
Home » radio labelled
Category Archives: radio labelled
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Talogreptide mesaroxetan


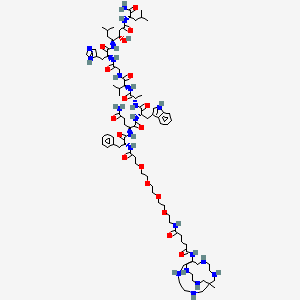
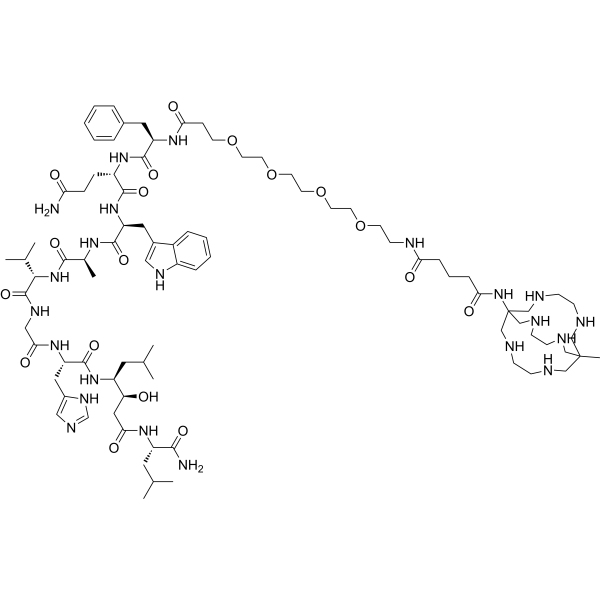
Talogreptide mesaroxetan
CAS 1801418-23-4
MF C86H140N22O18 MW1770.17
{MeCOSar}-PEG4-{d-Phe}-Gln-Trp-Ala-Val-Gly-His-{Sta}-Leu-NH2
(2S)-N-[(2S)-1-[[(2S)-1-[[(2S)-1-[[2-[[(2S)-1-[[(3S,4S)-1-[[(2S)-1-amino-4-methyl-1-oxopentan-2-yl]amino]-3-hydroxy-6-methyl-1-oxoheptan-4-yl]amino]-3-(1H-imidazol-5-yl)-1-oxopropan-2-yl]amino]-2-oxoethyl]amino]-3-methyl-1-oxobutan-2-yl]amino]-1-oxopropan-2-yl]amino]-3-(1H-indol-3-yl)-1-oxopropan-2-yl]-2-[[(2R)-2-[3-[2-[2-[2-[2-[[5-[(8-methyl-3,6,10,13,16,19-hexazabicyclo[6.6.6]icosan-1-yl)amino]-5-oxopentanoyl]amino]ethoxy]ethoxy]ethoxy]ethoxy]propanoylamino]-3-phenylpropanoyl]amino]pentanediamide
N-{21-[(8-methyl-3,6,10,13,16,19-hexaazabicyclo[6.6.6]icosan-1-yl)amino] -17,21-dioxo-4,7,10,13-tetraoxa-16-azahenicosan-1-oyl}-D-phenylalanyl-L-glutaminyl-L-tryptophyl-L-alanyl-Lvalylglycyl-L-histidyl-(3S,4S)-4-amino-3-hydroxy-6-methylheptanoyl-L-leucinamide
diagnostic imaging agent, antineoplastic, ZUN64K4H2X, SAR-BBN
Talogreptide mesaroxetan (CAS 1801418-23-4) is a synthetic peptide, a complex molecule used as a diagnostic imaging agent with potential antitumor effects, targeting G-protein coupled receptors (GRPr) often overexpressed in cancers, allowing for specific tumor visualization in PET scans, particularly for metastatic disease detection, known for its high specificity and contrast for imaging tumors like those expressing GRPr.
Key Characteristics:
- Type: A peptide-based diagnostic agent, often labeled with radioisotopes like Copper-64 ($^{64}$Cu) for Positron Emission Tomography (PET) imaging, notes Patsnap Synapse.
- Structure: It’s a modified peptide sequence incorporating elements like PEG4 and specific amino acids, MedchemExpress.com.
- Function: Binds strongly to GRPr, helping to highlight tumors and metastatic sites.
- Application: Used in research to create high-contrast PET scans for better tumor detection and monitoring, showing promise in visualizing lymph node metastasis.
In Simple Terms:
Imagine it as a “smart tracer” that seeks out specific cancer cells. When attached to a radioactive tag, it lights up tumors on a PET scan, helping doctors see cancer more clearly, notes Patsnap Synapse.
Syn
WO2024086891
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2024086891&_cid=P10-MIZEJM-53111-1
67Cu radioisotope
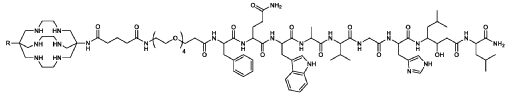
where R is CH3C(0)-;
(67CU-SAR-BBN)
Paper
Molecular Pharmaceutics (2015), 12(8), 2781-2790



AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
/////////Talogreptide mesaroxetan, diagnostic imaging agent, antineoplastic, ZUN64K4H2X, SAR-BBN
Gallium 68 PSMA-11
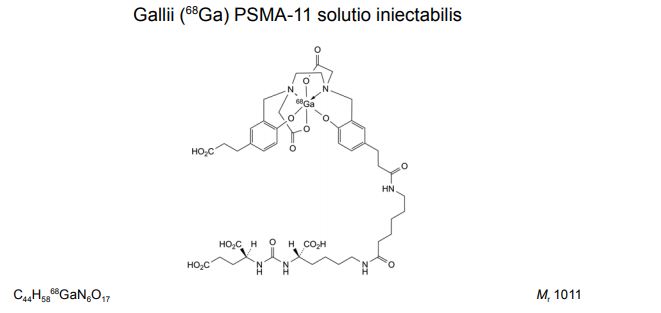

Gallium 68 PSMA-11
FDA APPROVED, 12/1/2020, Gallium 68 PSMA-11
For detection and localization of prostate cancer
Press Release
Drug Trials Snapshot

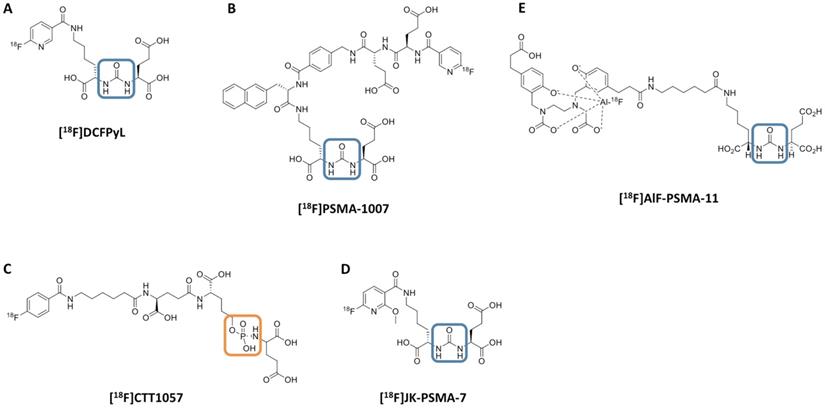
Chemical structure of 18F-labeled radiotracers. [18F]DCFPyL (A), [18F]PSMA-1007 (B), [18F]CTT1057 (C), (D) [18F]JK-PSMA-7 and (E) [18F]AIF-PSMA-11. The urea backbone of (A), (B), (D) and (E) is marked in blue, while the phosphoramidate of [18F]CTT1057 in (C) is highlighted in orange. Modified from Behr et al. [32], © by the Society of Nuclear Medicine and Molecular Imaging, Inc.
PSMA-11, also known as HBED-CC-PSMA or Psma-hbed-CC, is used to make gallium Ga 68-labeled PSMA-11, which has potential use as a tracer for PSMA-expressing tumors during positron emission tomography (PET). Upon intravenous administration of gallium Ga 68-labeled PSMA-11, the Glu-urea-Lys(Ahx) moiety targets and binds to PSMA-expressing tumor cells. Upon internalization, PSMA-expressing tumor cells can be detected during PET imaging. PSMA, a tumor-associated antigen and type II transmembrane protein, is expressed on the membrane of prostatic epithelial cells and overexpressed on prostate tumor cells

Name: PSMA-11
CAS#: 1366302-52-4
Chemical Formula: C44H62N6O17
Exact Mass: 946.4171
(3S,7S)-22-(3-(((2-((5-(2-Carboxyethyl)-2-hydroxybenzyl)(carboxymethyl)amino)ethyl)(carboxymethyl)amino)methyl)-4-hydroxyphenyl)-5,13,20-trioxo-4,6,12,19-tetraazadocosane-1,3,7-tricarboxylic acid
The Food and Drug Administration (FDA) has approved Gallium 68 PSMA-11 (Ga 68 PSMA-11), the first drug for positron emission tomography (PET) imaging of prostate-specific membrane antigen (PSMA) positive lesions in men with prostate cancer.
Ga 68 PSMA-11, a radioactive diagnostic agent, is indicated for patients with suspected prostate cancer metastasis who are potentially curable by surgery or radiation therapy. It is also indicated for patients with suspected prostate cancer recurrence based on elevated serum prostate-specific antigen (PSA) levels.
The approval was based on efficacy and safety data from 2 prospective clinical trials (Trial 1 and 2) with a total of 960 men with prostate cancer who each received 1 injection of Ga 68 PSMA-11. Trial 1 included 325 patients with biopsy-proven prostate cancer who underwent PET/CT or PET/MRI scans performed with Ga 68 PSMA-11. Results from the study showed that positive readings in the pelvic lymph nodes on Ga 68 PSMA-11 PET were associated with a clinically important rate of metastatic cancer confirmed by surgical pathology in those who proceeded to surgery.
In Trial 2, 635 patients with rising serum PSA levels after prostate surgery or radiotherapy received a single Ga 68 PSMA-11 PET/CT scan or PET/MR scan. Findings demonstrated that 74% of patients had at least 1 positive lesion detected by Ga 68 PSMA-11 PET, and local recurrence or metastasis of prostate cancer was confirmed in 91% of cases.
 This is the first drug approved for PET imaging of prostate-specific membrane antigen positive lesions in men with prostate cancer.
This is the first drug approved for PET imaging of prostate-specific membrane antigen positive lesions in men with prostate cancer.
REF
REFERENCES
1: Meißner S, Janssen JC, Prasad V, Brenner W, Diederichs G, Hamm B, Hofheinz F, Makowski MR. Potential of asphericity as a novel diagnostic parameter in the evaluation of patients with (68)Ga-PSMA-HBED-CC PET-positive prostate cancer lesions. EJNMMI Res. 2017 Oct 23;7(1):85. doi: 10.1186/s13550-017-0333-9. PubMed PMID: 29058157; PubMed Central PMCID: PMC5651532.
2: Verburg FA, Pfister D, Drude NI, Mottaghy FM, Behrendt F. PSA levels, PSA doubling time, Gleason score and prior therapy cannot predict measured uptake of [(68)Ga]PSMA-HBED-CC lesion uptake in recurrent/metastatic prostate cancer. Nuklearmedizin. 2017 Oct 18;56(6). doi: 10.3413/Nukmed-0917-17-07. [Epub ahead of print] PubMed PMID: 29044297.
3: Amor-Coarasa A, Kelly JM, Gruca M, Nikolopoulou A, Vallabhajosula S, Babich JW. Continuation of comprehensive quality control of the itG (68)Ge/(68)Ga generator and production of (68)Ga-DOTATOC and (68)Ga-PSMA-HBED-CC for clinical research studies. Nucl Med Biol. 2017 Oct;53:37-39. doi: 10.1016/j.nucmedbio.2017.07.006. Epub 2017 Jul 14. PubMed PMID: 28803001.
4: Janssen JC, Woythal N, Meißner S, Prasad V, Brenner W, Diederichs G, Hamm B, Makowski MR. [(68)Ga]PSMA-HBED-CC Uptake in Osteolytic, Osteoblastic, and Bone Marrow Metastases of Prostate Cancer Patients. Mol Imaging Biol. 2017 Dec;19(6):933-943. doi: 10.1007/s11307-017-1101-y. PubMed PMID: 28707038.
5: Damle NA, Tripathi M, Chakraborty PS, Sahoo MK, Bal C, Aggarwal S, Arora G, Kumar P, Kumar R, Gupta R. Unusual Uptake of Prostate Specific Tracer (68)Ga-PSMA-HBED-CC in a Benign Thyroid Nodule. Nucl Med Mol Imaging. 2016 Dec;50(4):344-347. Epub 2016 Mar 22. PubMed PMID: 27994690; PubMed Central PMCID: PMC5135692.
6: Behrendt F, Krohn T, Mottaghy F, Verburg FA. [(68)Ga]PSMA-HBED-CC PET/CT to differentiate between diffuse bone metastases of prostate cancer and osteopoikilosis. Nuklearmedizin. 2016 Dec 6;55(6):N64-N65. PubMed PMID: 27922151.
7: Krohn T, Birmes A, Winz OH, Drude NI, Mottaghy FM, Behrendt FF, Verburg FA. The reconstruction algorithm used for [(68)Ga]PSMA-HBED-CC PET/CT reconstruction significantly influences the number of detected lymph node metastases and coeliac ganglia. Eur J Nucl Med Mol Imaging. 2017 Apr;44(4):662-669. doi: 10.1007/s00259-016-3571-6. Epub 2016 Nov 29. PubMed PMID: 27900518.
8: Berliner C, Tienken M, Frenzel T, Kobayashi Y, Helberg A, Kirchner U, Klutmann S, Beyersdorff D, Budäus L, Wester HJ, Mester J, Bannas P. Detection rate of PET/CT in patients with biochemical relapse of prostate cancer using [(68)Ga]PSMA I&T and comparison with published data of [(68)Ga]PSMA HBED-CC. Eur J Nucl Med Mol Imaging. 2017 Apr;44(4):670-677. doi: 10.1007/s00259-016-3572-5. Epub 2016 Nov 28. PubMed PMID: 27896369.
9: Sathekge M, Lengana T, Modiselle M, Vorster M, Zeevaart J, Maes A, Ebenhan T, Van de Wiele C. (68)Ga-PSMA-HBED-CC PET imaging in breast carcinoma patients. Eur J Nucl Med Mol Imaging. 2017 Apr;44(4):689-694. doi: 10.1007/s00259-016-3563-6. Epub 2016 Nov 8. PubMed PMID: 27822700; PubMed Central PMCID: PMC5323468.
10: Rauscher I, Maurer T, Beer AJ, Graner FP, Haller B, Weirich G, Doherty A, Gschwend JE, Schwaiger M, Eiber M. Value of 68Ga-PSMA HBED-CC PET for the Assessment of Lymph Node Metastases in Prostate Cancer Patients with Biochemical Recurrence: Comparison with Histopathology After Salvage Lymphadenectomy. J Nucl Med. 2016 Nov;57(11):1713-1719. Epub 2016 Jun 3. PubMed PMID: 27261524.
11: Verburg FA, Behrendt FF, Mottaghy FM, Pfister D, Steib F, Knuechel R. Strong [(68)Ga]PSMA-HBED-CC accumulation in non-cancerous prostate tissue surrounding a PSMA-negative prostate carcinoma recurrence. Nuklearmedizin. 2016 Sep 26;55(5):N44-5. PubMed PMID: 27668299.
12: Kanthan GL, Izard MA, Emmett L, Hsiao E, Schembri GP. Schwannoma Showing Avid Uptake on 68Ga-PSMA-HBED-CC PET/CT. Clin Nucl Med. 2016 Sep;41(9):703-4. doi: 10.1097/RLU.0000000000001281. PubMed PMID: 27405039.
13: Noto B, Vrachimis A, Schäfers M, Stegger L, Rahbar K. Subacute Stroke Mimicking Cerebral Metastasis in 68Ga-PSMA-HBED-CC PET/CT. Clin Nucl Med. 2016 Oct;41(10):e449-51. doi: 10.1097/RLU.0000000000001291. PubMed PMID: 27355852.
14: Pfob CH, Ziegler S, Graner FP, Köhner M, Schachoff S, Blechert B, Wester HJ, Scheidhauer K, Schwaiger M, Maurer T, Eiber M. Biodistribution and radiation dosimetry of (68)Ga-PSMA HBED CC-a PSMA specific probe for PET imaging of prostate cancer. Eur J Nucl Med Mol Imaging. 2016 Oct;43(11):1962-70. doi: 10.1007/s00259-016-3424-3. Epub 2016 May 20. PubMed PMID: 27207281.
15: Amor-Coarasa A, Schoendorf M, Meckel M, Vallabhajosula S, Babich JW. Comprehensive Quality Control of the ITG 68Ge/68Ga Generator and Synthesis of 68Ga-DOTATOC and 68Ga-PSMA-HBED-CC for Clinical Imaging. J Nucl Med. 2016 Sep;57(9):1402-5. doi: 10.2967/jnumed.115.171249. Epub 2016 Apr 21. PubMed PMID: 27103024.
16: Prasad V, Steffen IG, Diederichs G, Makowski MR, Wust P, Brenner W. Biodistribution of [(68)Ga]PSMA-HBED-CC in Patients with Prostate Cancer: Characterization of Uptake in Normal Organs and Tumour Lesions. Mol Imaging Biol. 2016 Jun;18(3):428-36. doi: 10.1007/s11307-016-0945-x. PubMed PMID: 27038316.
17: Pfister D, Porres D, Heidenreich A, Heidegger I, Knuechel R, Steib F, Behrendt FF, Verburg FA. Detection of recurrent prostate cancer lesions before salvage lymphadenectomy is more accurate with (68)Ga-PSMA-HBED-CC than with (18)F-Fluoroethylcholine PET/CT. Eur J Nucl Med Mol Imaging. 2016 Jul;43(8):1410-7. doi: 10.1007/s00259-016-3366-9. Epub 2016 Mar 19. PubMed PMID: 26993315.
18: Kanthan GL, Coyle L, Kneebone A, Schembri GP, Hsiao E. Follicular Lymphoma Showing Avid Uptake on 68Ga PSMA-HBED-CC PET/CT. Clin Nucl Med. 2016 Jun;41(6):500-1. doi: 10.1097/RLU.0000000000001169. PubMed PMID: 26914565.
19: Kanthan GL, Hsiao E, Kneebone A, Eade T, Schembri GP. Desmoid Tumor Showing Intense Uptake on 68Ga PSMA-HBED-CC PET/CT. Clin Nucl Med. 2016 Jun;41(6):508-9. doi: 10.1097/RLU.0000000000001192. PubMed PMID: 26909712.
20: Eiber M, Weirich G, Holzapfel K, Souvatzoglou M, Haller B, Rauscher I, Beer AJ, Wester HJ, Gschwend J, Schwaiger M, Maurer T. Simultaneous (68)Ga-PSMA HBED-CC PET/MRI Improves the Localization of Primary Prostate Cancer. Eur Urol. 2016 Nov;70(5):829-836. doi: 10.1016/j.eururo.2015.12.053. Epub 2016 Jan 18. PubMed PMID: 26795686.
//////////Gallium 68 PSMA-11, FDA 2020, 2020 APPROVALS, RADIO ACTIVE
Borofalan (10B)

.png)
Borofalan (10B), ボロファラン (10B), 硼[10B]法仑
APPROVED JAPAN, 2020/3/25, Steboronine
Antineoplastic, Diagnostic aid, Radioactive agent
(2S)-2-amino-3-(4-(10B)dihydroxy(10B)phenyl)propanoic acid
| Formula | C9H12BNO4 |
|---|---|
| CAS | 80994-59-8 |
| Mol weight | 209.0069 |
- 4-(Borono-10B)-L-phenylalanine
- (10B)-4-Borono-L-phenylalanine
- Borofalan (10b)
- L-(p-[10B]Boronophenyl)alanine
- L-4-[10B]Boronophenylalanine
- p-[10B]Borono-L-phenylalanine
- L-Phenylalanine, 4-borono-10B-
Marketed Head and neck cancer - Originator Stella Pharma
- Developer Osaka University; Stella Pharma; Sumitomo Heavy Industries
- Class Antineoplastics; Borates; Propionic acids; Radiopharmaceuticals
- Mechanism of Action Ionising radiation emitters
- Phase IIGlioma
- Phase I Haemangiosarcoma; Malignant melanoma
- 20 May 2020 Launched for Head and neck cancer (Inoperable/Unresectable, Late-stage disease, Locally recurrent) in Japan (IV)
- 25 Mar 2020 Registered for Head and neck cancer (Inoperable/Unresectable, Late-stage disease, Locally recurrent) in Japan (IV) – First global approval
- 03 Mar 2020 Chemical structure information added
- Melting Point (Experimental)Value: 285-298 °C (decomp) Hattori, Yoshihide; Tetrahedron Letters 2008, VOL49(33), PG 4977-4980
- SP ROT -5.2 ° Conc: 0.50 g/100mL; Solv: hydrochloric acid 589.3 nm; Temp: 25 °C, Hattori, Yoshihide; Tetrahedron Letters 2008, VOL49(33), PG 4977-4980
Borofalan (10B)
4-[(10B)Borono]-L-phenylalanine
C9H1210BNO4 : 208.21
[80994-59-8]
With the development of atomic science, radiation therapy such as cobalt hexahydrate, linear accelerator, and electron beam has become one of the main methods of cancer treatment. However, traditional photon or electron therapy is limited by the physical conditions of the radiation itself. While killing the tumor cells, it also causes damage to a large number of normal tissues on the beam path. In addition, due to the sensitivity of tumor cells to radiation, traditional radiation therapy For the more radiation-resistant malignant tumors (such as: glioblastoma multiforme, melanoma), the treatment effect is often poor.
In order to reduce the radiation damage of normal tissues around the tumor, the concept of target treatment in chemotherapy has been applied to radiation therapy; and for tumor cells with high radiation resistance, it is currently actively developing with high relative biological effects (relative Biological effectiveness, RBE) radiation sources, such as proton therapy, heavy particle therapy, neutron capture therapy. Among them, neutron capture therapy combines the above two concepts, such as boron neutron capture therapy, by the specific agglomeration of boron-containing drugs in tumor cells, combined with precise neutron beam regulation, providing better radiation than traditional radiation. Cancer treatment options.
Boron Neutron Capture Therapy (BNCT) is a high-capture cross-section of thermal neutrons using boron-containing ( 10 B) drugs, with 10 B(n,α) 7 Li neutron capture and nuclear splitting reactions. Two heavy charged particles of 4 He and 7 Li are produced. The average energy of the two charged particles is about 2.33 MeV, which has high linear energy transfer (LET) and short range characteristics. The linear energy transfer and range of α particles are 150 keV/μm and 8 μm, respectively, while the 7 Li heavy particles are For 175 keV/μm, 5 μm, the total range of the two particles is equivalent to a cell size, so the radiation damage caused to the organism can be limited to the cell level, when the boron-containing drug is selectively aggregated in the tumor cells, with appropriate The sub-radiation source can achieve the purpose of locally killing tumor cells without causing too much damage to normal tissues.
Since the effectiveness of boron neutron capture therapy depends on the concentration of boron-containing drugs in the tumor cell position and the number of thermal neutrons, it is also called binary cancer therapy; thus, in addition to the development of neutron sources, The development of boron-containing drugs plays an important role in the study of boron neutron capture therapy.
4-( 10 B)dihydroxyboryl-L-phenylalanine (4-( 10 B)borono-L-phenylalanine, L- 10 BPA) is currently known to be able to utilize boron neutron capture therapy (boron neutron capture therapy) , BNCT) An important boron-containing drug for the treatment of cancer.
Therefore, various synthetic methods of L-BPA have been developed. As shown in the following formula (A), the prior art L-BPA synthesis method includes two methods of forming a bond (a) and a bond (b):

Among them, the method for synthesizing L-BPA by forming the bond (a) is to try to introduce a substituent containing a dihydroxylboryl group or a borono group into the skeleton of the phenylalanine, thereby the pair of the amide substituent. The position forms a carbon-boron bond to produce L-BPA.
J. Org. Chem. 1998, 63, 8019 discloses a method for the cross-coupling reaction of (S)-4-iodophenylalanine with a diboron compound by palladium-catalyzed amine end treatment. Amine-protected (S)-4-iodophenylalanine (eg (S)-N-tert-butoxycarbonyl-4-iodophenylalanine ((S)-N-Boc-4-) Iodophenylalanine)) is prepared by cross-coupling with a diboron compound such as bis(pinacolato diboron) to give (S)-N-tert-butoxycarbonyl-4-pentanoylboryl phenylalanine The amine-terminated (S)-4-boranyl ester phenylalanine of the acid ((S)-N-Boc-4-pinacolatoborono phenylalanine); afterwards, the protecting group on the amine end and the boronic end are removed. The above substituents complete the preparation of L-BPA.
However, since the selected 10 B-doped divaleryl diboron is not a commercially available compound, this method requires additional pretreatment of the preparation of the borating agent, resulting in a high process complexity and a long time consuming process. It is impossible to prepare a high yield of L-BPA. In addition, the carboxylic acid group of the protected (S)-4-iodophenylalanine at the amine end needs to be protected by a substituent to form a benzyl ester group to increase the process yield to 88%; however, The preparation of L-BPA in this manner also requires an additional step of deprotecting the carboxylic acid group, which in turn increases the process complexity of L-BPA.
Accordingly, the method provided in this document not only involves pre-treatment of the preparation of the borating agent, but also requires a large amount of process time and synthesis steps to complete the steps of protecting and deprotecting the carboxylic acid group, and is not advantageous as an industry. The main method of synthesizing L-BPA.
On the other hand, a method for synthesizing L-BPA by forming a bond (b) is a coupling reaction of an amino acid with a boron-containing benzyl fragment or a boron-containing benzaldehyde fragment. To synthesize L-BPA. Biosci. Biotech. Biochem. 1996, 60, 683 discloses an enantioselective synthesis of L-BPA which gives the hands of a cyclic ethers of boronic acid and L-proline The chiral derivatives from L-valine are subjected to a coupling reaction to produce L-BPA. However, this method requires the formation of a cyclic ether compound of boric acid from 4-boronobenzylbromide, followed by a coupling reaction with a chiral derivative of L-proline, and in the latter stage. The amino acid undergoes an undesired racemization in the synthesis step, so that the method requires an enzymatic resolution step to reduce the yield to obtain L-BPA having a certain optical purity.
Accordingly, the method provided in the literature still includes the steps of pretreatment of the preparation of the borating agent and post-treatment of the enzymatic resolution, so that the process involved in the method is complicated and takes a long time, and cannot be obtained. High yield of L-BPA.
In addition, L- 10 BPA (4-( 10 B)borono-L-phenylalanine, 4-( 10 B)dihydroxyboryl-L-phenylalanine) containing 10 boron is currently known to accumulate in tumor cells. The key factor is to use the thermal neutron beam to irradiate the boron element accumulated in the tumor cells to kill the tumor cells by capturing the high-energy particles generated by the reaction, thereby achieving the purpose of treating cancer. Therefore, 10 boron can promote the treatment of L- 10 BPA by boron neutron capture treatment.
However, the boron element present in nature contains about 19.9% of 10 boron and about 80.1% of 11 boron. Therefore, many researchers are still actively developing methods that can be applied to the synthesis of L-BPA, especially for the synthesis of 10- boron-rich L-BPA.
J.Org.Chem.1998,63,8019 additionally provides a method of synthesizing 10 boronated agents, since the method involves multiple steps, it is easy to greatly reduce the boron content of 10 10 boron enriched material in the manufacturing process. Therefore, the method provided in this document is not suitable for the synthesis of 10- boron-rich L-BPA.
Another example is the Biosci.Biotech.Biochem.1996,60,683, before the enzymatic resolution step is not performed, the method provided by the articles could not be obtained with a certain L-BPA optical purity; 10 and the method for preparing boronated agents when also relates to multi-step, resulting in conversion of boron-rich material 10 occurs during the manufacturing process. Therefore, the method provided in this document is also not suitable for the synthesis of 10- boron-rich L-BPA.
Furthermore, Bull. Chem. Soc. Jpn. 2000, 73, 231 discloses the use of palladium to catalyze 4-iodo-L-phenylalanine with 4,4,5,5-tetramethyl-1,3,2 A method in which a dioxonium pentoxide (common name: pinacolborane) is subjected to a coupling reaction. However, this document does not mention how to prepare articles 10 boron enriched L-BPA using this method, and 4,4,5,5-tetramethyl-1,3,2-dioxaborolane not a commercial 10 The compounds available in the literature are not suitable for the synthesis of 10- boron-rich L-BPA.
In addition, Synlett. 1996, 167 discloses a method for coupling a iodophenylborate with a zinc derivative of L-serine zinc derivatives, which involves first preparing phenyl iodoborate. The ester and the preparation of a zinc derivative of L-type serine acid, etc., result in a lower yield of the produced L-BPA. In addition, since the 10- boron-rich triiodide 10 boron and 1,3-diphenylpropane-1,3-diol selected for this method are not commercially available compounds, the methods provided in this document are also provided. Still not suitable for the synthesis of 10- boron-rich L-BPA.
SYN

Repub. Korean Kongkae Taeho Kongbo, 2018060319,
PAPER
Research and Development in Neutron Capture Therapy, Proceedings of the International Congress on Neutron Capture Therapy, 10th, Essen, Germany, Sept. 8-13, 2002 (2002), 1-8.
PAPER
European Journal of Pharmaceutical Sciences (2003), 18(2), 155-163
https://www.sciencedirect.com/science/article/abs/pii/S0928098702002567


PAPER
Tetrahedron Letters (2008), 49(33), 4977-4980
PATENT
WO 2004009135
PATENT
US 20130331599
PATENT
WO 2017028751
https://patents.google.com/patent/WO2017028751A1/en
Example 1
Before preparing (S)-N-tert-butoxycarbonyl-4-dihydroxyborylphenylalanine from (S)-N-tert-butoxycarbonyl-4-iodophenylalanine, it is necessary to reveal Process for preparing (S)-N-tert-butoxycarbonyl-4-iodophenylalanine by using (S)-4-iodophenylalanine as a starting material and a process for preparing 10 tributyl borate with 10 boric acid.
1. Preparation of (S)-N-tert-butoxycarbonyl-4-iodophenylalanine from (S)-4-iodophenylalanine
Please refer to the following reaction formula I, which is (S)-4-iodophenylalanine in a solvent of 1,4-dioxane (1,4-dioxane) and water (H 2 O) with hydrogen peroxide. Sodium (NaOH) and di-tert-butyl dicarbonate (Boc 2 O) are reacted to obtain a chemical reaction formula of (S)-N-tert-butoxycarbonyl-4-iodophenylalanine.

In the preparation process, two reaction vessels were selected for the reaction.
The specific operation process is as follows:
1. Set up a reaction using a 3L three-neck bottle.
2. (S)-4-iodo-L-phenylalanine (200.00 g, 687.10 mmol, 1.00 eq) was added to the reaction system.
3. Add 1,4-dioxane (1.00 L) and water (1.00 L) to the reaction system, respectively.
4. Sodium hydroxide (68.71 g, 1.72 mol, 2.50 eq) was added to the reaction system, the solution gradually became clear, and the temperature rose slightly to 19 °C.
5. When the system is cooled to 0-10 ° C, di-tert-butyl dicarbonate (254.93 g, 1.17 mol, 268.35 mL, 1.70 eq) is added to the reaction system, and the temperature of the reaction system is naturally raised to 10 to 30 ° C and Stir at room temperature (about 30 ° C) for 8 hours.
6. The reaction was detected using high performance liquid chromatography (HPLC) until the starting of the reaction.
7. The temperature of the control system is less than 40 ° C, and the 1,4-dioxane in the reaction solution is concentrated.
8. The reaction system was lowered to room temperature (about 25 ° C), 100 mL of water was added, and the pH was adjusted to 1.8-2 with hydrochloric acid (2M (ie, molarity, M)).
9. Extract three times with ethyl acetate (2 L).
10. Combine the organic phases and wash twice with saturated brine (1 L).
11. The organic phase was dried over sodium sulfate (200 g).
12. Continue drying in an oven (40-45 ° C) to give (S)-N-tert-butoxycarbonyl-4-iodo-L-phenylalanine (250.00 g, 626.28 mmol, HPLC analysis, yield 93.00 %, purity 98%).
The prepared (S) -N- tert-butoxycarbonyl-4-iodo-phenylalanine was -L- Hydrogen 1 nuclear magnetic resonance spectrum analysis (1 HNMR) as follows:
1 H NMR: (400 MHz DMSO-d 6 )
δ 7.49 (d, J = 7.8 Hz, 2H), 6.88 (d, J = 7.8 Hz, 2H), 5.80 (d, J = 5.9 Hz, 1H), 3.68 (d, J = 5.5 Hz, 1H), 3.00-2.90 (m, 1H), 2.87-2.75 (m, 1H), 1.35-1.15 (m, 9H).
Second, tributyl borate 10 was prepared from boronic acid 10
See the following reaction formulas II, 10 as boric acid (H 2 SO 4) is reacted with sulfuric acid in a solvent (butan-1-ol), and toluene (Toluene) in n-butanol, to obtain 10 tributyl borate (10 The chemical reaction formula of B(OBu) 3 ).

The specific operation process is as follows:
1. Set up a reaction device R1 using a 3L three-necked bottle, and configure a water separator on the device.
2. 10 boric acid (150.00 g, 2.46 mol, 1.00 eq) was added to the reaction R1 at room temperature (about 25 ° C).
3. Add n-butanol (1.00 L) to the reaction R1 at room temperature (about 25 ° C) and stir, and most of the boric acid cannot be dissolved.
4. Toluene (1.00 L) was added to the reaction R1 at room temperature (about 25 ° C) and stirred.
5. Concentrated sulfuric acid (4.82 g, 49.16 mmol, 2.62 mL, 0.02 eq) was added dropwise to the reaction at room temperature (about 25 ° C), at which time a large amount of solid remained undissolved.
6. The reaction system was heated to 130 ° C, and the water was continuously removed, stirred for 3.5 hours, and water (about 140 g) was formed in the water separator. The solids were all dissolved, and the solution changed from colorless to brown. .
7. TLC (DCM: MeOH = 5:1, Rf = 0.43, bromocresol green).
8. Distill off most of the toluene at atmospheric pressure.
9. After most of the toluene is distilled off, the temperature of the system is lowered to 20 to 30 ° C, and the reaction liquids of the two reactions are combined, and the apparatus is changed for distillation.
10. Oil bath external temperature 108-110 ° C pump distillation under reduced pressure, Kelvin thermometer 45 ° C, distilled n-butanol.
11. Oil bath external temperature 108-110 ° C oil pump distillation under reduced pressure, the residual butanol was distilled off.
12. Oil bath external temperature 118-120 ° C oil pump vacuum distillation, Kelvin thermometer 55 ° C, began to produce products.
13. The temperature is raised to 135-140 ° C oil pump vacuum distillation, the product is completely distilled.
14. The product is obtained as a colorless liquid 10 tributyl borate (830.00g, 3.62mol, yield 73.58%).
The results of the 1 H NMR analysis of the obtained tributyl 10 borate were as follows:
1 H NMR: (400 MHz CDCl 3 )
δ 3.82-3.68 (m, 6H), 1.57-1.42 (m, 6H), 1.34 (qd, J = 7.4, 14.9 Hz, 6H), 0.95-0.80 (m, 9H).
Three, -N- tert-butoxycarbonyl-4-iodo-phenylalanine was prepared (S) of (S) -N- tert-butoxycarbonyl-4-hydroxy-10-yl -L- phenylalanine boron
Please refer to the following reaction formula III, which is (S)-N-tert-butoxycarbonyl-4-iodophenylalanine with tributyl 10 borate, t-butyl magnesium chloride (t-BuMgCl) and bis (2-A) yl aminoethyl) ether (BDMAEE) reaction, to produce (S) -N- tert-butoxycarbonyl group -4- (10 B) dihydroxyboryl -L- phenylalanine chemical reaction.

In the preparation process, two reaction vessels were selected for the reaction.
The specific operation process is as follows:
1. Set up a reaction using a 3L three-neck bottle.
2. Tributyl 10 borate (187.60 g, 87.98 mmol, 3.20 eq) was placed in the reaction system at room temperature (about 22 ° C).
3. Sodium hydride (20.45 g, 511.24 mmol, purity 60%, 2.00 eq) was added to the reaction system at room temperature (about 22 ° C). The reaction solution was a suspension and stirred at room temperature (about 22 ° C). 5 minutes.
4. Bis(2-methylaminoethyl)ether (327.73 g, 2.04 mol, 8.00 eq) was added to the reaction at room temperature (about 22 ° C).
5. N-tert-Butoxycarbonyl-4-iodo-L-phenylalanine (100.00 g, 255.62 mmol, 1.00 eq) was added to the reaction system at room temperature (about 22 ° C), and a large amount of solid was not dissolved.
6. Lower the temperature of the reaction system to 0-5 ° C, add t-butyl magnesium chloride (1.7 M, 1.20 L, 2.04 mol, 8.00 eq) to the reaction, control the temperature between 0-10 ° C, the dropping time is about It is 1.5 hours.
7. After the completion of the charging, the temperature of the reaction system was naturally raised to room temperature (20 to 30 ° C) and stirred at this temperature for 12 hours.
8. Using high performance liquid chromatography (HPLC) to detect about 9.00% of the remaining material.
9. When the temperature of the reaction system was lowered to -5 to 0 ° C, it was quenched by dropwise addition of 500 mL of water.
10. Lower the temperature of the system to 0-5 ° C, add methyl tert-butyl ether (500 mL) to the reaction system and adjust the pH to 2.9-3.1 (using a pH meter) with 37% HCl (about 500 mL). Exothermic, the temperature of the control system is between 0-15 °C.
11. The aqueous phase obtained by liquid separation was extracted once with methyl tert-butyl ether (500 mL), and the obtained organic phases were combined to give an organic phase of about 1.1 L.
12. Slowly add a sodium hydroxide aqueous solution (1 M, 400 mL) to the obtained organic phase, exotherm during the dropwise addition, and control the system temperature between 0-15 °C.
13. After the completion of the dropwise addition, the pH of the system was about 10, and the pH was adjusted to between 12.10 and 12.6 with an aqueous sodium hydroxide solution (4M). (measured with a pH meter)
14. Dispensing.
15. The aqueous phase 1 obtained after liquid separation was extracted once with n-butanol (500 ml) to obtain aqueous phase 2.
16. Combine the aqueous phase 2 of the two reaction vessels.
17. Adjust the pH of the aqueous phase to 2.9-3.1 with 37% HCl, stir for about 40 minutes, and precipitate a large amount of solid.
18. Filtration gave a white solid which was washed once with dichloromethane (50 mL).
19. At 25 ° C, the precipitated solid was slurried with dichloromethane (150 mL) and stirred for 10 min.
20. A white solid was filtered to give (S) -N- tert-butoxycarbonyl group -4- (10 B) dihydroxyboryl -L- phenylalanine (75.00g, 240.82mmol, by HPLC analysis, a yield of 47.11% , purity 99%).
The prepared (S) -N- tert-butoxycarbonyl group -4- (10 B) results dihydroxyboryl -L- phenylalanine 1 HNMR was as follows:
1 H NMR: (400 MHz DMSO-d 6 )
Δ12.55 (br.s., 1H), 7.91 (s, 2H), 7.66 (d, J = 7.5 Hz, 2H), 7.17 (d, J = 7.5 Hz, 2H), 4.08-4.01 (m, 1H) ), 3.61-3.53 (m, 1H), 2.98 (dd, J = 4.2, 13.9 Hz, 1H), 2.79 (dd, J = 10.4, 13.5 Hz, 1H), 1.79-1.67 (m, 1H), 1.35- 1.17 (m, 9H).
Preparation of L- 10 BPA from (S)-N-tert-Butoxycarbonyl-4-dihydroxyboryl-L-phenylalanine
See the following reaction scheme IV, which is (S) -N- tert-butoxycarbonyl group -4- (10 B) of amine end dihydroxyboryl -L- phenylalanine deprotection of the chemical reaction, to obtain L- 10 BPA.

The specific operation process is as follows:
1. Set up a reaction using a 1L three-neck bottle.
2. room temperature (20-30 deg.] C) to (S) -N- tert-butoxycarbonyl group -4- (10 B) dihydroxyboryl -L- phenylalanine (67.00g, 217.31mmol, 1.00eq) was added the reaction In the system.
3. room temperature (20-30 deg.] C) water (23.75mL) and acetone (Acetone, 420.00mL) were added dropwise to the reaction flask, stirred (S) -N- tert-butoxycarbonyl group -4- (10 B) dihydroxy Boronyl-L-phenylalanine.
4. Concentrated hydrochloric acid (23.93 g, 656.28 mmol, 23.46 mL, 3.02 eq) was added dropwise to the reaction system at room temperature (20-30 ° C). After the addition was completed, the reaction system was heated to 55-60 ° C and stirred for 4.5 hours.
5. HPLC detection until the reaction of the starting material is completed.
6. The temperature is controlled below 40 ° C, and the acetone in the reaction system is concentrated.
7. Lower the concentrated system to below 15 °C, adjust the pH of the system to about 1.5 with sodium hydroxide solution (4M) (pH meter detection), stir for 40 minutes and continue to adjust the pH of the system to 6.15 using sodium hydroxide solution (4M). ~6.25, a large amount of white solid precipitated, which was filtered to give a white solid, and rinsed with acetone (200mL).
8. Obtained as a white solid L- 10 BPA (36.00 g, 171.17 mmol, HPLC, yield 78.77%, purity 99%).
The analytical results obtained by the L- 10 BPA 1 HNMR are as follows:
1 H NMR: (400 MHz D 2 O, CF 3 COOH)
δ 7.44 (d, J = 7.9 Hz, 1H), 7.03 (d, J = 7.9 Hz, 1H), 4.06 (dd, J = 5.7, 7.5 Hz, 1H), 3.11-3.01 (m, 1H), 2.98 -2.87 (m, 1H).
xample 6
Preparation of (S)-N-tert-butoxycarbonyl-4-dihydroxyboryl-L-phenylalanine from (S)-N-tert-butoxycarbonyl-4-iodophenylalanine
Please refer to the following reaction formula VII, which is a reaction of (S)-N-tert-butoxycarbonyl-4-iodophenylalanine with tributyl borate and t-butylmagnesium chloride (t-BuMgCl) to obtain (S The chemical reaction formula of -N-tert-butoxycarbonyl-4-dihydroxyboryl-L-phenylalanine.

The specific operation process is as follows:
1. Construct a reaction unit with a 250 mL three-neck bottle.
2. Tributyl borate (17.65 g, 76.68 mmol, 3.00 eq) was placed in a 250 mL reaction flask at 20-30 °C.
3. Sodium hydride (1.02 g, 25.56 mmol, 1.00 eq) was added to a 250 mL reaction vial at 20-30 °C.
4. (S)-N-tert-Butoxycarbonyl-4-iodo-L-phenylalanine (10.00 g, 25.56 mmol, 1.00 eq) was added to a 250 mL reaction vial at 20-30 °C.
5. Reduce the temperature of the reaction system to 0 ° C under nitrogen atmosphere, slowly add t-butyl magnesium chloride (1.7 M in THF, 120 mL, 8.00 eq) to the reaction, the dropping time is about 30 minutes, and the control temperature is 0. Between °C and 10 °C.
Stir at 20.20 ~ 30 ° C for 20 hours.
7. HPLC detection of the basic reaction of the raw materials, leaving only about 0.7% of the raw materials.
8. At a temperature of 0 ° C, 5 mL of water was added dropwise to the reaction to quench it. After complete quenching, stirring was continued for 10 minutes.
9. Cool down to 0 ° C, add methyl tert-butyl ether (50 mL) to the reaction and adjust the pH to 3 with 37% HCl (about 50 mL) (detected with a pH meter), adjust the pH during the process to exotherm, control the temperature at 0 Between °C and 15 °C.
12. The aqueous phase obtained by liquid separation was extracted once with methyl t-butyl ether (50 mL) and the organic phases were combined.
12. Add NaOH solution (1M, 55mL) to the obtained organic phase to adjust the pH to between 12.10-12.6. The process is exothermic and the temperature is controlled between 0 °C and 15 °C.
13. Liquid separation, the obtained aqueous phase was extracted once with n-butanol (50 mL), and most of the impurities were extracted and removed.
14. The aqueous phase obtained by liquid separation was adjusted to pH 3 with 37% HCl and stirred for about 30 minutes to precipitate a white solid.
15. Filtration gave a white solid which was washed once with dichloromethane (50 mL).
16. The precipitated solid was slurried with 25 mL of dichloromethane at 25 ° C and stirred for 10 minutes.
17. Filtration of (S)-N-tert-butoxycarbonyl-4-dihydroxyboryl-L-phenylalanine (6.8 g, HPLC, yield: 83.15%, purity 98%).
Example 7
Please continue to refer to Reaction Scheme VII. The specific operation process is as follows:
1. Construct a reaction unit with a 250 mL three-neck bottle.
2. Tributyl borate (8.82 g, 38.34 mmol, 3.00 eq) was added to a 250 mL reaction vial at 20-30 °C.
3. Sodium hydride (511.25 mg, 12.78 mmol, 1.00 eq) was added to a 250 mL reaction vial at 20-30 °C.
4. (S)-N-tert-Butoxycarbonyl-4-iodo-L-phenylalanine (5.00 g, 12.78 mmol, 1.00 eq) was added to a 250 mL reaction vial at 20-30 °C.
5. The temperature of the reaction system was lowered to 0 ° C under nitrogen atmosphere, and t-butyl magnesium chloride (1.7 M in THF, 60 mL, 8.00 eq) was added dropwise to the reaction, the dropwise addition time was about 30 minutes, and the control temperature was 0 ° C. -10 ° C between.
Stir at 6.20 ~ 30 ° C for 22 hours.
7. HPLC detection of the raw material reaction is completed.
8. At a temperature of 0 ° C, 2.5 mL of water was added dropwise to the reaction to quench it. After complete quenching, stirring was continued for 10 minutes.
9. Cool down to 0 ° C, add methyl tert-butyl ether (25 mL) to the reaction and adjust the pH to 3 with 37% HCl (about 25 mL) (detected with a pH meter), adjust the pH during the process to exotherm, control the temperature at 0 Between °C and 15 °C.
12. The aqueous phase obtained by liquid separation was extracted once with methyl t-butyl ether (25 mL) and the organic phases were combined.
12. Add NaOH solution (1M, 30mL) to the obtained organic phase to adjust the pH to between 12.10-12.6. The process is exothermic and the temperature is controlled between 0 °C and 15 °C.
13. Liquid separation, the obtained aqueous phase was extracted once with n-butanol (25 ml), and most of the impurities were extracted and removed.
14. The aqueous phase obtained by liquid separation was adjusted to pH 3 with 37% HCl and stirred for about 30 minutes to precipitate a white solid.
15. Filtration gave a white solid which was washed once with dichloromethane (25 mL).
16. The precipitated solid was slurried with 15 mL of dichloromethane at 25 ° C and stirred for 10 minutes.
17. Filtration gave (S)-N-tert-butoxycarbonyl-4-dihydroxyboryl-L-phenylalanine (3.4 g, obtained by HPLC, yield: 85.26%, purity 98%).
Bis(2-methylaminoethyl)ether is a complexing agent for Mg, which can reduce the occurrence of side reactions in the reaction. The reactions of Examples 6 and 7 were carried out without adding bis(2-methylaminoethyl)ether. The analysis showed that the iodine impurity in the reaction of Example 6 was about 17%, and the iodine impurity in the reaction of Example 7 was observed. About 28%. Therefore, it has been proved from the side that the addition of bis(2-methylaminoethyl)ether can protect the reaction from reducing iodine.
The BPA or 10 BPA obtained in the above examples were analyzed by chiral HPLC, and the ratio of the L-enantiomer to the D-enantiomer was 100:0.
The boron-containing drug L-BPA for neutron capture therapy disclosed in the present invention is not limited to the contents described in the above examples. The above-mentioned embodiments are only examples for convenience of description, and the scope of the claims should be determined by the claims.
PATENT
KR 2018060319
PATENT
WO 2019163790
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019163790
///////////Borofalan (10B), Borofalan, Steboronine, JAPAN 2020, 2020 APPROVALS, ボロファラン (10B), ボロファラン , 硼[10B]法仑 ,
B(C1=CC=C(C=C1)CC(C(=O)O)N)(O)O
Fluorodopa F 18, フルオロドパ (18F), флуородопа (18F) , فلورودوبا (18F) , 氟[18F]多巴 ,
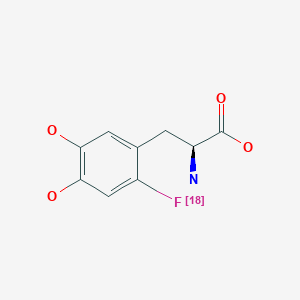
Fluorodopa F 18
2019/10/10, fda 2019,
| Formula |
C9H10FNO4
|
|---|---|
| Cas |
92812-82-3
|
| Mol weight |
215.1784
|
Diagnostic aid (brain imaging), Radioactive agent, for use in positron emission tomography (PET)
CAS 92812-82-3
フルオロドパ (18F)
Fluorodopa, also known as FDOPA, is a fluorinated form of L-DOPA primarily synthesized as its fluorine-18isotopologue for use as a radiotracer in positron emission tomography (PET).[1] Fluorodopa PET scanning is a valid method for assessing the functional state of the nigrostriatal dopaminergic pathway. It is particularly useful for studies requiring repeated measures such as examinations of the course of a disease and the effect of treatment
In October 2019, Fluorodopa was approved in the United States for the visual detection of certain nerve cells in adult patients with suspected Parkinsonian Syndromes (PS).[2][3]
The U.S. Food and Drug Administration (FDA) approved Fluorodopa F 18 based on evidence from one clinical trial of 56 patients with suspected PS.[2] The trial was conducted at one clinical site in the United States.[2]
PAPER
A one-pot two-step synthesis of 6-[18F]fluoro-L-DOPA ([18F]FDOPA) has been developed involving Cu-mediated radiofluorination of a pinacol boronate ester precursor. The method is fully automated, provides [18F]FDOPA in good activity yield (104 ± 16 mCi, 6 ± 1%), excellent radiochemical purity (>99%) and high molar activity (3799 ± 2087 Ci mmol−1), n = 3, and has been validated to produce the radiotracer for human use.
![Graphical abstract: One-pot synthesis of high molar activity 6-[18F]fluoro-l-DOPA by Cu-mediated fluorination of a BPin precursor](https://pubs.rsc.org/en/Image/Get?imageInfo.ImageType=GA&imageInfo.ImageIdentifier.ManuscriptID=C9OB01758E&imageInfo.ImageIdentifier.Year=2019 "Graphical abstract")

PATENT
KR 2019061368

The present invention relates to an L-dopa precursor compd., a method for producing the same, and a method for producing 18F-labeled L-dopa using the same. The method of prepg. 18F-labeled L-dopa I using the L-dopa precursor II [A = halogen-(un)substituted alkyl; W, X, Y = independently protecting group] can improve the labeling efficiency of 18F. After the labeling reaction, sepn. and purifn. steps of the product can be carried out continuously and it can be performed with on-column labeling (a method of labeling through the column). The final product I, 18 F-labeled L-dopa, can be obtained at a high yield relative to conventional methods. Further, it has an advantage that it is easy to apply various methods such as bead labeling.
PAPER
Science (Washington, DC, United States) (2019), 364(6446), 1170-1174.

PAPER
European Journal of Organic Chemistry (2018), 2018(48), 7058-7065.
PATENT
WO 2018115353
CN 107311877
References
- ^ Deng WP, Wong KA, Kirk KL (June 2002). “Convenient syntheses of 2-, 5- and 6-fluoro- and 2,6-difluoro-L-DOPA”. Tetrahedron: Asymmetry. 13 (11): 1135–1140. doi:10.1016/S0957-4166(02)00321-X.
- ^ Jump up to:a b c “Drug Trials Snapshots: Fluorodopa F 18”. U.S. Food and Drug Administration (FDA). 27 November 2019. Archived from the original on 27 November 2019. Retrieved 27 November 2019.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “Drug Approval Package: Fluorodopa F18”. U.S. Food and Drug Administration (FDA). 20 November 2019. Archived from the original on 27 November 2019. Retrieved 26 November 2019. This article incorporates text from this source, which is in the public domain.
 |
|
| Clinical data | |
|---|---|
| Other names | 6-fluoro-L-DOPA, FDOPA |
| License data |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| ChemSpider | |
| UNII | |
| CompTox Dashboard (EPA) | |
| Chemical and physical data | |
| Formula | C9H10FNO4 |
| Molar mass | 215.18 g/mol g·mol−1 |
| 3D model (JSmol) | |
//////////////////Fluorodopa F 18, フルオロドパ (18F), FDA 2019, флуородопа (18F) , فلورودوبا (18F) , 氟[18F]多巴 , radio labelled
N[C@@H](CC1=CC(O)=C(O)C=C1[18F])C(O)=O
