
Home » Parkinson’s disease
Category Archives: Parkinson’s disease
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
GNE-0877

GNE-0877
Maybe DNL-151 ?
CAS 1374828-69-9
Chemical Formula: C14H16F3N7
Molecular Weight: 339.31895
2-methyl-2-(3-methyl-4-(4-(methylamino)-5-(trifluoromethyl)pyrimidin-2-ylamino)-1H-pyrazol-1-yl)propanenitrile
Denali Therapeutics Inc, useful for treating Alzheimer’s disease, breast tumor, type I diabetes mellitus and Crohn’s disease
GNE-0877 is a highly potent and selective LRRK2 inhibitor. Leucine-rich repeat kinase 2 (LRRK2) has drawn significant interest in the neuroscience research community because it is one of the most compelling targets for a potential disease-modifying Parkinson’s disease therapy.
- Developer Denali Therapeutics Inc
- Class Antiparkinsonians; Small molecules
- Mechanism of Action LRRK2 protein inhibitors
- Phase I Parkinson’s disease
- 20 Dec 2017 Denali Therapeutics plans clinical studies for Parkinson’s disease
- 13 Nov 2017 Phase-I clinical trials in Parkinson’s disease (In volunteers) in Netherlands (unspecified route)
- 13 Nov 2017 Preclinical trials in Parkinson’s disease in USA (unspecified route) before November 2017
Denali Therapeutics is developing DNL-151 (phase 1, in July 2019), a lead from a program of small-molecule inhibitors of LRRK2 originally licensed from Genentech, for the treatment of Parkinson’s disease.
Leucine-rich repeat kinase 2 (LRRK2) is a complex signaling protein that is a key therapeutic target, particularly in Parkinson’s disease (PD). Combined genetic and biochemical evidence supports a hypothesis in which the LRRK2 kinase function is causally involved in the pathogenesis of sporadic and familial forms of PD, and therefore that LRRK2 kinase inhibitors could be useful for treatment (Christensen, K.V. (2017) Progress in medicinal chemistry 56:37-80). Inhibition of the kinase activity of LRRK2 is under investigation as a possible treatment for Parkinson’s disease (Fuji, R.N. et al (2015) Science Translational Medicine 7(273):ral5;
Taymans, J.M. et al (2016) Current Neuropharmacology 14(3):214-225). A group of LRRK2 kinase inhibitors have been studied (Estrada, A.A. et al (2015) Jour. Med. Chem. 58(17): 6733-6746; Estrada, A.A. et al (2013) Jour. Med. Chem. 57:921-936; Chen, H. et al (2012) Jour. Med. Chem. 55:5536-5545; Estrada, A.A. et al (2015) Jour. Med. Chem. 58:6733-6746; US 8354420; US 8569281; US8791130; US 8796296; US 8802674; US 8809331; US 8815882; US 9145402; US 9212173; US 9212186; WO 2011/151360; WO 2012/062783; and WO 2013/079493.
PATENT
WO2012062783 , assigned to Hoffmann-La Roche , but naming inventors specifically associated with both Genentech and BioFocus (which had an agreement with Genentech for drug discovery programs); the compound was also later identified in J.Med.Chem (57(3), 921-936, 2014) in an article from these two companies, with the lab code GNE-0877. So while this represents the first application in the name of Denali Therapeutics Inc that focuses on this compound, it is likely that it provides the structure of DNL-151 , a lead from a program of small-molecule inhibitors of leucine-rich repeat kinase 2 (LRRK2) originally licensed from Genentech, being developed for the oral treatment of Parkinson’s disease, and which had begun phase I trials by December 2017 (when this application was lodged).
PATENT
WO2019104086 ,
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019104086
claiming novel crystalline and amorphous forms of pyrimidinylamino-pyrazole compound, useful for treating Alzheimer’s disease, breast tumor, type I diabetes mellitus and Crohn’s disease.
Novel crystalline and amorphous forms of 2-methyl-2-(3-methyl-4-(4-(methylamino)-5-(trifluoromethyl)pyrimidin-2-ylamino)-1H-pyrazol-1-yl)propanenitrile (which is substantially pure form) and their anhydrous and solvates such as cyclohexanol solvate (designated as Forms B-D), processes for their preparation and compositions comprising them are claimed. The compound is disclosed to be leucine rich serine threonine kinase 2 inhibitor, useful for treating Gaucher disease, Alzheimer’s disease, motor neurone disease, Parkinson’s disease, prostate tumor, Lewy body dementia, mild cognitive impairment, breast tumor, type I diabetes mellitus and Crohn’s disease.
The present disclosure relates to crystalline polymorph or amorphous forms of a pyrimidinylamino-pyrazole kinase inhibitor, referred to herein as the Formula I compound and having the structure:
FORMULA I COMPOUND
The present disclosure includes polymorphs and amorphous forms of Formula I compound, (CAS Registry Number 1374828-69-9), having the structure:
and named as: 2-methyl-2-(3-methyl-4-(4-(methylamino)-5-(trifluoromethyl)pyrimidin-2-ylamino)-lH-pyrazol-l-yl)propanenitrile (WO 2012/062783; US 8815882; US 2012/0157427, each of which are incorporated by reference). As used herein, the Formula I compound includes tautomers, and pharmaceutically acceptable salts or cocrystals thereof. The Formula I compound is the API (Active Pharmaceutical Ingredient) in formulations for use in the treatment of neurodegenerative and other disorders, with pKa when protonated calculated at 6.7 and 2.1.
CRYSTALLIZATION
Initial polymorph screening experiments were performed using a variety of
crystallization or solid transition methods, including: anti-solvent addition, reverse anti-solvent addition, slow evaporation, slow cooling, slurry at room temperature (RT), slurry at 50 °C, solid vapor diffusion, liquid vapor diffusion, and polymer induced crystallization. By all these methods, the Form A crystal type was identified. Polarized light microscopy (PLM) images of Form A obtained from various polymorph screening methods were collected (Example 5).
Particles obtained via anti-solvent addition showed small size of about 20 to 50 microns (pm) diameter while slow evaporation, slow cooling (except for THF/isooctane), liquid vapor diffusion and polymer-induced crystallization resulted in particles with larger size. Adding isooctane into a dichloromethane (DCM) solution of the Formula I compound produced particles with the most uniform size. Crude Formula I compound crystallized from THF///-heptane and then was micronized. A crystallization procedure was developed to control particle size.
A total of four crystal forms (Forms A, B, C, and D) and an amorphous form E of Formula I compound were prepared, including 3 anhydrates (Form A, C, and D) and one solvate (Form B). Slurry competition experiments indicated that Form D was thermodynamically more stable when the water activity aw< 0.2 at RT, while Form C was more stable when aw> 0.5 at RT. The 24 hrs solubility evaluation showed the solubility of Form A, C and D in FLO at RT was 0.18, 0.14 and 0.11 mg/mL, respectively. DVS (dynamic vapor sorption) results indicated that Form A and D were non-hygroscopic as defined by less than 0.1% reversible water intake in DVS, while Form C was slightly hygroscopic. Certain characterization data and observations of the crystal forms are shown in Table 1.
Table 1 Characterization summary for crystal forms of Formula I compound
Differential Scanning Calorimetry (DSC) analysis of Forms A and C showed that Form C had higher melting point and higher heat of fusion (Table 1), suggesting that the two forms are monotropic with Form C being the more stable form. Competitive slurry experiments with 1 : 1 Form A and C in a variety of solvents always produced Form C confirming that Form C was
more stable than Form A. In accordance with this, Form C was produced even when the crystallization batch was seeded with seeds of Form A.
PATENT
WO-2019126383
Methods of making leucine-rich repeat kinase 2 (LRRK2)-inhibiting, pyrimidinyl-4-aminopyrazole compounds (eg 2-methyl-2-(3-methyl-4-((4-(methylamino)-5-(trifluoromethyl)pyrimidin-2-yl)amino)- lH-pyrazol-1-yl)propanenitrile), useful for treating LRRK2 mediated diseases such as Parkinson’s disease.
Example 1 Preparation of 2-(4-amino-3 -methyl- liT-pyrazol-l -yl)-2-methylpropanamide 5a
4a 5a
To a 20-L reactor containing dimethyl formamide (4.5 L) was charged 5-methyl-4-nitro-lH-pyrazole la (1.5 kg, 1.0 equiv). The solution was cooled to 0 °C and charged with finely ground K2CO3 (2.45 kg, 1.5 equiv) in three portions over ~l h. Methyl 2-bromo-2-methylpropanoate (3.2 kg, 1.5 equiv) was added dropwise to the mixture and then was allowed to warm to ~25 °C. The reaction mixture was maintained for 16 h and then quenched with water (15 L) and product was extracted with ethyl acetate. The combined organic layer was washed with water, and then with a brine. The organic layer was dried over anhydrous Na2S04, filtered, and concentrated under reduced pressure to give a light yellow solid. The crude product was purified by crystallization with petroleum ether (15 L), filtered, and dried to give methyl 2-m ethyl -2-(3 -methyl -4-nitro- l//-pyrazol- l -yl)propanoate 3a (2.25 kg, >99% purity by HPLC, 84 % yield) as an off-white solid. ¾ NMR (400 MHz, CDCb) 8.28 (s, 1H), 3.74 (s, 3H), 2.53 (s, 3H), 1.85 (s, 6H).
Methanol (23 L) and 2-methyl-2-(3-methyl-4-nitro-lif-pyrazol-l-yl)propanoate 3a (2.25 kg, 1.0 equiv) were charged into a 50-L reactor and cooled to approximately -20 °C. Ammonia gas was purged over a period of 5 h and then the reaction mixture warmed to 25 °C. After 16 h, the reaction mixture was concentrated under reduced pressure (~50 °C) to give the crude product. Ethyl acetate (23 L) was charged and the solution agitated in the presence of charcoal (0.1 w/w) and Celite® (0.1 w/w) at 45 °C. The mixture was filtered and concentrated under reduced pressure, and then the solid was slurried in methyl tert-butyl ether (MTBE, 11.3 L) at RT for 2 h. Filtration and drying at ~45 °C gave 2-m ethyl -2-(3 -m ethyl -4-ni tro- 1 //-pyrazol – 1 -yl)propanamide 4a (1.94 kg, >99% purity by HPLC, 92% yield).
Methanol (5 L) and 2-m ethyl-2-(3 -methyl -4-nitro-lif-pyrazol-l-yl)propanamide 4a (0.5 kg) were charged into a 10-L autoclave under nitrogen atmosphere, followed by slow addition of 10 % (50% wet) Pd/C (50 g). Hydrogen was charged (8.0 kg pressure/l 13 psi) and the reaction mixture agitated at 25 °C until complete. The mixture was filtered, concentrated under reduced
pressure and then slurried in MTBE (2.5 L) for 2 h at 25 °C. Filtration and drying under reduced pressure (45 °C) gave 2-(4-amino-3-methyl- l//-pyrazol- l -yl)-2-methyl propanamide 5a (0.43 kg, >99% purity by HPLC, 99% yield).
Example 2 Preparation of 2-(4-((4-chloro-5-(trifluoromethyl)pyrimidin-2-yl)amino)-3-methyl-lH-pyrazol-l-yl)-2-methylpropanamide 7a
DCM
Into a first reactor was charged /-BuOH (or alternatively 2-propanol) (15.5 vol) and 2-(4-amino-3 -methyl- li7-pyrazol-l-yl)-2-methylpropanamide 5a (15 kg), followed by zinc chloride (13.5 kg, 1.2 equiv) at room temperature and the suspension agitated ~2 h. Into a second reactor was charged dichloromethane (DCM, 26.6 vol) and 2,4-dichloro-5-trifluoromethyl pyrimidine 6a (19.6 kg, 1.1 equiv) and then cooled to 0 °C. The contents from first reactor were added portion-wise to the second reactor. After addition, the reaction mixture was agitated at 0 °C for ~l h and then Et3N (9.2 kg, 1.1 equiv) was slowly charged. After agitation for 1 h, the temperature was increased to 25 °C and monitored for consumption of starting material. The reaction mixture was quenched with 5% aqueous NaHCO, and then filtered over Celite®. The DCM layer was removed and the aqueous layer was back-extracted with DCM (3x). The combined organics were washed with water, dried (Na2S04), and concentrated. Methanol (2.5 vol) was charged and the solution was heated to reflux for 1 h, then cooled to 0 °C. After 1 h, the solids were filtered and dried under reduced pressure to give 2-(4-((4-chloro-5-(tri fluoromethyl)pyri mi din-2-yl)amino)-3 -methyl – l//-pyrazol- l -yl)-2-methyl propanamide 7a
(31.2 kg (wet weight)). 1H NMR (600 MHz, DMSO-de) 10.05 (br. s., 1H), 8.71 (d, J= 11 Hz, 1H), 7.95 (app. d, 1H), 7.18 (br. s., 1H), 6.78 (br. s., 1H), 2.14 (s, 3H), 1.67 (s, 6H).
Example 3 Preparation of 2-methyl-2-(3-methyl-4-((4-(methylamino)-5-(trifluoromethyl)pyrimidin-2-yl)amino)- lH-pyrazol- 1 -yl)propanamide 8a
A reactor was charged with anhydrous tetrahydrofuran (THF, 10 vol) and 2-(4-((4-chloro-5-(trifl uoromethyl )pyrimi din-2-yl)amino)-3 -methyl – l //-pyrazol- l -yl)-2-methylpropanamide 7a (21 kg) at room temperature with agitation. A solution of 2M
methylamine in THF (3.6 vol) was slowly charged to the reactor at 25 °C and maintained for ~3 h. The reaction mixture was diluted with 0.5 w/w aqueous sodium bicarbonate solution (10 w/w), and extracted with ethyl acetate (EtOAc, 4.5 w/w). The aqueous layer was extracted with EtOAc (4x), the organics were combined and then washed with H20 (7 w/w). The organic layer was dried over sodium sulfate, filtered and concentrated under reduced pressure. «-Heptane (3 w/v) was added to the residue, agitated, filtered and dried under reduced pressure to give 2-m ethyl -2-(3 -methyl -4-((4-(methyl ami no)-5-(trifl uoromethyl )pyri mi din-2-yl)amino)- l //-pyrazol-1 -yl)propanamide 8a (19.15 kg, 93% yield). ¾ NMR (600 MHz, DMSO-d6) 8.85 (m, 1H), 8.10 (s, 1H), 8.00 (m, 1H), 7.16 (br. s., 1H), 6.94 (m, 1H), 6.61 (br. s., 1H), 2.90 (d, J = 4.3 Hz, 3H), 2.18 (br. s., 3H), 1.65 (s, 6H).
Example 4 Preparation of 2-methyl-2-(3-methyl-4-((4-(methylamino)-5-(trifluoromethyl)pyrimidin-2-yl)amino)- lH-pyrazol- 1 -yl)propanenitrile 9a
To a reactor was charged 2-methyl-2-(3-methyl-4-((4-(methylamino)-5-(trifl uoromethyl )pyri mi din-2-yl)amino)- l //-pyrazol- l -yl)propan amide 8a (15 kg, 1 equiv) at room temperature followed by EtOAc (2 vol) and 6.7 vol T3P (50% w/w in EtOAc). The reaction mixture was heated to 75 °C over 1 h and then agitated for 16 h until consumption of starting material. The reaction mixture was cooled between -10 to -15 °C then added drop-wise 5N aqueous NaOH (7 vol) resulting in pH 8-9. The layers were separated and the aqueous layer back-extracted with EtOAc (2 x 4 vol). The combined organic extracts were washed with 5 %
aqueous NaHCO, solution, and then distilled to azeotropically remove water. The organics were further concentrated, charged with «-heptane (2 vol) and agitated for 1 h at room temperature. The solids were filtered, rinsed with «-heptane (0.5 vol) and then dried under vacuum (<50 °C). The dried solids were dissolved in EtOAc (1.5 vol) at 55 °C, and then «-heptane (3 vol) was slowly added followed by 5-10% of 9a seeds. To the mixture was slowly added «-heptane (7 vol) at 55 °C, agitated for 1 h, cooled to room temperature and then maintained for 16 h. The suspension was further cooled between 0-5 °C, agitated for 1 hour, filtered, and then rinsed the filter with chilled 1 :6.5 EtOAc/«-heptane (1 vol). The product was dried under vacuum at 50 °C to give 2-methyl-2-(3-methyl-4-((4-(methylamino)-5-(trifluoromethyl)pyrimidin-2-yl)amino)-1 //-pyrazol – 1 -yl )propaneni tri 1 e 9a (9.5 kg, first crop), 67% yield). ‘H NMR (600 MHz, DMSO-d6) 8.14 (s, 1H), 8.13 (br. s., 1H), 7.12 (br. s., 1H), 5.72 (br. s, 1H), 3.00 (d, J= 4.6 Hz, 3H),
2.23 (s, 3H), 1.96 (s, 3H).
Example 5 Preparation of methyl 2-(4-amino-3-methyl-lH-pyrazol-l-yl)-2-methylpropanoate 10a
Following the procedure of Example 1, a mixture of methanol and methyl 2-methyl-2-(3-methyl-4-nitro-lH-pyrazol-l-yl)propanoate 3a (0.5 kg) was charged into an autoclave under nitrogen atmosphere, followed by slow addition of 10 % (50% wet) Pd/C. Hydrogen was charged under pressure and the reaction mixture agitated at 25 °C until complete. The mixture was filtered, concentrated under reduced pressure and then slurried in MTBE for 2 h at 25 °C. Filtration and drying under reduced pressure gave methyl 2-(4-amino-3-methyl-lH-pyrazol-l-yl)-2-methylpropanoate 10a (LC-MS, M+l=l98).
Example 6 Preparation of methyl 2-(4-((4-chloro-5-(trifluoromethyl)pyrimidin-2-yl)amino)-3 -methyl- lH-pyrazol- 1 -yl)-2-methylpropanoate 11a
Following the procedure of Example 2, a mixture of methyl 2-(4-amino-3-methyl-lH-pyrazol-l-yl)-2-methylpropanoate 10a and DIPEA (1.2 equiv) in /-BuOH was warmed to 80 °C. Then a solution of 2,4-dichloro-5-trifluoromethyl pyrimidine 6a in /-BuOH was added slowly drop wise at 80 °C. After 15 minutes, LCMS showed the reaction was complete, including later eluting 59.9% of product ester 11a, earlier eluting 31.8% of undesired regioisomer (ester), and no starting material 10a. After completion of reaction, the mixture was cooled to room temperature and a solid was precipitated. The solid precipitate was filtered and dried to give methyl 2-(4-((4-chloro-5-(trifluoromethyl)pyrimi din-2 -yl)amino)-3-methyl-lH-pyrazol-l-yl)-2-methylpropanoate 11a (LC-MS, M+l=378).
PAPER
J.Med.Chem (57(3), 921-936, 2014
https://pubs.acs.org/doi/full/10.1021/jm401654j
2-Methyl-2-(3-methyl-4-((4-(methylamino)-5-(trifluoromethyl)pyrimidin-2-yl)amino)-1H-pyrazol-1-yl)propanenitrile (11)

aReagents and conditions: (a) NaH, methyl 2-bromo-2-methylpropanoate, DMF, 70%; (b) LiOH, THF-H2O, 90%; (c) (i) (COCl)2, CH2Cl2, (ii) R-NH2, THF; (d) Pd/C, H2, MeOH; (e) 26, Et3N, n-BuOH, 120 °C; (f) 26, TFA, 2-methoxyethanol, 70 °C; (g) POCl3, 90 °C, 42%.
GNE-9605
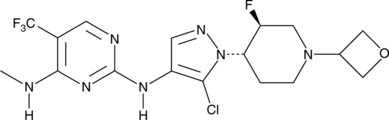
CAS № 1536200-31-3
GNE-9065 is an orally bioavailable and potent inhibitor of leucine-rich repeat kinase 2 (LRRK2; IC50 = 18.7 nM).1 It is selective for LRRK2 over 178 kinases, inhibiting only TAK1-TAB1 >50% at a concentration of 0.1 μM. GNE-9065 (10 and 50 mg/kg) inhibits LRRK2 Ser1292 autophosphorylation in BAC transgenic mice expressing human LRRK2 protein with the G2019S mutation found in families with autosomal Parkinson’s disease.
CNC1=C(C(F)(F)F)C=NC(NC2=C(Cl)N([C@H]3CCN(C4COC4)C[C@@H]3F)N=C2)=N1
N2-(5-Chloro-1-((trans)-3-fluoro-1-(oxetan-3-yl)piperidin-4-yl)-1H-pyrazol-4-yl)-N4-methyl-5-(trifluoromethyl)pyrimidine-2,4-diamine (20)

aReagents and conditions: (a) (±)-(cis)-tert-butyl 3-fluoro-4-hydroxypiperidine-1-carboxylate, PPh3, diisopropyl azodicarboxylate, THF; (b) TFA, DCM, 58% over two steps; (c) oxetan-3-one, DIPEA, NaBH(OAc)3, acetic acid, DCE, 85%; (d) LiHMDS then C2Cl6, THF, −78 °C, 65%; (e) iron dust, NH4Cl, EtOH, 90 °C; (f) 26, TFA, 2-methoxyethanol, 90 °C, 40%, two steps.
REFERENCES
1: Estrada AA, Chan BK, Baker-Glenn C, Beresford A, Burdick DJ, Chambers M, Chen H, Dominguez SL, Dotson J, Drummond J, Flagella M, Fuji R, Gill A, Halladay J, Harris SF, Heffron TP, Kleinheinz T, Lee DW, Pichon CE, Liu X, Lyssikatos JP, Medhurst AD, Moffat JG, Nash K, Scearce-Levie K, Sheng Z, Shore DG, Wong S, Zhang S, Zhang X, Zhu H, Sweeney ZK. Discovery of Highly Potent, Selective, and Brain-Penetrant Aminopyrazole Leucine-Rich Repeat Kinase 2 (LRRK2) Small Molecule Inhibitors. J Med Chem. 2014 Jan 15. [Epub ahead of print] PubMed PMID: 24354345.
/////////////DNL-151, DNL 151, DNL151, Alzheimer’s disease, breast tumor, type I diabetes mellitus, Crohn’s disease, phase 1, Parkinson’s disease, GNE0877, GNE 0877, GNE-0877, GNE-9605, GNE 9605, GNE9605, Genentech
CC(N1N=C(C)C(NC2=NC=C(C(F)(F)F)C(NC)=N2)=C1)(C)C#N
