
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
RISPERIDONE

Risperidone
EU APPROVED 2022/2/14, Okedi
- R-64,766
- R-64766
- RCN-3028
- RCN3028
Risperidone, R-64766, Risperdal M-Tab, Risperdal Consta, Rispolept, Belivon, Risperdal
| Formula | C23H27FN4O2 |
|---|---|
| CAS | 106266-06-2 |
| Mol weight | 410.4845 |
3-{2-[4-(6-fluoro-1,2-benzoxazol-3-yl)piperidin-1-yl]ethyl}-2-methyl-4H,6H,7H,8H,9H-pyrido[1,2-a]pyrimidin-4-one
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Risperidone tartrate | 0S6B72E3LK | 666179-92-6 | KSWIOGDSXUFKOC-LREBCSMRSA-N |
Risperidone
CAS Registry Number: 106266-06-2
CAS Name: 3-[2-[4-(6-Fluoro-1,2-benzisoxazol-3-yl)-1-piperidinyl]ethyl]-6,7,8,9-tetrahydro-2-methyl-4H-pyrido[1,2-a]pyrimidin-4-one
Manufacturers’ Codes: R-64766
Trademarks: Belivon (Organon); Risperdal (J & J)
Molecular Formula: C23H27FN4O2, Molecular Weight: 410.48
Percent Composition: C 67.30%, H 6.63%, F 4.63%, N 13.65%, O 7.80%
Literature References: Combined serotonin (5-HT2) and dopamine (D2) receptor antagonist. Prepn: L. E. J. Kennis, J. Vandenberk, EP196132; eidem,US4804663 (1986, 1989 both to Janssen). Pharmacology: P. A. J. Janssen et al.,J. Pharmacol. Exp. Ther.244, 685 (1988). Receptor binding studies: J. E. Leysen et al.,ibid.247, 661 (1988). HPLC determn in plasma: A. Avenoso et al.,J. Chromatogr. B746, 173 (2000). Clinical study in psychoses: Y. G. Gelders et al.,Pharmacopsychiatry23, 206 (1990); in autism: L. Scahill et al., N. Engl. J. Med.347, 314 (2002). Brief review: M. G. Livingston, Lancet343, 457-460 (1994). Review of pharmacology and therapeutic potential: S. Grant, A. Fitton, Drugs48, 253-273 (1994); B. Green, Curr. Med. Res. Opin.16, 57-65 (2000); of clinical experience in schizophrenia: H.-J. Möller, Expert Opin. Pharmacother.6, 803-818 (2005),
Properties: Crystals from DMF + 2-propanol, mp 170.0°. LD50 in male, female mice, rats, dogs (mg/kg): 29.7, 26.9, 34.3, 35.4, 14.1, 18.3 i.v.; 82.1, 63.1, 113, 56.6, 18.3, 18.3 orally (Janssen, 1988).
Melting point: mp 170.0°
Toxicity data: LD50 in male, female mice, rats, dogs (mg/kg): 29.7, 26.9, 34.3, 35.4, 14.1, 18.3 i.v.; 82.1, 63.1, 113, 56.6, 18.3, 18.3 orally (Janssen, 1988)
Therap-Cat: Antipsychotic.
Keywords: Antipsychotic; Benzisoxazoles; Serotonin-Dopamine Antagonist.
Risperidone, sold under the brand name Risperdal among others, is an atypical antipsychotic[2] used to treat schizophrenia and bipolar disorder.[2] It is taken either by mouth or by injection (subcutaneous or intramuscular).[2] The injectable versions are long-acting and last for 2-4 weeks.[6]
Common side effects include movement problems, sleepiness, dizziness, trouble seeing, constipation, and increased weight.[2][7] Serious side effects may include the potentially permanent movement disorder tardive dyskinesia, as well as neuroleptic malignant syndrome, an increased risk of suicide, and high blood sugar levels.[2][6] In older people with psychosis as a result of dementia, it may increase the risk of death.[2] It is unknown if it is safe for use in pregnancy.[2] Its mechanism of action is not entirely clear, but is believed to be related to its action as a dopamine and serotonin antagonist.[2]
Study of risperidone began in the late 1980s and it was approved for sale in the United States in 1993.[2][8][4] It is on the World Health Organization’s List of Essential Medicines.[9] It is available as a generic medication.[6] In 2019, it was the 149th most commonly prescribed medication in the United States, with more than 4 million prescriptions.[10][11]
Synthesis ReferenceUS4804663
SYN
| EP 0196132; ES 8705881; JP 1986221186; US 4804663 |
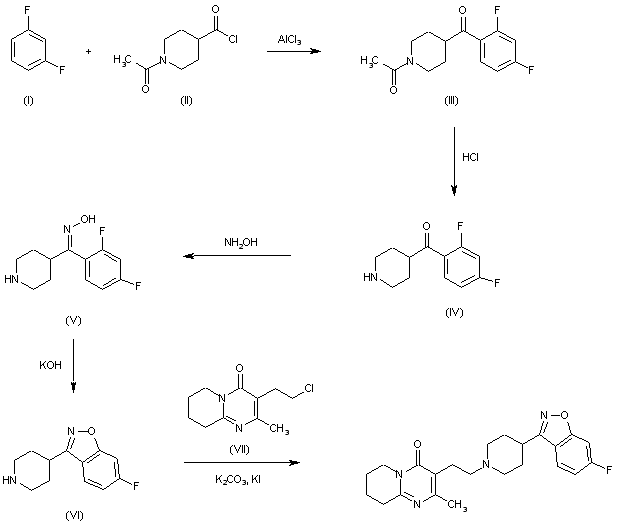
The Friedel-Crafts condensation of 1,3-difluorobenzene (I) with 1-acetylpiperidine-4-carbonyl chloride (II) by means of AlCl3 in dichloromethane gives 1-acetyl-4-(2,4-difluorobenzoyl)piperidine (III), which is hydrotyzed with refluxing 6N HCl to yield 4-(2,4-difluorobenzoyl)piperidine (IV). The reaction of (IV) with hydroxylamine in refluxing ethanol affords the corresponding oxime (V), which is cyclized by means of KOH in boiling water giving 6-fluoro-3-(4-piperidinyl)-1,2-benzisoxazole (VI). Finally, this compound is condensed with 3-(2-chloroethyl)-2-methyl-6,7,8,9-tetrahydro-4H-pyrido[1,2-a]pyrimidin-4-one (VII) by means of K2CO3 and Kl in a variety of solvents.
SYN
ES 2050069
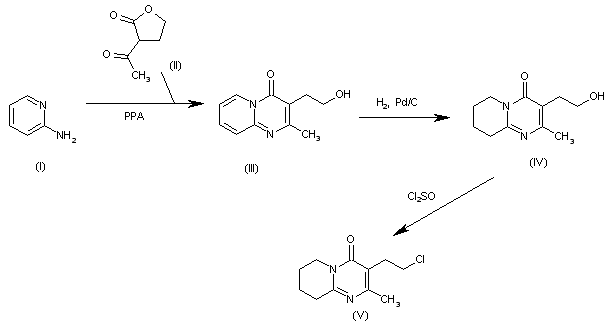
The intermediate 3-(2-chloroethyl)-2-methyl-6, 7,8,9-tetrahydro-4H-pyrido[1,2-a]pyrimidin-4-one (V) has been obtained as follows: The cyclization of 2-aminopyridine (I) with 3-acetyltetrahydrofuran-2-one (II) by means of polyphosphoric acid (PPA) at 160 C gives 3-(2-hydroxyethyl)-2-methyl-4H-pyrido[1,2-a]pyrimidin-4-one (III), which is hydrogenated with H2 over Pd/C in ethanol/water to yield the tetrahydro derivative (IV). Finally, the OH group of (IV) is treated with SOCl2 in dichloromethane to afford the target 2-chloroethyl intermediate (V).
SYN
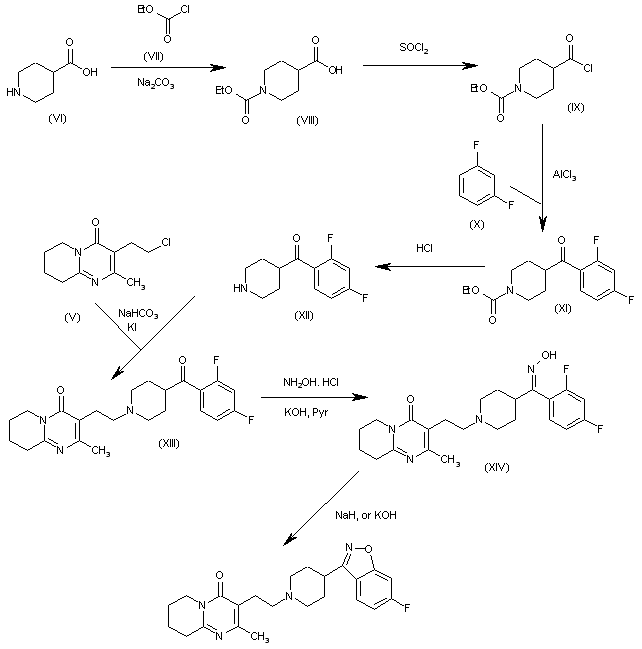
The condensation of piperidine-4-carboxylic acid (VI) with ethyl chloroformate (VII) by means of Na2CO3 in toluene/water gives 1-(ethoxycarbonyl)piperidine-4-carboxylic acid (VIII), which is treated with SOCl2 to yield the corresponding acyl chloride (IX). The Friedel-Crafts condensation of (IX) with refluxing 1,3-difluorobenzene (X) by means of AlCl3 gives 4-(2,4-difluorobenzoyl)piperidine-1-carboxylic acid ethyl ester (XI), which is treated with concentrated HCl at 100 C to yield 4-(2,4-difluorobenzoyl)piperidine (XII). The condensation of piperidine (XII) with the 2-chloroethyl intermediate (V) by means of KI and NaHCO3 in refluxing acetonitrile affords the adduct (XIII), which is treated with hydroxylamine hydrochloride and KOH in refluxing pyridine/ethanol to provide the corresponding oxime (XIV). Finally, this compound is cyclized by means of KOH in refluxing water or with NaH in refluxing THF to afford in both cases the target 1,2-benzisoxazole.
SYN
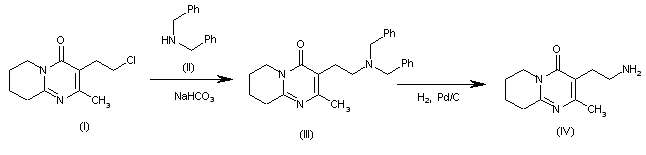
The intermediate 3-(2-aminoethyl)-2-methyl-6,7,8,9-tetrahydro-4H-pyrido[1,2-a]pyrimidin-4-one (IV) has been obtained as follows: The condensation of 3-(2-chloroethyl)-2-methyl-6,7,8,9-tetrahydro-4H-pyrido[1,2-a]pyrimidin-4-one (I) with dibenzylamine (II) by means of NaHCO3 in refluxing acetonitrile gives the tertiary amine (III), which is debenzylated by hydrogenation with H2 over Pd/C in warm ethanol to afford the target intermediate (IV).
SYN
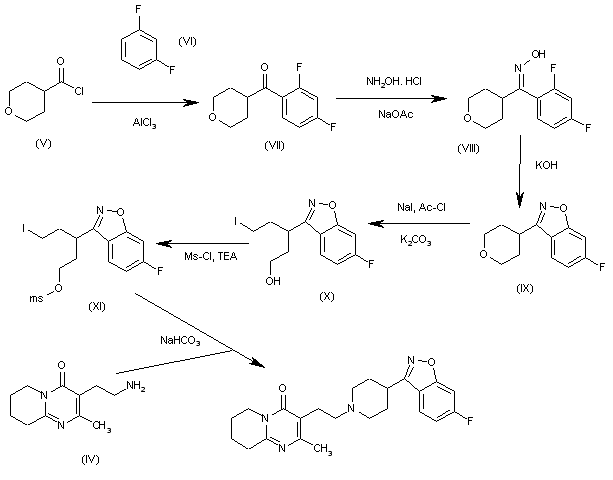
The condensation of tetrahydropyran-4-carbonyl chloride (V) with refluxing 1,3-difluorobenzene (VI) by means of AlCl3 gives 1-(2,4-difluorophenyl)-1-(tetrahydropyran-4-yl)methanone (VII), which is treated with hydroxylamine hydrochloride and sodium acetate in refluxing ethanol/water to yield the corresponding oxime (VIII). The cyclization of (VIII) by means of KOH in refluxing methanol affords 6-fluoro-3-(tetrahydropyran-4-yl)-1,2-benzisoxazole (IX), which is treated with NaI and Ac-Cl and then with K2CO3 in refluxing acetonitrile to provide the 5-iodopentanol derivative (X). The reaction of the OH group of (X) with Ms-Cl and TEA in dichloromethane gives the corresponding mesylate (XI), which is finally cyclized with the intermediate amine (IV) by means of NaHCO3 in refluxing acetonitrile to yield the target piperidine.
SYN

SYN
Eur. Pat. Appl. 196132

SYN
- Production Route of Risperidone
- (CAS NO.: ), with other name of 4H-Pyrido(1,2-a)pyrimidin-4-one, 6,7,8,9-tetrahydro-3-(2-(4-(6-fluoro-1,2-benzisoxazol-3-yl)-1-piperidinyl)ethyl)-2-methyl-, could be produced through many synthetic methods.Following is one of the synthesis routes:
The Friedel-Crafts condensation of 1,3-di (I) with 1-acetylpiperidine-4-carbonyl chloride (II) by means of AlCl3 in dichloromethane gives 1-acetyl-4-(2,4-difluorobenzoyl)piperidine (III), which is hydrotyzed with refluxing 6N HCl to yield 4-(2,4-difluorobenzoyl)piperidine (IV). The reaction of (IV) with hydroxylamine in refluxing ethanol affords the corresponding oxime (V), which is cyclized by means of KOH in boiling water giving 6-fluoro-3-(4-piperidinyl)-1,2-benzisoxazole (VI). Finally, this compound is condensed with 3-(2-chloroethyl)-2-methyl-6,7,8,9-tetrahydro-4H-pyrido[1,2-a]pyrimidin-4-one (VII) by means of K2CO3 and Kl in a variety of solvents.
- SYN
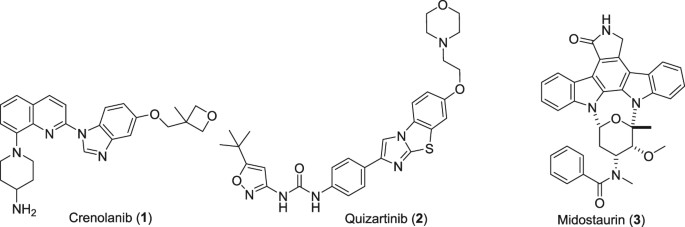
Piperidine-Based Nonfused Biheterocycles With C–N and C–C Coupling
Ruben Vardanyan, in Piperidine-Based Drug Discovery, 2017
Risperidone (15970)
Risperidone (7.2.1) (Risperdal) is the first second-generation antipsychotic that was specifically designed as a combined D2 and serotonin 5-HT(2A) receptor antagonist, thus following the pharmacological mechanism thought to be responsible for the antipsychotic effects. After its advent in the 1990s as the first novel second-generation antipsychotic, risperidone has achieved worldwide acceptance. It was initially approved for use in schizophrenia, mania of bipolar disorder, and irritability and aggression of autism. But it is also effectively used in other instances of psychosis, including schizoaffective disorder, depression with psychotic features, and psychosis secondary to general medical conditions. Risperidone may be effective in other conditions such as major depression, various anxiety disorders, delirium, dementia, for Alzheimer’s dementia, which occurs in 6–8% of persons older than 65 and increases to 30% among those 85 years or older, and substance abuse disorders [84–113].
Risperidone is proposed for inclusion in the WHO Model List of Essential Medications for treatment of schizophrenia, mania, and autism.
Risperidone (7.2.1) was synthesized starting from 1-acetyl-4-piperidine-carbonyl chloride (7.2.4), which was used to acylate 1,3-difluorobenzene (7.2.5) in dichloromethane using aluminum chloride as Lewis acid. The reaction gave 1-(4-(2,4-difluorobenzoyl)piperidin-1-yl)ethan-1-one (7.2.6). The protecting acetyl group of the last was removed off by hydrolysis in 6 N hydrochloric acid on reflux, which gave (2,4-difluorophenyl)(piperidin-4-yl)methanone (7.2.7). The obtained product was converted further to corresponding oxime (7.2.8) on reaction with hydroxylamine hydrochloride in ethanol in the presence of N,N-diethylenethanamine. Synthesized oxime (7.2.8) was cyclized to 6-fluoro-3-(piperidin-4-yl)benzo[d]isoxazole (7.2.9) on reflux with 50% potassium hydroxide solution in water. At the final stage the obtained product (7.2.9) was alkylated with 3-(2-chloroethyl)-2-methyl-6,7,8,9-tetrahydro-4H-pyrido[1,2-a]pyrimidin-4-one (7.2.10) on heating at 85–90°C in dimethylformamide in the presence of sodium carbonate and potassium iodide, which gave the desired product, risperidone (7.2.1) [114,115]. Later, another method of (7.2.7) → (7.2.1) transformation was proposed, which involved the reductive alkylation of (2,4-difluorophenyl)(piperidin-4-yl)methanone (7.2.7) with aldehyde (7.2.11) and sodium cyanoborohydride, which gave compound (7.2.12), coherently converted to oxime (7.2.13) and further to the desired compound, risperidone (7.2.1) [116] (Scheme 7.7).

///////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Medical uses
Risperidone is mainly used for the treatment of schizophrenia, bipolar disorder, and irritability associated with autism.[12]
Schizophrenia
Risperidone is effective in treating psychogenic polydipsia and the acute exacerbations of schizophrenia.[13][14]
Studies evaluating the utility of risperidone by mouth for maintenance therapy have reached varying conclusions. A 2012 systematic review concluded that evidence is strong that risperidone is more effective than all first-generation antipsychotics other than haloperidol, but that evidence directly supporting its superiority to placebo is equivocal.[15] A 2011 review concluded that risperidone is more effective in relapse prevention than other first- and second-generation antipsychotics with the exception of olanzapine and clozapine.[16] A 2016 Cochrane review suggests that risperidone reduces the overall symptoms of schizophrenia, but firm conclusions are difficult to make due to very low-quality evidence. Data and information are scarce, poorly reported, and probably biased in favour of risperidone, with about half of the included trials developed by drug companies. The article raises concerns regarding the serious side effects of risperidone, such as parkinsonism.[17] A 2011 Cochrane review compared risperidone with other atypical antipsychotics such as olanzapine for schizophrenia:[18]
| Summary |
|---|
| Risperidone seems to produce somewhat more extrapyramidal side effects and clearly more prolactin increase than most other atypical antipsychotics. It may also differ from other compounds in the occurrence of other adverse effects such as weight gain, metabolic problems, cardiac effects, sedation, and seizures. Nevertheless, the large proportion of participants leaving studies early and incomplete reporting of outcomes makes drawing firm conclusions difficult.[18] |
| showOutcomeFindings in wordsFindings in numbersQuality of evidence |
Long-acting injectable formulations of antipsychotic drugs provide improved compliance with therapy and reduce relapse rates relative to oral formulations.[19][20] The efficacy of risperidone long-acting injection appears to be similar to that of long acting injectable forms of first generation antipsychotics.[21]
Bipolar disorder
Second-generation antipsychotics, including risperidone, are effective in the treatment of manic symptoms in acute manic or mixed exacerbations of bipolar disorder.[22][23][24] In children and adolescents, risperidone may be more effective than lithium or divalproex, but has more metabolic side effects.[25] As maintenance therapy, long-acting injectable risperidone is effective for the prevention of manic episodes but not depressive episodes.[26] The long-acting injectable form of risperidone may be advantageous over long acting first generation antipsychotics, as it is better tolerated (fewer extrapyramidal effects) and because long acting injectable formulations of first generation antipsychotics may increase the risk of depression.[27]
Autism
Compared to placebo, risperidone treatment reduces certain problematic behaviors in autistic children, including aggression toward others, self-injury, challenging behaviour, and rapid mood changes.[28] The evidence for its efficacy appears to be greater than that for alternative pharmacological treatments.[29] Weight gain is an important adverse effect.[4][30] Some authors recommend limiting the use of risperidone and aripiprazole to those with the most challenging behavioral disturbances in order to minimize the risk of drug-induced adverse effects.[31] Evidence for the efficacy of risperidone in autistic adolescents and young adults is less persuasive.[32]
Other uses
Risperidone has shown promise in treating therapy-resistant obsessive–compulsive disorder, when serotonin reuptake inhibitors alone are not sufficient.[33]
Risperidone has not demonstrated a benefit in the treatment of eating disorders or personality disorders, except for limited evidence in schizotypal personality disorder.[34]
While antipsychotic medications such as risperidone have a slight benefit in people with dementia, they have been linked to higher incidence of death and stroke.[34] Because of this increased risk of death, treatment of dementia-related psychosis with risperidone is not FDA approved and carries a black box warning.[4]
Forms
Available forms of risperidone include tablet, oral dissolving tablet, oral solution, and powder and solvent for suspension for injection.[35]
Adverse effects
See also: List of adverse effects of risperidone
Common side effects include movement problems, sleepiness, dizziness, trouble seeing, constipation, and increased weight.[2][7] About 9 to 20% of people gained more than 7% of the baseline weight depending on the dose.[2] Serious side effects may include the potentially permanent movement disorder tardive dyskinesia, as well as neuroleptic malignant syndrome, an increased risk of suicide, and high blood sugar levels.[2][6] In older people with psychosis as a result of dementia, it may increase the risk of death.[2]
While atypical antipsychotics appear to have a lower rate of movement problems as compared to typical antipsychotics, risperidone has a high risk of movement problems among the atypicals.[36][37] Atypical antipsychotics however are associated with a greater amount of weight gain.[37]
Drug interactions
- Carbamazepine and other enzyme inducers may reduce plasma levels of risperidone.[4] If a person is taking both carbamazepine and risperidone, the dose of risperidone will likely need to be increased. The new dose should not be more than twice the patient’s original dose.[4]
- CYP2D6 inhibitors, such as SSRI medications, may increase plasma levels of risperidone and those medications.[4]
- Since risperidone can cause hypotension, its use should be monitored closely when a patient is also taking antihypertensive medicines to avoid severe low blood pressure.[4]
- Risperidone and its metabolite paliperidone are reduced in efficacy by P-glycoprotein inducers such as St John’s wort[38][39]
Discontinuation
The British National Formulary recommends a gradual withdrawal when discontinuing antipsychotic treatment to avoid acute withdrawal syndrome or rapid relapse.[40] Some have argued the additional somatic and psychiatric symptoms associated with dopaminergic super-sensitivity, including dyskinesia and acute psychosis, are common features of withdrawal in individuals treated with neuroleptics.[41][42][43][44] This has led some to suggest the withdrawal process might itself be schizomimetic, producing schizophrenia-like symptoms even in previously healthy patients, indicating a possible pharmacological origin of mental illness in a yet unknown percentage of patients currently and previously treated with antipsychotics. This question is unresolved, and remains a highly controversial issue among professionals in the medical and mental health communities, as well as the public.[45]
Dementia
Older people with dementia-related psychosis are at a higher risk of death if they take risperidone compared to those who do not. Most deaths are related to heart problems or infections.[4]
Pharmacology
Pharmacodynamics
See also: Atypical antipsychotic § Pharmacodynamics, and Antipsychotic § Comparison of medications
| Site | Ki (nM) | Action |
|---|---|---|
| 5-HT1A | 423 | Antagonist |
| 5-HT1B | 14.9 | Antagonist |
| 5-HT1D | 84.6 | Antagonist |
| 5-HT2A | 0.17 | Inverse agonist |
| 5-HT2B | 61.9 | Inverse agonist |
| 5-HT2C | 12.0 | Inverse agonist |
| 5-HT5A | 206 | Antagonist |
| 5-HT6 | 2,060 | Antagonist |
| 5-HT7 | 6.60 | Irreversible antagonist[47] |
| α1A | 5.0 | Antagonist |
| α1B | 9.0 | Antagonist |
| α2A | 16.5 | Antagonist |
| α2B | 108 | Antagonist |
| α2C | 1.30 | Antagonist |
| D1 | 244 | Antagonist |
| D2 | 3.57 | Antagonist |
| D2S | 4.73 | Antagonist |
| D2L | 4.16 | Antagonist |
| D3 | 3.6 | Inverse agonist |
| D4 | 4.66 | Antagonist |
| D5 | 290 | Antagonist |
| H1 | 20.1 | Inverse agonist |
| H2 | 120 | Inverse agonist |
| mACh | >10,000 | Negligible |
Risperidone pharmacodynamics excluding D-amino acid oxidase inhibition
Risperidone has been classified as a “qualitatively atypical” antipsychotic agent with a relatively low incidence of extrapyramidal side effects (when given at low doses) that has more pronounced serotonin antagonism than dopamine antagonism. Risperidone contains the functional groups of benzisoxazole and piperidine as part of its molecular structure. Although not a butyrophenone, it was developed with the structures of benperidol and ketanserin as a basis. It has actions at several 5-HT (serotonin) receptor subtypes. These are 5-HT2C, linked to weight gain, 5-HT2A, linked to its antipsychotic action and relief of some of the extrapyramidal side effects experienced with the typical neuroleptics.[48]
It has been found that D-amino acid oxidase, the enzyme that catalyses the breakdown of D-amino acids (e.g. D-alanine and D-serine — the neurotransmitters) is inhibited by risperidone.[49]
Risperidone acts on the following receptors:
Dopamine receptors: This drug is an antagonist of the D1 (D1, and D5) as well as the D2 family (D2, D3 and D4) receptors, with 70-fold selectivity for the D2 family. This drug has “tight binding” properties, which means it has a long half-life and like other antipsychotics, risperidone blocks the mesolimbic pathway, the prefrontal cortex limbic pathway, and the tuberoinfundibular pathway in the central nervous system. Risperidone may induce extrapyramidal side effects, akathisia and tremors, associated with diminished dopaminergic activity in the striatum. It can also cause sexual side effects, galactorrhoea, infertility, gynecomastia and, with chronic use reduced bone mineral density leading to breaks, all of which are associated with increased prolactin secretion.[48]
Serotonin receptors: Its action at these receptors may be responsible for its lower extrapyramidal side effect liability (via the 5-HT2A/2C receptors) and improved negative symptom control compared to typical antipsychotics such as haloperidol for instance. Its antagonistic actions at the 5-HT2C receptor may account, in part, for its weight gain liability.[medical citation needed]
Alpha α1 adrenergic receptors: This action accounts for its orthostatic hypotensive effects and perhaps some of the sedating effects of risperidone.[48]
Alpha α2 adrenergic receptors: Perhaps greater positive, negative, affective and cognitive symptom control.[50]
Histamine H1 receptors: effects on these receptors account for its sedation and reduction in vigilance. This may also lead to drowsiness and weight gain.[48]
Voltage-gated sodium channels: Because it accumulates in synaptic vesicles, Risperidone inhibits voltage-gated sodium channels at clinically used concentrations.[51]
Though this medication possesses similar effects to other typical and atypical antipsychotics, it does not possess an affinity for the muscarinic acetylcholine receptors. In many respects, this medication can be useful as an “acetylcholine release-promoter” similar to gastrointestinal drugs such as metoclopramide and cisapride.[medical citation needed]
Pharmacokinetics
Risperidone undergoes hepatic metabolism and renal excretion. Lower doses are recommended for patients with severe liver and kidney disease.[4] The active metabolite of risperidone, paliperidone, is also used as an antipsychotic.[52]
Society and culture

Risperdal (risperidone) 4 mg tablets (UK)
Legal status
Risperidone was approved by the United States Food and Drug Administration (FDA) in 1993 for the treatment of schizophrenia.[63] In 2003, the FDA approved risperidone for the short-term treatment of the mixed and manic states associated with bipolar disorder. In 2006, the FDA approved risperidone for the treatment of irritability in autistic children and adolescents.[64] The FDA’s decision was based in part on a study of autistic people with severe and enduring problems of violent meltdowns, aggression, and self-injury; risperidone is not recommended for autistic people with mild aggression and explosive behavior without an enduring pattern.[65] On 22 August 2007, risperidone was approved as the only drug agent available for treatment of schizophrenia in youths, ages 13–17; it was also approved that same day for treatment of bipolar disorder in youths and children, ages 10–17, joining lithium.
On 16 December 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Okedi, intended for the treatment of schizophrenia in adults for whom tolerability and effectiveness has been established with oral risperidone.[66] The applicant for this medicinal product is Laboratorios Farmacéuticos Rovi, S.A.[66]
Availability
Janssen’s patent on risperidone expired on 29 December 2003, opening the market for cheaper generic versions from other companies, and Janssen’s exclusive marketing rights expired on 29 June 2004 (the result of a pediatric extension). It is available under many brand names worldwide.[1]
Risperidone is available as a tablet, an oral solution, and an ampule, which is a depot injection.[1]
Lawsuits
On 11 April 2012, Johnson & Johnson (J&J) and its subsidiary Janssen Pharmaceuticals Inc. were fined $1.2 billion by Judge Timothy Davis Fox of the Sixth Division of the Sixth Judicial Circuit of the U.S. state of Arkansas.[67] The jury found the companies had downplayed multiple risks associated with risperidone (Risperdal). The verdict was later reversed by the Arkansas State Supreme court.[68]
In August 2012, Johnson & Johnson agreed to pay $181 million to 36 U.S. states in order to settle claims that it had promoted risperidone and paliperidone for off-label uses including for dementia, anger management, and anxiety.[69]
In November 2013, J&J was fined $2.2 billion for illegally marketing risperidone for use in people with dementia.[70]
In 2015, Steven Brill posted a 15-part investigative journalism piece on J&J in The Huffington Post, called “America’s most admired lawbreaker”, which was focused on J&J’s marketing of risperidone.[71][72]
J&J has faced numerous civil lawsuits on behalf of children who were prescribed risperidone who grew breasts (a condition called gynecomastia); as of July 2016 there were about 1,500 cases in Pennsylvania state court in Philadelphia, and there had been a February 2015 verdict against J&J with $2.5 million awarded to a man from Alabama, a $1.75M verdict against J&J that November, and in 2016 a $70 million verdict against J&J.[73] In October 2019, a jury awarded a Pennsylvania man $8 billion in a verdict against J&J.[74]
Names
Brand names include Risperdal, Risperdal Consta, Risperdal M-Tab, Risperdal Quicklets, Risperlet, Okedi, and Perseris.[75]
References
- ^ Jump up to:a b c Drugs.com International trade names for risperidone Archived 18 March 2016 at the Wayback Machine Page accessed 15 March 2016
- ^ Jump up to:a b c d e f g h i j k l m n o p q r “Risperidone”. The American Society of Health-System Pharmacists. Archived from the original on 2 December 2015. Retrieved 1 December 2015.
- ^ “Risperdal Consta 25 mg powder and solvent for prolonged-release suspension for injection – Summary of Product Characteristics (SmPC)”. (emc). 6 December 2018. Retrieved 29 January 2022.
- ^ Jump up to:a b c d e f g h i j “Risperdal- risperidone tablet Risperdal M-Tab- risperidone tablet, orally disintegrating Risperdal- risperidone solution”. DailyMed. Retrieved 31 December 2019.
- ^ “Okedi EPAR”. European Medicines Agency (EMA). 15 December 2021. Retrieved 2 March 2022.
- ^ Jump up to:a b c d Hamilton R (2015). Tarascon Pocket Pharmacopoeia 2015 Deluxe Lab-Coat Edition. Jones & Bartlett Learning. pp. 434–435. ISBN 9781284057560.
- ^ Jump up to:a b Hasnain M, Vieweg WV, Hollett B (July 2012). “Weight gain and glucose dysregulation with second-generation antipsychotics and antidepressants: a review for primary care physicians”. Postgraduate Medicine. 124 (4): 154–67. doi:10.3810/pgm.2012.07.2577. PMID 22913904. S2CID 39697130.
- ^ Schatzberg AF, Nemeroff CB (2009). The American Psychiatric Publishing textbook of psychopharmacology (4th ed.). Washington, D.C.: American Psychiatric Pub. p. 627. ISBN 9781585623099.
- ^ World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
- ^ “The Top 300 of 2019”. ClinCalc. Retrieved 16 October 2021.
- ^ “Risperidone – Drug Usage Statistics”. ClinCalc. Retrieved 16 October 2021.
- ^ “Respiridone”. The American Society of Health-System Pharmacists. Archived from the original on 13 April 2011. Retrieved 3 April 2011.
- ^ Leucht S, Cipriani A, Spineli L, Mavridis D, Orey D, Richter F, et al. (September 2013). “Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: a multiple-treatments meta-analysis”. Lancet. 382 (9896): 951–62. doi:10.1016/S0140-6736(13)60733-3. PMID 23810019. S2CID 32085212.
- ^ Osser DN, Roudsari MJ, Manschreck T (2013). “The psychopharmacology algorithm project at the Harvard South Shore Program: an update on schizophrenia”. Harvard Review of Psychiatry. 21 (1): 18–40. doi:10.1097/HRP.0b013e31827fd915. PMID 23656760. S2CID 22523977.
- ^ Barry SJ, Gaughan TM, Hunter R (June 2012). “Schizophrenia”. BMJ Clinical Evidence. 2012. PMC 3385413. PMID 23870705.
- ^ Glick ID, Correll CU, Altamura AC, Marder SR, Csernansky JG, Weiden PJ, et al. (December 2011). “Mid-term and long-term efficacy and effectiveness of antipsychotic medications for schizophrenia: a data-driven, personalized clinical approach”. The Journal of Clinical Psychiatry. 72 (12): 1616–27. doi:10.4088/JCP.11r06927. PMID 22244023.
- ^ Rattehalli RD, Zhao S, Li BG, Jayaram MB, Xia J, Sampson S (December 2016). “Risperidone versus placebo for schizophrenia” (PDF). The Cochrane Database of Systematic Reviews. 2016 (12): CD006918. doi:10.1002/14651858.CD006918.pub3. PMC 6463908. PMID 27977041.
- ^ Jump up to:a b Komossa K, Rummel-Kluge C, Schwarz S, Schmid F, Hunger H, Kissling W, Leucht S (January 2011). “Risperidone versus other atypical antipsychotics for schizophrenia”. The Cochrane Database of Systematic Reviews (1): CD006626. doi:10.1002/14651858.CD006626.pub2. PMC 4167865. PMID 21249678.
- ^ Leucht C, Heres S, Kane JM, Kissling W, Davis JM, Leucht S (April 2011). “Oral versus depot antipsychotic drugs for schizophrenia–a critical systematic review and meta-analysis of randomised long-term trials”. Schizophrenia Research. 127 (13): 83–92. doi:10.1016/j.schres.2010.11.020. PMID 21257294. S2CID 2386150.
- ^ Lafeuille MH, Dean J, Carter V, Duh MS, Fastenau J, Dirani R, Lefebvre P (August 2014). “Systematic review of long-acting injectables versus oral atypical antipsychotics on hospitalization in schizophrenia”. Current Medical Research and Opinion. 30 (8): 1643–55. doi:10.1185/03007995.2014.915211. PMID 24730586. S2CID 24814527.
- ^ Nielsen J, Jensen SO, Friis RB, Valentin JB, Correll CU (May 2015). “Comparative effectiveness of risperidone long-acting injectable vs first-generation antipsychotic long-acting injectables in schizophrenia: results from a nationwide, retrospective inception cohort study”. Schizophrenia Bulletin. 41 (3): 627–36. doi:10.1093/schbul/sbu128. PMC 4393684. PMID 25180312.
- ^ Muralidharan K, Ali M, Silveira LE, Bond DJ, Fountoulakis KN, Lam RW, Yatham LN (September 2013). “Efficacy of second generation antipsychotics in treating acute mixed episodes in bipolar disorder: a meta-analysis of placebo-controlled trials”. Journal of Affective Disorders. 150 (2): 408–14. doi:10.1016/j.jad.2013.04.032. PMID 23735211.
- ^ Nivoli AM, Murru A, Goikolea JM, Crespo JM, Montes JM, González-Pinto A, et al. (October 2012). “New treatment guidelines for acute bipolar mania: a critical review”. Journal of Affective Disorders. 140 (2): 125–41. doi:10.1016/j.jad.2011.10.015. PMID 22100133.
- ^ Yildiz A, Vieta E, Leucht S, Baldessarini RJ (January 2011). “Efficacy of antimanic treatments: meta-analysis of randomized, controlled trials”. Neuropsychopharmacology. 36 (2): 375–89. doi:10.1038/npp.2010.192. PMC 3055677. PMID 20980991.
- ^ Peruzzolo TL, Tramontina S, Rohde LA, Zeni CP (2013). “Pharmacotherapy of bipolar disorder in children and adolescents: an update”. Revista Brasileira de Psiquiatria. 35 (4): 393–405. doi:10.1590/1516-4446-2012-0999. PMID 24402215.
- ^ Gitlin M, Frye MA (May 2012). “Maintenance therapies in bipolar disorders”. Bipolar Disorders. 14 Suppl 2: 51–65. doi:10.1111/j.1399-5618.2012.00992.x. PMID 22510036. S2CID 21101054.
- ^ Gigante AD, Lafer B, Yatham LN (May 2012). “Long-acting injectable antipsychotics for the maintenance treatment of bipolar disorder”. CNS Drugs. 26 (5): 403–20. doi:10.2165/11631310-000000000-00000. PMID 22494448. S2CID 2786921.
- ^ Jesner OS, Aref-Adib M, Coren E (January 2007). “Risperidone for autism spectrum disorder”. The Cochrane Database of Systematic Reviews (1): CD005040. doi:10.1002/14651858.CD005040.pub2. PMID 17253538.
- ^ Kirino E (2014). “Efficacy and tolerability of pharmacotherapy options for the treatment of irritability in autistic children”. Clinical Medicine Insights. Pediatrics. 8: 17–30. doi:10.4137/CMPed.S8304. PMC 4051788. PMID 24932108.
- ^ Sharma A, Shaw SR (2012). “Efficacy of risperidone in managing maladaptive behaviors for children with autistic spectrum disorder: a meta-analysis”. Journal of Pediatric Health Care. 26 (4): 291–9. doi:10.1016/j.pedhc.2011.02.008. PMID 22726714.
- ^ McPheeters ML, Warren Z, Sathe N, Bruzek JL, Krishnaswami S, Jerome RN, Veenstra-Vanderweele J (May 2011). “A systematic review of medical treatments for children with autism spectrum disorders”. Pediatrics. 127 (5): e1312–21. doi:10.1542/peds.2011-0427. PMID 21464191. S2CID 2903864.
- ^ Dove D, Warren Z, McPheeters ML, Taylor JL, Sathe NA, Veenstra-VanderWeele J (October 2012). “Medications for adolescents and young adults with autism spectrum disorders: a systematic review”. Pediatrics. 130 (4): 717–26. doi:10.1542/peds.2012-0683. PMC 4074627. PMID 23008452.
- ^ Dold M, Aigner M, Lanzenberger R, Kasper S (April 2013). “Antipsychotic augmentation of serotonin reuptake inhibitors in treatment-resistant obsessive-compulsive disorder: a meta-analysis of double-blind, randomized, placebo-controlled trials”. The International Journal of Neuropsychopharmacology. 16 (3): 557–74. doi:10.1017/S1461145712000740. PMID 22932229.
- ^ Jump up to:a b Maher AR, Theodore G (June 2012). “Summary of the comparative effectiveness review on off-label use of atypical antipsychotics”. Journal of Managed Care Pharmacy. 18 (5 Suppl B): S1–20. doi:10.18553/jmcp.2012.18.s5-b.1. PMID 22784311.
- ^ Joint Formulary Committee. British National Formulary (online) London: BMJ Group and Pharmaceutical Press http://www.medicinescomplete.com [Accessed on 2 February 2020]
- ^ Divac N, Prostran M, Jakovcevski I, Cerovac N (2014). “Second-generation antipsychotics and extrapyramidal adverse effects”. BioMed Research International. 2014: 656370. doi:10.1155/2014/656370. PMC 4065707. PMID 24995318.
- ^ Jump up to:a b Pillay J, Boylan K, Carrey N, Newton A, Vandermeer B, Nuspl M, MacGregor T, Jafri SH, Featherstone R, Hartling L (March 2017). “First- and Second-Generation Antipsychotics in Children and Young Adults: Systematic Review Update”. Comparative Effectiveness Reviews (184): ES–24. PMID 28749632. Report 17-EHC001-EF. Bookshelf ID: NBK442352.
Compared with FGAs, SGAs may decrease the risk for experiencing any extrapyramidal symptom (EPS). FGAs probably cause lower gains in weight and BMI.
- ^ Wang, J. S.; Ruan, Y.; Taylor, R. M.; Donovan, J. L.; Markowitz, J. S.; Devane, C. L. (2004). “The Brain Entry of Risperidone and 9-hydroxyrisperidone Is Greatly Limited by P-glycoprotein”. The International Journal of Neuropsychopharmacology. 7 (4): 415–9. doi:10.1017/S1461145704004390. PMID 15683552.
- ^ Gurley BJ, Swain A, Williams DK, Barone G, Battu SK (July 2008). “Gauging the clinical significance of P-glycoprotein-mediated herb-drug interactions: comparative effects of St. John’s wort, Echinacea, clarithromycin, and rifampin on digoxin pharmacokinetics”. Molecular Nutrition & Food Research. 52 (7): 772–9. doi:10.1002/mnfr.200700081. PMC 2562898. PMID 18214850.
- ^ BMJ Group, ed. (March 2009). “4.2.1”. British National Formulary (57 ed.). United Kingdom: Royal Pharmaceutical Society of Great Britain. p. 192. ISSN 0260-535X.
Withdrawal of antipsychotic drugs after long-term therapy should always be gradual and closely monitored to avoid the risk of acute withdrawal syndromes or rapid relapse.
- ^ Chouinard G, Jones BD (January 1980). “Neuroleptic-induced supersensitivity psychosis: clinical and pharmacologic characteristics”. The American Journal of Psychiatry. 137 (1): 16–21. doi:10.1176/ajp.137.1.16. PMID 6101522.
- ^ Miller R, Chouinard G (November 1993). “Loss of striatal cholinergic neurons as a basis for tardive and L-dopa-induced dyskinesias, neuroleptic-induced supersensitivity psychosis and refractory schizophrenia”. Biological Psychiatry. 34 (10): 713–38. doi:10.1016/0006-3223(93)90044-E. PMID 7904833. S2CID 2405709.
- ^ Chouinard G, Jones BD, Annable L (November 1978). “Neuroleptic-induced supersensitivity psychosis”. The American Journal of Psychiatry. 135 (11): 1409–10. doi:10.1176/ajp.135.11.1409. PMID 30291.
- ^ Seeman P, Weinshenker D, Quirion R, Srivastava LK, Bhardwaj SK, Grandy DK, et al. (March 2005). “Dopamine supersensitivity correlates with D2High states, implying many paths to psychosis”. Proceedings of the National Academy of Sciences of the United States of America. 102 (9): 3513–8. Bibcode:2005PNAS..102.3513S. doi:10.1073/pnas.0409766102. PMC 548961. PMID 15716360.
- ^ Moncrieff J (July 2006). “Does antipsychotic withdrawal provoke psychosis? Review of the literature on rapid onset psychosis (supersensitivity psychosis) and withdrawal-related relapse”. Acta Psychiatrica Scandinavica. 114 (1): 3–13. doi:10.1111/j.1600-0447.2006.00787.x. PMID 16774655. S2CID 6267180.
- ^ National Institute of Mental Health. PDSD Ki Database (Internet) [cited 2013 Aug 10]. ChapelHill (NC): University of North Carolina. 1998-2013. Available from: “Archived copy”. Archived from the original on 8 November 2013. Retrieved 16 May 2016.
- ^ Smith C, Rahman T, Toohey N, Mazurkiewicz J, Herrick-Davis K, Teitler M (October 2006). “Risperidone irreversibly binds to and inactivates the h5-HT7 serotonin receptor”. Molecular Pharmacology. 70 (4): 1264–70. doi:10.1124/mol.106.024612. PMID 16870886. S2CID 1678887.
- ^ Jump up to:a b c d Brunton L, Chabner B, Knollman B. Goodman and Gilman’s The Pharmacological Basis of Therapeutics, Twelfth Edition. McGraw Hill Professional; 2010.
- ^ Abou El-Magd RM, Park HK, Kawazoe T, Iwana S, Ono K, Chung SP, et al. (July 2010). “The effect of risperidone on D-amino acid oxidase activity as a hypothesis for a novel mechanism of action in the treatment of schizophrenia”. Journal of Psychopharmacology. 24 (7): 1055–67. doi:10.1177/0269881109102644. PMID 19329549. S2CID 39050369.
- ^ Hecht EM, Landy DC (February 2012). “Alpha-2 receptor antagonist add-on therapy in the treatment of schizophrenia; a meta-analysis”. Schizophrenia Research. 134 (2–3): 202–6. doi:10.1016/j.schres.2011.11.030. PMID 22169246. S2CID 36119981.
- ^ Brauner, Jan M.; Hessler, Sabine; Groemer, Teja W.; Alzheimer, Christian; Huth, Tobias (2014). “Risperidone inhibits voltage-gated sodium channels”. European Journal of Pharmacology. 728: 100–106. doi:10.1016/j.ejphar.2014.01.062. PMID 24508524.
- ^ “The DrugBank database”. Archived from the original on 17 November 2011.
- ^ Parent M, Toussaint C, Gilson H (1983). “Long-term treatment of chronic psychotics with bromperidol decanoate: clinical and pharmacokinetic evaluation”. Current Therapeutic Research. 34 (1): 1–6.
- ^ Jump up to:a b Jørgensen A, Overø KF (1980). “Clopenthixol and flupenthixol depot preparations in outpatient schizophrenics. III. Serum levels”. Acta Psychiatrica Scandinavica. Supplementum. 279: 41–54. doi:10.1111/j.1600-0447.1980.tb07082.x. PMID 6931472.
- ^ Jump up to:a b Reynolds JE (1993). “Anxiolytic sedatives, hypnotics and neuroleptics.”. Martindale: The Extra Pharmacopoeia (30th ed.). London: Pharmaceutical Press. pp. 364–623.
- ^ Ereshefsky L, Saklad SR, Jann MW, Davis CM, Richards A, Seidel DR (May 1984). “Future of depot neuroleptic therapy: pharmacokinetic and pharmacodynamic approaches”. The Journal of Clinical Psychiatry. 45 (5 Pt 2): 50–9. PMID 6143748.
- ^ Jump up to:a b Curry SH, Whelpton R, de Schepper PJ, Vranckx S, Schiff AA (April 1979). “Kinetics of fluphenazine after fluphenazine dihydrochloride, enanthate and decanoate administration to man”. British Journal of Clinical Pharmacology. 7 (4): 325–31. doi:10.1111/j.1365-2125.1979.tb00941.x. PMC 1429660. PMID 444352.
- ^ Young D, Ereshefsky L, Saklad SR, Jann MW, Garcia N (1984). Explaining the pharmacokinetics of fluphenazine through computer simulations. (Abstract.). 19th Annual Midyear Clinical Meeting of the American Society of Hospital Pharmacists. Dallas, Texas.
- ^ Janssen PA, Niemegeers CJ, Schellekens KH, Lenaerts FM, Verbruggen FJ, van Nueten JM, et al. (November 1970). “The pharmacology of fluspirilene (R 6218), a potent, long-acting and injectable neuroleptic drug”. Arzneimittel-Forschung. 20 (11): 1689–98. PMID 4992598.
- ^ Beresford R, Ward A (January 1987). “Haloperidol decanoate. A preliminary review of its pharmacodynamic and pharmacokinetic properties and therapeutic use in psychosis”. Drugs. 33 (1): 31–49. doi:10.2165/00003495-198733010-00002. PMID 3545764.
- ^ Reyntigens AJ, Heykants JJ, Woestenborghs RJ, Gelders YG, Aerts TJ (1982). “Pharmacokinetics of haloperidol decanoate. A 2-year follow-up”. International Pharmacopsychiatry. 17 (4): 238–46. doi:10.1159/000468580. PMID 7185768.
- ^ Larsson M, Axelsson R, Forsman A (1984). “On the pharmacokinetics of perphenazine: a clinical study of perphenazine enanthate and decanoate”. Current Therapeutic Research. 36 (6): 1071–88.
- ^ “Electronic Orange Book”. Food and Drug Administration. April 2007. Archived from the original on 19 August 2007. Retrieved 24 May 2007.
- ^ “FDA approves the first drug to treat irritability associated with autism, Risperdal” (Press release). FDA. 6 October 2006. Archived from the original on 28 August 2009. Retrieved 14 August 2009.
- ^ Scahill L (July 2008). “How do I decide whether or not to use medication for my child with autism? Should I try behavior therapy first?”. Journal of Autism and Developmental Disorders. 38 (6): 1197–8. doi:10.1007/s10803-008-0573-7. PMID 18463973. S2CID 20767044.
- ^ Jump up to:a b “Okedi: Pending EC decision”. European Medicines Agency. 15 December 2021. Retrieved 18 December 2021. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “Companies belittled risks of Risperdal, slapped with huge fine” Archived 12 April 2012 at the Wayback Machine, Los Angeles Times, 11 April 2012.
- ^ Thomas K (20 March 2014). “Arkansas Court Reverses $1.2 Billion Judgment Against Johnson & Johnson”. The New York Times. Archived from the original on 5 November 2015.
- ^ “NY AG: Janssen pays $181M over drug marketing”. The Seattle Times. 30 August 2012. Archived from the original on 7 April 2016.
- ^ “Johnson & Johnson to Pay More Than $2.2 Billion to Resolve Criminal and Civil Investigations”. Department of Justice, Office of Public Affairs. 4 November 2013. Archived from the original on 5 March 2015. Retrieved 23 December 2020.
- ^ Ashbrook T (22 September 2015). “Johnson & Johnson And The Big Lies Of Big Pharma”. On Point. Archived from the original on 22 November 2016.
- ^ Brill S (September 2015). “America’s Most Admired Lawbreaker”. The Huffington Post. Archived from the original on 2 October 2015.
- ^ Feeley J (1 July 2016). “J&J Hit With $70 Million Risperdal Verdict Over Male Breasts”. Bloomberg News. Archived from the original on 7 May 2017.
- ^ “Jury says J&J must pay $8 billion in case over male breast growth linked to Risperdal”. Reuters. 9 October 2019. Retrieved 9 October 2019.
- ^ “Risperidone: MedlinePlus Drug Information”. medlineplus.gov. Retrieved 28 September 2020.
Further reading
- Dean L (2017). “Risperidone Therapy and CYP2D6 Genotype”. In Pratt VM, McLeod HL, Rubinstein WS, et al. (eds.). Medical Genetics Summaries. National Center for Biotechnology Information (NCBI). PMID 28520384. Bookshelf ID: NBK425795.
“Risperidone”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Risperdal, others[1] |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a694015 |
| License data | US DailyMed: Risperidone |
| Pregnancy category | AU: C |
| Routes of administration | By mouth, intramuscular, subcutaneous |
| Drug class | Atypical antipsychotic[2] |
| ATC code | N05AX08 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only)CA: ℞-onlyUK: POM (Prescription only) [3]US: ℞-only [4]EU: Rx-only [5] |
| Pharmacokinetic data | |
| Bioavailability | 70% (by mouth)[2] |
| Metabolism | Liver (CYP2D6 mediated to 9-hydroxyrisperidone)[2] |
| Elimination half-life | 20 hours (by mouth), 3–6 days (IM)[2] |
| Excretion | Urinary (70%) feces (14%)[2] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 106266-06-2 |
| PubChem CID | 5073 |
| PubChem SID | 475100 |
| IUPHAR/BPS | 96 |
| DrugBank | DB00734 |
| ChemSpider | 4895 |
| UNII | L6UH7ZF8HC |
| KEGG | D00426 |
| ChEBI | CHEBI:8871 |
| ChEMBL | ChEMBL85 |
| PDB ligand | 8NU (PDBe, RCSB PDB) |
| CompTox Dashboard (EPA) | DTXSID8045193 |
| ECHA InfoCard | 100.114.705 |
| Chemical and physical data | |
| Formula | C23H27FN4O2 |
| Molar mass | 410.493 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (verify) |
//////////////Risperidone, R-64,766, R-64766, RCN-3028, RCN3028

NEW DRUG APPROVALS
ONE TIME
$10.00
Lutetium Lu 177 vipivotide tetraxetan
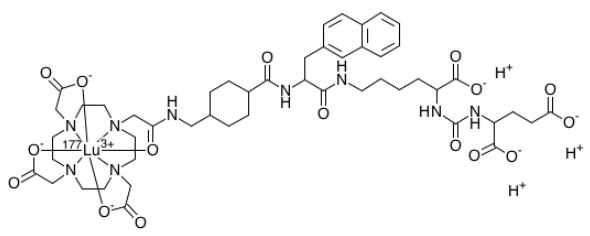


Lutetium Lu 177 vipivotide tetraxetan
FDA APPROVED 2022/3/23, Pluvicto
To treat prostate-specific membrane antigen-positive metastatic castration-resistant prostate cancer following other therapies
| Formula | C49H65N9O16. Lu. 3H |
|---|---|
| CAS | 1703749-62-5 |
| Mol weight | 1214.0819 |
| Antineoplastic, Radioactive agent | |
| Disease | Prostate cancer (PSMA positive) |
|---|
ルテチウム(177Lu)ビピボチドテトラキセタン;
UNII-G6UF363ECX, WHO 11429
G6UF363ECX
177Lu-Psma-617
Vipivotide tetraxetan Lu-177
177Lu-Labeled PSMA-617
2-[4-[2-[[4-[[(2S)-1-[[(5S)-5-carboxy-5-[[(1S)-1,3-dicarboxypropyl]carbamoylamino]pentyl]amino]-3-naphthalen-2-yl-1-oxopropan-2-yl]carbamoyl]cyclohexyl]methylamino]-2-oxoethyl]-7,10-bis(carboxylatomethyl)-1,4,7,10-tetrazacyclododec-1-yl]acetate;lutetium-177(3+)
(177Lu)Lutetium 2,2′,2”-[10-(2-{[(trans-4-{[(2S)-1-{[(5S)-5-carboxy-5-({[(1S)-1,3-dicarboxypropyl]carbamoyl}amino)pentyl]amino}-3-(2-naphthyl)-1-oxo-2-propanyl]carbamoyl}cyclohexyl)methyl]amino}-2- oxoethyl)-1,4,7,10-tetraazacyclododecane-1,4,7-triyl]triacetate (non-preferred name)
1983157-55-6[RN]
PSMA-617 LU-177
Lutetium Lu 177 Vipivotide Tetraxetan is a radioconjugate composed of PSMA-617, a human prostate-specific membrane antigen (PSMA)-targeting ligand, conjugated to the beta-emitting radioisotope lutetium Lu 177 (177Lu), with potential antineoplastic activity against PSMA-expressing tumor cells. Upon intravenous administration of lutetium Lu 177 vipivotide tetraxetan, vipivotide tetraxetan targets and binds to PSMA-expressing tumor cells. Upon binding, PSMA-expressing tumor cells are destroyed by 177Lu through the specific delivery of beta particle radiation. PSMA, a tumor-associated antigen and type II transmembrane protein, is expressed on the membrane of prostatic epithelial cells and overexpressed on prostate tumor cells.
Lutetium (177Lu) vipivotide tetraxetan, sold under the brand name Pluvicto, is a radiopharmaceutical medication used for the treatment of prostate-specific membrane antigen (PSMA)-positive metastatic castration-resistant prostate cancer (mCRPC).[2] Lutetium (177Lu) vipivotide tetraxetan is a targeted radioligand therapy.[2][3]
The most common adverse reactions include fatigue, dry mouth, nausea, anemia, decreased appetite, and constipation.[2]
Lutetium (177Lu) vipivotide tetraxetan is a radioconjugate composed of PSMA-617, a human prostate-specific membrane antigen (PSMA)-targeting ligand, conjugated to the beta-emitting radioisotope lutetium Lu 177 (177Lu), with potential antineoplastic activity against PSMA-expressing tumor cells.[4] Upon intravenous administration of lutetium Lu 177 vipivotide tetraxetan, vipivotide tetraxetan targets and binds to PSMA-expressing tumor cells.[4] Upon binding, PSMA-expressing tumor cells are destroyed by 177Lu through the specific delivery of beta particle radiation.[4] PSMA, a tumor-associated antigen and type II transmembrane protein, is expressed on the membrane of prostatic epithelial cells and overexpressed on prostate tumor cells.[4]
Lutetium (177Lu) vipivotide tetraxetan was approved for medical use in the United States in March 2022.[2][5]
///////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
History[edit]
Efficacy was evaluated in VISION (NCT03511664), a randomized (2:1), multicenter, open-label trial that evaluated lutetium (177Lu) vipivotide tetraxetan plus best standard of care (BSoC) (n=551) or BSoC alone (n=280) in men with progressive, prostate-specific membrane antigen (PSMA)-positive metastatic castration-resistant prostate cancer (mCRPC).[2] All participants received a GnRH analog or had prior bilateral orchiectomy.[2] Participants were required to have received at least one androgen receptor pathway inhibitor, and 1 or 2 prior taxane-based chemotherapy regimens.[2] Participants received lutetium (177Lu) vipivotide tetraxetan 7.4 GBq (200 mCi) every 6 weeks for up to a total of 6 doses plus BSoC or BSoC alone.[2]
The U.S. Food and Drug Administration granted the application for lutetium (177lu) vipivotide tetraxetan priority review and breakthrough therapy designations.[2]
References
- ^ “Highlights of prescribing information: PLUVICTOTM (lutetium Lu 177 vipivotide tetraxetan) injection, for intravenous use” (PDF). Advanced Accelerator Applications USA, Inc. Novartis. March 2022.
- ^ Jump up to:a b c d e f g h i j “FDA approves Pluvicto for metastatic castration-resistant prostate can”. U.S. Food and Drug Administration. 23 March 2022. Retrieved 23 March 2022.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Neels OC, Kopka K, Liolios C, Afshar-Oromieh A (December 2021). “Radiolabeled PSMA Inhibitors”. Cancers. 13 (24): 6255. doi:10.3390/cancers13246255. PMC 8699044. PMID 34944875.
- ^ Jump up to:a b c d “Lutetium Lu 177 Vipivotide Tetraxetan (Code C148145)”. NCI Thesaurus. 28 February 2022. Retrieved 23 March 2022. This article incorporates text from this source, which is in the public domain.
- ^ “Novartis Pluvicto approved by FDA as first targeted radioligand therapy for treatment of progressive, PSMA positive metastatic castration-resistant prostate cancer” (Press release). Novartis. 23 March 2022. Retrieved 23 March 2022.
External links
- “Lutetium lu 177 vipivotide tetraxetan”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Pluvicto |
| Other names | 177Lu-PSMA-617, Lutetium Lu 177 vipivotide tetraxetan (USAN US) |
| License data | US DailyMed: Pluvicto |
| Routes of administration | Intravenous |
| Drug class | Radiopharmaceutical |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1][2] |
| Identifiers | |
| CAS Number | 1703749-62-5 |
| PubChem CID | 122706785 |
| ChemSpider | 58828499 |
| UNII | G6UF363ECX |
| KEGG | D12335 |
| Chemical and physical data | |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| show |
////////////Lutetium Lu 177 vipivotide tetraxetan, ルテチウム(177Lu)ビピボチドテトラキセタン, FDA 2022, APPROVALS 2022, PROSTRATE CANCER, WHO 11429
C1CC(CCC1CNC(=O)CN2CCN(CCN(CCN(CC2)CC(=O)[O-])CC(=O)[O-])CC(=O)[O-])C(=O)NC(CC3=CC4=CC=CC=C4C=C3)C(=O)NCCCCC(C(=O)O)NC(=O)NC(CCC(=O)O)C(=O)O.[Lu+3]
Vipivotide tetraxetan (Synonyms: PSMA-617)
CAS No. : 1702967-37-0
Vipivotide tetraxetan (PSMA-617) is a high potent prostate-specific membrane antigen (PSMA) inhibitor, with a Ki of 0.37 nM.

NEW DRUG APPROVALS
one time
$10.00
CRENOLANIB

Crenolanib
- Molecular FormulaC26H29N5O2
- Average mass443.541 Da
1-(2-{5-[(3-Methyl-3-oxetanyl)methoxy]-1H-benzimidazol-1-yl}-8-quinolinyl)-4-piperidinamine
1-(2-{5-[(3-methyloxetan-3-yl)methoxy]-1H-benzimidazol-1-yl}quinolin-8-yl)piperidin-4-amine
1-[2-[5-[(3-methyl-3-oxetanyl)methoxy]-1H-benzimidazol-1-yl]-8-quinolinyl]-4-piperidinamine
4-Piperidinamine, 1-[2-[5-[(3-methyl-3-oxetanyl)methoxy]-1H-benzimidazol-1-yl]-8-quinolinyl]-
670220-88-9[RN]
9459
UNII-LQF7I567TQ
креноланиб
كرينولانيب
克拉尼布
CP-868,596-26 or AR-868,596-26
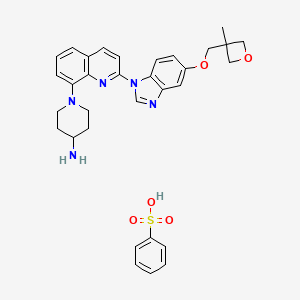
Crenolanib besylate
CAS#: 670220-93-6 (besylate)
Chemical Formula: C32H35N5O5S
Molecular Weight: 601.72
Crenolanib besylate (CP-868,596-26 or AR-868,596-26, 4-piperidinamine, 1-[2-[5-[(3-Methyl-3-oxetanyl) methoxy]-1H-benzimidazol-1-yl]- 8-quinolinyl]-, monobenzenesulfonate) is an investigational inhibitor being developed by AROG Pharmaceuticals, LLC. The compound is currently being evaluated for safety and efficacy in clinical trials for various types of cancer, including acute myeloid leukemia (AML),[1][2] gastrointestinal stromal tumor (GIST),[3] and glioma.[4] Crenolanib is an orally bioavailable benzamidazole that selectively and potently inhibits signaling of wild-type and mutant isoforms of class III receptor tyrosine kinases (RTK) FLT3 (FMS-like Tyrosine Kinase 3), PDGFR α (Platelet-Derived Growth Factor Receptor), and PDGFR β. Unlike most RTK inhibitors, crenolanib is a type I mutant-specific inhibitor that preferentially binds to phosphorylated active kinases with the ‘DFG in’ conformation motif.[5]
CN 109678849

PATENT
WO/2022/060421CRENOLANIB FOR TREATING TRK KINASE ASSOCIATED PROLIFERATIVE DISORDERS
PATENT
WO/2022/060422CRENOLANIB FOR TREATING PAIN
PAPER
https://www.nature.com/articles/s41598-018-21839-3
PAPER
Chembiochem : a European journal of chemical biology (2019), 20(14), 1783-1788.
PATENT
CN 109678849
PATENT
WO 2018118598
https://patents.google.com/patent/WO2018118598A1/en
PAT
US 20170121321
PAT
CN 107382984
https://patents.google.com/patent/CN107382984A/en
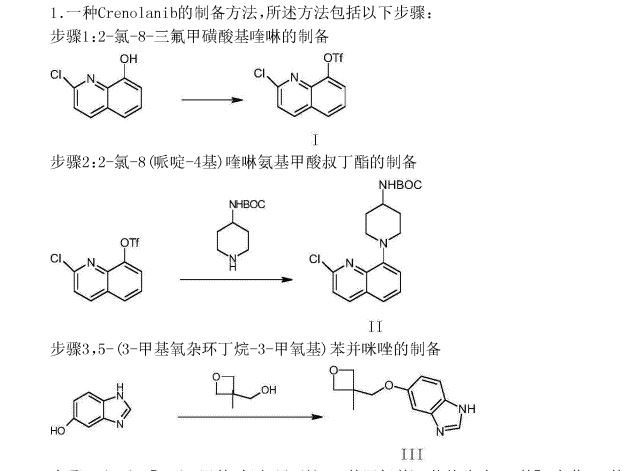
Embodiment is as follows:
The synthesis of the chloro- 8- trifluoromethanesulfonic acids base quinoline (Ι) of 2-
Compound 2- chloro-8-hydroxyquinolines 50g, DMF150ml, trifluoromethanesulfchloride chloride 53g, triethylamine 25g are added to 250ml In three-necked bottle, stir.Temperature control reacts 20~30h at 25~30 DEG C.After reaction completely, the solid of precipitation is filtered, filter cake is used Wash washing, 40 DEG C of forced air dryings, the chloro- 8- trifluoromethanesulfonic acids base benzimidazoles of gained off-white powder 2- in n-hexane 20ml × 3 83.39g yield 95.78%.
The synthesis of (base of piperidines -4) the quinoline t-butyl carbamates of 2- chloro- 8 (II)
BINAP 0.2g, toluene 70ml are added into 250ml three-necked bottles, temperature control stirs 1h at 20~25 DEG C.Added again into bottle The chloro- 8- trifluoromethanesulfonic acids base quinoline 10g of 2-, piperidin-4-yl t-butyl carbamate 6.41g, potassium carbonate 7.8g, stir lower by instead Answer liquid to be heated to 80 DEG C~100 DEG C, keep 20~30h of this thermotonus.TLC is detected, and whether reaction is complete.Reaction is complete, stops Only heat.20~30 DEG C are cooled to, dichloroethanes 50ml is added, adds diatomite to filter out the solid in reaction solution, filter cake second Acetoacetic ester 150ml is washed., 20~25 DEG C of stirring 8h.The solid separated out in solution is filtered out, filtrate is molten with 5% disodium hydrogen phosphate Liquid 2x50ml is washed.Organic phase is concentrated to dryness again, adds acetonitrile 50ml, 20~25 DEG C of 10~20h of stirring and crystallizing.Filtering analysis The solid gone out, 40 DEG C of forced air dryings obtain the tertiary fourth of yellow solid 10.69g, 2- chloro- 8 (piperidin-4-yl) benzimidazole carbamic acid Ester, yield 92.3%.
The synthesis of 5- (3- methy oxetane -3- methoxyl groups) benzimidazole (III)
Compound 3- methyl -3- oxetane methanols 30.77g, THF140ml, metallic sodium 6.95g are added to the necks of 250ml tri- In bottle, 66 DEG C of backflow 4h are heated under stirring, 55 DEG C is cooled to, then adds 5- hydroxybenzimidazole 40.4g, stir lower heat Backflow, react 20~24h.
Ethyl acetate 100ml is added into reaction bulb, 0.5h dissolvings are stirred at 30~50 DEG C, are then reduced to -5 DEG C, are added dropwise just Hexane 30ml, stirs 1h, and suction filtration obtains light yellow solid, 40 DEG C of dryings to constant weight, obtains 56.41 grams, yield 85.8%.
(1- { 2- [5- (3- methvl-oxetan -3- ylmethoxies)-benzimidazole -1- bases]-quinoline-8-yl }-piperazine The synthesis of pyridine -4- bases-t-butyl carbamate (IV)
II (50 grams), III (30.14 grams), potassium carbonate 80g, DIPHOS 4.3g, toluene 700ml, are added in 2L three-necked bottles, add Enter acid chloride 0.9g, stir.Stirring is lower to heat up, and temperature control reacts 24~30h at 80~100 DEG C.After the completion of reaction, it is cooled to 55 DEG C add dichloroethanes 700ml.10min is stirred, adds the solid in diatomite filtering reacting liquid, the filter cake chloroethenes of 500ml bis- Alkane rinses.Concentrate the filtrate to it is dry, add ethyl acetate 480ml, be heated to flowing back, be cooled to 20~25 DEG C of 10~20h of crystallization. The solid separated out is filtered, 50 DEG C of forced air dryings, obtains white solid, the amount of obtaining 70.51g, yield 93.90%.
(1- { 2- [5- (3- methvl-oxetan -3- ylmethoxies)-benzimidazole -1- bases]-quinoline-8-yl } -4- The synthesis of amino piperidine (V)
By compound (1- { 2- [5- (3- methvl-oxetan -3- ylmethoxies)-benzimidazole -1- bases]-quinoline -8- Base }-piperidin-4-yl-t-butyl carbamate 5g, caustic alcohol 2.8g, 2- methyltetrahydrofuran 30ml and water 0.08ml be added to In 100ml three-necked bottles, stir.The mixture is heated to flowing back, and stirs 3~4h under reflux.
TLC is detected, and reaction is complete.Stop heating, add purified water 60ml, extracting and demixing.Aqueous phase is extracted with 2 × 20ml of ethyl acetate Take, merge organic phase, washed with saturated nacl aqueous solution 20ml.Be concentrated under reduced pressure organic phase, and 30ml is added into condensate residue Ethyl acetate, in 20~25 DEG C of stirring and crystallizing 6h.The solid separated out is filtered, filtrate decompression is concentrated to dryness.Added into residue 24ml ethyl acetate, in 20~25 DEG C of 10~12h of stirring and crystallizing.Filter the solid separated out, dry white solid product, the amount of obtaining 3.68g, yield 90.3%.
1H NMR test (referring to accompanying drawing)
(d6-DMSO):δ 9.176 (s, 1H), δ 8.88-8.91 (d, 1H, J=8.7Hz), δ 8.51-8.53 (d, 1H, J= 9.0Hz), δ 8.13-8.15 (d, 1H, J=9.0Hz), δ 7.6 (d, 1H, J=7.5Hz), 7.49 (t, 1H, J=7.9Hz) 7.39 (d, 1H, J=2.4Hz), 7.29 (d, 1H, J=7.6Hz), 7.19 (dd, 1H, J=9.2hz, 2.5Hz) 4.56 (d, 2H, J= 5.6Hz), 4.34 (d, 2H, J=5.7Hz), 4.14 (s, 2H), 3.74 (d, 2H, J=10.1Hz), 2.77 (m, 3H), 1.91 (d, 2H, J=11.1Hz), 1.68 (m, 2H), 1.41 (s, 3H)


SET 2




///////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Background
Type III Receptor tyrosine kinase, including FLT3, PDGFRα and PDGFRβ, have been directly implicated in the pathogenesis of epithelial, mesenchymal, and hematological malignancies.[6]
Mutations of FLT3 comprise one of the most frequently identified types of genetic alterations in Angiomyolipoma.[7][8] Approximately one-third of AML patients present with a mutation in this gene.[9] The majority of these mutations result in constitutive activation of downstream signaling pathways and aberrant cell growth.[7] Mutations in FLT3 have also been reported in acute lymphoblastic leukemia (ALL)[10] and myelodysplastic syndrome (MDS).[11]
Activating mutations in PDGFRA have been detected in 5-12% of Gastrointestinal stromal tumor.[12] Fusion of PDGFRA has been found to be responsible for hematological malignances like hypereosinophilic syndrome.[13] The amplification of chromosome 4q12, the site of the PDGFRA gene[citation needed], has been identified in 13-29% of adult gliomas[citation needed] and in 29% to 36% of diffuse intrinsic pontine gliomas (DIPG)[citation needed], a subset of high-grade gliomas (HGG) in pediatric patients. Activation of PDGFRB, a third member of the type III RTK family, has been implicated in the development of chronic myelomonocytic leukemia due to the fusion of PDGFRB with the TEL gene.[13] Furthermore, PDGFB translocation to the COL1A1 gene locus has been identified to be responsible for dermatofibrosarcoma protuberans (DFSP).[13] In cancer cells, PDGFR promotes tumor development and migration via proto-oncogenic downstream mediators like AKT and MEK[citation needed]. In stromal fibroblasts, PDGFRα activation leads to local tissue invasion, production and secretion of VEGF, and elevated intratumoral interstitial pressure[citation needed]. In stromal pericytes, PDGFRβ activation mediates vascular stability.[13] Thus, either FLT3 or PDGF/PDGFR pathway is the primary driver of oncogenesis in the above malignancies and can be targeted by crenolanib therapy[citation needed].
Mechanism
FLT3: wild-type and mutant
Crenolanib inhibits both wild type FLT3 and its constitutively active mutations. In vitro studies have shown that crenolanib has low Kd for the FLT3 enzyme with constitutively activating internal tandem duplication (ITD) mutations and tyrosine kinase domain (TKD) mutations, D835H and D835Y, as compared to wild type. Crenolanib tightly binds to FLT3-ITD, FLT3-D835H and FLT3-D835Y with Kd of 0.74 nM, 0.4 nM, and 0.18 nM, respectively.[14] Crenolanib inhibits the phosphorylation of the FLT3-ITD receptor in transfected TF-1 cells and the FLT3-D835Y TKD mutation in transfected Ba/F3 cells at nanomolar IC50 concentrations of 1.3 nM and 8.8 nM, respectively.[15] Immunoblot experiments performed in the Molm14 FLT3-ITD positive cell line show that crenolanib inhibits downstream signaling of FLT3 at a concentration of 10 nM.[15] MTT assay measurements of crenolanib cytotoxicity evaluated in the FLT3-ITD expressing cell lines Molm14 and MV411, showed that crenolanib is toxic at IC50 concentrations of 7 nM and 8 nM, respectively.[15]
PDGFRα: wild-type and mutant
Crenolanib has been shown to inhibit PDGFRα with an IC50 of 0.4 ng/mL in porcine aortic epithelial cell lines. In Chinese hamster ovary (CHO) cells expressing PDGFRα, crenolanib inhibited the phosphorylation of wild type PDGFRα at an IC50 of 10 nM.[16] Additionally, crenolanib completely blocked PDGFRα phosphorylation and downstream AKT signaling at a concentration between 0.1 and 1 uM in Ink4a/Arf-/- mouse astrocytes transfected to stably co-express both human PDGFRα and PDGF AA.[17] The lung cancer cell line H1703, which is reported to have amplification of both PDGFRA (4q12) and PDGFC (4q32) genes on chromosome 4, and also overexpress PDGFRα, was sensitive to crenolanib with an IC50 of ~80 nM.[18] In CHO cells expressing an activating exon 18 (D842V) PDGFRα mutation, crenolanib was effective at an IC50 of 6nM and IC90 of 25nM. In addition, crenolanib also inhibited phosphorylation of the double mutants PDGFRα (V561D + D842V and T674I + D842V).[16]
PDGFRβ: wild-type
Crenolanib has been shown to inhibit PDGFRβ with an IC50 of 0.8 ng/mL in porcine aortic epithelial cell lines. Crenolanib inhibits the ability of recombinant PDGFRβ to phosphorylate a synthetic tyrosine substrate (poly-glutamic acid-tyrosine), with an IC50 of 0.4 ng/mL. Evaluation of the antitumor activity of crenolanib in a genetically engineered BSG DIPG mouse model showed that it is highly selective for PDGFRβ with an IC50 of 10 nM when measured by BrdU assay and 1.25 uM by MTT assay.
C-Kit: wild-type and mutant
Crenolanib has been shown to have IC50 and Kd values of 67 nM and 78 nM, respectively, for wild type c-KIT in in vitro assays[citation needed]. Similar assays show that crenolanib inhibits c-KIT activating mutations D816H and D816V with IC50 concentrations of 5.4 and 2.5 nM, respectively.[14][citation needed] Human bone marrow progenitor cell growth assays showed that crenolanib has modest effects on GM-CSF and BFUE driven colony formation at the IC50 concentration of 20 nM.[15]
Clinical
Phase I single-agent[19] and Phase Ib combination[20] studies have investigated the clinical pharmacology of crenolanib in patients with cancer. Pharmacokinetic and safety studies of Crenolanib administered alone or in combination with docetaxel with or without axitinib have been completed. Results suggest that Crenolanib is well tolerated as a single agent, and can also be safely combined with docetaxel and axitinib due to their non-overlapping toxicity profiles.
Clinical trials
- Clinical trial number NCT01229644 for “A Phase II Study of Crenolanib (CP-868,596), a Selective and Potent Inhibitor of PDGFR, for the Treatment of Adult Gliomas” at ClinicalTrials.gov
- Clinical trial number NCT01243346 for “Phase II Study of Crenolanib (CP-868,596), for the Treatment of Patients With Advanced Gastrointestinal Stromal Tumors With the D842-related Mutations and Deletions in the PDGFRA Gene” at ClinicalTrials.gov
- Clinical trial number NCT01393912 for “PDGFR Inhibitor Crenolanib in Children/Young Adults With Diffuse Intrinsic Pontine Glioma or Recurrent High-Grade Glioma” at ClinicalTrials.gov
- Clinical trial number NCT01522469 for “Phase II Study of Crenolanib in Subjects With Relapsed/Refractory AML With FLT3 Activating Mutations” at ClinicalTrials.gov
- Clinical trial number NCT01657682 for “A Phase II Study of Crenolanib in Relapsed/Refractory Acute Myeloid Leukemia Patients With FLT3 Activating Mutations” at ClinicalTrials.gov
References
- ^ “A Phase II Study of Crenolanib in Relapsed/Refractory Acute Myeloid Leukemia Patients With FLT3 Activating Mutations – Full Text View”. ClinicalTrials.gov. Retrieved 2014-04-08.
- ^ “Phase II Study of Crenolanib in Subjects With Relapsed/Refractory AML With FLT3 Activating Mutations – Full Text View”. ClinicalTrials.gov. Retrieved 2014-04-08.
- ^ “Phase II Study of Crenolanib (CP-868,596), for the Treatment of Patients With Advanced Gastrointestinal Stromal Tumors With the D842-related Mutations and Deletions in the PDGFRA Gene – Full Text View”. ClinicalTrials.gov. Retrieved 2014-04-08.
- ^ “PDGFR Inhibitor Crenolanib in Children/Young Adults With Diffuse Intrinsic Pontine Glioma or Recurrent High-Grade Glioma – Full Text View”. ClinicalTrials.gov. Retrieved 2014-04-08.
- ^ A. Ramachandran; H. Marshall; V. Jain. “CRENOLANIB, A NOVEL TYPE I, MUTANT -SPECIFIC INHIBITOR OF CLASS III RECEPTOR TYROSINE KINASES, PREFERENTIALLY BINDS TO PHOSPHORYLATED KINASES” (PDF). gistsupport.org. Retrieved 2014-04-08.
- ^ Lemmon, Mark A.; Schlessinger, Joseph (2010). “Cell Signaling by Receptor Tyrosine Kinases”. Cell. 141 (7): 1117–34. doi:10.1016/j.cell.2010.06.011. PMC 2914105. PMID 20602996.
- ^ Jump up to:a b Takahashi, S (2011-04-01). “Downstream molecular pathways of FLT3 in the pathogenesis of acute myeloid leukemia: biology and therapeutic implications”. J Hematol Oncol. 4: 13. doi:10.1186/1756-8722-4-13. PMC 3076284. PMID 21453545.
- ^ Cancer Genome Atlas Research Network; Ley, T. J.; Miller, C.; Ding, L.; Raphael, B. J.; Mungall, A. J.; Robertson, A.; Hoadley, K.; Triche Jr, T. J.; Laird, P. W.; Baty, J. D.; Fulton, L. L.; Fulton, R.; Heath, S. E.; Kalicki-Veizer, J.; Kandoth, C.; Klco, J. M.; Koboldt, D. C.; Kanchi, K. L.; Kulkarni, S.; Lamprecht, T. L.; Larson, D. E.; Lin, L.; Lu, C.; McLellan, M. D.; McMichael, J. F.; Payton, J.; Schmidt, H.; Spencer, D. H.; et al. (2013). “Genomic and Epigenomic Landscapes of Adult De Novo Acute Myeloid Leukemia”. New England Journal of Medicine. 368 (22): 2059–2074. doi:10.1056/NEJMoa1301689. ISSN 0028-4793. PMC 3767041. PMID 23634996.
- ^ “The Impact of FLT3 Mutations on the Development of Acute Myeloid Leukemias”. Hindawi.com. Retrieved 2014-04-08.
- ^ Xu, F; Taki, T; Yang, HW; Hanada, R; Hongo, T; Ohnishi, H; Kobayashi, M; Bessho, F; Yanagisawa, M; Hayashi, Y (2014-01-24). “Tandem duplication of the FLT3 gene is found in acute lymphoblastic leukaemia as well as acute myeloid leukaemia but not in myelodysplastic syndrome or juvenile chronic myelogenous leukaemia in children”. Br. J. Haematol. 105 (1): 155–62. doi:10.1111/j.1365-2141.1999.01284.x. PMID 10233379. S2CID 40898615.
- ^ Yokota, S; Kiyoi, H; Nakao, M; Iwai, T; Misawa, S; Okuda, T; Sonoda, Y; Abe, T; Kahsima, K; Matsuo, Y; Naoe, T (2014-01-24). “Internal tandem duplication of the FLT3 gene is preferentially seen in acute myeloid leukemia and myelodysplastic syndrome among various hematological malignancies. A study on a large series of patients and cell lines”. Leukemia. 11 (10): 1605–9. doi:10.1038/sj.leu.2400812. PMID 9324277.
- ^ Heinrich, M. C.; Corless, CL; Duensing, A; McGreevey, L; Chen, CJ; Joseph, N; Singer, S; Griffith, DJ; Haley, A; Town, A; Demetri, GD; Fletcher, CD; Fletcher, JA (2003). “PDGFRA Activating Mutations in Gastrointestinal Stromal Tumors”. Science. 299 (5607): 708–10. doi:10.1126/science.1079666. PMID 12522257. S2CID 11725958.
- ^ Jump up to:a b c d Östman, Arne; Heldin, Carl‐Henrik (2007). PDGF Receptors as Targets in Tumor Treatment. Advances in Cancer Research. Vol. 97. pp. 247–274. doi:10.1016/S0065-230X(06)97011-0. ISBN 9780120066971. PMID 17419949.
- ^ Jump up to:a b Muralidhara, C.; Ramachandran, A.; Jain, V. K. (2012). “Abstract 3683: Crenolanib, a novel Type I, mutant-specific inhibitor of Class III receptor tyrosine kinases, preferentially binds to phosphorylated kinases”. Cancer Research. 72 (8 Supplement): 3683. doi:10.1158/1538-7445.AM2012-3683.
- ^ Jump up to:a b c d Galanis, A.; Rajkhowa, T.; Muralidhara, C.; Ramachandran, A.; Levis, M. (2012). “Abstract 3660: Crenolanib: A next generation FLT3 inhibitor”. Cancer Research. 72 (8 Supplement): 3660. doi:10.1158/1538-7445.am2012-3660.
- ^ Jump up to:a b Heinrich, M. C.; Griffith, D.; McKinley, A.; Patterson, J.; Presnell, A.; Ramachandran, A.; Debiec-Rychter, M. (2012). “Crenolanib Inhibits the Drug-Resistant PDGFRA D842V Mutation Associated with Imatinib-Resistant Gastrointestinal Stromal Tumors”. Clinical Cancer Research. 18 (16): 4375–84. doi:10.1158/1078-0432.CCR-12-0625. PMID 22745105.
- ^ Yang, X.-L.; Mashimo, T.; Su, Y.; Vemireddy, V.; Guntipalli, P.; Ramachandran, A.; Chaudhary, P.; Mickey, B.; Hatanpaa, K.; Maher, E.; Bachoo, R. M. (2011). “Abstract 1111: Preclinical evaluation of CP868,596, a novel PDGFR Inhibitor for treatment of glioblastoma”. Cancer Research. 71 (8 Supplement): 1111. doi:10.1158/1538-7445.am2011-1111.
- ^ Peyton, M.; Chaudhary, P.; Ramachandran, A.; Minna, J. (2011). “Abstract 3601: CP-868,596, a highly potent and selective PDGFR TKI inhibits growth of PDGFR -driven lung cancer cells”. Cancer Research. 71 (8 Supplement): 3601. doi:10.1158/1538-7445.am2011-3601.
- ^ Lewis, N. L.; Lewis, L. D.; Eder, J. P.; Reddy, N. J.; Guo, F.; Pierce, K. J.; Olszanski, A. J.; Cohen, R. B. (2009). “Phase I Study of the Safety, Tolerability, and Pharmacokinetics of Oral CP-868,596, a Highly Specific Platelet-Derived Growth Factor Receptor Tyrosine Kinase Inhibitor in Patients with Advanced Cancers”. Journal of Clinical Oncology. 27 (31): 5262–9. doi:10.1200/jco.2009.21.8487. PMC 2773478. PMID 19738123.
- ^ Michael, M; Vlahovic, G; Khamly, K; Pierce, K J; Guo, F; Olszanski, A J (2010). “Phase Ib study of CP-868,596, a PDGFR inhibitor, combined with docetaxel with or without axitinib, a VEGFR inhibitor”. British Journal of Cancer. 103 (10): 1554–61. doi:10.1038/sj.bjc.6605941. PMC 2990584. PMID 20959830.
External links
- “PDGFR Inhibitor CP-868596 (Code C64639)”, National Cancer Institute Thesaurus.
- “PDGFR and Human Cancer” , AROG Pharmaceuticals LLC.
| Names | |
|---|---|
| IUPAC name1-(2-{5-[(3-methyloxetan-3-yl)methoxy]-1H-benzimidazol-1-yl}quinolin-8-yl)piperidin-4-amine | |
| Other namesCP-868,596; AR-868,596-26 | |
| Identifiers | |
| CAS Number | 670220-88-9 |
| 3D model (JSmol) | Interactive image |
| ChEBI | CHEBI:145365 |
| ChEMBL | ChEMBL2105728 ChEMBL2146086 |
| ChemSpider | 8541584 |
| IUPHAR/BPS | 7882 |
| KEGG | D10102 |
| PubChemCID | 10366136 |
| UNII | LQF7I567TQ |
| CompTox Dashboard (EPA) | DTXSID50985873 |
| showInChI | |
| showSMILES | |
| Properties | |
| Chemical formula | C26H29N5O2 |
| Molar mass | 443.551 g·mol−1 |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
| Infobox references |
//////////Crenolanib, UNII-LQF7I567TQ, креноланиб , كرينولانيب , 克拉尼布, CP-868,596-26, AR-868,596-26
CC1(COc2ccc3c(c2)ncn3c4ccc5cccc(N6CCC(N)CC6)c5n4)COC1.OS(=O)(=O)c7ccccc7

NEW DRUG APPROVALS
ONE TIME
$10.00
Ganaxolone
Ganaxolone
- Molecular FormulaC22H36O2
- Average mass332.520 Da
- CCD-1042
FDA APPROVED 3/18/2022, Ztalmy
To treat seizures in cyclin-dependent kinase-like 5 deficiency disorder
Ganaxolone, sold under the brand name Ztalmy, is a medication used to treat seizures associated with cyclin-dependent kinase-like 5 (CDKL5) deficiency disorder (CDD).[1][2]
Ganaxolone was approved for medical use in the United States in March 2022.[1]
Ganaxolone is the 3β-methylated synthetic analog of allopregnanolone; it belongs to a class of compounds referred to as neurosteroids. Ganaxolone is an allosteric modulator of GABAA receptors acting through binding sites which are distinct from the benzodiazepine binding site. It has activity in a broad range of animal models of epilepsy. Ganaxolone has been shown to be well tolerated in adults and children. In early phase II studies, Ganaxolone has been shown to have activity in adult patients with partial-onset seizures and epileptic children with history of infantile spasms. It is currently undergoing further development in infants with newly diagnosed infantile spasms, in women with catamenial epilepsy, and in adults with refractory partial-onset seizures.
Ganaxolone is in phase III clinical studies for the treatment of partial seizures in adults. Phase II clinical trials is ongoing for treatment of uncontrolled seizures in PCDH19 female pediatric epilepsy and Fragile X syndrome.
Ganaxolone was originally developed by CoCensys (aquired by Purdue Pharma). In 2003, Marinus Pharmaceuticals obtained the compound from Purdue Pharma.
In 2015, it was granted as orphan drug designation for the treatment of PCDH19 female epilepsy.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019209850&_cid=P10-L0YZTI-42413-1
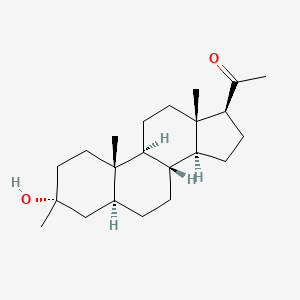
In an embodiment, the disclosure provides a method for using pregnenolone to make 21-OH ganaxolone and other intermediary compounds which are useful for preparing neurosteroid derivatives. The method of making 21-OH ganaxolone is shown below in Route 1.
Route 1
Referring to Route 1, Synthesis of 1-((3S,8R,10S,13S,14S,17S)-3-hydroxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)ethenone :
<a name="
Pregnenolone (3.17 g, 10 mmol) was dissolved in 30 mL of THF and 5 mL of acetic acid. To it, 10% W/C (0.3 g) was added. The resulting mixture was shaken under 60 psi hydrogen at 60°C overnight. It was filtered through a Celite ® pad and concentrated to give 3.2 g of the desired product (100%). 1 H NMR (400 MHz, CDCl3) δ 3.58 (tt, J = 11.0, 4.8 Hz, 1H), 2.50 (t, J = 9.0 Hz, 1H), 2.19 – 2.11 (m, 2H), 2.09 (s, 3H ), 2.06 – 1.93 (m, 2H), 1.85 – 1.75 (m, 1H), 1.74 -1.50 (m, 6H), 1.47 – 1.04 (m, 9H), 1.04 – 0.82 (m, 2H), 0.79 (s , 3H), 0.72 – 0.61 (m, 1H), 0.58 (d, J = 2.4 Hz, 3H).
[0107] Synthesis of (8R,10S,13S,14S,17S)-l7-acetyl-l0,l3-dimethyltetradecahydro-1H-cyclopenta[a]phenanthren-3(2H)-one:
To a solution of the above product (1-((3S,8R,10S,13S,14S,17S)-3-hydroxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)ethanone, 3.2 g, 10 mmol) in 40 mL of THF and 10 mL of acetic acid was added NaBr (1.03 g, 0.1 eq.). It was cooled in an ice bath and was followed by the dropwise addition of NaOCl (82 mL, 10-15%, 18 eq.) at such a rate that the internal temperature was maintained <40 °C. After addition, it was stirred at room temperature for 2h. Thin layer chromatography (TLC) indicated it was complete. The mixture was diluted with dichloromethane and layers were separated. The organic layer was washed with Na 2 S 2 O 3 (10% aq.), H 2 O, NaHCO 3 (sat.) and NaCl (sat.). Drying over Na 2SO 4 and concentration afforded 3.8 g of the crude product, which was recrystallized from CH 2 Cl 2 /Hex to give 2.57 g of the desired product (81%). 1 H NMR (400 MHz, CDC13): 2.51 (t, 1H), 2.2-2.4 (m, 3H), 2.1-2.2 (m, 1H), 2.10 (s, 3H), 1.98-2.01 (m, 2H) , 1.6-1.7 (m, 4H), 1.55-1.6 (m, 1H), 1.3-1.4 (m, 7H), 1.1-1.2 (m, 2H), 0.99 (s, 3H), 0.95-0.98 (m, 1H), 0.75-0.78 (m, 1H), 0.62 (s, 3H).
Synthesis of 1-((2’R,8R,10S,13S,14S,17S)-10,11-dimethylhexadecahydrospiro[cyclopenta[a]phenanthrene-3,2′-oxiran]-17-yl)ethanone.<a name="
Under argon, trimethyl sulfoxonium iodide (2.6 g, 1.7 eq.) and sodium t-butoxide (1.18 g, 1.75 eq.) in DMSO (20 mL) was heated at 65 °C for 2h. After it was cooled to RT, the above di-ketone ((8R, 10S, 13 S, 14S, 17S)-17-acetyl- 10,13 -dimethyl tetradecahy dro-1H-cyclopenta[a]phenanthren-3(2H) -one, 2.2 g, 7 mmol) was added scoop-wise so that the internal temperature was maintained between 25-35 °C. The resulting mixture was stirred at RT for 2h. After TLC indicated it was complete, it was quenched with 30 mL of H 2 O, stirred for 10 min and was kept in fridge overnight. The precipitate was filtered, washed with 20 mL of (4:1 of H 2 O /MeOH), dried to give 94% of the desired product (W = 2.17 g). 1H NMR (400 MHz, CDC13) δ 2.63 (s, 2H), 2.53 (t, J = 8.9 Hz, 1H), 2.20 – 2.13 (m, 1H), 2.11 (s, 3H), 2.10 – 1.95 (m, 2H), 1.87 (dd, J = 13.9, 13.1 Hz, 1H), 1.76 – 1.59 (m, 4H), 1.58 – 1.48 (m, 1H), 1.48 – 1.24 (m, 5H), 1.24 – 1.07 (m, 3H), 1.02 – 0.87 (m, 2H), 0.86 (dd, J = 3.7, 2.2 Hz, 1H), 0.84 (s, 3H), 0.81 – 0.74 (m, 1H), 0.61 (s, 3H).
[0109] Synthesis of 1-((3R,8R,10S,13S,14S,17S)-3-hydroxy-3,10,13-trimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)ethanone (ganaxolone) .
To a solution of the above epoxide (1.5 g, 4.56 mmol) in 15 mL of THF and 15 mL of MeOH were added Nal (1.02 g, 1.5 eq.) and HO Ac (0.6 mL, 2.2 eq.). The resulting mixture was heated at 65°C for 2h. After TLC indicated that the epoxy was completely converted to an iodo compound, it was cooled to RT. Sodium acetate (1.02 g, 2.7 eq.) and 150 mg of 10% Pd/C were added and the mixture was transferred to a hydrogenation bottle with the aid of MeOH (10 mL) and was hydrogenated under 50 psi hydrogen over the weekend. It was filtered through<a name="Celite ® and the filtrate was concentrated. The residue was then partitioned between dichloromethane and water. The aqueous solution was extracted twice with CH 2 Cl 2 and the combined organic layers were washed with brine, dried over Na 2 SO 4 and concentrated. The Biotage flash purification with 10-35% EtOAc in hexane to give 0.5 g of the desired product (33%).
The synthesis was repeated with 1.1 g of the epoxy and 1 g of the product was obtained (90%).
Both lots of product were combined and recrystallized with CH 2 Cl 2 and hexane to give 0.522 g of the product with 96.6% purity by HPLC. 1 H NMR (400 MHz, Chloroform-d) δ 2.51 (t, J = 8.9 Hz, 1H), 2.18 – 2.10 (m, 1H), 2.09 (s, 3H), 2.01 – 1.93 (m, 1H), 1.72 – 1.57 (m, 4H), 1.57 – 1.41 (m, 5H), 1.41 – 1.30 (m, 3H), 1.30 – 1.20 (m, 3H), 1.18 (s, 3H), 1.17 – 1.09 (m, 2H) , 1.00 – 0.85 (m, 1H), 0.78 (ddd, J = 10.6, 7.7, 5.4 Hz, 1H), 0.73 (d, J = 0.6 Hz, 3H), 0.58 (s, 3H). UV: Absorbances at 206.2 nm. TLC: (Silica Gel plates) 20% EtOAc/Hexane; R f = 0.50. HPLC: Sunfire C18 5m 250 x 4.6mm; flow 1.0 mL/min; Waters 996 PDA detection at 210 nm; solvent 80% Acetonitrile in H 2 O (0.1% formic acid) over 30 min; retention time 8.24 min; 96.6%.
SYN
https://patents.google.com/patent/WO2016164763A1/en
SYN
US3953429.
 J. Med. Chem. 1997, 40, 61-72.
J. Med. Chem. 1997, 40, 61-72.https://pubs.acs.org/doi/10.1021/jm960021x
Two naturally occurring metabolites of progesterone, 3α-hydroxy-5α- and 5β-pregnan-20-one (1 and 2), are potent allosteric modulators of the GABAA receptor. Their therapeutic potential as anxiolytics, anticonvulsants, and sedative/hypnotics is limited by rapid metabolism. To avoid these shortcomings, a series of 3β-substituted derivatives of 1 and 2 was prepared. Small lipophilic groups generally maintain potency in both the 5α- and 5β-series as determined by inhibition of [35S]TBPS binding. In the 5α-series, 3β-ethyl, -propyl, -trifluoromethyl and -(benzyloxy)methyl, as well as substituents of the form 3β-XCH2, where X is Cl, Br, or I or contains unsaturation, show limited efficacy in inhibiting [35S]TBPS binding. In the 5β-series, the unsubstituted parent 2 is a two-component inhibitor, whereas all of the 3β-substituted derivatives of 2 inhibit TBPS via a single class of binding sites. In addition, all of the 3-substituted 5β-sterols tested are full inhibitors of [35S]TBPS binding. Electrophysiological measurements using α1β2γ2L receptors expressed in oocytes show that 3β-methyl- and 3β-(azidomethyl)-3α-hydroxy-5α-pregnan-20-one (6 and 22, respectively) are potent full efficacy modulators and that 3α-hydroxy-3β-(trifluoromethyl)-5α-pregnan-20-one (24) is a low-efficacy modulator, confirming the results obtained from [35S]TBPS binding. These results indicate that modification of the 3β-position in 1 and 2 maintains activity at the neuroactive steroid site on the GABAA receptor. In animal studies, compound 6 (CCD 1042) is an orally active anticonvulsant, while the naturally occurring progesterone metabolites 1 and 2 are inactive when administered orally, suggesting that 3β-substitution slows metabolism of the 3-hydroxyl, resulting in orally bioavailable steroid modulators of the GABAA receptor.
PATENT
WO9303732A1.,
https://patents.google.com/patent/WO1993003732A1/nl
SYN
| GB 1380248 |
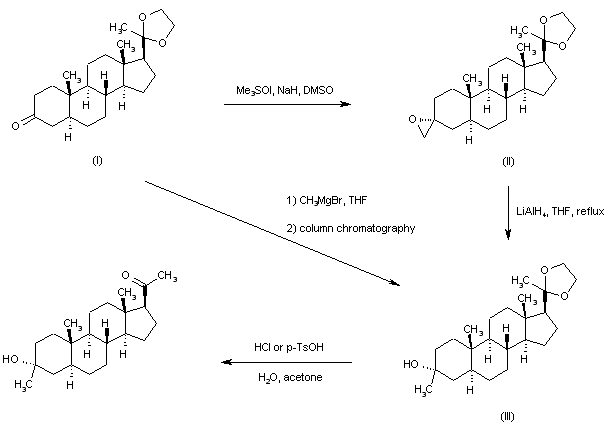
Addition of the sulfur ylide generated from trimethylsulfoxonium iodide and NaH to the 20-ethylene ketal of pregnane-3,20-dione (I) furnished the spiro oxirane derivative (II). This was reduced to the tertiary alcohol (III) by means of LiAlH4 in refluxing THF. Then, acid hydrolysis of the ethylene ketal function of (III) provided the title compound. Alternatively, the intermediate ketal (III) was prepared by addition of methylmagnesium bromide to ketone (I), followed by chromatographic separation of the resultant mixture of 3-alpha and 3-beta methyl adducts.
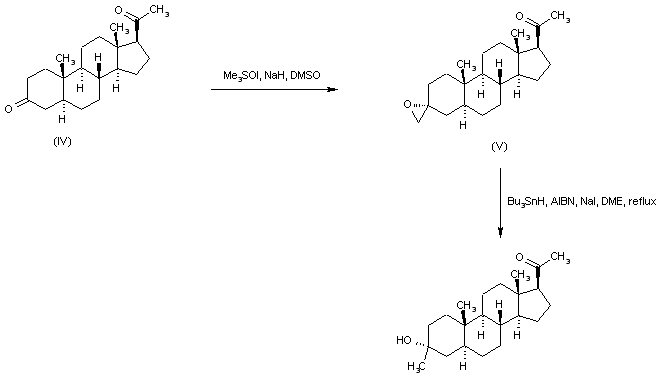
Starting from the unprotected diketone (IV), selective addition of dimethyloxosulfonium methylide to the 3 keto group furnished oxirane (V). This was then reduced to the title alcohol by treatment with tributylstannyl hydride and AIBN.
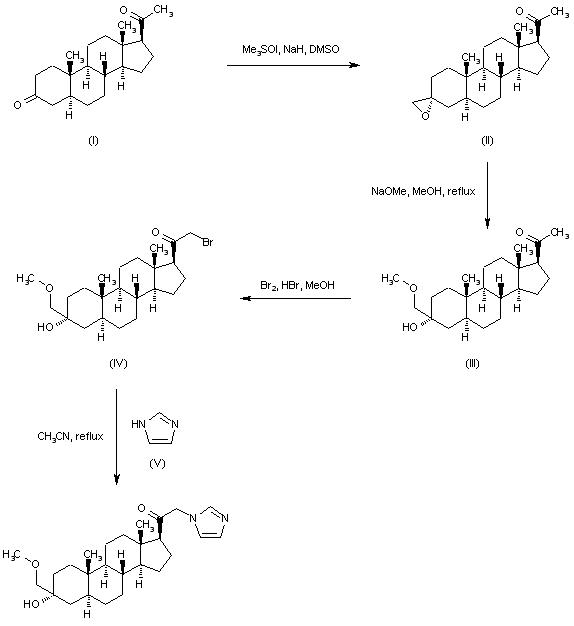
Regioselective addition of dimethylsulfoxonium methylide to 5-alpha-pregnane-3,20-dione (I) gave the epoxide (II). Opening of the epoxide ring of (II) with sodium methoxide produced the hydroxy ether (III). Bromination of (III) with Br2 in the presence of a catalytic amount of HBr afforded bromo ketone (IV). This was then condensed with imidazole (V) in refluxing acetonitrile to furnish the title compound.
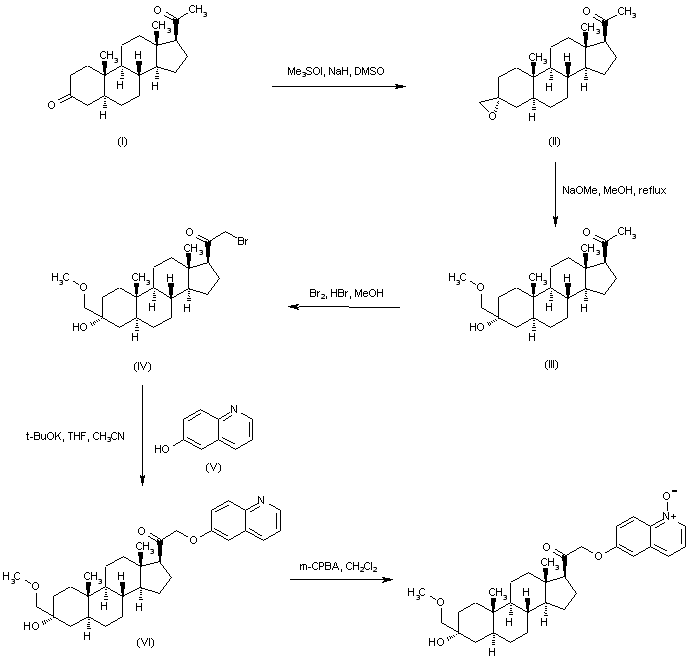
Regioselective addition of dimethylsulfoxonium methylide to 5-alpha-pregnane-3,20-dione (I) gave the epoxide (II). Opening of the epoxide ring of (II) with sodium methoxide produced the hydroxy ether (III). Bromination of (III) with Br2 in the presence of a catalytic amount of HBr afforded bromo ketone (IV). This was then condensed with 6-hydroxyquinoline (V) in the presence of potassium tert-butoxide to furnish the quinolinyl ether (VI). The quinoline ring was then oxidized with m-chloroperbenzoic acid, yielding the title N-oxide.
3. WO9318053A1.
4. WO9427608A1.
 WO2011019821A2 / US8362286B2.
WO2011019821A2 / US8362286B2.///////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Pharmacology
Mechanism of action
The exact mechanism of action for ganaxolone is unknown; however, results from animal studies suggest that it acts by blocking seizure propagation and elevating seizure thresholds.[3][4]
Ganaxolone is thought to modulate both synaptic and extrasynaptic GABAA receptors to normalize over-excited neurons.[2] Ganaxolone’s activation of the extrasynaptic receptor is an additional mechanism that provides stabilizing effects that potentially differentiates it from other drugs that increase GABA signaling.[2]
Ganaxolone binds to allosteric sites of the GABAA receptor to modulate and open the chloride ion channel, resulting in a hyperpolarization of the neuron.[2] This causes an inhibitory effect on neurotransmission, reducing the chance of a successful action potential (depolarization) from occurring.[2][3][4]
Chemistry
ResearchGanaxolone is a synthetic pregnane steroid. Other pregnane neurosteroids include alfadolone, alfaxolone, allopregnanolone (brexanolone), hydroxydione, minaxolone, pregnanolone (eltanolone), and renanolone, among others.
Ganaxolone is being investigated for potential medical use in the treatment of epilepsy. It is well tolerated in human trials, with the most commonly reported side effects being somnolence (sleepiness), dizziness, and fatigue.[5] Trials in adults with focal onset seizures and in children with infantile spasms have recently been completed.[6][7] There are ongoing studies in patients with focal onset seizures, PCDH19 pediatric epilepsy, and behaviors in Fragile X syndrome.[6][7]
Ganaxolone has been shown to protect against seizures in animal models,[3][4] and to act a positive allosteric modulator of the GABAA receptor.[2][8]
Clinical trials
The most common adverse events reported across clinical trials have been somnolence (sleepiness), dizziness, and fatigue.[5] In 2015, the MIND Institute at the University of California, Davis, announced that it was conducting, in collaboration with Marinus Pharmaceuticals, a randomized, placebo-controlled, Phase 2 clinical trial evaluating the effect of ganaxolone on behaviors associated with Fragile X syndrome in children and adolescents.[9][10][11]
References
- ^ Jump up to:a b c https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/215904s000lbl.pdf
- ^ Jump up to:a b c d e f Carter RB, Wood PL, Wieland S, Hawkinson JE, Belelli D, Lambert JJ, White HS, Wolf HH, Mirsadeghi S, Tahir SH, Bolger MB, Lan NC, Gee KW (March 1997). “Characterization of the anticonvulsant properties of ganaxolone (CCD 1042; 3alpha-hydroxy-3beta-methyl-5alpha-pregnan-20-one), a selective, high-affinity, steroid modulator of the gamma-aminobutyric acid(A) receptor”. The Journal of Pharmacology and Experimental Therapeutics. 280 (3): 1284–95. PMID 9067315.
- ^ Jump up to:a b c Kaminski RM, Livingood MR, Rogawski MA (July 2004). “Allopregnanolone analogs that positively modulate GABA receptors protect against partial seizures induced by 6-Hz electrical stimulation in mice”. Epilepsia. 45 (7): 864–7. doi:10.1111/j.0013-9580.2004.04504.x. PMID 15230714. S2CID 21974013.
- ^ Jump up to:a b c Reddy DS, Rogawski MA (May 2010). “Ganaxolone suppression of behavioral and electrographic seizures in the mouse amygdala kindling model”. Epilepsy Research. 89 (2–3): 254–60. doi:10.1016/j.eplepsyres.2010.01.009. PMC 2854307. PMID 20172694.
- ^ Jump up to:a b Monaghan EP, Navalta LA, Shum L, Ashbrook DW, Lee DA (September 1997). “Initial human experience with ganaxolone, a neuroactive steroid with antiepileptic activity”. Epilepsia. 38 (9): 1026–31. doi:10.1111/j.1528-1157.1997.tb01486.x. PMID 9579942. S2CID 27584114.
- ^ Jump up to:a b Nohria V, Giller E (January 2007). “Ganaxolone”. Neurotherapeutics. 4 (1): 102–5. doi:10.1016/j.nurt.2006.11.003. PMC 7479704. PMID 17199022.
- ^ Jump up to:a b Pieribone VA, Tsai J, Soufflet C, Rey E, Shaw K, Giller E, Dulac O (October 2007). “Clinical evaluation of ganaxolone in pediatric and adolescent patients with refractory epilepsy”. Epilepsia. 48 (10): 1870–4. doi:10.1111/j.1528-1167.2007.01182.x. PMID 17634060. S2CID 24656918.
- ^ Reddy DS, Rogawski MA (December 2000). “Chronic treatment with the neuroactive steroid ganaxolone in the rat induces anticonvulsant tolerance to diazepam but not to itself”. The Journal of Pharmacology and Experimental Therapeutics. 295 (3): 1241–8. PMID 11082461.
- ^ “Fragile X Research and Treatment Center: Clinical Research Studies” (PDF). UC Davis MIND Institute. 10 February 2015. Archived from the original (PDF) on 5 June 2015. Retrieved 27 January 2016.
- ^ “Ganaxolone Treatment in Children With Fragile X Syndrome”. Clinicaltrials.gov. 7 November 2012. Retrieved 27 January 2016.
- ^ “UC Davis Health System. UC Davis researchers win $3 million grant from U.S. Congress to study fragile X” (Press release). UC Davis Health System. 8 February 2011. Archived from the original on 3 February 2016. Retrieved 27 January 2016.
External links
- “Ganaxolone”. Drug Information Portal. U.S. National Library of Medicine.
 | |
| Clinical data | |
|---|---|
| Trade names | Ztalmy |
| Other names | GNX; CCD-1042; 3β-Methyl-5α-pregnan-3α-ol-20-one; 3α-Hydroxy-3β-methyl-5α-pregnan-20-one |
| License data | |
| Routes of administration | By mouth |
| Drug class | Neurosteroid |
| ATC code | |
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.210.937 |
| Chemical and physical data | |
| Formula | C22H36O2 |
| Molar mass | 332.528 g·mol−1 |
| 3D model (JSmol) | |
| | |
////////////Ganaxolone, ZTALMY, FDA 2022, APPROVALS 2022, CCD 1042
[H][C@@]12CC[C@H](C(C)=O)[C@@]1(C)CC[C@@]1([H])[C@@]2([H])CC[C@@]2([H])C[C@](C)(O)CC[C@]12C
https://doi.org/10.1021/acs.jmedchem.3c02374
J. Med. Chem. 2024, 67, 4376−4418
Ganaxolone (Ztalmy). Ganaxolone (10), a first-in class medication, is a neuroactive steroid gamma-aminobutyric acid (GABA) A receptor positive modulator indicated for the treatment of seizures associated with cyclin-dependent kinase like 5 (CDKL5) deficiency disorder (CDD) in patients 2 years
of age and older.
CDKL5 deficiency disorder is a rare neurodevelopmental condition resulting from pathogenic variants in the CDKL5 gene with an incidence rate ranging from 1 in 40,000 to 1 in 60,000 newborns.72 While CDKL5
deficiency is rare, it represents one of the most common forms of genetic epilepsy. 73,74
The synthesis of ganaxolone was previously described in the literature in 1976.75 However, the synthesis illustrated in Scheme 19 was recently published and outlines a manufactory process for ganaxolone.
The approach began with protection of the ketone functional group in pregnanolone 10.1, a naturally occurring steroid. Using acid catalysis, the ketal was formed with refluxing ethylene glycol in toluene, and after neutralizing the acidic solution, ketal 10.2 was obtained in 88% yield. Multiple options for the oxidation of 10.2 to form ketone 10.3 were provided such as Swern, Dess-Martin, and TPAP conditions. The oxidative procedure described in Scheme 19 illustrates the use of calcium hypochlorite and TEMPO to
form ketone 10.3. Next, methylation was accomplished by addition of MeMgBr in the presence of LiCl and FeCl3 to provide tertiary alcohol 10.4. Finally, the acetal protecting group was removed by treatment with iodine in DCM and acetone to provide ganaxolone 10 in 98% yield over the last two steps.
(70) Marinus Pharmaceuticals Inc.: ZTALMY® (ganaxolone)
[package insert]. https://www.accessdata.fda.gov/drugsatfda_docs/
label/2022/215904s000lbl.pdf, (accessed June 6, 2023).
(71) Carter, R. B.; Wood, P. L.; Wieland, S.; Hawkinson, J. E.;
Belelli, D.; Lambert, J. J.; White, S. H.; Wolf, H. H.; Mirsadeghi, S.;
Tahir, S. H.; Bolger, M. B.; Lan, N. C.; Gee, K.-W. Characterization of
the anticonvulsant properties of ganaxolone (CCD 1042; 3-alpha
hydroxy-3beta-methyl-5alpha-pregnan-20-one), a selective, high-affin
ity, steroid modulator of the gamma-aminobutyric acid(A) receptor. J.
Pharmacol. Exp. Ther. 1997, 280, 1284−1295.
(72) Jakimiec, M.; Paprocka, J.; Smigiel, R. CDKL5 deficiency
disorder-A complex epileptic encephalopathy. Brain Sci. 2020, 10,
107.
(73) Cook, M. C.; Lawrence, R.; Phillipps, G. H.; Hunter, A. C.;
Newall, C. E.; Stephenson, L.; Weir, N. G. Anaesthetic steroids of the
androstance and pregnane series. U.S. Patent US 3,953,429, 1976.
(74) Hogenkamp, D. J.; Tahir, S. H.; Hawkinson, J. E.; Upasani, R.
B.; Alauddin, M.; Kimbrough, C. L.; Acosta-Burruel, M.; Whittemore,
E. R.; Woodward, R. M.; Lan, N. C.; et al. Synthesis and in vitro
activity of 3 beta-substituted-3 alpha-hydroxypregnan-20-ones:
allosteric modulators of the GABA-A receptor. J. Med. Chem. 1997,
40, 61−72.
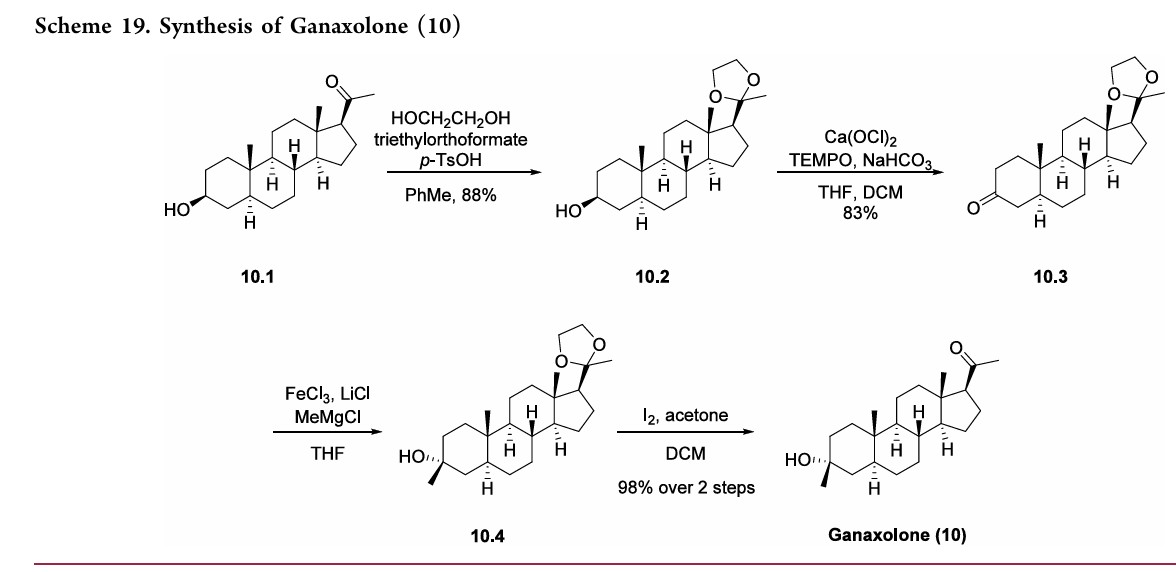
Ciltacabtagene autoleucel
Ciltacabtagene autoleucel
FDA APPROVED, 2022/2/28,
| Carvykti |
Treatment of multiple myeloma
- JNJ-68284528
- LCAR-B38M CAR-T cells
Ciltacabtagene autoleucel is a BCMA-directed CAR T-cell therapy used in the treatment of relapsed or refractory multiple myeloma in previously treated patients.
U.S. FDA Approves CARVYKTI™ (ciltacabtagene autoleucel), Janssen’s First Cell Therapy, a BCMA-Directed CAR-T Immunotherapy for the Treatment of Patients with Relapsed or Refractory Multiple Myeloma
In the pivotal clinical study, 98 percent of patients with relapsed or refractory multiple myeloma responded to a one-time treatment with ciltacabtagene autoleucel and 78 percent of patients who responded experienced a stringent complete response
HORSHAM, Pa., February 28, 2022 – The Janssen Pharmaceutical Companies of Johnson & Johnson announced today the U.S. Food and Drug Administration (FDA) has approved CARVYKTI™ (ciltacabtagene autoleucel; cilta-cel) for the treatment of adults with relapsed or refractory multiple myeloma (RRMM) after four or more prior lines of therapy, including a proteasome inhibitor, an immunomodulatory agent, and an anti-CD38 monoclonal antibody.1 The approval is based on data from the pivotal CARTITUDE-1 study, which included patients who had received a median of six prior treatment regimens (range, 3-18), and had previously received a proteasome inhibitor, an immunomodulatory agent and an anti-CD38 monoclonal antibody.1 In December 2017, Janssen entered into an exclusive worldwide license and collaboration agreement with Legend Biotech USA, Inc. to develop and commercialize ciltacabtagene autoleucel.
CARVYKTI™ is a chimeric antigen receptor T-cell (CAR-T) therapy featuring two B-cell maturation antigen (BCMA)-targeting single domain antibodies.1 In the pivotal CARTITUDE-1 study, one-time treatment with ciltacabtagene autoleucel resulted in deep and durable responses, with 98 percent (95 percent Confidence Interval [CI], 92.7-99.7) of patients with RRMM responding to therapy (98 percent overall response rate [ORR] (n=97).1 Notably, 78 percent (95 percent CI, 68.8-86.1) of the patients achieving this level of response (n=76) experienced a stringent complete response (sCR), a measure in which a physician is unable to observe any signs or symptoms of disease via imaging or other tests after treatment.1 At a median of 18 months follow-up, median duration of response (DOR) was 21.8 months.1
CARVYKTI™ is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called the CARVYKTI™ REMS Program.1 The Safety Information for CARVYKTI™ includes a Boxed Warning regarding Cytokine Release Syndrome (CRS), Immune Effector Cell-Associated Neurotoxicity Syndrome (ICANS), Parkinsonism and Guillain-Barré syndrome, hemophagocytic lymphohistiocytosis/macrophage activation syndrome (HLH/MAS), and prolonged and/or recurrent cytopenias.1 Warnings and Precautions include prolonged and recurrent cytopenias, infections, hypogammaglobulinemia, hypersensitivity reactions, secondary malignancies, and effects on ability to drive and use machines.1 The most common adverse reactions (≥20 percent) are pyrexia, CRS, hypogammaglobulinemia, hypotension, musculoskeletal pain, fatigue, infections-pathogens unspecified, cough, chills, diarrhea, nausea, encephalopathy, decreased appetite, upper respiratory tract infection, headache, tachycardia, dizziness, dyspnea, edema, viral infections, coagulopathy, constipation, and vomiting.1
“We are committed to harnessing our science, deep disease understanding and capabilities to bring forward cell therapies like CARVYKTI as we continue to focus on our ultimate goal of delivering a cure for multiple myeloma,” said Peter Lebowitz, M.D., Ph.D., Global Therapeutic Area Head, Oncology, Janssen Research & Development, LLC. “We extend our sincere gratitude to the patients, their families and the teams of researchers and study centers who have participated in the clinical study of CARVYKTI and enabled today’s approval.”
Multiple myeloma is an incurable blood cancer that affects a type of white blood cell called plasma cells, which are found in the bone marrow. 2 Despite the development of additional treatment options in recent years, most people living with multiple myeloma face poor prognoses after experiencing disease progression following treatment with three major therapy classes, which include an immunomodulatory agent, a proteasome inhibitor and an anti-CD38 monoclonal antibody. 3
“The responses in the CARTITUDE-1 study showed durability over time and resulted in the majority of heavily pretreated patients achieving deep responses after 18-month follow-up,” said Sundar Jagannath, M.D.†, Director of the Center of Excellence for Multiple Myeloma and Professor of Medicine, Hematology and Medical Oncology, at The Tisch Cancer Institute at the Icahn School of Medicine at Mount Sinai, and principal study investigator. “The approval of cilta-cel provides physicians an immunotherapy treatment option that offers patients an opportunity to be free from anti-myeloma therapies for a period of time.”
As a personalized medicine, CARVYKTI™ treatment requires extensive training, preparation, and certification to ensure a positive experience for patients. Through a phased approach, Janssen and Legend Biotech will activate a limited network of certified treatment centers as the company works to scale its production capacity and increase the availability of CARVYKTI™ throughout the U.S. in 2022 and beyond, to ensure that we can provide CARVYKTI™ treatment to oncologists and their patients in a reliable and timely manner.
“This approval of Janssen’s first cell therapy is a testament to our continuing commitment in oncology to deliver new therapeutic options and drive toward our vision of the elimination of cancer,” said Mathai Mammen, M.D., Ph.D., Executive Vice President, Pharmaceuticals, Janssen Research & Development, LLC, Johnson & Johnson. “Today’s approval underscores our determination to develop therapies that can help patients living with what remains an intractable blood cancer today and at the same time offer hope for the future.”
The longer-term efficacy and safety profile of ciltacabtagene autoleucel is being assessed in the ongoing CARTITUDE-1 study. Two-year follow-up results recently presented at the American Society of Hematology (ASH) 2021 Annual Meeting showed that 98 percent of patients treated with ciltacabtagene autoleucel for RRMM responded to therapy (98 percent overall response rate [ORR] (n=97), and a majority of patients achieving sustained depth of response with 83 percent of patients achieving an sCR at the 22-month follow-up.4
About CARVYKTI™ (ciltacabtagene autoleucel)
CARVYKTI™ is a BCMA-directed, genetically modified autologous T-cell immunotherapy, which involves reprogramming a patient’s own T-cells with a transgene encoding a chimeric antigen receptor (CAR) that identifies and eliminates cells that express the B-cell maturation antigen (BCMA). BCMA is primarily expressed on the surface of malignant multiple myeloma B-lineage cells, as well as late-stage B-cells and plasma cells. The CARVYKTI™ CAR protein features two BCMA-targeting single domain antibodies designed to confer high avidity against human BCMA. Upon binding to BCMA-expressing cells, the CAR promotes T-cell activation, expansion, and elimination of target cells.1
In December 2017, Janssen Biotech, Inc. entered into an exclusive worldwide license and collaboration agreement with Legend Biotech USA, Inc. to develop and commercialize ciltacabtagene autoleucel.
In April 2021, Janssen announced the submission of a Marketing Authorisation Application to the European Medicines Agency seeking approval of CARVYKTI™ for the treatment of patients with relapsed and/or refractory multiple myeloma. In addition to a U.S. Breakthrough Therapy Designation granted in December 2019, ciltacabtagene autoleucel received a Breakthrough Therapy Designation in China in August 2020. Janssen also received an Orphan Drug Designation for CARVYKTI™ from the U.S. FDA in February 2019, and from the European Commission in February 2020.
About the CARTITUDE-1 Study
CARTITUDE-1 (NCT03548207) is an ongoing Phase 1b/2, open-label, multi-center study evaluating ciltacabtagene autoleucel for the treatment of patients with relapsed or refractory multiple myeloma, who previously received a proteasome inhibitor (PI), an immunomodulatory agent (IMiD) and an anti-CD38 monoclonal antibody, and who had disease progression on or after the last regimen. All patients in the study had received a median of six prior treatment regimens (range, 3-18). Of the 97 patients enrolled in the trial, 99 percent were refractory to the last line of treatment and 88 percent were triple-class refractory, meaning their cancer did not respond, or no longer responds, to an IMiD, a PI and an anti-CD38 monoclonal antibody.1
About Multiple Myeloma
Multiple myeloma is an incurable blood cancer that affects some white blood cells called plasma cells, which are found in the bone marrow.3 When damaged, these plasma cells rapidly spread and replace normal cells in the bone marrow with tumors. In 2022, it is estimated that more than 34,000 people will be diagnosed with multiple myeloma, and more than 12,000 people will die from the disease in the U.S.5 While some people diagnosed with multiple myeloma initially have no symptoms, most patients are diagnosed due to symptoms that can include bone fracture or pain, low red blood cell counts, tiredness, high calcium levels, kidney problems or infections.2
//////////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Ciltacabtagene autoleucel, sold under the brand name Carvykti, is a medication used to treat multiple myeloma.[1][2]
The most common adverse reactions include pyrexia, cytokine release syndrome, hypogammaglobulinemia, musculoskeletal pain, fatigue, infections, diarrhea, nausea, encephalopathy, headache, coagulopathy, constipation, and vomiting.[2]
Ciltacabtagene autoleucel is a B-cell maturation antigen (BCMA)-directed genetically modified autologous chimeric antigen receptor (CAR) T-cell therapy.[1][2] Each dose is customized using the recipient’s own T-cells, which are collected and genetically modified, and infused back into the recipient.[1][2]
Ciltacabtagene autoleucel was approved for medical use in the United States in February 2022.[2][3][4]
Medical uses
Ciltacabtagene autoleucel is indicated for the treatment of adults with relapsed or refractory multiple myeloma after four or more prior lines of therapy, including a proteasome inhibitor, an immunomodulatory agent, and an anti-CD38 monoclonal antibody.[1][2]
History
The safety and efficacy of ciltacabtagene autoleucel were evaluated in CARTITUDE-1 (NCT03548207), an open label, multicenter clinical trial evaluating ciltacabtagene autoleucel in 97 participants with relapsed or refractory multiple myeloma who received at least three prior lines of therapy which included a proteasome inhibitor, an immunomodulatory agent, and an anti-CD38 monoclonal antibody and who had disease progression on or after the last chemotherapy regimen; 82% had received four or more prior lines of antimyeloma therapy.[1][2]
The U.S. Food and Drug Administration (FDA) granted the application for ciltacabtagene autoleucel priority review, breakthrough therapy, and orphan drug designations.[2]
References
- ^ Jump up to:a b c d e f “Carvykti- ciltacabtagene autoleucel injection, suspension”. DailyMed. 9 March 2022. Retrieved 16 March 2022.
- ^ Jump up to:a b c d e f g h “FDA approves ciltacabtagene autoleucel for relapsed or refractory multiple myeloma”. U.S. Food and Drug Administration (FDA). 7 March 2022. Retrieved 16 March 2022. This article incorporates text from this source, which is in the public domain.
- ^ “Carvykti”. U.S. Food and Drug Administration (FDA). 8 March 2022. Retrieved 16 March 2022.
- ^ “U.S. FDA Approves Carvykti (ciltacabtagene autoleucel), Janssen’s First Cell Therapy, a BCMA-Directed CAR-T Immunotherapy for the Treatment of Patients with Relapsed or Refractory Multiple Myeloma”. Janssen Pharmaceutical Companies (Press release). 1 March 2022. Retrieved 16 March 2022.
External links
- “Ciltacabtagene autoleucel”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Carvykti |
| Other names | JNJ-68284528 |
| License data | US DailyMed: Ciltacabtagene_autoleucel |
| Routes of administration | Intravenous |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1] |
| Identifiers | |
| DrugBank | DB16738 |
| UNII | 0L1F17908Q |
//////////Ciltacabtagene autoleucel, JNJ 68284528, Carvykti, FDA 2022, APPROVALS 2022, JNJ-68284528, LCAR-B38M CAR-T cells

NEW DRUG APPROVALS
ONE TIME
$10.00
TRIAMCINOLONE

TRIAMCINOLONE
- Molecular FormulaC21H27FO6
- Average mass394.434 Da
(11β,16α)-9-Fluoro-11,16,17,21-tetrahydroxypregna-1,4-diene-3,20-dione
(8S,9R,10S,11S,13S,14S,16R,17S)-9-Fluor-11,16,17-trihydroxy-17-(hydroxyacetyl)-10,13-dimethyl-6,7,8,9,10,11,12,13,14,15,16,17-dodecahydro-3H-cyclopenta[a]phenanthren-3-on
(8S,9R,10S,11S,13S,14S,16R,17S)-9-fluoro-11,16,17-trihydroxy-17-(hydroxyacetyl)-10,13-dimethyl-6,7,8,9,10,11,12,13,14,15,16,17-dodecahydro-3H-cyclopenta[a]phenanthren-3-one
(8S,9R,10S,11S,13S,14S,16R,17S)-9-fluoro-11,16,17-trihydroxy-17-(hydroxyacétyl)-10,13-diméthyl-6,7,8,9,10,11,12,13,14,15,16,17-dodécahydro-3H-cyclopenta[a]phénanthrén-3-one
(8S,9R,10S,11S,13S,14S,16R,17S)-9-Fluoro-17-glycoloyl-11,16,17-trihydroxy-10,13-dimethyl-6,7,8,9,10,11,12,13,14,15,16,17-dodecahydro-3H-cyclopenta[a]phenanthren-3-one
124-94-7[RN]
16a-Hydroxy-9a-fluoroprednisolone
1ZK20VI6TY
204-718-7[EINECS]
755
9a-Fluoro-16a-hydroxyprednisolone
TU3850000
トリアムシノロン[Japanese]
去炎松[Chinese]
Triamcinolone
CAS Registry Number: 124-94-7
CAS Name: (11b, 16a)-9-Fluoro-11,16,17,21-tetrahydroxypregna-1,4-diene-3,20-dione
Additional Names: D1-9a-fluoro-16a-hydroxyhydrocortisone; 9a-fluoro-16a-hydroxyprednisolone; D1-16a-hydroxy-9a-fluorohydrocortisone; 16a-hydroxy-9a-fluoroprednisolone
Manufacturers’ Codes: CL-19823
Trademarks: Aristocort (Lederle); Kenacort (BMS); Ledercort (tabl.) (Lederle); Omcilon (BMS); Tricortale (Bergamon); Volon (BMS)
Molecular Formula: C21H27FO6, Molecular Weight: 394.43
Percent Composition: C 63.95%, H 6.90%, F 4.82%, O 24.34%
Literature References: Prepn: Bernstein et al.,J. Am. Chem. Soc.78, 5693 (1956); 81, 1689 (1959); Thoma et al.,ibid.79, 4818 (1957); Bernstein et al., Allen et al.,US2789118; US3021347 (1957, 1962, both to Am. Cyanamid). Comprehensive description: K. Florey, Anal. Profiles Drug Subs.1, 367-396, 423-442 (1972); D. H. Sieh, ibid.11, 593-614, 651-661 (1982).
Properties: Crystals, mp 269-271°. mp also reported as 260-262.5°. [a]D25 +75° (acetone). uv max: 238 nm (e 15800).
Melting point: mp 269-271°; mp also reported as 260-262.5°
Optical Rotation: [a]D25 +75° (acetone)
Absorption maximum: uv max: 238 nm (e 15800)
………………………………
Derivative Type: 16,21-Diacetate
CAS Registry Number: 67-78-7
CAS Name: (11b,16a)-16,21-Bis(acetyloxy)-9-fluoro-11,17-dihydroxypregna-1,4-diene-3,20-dione
Additional Names: 16a,21-diacetoxy-9a-fluoro-11b,17a-dihydroxy-1,4-pregnadiene-3,20-dione
Trademarks: Cenocort (Central Pharm.); CINO-40 (Tutag); Tracilon (Savage)
Molecular Formula: C25H31FO8, Molecular Weight: 478.51
Percent Composition: C 62.75%, H 6.53%, F 3.97%, O 26.75%
Properties: Solvated crystals, mp 186-188° (with effervescence, mp 235° after drying). [a]D25 +22° (chloroform). uv max: 239 nm (e 15200).
Melting point: Solvated crystals, mp 186-188° (with effervescence, mp 235° after drying)
Optical Rotation: [a]D25 +22° (chloroform)
Absorption maximum: uv max: 239 nm (e 15200)
Therap-Cat: Glucocorticoid., Therap-Cat-Vet: Glucocorticoid., Keywords: Glucocorticoid.
///////////////////////
Triamcinolone Acetonide
CAS Registry Number: 76-25-5
CAS Name: (11b,16a)-9-Fluoro-11,21-dihydroxy-16,17-[1-methylethylidenebis(oxy)]pregna-1,4-diene-3,20-dione
Additional Names: 9a-fluoro-11b,16a,17,21-tetrahydroxypregna-1,4-diene-3,20-dione cyclic 16,17-acetal with acetone; 9a-fluoro-16a-hydroxyprednisolone acetonide; triamcinolone 16a,17-acetonide; 9a-fluoro-11b,21-dihydroxy-16a,17a-isopropylidenedioxy-1,4-pregnadiene-3,20-dione; 9a-fluoro-16a,17-isopropylidenedioxyprednisolone
Trademarks: Adcortyl (BMS); Azmacort (Aventis); Delphicort (Lederle); Extracort (Basotherm); Ftorocort (Gedeon Richter); Kenacort-A (BMS); Kenalog (Apothecon); Ledercort Cream (Lederle); Nasacort (Aventis); Respicort (Mundipharma); Rineton (Sanwa); Solodelf (Cyanamid); Tramacin (J & J); Triam (Lichtenstein); Tricinolon (Kaken); Vetalog (Solvay); Volon A (BMS); Volonimat (BMS)
Molecular Formula: C24H31FO6, Molecular Weight: 434.50
Percent Composition: C 66.34%, H 7.19%, F 4.37%, O 22.09%
Literature References: Prepd by stirring a suspension of triamcinolone in acetone in the presence of a trace of perchloric acid: Fried et al.,J. Am. Chem. Soc.80, 2338 (1958); Bernstein et al.,ibid.81, 1689 (1959); Bernstein, Allen, US2990401 (1961 to Am. Cyanamid). Alternate synthesis using 2,3-dibromo-5,6-dicyanoquinone: Hydorn, US3035050 (1962 to Olin Mathieson). Clinical trial in chronic asthma: I. L. Bernstein et al.,Chest81, 20 (1982). Comprehensive description: K. Florey, Anal. Profiles Drug Subs.1, 397-421 (1972); D. H. Sieh, ibid.11, 615-649 (1982).
Properties: Crystals, mp 292-294°. [a]D23 +109° (c = 0.75 in chloroform). uv max (abs alc.): 238 nm (e 14600). Sparingly sol in methanol, acetone, ethyl acetate.
Melting point: mp 292-294°
Optical Rotation: [a]D23 +109° (c = 0.75 in chloroform)
Absorption maximum: uv max (abs alc.): 238 nm (e 14600)
………………..
Derivative Type: 21-Acetate
Properties: Crystals, mp 268-270°. [a]D23 +92° (c = 0.59 in chloroform).
Melting point: mp 268-270°
Optical Rotation: [a]D23 +92° (c = 0.59 in chloroform)
Derivative Type: 21-Disodium phosphate
CAS Registry Number: 1997-15-5
Trademarks: Aristosol (Lederle)
Molecular Formula: C24H30FNa2O9P, Molecular Weight: 558.44
Percent Composition: C 51.62%, H 5.41%, F 3.40%, Na 8.23%, O 25.79%, P 5.55%
………………….
Derivative Type: 21-Hemisuccinate
Trademarks: Solutedarol (Specia)
Molecular Formula: C28H35FO9, Molecular Weight: 534.57
Percent Composition: C 62.91%, H 6.60%, F 3.55%, O 26.94%
Therap-Cat: Glucocorticoid; antiasthmatic (inhalant); antiallergic (nasal).
Therap-Cat-Vet: Glucocorticoid.
Keywords: Antiallergic (Steroidal, Nasal); Antiasthmatic (Steroidal, Inhalant); Glucocorticoid.
//////////////////////////
Title: Triamcinolone Benetonide
CAS Registry Number: 31002-79-6
CAS Name: (11b,16a)-21-[3-(Benzoylamino)-2-methyl-1-oxopropoxy]-9-fluoro-11-hydroxy-16,17-[(1-methylethylidene)bis(oxy)]pregna-1,4-diene-3,20-dione
Additional Names: 9-fluoro-11b,16a,17,21-tetrahydroxypregna-1,4-diene-3,20-dione cyclic 16,17-acetal with acetone 21-ester with N-benzoyl-2-methyl-b-alanine; 9a-fluoro-16a-hydroxyprednisolone 16a,17a-acetonide 21-(b-benzoylamino)isobutyrate; triamcinolone acetonide b-benzoylaminoisobutyrate; TBI
Trademarks: Tibicorten (Stiefel)
Molecular Formula: C35H42FNO8, Molecular Weight: 623.71
Percent Composition: C 67.40%, H 6.79%, F 3.05%, N 2.25%, O 20.52%
Literature References: Prepn: C. Cavazza et al.,DE2047218; eidem,US3749712 (1971, 1973 both to Sigma-Tau). Pharmacology: E. T. Ordonez, Arzneim.-Forsch.21, 248 (1971). Percutaneous absorption by rats and rabbits: W. H. Down et al.,Toxicol. Lett.1, 95 (1977). Clinical study: D. J. Tazelaar, J. Int. Med. Res.5, 338 (1977). HPLC analysis: S. Muck et al.,Boll. Chim. Farm.120, 240 (1981). For structure see Triamcinolone Acetonide.
Properties: Crystalline powder, mp 203-207°. [a]D20 +96 ±3° (c = 1 in ethanol). Sol in methanol, acetone, ethanol, dioxane, pyridine, DMF, chloroform. Insol in water.
Melting point: mp 203-207°
Optical Rotation: [a]D20 +96 ±3° (c = 1 in ethanol)
Therap-Cat: Glucocorticoid; anti-inflammatory (topical).
Keywords: Glucocorticoid
////////////////////////
Triamcinolone Hexacetonide
CAS Registry Number: 5611-51-8
CAS Name: (11b,16a)-21-(3,3-dimethyl-1-oxobutoxy)-9-fluoro-11-hydroxy-16,17-[(1-methylethylidene)bis(oxy)]pregna-1,4-diene-3,20-dione
Additional Names: 9-fluoro-11b,16a,17,21-tetrahydroxypregna-1,4-diene-3,20-dione cyclic 16,17-acetal with acetone, 21-(3,3-dimethylbutyrate); 21-tert-butylacetate-9a-fluoro-11b-hydroxy-16a,17a-(isopropylidenedioxy)pregna-1,4-diene-3,20-dione; 21-(3,3-dimethylbutyryloxy)-9a-fluoro-11b-hydroxy-16a,17a-(isopropylidenedioxy)pregna-1,4-diene-3,20-dione; triamcinolone acetonide tert-butyl acetate; TATBA
Manufacturers’ Codes: CL-34433
Trademarks: Aristospan (Fujisawa); Hexatrione (Lederle); Lederlon (Lederle); Lederspan (Lederle)
Molecular Formula: C30H41FO7, Molecular Weight: 532.64
Percent Composition: C 67.65%, H 7.76%, F 3.57%, O 21.03%
Literature References: The hexacetonide ester of the potent glucocorticoid, triamcinolone, q.v. Prepn of syringeable suspension: Nash, Naeger, US3457348 (1969 to Am. Cyanamid). Anti-inflammatory activity in rabbits: I. M. Hunneyball, Agents Actions11, 490 (1981). Early clinical studies: Bilka, Minn. Med.50, 483 (1967); Layman, Peterson, ibid. 669. Clinical studies of intra-articular therapy in arthritis: R. C. Allen et al.,Arthritis Rheum.29, 997 (1986); M. Talke, Fortschr. Med.104, 742 (1986). Toxicity study: Tonelli, Steroids8, 857 (1966). Comprehensive description: V. Zbinovsky, G. P. Chrekian, Anal. Profiles Drug Subs.6, 579-595 (1977). For structure see Triamcinolone Acetonide.
Properties: Fine, white, needle-like crystals, mp 295-296° (dec), also reported as mp 271-272° (dec). uv max (ethanol): 238 nm (e 15500). [a]D25 +90±2° (c = 1.13% in chloroform). Soly in g/100 ml at 25°: chloroform and dimethylacetamide >5; ethyl acetate 0.77, methanol 0.59, diethyl carbonate 0.50, glycerin 0.42, propylene glycol 0.13; absolute alcohol 0.03; water 0.0004.
Melting point: mp 295-296° (dec); mp 271-272° (dec)
Optical Rotation: [a]D25 +90±2° (c = 1.13% in chloroform)
Absorption maximum: uv max (ethanol): 238 nm (e 15500)
Therap-Cat: Anti-inflammatory.
Keywords: Glucocorticoid.
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Triamcinolone acetonide | F446C597KA | 76-25-5 | YNDXUCZADRHECN-JNQJZLCISA-N |
| Triamcinolone diacetate | A73MM2Q32P | 67-78-7 | XGMPVBXKDAHORN-RBWIMXSLSA-N |
| Triamcinolone hexacetonide | I7GT1U99Y9 | 5611-51-8 | TZIZWYVVGLXXFV-FLRHRWPCSA-N |
Triamcinolone is a glucocorticoid used to treat a wide variety of inflammatory conditions of organ systems and tissues.
Triamcinolone is a glucocorticoid used to treat certain skin diseases, allergies, and rheumatic disorders among others.[6] It is also used to prevent worsening of asthma and COPD.[6] It can be taken in various ways including by mouth, injection into a muscle, and inhalation.[6]
Common side effects with long-term use include osteoporosis, cataracts, thrush, and muscle weakness.[6] Serious side effects may include psychosis, increased risk of infections, adrenal suppression, and bronchospasm.[6] Use in pregnancy is generally safe.[7] It works by decreasing inflammation and immune system activity.[6]
Triamcinolone was patented in 1956 and came into medical use in 1958.[8] It is available as a generic medication.[9] In 2019, it was the 107th most commonly prescribed medication in the United States, with more than 6 million prescriptions.[10][11]
PATENT
Skin is the layer of usually soft, flexible outer tissue covering the body of a vertebrate animal, with three main functions: protection, regulation, and sensation. Skin diseases are the medical condition that affects the skin, hair, nails and related muscle and glands.
Skin disorders vary greatly in symptoms and severity. They can be temporary or permanent, and may be painless or painful. Some have situational causes, while others may be genetic. Some skin conditions are minor, and others can be lifethreatening.
There are many different types of skin disorders which include rashes, dermatoses or skin eruptions. Such rashes, dermatoses or skin eruptions include acute, inflammatory reactions of the skin caused by an allergic or irritant reaction, other forms of eczema, lichen simplex chronicus. Chronic nature includes seborrheic dermatitis, psoriasis, and atopic dermatitis or caused by infection, irritation or aggravation of another condition such as occurs with acne, other rashes, dermatoses or skin eruptions, inflammatory and pruritic manifestations of corticosteroid-responsive dermatoses, contact dermatitis, impetigo, urticarial and scabies.
Typical symptoms of the skin disorders include but not limited to raised bumps that are red or white, a rash, which might be painful or itchy, scaly or rough skin peeling skin, ulcers, open sores or lesions, dry, cracked skin, discolored patches of skin, fleshy bumps, warts, or other skin growths, changes in mole color or size a loss of skin pigment, excessive flushing or the like.
Atopic dermatitis (AD), also known as eczema or atopic eczema, is a type of inflammation of the skin (dermatitis). Atopic dermatitis (AD) is common worldwide. People of all ages from newborns to adults and older live with this condition. Symptoms range from excessively dry, itchy skin to painful, itchy rashes that cause sleepless nights and interfere with everyday life.
Topical corticosteroids have been the mainstay of treatment for atopic dermatitis over the past years, further the cure for atopic dermatitis involves Lifestyle modification, balanced diet intake, self-care measures, phototherapy, wet wrap therapy, use of medications like tacrolimus, pimecrolimus, crisaborole, dupilumab, ciclosporin, methotrexate, interferon gamma- lb, mycophenolate mofetil, and azathioprine or the like.
Triamcinolone Acetonide is a synthetic corticosteroid. Chemically it is [Pregna-1, 4-diene-3, 20-dione, 9-fluoro-l l, 21 -dihydroxy- 16, 17-[(1 methylethylidene) bis-(oxy)]-, (HP, 16a)-] with the empirical formula C24H31FO6 and molecular weight 434.50. Triamcinolone Acetonide is represented by compound of structural formula I
Triamcinolone Acetonide topical cream and ointment with strengths 0.025%, 0.1% and 0.5% (containing 0.25 mg/gm, 1 mg/gm & 5 mg/gm Triamcinolone Acetonide respectively) were approved in USA prior to Jan 1, 1982 under the trade name “Triamcinolone Acetonide” and were indicated for the relief of the inflammatory and pruritic manifestations of corticosteroid-responsive dermatoses.
The commercially available products or product known in the prior art produces side effects such as burning, itching, irritation, or dryness of skin at site of application, folliculitis, hypertrichosis, acneiform eruptions, hypopigmentation, perioral dermatitis, allergic contact dermatitis, maceration of the skin, secondary infection, skin atrophy, striae and miliaria.
Pediatric patients may demonstrate greater susceptibility to topical triamcinolone -induced HPA axis suppression and Cushing’s syndrome than mature patients because of a larger skin surface area to body weight ratio. Hypothalamic -pituitary-adrenal (HPA) axis suppression, Cushing’s syndrome and intracranial hypertension have been reported in children receiving topical triamcinolone. Manifestations of adrenal suppression in children include linear growth retardation, delayed weight gain, low plasma cortisol levels, and absence of response to ACTH stimulation. Manifestations of intracranial hypertension include bulging fontanelles, headaches, and bilateral papilledema. Chronic corticosteroid therapy may interfere with the growth and development of children.
Making low dose compositions can present technical and economic challenges that are not present for higher dose formulations.
Examples
The following table 1 shows cream formulation containing lOO.OOmcg per gm, 50.00mcg per gm and 25.00mcg per gm of Triamcinolone Acetonide
Table – 1: cream
Drug Strength IQOmcg/gm 50mcg/gm 25mcg/gm
lOO.OOmcg per gm and for lOOgm, it is lO.OOmg*
50.00mcg per gm and for lOOgm, it is 5.00mg*
25.00mcg per gm and for lOOgm, it is 2.50mg**
Manufacturing process:
a) Dispensing following excipients – isopropyl myristate, glyceryl monostearate and white soft paraffin in vessel I;
b) Dispensing the following excipients – polysorbate 40 and purified water in vessel II;
c) Dispensing the following excipients methyl paraben, propylene glycol in vessel III; wherein methyl paraben is dissolved in propylene glycol to form a clear solution;
d) Dispensing the following active & excipients triamcinolone acetonide or salt thereof, propylene glycol in vessel IV; wherein triamcinolone acetonide or salt thereof is dissolved in propylene glycol to form clear solution;
e) Adding content of step (c) into content of step (b) and stirring to form uniform and homogeneous emulsion;
f) Heating content of step (b) and step (a) at about 75 °C and stirring to form a homogenous uniform emulsion;
g) Cooling the above emulsion gradually to temperature of about 25 °C – 30°C h) Adding the content of step (d) to the primary emulsion of (f) with constant stirring; and
i) Making up the volume of the emulsion with purified water to the required quantity.
SYN
DOI: 10.1021/ja01516a043

CLIP
Corticosteroids
R.S. Vardanyan, V.J. Hruby, in Synthesis of Essential Drugs, 2006
Triamcinolone
Triamcinolone, 9a-fluoro-11b,16a,17,21-tetrahydroxypregna-1, 4-dien-3,20-dione (27.1.61), differs from dexamethsone in terms of chemical structure in that the a methyl group at C16 is replaced with a hydroxyl group. It is synthesized from the 21-O-acetate of hydrocortisone 27.1.17. In the first stage, both carbonyl groups of this compound undergo ketalization by ethylene glycol. Next, the hydroxyl group in the resulting diketal 27.1.53 is replaced with chlorine using thionyl chloride, and the product undergoes dehydrochlorination using an alkaline, during which the 21-O-acetyl group also is hydrolyzed. Acetylating the hydroxyl group once again with acetic anhydride gives a triene 27.1.54. Reacting this with osmium tetroxide gives the vicinal diol 27.1.55. The secondary hydroxyl group at C16 of this product undergoes acetylation by acetic anhydride in pyridine, which forms the diacetate 27.1.56. Treating the product with N-bromoacetamide in chloric acid gives a bromohydrin (27.1.57), which upon reaction with potassium acetate is transformed to an epoxide (27.1.58). Opening of the epoxide ring, using hydrofluoric acid, gives the corresponding 9-fluoro-11-hydroxy derivative 27.1.59. Upon microbiological dehydrogenation, the C1–C2 bond is oxidized to a double bond, forming triamcinolone acetate (27.1.60), the acetyl group of which is hydrolyzed, forming the desired triamcinolone (27.1.61) [30–32].

Triamcinolone is similar to dexamethasone in terms of pharmacological action, and it is better tolerated in some cases. Synonyms of this drug are ledercort, cenocort, delsolon, and others.
SYN
Drugs for Treating Respiratory System Diseases
Ruben Vardanyan, Victor Hruby, in Synthesis of Best-Seller Drugs, 2016
Triamcinolone–Nasacort
The synthesis of triamcinolone (23.2.1) starts from ketalization of cortisol 21-acetate (23.2.8) using ethylene glycol. Dehydration of the obtained compound (23.2.9) for creation of a double bond in position 16,17 of the steroid skeleton through the series of sequential reactions of chlorination, dehydrochlorination, hydrolysis, and acetylation produces 21-acetoxy-4,9(11),16-pregnatriene-3,20-dione (23.2.10), treatment of which with osmium tetroxide in benzene and pyridine produced diol (23.2.11), the secondary hydroxyl group of which, in position 16, was acetylated with acetic anhydride in pyridine to produce the diacetate (23.2.12). The obtained compound in dioxane and water was treated with N-bromoacetamide and 10% perchloric acid to yield bromohydrine (23.2.13). Dehydrobromination of the bromohydrine (23.2.13) with anhydrous potassium acetate in refluxing ethanol produced the epoxy-derivative (23.2.14). Opening of the epoxide ring in (23.2.14) with anhydrous hydrogen fluoride in chloroform produced (23.2.15). Microbiological dehydrogenation of the obtained product with Corynebacterium simplex produced crude diacetate (23.2.16), saponification of which produced triamcinolone (23.2.1) [108-110] (Scheme 23.7.).

Scheme 23.7. Synthesis of triamcinolone.
Triamcinolone is commonly used in the treatment of respiratory inflammation and improves airway reactivity, decreasing respiratory problems. Strangely, there are only few reviews of the pharmacotherapy of triamcinolone [111-113].
SYN
| CAS-RN | Formula | Chemical Name | CAS Index Name |
|---|---|---|---|
| 426-39-1 | C25H33FO8 | 16α,21-diacetoxy-11β,17-dihydroxy-3,20-dioxo-9-fluoro-4-pregnene | Pregn-4-ene-3,20-dione, 16,21-bis(acetyloxy)-9-fluoro-11,17-dihydroxy-, (11β,16α)- |
| 96670-24-5 | C25H30O8 | 16α,21-diacetoxy-3,20-dioxo-17-hydroxy-9β,11β-epoxy-1,4-pregnadiene | 9β-Pregna-1,4-diene-3,20-dione, 9,11β-epoxy-16α,17,21-trihydroxy-, 16,21-diacetate |
SYN
https://patents.google.com/patent/WO2016120891A1/en
Glucocorticoids have a number of diverse effects in different body tissues. Glucocorticoids, in topical, oral and inhaled formulations, are useful for their anti-inflammatory and immunosuppressive properties. Several glucocorticoids such as budesonide and ciclesonide are used for treatment of several disorders.
The synthesis and purification of glucocorticoids have been disclosed at different instances. However, most of these synthetic procedures involve toxic solvents or long reaction times and are ineffective for large scale synthesis. For instance, US 3,92,9768 discloses a process for preparation of budesonide by reacting 16, 17-dihydroxy compound with aldehyde in solvents such as dioxane, methylene chloride or their combinations.
DE 4129535 discloses a process for the synthesis of Ciclesonide which involves the intermediate 16A, 17-[(7?,S)-cyclohexylmethylenedioxy]-l 13, 21-dihydroxy-pregna-l 4- dien-3,20-one which is obtained by an acid catalysed reaction of 11 , 16 , 17, 21-tetra hydroxypregna-l,4-dien-3,20-one with cyclohexane aldehyde.
WO 02/38584 discloses the synthesis of Ciclesonide by reacting corresponding 16, 17-ketals with a cyclohexane aldehyde in the presence of 70% perchloric acid, 1-nitropropane as solvent. However, perchloric acid is a dangerous solvent and can cause serious accidents with fatal consequences.
US Patent No. 6169178 relates to a process for the preparation of budesonide and of 16, 17- acetals of pregnane derivatives structurally co-related thereto comprising treating 16, 17-dios or of 16, 17-ketals or cyclic acetals with aldehydes in the presence of aqueous hydrobromic acid or hydroiodic acid used as reaction catalyst or solvents. However, hydroiodic and other hydrohalic solvents are corrosive, light sensitive and expensive. Further, these acids also post environmental problems. Notwithstanding the use of hydrohalo acids requires use of special equipment since they are extremely corrosive and consequently increase the cost of production.
US 5,55,6964 discloses a process for the preparation of budesonide by reacting 16 – Hydroxy Prednisolone in acetonitrile in the presence of /^-toluene sulfonic acid as a catalyst. There are certain other patents that use alkyl sulfonic acid instead of aryl sulfonic acid for the synthesis of budesonide or similar compounds. However, sulfonic acids are hazardous solvents and FDA has expressed significant concern over the presence or traces of sulfonic acid in pharmaceutical products. Hence, there is a need to have a process for the synthesis 16, 17- acetals of pregnane compounds that is industrially scalable and which does not involve the use of harmful solvents.


Example- 1: Process for preparation of 16-HPN from 3TR
Stage-I


Stage- 1 Stage-I I

Stage-IV

1 6-HPN acetate 1 6-HPN
Scheme 2: Synthesis of 16HPN from 3TR
Stage-I (oxidation)
Charge 750L of acetone (50 volume), 39L of purified water (2.60 volume) and 15 Kg of 3TR (40.93mol) in a SS Reactor at ambient temperature. Cool to -7°C to -5°C than added 6.0L of formic acid (159.03 mol) and 9.0 kg of potassium permanganate (56.95 mol). Maintain at – 5°Cto -3°C for 30 minutes. In-process check by TLC, 3TR should be less than 1.0%. Added 1.5kg sodium metabisulphite (7.89 mol solution in 12L of purified water at -5°C to -3°C then added 3.0 kg of hyflow super cell at 15°C (+2°C) and filter through 10.0 kg of hyflowbed at 27°C(+3°C) and wash with 150L of acetone Added 1.5 kg of activated charcoal, Stir and filter through hyflow bed and wash with 60L of acetone. Total filtrate was distilled under reduced pressure, while maintaining temperature below 45°C. Added 81L of purified water and cool to 5°C+5°C. Filter through centrifuge and wash with 156L of purified water. Wet material is dry at 60°+5°C till moisture less than 0.50%, Yield=15 kg, HPLC purity=98%.
Stage-II (Bromination)
Charge 75L of tetrahydrofuran, 16L of purified water and 15.0 kg of Stage-I (37.46 mol) in a glass reactor. Cool to -6°C (+2°C) and added 7.50 kg of dibromantin (26.23 mol) and 0.60L of perchloric acid (9.38 mol) and maintain at -6°C (+2°C) for one hour. In-process check by TLC, stage-I should be less than 0.50%. Reaction mass is quench in 390L of purified water at ~5°C. Raised the temperature to 25°C and maintained for 01 hour, filter through centrifuge and wash with 828L of purified water or till neutral pH. Wet material is dry at 40°C+5°C till moisture content should be less than 10%, Yield=21.0kg, HPLC purity=97%.
Stage-Ill (Debromination)
Charge 68.0L of N, N-dimethyl formamide(3.238volume) and 21.0kg of stage-II (42.22 mol) in glass reactor, start argon gas purging and cool to -5°C. Charge 13.0L of N,N- dimethylformamide (0.619volume) , 9.70L of dimethylsulfoxide(0.462volume), 1.62kg of chromium chloride hexahydrate (6.51 mol) and 1.94 kg of zinc dust (0.703 mol). Cool to – 10°Cand added 5.50L of thioglycolic acid (79.21 mol). Maintain for one hour while maintaining temperature around -10°C. In-process check by TLC, stage-II should be less than 1.0%. Added 310 L of purified water and cool to 0°C. Filter through centrifuge and wash with 1600L of purified water. Wet material is dry at 60°C+ (5°C) till moisture content less than 6.0%, Yield=15.0kg, HPLC Purity=90%.
Charge 150L of methylene chloride (10 volume), 150L of methanol (10 volume.) and 15.0kg (30.16 mol) of stage-Ill in a SS Reactor. Heat to clear solution then added 3.0 kg of activated charcoal (20%) and reflux for 04 hours, Filter through hyflow bed and wash with 75L of methylene chloride (5 volume), and 75L of methanol (5 volume) mixture. Total filtrate is distilled till last drop and added 75L (5 volume) of methylene dichloride, reflux for 04 hours than cool to 40°C+(5°C), Filter through centrifuge and wash with 15L (one volume) of methylene chloride. Wet material is dry at 60°C (+5°C) till moisture contents less than 1.0% (Yield =13.0kg, HPLC Purity=96%). Further charge 65.0L (5volume) of ethyl acetate and 13.0 kg (1.0 mol) of purified material. Heat to reflux and maintain for 04 hours under reflux, then cool to 40°C. Filter through centrifuge and wash with 13.0L (one volume) of ethyl acetate. Wet material is dry at 60°C (+5°C) till moisture contents less than 0.50%, Yield=12.0kg, HPLC Purity=98.6%.
Stage-IV (Deacetylation)
Charge 5.83L of methanol (10 volume) and 5.83L of methylene chloride (10 volume) in a glass flask and added 583 gm of 16-HPN acetate(1.397mol) at RT. Start argon gas purging and cool to 0°C to 5°C under argon purging. Prepare 11.66 gm of sodium hydroxide (0.2915mol) solution in 0.583L of methanol (one volume) under argon purging and cool to 0°Cto 5°C. Sodium hydroxide solution is charge in reaction mass at 0°C to 5°C. Maintained the reaction mass at 0°C to 5°C for one hour, In-process check by TLC against 16-HPN acetate it should be nil. Adjust pH to neutral by 21.40ml of acetic acid (0.3742 mol); distill under reduced pressure while maintaining temperature below 40°C, till dry. Cool to ambient temperature and added 1.166L of purified water (02 volume). Cool to 0°C and maintain for one hour. Filter and wash with 300ml of purified water. Dry at 60°C (+5°C) till moisture content less than 1.0%, Yield=490gm (93.50%), HPLC Purity=98.97%, Single impurity= 0.40%. Example 2: Process of synthesis of Budesonide from 16-HPN

16-HPN Budesonide
Charge 800 ml of aqueous hydrochloric acid (8 volume) in a glass flask, start nitrogen gas purging and Cool to -5°C and maintain for 15 min. then added 100 gm of stage-I (0.27 mol) at -5°C and stir for 15 min., added 30 ml of N-butyraldehyde (0.33 mol) while maintaining temperature -5°C to 0°C in around 30 minutes and maintain at 0°C to 5°C for 150 min. under stirring. In-process check by TLC against stage-I, it should be nil. Reaction mass is quench in 1200 ml of purified water (12 volume) at 5°C to 10°C and stir for 15 min. Added solution of 100 kg of sodium bicarbonate (1.19 mol) and 1 ml of purified water (10 volume) in reaction mass at 5°C to 10°C. Stir at 5°C to 10°C for 15 min. Filter and wash with purified water till neutral pH. Wet material is dry at 50°C (+5°C) till moisture contents less than 1.0 %, Yield =110 gm (96.49%), HPLC purity=96.45%, single impurity=1.29%, Epimer-A=47.76%, Epimer-B=49.69%.
(Purification)
Charge 2.5 L of methanol (25 volume) in a Glass flask and added 100 gm of above mentioned crude product. Dissolved at 25°C+5°C till clear solution, added 10 gm of activated charcoal and stir for 30 min. than filter through hyflow bed and wash with 200 ml of methanol (2 volume). Combined filtrates charged in a Glass flask and cool to 10°C to 15°C and added 5.40 L of purified water (54 volume) at 5°Cto 10°C, stir for 15min., filter and wash with purified water. Wet material is dry at 50°C (+5°C) under vacuum till moisture content less than 0.50%, Output=90.0gm, HPLC purity=99.66%, single impurity=0.1%, Epimer-A=44.47%, Epimer-B=55.01%.
Example 2.1: Scale-up process of manufacturing of Budesonide from 16-HPN
Charge 40 L of aqueous hydrochloric acid (8 volume) in a glass flask, start nitrogen gas purging and Cool to – 10°C and maintain for 15 min. then added 5.0 kg of stage-I (13.315 mol) at – 10°C and stir for 45 min. added 1.5 L of N-butyraldehyde (16.68 mol) while maintaining temperature -7°C to – 11°C in around 30 minutes and maintain at -2°C to -6°C for 60 min. under stirring In-process check by TLC against stage-I, it should be nil. Reaction mass is quench in 60 L of purified water (12 volume) at 5°C to 10°C and stir for 15 min. Added solution of 5.0 kg of sodium bicarbonate (59.525 mol) and 50L of purified water (10 volume) in reaction mass at 5°Cto 10°C. Stir at 5°C to 10°C for 15 min. Filter and wash with purified water till neutral pH. Wet material is dry at 50°C (+5°C) till moisture contents less than 1.0 %, Yield =5.293 kg (94.46%), HPLC purity=95.45%, single impurity=1.45%, Epimer-A=53.51 %, Epimer-B=43.78% Effect of temperature and its variation on epimer ratio (A and B) with respect to batch size (From lab to commercial batch)
Example 3: Process for synthesis ofCiclesonide from 16HPN
Preparation of cyclohexane carboxaldehydemetabisulphite complex
200gm of Cyclohexane carboxaldehyde (1.786 mol) was dissolved in 3.0L of denatured sprit (15 volume) and a solution of 190gm of sodium metabisulphite (1.827 mol) in 300ml of purified water (1.5 volume) was added. The resulting precipitate was filtered and washed with 1.0L of denatured sprit(5.0 volume) and dried under vacuum at 50°C, till moisture content less than 6.00%, Yield=400gm (97 %)
Stage I: Preparation of stage-I from 16-HPN

Cyclohexane carboxaldehyde
sodium metabisulphite complex
170gm of 16-HPN (0.4528 mol) was suspended in 3.40L of dichloromethane (20 volume) and treated with 340ml of 70% perchloric acid. (5.65 mol) and 110.5gm of cyclohexane carboxaldehyde metabisulphite complex (0.512 mol) was added in lots while maintaining the temperature between 0°Cto 5°C. The reaction mass was stirred at 0°C to 5°C for 03 hours. In- process check by TLC 16-HPN should be nil and then neutralized with 10% aqueous sodium bicarbonate solution. The organic layer was separated and concentrated under vacuum to obtain a residue which was stripped with methanol (1.0 volume). The solvent was concentrated and the residue was dissolved by refluxing in methanol (5.0 volume). The clear solution was cooled to 0°C to 5.0°C and the resulting solid was filtered and dried at 50°C till moisture content less than 0.50%, Yield=170.0gm (80.0%), HPLC purity=91.68%.
Stage -II Preparation of Ciclesonide from Stage -I

Stage-I Ciclesonide
158gm of stage-I (0.34mol) was suspended in 1.58L of methylene chloride (10.0 volume) at 25°C to 30°C. The reaction mass was chilled to 0°C to 5°C and 81.0ml of triethylamine(0.581 mol) was added, followed by the addition of 79.0ml of isobutyryl chloride [0.75 mol; diluted with 79.0 ml of methylene chloride (0.50 volume)] slowly at 0° to 5°C and maintained at same temperature for 60min. In-process check by TLC, Stage-I should be nil. The reaction mass was diluted with 2.53L of purified water (16.0 volume) , the organic layer was separated and washed with purified water till neutral pH, than organic layer was separated and concentrated under vacuum to obtained a residue. The residue was dissolved by refluxing in 948ml of methanol (6.0 volume); the clear solution was cooled to 0°C to 5°C under stirring and filtered. The product was dried under vacuum at ~50°C till moisture contents comes less than 0.50%, Yield=158.0 gm (87.0%), HPLC purity=95.74%.
(Purification)
120gm of Ciclesonide crude was dissolved by refluxing in 600ml of methanol. The clear solution was chilled to 20°C under stirring and filtered. The product was dried under vacuum at 90°C till moisture content less than 0.50%. Yield=105 gm (87.50%), HPLC purity=99.7 %.
Example 4: Process for synthesis of Desonide from 16HPN acetate
Stage-I : Preparation of Desonide acetate from 16 HPN acetate

Desonide acetate
16HPN acetate 190.0 ml of acetone (7.0 volume) was charged in a glass flask under nitrogen blanketing than added 27 gm of 16HPN acetate (0.0645mol) at ambient temperature. Temperature raised to 28°C (+2°C) and stir for 20 minutes. 1.35 ml of perchloric acid 70% (0.02 lmol) was added at 28°C (+2°C) and stir for 30 minutes. Temperature further raised to 35°C and stir for 60 minutes. In-process check by TLC against 16HPN acetate, it should be nil. Reaction mass cooled to 10°C, filtered and washed with purified water till neutral pH (~7) and finally washed with acetone. Wet material dried at 50°C+5°C till moisture content less than 0.50% to get stage-I. Yield =23gm (77.76%), HPLC Purity=98.28%
Stage-II: Preparation of Desonide from Desonide acetate

Desonide
Desonide acetate
200 ml of methanol (10 volume) and 200ml of methylene dichloride (10 volume) was charged in a glass flask and start argon gas purging. 20 gm of stage- 1st (0.0436mol) was added at ambient temperature. Cool to 0°C+5°C. 0.40gm of sodium hydroxide (O.Olmol) solution in 20ml of methanol (l.Ovolume) was added at 0°C+5°C. Stir at 0°C+5°C for 120 minutes. In-process check by TLC against stage- 1st it should be nil. Adjust pH to neutral (~7) by 2.0ml of acetic acid at 0°C+5°C. Distilled the solvent from reaction mass under vacuum while maintaining temperature below 40°C till the volume get reduced to 3 to 4 volume of the input. Cool to 0°C and further added 60ml of purified water and stir for 30 minutes. Filtered, washed with purified water till neutral pH (~7). Wet material dried at 50°C+5°C till moisture content less than 0.50% to get crude Desonide. Yield =14.70gm (80.92%), HPLC Purity=88.15%.
(Purification)
140 ml of methanol (10 volume) and 140 ml of methylene chloride (10 volume) was charged in a glass flask and added 14.0 gm of crude material (0.034mol) than stir till clear solution. Added 1.5 gm of activated charcoal and stir for 30 minutes than filtered through hyflow supercel bed and washed with 30ml of methanol and 30ml of methylene chloride mixture. Combined filtrate and distilled the solvent from reaction mass under vacuum while maintaining temperature below 40°C till the volume reduced to 3 to 4 volume of the input. Cool to 0°C. Filtered the reaction mass and washed with 10ml of precooled methanol. Wet material was dried at 50°C+5°C till moisture content less than 0.50% to get Desonide. Yield=8.60gm, HPLC Purity= 99.43%

lOOgm of 3TR (0.27 mol.)was suspended in 1300ml (13 volume) acetone. Cooled it to -5°C to -10°C than added 4.0 ml (0.062 mol.) perchloric acid solution and 50gm of dibromantin. Maintained the reaction at same temperature for 02 hours. In-process check by TLC against 3TR it should be nil. Added lOOgm of potassium carbonate solution (0.723 mol.) in 5 lots and reaction was maintained at 35°C+2°C. In-process check by TLC against step-I reaction mass, it should be nil. Cooled to 0°C (+5°C) and adjust pH neutral (~7) by 36ml of acetic acid (0.63 mol.). Added 3.0L of purified water (30 volume). Filter and washed with purified water till neutral pH (~7). Wet material was dried at 45°C (+2°C) till moisture content less than 0.50%. Yield =87gm, (83.36%), HPLC Purity=97.883%.
Stage – II:

80gm of stage-I (0.21 mol) was dissolved in 4.0L of acetone (50 volume) and 208ml of purified water (2.6 volume). Cool to -5°C (+2°C) than added 32ml of formic acid (0.85 mol.) and 48gm of potassium permagnate (0.30 mol.) at -5°C (+2°C). Reaction was maintained at – 5°C+2°Cfor one hour. In-process check by TLC against stage-I it should be nil. Added 8gm of sodium metabisulphite (0.042 mol.) In 80 ml purified water (01 volume) solution at -5°C (+2°C). Temperature raised up to 27°C and filtered through hyflow bed and washed with acetone. Acetone was distilled under vacuum till 3 to 4volume of stage-I than cool to 0°C to 5°C and added 480ml of purified water stir and filter and washed with purified water to get wet stage-II. Wet material was dried at 50°C (+5°C) till moisture content less than 3.0%. Yield =78.30gm, (89.88%), HPLC Purity=99.178%. Stage -III:

Stage-ll Stage-
300ml of hydrofluoric acid (12.60mol) was cooled at -25°C to -30°C than added 75gm of stage-II (0.180mol). Reaction was maintained at -25°C to -30°C for 04 hours. In-process check by TLC against stage-II, it should be nil. Reaction mass was cooled to -50°C than added 45ml of acetone (0.60volume) at -45°C to -50°C. Reaction was maintained at -45°C to -50°C for 02 hours. In-process check by TLC against before acetone reaction mass. Added 565ml of purified water at 0°C and 340ml of liq. ammonia at ~20°C than reaction mass was quenched in 410ml of liq. ammonia and 735ml of purified water solution at 15°C (+2°C), stir and filter and washed with purified water till neutral pH. Wet material was dried at 45°C to 50°C, Yield =78.50gm, (91.48%), HPLC Purity=91.593%.
(Purification)
76 gm of stage-Ill Crude (0.16 mol.) was dissolved in 760ml of methylene chloride (lOvolume) and 760ml of methanol (lOvolume) mixture at ambient temperature. Stir till clear solution and added 7.6gm of activated charcoal (0. lOvolume) than stir for 30minutes, filter through hyflow bed and washed with methanol (one volume) and methylene chloride (one volume) mixture. Total filtrate was distilled under vacuum till 3 to 4 volume of input. Cool to 0°C to 5°C and stir for 02 hours. Filtered and washed with minimum precooled methanol, Wet material was dried 45°C to 50°C till moisture contents less than 0.50%, Yield=62gm, HPLC Purity=98.633%.
Stage – IV (Process for synthesis of Triamcinolone acetonide from Stage – III):

Stage- Ill Triamcinolone acetonide
60gm of stage-Ill (0.13 mol) was dissolved in 600ml of methanol (lOvolume) and 600ml of methylene chloride (lOvolume) mixture under argon bubbling. Cool to -5°C+2°C and added 1.2gm of sodium hydroxide (0.03mol.) solution in 60ml of methanol (Olvolume) at -5°C (+2°C). Reaction maintained at -5°C (+2°C) for 03 hours. In-process check by TLC against stage-Ill, it should be nil. Adjust pH neutral (~7) by adding 1.8ml of acetic acid at -5°C (+2°C). Reaction mass was distilled at below 40°C under vacuum till 3 to 4 volume of input. Cool to 30°C and added 120ml of purified water, stir for one hour than filter and washed with purified water till neutral pH (~7). Wet material was dried at 45°C to 50°C till moisture content less than 0.50%, Yield =52gm, (95.04%), HPLC Purity=99.21%
(Purification)
50gm of crude material (0.12 mol.) was dissolved in 1100ml of acetone (22volume) and 100ml of purified water (02volume) at 50°C than added 2.5gm of activated charcoal and stir for one hour at same temperature, Filter through hyflow bed and washed with 120ml of acetone (2.40volume). Filtrate was distilled below 40°C under vacuum till 3 to 4 volume of input. Cool to 0°C to 5°Cand maintained for one hour at same temperature. Filter and washed with water. Wet material was dried at 45°C to 50°C till moisture content less than 0.50%, Yield=43gm, HPLC Purity=99.40%.
Example 6: Process for synthesis of Flunisolide from 16HPN acetate Stage -I (Preparation of Desonide acetate from 16HPN acetate):

1 6 H PN acetate eson e acetate
140ml of acetone (7 volume) was charged in glass flask and start argon blanketing than added 20 gm of 16-HPN acetate (0.048mol) at ambient temperature. Cooled to 28°C (+2°C). 1.0ml of perchloric acid 70% (0.016mol) was added at 28°C (+2°) C and stirred for 30 minutes. Temperature raised up to 35°Cand stirred for 60 minutes. In-process check by TLC against 16-HPN acetate, it should be nil. Reaction mass was cooled to 10°C (+2°C). Reaction mass was filtered and washed with purified water till neutral pH (~7) to get wet material. Wet material was dried at 50°C+5°C till moisture content less than 0.50% to get stage-lst. Yield=17.40gm, (79.40%), HPLC Purity=98.241%.
Stage -II (Preparation of Desonide from Desonide acetate):

170ml of methanol (lOvolume) and 170ml of methylene chloride (lOvolume) was charged in a glass flask and start inert atmosphere. 17gm of stage-lst (0.037mol) was added at ambient temperature. Cooled to -5°C. 0.4gm of sodium hydroxide (O.Olmol) solution in 17ml of methanol was added at 0°C (+5°C). Reaction mass was stirred for 02 hours at 0°C (+5°C). In- process check by TLC against stage- 1st it should be nil. Neutral pH (~7) was adjusted by acetic acid. Reaction mass was distilled under vacuum at below 40°C till ~ 100ml. Concentrated mass was cooled to 0°C (+5°C) and stir for one hour. Reaction mass was filtered and washed with precooled methanol to get wet material. Wet material was dried at 50°C (+5°C) till moisture content less than 0.50% to get stage-2nd. Yield=14.0gm, (90.67%), HPLC Purity=99.426%, Single impurity=0.136%.
Stage -III (Preparation of Flunisolide acetate from Desonide):

Desonide Flunisolide acetate
50ml of isopropenyl acetate (5 volume) was charged in a glass flask and added lOgm of stage-2nd (0.024mol) at ambient temperature than heated to 65°C and added 1.5ml of methane sulphonic acid (0.023mol) and temperature raised up to 80°C and stir for one hour. In-process check by TLC against stage-2, it should be nil. Reaction mass cooled to 25°C and adjust pH neutral (~7) by triethylamine. Reaction mass was distilled under vacuum till last drop and degases with acetonitrile. 90ml of acetonitrile (09 volume) was added and cooled to -5°C and than further added 10ml of purified water. lOgm of selectfluor(0.028mol) was added in two lots at 0°C(+5°C) in 02 volume of acetonitrile. Reaction mass was stirred at 10°C to 15°C for 12 hours. In-process check by TLC against before selectfluor reaction mass it should be nil. Adjust pH neutral (~7) by liq. ammonia solution at 0°C+5°C. Reaction mass was quenched in 500ml of purified water (lOOvolume) at ambient temperature. Reaction mass was filtered and washed with purified water till neutral pH (~7). Wet material was dried at 45°C+5°C till moisture content less than 0.50% to get stage-3rd. Yield=8.60gm, (75.17%), HPLC Purity= 94.12%.
Stage -IV (Preparation of Flunisolide from Flunisolide acetate):

Flunisolide acetate Flunisolide
80ml of methanol (lOvolume) and 80ml of methylene chloride (lOvolume) was charged in a glass flask under inert atmosphere at ambient temperature than added 8.0gm of stage-3r (0.017mol) at ambient temperature. Cooled to -5°C and added 0.16gm of sodium hydroxide (0.004mol) solution in 8ml of methanol at -5°C(+5°C) and stir for 02 hours at -5°C(+5°C). In-process check by TLC against stage-3 ‘ it should be nil. Adjust pH neutral(~7) by acetic acid and reaction mass was distilled under vacuum at below 40°C(+5°C) till ~40ml of volume. Cool to 0°C to 5°C and stir for one hour. Reaction mass was filtered and washed with precooled methanol to get wet material. Wet material was dried at 45°C (+5°C) till moisture content less than 0.50% to get Flunisolide crude. Yield=6.0gm, (82.30%), HPLC Purity=86.50%.
(Purification)
6.0gm of crude Flunisolide(0.014mol) was dissolved in 65ml of ethyl acetate (10.83volume) and 35ml of n-hexane (5.83volume) mixture and clear solution was passed through 60gm of silica gel column. Column was washed with 975ml of ethyl acetate (162.5volume) and 525ml of ft-hexane (87.5volume) mixture. Eluted fraction was distilled under vacuum till 3 to 4 volume of input than cooled it to 0°C and filter to get wet material. Wet material was dried at 50°C (+5°C) till moisture content less than 0.50% to get Flunisolide. Yield=4.28gm, HPLC Purity=95.60%.
Example 7: Process for synthesis of Triamcinolone from 3TR
S

lOOgm of 3TR (0.27mol) was suspended in 1300ml (13 volume) acetone. Cool to -5°C to- 10°C than added 4.0 ml (0.062mol) perchloric acid solution and 50gm of dibromantin. Reaction maintained at same temperature for 02 hours. In-process check by TLC against 3TR, it should be nil. Added lOOgm of potassium carbonate solution (0.723 mol) in 5 lots and reaction was maintained at 35°C (+2°C). In-process check by TLC against step-I reaction mass, it should be nil. Cool to 0°C+5°Cand adjust pH neutral (~7) by 36ml of acetic acid (0.63 mol). Added 3.0L of purified water (30 volume). Filter and washed with purified water till neutral pH (~7). Wet material was dried at 45°C (+2°C) till moisture content less than 0.50% to get stage-I. Yield=85.30gm, (81.74%), HPLC Purity=96.54%. Stage -II:

80gm of stage-I (0.21 mol) was dissolved in 4.0L of acetone (50 volume) and 208ml of purified water (2.6 volume). Cool to -5°C (+2°C) than added 32ml of formic acid (0.85 mol.) and 48gm of potassium per magnate (0.30 mol) at -5°C (+2°C). Reaction was maintained at same temperature for one hour. In-process check by TLC against stage-I, it should be nil. Added sodiummetabisulphite solution (8 gm in 80 ml of water) at -5°C+2°C. Temperature was raised up to 27°C and filtered through hyflow bed and washed with acetone. Acetone was distilled under vacuum till 3 to 4 volume of stage-I than further cooled to 0°C to 5°C and added 480ml of purified water, stirred, filter and washed with purified water to get wet stage- II. Wet material was dried at 50°C (+5°C) till moisture content less than 3.0% to get stage-II. Yield=82gm, (94.13%), HPLC Purity=97.75%.
Stage -III:

Stage-II Triamcinolone acetate
160ml of hydrofluoric acid (70%) (6.72mol) was cooled at -25°C to -30°C than added 40gm of stage-II (0.096mol). Reaction was maintained at -25°C to -30°C for 04 hours. In-process check by TLC against stage-II, it should be nil. Added 280ml of purified water at 0°C and 650ml of liq. ammonia at 20°C than reaction mass was quenched in 200ml of liq. ammonia and 500ml of purified water solution at 15°C(+2°C), stirred, filtered and washed with purified water till neutral pH(~7). Wet material was dried at 45°C to 50°C to get stage-Ill Yield=40gm, (95.42%), HPLC Purity=88.71%
(Purification)
40gm of stage-Ill crude (0.0916 mol) was refluxed in 160ml of acetone. Cool to 0°C. Filtered and washed with minimum precooled acetone. Wet material was dried at 50°C+5°C till moisture content comes less than 0.50% to get stage-Ill. Yield=24.9gm HPLC Purity=95.17%.

24gm of stage-Ill (0.055mol) was dissolved in 240ml of methanol (lOvolume) and 240ml of methylene chloride (lOvolume) mixture under argon bubbling. Cool to -5°C+2°C and added 0.48gm of sodium hydroxide (0.012mol) solution in 24ml of methanol (Olvolume) at – 5°C+2°C. Reaction was maintaining at -5°C (+2°C) for 03hours. In-process check by TLC against stage-Ill, it should be nil. Adjust pH neutral by adding 0.70ml of acetic acid at -5°C (+2°C). Reaction mass distilled at below 40°C under vacuum till 04-05 volume of input. Cooled to 0°C+5°Cand stir for one hour than filtered and washed with minimum precooled methanol. Wet material was dried at 45°C to 50°C till moisture content less than 0.50%. Yield=18.50gm, (85.29%), HPLC Purity=98.60%.
Example 8: Process for synthesis of Triamcinolone Hexacetonide from 3TR
S

lOOgm of 3TR (0.27288 mol) was suspended in 1300ml (13 volume) acetone. Cool to -5°C to -10°C than added 4.0 ml (0.0625 mol) perchloric acid solution and 50gm of dibromantin. Reaction was maintained at same temperature for 02 hours. In-process check by TLC against 3TR, it should be nil. Added lOOgm of potassium carbonate solution (0.723 mol) in 5 lots and reaction was maintained at 35°C (+2°C). In-process check by TLC against step-I reaction mass, it should be nil. Cool to 0°C (+5°C) and adjust pH neutral (~7) by 36ml of acetic acid (0.63 mol). Added 3.0L of purified water (30 volume). Filter and washed with purified water till neutral pH. Wet material was dried at45°C(+2°C) till moisture content less than 0.50% to get stage-lst. Yield =87gm, (83.36%), HPLC Purity=97.883%. Stage-II :

80gm of stage-I (0.21 mol) was dissolved in 4.0L of acetone (50 volume) and 208ml of purified water (2.6 volume). Cool to -5°C than added 32ml of formic acid (0.85 mol.) and 48gm of potassium permanganate (0.30 mol) at -5°C+2°C. Reaction maintained at -5°C (+2°C) for one hour. In-process check by TLC against stage-I, it should be nil. Added sodium metabisulphite solution (8 gm in 80 ml water) at -5°C (+2°C). Temperature raised up to 27°Cand filtered through hyflow bed and washed with acetone. Acetone was distilled under vacuum till 3 to 4 volume of stage-I than cooled to 0°C to 5°C and added 480ml of purified water, stirred, filtered and washed with purified water to get wet stage-II. Wet material was dried at 50°C (+5°C) till moisture content less than 3.0% to get stage-2nd. Yield=78.30gm, (89.88%), HPLC Purity=99.18%.
Stage – III:

300ml of hydrofluoric acid (12.60mol) was cooled at -25°C to -30°C than added 75gm of stage-II (0.180mol). Reaction was maintained at -25°C to -30°C for 04 hours. In-process check by TLC against stage-II. It should be nil. Reaction mass was cooled to -50°C than added 45ml of acetone (0.60volume) at -45°C to -50°C. Reaction maintained at -45°Cto – 50°C for 02 hours. In-process check by TLC against reaction input, it should be nil. Added 565ml of purified water at 0°C and 340ml of liq. ammonia at 20°C than reaction mass was quenched in 410ml of liq. ammonia and 735ml of purified water solution at 15°C(+2°C), stirred, filtered and washed with purified water till neutral pH (~7). Wet material was dried at 45°C to 50°Cto get stage-3rd. Yield=78.50gm, (91.48%), HPLC Purity=91.59%.
(Purification)
76 gm of stage-Ill Crude (0.16 mol) was dissolved in 760ml of methylene chloride (01 volume) and 760ml of methanol (lOvolume) mixture at ambient temperature. Stirred till clear solution and added 7.6gm of activated charcoal (0. lOvolume) than further stir for 30 minutes and filtered through hyflow bed and washed with methanol (one volume) and methylene chloride (one volume) mixture. Total filtrate was distilled under vacuum till 3 to 4 volume of input. Cooled to 0°C to 5°Cand stir for 02 hours. Filtered and washed with minimum precooled methanol. Wet material was dried at 45°C to 50°C till moisture content less than 0.50% to get purified stage-3rd. Yield=62gm, HPLC Purity=98.633%
Stage -IV : (Preparation of Triamcinolone acetonide from Stage – III)

Stage- Ill Triamcinolone acetonide
60gm of stage-Ill (0.1259 mol) dissolved in 600ml of methanol (lOvolume) and 600ml of methylene chloride (lOvolume) mixture under inert atmosphere. Cool to -5°C and added 1.2gm of sodium hydroxide (0.03mol) solution in 60ml of methanol (Olvolume) at -5°C (+2°C). Reaction maintained at -5°C+2°C for 03 hours. In-process check by TLC against stage-Ill, it should be nil. Adjust pH neutral (~7) by adding 1.8ml of acetic acid at -5°C+2°C. Reaction mass was distilled below 40°C under vacuum till 3 to 4 volume of input. Cool to
30°C and added 120ml of purified water, stir for one hour than filtered and washed with purified water till neutral pH (~7). Wet material was dried at 45°C to 50°C till moisture content less than 0.50% to get stage-4111 (Triamcinolone acetonide). Yield=52gm, (95.04%), HPLC Purity=99.21%.
(Purification)
50gm of crude material (0.12 mol) dissolved in 1100ml of acetone (22volume) and 100ml of purified water (02volume) at 50°C than added 2.5gm of activated charcoal and stirred for one hour at same temperature. Filter through hyflow bed and washed with 120ml acetone (2.40volume). Filtrate was distilled below 40°C under vacuum till 3 to 4 volume of input. Cool to 0°C to 5°C and maintained for one hour at same temperature. Filtered and washed with water. Wet material was dried at 45°C to 50°C till moisture content less than 0.50% to get purified stage-4th. Yield =43gm, HPLC Purity=99.40%
-V: (Preparation of Triamcinolone Hexacetonide from Triamcinolone acetonide):

50ml of pyridine (lOvolume) charged in a glass flask and added lOgm of Triamcinolone acetonide (0.023mol) at ambient temperature. Heated to 80°C to 90°C than added 10ml of 3, 3-dimethyl butyryl chloride (O.l lmol) at 80°C to 90°C. Stirred at 80°C to 90°C for 02 hours. In-process check by TLC against Triamcinolone acetonide, it should be nil. Reaction mass cooled to ambient temperature and reaction mass was quenched in 1000ml of purified water (lOOvolume) at ambient temperature, stir for one hour than filtered and washed with purified water till neutral pH (~7). Wet material was dried at 50°C (+5°C) till moisture content less than 1.0% to get stage-5th (Triamcinolone Hexacetonide). Yield=12gm, (97.90%), HPLC Purity=98.63%.
(Purification)
120ml of methanol and 120ml of methylene chloride charged in a glass flask and added 12gm of crude material, stir till clear solution than added 1.2gm of activated charcoal and stir for 30 minutes. Filtered through hyflow bed and washed with 12ml of methanol and 12ml of methylene chloride mixture. Total filtrate was distilled under vacuum at below 40°C till 5 to 6 volume of crude. Cooled to 0°C+5°C and stir for one hour. Filtered and washed with 12ml of precooled methanol. Wet material was dried at 40°C+5°C till moisture content less than 0.50% to get TrimcinolneHexacetonide. Yield=8.8gm, HPLC Purity=99.625%//////////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Medical uses

Aristocort brand triamcinolone cream
Triamcinolone is used to treat a number of different medical conditions, such as eczema, alopecia areata, lichen sclerosus, psoriasis, arthritis, allergies, ulcerative colitis, lupus, sympathetic ophthalmia, temporal arteritis, uveitis, ocular inflammation, keloids, urushiol-induced contact dermatitis, aphthous ulcers (usually as triamcinolone acetonide), central retinal vein occlusion, visualization during vitrectomy and the prevention of asthma attacks.[12][13][14]
The derivative triamcinolone acetonide is the active ingredient in various topical skin preparations (cream, lotion, ointment, aerosol spray) designed to treat skin conditions such as rash, inflammation, redness, or intense itching due to eczema[15] and dermatitis.[16]
Contraindications
Contraindications for systemic triamcinolone are similar to those of other corticoids. They include systemic mycoses (fungal infections) and parasitic diseases, as well as eight weeks before and two weeks after application of live vaccines. For long-term treatment, the drug is also contraindicated in people with peptic ulcers, severe osteoporosis, severe myopathy, certain viral infections, glaucoma, and metastasizing tumours.[17]
There are no contraindications for use in emergency medicine.[4]
Side effects
Further information: Glucocorticoid § Side effects
Side effects of triamcinolone are similar to other corticoids. In short-term treatment up to ten days, it has very few adverse effects; however, sometimes gastrointestinal bleeding is seen, as well as acute infections (mainly viral) and impaired glucose tolerance.[4]
Side effects of triamcinolone long-term treatment may include coughing (up to bronchospasms), sinusitis, metabolic syndrome–like symptoms such as high blood sugar and cholesterol, weight gain due to water retention, and electrolyte imbalance, as well as cataract, thrush, osteoporosis, reduced muscle mass, and psychosis.[5][6][17] Triamcinolone injections can cause bruising and joint swelling.[5] Symptoms of an allergic reaction include rash, itch, swelling, severe dizziness, trouble breathing,[18] and anaphylaxis.[17]
Overdose
No acute overdosing of triamcinolone has been described.[17]
Interactions
Drug interactions are mainly pharmacodynamic, that is, they result from other drugs either adding to triamcinolone’s corticoid side effects or working against its desired effects. They include:[4][17]
- Atropin and other anticholinergics can substantially increase pressure in the eyes.
- Antidiabetic drugs can become less effective because triamcinolone causes diabetes-like symptoms.
- Aspirin and other NSAIDs, as well as anticoagulants such as warfarin, add to the risk of gastrointestinal bleeding.
- Diuretics that excrete potassium (such as loop diuretics and thiazides) can increase the risk of hypokalemia and thus lead to abnormal heart rhythm.
- Cardiac glycosides may have more adverse effects due to reduced potassium levels in the blood.
- The risk for blood count changes is increased when combining triamcinolone with ACE inhibitors.
Triamcinolone and other drugs can also influence each other’s concentrations in the body, amounting to pharmacokinetic interactions such as:[4][17]
- Rifampicin, phenytoin, carbamazepine and other inducers of the liver enzyme CYP3A4[19] speed up metabolization of triamcinolone and can therefore reduce its effectiveness.
- Conversely, CYP3A4 inhibitors such as ketoconazole and itraconazole can increase its concentrations in the body and the risk for adverse effects.
- Blood concentrations of ciclosporin can be increased.
Pharmacology
Mechanism of action
Further information: Glucocorticoid § Mechanism of action
Triamcinolone is a glucocorticoid that is about five times as potent as cortisol, but has very little mineralocorticoid effects.[4]
Pharmacokinetics
When taken by mouth, the drug’s bioavailability is over 90%. It reaches highest concentrations in the blood plasma after one to two hours and is bound to plasma proteins to about 80%. The biological half-life from the plasma is 200 to 300 minutes; due to stable complexes of triamcinolone and its receptor in the intracellular fluid, the total half-life is significantly longer at about 36 hours.[4][5]
A small fraction of the substance is metabolized to 6-hydroxy- and 20-dihydro-triamcinolone; most of it probably undergoes glucuronidation, and a smaller part sulfation. Three quarters are excreted via the urine, and the rest via the faeces.[4][17]
Due to corticoids’ mechanism of action, the effects are delayed as compared to plasma concentrations. Depending on the route of administration and the treated condition, the onset of action can be from two hours up to one or two days after application; and the drug can act much longer than its elimination half-life would suggest.[4][5]
Chemistry
Triamcinolone is a synthetic pregnane corticosteroid and derivative of cortisol (hydrocortisone) and is also known as 1-dehydro-9α-fluoro-16α-hydroxyhydrocortisone or 9α-fluoro-16α-hydroxyprednisolone as well as 9α-fluoro-11β,16α,17α,21-tetrahydroxypregna-1,4-diene-3,20-dione.[20][21]
The substance is a light-sensitive, white to off-white, crystalline powder, or has the form of colourless, matted crystals. It has no odour or is nearly odourless. Information on the melting point varies, partly due to the substance’s polymorphism: 260 to 263 °C (500 to 505 °F), 264 to 268 °C (507 to 514 °F), or 269 to 271 °C (516 to 520 °F) can be found in the literature.[4]
Solubility is 1:500 in water and 1:240 in ethanol; it is slightly soluble in methanol, very slightly soluble in chloroform and diethylether, and practically insoluble in dichloromethane. The specific rotation is {\displaystyle [\alpha ]_{D}^{20}}![{\displaystyle [\alpha ]_{D}^{20}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/b67b300e0d318e964fcdab92c872ed89d1d4f43f)
Society and culture
In 2010, TEVA and Perrigo launched the first generic inhalable triamcinolone.[22]
According to Chang et al. (2014), “Triamcinolone acetonide (TA) is classified as an S9 glucocorticoid in the 2014 Prohibited List published by the World Anti-Doping Agency, which caused it to be prohibited in international athletic competition when administered orally, intravenously, intramuscularly or rectally”.[23]
See also
- Glucocorticoid (a chart comparing various glucocorticoids)
References
- ^ “Kenalog Intra-articular / Intramuscular Injection – Summary of Product Characteristics (SmPC)”. (emc). 10 June 2020. Retrieved 20 August 2020.
- ^ “Nasacort Allergy 55 micrograms/dose Nasal Spray suspension – Summary of Product Characteristics (SmPC)”. (emc). 30 August 2018. Retrieved 20 August 2020.
- ^ “Adcortyl Intra-Articular/Intradermal Injection 10mg/ml – Summary of Product Characteristics (SmPC)”. (emc). 11 December 2017. Retrieved 20 August 2020.
- ^ Jump up to:a b c d e f g h i j k l m n Dinnendahl V, Fricke U, eds. (2004). Arzneistoff-Profile (in German). Vol. 10 (19 ed.). Eschborn, Germany: Govi Pharmazeutischer Verlag. Triamcinolon. ISBN 978-3-7741-9846-3.
- ^ Jump up to:a b c d e f Triamcinolone (systemic) Professional Drug Facts. Accessed 2020-08-19.
- ^ Jump up to:a b c d e f g “Triamcinolone Monograph for Professionals”. Drugs.com. American Society of Health-System Pharmacists. Retrieved 3 March 2019.
- ^ “Triamcinolone Use During Pregnancy”. Drugs.com. Retrieved 3 March 2019.
- ^ Fischer J, Ganellin CR (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 486. ISBN 978-3-527-60749-5.
- ^ Vallerand, April Hazard (2018). Davis’s Drug Guide for Nurses. F.A. Davis. p. 365. ISBN 978-0-8036-7000-6.
- ^ “The Top 300 of 2019”. ClinCalc. Retrieved 16 October 2021.
- ^ “Triamcinolone – Drug Usage Statistics”. ClinCalc. Retrieved 16 October 2021.
- ^ Triamcinolone – Drugs.com
- ^ Triamcinolone Inhalation – Drugs.com
- ^ Alcon Receives FDA Approval of Triesence Injectable Triamcinolone Suspension for Use in Eye Surgery – Drugs.com
- ^ Chong M, Fonacier L (December 2016). “Treatment of Eczema: Corticosteroids and Beyond”. Clinical Reviews in Allergy & Immunology. 51 (3): 249–262. doi:10.1007/s12016-015-8486-7. PMID 25869743. S2CID 44337035.
- ^ Eichenfield LF, Tom WL, Berger TG, Krol A, Paller AS, Schwarzenberger K, et al. (July 2014). “Guidelines of care for the management of atopic dermatitis: section 2. Management and treatment of atopic dermatitis with topical therapies”. Journal of the American Academy of Dermatology. 71 (1): 116–32. doi:10.1016/j.jaad.2014.03.023. PMC 4326095. PMID 24813302.
Topical corticosteroids (TCS) are used in the management of AD in both adults and children and are the mainstay of anti-inflammatory therapy.
- ^ Jump up to:a b c d e f g Haberfeld H, ed. (2020). Austria-Codex (in German). Vienna: Österreichischer Apothekerverlag. Volon 4 mg-Tabletten.
- ^ “Drugs and Treatments – Nasacort AQ Nasl – Patient Handout”. WebMD. Retrieved 2008-03-24.
- ^ Moore CD, Roberts JK, Orton CR, et al. (2012). “Metabolic Pathways of Inhaled Glucocorticoids by the CYP3A Enzymes”. Drug Metab. Dispos. 41 (2): 379–389. doi:10.1124/dmd.112.046318. PMC 3558858. PMID 23143891.
- ^ Elks J (14 November 2014). The Dictionary of Drugs: Chemical Data: Chemical Data, Structures and Bibliographies. Springer. pp. 1228–. ISBN 978-1-4757-2085-3.
- ^ Index Nominum 2000: International Drug Directory. Taylor & Francis. January 2000. pp. 1054–. ISBN 978-3-88763-075-1.
- ^ Perrigo Announces Launch Of Generic Version Of Nasacort AQ – CBS Detroit
- ^ Chang CW, Huang TY, Tseng YC, Chang-Chien GP, Lin SF, Hsu MC (November 2014). “Positive doping results caused by the single-dose local injection of triamcinolone acetonide”. Forensic Science International. 244: 1–6. doi:10.1016/j.forsciint.2014.07.024. PMID 25126738.
External links
- “Triamcinolone”. Drug Information Portal. U.S. National Library of Medicine.
- “Triamcinolone Topical”. MedlinePlus.
- “Triamcinolone Nasal Spray”. MedlinePlus.
- “Triamcinolone Acetonide Cream”. HealthClubFinder.
///////////////TRIAMCINOLONE, TU3850000, トリアムシノロン , 去炎松 , Glucocorticoid
[H][C@@]12C[C@@H](O)[C@](O)(C(=O)CO)[C@@]1(C)C[C@H](O)[C@@]1(F)[C@@]2([H])CCC2=CC(=O)C=C[C@]12C

NEW DRUG APPROVALS
ONE TIME
$10.00
CDSCO INDIA APPROVED 20.01.2022
Triamcinolone Hexacetonide injectable suspension
20mg/ml
For intraarticular, intra-synovial or
periarticular use in adults and adolescents for
the symptomatic treatment of subacute and
chronic inflammatory joint diseases including
rheumatoid arthritis and Juvenile Idiopathic
Arthritis (JIA), Osteoarthritis and posttramautic arthritis, Synovitis, tendinitis,
bursitis and epicondylitis.
Triamcinolone hexacetonide (brand name Aristospan; also known as triamcinolone acetonide 21-tebutate) is a synthetic glucocorticoid corticosteroid.[1][2][3]
References
- ^ Elks J (14 November 2014). The Dictionary of Drugs: Chemical Data: Chemical Data, Structures and Bibliographies. Springer. pp. 1228–. ISBN 978-1-4757-2085-3.
- ^ Index Nominum 2000: International Drug Directory. Taylor & Francis. 2000. p. 1657. ISBN 978-3-88763-075-1.
- ^ Morton IK, Hall JM (6 December 2012). Concise Dictionary of Pharmacological Agents: Properties and Synonyms. Springer Science & Business Media. pp. 280–. ISBN 978-94-011-4439-1.
UPDATE
| Clinical data | |
|---|---|
| Trade names | Aristospan |
| Other names | Triamcinolone acetonide 21-tebutate; Triamcinolone acetonide 21-(tert-butylacetate); 9α-Fluoro-11β,16α,17α,21-tetrahydroxypregna-1,4-diene-3,20-dione cyclic 16,17-acetal with acetone, 21-(3,3-dimethylbutyrate); 9α-Fluoro-11β-hydroxy-16α,17α-((1-methylethylidene)bis(oxy))pregna-1,4-diene-3,20-dione 21-(3,3-dimethylbutyrate) |
| Drug class | Corticosteroid; Glucocorticoid |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 5611-51-8 |
| PubChem CID | 21826 |
| ChemSpider | 20516 |
| UNII | I7GT1U99Y9 |
| ChEBI | CHEBI:9670 |
| ChEMBL | ChEMBL1200878 |
| CompTox Dashboard (EPA) | DTXSID0048634 |
| ECHA InfoCard | 100.024.575 |
| Chemical and physical data | |
| Formula | C30H41FO7 |
| Molar mass | 532.649 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
ENSITRELVIR
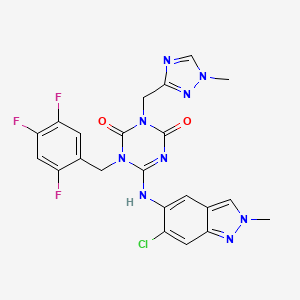
Ensitrelvir
S-217622, S 217622, Xocova, SHIONOGI,
6-[(6-chloro-2-methylindazol-5-yl)amino]-3-[(1-methyl-1,2,4-triazol-3-yl)methyl]-1-[(2,4,5-trifluorophenyl)methyl]-1,3,5-triazine-2,4-dione
CAS 2647530-73-0
| C22H17ClF3N9O2531.9 | |
| Synonyms | BDBM513874bioRxiv20220126.477782, S-217622 |
|---|
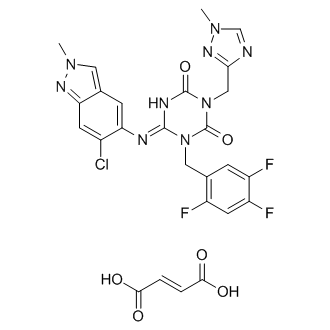
Ensitrelvir fumarate
CAS No. : 2757470-18-9
C22 H17 Cl F3 N9 O2 . C4 H4 O4
1,3,5-Triazine-2,4(1H,3H)-dione, 6-[(6-chloro-2-methyl-2H-indazol-5-yl)imino]dihydro-3-[(1-methyl-1H-1,2,4-triazol-3-yl)methyl]-1-[(2,4,5-trifluorophenyl)methyl]-, (6E)-, (2E)-2-butenedioate (1:1)
| Formula: | C26H21ClF3N9O6 |
|---|---|
| M. Wt. : | 647.95 |
FDA 2026, APPROVALS 2026, 5/29/2026, Xocova
To use as post-exposure prophylaxis of coronavirus disease 2019 (COVID-19) following contact with an individual who has COVID-19
A Phase 1 study of S-217622 in healthy adult participants (jRCT2031210202)
Japan Registry of Clinical Trials Web Site 2021, July 16
PMDA APPROVED 2022/11/22, Xocova
Ensitrelvir[1] (code name S-217622, brand name Xocova)[2] is an antiviral drug developed by Shionogi in partnership with Hokkaido University, which acts as an orally active 3C-like protease inhibitor for the treatment of COVID-19 infection.[3][4] It is taken by mouth, and has been successfully tested against the recently emerged Omicron variant.[5]
About S-217622
S-217622, a therapeutic drug for COVID-19, is a 3CL protease inhibitor created through joint research between Hokkaido University and Shionogi. SARS-CoV-2 has an enzyme called 3CL protease, which is essential for the replication of the virus. S-217622 suppresses the replication of SARS-CoV-2 by selectively inhibiting 3CL protease. Shionogi has already been submitting the non-clinical, manufacturing/CMC data, and clinical trial data obtained so far to the PMDA. Currently the Phase 3 part of a Phase 2/3 clinical trial in patients with mild/moderate symptoms and the Phase 2b/3 part in patients with asymptomatic/only mild symptoms are in progress.
SYN
J.Med.Chem.2024,67,4376−4418
Ensitrelvir fumaric acid (3), also referred to as S-217622, is an oral noncovalent SARS-CoV-2 main protease (Mpro) inhibitordeveloped by Shionogi & Co. that was approved by the japan Pharmaceuticals and Medical Devices Agency (PMDA)for the treatment of disease caused by SARS-CoV-2 (COVID
19) infection. Dosed once daily for 5 days, ensitrelvirsuppresses the replication of SARS-CoV-2 in infected patients as a result of its inhibition of the viral mpro.25,26
Ensitrelvir retains potent inhibitory activity against many of the most common M mutants and exhibits antiviral activity against a wide variety of circulating SARS-CoV-2 variants. 27is the second Mpro
Ensitrelvir inhibitor approved for the treatment of 28 disease caused by COVID-19. Unlike the first approved treatment, Paxlovid, ensitrelvir does not require coadministration with a CYP3A4 inhibitor to attenuate metabolism in vivo.
Furthermore, crystal structures of ensitrelvir in complex with the main proteases of three other human-infecting coronaviruses (MERS-CoV, SARS-CoV, and HCoV-NL63)
A convergent, kilogram-scale synthesis of ensitrelvir suitable for manufacturing has been described in the literature by researchers at Shionogi. 30 The synthetic approach involved the union of two key building blocks indazole 3.7 and 1,3,5triazinone 3.14, each necessitating development of a scale worthy route. The preparation of triazinone 3.14 necessitated construction of a triazolyl methylene chloride subunit which
began with the reduction of triazole ester 3.1 with aluminum hydride 3.2 (a less pyrophoric alternative to LAH yet still required aqueous Rochelle salt quench to chelate excess aluminum) 31to provide alcohol 3.3, which was then convertedto the corresponding chloride and isolated as the triazole HCl
salt 3.4 (Scheme 6). Assembly of indazole intermediate 3.7began with regioselective nitration of benzaldehyde 3.5followed by treatment with hydrazine hydrate in aqueous EtOHtoprovide indazole3.6(Scheme7).Fascinatingly, the Shionogi team isolated a variety of byproducts during the
conversionof3.5to3.6whichsupportedtheirhypothesisforareaction mechanism that likely equilibrated through a dibenzylidenehydrazine intermediateenroute tothedesired
indazole3.6.Ascreenofelectrophilicmethyl sourcesrevealed thatMeerwein’s salt facilitatedthebest conversionof 3.6to the correspondingN2-monomethylated indazole; subsequenthydrogenative nitro reduction furnished the key indazole intermediate 3.7.Construction of the ensitrelvir core started with reaction of carboximidamide 3.8 with t-butyl isocyanate followed by N,N′carbonyldiimidazole (CDI) to secure 1,3,5-triazinone 3.10(Scheme 8). Subsequent N-alkylation with bromide 3.11provided benzyl triazinone 3.12. Substitution of the pyrazolewith m-cresol was accomplished under acidic conditions. The
authors report that m-cresol was identified as a leaving group that facilitated introduction of indazole 3.7 with a minimal number of byproducts in a later step of the synthesis. The TFA-mediated reaction concomitantly removed the N-tertbutyl group providing compound 3.13 in 91% yield. Nalkylation with chloride 3.4 in the presence of a base resulted in intermediate 3.14 which was then treated with building
block 3.7 in the presence of anhydrous acetic acid. Isolation of ensitrelvir fumaric acid was achieved by exposure to fumaric acid in aqueous acetone.
(25) Yotsuyanagi, H.; Ohmagari, N.; Doi, Y.; Imamura, T.;
Sonoyama, T.; Ichihashi, G.; Sanaki, T.; Tsuge, Y.; Uehara, T.;
Mukae, H. A phase 2/3 study of S-217622 in participants with SARS
CoV-2 infection (Phase 3 part). Medicine 2023, 102, No. e33024.
(26) Mukae, H.; Yotsuyanagi, H.; Ohmagari, N.; Doi, Y.; Imamura,
T.; Sonoyama, T.; Fukuhara, T.; Ichihashi, G.; Sanaki, T.; Baba, K.;
Takeda, Y.; Tsuge, Y.; Uehara, T. A randomized phase 2/3 study of
ensitrelvir, a novel oral SARS-CoV-2 3C-like protease inhibitor, in
Japanese patients with mild-to-moderate COVID-19 or asymptomatic
SARS-CoV-2 infection: results of the phase 2a part. Antimicrob. Agents
Chemother. 2022, 66, No. 00697.
(27) Kawashima, S.; Matsui, Y.; Adachi, T.; Morikawa, Y.; Inoue, K.;
Takebayashi, S.; Nobori, H.; Rokushima, M.; Tachibana, Y.; Kato, T.
Ensitrelvir is effective against SARS-CoV-2 3CL protease mutants
circulating globally. Biochem. Biophys. Res. Commun. 2023, 645, 132−
136.
(28) Unoh, Y.; Uehara, S.; Nakahara, K.; Nobori, H.; Yamatsu, Y.;
Yamamoto, S.; Maruyama, Y.; Taoda, Y.; Kasamatsu, K.; Suto, T.;
et al. Discovery of S-217622, a noncovalent oral SARS-CoV-2 3CL
protease inhibitor clinical candidate for treating COVID-19. J. Med.
Chem. 2022, 65, 6499−6512
(29) Lin, C.; Jiang, H.; Li, W.; Zeng, P.; Zhou, X.; Zhang, J.; Li, J.
Structural basis for the inhibition of coronaviral main proteases by
ensitrelvir. Structure 2023, 31, 1016.
(30) Kawajiri, T.; Kijima, A.; Iimuro, A.; Ohashi, E.; Yamakawa, K.;
Agura, K.; Masuda, K.; Kouki, K.; Kasamatsu, K.; Yanagisawa, S.; et al.
Development of a manufacturing process toward the convergent
synthesis of the COVID-19 antiviral Ensitrelvir. ACS Cent. Sci. 2023,
9, 836−843.
(31) Gugelchuk, M.; Silva, III, L. F.; Vasconcelos, R. S.; Quintiliano,
S. A. P. Sodium bis(2-methoxyethoxy)aluminum hydride. In
Encyclopedia of Reagents for Organic Synthesis; Charette, A., Bode, J.,
Rovis, T., Shenvi, R., Eds.; John Wiley & Sons, Ltd., 2007.


Syn
Discovery of S-217622, a Non-Covalent Oral SARS-CoV-2 3CL Protease Inhibitor Clinical Candidate for Treating COVID-19
View ORCID ProfileYuto Unoh, View ORCID ProfileShota Uehara, View ORCID ProfileKenji Nakahara, View ORCID ProfileHaruaki Nobori, Yukiko Yamatsu, View ORCID ProfileShiho Yamamoto, View ORCID ProfileYuki Maruyama, View ORCID ProfileYoshiyuki Taoda, View ORCID ProfileKoji Kasamatsu, View ORCID ProfileTakahiro Suto, Kensuke Kouki, View ORCID ProfileAtsufumi Nakahashi, Sho Kawashima, View ORCID ProfileTakao Sanaki, Shinsuke Toba, Kentaro Uemura, Tohru Mizutare, View ORCID ProfileShigeru Ando, View ORCID ProfileMichihito Sasaki, View ORCID ProfileYasuko Orba, View ORCID ProfileHirofumi Sawa, View ORCID ProfileAkihiko Sato, View ORCID ProfileTakafumi Sato, View ORCID ProfileTeruhisa Kato, View ORCID ProfileYuki Tachibana
doi: https://doi.org/10.1101/2022.01.26.477782
https://www.biorxiv.org/content/10.1101/2022.01.26.477782v1.full
The coronavirus disease 2019 (COVID-19) pandemic, caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has resulted in millions of deaths and threatens public health and safety. Despite the rapid global spread of COVID-19 vaccines, effective oral antiviral drugs are urgently needed. Here, we describe the discovery of S-217622, the first oral non-covalent, non-peptidic SARS-CoV-2 3CL protease inhibitor clinical candidate. S-217622 was discovered via virtual screening followed by biological screening of an in-house compound library, and optimization of the hit compound using a structure-based drug-design strategy. S-217622 exhibited antiviral activity in vitro against current outbreaking SARS-CoV-2 variants and showed favorable pharmacokinetic profiles in vivo for once-daily oral dosing. Furthermore, S-217622 dose-dependently inhibited intrapulmonary replication of SARS-CoV-2 in mice, indicating that this novel non-covalent inhibitor could be a potential oral agent for treating COVID-19.
Chemistry

The synthetic scheme for compound 1 is described in Scheme 1. Starting from the pyrazole derivative 4, cyclization with Ethyl isocyanatoacetate and CDI was conducted, giving 5 in 90% yield. Then, an alkylation with 5-bromomethyl-1,2,3-trifluorobenzene followed by introduction of a 4-difluoromethoxy-2-methylaniline unit, to give 7 (40% in 2 steps). The ester group in 7 was hydrolyzed and then amidated with methylamine, yielding 1 (58% in 2 steps). Compound 2 was synthesized similarly as shown in Scheme 2.
S-217622 (3) was synthesized as described in Scheme 3. Starting from known compound 9,21 an alkylation with 1-(bromomethyl)-2,4,5-trifluorobenzene gave 10 in 93% yield. Then, the 3-tert-Bu group was removed and the triazole unit was introduced, and the substitution of the SEt moiety with the indazole unit finally gave S-217622 (3).
21 Kai, H.; Kameyama, T.; Horiguchi, T.; Asahi, K.; Endoh, T.; Fujii, Y.; Shintani, T.; Nakamura, K.; Matsumoto, S.; Hasegawa, T.; Oohara, M.; Tada, Y.; Maki, T.; Iida, A. Preparation of triazine derivatives and pharmaceutical compound that contains same and exhibits analgesic activity. WO 2012020749 A1, Feb 16, 2012

Scheme 1.
Reagents and Conditions: (a) ethyl isocyanato-acetate, DBU, CDI, DMA, –10 °C to rt, 90%; (b) 5-bromomethyl-1,2,3-trifluorobenzene, N,N-diisopropylethylamine, DMA, 60 °C; (c) 4-difluoromethoxy-2-methylaniline, tert-butanol, 100 °C, 40% in 2 steps; (d) (i) NaOH aq., THF/MeOH, rt; (ii) methylamine, HATU, N,N-diisopropylethylamine, THF, rt., 58% in 2 steps.

Scheme 2.
Reagents and Conditions: (a) 6-chloro-2-methyl-2H-indazol-5-amine, tert-amyl alcohol, 100 °C, 44% in 2 steps from 5; (b) (i) NaOH aq., THF/MeOH, rt; (ii) methylamine, HATU, N,N-diisopropylethylamine, THF, rt., 29% in 2 steps.

Scheme 3.
Reagents and Conditions: (a) 1-(bromomethyl)-2,4,5-trifluorobenzene, K2CO3, MeCN, 80 °C, 93%; (b) TFA, rt, 97%; (c) 3-(chloromethyl)-1-methyl-1H-1,2,4-triazole hydrochloride, K2CO3, DMF, 60 °C, 45%; (d) 6-chloro-2-methyl-2H-indazol-5-amine, LHMDS, THF, 0 °C to rt., 25%.
(6E)-6-[(6-Chloro-2-methyl-2H-indazol-5-yl)imino]-3-[(1-methyl-1H-1,2,4-triazol-3-yl)methyl]-1-(2,4,5-trifluorobenzyl)-1,3,5-triazinane-2,4-dione (3, S-217622)
To a solution of 12 (300 mg, 0.727 mmol) and 6-chloro-2-methyl-2H-indazol-5-amine (172 mg, 0.946 mmol) in THF (6 mL) was added LHMDS (1M in THF; 1.46 mL, 1.46 mmol) dropwisely at 0 °C. The reaction mixture was stirred at 0 °C for 2.5 h and then at rt for 40 min. The reaction was quenched with aqueous NH4Cl solution, and the aqueous layer was extracted with EtOAc. The organic layer was washed with brine, dried over MgSO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (CHCl3/MeOH gradient, 0-20% MeOH). The solid was recrystallized from acetone/H2O to afford 3 (S-217622) (95.3 mg, 25%) as a pale brown solid. 1H NMR (400 MHz, DMSO-d6, DCl in D2O) δ 3.90 (3H, s), 4.15 (3H, s), 5.04 (2H, s), 5.26 (2H, s), 7.44 (1H, m), 7.52-7.65 (2H, m), 7.73 (1H, s), 8.40 (1H, s), 9.31 (1H, s). 13C NMR (100 MHz, DMSO-d6, DCl in D2O) δ 37.34, 38.04, 40.06, 40.29, 106.16 (dd, J = 28.2, 21.6 Hz), 116.46-116.70, 116.70, 120.54-120.76, 120.76, 125.93, 129.10, 132.35, 143.84, 145.98, 146.38 (ddd, J = 241.4, 12.5, 3.7 Hz), 146.60, 148.52 (td, J = 247.7, 13.6 Hz), 150.43, 150.50, 155.22 (ddd, J = 244.3, 10.3, 2.2 Hz), 155.58. HRMS-ESI (m/z): [M + H]+ calcd for [C22H18 F3ClN9O2]+ 532.1219; found 532.1221.
Preparation of Compound 3 (S-217622) fumaric acid co-crystal
A mixture of 3 (S-217622) (1.17 g, 2.2 mmol) and fumaric acid (278 mg, 2.4 mmol) in EtOAc (5.9 mL) was stirred at room temperature for 45 min. The suspension was filtrated to afford 3 (S-217622) fumaric acid co-crystal (1.37 g, 95 %) as a white solid. 1H NMR (400 MHz, pyridine-d5) δ 3.64 (s, 3H), 3.99 (s, 3H), 5.56 (s, 2H), 5.61 (s, 2H), 7.16-7.25 (m, 2H), 7.44 (s, 2H), 7.81 (s, 1H), 7.89 (s, 1H), 7.89-7.97 (m, 1H), 8.32 (s, 1H).
Notes
SHIONOGI has applied for a patent covering 1, 2, and 3 (S-217622). Y.U., S.U., K.N., H.N., Y.Y., S.Y., Y.M., Y.T., K.K., T.S., K.K., A.N., S.K., T.S., S.T., K.U., T.M., S.A., A.S., T.S., T.K., and Y.T. are employees of SHIONOGI & Co., Ltd. S.U., K.N., H.N., Y.M., Y.T., K.K., T.S., K.K., S.K., TS, S.T., K.U., T.S., and T.K. are shareholders in SHIONOGI & Co., Ltd. M.S., Y.O., and H.S. are financially supported by the joint research fund from SHIONOGI & Co., Ltd.
- Supporting information[supplements/477782_file02.pdf]
see spectrum at end of page
///////////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Oral antiviral medications, in addition to vaccines, are expected to play an important role in treating coronavirus disease 2019 (COVID-19), which is caused by infection with the severe acute respiratory disease coronavirus-2 (SARS-CoV-2).
These drugs must have significant antiviral activity, as well as target specificity, oral bioavailability, and metabolic stability. Although several antiviral compounds have been reported as possible SARS-CoV-2 inhibitors in vitro, only a few of these drugs have been shown to be effective in vivo.
Ensitrelvir, a novel SARS-CoV-2 antiviral
Ensitrelvir (code name S-217622, brand name Xocova), is a new inhibitor of the SARS-CoV-2 major protease (Mpro), also known as 3C-like protease, has been shown to reduce the viral load and help alleviate the severity of SARS-CoV-2 in infected hamsters. In cells, low nanomolar to sub-micromolar doses of S-217622 suppress viral growth. In hamsters, oral treatment of S-217622 showed excellent pharmacokinetic qualities and hastened recovery from acute SARS-CoV-2 infection.
S-217622 also demonstrated antiviral effectiveness against SARS-CoV-2 variants of concern (VOCs), such as the highly pathogenic Delta variant and the newly discovered Omicron variant. Overall, these findings show that S-217622, which is an antiviral drug that is currently being tested in Phase II/III clinical trials, has impressive antiviral efficiency and effectiveness against SARS-CoV-2 and could be a viable oral treatment option for COVID-19.
History
It has reached Phase III clinical trials.[3] The Japanese government is reportedly considering allowing Shionogi permission to apply for approval for medical use before the final steps of trials are completed, potentially speeding up the release for sale. This conditional early approval system has previously been used in Japan to accelerate the progression to market of other antiviral drugs targeting COVID-19, including remdesivir and molnupiravir.[6] In a study of 428 patients, viral load was reduced, but symptoms were not significantly reduced. [7]
It became the first Japanese domestic pill to treat COVID-19, third to be regulatorally approved in Japan; in February 2022.[8]

NEW DRUG APPROVALS
ONE TIME
$10.00
References
- ^ World Health Organization (2021). “International Nonproprietary Names for Pharmaceutical Substances. Proposed INN: List 126” (PDF). WHO Drug Information. 35 (4): 1135.
- ^ Xocova: Powerful New Japanese Pill for Coronavirus Treatment. BioPharma Media, February 2022
- ^ Jump up to:a b Unoh Y, Uehara S, Nakahara K, Nobori H, Yamatsu Y, Yamamoto S, et al. (January 2022). “Discovery of S-217622, a Non-Covalent Oral SARS-CoV-2 3CL Protease Inhibitor Clinical Candidate for Treating COVID-19”. bioRxiv. doi:10.1101/2022.01.26.477782. S2CID 246367525.
- ^ “Shionogi presents positive Ph II/III results for COVID-19 antiviral S-217622”. thepharmaletter.com. 31 January 2022.
- ^ Shionogi’s new COVID pill appears to ease omicron symptoms. Nikkei Asia, 21 December 2021
- ^ Japan to consider early approval for Shionogi COVID-19 pill. Japan Times, 8 February 2022
- ^ https://www.reuters.com/business/healthcare-pharmaceuticals/japans-shionogi-seeks-approval-oral-covid-19-drug-2022-02-25/[bare URL]
- ^ “Japan’s Shionogi seeks approval for COVID-19 pill”. Reuters. Reuters. 25 February 2022.
| Clinical data | |
|---|---|
| Other names | S-217622 |
| Identifiers | |
| showIUPAC name | |
| PubChem CID | 162533924 |
| Chemical and physical data | |
| Formula | C22H17ClF3N9O2 |
| Molar mass | 531.88 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
Journal reference:
- Sasaki, M., Tabata, K., Kishimoto, M., et al. (2022). Oral administration of S-217622, a SARS-CoV-2 main protease inhibitor, decreases the viral load and accelerates recovery from clinical aspects of COVID-19. bioRxiv. doi:10.1101/2022.02.14.480338. https://www.biorxiv.org/content/10.1101/2022.02.14.480338v1.full.
///////////Ensitrelvir, S-217622, S 217622, Xocova, SHIONOGI, CORONA VIRUS, covid 19



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}