereofPublication Number: KR-20140010137-APriority Date: 2011-03-14
- NEW OCTAHYDROTHYENOCHINOLINE DERIVATIVE, PHARMACEUTICAL COMPOSITION CONTAINING A DERIVATIVE AND THEIR APPLICATIONPublication Number: RU-2013145799-APriority Date: 2011-03-14
- Novel octahydrothienoquinoline derivatives, pharmaceutical compositions comprising said derivatives and their usesPublication Number: CN-103443106-BPriority Date: 2011-03-14Grant Date: 2015-09-30
- New octahydrothienoquinoline derivative, pharmaceutical composition comprising the derivative, and use thereofPublication Number: DK-2687532-T3Priority Date: 2011-03-14Grant Date: 2017-02-20
- Novel octahydrothienoquinoline derivative, pharmaceutical composition comprising derivative, and use of thesePublication Number: EP-2687532-A1Priority Date: 2011-03-14
- Novel octahydrothienoquinoline derivative, pharmaceutical composition comprising derivative, and use of thesePublication Number: EP-2687532-B1Priority Date: 2011-03-14Grant Date: 2016-12-14
- Novel octahydrothienoquinoline derivative, pharmaceutical composition comprising the derivative, and use thereofPublication Number: ES-2613658-T3Priority Date: 2011-03-14Grant Date: 2017-05-25
- Novel dopamine D2 receptor agonistPublication Number: JP-2014074013-APriority Date: 2012-09-12
- Novel dopamine D2 receptor agonistPublication Number: JP-6177061-B2Priority Date: 2012-09-12Grant Date: 2017-08-09
- Novel octahydrothienoquinoline derivative, pharmaceutical composition comprising derivative, and use of thesePublication Number: AU-2012227428-A1Priority Date: 2011-03-14
- Novel octahydrothienoquinoline derivative, pharmaceutical composition comprising derivative, and use of thesePublication Number: AU-2012227428-B2Priority Date: 2011-03-14Grant Date: 2016-05-05
- Novel octahydrothienoquinoline derivatives, pharmaceutical compositions comprising said derivatives and their usesPublication Number: CN-103443106-APriority Date: 2011-03-14
- Succinate of octahydrothienoquinoline compound, and crystals thereofPublication Number: WO-2022009815-A1Priority Date: 2020-07-06
- Succinate of octahydrothienoquinoline compound and its crystalPublication Number: CN-115803329-APriority Date: 2020-07-06
- Succinate salts of octahydrothienoquinoline compounds and crystals thereofPublication Number: KR-20230035050-APriority Date: 2020-07-06
- Succinate of octahydrothienoquinoline compound, and crystals thereofPublication Number: EP-4177257-A1Priority Date: 2020-07-06
- Succinate salts of octahydrothienoquinoline compound and crystals thereofPublication Number: US-2023286998-A1Priority Date: 2020-07-06
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2022009815&_cid=P22-MHO952-66657-1
[0018]Example 11-{[(4aR,6R,8aR)-2-amino-3-cyano-8-methyl-4,4a,5,6,7,8,8a,9-octahydrothieno[3,2-g]quinolin-6-yl]carbonyl}-3-[2-(dimethylamino)ethyl]-1-propylurea sesquisuccinate monohydrate (Form I crystals of salt (A-1)) 102.8 g of acetone was added to 1-{[(4aR,6R,8aR)-2-amino-3-cyano-8-methyl-4,4a,5,6,7,8,8a,9-octahydrothieno[3,2-g]quinolin-6-yl]carbonyl}-3-[2-(dimethylamino)ethyl]-1-propylurea (22.00 g), the mixture was suspended, and the suspension was heated and stirred at an external temperature of 52°C to dissolve the suspension. Activated carbon (2.2 g) was added to this solution and stirred for 10 minutes. This suspension was hot filtered and washed with 35.2 g of acetone. 220.0 g of acetone was then added, and the reaction solution was heated to an external temperature of 52°C and stirred. Next, 44.0 g of water was added to the reaction solution. Separately, 8.73 g of succinic acid was dissolved in a mixed solution of 156.1 g of acetone and 19.8 g of water. This succinic acid solution was added dropwise to the reaction solution over approximately 10 minutes. The dropping funnel was washed with a mixed solution of 17.4 g of acetone and 2.2 g of water and then added dropwise to the reaction solution. The reaction solution was stirred at an internal temperature of 50°C for 1 hour and cooled to 15°C over 30 minutes. The reaction solution was stirred at an external temperature of 10°C for 2 hours, and the crystals were collected by filtration. The crystals were washed twice with 52.8 g of acetone. The obtained wet crystals were dried under reduced pressure at 50°C for 37 hours and then returned to room temperature under reduced pressure over 3 hours. The crystals were stored under air for 24 hours to obtain crystals (27.75 g) of the title compound.
1 H-NMR (DMSO-d6) (δ (ppm)): 0.85 (3H, t, J = 7.4Hz), 1.32 (1H, ddd, J=12.2Hz, 12.2Hz, 12.2Hz), 1.42-1.57 (2H, m), 1.57-1.70 (1H, m ), 1.89-2.00 (2H, m), 2.20-2.13 (1H, m), 2.13-2.28 (2H, m), 2.21 (3H, s), 2.24 (6H, s ), 2.35-2.48 (1H, m), 2.40 (6H, s), 2.46 (2H, t, J = 6.4Hz), 2.81-2.96 (2H, m), 3.00-3 .12 (1H, m), 3.21-3.33 (2H, m), 3.47-3.66 (2H, m), 6.99 (2H, s), 8.50-8.90 (1H, br).
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2012124649&_cid=P22-MHO8UB-55660-1
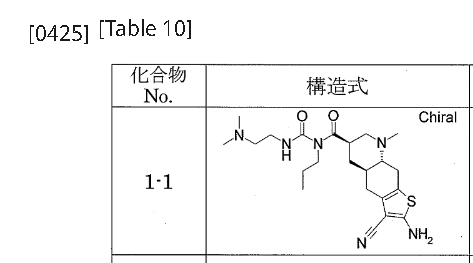
[0422]Example 1-11-{[(4aR,6R,8aR)-2-amino-3-cyano-8-methyl-4H,4aH,5H,6H,7H,8H,8aH,9H-thieno[3,2-g]quinolin-6-yl]carbonyl}-3-[2-(dimethylamino)ethyl]-1-propylurea (Compound 1-1) To a mixture of 1-{[(3R,4aR,8aR)-1-methyl-6-oxodecahydroquinolin-3-yl]carbonyl}-3-[2-(dimethylamino)ethyl]-1-propylurea (Reference Example 10-1) (1.602 g) and ethanol (44 mL) were added malononitrile (435 mg), morpholine (0.572 mL), and then elemental sulfur (282 mg) with stirring at room temperature, and the mixture was heated to 55°C and stirred for 1.5 hours. After cooling to room temperature, the reaction mixture was concentrated under reduced pressure, and the residue was purified by column chromatography on aminopropyl silica gel (eluent: 0%-5% methanol/ethyl acetate, gradient elution) to give the title compound (1.479 g) as a solid.
1 H-NMR (CDCl
3 ) δ ppm: 0.94(3H, t, J=7.4Hz), 1.45-1.85(4H, m), 1.95-2.15(2H, m), 2.15-2.30(7H, m), 2.30-2.55(7H, m), 2.60-2.75(1H, m), 2.90-3.00(2H, m), 3.00-3.10(1H, m), 3.35-3.45(2H, m), 3.60-3.85(2H, m), 4.65(2H, s), 9.27(1H, br)[α]
D 29 =-105.54°(c=0.30, MeOH)
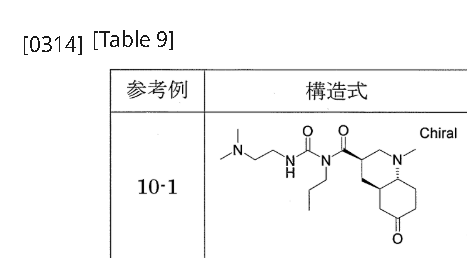
[0311]Reference Example 10-11-{[(3R,4aR,8aR)-1-methyl-6-oxodecahydroquinolin-3-yl]carbonyl}-3-[2-(dimethylamino)ethyl]-1-propylurea 1-{[(3’R,4’aR,8’aR)-1′-methyloctahydro-1’H-spiro[1,3-dioxolane-2,6′-quinoline]-3′-yl]carbonyl}-3-[2-(dimethylamino)ethyl]-1-propylurea (Reference Example 8-1) (2.366 g) was added to 2 mol/L hydrochloric acid (30 mL), and the mixture was stirred at room temperature for 2 hours. The reaction mixture was washed with diethyl ether, and then potassium carbonate was added to the aqueous layer to make it alkaline. The mixture was extracted with a methylene chloride/methanol mixed solvent (methylene chloride:methanol = 9:1). The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure to give the title compound (1.605 g).
1 H-NMR (CDCl
3 ) δ ppm: 0.94 (3H, t, J=7.4 Hz), 1.45-1.90 (6H, m), 1.95-2.05 (1H, m), 2.10-2.55 (17H, m), 2.90-3.10 (2H, m), 3.30-3.45 (2H, m), 3.60-3.80 (2H, m), 9.22 (1H, brs).[α]
D 28 =-37.56° (c=0.38, MeOH).
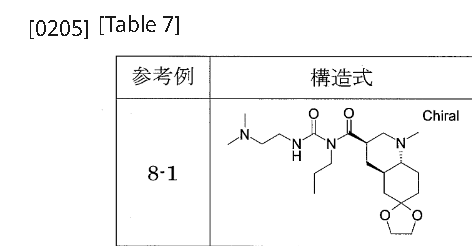
[0198]Reference Example 8-1To a mixture of phenyl 1-{[(3’R,4’aR,8’aR)-1′-methyloctahydro-1’H-spiro[1,3-dioxolane-2,6′-quinoline]-3′-yl]carbonyl}-3-[2-(dimethylamino)ethyl]-1-propylurea N-{[(3’R,4’aR,8’aR)-1′-methyloctahydro-1’H-spiro[1,3-dioxolane-2,6′-quinoline]-3′-yl]carbonyl}-N-propylcarbamate (Reference Example 6-1) (2.401 g) and 2-propanol (30 mL), N,N-dimethylethylenediamine (1.26 mL) was added with stirring at room temperature, and the mixture was heated to 53°C and stirred for 13 hours. After cooling to room temperature, the reaction mixture was concentrated under reduced pressure. The residue was purified by aminopropyl silica gel column chromatography (eluent: 0%-100% ethyl acetate/hexane, gradient elution) to give the title compound (2.383 g).
1 H-NMR (CDCl
3 ) δ ppm: 0.92(3H, t, J=7.4Hz), 1.35-1.50(3H, m), 1.50-1.90(8H, m), 2.00-2.15(1H, m), 2.26(6H, s), 2.31(3H, s), 2.37(1H, t, J=11.2Hz), 2.46(2H, t, J=6.4Hz), 2.85-3.10(2H, m), 3.35-3.45(2H, m), 3.60-3.70(1H, m), 3.70-3.80(1H, m), 3.90-4.00(4H, m), 9.33(1H, br)[α]
D 28 =-6.62°(c=0.31, MeOH)
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
///////matsupexole, dopamine receptor agonist, Phase 2, Parkinson’s disease, K4UEG65HTX
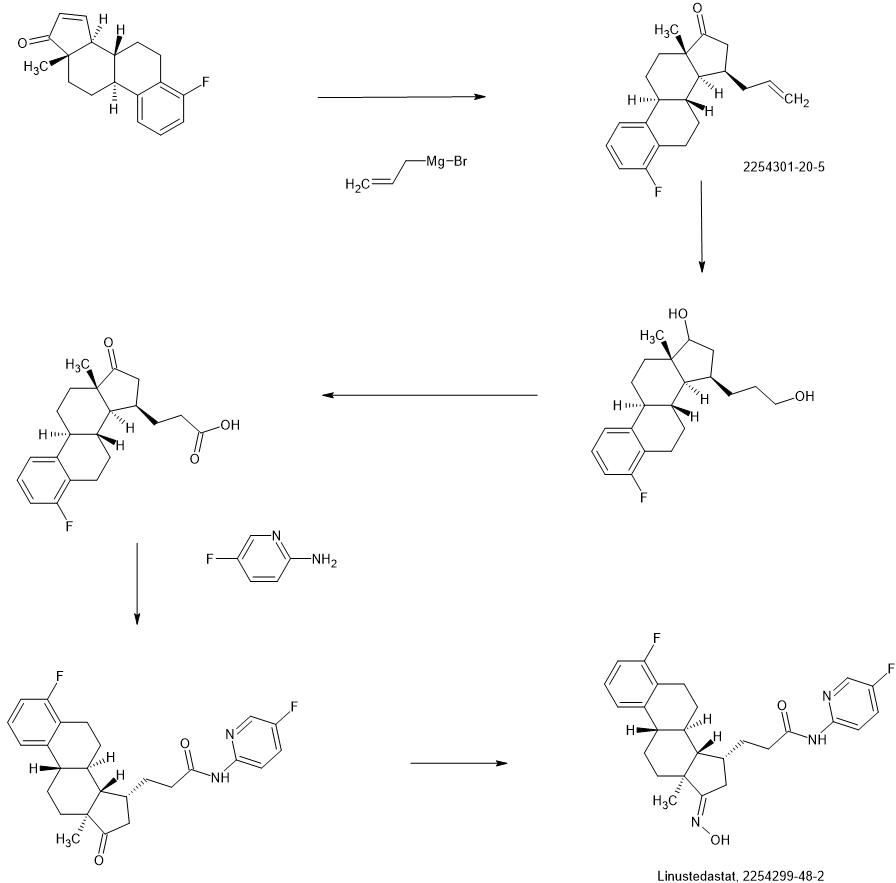
Linustedastat
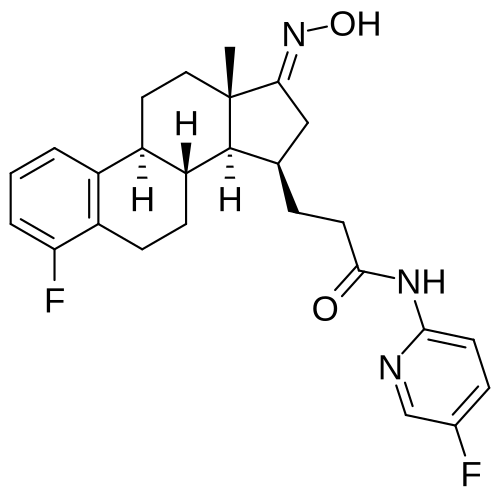
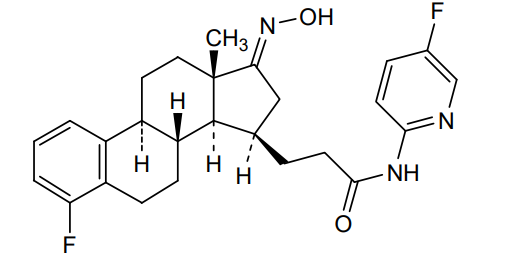
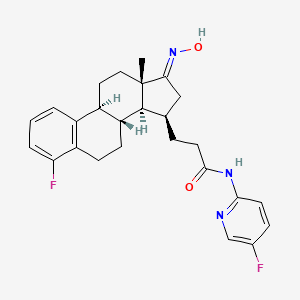
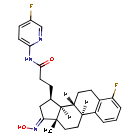
Linustedastat
CAS 2254299-48-2
MFC26H29F2N3O2 MW 453.5 g/mol
FOR-6219, OG-6219, FOR 6219, OG 6219, PP3PLL7GZY, Phase 2, Endometriosis
3-[(8R,9S,13S,14S,15R,17E)-4-fluoro-17-hydroxyimino-13-methyl-7,8,9,11,12,14,15,16-octahydro-6H-cyclopenta[a]phenanthren-15-yl]-N-(5-fluoro-2-pyridinyl)propanamide
- (15beta,17E)-4-Fluoro-N-(5-fluoro-2-pyridinyl)-17-(hydroxyimino)estra-1,3,5(10)-triene-15-propanamide
- 3-[(17E)-4-fluoro-17-(hydroxyimino)estra-1,3,5(10)-trien-15beta-yl]-N-(5-fluoropyridin-2-yl)propanamide
- Estra-1,3,5(10)-triene-15-propanamide, 4-fluoro-N-(5-fluoro-2-pyridinyl)-17-(hydroxyimino)-, (15beta,17E)-
3-[(17E)-4-fluoro-17-(hidroxiimino)estra-1,3,5(10)-trien-15β-il]-N-(5-fluoropiridin-2-il)propanamida
inhibidor de la hidroxiesteroide 17-beta deshidrogenasa 1(HSD17B1)
- OriginatorHormos Medical; Solvay Pharmaceuticals B.V.; University of Turku
- DeveloperOrganon
- ClassSmall molecules
- Mechanism of ActionEstradiol dehydrogenase inhibitors
- Phase IIEndometriosis
- 02 Jul 2025Efficacy data from the phase II ELENA trial in Endometriosis released by Organon
- 28 May 2025Organon completes a phase-II clinical trials in Endometriosis (In adults) in Latvia, Sweden, Poland, Italy, France, Hungary, Germany, Czech Republic, Czech Republic, Bulgaria, Belgium, USA (PO) (NCT05560646)
- 28 Nov 2023No recent reports of development identified for phase-I development in Endometriosis(In volunteers) in United Kingdom (PO)
Linustedastat (developmental code names FOR-6219 and OG-6219) is a 17β-hydroxysteroid dehydrogenase 1 (17β-HSD1; HSD17B1) inhibitor which is under development for the treatment of endometriosis.[1][2][3][4][5] It is a steroidal compound derived from estrone and works by preventing the formation of the more potent estrogen estradiol from the minimally active precursor estrone.[1][2][5] This in turn results in antiestrogenic effects that may be useful in the treatment of estrogen-dependent conditions.[1][2][5] As of November 2023, the drug is in phase 2 clinical trials for endometriosis.[1][2] It is also under preclinical investigation for treatment of breast cancer and endometrial cancer.[5]
A Study to Investigate Efficacy and Safety of OG-6219 BID in 3 Dose Levels Compared With Placebo in Participants Aged 18 to 49 With Moderate to Severe Endometriosis-related Pain
CTID: NCT05560646
Phase: Phase 2
Status: Completed
Date: 2025-05-29
Pat
WO2018224736
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018224736&_cid=P21-MHFVBM-49409-1
Compound 26
3-((13S,15R,E)-3-fluoro-17-(hydroxyimino)-13-methyl-7,8,9,11,12,13,14,15,16,17-decahydro-6H-cyclopenta[a]phenanthren-15-yl)-N-(5-fluoropyridin-2-yl)propanamide
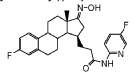
Example 26 was prepared in 94% yield from the compound 25 by the same method as with Example 2 in three hours reaction time.
1H NMR (200 MHz, DMSO-d6): 1.03 (s, 3 H), 1.12 – 2.48 (m, 15 H), 2.57 – 2.78 (m, 1 H), 2.80 – 2.95 (m, 2 H), 6.79 – 7.01 (m, 2 H), 7.18 – 7.38 (m, 1 H), 7.72 (td, 1 H), 8.15 (dd, 1 H), 8.31 (d, 1 H), 10.18 (s, 1 H), 10.64 (s, 1 H). MS m/z (TOF ES+): 454 (M+1).
SYNTHESIS
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
| Clinical data | |
|---|---|
| Other names | FOR-6219; OG-6219; 3-[(17E)-4-Fluoro-17-(hydroxyimino)estra-1,3,5(10)-trien-15β-yl]-N-(5-fluoropyridin-2-yl)propanamide |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2254299-48-2 |
| PubChem CID | 171390018 |
| UNII | PP3PLL7GZY |
| KEGG | D13078 |
| Chemical and physical data | |
| Formula | C26H29F2N3O2 |
| Molar mass | 453.534 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- “FOR 6219”. AdisInsight. 28 November 2023. Retrieved 15 August 2024.
- “Delving into the Latest Updates on Linustedastat with Synapse”. Synapse. 3 August 2024. Retrieved 15 August 2024.
- Barra F, Romano A, Grandi G, Facchinetti F, Ferrero S (June 2019). “Future directions in endometriosis treatment: discovery and development of novel inhibitors of estrogen biosynthesis”. Expert Opin Investig Drugs. 28 (6): 501–504. doi:10.1080/13543784.2019.1618269. hdl:11380/1201688. PMID 31072144.
- Perrone U, Evangelisti G, Laganà AS, Bogliolo S, Ceccaroni M, Izzotti A, Gustavino C, Ferrero S, Barra F (December 2023). “A review of phase II and III drugs for the treatment and management of endometriosis”. Expert Opin Emerg Drugs. 28 (4): 333–351. doi:10.1080/14728214.2023.2296080. PMID 38099328.
- Rižner TL, Romano A (2023). “Targeting the formation of estrogens for treatment of hormone dependent diseases-current status”. Front Pharmacol. 14 1155558. doi:10.3389/fphar.2023.1155558. PMC 10175629. PMID 37188267.
Several compounds with inhibitory action on the enzyme HSD17B1 have been developed and one steroidal compound, a competitive HSD17B1 inhibitor (OG-6219) recently entered the clinical phase for endometriosis […] and it is in the preclinical phase for endometrial and breast cancer (Husen et al., 2006a; Husen et al., 2006b; Konings et al., 2018b; Jarvensivu et al., 2018; Xanthoulea et al., 2021). […] Only the C15 estrone derivative developed by Organon Finland, former Forendo pharma (compound FOR-6219/OR-6219) reached the clinical phase for endometriosis with three clinical trials registered in the database Clinical Trails (Table 2). Phase 1 and 1b trials NCT04686669 and NCT03709420 determined the bio-availability of the compound administered orally as gelatine capsule in 12 subjects (NCT04686669) and then the safety, tolerability, food interactions, the pharmacokinetics and pharmacodynamics of escalating doses of the drug in 87 subjects (NCT03709420). The phase 2 randomized, double-blind, Elena study (NCT05560646) is currently recruiting patients and aims at evaluating the efficacy and safety of OG-6219 in women with moderate to severe endometriosis […]
External links
//////////Linustedastat, FOR-6219, OG-6219, FOR 6219, OG 6219, PP3PLL7GZY, Phase 2, Endometriosis
Iodofalan (131I)
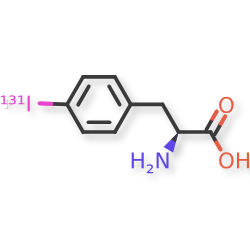
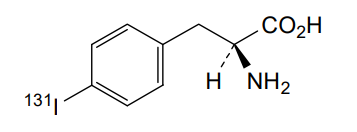
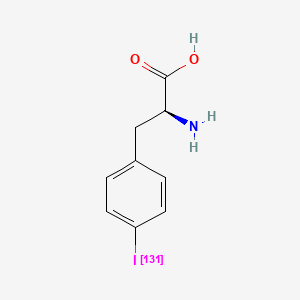
Iodofalan (131I)
CAS 76641-05-9
MFC9H10131INO2
Molecular FormulaC9H10INO2
Molecular Weight295.09
4-(131I)iodo-L-phenylalanine
(2S)-2-amino-3-(4-iodophenyl)propanoic acid
radiopharmaceutical, antineoplastic, Phase 2, Glioblastoma, 606VTF676Y, 131I-TLX-101, ACD 101
- 4-Iodophenylalanine I-131
- 4-(131I)Iodo-L-phenylalanine
- 4-Iodo-L-phenylalanine-131I
- ACD-101
- L-Phenylalanine, 4-(iodo-131I)-
- OriginatorTherapeia
- DeveloperTelix Pharmaceuticals; Therapeia
- ClassAmino acids; Antineoplastics; Radioisotopes; Radiopharmaceutical diagnostics; Radiopharmaceuticals; Small molecules
- Mechanism of ActionApoptosis stimulants; Positron-emission tomography enhancers
- Orphan Drug StatusYes – Glioblastoma
- Phase IIGlioblastoma
- 14 Oct 2025Telix Pharmaceuticals receives IND approval for TLX 101 in Glioblastoma
- 27 Jul 2025Telix Pharmaceuticals plans a phase III IPAX BrIGHT trial for Glioblastoma (Monotherapy, Combination therapy, Recurrent, Second-line therapy or greater) in Australia(IV) (NCT07100730)(EudraCT2025-521785-10) in September 2025
- 16 Apr 2025Telix has submitted for ethics approval a registration-enabling study of TLX101 in recurrent glioblastoma.
Iodofalan (131I) is a radiopharmaceutical that has garnered significant attention in oncological research due to its targeted therapeutic potential. This compound, which includes the radioactive isotope Iodine-131, has been explored for its efficacy in treating certain types of cancers, particularly those associated with the thyroid. Various research institutions worldwide have been studying Iodofalan (131I) to better understand its clinical benefits, optimize its usage, and minimize potential side effects. As a drug type, Iodofalan (131I) is categorized as a targeted radiopharmaceutical therapy, which leverages the properties of radioactive isotopes to destroy cancer cells with precision. Currently, its primary indications include differentiated thyroid cancer and non-resectable metastatic thyroid cancer, among other investigational uses.
Iodofalan (131I) Mechanism of Action
The mechanism of action for Iodofalan (131I) centers on the properties of Iodine-131, a beta-emitting isotope. When administered, Iodofalan (131I) is selectively absorbed by thyroid cells. This selectivity is due to the thyroid gland’s natural ability to uptake iodine, a key element required for the production of thyroid hormones. Cancerous thyroid tissues retain this ability, making them ideal targets for Iodofalan (131I) therapy.
Once absorbed by the thyroid cancer cells, the radioactive decay of Iodine-131 begins. This decay process emits beta particles, which possess sufficient energy to destroy nearby cells. The radiation from these beta particles causes direct DNA damage, leading to cell death. Additionally, the gamma radiation emitted by Iodine-131 can be used diagnostically to track the distribution and uptake of the compound in the body via imaging techniques such as SPECT (Single Photon Emission Computed Tomography).
The dual role of Iodofalan (131I) in both treatment and diagnostic contexts underscores its importance in managing thyroid cancers. By delivering a localized radiation dose to thyroid cancer cells, Iodofalan (131I) minimizes damage to surrounding healthy tissues, which is a significant advantage over traditional external beam radiotherapy.
What is the indication of Iodofalan (131I)?
The primary indication for Iodofalan (131I) is the treatment of differentiated thyroid cancer, a category that includes papillary and follicular thyroid cancers. These subtypes are characterized by their ability to absorb iodine, making them particularly amenable to radioiodine therapy. Iodofalan (131I) is typically used in cases where the thyroid cancer is not amenable to surgical removal or has metastasized to other parts of the body. In such scenarios, the radiopharmaceutical offers a non-invasive therapeutic option that can target and destroy cancer cells even in distant metastatic sites.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US42129729&_cid=P21-MHE8B5-15309-1
EXAMPLE 1
EXAMPLE 2
General synthesis of 3,4-[124I]iodo-L-phenylalanine (m, p-IPA-124), 3,4-[125I]iodo-L-phenylalanine (m,p-IPA-125) and 3,4-[131I]iodo-L-phenylalanine (m,p-IPA-131) by non-isotopic radioiodo-debromination
PAT
- Pharmaceutical combinations and uses thereofPublication Number: US-2024197715-A1Priority Date: 2022-11-18
- Pharmaceutical combinations and uses thereofPublication Number: WO-2024105610-A1Priority Date: 2022-11-18
- Iodine-labeled homoglutamic acid and glutamic acid derivativesPublication Number: US-2013034497-A1Priority Date: 2009-11-17
- MALIGNAS NEOPLASIAS THERAPY.Publication Number: ES-2341575-T3Priority Date: 2005-11-25Grant Date: 2010-06-22
- Therapy of malignant neoplasiasPublication Number: US-2007128108-A1Priority Date: 2005-11-18
- Therapy of malignant neoplasias
- Publication Number: US-9682158-B2
- Priority Date: 2005-11-18
- Grant Date: 2017-06-20
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
//////////Iodofalan (131I), radiopharmaceutical, antineoplastic, Phase 2, Glioblastoma, 606VTF676Y, 131I-TLX-101, ACD 101
Glovadalen



Glovadalen
CAS 2576359-31-2
MF C24H27Cl2N3O3 MW 476.4 g/mol
2-(3,5-dichloro-1-methyl-1H-indazol-4-yl)-1-[(1S,3R)-3-(hydroxymethyl)-5-(2-hydroxypropan-2-yl)-1-methyl-3,4-dihydroisoquinolin-2(1H)-yl]ethan-1-one,
2-(3,5-dichloro-1-methylindazol-4-yl)-1-[(1S,3R)-3-(hydroxymethyl)-5-(2-hydroxypropan-2-yl)-1-methyl-3,4-dihydro-1H-isoquinolin-2-yl]ethanone
dopamine D1 receptor positive allosteric modulator, Phase 2, Parkinson’s disease, UCB-0022, UCB 0022, H8T5VKH4CZ
- OriginatorUCB Biopharma
- ClassAlcohols; Antiparkinsonians; Benzene derivatives; Chlorinated hydrocarbons; Isoquinolines; Ketones; Neuroprotectants; Propanols; Pyrazoles; Small molecules
- Mechanism of ActionDopamine D1 receptor modulators
- Phase IIParkinson’s disease
- 27 Aug 2025Chemical structure information added.
- 21 May 2025UCB Biopharma SRL initiate a phase I trial in healthy volunteers (PO) (NCT06970301)
- 11 Apr 2025UCB Pharma completes a phase-II ATLANTIS trial in Parkinson’s disease (In adults, In the elderly, Adjunctive treatment) in USA (PO) (NCT06055985)
Glovadalen (developmental code name UCB-0022) is a dopamine D1 receptor positive allosteric modulator which is under development for the treatment of Parkinson’s disease.[1][2][3][4][5][6] It has been found to potentiate the capacity of dopamine to activate the D1 receptor by 10-fold in vitro with no actions on other dopamine receptors.[5][6] As of May 2024, glovadalen is in phase 2 clinical trials for this indication.[1][2][5] The drug is under development by UCB Biopharma.[1][4][5] It is described as an orally active, centrally penetrant small molecule.[1][5][6]
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021001288&_cid=P21-MH738G-96748-1

1. Preparation of intermediate of formula (ID- 2-(3,5-dichloro-1-methyl-indazol-4- vDacetic acid

1.1. Preparation of intermediate (Xlb) -1-methyl-5-nitro-indazole
5-Nitro-1H-indazole (Xla) (3.00 kg, 18.4 mol) and DMF (30.0 L) are charged into a 50 L three-neck round-bottom flask at 15-30°C. KOH (2.06 Kg, 36.7 mol) is added in one portion into the reactor at 0-5°C. The mixture is stirred at 0-50°C for 1h. Methyl iodide (2.87 kg, 20.2 mol) is then added at 0-5°C and the mixture is stirred for 3h at 15-30°C. The reaction mixture is added into water (30 L) at 0-10°C and the mixture is stirred for 10 min then filtered. The filter cake is washed with water (5 L) and dried. This overall procedure is carried out on 4 batches of the same size in parallel. The solids obtained from the four batches are combined to give 1-methyl-5-nitro-indazole (Xlb) as a brown solid (10.0 kg, 42.3 mol, 75% purity (LC/MS), 57.5% yield) which is used in the next step without further purification.
1H NMR (400 MHz, CDCIs) d 8.65 (s, 1H), 8.21 (d, J = 9.17 Hz, 1 H), 8.13 (s, 1 H), 7.39 (d, J = 9.17 Hz, 1 H), 4.08 (s, 3 H).
1.2. Preparation of intermediate (Xa)- tert-butyl 2-(1-methyl-5-nitro-indazol-4- yl)acetate
t-BuOK (4.43 kg, 39.5 mol) and THF (30 L) are charged into a 50 L three-neck round-bottom flask and the mixture is cooled to -45 / -35°C under nitrogen and stirring. 1-Methyl-5-nitro-indazole (Xlb) (3.50 kg, 19.7 mol) is then added in portions at -45 / -35°C. Tert-butyl 2-chloroacetate (3.57 kg, 23.7 mol) is added dropwise at the same temperature and the mixture is stirred at 1h. The mixture is warmed up to 15-30°C and stirred for 5h. The reaction is quenched by the addition of a saturated ammonium chloride solution (9 L) and water (2 L) is added. The organic layer is separated and the aqueous layer is extracted with ethyl acetate (2 x 5 L). The organic phases are combined, washed with brine (2 L), dried over Na2SC>4, filtered and concentrated under vacuum. The crude product is purified by recrystallization with ethyl acetate (5 L). This overall procedure is carried out on 2 batches of the same size in parallel. The solids obtained from the two batches are combined and dried together to give tert-butyl 2-(1-methyl-5-nitro-indazol-4-yl)acetate as a yellow solid (Xa) (5.30 kg, 17.7 mol, 97.6% purity (LC/MS), 44.9% yield).
1H NMR (400 MHz, CDCIs) d 8.18-8.20 (m, 2H), 7.37 (d, J = 9.21 Hz, 1 H), 4.27 (s, 2 H), 4.14 (s, 3 H), 1.44 (s, 9 H).
1.3. Preparation of intermediate (Xb) – tert-butyl 2-(5-amino-1-methyl-indazol-4- yl)acetate
Tert-butyl 2-(1-methyl-5-nitro-indazol-4-yl)acetate (Xa) (7.30 kg, 25.0 mol) and MeOH (76 L) are charged into a reactor. Argon is purged and Pd/C (50%, 760 g) is added. Hydrogen is added three times and the mixture is stirred at 50°C under hydrogen atmosphere (50 psi) for 3h. The reaction mixture is filtered and the solid is washed with MeOH (5 L). The mixture is concentrated to give tert-butyl 2-(5-amino-1-methyl-indazol-4-yl)acetate (Xb) as a brown oil (6.50 kg, 23.9 mol, 96.2% purity (LC/MS), 95.4% yield) which is used in the next step without further purification.
1H NMR (400 MHz, CDCI3) d 7.72 (s, 1H), 7.27 (d, J = 8.80 Hz, 1 H), 6.91 (d, J = 8.80 Hz, 1 H), 4.60 (s, 2 H), 3.93 (s, 3 H), 3.68 (s, 2H), 1.38 (s, 9 H).
1.4. Preparation of intermediate (Xc)- 2-(5-chloro-1-methyl-indazol-4-yl)acetic acid
Tert-butyl 2-(5-amino-1-methyl-indazol-4-yl)acetate (Xb) (2.00 kg, 7.65 mol) and concentrated HCI (10.0 L, 12M) are charged into a 50 L three-neck round bottom flask and the mixture is cooled to -10/-5°C and stirred. A water solution (5 L) of sodium nitrite (686 g, 9.95 mol) is added dropwise at -10/-5°C and stirred for 30 min. CuCI (833 g, 8.42 mol) and concentrated HCI (10.0 L, 12M) are charged into a 20 L three-neck round bottom flask and the mixture is stirred for 30 min. at -10/-5°C, then added into the other reactor. The mixture is stirred at -10/-5°C for 1 h, then at 10-30°C for 16h. The reaction mixture is filtered and the solid washed with water. This overall procedure is carried out on 3 batches of the same size in parallel. The solids obtained from the three batches are combined and dried together to give 2-(5-chloro-1-methyl-indazol-4-yl)acetic acid (Xc) as a yellow solid (4.00 kg, 16.3 mol, 92% purity (LC/MS), 71.3% yield) which is used in the next step without further purification.
1.5. Preparation of 2-(3,5-dichloro-1-methyl-indazol-4-yl)acetic acid (II)
2-(5-Chloro-1-methyl-indazol-4-yl)acetic acid (Xc) (1.30 kg, 5.79 mol) and DMF (6.5 L) are charged into a 50 L three-neck round bottom flask at 20°C. N-Chlorosuccinimide (772 g, 5.79 mol) is added portionwise at 20°C and the mixture is stirred at 20°C for 2h. The reaction mixture is poured into water (25 L) and filtered. The crude product is triturated with isopropyl etherethyl acetate (3:1) (7.0 L) at 20°C for 2h then filtered and dried. This overall procedure is carried out on 3 batches of the same size in parallel. The solids obtained from the three batches are combined to give 2-(3,5-dichloro-1-methyl-indazol-4-yl)acetic acid (II) (2.1 kg, 7.9 mol, 97.5% purity (LC/MS), 46% yield).
1H NMR (400 MHz, CDCI3) d 12.67 (s, 1 H), 7.68 (d, J = 9.05 Hz, 1 H), 7.53 (d, J = 9.05 Hz, 1 H), 4.20 (s, 2 H), 4.02 (s, 3 H).
2. Preparation of compound of formula (I)
2-(3,5-dichloro-1-methyl-indazol-4-yl)-1-[(1S,3R)-3-(hydroxymethyl)-5-(1-hydroxy-1- methyl-ethyl)-1-methyl-3,4-dihydro-1H-isoquinolin-2-yl]ethanone

2.1. Preparation of intermediate (IX).
(2R)-2-amino-3-(2-bromophenyl)propan-1-ol – a6
(2R)-2-amino-3-(2-bromophenyl)propanoic acid a5 (34.0 kg, 139 mol) and THF (238 L) are charged into a reactor. Sodium borohydride (15.6 kg, 413 mol) is added slowly at 20-30°C. A solution of iodine (35.3 kg, 139 mol) in dry THF (20.0 L) is added slowly at 0-10°C and the reaction mixture is stirred at 70°C for 12h. The reaction was quenched with methanol (70.0 L) at 0°C and heated to 80°C for 30 min. The mixture was cooled down, concentrated under vacuum and the residue was suspended in NaOH (30.0 L, 2N), then filtered. The filter cake was dried under vacuum to give (2R)-2-amino-3-(2-bromophenyl)propan-1-ol a6 as a white solid (31.0 kg, 135 mol, 96.7% yield) which is used in the next step without further purification. 1H NMR (400 MHz, CDCIs) d 7.57 (d, J = 7.7 Hz, 1H), 7.21 – 7.29 (m, 2H), 7.07 – 7.15 (m, 1H), 3.66 (dd, J = 10.5, 3.6 Hz, 1 H), 3.41 (dd, J = 10.5, 7.2 Hz, 1 H), 3.18 – 3.29 (m, 1 H), 2.95 (dd, J = 13.5, 5.5 Hz, 1 H), 2.70 (dd, J = 13.5, 8.2 Hz, 1H), 1.51 – 1.91 (m, 3H).
2.2. Preparation of intermediate of formula (VIII).
(4R)-4-[(2-bromophenyl)methyl]oxazolidin-2-one – a7
(2R)-2-amino-3-(2-bromophenyl)propan-1-ol a6 (31.0 kg, 135 mol) and dichloromethane (220 L) are charged into a reactor. Triphosgene (13.9 kg, 47.1 mol) is added at room temperature then N,N-diisopropylethylamine (39.1 kg, 303 mol) is slowly added at 0-10°C. The reaction mixture is stirred at 0-10°C for 1h then washed with water (50.0 L) twice, dried with anhydrous sodium sulfate and filtered to give (4R)-4-[(2-bromophenyl)methyl]oxazolidin-2-one a7 as a solution in dichloromethane which is used directly in the next step.
2.3. Preparation of intermediate (VII).
(10aR)-9-bromo-1 ,5, 10, 10a-tetrahydrooxazolo[3,4-b]isoquinolin-3-one a8
A solution of (4R)-4-[(2-bromophenyl)methyl]oxazolidin-2-one a7 (135 mol) in dichloromethane (220 L) is charged into a reactor and cooled down to 0-5°C. Trimethylsilyl triflate (35.9 kg, 162 mol) and paraformaldehyde (13.3 kg, 148 mol) are added at 0-5°C, then stirred for 2h at 15-20°C. Water (170 L) is added into the mixture which is then extracted twice with dichloromethane (50.0 L). the organic layer is dried with anhydrous sodium sulfate, filtered and concentrated under vacuum. A mixture of petroleum etherethyl acetate (1 :1, 45.0 L) is added and the mixture is stirred at room temperature for 6h and filtered. The solid was dried to get (10aR)-9-bromo-1,5,10,10a-tetrahydrooxazolo[3,4-b]isoquinolin-3-one a8 as an off-white solid (29.0 kg, 80.2% yield).
1H NMR (400 MHz, CDCI3) d 7.45 – 7.52 (m, 1H), 7.08 – 7.14 (m, 2H), 4.83 (d, J = 17.0 Hz, 1H), 4.62 (t, J = 8.4 Hz, 1H), 4.36 (d, J = 17.0 Hz, 1H), 4.21 (dd, J = 8.6, 4.9 Hz, 1 H), 3.91 -3.99 (m, 1H), 3.25 (dd, J= 16.3, 4.2 Hz, 1 H), 2.67 (dd, J = 16.1 , 11.0 Hz, 1H).
2.4. Preparation of intermediates (VI)
2.4.1. [(3R)-5-bromo-1,2,3,4-tetrahydroisoquinolin-3-yl]methanol a9
Ethanol (120 L) and water (60.0 L) are mixed into a reactor. (10aR)-9-bromo-1,5,10,10a-tetrahydrooxazolo[3,4-b]isoquinolin-3-one a8 (29.7 kg, 111 mol) is added then sodium hydroxide (13.3 kg, 332 mol) is slowly added at 15-20°C. The reaction mixture is stirred at 90°C for 2h then cooled down to room temperature. Water (300 L) is added into the mixture which is centrifugated. The centrifugal cake is dried in circulation oven to give [(3R)-5-bromo- 1,2,3,4-tetrahydroisoquinolin-3-yl]methanol a9 as a white solid (23.7 kg, 88.3% yield) which is used in the next step without further purification.
1H NMR (400 MHz, CDCIs) d 7.37 – 7.47 (m, 1H), 6.95 – 7.08 (m, 2H), 4.00 – 4.10 (m, 2H), 3.85 (dd, J = 10.9, 3.7 Hz, 1 H), 3.57 (dd, J = 10.9, 7.9 Hz, 1 H), 3.06 (ddt, J = 11.3, 7.6, 4.1 , 4.1 Hz, 1H), 2.79 (dd, J= 17.1, 4.4 Hz, 1H), 2.40 (dd, J= 17.1, 10.9 Hz, 1H), 1.93 (br s, 2H).
2.4.2. [(3R)-5-bromo-1 ,2,3,4-tetrahydroisoquinolin-3-yl]methoxy-tert-butyl-dimethyl- silane a10
[(3R)-5-bromo-1,2,3,4-tetrahydroisoquinolin-3-yl]methanol a9 (23.7 kg, 97.8 mol) and dichloromethane (240 L) are charged into a reactor. DMAP (120 g, 0.98 mol) and imidazole (13.3 kg, 196 mol) are added. Tert-butyldimethylsilyl chloride (TBSCI) (17.7 kg, 117 mol) is slowly added at 15-20°C and the mixture is stirred for 12h. Ammonium chloride (100 L) is added into the mixture. The organic phase was separated, washed with water (50.0 L), dried with anhydrous sodium sulfate, filtered and concentrated under vacuum to give [(3R)-5-bromo-1 ,2,3,4-tetrahydroisoquinolin-3-yl]methoxy-tert-butyl-dimethyl-silane a10 as a yellow oil (37.6 kg, 86% purity, 93% yield) which is used in the next step without further purification.
1H NMR (400 MHz, CDCI3) d 7.36 – 7.45 (m, 1H), 7.01 (d, J = 4.6 Hz, 1H), 4.01 – 4.13 (m, 2H), 3.84 (dd, J = 9.9, 3.7 Hz, 1 H), 3.64 (dd, J = 9.8, 7.2 Hz, 1 H), 2.96 – 3.08 (m, 1 H), 2.75 (dd, J = 17.0, 4.2 Hz, 1 H), 2.44 (dd, J = 17.0, 10.8 Hz, 1H), 1.76 – 2.20 (m, 2H), 0.89 – 0.97 (m, 9H), 0.08 – 0.14 (m, 6H).
2.5. Preparation of intermediate (V).
[(3R)-5-bromo-3,4-dihydroisoquinolin-3-yl]methoxy-tert-butyl-dimethyl-silane a11
[(3R)-5-bromo-1 ,2,3,4-tetrahydroisoquinolin-3-yl]methoxy-tert-butyl-dimethyl-silane a10 (3.42 kg, 8.31 mol) and THF (30.0 L) are charged into a reactor. N-Chlorosuccinimide (NCS) (1.17 kg, 8.73 mol) is slowly added at room temperature and the mixture is stirred at 25°C for 30 min. A solution of KOH (1.52 kg, 27.1 mol) in dry methanol (7.00 L) is slowly added at room temperature and the reaction is stirred at 25°C for 1h. The reaction is quenched with water (10.0 L) and extracted with petroleum etherethyl acetate (1:2, 5.00 L). The organic layer is separated, washed with brine (10.0 L), dried with anhydrous sodium sulfate and filtered. This overall procedure is carried out on 10 batches of the same size in parallel and the 10 reaction filtrates are combined and concentrated under vacuum to give [(3R)-5-bromo-3,4-dihydroisoquinolin-3-yl]methoxy-tert-butyl-dimethyl-silane a11 as a brown oil (28.0 kg, crude) which is used in the next step without further purification.
1H NMR (400 MHz, CDC ) d 8.24 (d, J = 2.6 Hz, 1H), 7.58 (dd, J = 7.8, 1.2 Hz, 1 H), 7.12 -7.25 (m, 2H), 4.03 (dd, J = 9.5, 4.0 Hz, 1 H), 3.67 – 3.77 (m, 2H), 3.07 (dd, J = 17.0, 6.2 Hz, 1H), 2.68 (dd, J = 17.1, 10.9 Hz, 1 H), 0.88 – 0.91 (m, 9H), 0.07 (d, J= 1.5 Hz, 6H).
2.6. Preparation of intermediates of formula (IV)
2.6.1. [(1S,3R)-5-bromo-1-methyl-1,2,3,4-tetrahydroisoquinolin-3-yl]methoxy-tert-butyl- dimethyl-silane (IVa)
[(3R)-5-bromo-3,4-dihydroisoquinolin-3-yl]methoxy-tert-butyl-dimethyl-silane a11 (3.10 kg, 8.75 mol) and THF (20.0 L) are charged into a reactor. The mixture is cooled down to 0°C and methylmagnesium chloride (3M, 11.6 L) is added. The mixture is stirred at 20°C for 12h. The reaction is quenched with a saturated solution of ammonium chloride. The phases are separated and the aqueous layer is extracted twice with petroleum ether: ethyl acetate (3:1, 5.00 L). The combined organic phases are washed with brine (10.0 L), dried over anhydrous sodium sulfate and filtered. This overall procedure is carried out on 9 batches of the same size in parallel and the nine reaction filtrates are combined and concentrated under vacuum. The crude mixture is purified by silica gel chromatography with petroleum ether : ethyl acetate (10:1) to give [(1S,3R)-5-bromo-1 -methyl-1, 2, 3, 4-tetrahydroisoquinolin-3-yl]methoxy-tert-butyl-dimethyl-silane (IVa) as a brown oil (4.60 kg, 99.7% purity, 15.7% yield).
1H NMR (400 MHz, DMSO-de) d 7.41 (dd, J=7.7, 0.9 Hz, 1H), 7.12 – 7.18 (m, 1H), 7.03 – 7.11 (m, 1H), 4.12 (q, J= 6.8 Hz, 1H), 3.62 (d, J= 5.7 Hz, 2H), 3.07 – 3.17 (m, 1H), 2.67 – 2.76 (m, 1H), 2.26 (dd, J=16.9, 10.0 Hz, 1H), 2.12 (br s, 1 H), 1.32 (d, J= 6.8 Hz, 3H), 0.84 – 0.93 (m, 9H), 0.07 (d, J=0.9 Hz, 6H).
2.6.2. tert-butyl (1S,3R)-5-bromo-3-[[tert-butyl(dimethyl)silyl]oxymethyl]-1-methyl-3,4- dihydro-1 H-isoquinoline-2-carboxylate (IVb)
[(1S,3R)-5-bromo-1-methyl-1,2,3,4-tetrahydroisoquinolin-3-yl]methoxy-tert-butyl-dimethyl-silane (IVa) (1.85 kg, 4.99 mol) and dichloromethane (13.0 L) are charged in a reactor. N,N-diisopropylethylamine (1.94 kg, 14.9 mol) and di-tert-butyl dicarbonate (1.14 kg, 5.24 mol) are added at room temperature and the mixture is stirred for 12h. The reaction mixture is washed twice with a saturated ammonium chloride solution (10.0 L), the organic layer is dried with anhydrous sodium sulfate and filtered. This overall procedure is carried out on 2 batches of the same size in parallel and the two reaction filtrates are combined and concentrated under vacuum. The crude mixture is purified by silica gel chromatography with petroleum ether ethyl acetate (30:1) to give tert-butyl (1S,3R)-5-bromo-3-[[tert-butyl(dimethyl)silyl]oxymethyl]-1 -methyl-3, 4-dihydro-1 H-isoquinoline-2-carboxylate (IVb) as a yellow oil (4.00 kg, 99.5% purity, 85.2% yield).
1H NMR (400 MHz, DMSO-de) d 7.50 (d, J = 7.9 Hz, 1 H), 7.22 (br d, J = 6.7 Hz, 1 H), 7.06 -7.18 (m, 1 H), 4.84 (br s, 1 H), 4.12 (br s, 1H), 3.46 (br d, J = 15.4 Hz, 2H), 2.94 (br dd, J = 15.8, 5.2 Hz, 1H), 2.71 (br t, J = 9.5 Hz, 1 H), 1.45 (s, 9 H), 1.28 (br s, 3H), 0.81 (s, 9H), -0.08 (s, 6H).
2.6.3. tert-butyl (1S,3R)-3-[[tert-butyl(dimethyl)silyl]oxymethyl]-5-(1-hydroxy-1 -methyl- ethyl)-1 -methyl-3, 4-dihydro-1H-isoquinoline-2-carboxylate (IVc)
A solution of tert-butyl (1S,3R)-5-bromo-3-[[tert-butyl(dimethyl)silyl]oxymethyl]-1-methyl-3,4-dihydro-1 H-isoquinoline-2-carboxylate (IVb) (42.5 g, 90.3 mmol) in dry THF (0.5 M solution) and a commercial solution of n-Buthylithium in Hexanes (1.6 M solution) were pumped at respectively 6.0 ml/min (1.0 equiv) and 2.46 mL/min (1.3 equiv.) and were mixed in a glass microchip cooled at -40°C. The mixed flow stream was pumped through the reaction zone 1 of the microchip (0.3 ml_) and was then combined with a solution of dry acetone (13.5 M) pumped at 6.0 mL/min (27 equiv.). The resulting stream was then passed through the reaction zone 2 of the microchip (0.7 ml_) at -40 °C. Finally, the global flow stream exiting the reactor was collected and quenched at room temperature in a saturated solution of aqueous ammonium chloride. When all the feed solutions were consumed, a bilayer reaction mixture was obtained. The aqueous layer was separated from the organic layer, and then extracted twice with ethyl acetate. The combined organic layers were washed with brine, dried over anhydrous sodium sulfate and concentrated under vacuum. A yellow oil was obtained (46.5 g) and was purified by SFC chromatography on a GreenSep Nitro column (10m, 5×22.3 using CO298 %/EtOH 2% eluent). The solvent was removed under vacuum to yield to a white solid, tert-butyl (1S,3R)-3-[[tert-butyl(dimethyl)silyl]oxymethyl]-5-(1-hydroxy-1-methyl-ethyl)-1-methyl-3, 4-dihydro-1H-isoquinoline-2-carboxylate (IVc) (25 g, 56 mmol, 62 % yield).
UPLC_MS basic 1 pic @ 3.83 min (ES+): 350 (M-Boc+H)+, 332 (M-Boc-H20+H)+, 100 % purity.
1H NMR (400 MHz, DMSO-de) d 7.44 (d, J = 7.9 Hz, 1H), 7.19 (dt, J = 8.1 , 5.2 Hz, 1 H), 7.09 (t, J = 9.0 Hz, 1H), 4.99 (s, 1 H), 4.87 (dq, J = 13.4, 6.4 Hz, 1 H), 4.11 (s, 1H), 3.96 (t, J = 14.9 Hz, 1 H), 3.48 (dd, J = 9.4, 4.1 Hz, 1H), 2.98 (dd, J = 16.5, 5.0 Hz, 1H), 2.89 (t, J = 9.6 Hz, 1H), 1.65 (s, 3H), 1.58 (s, 3H), 1.55 (d, J = 2.5 Hz, 9H), 1.34 (dd, J = 20.5, 6.6 Hz, 3H), 0.90 (s, 9H), 0.08 (d, J = 7.2 Hz, 3H), -0.00 (s, 3H).
2.7. Preparation of intermediate (III) 2-[(1S,3R)-3-(hydroxymethyl)-1-methyl- 1.2.3.4-tetrahydroisoquinolin-2-ium-5-yl]propan-2-ol chloride
2.7.1. tert-butyl-dimethyl-[[(1S,3R)-1-methyl-5-(1-methyl-1-trimethylsilyloxy-ethyl)- 1.2.3.4-tetrahydroisoquinolin-3-yl]methoxy]silane- a15
Tert-butyl (1S,3R)-3-[[tert-butyl(dimethyl)silyl]oxymethyl]-5-(1-hydroxy-1-methyl-ethyl)-1-methyl-3, 4-dihydro-1H-isoquinoline-2-carboxylate (IVc) (148 g, 87% purity, 287 mmol) is dissolved in 1000 ml_ dichloromethane and transferred to a 2 liter double walled reactor. 2,6-Lutidine (100 ml_, 860 mmol) is added and the jacket temperature is set at-2°C. Trimethylsilyl trifluoromethanesulfonate (154 g, 129 ml_, 692 mmol) is added over 40 min via an addition funnel. Two hours after the start of addition, the reaction is quenched by adding 650 ml_ of an aqueous citric acid solution (1M) and the temperature of the mixture is brought back to 20°C. One hour after the start of the quench, the layers are separated. The organic layer is washed twice with 350 ml_ of an aqueous solution of citric acid (1M). The organic layer is stirred with 750 ml_ of aqueous sodium carbonate (10% w/w) for 10 min before separation of the layers. The organic layer is dried over anhydrous sodium sulfate. The organic layer is then filtered and the filtrate is concentrated under vacuum at 40°C providing a yellow oil (128 g) of tert-butyl-dimethyl-[[(1S,3R)-1-methyl-5-(1-methyl-1-trimethylsilyloxy-ethyl)-1 ,2,3,4-tetrahydroisoquinolin-3-yl]methoxy]silane a15 which is used in the next step without further purification.
1H NMR (400 MHz, CDC ) d 7.19 (d, J = 7.7 Hz, 1 H), 7.07 (t, J = 7.7 Hz, 1 H), 7.00 (d, J = 7.6 Hz, 1 H), 4.24 (q, J = 6.8 Hz, 1 H), 3.75 (dd, J = 9.7, 4.4 Hz, 1H), 3.60 (dd, J = 9.7, 7.0 Hz, 1H), 3.54 (dd, J = 16.3, 3.5 Hz, 1H), 3.15 (ddt, J = 10.9, 7.4, 4.0 Hz, 1 H), 2.52 (dd, J = 16.3, 10.9 Hz, 1H), 1.66 (d, J = 14.6 Hz, 6H), 1.52 – 1.43 (m, 3H), 0.92 (q, J = 1.2 Hz, 9H), 0.14 (q, J = 1.2 Hz, 2H), 0.09 (d, J = 1.1 Hz, 6H), 0.00 (q, J = 1.2, 0.8 Hz, 9H).
2.7.2. 2-[(1S,3R)-3-(hydroxymethyl)-1-methyl-1,2,3,4-tetrahydroisoquinolin-2-ium-5- yl]propan-2-ol chloride Intermediate (III)
In a three-neck round bottom flask equipped with a mechanical stirrer, tert-butyl-dimethyl-[[(1S,3R)-1-methyl-5-(1-methyl-1-trimethylsilyloxy-ethyl)-1,2,3,4-tetrahydroisoquinolin-3-yljmethoxyjsilane a15 (20.0 g, 47.4 mmol) is dissolved in 220 ml_ of isopropanol. To this solution, 42.3 ml_ of hydrochloric acid in iso-propanol (5-6 M, around 5 eq.) are added. 45 min after addition of hydrochloric acid, a 100 mg of seeds of the desired product are introduced. After 7 hours at room temperature, the reaction mixture is filtered over a sintered glass filter. The filtercake is washed with 40 ml_ isopropanol and dried under vacuum at room temperature overnight. 11.1 g of 2-[(1S,3R)-3-(hydroxymethyl)-1 -methyl-1 , 2,3,4-tetrahydroisoquinolin-2-ium-5-yl]propan-2-ol chloride (III) are obtained as a pinkish solid. The yield over the two deprotection steps is 91%.
1H NMR (400 MHz, CD3OD) d 7.46 (dd, J = 7.8, 1.3 Hz, 1H), 7.28 (t, J = 7.8 Hz, 1H), 7.21 (dd, J = 7.8, 1.3 Hz, 1H), 4.63 (q, J = 6.9 Hz, 1H), 3.97 (dd, J = 11.7, 3.8 Hz, 1 H), 3.88 (dd, J = 17.2, 4.3 Hz, 1H), 3.78 (dd, J = 11.8, 6.1 Hz, 1H), 3.66 – 3.56 (m, 1 H), 3.14 (dd, J = 17.2, 11 .4 Hz, 1 H), 1 .73 (d, J = 6.8 Hz, 3H), 1 .64 (d, J = 4.8 Hz, 6H). OH and NH protons are not observed.
2.8. Preparation of compound of formula (I).
2-(3,5-dichloro-1 -methyl-indazol-4-yl)-1 -[(1 S,3R)-3-(hydroxymethyl)-5-(1 – hydroxy-1 -methyl-ethyl)-1 -methyl-3, 4-dihydro-1 H-isoquinolin-2-yl]ethanone
In a 100 ml. Easymax reactor equipped with a mechanical stirrer, 2-(3,5-dichloro-1 -methyl-indazol-4-yl)acetic acid (II) (4.00 g, 15.4 mmol), 2-[(1S,3R)-3-(hydroxymethyl)-1-methyl-1 ,2,3,4-tetrahydroisoquinolin-2-ium-5-yl]propan-2-ol chloride (III) (4.46 g, 16.4 mmol) and 48 mL of DMF are charged. The suspension is stirred at 20°C and then cooled by setting the jacket temperature to -2°C. Once the temperature of the mixture is below 3°C, N,N-diisopropylethylamine (9.5 mL, 54 mmol) is added. (2-(1 H-benzotriazol-1 -yl)-1 , 1 ,3,3-tetramethyluronium hexafluorophosphate (6.4 g, 17 mmol) is added in four portions over 1 hour. The mixture is stirred for 1 h 45 before setting the jacket temperature at 15°C. 16 mL of water are then added over the course of a few minutes. 15 min later, 30 mg of solid product are added as seeds to initiate the crystallization. The jacket temperature is set at 20°C. Half an hour later, 16 mL of water are added over 17 min. Stirring of the suspension is pursued for 2 h 15 at 20°C before being filtered on sintered glass. The filtercake is washed with two portions of 20 mL of water and then dried at 50°C overnight under vacuum yielding 6.03 g of 2-(3,5-dichloro-1-methyl-indazol-4-yl)-1-[(1 S,3R)-3-(hydroxymethyl)-5-(1 -hydroxy-1 -methyl-ethyl)-1 -methyl-3, 4-dihydro-1 H-isoquinolin-2-yl]ethanone (I) (crude material).
A recristallization is carried out on 5.00 g of the crude material obtained by first suspending in 50 mL acetonitrile. The jacket temperature is set to 70°C. Once the solid has dissolved and the mass temperature has reached 66°C, 720 mI of water are added. The mass temperature is then cooled to 59°C and 125 mg of solid product is added as seeding material. The mass temperature is then decreased to 55°C over 25 min at which stage crystallization is occurring. The jacket temperature is then decreased over two hours from 58°C down to 20°C. After 50 min, the suspension is filtered and the filtercake is washed with 7.5 mL acetonitrile. The filtercake is then dried under vacuum at 45°C overnight and 2 hours at 50°C providing 4.04 g of 2-(3,5-dichloro-1 -methyl-indazol-4-yl)-1-[(1S,3R)-3-(hydroxymethyl)-5-(1-hydroxy-1-methyl-ethyl)-1 -methyl-3, 4-dihydro-1 H-isoquinolin-2-yl]ethanone (I) as an off-white powder (hydrate form) Yield = 64%.
1H NMR (400 MHz, DMSO-cfe) d 7.65 (dd, J = 9.0, 2.2 Hz, 1H), 7.52 (dd, J = 9.0, 2.1 Hz, 1 H), 7.37 (ddd, J = 19.6, 7.6, 1 .7 Hz, 1 H), 7.25 – 7.03 (m, 2H), 5.30 (q, J = 6.5 Hz, 0.3H), 5.16 -4.99 (m, 1 .7H), 4.99 – 4.84 (m, 0.7H), 4.63 – 4.30 (m, 3.3H), 4.17 – 3.93 (m, 4H), 3.28 (dt, J = 10.5, 5.1 Hz, 1.3H), 3.10 – 2.85 (m, 1.7H), 1.56 (dd, J = 13.2, 6.9 Hz, 6.7H), 1.24 (d, J = 6.5 Hz, 2.3H).
PAT
- A Substituted Tetrahydroisoquinoiline Derivative as a D1 Positive Allosteric ModulatorPublication Number: US-2022259179-A1Priority Date: 2019-07-01
- Substituted tetrahydroisoquinoline derivatives as D1 positive allosteric modulatorsPublication Number: CN-113993857-BPriority Date: 2019-07-01Grant Date: 2024-01-02
- Substituted tetrahydroisoquinoline derivatives as D1 positive allosteric modulatorsPublication Number: CN-117700395-APriority Date: 2019-07-01
- Substituted tetrahydroisoquinoline derivatives as D1 positive allosteric modulatorsPublication Number: JP-7510444-B2Priority Date: 2019-07-01Grant Date: 2024-07-03
- Substituted Tetrahydroisoquinoline Derivatives as Positive Allosteric Modulators of D1Publication Number: CN-113993857-APriority Date: 2019-07-01
- A substituted tetrahydroisoquinoline derivative as a d1 positive allosteric modulatorPublication Number: EP-3993794-A1Priority Date: 2019-07-01
- D1 Substituted tetrahydroisoquinoline derivatives as positive allosteric modulatorsPublication Number: KR-20220029686-APriority Date: 2019-07-01
- A SUBSTITUTED TETRAHYDROISOQUINOLINE DERIVATIVE AS A POSITIVE ALOSTERIC MODULATOR OF D1Publication Number: PE-20221020-A1Priority Date: 2019-07-01
- Substituted Tetrahydroisoquinoline Derivatives as D1 Positive Allosteric ModulatorsPublication Number: JP-2022539152-APriority Date: 2019-07-01
- A substituted tetrahydroisoquinoline derivative as a d1 positive allosteric modulatorPublication Number: WO-2021001288-A1Priority Date: 2019-07-01
- A substituted tetrahydroisoquinoline derivative as a d1 positive allosteric modulatorPublication Number: TW-202115010-APriority Date: 2019-07-01
- A substituted tetrahydroisoquinoline derivative as a d1 positive allosteric modulatorPublication Number: WO-2021001288-A9Priority Date: 2019-07-01
- A substituted tetrahydroisoquinoline derivative as a D1 positive allosteric modulatorPublication Number: AU-2020299953-A1Priority Date: 2019-07-01
- A substituted tetrahydroisoquinoline derivative as a d1 positive allosteric modulatorPublication Number: CA-3139571-A1Priority Date: 2019-07-01
- A Substituted Tetrahydroisoquinoline Derivative As A D1 Positive Allosteric ModulatorPublication Number: US-2024059665-A1Priority Date: 2020-12-18
- Prodrugs of 2-(3,5-Dichloro-1-methyl-indazol-4-yl)-1-[(1S,3R)-3-(hydroxymethyl)-5-(1-hydroxy-1-methyl-ethyl)-1-methyl-3,4-dihydro-1H-isoquinolin-2-yl]ethanonePublication Number: US-2024083925-A1Priority Date: 2020-12-18
- A substituted tetrahydroisoquinoline derivative as a d1 positive allosteric modulatorPublication Number: AU-2021403603-A9Priority Date: 2020-12-18
- A substituted tetrahydroisoquinoline derivative as a d1 positive allosteric modulatorPublication Number: EP-4263517-B1Priority Date: 2020-12-18Grant Date: 2024-10-02
- Prodrugs of 2-(3,5-dichloro-1-methyl-indazol-4-yl)-1-[(1s,3r)-3-(hydroxymethyl)-5-(1-hydroxy-1-methyl-ethyl)-1-methyl-3,4-dihydro-1h-isoquinolin-2-yl]ethanonePublication Number: EP-4263519-B1Priority Date: 2020-12-18Grant Date: 2024-10-02
- Prodrugs of 2-(3,5-dichloro-1-methyl-indazol-4-yl)-1-[(1s,3r)-3-(hydroxymethyl)-5-(1-hydroxy-1-methyl-ethyl)-1-methyl-3,4-dihydro-1h-isoquinolin-2-yl]ethanonePublication Number: EP-4263519-A1Priority Date: 2020-12-18
- amorphous solid dispersionPublication Number: JP-2023553457-APriority Date: 2020-12-18
- Substituted tetrahydroisoquinoline derivatives as D1-positive allosteric modulatorsPublication Number: JP-2023553671-APriority Date: 2020-12-18
- 2-(3,5-dichloro-1-methyl-indazol-4-yl)-1-[(1S,3R)-3-(hydroxymethyl)-5-(1-hydroxy-1-methyl-ethyl)- Prodrug of 1-methyl-3,4-dihydro-1H-isoquinolin-2-yl]ethanonePublication Number: JP-2024500391-APriority Date: 2020-12-18
- Amorphous solid dispersionsPublication Number: US-2024000769-A1Priority Date: 2020-12-18
- A substituted tetrahydroisoquinoline derivative as a d1 positive allosteric modulatorPublication Number: IL-303693-APriority Date: 2020-12-18
- Substituted tetrahydroisoquinoline derivatives as D1 positive allosteric modulatorsPublication Number: KR-20230121849-APriority Date: 2020-12-18
- amorphous solid dispersionPublication Number: KR-20230121867-APriority Date: 2020-12-18
- Amorphous solid dispersionsPublication Number: EP-4262756-A1Priority Date: 2020-12-18
- A substituted tetrahydroisoquinoline derivative as a d1 positive allosteric modulatorPublication Number: EP-4263517-A1Priority Date: 2020-12-18
- Prodrugs of 2-(3,5-dichloro-1-methyl-indazol-4-yl)-1-[(1s,3r)-3-(hydroxymethyl)-5-(1-hydroxy-1-methyl-ethyl)-1-methyl-3,4-dihydro-1h-isoquinolin-2-yl]ethanonePublication Number: CA-3203281-A1Priority Date: 2020-12-18
- Substituted tetrahydroisoquinoline derivatives useful as D1 positive allosteric modulatorsPublication Number: CN-116583280-APriority Date: 2020-12-18
- 2-(3,5-Dichloro-1-methyl-indazol-4-yl)-1-[(1S,3R)-3-(hydroxymethyl)-5-(1-hydroxy-1-methyl Prodrug of -ethyl)-1-methyl-3,4-dihydro-1H-isoquinolin-2-yl]ethanonePublication Number: CN-116601161-APriority Date: 2020-12-18
- Amorphous Solid DispersionPublication Number: CN-116685308-APriority Date: 2020-12-18
- Amorphous solid dispersionsPublication Number: IL-303688-APriority Date: 2020-12-18
- Amorphous solid dispersionsPublication Number: WO-2022129267-A1Priority Date: 2020-12-18
- A substituted tetrahydroisoquinoline derivative as a d1 positive allosteric modulatorPublication Number: WO-2022129268-A1Priority Date: 2020-12-18
- Prodrugs of 2-(3,5-dichloro-1-methyl-indazol-4-yl)-1-[(1s,3r)-3-(hydroxymethyl)-5-(1-hydroxy-1-methyl-ethyl)-1-methyl-3,4-dihydro-1h-isoquinolin-2-yl]ethanonePublication Number: WO-2022129356-A1Priority Date: 2020-12-18
- Amorphous solid dispersionsPublication Number: AU-2021401128-A1Priority Date: 2020-12-18
- A substituted tetrahydroisoquinoline derivative as a d1 positive allosteric modulatorPublication Number: AU-2021403603-A1Priority Date: 2020-12-18

AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
| Clinical data | |
|---|---|
| Other names | UCB-0022; UCB0022 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2576359-31-2 |
| PubChem CID | 155460962 |
| IUPHAR/BPS | 13232 |
| UNII | H8T5VKH4CZ |
| Chemical and physical data | |
| Formula | C24H27Cl2N3O3 |
| Molar mass | 476.40 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- “UCB 0022”. AdisInsight. Springer Nature Switzerland AG. 28 May 2024. Retrieved 10 August 2024.
- “Delving into the Latest Updates on Glovadalen with Synapse”. Synapse. 8 August 2024. Retrieved 10 August 2024.
- McFarthing K, Buff S, Rafaloff G, Fiske B, Mursaleen L, Fuest R, et al. (2023). “Parkinson’s Disease Drug Therapies in the Clinical Trial Pipeline: 2023 Update”. Journal of Parkinson’s Disease. 13 (4): 427–439. doi:10.3233/JPD-239901. PMC 10357160. PMID 37302040.
Our analysis of dopaminergic therapies shows a continued emphasis on DA agonists and levodopa reformulation. These include Cerevel’s tavapadon, a D1/D5 receptor partial agonist and UCB0022, a positive allosteric modulator of the D1 receptor, as well as approaches to sub-cutaneously deliver levodopa/carbidopa such as Abbvie’s ABBV-951 and Neuroderm’s ND0612.
- “Glovadalen”. IUPHAR/BPS Guide to PHARMACOLOGY. Retrieved 10 August 2024.
- “UCB0022”. ALZFORUM. 3 May 2024. Retrieved 10 August 2024.
- Vermeiren C, Ates A, Bouzom F, Delaunois A, Gillard M, Kenda B, et al. (7 September 2022). “Preclinical characterization of UCB0022, an oral, brain penetrant, selective, clinical-stage positive allosteric modulator of the dopamine 1 receptor (D1 PAM)”. Movement Disorders. 37 (Suppl 2 [2022 International Congress September 15-18, 2022. Madrid, Spain]). Retrieved 10 August 2024.
////////Glovadalen, dopamine D1 receptor positive allosteric modulator, Phase 2, Parkinson’s disease, UCB-0022, UCB 0022, H8T5VKH4CZ
Tebapivat
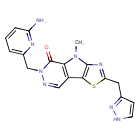
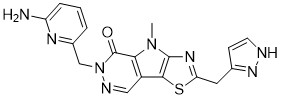
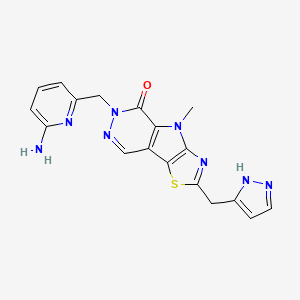
Tebapivat
CAS 2283422-04-6
WeightAverage: 392.44
Monoisotopic: 392.116778341
Chemical FormulaC18H16N8OS
10-[(6-aminopyridin-2-yl)methyl]-7-methyl-4-(1H-pyrazol-5-ylmethyl)-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one
6-[(6-aminopyridin-2-yl)methyl]-4-methyl-2-[(1H-pyrazol-3-yl)methyl]-4,6-dihydro-5H-[1,3]thiazolo[5′,4′:4,5]pyrrolo[2,3-d]pyridazin-5-one
6-[(6-aminopyridin-2-yl)methyl]-4-methyl-2-[(1H-pyrazol-3-yl)methyl]-4,6-dihydro-5H-[1,3]thiazolo[5′,4′:4,5]pyrrolo[2,3-d]pyridazin-5-one
- AG946
- CS-0115951
- HY-135884
- ORG4KGP5ZS
- OriginatorAgios Pharmaceuticals
- ClassAntianaemics; Small molecules
- Mechanism of ActionPyruvate kinase stimulants
- Orphan Drug StatusYes – Myelodysplastic syndromes
- Phase IIAnaemia; Sickle cell anaemia
- 01 May 2025Phase-II clinical trials in Sickle cell anaemia in USA (PO) (NCT06924970)
- 01 May 2025Agios plans to initiate a phase II clinical trial for Sickle cell disease(PO) in mid-2025.
- 21 Feb 2025Agios Pharmaceuticals completes a phase I bioavailability trial (In volunteers) in USA (PO, capsule) (NCT06745271)
Tebapivat is under investigation in clinical trial NCT05490446 (A Study of Tebapivat (AG-946) in Participants With Anemia Due to Lower-risk Myelodysplastic Syndromes (LR-MDS)).
Tebapivat is an orally available activator of the red cell isoform of pyruvate kinase (PK-R; PKR), with potential to improve hemolytic anemia and related-symptoms in patients with pyruvate kinase deficiency (PKD). Upon oral administration, tebapivat binds to and activates PKR, thereby enhancing glycolytic pathway activity in red blood cells (RBCs), improving adenosine triphosphate (ATP) levels and reducing 2,3-diphosphoglycerate (2,3-DPG) levels. This may result in increased oxygen affinity, improved RBC deformability, decreased sickle RBC hemolysis, increased hemoglobin (Hb) levels and improved RBC membrane function. Mutations in PKR cause deficiency in PKR which prevents adequate RBC glycolysis, leading to a build-up of the upstream glycolytic intermediate 2,3-DPG and deficiency in the PKR product ATP.
SCHEME
COUPLER
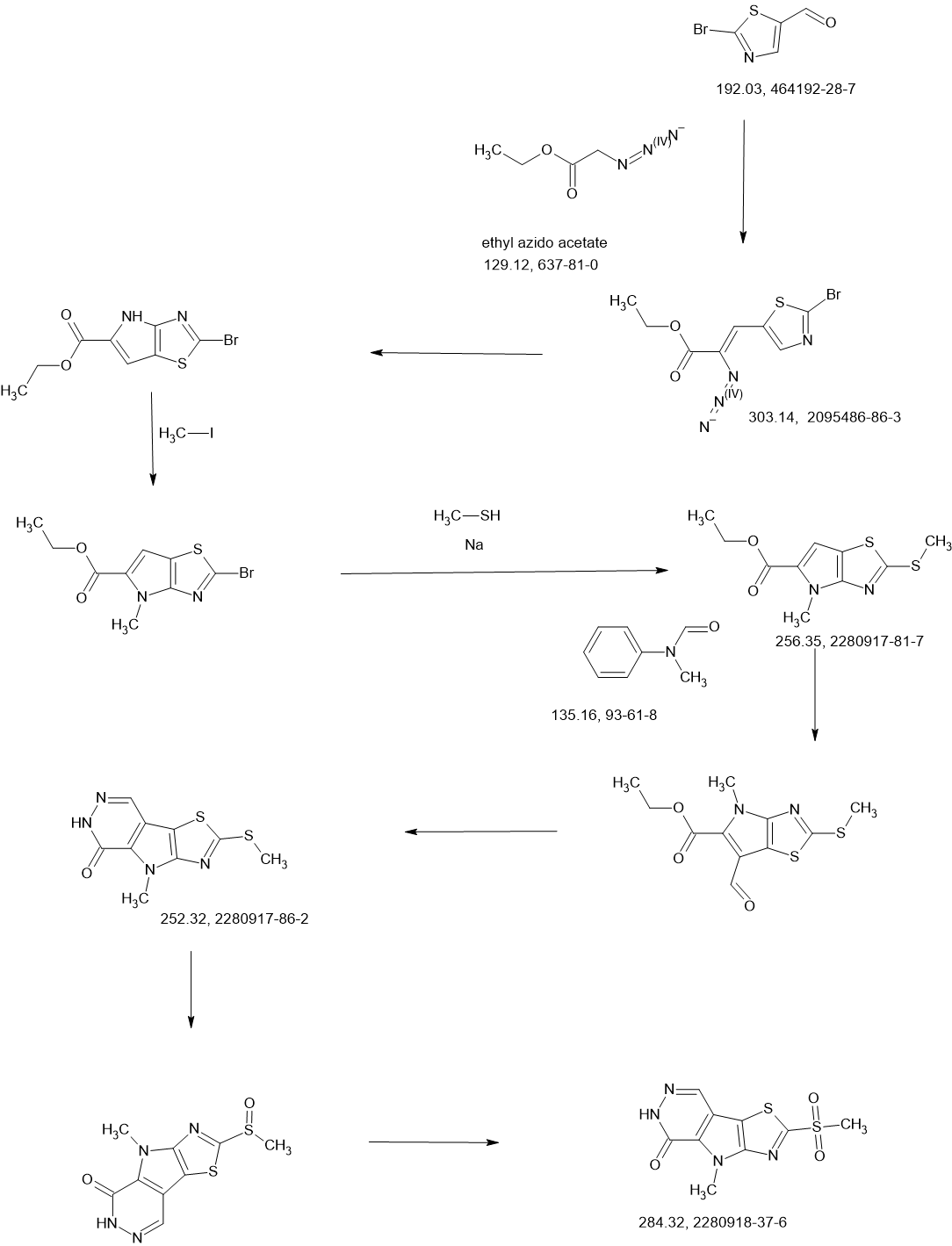
COUPLER
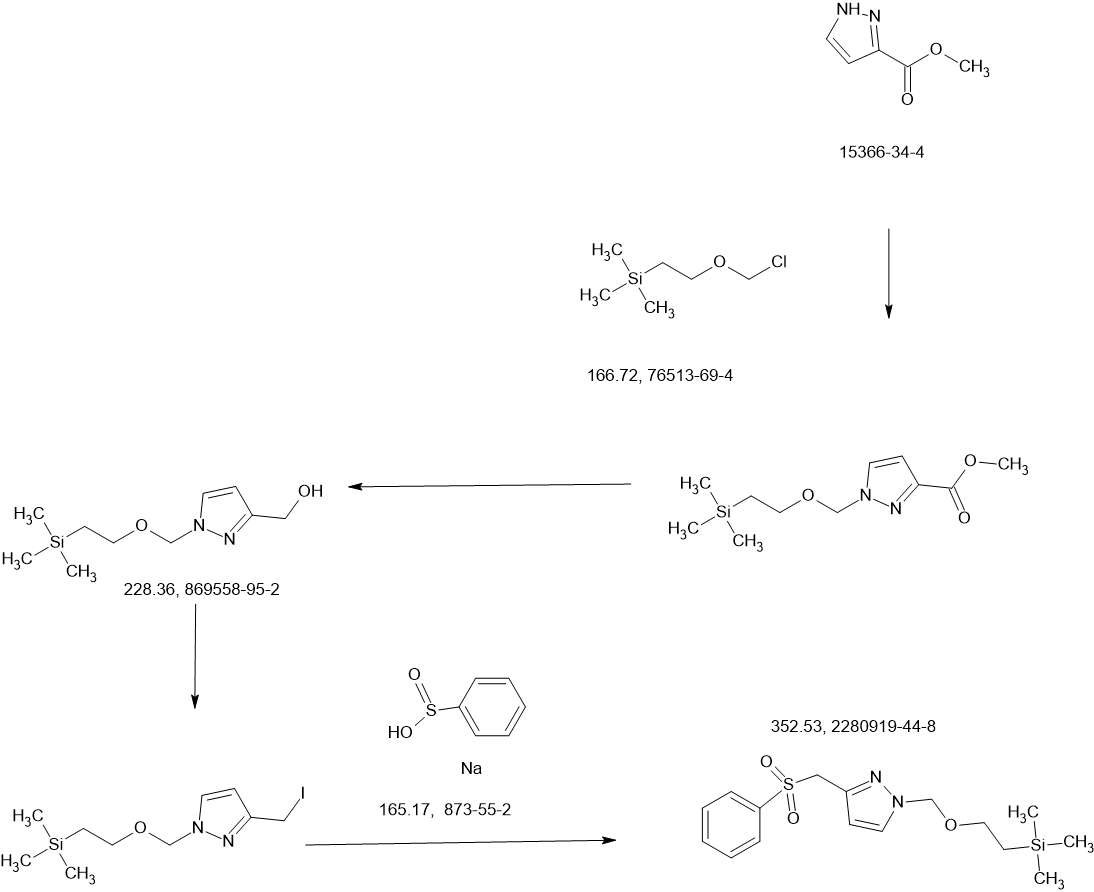
MAIN
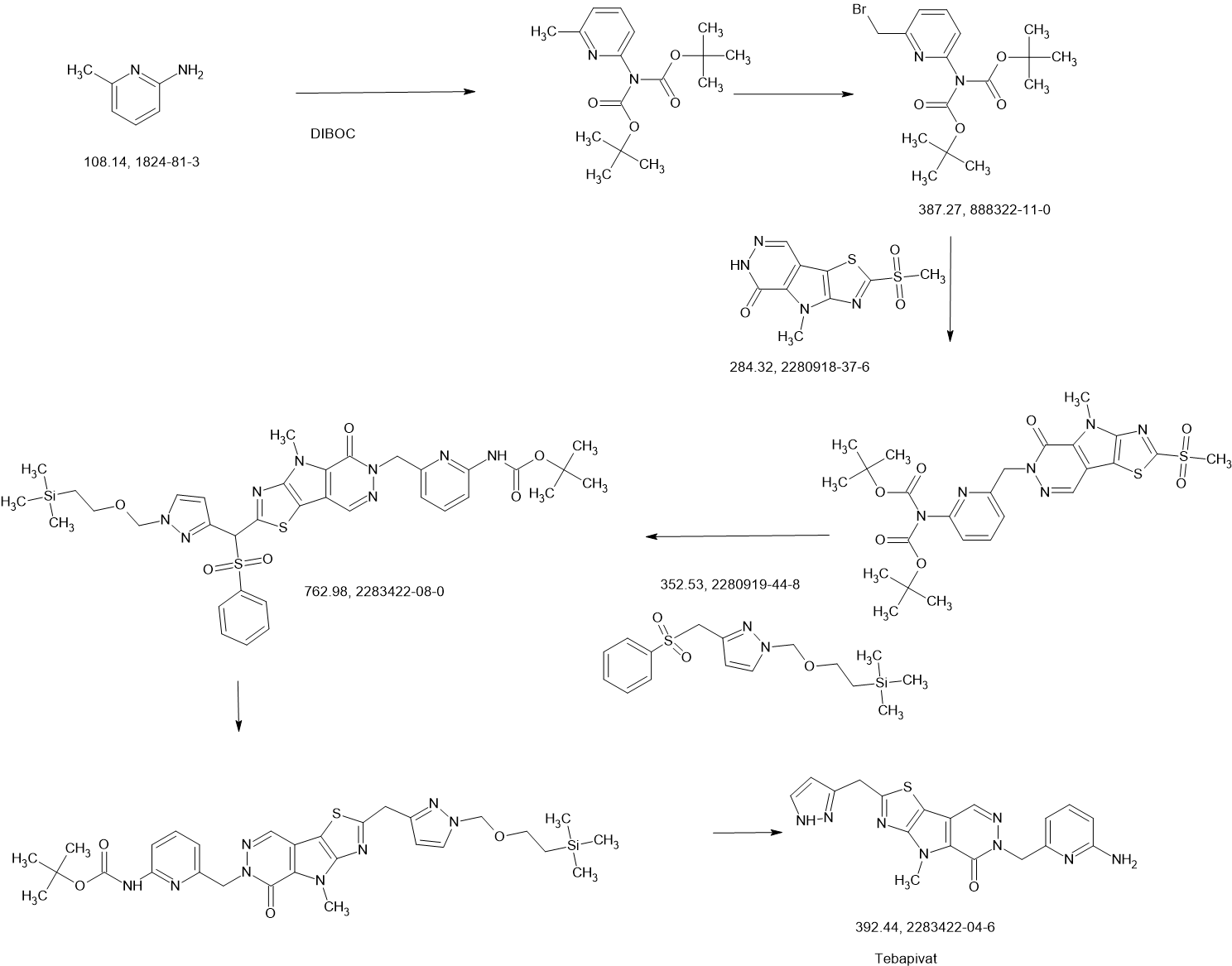
PATENT
Agios Pharmaceuticals, Inc.
WO2019035864
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019035864&_cid=P22-MDGSEF-03229-1
Example 8A. Synthesis of 2-((1H-pyrazol-3-yl)methyl)-6-((6-aminopyridin-2-yl)methyl)- 4-methyl-4H-thiazolo[5′,4′:4,5]pyrrolo[2,3-d]pyridazin-5(6H)-one and 6-((6- aminopyridin-2-yl)methyl)-4-methyl-2-(1H-pyrazole-3-carbonyl)-4H- thiazolo[5′,4′:4,5]pyrroIo[2,3-d]pyridazin-5(6H)-one
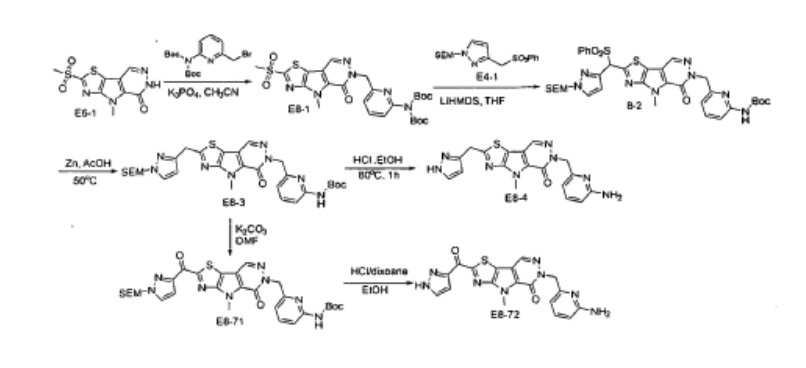


Step F. Synthesis of 6-((6-aminopyridin-2-yl)methyl)-4-methyl-2-(1H-pyrazole-3- carbonyl)-4H-thiazolo[5′,4′:4,5]pyrrolo[2,3-d]pyridazin-5(6H)-one To a solution of tert- butyl (6-((4-methyl-5-oxo-2-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazole-3-carbonyl)- 4H-thiazolo[5′,4′:4,5]pyrrolo[2,3-d]pyridazin-6(5H)-yl)methyl)pyridin-2-yl)carbamate (20 mg, 0.03 mmol) in EtOH (1 mL) was added HCl (1 mL, 4 mol/L in dioxane). The mixture was stirred at 80 °C for lhr and cooled down. The precipitate was collected by filtration and neutralized with sat. NaHCO3, washed with water and dried to afford 5 mg of 6-((6- aminopyridin-2-yl)methyl)-4-methyl-2-(1H-pyrazole-3-carbonyl)-4H- thiazolo[5′,4′:4,5]pyrrolo[2,3-d]pyridazin-5(6H)-one. LC-MS (ESI): m/z 407 (M+H)+. 1H NMR (400 MHz, DMSO-d6) δ: 8.75 (s, 1H), 7.96 (s, 1H), 7.50 (s, 1H), 7.31-7.22 (m, 1H), 6.31 (d, 1H), 6.14 (d, 1H), 5.91 (s, 2H), 5.23 (s, 2H), 4.38 (s, 3H).
PATENT
WO2023091414
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023091414&_cid=P22-MDGSRV-15431-1
PATENT
WO2019035863
WO2019035865
WO2019035864
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
/////////Tebapivat, 2283422-04-6, AG946, CS-0115951, HY-135884, AG 946, CS 0115951, HY 135884, ORG4KGP5ZS, AGIOS, Orphan Drug, PHASE 2,
Alflutinib, Furmonertinib, Firmonertinib



FIRMOMERTINIB, Furmonertinib, Alflutinib
CAS 1869057-83-9
, AST 2818, UNII-A49A7A5YN4
N-[2-[[2-(Dimethylamino)ethyl]methylamino]-5-[[4-(1-methyl-1H-indol-3-yl)-2-pyrimidinyl]amino]-6-(2,2,2-trifluoroethoxy)-3-pyridinyl]-2-propenamide
N-[2-[2-(dimethylamino)ethyl-methylamino]-5-[[4-(1-methylindol-3-yl)pyrimidin-2-yl]amino]-6-(2,2,2-trifluoroethoxy)pyridin-3-yl]prop-2-enamide
C28H31F3N8O2 568.6 g/mol
2-Propenamide, N-[2-[[2-(dimethylamino)ethyl]methylamino]-5-[[4-(1-methyl-1H-indol-3-yl)-2-pyrimidinyl]amino]-6-(2,2,2-trifluoroethoxy)-3-pyridinyl]-
Alflutinib is under investigation in clinical trial NCT03452592 (Efficacy and Safety of Alflutinib in Locally Advanced or Metastatic Non-small Cell Lung Cancer Patients With T790M).
Firmonertinib is an orally available selective inhibitor of the epidermal growth factor receptor (EGFR) mutant form T790M, with potential antineoplastic activity. Upon administration, firmonertinib specifically binds to and inhibits the tyrosine kinase activity of EGFR T790M, a secondarily acquired resistance mutation. This prevents EGFR T790M-mediated signaling and leads to cell death in EGFR T790M-expressing tumor cells. EGFR, a receptor tyrosine kinase that is mutated in many tumor cell types, plays a key role in tumor cell proliferation and tumor vascularization. Compared to some other EGFR inhibitors, alflutinib may have therapeutic benefits in tumors with T790M-mediated drug resistance.
FIRMONERTINIB is a small molecule drug with a maximum clinical trial phase of III (across all indications) and has 4 investigational indications.
SCHEME

CONTD……..

REF
https://patentscope.wipo.int/search/en/detail.jsf?docId=US201062358&_cid=P22-MBFXFH-62339-1
Example 3: N-{2-{[2-(dimethylamino)ethyl](methyl)amino}-6-(2,2,2-trifluoroethoxyl)-5-{[4-(1-methyl-H-indol-3-yl)pyrimidin-2-yl]amino}pyridin-3-yl}acrylamide
Step 1: Synthesis of N2-methyl-N2-[2-(dimethylamino)ethyl]-6-(2,2,2-trifluoroethoxyl)-N5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-yl]-3-nitropyridin-2,5-diamine
| The compound was synthesized in the same manner as those in Step 1 of Example 1 with a yield of 86%. MS m/z: 545 [M+1]. |
Step 2: Synthesis of N2-methyl-N2-[2-(dimethylamino)ethyl]-6-(2,2,2-trifluoroethoxyl)-N5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-yl]pyridin-2,3,5-triamine
| The compound was synthesized in the same manner as those in Step 2 of Example 2 with a yield of 56%. MS m/z: 515 [M+1]. |
Step 3: Synthesis of N-{2-{[2-(dimethylamino)ethyl](methyl)amino}-6-(2,2,2-trifluoroethoxyl)-5-{[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-yl]amino}pyridin-3-yl}acrylamide
| The compound was synthesized in the same manner as those in Step 3 of Example 1 with a yield of 23%. MS m/z: 569 [M+1]. |
PATENT
CN110606842
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN280196686&_cid=P22-MBFXJY-67679-1
Patent application CN105315259A protects the compound of formula I and discloses its preparation method as follows:



| Example 1: Preparation of 6-chloro-3-nitro-2-(2,2,2-trifluoroethoxy)pyridine (XI-1) |
| Add toluene (24.0L) to the reactor, then add 2,6-dichloro-3-nitropyridine (3000g, 15.54mol), adjust the internal temperature between -20℃ and -10℃, and add sodium hydrogen (933g, 23.33mol) in batches. Add 2,2,2-trifluoroethanol (1586g, 16.00mol) toluene (6.0L) solution dropwise. React for 2h, and monitor the reaction end point by TLC and HPLC. After the reaction is completed, add 10% ammonium chloride solution (6.0L) dropwise. Let stand and separate. Wash the organic phase with water (6.0L) and concentrate under reduced pressure. Add ethyl acetate (0.3L), heat to 40-50℃, add n-heptane (2.7L) dropwise, cool to -15 to -5℃ after dripping, and continue crystallization for 3 hours, and filter with suction. Obtain 3017g of product solid, with a yield of 75.65%. |
| 1H NMR(500MHz,DMSO-d6)δ8.60(d,J=8.0Hz,1H),7.50(d,J=8.5Hz,lH),5.13(q,J=9.0Hz,2H); |
| 13C NMR(126MHz,DMSO-d6)δ153.20,151.09,139.34,132.67,123.38(q,J=277.2Hz),119.14,63.34(q,J=36Hz); |
| MS m/z:256.99[M+1]。 |
| Example 2: Preparation of 6-chloro-3-amino-2-(2,2,2-trifluoroethoxy)pyridine (X-1) |
| At room temperature, add acetonitrile (21.0L) and water (21.0L) to the reactor, start stirring, add 6-chloro-3-nitro-2-(2,2,2-trifluoroethoxy)pyridine (3017.0g, 11.76mol) obtained in Example 1, and add hydrosulfite (15.1Kg, 70.54mol). Control the temperature at 27-33°C to react for 2 hours. Add 36% concentrated hydrochloric acid (11.9Kg, 117.60mol) dropwise, and continue to react for 1.5 hours. Add solid sodium bicarbonate (12.8Kg, 12.96mol). Filter, separate the mother liquor, wash the organic phase with saturated brine (21.0L), and concentrate under reduced pressure to obtain an oily substance. Theoretically calculated for the next step reaction. |
| 1H NMR(500MHz,DMSO-d6)δ7.03(d,J=8.0Hz,1H),6.90(d,J=8.0Hz,1H),5.21(s,2H),4.93(q,J=9.0Hz,2H); |
| 13C NMR(126MHz,DMSO-d6)δ148.16,131.72,130.55,123.93(q,J=278.5Hz),121.02,118.42,61.72(q,J=34.0Hz); |
| MS m/z:227.01[M+1]。 |
| Example 3: Preparation of 6-chloro-3-(2,2,2-trifluoroacetamido)-2-(2,2,2-trifluoroethoxy)pyridine (IX-1) |
| At room temperature, dichloromethane (10.4 L) was added to the reaction kettle, stirring was started, 6-chloro-3-amino-2-(2,2,2-trifluoroethoxy)pyridine (2664 g, 11.76 mol) obtained in Example 2 was added, diisopropylethylamine (2279 g, 17.64 mol) was added, the temperature was controlled at -15 to -10°C, a dichloromethane (5.2 L) solution of trifluoroacetic anhydride (2963 g, 14.11 mol) was added dropwise, and stirring was continued for 20 minutes after the addition was completed. Water (13.0 L) was added dropwise, the layers were separated, the organic phase was concentrated under reduced pressure, and the next step reaction was theoretically calculated. |
| 1 H NMR(400MHz,DMSO-d6)δ11.23(s,7H),7.95(d,J8.0Hz,1H),7.34(d,J8.0Hz,1H),5.03(q,J8.9Hz,2H) |
| 13C NMR(101MHz,DMSO-d6)δ155.74(q,J=46.6Hz),155.60,145.37,140.24,124.01(q,J=278.8Hz),119.07,118.30,116.19(q,J=289.9Hz),62.99(q,J=35.4Hz); |
| MS m/z.322.99[M+1]。 |
| Example 4: Preparation of 6-chloro-5-nitro-3-(2,2,2-trifluoroacetamido)-2-(2,2,2-trifluoroethoxy)pyridine (VIII-1) |
| At room temperature, concentrated sulfuric acid (11.7 L) was added to the reaction kettle, stirring was started, 6-chloro-3-(2,2,2-trifluoroacetamido)-2-(2,2,2-trifluoroethoxy)pyridine (3.9 Kg, 11.76 mol) obtained in Example 3 was added, and potassium nitrate solid (1783.4 g, 17.64 mol) was added in batches. After the addition, stirring was continued for about 40 minutes. After monitoring the reaction, the temperature was lowered to control the internal temperature at 10-25°C, and dichloromethane (27.3 L) was added dropwise. Stirring was continued, stirring was continued for 45 minutes, and the layers were separated. The organic phase was taken and washed once with water (11.7 L). The organic phase was concentrated under reduced pressure and theoretically calculated for the next step reaction. |
| 1H NMR(500MHz,DMSO-d6)δ11.58(s,1H),8.78(s,1H),5.17(q,J=8.7Hz,2H); |
| 13C NMR(126MHz,DMSO-d6)δ155.89,155.43(q,J=37.8Hz),138.84,138.57,135.05,123.22(q,J=273.4Hz),118.47,115.51(q,J=278.5Hz),63.65(q,J=35.3Hz); |
| MS m/z:367.98[M+1]。 |
| Example 5: Preparation of 6-chloro-5-nitro-3-amino-2-(2,2,2-trifluoroethoxy)pyridine (VII-1) |
| At room temperature, methanol (13.0 L) was added to the reactor, 6-chloro-5-nitro-3-(2,2,2-trifluoroacetamido)-2-(2,2,2-trifluoroethoxy)pyridine (4322 g, 11.76 mol) obtained in Example 4 was added, p-toluenesulfonic acid monohydrate (3355 g, 17.64 mol) was added, the temperature was controlled at 60-65°C for 15 hours, and the methanol was removed under reduced pressure. Methyl tert-butyl ether (13.0 L) and water (6.5 L) were added, and the pH was adjusted to 7-8 with potassium carbonate. Layering was performed, the organic phase was washed once with water (8.6 L), separated, and concentrated under reduced pressure. n-heptane (21.5 L) was added, the temperature was controlled at 60-65°C and stirred for 1 hour, cooled to room temperature, filtered, and the filter cake was dried with air at 50°C for 18 hours to obtain 1475 g of the product. |
| The total yield of the five-step reaction from Example 1 to Example 5 is 34.9%. |
| 1H NMR(500 MHz,DMSO-d6)δ7.62(s,1H),5.92(s,2H),5.05(q,J=8.9Hz,2H). |
| 13C NMR(126MHz,DMSO-d6)δ149.30,139.53,132.84,123.46,123.44(q,J=278.5Hz),116.25,62.52(q,J=35.3Hz); |
| MS m/z:272.00[M+1]。 |
| Example 6: Preparation of 2-chloro-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (V-1) |
| Toluene (50 mL) was added to a 100 mL reaction bottle, and the compound of formula VII-1, 6-chloro-5-nitro-3-amino-2-(2,2,2-trifluoroethoxy)pyridine (5.0 g, 18.4 mmol), the compound of formula VI, 3-(2-chloropyrimidin-4-yl)-1-methyl-1H-indole (5.8 g, 23.8 mmol), p-toluenesulfonic acid monohydrate (1.8 g, 9.2 mmol) were added in sequence, and the reaction mixture was heated to 110-115°C and reacted for 24 hours. The temperature was lowered to 22°C, filtered by suction, and the filter cake was dried at 50°C for 20 hours to obtain the compound of formula V-1, 2-chloro-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (10.4 g, 74.7 HPLC area% purity). According to the HPLC purity conversion, the next step reaction was carried out. |
| 1H NMR(400MHz,DMSO-d6)δ9.43(s,1H),8.76(s,1H),8.46-8.45(d,J=5.4Hz,1H),8.39(s,1H),8.38-8.36(d,J=7.8Hz,1H),7.57-7.55(d,J=8.2Hz,1H),7.41-7.40(d,J=5.4Hz,1H),7.31-7.27(t,J=7.5Hz,1H),7.20-7.16(t,J=7.5Hz,1H),5.23-5.16(q,J=8.8Hz,2H),3.90(s,3H); |
| MS m/z:479.08[M+1]。 |
| Example 7: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (IV) |
| Add N,N-dimethylformamide (30 mL) to a 250 mL reaction bottle, add the compound of formula V-1 obtained in Example 6, 2-chloro-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (10.4 g, 16.22 mmol), stir, add potassium carbonate (4.48 g, 32.44 mol), N,N,N’-trimethylethylenediamine (2.48 g, 24.33 mol) in sequence, heat the reaction mixture to 77-82°C, keep warm for 1-1.5 hours. Add water (60 mL), and cool to room temperature after addition. Filter by suction, transfer the filter cake to a 50 L reactor, add acetonitrile (40 mL), and heat to reflux for 2 hours. The mixture was cooled to room temperature and filtered with suction. The filter cake was dried at 50°C for 18 hours to give a compound of formula IV, 2-[2-(dimethylaminoethyl)methylamino]-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (6.7 g). The total yield of the two-step reaction with Example 6 was 66.8%. |
| 1H NMR(500MHz,DMSO-d6)δ8.62(s,1H),8.41(s,1H),8.26(s,2H),8.24(s,1H),7.48(d,J=8.2Hz,1H),7.21(t,J=7.6Hz,1H),7.16(d,J=5.3Hz,1H),7.05(t,J=7.3Hz,1H),5.04(q,J=8.9Hz,2H),3.84(s,3H),3.69(t,J=6.9Hz,2H),2.89(s,3H),2.55(t,J=6.9Hz,2H),2.17(s,6H); |
| 13C NMR(126MHz,DMSO-d6)δ162.15,160.55,156.99,154.98,148.42,137.53,132.83,132.68,125.50,123.58(q,J=279.7Hz),124.38,122.11,122.06,120.67,113.38,112.27,110.30,107.11,62.14(q,J=35.3Hz),56.10,49.51,45.34,45.33,39.35,32.98。 |
| MS m/z.:545.22[M+1]。 |
| Example 8: Preparation of 2-chloro-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine p-toluenesulfonate (V-1′) |
| Toluene (7.43 L) was added to a 20 L reactor, and compound VII-1 6-chloro-5-nitro-3-amino-2-(2,2,2-trifluoroethoxy)pyridine (743.0 g, 2.74 mol), compound VI 3-(2-chloropyrimidin-4-yl)-1-methyl-1H-indole (866.7 g, 3.56 mol), p-toluenesulfonic acid monohydrate (780.7 g, 4.10 mol) were added in sequence, stirred, and the reaction mixture was heated to 110-115°C and reacted for 36 hours. The temperature was controlled at 15-30°C, tetrahydrofuran (3.72 L) was added and stirred for 30 minutes. Filtered by suction, the filter cake was transferred to a 50 L reactor, tetrahydrofuran (4.46 L) was added, and heated to reflux for 3 hours. The temperature was lowered to 15-25°C, filtered, and the filter cake was dried at 50°C for 17 hours to obtain 2-chloro-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine p-toluenesulfonate (1719 g, 85.96 HPLC area% purity). The purity was calculated according to HPLC and used for the next step reaction. |
| Melting point: 216.0-218.3℃ |
| 1H NMR(500MHz,DMSO-d6)δ9.70(s,1H),9.21(s,1H),8.62(s,1H),8.40(d,J=6.2Hz,1H),8.24(d,J=7.8Hz,1H),7.59(d,J=8.3Hz,1H),7.50(d,J=6.5Hz,1H),7.49(d,J=8.3Hz,2H),7.32(t,J=7.6Hz,1H),7.18(t,J=7.5Hz,1H),7.12(d,J=7.9Hz,2H),5.17(q,J=8.8Hz,2H),3.91(s,3H),2.29(d,J=5.2Hz,3H); |
| 13C NMR(126MHz,DMSO-d6)δ166.66,157.35,155.72,147.40,140.87,139.90,139.72,138.59,135.83,130.09,129.99,129.98,129.97,127.39,127.38,127.37,127.15,125.22(q,J=278.5Hz),124.97,123.85,123.69,113.63,112.97,110.27,63.58(q,J=35.3Hz),35.57,22.81。 |
| Example 9: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (IV) |
| Add N,N-dimethylformamide (5.14L) to a 50L reactor, add 2-chloro-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine p-toluenesulfonate (1714.0g, 2.261mol) obtained in Example 8, stir, add potassium carbonate (624.7g, 4.52mol), N,N,N’-trimethylethylenediamine (346.2g, 3.39mol) in sequence, heat the reaction mixture to 77-82°C, keep warm for 1-1.5 hours. Add water (10.28L), and cool to room temperature after adding. Filter by suction, transfer the filter cake to a 50L reactor, add acetonitrile (6.86L), and heat to reflux for 2 hours. The temperature was lowered to 15-25°C, filtered with suction, and the filter cake was dried at 50°C for 18 hours to obtain the compound of formula IV, 2-[2-(dimethylaminoethyl)methylamino]-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (1142 g). The total yield of the two-step reaction with Example 8 was 76.54%. |
| 1H NMR(500MHz,DMSO-d6)δ8.62(s,1H),8.41(s,1H),8.26(s,2H),8.24(s,1H),7.48(d,J=8.2Hz,1H),7.21(t,J=7.6Hz,1H),7.16(d,J=5.3Hz,1H),7.05(t,J=7.3Hz,1H),5.04(q,J=8.9Hz,2H),3.84(s,3H),3.69(t,J=6.9Hz,2H),2.89(s,3H),2.55(t,J=6.9Hz,2H),2.17(s,6H); |
| 13C NMR(126MHz,DMSO-d6)δ162.15,160.55,156.99,154.98,148.42,137.53,132.83,132.68,125.50,123.58(q,J=279.7Hz),124.38,122.11,122.06,120.67,113.38,112.27,110.30,107.11,62.14(q,J=35.3Hz),56.10,49.51,45.34,45.33,39.35,32.98。 |
| MS m/z:545.22[M+1]。 |
| Example 10: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (IV) |
| Acetonitrile (10 mL) was added to a 50 L reactor, and 2-chloro-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine p-toluenesulfonate (1.0 g, 1.5 mmol) obtained in Example 8 was added, and stirred. Potassium carbonate (577 mg, 3 mmol) and N,N,N’-trimethylethylenediamine (320 mg, 2.25 mmol) were added in sequence. The reaction mixture was heated to 77-82°C and kept for 1-2 hours. Water (10 mL) was added and the temperature was cooled to room temperature after the addition. The product was filtered to give a compound of formula IV, 2-[2-(dimethylaminoethyl)methylamino]-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (629 mg) with a purity of 95.94%. The total yield of the two-step reaction with Example 8 was 77%. |
| Example 11: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (III’) |
| Add the compound of formula IV 2-[2-(dimethylaminoethyl)methylamino]-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (4.0 g, 7.34 mmol) to a 100 mL reaction bottle at room temperature, add tetrahydrofuran (27 mL) and water (13 mL), and stir for 10 to 20 minutes. Add hydrosulfite (9.6 g, 44.1 mmol) to the reactor in batches. After addition, continue stirring for 10 to 20 minutes. Control the temperature of the reactor to 30 to 35 ° C for reaction. The purity of the product compound of formula III’ was 64.68% after sampling the liquid phase after 2 hours of reaction. The reaction was continued until 17 hours after the reaction. 40 mL of water was added to the reaction solution, and the layers were separated by standing. The tetrahydrofuran phase was taken, and the aqueous phase was extracted twice with 100 mL of dichloromethane. The organic phases were combined, washed with saturated brine, separated by standing, and concentrated under reduced pressure to obtain 3.2 g of solid with a purity of 62.32%. |
| 1H NMR(500MHz,DMSO)δ10.67(s,1H),10.36(s,1H),8.82(s,1H),8.18(s,1H),8.01(s,1H),7.59(d,J=8.2Hz,1H),7.45(d,J=6.8Hz,1H),7.32(t,J=7.5Hz,1H),7.24(s,1H),4.97(q,J=8.7Hz,2H),3.93(s,3H),3.75(s,2H),3.41(s,2H),3.10(s,3H),2.78(s,6H); |
| MS m/z:515.24[M+1]。 |
| Example 12: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (III’) |
| In a 100mL single-mouth bottle, there is 2-[2-(dimethylaminoethyl)methylamino]-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (2.0g, 3.67mmol), palladium carbon (200mg), ethanol (20mL), hydrogen balloon replacement twice, hydrogen gas, magnetic stirring, room temperature overnight (17 hours). After the reaction is completed, suction filtration, the filtrate is taken, and it is concentrated to dryness under reduced pressure to obtain 2.1g of product with a purity of 56.93%. |
| Example 13: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (III’) |
| At room temperature, add the compound of formula IV 2-[2-(dimethylaminoethyl)methylamino]-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (1317.0 g, 2.42 mol) to a 50 L reactor, add tetrahydrofuran (8.8 L) and water (4.3 L), and stir for 10 to 20 minutes. Add hydrosulfite (2970.0 g, 14.52 mol) to the reactor in batches. After adding, continue stirring for 10 to 20 minutes. Control the temperature of the reactor to 40-45 ° C and react for 2 hours. Add concentrated hydrochloric acid (5882.2 g, 58.08 mol) to the reactor. After the addition is complete, heat to 42 to 47 ° C and react for 15 hours. Add 30% sodium hydroxide (2323.2g, 58.08mol) aqueous solution dropwise, and then add solid sodium bicarbonate (1219.7g, 14.52mol) in batches to adjust the pH value to 6-8. After stirring for 20 minutes, filter with suction, let the filtrate stand and separate. The organic phase is concentrated under reduced pressure to obtain 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine, with a purity of 97.1%. Calculated based on the theoretical yield of 100%, it is directly used in the next step reaction. |
| 1H NMR(500MHz,DMSO)δ10.67(s,1H),10.36(s,1H),8.82(s,1H),8.18(s,1H),8.01(s,1H),7.59(d,J=8.2Hz,1H),7.45(d,J=6.8Hz,1H),7.32(t,J=7.5Hz,1H),7.24(s,1H),4.97(q,J=8.7Hz,2H),3.93(s,3H),3.75(s,2H),3.41(s,2H),3.10(s,3H),2.78(s,6H); |
| MS m/z:515.24[M+1]。 |
| Example 14: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine dihydrochloride (III-1) |
| To the 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine obtained in Example 13, THF (5.3 L) and ethanol (4.0 L) were added, the temperature was raised to 50-70°C, and concentrated hydrochloric acid (617.8 g, 6.1 mol) was added dropwise. After the addition was completed, the mixture was cooled to room temperature and stirred for 12 hours. Filtered by suction, the filter cake was dried by air at 50°C to obtain 1507.4 g of a crude product. Methanol (6.0 L) and ethanol (4.5 L) were added to a 20 L reaction bottle, and the above crude product was added, the temperature was raised to 55-60 ° C, hot slurry was added for 1-2 hours, the temperature was lowered to room temperature, and suction was filtered to obtain 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine dihydrochloride (1335.6 g), the liquid phase purity was 99.80%, and the total yield of the two-step reaction with Example 13 was 94.0%. Melting point: 236.6-240.8 ° C. |
| 1H NMR(500MHz,DMSO-d6)δ10.67(s,1H),10.36(s,1H),8.82(s,1H),8.18(s,1H),8.01(s,1H),7.59(d,J=8.2Hz,1H),7.45(d,J=6.8Hz,1H),7.32(t,J=7.5Hz,1H),7.24(s,1H),4.97(q,J=8.7Hz,1H),3.93(s,3H),3.75(s,2H),3.41(s,2H),3.10(s,3H),2.78(s,6H); |
| 13C NMR(126MHz,DMSO-d6)δ166.81,153.27,152.17,150.76,138.61,138.16,138.15,125.46,124.94,123.83(q.J=278.5Hz),123.42,123.41,122.60,122.59,120.52,111.34,111.17,106.29,62.14(q,J=35.3Hz),53.53,46.28,42.27,42.26,40.92,33.67。 |
| Example 15: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine dihydrochloride (III-1) |
| At room temperature, add the compound of formula IV 2-[2-(dimethylaminoethyl)methylamino]-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (1136.0 g, 2.09 mol) to a 50 L reactor, add acetonitrile (7.95 L) and water (7.95 L), and stir for 10 to 20 minutes. Add hydrosulfite (2563.9 g, 12.50 mol) to the reactor in batches. After adding, continue stirring for 10 to 20 minutes. Control the temperature of the reactor to 35 to 40 ° C and react for 3 hours. Add concentrated hydrochloric acid (2505.3 g, 25.08 mol) to the reactor. After the addition is complete, heat to 35 to 45 ° C and react for 18 hours. 30% sodium hydroxide (1003.2 g, 25.08 mol) aqueous solution was added dropwise to adjust the pH value to 6-8. Solid sodium bicarbonate (1053.5 g, 12.54 mol) was added to adjust the pH value to 7-8. After stirring for 40 minutes, the mixture was filtered, the filtrate was allowed to stand, the layers were separated, and the organic phase was concentrated under reduced pressure. The purity of the liquid phase was detected to be 97.60%. |
| Add ethanol (5.68 L) to the product of the previous step, raise the temperature to 50-70°C, and drop concentrated hydrochloric acid (522 g, 5.23 mol). After the dropwise addition is completed, cool to room temperature and stir for 15 hours. Filter by suction, and air dry the filter cake at 50°C to obtain 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine dihydrochloride (780 g), with a liquid phase purity of 98.74%. |
| Example 16: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-(3-chloro-propionamido)-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine hydrochloride (II-1) |
| 2-[2-(Dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine dihydrochloride (1543.5 g, 2.63 mol) was added to a 50 L reactor, and dichloromethane (13.1 L) and triethylamine (532.2 g, 5.26 mol) were added. The mixture was stirred and cooled to -10 to -5 °C, and a solution of 3-chloropropionyl chloride (501.5 g, 3.95 mol) in dichloromethane (10.0 L) was added dropwise. After the addition is completed, keep warm and stir for 10 to 20 minutes, filter with suction, and the filter cake is formula II-12-[2-(dimethylaminoethyl)methylamino]-3-(3-chloro-propionamide)-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine hydrochloride wet product (2683.5g), which is calculated based on the theoretical yield of 100% and is directly used in the next reaction. |
| Melting point: 233.2-238.7℃ |
| 1H NMR(500MHz,DMSO-d6)δ10.18(s,1H),8.57(s,1H),8.42(s,1H),8.27(t,J=6.6Hz,2H),8.17(s,1H),7.51(d,J=8.1Hz,1H),7.26-7.22(m,1H),7.22-7.17(m,2H),4.99(q,J=9.1Hz,2H),3.91(d,J=6.3Hz,2H),3.89(s,3H),3.55(s,2H),3.13(s,2H),3.02(t,J=6.1Hz,2H),2.85(s,3H),2.64(s,6H); |
| 13C NMR(126MHz,DMSO-d6)δ168.41,161.88,160.22,157.34,148.05,146.73,137.62,133.25,130.86,125.43,124.09(q,J=279.2Hz),122.04,121.74,120.88,118.51,116.60,112.33,110.40,107.09,61.65(q,J=35.3Hz),54.90,40.96,40.95,40.60,38.71,32.96,32.95,32.94。 |
| Example 17: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I, crude product) |
| The wet product (2683.5 g) of Formula II 2-[2-(dimethylaminoethyl)methylamino]-3-(3-chloro-propionamide)-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine hydrochloride obtained in Example 16 was added to a 20L reactor, and acetonitrile (16.8L) and triethylamine (1329.3g, 13.15mol) were added, stirred, and heated to reflux for 4 hours. Cooled to room temperature, purified water (4.20L) was added, stirred at room temperature for 3-4 hours, and filtered. The filter cake was transferred to a 50L reactor, dichloromethane (17L) was added, and the pH value was adjusted to 7-8 with saturated sodium bicarbonate aqueous solution (17L). Liquid separation, the organic phase was transferred to a 20L reactor, activated carbon (84.3g) was added, refluxed for 1 hour, cooled to 20-30°C, and filtered. The filtrate was concentrated to dryness under reduced pressure to obtain the compound of formula I 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (1390g), with a total yield of 92.9% and a purity of 99.21% for the two-step reaction with Example 16. |
| 1H NMR(500MHz,DMSO-d6)δ9.96(s,1H),8.71(s,1H),8.44(s,1H),8.29(d,J=5.3Hz,1H),8.26(d,J=7.7Hz,1H),8.13(s,1H),7.51(d,J=8.2Hz,1H),7.24(t,J=7.2Hz,1H),7.20(d,J=5.3Hz,1H),7.15(t,J=7.2Hz,1H),6.51(dd,J=17.0,10.2Hz,1H),6.28(dd,J=17.0,1.8Hz,1H),5.78(dd,J=10.2,1.8Hz,1H),5.00(q,J=9.1Hz,2H),3.89(s,3H),3.18(t,J=6.5Hz,2H),2.87(s,3H),2.48(t,J=6.5Hz,2H),2.22(s,6H); |
| 13C NMR(126MHz,DMSO-d6)δ163.40,161.84,160.26,157.35,148.07,147.15,137.60,133.23,131.61,130.07,126.67,125.41,124.03(q,J=278.5Hz),122.00,121.68,120.80,118.39,116.13,112.36,110.37,107.02,61.29(q,J=35.3Hz),56.57,52.44,45.60,45.59,38.54,32.93; |
| MS m/z:569.25[M+1]。 |
| Example 18: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (III’) |
| At room temperature, add the compound of formula IV 2-[2-(dimethylaminoethyl)methylamino]-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (20.0 g, 36.73 mmol) to a 1L reaction bottle, add tetrahydrofuran (134 mL) and water (66 mL), and stir for 10 to 20 minutes. Add hydrosulfite (47.9 g, 220.38 mmol) to the reaction bottle in batches. After addition, continue stirring for 10 to 20 minutes. Control the internal temperature to 35-40°C and react for 3 hours. Add concentrated hydrochloric acid (89.3 g, 881.52 mmol) to the reaction bottle. After the addition is complete, heat to 42 to 47°C and react for 17 hours. 30% sodium hydroxide (35.26 g, 881.52 mmol) aqueous solution was added dropwise, and solid sodium bicarbonate (18.5 g, 220.38 mmol) was added in batches to adjust the pH value to 6-8. After stirring for 30 minutes, the mixture was filtered, and the filtrate was allowed to stand and separated. The organic phase was concentrated to dryness under reduced pressure to obtain 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (19.2 g) with a purity of 95.8% and a yield of 97.12%. |
| Example 19: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-(3-chloro-propionamido)-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine hydrochloride (II-1) |
| Add 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (5 g, 9.72 mmol) to a 250 mL reaction bottle, add dichloromethane (42 mL), stir, protect with argon, cool to -5 to 0°C, and add 3-chloropropionyl chloride (1.851 g) and dichloromethane (33 mL) dropwise. After the addition is complete, the mixture is stirred for 10-20 minutes at a temperature maintained at room temperature. After the reaction is complete, the mixture is concentrated under reduced pressure to obtain 2-[2-(dimethylaminoethyl)methylamino]-3-(3-chloro-propionamido)-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine hydrochloride (7.0 g) with a purity of 85.67%. Melting point: 233.5-238.9°C. |
| 1H NMR(500MHz,DMSO-d6)δ10.18(s,1H),8.57(s,1H),8.42(s,1H),8.27(t,J=6.6Hz,2H),8.17(s,1H),7.51(d,J=8.1Hz,1H),7.26-7.22(m,1H),7.22-7.17(m,2H),4.99(q,J=9.1Hz,2H),3.91(d,J=6.3Hz,2H),3.89(s,3H),3.55(s,2H),3.13(s,2H),3.02(t,J=6.1Hz,2H),2.85(s,3H),2.64(s,6H); |
| 13C NMR(126MHz,DMSO-d6)δ168.41,161.88,160.22,157.34,148.05,146.73,137.62,133.25,130.86,125.43,124.09(q,J=279.2Hz),122.04,121.74,120.88,118.51,116.60,112.33,110.40,107.09,61.65(q,J=35.3Hz),54.90,40.96,40.95,40.60,38.71,32.96,32.95,32.94。 |
| Example 20: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I, crude product) |
| The 2-[2-(dimethylaminoethyl)methylamino]-3-(3-chloro-propionamido)-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine hydrochloride obtained in Example 19 was added to a 250 mL reaction bottle, and acetonitrile (45 mL) and triethylamine (4.9 g) were added. The mixture was stirred magnetically and protected by argon. The temperature was raised to reflux in an oil bath. The reaction was allowed to react for 6 h. Water (23 mL) was added dropwise, and the mixture was naturally cooled to room temperature in an oil bath. The mixture was filtered with suction, and the filter cake was transferred to a 500 mL reaction bottle. Dichloromethane (100 mL) was added, and the pH value was adjusted to 7-8 with saturated aqueous sodium bicarbonate solution (100 mL). The liquids were separated and the organic phase was concentrated under reduced pressure. The solid was dried in an oven at 50°C to give 2-[2-(dimethylaminoethyl)methylamino]-3-(3-chloro-propionamide)-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (4.1 g) with a purity of 97.7%. The total yield of the two-step reaction with Example 19 was 74.17%. |
| Comparative Example 1: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I, crude product) |
| 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (1 g, 1.94 mmol) was added to a 50 mL multi-necked flask, tetrahydrofuran (10 mL) was used as the solvent, argon was replaced three times, and stirring was maintained at 0-5°C under argon protection, and 3-chloropropionyl chloride (0.37 g, 2.92 mmol), the addition was completed in 15 minutes, and the mixture was stirred at 0-5°C for 1 hour. Sodium hydroxide (0.31 g, 7.77 mmol) and water (1 mL) were added to the reaction solution, and the temperature was raised to 65°C and stirred for 15 hours. Saturated ammonium chloride solution (10 mL) was added, and the liquids were separated. The organic phase was washed with saturated sodium bicarbonate solution (10 mL). The liquids were separated and the organic phase was concentrated to dryness to obtain 1.04 g of a yellow solid with a yield of 94.9% and a purity of 87.35%. |
| Comparative Example 2: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I, crude product) |
| Add 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (5.0 g) to a 250 mL reaction bottle, add acetone (50 mL) and potassium carbonate (940 mg), stir, protect with argon, cool to -50°C, and add 3-chloropropionyl chloride (1.481 g) dropwise. After the addition is completed, the temperature is raised to -20°C and stirred for 30 minutes. A solution of sodium hydroxide (350 mg) and water (60 ml) is added dropwise over 10 minutes. The mixture is stirred at room temperature for 3 to 4 hours. The mixture is filtered and the filter cake is dried in an oven at 50°C to obtain a compound of formula II-1′, 2-[2-(dimethylaminoethyl)methylamino]-3-(3-chloro-propionamide)-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (4.28 g) with a purity of 64.18%. |
| 1H NMR(400MHz,DMSO-d 6 )δ10.32(s,1H),10.21(s,1H),8.54(s,1H),8.43(s,1H)8.29-8.28(d,J=5.1Hz,1H),8.28-8.26(d,J=6.2Hz,1H),8.19(s,1H),7.54-7.52(d,J=8.0Hz,1H),7.27-7.18(m,3H),5.77(s,2H),5.00(q,J=9.1Hz,1H),3.92(t,J=6.2Hz,1H),3.63(t,J=5.7Hz,2H),3.28(t,J=5.7Hz,2H),3.06-3.03(t,J=6.2Hz,2H),2.85(s,3H),2.74(s,6H). |
| MS m/z:605.23[M+1]。 |
| Add 2-[2-(dimethylaminoethyl)methylamino]-3-(3-chloro-propionamido)-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (4.28 g) to a 250 mL reaction bottle, add acetonitrile (45 ml) and triethylamine (3.606 g), stir magnetically, protect with argon, heat in an oil bath to reflux, and react for 6 h. Water (23 ml) was added dropwise, the temperature was naturally lowered in an oil bath and stirring was continued overnight (16 h), filtered with suction, and the solid was dried to obtain 2-[2-(dimethylaminoethyl)methylamino]-3-(3-chloro-propionamido)-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (3.3 g) with a purity of 95.13% and a two-step yield of 59.42%. |
| Example 21: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| The crude product (1390 g) of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine was transferred to a 50L reactor, acetone (25.0L) was added, argon was replaced 3 times, the temperature was raised to 45-50°C, all the solids were dissolved, and purified water (6.95L) was added dropwise. After the addition was completed, the mixture was cooled to 20-25°C and stirred for 2 hours. The mixture was filtered and the filter cake was vacuum dried at 50°C for 24 hours to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (895g). The reaction yield is 66.7% and the purity is 99.89%. |
| Example 22: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| In a 250mL three-necked flask, add 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine crude product (5.0g), add ethyl acetate 100mL, heat to 70-75°C in an oil bath to dissolve all the solids, then cool naturally to 25°C in an oil bath, filter and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (3.1g) with a purity of 99.73% and a yield of 62.0%. |
| Example 23: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| In a 250mL three-necked flask, add crude 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (5.0g), add ethyl acetate 100mL, heat to 70-75°C in an oil bath, dissolve all the solids, continue to stir for 30min, and drop 150mL of n-heptane. After the drop is complete, cool to 25°C in an oil bath, filter by suction, and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (4.0g), with a purity of 99.32% and a yield of 80%. |
| Example 24: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| In a 250mL three-necked flask, add crude 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (5.0g), add acetonitrile 75mL, heat in an oil bath to 77-82°C, dissolve all the solids, and drop 25mL of water. After dripping, naturally cool to 25°C in the oil bath, filter with suction, and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (4.3g), with a purity of 99.64% and a yield of 86%. |
| Example 25: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| In a 250mL three-necked flask, add crude 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (5.0g), add acetonitrile 75mL, heat in an oil bath to 77-82°C, dissolve all the solids, and continue to stir for 30min. Cool naturally to 25°C in the oil bath, filter with suction, and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (4.0g), with a purity of 99.45% and a yield of 80%. |
| Example 26: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| In a 250mL three-necked flask, add crude 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (5.0 g), add 20mL of tetrahydrofuran, heat in an oil bath to 45-50°C to dissolve all the solids, continue to stir and maintain the temperature for 30 minutes, and add 40mL of n-heptane dropwise. After the addition was completed, the mixture was naturally cooled to 25°C in an oil bath, filtered and dried to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (4.23 g) with a purity of 99.51% and a yield of 84.6%. |
| Example 27: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| In a 250mL three-necked flask, add crude 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (5.0g), add 100mL of isopropanol, heat to 50°C in an oil bath, dissolve all the solids, and continue to stir for 30 minutes. Cool naturally to 22°C in the oil bath, filter with suction, and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (4.25g), with a purity of 99.51% and a yield of 85%. |
| Example 28: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| In a 250mL three-necked flask, add crude 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (5.0g), add 75mL of methanol, and heat to 55-60°C in an oil bath to dissolve all the solids. Cool naturally to 17°C in the oil bath, stir overnight, filter with suction, and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (3.55g), with a purity of 99.63% and a yield of 71%. |
| Example 29: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| In a 250mL three-necked flask, add crude 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (5.0 g), add 50mL of dichloromethane, heat in an oil bath to 40°C to dissolve all the solids, continue to stir and maintain the temperature for 30 minutes, and add 100mL of n-heptane dropwise. The mixture was naturally cooled to 15°C in an oil bath, stirred overnight, filtered and dried to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (3.78 g) with a purity of 99.56% and a yield of 75.6%. |
| Example 30: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| In a 250mL three-necked flask, add crude 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (5.0g), add 100mL of toluene, heat to 65°C in an oil bath, dissolve all the solids, and continue to stir for 30 minutes. Cool naturally to 20°C in the oil bath, filter with suction, and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (3.27g), with a purity of 99.57% and a yield of 65.4%. |
| Example 31: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| In a 250mL three-necked flask, add 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine crude product (5.0g), add DMF50mL, heat to 80°C in an oil bath, dissolve all the solids, continue to stir for 30min, and drop 25mL of water. Naturally cool to 20°C in the oil bath, filter with suction, and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (3.84g), with a purity of 99.77% and a yield of 76.8%. |
| Example 32: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| To a 25 mL single-necked bottle, add 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine crude product (1.0 g), add tetrahydrofuran (6 mL), protect with argon, heat in an oil bath to 40-45°C until all the solution is dissolved, continue to stir and keep warm for 30 min, cool naturally to 22°C in an oil bath, filter and obtain a solid. The solid was transferred to a crystallization dish and dried to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (622 mg) with a purity of 99.83% and a yield of 62.2%. |
| Example 33: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| To a 50 mL single-mouth bottle, add 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine crude product (1.0 g), add acetone (15 mL), protect with argon, heat in an oil bath to 45-50° C. until all the solution is dissolved, and then continue to stir and keep warm for 30 min, cool naturally to 22° C. in an oil bath, filter and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (537 mg) with a purity of 99.83% and a yield of 53.7%. |
| Example 34: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| To a 50 mL single-mouth bottle, add 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine crude product (1.0 g), add tetrahydrofuran (8 mL), protect with argon, heat in an oil bath to 40-45° C. until all the solution is dissolved, and continue to stir and keep warm for 30 min. Add water (16 mL) dropwise, cool naturally to 21° C. in an oil bath, filter and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (880 mg) with a purity of 99.68% and a yield of 88.0%. |
| Example 35: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| To a 100 mL single-mouth bottle, add crude 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (1.0 g), add ethanol (35 mL), protect with argon, heat in an oil bath to 75-80° C. until all the solution is dissolved, add water (10 mL) dropwise over 10 min, cool naturally to 20° C. in an oil bath, filter with suction, and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (915 mg), with a yield of 91.5% and a purity of 99.49%. |
| Example 36: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| To a 50 mL single-mouth bottle, add 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine crude product (1.0 g), add xylene (20 mL), protect with argon, heat in an oil bath to 80° C. until all the solution is dissolved, cool naturally to 20° C. in an oil bath, filter with suction, and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (798 mg), with a yield of 79.8% and a purity of 99.48%. |
| Example 37: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| In a 250mL three-necked flask, add 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine crude product (5.0g), add ethanol 125mL, heat in an oil bath to 75-80°C to dissolve all the solids, continue to stir and keep warm for 30min, then cool naturally to 25°C in an oil bath, filter and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (3.7g) with a purity of 99.66% and a yield of 74%. |
| Example 38: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| To a 100 mL single-mouth bottle, add crude 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (1.0 g), add methanol (35 mL), protect with argon, heat in an oil bath to 80° C. until all the solution is dissolved, add water (10 mL) dropwise over 10 min, cool naturally to 20° C. in an oil bath, filter and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (912 mg), with a yield of 91.2% and a purity of 99.53%. |
PATENT
CN110606842
WO2019238103
PAPER
https://www.nature.com/articles/s41401-020-0389-3
| NCT Number | Sponsor | Condition | Start Date | Phase |
|---|---|---|---|---|
| NCT02973763 | Allist Pharmaceuticals, Inc. | NSCLC | December 30, 2016 | Phase 1 |
| NCT03787992 | Allist Pharmaceuticals, Inc. | Locally Advanced or Metastatic EGFR Sensitising Mutation Positive Non-small Cell Lung Cancer | May 30, 2019 | Phase 3 |
| NCT03452592 | Allist Pharmaceuticals, Inc. | Advanced NSCLC Patients With T790M | April 30, 2018 | Phase 2 |
- [1]. Y. Shi, et al. P2.03-028 Third Generation EGFR Inhibitor AST2818 (Alflutinib) in NSCLC Patients with EGFR T790M Mutation: A phase1/2 Multi-Center Clinical Trial.[2]. Alexander I. Spira, et al. FURVENT: Phase 3 trial of furmonertinib vs chemotherapy as first-line treatment for advanced NSCLC with EGFR exon 20 insertion mutations (FURMO-004). Journal of Clinical Oncology. Volume 42, Number 16_suppl.
/////////FIRMOMERTINIB, Furmonertinib, Alflutinib, AST 2818, UNII-A49A7A5YN4, PHASE 2, CANCER
Ervogastat


Ervogastat
CAS 2186700-33-2
Non-alcoholic Steatohepatitis (NASH) with Liver Fibrosis (FAST TRACK – U.S.)
- 2-[5-[(3-Ethoxy-2-pyridinyl)oxy]-3-pyridinyl]-N-[(3S)-tetrahydro-3-furanyl]-5-pyrimidinecarboxamide
- (S)-2-(5-((3-Ethoxypyridin-2-yl]oxy]pyridin-3-yl)-N-(tetrahydrofuran-3-yl)pyrimidine-5-carboxamide
- PF 06865571
- BSOIY5AKQW
407.4 g/mol, C21H21N5O4
2-[5-(3-ethoxypyridin-2-yl)oxypyridin-3-yl]-N-[(3S)-oxolan-3-yl]pyrimidine-5-carboxamide
- OriginatorPfizer
- ClassAmides; Ethers; Furans; Hepatoprotectants; Pyridines; Pyrimidines; Small molecules
- Mechanism of ActionDiacylglycerol O-acyltransferase inhibitors
Phase IINon-alcoholic fatty liver disease; Non-alcoholic steatohepatitis
- 08 Jan 2025Chemical structure information added.
- 21 Feb 2024Pfizer completes a phase II trial in Non-alcoholic steatohepatitis (Combination therapy) in Slovakia, Japan, Bulgaria, Canada, China, Hong Kong, India, Poland, Puerto Rico, South Korea, Taiwan (PO) (NCT04321031) (EudraCT2019-004775-39)
- 21 Feb 2024Pfizer completes a phase II trial in Non-alcoholic steatohepatitis (Monotherapy) in Slovakia, Japan, Bulgaria, Canada, China, Hong Kong, India, Poland, Puerto Rico, South Korea, Taiwan (PO) (NCT04321031) (EudraCT2019-004775-39)
Ervogastat is an experimental small-molecule drug and selective diacylglycerol O-acyltransferase 2 inhibitor developed by Pfizer for non-alcoholic steatohepatitis.[1] Its development was previously halted by the company but resumed in 2022.[2]
Scheme
SIDE CHAIN

MAIN

https://doi.org/10.1021/acs.jmedchem.2c01200

SYN
WO2023026180
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023026180&_cid=P11-MB5XF4-40032-1
Preparation of Intermediates and Examples
Preparation of Intermediate 1 and Example 1 (Forms 1 and 2) were described in WO2018/033832 and are reproduced below.
Intermediate 1 : 2-(5-((3-ethoxypyridin-2-yl)oxy)pyridin-3-yl)pyrimidine-5-carboxylic acid
Step 1 : 3-Ethoxypyridine
Cesium carbonate (12 mol, 1.5 equiv) and ethyl iodide (9.7 mol, 1.2 equiv) were added to a solution of 3-hydroxypyrdine (8.10 mol, 1.0 equiv) in acetone (12 L) at 15 °C. The reaction mixture was stirred at room temperature for 24 hours. The reaction mixture was filtered and the organic layer was concentrated to give crude product. Ethyl acetate (20 L) was added and washed with water (3×5 L). The organic layer was dried over sodium sulfate, filtered and concentrated to give 3-ethoxypyridine (620 g, 62%) as an oil. 1H NMR (400 MHz, CDCh) 5 1.44 (t, 3H), 4.07 (q, 2H), 7.15-7.23 (m, 2H), 8.20 (dd, 1 H), 8.30 (d, 1 H).
Step 2: 3-Ethoxypyridine-1 -oxide
m-Chloroperoxybenzoic acid (6.5 mol, 1.3 equiv) was added to a solution of 3-ethoxypyridine (5.0 mol, 1.0 equiv) in dichloromethane (12 L) at 10 °C. The reaction mixture was stirred at room temperature for 24 hours. Sodium thiosulfate (4 kg, in 5 L of water) was added. The reaction mixture was stirred at 15 °C for 2 hours. Another portion of sodium thiosulfate (1.5 kg, in 5 L of water) was added. The reaction mixture was stirred at 15 °C for 1 hour. The mixture was extracted with dichloromethane (16×10 L). The combined organic layers were concentrated to give crude product. The crude product was purified by silica gel column chromatography (dichloromethane:methanol; 100:1-10:1) to give the title compound (680 g, 97%) as brown oil. This was further purified by trituration with petroleum ether (4 L) at room temperature for 24 hours to give 3-ethoxypyridine-1 -oxide (580 g, 83%) as yellow solid. 1H NMR (400 MHz, CDCh) 5 1.41 (t, 3H), 4.02 (q, 2H), 6.84 (dd, 1 H), 7.12 (dd, 1 H), 7.85 (d, 1 H), 7.91-7.95 (m, 1 H).
Step 3: 2-((5-Bromopyridin-3-yl)oxy)-3-ethoxypyridine
This reaction was carried out in five parallel batches.
Diisopropylethylamine (2.69 mol, 3.7 equiv) and bromotripyrrolidinophosphonium hexafluorophosphate (0.93 mol, 1.3 equiv) were added to a stirred solution of 3-ethoxypyridine-1-oxide (0.72 mol, 1.0 equiv) and 3-bromo-5-hydroxypyridine (0.72 mol, 1.0 equiv) in tetrahydrofuran (2500 mL) at room temperature. The reaction mixture was stirred at room temperature for 2 days then the separate batches were combined to a single batch. The resulting suspension was concentrated to dryness and dissolved in dichloromethane (25 L). The organic layer was washed with 1 N sodium hydroxide (15 L), water (3×20 L), and brine (20 L). The organic layer was dried over sodium sulfate, filtered and concentrated to give an oil. The crude oil was purified by silica gel column chromatography (petroleum ether : ethyl acetate; 10:1-1 :1) to give crude product as brown solid. This solid was triturated with methyl tert-butyl ether: petroleum ether (1 :10; 11 L) to afford 2-((5-bromopyridin-3-yl)oxy)-3-ethoxypyridine (730 g, 69%) as off yellow solid. 1H NMR (400 MHz, CDCh) 5 1.49 (t, 3H), 4.16 (q, 2H), 7.04 (dd, 1 H), 7.25 (dd, 1 H), 7.68-7.73 (m, 2H), 8.44 (d, 1 H), 8.49 (d, 1 H). MS (ES+) 297.1 (M+H).
Step 4: Ethyl 2-(5-((3-ethoxypyridin-2-yl)oxy)pyridin-3-yl)pyrimidine-5-carboxylate
A solution of 2-((5-bromopyridin-3-yl)oxy)-3-ethoxypyridine (300 mmol, 1.0 equiv) in tetrahydrofuran (1.3 L) was degassed with nitrogen for 30 minutes. Turbo Grignard
(390 mmol, 1.3 equiv, 1.3 M in tetrahydrofuran) was added at room temperature at a rate to maintain the internal temperature below 30 °C. The reaction mixture was allowed to cool to room temperature and stirred for 3 hours. The reaction was cooled to 10 °C and zinc chloride (390 mmol, 1.3 equiv, 1.9 M in 2-methyltetrahydrofuran) was added at a rate to maintain the temperature below 15 °C. The resulting suspension was warmed to room temperature until all the precipitate was dissolved and then cooled back to 10 °C. Ethyl 2-chloropyrimidine-5-carboxylate (360 mmol, 1.2 equiv) and dichloro[bis(2-(diphenylphosphino)phenyl)ether]palladium(ll) (6.00 mmol, 0.02 equiv) were added as solids. The resulting suspension was degassed with nitrogen for 30 minutes then heated to 50 °C for 16 hours. The reaction was worked up under aqueous conditions then treated sequentially with ethylenediaminetetraacetic acid disodium salt, thiosilica, and charcoal to remove metal impurities. The crude compound was recrystallized from methanol (450 mL) to yield ethyl 2-(5-((3-ethoxypyridin-2-yl)oxy)pyridin-3-yl)pyrimidine-5-carboxylate (77 g, 70%) as a pale, yellow solid. 1H NMR (400 MHz, CDCI3) 5 1.44 (t, 3H), 1.50 (t, 3H), 4.19 (q, 2H), 4.46 (q, 2H), 7.00-7.04 (m, 1 H), 7.25 (s, 1 H), 7.71 (d, 1 H), 8.59 (s, 1 H), 8.66 (d, 1 H), 9.32 (s, 2H), 9.55 (s, 1 H).
Step 5: 2-(5-((3-ethoxypyridin-2-yl)oxy)pyridin-3-yl)pyrimidine-5-carboxylic acid
Sodium hydroxide (307 mmol, 1.5 equiv, 4M aqueous) and methanol (50 mL) were added to a suspension of 2-(5-((3-ethoxypyridin-2-yl)oxy)pyridin-3-yl)pyrimidine-5-carboxylate (205 mmol, 1.0 equiv) in tetrahydrofuran (300 mL). The resulting solution was stirred at room temperature for 3 hours. The reaction mixture was diluted with water (400 mL) and extracted with 2:1 diethyl ether: heptanes (2x 300 mL). The aqueous layer was acidified to pH of 4 with 4M hydrochloric acid. The resulting suspension was stirred at room temperature for 1 hour. The solid was filtered, washed with water, and dried to yield 2-(5-((3-ethoxypyridin-2-yl)oxy)pyridin-3-yl)pyrimidine-5-carboxylic acid (69 g, 100%) as a pale, yellow solid. 1H NMR (400 MHz, DMSO-de) 51.37 (t, 3H), 4.18 (q, 2H), 7.19 (dd, 1 H), 7.58 (dd, 1 H), 7.70 (dd, 1 H), 8.35-8.40 (m, 1 H), 8.66 (d, 1 H), 9.33 (s, 2H), 9.41 (d, 1 H), 13.9 (br. s, 1 H).
Example 1 : (S)-2-(5-((3-ethoxypyridin-2-yl)oxy)pyridin-3-yl)-/V-(tetrahydrofuran-3-yl)pyrimidine-5-carboxamide
Preparation of Form 1 of (S)-2-(5-((3-ethoxypyridin-2-yl)oxy)pyridin-3-yl)-/\/- (tetrahydrofuran-3-yl)pyrimidine-5-carboxamide
Oxalyl chloride (13.8 mL, 160 mmol, 1.2 equiv) and dimethylformamide (0.510 mL, 6.65 mmol, 0.05 equiv) were added to a suspension of 2-(5-((3-ethoxypyridin-2-yl)oxy)pyridin-3-yl)pyrimidine-5-carboxylic acid (45.0 g, 133 mmol, 1.0 equiv) in dichloromethane (500 mL). The suspension was stirred for 2 hours when a solution was achieved. The reaction mixture was concentrated to yield crude acid chloride as a red solid. A solution of (S)-tetrahydrofuran-3-amine (12.2 g, 140 mmol, 1.05 equiv) and diisopropylethylamine (51.0 mL, 293 mmol, 2.2 equiv) in tetrahydrofuran (100 mL) was added dropwise to a solution of the crude acid chloride in dichloromethane (200 mL) at 0 °C. The reaction was allowed to warm to room temperature and stirred for 16 hours. Water (1.0 L) and ethyl acetate (600 mL) were added and the organic layer was separated, washed with saturated sodium bicarbonate, dried over magnesium sulfate, and filtered. The filtrate was treated with activated charcoal (20 g) was stirred at 65 °C for 20 minutes. The suspension was filtered warm and filtrate was concentrated to a pale, yellow solid which was recrystallized from methanol in ethyl acetate (1 :4, 1 L) to yield (S)-2-(5-((3-ethoxypyridin-2-yl)oxy)pyridin-3-yl)-/\/-(tetrahydrofuran-3-yl)pyrimidine-5-carboxamide (43.5 g, 81%) as a colorless solid. The title compound was combined with previous batches (108.7 g, 266.8 mmol) prepared in the same manner and slurried with ethyl acetate (1 .0 L) at 80 °C for 4 hours. The suspension was allowed to cool to room temperature and stirred for 4 days. The solid was filtered, washed with ethyl acetate (3×200 mL) and dried under high vacuum at 50 °C for 24 hours to yield (S)-2-(5-((3-ethoxypyridin-2-yl)oxy)pyridin-3-yl)-/\/-(tetrahydrofuran-3-yl)pyrimidine-5-carboxamide (100.5 g, 92%) as a colorless solid. 1H NMR (300 MHz, DMSO-de) 5 1.38 (t, 3H), 1.89-1.98 (m, 1 H), 2.15-2.26 (m, 1 H), 3.65 (dd, 1 H), 3.70-3.78 (m, 1 H), 3.85-3.92 (m, 2H), 4.18 (q, 2H), 4.46-4.55 (m, 1 H), 7.18 (dd, 1 H), 7.58 (dd, 1 H), 7.69 (dd, 1 H), 8.37 (dd, 1 H), 8.64 (d, 1 H), 8.95 (d, 1 H), 9.28 (s, 2H), 9.39 (d, 1 H). MS (ES+) 408.4 (M+H). Melting point 177.5 °C. Elemental analysis for C21H21N5O4: calculated C, 61.91 ; H, 5.20; N, 17.19; found C, 61.86; H, 5.18; N, 17.30.
PATENT
WO2020234726
WO2020044266
WO2018033832
WO2021171164
WO2016036636 EG 1
References
- ^ Futatsugi, Kentaro; Cabral, Shawn; Kung, Daniel W.; Huard, Kim; Lee, Esther; Boehm, Markus; Bauman, Jonathan; Clark, Ronald W.; Coffey, Steven B.; Crowley, Collin; Dechert-Schmitt, Anne-Marie; Dowling, Matthew S.; Dullea, Robert; Gosset, James R.; Kalgutkar, Amit S.; Kou, Kou; Li, Qifang; Lian, Yajing; Loria, Paula M.; Londregan, Allyn T.; Niosi, Mark; Orozco, Christine; Pettersen, John C.; Pfefferkorn, Jeffrey A.; Polivkova, Jana; Ross, Trenton T.; Sharma, Raman; Stock, Ingrid A.; Tesz, Gregory; Wisniewska, Hanna; Goodwin, Bryan; Price, David A. (24 November 2022). “Discovery of Ervogastat (PF-06865571): A Potent and Selective Inhibitor of Diacylglycerol Acyltransferase 2 for the Treatment of Non-alcoholic Steatohepatitis”. Journal of Medicinal Chemistry. 65 (22): 15000–15013. doi:10.1021/acs.jmedchem.2c01200. PMID 36322383. S2CID 253257260.
- ^ “With the right partner, Pfizer gains fast-track tag for previously shelved NASH drug”. Retrieved 20 November 2023.
| Clinical data | |
|---|---|
| Other names | PF-06865571 |
| Legal status | |
| Legal status | Investigational |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2186700-33-2 |
| PubChem CID | 134262752 |
| ChemSpider | 114929473 |
| UNII | BSOIY5AKQW |
| ChEMBL | ChEMBL4760665 |
| Chemical and physical data | |
| Formula | C21H21N5O4 |
| Molar mass | 407.430 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
////////////Ervogastat, PF 06865571, fast track, BSOIY5AKQW, PFIZER, PHASE 2,

{kind=link}
{kind=link}
{kind=link}
{kind=link}