
Home » Posts tagged 'PHASE 3'
Tag Archives: PHASE 3
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Gedatolisib
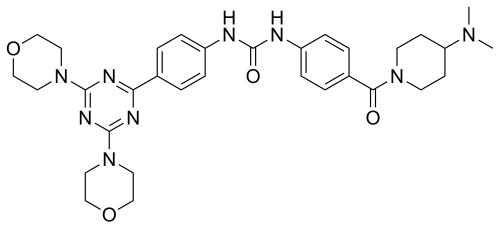
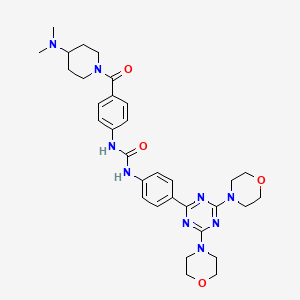
Gedatolisib
Approvals 3026, FDA 2026, 7/14/2026, Revtorpyk
| PF-05212384; PF-5212384; PKI-587 CAS 1197160-78-3 Chemical Formula: C32H41N9O4 Molecular Weight: 615.72 1-(4-{[4-(Dimethylamino)-1-piperidinyl]carbonyl}phenyl)-3-{4-[4,6-di(4-morpholinyl)-1,3,5-triazin-2-yl]phenyl}urea 3-{4-[bis(morpholin-4-yl)-1,3,5-triazin-2-yl]phenyl}-1-{4-[4-(dimethylamino)piperidine-1-carbonyl]phenyl}urea N-[4-[[4-(Dimethylamino)-1-piperidinyl]carbonyl]phenyl]-N’-[4-[4,6-di(4-morpholinyl)-1,3,5-triazin-2-yl]phenyl]urea гедатолисиб [Russian] [INN] غيداتوليسيب [Arabic] [INN] 吉达利塞 [Chinese] [INN] |
1-(4-(4-(Dimethylamino)piperidine-1-carbonyl)phenyl)-3-(4-(4,6-dimorpholino-1,3,5-triazin-2-yl)phenyl)urea
In combination with fulvestran, to treat hormone receptor-positive, human epidermal growth factor receptor 2-negative, locally advanced or metastatic breast cancer without a PIK3CA mutation detected following progression on or after treatment with at least one line of endocrine therapy in the metastatic setting
Gedatolisib, sold under the brand name Revtorpyk, is an anti-cancer drug used for the treatment of breast cancer.[1] It is under development by Celcuity, Inc. Gedatolisib is a kinase inhibitor.[1] The mechanism of action is accomplished by binding the different p110 catalytic subunit isoforms of PI3K and the kinase site of mTOR.[2] Gedatolisib is administered by intravenous infusion.[1]
Gedatolisib was approved for medical use in the United States in July 2026.[1][3]
Medical uses
Gedatolisib is indicated in combination with fulvestrant, with or without palbociclib, for the treatment of adults with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative locally advanced or metastatic breast cancer without a PIK3CA mutation detected following progression on or after treatment with at least one line of endocrine therapy in the metastatic setting.[1]
- Synthesis of New Dialkyl 2,2′-[Carbonylbis(azanediyl)]dibenzoates via Curtius RearrangementDOI: 10.1055/s-0040-1706643Publication Date: 2021Publication Name: Synthesis
- A New One-Pot Three-Component Synthesis of 4-Aryl-6-cycloamino-1,3,5-triazin-2-amines under Microwave IrradiationDOI: 10.1055/a-1401-2795Publication Date: 2021Publication Name: Synthesis
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US42900900&_cid=P22-MRVGV9-51327-1
Example 76
Preparation of 1-(4-(4-(dimethylamino)piperidine-1-carbonyl)phenyl)-3-(4-(4,6-dimorpholino-1,3,5-triazin-2-yl)phenyl)urea
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2010096619&_cid=P22-MRVGV9-51327-1
Scheme 1
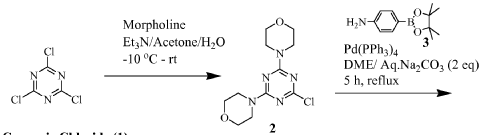
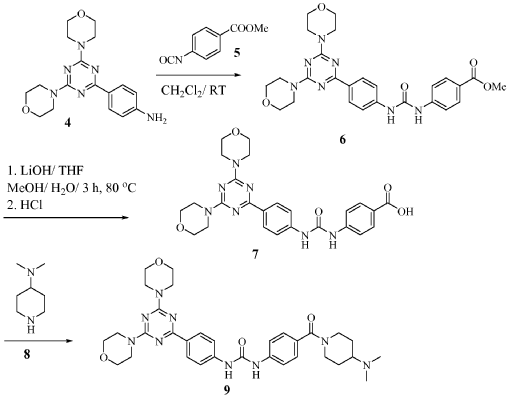
Preparation of 1-(4-(4-(dimethylamino) piperidine-1-carbonyl)phenyl-3-(4-(4,6- dimorpholino-1 ,3,5-triazine-2-yl)phenyl) urea (9)
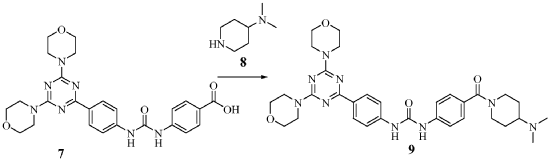
To a slurry of 4-(3-(4-(4,6-dimorpholino-1 ,3,5-triazine-2- yl)phenyl)ureido)benzoic acid (7, 45.5 g, 0.09 mol) in dry THF (1.6 L) heated to 50 0C was added N,N’-carbonyl diimidazole (28 g, 0.17 mol). The reaction mixture was heated for 2 hours and followed by dimethylaminopiperidine (8, 23.5 g, 0.18 mol) and stirred at 53 0C for 16 hours. The reaction mixture was cooled to the room temperature and filtered. The cake was washed with 2-propanol and air-dried to give 97 % pure white powder in 88% yield (49.2 g, 0.08 mol). To the solids stirred in dimethyl acetamide (DMAC, 165 ml) at 70° C for 1 hour was added 2-propanol (640 ml) and the mixture was stirred at 65 0C for additional 1 hour. The solids were filtered, washed with 2-propanol and dried in a vacuum oven at 700C for 16 hour to give crystalline white powder (45 g) with >99% purity. The above-mentioned work up process and crystallization procedure gave a Pd residue of 20 ppm. Alternate procedures for the formation of 1-(4-(4-(dimethylamino) piperidine-1 – carbonyl)phenyl-3-(4-(4,6-dimorpholino-1 ,3,5-triazine-2-yl)phenyl) urea (9)
To the solution of 4-(4,6-dimorpholin-4-yl-1 ,3,5-triazin-2-yl) aniline (4, 18 g, 0.052 mol) in dichloromethane (300 ml) was added methyl 4-isocyanato benzoate (5, 10.5 g, 0.061 mol) and the reaction mixture was stirred for 5 hours. The separated solids were filtered, washed with ether and air dried to give beige solids (21 g, 0.04 mol). Yield 77%. 90 % pure by HPLC; Mass: 520.1 (M+H). Preparation of 4-(3-(4-(4,6-dimorpholino-1 ,3,5-triazine-2-yl)phenyl)ureido) benzoic acid (7)
The mixture of methyl 4-(3-(4-(4,6-dimorpholino-1 ,3,5-triazine-2-yl)ureido)benzoate (6, 21 g, 0.04mol) and lithium hydroxide monohydrate (3.8 g, 0.09 mol) in THF (120 ml), MeOH (60 ml), and water (60 ml) was heated at 80 0C for 3 hours. The dark brown solution was cooled to room temperature and made acidic with concentrated HCI. The solids were filtered, washed with water, washed with acetone , washed with ether, and dried in a vacuum oven at 60 0C for 48 hours to give off white solids of 4-(3-(4-(4,6-dimorpholino-1 ,3,5-triazine-2-yl)phenyl)ureido) benzoic acid (19.2 g, 0.038 mol). Mass: 506.3 (M+H)+; Yield.94%. 1 -(4-(4-(dimethylamino) piperidine-1 -carbonyl)phenyl-3-(4-(4,6-dimorpholino- 1 ,3,5-triazine-2-yl)phenyl) urea (9)
The suspension of 4-(3-(4-(4,6-dimorpholino-1 ,3,5-triazine-2-yl)phenyl)ureido) benzoic acid (7, 17 g, 33.66 mmol) and N-(3-dimethylaminopropyl)ethyl carbodiimide hydrochloride (9.5 g, 49.5 mmol) in THF (200 ml) and acetonitrile (50 ml) was stirred for 10 min and followed by addition of 1-hydroxybenzotriazole hydrate (6.4 g, 47.88 mmol). The reaction mixture was stirred for 30 min and 4-dimethylaminopiperidine (8, 8.86 g, 69.2 mmol) was added by drops. After being stirred for additional 16 hours, the reaction mixture was concentrated to min. The solids were filtered and washed thoroughly with water (very fine suspension). The cake was slurred in hot ethanol, filtered and dried in a vacuum oven at 68 0C for 16 hours to give off white solids (10.3 g, 16.77 mmol). M. p. 238-240 0C. 99 % pure. Mass: 616.3 (M+H)+; Yield 50 %.
PATENT
WO 2009143317
WO 2010096619
WO 2012148540
WO 2014151147
PATENT
US 20170119778
PAPER
Journal of Medicinal Chemistry (2010), 53(6), 2636-2645
http://pubs.acs.org/doi/abs/10.1021/jm901830p
J. Med. Chem., 2010, 53 (6), pp 2636–2645
DOI: 10.1021/jm901830p
Abstract
The PI3K/Akt signaling pathway is a key pathway in cell proliferation, growth, survival, protein synthesis, and glucose metabolism. It has been recognized recently that inhibiting this pathway might provide a viable therapy for cancer. A series of bis(morpholino-1,3,5-triazine) derivatives were prepared and optimized to provide the highly efficacious PI3K/mTOR inhibitor 1-(4-{[4-(dimethylamino)piperidin-1-yl]carbonyl}phenyl)-3-[4-(4,6-dimorpholin-4-yl-1,3,5-triazin-2-yl)phenyl]urea 26 (PKI-587). Compound 26 has shown excellent activity in vitro and in vivo, with antitumor efficacy in both subcutaneous and orthotopic xenograft tumor models when administered intravenously. The structure−activity relationships and the in vitro and in vivo activity of analogues in this series are described.
Preparation of 1-(4-{[4-(Dimethylamino)piperidin-1-yl]carbonyl}phenyl)-3-[4-(4,6-dimorpholin-4- yl-1,3,5-triazin-2-yl)phenyl]urea (26)
MS (ESI) m/z = 616.7. HRMS: calcd for C32H41N9O4 + H+, 616.335 43; found (ESI-FTMS, [M + H]+), 616.334 24. Purity by analytical HPLC 99.3%. (Prodigy ODS3, 0.46 cm × 15 cm, 20 min gradient acetonitrile in water, trifluoroacetic acid, detector wavelengths, 215 and 254 nm.) 1H NMR (DMSO-d6) δ 1.29−1.36 (m, 6H), 2.6 (m, 4H), 2.9 (m,1H), 3.3 (m, 4H), 3.6 (m, 8H), 3.7 (m, 8H), 7.3 (d, J = 8.3 Hz, 2H), 7.51−7.57 (m, 4H), 8.3 (d, J = 8.3 Hz 2H), 8.9 (s, 1H), 9.0 (s, 1H) ppm. Anal. Calcd for C32H41N9O4: C 62.42%, H 6.71%, N 20.47%. Found: C 62.34%, H 6.67%, N 20.39%.
PAPER
Bioorganic & Medicinal Chemistry Letters (2011), 21(16), 4773-4778.
http://www.sciencedirect.com/science/article/pii/S0960894X11008468
PAPER
New and Practical Synthesis of Gedatolisib
http://pubs.acs.org/doi/10.1021/acs.oprd.7b00298
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.7b00298
Abstract
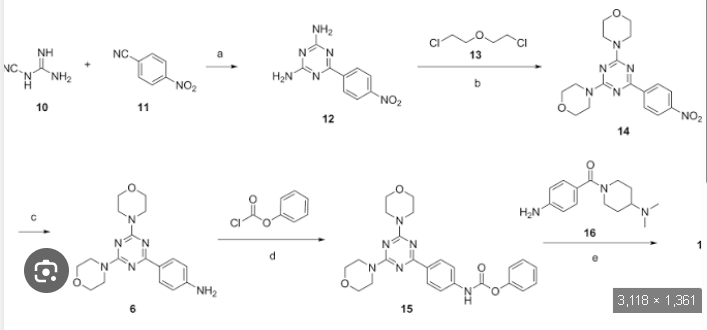
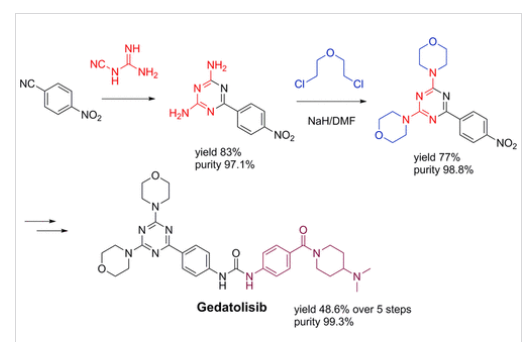
A new, practical, and convergent synthetic route of gedatolisib, an antitumor agent, is developed on a hectogram scale which avoids the Pd coupling method. The key step is adopting 6-(4-nitrophenyl)-1,3,5-triazine-2,4-diamine and 2,2′-dichlorodiethyl ether to prepare the key 4,4′-(6-(4-nitrophenyl)-1,3,5-triazine-2,4-diyl)dimorpholine in 77% yield and 98.8% purity. Gedatolisib is obtained in 48.6% yield over five simple steps and 99.3% purity (HPLC). Purification methods of the intermediates and the final product involved in the route are given.
off-white solid. 1H NMR (400 MHz, DMSO-d6): δ 1.46 (brs, 2H), 1.89 (brs, 2H), 2.29 (s, 6H), 2.94 (brs, 2H), 3.76 (m, 8H), 3.89 (m, 8H), 7.09 (d, J = 8.4 Hz, 2H), 7.20 (d, J = 8.4 Hz, 2H), 7.50 (d, J = 8.7 Hz, 2H), 8.28 (s, 1H), 8.31 (d, J = 8.6 Hz, 2H), 8.48 (s, 1H). ESI-MS (m/z) 615.9 (M + H). HPLC conditions: Column: Agilent Eclipse XDB-C18 (250 mm × 4.6 mm × 5 μm); Detection: 254 nm; Flow rate: 0.8 mL/min; Temperature: 30 °C; Injection load: 1 μL; Solvent: MeOH; Concentration: 0.5 mg/mL; Run time: 20 min; Mobile phase A: water; Mobile phase B: MeOH/TEA = 100:0.1; Gradient program: time (min): 20; % of mobile phase A: 10; % of mobile phase B: 90; tR = 2.598 min, purity: 99.34%
- Zhao, X.; Tan, Q.; Zhang, Z.; Zhao, Y. Med. Chem. Res. 2014, 23, 5188– 5196 DOI: 10.1007/s00044-014-1084-z
- Khafizova, G.; Potoski, J. R. PCT Int. Appl. WO 2010096619, 2010.
- Venkatesan, A. M.; Chen, Z.; Dehnhardt, C. M.; Dos Santos, O.; Delos Santos, E. G.; Zask, A.; Verheijen, J. C.; Kaplan, J. A.; Richard, D. J.; Ayral-Kaloustian, S.; Mansour, T. S.; Gopalsamy, A.; Curran, K. J.; Shi, M. PCT Int. Appl. WO 2009143317, 2009.
REFERENCES
1: Gedaly R, Galuppo R, Musgrave Y, Angulo P, Hundley J, Shah M, Daily MF, Chen C, Cohen DA, Spear BT, Evers BM. PKI-587 and sorafenib alone and in combination on inhibition of liver cancer stem cell proliferation. J Surg Res. 2013 Nov;185(1):225-30. doi: 10.1016/j.jss.2013.05.016. Epub 2013 May 25. PubMed PMID: 23769634.
2: Gedaly R, Angulo P, Hundley J, Daily MF, Chen C, Evers BM. PKI-587 and sorafenib targeting PI3K/AKT/mTOR and Ras/Raf/MAPK pathways synergistically inhibit HCC cell proliferation. J Surg Res. 2012 Aug;176(2):542-8. doi: 10.1016/j.jss.2011.10.045. Epub 2011 Nov 21. PubMed PMID: 22261591.
3: Dehnhardt CM, Venkatesan AM, Chen Z, Delos-Santos E, Ayral-Kaloustian S, Brooijmans N, Yu K, Hollander I, Feldberg L, Lucas J, Mallon R. Identification of 2-oxatriazines as highly potent pan-PI3K/mTOR dual inhibitors. Bioorg Med Chem Lett. 2011 Aug 15;21(16):4773-8. doi: 10.1016/j.bmcl.2011.06.063. Epub 2011 Jun 21. PubMed PMID: 21763134.
4: Mallon R, Feldberg LR, Lucas J, Chaudhary I, Dehnhardt C, Santos ED, Chen Z, dos Santos O, Ayral-Kaloustian S, Venkatesan A, Hollander I. Antitumor efficacy of PKI-587, a highly potent dual PI3K/mTOR kinase inhibitor. Clin Cancer Res. 2011 May 15;17(10):3193-203. doi: 10.1158/1078-0432.CCR-10-1694. Epub 2011 Feb 15. PubMed PMID: 21325073.
5: Venkatesan AM, Chen Z, dos Santos O, Dehnhardt C, Santos ED, Ayral-Kaloustian S, Mallon R, Hollander I, Feldberg L, Lucas J, Yu K, Chaudhary I, Mansour TS. PKI-179: an orally efficacious dual phosphatidylinositol-3-kinase (PI3K)/mammalian target of rapamycin (mTOR) inhibitor. Bioorg Med Chem Lett. 2010 Oct 1;20(19):5869-73. doi: 10.1016/j.bmcl.2010.07.104. Epub 2010 Jul 30. PubMed PMID: 20797855.
6: Venkatesan AM, Dehnhardt CM, Delos Santos E, Chen Z, Dos Santos O, Ayral-Kaloustian S, Khafizova G, Brooijmans N, Mallon R, Hollander I, Feldberg L, Lucas J, Yu K, Gibbons J, Abraham RT, Chaudhary I, Mansour TS. Bis(morpholino-1,3,5-triazine) derivatives: potent adenosine 5′-triphosphate competitive phosphatidylinositol-3-kinase/mammalian target of rapamycin inhibitors: discovery of compound 26 (PKI-587), a highly efficacious dual inhibitor. J Med Chem. 2010 Mar 25;53(6):2636-45. doi: 10.1021/jm901830p. PubMed PMID: 20166697.
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma



AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

References
- PRMT5 inhibitors and uses thereofPublication Number: US-12448388-B2Grant Date: 2025-10-21
- KRAS G12D modulating compoundsPublication Number: US-12448400-B2Grant Date: 2025-10-21
- TRIAZINE COMPOUNDS AS P13 KINASE AND MTOR INHIBITORSPublication Number: PT-2294072-TPriority Date: 2008-05-23
- TRIASINE UNITS AS P13 KINASE INHIBITORS AND MOTORPublication Number: ME-01111-BPriority Date: 2008-05-23
- HER2 mutation inhibitorsPublication Number: US-12447153-B2Grant Date: 2025-10-21
- Anti-hiv compoundsPublication Number: US-2025326779-A1
- Aryl aminopyrimidines as dual MerTK and TYRO3 inhibitors and methods thereofPublication Number: US-12448365-B2Grant Date: 2025-10-21
References
- https://celcuity.com/revtorpyk/REVTORPYK_PI_2026.pdf
- Dehnhardt CM, Venkatesan AM, Chen Z, Delos-Santos E, Ayral-Kaloustian S, Brooijmans N, et al. (August 2011). “Identification of 2-oxatriazines as highly potent pan-PI3K/mTOR dual inhibitors”. Bioorganic & Medicinal Chemistry Letters. 21 (16): 4773–8. doi:10.1016/j.bmcl.2011.06.063. PMID 21763134.
- “FDA approves gedatolisib with fulvestrant, with or without palbociclib, for HR-positive, HER2-negative locally advanced or metastatic breast cancer”. U.S. Food and Drug Administration (FDA). 14 July 2026. Retrieved 20 July 2026.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - Sabatini DM (November 2017). “Twenty-five years of mTOR: Uncovering the link from nutrients to growth”. Proceedings of the National Academy of Sciences of the United States of America. 114 (45): 11818–11825. Bibcode:2017PNAS..11411818S. doi:10.1073/pnas.1716173114. PMC 5692607. PMID 29078414.
- Tian T, Li X, Zhang J (February 2019). “mTOR Signaling in Cancer and mTOR Inhibitors in Solid Tumor Targeting Therapy”. International Journal of Molecular Sciences. 20 (3): 755. doi:10.3390/ijms20030755. PMC 6387042. PMID 30754640.
- Hua H, Kong Q, Zhang H, Wang J, Luo T, Jiang Y (July 2019). “Targeting mTOR for cancer therapy”. Journal of Hematology & Oncology. 12 (1) 71. doi:10.1186/s13045-019-0754-1. PMC 6612215. PMID 31277692.
- Vanhaesebroeck B, Perry MW, Brown JR, André F, Okkenhaug K (October 2021). “PI3K inhibitors are finally coming of age”. Nature Reviews. Drug Discovery. 20 (10): 741–769. doi:10.1038/s41573-021-00209-1. PMC 9297732. PMID 34127844. S2CID 235437841.
- Millis SZ, Ikeda S, Reddy S, Gatalica Z, Kurzrock R (December 2016). “Landscape of Phosphatidylinositol-3-Kinase Pathway Alterations Across 19 784 Diverse Solid Tumors”. JAMA Oncology. 2 (12): 1565–1573. doi:10.1001/jamaoncol.2016.0891. PMID 27388585.
- Anderson EJ, Mollon LE, Dean JL, Warholak TL, Aizer A, Platt EA, et al. (2020). “A Systematic Review of the Prevalence and Diagnostic Workup of PIK3CA Mutations in HR+/HER2- Metastatic Breast Cancer”. International Journal of Breast Cancer. 2020 3759179. doi:10.1155/2020/3759179. PMC 7322582. PMID 32637176.
- Clinical trial number NCT01420081 for “A Study Of Two Dual PI3K/mTOR Inhibitors, PF-04691502 And PF-05212384 In Patients With Recurrent Endometrial Cancer” at ClinicalTrials.gov
- Clinical trial number NCT01925274 for “A Study Of PF-05212384 Plus Irinotecan Vs Cetuximab Plus Irinotecan In Patients With KRAS And NRAS Wild Type Metastatic Colorectal Cancer” at ClinicalTrials.gov
- Clinical trial number NCT02438761 for “PF-05212384 (PKI-587) for t-AML/MDS or de Novo Relapsed or Refractory Acute Myeloid Leukemia (AML)” at ClinicalTrials.gov
- Clinical trial number NCT03698383 for “Phase II Study of Herzuma® Plus Gedatolisib in Patients With HER-2 Positive Metastatic Breast Cancer” at ClinicalTrials.gov
- Clinical trial number NCT03911973 for “Gedatolisib Plus Talazoparib in Advanced Triple Negative or BRCA1/2 Positive, HER2 Negative Breast Cancers” at ClinicalTrials.gov
- Clinical trial number NCT03065062 for “Study of the CDK4/6 Inhibitor Palbociclib (PD-0332991) in Combination With the PI3K/mTOR Inhibitor Gedatolisib (PF-05212384) for Patients With Advanced Squamous Cell Lung, Pancreatic, Head & Neck and Other Solid Tumors” at ClinicalTrials.gov
- Clinical trial number NCT02626507 for “Phase I Study of Combination of Gedatolisib With Palbociclib and Faslodex in Patients With ER+/HER2- Breast Cancer” at ClinicalTrials.gov
- “Celcuity Announces FDA Approval of Revtorpyk (gedatolisib) for the Treatment of HR+/HER2-, PIK3CA Wild-Type Locally Advanced or Metastatic Breast Cancer” (Press release). Celcuity. 14 July 2026. Retrieved 20 July 2026 – via GlobeNewswire.
- World Health Organization (2015). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 73”. WHO Drug Information. 29 (1). hdl:10665/331088.
External links
- Clinical trial number NCT05501886 for “Gedatolisib Plus Fulvestrant With or Without Palbociclib vs Standard-of-Care for the Treatment of Patients With Advanced or Metastatic HR+/HER2- Breast Cancer (VIKTORIA-1) (VIKTORIA-1)” at ClinicalTrials.gov
 | |
| Clinical data | |
|---|---|
| Trade names | Revtorpyk |
| Other names | PF-05212384; PKI-587 |
| AHFS/Drugs.com | revtorpyk |
| License data | US DailyMed: Gedatolisib |
| Routes of administration | Intravenous infusion |
| Drug class | Antineoplastic |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1197160-78-3 |
| PubChem CID | 44516953 |
| IUPHAR/BPS | 7940 |
| DrugBank | DB11896 |
| ChemSpider | 24644946 |
| UNII | 96265TNH2R |
| KEGG | D10635 |
| ChEMBL | ChEMBL592445 |
| CompTox Dashboard (EPA) | DTXSID40152557 |
| Chemical and physical data | |
| Formula | C32H41N9O4 |
| Molar mass | 615.739 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
/////////Gedatolisib, anax labs, approvals 3026, FDA 2026, PF 05212384, PF 5212384, PKI-587, PF-05212384; PF-5212384; PKI 587, gedatolisib, antitumor agent, PHASE 3, PFIZER, гедатолисиб , غيداتوليسيب , 吉达利塞 , 96265TNH2R
O=C(NC1=CC=C(C2=NC(N3CCOCC3)=NC(N4CCOCC4)=N2)C=C1)NC5=CC=C(C(N6CCC(N(C)C)CC6)=O)C=C5
Journal of Medicinal Chemistry (2017), 60(17), 7524-7538 PQR 309
Admilparant
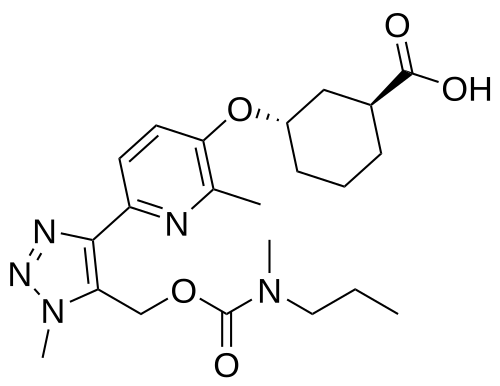
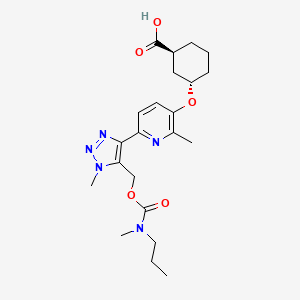
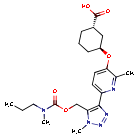
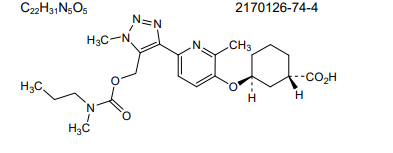
Admilparant, (BMS-986278)
CAS 2170126-74-4
MF C22H31N5O5 MW 445.5 g/mol
(1S,3S)-3-({2-methyl-6-[1-methyl-5-({[methyl(propyl)carbamoyl]oxy}methyl)-1H-1,2,3-triazol-4-l]pyridin-3-yl}oxy)cyclohexane-1-carboxylic acid
lysophosphatidic acid receptor 1 (LPA1) antagonist
- 4UN9AOU6G8
- BMS986278
- (1S,3S)-3-((2-Methyl-6-(1-methyl-5-(((methyl(propyl)carbamoyl)oxy)methyl)-1H-1,2,3-triazol-4-yl)pyridin-3-yl)oxy)cyclohexane-1-carboxylic acid
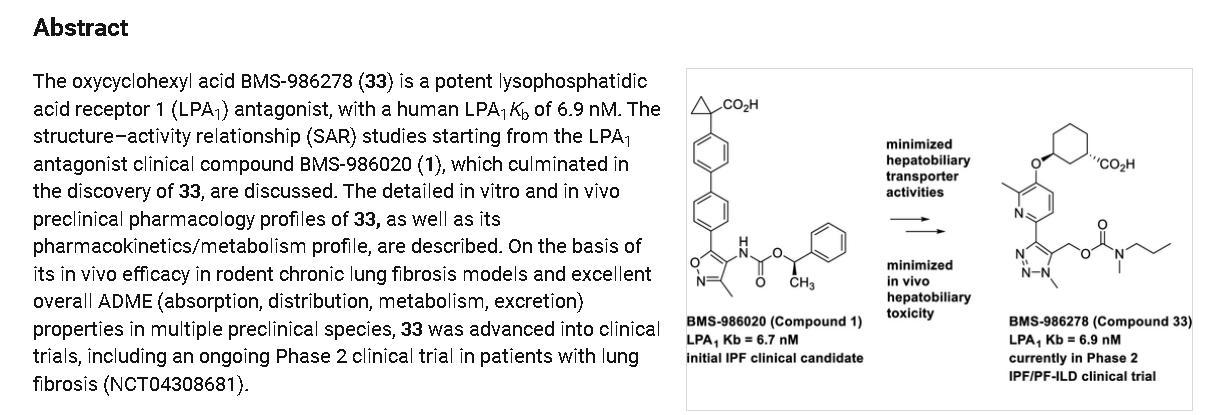
Admilparant is an investigational new drug being developed by Bristol-Myers Squibb for the treatment of idiopathic pulmonary fibrosis (IPF) and progressive pulmonary fibrosis (PPF). It is a first-in-class lysophosphatidic acid receptor 1 (LPA1) antagonist.[1][2]
As of 2024, admilparant is in Phase III clinical trials for both IPF and PPF.[2][3]
SYN
Publication Name: Journal of Medicinal Chemistry, Publication Date: 2021-10-28, PMID: 34709814
DOI: 10.1021/acs.jmedchem.1c01256
(1S,3S)-3-((2-Methyl-6-(1-methyl-5-(((methyl(propyl)carbamoyl)-oxy)methyl)-1H-1,2,3-triazol-4-yl)pyridin-3-yl)oxy)cyclohexane-1-carboxylic Acid (33). Compound 33 was prepared using the same
synthetic sequence as 25, except that intermediate 42 was reacted with
N-methylpropan-1-amine instead of 1-cyclobutyl-N-methylmethanamine. 1H NMR (500 MHz, DMSO-d6, 100 °C) δ 11.99−11.46 (m,1H), 7.82 (d, J = 8.3 Hz, 1H), 7.43 (d, J = 8.8 Hz, 1H), 5.65 (s, 2H),
4.89−4.62 (m, 1H), 4.10 (s, 3H), 3.12 (br t, J = 7.2 Hz, 2H), 2.79 (s,3H), 2.69 (tt, J = 9.4, 4.4 Hz, 1H), 2.44 (s, 3H), 2.03 (dt, J = 13.8, 4.5Hz, 1H), 1.92−1.86 (m, 1H), 1.86−1.79 (m, 2H), 1.74−1.68 (m, 1H),
1.68−1.58 (m, 2H), 1.58−1.51 (m, 1H), 1.43 (dq, J = 14.4, 7.1 Hz,2H), 0.76 (br t, J = 7.3 Hz, 3H). 13C NMR (126 MHz, DMSO-d6, 100°C) δ 175.4, 154.7, 150.1, 147.7, 143.9, 141.4, 129.6, 120.0, 118.6, 71.8,
54.5, 49.5, 37.4, 34.4, 33.4, 31.6, 28.7, 27.2, 19.8, 19.4, 18.6, 10.1. m/z446 [M + H]+
. HPLC/UV purity: 99.9% using the following reverse phase chromatographic conditions: Agilent HPLC; Phenomenex Kinetex-C-18; 100 (L) × 4.6 mm2 (i.d.) column; 2.6 μm particle size; wavelength, 220−380 nm; flow rate, 1.0 mL/min; temperature, 35°C; injection volume, 4 μL of 0.25 mg/mL in 1:1 MeCN:H2O; mobilephase A, H2O−0.05% TFA; mobile phase B, MeCN−0.05% TFA; gradient elution, starting at 10−80% B over 10 min and ending at 95% Bafter an additional 4 min; retention time = 8.28 min. Stereoisomeric purity was >99.5% using the following chiral chromatographic conditions: UPC2 Analytical SFC, ChromegaChiral CC4; 250 (L) ×4.6 mm2 (i.d.); 5 μm column; flow rate, 3 mL/min; temperature, 40 °C;injection volume, 10 μL of 0.25 mg/mL in MeCN:MeOH (1:1);mobile phase, 30% MeOH and 70% CO2 at 120 bar retention time =6.05 min. Accurate mass, [M + H]+ at m/z = 446.2398 (−2.03 ppmfrom theoretical for C22H32N5O5). [α]20D = +28.24° (MeOH, c = 0.51).
Elem. Anal. (theoretical): C, 59.31; H, 7.01; N, 15.72. Found: C, 59.35;H, 6.78; N, 15.69. UV (MeOH) at 254 nm (ε = 17,856), 290 nm (ε =7,519), and 296 nm (ε = 8,288). Concentration: adjusted for purity,
0.05154840 g/L or 0.0001157047 mol/L. Melting point = 152−154°C. Accurate mass, [M + H]+ at m/z 466.2398 (−2.03 ppm fromtheoretical for C22H32N5O5).
synthetic sequence as 25, except that intermediate 42 was reacted with N-methylpropan-1-amine instead of 1-cyclobutyl-N-methylmethanamine
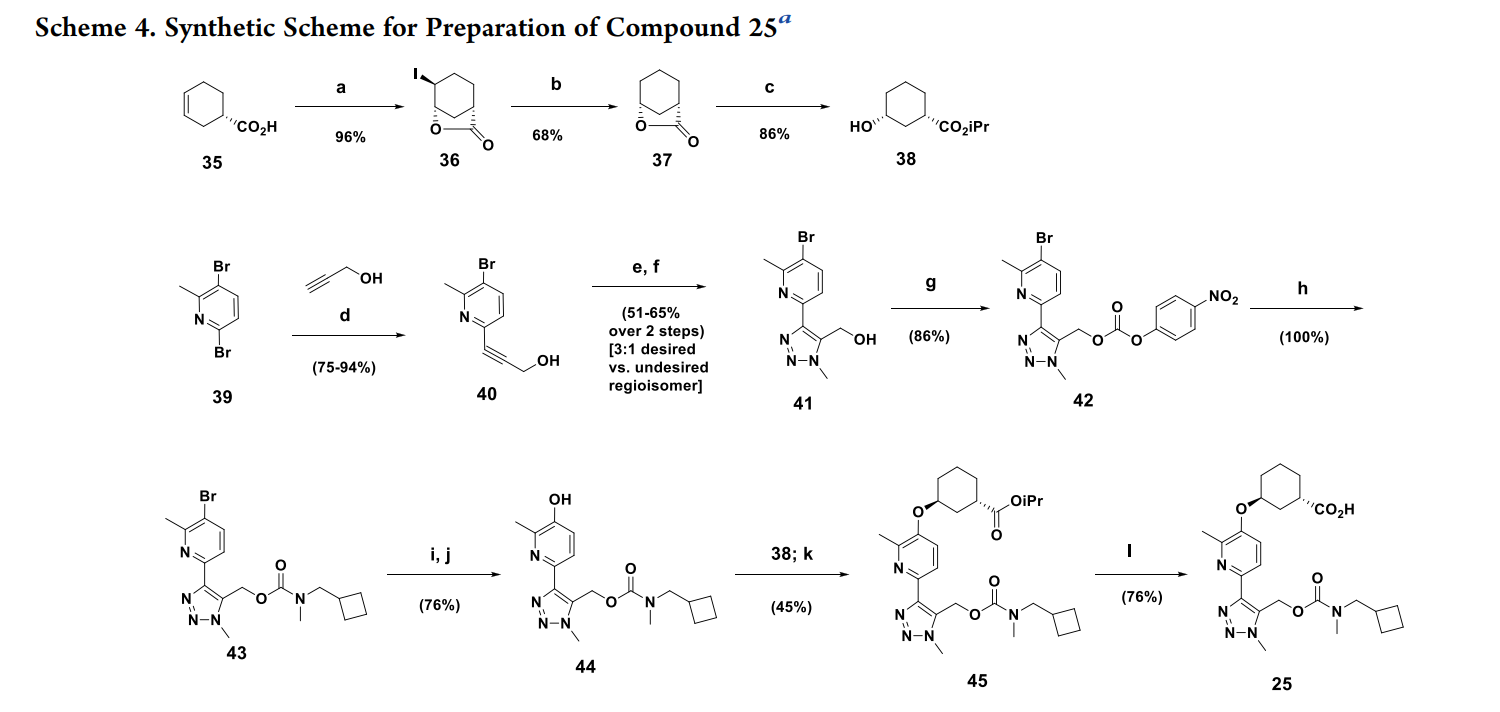
a
Reagents and conditions: (a) I2 (1.1 equiv)/KI (2.5 equiv)/NaHCO3 (3 equiv)/water (96%); (b) H2 (50 psi)/ Pd/C (cat)/Et3N (2 equiv)/EtOAc (68%); (c) CH3COCl (2.5 equiv)/iPrOH (87−95%); d) (Ph3P)2PdCl2 (5%)/ Et3N/CuI (5%)/RT (75−94%); (e) Ru(II)-(Ph3P)2(Me5Cyp)Cl (5%)/TMSCH2N3/dioxane 50 °C/15 h; (f) Bu4NF/0 °C to RT (51−65% over 2 steps; 3:1 desired:undesired regioisomer); (g) 4-nitrophenyl chloroformate/pyridine/CH2Cl2 (86%); (h) N-cyclobutyl N-methylamine/iPr2NEt/CH2Cl2 (100%); (i) B2(pin)2/KOAc/PdCl2(dppf)/THF/80 °C; (j) NaH2BO4/H2O/RT (76% over 2 steps); (k) 38; 1,1′-(azodicarbonyl)dipiperidine/Bu3P/toluene/50 °C (45%); (l)LiOH/H2O/MeOH (76%).
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US208146892&_cid=P20-MFS2PF-83792-1
PATENT
- Carbamoyloxymethyl triazole cyclohexyl acids as lpa antagonistsPublication Number: US-2022249443-A1Priority Date: 2016-06-21
- Carbamoyloxymethyl triazole cyclohexyl acids as LPA antagonistsPublication Number: US-RE49352-EPriority Date: 2016-06-21Grant Date: 2023-01-03
- Carbamoyloxymethyl triazole cyclohexyl acids as LPA antagonistsPublication Number: AU-2021209334-B2Priority Date: 2016-06-21Grant Date: 2023-06-01
- Carbamoyloxymethyltriazole cyclohexylates as LPA antagonistsPublication Number: JP-7312295-B2Priority Date: 2016-06-21Grant Date: 2023-07-20
- Carbamoyloxymethyl triazole cyclohexyl acids as lpa antagonistsPublication Number: US-2023390249-A1Priority Date: 2016-06-21
- Carbamoyloxymethyltriazolylcyclohexanes as LPA antagonistsPublication Number: CN-109963843-BPriority Date: 2016-06-21Grant Date: 2022-03-11
- Carbamoyloxymethyltriazole cyclohexyl acid as LPA antagonistPublication Number: CN-114601830-APriority Date: 2016-06-21
- Carbamoyloxymethyl triazole cyclohexyl acid as an LPA antagonistPublication Number: KR-102377340-B1Priority Date: 2016-06-21Grant Date: 2022-03-21
- Carbamoyloxymethyl triazole cyclohexyl acids as lpa antagonistsPublication Number: KR-20220038537-APriority Date: 2016-06-21
- Carbamoyloxymethyl triazole cyclohexyl acids as lpa antagonistsPublication Number: KR-102463621-B1Priority Date: 2016-06-21Grant Date: 2022-11-03

AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- “Admilparant (BMS-986278): Idiopathic Pulmonary Fibrosis Likelihood of Approval”. Pharmaceutical Technology. 25 December 2023. Retrieved 2024-11-23.
- Corte TJ, Behr J, Cottin V, Glassberg MK, Kreuter M, Martinez FJ, et al. (October 2024). “Efficacy and Safety of Admilparant, an LPA1 Antagonist in Pulmonary Fibrosis: A Phase 2 Randomized Clinical Trial”. American Journal of Respiratory and Critical Care Medicine. 211 (2): 230–238. doi:10.1164/rccm.202405-0977OC. PMID 39393084.
- Splete H (16 September 2024). “Admilparant Affects Biomarkers in Pulmonary Fibrosis”. Medscape. Retrieved 2024-11-23.
| Clinical data | |
|---|---|
| Other names | BMS-986278 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2170126-74-4 |
| PubChem CID | 132232205 |
| DrugBank | DB18011 |
| ChemSpider | 115009679 |
| UNII | 4UN9AOU6G8 |
| KEGG | D12657 |
| ChEMBL | ChEMBL5087506 |
| Chemical and physical data | |
| Formula | C22H31N5O5 |
| Molar mass | 445.520 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- Zhou Y, Zhang Y, Zhao D, Yu X, Shen X, Zhou Y, Wang S, Qiu Y, Chen Y, Zhu F: TTD: Therapeutic Target Database describing target druggability information. Nucleic Acids Res. 2024 Jan 5;52(D1):D1465-D1477. doi: 10.1093/nar/gkad751. [Article]
/////////Admilparant, BMS 986278, PHASE 3, Bristol-Myers Squibb, idiopathic pulmonary fibrosis, 4UN9AOU6G8
Ebselen
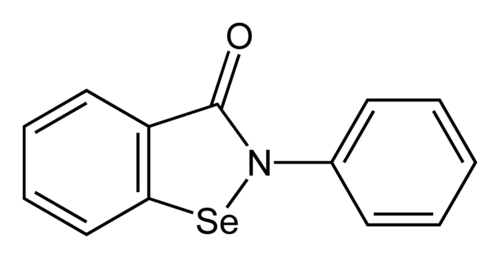
Ebselen
274.19 g/mol,
C13H9NOSe
2-phenyl-1,2-benzoselenazol-3-one
- CAS 60940-34-3
- 2-phenyl-1,2-benzoselenazol-3-one
- 2-Phenyl-1,2-benzisoselenazol-3(2H)-one
- Ebselene
- PZ 51, DR3305, and SPI-1005
- 40X2P7DPGH
Ebselen is a benzoselenazole that is 1,2-benzoselenazol-3-one carrying an additional phenyl substituent at position 2. Acts as a mimic of glutathione peroxidase. It has a role as a neuroprotective agent, an apoptosis inducer, an anti-inflammatory drug, an antioxidant, a hepatoprotective agent, a genotoxin, a radical scavenger, an enzyme mimic, an EC 1.3.1.8 [acyl-CoA dehydrogenase (NADP(+))] inhibitor, an EC 1.8.1.12 (trypanothione-disulfide reductase) inhibitor, an EC 1.13.11.33 (arachidonate 15-lipoxygenase) inhibitor, an EC 1.13.11.34 (arachidonate 5-lipoxygenase) inhibitor, an EC 2.5.1.7 (UDP-N-acetylglucosamine 1-carboxyvinyltransferase) inhibitor, an EC 2.7.10.1 (receptor protein-tyrosine kinase) inhibitor, an EC 3.5.4.1 (cytosine deaminase) inhibitor, an EC 5.1.3.2 (UDP-glucose 4-epimerase) inhibitor, a ferroptosis inhibitor, an antifungal agent, an EC 3.4.22.69 (SARS coronavirus main proteinase) inhibitor, an anticoronaviral agent, an antibacterial agent, an antineoplastic agent and an EC 3.1.3.25 (inositol–phosphate phosphatase) inhibitor.
Ebselen (also called PZ 51, DR3305, and SPI-1005), is a synthetic organoselenium molecule under preliminary investigation as a drug candidate.[1] It belongs to the class of compounds related to benzene and its derivatives.[1] It is being developed by the Seattle biotechnology company, Sound Pharmaceuticals, Inc.[1] It has also been reported to target tubulin, blocking its polymerization.[2]
Ebselen has been investigated for the treatment and basic science of Meniere’s Disease, Type 2 Diabetes Mellitus, and Type 1 Diabetes Mellitus.
Ebselen has been entered into clinical trials as a lead compound intended for the potential treatment of various diseases.[3] Its most advanced clinical trial is a Phase III study in people with Meniere’s disease, completed in July 2024.[4]
In vitro, ebselen is a mimic of glutathione peroxidase and reacts with peroxynitrite.[5] It is purported to have antioxidant and anti-inflammatory properties.[1][5]
Synthesis
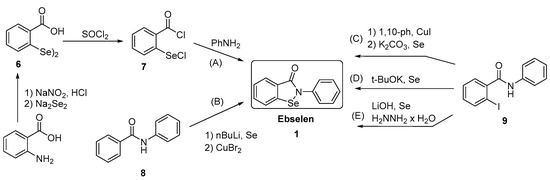
Generally, synthesis of the characteristic scaffold of ebselen, the benzoisoselenazolone ring system, can be achieved either through reaction of primary amines (RNH2) with 2-(chloroseleno)benzoyl chloride (Route I),[6] by ortho-lithiation of benzanilides followed by oxidative cyclization (Route II) mediated by cupric bromide (CuBr2),[7] or through the efficient Cu-catalyzed selenation / heterocyclization of o-halobenzamides, a methodology developed by Kumar et al.[8] (Route III).

SYN
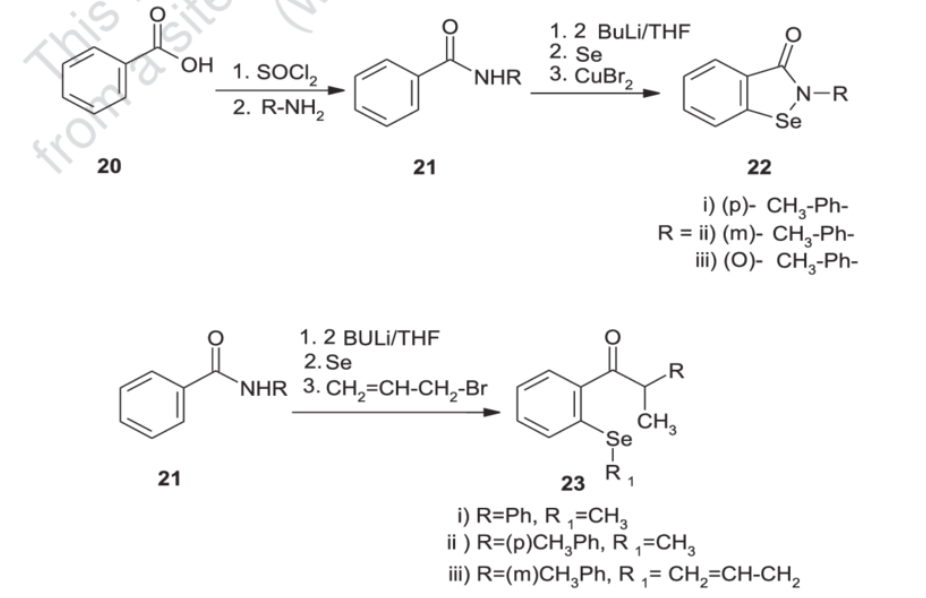
Synthesis of ebselen from benzoic acid by ortholithiation of benzanilide SOCl 2 =Thionyl chloride, R-NH 2 =Substituted aryl mine, BuLi/THF=n-butyllithium/ tetrahydrofuran, CuBr 2 =Cupper bromide, CH 2 =CH- CH 2 -Br = Allyl bromide.
SYN
New Chiral Ebselen Analogues with Antioxidant and Cytotoxic Potential
Molecules, March 2017, 22(3):492
SYN
https://pubs.acs.org/doi/10.1021/ol102027j
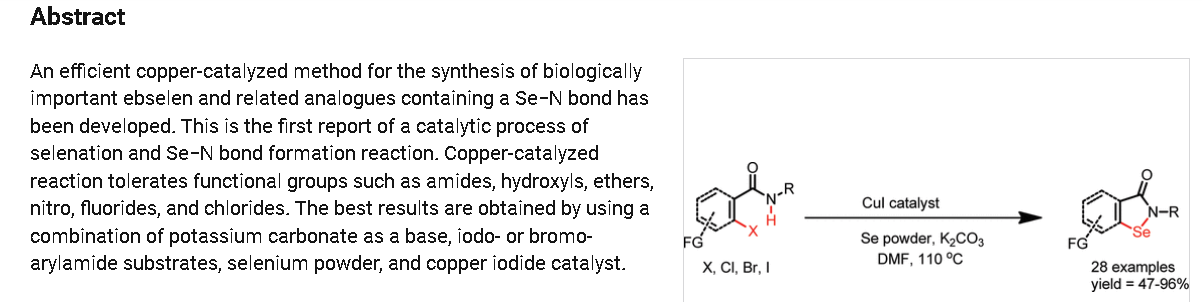

2-Phenylbenzo[d][1,2]selenazol-3(2H)-one (1) from 2-Iodo-N-phenylbenzamide (Typical
Procedure): Copper iodide (114 mg, 0.6 mmol) and 1,10-phenanthroline (108 mg, 0.6 mmol)
were added into DMF (3 mL) in a single neck flask. Resulted brownish solution was stirred for
15 min and then 2-iodo-N-phenylbenzamide1 (0.97 g, 3.0 mmol), selenium powder (0.29 g, 3.6
mmol), and potassium carbonate powder (0.65 g, 4.7 mmol) were added sequentially to same reaction flask. Brown colored reaction mixture was refluxed at 110oC using refluxing condenser
under nitrogen atmosphere. Progress of reaction was monitored by TLC. Reaction mixture was
refluxed for 8h. After this, reaction mixture poured over brine solution (60 mL) and stirred for 3
h. Product was precipitated as white solid which was collected by filtration over Buchner funnel,
product was washed with water (15 mL x 2), dried in air, dissolved in ethyl acetate, concentrated
over rotary evaporator, resulted brown solid which was purified by column chromatography
using hexane/ ethyl acetate (8:2) over silica gel. Yield 0.69 g (84%), mp 182-183 °C (180-181
°C).14,15 1H NMR (400 MHz, DMSO-d6) 8.09 (d, J = 8.0 Hz, 1H), 7.91 (d, J = 8.0 Hz, 1H),
7.71-7.62 (m, 3H), 7.51-7.43 (m, 3H), 7.28 (t, J = 8.0 Hz, 1H). 1H NMR (400 MHz, CDCl3)
8.12 (d, 7.6 Hz, 1H), 7.68-7.62 (m, 4H), 7.52-7.41 (m, 3H), 7.29 (m, 1H). IR (plate): 3057, 2921,
1598, 1443, 1346, 1263, 1028 cm-1; ESMS m/z: 276 (M+H+).
2-Phenylbenzo[d][1,2]selenazol-3(2H)-one (1) from 2-Iodo-N-phenylbenzamide at 74 mmol
scale: Reaction was carried out at 74 mmol scale using 2-iodo-N-phenylbenzamide (24.00 g,
74.3 mmol), selenium powder (7.04 g, 89.1 mmol), CuI (2.83 g, 14.9 mmol), 1,10
phenanthroline (2.69 g, 14.9 mmol), and anhydrous potassium carbonate powder (15.40 g, 111.4
mmol) in DMF (50 mL) and procedure and workup followed are similar to 3.6 mmol scale
reaction. Yield 16.28 g (80%), Figure S1.
2-Phenylbenzo[d][1,2]selenazol-3(2H)-one (1) from 2-Bromo-N-phenylbenzamide: Ebselen 1
was prepared from 2-bromo-N-phenylbenzamide2 (1.00 g, 3.6 mmol), selenium powder (0.34 g,
4.3 mmol), K2CO3 powder (0.74 g, 5.4 mmol), CuI (137 mg, 0.7 mmol), and 1,10-phenanthroline
(130 mg, 0.7 mmol) in DMF (3 mL). Reaction mixture was refluxed for 16 h at 110oC. Progress of reaction was monitored by TLC. After completion of reaction, mixture was poured into brine
solution (60 mL) and the resulted white precipitate was washed with water (20 mL x 2), and
dried in air. Purification by column chromatography on silica gel using CH2Cl2 provided white
crystalline solid (0.77 g, 78%).
2-Phenylbenzo[d][1,2]selenazol-3(2H)-one (1) from 2-Chloro-N-phenylbenzamide: Reaction
was carried out at 4 mmol scale using 2-chloro-N-phenylbenzamide3 (1.00 g, 4.3 mmol), CuI
(172 mg, 0.9 mmol), 1,10-phenanthroline (162 mg, 0.9 mmol), selenium powder (0.41 g, 5.2
mmol), K2CO3 (0.89 g, 6.4 mmol) in DMF (4 mL). Reaction mixture was refluxed for 24 h at
110oC. Workup procedure is similar as followed for bromo substrate. Yield 0.55 g (47%).
History
The first patent for 2-phenyl-1,2-benzoselenazol-3(2H)-one was filed in 1980 and granted in 1982.[9]
Research
Ebselen is in preliminary clinical development for the potential treatment of hearing loss and depression, among other medical indications.[3][10]
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- “Ebselen”. DrugBank. 29 January 2025. Retrieved 4 February 2025.
- Baksheeva VE, La Rocca R, Allegro D, Derviaux C, Pasquier E, Roche P, Morelli X, Devred F, Golovin AV, Tsvetkov PO (2025). “NanoDSF Screening for Anti-tubulin Agents Uncovers New Structure–Activity Insights”. Journal of Medicinal Chemistry. doi:10.1021/acs.jmedchem.5c01008.
- “Ebselen pipeline”. Sound Pharmaceuticals, Inc. 2025. Retrieved 4 February 2025.
- “SPI-1005 for the Treatment of Meniere’s Disease (STOPMD-3)”. ClinicalTrials.gov, US National Library of Medicine. 1 August 2024. Retrieved 4 February 2025.
- Schewe T (October 1995). “Molecular actions of ebselen – an antiinflammatory antioxidant”. General Pharmacology. 26 (6): 1153–69. doi:10.1016/0306-3623(95)00003-J. PMID 7590103.
- Kamigata N, Iizuka H, Izuoka A, Kobayashi M (July 1986). “Photochemical Reaction of 2-Aryl-1, 2-benzisoselenazol-3 (2 H)-ones”. Bulletin of the Chemical Society of Japan. 59 (7): 2179–83. doi:10.1246/bcsj.59.2179.
- Engman L, Hallberg A (1989-06-01). “Expedient synthesis of ebselen and related compounds”. The Journal of Organic Chemistry. 54 (12): 2964–2966. doi:10.1021/jo00273a035. ISSN 0022-3263.
- Balkrishna SJ, Bhakuni BS, Chopra D, Kumar S (December 2010). “Cu-catalyzed efficient synthetic methodology for ebselen and related Se-N heterocycles”. Organic Letters. 12 (23): 5394–7. doi:10.1021/ol102027j. PMID 21053969.
- DE3027073A1, Etschenberg, Eugen Dr; Renson, Marcel Prof Dipl-Chem Jupille & Winkelmann, Johannes Dr 5000 Köln, “2-phenyl-1,2-benzisoselenazol-3(2h)-on enthaltende pharmazeutische praeparate und ihre verwendung”, issued 1982-02-18
- “Ebselen search: list of clinical trials sponsored by Sound Pharmaceuticals”. ClinicalTrials.gov, US National Library of Medicine. 2025. Retrieved 4 February 2025.
External links
| Names | |
|---|---|
| Preferred IUPAC name2-Phenyl-1,2-benzoselenazol-3(2H)-one | |
| Identifiers | |
| CAS Number | 60940-34-3 |
| 3D model (JSmol) | Interactive imageInteractive image |
| ChEBI | CHEBI:77543 |
| ChEMBL | ChEMBL51085 |
| ChemSpider | 3082 |
| ECHA InfoCard | 100.132.190 |
| PubChem CID | 3194 |
| UNII | 40X2P7DPGH |
| CompTox Dashboard (EPA) | DTXSID7045150 |
| InChI | |
| SMILES | |
| Properties | |
| Chemical formula | C13H9NOSe |
| Molar mass | 274.17666 |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
References
- Zhou Y, Zhang Y, Zhao D, Yu X, Shen X, Zhou Y, Wang S, Qiu Y, Chen Y, Zhu F: TTD: Therapeutic Target Database describing target druggability information. Nucleic Acids Res. 2024 Jan 5;52(D1):D1465-D1477. doi: 10.1093/nar/gkad751. [Article]
////////Ebselen, Ebselene, PZ 51, DR 3305, SPI 1005, PHASE 3, 40X2P7DPGH, Meniere’s Disease, Type 2 Diabetes Mellitus, Type 1 Diabetes Mellitus
Sergliflozin Etabonate

Sergliflozin Etabonate
408504-26-7 Cas no
Ethyl [(2R,3S,4S,5R,6S)-3,4,5-trihydroxy-6-[2-[(4-methoxyphenyl)methyl]phenoxy]oxan-2-yl]methyl carbonate
2-(4-methoxybenzyl)phenyl 6-O-ethoxycarbonyl-beta-D-glucopyranoside
ethyl [(2R,3S,4S,5R,6S)-3,4,5-trihydroxy-6-[2-[(4-methoxyphenyl)methyl]phenoxy]tetrahydropyran-2-yl]methyl carbonate
ethyl [(2R,3S,4S,5R,6S)-3,4,5-trihydroxy-6-{2-[(4-methoxyphenyl)methyl]phenoxy}oxan-2-yl]methyl carbonate
PHASE 2……….TYPE 3 DIABETES AND OBESITY
A SGLT-2 inhibitor potentially for the treatment of type 2 diabetes and obesity.
GW-869682; GW-869682X; KGT-1251
- etabonate de sergliflozine
- etabonato de sergliflozina
MW 448.4, C23H28O9
KISSEI INNOVATOR
GSK DEVELOPER
Sergliflozin Etabonate is a benzylphenol glucoside and selective sodium-glucose co-transporter subtype 2 (SGLT2) inhibitor with antihyperglycemic activity. Its prodrug form, sergliflozin etabonate, is orally available and is converted to sergiflozin upon absorption.
Sergliflozin etabonate (INN/USAN,[1][2] codenamed GW869682X) is an investigational anti-diabetic drug being developed by GlaxoSmithKline. It did not undergo further development after phase II
Sergliflozin inhibits subtype 2 of the sodium-glucose transport proteins (SGLT2), which is responsible for at least 90% of the glucose reabsorption in the kidney. Blocking this transporter causes blood glucose to be eliminated through the urine.[3][4]
Chemistry
Etabonate refers to the ethyl carbonate group. The remaining structure, which is the active substance, is called sergliflozin.

Sergliflozin
[PDF] Design, Syntheses, and SAR Studies of Carbocyclic Analogues of …onlinelibrary.wiley.com974 × 740Search by imageDesign, Syntheses, and SAR Studies of Carbocyclic Analogues of Sergliflozin as Potent SodiumDependent Glucose Cotransporter 2 In
Sergliflozin Etabonate is a benzylphenol glucoside and selective sodium-glucose co-transporter subtype 2 (SGLT2) inhibitor with antihyperglycemic activity. Its prodrug form, sergliflozin etabonate, is orally available and is converted to sergiflozin upon absorption.

sergliflozin and prodrugs of sergliflozin, in particular sergliflozin etabonate, including hydrates and solvates thereof, and crystalline forms thereof. Methods for its manufacture are described in the patent applications EP 1344780 and EP 1489089 for example.
The compounds are described in EP 1 329 456 A1 and a crystalline form ofSergliflozin etabonate is described in EP 1 489 089 A1.
PATENT
US6872706B2
https://patentscope.wipo.int/search/en/detail.jsf?docId=US40677423&_cid=P20-MF4ZUQ-42384-1
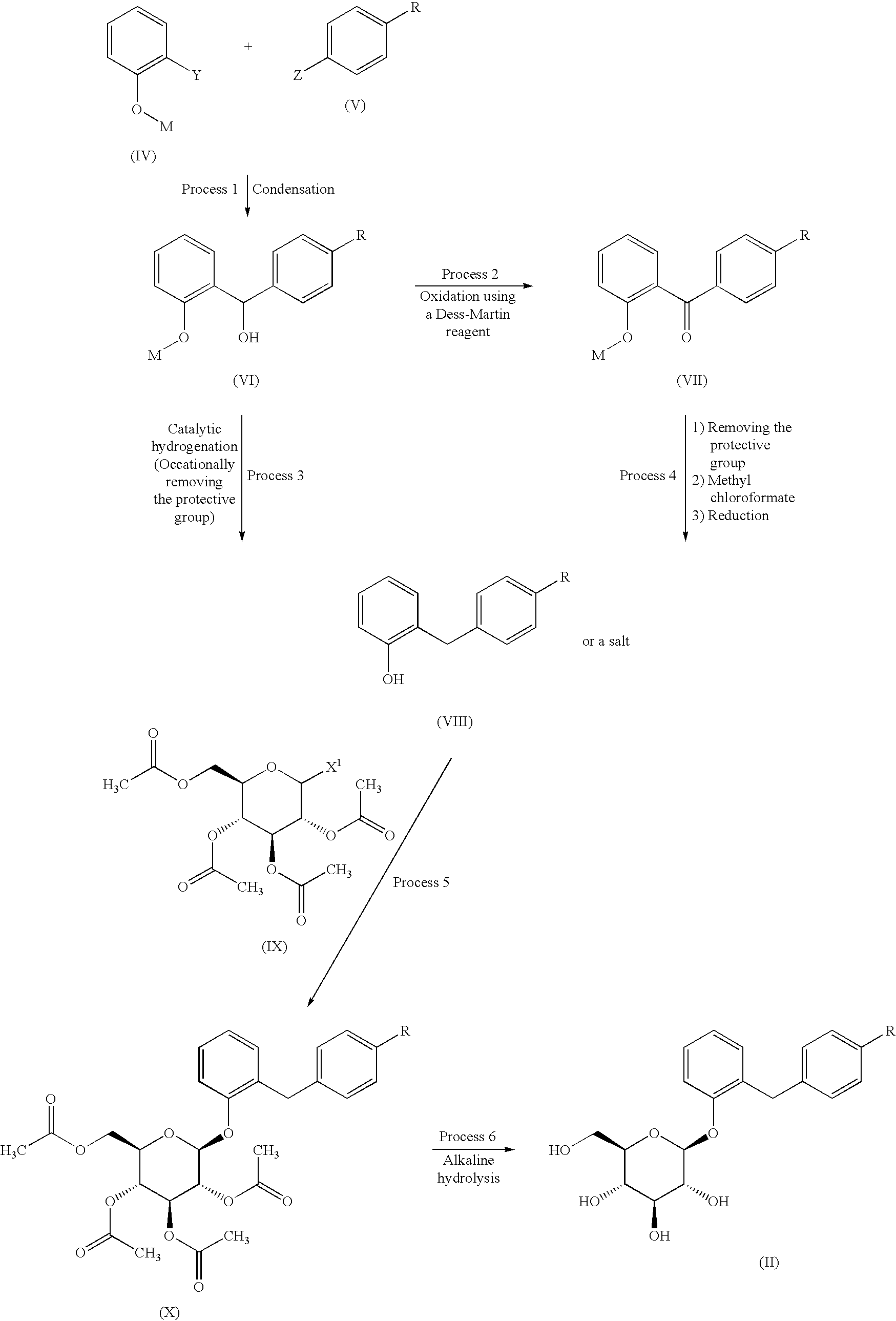
PATENT
WO2001068660A1
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2001068660&_cid=P20-MF4ZXC-45172-1
SYN
Heterocycles 2016, 92, 1599
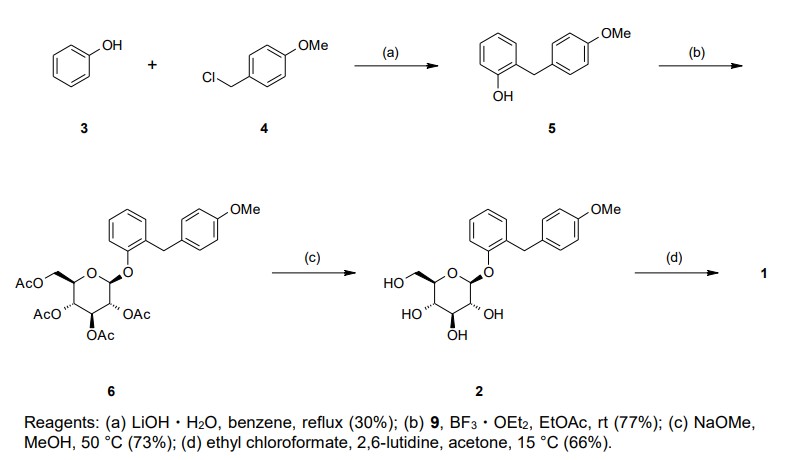
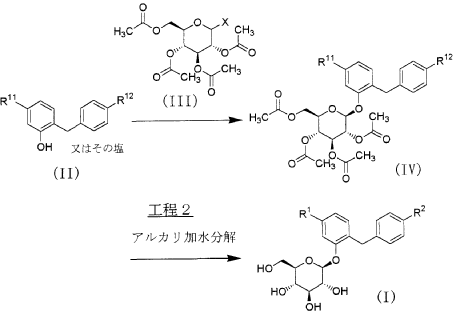
Our initial synthetic route of Serglifrozin etabonate (1) in early development consisted of six steps,
including synthesis of tetra-O-acetyl-D-glucopyranosyl trichloroacetimidate (9), as shown in Scheme 1
and Scheme 2 The first step is the coupling reaction of phenol (3) and 4-methoxybenzyl chloride (4) in the presence of
lithium hydroxide monohydrate (LiOH·H2O) to provide the aglycon 5 in a 30% yield following
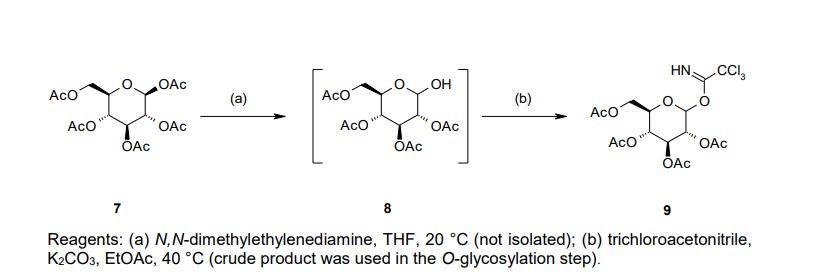
chromatographic purification (Scheme 1). We prepared 9 separately by mono-deacetylation of
penta-O-acetyl-β-D-glucopyranose (7) with N,N-dimethylethylenediamine in THF followed by reaction of
the crude product of 8 with trichloroacetonitrile in the presence of potassium carbonate (K2CO3) in ethyl
acetate (EtOAc) (Scheme 2). Next, we carried out glycosylation of 5 with 9 in the presence of boron
trifluoride diethyl etherate (BF3·OEt2) in EtOAc to produce 6 in a 77% yield. The obtained 6 was
deacetylated with sodium methoxide (NaOMe) in MeOH to produce Serglifrozin (2) in a 73% yield, and
reaction of the isolated 2 with ethyl chloroformate in the presence of 2,6-lutidine in acetone provided 1 in
a 66% yield. The overall yield from 3 was 11%. While this route was capable of supplying small
amounts of 1, it suffered from several disadvantages.
The coupling reaction between 3 and 4 provided the aglycon 5 in low yield (30%); thus, chromatographic
purification was required to obtain highly pure 5. The trichloroimidation reaction of 8 is too hazardous
for large-scale manufacturing, because an excess amount of trichloroacetonitrile, a volatile and highly
toxic reagent, is required to obtain the trichloroacetimidate 9. Furthermore, 9 is too unstable to use
conveniently in large-scale manufacturing. Trichloroacetamide, a sublimation compound, is formed as a
by-product from the glycosylation of 5 with 9. Thus, the vacuum line and the vacuum pump of the
manufacturing equipment would be polluted by trichloroacetamide.
Because of these issues, this synthetic method is unsuitable for large-scale manufacturing. Therefore,
we investigated alternative processes for the preparation of 1, suitable for large-scale manufacturing. An improved synthetic method for 1 was achieved in a five-step procedure without purification of 6
(intermediate), as shown in Scheme 3.
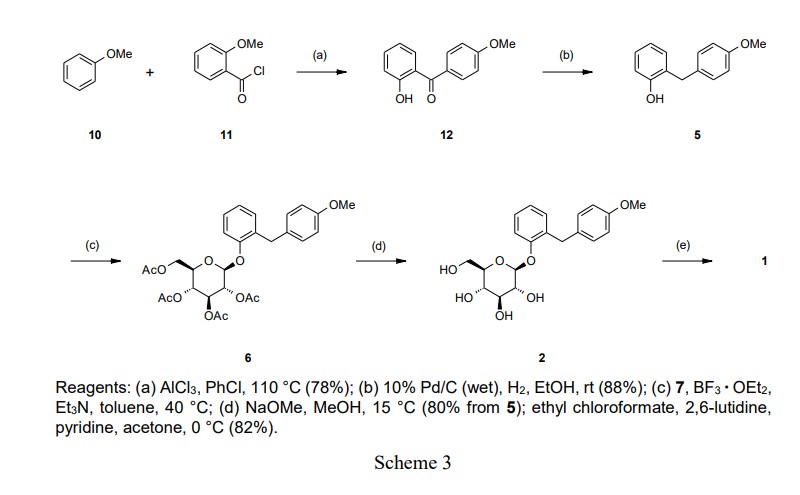
The Friedel-Crafts acylation of anisole (10) with 2-methoxybenzoyl chloride (11) in the presence of
aluminum chloride (AlCl3) at 110 °C provided benzophenone (12), which was selectively demethylated
on the methoxy group at the 2-position. The crude product of 12 was crystallized from MeOH to
provide highly pure 12 in a 78% yield. Hydrogenation of 12 in EtOH with 0.3–0.4 MPa H2 at room
temperature in the presence of 10% Pd/C provided 5. The crude product of 5 was crystallized from
toluene/n-heptane to provide highly pure 5 in an 88% yield.
The key step of the synthesis was the formation of the O-glycosylated product 6. In the initial synthesis,
it was necessary to isolate 6 to remove trichloroacetamide. Consequently, 2 was provided in a 56%
yield from 5. To obtain 6 efficiently without using the trichloroacetimidate (9), we evaluated several
conditions for the direct O-glycosylation of 5 with 7. The results are summarized in Table 1. The
O-glycosylation of 5 with 7 (200 mol%) in the presence of boron trifluoride diethyl etherate (BF3·OEt2;
100 mol%) in dichloromethane (DCM) at room temperature provided the crude product of 6 with a good
yield (80%) and β-selectivity (94/6), and then the deacetylation of the crude product of 6 in the presence
of sodium methoxide (NaOMe) in MeOH proceeded almost quantitatively to provide 2 in a 71% isolated
yield from 5 (run 1). Using this method, it was not necessary to isolate 6 because the excess amount of
7 was converted to glucose and removed to the aqueous layers in the deacetylation step. Use of DCM is
undesirable for large-scale manufacturing because quenching of O-glycosylation with water is highly
exothermic and washing of the DCM layer with water is a complicated procedure. Additionally, it is
strongly desirable to avoid using DCM in a manufacturing process due to environmental issues. For the reasons mentioned above, we attempted to use toluene as an alternative solvent. The O-glycosylation in
the presence of BF3·OEt2 (100 mol%) in toluene at 30 °C did not proceed completely, and the yield of 6
was lower than run 1 (run 2). We concluded that the lower solubility of 7 in toluene, compared with
DCM, caused the low yield. Because it was difficult to increase the amount of toluene from the
perspective of manufacturing efficiency, we tried to improve its solubility by optimizing the reagent
equivalent. Fortunately, we found that an excess amount of BF3·OEt2 enhanced the solubility of 7 in
toluene, and using 300 mol% of BF3·OEt2 in toluene provided 6 in a good yield (80%), similar to that
when using DCM (run 3). In contrast, reducing the amount of 7 provided 6 in an insufficient yield, and
2 was consequently provided in a lower yield (60%) (run 4). To achieve higher β-selectivity and an
increased yield, triethylamine (Et3N) was added to the O-glycosylation of 5 with 7 in the presence of
BF3·OEt2, according to the method of Lee et al.
9 Addition of Et3N (30 mol%) at 30 °C resulted in both
higher yield (89%) and higher β-selectivity (97/3) to provide 2 with a 79% isolated yield (run 5).
Increasing the amount of Et3N to 60 mol% at 30 °C resulted in a lower yield (85%) of 6 compared with
run 5, and the yield of 2 decreased (74%) (run 6). Increasing the reaction temperature to 40 °C in the
presence of 60 mol% of Et3N achieved the best results for both high yield (90%) and high β-selectivity
(99/1) to provide 2 in an 80% yield (run 7).
6-O-Ethoxycarbonyl-2-[(4-methoxyphenyl)methyl]phenyl-β-D-glucopyranoside (1). Ethyl
chloroformate (407 mg, 3.75 mol) was added drop-wise to the mixture of 2 (1.13 g, 3.0 mmol) and
2,6-lutidine (563 mg, 5.25 mmol) in acetone (4 mL) while maintaining the temperature between 12 and
18 °C. The reaction mixture was stirred at 15 °C for 23 h. Water (5 mL) was added drop-wise while
maintaining the temperature below 30 °C, and EtOAc (10 mL) was then added to the mixture. The
biphasic solution was transferred to a separating funnel for phase separation. The aqueous layer was
extracted with EtOAc (5 mL). The EtOAc layers were combined, washed successively with an aqueous
solution of 10% citric acid (5 mL × 2), an aqueous solution of 10% NaCl (5 mL), an aqueous solution of
5% NaHCO3 (5 mL × 2), and an aqueous solution of 10% NaCl (5 mL). They were then dried over
Na2SO4 and the filtrate was concentrated under reduced pressure. EtOH was added to the residue, and
the weight was adjusted to 7.2 g. The mixture was heated to 65 °C to dissolve solids. The solution was
cooled to 55 °C and seeded with 1. The solution was aged for 1 h at 50 °C, during which time the
product began to crystallize. After the slurry was cooled to 25 °C, n-heptane (11 mL) was added
drop-wise to the slurry at 25 °C followed by stirring for 1 h at 25 °C. The slurry was cooled to 3 °C and
then stirred for 2 h at 3 °C. The slurry was filtered, and the wet cake was washed with a mixed solvent
of EtOH (1.5 mL) and n-heptane (3 mL). The precipitate was dried in vacuo at 70 °C to give 888 mg
(66% yield) of 1 as a white solid. [α]
20
D -43.5 (c 1.0, DMSO). IR (KBr) cm-1
: 3495, 1744, 1514, 1488,
1454, 1467, 1411, 1372, 1340, 1266. 1H-NMR (CDCl3) δ: 1.27 (3H, t, J=7.0 Hz), 2.00 (1H, d, J=1.6
Hz), 3.46–3.54 (3H, m), 3.56–3.61 (2H, m), 3.72 (1H, d, J=2.1 Hz), 3.75 (3H, s), 3.82 (1H, d, J=15.5 Hz),
4.03 (1H, d, J=15.5 Hz), 4.11–4.22 (2H, m), 4.42 (2H, d, J=3.8 Hz), 4.69 (1H, d, J=7.4 Hz), 6.79–6.83
(2H, m), 6.97–7.02 (2H, m), 7.04–7.07 (2H, m), 7.15–7.22 (2H, m). 13C-NMR (CDCl3) δ: 14.2 (q), 36.1
(t), 55.4 (q), 64.4 (t), 66.4 (t), 69.6 (d), 73.4 (d), 73.8 (d), 75.7 (d), 100.8 (d), 114.1 (d×2), 114.4 (d), 122.7
(d), 128.0 (d), 129.2 (d×2), 130.0 (s), 131.1 (d), 133.4 (s), 155.2 (s), 155.4 (s), 157.8 (s). HRMS (ESI)
m/z: 466.2070 [M+NH4]
+
(Calcd for C23H32NO9: 466.2072)
6-O-Ethoxycarbonyl-2-[(4-methoxyphenyl)methyl]phenyl-β-D-glucopyranoside (1). Ethyl
chloroformate (21.6 g, 0.199 mol) was added drop-wise to the mixture of 2 (65.0 g, 0.173 mol),
2,6-lutidine (27.8 g, 0.259 mol) and pyridine (0.33 g, 4.2 mmol) in acetone (210 mL), maintaining the
temperature between -1 and 5 °C. The reaction mixture was stirred at 0 °C for 2 h. The reaction was
monitored by HPLC.15 Water (200 mL) was added drop-wise, maintaining the temperature below 30 °C,
and then EtOAc (220 mL) was added to the mixture. The biphasic solution was transferred to a
separating funnel for phase separation. The aqueous layer was extracted with EtOAc (140 mL). The
EtOAc layers were combined, washed successively with an aqueous solution of 10% citric acid (180 mL
× 2), an aqueous solution of 10% NaCl (66 g), an aqueous solution of 5% NaHCO3 (65 g × 2), and an aqueous solution of 10% NaCl (100 g), and then dried over Na2SO4 (65 g). After acetic acid (10 g,
0.167 mol) was added to the filtrate, the mixture was concentrated under reduced pressure. The residue
was dissolved in EtOH (660 mL) at 65 °C. The solution was concentrated under reduced pressure until
more than 330 mL distillate had been collected. EtOH was added to the residue, and the weight was
adjusted to 370 g. n-Heptane (120 mL) was added, and the resulting slurry was heated to 65 °C to
dissolve solids. The solution was cooled to 55 °C and seeded with 1. The solution was aged for 1 h at
50 °C, during which time the product began to crystallize. n-Heptane (480 mL) was added drop-wise to
the slurry, maintaining the temperature between 50 and 60 °C, and the slurry was stirred for 0.5 h at 55 °C.
The slurry was allowed to cool slowly over 2.5 h to 30 °C, then cooled to 3 °C, and then stirred for 1.5 h
at 3 °C. The slurry was filtered, and the wet cake was washed with a mixed solvent of EtOH (80 mL)
and n-heptane (180 mL). The precipitate was dried in vacuo at 70 °C to give 63.6 g (82% yield) of 1 as
a white solid.
REFERENCES (AND NOTES)
- W. N. Washburn, Expert Opin. Ther. Patents, 2009, 19, 1485.
- A. M. Pajor and E. M. Wright, J. Biol. Chem., 1992, 267, 3557.
- E. M. Wright, Am. J. Physiol. Renal Physiol., 2001, 280, F10.
- Y. Kanai, W. S. Lee, G. You, D. Brown, and M. A. Hediger, J. Clin. Invest., 1994, 93, 397.
- H. Fujikura, N. Fushimi, T. Nishimura, K. Tatani, and M. Isaji, PCT, WO 02/28872 (2002).
- H. Fujikura, N. Fushimi, T. Nishimura, K. Tatani, K. Katsuno, M. Hiratochi, Y. Tokutake, and M.
Isaji, PCT, WO 01/688660 (2001). - K. Katsuno, Y. Fujimori, Y. Takemura, M. Hiratochi, F. Itoh, Y. Komatsu, H. Fujikura, and M. Isaji,
J. Pharmacol. Exp. Ther., 2007, 320, 323. - M. Isaji, Curr. Opin. Investig. Drugs, 2007, 8, 285.
- S. Y. Lee, S. E. Rho, K. Y. Min, T. B. Kim, and H. K. Kim, J. Carbohydr. Chem., 2001, 20, 503.
- M. Yamaguchi, A. Horiguchi, A. Fukuda, and T. Minami, J. Chem. Soc., Perkin Trans. 1, 1990,
1079. - K. Ishihara, H. Kurihara, and H. Yamamoto, J. Org. Chem., 1993, 58, 3791.
- I. T. Akimova, A. V. Kaminsky, and V. I. Svistunova, Chem. Heterocycl. Compd., 2005, 41, 1374.
- B. N. Cook, S. Bhakta, T. Biegel, K. G. Bowman, J. I. Armstrong, S. Hemmerich, and C. R. Bertozzi,
J. Am. Chem. Soc., 2000, 122, 8612. - HPLC conditions: column, Inertsil ODS-3 (5 µm) 4.6 mm × 250 mm (GL Science Inc.); mobile
phase, isocratic elution with acetonitrile / 0.02 M KH2PO4, pH 3 = 6/4; flow rate, 1.0 mL/min;
column oven temperature, 40 °C; wave length, 225 nm; retention times, 5 = 16 min, α-anomer of 5 =18 min. - HPLC conditions: column, Inertsil ODS-3 (5 µm) 4.6 mm × 250 mm (GL Science Inc.); mobile
phase, gradient elution with 5 min 4/6 → 15 min 6/4 → 30 min 6/4 of acetonitrile/0.02 M KH2PO4,
pH 3; flow rate, 1.0 mL/min; column oven temperature, 40 °C; wavelength, 225 nm; retention times,
1 = 17 min, 2,6- and 4,6-bis-O-ethoxycarbonyl derivatives = 24 min, 3,6-bis-O-ethoxycarbonyl
derivative = 25 min.
SYN
Synthesis 2024, 56, 906–943
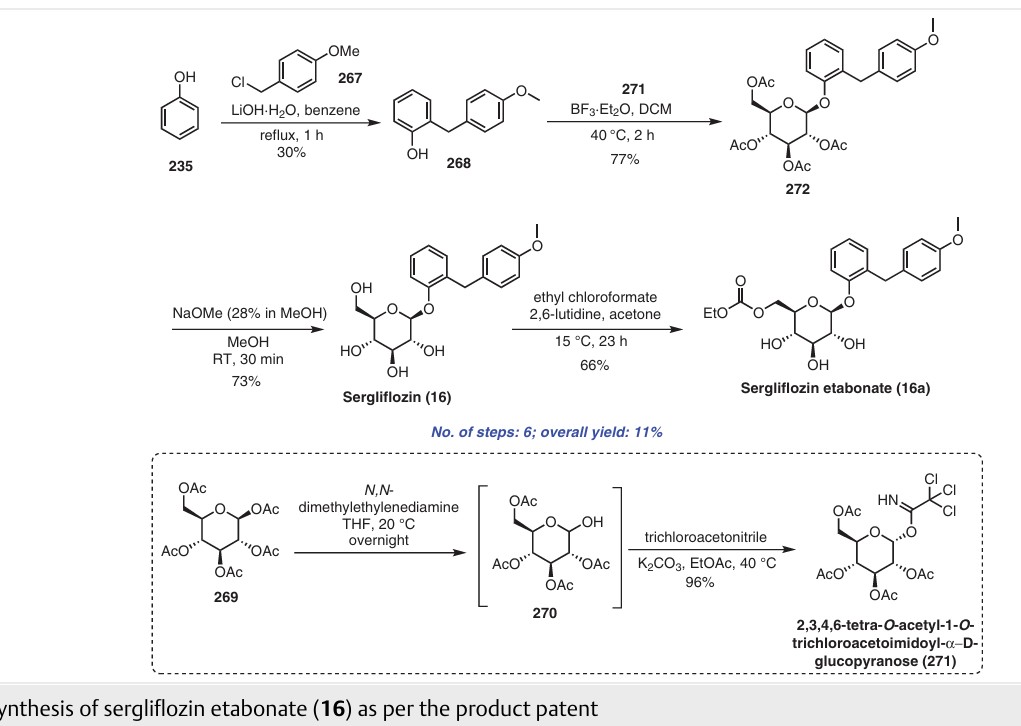
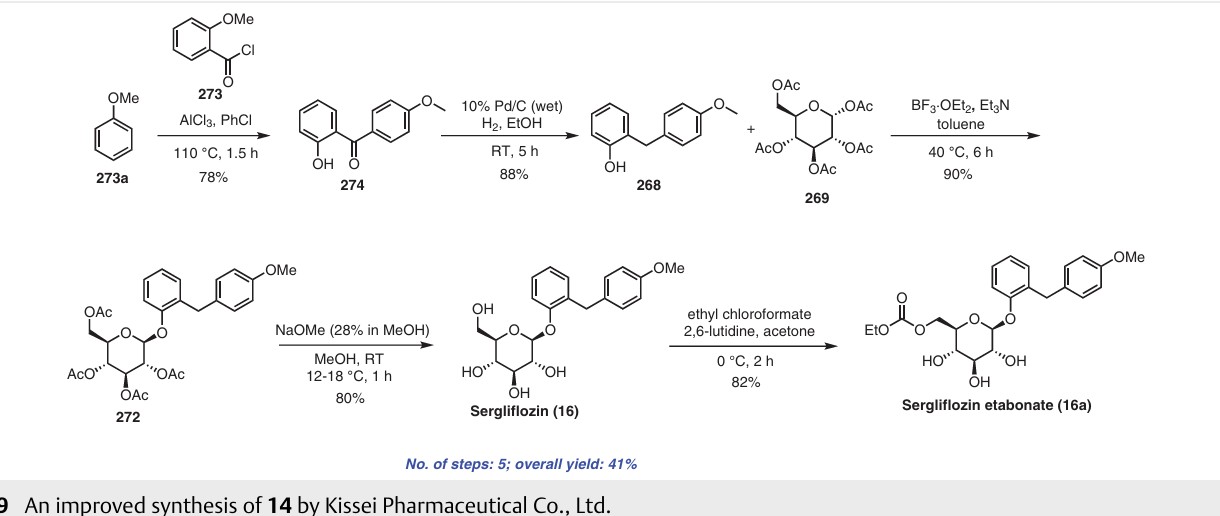
Sergliflozin etabonate (16), also known as GW869682X, was developed collaboratively by GlaxoSmithKline and Kissei Pharmaceutical (Japan). Unfortunately, it did not pass phase III trials. It belongs to the class of sodium–glucose linked transporter 2 (SGLT2) inhibitors and acts as a prodrug of sergliflozin, with the ethyl carbonate group referred to as etabonate. When compared to phlorizin, sergliflozin etabonate demonstrated significantly higher activity against SGLT2 than SGLT1. The initial synthetic route for the preparation of sergliflozin was described and patented by Kissei Pharmaceutical Co., Ltd. This particular route for Oaryl-glycoside-type derivatives was registered in the United States under patent application number US6872706B2.73 The first reported synthesis of sergliflozin etabonate
(16), which involves six steps, can be found in the patents US6872706B2 73a and WO2001068660A1 (Scheme 48).73b Compound 271 was prepared in a high yield of 96% follow ing a literature procedure. The selective monodeacetylationof penta-O-acetyl-b-D-glucopyranose, compound 269, was
achieved using N,N-dimethylethylenediamine in THF, resulting in the formation of compound 270. Subsequently, a reaction with trichloroacetonitrile and potassium carbonate led to the synthesis of intermediate 271 in excellent yield. To prepare the aglycone intermediate 268, phenol (235) was condensed with 4-methoxybenzyl chloride (267) using LiOH under reflux conditions. Further,O-glycosyla
tion of compound 268 with 271 was accomplished using boron trifluoride–diethyl etherate (BF3·OEt2), yielding intermediate 272. Removal of the acetyl groups from intermediate 272 was carried out using NaOMe in methanol to obtain sergliflozin (16a) in a yield of 73%. Finally, sergliflozin etabonate (16) was obtained by reacting compound 16a with ethyl chloroformate and 2,6-lutidine, resulting in a yield of
66%. The overall yield of sergliflozin etabonate (16a) was calculated to be 11%. It is important to note that the trichloroimidation reaction used in the synthesis of trichloroacetimidate 271 is considered hazardous and is not recommended for commercial use due to the highly toxic reagent, trichloroaceto
nitrile. Additionally, the process poses challenges in effectively removing the unwanted by-product, trichloroacetamide, formed during the preparation.A recently published approach presents an alternative synthesis of sergliflozin etabonate (16) that avoids the use of a trichloroacetimidate intermediate (Scheme 49).74a The five-step synthesis of compound 16a commenced from
readily available anisole (273a). An efficient Friedel–Crafts reaction was performed on anisole (273a) using the acid chloride 273 in the presence of aluminum chloride in chlorobenzene, leading to formation of benzophenone 274. Notably, demethylation of 274 was also observed under these
conditions. Next, ketone group reduction was achieved us ing 10% Pd/C and ethanol under 0.3–0.4 MPa of H2, providing compound 268 in 88% yield and high purity. Subsequently, O-glycosylation of 268 with penta-acetylated com pound 269 was carried out using BF3·Et2O and triethylamine, resulting in the formation of 272 in 90% yield with a high b-selectivity (99:1).74b Deacetylation of compound 272 was performed using NaOMe in methanol, affording sergliflozin (16a) in 80% yield. Further reaction with
ethyl chloroformate in the presence of 2,6-lutidine resulted in sergliflozin etabonate (16). The overall yield of compound 16 was calculated to be 41%. This novel synthetic route offers a promising alternative to the traditional method and demonstrates improved efficiency in the preparation of sergliflozin etabonate (16)
(73) (a) Fujikura, H.; Fushimi, N. US6872706B2, 2005. (b) Fujikura, H.; Fushimi, N.; Nishimura, T.; Tatani, K.; Katsuno, K.; Hiratochi, M.; Tokutake, Y.; Isaji, M. WO2001068660A1, 2001.
(74) (a) Kobayashi, M.; Isawa, H.; Sonehara, J.; Kubota, M. Heterocycles 2016, 92, 1599. (b) Lee, Y. S.; Rho, S. E.; Min, K. Y.; Kim, T. B.; Kim, H. K. J. Carbohydr. Chem. 2001, 20, 503.
| Patent | Submitted | Granted |
|---|---|---|
| Progression Inhibitor For Disease Attributed To Abnormal Accumulation Of Liver Fat [US2008045466] | 2008-02-21 | |
| NOVEL SUBSTITUTED TETRAHYDRONAPHTHALENES, PROCESS FOR THE PREPARATION THEREOF AND THE USE THEREOF AS MEDICAMENTS [US2010249097] | 2010-09-30 | |
| (CARBOXYLALKYLENEPHENYL)PHENYLOXAMIDES, METHOD FOR THE PRODUCTION THEREOF AND USE OF SAME AS A MEDICAMENT [US2010261645] | 2010-10-14 | |
| (CYCLOPROPYLPHENYL)PHENYLOXAMIDES, METHOD FOR THE PRODUCTION THEREOF, AND USE OF SAME AS A MEDICAMENT [US8148375] | 2010-10-14 | 2012-04-03 |
| Crystals of glucopyranosyloxybenzyl benzene derivative [US7371730] | 2005-06-02 | 2008-05-13 |
| CERTAIN CRYSTALLINE DIPHENYLAZETIDINONE HYDRATES, PHARMACEUTICAL COMPOSITIONS THEREOF AND METHODS FOR THEIR USE [US8003636] | 2009-08-13 | 2011-08-23 |
| NOVEL DIPHENYLAZETIDINONE SUBSTITUTED BY PIPERAZINE-1-SULFONIC ACID AND HAVING IMPROVED PHARMACOLOGICAL PROPERTIES [US2009264402] | 2009-10-22 | |
| Arylaminoaryl-alkyl-substituted imidazolidine-2,4-diones, process for preparing them, medicaments comprising these compounds, and their use [US7759366] | 2009-08-27 | 2010-07-20 |
| Glucopyranosyloxybenzylbenzene derivatives and medicinal compositions containing the same [US2005065098] | 2005-03-24 | |
| Glucopyranosyloxybenzylbenzene derivatives and medicinal compositions containing the same [US6872706] | 2004-01-29 | 2005-03-29 |
| Patent | Submitted | Granted |
|---|---|---|
| PROGRESSION INHIBITOR FOR DISEASE ATTRIBUTED TO ABNORMAL ACCUMULATION OF LIVER FAT [US2009286751] | 2009-11-19 | |
| THERAPEUTIC USES OF SGLT2 INHIBITORS [US2011077212] | 2011-03-31 | |
| PHARMACEUTICAL COMPOSITION COMPRISING A SGLT2 INHIBITOR IN COMBINATION WITH A DPP-IV INHIBITOR [US2011098240] | 2011-04-28 | |
| Substituted imidazoline-2,4-diones, process for preparation thereof, medicaments comprising these compounds and use thereof [US2011112097] | 2011-05-12 | |
| Heterocycle-substituted imidazolidine-2,4-diones, process for preparation thereof, medicaments comprising them and use thereof [US2011046105] | 2011-02-24 | |
| Arylchalcogenoarylalkyl-substituted imidazolidine-2,4-diones, process for preparation thereof, medicaments comprising these compounds and use thereof [US2011046185] | 2011-02-24 | |
| Arylchalcogenoarylalkyl-substituted imidazolidine-2,4-diones, process for preparation thereof, medicaments comprising these compounds and use thereof [US2011053947] | 2011-03-03 | |
| Novel aromatic fluoroglycoside derivatives, pharmaceuticals comprising said compounds and the use thereof [US2011059910] | 2011-03-10 | |
| Novel phenyl-substituted imidazolidines, process for preparation thereof, medicaments comprising said compounds and use thereof [US2011178134] | 2011-07-21 | |
| HETEROCYCLIC COMPOUNDS, PROCESSES FOR THEIR PREPARATION, MEDICAMENTS COMPRISING THESE COMPOUNDS, AND THE USE THEREOF [US2011183998] | 2011-07-28 |
| Systematic (IUPAC) name | |
|---|---|
| 2-(4-methoxybenzyl)phenyl 6-O-(ethoxycarbonyl)-β-D-glucopyranoside | |
| Clinical data | |
| Routes of administration | Oral |
| Identifiers | |
| CAS Number | 408504-26-7 |
| ATC code | None |
| PubChem | CID: 9824918 |
| IUPHAR/BPS | 4587 |
| ChemSpider | 21234810 |
| ChEMBL | CHEMBL450044 |
| Chemical data | |
| Formula | C23H28O9 |
| Molecular mass | 448.463 g/mol |
References
- World Health Organization (2008). “International Nonproprietary Names for Pharmaceutical Substances (INN). Recommended International Nonproprietary Names: List 59” (PDF). WHO Drug Information. 22 (1): 66. Archived from the original (PDF) on February 19, 2009.
- “Statement on a nonproprietary name adopted by the USAN council: Sergliflozin etabonate” (PDF). American Medical Association. Retrieved 2008-08-10.
- Katsuno K, Fujimori Y, Takemura Y, et al. (January 2007). “Sergliflozin, a novel selective inhibitor of low-affinity sodium glucose cotransporter (SGLT2), validates the critical role of SGLT2 in renal glucose reabsorption and modulates plasma glucose level”. J Pharmacol Exp Ther. 320 (1): 323–30. doi:10.1124/jpet.106.110296. PMID 17050778. S2CID 8306408.
- “Prous Science: Molecule of the Month November 2007”. Archived from the original on 2007-11-05. Retrieved 2008-10-28.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
////////// etabonate, Sergliflozin etabonate, Sergliflozin, PHASE 3, GW869682X, GSK, KISSEI, GW-869682; GW-869682X; KGT-1251
CCOC(=O)OCC1C(C(C(C(O1)OC2=CC=CC=C2CC3=CC=C(C=C3)OC)O)O)O
CCOC(=O)OCC1C(C(C(C(O1)Oc2ccccc2Cc3ccc(cc3)OC)O)O)O
Baxdrostat
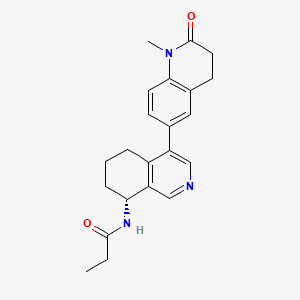
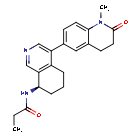
Baxdrostat
cas 1428652-17-8
APPROVALS 2026, FDA 2026, 5/15/2026, Baxfendy
To treat hypertension in combination with other antihypertensive drugs
- NF3P9Z8J5Y
- CIN-107
- RO6836191
- 363.5 g/mol
WeightAverage: 363.461
Monoisotopic: 363.194677057
Chemical FormulaC22H25N3O2
N-[(8R)-4-(1-methyl-2-oxo-3,4-dihydroquinolin-6-yl)-5,6,7,8-tetrahydroisoquinolin-8-yl]propanamide
- (+)-(R)-N-(4-(1-Methyl-2-oxo-1,2,3,4-tetrahydroquinolin-6-yl)-5,6,7,8-tetrahydroisoquinolin-8-yl)propionamide
- N-((8R)-5,6,7,8-Tetrahydro-4-(1,2,3,4-tetrahydro-1-methyl-2-oxo-6-quinolinyl)-8-isoquinolinyl)propanamide
- N-[(8R)-4-(1-methyl-2-oxo-3,4-dihydroquinolin-6-yl)-5,6,7,8-tetrahydroisoquinolin-8-yl]propanamide
- Propanamide, N-((8R)-5,6,7,8-tetrahydro-4-(1,2,3,4-tetrahydro-1-methyl-2-oxo-6-quinolinyl)-8-isoquinolinyl)-
Baxdrostat is an investigational drug that is being evaluated for the treatment of hypertension.[1] It is an aldosterone synthase inhibitor.[2][3]
Baxdrostat is under investigation in clinical trial NCT06344104 (A Phase III Study to Investigate the Efficacy and Safety of Baxdrostat in Asian Participants With Uncontrolled Hypertension on Two or More Medications Including Participants With Resistant Hypertension).
LIT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US76841362&_cid=P21-MEZ3MG-55484-1
Example 3-1
(+)-(R)—N-(4-(1-Methyl-2-oxo-1,2,3,4-tetrahydroquinolin-6-yl)-5,6,7,8-tetrahydroisoquinolin-8-yl)propionamide
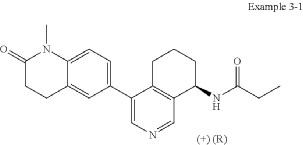
In analogy to the procedures described for the preparation of intermediate A-2 [E] and for the preparation of intermediate B-1, Suzuki reaction of (+)-(R)-4-bromo-5,6,7,8-tetrahydroisoquinolin-8-amine (intermediate B-3b) with 1-methyl-6-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-3,4-dihydro-1H-quinolin-2-one (intermediate A-1) gave (R)-6-(8-amino-5,6,7,8-tetrahydroisoquinolin-4-yl)-1-methyl-3,4-dihydroquinolin-2(1H)-one and after subsequent reaction with propionyl chloride the title compound as colorless solid. MS: 364.2 (M+H +).
Pat
CN 117247371
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN418385740&_cid=P12-MEZHY3-66430-1
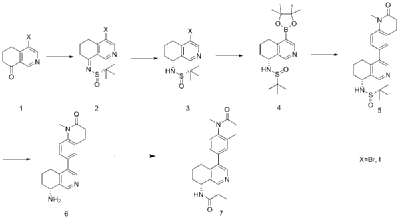
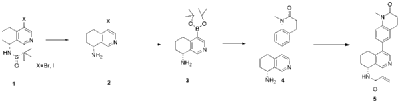
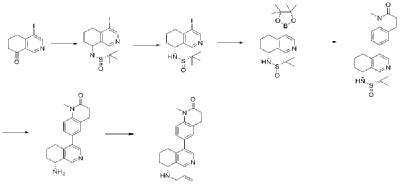
| Example 1 |
| Step A |
| Dissolve 4-bromo-6,7-dihydroisoquinolin-8(5H)-one (1.56 g, 6.9 mmol) and (S)-tert-butylsulfenamide (2.51 g, 20.7 mmol) in 20 mL of tetrahydrofuran. Add ethyl titanate (10.08 mL, 48.28 mmol). Heat to 65°C and stir for 48 hours. Cool to room temperature, add ethyl acetate and water, stir for 15 minutes, and remove the resulting solid by filtration. Separate the liquids, dry the organic phase over anhydrous sodium sulfate, filter, and evaporate to dryness under reduced pressure to obtain the crude product (S,Z)-N-(4-bromo-6,7-dihydroisoquinolin-8(5H)-tert-butylsulfenimide), which is used directly in the next step. |
| Step B |
| Compound (S,Z)-N-(4-bromo-6,7-dihydroisoquinoline-8(5H)-tert-butylsulfonyl imide) (1.98 g, 6 mmol) was dissolved in 15 mL of tetrahydrofuran and cooled to -45°C. Sodium borohydride (0.34 g, 9.0 mmol) was added, and the mixture was allowed to return to room temperature and stirred for 18 hours. The mixture was quenched with ice water and extracted with dichloromethane. The resulting organic phase was washed with saturated brine, dried over anhydrous sodium sulfate, and evaporated to dryness under reduced pressure. The residue was purified by column chromatography to obtain compound (S)-N-(4-bromo-6,7-dihydroisoquinoline-8(5H))-tert-butylsulfonyl imide (755 mg, 38% yield). LC/MS (ESI): m/z = 331.2 [M+H] + . |
| Step C |
| To a mixture of (S)-N-(4-bromo-6,7-dihydroisoquinoline-8(5H))-tert-butylsulfonimide (0.66 g, 2 mmol), pinacol diboronate (1.05 g, 2.1 mmol), and AcOK (0.578 g, 6 mmol) in toluene (10 mL) was added Pd(dppf)Cl 2 (0.144 g, 0.2 mmol). The mixture was degassed and stirred at 130 ° C for 3 hours. The reaction mixture was filtered and concentrated to give a residue. EtOAc (15 mL) and water (10 mL) were added to the residue. The organic phase was washed with brine (50 mL), dried over anhydrous sodium sulfate, filtered and concentrated to give a residue. The residue was purified by column chromatography (SiO 2 ) and eluted with 30-40% ethyl acetate in petroleum ether to afford (S)-N-tert-butylsulfonamido-6,7-dihydroisoquinolin-8(5H)-4-boronic acid pinacol ester (0.45 g, 60% yield). LC/MS (ESI): m/z = 378.3 [M+H] + . |
| Step D |
| To a reaction flask, add 6-bromo-1-methyl-3,4-dihydroquinolin-2(1H)-one (0.29 g, 1.2 mmol), (S)-N-tert-butylsulfonamido-6,7-dihydroisoquinolin-8(5H)-4-boronic acid pinacol ester (0.42 g, 1.26 mmol), bistriphenylphosphine palladium dichloride (84 mg, 0.12 mmol), cuprous iodide (38 mg, 0.2 mmol), triethylamine (1.01 g, 10.0 mmol), and 15 mL of N,N-dimethylformamide. The atmosphere was purged with nitrogen three times and the reaction was stirred at 90°C overnight. After cooling to room temperature, the reaction mixture was diluted with ethyl acetate and water, and extracted with ethyl acetate. The resulting organic phase was washed with water and saturated brine, dried over anhydrous sodium sulfate, and evaporated to dryness under reduced pressure. The residue was purified by column chromatography to afford (S)-2-methyl-N-((R)-4-(1-methyl-2-oxo-1,2,3,4-tetrahydroquinolin-6-yl)-5,6,7,8-tetrahydroisoquinolin-8-yl)tert-butylsulfonimide (0.37 g, 74% yield) as a yellow solid. LC/MS (ESI): m/z = 411.5 [M+H] + . |
| Step E |
| Compound (S)-2-methyl-N-((R)-4-(1-methyl-2-oxo-1,2,3,4-tetrahydroquinolin-6-yl)-5,6,7,8-tetrahydroisoquinolin-8-yl)tert-butylsulfonimide (0.33 g, 0.80 mmol) was dissolved in 1 mL of dichloromethane, and 1 mL of trifluoroacetic acid was added. The mixture was stirred and reacted for 1 hour. The reaction solution was concentrated under reduced pressure. The residue was purified by reverse preparative column chromatography to obtain compound (R)-6-(8-amino-5,6,7,8-tetrahydroisoquinolin-4-yl)-1-methyl-3,4-dihydroquinolin-2(1H)-one (0.24 g, 97% yield). LC/MS (ESI): m/z = 307.1 [M+H] + . |
| Step F |
| To a reaction flask, add (R)-6-(8-amino-5,6,7,8-tetrahydroisoquinolin-4-yl)-1-methyl-3,4-dihydroquinolin-2(1H)-one (100 mg, 0.33 mmol), triethylamine (51 mg, 0.5 mmol), and 4 ml of tetrahydrofuran. After cooling in an ice-water bath, slowly add a solution of propionyl chloride (46.25 mg, 0.5 mmol) in 0.5 ml of tetrahydrofuran dropwise. Stirring is continued for 4 hours after addition. The reaction mixture is quenched with methanol and evaporated to dryness under reduced pressure. The residue is purified by column chromatography to obtain the target compound, Baxdrostat (46 mg, 38% yield). LC/MS(ESI):m/z=363.1[M+H]+.H NMR(400MHz, CDCl3)ppm 1.22(t,3H)1.79(s,3H)2.07(s,1H)2.28(q,2H)2.43-2.68(m,2H)2.71(t,2H)2.82-3.12(m,2H) 3.40(s,3H)5.34(d,1H)5.78(d,1H)7.05(d,1H)7.09(s,1H)7.17(d,1H)8.28(s,1H)8.49(s,1H) |
| Example 2 |
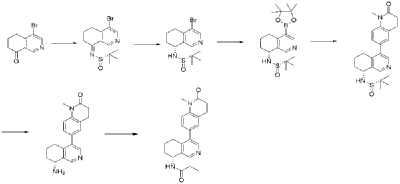
| Step A |
| Compound (S)-N-(4-bromo-6,7-dihydroisoquinolin-8(5H))-tert-butylsulfonylimide (1.65 g, 5 mmol) was dissolved in 20 mL of dichloromethane, and 20 mL of trifluoroacetic acid was added. The mixture was stirred and reacted for 1 hour. The reaction solution was concentrated under reduced pressure. The residue was purified by reverse-phase preparative column chromatography to obtain compound (R)-4-bromo-5,6,7,8-tetrahydroisoquinolin-8-amine (1.07 g, 94% yield). LC/MS (ESI): m/z = 226.0 [M+H] + . |
| Step B |
| To a mixture of (R)-4-bromo-5,6,7,8-tetrahydroisoquinolin-8-amine (0.86 g, 3.8 mmol), pinacol diboron (2 g, 4 mmol), AcOK (1.10 g, 11.4 mmol) in toluene (10 mL) was added Pd(dppf)Cl 2 (0.27 g, 0.38 mmol). The mixture was degassed and stirred at 130 ° C for 3 hours. The reaction mixture was filtered and concentrated to give a residue. EtOAc (10 mL) and water (10 mL) were added to the residue. The organic phase was washed with brine (10 mL), dried over anhydrous sodium sulfate, filtered and concentrated to give a residue. The residue was purified by column chromatography (SiO 2 ) and eluted with 30-40% ethyl acetate in petroleum ether to afford (R)-8-amino-5,6,7,8-tetrahydroisoquinoline-4-boronic acid pinacol ester (0.68 g, 65% yield). LC/MS (ESI): m/z = 274.1 [M+H] + . |
| Step C |
| To a reaction flask, add 6-bromo-1-methyl-3,4-dihydroquinolin-2(1H)-one (0.72 g, 3.0 mmol), (R)-8-amino-5,6,7,8-tetrahydroisoquinolin-4-boronic acid pinacol ester (0.99 g, 3.6 mmol), bistriphenylphosphine palladium dichloride (210 mg, 0.3 mmol), and potassium phosphate monohydrate (204 mg, 0.9 mmol). Dissolve the mixture in dioxane and water (9:1, 30 mL). Replace the atmosphere with nitrogen three times and allow the mixture to react overnight at 90°C with stirring. Cool to room temperature, dilute the reaction solution with ethyl acetate and water, and extract with ethyl acetate. The resulting organic phase is then washed with water and saturated brine, dried over anhydrous sodium sulfate, and evaporated to dryness under reduced pressure. The residue was purified by column chromatography to obtain (R)-6-(8-amino-5,6,7,8-tetrahydroisoquinolin-4-yl)-1-methyl-3,4-dihydroquinolin-2(1H)-one (0.81 g, 88% yield). LC/MS (ESI): m/z = 307.1 [M+H] + . The target compound, Baxdrostat, was then prepared using a method similar to the last step in Example 1. |
| Example 3 |
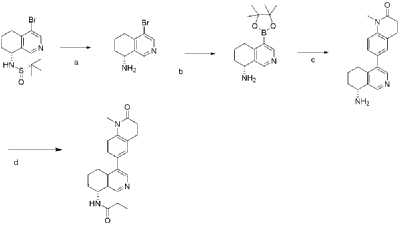
| Step A |
| 4-Bromo-6,7-dihydroisoquinolin-8(5H)-one (1.88 g, 6.9 mmol) and (S)-tert-butylsulfenamide (2.51 g, 20.7 mmol) were dissolved in 20 mL of tetrahydrofuran. Ethyl titanate (10.08 mL, 48.28 mmol) was added and the mixture was heated to 65°C with stirring for 48 hours. After cooling to room temperature, ethyl acetate and water were added and stirred for 15 minutes. The resulting solid was removed by filtration. The organic phase was separated and dried over anhydrous sodium sulfate, filtered, and evaporated to dryness under reduced pressure to obtain the crude product (S,Z)-N-(4-bromo-6,7-dihydroisoquinolin-8(5H)-tert-butylsulfenimide), which was used directly in the next step. LC/MS (ESI): m/z = 376.2 [M+H] + . |
| Step B |
| Compound (S,Z)-N-(4-iodo-6,7-dihydroisoquinoline-8(5H)-tert-butylsulfonyl imide) (2.26 g, 6 mmol) was dissolved in 15 mL of tetrahydrofuran and cooled to -45°C. Sodium borohydride (0.36 g, 9.0 mmol) was added, and the mixture was allowed to return to room temperature and stirred for 18 hours. The mixture was quenched with ice water and extracted with dichloromethane. The resulting organic phase was washed with saturated brine, dried over anhydrous sodium sulfate, and evaporated to dryness under reduced pressure. The residue was purified by column chromatography to obtain compound (S)-N-(4-iodo-6,7-dihydroisoquinoline-8(5H))-tert-butylsulfonyl imide (1.04 g, 46% yield). LC/MS (ESI): m/z = 378.0 [M+H] + . |
| Step C |
| To a mixture of (S)-N-(4-iodo-6,7-dihydroisoquinoline-8(5H))-tert-butylsulfonimide (0.76 g, 2 mmol), pinacol diboronate (1.05 g, 2.1 mmol), and AcOK (0.578 g, 6 mmol) in toluene (10 mL) was added Pd(dppf)Cl 2 (0.144 g, 0.2 mmol). The mixture was degassed and stirred at 130 ° C for 3 hours. The reaction mixture was filtered and concentrated to give a residue. EtOAc (15 mL) and water (10 mL) were added to the residue. The organic phase was washed with brine (50 mL), dried over anhydrous sodium sulfate, filtered and concentrated to give a residue. The residue was purified by column chromatography (SiO 2 ) and eluted with 30-40% ethyl acetate in petroleum ether to afford (S)-N-tert-butylsulfonamido-6,7-dihydroisoquinolin-8(5H)-4-boronic acid pinacol ester (0.51 g, 68% yield). LC/MS (ESI): m/z = 378.2 [M+H] + . |
| The next three steps were carried out in the same manner as in Example 1 to prepare the target compound Baxdrostat. |
LIT
https://medicalxpress.com/news/2025-08-stubborn-high-blood-pressure-experimental.html
A new treatment has been shown to significantly lower blood pressure in people whose levels stay dangerously high, despite taking several existing medicines, according to the results of a Phase III clinical trial led by a UCL Professor. Globally, around 1.3 billion people have high blood pressure (hypertension), and in around half of cases the condition is uncontrolled or treatment resistant. These individuals face a much greater risk of heart attack, stroke, kidney disease, and early death. In the UK the number of people with hypertension is around 14 million.
The international BaxHTN trial, led by Professor Bryan Williams (UCL Institute of Cardiovascular Science), assessed the new drug baxdrostat—which is taken as a tablet—with participation from nearly 800 patients across 214 clinics worldwide.
Results were presented at the European Society of Cardiology (ESC) Congress 2025 in Madrid and were simultaneously published in the New England Journal of Medicine.
The trial results showed that, after 12 weeks, patients taking baxdrostat (1 mg or 2 mg once daily in pill form) saw their blood pressure fall by around 9-10 mmHg more than placebo—a reduction large enough to cut cardiovascular risk. About four in 10 patients reached healthy blood pressure levels, compared with fewer than two in 10 on placebo.
Principal Investigator, Professor Williams, who is presenting the results at ESC, said, “Achieving a nearly 10 mmHg reduction in systolic blood pressure with baxdrostat in the BaxHTN Phase III trial is exciting, as this level of reduction is linked to substantially lower risk of heart attack, stroke, heart failure and kidney disease.”
How baxdrostat works
Blood pressure is strongly influenced by a hormone called aldosterone, which helps the kidneys regulate salt and water balance.
Some people produce too much aldosterone, causing the body to hold onto salt and water. This aldosterone dysregulation pushes blood pressure up and makes it very difficult to control.
Addressing aldosterone dysregulation has been a key effort in research over many decades, but it has been so far difficult to achieve.
Baxdrostat works by blocking aldosterone production, directly addressing this driver of high blood pressure (hypertension).
Professor Williams, Chair of Medicine at UCL, said, “These findings are an important advance in treatment and in our understanding of the cause of difficult-to-control blood pressure.
“Around half of people treated for hypertension do not have it controlled, however this is a conservative estimate and the number is likely higher, especially as the target blood pressure we try to reach is now much lower than it was previously.
“In patients with uncontrolled or resistant hypertension, the addition of baxdrostat 1mg or 2mg once daily to background antihypertensive therapy led to clinically meaningful reductions in systolic blood pressure, which persisted for up to 32 weeks with no unanticipated safety findings.
“This suggests that aldosterone is playing an important role in causing difficult to control blood pressure in millions of patients and offers hope for more effective treatment in the future.”
Historically, higher-income Western countries were reported to have far higher levels of hypertension. However, largely due to changing diets (adding less salt to food), the numbers of people living with the condition is now far higher in Eastern and lower-income countries. More than half of those affected live in Asia, including 226 million people in China and 199 million in India.
Professor Williams added, “The results suggest that this drug could potentially help up to half a billion people globally—and as many as 10 million people in the UK alone, especially at the new target level for optimal blood pressure control.”
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
| Identifiers | |
|---|---|
| IUPAC name | |
| CAS Number | 1428652-17-8 |
| PubChem CID | 71535962 |
| IUPHAR/BPS | 12362 |
| ChemSpider | 76804781 |
| UNII | NF3P9Z8J5Y |
| KEGG | D12789 |
| ChEMBL | ChEMBL4113975 |
| Chemical and physical data | |
| Formula | C22H25N3O2 |
| Molar mass | 363.461 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
PATENTS
- New bicyclic dihydroquinoline-2-one derivativesPublication Number: TW-201319054-APriority Date: 2011-09-23
- New bicyclic dihydroquinoline-2-one derivativesPublication Number: TW-I576342-BPriority Date: 2011-09-23Grant Date: 2017-04-01
- New bicyclic dihydroquinoline-2-one derivativesPublication Number: US-2013079365-A1Priority Date: 2011-09-23
- Bicyclic dihydroquinoline-2-one derivativesPublication Number: US-9353081-B2Priority Date: 2011-09-23Grant Date: 2016-05-31
- New bicyclic dihydroquinoline-2-one derivativesPublication Number: WO-2013041591-A1Priority Date: 2011-09-23
- Novel bicyclic dihydroquinolin-2-one derivativesPublication Number: JP-2014526539-APriority Date: 2011-09-23
- Novel bicyclic dihydroquinolin-2-one derivativesPublication Number: JP-6012737-B2Priority Date: 2011-09-23Grant Date: 2016-10-25
- New bicyclic dihydroquinoline-2-one derivativesPublication Number: KR-101723276-B1Priority Date: 2011-09-23Grant Date: 2017-04-04
- New bicyclic dihydroquinoline-2-one derivativesPublication Number: KR-20140076591-APriority Date: 2011-09-23
- New bicyclic dihydroquinoline-2-one derivativesPublication Number: NZ-620652-APriority Date: 2011-09-23
- New bicyclic dihydroquinolin-2-one derivativesPublication Number: CN-103827101-APriority Date: 2011-09-23
- Bicyclic dihydroquinolin-2-one derivativesPublication Number: CN-103827101-BPriority Date: 2011-09-23Grant Date: 2016-12-07
- New bicyclic dihydroquinoline-2-one derivativesPublication Number: EP-2758388-A1Priority Date: 2011-09-23
- New bicyclic dihydroquinoline-2-one derivativesPublication Number: EP-2758388-B1Priority Date: 2011-09-23Grant Date: 2018-02-21
- NEW BICYCLIC DERIVATIVES OF DIHYDROKINOLIN-2-ONPublication Number: HR-P20180592-T1Priority Date: 2011-09-23
- Methods of using aldosterone synthase inhibitorsPublication Number: US-2024277698-A1Priority Date: 2021-06-24
- How to use aldosterone synthase inhibitorsPublication Number: CN-117545482-APriority Date: 2021-06-24
- New bicyclic dihydroquinoline-2-one derivativesPublication Number: AU-2012311582-A1Priority Date: 2011-09-23
- New bicyclic dihydroquinoline-2-one derivativesPublication Number: AU-2012311582-B2Priority Date: 2011-09-23Grant Date: 2017-07-06
- Bicyclic dihydroquinoline-2-one derivativesPublication Number: CA-2845170-CPriority Date: 2011-09-23Grant Date: 2019-08-13
References
- “Baxdrostat – CinCor Pharma”. AdisInsight. Springer Nature Switzerland AG.
- Dogra S, Shah S, Gitzel L, Pusukur B, Sood A, Vyas AV, Gupta R (July 2023). “Baxdrostat: A Novel Aldosterone Synthase Inhibitor for Treatment Resistant Hypertension”. Current Problems in Cardiology. 48 (11): 101918. doi:10.1016/j.cpcardiol.2023.101918. PMID 37399857. S2CID 259320969.
- Awosika A, Cho Y, Bose U, Omole AE, Adabanya U (October 2023). “Evaluating phase II results of Baxdrostat, an aldosterone synthase inhibitor for hypertension”. Expert Opinion on Investigational Drugs. 32 (11): 985–995. doi:10.1080/13543784.2023.2276755. PMID 37883217. S2CID 264517675.
- The selective aldosterone synthase inhibitor Baxdrostat significantly lowers blood pressure in patients with resistant hypertensionPublication Name: Frontiers in EndocrinologyPublication Date: 2022-12-09PMCID: PMC9780529PMID: 36568122DOI: 10.3389/fendo.2022.1097968
- Results from a phase 1, randomized, double-blind, multiple ascending dose study characterizing the pharmacokinetics and demonstrating the safety and selectivity of the aldosterone synthase inhibitor baxdrostat in healthy volunteersPublication Name: Hypertension research : official journal of the Japanese Society of HypertensionPublication Date: 2022-10-20PMCID: PMC9747611PMID: 36266539DOI: 10.1038/s41440-022-01070-4
- Preclinical and Early Clinical Profile of a Highly Selective and Potent Oral Inhibitor of Aldosterone Synthase (CYP11B2)Publication Name: Hypertension (Dallas, Tex. : 1979)Publication Date: 2017-01PMCID: PMC5142369PMID: 27872236DOI: 10.1161/hypertensionaha.116.07716
| Clinical data | |
|---|---|
| Trade names | Baxfendy |
| ATC code | C02KN02 (WHO) |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1428652-17-8 |
| PubChem CID | 71535962 |
| IUPHAR/BPS | 12362 |
| ChemSpider | 76804781 |
| UNII | NF3P9Z8J5Y |
| KEGG | D12789 |
| ChEMBL | ChEMBL4113975 |
| Chemical and physical data | |
| Formula | C22H25N3O2 |
| Molar mass | 363.461 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
/////Baxdrostat, PHASE 3, NF3P9Z8J5Y, CIN 107, RO 6836191,
DAZDOTUFTIDE


DAZDOTUFTIDE
- TRS-01
- CAS 2522933-44-2
- 4-((E)-(5-(2-(2-((S)-2-((S)-1-(L-Threonyl-L-lysyl)pyrrolidine-2-carboxamido)-5-guanidinopentanamido)acetamido)-2-carboxyethyl)-2-hydroxyphenyl)diazenyl)phenyl (2-(trimethylammonio)ethyl) phosphate
- L-Tyrosine, L-threonyl-L-lysyl-L-prolyl-L-arginylglycyl-3-((1E)-2-(4-((hydroxy(2-(trimethylammonio)ethoxy)phosphinyl)oxy)phenyl)diazenyl)-, inner salt
- [4-[[5-[(2S)-2-[[2-[[(2S)-2-[[(2S)-1-[(2S)-6-amino-2-[[(2S,3R)-2-amino-3-hydroxybutanoyl]amino]hexanoyl]pyrrolidine-2-carbonyl]amino]-5-(diaminomethylideneamino)pentanoyl]amino]acetyl]amino]-2-carboxyethyl]-2-hydroxyphenyl]diazenyl]phenyl] 2-(trimethylazaniumyl)ethyl phosphate
C43H68N13O13P
1006.1 g/mol
L-Tyrosine, L-threonyl-L-lysyl-L-prolyl-L-arginylglycyl-3-[(1E)-2-[4-[[hydroxy[2-(trimethylammonio)ethoxy]phosphinyl]oxy]phenyl]diazenyl]-, inner salt
SQ
| 1 | TKPRGY |
Protein/Peptide Sequence, Sequence Length: 6
modified (modifications unspecified)
- OriginatorTarsius Pharma
- DeveloperTarsier Pharma
- ClassAnti-inflammatories; Eye disorder therapies; Small molecules
- Mechanism of ActionImmunomodulators
- Orphan Drug StatusYes – Uveitis
- Phase IIIUveitis
- Phase I/IIOcular inflammation
- PreclinicalDiabetic macular oedema; Diabetic retinopathy; Dry age-related macular degeneration
- 16 Jan 2024Tarsier Pharma receives an agreement from the US FDA under Special Protocol Assessment for Tarsier-04 phase III trial for TR S01 eye drops for Uveitis
- 13 Nov 2023Tarsier Pharma announces successful outcome of a Type C meeting with the US FDA supporting the advancement of TRS 01 eye drop for Uveitis
- 13 Nov 2023Tarsier Pharma plans a Tarsier-04 phase III registrational trial of TR S01 for Uveitis in USA

| Molecular Formula | C43H68N13O13P.C2HF3O2 |
| Molecular Weight | 1120.0764 |
TRS-01 trifluoroacetate
I35XEI0JIK
CAS 2522933-45-3
4-((E)-(5-(2-(2-((S)-2-((S)-1-(L-Threonyl-L-lysyl)pyrrolidine-2-carboxamido)-5-guanidinopentanamido)acetamido)-2-carboxyethyl)-2-hydroxyphenyl)diazenyl)phenyl (2-(trimethylammonio)ethyl) phosphate, trifluoroacetate salt
Ocular inflammation, an inflammation of any part of the eye, is one of the most common ocular diseases. Ocular inflammation refers to a wide range of inflammatory disease of the eye, one of them is uveitis. These diseases are prevalent in all age groups and may be associated with systemic diseases such as Crohn’s disease, Behcet disease, Juvenile idiopathic arthritis and others. The inflammation can also be associated with other common eye symptoms such as dry eye and dry macular degeneration. Several drugs have the known side effect of causing uveitis and/or dry eye. The most common treatment for ocular inflammation, is steroids and specifically corticosteroids. However, these treatments have several known and sometimes severe side effects.
Phosphorylcholine (PC) is a small zwitterionic molecule secreted by helminths which permits helminths to survive in the host inducing a situation of immune tolerance as well as on the surface of some bacteria and apoptotic cells. Tuftsin-PhosphorylCholine (TRS) is bi-specific small molecule with immunomodulatory activities. TRS (Thr-Lys-Pro-Arg-Gly-Tyr-PC) is an immunomodulating peptide derivative.
Currently, TRS has been synthesized by post-synthesis modification of Thr-Lys-Pro-Arg-Gly-Tyr, so as to couple the PC moiety to the phenol ring of tyrosine. However, this synthetic approach results in very low yield, thus making the synthesis of TRS ineffective and costly. New simple and efficient methods of synthesizing TRS are highly required.
SCHEME

PATENT
WO2022224259
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2022224259&_cid=P11-MAOYY3-78105-1
EXAMPLES
EXAMPLE 1
CONJUGATION OF PHOSPHORYLCHOLINE TO BOC-TYR
[0151] 1) Preparation of diazonium salt
[0152] 4-Aminophenyl (2-(trimethylammonio)ethyl) phosphate (50 mg, 0.18 mmol)) was dissolved in 1M aqueous HC1 (1 mL), cooled in an ice-water bath and sodium nitrite (12.6 mg, 0.18 mmol) was added in a single batch. The resulting solution was stirred at 0°C for 30 min.
[0153] 2) Azo coupling
[0154] A new mixture was prepared with BOC-L-tyrosine (107 mg, 0.38 mmol) in NaHC03(lM)+NaOH buffer (pH 10) (3.3 mL) + acetonitrile (1.2 mL). The mixture was cooled in an ice-water bath. The diazonium salt mixture was added drop-wise. A red solution was formed. Stirring of this was continued at 0 °C for 6 minutes. The reaction mixture was acidified with IN aqueous HC1 to pH=~3.
[0155] The obtained solution was lyophilized overnight, and subsequently purified (e.g. by preparative MPLC), to obtain the compound:
, wherein R is Boc.
EXAMPLE 2
PREPARATION OF AN EXEMPLARY COMPOUND OF THE INVENITON
Preparation of diazonium salt:
Fmoc-Tyr-PPC
(compound 10)
[0156] 4-Aminophenyl (2-(trimethylammonio)ethyl) phosphate (250 mg, 0.912 mmol)) was dissolved in 1M aqueous HC1 (5 mL), cooled in an ice-water bath and sodium nitrite (62.9 mg, 0.912 mmol) was added in a single batch. The resulting solution was stirred at 0°C for 30 min. Azo coupling, a new mixture was prepared with Fmoc-Tyr-OH (739 mg, 1.832 mmol) in saturated NaHC03 (17 mL) + acetonitrile (12.5 mL). The resulting suspension/solution was cooled in an ice-water bath. The diazonium salt mixture was added drop-wise. Stirred at 0°C. The reaction mixture slowly turned yellow. After 5.5 h LCMS showed complete conversion. The reaction mixture was acidified with IN HC1 to pH~6, the yellowish suspension turned into a clear orange solution, which was lyophilized. This afforded 2.10 g. Dissolved in a mixture of DMSO/H20/MeCN (-1:1:1) and purified in 5 runs by acidic preparative MPLC. The fractions were combined and lyophilized overnight, to obtain the desired product (compound 10).
EXAMPLE 3
SPPS SYNTHESIS OF TRS
[0157] While facing difficulties with protection of the hydroxy group of compound 10, the inventors explored a novel strategy for SPPS synthesis of TRS :
[0158] The inventors initiated the SPPS synthesis by implementing the N-protected (Fmoc) phosphorylcholine modified tyrosine (e.g. compound 10) 200 mg of compound 10 were loaded onto the CTC resin. In brief, 2-Chlorotrityl chloride resin (1.0 – 1.2 mmol/g, 200 – 400 mesh) (450 mg, 1.441 mmol) was allowed to swell in dichloromethane (12 mL) by rocking for 30 min. The solvent was removed and a solution of (S,E)-4-((5-(2-((((9//-f1uoren-9-yl)methoxy)carbonyl)amino)-2-carboxyethyl)-2-hydroxyphenyl)diazenyl)phenyl(2-(trimethylammonio)-ethyl) phosphate (200 mg, 0.290 mmol) in dichloromethane (12 mL) containing DIPEA (0.177 mL, 1.016 mmol) (substrate did not dissolve in DCM, after addition of DIPEA a solution was obtained) was added.
[0159] After 17 h the solvent was removed and the resin was washed with dichloromethane (3×10 mL, each washing step > 2 minutes). The capping solution (CH2C12:MeOH: DIPEA 9: 1:0.5) was added (10.5 mL) and the resin was rocked for 1 hour. Then the resin was washed with dichloromethane (3×10 mL) and dried in vacuo.
[0160] This resin was then split into equal portions in order to investigate a number of conditions for the subsequent chemistry in parallel, aimed at preventing the formation of the previously found tyrosine O-acylation, as witnessed by the isolation of compound 13 (see Scheme 2). The different reaction conditions were outlined in Table 1 (see below).
Scheme 2: Solid phase peptide synthesis
Table 1: exemplary coupling conditions tested
[0161] As shown in Table 1, various coupling conditions have been tested. Entries a-c resulted in the formation of a substantial amount of the byproduct (13). An improvement was obtained by using Fmoc-Gly-OSu in DMF (entry d). In this case the formation of byproduct (13) was reduced to only 3% relative to the desired compound 12. Nonetheless, neither of these methods was capable of suppressing the formation of 13 completely, therewith still posing a risk for further peptide synthesis, as this may lead to the accumulation of byproducts (compound 13).
[0162] Surprisingly, the inventors found that the byproduct (or phenolic ester byproduct, represented by compound 13 in Scheme 3) can be cleaved under standard Fmoc deprotection conditions with piperidine or with DBU in DMF, affording compound 15 cleanly, as illustrated below:
/////////DAZDOTUFTIDE, PHASE 3, TRS-01, TRS 01
CEFILAVANCIN



CEFILAVANCIN, TD-1792
CAS 722454-12-8
C87H96Cl3N16O28S2, 1984.28
F76229E21M
Vancomycin, 26-[[[3-[[(Z)-[1-(2-amino-5-chloro-4-thiazolyl)-2-[[(6R,7R)-2-carboxy-8-oxo-3-(pyridiniomethyl)-5-thia-1-azabicyclo[4.2.0]oct-2-en-7-yl]amino]-2-oxoethylidene]amino]oxy]propyl]amino]carbonyl]-26-decarboxy-
1-{[(6R,7R)-7-[(2Z)-2-(2-amino-5-chloro-1,3-thiazol-4-yl)-2-[(3-{[(1S,2R,18R,19R,22S,25R,28R,40S)-48-{[(2S,3R,4S,5S,6R)-3-{[(2S,4S,5S,6S)-4-amino-5-hydroxy-4,6-dimethyloxan-2-yl]oxy}-4,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy}-22-(carbamoylmethyl)-5,47-dichloro-2,18,32,35,37-pentahydroxy-19-[(2R)-4-methyl-2-(methylamino)pentanamido]-20,23,26,42,44-pentaoxo-7,13-dioxa-21,24,27,41,43-pentaazaoctacyclo[26.14.2.2^{3,6}.2^{14,17}.1^{8,12}.1^{29,33}.0^{10,25}.0^{34,39}]pentaconta-3,5,8,10,12(48),14,16,29(45),30,32,34(39),35,37,46,49-pentadecaen-40-yl]formamido}propoxy)imino]acetamido]-2-carboxylato-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-en-3-yl]methyl}pyridin-1-ium
Phase III Skin and soft tissue infections
- OriginatorGlaxoSmithKline; Theravance
- DeveloperR-Pharm; Theravance Biopharma
- ClassAcetamides; Antibacterials; Azabicyclo compounds; Beta-lactams; Cephalosporins; Peptide antibiotics; Pyridines; Thiazoles
- Mechanism of ActionCell wall inhibitors
BUILDING BLOCK
Vancomycin,

| Formula | C66H75Cl2N9O24 |
| Molar mass | 1449.27 g·mol−1 |
Cefilavancin (TD-1792) is an experimental antibiotic medication developed for the treatment of bacterial infections such as drug-resistant strains of Staphylococcus aureus. It is a prodrug which is also a codrug, injected intravenously and then cleaved inside the body to two active components, one of which is a modified form of vancomycin and the other a cephalosporin antibiotic. In clinical trials cefilavancin has shown similar efficacy with reduced side effects compared to vancomycin itself.[1][2][3][4][5][6][7][8]
- 31 Jan 2020Cefilavancin is still in phase III trials for Skin and soft tissue infection in Russia and Georgia (R-Pharm pipeline, January 2020)
- 17 Jun 2015Phase II development is ongoing the USA
- 02 Jun 2014Theravance Biopharma is formed as a spin-off of Theravance
SCHEME

SYN
WO2003031449
https://patentscope.wipo.int/search/en/WO2003031449
cheme A
REF
Li, Huijuan; ET AL, Medicine (Philadelphia, PA, United States) (2022), 101(34), e30120
References
- ^ Long DD, Aggen JB, Chinn J, Choi SK, Christensen BG, Fatheree PR, et al. (October 2008). “Exploring the positional attachment of glycopeptide/beta-lactam heterodimers”. The Journal of Antibiotics. 61 (10): 603–614. doi:10.1038/ja.2008.80. PMID 19168974.
- ^ Tyrrell KL, Citron DM, Warren YA, Goldstein EJ (April 2012). “In vitro activity of TD-1792, a multivalent glycopeptide-cephalosporin antibiotic, against 377 strains of anaerobic bacteria and 34 strains of Corynebacterium species”. Antimicrobial Agents and Chemotherapy. 56 (4): 2194–2197. doi:10.1128/AAC.06274-11. PMC 3318369. PMID 22290981.
- ^ Stryjewski ME, Potgieter PD, Li YP, Barriere SL, Churukian A, Kingsley J, et al. (November 2012). “TD-1792 versus vancomycin for treatment of complicated skin and skin structure infections”. Antimicrobial Agents and Chemotherapy. 56 (11): 5476–5483. doi:10.1128/aac.00712-12. PMC 3486540. PMID 22869571.
- ^ Douglas EJ, Laabei M (September 2023). “Staph wars: the antibiotic pipeline strikes back”. Microbiology. 169 (9). Reading, England. doi:10.1099/mic.0.001387. PMC 10569064. PMID 37656158.
- ^ Surur AS, Sun D (2021). “Macrocycle-Antibiotic Hybrids: A Path to Clinical Candidates”. Frontiers in Chemistry. 9: 659845. Bibcode:2021FrCh….9..317S. doi:10.3389/fchem.2021.659845. PMC 8120311. PMID 33996753.
- ^ Saxena D, Maitra R, Bormon R, Czekanska M, Meiers J, Titz A, et al. (December 2023). “Tackling the outer membrane: facilitating compound entry into Gram-negative bacterial pathogens”. npj Antimicrobials and Resistance. 1 (1): 17. doi:10.1038/s44259-023-00016-1. PMC 11721184. PMID 39843585.
- ^ Koh AJ, Thombare V, Hussein M, Rao GG, Li J, Velkov T (2023). “Bifunctional antibiotic hybrids: A review of clinical candidates”. Frontiers in Pharmacology. 14: 1158152. doi:10.3389/fphar.2023.1158152. PMC 10313405. PMID 37397488.
- ^ Homer JA, Johnson RM, Koelln RA, Moorhouse AD, Moses JE (2024). “Strategic re-engineering of antibiotics”. Nature Reviews Bioengineering. doi:10.1038/s44222-024-00250-w.
| Clinical data | |
|---|---|
| Other names | TD-1792 |
| Routes of administration | Intravenous |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 722454-12-8 |
| PubChem CID | 76960417 |
| DrugBank | DB05735 |
| ChemSpider | 34990483 |
| UNII | F76229E21M |
| ChEMBL | ChEMBL4297645 |
| Chemical and physical data | |
| Formula | C87H95Cl3N16O28S2 |
| Molar mass | 1983.27 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
////////////CEFILAVANCIN, TD-1792, TD 1792, F76229E21M, цефилаванцин, 头孢拉凡星, سيفيلافانسين , GlaxoSmithKline, Theravance, PHASE 3
BOFUTRELVIR
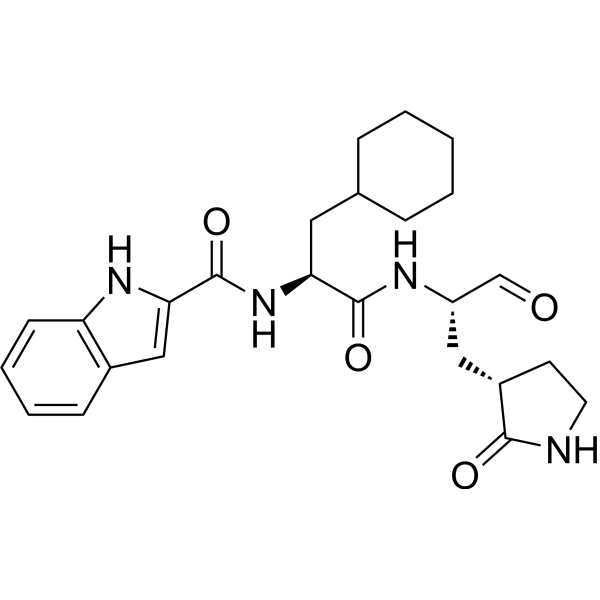
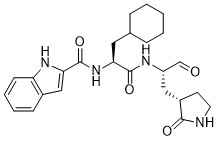
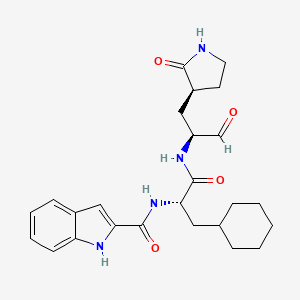
BOFUTRELVIR
Cas 2103278-86-8
| Molecular Weight | 452.55 |
|---|---|
| Formula | C25H32N4O4 |
UNII-T5UX5SKK2S; Mpro inhibitor 11A; 2103278-86-8; T5UX5SKK2S, DC-402234, DC402234, MPI-10
IUPAC/Chemical Name: N-[(2S)-3-cyclohexyl-1-oxo-1-[[(2S)-1-oxo-3-[(3S)-2-oxopyrrolidin-3-yl]propan-2-yl]amino]propan-2-yl]-1H-indole-2-carboxamide
N-[(2S)-3-cyclohexyl-1-oxo-1-[[(2S)-1-oxo-3-[(3S)-2-oxopyrrolidin-3-yl]propan-2-yl]amino]propan-2-yl]-1H-indole-2-carboxamide
Bofutrelvir has an additive antiviral effect when combined with Remdesivir
FB2001
Bofutrelvir (FB2001) is a SARS-CoV-2 main protease Mpro inhibitor with an IC50 value of 53 nM and an EC50 value of 0.53 μM. Bofutrelvir exhibits potent antiviral efficacy against several current SARS-CoV-2 variants with EC50 values of 0.26-0.42 μM. Bofutrelvir has an additive antiviral effect when combined with Remdesivir.
Bofutrelvir is a small molecule inhibitor of the severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) main protease (Mpro; 3C-like protease; 3CL protease; 3CLpro; nsp5 protease), with potential antiviral activity against SARS-CoV-2. Upon intravenous administration or inhalation into the lungs, bofutrelvir selectively targets, binds to, and inhibits the activity of SARS-CoV-2 Mpro. This inhibits the proteolytic cleavage of viral polyproteins, thereby inhibiting the formation of viral proteins including helicase, single-stranded-RNA-binding protein, RNA-dependent RNA polymerase, 20-O-ribose methyltransferase, endoribonuclease and exoribonuclease. This prevents viral transcription and replication. Bofutrelvir may have antiviral activity in the brain.
- Originator Frontier Biotechnologies
- Class Amides; Antivirals; Indoles; Pyrrolidinones; Small molecules
- Mechanism of Action Coronavirus 3C-like proteinase inhibitors
Highest Development Phases
- Phase II/III COVID 2019 infections
Most Recent Events
- 28 Apr 2024No recent reports of development identified for phase-I development in COVID-2019-infections in USA (IV, Infusion)
- 04 Jan 2023Phase-II/III clinical trials in COVID-2019 infections in China (Inhalation) (NCT05675072)
- 30 Dec 2022Frontier Biotechnologies completes a phase I trial in COVID-2019 infections in China (Inhalation) (NCT05583812)
- N-[(2S)-3-cyclohexyl-1-oxo-1-({(2S)-1-oxo-3-[(3S)-2-oxopyrrolidin-3-yl]propan-2-yl}amino)propan-2-yl]-1H-indole-2-carboxamide is a secondary carboxamide resulting from the formal condensation of the carboxy group of 1H-indole-2-carboxylic acid with the primary amino group of 3-cyclohexyl-N-{(2S)-1-oxo-3-[(3S)-2-oxopyrrolidin-3-yl]propan-2-yl}-L-alaninamide. It is an inhibitor of SARS coronavirus main proteinase and inhibits SARS-CoV-2 replication in cell culture (EC50 = 0.53 muM). It has a role as an EC 3.4.22.69 (SARS coronavirus main proteinase) inhibitor and an anticoronaviral agent. It is an indolecarboxamide, a member of pyrrolidin-2-ones, an aldehyde, a secondary carboxamide and an oligopeptide.
SCHEME
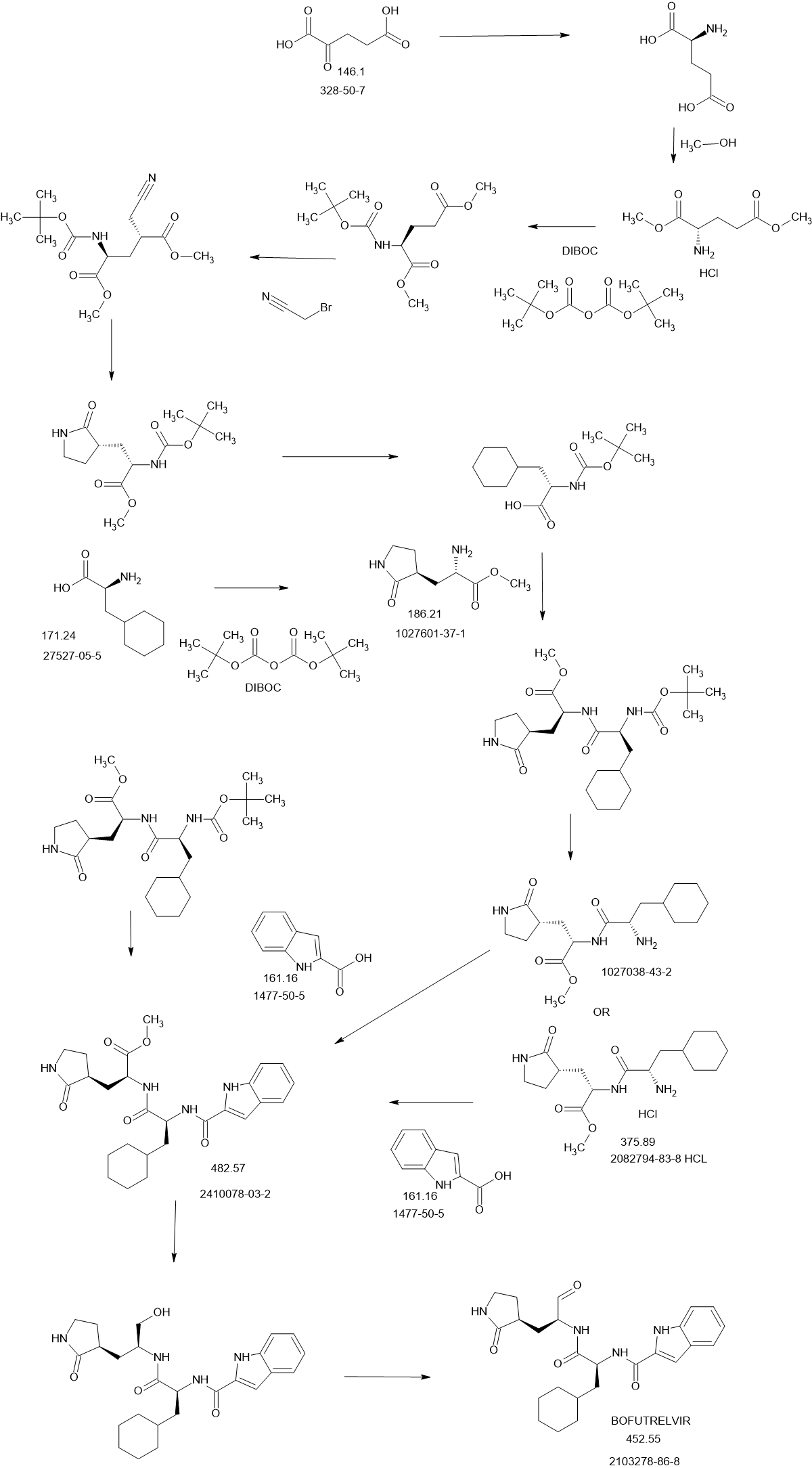
PATENTS
CN110818691
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN289596961&_cid=P11-M9Z1Y3-09353-1
| Synthesis of compound 1-2: |
| Under argon protection, N-tert-butyloxycarbonyl-L-glutamic acid dimethyl ester (1-1) (6g, 21.8mmol) was dissolved in 60mL of anhydrous tetrahydrofuran, and a tetrahydrofuran solution of LiHMDS (1M in THF) (47mL, 47mmol) was slowly dripped at -78℃, and the temperature was kept stable at -78℃ during the dripping process, which lasted for about 1 hour. After the dripping was completed, it was stirred at -78℃ for 1 hour. Bromoacetonitrile (2.79g, 23.3mmol) was dissolved in 20ml of tetrahydrofuran, and then the solution was slowly dripped into the reaction system, and the dripping process lasted for 1 to 2 hours. The temperature was controlled at -78℃ and the reaction was continued for 3 hours. After the reaction was completed, NH4Cl solution was added to the reaction solution to quench the reaction, and the mixture was stirred for 10min and then warmed to room temperature. 40mL of saturated sodium chloride solution was poured in and stirred thoroughly, and the reaction system was seen to be stratified. The organic layer was separated, and the aqueous phase was extracted with ethyl acetate (EA). The organic layers were combined, dried over anhydrous sodium sulfate, concentrated, and subjected to column chromatography (Flash, PE:EA=1:5) to obtain 3.9 g of a light yellow oil 1-2 with a yield of 58%. |
| Synthesis of compound 1-3: |
| Dissolve 1-2 (1 g, 3.15 mmol) in 25 mL of anhydrous methanol, stir to 0°C in an ice bath, and then add cobalt dichloride hexahydrate (450 mg, 1.89 mmol). After 10 min, add sodium borohydride (715 mg, 18.9 mmol) in small portions. The reaction solution continues to react in an ice bath for 1 h and then returns to room temperature. After 15 h, quench with 5 mL of saturated NH4Cl solution and continue stirring for 10 min. After filtering out the solid, evaporate the filtrate to dryness, extract with 20 mL of water and 30×3 mL of ethyl acetate, combine the organic phases, and add anhydrous Na 2 SO 4 After drying for 1 h, the residue was concentrated under reduced pressure and separated by column chromatography [PE:EA=1:2] to obtain 460 mg of a white powdery solid with a yield of 51%. |
| Synthesis of compound 1-4: |
| Compound 1-3 (2.6 g) was dissolved in a dichloromethane solution of trifluoroacetic acid (1/1, v/v), stirred at room temperature for 1 hour, concentrated, added with 100 ml of dichloromethane, washed with saturated sodium carbonate solution, and the organic layer was dried over anhydrous sodium sulfate and concentrated to obtain an oily substance 1-4 (2.7 g) with a yield of 99%. |
| Synthesis of compound 1-5: |
| Boc-cyclohexylalanine (1.26 g, 5 mmol), EDCI (1.36 g, 6 mmol), and HOBt (0.822 g, 6 mmol) were added to 80 ml of dichloromethane solution and stirred at room temperature for 30 min. Compound 1-4 (0.896 g, 5 mmol) was then added, and 1.2 equivalents of triethylamine were added dropwise, and stirred at room temperature. After TLC monitoring (ultraviolet), dichloromethane was used for extraction after the reaction was complete, and the mixture was washed with dilute hydrochloric acid, saturated sodium bicarbonate solution, and saturated sodium chloride. The organic layers were combined and dried over anhydrous sodium sulfate, and concentrated to obtain 1.2 g of a white viscous solid with a yield of 60%. |
| Synthesis of compound 1-6: |
| Compound 1-5 (2.5 g) was dissolved in a dichloromethane solution of trifluoroacetic acid (1/1, v/v), stirred at room temperature for 1 hour, concentrated, added with 100 ml of dichloromethane, washed with saturated sodium carbonate solution, and the organic layer was dried over anhydrous sodium sulfate and concentrated to obtain an oily substance 1-6 (2.61 g) with a yield of 99%. |
| Synthesis of compound 1-7: |
| Indole 2-carboxylic acid (0.795 g, 5 mmol), EDCI (1.36 g, 6 mmol), and HOBt (0.822 g, 6 mmol) were added to 80 ml of dichloromethane solution and stirred at room temperature for 30 min. Compound 1-6 (2.2 g, 5 mmol) was then added, and 1.2 equivalents of triethylamine were added dropwise, and stirred at room temperature. After TLC monitoring (ultraviolet), dichloromethane was used for extraction after the reaction was complete, and the mixture was washed with dilute hydrochloric acid, saturated sodium bicarbonate solution, and saturated sodium chloride. The organic layers were combined and dried over anhydrous sodium sulfate, and concentrated to obtain 1.3 g of a white viscous solid with a yield of 60%. |
| Synthesis of compound 1-8: |
| Dissolve 1-7 (243 mg, 0.51 mmol) in 20 ml of methanol, slowly add sodium borohydride (107 mg, 2.9 mmol) in batches, and stir at room temperature for about 2 hours to complete the reaction. After the reaction is completed, add about 20 ml of saturated brine to quench the reaction, concentrate the methanol in the reaction system, and add dichloromethane for extraction. The organic phase is washed with saturated brine, dried over anhydrous sodium sulfate, and concentrated to obtain a white solid substance 1-8, which can be directly used in the next step. |
| Synthesis of compound 1-9: |
| Dissolve the intermediate 1-8 (129 mg, 0.29 mmol) in 20 ml of dichloromethane, add Dess-Martin oxidant (147 mg, 0.35 mmol) and solid sodium bicarbonate (29 mg, 0.35 mmol), and stir at room temperature. After the reaction is complete by TLC monitoring (ultraviolet), filter the reaction system, extract the filtrate with saturated sodium bicarbonate, and the organic layer is purified by saturated sodium salt. |
| The product was washed with water, dried over anhydrous sodium sulfate and concentrated. The product was separated and purified by flash column chromatography (CH2Cl2:MeOH=20:1) to obtain 77 mg of compound 1 as a white solid powder with a yield of 60%. |
| Synthesis of compound 1-10: |
| Compound 1-9 (129 mg, 0.29 mmol) was dissolved in dichloromethane solvent, acetic acid (19.2 mg, 0.32 mmol) and benzyl isocyanate (37.6 mg, 0.32 mmol) were added to react to obtain compound 1-10. Flash column chromatography (CH 2 Cl 2 :MeOH=20:1) to separate and purify to obtain 126 mg of white solid powder compound 1-10 with a yield of 70%. |
| Synthesis of compound 1-11: |
| Compound 1-10 (187 mg, 0.3 mmol) was dissolved in methanol solvent, LiOH (0.6 mmol) was added and stirred to obtain compound 1-11. 2 Cl 2 :MeOH=20:1) to separate and purify to obtain 148 mg of white solid powder compound 1-11 with a yield of 85%. |
| Synthesis of compound 1-12: |
| Compound 1-11 (174 mg, 0.3 mmol) was dissolved in dichloromethane solvent, Dess-Martin oxidant (152 mg, 0.36 mmol) was added, sodium bicarbonate (30 mg, 0.36 mmol) was added, and stirred to obtain a white solid powder compound 1-12 of 140 mg in total, with a yield of 80%. |
| 1 H NMR(500MHz,Chloroform)δ9.76(s,1H),7.73(s,1H),7.39(s,1H),7.32–7.26(m,2H),7.22(s,1H),7 .20–7.10(m,3H),7.01(s,1H),6.82(s,1H),6.68(s,1H),6.14(s,1H),5.57(s,1H),5.43(s,1H),4.3 8(s,1H),4.32(d,J=19.2Hz,2H),3.45(s,1H),3.35(s,1H),3.06(s,1H),2.20(dd,J=15.4,2.3Hz,4H ),2.12–2.03(m,2H),1.92(s,1H),1.77(s,1H),1.73–1.67(m,3H),1.66–1.53(m,6H),1.37(s,1H).; |
PATENT
WO2020030143
bioRxiv (2020), 1-17
- [1]. Ullrich S, Nitsche C. The SARS-CoV-2 main protease as drug target. Bioorg Med Chem Lett. 2020 Sep 1;30(17):127377. [Content Brief]
- [2]. Shang W, et al. In vitro and in vivo evaluation of the main protease inhibitor FB2001 against SARS-CoV-2. Antiviral Res. 2022 Dec;208:105450. [Content Brief]
///BOFUTRELVIR, FB2001, FB 2001, Phase 3, COVID 2019, T5UX5SKK2S, Mpro inhibitor, DC-402234, DC402234, MPI-10
Tibremciclib


Tibremciclib
cas 2397678-18-9, GTPL12881
CRB7BT5JDQ
518.6 g/mol, C28H32F2N8
N-[5-[(4-ethylpiperazin-1-yl)methyl]pyridin-2-yl]-5-fluoro-4-[(1R)-6-fluoro-1-methyl-1,2,3,4-tetrahydropyrido[1,2-a]benzimidazol-8-yl]pyrimidin-2-amine
Tibremciclib is a CDK4 inhibitor with antineoplastic activity[1].
- Originator Betta Pharmaceuticals Co Ltd
- Class Antineoplastics; Small molecules
- Mechanism of Action Cyclin-dependent kinase 4 inhibitors; Cyclin-dependent kinase 6 inhibitors
- Phase III Breast cancer; Solid tumours
13 Sep 2024 Efficacy and adverse event data from a phase III trial in Breast cancer presented at the 49th European Society for Medical Oncology Congress 2024 (ESMO-2024)
- 30 Jun 2023Phase-III clinical trials in Breast cancer (Metastatic disease, Late-stage disease, Combination therapy, Second-line therapy or greater) in China (PO) (NCT05433480)
- 02 Jun 2023Efficacy, adverse events and PK data from a phase I trial in Solid tumours presented at the 59th Annual Meeting of the American Society of Clinical Oncology (ASCO-2023)
Cyclin-dependent kinases (CDKs) are a class of serine / threonine protein kinases that participate in the regulation of the cell cycle, transcription initiation, and control of certain specific metabolic cascades. Different CDKs and cyclins form CDK-cyclin complexes. If the CDK activity is dysregulated, it will directly or indirectly cause uncontrolled cell proliferation, genomic instability (increased DNA mutation, chromosome deletion, etc.) and chromosomal instability (change in chromosome number). )Wait.
The CDKs family has identified more than 20 subtypes. CDK1, CDK2, CDK4, and CDK6 are involved in cell cycle regulation; CDK7, CDK8, CDK9, and CDK11 are involved in transcription regulation; and other kinases include CDK3 and CDK5. Among them, CDK4 / 6 (cyclin-dependent kinases 4 and 6) is a key factor in regulating the cell cycle. Cancer-related cell cycle mutations mainly exist in the G1 and G1 / S phase transformation. CDK4 / 6 binds to CyclinD A complex with kinase activity is formed and phosphorylation of the tumor suppressor gene Rb product pRb releases the bound transcription factor E2F to initiate transcription of genes related to the S phase, prompting cells to pass the checkpoint and transfer from the G1 phase to the S phase. The specific activation of CDK4 / 6 is closely related to the proliferation of some tumors. About 80% of human tumors have abnormalities in the cyclin D-CDK4 / 6-INK4-Rb pathway. CDK4 / 6 inhibitors block the cell cycle in the G1 phase, thereby inhibiting tumor proliferation.
The development of drugs targeting CDK4 / 6 kinases is a significant area. The advantages of anti-tumor targets are: (1) Most proliferating cells rely on CDK2 or CDK4 / 6 to proliferate, but CDK4 / 6 inhibitors do not show Cytotoxicity of “pan-CDK inhibitors”, such as bone marrow suppression and intestinal response; (2) Preclinical experiments show that if the level of cyclin D or the inactivation of P16INK4a can increase the sensitivity of cells to drugs, due to tumors Compared with normal cells, cells have the above phenomenon, so the targeting of drugs is increased to a certain extent.
PCT International Application PCT / CN2017 / 117950 describes a class of benzimidazole derivatives that are used as CDK4 / 6 protein kinase inhibitors, and most of these compounds effectively inhibit CDK4 and CDK6. Because there are still unmet needs in the treatment options for kinase-mediated diseases, here we further screen the salt forms and crystal forms of benzimidazole derivatives to meet the medical needs of patients.
SCHEME
SIDE CHAIN

SIDE CHAIN

MAIN

Patent
Betta Pharmaceuticals Co., Ltd., WO2019242719
https://patents.google.com/patent/WO2019242719A1/en


Synthesis of 1-A1-01 (Step 1)
In a 50L reactor, add 20L of dichloromethane (DCM), 1-A1-S1 (300g), and triethylamine (390g). While stirring, lower the temperature to below -5 ° C, and add benzyl chloroformate / Cbz- Cl (570 g) was added dropwise for 5 hours, and the temperature was naturally raised to room temperature. TLC (ethyl acetate: n-hexane = 1: 3) was monitored until the reaction was completed. Water (1.5 L) was added, and concentrated hydrochloric acid (80 mL) was slowly added dropwise to control the pH to 1-2. The solution was allowed to stand and separate. The organic phase was washed with 15 L of water, dried over anhydrous sodium sulfate for 0.5 hours, filtered to remove the desiccant, and collected the filtrate. And concentrated to obtain 730 g of light yellow oily liquid, which is crude 1-A1-01, yield 95.4%
Synthesis of 1-A1-02 (Step 2)
720mL of DCM, N, N-dimethylsulfoxide (90g) was added to a 20L reaction flask, protected by nitrogen, and the temperature was lowered below -65 ° C under stirring, and oxalyl chloride (106g) was added dropwise. The addition was completed in 2 hours. Stir for 20 minutes under heat preservation; add 1-A1-01’s dichloromethane solution (143g / 500mL DCM) dropwise. After 40 minutes, the addition is complete and the reaction is held for 15 minutes. Controlled at this temperature, TEA was added dropwise. After the addition was completed for 2 hours, the temperature was naturally raised to -20 ° C. 250 L of water was added to the system. The pH of the system was adjusted to 1-2 with hydrochloric acid. × 2) Washed, dried over anhydrous sodium sulfate, filtered to remove the desiccant, collected the filtrate and concentrated to obtain 432 g of a yellow oily liquid, which is the crude product 1-A1-02, which was directly used in the next reaction.
Synthesis of 1-A1-03 (Step 3)
In a stirred state, 400 mL of tetrahydrofuran (THF) and potassium tert-butoxide (215 g) were sequentially added to a 1 L reaction kettle, the temperature was lowered to 5-15 ° C., and triethyl phosphoryl acetate (430 g) was added dropwise. The dropwise addition was completed in 50 minutes. At a controlled temperature of 15 ° C, a tetrahydrofuran solution of 1-A1-02 (431 g / 100 mL of THF) was added dropwise. After the dropwise addition was completed for 1 hour, TLC (ethyl acetate: n-hexane = 1: 3) was monitored to complete the reaction, and the system was added. Saturated aqueous sodium chloride solution (1.5L), allowed to stand and separate, and collected the tetrahydrofuran phase; the aqueous phase was extracted with dichloromethane (2L), and the organic phases were combined and dried over anhydrous sodium sulfate for 0.5 hours, and the drying agent was removed by filtration. The filtrate was collected and concentrated, and the concentrate was purified by column chromatography to obtain 390 g of a pale yellow oily liquid, which was 1-A1-03 product.
Synthesis of 1-A1-041 (step 4)
In a 5L reactor, an aqueous solution of sodium hydroxide (301 g / 1.5 L of water) was added to a tetrahydrofuran (601 g / 2.3 L of THF) solution of 1-A1-03, and the mixture was heated to reflux for 3-4 hours to stop the reaction. The temperature was lowered to 40-50 ° C, and the layers were left to stand. The organic phase (THF) was collected and concentrated to a large amount of solids; the solids were dissolved by adding water (20L), and the aqueous phase was sequentially treated with methyl tert-butyl ether (2L) and ethyl acetate. Ester (2L), methyl tert-butyl ether (2L) washing; the aqueous phase was adjusted to pH 1-2 with concentrated hydrochloric acid, extracted twice with ethyl acetate (1.5L, 3L), the organic phases were combined, and anhydrous sulfuric acid was used Sodium was dried for 0.5 hours; the desiccant was removed by filtration, and the filtrate was collected and concentrated to a large amount of solids. The solids were added with isopropyl ether (3L) and slurried for 2 hours. The solids were collected by filtration and the solids were rinsed with isopropyl ether (1L). The solid was air-dried at 50 ° C for 3-4 hours to obtain 331 g of a pale yellow solid, which is a 1-A1-041 product with a yield of 52.7%.
Synthesis of 1-051 (step 5)
In a stirred state, 1-A1-041 (600g), methanol (25L), and concentrated sulfuric acid were added to a 50L reactor, and the reaction was heated under reflux for 3-4 hours. After the reaction was completed, the temperature was reduced to room temperature. Dichloromethane (15L) was added to the concentrate, and the pH was adjusted to 9-10 with an aqueous solution of potassium carbonate. The organic phase was collected by stirring, standing, and separating. The organic phase was dried over anhydrous sodium sulfate for 0.5 hours. The desiccant was removed by filtration and the filtrate was collected. And concentrated to obtain 6.37 kg of off-white solid, which is 1-A1-051 product, with a yield of 97.3%.
Synthesis of 1-A1 (step 6)
In a 2L hydrogenation kettle, add 1-A1-051 (500g), methanol (1.8L), and palladium on carbon. The system replaces nitrogen 3 times and hydrogen 3 times in sequence. The system maintains a hydrogen atmosphere, and the temperature is increased to 85 ° C and the pressure is 3.0. The reaction was carried out at Mpa for 3 hours, and the reaction was completed. The temperature was lowered to room temperature, the palladium on carbon was removed by filtration, and the organic phase was collected and concentrated until a large amount of light yellow solid appeared. Isopropyl ether (3L) was added to freeze (-20 ° C) for crystallization, and the solid product was collected by filtration. Ether (500 mL) was rinsed to obtain 234 g of a pale yellow solid, which was a 1-A1 product with a yield of 90.5%.
Synthesis of 1-A2 (Step 7)
In a stirred state, 1-A1 (200g), 4-bromo-2,6-difluoroaniline (410g), and toluene (1.2L) were added to a 50L reactor, and phosphorus oxychloride (413g) was added dropwise to the system. The addition was completed in 1 hour. Triethylamine was added dropwise under an ice bath, and the addition was completed in 1 hour. The temperature was raised to 110 ° C, and the reaction was performed for 1 hour. Reduce the temperature of the system to 2-10 ° C, add 1L of water, adjust the pH = 9-10 with saturated potassium carbonate aqueous solution, extract twice with ethyl acetate (1.5L, 1L), and combine the organic phases with 2L saturated sodium chloride aqueous solution. Wash, dry with anhydrous sodium sulfate for 0.5 hours, remove the desiccant by filtration, collect the filtrate and concentrate to the appearance of a solid product, add isopropyl ether (1L) to beat the solid for 10 minutes, filter, and collect 460 g of a yellow solid as a 1-A2 product.
Synthesis of 1-A3 (step eight)
Under stirring, 1-A2 (450g), N, N-dimethylformamide (2L), and cesium carbonate (700g) were added to the reaction kettle, and the reaction was heated to 110 ° C for 24 hours, and the reaction was detected by TLC. Ethyl acetate (3L) was added to the system, and solid impurities were removed by filtration. The filtrate was washed with a saturated sodium chloride aqueous solution (1L × 5), and the organic phase was dried over anhydrous sodium sulfate for 0.5 hours, concentrated to the appearance of a large amount of solid, Butyl ether (1L × 2) was beaten for 30 minutes, and 382 g of pale yellow solid product was obtained by filtration, that is, 1-A3, and the yield was 90.10%.
Synthesis of 1-01 (step 9)
With stirring, 1-A3 (380 g), pinacol diborate (400 g), potassium acetate (340 g), palladium acetate (6 g), tricyclohexyl phosphorus (7 g), and 1,4-dioxane were sequentially added. The ring was added to the reaction kettle, protected by nitrogen, and heated to 90 ° C for 2 hours. TLC was monitored until the reaction was complete. The temperature was reduced to room temperature, and the filtrate was concentrated to remove a large amount of 1,4-dioxane. The concentrate was purified by n-hexane and dichloromethane column chromatography, and n-hexane (1.2 L) was slurried for 1 hour to obtain 334 g of a gray solid. That is 1-01, and the yield is 70.10%.
Synthesis of 1-02 (step 10)
Under stirring, take 1-01 (128g), 1,4-dioxane (1L), 1-S3 (85g), potassium carbonate (110g), and purified water and add them to a 2L three-necked flask in sequence. [1,1′-Bis (diphenylphosphine) ferrocene] palladium dichloromethane complex (Pd (dppf) Cl 2 .DCM) was added. The temperature was raised to 60 ° C. After 4 hours of reaction, the reaction was complete. The reaction solution was cooled to room temperature, and concentrated under reduced pressure to remove most of 1,4-dioxane. Dichloromethane (1.5 L) and purified water (1.1 L) were added, stirred, and allowed to stand and separate. The layers were separated, and water was added. The phases were extracted with dichloromethane (10 L), the organic phases were combined, washed with 0.5% dilute hydrochloric acid (1 L x 2), saturated aqueous sodium chloride solution (1 L), and the layers were separated. The organic phase was dried over anhydrous sodium sulfate (500 g), filtered to remove the drying agent, and the filtrate was concentrated under reduced pressure. Ethyl acetate (0.5 L) was added to the concentrate and the mixture was stirred for 30 minutes to precipitate a solid. After filtration, the obtained solid was rinsed with ethyl acetate (0.5 L) and dried under vacuum at 45 ° C for 3 hours to obtain 120 g of a yellow solid.
Synthesis of 1-03 (step 11)
Under stirring, take 1-02 (100g), 1,4-dioxane (1L), 1-C2 (80g), and cesium carbonate (163g) into a 2L three-necked bottle in sequence, protected by nitrogen, and add palladium acetate ( 2g) and 4,5-bisdiphenylphosphine-9,9-dimethylxanthracene (Xantphos) (4g), heated to 85 ° C. until the reaction was complete. The reaction solution was cooled to room temperature and filtered to obtain a solid product. The solid was rinsed with ethyl acetate, and then added to a mixed system of dichloromethane (1.5 L) and purified water (1.1 L), stirred, allowed to stand, and separated into layers. The aqueous phase was extracted with dichloromethane (700 mL). The organic phases were combined and washed with purified water (700 mL x 2). The organic phase was dried by adding anhydrous sodium sulfate (700 g), filtered to remove the desiccant, and the filtrate was concentrated. Methanol (0.5 L) was added, heated to 55-65 ° C. and stirred for 0.5 hours, lowered to room temperature, and filtered. The solid product was filtered and rinsed with 500 mL of ethyl acetate. The solid was dried under vacuum at 45 ° C for 8 hours to obtain 111.79 g of a pale yellow solid 1-03.
Synthesis of compound II (step twelve)
Under stirring, take 1-03 (500g) and anhydrous methanol (3.8L), add them to a 10L reactor in sequence, and heat to 65 ° C. After the reaction system is clarified for 0.5 hours, add L-tartaric acid in methanol (150.89) dropwise. g of tartaric acid is dissolved in 500mL of anhydrous methanol), and the dropping time is controlled to be 45 to 60 minutes. After the addition is complete, the reaction is kept at 65 ° C for 4 hours. ), Control the dropwise addition time to 30 to 45 minutes. After the dropwise addition is complete, hold the reaction at 65 ° C for 1 hour. Continue to dropwise add L-tartaric acid in methanol (36.55g of tartaric acid dissolved in 250mL of anhydrous methanol) and control the dropwise addition time to 30. To 45 minutes, the dropwise addition was completed. The temperature was kept at 65 ° C for 1.5 hours, and the heating was stopped. The temperature was naturally lowered to 20-30 ° C, filtered, the filter cake was rinsed with methanol (400mL × 2), and dried at 45 ° C under vacuum for 36 hours. 530.64 g of crystalline powder was Compound II, which was identified by X-ray powder diffraction, and showed that the crystal form was Form A of Compound II.
WO2022199656
WO2023131179
///Tibremciclib, GTPL12881, BETTA, PHASE 3, CANCER
ATICAPRANT


ATICAPRANT
1174130-61-0
BENZAMIDE, 4-(4-(((2S)-2-(3,5-DIMETHYLPHENYL)-1-PYRROLIDINYL)METHYL)PHENOXY)-3-FLUORO-
C26H27FN2O2, 418.512
- 4-[4-[[(2S)-2-(3,5-Dimethylphenyl)-1-pyrrolidinyl]methyl]phenoxy]-3-fluorobenzamide (ACI)
- (S)-4-(4-((2-(3,5-Dimethylphenyl)pyrrolidin-1-yl)methyl)phenoxy)-3-fluorobenzamide
- 4-(4-{[(2S)-2-(3,5-dimethylphenyl)pyrrolidin-1-yl]methyl}phenoxy)-3-fluorobenzamide
- Aticaprant
- CERC 501
- JNJ 67953964
- JNJ 67953964AAA
- LY 2456302
- S-Aticaprant
- CERC-501
- JSPA 0658 JSPA-0658 JSPA0658
- LY 2456302 LY-2456302 , LY2456302
- OriginatorEli Lilly and Company
- DeveloperAvalo Therapeutics; Eli Lilly and Company; Johnson & Johnson Innovative Medicine
- ClassAntidepressants; Benzamides; Benzene derivatives; Drug withdrawal therapies; Fluorinated hydrocarbons; Pyrrolidines; Smoking cessation therapies
- Mechanism of ActionOpioid kappa receptor antagonists
- Phase III Major depressive disorder
- DiscontinuedAlcoholism; Cocaine-related disorders; Smoking withdrawal
- 26 Jun 2024Janssen Research & Development initiates a phase III VENTURA-7 trial for Major depressive disorder (Adjunctive treatment) in USA (PO, Tablet) (NCT06514742) (EudraCT2024-511557-21-00)
- 01 Oct 2023Janssen Pharmaceuticals is now called Johnson & Johnson Innovative Medicine (Janssen Pharmaceuticals website, October 2023)
- 19 May 2023Chemical structure information added
Aticaprant, also known by its developmental codes JNJ-67953964, CERC-501, and LY-2456302, is a κ-opioid receptor (KOR) antagonist which is under development for the treatment of major depressive disorder.[2][3][4] A regulatory application for approval of the medication is expected to be submitted by 2025.[2] Aticaprant is taken by mouth.[1]
Side effects of aticaprant include itching, among others.[4][5] Aticaprant acts as a selective antagonist of the KOR, the biological target of the endogenous opioid peptide dynorphin.[3] The medication has decent selectivity for the KOR over the μ-opioid receptor (MOR) and other targets, a relatively long half-life of 30 to 40 hours, and readily crosses the blood–brain barrier to produce central effects.[4][6]
Aticaprant was originally developed by Eli Lilly, was under development by Cerecor for a time, and is now under development by Janssen Pharmaceuticals.[2] As of July 2022, it is in phase 3 clinical trials for major depressive disorder.[2] Like other kappa opioid antagonists currently under clinical investigation for the treatment of major depression, its efficacy may be compromised by the countervailing activation of pro-inflammatory cytokines in microglia within the CNS.[7]
Aticaprant was also under development for the treatment of alcoholism, cocaine use disorder, and smoking withdrawal, but development for these indications was discontinued.[2]
Pharmacology
Pharmacodynamics
Aticaprant is a potent, selective, short-acting (i.e., non-“inactivating”) antagonist of the KOR (Ki = 0.81 nM vs. 24.0 nM and 155 nM for the μ-opioid receptor (MOR) and δ-opioid receptor (DOR), respectively; approximately 30-fold selectivity for the KOR).[8][9][10] The drug has been found to dose-dependently block fentanyl-induced miosis at 25 mg and 60 mg in humans (with minimal to no blockade at doses of 4 to 10 mg), suggesting that the drug significantly occupies and antagonizes the MOR at a dose of at least 25 mg but not of 10 mg or less.[10] However, a more recent study assessing neuroendocrine effects of the drug in normal volunteers and subjects with a history of cocaine dependence reported observations consistent with modest MOR antagonism at the 10 mg dose.[11] In animal models of depression, aticaprant has been found to have potent synergistic efficacy in combination with other antidepressants such as citalopram and imipramine.[12]
Positron emission tomography imaging revealed that brain KORs were almost completely saturated by the drug 2.5 hours following a single dose of 10 mg, which supported the 4 mg to 25 mg dosages that aticaprant is being explored at in clinical trials.[13][14] Occupancy was 35% for a 0.5 mg dose and 94% for a 10 mg dose.[15][14] At 24 hours post-dose, receptor occupancy was 19% for 0.5 mg and 82% for 25 mg.[15][14] No serious side effects were observed, and all side effects seen were mild to moderate and were not thought to be due to aticaprant.[14]
Pharmacokinetics
The oral bioavailability of aticaprant is 25%.[1] The drug is rapidly absorbed, with maximal concentrations occurring 1 to 2 hours after administration.[1] It has an elimination half-life of 30 to 40 hours in healthy subjects.[1] The circulating levels of aticaprant increase proportionally with increasing doses.[1] Steady-state concentrations are reached after 6 to 8 days of once-daily dosing.[1] Aticaprant has been shown to reproducibly penetrate the blood–brain barrier.[13][14]
History
Aticaprant was originally developed by Eli Lilly under the code name LY-2456302.[2] It first appeared in the scientific literature in 2010 or 2011.[16][17] The compound was first patented in 2009.[18]
In February 2015, Cerecor Inc. announced that they had acquired the rights from Eli Lilly to develop and commercialize LY-2456302 (under the new developmental code CERC-501).[19]
As of 2016, aticaprant has reached phase II clinical trials as an augmentation to antidepressant therapy for treatment-resistant depression.[20][12] A phase II study of aticaprant in heavy smokers was commenced in early 2016 and results of the study were expected before the end of 2016.[14] Aticaprant failed to meet its main endpoint for nicotine withdrawal in the study.[21]
In August 2017, it was announced that Cerecor had sold its rights to aticaprant to Janssen Pharmaceuticals.[22][21] Janssen was also experimenting with esketamine for the treatment of depression as of 2017.[21]
Research
In addition to major depressive disorder, aticaprant was under development for the treatment of alcoholism, cocaine use disorder, and smoking withdrawal.[2] However, development for these indications was discontinued.[2]
See also
κ-Opioid receptor § Antagonists
SCHEME

SYNTHESIS
WO/2024/178082COMPOSITION OF OPIOID RECEPTOR MODULATOR AND MDMA FOR USE THEREOF
WO/2024/173843QUINOLINE DERIVATIVES WHICH ACT AS KAPPA-OPIOID RECEPTOR ANTAGONISTS
20240238245COMPOSITIONS AND METHODS FOR THE TREATMENT OF DEPRESSION
20240189274Compositions And Methods For The Treatment Of Depression
WO/2024/102802ZELATRIAZIN FOR THE TREATMENT OF DEPRESSION
WO/2024/100285TREATMENT OF A COGNITIVE DISORDER WITH AN AGENT THAT INCREASES THE..
117615757Compositions and methods for treating depression
117142999Racemization method of drug intermediate
20230348377PURE FORMS OF CRYSTALLINE ATICAPRANT
WO/2023/170550POLYMORPH FORMS OF ATICAPRANT FOR USE IN TREATING MAJOR DEPRESSIVE DISORDER
WO/2023/170547PURE FORMS OF CRYSTALLINE ATICAPRANT
20230277499Forms of aticaprant
20230277500COMPOSITIONS COMPRISING ATICAPRANT
WO/2023/164385NEUROACTIVE STEROIDS FOR TREATMENT OF GASTROINTESTINAL DISEASES OR CONDITIONS
20090186873Kappa selective opioid receptor antagonist
WO/2009/094260KAPPA SELECTIVE OPIOID RECEPTOR ANTAGONIST
20100197669Kappa selective opioid receptor antagonist
2252581KAPPA SELECTIVE OPIOID RECEPTOR ANTAGONIST
201500053151-substituted 4-arylpiperazine as kappa opioid receptor antagonists
WO/2013/0864961-SUBSTITUTED 4-ARYLPIPERAZINE AS KAPPA OPIOID RECEPTOR ANTAGONISTS
101925576Kappa selective opioid receptor antagonist
PAPERS
ACS Omega (2020), 5(41), 26938-26945 https://pubs.acs.org/doi/full/10.1021/acsomega.0c04329


REF https://pubs.acs.org/doi/suppl/10.1021/acsomega.0c04329/suppl_file/ao0c04329_si_001.pdf
N-Methoxy-N-methyl-4-chlorobutyramide (S1). To a mixture of N,O-dimethylhydroxylamine hydrochloride (95.0 mmol, 9.27 g) in CH2Cl2 (150 mL) was
added 2 M NaOH (300 mmol, 150 mL) and 4-chlorobutyryl chloride (100 mmol,
11.2 mL) at 0 ˚C. The mixture was stirred for 42 h at room temperature. The
organic phase was separated, and the aqueous phase was extracted with CH2Cl2 (2 × 50 mL). The combined organic phase was washed with 2 M NaOH (100 mL), dried over Na2SO4, filtered, and concentrated
to afford the title comlund in 75% yield as a colorless liquid.
1H NMR (400 MHz, CDCl3) : 2.08-2.15
(m, 2H), 2.63 (t, J = 7.0 Hz, 2H), 3.19 (s, 3H), 3.64 (t, J = 6.3 Hz, 2H), 3.71 (s, 3H).
13C{
1H} NMR (100
MHz, CDCl3) : 27.1, 28.6, 32.1, 44.6, 61.1. IR (max/cm-1
): 2965, 2940, 2821, 1656, 14421, 1417, 1387,
1178, 1107, 997. HRMS (ESI+): calculated for [M+Na]+
: 188.0449, found: 188.0450.
4-Chloro-1-(3,5-dimethylphenyl)butan-1-one (S2). To a mixture of N-methoxy-N-methyl-4-chlorobutyramide (S1, 65.0 mmol, 10.8 g) in anhydrous Et2O
(100 mL) was added dropwise 3,5-dimethylphenylmagnesium bromide (ca. 1 M
in Et2O, ca. 130 mmol, prepared from 1-bromo-3,5-dimethylbenzene (130 mmol,
17.7 mL) and Mg turnings (169 mmol, 4.11 g) in anhydrous Et2O (130 mL)) over 1 h at -40 ˚C under Ar.
The reaction mixture was stirred at room temperature for 20 h. After cooling to 0 ˚C, saturated NH4Cl
solution (200 mL) was added. The organic phase was separated, washed with water (100 mL) and brine
(100 mL), dried over Na2SO4, and filtered. After concentration, the residue was purified by column chromatography (silica gel, hexane/EtOAc as eluent) to afford the title compound in 91% yield as a greenish
yellow liquid.
1H NMR (400 MHz, CDCl3) : 2.18-2.25 (m, 2H), 2.38 (s, 6H), 3.15 (t, J = 7.0 Hz, 2H),
3.67 (t, J = 6.3 Hz, 2H), 7.21 (s, 1H), 7.58 (s, 2H). 13C{
1H} NMR (100 MHz, CDCl3) : 21.2, 26.8, 35.4,
44.7, 125.8, 134.8, 136.8, 138.3, 199.4. IR (max/cm-1
): 3047, 3006, 2961, 2920, 2868, 1443, 1411, 1322,
1303, 1181, 1159, 844, 785, 687. HRMS (APCI+): calculated for [M+H]+
: 211.0884, found: 211.0884.
(RS)-N-(4-Chloro-1-(3,5-dimethylphenyl)butylidene)-tertbutanesulfinamide (S3). Ti(OEt)4 (100 mol, 21.0 mL) was added to a mixture
of (RS)-tert-butanesulfinamide (1.0 M in THF, 50 mmol, 50 mL) and 4-chloro1-(3,5-dimethylphenyl)butan-1-one (S2, 50.0 mmol, 10.5 g) under N2. The mixture was refluxed for 48 h. After cooling to room temperature, brine (100 mL)
was added, and the resulting mixture was filtered over Celite using EtOAc (ca.
300 mL). The organic was separated, dried over Na2SO4, and filtered. After concentration under reduced
pressure, the residue was purified by column chromatography (silica gel, hexane/EtOAc as eluent) to
afford the title compound in 57% yield as a brown viscous liquid.
1H NMR (400 MHz, CDCl3) : 1.33
(s, 9H), 2.10-2.22 (m, 2H), 2.36 (s, 6H), 3.27 (s, 1H), 3.43 (s, 1H), 3.64 (t, J = 6.5 Hz, 2H), 7.13 (s, 1H),
7.47 (s, 2H).
13C{
1H} NMR (100 MHz, CDCl3) : 21.3, 22.7, 30.2, 31.6, 44.7, 57.7, 125.2, 133.4, 137.6,
138.2, 178.6. IR (max/cm-1
): 3046, 2958, 2922, 2866, 1599, 1577, 1455, 1361, 1320, 1308, 1069, 856.
HRMS (ESI+): calculated for [M+H]
+
: 314.1340, found: 314.1344. []D
20 +11.0 (c = 1.01, CH2Cl2).
(RS,S)-1-tert-Butylsulfinyl-2-(3,5-dimethylphenyl)pyrrolidine (S4). To a solution of (RS)-N-(4-chloro-1-(3,5-dimethylphenyl)butylidene)-tert-butanesulfinamide
(S3, 25.6 mmol, 8.06 g) in anhydrous THF (100 mL) at -78 °C was added LiBEt3H
(28 mmol, 0.5 M in THF, 28.2 mL) under Ar. The reaction was stirred at -78 °C for
1 h, subsequently allowed to warm up to room temperature and stirred for additional
20 h. Saturated NaHCO3 solution (80 mL) was slowly added. The mixture was filtered and extracted
with EtOAc (3 × 100 mL). The combined organic phase was dried over Na2SO4 and filtered. After
concentration, the residue was purified by column chromatography (silica gel, hexane/EtOAc as eluent)
to afford the title compound in 72% yield as pale yellow solid. mp.: 56 ˚C. 1H NMR (400 MHz, CDCl3)
: 1.12 (s, 9H), 1.74-1.90 (m, 3H), 1.93-2.02 (m, 1H), 2.18-2.27 (m, 1H), 2.30 (s, 6H), 2.94-3.02 (m, 1H),
3.85-3.91 (m, 1H), 4.55-4.59 (m, 1H), 6.88 (s, 1H), 6.90 (s, 2H).
13C{
1H} NMR (100 MHz, CDCl3) :
21.3, 23.8, 26.3, 36.0, 42.1, 57.2, 69.2, 125.0, 128.7, 137.7, 143.2. IR (max/cm-1
): 3023, 2957, 2920,
2866, 1607, 1471, 1360, 1061, 957, 847. HRMS (ESI+): calculated for [M+Na]+
: 302.1549, found:
302.1548. []D
20
-137 (c = 0.49, CH2Cl2)
(S)-2-(3,5-Dimethylphenyl)pyrrolidine hydrochloride (1j•HCl). To a solution
of (RS,S)-1-tert-butylsulfinyl-2-(3,5-dimethylphenyl)pyrrolidine (S4, 14.7 mmol,
4.12 g) in dioxane (250 mL) was added dropwise HCl (ca. 150 mmol, 4 M in dioxane, 38 mL). The mixture was stirred for 1 h at room temperature under N2, and
then the mixture was concentrated under reduced pressure. Then, Et2O (200 mL) was added to the residue
and the mixture was cooled to 0 ˚C. The precipitate was collected by filtration, washed with Et2O (40
mL), and dried under reduced pressure to afford the title compound in 94% yield as white solid. mp.: 198
˚C. 1H NMR (400 MHz, D2O) : 2.00-2.15 (m, 3H), 2.18 (s, 6H), 2.27-2.35 (m, 1H), 3.27-3.36 (m, 2H),
4.45 (t, J = 8.0 Hz, 1H), 6.97 (s, 2H), 7.01 (s, 1H). 13C{
1H} NMR (100 MHz, D2O) : 20.9, 24.19, 30.9,
46.0, 63.8, 119.79, 125.6, 131.4, 135.3, 140.1. IR (max/cm-1
): 3033, 3012, 2970, 2855, 2743, 2571, 2480,
1608, 1590, 1414, 850. HRMS (ESI+): calculated for [M-Cl]
+
: 176.1434, found: 176.1435. []D
20 +7.1
(c = 1.01, MeOH).
(S)-2-(3,5-Dimethylphenyl)pyrrolidine (1j). To a suspension of (S)-2-(3,5-dimethylphenyl)pyrrolidine hydrochloride (1j•HCl, 13.5 mmol, 2.86 g) in anhydrous Et2O
(200 mL) was added a saturated solution of NaHCO3 (200 mL). The resulting mixture
was stirred for 20 min at room temperature. The organic was separated and the aqueous
phase was extracted with Et2O (2 × 100 mL). The combined organic phase was dried over MgSO4 and
filtered. The solvent was removed under reduced pressure to afford the title compound as a pale yellow
liquid in 99% yield.
1H NMR (400 MHz, CDCl3) : 1.60-1.71 (m, 1H), 1.78-1.96 (m, 2H), 1.98 (s, 1H),
2.11-2.19 (m, 1H), 2.30 (s, 6H), 2.95-3.02 (m, 1H), 3.17-3.23 (m, 1H), 4.03 (t, J = 7.7 Hz, 1H), 6.87 (s,
1H), 6.97 (s, 2H). 13C{
1H} NMR (100 MHz, CDCl3) : 21.3, 25.5, 34.2, 46.9, 62.6, 124.2, 128.4, 137.8,
144.7. IR (max/cm-1
): 3332, 3010, 2960, 2915, 2869, 1605, 1458, 1101, 845. HRMS (ESI+): calculated
for [M+H]+
: 176.1434, found: 176.1436. []D
20
-30.5 (c = 1.01, MeOH). Chiral HPLC (ChiralPak ODH, 4.6 mm × L 250 mm, hexane:2-propanol = 90:10, 0.5 mL/min, = 254 nm): tR/min = 18.7 (1%),
19.8 (99%).

3-Fluoro-4-(4-formylphenoxy)benzonitrile2
(S5). A mixture of 3,4-
difluorobenzonitrile (35.0 mmol, 4.87 g), 4-hydroxybenzaldehyde (35.0
mmol, 4.27 g), and K2CO3 (70.0 mmol, 9.67 g) in N,N-dimethylacetamide
(90 mL) was stirred at 100 ˚C for 2 h under N2. After cooling, the reaction
mixture was poured into ice water. White precipitate was collected by filtration, washed with water, and dried under reduced pressure to afford the title compound as pale yellow
solid in 82% yield. mp.: 101 ˚C. 1H NMR (400 MHz, CDCl3) : 7.11-7.15 (m, 2H), 7.20 (t, J = 8.2 Hz,
1H), 7.49-7.51 (m, 1H), 7.54 (dd, J = 9.7, 1.9 Hz, 1H), 7.91-7.94 (m, 2H), 9.98 (s, 1H).
13C{
1H} NMR
(100 MHz, CDCl3) : 109.1 (d, 3
JC-F = 8.2 Hz), 117.1 (d, 4
JC-F = 2.5 Hz), 117.9, 121.3 (d, 2
JC-F = 21.3 Hz),
122.5 (d, 4
JC-F = 1.6 Hz), 129.6 (d, 3
JC-F = 4.1 Hz), 132.1, 132.7, 147.0 (d, 2
JC-F = 11.5 Hz), 153.6 (d, 1
JCF = 254.8 Hz), 160.7, 190.4. IR (max/cm-1
): 3100, 3060, 2846, 2812, 2761, 2232, 1697, 1687, 1585, 1497,
1277, 1216, 1166, 1114, 836. HRMS (APCI+): calculated for [M+H]+
: 242.0612, found: 242.0616.
3-Fluoro-4-(4-formylphenoxy)benzamide2
(2f). To a mixture of 3-
fluoro-4-(4-formylphenoxy)benzonitrile (S5, 26.0 mmol, 6.27 g) and
K2CO3 (13.0 mmol, 1.80 g) in DMSO (24 mL) was added dropwise 35%
H2O2 (ca. 29 mmol, 3.1 mL) at 10 ˚C over 5 min. The reaction mixture
was stirred at room temperature for 2 h. The reaction mixture was
poured into ice water. White precipitate was collected by filtration, washed with water, and dried under
reduced pressure to afford the title compound as white solid in 92% yield. mp. 129 ˚C. 1H NMR (400
MHz, (D3C)2SO) : 9.96 (s, 1H), 8.12 (s, 1H), 7.96 (d, J = 8.2 Hz, 2H), 7.93 (dd, J = 1.9, 10.0 Hz, 1H),
7.85-7.82 (m, 1H), 7.58 (s, 1H), 7.42 (t, J = 8.2 Hz, 1 H), 7.20 (d, J = 8.2 Hz, 2H).
13C{
1H} NMR (100
MHz, (D3C)2SO) : 116.6 (d, 2
JC-F = 19.7 Hz), 116.9, 122.6, 125.1 (d, 4
JC-F = 3.3 Hz), 131.9 (d, 2
JC-F =
21.3 Hz), 132.1, 132.7 (d, 3
JC-F = 5.7 Hz), 143.7 (d, 3
JC-F = 12.3 Hz), 153.1 (d, 1
JC-F = 248.2 Hz), 161.3,
165.8, 191.5. IR (max/cm-1
): 3356, 3185, 2844, 1668, 1598, 1504, 1433, 1382, 1269, 1218, 1156, 1128,
- HRMS (ESI+): calculated for [M+Na]
+
: 282.0537, found: 282.0541. HRMS (APCI+): calculated
for [M+H]+
: 260.0717, found: 260.0716.
NEXT
Reaction Chemistry & Engineering (2022), 7(8), 1779-1785
Journal of Medicinal Chemistry (2011), 54(23), 8000-8012
| Clinical data | |
|---|---|
| Other names | JNJ-67953964; CERC-501; LY-2456302 |
| Routes of administration | By mouth[1] |
| Pharmacokinetic data | |
| Bioavailability | 25%[1] |
| Elimination half-life | 30–40 hours[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1174130-61-0 |
| PubChem CID | 44129648 |
| IUPHAR/BPS | 9194 |
| DrugBank | DB12341 |
| ChemSpider | 28424203 |
| UNII | DE4G8X55F5 |
| KEGG | D11831 |
| ChEMBL | ChEMBL1921847 |
| CompTox Dashboard (EPA) | DTXSID90151777 |
| Chemical and physical data | |
| Formula | C26H27FN2O2 |
| Molar mass | 418.512 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
References
^ Jump up to:a b c d e f g h i Li W, Sun H, Chen H, Yang X, Xiao L, Liu R, et al. (2016). “Major Depressive Disorder and Kappa Opioid Receptor Antagonists”. Translational Perioperative and Pain Medicine. 1 (2): 4–16. PMC 4871611. PMID 27213169.
- ^ Jump up to:a b c d e f g h “CERC 501”. Adis Insight. 30 January 2018.
- ^ Jump up to:a b Browne CA, Wulf H, Lucki I (2022). “Kappa Opioid Receptors in the Pathology and Treatment of Major Depressive Disorder”. In Liu-Chen LY, Inan S (eds.). The Kappa Opioid Receptor. Handbook of Experimental Pharmacology. Vol. 271. pp. 493–524. doi:10.1007/164_2020_432. ISBN 978-3-030-89073-5. PMID 33580854. S2CID 231908782.
- ^ Jump up to:a b c Reed B, Butelman ER, Kreek MJ (2022). “Kappa Opioid Receptor Antagonists as Potential Therapeutics for Mood and Substance Use Disorders”. In Liu-Chen LY, Inan S (eds.). The Kappa Opioid Receptor. Handbook of Experimental Pharmacology. Vol. 271. pp. 473–491. doi:10.1007/164_2020_401. ISBN 978-3-030-89073-5. PMID 33174064. S2CID 226305229.
- ^ Krystal AD, Pizzagalli DA, Smoski M, Mathew SJ, Nurnberger J, Lisanby SH, et al. (May 2020). “A randomized proof-of-mechanism trial applying the ‘fast-fail’ approach to evaluating κ-opioid antagonism as a treatment for anhedonia”. Nature Medicine. 26 (5): 760–768. doi:10.1038/s41591-020-0806-7. PMC 9949770. PMID 32231295. S2CID 256839849.
- ^ Dhir A (January 2017). “Investigational drugs for treating major depressive disorder”. Expert Opinion on Investigational Drugs. 26 (1): 9–24. doi:10.1080/13543784.2017.1267727. PMID 27960559. S2CID 45232796.
- ^ Missig G, Fritsch EL, Mehta N, Damon ME, Jarrell EM, Bartlett AA, et al. (January 2022). “Blockade of kappa-opioid receptors amplifies microglia-mediated inflammatory responses”. Pharmacology, Biochemistry, and Behavior. 212: 173301. doi:10.1016/j.pbb.2021.173301. PMC 8748402. PMID 34826432.
- ^ Rorick-Kehn LM, Witkin JM, Statnick MA, Eberle EL, McKinzie JH, Kahl SD, et al. (February 2014). “LY2456302 is a novel, potent, orally-bioavailable small molecule kappa-selective antagonist with activity in animal models predictive of efficacy in mood and addictive disorders”. Neuropharmacology. 77: 131–144. doi:10.1016/j.neuropharm.2013.09.021. PMID 24071566. S2CID 3230414.
- ^ Lowe SL, Wong CJ, Witcher J, Gonzales CR, Dickinson GL, Bell RL, et al. (September 2014). “Safety, tolerability, and pharmacokinetic evaluation of single- and multiple-ascending doses of a novel kappa opioid receptor antagonist LY2456302 and drug interaction with ethanol in healthy subjects”. Journal of Clinical Pharmacology. 54 (9): 968–978. doi:10.1002/jcph.286. PMID 24619932. S2CID 14814449.
- ^ Jump up to:a b Rorick-Kehn LM, Witcher JW, Lowe SL, Gonzales CR, Weller MA, Bell RL, et al. (October 2014). “Determining pharmacological selectivity of the kappa opioid receptor antagonist LY2456302 using pupillometry as a translational biomarker in rat and human”. The International Journal of Neuropsychopharmacology. 18 (2): pyu036. doi:10.1093/ijnp/pyu036. PMC 4368892. PMID 25637376.
- ^ Reed B, Butelman ER, Fry RS, Kimani R, Kreek MJ (March 2018). “Repeated Administration of Opra Kappa (LY2456302), a Novel, Short-Acting, Selective KOP-r Antagonist, in Persons with and without Cocaine Dependence”. Neuropsychopharmacology. 43 (4): 928. doi:10.1038/npp.2017.245. PMC 5809790. PMID 29422497.
- ^ Jump up to:a b Urbano M, Guerrero M, Rosen H, Roberts E (May 2014). “Antagonists of the kappa opioid receptor”. Bioorganic & Medicinal Chemistry Letters. 24 (9): 2021–2032. doi:10.1016/j.bmcl.2014.03.040. PMID 24690494.
- ^ Jump up to:a b “Publication Reports Human Brain Penetration and Target Engagement of Cerecor’s Oral Kappa Opioid Receptor Antagonist, CERC-501”. BusinessWire. 11 December 2015.
- ^ Jump up to:a b c d e f Naganawa M, Dickinson GL, Zheng MQ, Henry S, Vandenhende F, Witcher J, et al. (February 2016). “Receptor Occupancy of the κ-Opioid Antagonist LY2456302 Measured with Positron Emission Tomography and the Novel Radiotracer 11C-LY2795050”. The Journal of Pharmacology and Experimental Therapeutics. 356 (2): 260–266. doi:10.1124/jpet.115.229278. PMC 4727157. PMID 26628406.
- ^ Jump up to:a b Placzek MS (August 2021). “Imaging Kappa Opioid Receptors in the Living Brain with Positron Emission Tomography”. In Liu-Chen LY, Inan S (eds.). The Kappa Opioid Receptor. Handbook of Experimental Pharmacology. Vol. 271. pp. 547–577. doi:10.1007/164_2021_498. ISBN 978-3-030-89073-5. PMID 34363128. S2CID 236947969.
- ^ Zheng MQ, Nabulsi N, Kim SJ, Tomasi G, Lin SF, Mitch C, et al. (March 2013). “Synthesis and evaluation of 11C-LY2795050 as a κ-opioid receptor antagonist radiotracer for PET imaging”. Journal of Nuclear Medicine. 54 (3): 455–463. doi:10.2967/jnumed.112.109512. PMC 3775344. PMID 23353688.
- ^ Mitch CH, Quimby SJ, Diaz N, Pedregal C, de la Torre MG, Jimenez A, et al. (December 2011). “Discovery of aminobenzyloxyarylamides as κ opioid receptor selective antagonists: application to preclinical development of a κ opioid receptor antagonist receptor occupancy tracer”. Journal of Medicinal Chemistry. 54 (23): 8000–8012. doi:10.1021/jm200789r. PMID 21958337.
- ^ “WO2009094260A1 – Kappa selective opioid receptor antagonist”. Google Patents. 13 January 2009. Retrieved 29 August 2022.
- ^ “Cerecor Bolsters Clinical Pipeline with Acquisition of Phase 2-ready Kappa Opioid Receptor Antagonist from Eli Lilly and Company”. cerecor.com. February 20, 2015. Archived from the original on 2015-02-23. Retrieved March 18, 2015.
- ^ Rankovic Z, Hargreaves R, Bingham M (2012). Drug Discovery for Psychiatric Disorders. Royal Society of Chemistry. pp. 314–317. ISBN 978-1-84973-365-6.
- ^ Jump up to:a b c Bushey R (August 2017). “J&J Adds New Depression Drug to Portfolio”. Drug Discovery and Development Magazine.
- ^ “Cerecor Announces Divestiture of CERC-501 to Janssen Pharmaceuticals, Inc”. Marketwired. August 2017. Archived from the original on 2017-09-01. Retrieved 2017-09-01.
Further reading
- Carlezon WA, Krystal AD (October 2016). “Kappa-Opioid Antagonists for Psychiatric Disorders: From Bench to Clinical Trials”. Depression and Anxiety. 33 (10): 895–906. doi:10.1002/da.22500. PMC 5288841. PMID 27699938.
- Li W, Sun H, Chen H, Yang X, Xiao L, Liu R, et al. (2016). “Major Depressive Disorder and Kappa Opioid Receptor Antagonists”. Translational Perioperative and Pain Medicine. 1 (2): 4–16. PMC 4871611. PMID 27213169.
- Dhir A (January 2017). “Investigational drugs for treating major depressive disorder”. Expert Opinion on Investigational Drugs. 26 (1): 9–24. doi:10.1080/13543784.2017.1267727. PMID 27960559. S2CID 45232796.
- Reed B, Butelman ER, Kreek MJ (2017). “Endogenous opioid system in addiction and addiction-related behaviors”. Current Opinion in Behavioral Sciences. 13: 196–202. doi:10.1016/j.cobeha.2016.12.002. ISSN 2352-1546. S2CID 53149180.
- Rakesh G, Pae CU, Masand PS (August 2017). “Beyond serotonin: newer antidepressants in the future”. Expert Review of Neurotherapeutics. 17 (8): 777–790. doi:10.1080/14737175.2017.1341310. PMID 28598698. S2CID 205823807.
- Helal MA, Habib ES, Chittiboyina AG (December 2017). “Selective kappa opioid antagonists for treatment of addiction, are we there yet?”. European Journal of Medicinal Chemistry. 141: 632–647. doi:10.1016/j.ejmech.2017.10.012. PMID 29107424.
- McHugh KL, Kelly JP (2018). “Modulation of the central opioid system as an antidepressant target in rodent models”. The Opioid System as the Interface between the Brain’s Cognitive and Motivational Systems. Progress in Brain Research. Vol. 239. pp. 49–87. doi:10.1016/bs.pbr.2018.07.003. ISBN 9780444641670. PMID 30314569.
- Bailey SJ, Husbands SM (June 2018). “Targeting opioid receptor signaling in depression: do we need selective κ opioid receptor antagonists?”. Neuronal Signaling. 2 (2): NS20170145. doi:10.1042/NS20170145. PMC 7373229. PMID 32714584.
- Chavkin C (August 2018). “Kappa-opioid antagonists as stress resilience medications for the treatment of alcohol use disorders”. Neuropsychopharmacology. 43 (9): 1803–1804. doi:10.1038/s41386-018-0046-4. PMC 6046055. PMID 29752444.
- Krystal AD, Pizzagalli DA, Mathew SJ, Sanacora G, Keefe R, Song A, et al. (December 2018). “The first implementation of the NIMH FAST-FAIL approach to psychiatric drug development”. Nature Reviews. Drug Discovery. 18 (1): 82–84. doi:10.1038/nrd.2018.222. PMC 6816017. PMID 30591715.
- Lazar MA, McIntyre RS (2019). “Novel Therapeutic Targets for Major Depressive Disorder”. Neurobiology of Depression. pp. 383–400. doi:10.1016/B978-0-12-813333-0.00034-2. ISBN 9780128133330. S2CID 86782597.
- Browne CA, Lucki I (September 2019). “Targeting opioid dysregulation in depression for the development of novel therapeutics”. Pharmacology & Therapeutics. 201: 51–76. doi:10.1016/j.pharmthera.2019.04.009. PMC 6859062. PMID 31051197.
- Banks ML (2020). “The Rise and Fall of Kappa-Opioid Receptors in Drug Abuse Research”. In Nader MA, Hurd YL (eds.). Substance Use Disorders. Handbook of Experimental Pharmacology. Vol. 258. pp. 147–165. doi:10.1007/164_2019_268. ISBN 978-3-030-33678-3. PMC 7756963. PMID 31463605.
- Browne CA, Jacobson ML, Lucki I (2020). “Novel Targets to Treat Depression: Opioid-Based Therapeutics”. Harvard Review of Psychiatry. 28 (1): 40–59. doi:10.1097/HRP.0000000000000242. PMID 31913981. S2CID 210120636.
- Jacobson ML, Browne CA, Lucki I (January 2020). “Kappa Opioid Receptor Antagonists as Potential Therapeutics for Stress-Related Disorders”. Annual Review of Pharmacology and Toxicology. 60: 615–636. doi:10.1146/annurev-pharmtox-010919-023317. PMID 31914893. S2CID 210121357.
- Mercadante S, Romualdi P (2020). “The Therapeutic Potential of Novel Kappa Opioid Receptor-based Treatments”. Current Medicinal Chemistry. 27 (12): 2012–2020. doi:10.2174/0929867326666190121142459. PMID 30666905. S2CID 58558833.
External links
Aticaprant – Eli Lilly and Company/Janssen Pharmaceuticals – AdisInsight
//////ATICAPRANT, CERC-501, JSPA 0658, JSPA-0658, JSPA0658, LY 2456302, LY-2456302, LY2456302, Phase 3, ELI LILLY, Major depressive disorder, JNJ-67953964, WHO 10582

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}