DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
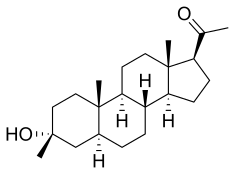
Ganaxolone
- Molecular FormulaC22H36O2
- Average mass332.520 Da
- CCD-1042
FDA APPROVED 3/18/2022, Ztalmy
To treat seizures in cyclin-dependent kinase-like 5 deficiency disorder
Ganaxolone, sold under the brand name Ztalmy, is a medication used to treat seizures associated with cyclin-dependent kinase-like 5 (CDKL5) deficiency disorder (CDD).[1][2]
Ganaxolone was approved for medical use in the United States in March 2022.[1]
Ganaxolone is the 3β-methylated synthetic analog of allopregnanolone; it belongs to a class of compounds referred to as neurosteroids. Ganaxolone is an allosteric modulator of GABAA receptors acting through binding sites which are distinct from the benzodiazepine binding site. It has activity in a broad range of animal models of epilepsy. Ganaxolone has been shown to be well tolerated in adults and children. In early phase II studies, Ganaxolone has been shown to have activity in adult patients with partial-onset seizures and epileptic children with history of infantile spasms. It is currently undergoing further development in infants with newly diagnosed infantile spasms, in women with catamenial epilepsy, and in adults with refractory partial-onset seizures.
Ganaxolone is in phase III clinical studies for the treatment of partial seizures in adults. Phase II clinical trials is ongoing for treatment of uncontrolled seizures in PCDH19 female pediatric epilepsy and Fragile X syndrome.
Ganaxolone was originally developed by CoCensys (aquired by Purdue Pharma). In 2003, Marinus Pharmaceuticals obtained the compound from Purdue Pharma.
In 2015, it was granted as orphan drug designation for the treatment of PCDH19 female epilepsy.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019209850&_cid=P10-L0YZTI-42413-1
In an embodiment, the disclosure provides a method for using pregnenolone to make 21-OH ganaxolone and other intermediary compounds which are useful for preparing neurosteroid derivatives. The method of making 21-OH ganaxolone is shown below in Route 1.
Route 1
Referring to Route 1, Synthesis of 1-((3S,8R,10S,13S,14S,17S)-3-hydroxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)ethenone :
<a name="
Pregnenolone (3.17 g, 10 mmol) was dissolved in 30 mL of THF and 5 mL of acetic acid. To it, 10% W/C (0.3 g) was added. The resulting mixture was shaken under 60 psi hydrogen at 60°C overnight. It was filtered through a Celite ® pad and concentrated to give 3.2 g of the desired product (100%). 1 H NMR (400 MHz, CDCl3) δ 3.58 (tt, J = 11.0, 4.8 Hz, 1H), 2.50 (t, J = 9.0 Hz, 1H), 2.19 – 2.11 (m, 2H), 2.09 (s, 3H ), 2.06 – 1.93 (m, 2H), 1.85 – 1.75 (m, 1H), 1.74 -1.50 (m, 6H), 1.47 – 1.04 (m, 9H), 1.04 – 0.82 (m, 2H), 0.79 (s , 3H), 0.72 – 0.61 (m, 1H), 0.58 (d, J = 2.4 Hz, 3H).
[0107] Synthesis of (8R,10S,13S,14S,17S)-l7-acetyl-l0,l3-dimethyltetradecahydro-1H-cyclopenta[a]phenanthren-3(2H)-one:
To a solution of the above product (1-((3S,8R,10S,13S,14S,17S)-3-hydroxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)ethanone, 3.2 g, 10 mmol) in 40 mL of THF and 10 mL of acetic acid was added NaBr (1.03 g, 0.1 eq.). It was cooled in an ice bath and was followed by the dropwise addition of NaOCl (82 mL, 10-15%, 18 eq.) at such a rate that the internal temperature was maintained <40 °C. After addition, it was stirred at room temperature for 2h. Thin layer chromatography (TLC) indicated it was complete. The mixture was diluted with dichloromethane and layers were separated. The organic layer was washed with Na 2 S 2 O 3 (10% aq.), H 2 O, NaHCO 3 (sat.) and NaCl (sat.). Drying over Na 2SO 4 and concentration afforded 3.8 g of the crude product, which was recrystallized from CH 2 Cl 2 /Hex to give 2.57 g of the desired product (81%). 1 H NMR (400 MHz, CDC13): 2.51 (t, 1H), 2.2-2.4 (m, 3H), 2.1-2.2 (m, 1H), 2.10 (s, 3H), 1.98-2.01 (m, 2H) , 1.6-1.7 (m, 4H), 1.55-1.6 (m, 1H), 1.3-1.4 (m, 7H), 1.1-1.2 (m, 2H), 0.99 (s, 3H), 0.95-0.98 (m, 1H), 0.75-0.78 (m, 1H), 0.62 (s, 3H).
Synthesis of 1-((2’R,8R,10S,13S,14S,17S)-10,11-dimethylhexadecahydrospiro[cyclopenta[a]phenanthrene-3,2′-oxiran]-17-yl)ethanone.<a name="
Under argon, trimethyl sulfoxonium iodide (2.6 g, 1.7 eq.) and sodium t-butoxide (1.18 g, 1.75 eq.) in DMSO (20 mL) was heated at 65 °C for 2h. After it was cooled to RT, the above di-ketone ((8R, 10S, 13 S, 14S, 17S)-17-acetyl- 10,13 -dimethyl tetradecahy dro-1H-cyclopenta[a]phenanthren-3(2H) -one, 2.2 g, 7 mmol) was added scoop-wise so that the internal temperature was maintained between 25-35 °C. The resulting mixture was stirred at RT for 2h. After TLC indicated it was complete, it was quenched with 30 mL of H 2 O, stirred for 10 min and was kept in fridge overnight. The precipitate was filtered, washed with 20 mL of (4:1 of H 2 O /MeOH), dried to give 94% of the desired product (W = 2.17 g). 1H NMR (400 MHz, CDC13) δ 2.63 (s, 2H), 2.53 (t, J = 8.9 Hz, 1H), 2.20 – 2.13 (m, 1H), 2.11 (s, 3H), 2.10 – 1.95 (m, 2H), 1.87 (dd, J = 13.9, 13.1 Hz, 1H), 1.76 – 1.59 (m, 4H), 1.58 – 1.48 (m, 1H), 1.48 – 1.24 (m, 5H), 1.24 – 1.07 (m, 3H), 1.02 – 0.87 (m, 2H), 0.86 (dd, J = 3.7, 2.2 Hz, 1H), 0.84 (s, 3H), 0.81 – 0.74 (m, 1H), 0.61 (s, 3H).
[0109] Synthesis of 1-((3R,8R,10S,13S,14S,17S)-3-hydroxy-3,10,13-trimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)ethanone (ganaxolone) .
To a solution of the above epoxide (1.5 g, 4.56 mmol) in 15 mL of THF and 15 mL of MeOH were added Nal (1.02 g, 1.5 eq.) and HO Ac (0.6 mL, 2.2 eq.). The resulting mixture was heated at 65°C for 2h. After TLC indicated that the epoxy was completely converted to an iodo compound, it was cooled to RT. Sodium acetate (1.02 g, 2.7 eq.) and 150 mg of 10% Pd/C were added and the mixture was transferred to a hydrogenation bottle with the aid of MeOH (10 mL) and was hydrogenated under 50 psi hydrogen over the weekend. It was filtered through<a name="Celite ® and the filtrate was concentrated. The residue was then partitioned between dichloromethane and water. The aqueous solution was extracted twice with CH 2 Cl 2 and the combined organic layers were washed with brine, dried over Na 2 SO 4 and concentrated. The Biotage flash purification with 10-35% EtOAc in hexane to give 0.5 g of the desired product (33%).
The synthesis was repeated with 1.1 g of the epoxy and 1 g of the product was obtained (90%).
Both lots of product were combined and recrystallized with CH 2 Cl 2 and hexane to give 0.522 g of the product with 96.6% purity by HPLC. 1 H NMR (400 MHz, Chloroform-d) δ 2.51 (t, J = 8.9 Hz, 1H), 2.18 – 2.10 (m, 1H), 2.09 (s, 3H), 2.01 – 1.93 (m, 1H), 1.72 – 1.57 (m, 4H), 1.57 – 1.41 (m, 5H), 1.41 – 1.30 (m, 3H), 1.30 – 1.20 (m, 3H), 1.18 (s, 3H), 1.17 – 1.09 (m, 2H) , 1.00 – 0.85 (m, 1H), 0.78 (ddd, J = 10.6, 7.7, 5.4 Hz, 1H), 0.73 (d, J = 0.6 Hz, 3H), 0.58 (s, 3H). UV: Absorbances at 206.2 nm. TLC: (Silica Gel plates) 20% EtOAc/Hexane; R f = 0.50. HPLC: Sunfire C18 5m 250 x 4.6mm; flow 1.0 mL/min; Waters 996 PDA detection at 210 nm; solvent 80% Acetonitrile in H 2 O (0.1% formic acid) over 30 min; retention time 8.24 min; 96.6%.
SYN
https://patents.google.com/patent/WO2016164763A1/en
SYN
US3953429.
 J. Med. Chem. 1997, 40, 61-72.
J. Med. Chem. 1997, 40, 61-72.https://pubs.acs.org/doi/10.1021/jm960021x
Two naturally occurring metabolites of progesterone, 3α-hydroxy-5α- and 5β-pregnan-20-one (1 and 2), are potent allosteric modulators of the GABAA receptor. Their therapeutic potential as anxiolytics, anticonvulsants, and sedative/hypnotics is limited by rapid metabolism. To avoid these shortcomings, a series of 3β-substituted derivatives of 1 and 2 was prepared. Small lipophilic groups generally maintain potency in both the 5α- and 5β-series as determined by inhibition of [35S]TBPS binding. In the 5α-series, 3β-ethyl, -propyl, -trifluoromethyl and -(benzyloxy)methyl, as well as substituents of the form 3β-XCH2, where X is Cl, Br, or I or contains unsaturation, show limited efficacy in inhibiting [35S]TBPS binding. In the 5β-series, the unsubstituted parent 2 is a two-component inhibitor, whereas all of the 3β-substituted derivatives of 2 inhibit TBPS via a single class of binding sites. In addition, all of the 3-substituted 5β-sterols tested are full inhibitors of [35S]TBPS binding. Electrophysiological measurements using α1β2γ2L receptors expressed in oocytes show that 3β-methyl- and 3β-(azidomethyl)-3α-hydroxy-5α-pregnan-20-one (6 and 22, respectively) are potent full efficacy modulators and that 3α-hydroxy-3β-(trifluoromethyl)-5α-pregnan-20-one (24) is a low-efficacy modulator, confirming the results obtained from [35S]TBPS binding. These results indicate that modification of the 3β-position in 1 and 2 maintains activity at the neuroactive steroid site on the GABAA receptor. In animal studies, compound 6 (CCD 1042) is an orally active anticonvulsant, while the naturally occurring progesterone metabolites 1 and 2 are inactive when administered orally, suggesting that 3β-substitution slows metabolism of the 3-hydroxyl, resulting in orally bioavailable steroid modulators of the GABAA receptor.
PATENT
WO9303732A1.,
https://patents.google.com/patent/WO1993003732A1/nl
SYN
| GB 1380248 |
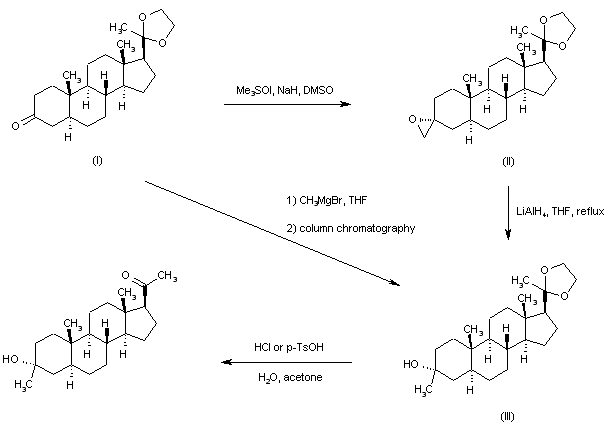
Addition of the sulfur ylide generated from trimethylsulfoxonium iodide and NaH to the 20-ethylene ketal of pregnane-3,20-dione (I) furnished the spiro oxirane derivative (II). This was reduced to the tertiary alcohol (III) by means of LiAlH4 in refluxing THF. Then, acid hydrolysis of the ethylene ketal function of (III) provided the title compound. Alternatively, the intermediate ketal (III) was prepared by addition of methylmagnesium bromide to ketone (I), followed by chromatographic separation of the resultant mixture of 3-alpha and 3-beta methyl adducts.
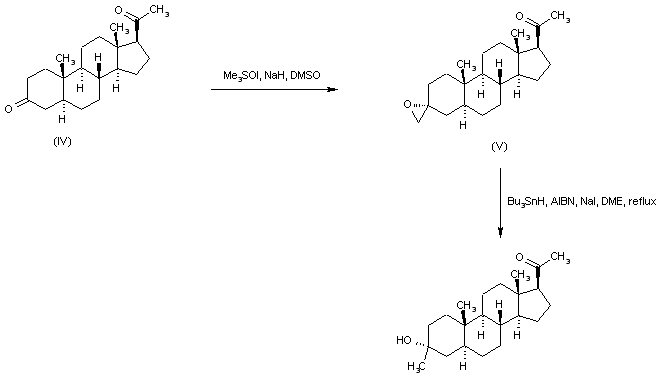
Starting from the unprotected diketone (IV), selective addition of dimethyloxosulfonium methylide to the 3 keto group furnished oxirane (V). This was then reduced to the title alcohol by treatment with tributylstannyl hydride and AIBN.
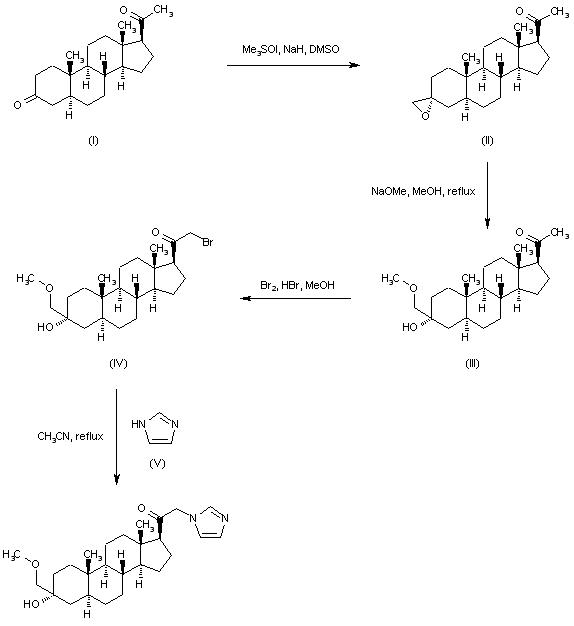
Regioselective addition of dimethylsulfoxonium methylide to 5-alpha-pregnane-3,20-dione (I) gave the epoxide (II). Opening of the epoxide ring of (II) with sodium methoxide produced the hydroxy ether (III). Bromination of (III) with Br2 in the presence of a catalytic amount of HBr afforded bromo ketone (IV). This was then condensed with imidazole (V) in refluxing acetonitrile to furnish the title compound.
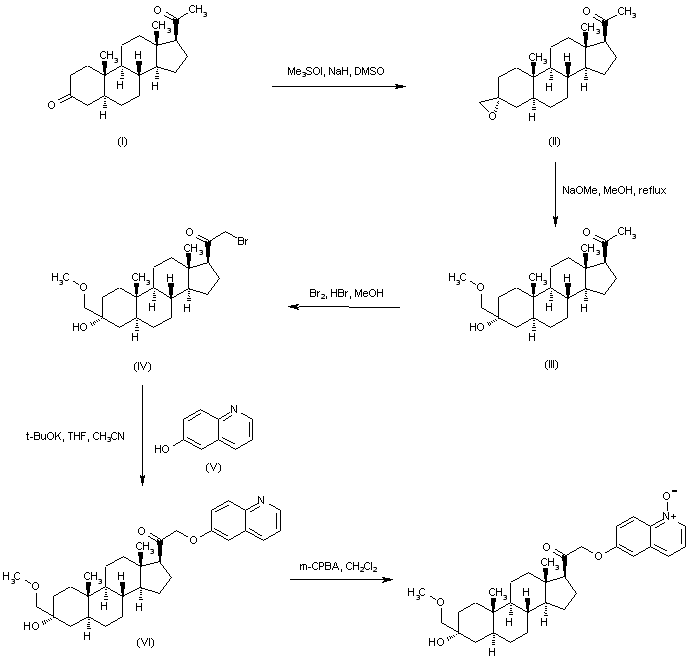
Regioselective addition of dimethylsulfoxonium methylide to 5-alpha-pregnane-3,20-dione (I) gave the epoxide (II). Opening of the epoxide ring of (II) with sodium methoxide produced the hydroxy ether (III). Bromination of (III) with Br2 in the presence of a catalytic amount of HBr afforded bromo ketone (IV). This was then condensed with 6-hydroxyquinoline (V) in the presence of potassium tert-butoxide to furnish the quinolinyl ether (VI). The quinoline ring was then oxidized with m-chloroperbenzoic acid, yielding the title N-oxide.
3. WO9318053A1.
4. WO9427608A1.
 WO2011019821A2 / US8362286B2.
WO2011019821A2 / US8362286B2.///////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Pharmacology
Mechanism of action
The exact mechanism of action for ganaxolone is unknown; however, results from animal studies suggest that it acts by blocking seizure propagation and elevating seizure thresholds.[3][4]
Ganaxolone is thought to modulate both synaptic and extrasynaptic GABAA receptors to normalize over-excited neurons.[2] Ganaxolone’s activation of the extrasynaptic receptor is an additional mechanism that provides stabilizing effects that potentially differentiates it from other drugs that increase GABA signaling.[2]
Ganaxolone binds to allosteric sites of the GABAA receptor to modulate and open the chloride ion channel, resulting in a hyperpolarization of the neuron.[2] This causes an inhibitory effect on neurotransmission, reducing the chance of a successful action potential (depolarization) from occurring.[2][3][4]
Chemistry
ResearchGanaxolone is a synthetic pregnane steroid. Other pregnane neurosteroids include alfadolone, alfaxolone, allopregnanolone (brexanolone), hydroxydione, minaxolone, pregnanolone (eltanolone), and renanolone, among others.
Ganaxolone is being investigated for potential medical use in the treatment of epilepsy. It is well tolerated in human trials, with the most commonly reported side effects being somnolence (sleepiness), dizziness, and fatigue.[5] Trials in adults with focal onset seizures and in children with infantile spasms have recently been completed.[6][7] There are ongoing studies in patients with focal onset seizures, PCDH19 pediatric epilepsy, and behaviors in Fragile X syndrome.[6][7]
Ganaxolone has been shown to protect against seizures in animal models,[3][4] and to act a positive allosteric modulator of the GABAA receptor.[2][8]
Clinical trials
The most common adverse events reported across clinical trials have been somnolence (sleepiness), dizziness, and fatigue.[5] In 2015, the MIND Institute at the University of California, Davis, announced that it was conducting, in collaboration with Marinus Pharmaceuticals, a randomized, placebo-controlled, Phase 2 clinical trial evaluating the effect of ganaxolone on behaviors associated with Fragile X syndrome in children and adolescents.[9][10][11]
References
- ^ Jump up to:a b c https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/215904s000lbl.pdf
- ^ Jump up to:a b c d e f Carter RB, Wood PL, Wieland S, Hawkinson JE, Belelli D, Lambert JJ, White HS, Wolf HH, Mirsadeghi S, Tahir SH, Bolger MB, Lan NC, Gee KW (March 1997). “Characterization of the anticonvulsant properties of ganaxolone (CCD 1042; 3alpha-hydroxy-3beta-methyl-5alpha-pregnan-20-one), a selective, high-affinity, steroid modulator of the gamma-aminobutyric acid(A) receptor”. The Journal of Pharmacology and Experimental Therapeutics. 280 (3): 1284–95. PMID 9067315.
- ^ Jump up to:a b c Kaminski RM, Livingood MR, Rogawski MA (July 2004). “Allopregnanolone analogs that positively modulate GABA receptors protect against partial seizures induced by 6-Hz electrical stimulation in mice”. Epilepsia. 45 (7): 864–7. doi:10.1111/j.0013-9580.2004.04504.x. PMID 15230714. S2CID 21974013.
- ^ Jump up to:a b c Reddy DS, Rogawski MA (May 2010). “Ganaxolone suppression of behavioral and electrographic seizures in the mouse amygdala kindling model”. Epilepsy Research. 89 (2–3): 254–60. doi:10.1016/j.eplepsyres.2010.01.009. PMC 2854307. PMID 20172694.
- ^ Jump up to:a b Monaghan EP, Navalta LA, Shum L, Ashbrook DW, Lee DA (September 1997). “Initial human experience with ganaxolone, a neuroactive steroid with antiepileptic activity”. Epilepsia. 38 (9): 1026–31. doi:10.1111/j.1528-1157.1997.tb01486.x. PMID 9579942. S2CID 27584114.
- ^ Jump up to:a b Nohria V, Giller E (January 2007). “Ganaxolone”. Neurotherapeutics. 4 (1): 102–5. doi:10.1016/j.nurt.2006.11.003. PMC 7479704. PMID 17199022.
- ^ Jump up to:a b Pieribone VA, Tsai J, Soufflet C, Rey E, Shaw K, Giller E, Dulac O (October 2007). “Clinical evaluation of ganaxolone in pediatric and adolescent patients with refractory epilepsy”. Epilepsia. 48 (10): 1870–4. doi:10.1111/j.1528-1167.2007.01182.x. PMID 17634060. S2CID 24656918.
- ^ Reddy DS, Rogawski MA (December 2000). “Chronic treatment with the neuroactive steroid ganaxolone in the rat induces anticonvulsant tolerance to diazepam but not to itself”. The Journal of Pharmacology and Experimental Therapeutics. 295 (3): 1241–8. PMID 11082461.
- ^ “Fragile X Research and Treatment Center: Clinical Research Studies” (PDF). UC Davis MIND Institute. 10 February 2015. Archived from the original (PDF) on 5 June 2015. Retrieved 27 January 2016.
- ^ “Ganaxolone Treatment in Children With Fragile X Syndrome”. Clinicaltrials.gov. 7 November 2012. Retrieved 27 January 2016.
- ^ “UC Davis Health System. UC Davis researchers win $3 million grant from U.S. Congress to study fragile X” (Press release). UC Davis Health System. 8 February 2011. Archived from the original on 3 February 2016. Retrieved 27 January 2016.
External links
- “Ganaxolone”. Drug Information Portal. U.S. National Library of Medicine.
 | |
| Clinical data | |
|---|---|
| Trade names | Ztalmy |
| Other names | GNX; CCD-1042; 3β-Methyl-5α-pregnan-3α-ol-20-one; 3α-Hydroxy-3β-methyl-5α-pregnan-20-one |
| License data | |
| Routes of administration | By mouth |
| Drug class | Neurosteroid |
| ATC code | |
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.210.937 |
| Chemical and physical data | |
| Formula | C22H36O2 |
| Molar mass | 332.528 g·mol−1 |
| 3D model (JSmol) | |
| | |
////////////Ganaxolone, ZTALMY, FDA 2022, APPROVALS 2022, CCD 1042
[H][C@@]12CC[C@H](C(C)=O)[C@@]1(C)CC[C@@]1([H])[C@@]2([H])CC[C@@]2([H])C[C@](C)(O)CC[C@]12C
https://doi.org/10.1021/acs.jmedchem.3c02374
J. Med. Chem. 2024, 67, 4376−4418
Ganaxolone (Ztalmy). Ganaxolone (10), a first-in class medication, is a neuroactive steroid gamma-aminobutyric acid (GABA) A receptor positive modulator indicated for the treatment of seizures associated with cyclin-dependent kinase like 5 (CDKL5) deficiency disorder (CDD) in patients 2 years
of age and older.
CDKL5 deficiency disorder is a rare neurodevelopmental condition resulting from pathogenic variants in the CDKL5 gene with an incidence rate ranging from 1 in 40,000 to 1 in 60,000 newborns.72 While CDKL5
deficiency is rare, it represents one of the most common forms of genetic epilepsy. 73,74
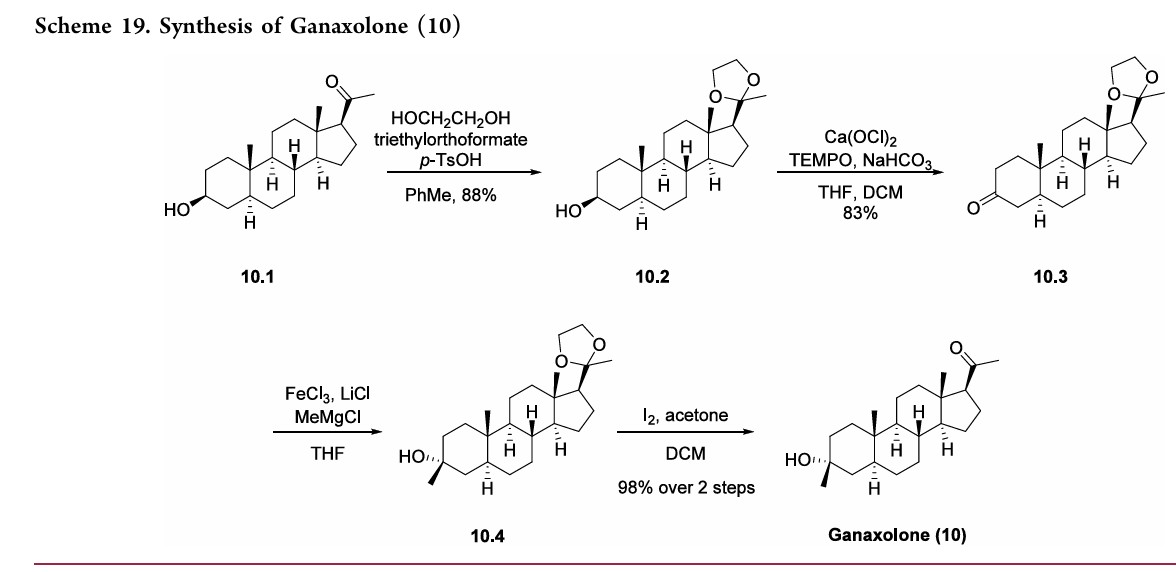
The synthesis of ganaxolone was previously described in the literature in 1976.75 However, the synthesis illustrated in Scheme 19 was recently published and outlines a manufactory process for ganaxolone.
The approach began with protection of the ketone functional group in pregnanolone 10.1, a naturally occurring steroid. Using acid catalysis, the ketal was formed with refluxing ethylene glycol in toluene, and after neutralizing the acidic solution, ketal 10.2 was obtained in 88% yield. Multiple options for the oxidation of 10.2 to form ketone 10.3 were provided such as Swern, Dess-Martin, and TPAP conditions. The oxidative procedure described in Scheme 19 illustrates the use of calcium hypochlorite and TEMPO to
form ketone 10.3. Next, methylation was accomplished by addition of MeMgBr in the presence of LiCl and FeCl3 to provide tertiary alcohol 10.4. Finally, the acetal protecting group was removed by treatment with iodine in DCM and acetone to provide ganaxolone 10 in 98% yield over the last two steps.
(70) Marinus Pharmaceuticals Inc.: ZTALMY® (ganaxolone)
[package insert]. https://www.accessdata.fda.gov/drugsatfda_docs/
label/2022/215904s000lbl.pdf, (accessed June 6, 2023).
(71) Carter, R. B.; Wood, P. L.; Wieland, S.; Hawkinson, J. E.;
Belelli, D.; Lambert, J. J.; White, S. H.; Wolf, H. H.; Mirsadeghi, S.;
Tahir, S. H.; Bolger, M. B.; Lan, N. C.; Gee, K.-W. Characterization of
the anticonvulsant properties of ganaxolone (CCD 1042; 3-alpha
hydroxy-3beta-methyl-5alpha-pregnan-20-one), a selective, high-affin
ity, steroid modulator of the gamma-aminobutyric acid(A) receptor. J.
Pharmacol. Exp. Ther. 1997, 280, 1284−1295.
(72) Jakimiec, M.; Paprocka, J.; Smigiel, R. CDKL5 deficiency
disorder-A complex epileptic encephalopathy. Brain Sci. 2020, 10,
107.
(73) Cook, M. C.; Lawrence, R.; Phillipps, G. H.; Hunter, A. C.;
Newall, C. E.; Stephenson, L.; Weir, N. G. Anaesthetic steroids of the
androstance and pregnane series. U.S. Patent US 3,953,429, 1976.
(74) Hogenkamp, D. J.; Tahir, S. H.; Hawkinson, J. E.; Upasani, R.
B.; Alauddin, M.; Kimbrough, C. L.; Acosta-Burruel, M.; Whittemore,
E. R.; Woodward, R. M.; Lan, N. C.; et al. Synthesis and in vitro
activity of 3 beta-substituted-3 alpha-hydroxypregnan-20-ones:
allosteric modulators of the GABA-A receptor. J. Med. Chem. 1997,
40, 61−72.