
Home » Posts tagged 'Orphan Drug Designation'
Tag Archives: Orphan Drug Designation
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
IMIPRIDONE

![7-Benzyl-4-(2-methylbenzyl)-1,2,6,7,8,9-hexahydroimidazo[1,2-A]pyrido[3,4-E]pyrimidin-5(4H)-one.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=73777259&t=l)
IMIPRIDONE
CAS No. : 1616632-77-9
Molecular Weight, 386.4964
Related CAS #: 41276-02-2 (TIC10 isomer) 1616632-77-9 (free base) 1638178-82-1 (HCl) 1777785-71-3 (HBr) 2007141-57-1 (2HBr)
TIC 10, 0NC 201, OP 10
Synonym: ONC201; ONC 201; ONC-201; NSC350625; NSC-350625; NSC 350625; TIC10; TIC 10; TIC-10; TRAIL inducing compound 10; imipridone
7-benzyl-4-(2-methylbenzyl)-1,2,6,7,8,9-hexahydroimidazo[1,2-a]pyrido[3,4-e]pyrimidin-5(4H)-one
2,4,6,7,8,9-Hexahydro-4-((2-methylphenyl)methyl)-7-phenylmethyl)imidazo)(1,2-a)pyrido(3,4-e)pyrimidin-5(1H)-one
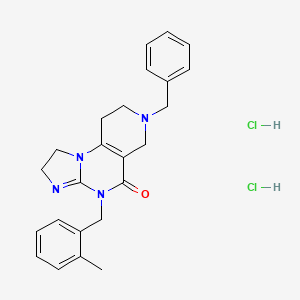
ONC-201 Dihydrochloride
459.4
UNII-53VG71J90J
53VG71J90J
Q27896336
1638178-82-1
- A TRAIL-dependent antitumor agent.
TIC10 (ONC-201) is a potent, orally active, and stable tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) inducer which acts by inhibiting Akt and ERK, consequently activating Foxo3a and significantly inducing cell surface TRAIL. TIC10 can cross the blood-brain barrier.
ONC-201, also known as TIC10, is a potent, orally active, and stable small molecule that transcriptionally induces TRAIL in a p53-independent manner and crosses the blood-brain barrier. TIC10 induces a sustained up-regulation of TRAIL in tumors and normal cells that may contribute to the demonstrable antitumor activity of TIC10. TIC10 inactivates kinases Akt and extracellular signal-regulated kinase (ERK), leading to the translocation of Foxo3a into the nucleus, where it binds to the TRAIL promoter to up-regulate gene transcription. TIC10 is an efficacious antitumor therapeutic agent that acts on tumor cells and their microenvironment to enhance the concentrations of the endogenous tumor suppressor TRAIL.
Akt/ERK Inhibitor ONC201 is a water soluble, orally bioavailable inhibitor of the serine/threonine protein kinase Akt (protein kinase B) and extracellular signal-regulated kinase (ERK), with potential antineoplastic activity. Upon administration, Akt/ERK inhibitor ONC201 binds to and inhibits the activity of Akt and ERK, which may result in inhibition of the phosphatidylinositol 3-kinase (PI3K)/Akt signal transduction pathway as well as the mitogen-activated protein kinase (MAPK)/ERK-mediated pathway. This may lead to the induction of tumor cell apoptosis mediated by tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL)/TRAIL death receptor type 5 (DR5) signaling in AKT/ERK-overexpressing tumor cells. The PI3K/Akt signaling pathway and MAPK/ERK pathway are upregulated in a variety of tumor cell types and play a key role in tumor cell proliferation, differentiation and survival by inhibiting apoptosis. In addition, ONC201 is able to cross the blood-brain barrier.
SYN
Organic & Biomolecular Chemistry, 19(39), 8497-8501; 2021
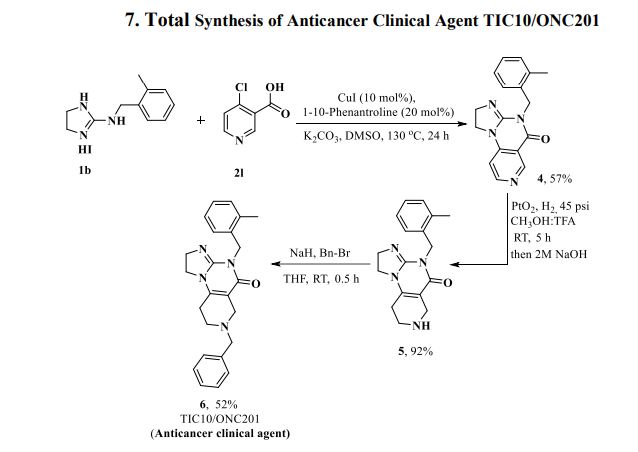
Herein, we present a copper-catalyzed tandem reaction of 2-aminoimidazolines and ortho-halo(hetero)aryl carboxylic acids that causes the regioselective formation of angularly fused tricyclic 1,2-dihydroimidazo[1,2-a]quinazolin-5(4H)-one derivatives. The reaction involved in the construction of the core six-membered pyrimidone moiety proceeded via regioselective N-arylation–condensation. The presented protocol been successfully applied to accomplish the total synthesis of TIC10/ONC201, which is an active angular isomer acting as a tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL): a sought after anticancer clinical agent.
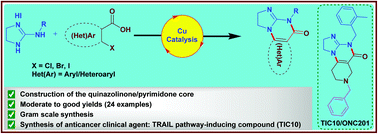

7-Benzyl-4-(2-methylbenzyl)-1,2,6,7,8,9-hexahydroimidazo[1,2-a]pyrido[3,4-e]pyrimidin-5(4H)-one (6): Pale orange semi-solid, 202 mg (0.521 mmol), 52 % Rf = 0.25 (CH3OH/CHCl3 5:95); IR 1490, 1610, 1644, 2882, 2922 cm-1 ; 1H-NMR (500 MHz, CDCl3) δ = 2.39 (s, 3H), 2.54 (t, J = 5.5 Hz, 2H), 2.72 (t, J = 5.7 Hz, 2H), 3.31 (s, 2H), 3.67 (s, 2H), 3.84-3.91 (m, 4H), 5.04 (s, 2H), 7.02-7.04 (m, 1H), 7.08-7.12 (m, 3H), 7.26- 7.34 (m, 5H). 13C{1H}-NMR (101 MHz, CDCl3) δ = 19.3, 26.8, 43.4, 46.9, 48.2, 49.6, 50.45, 62.3, 102.1, 125.2, 125.9, 126.8, 127.4, 128.45, 129.2, 130.2, 134.2, 135.6, 137.9, 145.7, 153.3, 161.4; MS (ESI, m/z): [M+H]+ 387; HRMS (ESI, m/z): calcd for C24H27N4O [M+H]+ found 387.2183.


PATENT
https://patents.google.com/patent/WO2017132661A2/en
Scheme 1.


Scheme 2.



NEW DRUG APPROVALS
ONE TIME
$10.00
//////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
CLIP
https://mdanderson.elsevierpure.com/en/publications/discovery-and-clinical-introduction-of-first-in-class-imipridone-Discovery and clinical introduction of first-in-class imipridone ONC201
Abstract
ONC201 is the founding member of a novel class of anti-cancer compounds called imipridones that is currently in Phase II clinical trials in multiple advanced cancers. Since the discovery of ONC201 as a p53-independent inducer of TRAIL gene transcription, preclinical studies have determined that ONC201 has anti-proliferative and pro-apoptotic effects against a broad range of tumor cells but not normal cells. The mechanism of action of ONC201 involves engagement of PERK-independent activation of the integrated stress response, leading to tumor upregulation of DR5 and dual Akt/ERK inactivation, and consequent Foxo3a activation leading to upregulation of the death ligand TRAIL. ONC201 is orally active with infrequent dosing in animals models, causes sustained pharmacodynamic effects, and is not genotoxic. The first-in-human clinical trial of ONC201 in advanced aggressive refractory solid tumors confirmed that ONC201 is exceptionally well-tolerated and established the recommended phase II dose of 625 mg administered orally every three weeks defined by drug exposure comparable to efficacious levels in preclinical models. Clinical trials are evaluating the single agent efficacy of ONC201 in multiple solid tumors and hematological malignancies and exploring alternative dosing regimens. In addition, chemical analogs that have shown promise in other oncology indications are in pre-clinical development. In summary, the imipridone family that comprises ONC201 and its chemical analogs represent a new class of anti-cancer therapy with a unique mechanism of action being translated in ongoing clinical trials.
////////////IMIPRIDONE, TIC 10, ONC 201, NSC 350625, OP 10, Fast Track Designation, Orphan Drug Designation, Rare Pediatric Disease Designation, PHASE 3, GLIOMA, CHIMERIX
O=C1N(CC2=CC=CC=C2C)C3=NCCN3C4=C1CN(CC5=CC=CC=C5)CC4
Tebentafusp-tebn

Tebentafusp-tebn
- IMCGP100
UNIIN658GY6L3E
CAS number1874157-95-5
FDA APPROVED 1/25/2022, Kimmtrak, To treat unresectable or metastatic uveal melanoma
Immunocore Limited
- T cell receptor α chain (synthetic human) fusion protein with T cell receptor β chain (synthetic human) fusion protein with immunoglobulin, anti-(human CD3 antigen) (synthetic scFv fragment)
- Protein Sequence
- Sequence Length: 695, 500, 195
Sequence:
1AIQMTQSPSS LSASVGDRVT ITCRASQDIR NYLNWYQQKP GKAPKLLIYY51TSRLESGVPS RFSGSGSGTD YTLTISSLQP EDFATYYCQQ GNTLPWTFGQ101GTKVEIKGGG GSGGGGSGGG GSGGGGSGGG SEVQLVESGG GLVQPGGSLR151LSCAASGYSF TGYTMNWVRQ APGKGLEWVA LINPYKGVST YNQKFKDRFT201ISVDKSKNTA YLQMNSLRAE DTAVYYCARS GYYGDSDWYF DVWGQGTLVT251VSSGGGGSDG GITQSPKYLF RKEGQNVTLS CEQNLNHDAM YWYRQDPGQG301LRLIYYSWAQ GDFQKGDIAE GYSVSREKKE SFPLTVTSAQ KNPTAFYLCA351SSWGAPYEQY FGPGTRLTVT EDLKNVFPPE VAVFEPSEAE ISHTQKATLV401CLATGFYPDH VELSWWVNGK EVHSGVCTDP QPLKEQPALN DSRYALSSRL451RVSATFWQDP RNHFRCQVQF YGLSENDEWT QDRAKPVTQI VSAEAWGRAD
Sequence:
1AQQGEEDPQA LSIQEGENAT MNCSYKTSIN NLQWYRQNSG RGLVHLILIR51SNEREKHSGR LRVTLDTSKK SSSLLITASR AADTASYFCA TDGSTPMQFG101KGTRLSVIAN IQKPDPAVYQ LRDSKSSDKS VCLFTDFDSQ TNVSQSKDSD151VYITDKCVLD MRSMDFKSNS AVAWSNKSDF ACANAFNNSI IPEDT
Sequence Modifications
| Type | Location | Description |
|---|---|---|
| bridge | Cys-23 – Cys-88 | disulfide bridge |
| bridge | Cys-153 – Cys-227 | disulfide bridge |
| bridge | Cys-281 – Cys-349 | disulfide bridge |
| bridge | Cys-401 – Cys-466 | disulfide bridge |
| bridge | Cys-427 – Cys-157′ | disulfide bridge |
| bridge | Cys-23′ – Cys-89′ | disulfide bridge |
| bridge | Cys-132′ – Cys-182′ | disulfide bridge |
Tebentafusp, sold under the brand name Kimmtrak, is an anti-cancer medication used to treat uveal melanoma (eye cancer).[1][2]
The most common side effects include cytokine release syndrome, rash, pyrexia (fever), pruritus (itching), fatigue, nausea, chills, abdominal pain, edema, hypotension, dry skin, headache, and vomiting.[1][2]
Tebentafusp is a bispecific gp100 peptide-HLA-directed CD3 T cell engager.[1][2] It was approved for medical use in the United States in January 2022.[1][2]
Tebentafusp is a bispecific gp100 peptide-HLA-directed CD3 T cell engager used to treat unresectable or metastatic uveal melanoma.
Tebentafusp is a gp100 peptide-HLA-directed CD3 T cell engager.5 It is a bispecific, fusion protein and first-in-class drug of immune-mobilizing monoclonal T cell receptors against cancer (ImmTACs), a recently developed cancer immunotherapy with a novel mechanism of action. ImmTACs bind to target cancer cells that express a specific antigen of interest and recruit cytotoxic T cells to lyse the cells, such as melanocytes.1,2
Uveal melanoma is a rare ocular tumour with often poor prognosis and limited treatment options. Even after surgical ablation or removal of the ocular tumour, almost 50% of patients with uveal melanoma develop metastatic disease.1 On January 26, 2022, tebentafusp was first approved by the FDA for the treatment of HLA-A*02:01-positive adults with unresectable or metastatic uveal melanoma. This approval marks the first bispecific T cell engager to be approved by the FDA to treat a solid tumour and being the first and only therapy for the treatment of unresectable or metastatic uveal melanoma to be approved by the FDA.5
FDA approves tebentafusp-tebn for unresectable or metastatic uveal melanoma
On January 25, 2022, the Food and Drug Administration approved tebentafusp-tebn (Kimmtrak, Immunocore Limited), a bispecific gp100 peptide-HLA-directed CD3 T cell engager, for HLA-A*02:01-positive adult patients with unresectable or metastatic uveal melanoma.
Efficacy was evaluated in IMCgp100-202 (NCT03070392), a randomized, open-label, multicenter trial of 378 patients with metastatic uveal melanoma. Patients were required to be HLA-A*02:01 genotype positive identified by a central assay. Patients were excluded if prior systemic therapy or localized liver-directed therapy were administered. Prior surgical resection of oligometastatic disease was permitted. Patients with clinically significant cardiac disease or symptomatic, untreated brain metastases were excluded.
Patients were randomized (2:1) to receive tebentafusp-tebn (N=252) or investigator’s choice (N=126) of either pembrolizumab, ipilimumab, or dacarbazine. Tebentafusp-tebn was administered weekly by intravenous infusion at 20 mcg on day 1, 30 mcg on day 8, 68 mcg on day 15 and every subsequent week until disease progression or unacceptable toxicity. The main efficacy outcome measure was overall survival (OS). An additional efficacy outcome was investigator-assessed progression-free survival (PFS) per RECIST 1.1. Median OS was 21.7 months (95% CI: 18.6, 28.6) for patients treated with tebentafusp-tebn and 16 months (95% CI: 9.7, 18.4) in the investigator’s choice arm (HR=0.51, 95% CI: 0.37, 0.71, p<0.0001) PFS was 3.3 months (95% CI: 3, 5) for those receiving tebentafusp-tebn and 2.9 months (95% CI: 2.8, 3) in the investigator’s choice arm (HR=0.73, 95% CI: 0.58, 0.94, p=0.0139).
The most common adverse reactions (≥30%) were cytokine release syndrome, rash, pyrexia, pruritus, fatigue, nausea, chills, abdominal pain, edema, hypotension, dry skin, headache, and vomiting. The most common laboratory abnormalities (≥50%) were decreased lymphocyte count, increased creatinine, increased glucose, increased aspartate aminotransferase, increased alanine aminotransferase, decreased hemoglobin, and decreased phosphate.
The recommended tebentafusp-tebn dose administered intravenously is:
- 20 mcg on day 1,
- 30 mcg on day 8,
- 68 mcg on day 15, and
- 68 mcg once weekly thereafter.
View full prescribing information for Kimmtrak.
This review was conducted under Project Orbis, an initiative of the FDA Oncology Center of Excellence. Project Orbis provides a framework for concurrent submission and review of oncology drugs among international partners. For this review, FDA collaborated with the Australian Therapeutic Goods Administration (TGA), Health Canada, and the United Kingdom’s Medicines and Healthcare product Regulatory Agency (MHRA). The application reviews may be ongoing at the other regulatory agencies.
This review used the Real-Time Oncology Review (RTOR) pilot program, which streamlined data submission prior to the filing of the entire clinical application, and the Assessment Aid, a voluntary submission from the applicant to facilitate the FDA’s assessment.
This application was granted priority review, breakthrough designation and orphan drug designation. A description of FDA expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.
//////////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
| Clinical data | |
|---|---|
| Trade names | Kimmtrak |
| Other names | IMCgp100, tebentafusp-tebn |
| License data | US DailyMed: Tebentafusp |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1][2] |
| Identifiers | |
| CAS Number | 1874157-95-5 |
| DrugBank | DB15283 |
| UNII | N658GY6L3E |
Medical uses
Tebentafusp is indicated for HLA-A*02:01-positive adults with unresectable or metastatic uveal melanoma.[1][2]
History
Efficacy was evaluated in IMCgp100-202 (NCT03070392), a randomized, open-label, multicenter trial of 378 participants with metastatic uveal melanoma.[2] Participants were required to be HLA-A*02:01 genotype positive identified by a central assay.[2] Participants were excluded if prior systemic therapy or localized liver-directed therapy were administered.[2] Prior surgical resection of oligometastatic disease was permitted.[2] Participants with clinically significant cardiac disease or symptomatic, untreated brain metastases were excluded.[2]
The U.S. Food and Drug Administration (FDA) granted Immunocore‘s application for tebentafusp priority review, breakthrough therapy, and orphan drug designations.[2]
References
- ^ Jump up to:a b c d e f https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/761228s000lbl.pdf
- ^ Jump up to:a b c d e f g h i j k l “FDA approves tebentafusp-tebn for unresectable”. U.S. Food and Drug Administration (FDA). 25 January 2022. Retrieved 28 January 2022.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain.
External links
- “Tebentafusp”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03070392 for “Safety and Efficacy of IMCgp100 Versus Investigator Choice in Advanced Uveal Melanoma” at ClinicalTrials.gov
/////////////////Tebentafusp-tebn, Kimmtrak, priority review, breakthrough designation, orphan drug designation, Immunocore Limited, IMCGP100, APPROVALS 2022, FDA 2022

NEW DRUG APPROVALS
ONE TIME
$10.00
ZY 19489, MMV 253
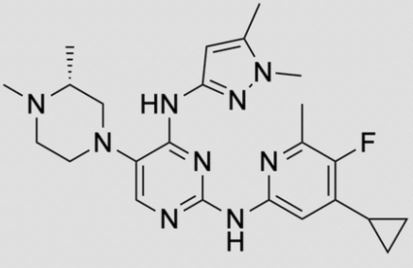

![2-N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-5-[(3R)-3,4-dimethylpiperazin-1-yl]-4-N-(1,5-dimethylpyrazol-3-yl)pyrimidine-2,4-diamine.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=92045019&t=l)
ZY 19489, MMV 253
C24 H32 FN9, 465.5
CAS 1821293-40-6
MMV253, GTPL10024, MMV674253
N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-5-((3R)-2-((1,5-dimethyl-1H-pyrazol-3-yl)amino)-3,4-dimethylpiperazin-1-yl)pyrimidin-2-amine
2-N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-5-[(3R)-3,4-dimethylpiperazin-1-yl]-4-N-(1,5-dimethylpyrazol-3-yl)pyrimidine-2,4-diamine
- N2-(4-Cyclopropyl-5-fluoro-6-methyl-2-pyridinyl)-5-[(3R)-3,4-dimethyl-1-piperazinyl]-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-2,4-pyrimidinediamine
- (R)-N2-(4-Cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-1-yl)pyrimidine-2,4-diamine

SYN
IN 201721031453
The invention relates to triaminopyrimidine compd. of formula I, pharmaceutically acceptable salts thereof, hydrates, solvates, polymorphs, optically active forms thereof, in solid state forms useful for preventing or treating malaria. The invention also relates to a process for prepn. of triaminopyrimidine compd. and intermediates thereof. Compd. I was prepd. by condensation of 5-bromouracil with tert-Bu (R)-2-methylpiperazine-1-carboxylate to give tert-Bu (R)-4-(2,4-dichloropyrimidin-5-yl)-2-methylpiperazine-1-carboxylate, which underwent chlorination followed by condensation with 1,5-dimethyl-1H-pyrazol-3-amine followed by condensation with 4-cyclopropyl-5-fluoro-6-methylpyridin-2-amine hydrochloride to give (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-5-(3-methylpiperazin-1-yl)pyrimidine-2,4-diamine, which underwent Boc-deprotection followed by methylation to give I.
SYN
WO 2019049021
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019049021
Malaria is caused by protozoan parasites of the genus Plasmodium that infect and destroy red blood cells, leading to fever, severe anemia, cerebral malaria and, if untreated, death.
International (PCT) Publication No. WO 2015/165660 (the WO ‘660) discloses triaminopyrimidine compounds, intermediates, pharmaceutical compositions and methods for use for preventing or treating malaria. The WO ‘660 discloses a process for preparation of 4-cyclopropyl-5-fluoro-6-methylpyridin-2-amine (compound 5) as depicted in scheme-1.
Scheme 1
WO ‘660 discloses a process for preparation of triaminopyrimidine compounds depicted in scheme-2.

WO ‘660 discloses the preparation of compounds 8 and 4 by using microwave technique using Biotage microwave vial. WO ‘660 in example- 13, discloses the isolation of compound 1 by concentration of reaction mixture to obtain crude product, which was purified through reverse phase HPLC GILSON instrument to obtain pure solid compound 1 in 40.8% yield, without providing the purity of the solid compound 1. The process disclosed in WO ‘660 is not industrially advantageous as it requires microwave conditions as well as chromatographic purification and provides compound 1 with lower yields. The compound 1 prepared may not be suitable for pharmaceutical preparations based on various regulatory requirements.
Polymorphism, the occurrence of different crystalline forms, is a property of some molecules. A single molecule can exist in different crystalline forms having distinct physical properties like melting point, thermal behaviors (e.g. measured by thermogravimetric analysis – TGA, or different scanning calorimetry – DSC, Powder x-ray diffraction pattern – PXRD, infrared absorption – IR). One or more these techniques may be used to distinguish different polymorphic forms of a compound.
Different salts and solid states (e.g. solvates, hydrates) of an active pharmaceutical ingredient may possess different physio-chemical properties. Such variation in the properties of different salts and solid states forms may provide a basis for improving formulation, for example, by facilitating better processing or handling characteristics, changing the dissolution profile in a favorable direction, or improving stability (both chemical and polymorph) and shelf-life. These variations in the properties of different salts and solid states forms may offer improvements to the final dosage form for example, to improve bioavailability. Different salts and solid state forms of an active pharmaceutical ingredient may also give rise to a variety of polymorphs or crystalline forms or amorphous form, which may in turn provide additional opportunities to assess variations in the properties and characteristics of an active pharmaceutical ingredient.
In view of the above, the present invention provides a process for the preparation of triaminopyrimidine compound 1 or pharmaceutically acceptable salts thereof or hydrates or solvates or polymorphs or optically active forms thereof, which is industrially scalable, environment friendly and efficient so as to obtain compounds of the invention in higher yields and purity.
The process for the preparation of triaminopyrimidine compound 1 or intermediates thereof of the present invention, takes the advantage by using appropriate solvent systems and isolation techniques as well as purification techniques, thereby to overcome problems of lower yields, chromatography purifications and microwave reactions of the prior art.
SUMMARY OF THE INVENTION
The present invention provides solid state forms of triaminopyrimidine compound
1,
1
Examples: Preparation of Intermediates
Example-1: Preparation of 6-chloro-4-cyclopropyl-3-fluoro-2-methylpyridine
In a 250 mL 4N round bottom flask, process water (30 ml) and cyclopropanecarboxylic acid (14.19 g, 164.88 mmol) were added at 25 to 35°C and started stirring. Sulphuric acid (4.4 ml, 82.44 mmol) was charged to the reaction mixture. Silver nitrate (4.18 g, 24.73 mmol), 6-Chloro-3-fluoro-2-methylpyridine (6 g, 41.22 mmol) were charged to the reaction mixture. Aqueous solution of ammonium persulphate (65.85 g, 288.54 mmol in 90 mL water) was added to the reaction mixture in 30 to 60 min at temperature NMT 60 °C. After the completion of the reaction as monitored by HPLC, toluene (30 ml) was added to the reaction mixture and stirred for 15 min. The reaction mixture filtered, separated layers from filtrate and extracted aqueous layer using toluene (30 mL). The organic layer was washed with aqueous sodium carbonate solution (30 mL) and water. The organic layer was distilled completely under vacuum at 60 °C to obtain 3.37 g syrupy mass as titled compound.
Example-2: Preparation of 6-chloro-4-cyclopropyl-3-fluoro-2-methylpyridine
In a suitable glass assembly, process water (7.5 L) and cyclopropanecarboxylic acid (3.55 Kg, 41.24 mol) were added at 25 to 35 °C and stirred. Sulphuric acid (2.02 Kg, 20.59 mol), silver nitrate (1.05 Kg, 6.21 mol), 6-chloro-3-fluoro-2-methylpyridine (1.5 Kg, 10.3 mol) were added to the reaction mixture. Aqueous solution of ammonium persulphate (16.46 g, 72.13 mmol in 22.5 L water) was added to the reaction mixture at 55 to 60 °C and maintained. After the completion of the reaction as monitored by HPLC, toluene (7.5 L) was added to the reaction mixture and stirred for 15 min. The reaction mixture was filtered, organic layer was separated and aqueous layer was extracted using toluene (6 L), filtered the reaction mixture and washed the solid with toluene (1.5 L). The combined organic layer was washed with 20% sodium carbonate solution (9 L) and water. The organic layer was concentrated completely under vacuum at 60 °C to obtain 880 g (86.50%) syrupy mass of titled compound.
Example-3: Preparation of N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-l,l-diphenyl-methanimine
In a 100 mL 3N round bottom flask, 6-chloro-4-cyclopropyl-3-fluoro-2-methylpyridine (2.69 g, 14.48 mmol) and toluene (30 mL) were added at 25 to 35 °C. Diphenylmethanimine (3.15 g, 17.38 mmol) was charged to the reaction mixture and stirred for 5-10 min under nitrogen purging. Racemic BINAP (270 mg, 0.43 mmol) and palladium acetate (98 mg, 0.43 mmol) were added to the reaction mixture. Sodium-ie/ -butoxide (2.78 g, 28.96 mmol) was added to the reaction mixture and heated to 100 to 110° C under nitrogen. After the completion of the reaction as monitored by HPLC, the reaction mixture was cooled to 25 to 35 °C and filtered over hyflo bed and washed with toluene. The filtrate containing titled compound was preserved for next stage of reaction.
Example-4: Preparation of N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-l,l-diphenyl-methanimine
In a suitable assembly, 6-chloro-4-cyclopropyl-3-fluoro-2-methylpyridine (880) and toluene (7.5 L) were added at 25 to 35 °C. Diphenylmethanimine (787 g, 4.34 mmol) and BOC anhydride (237 g, 1.086 mol) was added to the reaction mixture and stirred for 5-10 min under nitrogen purging. Racemic BINAP (67.6 g, 0.108 mmol) and palladium acetate (24.4 g, 0.108 mol) were added to the reaction mixture. S odium- ieri-butoxide (870 g, 9.05 mol) was added to the reaction mixture and heated to 100 to 110 °C under nitrogen. After the completion of the reaction as monitored by HPLC, the reaction mixture was cooled to 25 to 35 °C, water (6 L) was added. The reaction mixture was filtered over hyflo bed and washed with toluene. The filtrate containing titled compound was preserved for next stage of reaction.
Example-5: Preparation of 4-cyclopropyl-5-fluoro-6-methylpyridin-2-amine hydrochloride monohydrate
In a 100 mL 3N round bottom flask, N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-l,l-diphenylmethanimine in toluene as obtained in example-3 was added water (25 mL) at 25 to 35° C. The cone. HCl (3 mL) was charged to the reaction mixture and heated to 40 to 50 °C. After the completion of the reaction as monitored by HPLC, the reaction mixture was cooled to 25 to 35 °C. Layers were separated. The aqueous layer was treated with methylene dichloride and pH was adjusted to 7.5 to 8.5 using sodium carbonate solution, stirred for 15 min and layers were separated. Aqueous layer was extracted with methylene dichloride, charcoaled and acidified to pH 3 to 4 with aqueous HCl. The solvent was distilled completely and acetonitrile (9 mL) and ethyl acetate (9 mL) was added. The reaction mixture was stirred for 1 hour at 25 to 35° C. The product was filtered and washed with ethyl acetate. The product was dried at 50° C for 4 hours under vacuum to obtain 1.62 g title compound as monohydrate yellow crystalline solid having 99.51% HPLC purity.
Example-6: Preparation of 4-cyclopropyl-5-fluoro-6-methylpyridin-2-amine hydrochloride monohydrate
In a suitable glass assembly, N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-l,l-diphenylmethanimine in toluene as obtained in example-4 was added water (6 L) at 25 to 35° C. The cone. HCl (750 mL) was charged to the reaction mixture and heated to 40 to 50 °C. After the completion of the reaction as monitored by HPLC, the reaction mixture was cooled to 25 to 35 °C. Layers were separated. The aqueous layer was treated with methylene dichloride (3 L) and pH was adjusted to 7.5 to 8.5 using sodium carbonate solution, stirred for 15 min and layers were separated. Aqueous layer was extracted with methylene dichloride (3 L), charcoaled and acidified to pH 3 to 4 with aqueous HCl. The solvent was distilled completely and acetonitrile (1.5 L) and ethyl acetate (1.5 L) were added. The reaction mixture was stirred for 1 hour at 25 to 35° C. The product was filtered and washed with ethyl acetate. The product was dried at 50° C for 4 hours under vacuum to obtain 489 g (96.80%) title compound as monohydrate yellow crystalline solid having 99.51% HPLC purity. The crystalline compound is characterized by Powder x-ray diffraction pattern (FIG.5), Differential scanning calorimetry (FIG.6) and Thermogravimetric analysis (FIG.7).
Example 7: Preparation of 2,3-dibromobutanenitrile
In a 2 L round bottom flask, dichloromethane (550 mL) and 2-butenenitrile 110 g
(1.64 mol) were cooled to 20 to 25 °C. A solution of bromine 275 g (1.72 mol) in dichloromethane (220 mL) was dropwise added at 20 to 25 °C. Hydrobromic acid 1.43 ml (0.0082 mol) in acetic acid (33%) solution was added into the reaction mixture and stirred for 4 hours. After the completion of reaction, Na2S203 (550 mL) 4% aqueous solution was added and the reaction mixture was stirred for 15 min. The separated organic layer was distilled under vacuum completely to obtain 364.2 g (97.9%) of title compound as an oil.
Example 8: Preparation of l,5-dimethyl-lH-pyrazol-3-amine
In a 5 L round bottom flask, water (1. 36 L), sodium hydroxide 340 g (8.99 mol) were added and the reaction mixture was cooled to 0 to 5°C. A solution of methyl hydrazine sulphate 237.8 g (1.65 mol) in 680 mL water was added dropwise to the reaction mixture and stirred below 10 °C. 2,3-dibromobutanenitrile 340 g (1.5 mol) prepared in example-7 was added and the reaction mixture was stirred below 10 °C for 2 hours. After the completion of reaction, toluene (630 mL) was added and the reaction mixture was stirred for 15 min. The aqueous layer was separated and the organic layer was removed. The aqueous layer was extracted with dichloromethane (5.1 L). The combined organic layer was distilled completely under vacuum to obtain residue. Diisopropyl ether (680 mL) was added and the reaction mixture was stirred at 0 to 5 °C for 1 hour. The reaction mixture was filtered, washed with diisopropyl ether and dried to obtained 121.5 g (72.93%) of title compound having 95.63% purity.
Examples: Preparation of triaminopyrimidine compounds
Example-9: Preparation of tert-butyl (R)-4-(2,4-dioxo-l,2,3,4-tetrahydro- pyrimidin-5-yl)-2-methylpiperazine-l-carboxylate
In 2 L four neck round bottom flask, 1.25 Kg (6.545 mol) 5-bromouracil, 1.87 Kg (9.360 mol) tert-butyl (R)-2-methylpiperazine-l-carboxylate and 5L pyridine were added at 25 to 35° C. The reaction mass was stirred for 15 hours at 115 to 120°C. After completion, the reaction mass was cooled to 25 to 35°C. 12.5 L water was added and stirred for 1 hour. The reaction mass was filtered, washed with 2.5 L water and dried to obtain 1.37 Kg (67.4%) of title compound.
Example-10: Preparation of tert-butyl (R)-4-(2,4-dichloropyrimidin-5-yl)-2-methylpiperazine- 1 -carboxylate
In 20 L four neck round bottom flask, 1.36 Kg (4.382 mmol) tert-butyl (R)-4-(2,4-dioxo-1, 2,3, 4-tetrahydropyrimidin-5-yl)-2-methylpiperazine-l -carboxylate and 6.8 L phosphorus oxychloride were added at 25 to 35° C. 26.5 mL pyridine (0.329 mol) was added and the reaction mass was heated to 105 to 110 °C and stirred for 4 hours. After the completion of the reaction, phosphorus oxychloride was distilled completely at atmospheric pressure. 2.72 L acetone was added and the reaction mixture was quenched into 4.08 L water. Acetone was removed by distillation under vacuum. 20% sodium carbonate solution was added to adjust pH 7.5-8.5 of the reaction mixture. 1.14 Kg (5.258 mol) di-tert-butyl dicarbonate and 9.52 L ethyl acetate were added and stirred for 2 hours at 25 to 35 °C. After the completion of the reaction, the organic layer was separated and aqueous layer was extracted with 6.8 L ethyl acetate. The combined ethyl layers were distilled to remove ethyl acetate completely under vacuum to obtain residue. 1.36 L isopropyl alcohol was added to the residue and isopropyl alcohol was removed completely. 4.08 L isopropyl alcohol and 6.8 L water were added to the residue and stirred for 1 hour. The reaction mass was filtered, washed with water and dried to obtain 1.25 Kg of title compound.
Example-11: Preparation of tert-butyl (R)-4-(2-chloro-4-[(l,5-dimethyl-lH-pyrazol-3-yl)amino)pyrimidin-5-yl]-2-methylpiperazine-l-carboxylate
In 20 L round bottom flask, 640 g (1.843 mol) tert-butyl (R)-4-(2, 4-dichloropyrimidin-5-yl)-2-methylpiperazine-l -carboxylate, 225.3 g (2.027 g) 1,5-dimethyl-lH-pyrazol-3-amine and 9.6L toluene were added at 25 to 35°C. 1.2 Kg (3.686 mol) cesium carbonate was added. The reaction mixture was purged for 15 min under nitrogen. 12.41 g (0.0553 mol) palladium acetate and 34.43 g (0.0553 mol) racemic 2,2′-bis(diphenylphosphino)-l,l’-binaphthyl were added and the reaction mass was maintained for 16 hours at 110 to 115 °C under nitrogen. After the completion of the reaction, the reaction mixture was filtered through a celite bed and washed the bed with 1.28 L toluene. Toluene was distilled completely and 2.56 L dichlromethane was added. The compound was adsorbed by 1.92 Kg silica gel (60-120 mesh). The dichloromethane was distilled completely under vacuum and 12.8 L mixture of ethyl acetate and hexane was added to the residue and stirred for 2 hours. The silica gel was filtered and the filtrate was distilled completely under vacuum to obtain 595 g title compound.
Example-12: Preparation of tert-butyl (R)-4-(2-((4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)amino)-4-((l,5-dimethyl-lH-pyrazol-3-yl)amino) pyrimidin-5-yl)-2-methylpiperazine-l-carboxylate
In 20 L round bottom flask, 595 g (1.40 mol) tert-butyl (R)- 4-(2-chloro-4-[(l,5-dimethyl-lH-pyrazol-3-yl)amino)pyrimidin-5-yl]-2-methylpiperazine-l-carboxylate, 305 g (1.38 mol) 4-cyclopropyl-5-fluoro-6-methylpyridin-2-amine hydrochloride and 11.5 L toluene were added at 25 to 35°C. 1.08 Kg (3.32 mol) cesium carbonate was added. The reaction mixture was purged for 15 min under nitrogen. 17.21 g (27.6 mmol) palladium acetate and 6.21 g (27.6 mmol) racemic 2,2′-bis(diphenylphosphino)-l, -binaphthyl were added. The reaction mass was stirred for 6 hours at 110 tol l5 °C under nitrogen. After the completion of the reaction, the reaction mixture was filtered through a celite bed and washed with toluene. The filtrate was used as such in the next step without further treatment.
Example-13: Preparation of tert-butyl (R)-4-(2-((4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)amino)-4-((l,5-dimethyl-lH-pyrazol-3-yl)amino) pyrimidin-5-yl)-2-methylpiperazine-l-carboxylate
In 500 mL four neck round bottom flask, 7.5 g (17.77 mmol) (R)-tert-butyl 4-(2-chloro-4-[(l,5-dimethyl-lH-pyrazol-3-yl)amino)pyrimidin-5-yl]-2-methylpiperazine-l-carboxylate, 3.92 g (17.77 mmol) 4-cyclopropyl-5-fluoro-6-methylpyridin-2-amine hydrochloride compound and 150 mL toluene were added at 25 to 35 °C. 20 g (61.3 mmol) cesium carbonate was added. The reaction mixture was purged for 15 min under nitrogen. Then, 130 mg (0.58 mmol) palladium acetate and 360 mg (0.58 mmol) racemic 2,2′-bis(diphenylphosphino)-l,l’-binaphthyl were added. The reaction mass was stirred for 18 hours at 110 to 115° C under nitrogen. After completion, the reaction mixture was filtered through a celite bed and washed with toluene. The filtrate was used as such in the next step without further treatment.
2 4
Example-14: (R)-N -(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N -(1, 5-dimethyl-lH-pyrazol-3-yl)-5-(3-methylpiperazin-l-yl)pyrimidine-2,4-diamine
In 50 L glass assembly, the filtrate containing tert-butyl (R)-4-(2-((4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)amino)-4-((l,5-dimethyl-lH-pyrazol-3-yl)amino) pyrimidin-5-yl)-2-methylpiperazine-l-carboxylate from example 13 was taken. 11.5 L water and 1.28 L Cone. HC1 were added at 25 to 35 °C. The reaction mass was stirred for 2 hours at 50 to 55 °C. After the completion of the reaction, reaction mixture was cooled to room temperature and filtered over celite bed and washed with water. The separated the aqueous layer from filtrate was basified by using 20% sodium carbonate solution and extracted with 12.8 L methylene dichloride. The organic layer was distilled completely under vacuum to obtain residue. 9.6 L acetonitrile was added to the residue and heated to reflux for 30 min. The reaction mixture was cooled and stirred at 25 to 35 °C for 1 hour. The reaction mixture was filtered, washed with 640 mL acetonitrile and dried to obtain 360 g titled compound.
2 4
Example-15: (R)-N -(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N -(1,5-dimethyl-lH-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-l-yl)pyrimidine-2,4-diamine
In 250 mL four neck round bottom flask, 4.7 g (10.4 mmol) (R)-N -(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(l,5-dimethyl-lH-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-l-yl)pyrimidine-2,4-diamine was dissolved in 56 mL ethanol. 1.89 g (23.32 mmol) formaldehyde and 1.44 g (22.90 mmol) sodium cyanoborohydride were added. Adjusted pH 5-6 using acetic acid and stirred the reaction mass at 25 to 35 °C for 2 hours. After completion, ethanol was distilled completely under vacuum. 47 mL water was added to the residue. The reaction mass was basified by 20% sodium carbonate solution and extracted with methylene dichloride. Both the organic layers were combined and distilled completely under vacuum. 94 mL acetonitrile was added to the residue and heated to reflux for 15 min. The reaction mass was cooled to 25 to 35° C and stirred for 1 hour. The reaction mass was filtered, washed with 5 mL acetonitrile and dried to obtain 3.7 g title compound as crystalline solid, having HPLC purity of about 99.61%.
2 4
Example-16: (R)-N -(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N -(1,5-dimethyl-lH-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-l-yl)pyrimidine-2,4-diamine
In 20 L round bottom flask, 725 g (1.60 mol) (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(l,5-dimethyl-lH-pyrazol-3-yl)-5-(3,4-dimethylpiperazine-l-yl)pyrimidine-2,4-diamine was dissolved in 6.52 L dichloromethane. 261.5 g (3.2 mol) formaldehyde and 510.4 g (2.4 mol) sodium triacetoxyborohydride were added and stirred the reaction mixture at 25 to 35 °C for 2 hours. After the completion of the reaction, 3.63 L water was added into the reaction mixture. The reaction mixture was basified by 20% sodium carbonate solution and the organic layer was separated. The aqueous layer was extracted with 1.45 L methylene dichloride. The combined organic layers were distilled completely under vacuum. 14.5 L acetonitrile was added to the residue and heated to reflux for 15 min. The reaction mixture was cooled to 25 to 35° C and stirred for 1 hour. The reaction mass was filtered, washed with 1.45 L acetonitrile and dried to obtain 632 g of title compound as crystalline solid having 99.01% HPLC purity. The crystalline compound is characterized by Powder x-ray diffraction pattern (FIG.l) and Differential Scanning Calorimetry (FIG.2).
2 4
Example-17: Preparation of (R)-N -(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N -(l,5-dimethyl-lH-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-l-yl)pyrimidine-2,4-diamine In a 10 mL round bottom flask, 300 mg (0.644 mmol) (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(l,5-dimethyl-lH-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-l-yl)pyrimidine-2,4-diamine, 2.7 mL acetonitrile and 0.3 mL water were added and the reaction mixture was heated to reflux for 15 min. The reaction mixture was cooled to 25 to 35 °C and stirred for 1 hour. The reaction mass was filtered, washed with acetonitrile and dried to obtain 201 mg (67%) title compound as crystalline solid. The crystalline compound is characterized by Powder x-ray diffraction pattern (FIG.3) and Differential Scanning Calorimetry (FIG.4).
SYN
WO 2015165660
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015165660
Example 13
Synthetic scheme 1
Synthetic scheme 2
(R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-1-yl)pyrimidine-2,4-diamine
In a 50 mL round-bottomed flask (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-5-(3-methylpiperazin-1-yl)pyrimidine-2,4-diamine hydrochloride (190 mg, 0.42 mmol, Example 2) was taken in DCM (2 mL) to give a yellow suspension. To this Hunig’s Base (0.184 mL, 1.05 mmol) was added and the suspension turned clear. After 10 minutes, it turned into a white suspension. After another 10 minutes, the mixture was concentrated to dryness. Resultant residue was dissolved in ethanol (absolute, 99.5%) (3 mL) and formaldehyde (0.042 mL, 0.63 mmol) was added and stirred for 10 minutes. White suspension slowly cleared to yellow solution. To this clear solution sodium cyanoborohydride (26.4 mg, 0.42 mmol) was added in one portion to get white suspension. After 30 minutes LCMS showed completion of reaction. The reaction mixture was concentrated and the crude was purified through reverse phase HPLC GILSON instrument to get the pure solid of (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-1-yl)pyrimidine-2,4-diamine (80 mg, 40.8 %).1H NMR (300
MHz, DMSO-d6) δ ppm 0.67 – 0.78 (m, 2 H) 1.00 (d, J=6.22 Hz, 3 H) 1.02 – 1.08 (m, 2 H) 1.96 – 2.10 (m, 1 H) 2.23 (s, 7 H) 2.30 – 2.38 (m, 4 H) 2.73 – 2.96 (m, 4 H) 3.33 (s, 3 H) 6.83 (s, 1 H) 7.67 (d, J=5.09 Hz, 1 H) 8.00 (s, 1 H) 8.03 (s, 1 H) 9.26 (s,1 H) MS (ES+), (M+H)+ = 466.45 for C21H32FN9.
SYN
Nature Communications (2015), 6, 6715.
https://www.nature.com/articles/ncomms7715
Hameed P., S., Solapure, S., Patil, V. et al. Triaminopyrimidine is a fast-killing and long-acting antimalarial clinical candidate. Nat Commun 6, 6715 (2015). https://doi.org/10.1038/ncomms7715
The widespread emergence of Plasmodium falciparum (Pf) strains resistant to frontline agents has fuelled the search for fast-acting agents with novel mechanism of action. Here, we report the discovery and optimization of novel antimalarial compounds, the triaminopyrimidines (TAPs), which emerged from a phenotypic screen against the blood stages of Pf. The clinical candidate (compound 12) is efficacious in a mouse model of Pf malaria with an ED99 <30 mg kg−1 and displays good in vivo safety margins in guinea pigs and rats. With a predicted half-life of 36 h in humans, a single dose of 260 mg might be sufficient to maintain therapeutic blood concentration for 4–5 days. Whole-genome sequencing of resistant mutants implicates the vacuolar ATP synthase as a genetic determinant of resistance to TAPs. Our studies highlight the potential of TAPs for single-dose treatment of Pf malaria in combination with other agents in clinical development.

(A) Pyridine, microwave, 150 °C, 45 min. (B) (i) POCl3, reflux, 6 h (ii) sodium carbonate, di-tert-butyl dicarbonate, room temperature, 16 h. (C) N,N-Diisopropylethylamine (DIPEA), ethanol, microwave, 110 °C, 1 h. (D) (i) Potassium tert-butoxide, 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl (BINAP), pd2(dba)3, toluene, reflux, 12 h. (E) HCl (4 N) in dioxane, 15–30 min. (F) Compound 9, DIPEA, dichloromethane, formaldehyde (HCHO), sodium cyanoborohydride, 15 min.
Synthesis of (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1, 5-dimethyl-1H-pyrazol-3-yl)-5-(3, 4-dimethylpiperazin-1-yl)pyrimidine-2,4-diamine (12). (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-5-(3-methylpiperazin-1-yl)pyrimidine-2,4-diamine hydrochloride (compound 9, 190 mg, 0.42 mmol) was taken in dichloromethane (2 ml) to give a yellow suspension. To this Hunig’s Base (0.184 ml, 1.05 mmol) was added and the suspension turned clear. After 10 min of stirring, reaction mixture turned into a white suspension and then it was concentrated to dryness. Resultant residue was dissolved in ethanol (absolute, 99.5%) (3 ml), and formaldehyde (0.042 ml, 0.63 mmol) was added and stirred for 10 min. To this clear solution, sodium cyanoborohydride (26.4 mg, 0.42 mmol) was added in one portion to get a white suspension. The reaction mixture was concentrated and the crude product was purified through reverse-phase chromatography to get the pure off-white solid of (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1, 5-dimethyl-1H-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-1-yl)pyrimidine-2,4-diamine (80 mg, 40.8%). Yield: 40.8%, purity: >95% by HPLC (ultraviolet at 220 and 254 nm). 1H NMR (300 MHz, DMSO-d6) δ 9.26 (s,1H), 8.03 (s, 1H) 8.00 (s, 1H) 7.67 (d, J=5.1 Hz, 1H) 6.83 (s, 1H) 3.33 (s, 3H) 2.96–2.73 (m, 4H) 2.75–2.50 (m, 1H) 2.38–2.30 (m, 4H) 2.23 (s, 7H) 2.10–1.96 (m, 1H),1.08–1.02 (m, 2H) 1.00 (d, J=6.2 Hz, 3H) 0.78–0.67 (m, 2H). 13C-NMR (126 MHz, DMO-d6) δ 155.30, 154.67, 152.10, 150.93, 148.98, 146.81. 145.29, 141.95, 140.31, 138.81, 124.91, 106.20, 97.07, 58.78, 51.87, 42.16, 35.28, 17.23. 10.99 and 8.77, HRMS (ESI): m/z calculated for C24H32FN9+H [M+H]: 466.2765. Found: 466. 2838. Traces of LC-MS, HRMS, 1H NMR and 13C-NMR of compound 12 are shown in Supplementary Figs 1–3.


| Product vision |
|
| MoA |
|
| Key features |
|
| Challenges |
|
| Status |
|
| Next milestone |
|
| Previously |
|
Zydus receives Orphan Drug Designation from USFDA for ZY-19489, a novel compound to treat malaria;
ZY19489 is a novel antimalarial compound active against all current clinical strains of P. falciparum and P. vivax, including drug-resistant strains.
Zydus Cadila listed as Cadila Healthcare Limited announced that its antimalarial compound ZY19489 (MMV253), currently in development together with Medicines for Malaria Venture (MMV), a leading product development partnership (PDP) in antimalarial drug research, has received Orphan Drug Designation from the USFDA.
Orphan drug designation provides eligibility for certain development incentives, including tax credits for qualified clinical testing, prescription drug user fee exemptions, and seven-year marketing exclusivity upon FDA approval.
The company said that the Phase I study of ZY19489 has demonstrated a long half-life and potential for a single-dose cure for malaria. In a separate malaria challenge trial, potent antimalarial activity has been demonstrated following single-dose oral administration of ZY19489.
“As a global community facing threats from rapidly mutating malaria strains and the rise in artemisinin resistance cases, we have to be prepared with novel therapeutic drugs. ZY-19489 is a potential single dose radical cure for P. falciparum and P. vivax malaria which is a major global health risk today,” Pankaj R. Patel, Chairman, Zydus Group, said.
“ZY19489 is a potent, first in class molecule, originally discovered and elaborated in India” said Dr. Timothy Wells, Chief Scientific Officer, MMV. “It has tremendous potential as part of a new generation of treatments and is fully active against drug resistant strains of malaria which are increasingly a concern.”
Artemisinin resistance is seen as a mounting challenge to the global fight against malaria. ZY19489 is being developed to provide an effective alternative to the current front-line antimalarial drugs for the treatment of P. falciparum and P. vivax malaria, as artemisinin-based combination therapies (ACTs) are under threat of resistance.
As per the World Malaria Report 2021, there were an estimated 241 million cases of malaria worldwide and the estimated number of malaria deaths stood at 627,000 in 2020. A major health concern, it is estimated that a child dies from malaria every minute. About 96% of malaria deaths globally were in 29 countries. India accounted for about 82% of all malaria deaths in the WHO South-East Asia Region.
////////////ZY 19489, MMV 253, Orphan Drug Designation, PHASE 1, ZYDUS CADILA, ANTIMALARIAL
Cn1nc(Nc2nc(Nc3cc(C4CC4)c(F)c(C)n3)ncc2N2C[C@@H](C)N(C)CC2)cc1C
CC1CN(CCN1C)C2=CN=C(N=C2NC3=NN(C(=C3)C)C)NC4=NC(=C(C(=C4)C5CC5)F)C
Avalglucosidase alfa
QQGASRPGPR DAQAHPGRPR AVPTQCDVPP NSRFDCAPDK AITQEQCEAR GCCYIPAKQG
LQGAQMGQPW CFFPPSYPSY KLENLSSSEM GYTATLTRTT PTFFPKDILT LRLDVMMETE
NRLHFTIKDP ANRRYEVPLE TPRVHSRAPS PLYSVEFSEE PFGVIVHRQL DGRVLLNTTV
APLFFADQFL QLSTSLPSQY ITGLAEHLSP LMLSTSWTRI TLWNRDLAPT PGANLYGSHP
FYLALEDGGS AHGVFLLNSN AMDVVLQPSP ALSWRSTGGI LDVYIFLGPE PKSVVQQYLD
VVGYPFMPPY WGLGFHLCRW GYSSTAITRQ VVENMTRAHF PLDVQWNDLD YMDSRRDFTF
NKDGFRDFPA MVQELHQGGR RYMMIVDPAI SSSGPAGSYR PYDEGLRRGV FITNETGQPL
IGKVWPGSTA FPDFTNPTAL AWWEDMVAEF HDQVPFDGMW IDMNEPSNFI RGSEDGCPNN
ELENPPYVPG VVGGTLQAAT ICASSHQFLS THYNLHNLYG LTEAIASHRA LVKARGTRPF
VISRSTFAGH GRYAGHWTGD VWSSWEQLAS SVPEILQFNL LGVPLVGADV CGFLGNTSEE
LCVRWTQLGA FYPFMRNHNS LLSLPQEPYS FSEPAQQAMR KALTLRYALL PHLYTLFHQA
HVAGETVARP LFLEFPKDSS TWTVDHQLLW GEALLITPVL QAGKAEVTGY FPLGTWYDLQ
TVPIEALGSL PPPPAAPREP AIHSEGQWVT LPAPLDTINV HLRAGYIIPL QGPGLTTTES
RQQPMALAVA LTKGGEARGE LFWDDGESLE VLERGAYTQV IFLARNNTIV NELVRVTSEG
AGLQLQKVTV LGVATAPQQV LSNGVPVSNF TYSPDTKVLD ICVSLLMGEQ FLVSWC
(Disulfide bridge:26-53, 36-52, 47-71, 477-502, 591-602, 882-896)
Avalglucosidase alfa
アバルグルコシダーゼアルファ (遺伝子組換え)
Avalglucosidase alfa (USAN/INN);
Avalglucosidase alfa (genetical recombination) (JAN);
Avalglucosidase alfa-ngpt
To treat late-onset Pompe disease
| Formula | C4490H6818N1197O1299S32 |
|---|---|
| CAS | 1802558-87-7 |
| Mol weight | 99375.4984 |
FDA APPROVED Nexviazyme, 2021/8/6, Enzyme replacement therapy product
Treatment of Pompe disease
Biologic License Application (BLA): 761194
Company: GENZYME CORP
https://www.fda.gov/news-events/press-announcements/fda-approves-new-treatment-pompe-diseaseFor Immediate Release:August 06, 2021
Today, the U.S. Food and Drug Administration approved Nexviazyme (avalglucosidase alfa-ngpt) for intravenous infusion to treat patients 1 year of age and older with late-onset Pompe disease.
Patients with Pompe disease have an enzyme deficiency that leads to the accumulation of a complex sugar, called glycogen, in skeletal and heart muscles, which cause muscle weakness and premature death from respiratory or heart failure. Normally, glycogen—the stored form of glucose—breaks down to release glucose into the bloodstream to be used as fuel for the cells.
“Pompe disease is a rare genetic disease that causes premature death and has a debilitating effect on people’s lives,” said Janet Maynard, M.D., deputy director of the Office of Rare Diseases, Pediatrics, Urologic and Reproductive Medicine in the FDA’s Center for Drug Evaluation and Research. “Today’s approval brings patients with Pompe disease another enzyme replacement therapy option for this rare disease. The FDA will continue to work with stakeholders to advance the development of additional new, effective and safe therapies for rare diseases, including Pompe disease.”
Nexviazyme, an enzyme replacement therapy, is an intravenous medication that helps reduce glycogen accumulation. The effectiveness of Nexviazyme for the treatment of Pompe disease was demonstrated in a study of 100 patients who were randomized to take Nexviazyme or another FDA-approved enzyme replacement therapy for Pompe disease. Treatment with Nexviazyme improved lung function similar to the improvement seen with the other therapy.
The most common side effects included headache, fatigue, diarrhea, nausea, joint pain (arthralgia), dizziness, muscle pain (myalgia), itching (pruritus), vomiting, difficulty breathing (dyspnea), skin redness (erythema), feeling of “pins and needles” (paresthesia) and skin welts (urticaria). Serious reactions included hypersensitivity reactions like anaphylaxis and infusion-associated reactions, including respiratory distress, chills and raised body temperature (pyrexia). Patients susceptible to fluid volume overload or with compromised cardiac or respiratory function may be at risk for serious acute cardiorespiratory failure.
The FDA granted this application Fast Track, Priority Review and Breakthrough Therapy designations. Nexviazyme also received an orphan drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases. The FDA granted the approval of Nexviazyme to Genzyme Corporation.
###
NEW DRUG APPROVALS
one time
$10.00
FDA grants priority review for avalglucosidase alfa, a potential new therapy for Pompe disease
- The FDA decision date for avalglucosidase alfa, an investigational enzyme replacement therapy, is set for May 18, 2021
- Regulatory submission based on positive data from two trials in patients with late-onset and infantile-onset Pompe disease, respectively
- Avalglucosidase alfa received FDA Breakthrough Therapy and Fast Track designations for the treatment of people with Pompe Disease
- Pompe disease, a rare degenerative muscle disorder, affects approximately 3,500 people in the U.S.
- Milestone reinforces 20+year commitment to Pompe disease community
PARIS – November 18, 2020 – The U.S. Food and Drug Administration (FDA) has accepted for priority review the Biologics License Application (BLA) for avalglucosidase alfa for long-term enzyme replacement therapy for the treatment of patients with Pompe disease (acid α-glucosidase deficiency). The target action date for the FDA decision is May 18, 2021.
Avalglucosidase alfa is an investigational enzyme replacement therapy designed to improve the delivery of acid alpha-glucosidase (GAA) enzyme to muscle cells, and if approved, would offer a potential new standard of care for patients with Pompe disease.
In October, the European Medicines Agency accepted for review the Marketing Authorization Application for avalglucosidase alfa for long-term enzyme replacement therapy for the treatment of patients with Pompe disease. The Medicines and Healthcare Products Regulatory Agency in the UK has granted Promising Innovative Medicine designation for avalglucosidase alfa.
“The hallmarks of Pompe disease are the relentless and debilitating deterioration of the muscles, which causes decreased respiratory function and mobility,” said Karin Knobe, Head of Development for Rare Diseases and Rare Blood Disorders at Sanofi. “Avalglucosidase alfa is specifically designed to deliver more GAA enzyme into the lysosomes of the muscle cells. We have been greatly encouraged by positive clinical trial results in patients with late-onset and infantile-onset Pompe disease.”
Pompe disease is a rare, degenerative muscle disorder that can impact an individual’s ability to move and breathe. It affects an estimated 3,500 people in the U.S. and can manifest at any age from infancy to late adulthood.i
The BLA is based on positive data from two trials:
- Pivotal Phase 3, double-blind, global comparator-controlled trial (COMET), which evaluated the safety and efficacy of avalglucosidase alfa compared to alglucosidase alfa (standard of care) in patients with late-onset Pompe disease. Results from this trial were presented during a Sanofi-hosted virtual scientific session in June 2020 and in October 2020 at World Muscle Society and the American Association of Neuromuscular and Electrodiagnostic Medicine.
- The Phase 2 (mini-COMET) trial evaluated the safety and exploratory efficacy of avalglucosidase alfa in patients with infantile-onset Pompe disease previously treated with alglucosidase alfa. Results from this trial were presented at the WORLDSymposium, in February 2020.
Delivery of GAA to Clear Glycogen
Pompe disease is caused by a genetic deficiency or dysfunction of the lysosomal enzyme GAA, which results in build-up of complex sugars (glycogen) in muscle cells throughout the body. The accumulation of glycogen leads to irreversible damage to the muscles, including respiratory muscles and the diaphragm muscle supporting lung function, and other skeletal muscles that affect mobility.
To reduce the glycogen accumulation caused by Pompe disease, the GAA enzyme must be delivered into the lysosomes within muscle cells. Research led by Sanofi has focused on ways to enhance the delivery of GAA into the lysosomes of muscle cells by targeting the mannose-6-phosphate (M6P) receptor that plays a key role in the transport of GAA.
Avalglucosidase alfa is designed with approximately 15-fold increase in M6P content, compared to standard of care alglucosidase alfa, and aims to help improve cellular enzyme uptake and enhance glycogen clearance in target tissues.ii The clinical relevance of this difference has not been confirmed.
Avalglucosidase alfa is currently under clinical investigation and its safety and efficacy have not been evaluated by any regulatory authority worldwide.
| About Sanofi Sanofi is dedicated to supporting people through their health challenges. We are a global biopharmaceutical company focused on human health. We prevent illness with vaccines, provide innovative treatments to fight pain and ease suffering. We stand by the few who suffer from rare diseases and the millions with long-term chronic conditions. With more than 100,000 people in 100 countries, Sanofi is transforming scientific innovation into healthcare solutions around the globe. Sanofi, Empowering Life |
/////////Avalglucosidase alfa, FDA 2021, Nexviazyme, APPROVALS 2021, PEPTIDE, Enzyme replacement therapy , Pompe disease, アバルグルコシダーゼアルファ (遺伝子組換え), Fast Track, Priority Review, Breakthrough Therapy, orphan drug designation, genzyme, sanofi
BELUMOSUDIL

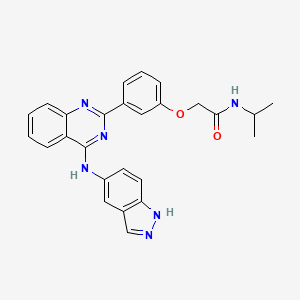
BELUMOSUDIL
MW 452.5
911417-87-3, SLx-2119, KD-025, KD 025, WHO 11343
2-[3-[4-(1H-indazol-5-ylamino)quinazolin-2-yl]phenoxy]-N-propan-2-ylacetamide
2-(3-(4-(lH-indazol-5-ylamino)quinazolin-2-yl)phenoxy)-N-isopropylacetamide
Belumosudil mesylate
KD025 mesylate
2109704-99-4
UPDATE FDA APPROVED 7/16/2021 To treat chronic graft-versus-host disease after failure of at least two prior lines of systemic therapy, Rezurock
New Drug Application (NDA): 214783
Company: KADMON PHARMA LLC
200 MG TABLET
FDA approves belumosudil for chronic graft-versus-host disease
On July 16, 2021, the Food and Drug Administration approved belumosudil (Rezurock, Kadmon Pharmaceuticals, LLC), a kinase inhibitor, for adult and pediatric patients 12 years and older with chronic graft-versus-host disease (chronic GVHD) after failure of at least two prior lines of systemic therapy.
Efficacy was evaluated in KD025-213 (NCT03640481), a randomized, open-label, multicenter dose-ranging trial that included 65 patients with chronic GVHD who were treated with belumosudil 200 mg taken orally once daily.
The main efficacy outcome measure was overall response rate (ORR) through Cycle 7 Day 1 where overall response included complete response (CR) or partial response (PR) according to the 2014 criteria of the NIH Consensus Development Project on Clinical Trials in Chronic Graft-versus-Host Disease. The ORR was 75% (95% CI: 63, 85); 6% of patients achieved a CR, and 69% achieved a PR. The median time to first response was 1.8 months (95% CI: 1.0, 1.9). The median duration of response, calculated from first response to progression, death, or new systemic therapies for chronic GVHD, was 1.9 months (95% CI: 1.2, 2.9). In patients who achieved response, no death or new systemic therapy initiation occurred in 62% (95% CI: 46, 74) of patients for at least 12 months since response.
The most common adverse reactions (≥ 20%), including laboratory abnormalities, were infections, asthenia, nausea, diarrhea, dyspnea, cough, edema, hemorrhage, abdominal pain, musculoskeletal pain, headache, phosphate decreased, gamma glutamyl transferase increased, lymphocytes decreased, and hypertension.
The recommended dosage of belumosudil is 200 mg taken orally once daily with food.
View full prescribing information for Rezurock.
This review was conducted under Project Orbis, an initiative of the FDA Oncology Center of Excellence. Project Orbis provides a framework for concurrent submission and review of oncology drugs among international partners. For this review, FDA collaborated with Australia’s Therapeutic Goods Administration, Health Canada, Switzerland’s Swissmedic, and the United Kingdom’s Medicines and Healthcare products Regulatory Agency.
This review used the Real-Time Oncology Review (RTOR) pilot program, which streamlined data submission prior to the filing of the entire clinical application, and the Assessment Aid, a voluntary submission from the applicant to facilitate the FDA’s assessment. The FDA approved this application 6 weeks ahead of the FDA goal date.
This application was granted priority review and breakthrough therapy designation. A description of FDA expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.
Belumosudil mesylate is an orally available rho kinase 2 (ROCK 2) inhibitor being developed at Kadmon. In 2020, the drug candidate was submitted for a new drug application (NDA) in the U.S., under a real-time oncology review pilot program, for the treatment of chronic graft-versus-host disease (cGVHD). The compound is also in phase II clinical development for the treatment of idiopathic pulmonary fibrosis and diffuse cutaneous systemic sclerosis. Formerly, the company had also been conducting clinical research for the treatment of psoriasis and non-alcoholic steatohepatitis (NASH); however, no further development has been reported for these indications. Originally developed by Nano Terra, the product was licensed to Kadmon on an exclusive global basis in 2011. In 2019, Kadmon entered into a strategic partnership with BioNova Pharmaceuticals and established a joint venture, BK Pharmaceuticals, to exclusively develop and commercialize KD-025 for the treatment of graft-versus-host disease in China. The compound has been granted breakthrough therapy designation in the U.S. for the treatment of cGVHD and orphan drug designations for cGVHD and systemic sclerosis. In the E.U. belumosudil was also granted orphan drug status in the E.U. for the treatment of cGVHD.
Kadmon , under license from NT Life Sciences , is developing belumosudil as mesylate salt, a ROCK-2 inhibitor, for treating IPF, chronic graft-versus-host disease, hepatic impairment and scleroderma. In July 2021, belumosudil was reported to be in pre-registration phase.
Belumosudil (formerly KD025 and SLx-2119) is an experimental drug being explored for the treatment of chronic graft versus host disease (cGvHD), idiopathic pulmonary fibrosis (IPF), and moderate to severe psoriasis. It is an inhibitor of Rho-associated coiled-coil kinase 2 (ROCK2; ROCK-II).[1] Belumosudil binds to and inhibits the serine/threonine kinase activity of ROCK2. This inhibits ROCK2-mediated signaling pathways which play major roles in pro- and anti-inflammatory immune cell responses. A genomic study in human primary cells demonstrated that the drug also has effects on oxidative phosphorylation, WNT signaling, angiogenesis, and KRAS signaling.[2] Originally developed by Surface Logix, Inc,[1] Belumosudil was later acquired by Kadmon Corporation. As of July 2020 the drug was in completed or ongoing Phase II clinical studies for cGvHD, IPF and psoriasis.[3]
cGvHD is a complication that can follow stem cell or hematopoietic stem cell transplantation where the transplanted cells (graft) attack healthy cells (host). This causes inflammation and fibrosis in multiple tissues. Two cytokines controlled by the ROCK2 signaling pathway, IL-17 and IL-21, have a major role in the cGvHD response. In a 2016 report using both mouse models and a limited human clinical trial ROCK2 inhibition with belumosudil targeted both the immunologic and fibrotic components of cGvHD and reversed the symptoms of the disease.[4] In October 2017 KD025 was granted orphan drug status in the United States for treatment of patients with cGvHD.[5]
IPF is a progressive fibrotic disease where the lining of the lungs become thickened and scarred.[6] Increased ROCK activity has been found in the lungs of humans and animals with IPF. Treatment with belumosudil reduced lung fibrosis in a bleomycin mouse model study.[7] Belumosudil may have a therapeutic benefit in IPF by targeting the fibrotic processes mediated by the ROCK signaling pathway.
Psoriasis is an inflammatory skin condition where patients experiences eruptions and remissions of thickened, erythematous, and scaly patches of skin. Down-regulation of pro-inflammatory responses was observed with KD025 treatment in Phase 2 clinical studies in patients with moderate to severe psoriasis.[8]
“Substance Name:Substance Name: Belumosudil [USAN]”.
PATENT
| WO2012040499 |
https://patents.google.com/patent/WO2012040499A2/en
PATENT
| CN106916145 |
https://patents.google.com/patent/CN106916145A/en

WO 2014055996, WO 2015157556



Patent
WO-2021129589
Novel crystalline polymorphic forms (N1, N2 and N15) of KD-025 (also known as belumosudil ), useful as a Rho A kinase 2 (ROCK-2) inhibitor for treating multiple sclerosis, psoriasis, rheumatoid arthritis, idiopathic pulmonary fibrosis (IPF), atherosclerosis, non-alcoholic fatty liver and systemic sclerosis. Represents the first filing from Sunshine Lake Pharma or its parent HEC Pharm that focuses on belumosudil.KD-025 is a selective ROCK2 (Rho-associated protein kinase 2, Rho-related protein kinase 2) inhibitor. It has multiple clinical indications such as the treatment of multiple sclerosis, psoriasis, rheumatoid arthritis, and Primary pulmonary fibrosis, atherosclerosis, non-alcoholic fatty liver, etc., among which many indications are in clinical phase I, and psoriasis and systemic sclerosis are in clinical phase II.
The structure of KD-025 is shown in the following formula (1).
Example 1 Preparation method of crystal form N1 of KD-025[0222]300mg of KD-025 solid was suspended and stirred in 10mL methanol at room temperature. After 22h, it was filtered, suction filtered and placed in a drying oven at 50°C under vacuum overnight to obtain 262mg of powder. The obtained crystal was detected by XPRD and confirmed to be KD-025 crystal form N1; its X-ray powder diffraction pattern was basically the same as that of Fig. 1, its DSC pattern was basically the same as that of Fig. 2, and the TGA pattern was basically the same as that of Fig. 3.
PATENT
WO2006105081 ,
Belumosudil product pat,
protection in the EU states until March 2026, expires in the US in May 2029 with US154 extension.
Example 82
2-(3-(4-(lH-indazol-5-ylamino)quinazolin-2-yl)phenoxy)-N-isopropylacetamide
[0257] A suspension of 2-(3-(4-(lH-indazol-5-ylamino)qumazolin-2-yl)ρhenoxy)acetic acid (70 mg, 0.14 mmol), PyBOP® (40 mg, 0.077 mmol), DlEA (24 μL, 0.14 mmol) in dry CH2Cl2 : DMF (2 : 0.1 mL) was stirred at RT for 15 minutes. To this solution of activated acid was added propan-2-amine (5.4 mg, 0.091 mmol). After 30 minutes, 1.0 equivalent of DIEA and 0.55 equivalents of PyBOP® were added. After stirring the solution for 15 minutes, 0.65 equivalents of propan-2-aminewere added and the mixture was stirred for an additional 30 minutes. The solvent was removed in vacuo and the crude product was purified using prep HPLC (25-50 90 rnins) to afford 2-(3-(4-(lH-indazol-5-ylamino)quinazolin-2-yl)phenoxy)-N-isopropylacetamide. (40 mg, 0.086 mmol, 61 %).
References
- ^ Jump up to:a b Boerma M, Fu Q, Wang J, Loose DS, Bartolozzi A, Ellis JL, et al. (October 2008). “Comparative gene expression profiling in three primary human cell lines after treatment with a novel inhibitor of Rho kinase or atorvastatin”. Blood Coagulation & Fibrinolysis. 19 (7): 709–18. doi:10.1097/MBC.0b013e32830b2891. PMC 2713681. PMID 18832915.
- ^ Park J, Chun KH (5 May 2020). “Identification of novel functions of the ROCK2-specific inhibitor KD025 by bioinformatics analysis”. Gene. 737: 144474. doi:10.1016/j.gene.2020.144474. PMID 32057928.
- ^ “KD025 – Clinical Trials”. ClinicalTrials.gov. Retrieved 25 July 2020.
- ^ Flynn R, Paz K, Du J, Reichenbach DK, Taylor PA, Panoskaltsis-Mortari A, et al. (April 2016). “Targeted Rho-associated kinase 2 inhibition suppresses murine and human chronic GVHD through a Stat3-dependent mechanism”. Blood. 127 (17): 2144–54. doi:10.1182/blood-2015-10-678706. PMC 4850869. PMID 26983850.
- ^ Shanley M (October 6, 2017). “Therapy to Treat Transplant Complications Gets Orphan Drug Designation”. RareDiseaseReport. Retrieved 25 July 2018.
- ^ “Pulmonary Fibrosis”. The Mayo Clinic. Retrieved July 25, 2018.
- ^ Semedo D (June 5, 2016). “Phase 2 Study of Molecule Inhibitor for Idiopathic Pulmonary Fibrosis Begins”. Lung Disease News. BioNews Services, LLC. Retrieved 25 July 2018.
- ^ Zanin-Zhorov A, Weiss JM, Trzeciak A, Chen W, Zhang J, Nyuydzefe MS, et al. (May 2017). “Cutting Edge: Selective Oral ROCK2 Inhibitor Reduces Clinical Scores in Patients with Psoriasis Vulgaris and Normalizes Skin Pathology via Concurrent Regulation of IL-17 and IL-10”. Journal of Immunology. 198 (10): 3809–3814. doi:10.4049/jimmunol.1602142. PMC 5421306. PMID 28389592.
| Clinical data | |
|---|---|
| Routes of administration |
Oral administration (tablets or capsules) |
| ATC code | None |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 911417-87-3 |
| PubChem CID | 11950170 |
| UNII | 834YJF89WO |
| CompTox Dashboard (EPA) | DTXSID80238425 |
| Chemical and physical data | |
| Formula | C26H24N6O2 |
| Molar mass | 452.518 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
////////////BELUMOSUDIL, SLx-2119, KD-025, KD 025, WHO 11343, PHASE 2, cGvHD, IPF, psoriasis, Breakthrough Therapy, Orphan Drug Designation
CC(C)NC(=O)COC1=CC=CC(=C1)C2=NC3=CC=CC=C3C(=N2)NC4=CC5=C(C=C4)NN=C5
NEW DRUG APPROVALS
ONE TIME
$10.00
Idecabtagene vicleucel
Idecabtagene vicleucel
CAS 2306267-75-2
STN: BLA 125736
An autologous T lymphocyte-enriched cell transduced ex vivo with an anti-BCMA CAR lentiviral vector encoding a chimeric antigen receptor CAR, comprising a CD8 hinge and TM domain, 4-1BB costimulatory domain and CD3ζ signaling domain, targeting human B cell maturation antigen for cancer immunotherapy (Celgene Corp., NJ)
- Bb2121
| Name | Idecabtagene vicleucel (USAN); Abecma (TN) |
|---|---|
| Product | ABECMA (Celgene Corporation) |
| CAS | 2306267-75-2 |
| Efficacy | Antineoplastic, Anti-BCMA CAR-T cell |
| Disease | Multiple myeloma [DS:H00010] |
| Comment | Cellular therapy product |
USFDA 2021/4/21 APPROVED
Dendritic cells (DCs) are antigen-presenting cells (APCs) that process antigens and display them to other cells of the immune system. Specifically, dendritic cells are capable of capturing and presenting antigens on their surfaces to activate T cells such as cytotoxic T cells (CTLs). Further, activated dendritic cells are capable of recruiting additional immune cells such as macrophages, eosinophils, natural killer cells, and T cells such as natural killer T cells.
Despite major advances in cancer treatment, cancer remains one of the leading causes of death globally. Hurdles in designing effective therapies include cancer immune evasion, in which cancer cells escape destructive immunity, as well as the toxicity of many conventional cancer treatments such as radiation therapy and chemotherapy, which significantly impacts a patient’s ability to tolerate the therapy and/or impacts the efficacy of the treatment.
Given the important role of dendritic cells in immunity, derailed dendritic cell functions have been implicated in diseases such as cancer and autoimmune diseases. For example, cancer cells may evade immune detection and destruction by crippling dendritic cell functionality through prevention of dendritic cell recruitment and activation. In addition, dendritic cells have been found in the brain during central nervous system inflammation and may be involved in the pathogenesis of autoimmune diseases in the brain.
One mechanism by which cancers evade immune detection and destruction is by crippling dendritic cell functionality through prevention of dendritic cell (DC) recruitment and activation. Accordingly, there remains a need for cancer therapies that can effectively derail tumor evasion and enhance anti-tumor immunity as mediated, for example, by dendritic cells.
NEW DRUG APPROVALS
ONE TIME
$10.00
DESCRIPTION
ABECMA is a BCMA-directed genetically modified autologous T cell immunotherapy product consisting of a patient’s own T cells that are harvested and genetically modified ex vivo through transduction with an anti-BCMA02 chimeric antigen receptor (CAR) lentiviral vector (LVV). Autologous T cells transduced with the anti-BCMA02 CAR LVV express the anti-BCMA CAR on the T cell surface. The CAR is comprised of a murine extracellular single-chain variable fragment (scFv) specific for recognizing B cell maturation antigen (BCMA) followed by a human CD8α hinge and transmembrane domain fused to the T cell cytoplasmic signaling domains of CD137 (4-1BB) and CD3ζ chain, in tandem. Binding of ABECMA to BCMA-expressing target cells leads to signaling initiated by CD3ζ and 4-1BB domains, and subsequent CAR-positive T cell activation. Antigen-specific activation of ABECMA results in CAR-positive T cell proliferation, cytokine secretion, and subsequent cytolytic killing of BCMA-expressing cells.
ABECMA is prepared from the patient’s peripheral blood mononuclear cells (PBMCs), which are obtained via a standard leukapheresis procedure. The mononuclear cells are enriched for T cells, through activation with anti-CD3 and anti-CD28 antibodies in the presence of IL-2, which are then transduced with the replication-incompetent lentiviral vector containing the anti-BCMA CAR transgene. The transduced T cells are expanded in cell culture, washed, formulated into a suspension, and cryopreserved. The product must pass a sterility test before release for shipping as a frozen suspension in one or more patient-specific infusion bag(s). The product is thawed prior to infusion back into the patient [see DOSAGE AND ADMINISTRATION and HOW SUPPLIED/Storage And Handling].
The ABECMA formulation contains 50% Plasma-Lyte A and 50% CryoStor® CS10, resulting in a final DMSO concentration of 5%.
FDA approves idecabtagene vicleucel for multiple myeloma
On March 26, 2021, the Food and Drug Administration approved idecabtagene vicleucel (Abecma, Bristol Myers Squibb) for the treatment of adult patients with relapsed or refractory multiple myeloma after four or more prior lines of therapy, including an immunomodulatory agent, a proteasome inhibitor, and an anti-CD38 monoclonal antibody. This is the first FDA-approved cell-based gene therapy for multiple myeloma.
Idecabtagene vicleucel is a B-cell maturation antigen (BCMA)-directed genetically modified autologous chimeric antigen receptor (CAR) T-cell therapy. Each dose is customized using a patient’s own T-cells, which are collected and genetically modified, and infused back into the patient.
Safety and efficacy were evaluated in a multicenter study of 127 patients with relapsed and refractory multiple myeloma who received at least three prior lines of antimyeloma therapies; 88% had received four or more prior lines of therapies. Efficacy was evaluated in 100 patients who received idecabtagene vicleucel in the dose range of 300 to 460 x 106 CAR-positive T cells. Efficacy was established based on overall response rate (ORR), complete response (CR) rate, and duration of response (DOR), as evaluated by an Independent Response committee using the International Myeloma Working Group Uniform Response Criteria for Multiple Myeloma.
The ORR was 72% (95% CI: 62%, 81%) and CR rate was 28% (95% CI 19%, 38%). An estimated 65% of patients who achieved CR remained in CR for at least 12 months.
The idecabtagene vicleucel label carries a boxed warning for cytokine release syndrome (CRS), neurologic toxicities, hemophagocytic lymphohistiocytosis/ macrophage activation syndrome, and prolonged cytopenias. The most common side effects of idecabtagene vicleucel include CRS, infections, fatigue, musculoskeletal pain, and hypogammaglobulinemia.
Idecabtagene vicleucel is approved with a risk evaluation and mitigation strategy requiring that healthcare facilities that dispense the therapy must be specially certified to recognize and manage CRS and nervous system toxicities. To evaluate long-term safety, the FDA is requiring the manufacturer to conduct a post-marketing observational study involving patients treated with idecabtagene vicleucel.
The recommended dose range for idecabtagene vicleucel is 300 to 460 × 106 CAR-positive T cells. View full prescribing information for Abecma.
This application was granted breakthrough therapy designation and orphan drug designation. A description of FDA expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.
FDA D.I.S.C.O. Burst Edition: FDA approval of ABECMA (idecabtagene vicleucel) the first FDA approved cell-based gene therapy for the treatment of adult patients with relapsed or refractory multiple myeloma
Welcome back to the D.I.S.C.O., FDA’s Drug Information Soundcast in Clinical Oncology, Burst Edition, brought to you by FDA’s Division of Drug Information in partnership with FDA’s Oncology Center of Excellence. Today we have another quick update on a recent FDA cancer therapeutic approval.
On March 26, 2021, the FDA approved idecabtagene vicleucel (brand name Abecma) for the treatment of adult patients with relapsed or refractory multiple myeloma after four or more prior lines of therapy, including an immunomodulatory agent, a proteasome inhibitor, and an anti-CD38 monoclonal antibody. This is the first FDA-approved cell-based gene therapy for multiple myeloma.
Idecabtagene vicleucel is a B-cell maturation antigen-directed genetically modified autologous chimeric antigen receptor T-cell therapy. Each dose is customized using a patient’s own T-cells, which are collected and genetically modified, and infused back into the patient.
Safety and efficacy were evaluated in a multicenter study of 127 patients with relapsed and refractory multiple myeloma who received at least three prior lines of antimyeloma therapies, 88% of whom had received four or more prior lines of therapies. Efficacy was evaluated in 100 patients who received idecabtagene vicleucel and was established based on overall response rate, complete response rate, and duration of response, as evaluated by an Independent Response committee using the International Myeloma Working Group Uniform Response Criteria for Multiple Myeloma.
The overall response rate was 72% and complete response rate was 28%. An estimated 65% of patients who achieved complete response remained in complete response for at least 12 months.
The idecabtagene vicleucel label carries a boxed warning for cytokine release syndrome, neurologic toxicities, hemophagocytic lymphohistiocytosis/ macrophage activation syndrome, and prolonged cytopenias. Idecabtagene vicleucel is approved with a risk evaluation and mitigation strategy requiring that healthcare facilities dispensing the therapy must be specially certified to recognize and manage cytokine release syndrome and nervous system toxicities. To evaluate long-term safety, the FDA is requiring the manufacturer to conduct a post-marketing observational study involving patients treated with idecabtagene vicleucel.
Full prescribing information for this approval can be found on the web at www.fda.gov, with key word search “Approved Cellular and Gene Therapy Products”.
Health care professionals should report serious adverse events to FDA’s MedWatch Reporting System at www.fda.gov/medwatch.
Follow the Division of Drug Information on Twitter @FDA_Drug_InfoExternal Link Disclaimer and the Oncology Center of Excellence @FDAOncologyExternal Link Disclaimer. Send your feedback via email to FDAOncology@fda.hhs.gov. Thanks for tuning in today to the DISCO Burst Edition.
PAT
WO 2019148089
In various aspects, the present invention relates to XCR1 binding agents having at least one targeting moiety that specifically binds to XCR1. In various embodiments, these XCR1 binding agents bind to, but do not functionally modulate ( e.g . partially or fully neutralize) XCR1. Therefore, in various embodiments, the present XCR1 binding agents have use in, for instance, directly or indirectly recruiting a XCR1-expressing cell to a site of interest while still allowing the XCR1-expressing cell to signal via XCR1 (i.e. the binding of the XCR1 binding agent does not reduce or eliminate XCR1 signaling at the site of interest). In various embodiments, the XCR-1 binding agent functionally modulates XCR1. In an embodiment, the targeting moiety is a single domain antibody (e.g. VHH, HUMABODY, scFv, on antibody). In various embodiments, the XCR1 binding agent further comprises a signaling agent, e.g., without limitation, an interferon, an interleukin, and a tumor necrosis factor, that may be modified to attenuate activity. In various embodiments, the XCR1 binding agent comprises additional targeting moieties that bind to other targets (e.g. antigens, receptor) of interest. In an embodiment, the other targets (e.g. antigens, receptor) of interest are present on tumor cells. In another embodiment, the other targets (e.g. antigens, receptor) of interest are present on immune cells. In some embodiments, the present XCR1 binding agent may directly or indirectly recruit an immune cell (e.g. a dendritic cell) to a site of action (such as, by way of non-limiting example, the tumor microenvironment). In some embodiments, the present XCR1 binding agent facilitates the presentation of antigens (e.g., tumor antigens) by dendritic cells.
In various embodiments, the present XCR binding agent or targeting moiety of the present chimeric proteins comprises the heavy chain of SEQ ID NO: 223 and/or the light chain of SEQ ID NO: 224, or a variant thereof (e.g. an amino acid sequence having at least about 90%, or at least about 93%, at least about 95%, at least about 97%, at least about 98%, at least about 99%, identity with SEQ ID NO: 223 and/or SEQ ID NO: 224).
In various embodiments, the present XCR binding agent or targeting moiety of the present chimeric proteins comprises a heavy chain CDR 1 of SHNLH (SEQ ID NO: 225), heavy chain CDR 2 of AIYPGNGNTAYNQKFKG (SEQ ID NO: 226), and heavy chain CDR 3 of WGSVVGDWYFDV (SEQ ID NO: 227) and/or a light chain CDR 1 of RSSLGLVHRNGNTYLH (SEQ ID NO: 228), light chain CDR 2 of KVSHRFS (SEQ ID NO: 229), and light chain CDR 3 of SQSTFIVPWT (SEQ ID NO: 230), or a variant thereof (e.g. with four or fewer amino acid substitutions, or with three or fewer amino acid substitutions, or with two or fewer amino acid substitutions, or with one amino acid substitution).
In various embodiments, the present XCR binding agent or targeting moiety of the present chimeric proteins comprises a heavy chain CDR 1 of SHNLH (SEQ ID NO: 225), heavy chain CDR 2 of AIYPGNGNTAYNQKFKG (SEQ ID NO: 226), and heavy chain CDR 3 of WGSVVGDWYFDV (SEQ ID NO: 227).
Illustrative Disease Modifying Therapies
EXAMPLES
Example 1. Identification and Characterization of Human XCR1 Ab AFNs
As used in this Example and associated figures,“AFN” is a chimera of the anti-Xcr1 5G7 antibody and human IFNa2 with an R149A mutation.
AFNs were made based on the 5G7 anti-hXcr1 Ab using the intact (full) Ab or a scFv format.
The 5G7 heavy chain is:
QAYLQQSGAELVRPGASVKMSCKASGYTFTSHNLHWVKQTPRQGLQWIGAIYPGNGNTAYNQKFKGKATLTVD
KSSSTAYMQLSSLTSDDSAVYFCARWGSVVGDWYFDVWGTGTTVTVSSASTKGPSVFPLAPCSRSTSESTAAL
GCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSWTVPSSNFGTQTYTCNVDHKPSNTKVDKTVE
RKCCVECPPCPAPPAAAPSVFLFPPKPKDTLMISRTPEVTCVWDVSHEDPEVQFNWYVDGVEVHNAKTKPREE
QFNSTFRVVSVLTWHQDWLNGKEYKCKVSNKGLPAPIEKTISKTKGQPREPQVYTLPPSREEMTKNQVSLTCLV
KGFYPSDIAVEWESNGQPENNYKTTPPMLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLS
LSPGK (SEQ ID NO: 223)
The 5G7 light chain is:
DWMTQTPLSLPVTLGNQASIFCRSSLGLVHRNGNTYLHWYLQKPGQSPKLLIYKVSHRFSGVPDRFSGSGSGT DFTLKISRVEAEDLGVYFCSQSTHVPWTFGGGTKLEIKRTVAAPSVFIFPPSDEQLKSGTASWCLLNNFYPREAK VQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC (SEQ ID NO: 224)
5G7 Heavy chain CDR 1 is SHNLH (SEQ ID NO: 225), Heavy chain CDR 2 is AIYPGNGNTAYNQKFKG (SEQ ID NO: 226), Heavy chain CDR 3 is WGSVVGDWYFDV (SEQ ID NO: 227). 5G7 Light chain CDR 1 is RSSLGLVHRNGNTYLH (SEQ ID NO: 228), Light chain CDR 2 is KVSHRFS (SEQ ID NO: 229), and Light chain CDR 3 is SQSTHVPWT (SEQ ID NO: 230).
The sequence of hulFNa2(R149A) is:
CDLPQTHSLGSRRTLMLLAQMRKISLFSCLKDRHDFGFPQEEFGNQFQKAETIPVLHEMIQQIFNLFSTKDSSAA WDETLLDKFYTELYQQLNDLEACVIQGVGVTETPLMKEDSILAVRKYFQRITLYLKEKKYSPCAWEVVRAEIMASF SLSTNLQESLRSKE (SEQ ID NO: 231).
In case of the intact Ab AFN, the 5G7 Ab heavy chain was fused to h I FN a2_R149A (human IFNal with a R149A mutation) via a flexible (GGS)2oG-linker and co-expressed with the 5G7 Ab light chain (sequences shown below). 5G7 scFv-AFN was constructed by linking the Ab VL and VH domains via a (GGGS)4 linker and followed by a (GGS)2o-linker and the sequence encoding hlFNa2_R149A. Recombinant proteins, cloned in the pcDNA3.4 expression-vector, were produced in ExpiCHO cells (Thermo Fisher Scientific) and purified on HisPUR spin plates (Thermo Fisher Scientific) according to the manufacturer’s instructions.
To test binding of the AFNs, parental HL1 16 and HL1 16 cells stably expressing hXcrl (HL116-hXcr1) were incubated with a serial dilution AFN for two hours at 4°C. Binding was detected using THE™ HIS antibody-FITC (GenScript) and measured on a MACSQuant X instrument (Miltenyi Biotec) and analysed using the FlowLogic software (Miltenyi Biotec). Data in Figures 1A and 1 B clearly show that both 5G7 Ab-AFN and 5G7 scFv bind specifically to hXcrl expressing cells.
Biological activity was measured on parental HL1 16 cells (an IFN responsive cell-line stably transfected with a p6-16 luciferase reporter) and the derived HL116-hXcr1 cells. Cells were seeded overnight and stimulated for 6 hours with a serial dilution 5G7 AFNs. Luciferase activity was measured on an EnSight Multimode Plate Reader (Perkin Elmer). Data in Figures 2A and 2B clearly illustrate that 5G7 AFNs, in the intact Ab format or as scFv, are clearly more active on cells expressing hXcrl compared to parental cells, illustrating that it is possible to restore signaling of an IFNa2 mutant by specific targeting to hXcrl .
Example 2. Identification and Characterization of Mouse Xcr1 Ab AFNs
As used in this Example and associated figures,“AFN” is a chimera of the anti-Xcr1 MAARX10 antibody and human IFNa2 with Q124R mutation.
Similar to the anti-human Xcr1 Ab, AFNs based on the MARX10 anti-mouse Xcr1 Ab were made, as intact Ab or as scFv. In case of the intact Ab AFN, the MARX10 Ab heavy chain was fused to hlFNa2_Q124R (human IFNa2 with Q124R mutation) via a flexible (GGS)2oG-linker and co-expressed with the MARX10 Ab light chain. scFv-AFN was constructed by linking the Ab VL and VH domains, in VH-VL (scFv(1 )) or VL-VH (scFv(2)) orientation, via a (GGGS)4 linker and followed by a (GGS)2o-linker and h I FN a2_Q 124R.
Selectivity of AFNs (produced and purified as described above for the human Xcr1 Ab AFNs) was tested by comparing binding at 2.5 pg/ml to MOCK or mouse Xcr1 transfected Hek293T cells. Binding was detected using THE™ HIS antibody-FITC (GenScript) and measured on a MACSQuant X instrument (Miltenyi Biotec) and analysed using the FlowLogic software (Miltenyi Biotec). Data in Figure 3 clearly show that all three specifically bind to mXcrl expressing cells.
REF
New England Journal of Medicine (2021), 384(8), 705-716
https://www.rxlist.com/abecma-drug.htm#indications
///////////Idecabtagene vicleucel, breakthrough therapy designation, orphan drug designation, FDA 2021, APPROVALS 2021, Bb2121, Bb , ABECMA
Manufacturer: Celgene Corporation, a Bristol-Myers Squibb Company
Indications:
- Treatment of adult patients with relapsed or refractory multiple myeloma after four or more prior lines of therapy including an immunomodulatory agent, a proteasome inhibitor, and an anti-CD38 monoclonal antibody.
Product Information
- Package Insert – ABECMA
- Demographic Subgroup Information – idecabtagene vicleucel [ABECMA]
Refer to Section 1.1 of the Clinical Review Memo for information about participation in the clinical trials and any analysis of demographic subgroup outcomes that is notable.
Supporting Documents
Fosdenopterin hydrobromide

Fosdenopterin hydrobromide
FDA APPR 2021/2/26, NULIBRY
BBP-870/ORGN001
a cyclic pyranopterin monophosphate (cPMP) substrate replacement therapy, for the treatment of patients with molybdenum cofactor deficiency (MoCD) Type A.
| ホスデノプテリン臭化水素酸塩水和物; |
| Formula | C10H14N5O8P. 2H2O. HBr |
|---|---|
| CAS | 2301083-34-9DIHYDRATE |
| Mol weight | 480.1631 |
2301083-34-9
(1R,10R,12S,17R)-5-amino-11,11,14-trihydroxy-14-oxo-13,15,18-trioxa-2,4,6,9-tetraza-14λ5-phosphatetracyclo[8.8.0.03,8.012,17]octadeca-3(8),4-dien-7-one;dihydrate;hydrobromide
1,3,2-DIOXAPHOSPHORINO(4′,5′:5,6)PYRANO(3,2-G)PTERIDIN-10(4H)-ONE, 8-AMINO-4A,5A,6,9,11,11A,12,12A-OCTAHYDRO-2,12,12-TRIHYDROXY-, 2-OXIDE, HYDROBROMIDE, HYDRATE (1:1:2), (4AR,5AR,11AR,12AS)-
| CYCLIC PYRANOPTERIN MONOPHOSPHATE MONOHYDROBROMIDE DIHYDRATE |
(4aR,5aR,11aR,12aS)-8-Amino-2,12,12-trihydroxy-4a,5a,6,7,11,11a,12,12aoctahydro-2H-2lambda5-(1,3,2)dioxaphosphinino(4′,5′:5,6)pyrano(3,2-g)pteridine-2,10(4H)-dione, hydrobromide (1:1:2)
1,3,2-Dioxaphosphorino(4′,5′:5,6)pyrano(3,2-g)pteridin-10(4H)-one, 8-amino-4a,5a,6,9,11,11a,12,12a-octahydro-2,12,12-trihydroxy-, 2-oxide, hydrobromide, hydrate (1:1:2), (4aR,5aR,11aR,12aS)-
1,3,2-Dioxaphosphorino(4′,5′:5,6)pyrano(3,2-g)pteridin-10(4H)-one, 8-amino-4a,5a,6,9,11,11a,12,12a-octahydro-2,12,12-trihydroxy-, 2-oxide,hydrobromide, hydrate (1:1:2), (4aR,5aR,11aR,12aS)-
ALXN1101 HBr, UNII-X41B5W735T, X41B5W735T, D11780



C10H14N5O8P, Average: 363.223
150829-29-1
- ALXN-1101
- WHO 11150
- Synthesis ReferenceClinch K, Watt DK, Dixon RA, Baars SM, Gainsford GJ, Tiwari A, Schwarz G, Saotome Y, Storek M, Belaidi AA, Santamaria-Araujo JA: Synthesis of cyclic pyranopterin monophosphate, a biosynthetic intermediate in the molybdenum cofactor pathway. J Med Chem. 2013 Feb 28;56(4):1730-8. doi: 10.1021/jm301855r. Epub 2013 Feb 19.
Fosdenopterin (or cyclic pyranopterin monophosphate, cPMP), sold under the brand name Nulibry, is a medication used to reduce the risk of death due to a rare genetic disease known as molybdenum cofactor deficiency type A (MoCD-A).[1]
Adverse effects
The most common side effects include complications related to the intravenous line, fever, respiratory infections, vomiting, gastroenteritis, and diarrhea.[1]
Mechanism of action
People with MoCD-A cannot produce cyclic pyranopterin monophosphate (cPMP) in their body.[1] Fosdenopterin is an intravenous medication that replaces the missing cPMP.[1][2] cPMP is a precursor to molybdopterin, which is required for the enzyme activity of sulfite oxidase, xanthine dehydrogenase/oxidase and aldehyde oxidase.[3]
History
Fosdenopterin was developed by José Santamaría-Araujo and Guenter Schwarz at the German universities TU Braunschweig and the University of Cologne.[4][5]
The effectiveness of fosdenopterin for the treatment of MoCD-A was demonstrated in thirteen treated participants compared to eighteen matched, untreated participants.[1][6] The participants treated with fosdenopterin had a survival rate of 84% at three years, compared to 55% for the untreated participants.[1]
The U.S. Food and Drug Administration (FDA) granted the application for fosdenopterin priority review, breakthrough therapy, and orphan drug designations along with a rare pediatric disease priority review voucher.[1] The FDA granted the approval of Nulibry to Origin Biosciences, Inc., in February 2021.[1] It is the first medication approved for the treatment of MoCD-A.[1]
References
- ^ Jump up to:a b c d e f g h i j “FDA Approves First Treatment for Molybdenum Cofactor Deficiency Type A”. U.S. Food and Drug Administration (FDA) (Press release). 26 February 2021. Retrieved 26 February 2021.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ DrugBank DB16628 . Accessed 2021-03-05.
- ^ Santamaria-Araujo JA, Fischer B, Otte T, Nimtz M, Mendel RR, Wray V, Schwarz G (April 2004). “The tetrahydropyranopterin structure of the sulfur-free and metal-free molybdenum cofactor precursor”. The Journal of Biological Chemistry. 279 (16): 15994–9. doi:10.1074/jbc.M311815200. PMID 14761975.
- ^ Schwarz G, Santamaria-Araujo JA, Wolf S, Lee HJ, Adham IM, Gröne HJ, et al. (June 2004). “Rescue of lethal molybdenum cofactor deficiency by a biosynthetic precursor from Escherichia coli”. Human Molecular Genetics. 13 (12): 1249–55. doi:10.1093/hmg/ddh136. PMID 15115759.
- ^ Tedmanson S (5 November 2009). “Doctors risk untried drug to stop baby’s brain dissolving”. TimesOnline.
- ^ Schwahn BC, Van Spronsen FJ, Belaidi AA, Bowhay S, Christodoulou J, Derks TG, et al. (November 2015). “Efficacy and safety of cyclic pyranopterin monophosphate substitution in severe molybdenum cofactor deficiency type A: a prospective cohort study”. Lancet. 386 (10007): 1955–63. doi:10.1016/S0140-6736(15)00124-5. PMID 26343839. S2CID 21954888.
External links
- “Fosdenopterin”. Drug Information Portal. U.S. National Library of Medicine.
Molybdenum cofactor deficiency (MoCD) is an exceptionally rare autosomal recessive disorder resulting in a deficiency of three molybdenum-dependent enzymes: sulfite oxidase (SOX), xanthine dehydrogenase, and aldehyde oxidase.1 Signs and symptoms begin shortly after birth and are caused by a build-up of toxic sulfites resulting from a lack of SOX activity.1,5 Patients with MoCD may present with metabolic acidosis, intracranial hemorrhage, feeding difficulties, and significant neurological symptoms such as muscle hyper- and hypotonia, intractable seizures, spastic paraplegia, myoclonus, and opisthotonus. In addition, patients with MoCD are often born with morphologic evidence of the disorder such as microcephaly, cerebral atrophy/hypodensity, dilated ventricles, and ocular abnormalities.1 MoCD is incurable and median survival in untreated patients is approximately 36 months1 – treatment, then, is focused on improving survival and maintaining neurological function.
The most common subtype of MoCD, type A, involves mutations in MOCS1 wherein the first step of molybdenum cofactor synthesis – the conversion of guanosine triphosphate into cyclic pyranopterin monophosphate (cPMP) – is interrupted.1,3 In the past, management strategies for this disorder involved symptomatic and supportive treatment,5 though efforts were made to develop a suitable exogenous replacement for the missing cPMP. In 2009 a recombinant, E. coli-produced cPMP was granted orphan drug designation by the FDA, becoming the first therapeutic option for patients with MoCD type A.1
Fosdenopterin was approved by the FDA on Februrary 26, 2021, for the reduction of mortality in patients with MoCD type A,5 becoming the first and only therapy approved for the treatment of MoCD. By improving the three-year survival rate from 55% to 84%,7 and considering the lack of alternative therapies available, fosdenopterin appears poised to become a standard of therapy in the management of this debilitating disorder.
Fosdenopterin replaces an intermediate substrate in the synthesis of molybdenum cofactor, a compound necessary for the activation of several molybdenum-dependent enzymes including sulfite oxidase (SOX).1 Given that SOX is responsible for detoxifying sulfur-containing acids and sulfites such as S-sulfocysteine (SSC), urinary levels of SSC can be used as a surrogate marker of efficacy for fosdenopterin.7 Long-term therapy with fosdenopterin has been shown to result in a sustained reduction in urinary SSC normalized to creatinine.7
Animal studies have identified a potential risk of phototoxicity in patients receiving fosdenopterin – these patients should avoid or minimize exposure to sunlight and/or artificial UV light.7 If sun exposure is necessary, use protective clothing, hats, and sunglasses,7 in addition to seeking shade whenever practical. Consider the use of a broad-spectrum sunscreen in patients 6 months of age or older.8
Molybdenum cofactor deficiency (MoCD) is a rare autosomal-recessive disorder in which patients are deficient in three molybdenum-dependent enzymes: sulfite oxidase (SOX), xanthine dehydrogenase, and aldehyde dehydrogenase.1 The loss of SOX activity appears to be the main driver of MoCD morbidity and mortality, as the build-up of neurotoxic sulfites typically processed by SOX results in rapid and progressive neurological damage. In MoCD type A, the disorder results from a mutation in the MOCS1 gene leading to deficient production of MOCS1A/B,7 a protein that is responsible for the first step in the synthesis of molybdenum cofactor: the conversion of guanosine triphosphate into cyclic pyranopterin monophosphate (cPMP).1,4
Fosdenopterin is an exogenous form of cPMP, replacing endogenous production and allowing for the synthesis of molybdenum cofactor to proceed.7
- Mechler K, Mountford WK, Hoffmann GF, Ries M: Ultra-orphan diseases: a quantitative analysis of the natural history of molybdenum cofactor deficiency. Genet Med. 2015 Dec;17(12):965-70. doi: 10.1038/gim.2015.12. Epub 2015 Mar 12. [PubMed:25764214]
- Schwahn BC, Van Spronsen FJ, Belaidi AA, Bowhay S, Christodoulou J, Derks TG, Hennermann JB, Jameson E, Konig K, McGregor TL, Font-Montgomery E, Santamaria-Araujo JA, Santra S, Vaidya M, Vierzig A, Wassmer E, Weis I, Wong FY, Veldman A, Schwarz G: Efficacy and safety of cyclic pyranopterin monophosphate substitution in severe molybdenum cofactor deficiency type A: a prospective cohort study. Lancet. 2015 Nov 14;386(10007):1955-63. doi: 10.1016/S0140-6736(15)00124-5. Epub 2015 Sep 3. [PubMed:26343839]
- Iobbi-Nivol C, Leimkuhler S: Molybdenum enzymes, their maturation and molybdenum cofactor biosynthesis in Escherichia coli. Biochim Biophys Acta. 2013 Aug-Sep;1827(8-9):1086-101. doi: 10.1016/j.bbabio.2012.11.007. Epub 2012 Nov 29. [PubMed:23201473]
- Mendel RR: The molybdenum cofactor. J Biol Chem. 2013 May 10;288(19):13165-72. doi: 10.1074/jbc.R113.455311. Epub 2013 Mar 28. [PubMed:23539623]
- FDA News Release: FDA Approves First Treatment for Molybdenum Cofactor Deficiency Type A [Link]
- OMIM: MOLYBDENUM COFACTOR DEFICIENCY, COMPLEMENTATION GROUP A (# 252150) [Link]
- FDA Approved Drug Products: Nulibry (fosdenopterin) for intravenous injection [Link]
- Health Canada: Sun safety tips for parents [Link]
SYN
Journal of Biological Chemistry (1995), 270(3), 1082-7.
https://linkinghub.elsevier.com/retrieve/pii/S0021925818829696
PATENT
WO 2005073387
PATENT
WO 2012112922
PAPER

Journal of Medicinal Chemistry (2013), 56(4), 1730-1738
https://pubs.acs.org/doi/10.1021/jm301855r

Cyclic pyranopterin monophosphate (1), isolated from bacterial culture, has previously been shown to be effective in restoring normal function of molybdenum enzymes in molybdenum cofactor (MoCo)-deficient mice and human patients. Described here is a synthesis of 1 hydrobromide (1·HBr) employing in the key step a Viscontini reaction between 2,5,6-triamino-3,4-dihydropyrimidin-4-one dihydrochloride and d-galactose phenylhydrazone to give the pyranopterin (5aS,6R,7R,8R,9aR)-2-amino-6,7-dihydroxy-8-(hydroxymethyl)-3H,4H,5H,5aH,6H,7H,8H,9aH,10H-pyrano[3,2-g]pteridin-4-one (10) and establishing all four stereocenters found in 1. Compound 10, characterized spectroscopically and by X-ray crystallography, was transformed through a selectively protected tri-tert-butoxycarbonylamino intermediate into a highly crystalline tetracyclic phosphate ester (15). The latter underwent a Swern oxidation and then deprotection to give 1·HBr. Synthesized 1·HBr had in vitro efficacy comparable to that of 1 of bacterial origin as demonstrated by its enzymatic conversion into mature MoCo and subsequent reconstitution of MoCo-free human sulfite oxidase–molybdenum domain yielding a fully active enzyme. The described synthesis has the potential for scale up.
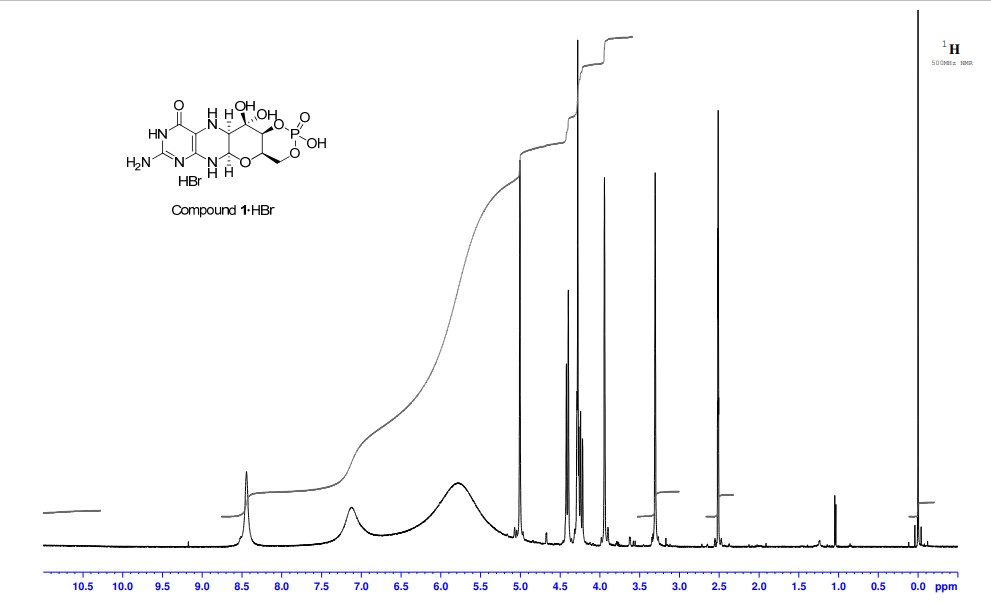
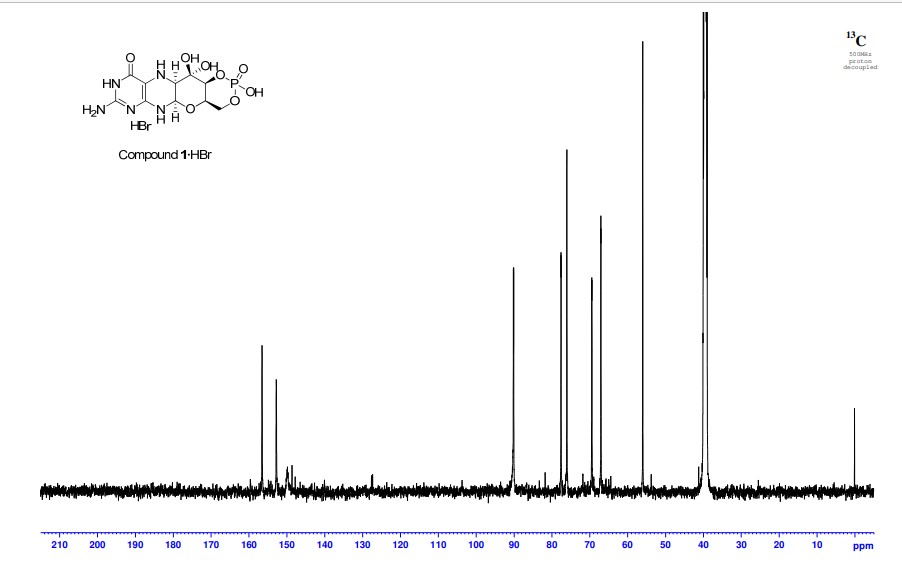





PAPER
European Journal of Organic Chemistry (2014), 2014(11), 2231-2241.
https://chemistry-europe.onlinelibrary.wiley.com/doi/abs/10.1002/ejoc.201301784
Abstract
The first synthesis of an oxygen‐stable analogue of the natural product cyclic pyranopterin monophosphate (cPMP) is reported. In this approach, the hydropyranone ring is annelated to pyrazine by a sequence comprising ortho‐lithiation/acylation of a 2‐halopyrazine, followed by nucleophilic aromatic substitution. The tetrose substructure is introduced from the chiral pool, from D‐galactose or D‐arabitol.

Abstract
Molybdenum cofactor (Moco) deficiency is a lethal hereditary metabolic disease. A recently developed therapy requires continuous intravenous supplementation of the biosynthetic Moco precursor cyclic pyranopterin monophosphate (cPMP). The limited stability of the latter natural product, mostly due to oxidative degradation, is problematic for oral administration. Therefore, the synthesis of more stable cPMP analogues is of great interest. In this context and for the first time, the synthesis of a cPMP analogue, in which the oxidation‐labile reduced pterin unit is replaced by a pyrazine moiety, was achieved starting from the chiral pool materials D‐galactose or D‐arabitol. Our synthesis, 13 steps in total, includes the following key transformations: i) pyrazine lithiation, followed by acylation; ii) closure of the pyrane ring by nucleophilic aromatic substitution; and iii) introduction of phosphate.
Patent
https://patents.google.com/patent/US9260462B2/en
Molybdenum cofactor (Moco) deficiency is a pleiotropic genetic disorder. Moco consists of molybdenum covalently bound to one or two dithiolates attached to a unique tricyclic pterin moiety commonly referred to as molybdopterin (MPT). Moco is synthesized by a biosynthetic pathway that can be divided into four steps, according to the biosynthetic intermediates precursor Z (cyclic pyranopterin monophosphate; cPMP), MPT, and adenylated MPT. Mutations in the Moco biosynthetase genes result in the loss of production of the molybdenum dependent enzymes sulfite-oxidase, xanthine oxidoreductase, and aldehyde oxidase. Whereas the activities of all three of these cofactor-containing enzymes are impaired by cofactor deficiency, the devastating consequences of the disease can be traced to the loss of sulfite oxidase activity. Human Moco deficiency is a rare but severe disorder accompanied by serious neurological symptoms including attenuated growth of the brain, untreatable seizures, dislocated ocular lenses, and mental retardation. Until recently, no effective therapy was available and afflicted patients suffering from Moco deficiency died in early infancy.
It has been found that administration of the molybdopterin derivative precursor Z, a relatively stable intermediate in the Moco biosynthetic pathway, is an effective means of therapy for human Moco deficiency and associated diseases related to altered Moco synthesis (see U.S. Pat. No. 7,504,095). As with most replacement therapies for illnesses, however, the treatment is limited by the availability of the therapeutic active agent.
Scheme 3.

Scheme 4.

(I).
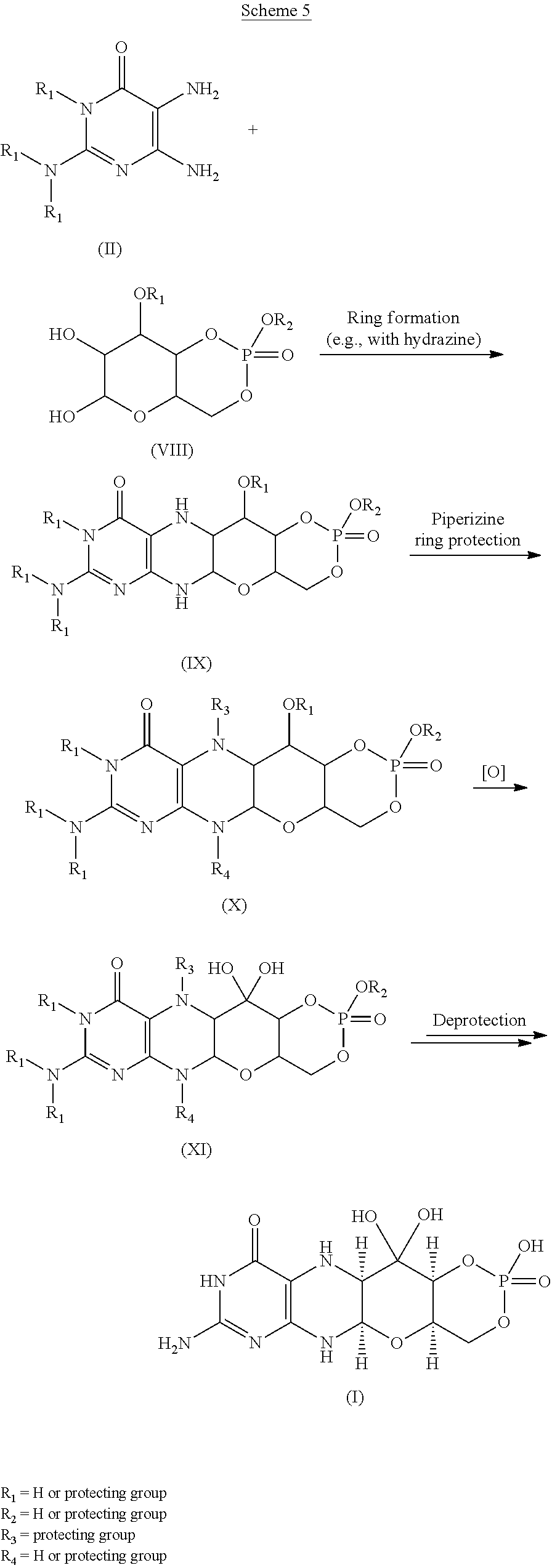
Scheme 6.

(I).

Scheme 8.

(I).

Scheme 10.

EXAMPLESExample 1Preparation of Precursor Z (cPMP)


Experimental
Air sensitive reactions were performed under argon. Organic solutions were dried over anhydrous MgSO4 and the solvents were evaporated under reduced pressure. Anhydrous and chromatography solvents were obtained commercially (anhydrous grade solvent from Sigma-Aldrich Fine Chemicals) and used without any further purification. Thin layer chromatography (t.l.c.) was performed on glass or aluminum sheets coated with 60 F254 silica gel. Organic compounds were visualized under UV light or with use of a dip of ammonium molybdate (5 wt %) and cerium(IV) sulfate 4H2O (0.2 wt %) in aq. H2SO4 (2M), one of I2 (0.2%) and KI (7%) in H2SO4 (1M), or 0.1% ninhydrin in EtOH. Chromatography (flash column) was performed on silica gel (40-63 μm) or on an automated system with continuous gradient facility. Optical rotations were recorded at a path length of 1 dm and are in units of 10−1 deg cm2 g−1; concentrations are in g/100 mL. 1H NMR spectra were measured in CDCl3, CD3OD (internal Me4Si, δ 0 ppm) or D2O(HOD, δ 4.79 ppm), and 13C NMR spectra in CDCl3 (center line, δ 77.0 ppm), CD3OD (center line, δ 49.0 ppm) or DMSO d6 (center line δ 39.7 ppm), D2O (no internal reference or internal CH3CN, δ 1.47 ppm where stated). Assignments of 1H and 13C resonances were based on 2D (1H—1H DQF-COSY, 1H—13C HSQC, HMBC) and DEPT experiments. 31P NMR were run at 202.3 MHz and are reported without reference. High resolution electrospray mass spectra (ESI-HRMS) were recorded on a Q-TOF Tandem Mass
Spectrometer. Microanalyses were performed by the Campbell Microanalytical Department, University of Otago, Dunedin, New Zealand.
A. Preparation of (5aS,6R,7R,8R,9aR)-2-amino-6,7-dihydroxy-8-(hydroxymethyl)-3H,4H,5H,5aH,6H,7H,8H,9aH,10H-pyrano[3,2-g]pteridin-4-one mono hydrate (1)
2,5,6-Triamino-3,4-dihydropyrimidin-4-one dihydrochloride (Pfleiderer, W.; Chem. Ber. 1957, 90, 2272; Org. Synth. 1952, 32, 45; Org. Synth. 1963, Coll. Vol. 4, 245, 10.0 g, 46.7 mmol), D-galactose phenylhydrazone (Goswami, S.; Adak, A. K. Tetrahedron Lett. 2005, 46, 221-224, 15.78 g, 58.4 mmol) and 2-mercaptoethanol (1 mL) were stirred and heated to reflux (bath temp 110° C.) in a 1:1 mixture of MeOH—H2O (400 mL) for 2 h. After cooling to ambient temperature, diethyl ether (500 mL) was added, the flask was shaken and the diethyl ether layer decanted off and discarded. The process was repeated with two further portions of diethyl ether (500 mL) and then the remaining volatiles were evaporated. Methanol (40 mL), H2O (40 mL) and triethylamine (39.4 mL, 280 mmol) were successively added and the mixture seeded with a few milligrams of 1. After 5 min a yellow solid was filtered off, washed with a little MeOH and dried to give 1 as a monohydrate (5.05 g, 36%) of suitable purity for further use. An analytical portion was recrystallized from DMSO-EtOH or boiling H2O. MPt 226 dec. [α]D 20 +135.6 (c1.13, DMSO). 1H NMR (DMSO d6): δ 10.19 (bs, exchanged D2O, 1H), 7.29 (d, J=5.0 Hz, slowly exchanged D2O, 1H), 5.90 (s, exchanged D2O, 2H), 5.33 (d, J=5.4 Hz, exchanged D2O, 1H), 4.66 (ddd, J˜5.0, ˜1.3, ˜1.3 Hz, 1H), 4.59 (t, J=5.6 Hz, exchanged D2O, 1H), 4.39 (d, J=10.3 Hz, exchanged D2O, 1H), 3.80 (bt, J˜1.8 Hz, exchanged D2O, 1H), 3.70 (m, 1H), 3.58 (dd, J=10.3, 3.0 Hz, 1H), 3.53 (dt, J=10.7, 6.4 Hz, 1H), 3.43 (ddd, J=11.2, 5.9, 5.9 Hz, 1H), 3.35 (t, J=6.4 Hz, 1H), 3.04 (br m, 1H). 13C NMR (DMSO d6 center line 6 39.7): δ 156.3 (C), 150.4 (C), 148.4 (C), 99.0 (C), 79.4 (CH), 76.5 (CH), 68.9 (CH), 68.6 (CH), 60.6 (CH2), 53.9 (CH). Anal. calcd. for C10H15N5O5H2O 39.60; C, 5.65; H, 23.09; N. found 39.64; C, 5.71; H, 22.83; N.
B. Preparation of Compounds 2 (a or b) and 3 (a, b or c)
Di-tert-butyl dicarbonate (10.33 g, 47.3 mmol) and DMAP (0.321 g, 2.63 mmol) were added to a stirred suspension of 1 (1.5 g, 5.26 mmol) in anhydrous THF (90 mL) at 50° C. under Ar. After 20 h a clear solution resulted. The solvent was evaporated and the residue chromatographed on silica gel (gradient of 0 to 40% EtOAc in hexanes) to give two product fractions. The first product to elute was a yellow foam (1.46 g). The product was observed to be a mixture of two compounds by 1H NMR containing mainly a product with seven Boc groups (2a or 2b). A sample was crystallized from EtOAc-hexanes to give 2a or 2b as a fine crystalline solid. MPt 189-191° C. [α]D 20 −43.6 (c 0.99, MeOH). 1H NMR (500 MHz, CDCl3): δ 5.71 (t, J=1.7 Hz, 1H), 5.15 (dt, J=3.5, ˜1.0, 1H), 4.97 (t, J=3.8, 1H), 4.35 (br t, J=˜1.7, 1H), 4.09-3.97 (m, 3H), 3.91 (m, 1H), 1.55, 1.52, 1.51, 1.50, 1.45 (5s, 45H), 1.40 (s, 18H). 13C NMR (125.7 MHz, CDCl3): δ 152.84 (C), 152.78 (C), 151.5 (C), 150.9 (C), 150.7 (2×C), 150.3 (C), 149.1 (C), 144.8 (C), 144.7 (C), 118.0 (C), 84.6 (C), 83.6 (C), 83.5 (C), 82.7 (3×C), 82.6 (C), 76.3 (CH), 73.0 (CH), 71.4 (CH), 67.2 (CH), 64.0 (CH2), 51.4 (CH), 28.1 (CH3), 27.8 (2×CH3), 27.7 (CH3), 27.6 (3×CH3). MS-ESI+ for C45H72N5O19 +, (M+H)+, Calcd. 986.4817. found 986.4818. Anal. calcd. for C45H71N5O19H2O 54.39; C, 7.39; H, 6.34; N. found 54.66; C, 7.17; H, 7.05; N. A second fraction was obtained as a yellow foam (2.68 g) which by 1H NMR was a product with six Boc groups present (3a, 3b or 3c). A small amount was crystallized from EtOAc-hexanes to give colorless crystals. [α]D 2O −47.6 (c, 1.17, CHCl3). 1H NMR (500 MHz, CDCl3): δ 11.10 (br s, exchanged D2O, 1H), 5.58 (t, J=1.8 Hz, 1H), 5.17 (d, J=3.4 Hz, 1H), 4.97 (t, J=3.9 Hz, 1H), 4.62 (s, exchanged D2O, 1H), 4.16 (dd, J=11.3, 5.9 Hz, 1H), 4.12 (dd, J=11.3, 6.4 Hz, 1H), 3.95 (dt, J=6.1, 1.1 Hz, 1H), 3.76 (m, 1H), 1.51, 1.50, 1.49, 1.48, 1.46 (5s, 54H). 13C NMR (125.7 MHz, CDCl3): δ 156.6 (C), 153.0 (C), 152.9 (C), 151.9 (C), 150.6 (C), 149.4 (2×C), 136.2 (C), 131.8 (C), 116.9 (C), 85.0 (2×C), 83.3 (C), 82.8 (C), 82.49 (C), 82.46 (C), 73.3 (CH), 71.5 (CH), 67.2 (CH), 64.5 (CH2), 51.3 (CH), 28.0, 27.72, 27.68, 27.6 (4×CH3). MS-ESI+ for C40H64N5O17 +, (M+H)+calcd. 886.4287. found 886.4289.
C. Preparation of Compound 4a, 4b or 4c
Step 1—The first fraction from B above containing mainly compounds 2a or 2b (1.46 g, 1.481 mmol) was dissolved in MeOH (29 mL) and sodium methoxide in MeOH (1M, 8.14 mL, 8.14 mmol) added. After leaving at ambient temperature for 20 h the solution was neutralized with Dowex 50WX8 (H+) resin then the solids filtered off and the solvent evaporated.
Step 2—The second fraction from B above containing mainly 3a, 3b or 3c (2.68 g, 3.02 mmol) was dissolved in MeOH (54 mL) and sodium methoxide in MeOH (1M, 12.10 mL, 12.10 mmol) added. After leaving at ambient temperature for 20 h the solution was neutralized with Dowex 50WX8 (H+) resin then the solids filtered off and the solvent evaporated.
The products from step 1 and step 2 above were combined and chromatographed on silica gel (gradient of 0 to 15% MeOH in CHCl3) to give 4a, 4b or 4c as a cream colored solid (1.97 g). 1H NMR (500 MHz, DMSO d6): δ 12.67 (br s, exchanged D2O, 1H), 5.48 (d, J=5.2 Hz, exchanged D2O, 1H), 5.43 (t, J=˜1.9 Hz, after D2O exchange became a d, J=1.9 Hz, 1H), 5.00 (br s, exchanged D2O, 1H), 4.62 (d, J=5.7 Hz, exchanged D2O, 1H), 4.27 (d, J=6.0 Hz, exchanged D2O, 1H), 3.89 (dt, J=5.2, 3.8 Hz, after D2O became a t, J=3.9 Hz, 1H), 3.62 (dd, J=6.0, 3.7 Hz, after D2O exchange became a d, J=3.7 Hz, 1H), 3.52-3.39 (m, 4H), 1.42 (s, 9H), 1.41 (s, 18H). 13C NMR (125.7 MHz, DMSO d6): δ 157.9 (C), 151.1, (C), 149.8 (2×C), 134.6 (C), 131.4 (C), 118.8 (C), 83.5 (2×C), 81.3 (C), 78.2 (CH), 76.5 (CH), 68.1 (CH), 66.8 (CH), 60.6 (CH2), 54.4 (CH), 27.9 (CH3), 27.6 (2×CH3). MS-ESI+ for C25H40N5O11 +, (M+H)+ calcd. 586.2719. found 586.2717.
D. Preparation of Compound 5a, 5b or 5c
Compound 4a, 4b or 4c (992 mg, 1.69 mmol) was dissolved in anhydrous pyridine and concentrated. The residue was dissolved in anhydrous CH2Cl2 (10 mL) and pyridine (5 mL) under a nitrogen atmosphere and the solution was cooled to −42° C. in an acetonitrile/dry ice bath. Methyl dichlorophosphate (187 μL, 1.86 mmol) was added dropwise and the mixture was stirred for 2 h 20 min. Water (10 mL) was added to the cold solution which was then removed from the cold bath and diluted with ethyl acetate (50 mL) and saturated NaCl solution (30 mL). The organic portion was separated and washed with saturated NaCl solution. The combined aqueous portions were extracted twice further with ethyl acetate and the combined organic portions were dried over MgSO4 and concentrated. Purification by silica gel flash column chromatography (eluting with 2-20% methanol in ethyl acetate) gave the cyclic methyl phosphate 5a, 5b or 5c (731 mg, 65%). 1H NMR (500 MHz, CDCl3,): δ 11.72 (bs, exchanged D2O, 1H), 5.63 (t, J=1.8 Hz, 1H), 5.41 (s, exchanged D2O, 1H), 4.95 (d, J=3.2 Hz, 1H), 4.70 (dt, J=12.4, 1.8 Hz, 1H), 4.42 (dd, J=22.1, 12.1 Hz, 1H). 4.15 (q, J=3.7 Hz, 1H), 3.82 (s, 1H), 3.75 (s, 1H), 3.58 (d, J=11.7 Hz, 3H), 2.10 (bs, exchanged D20, 1H+H2O), 1.50 (s, 9H), 1.46 (s, 18H). 13C NMR (125.7 MHz, CDCl3, centre line δ 77.0): δ 157.5 (C), 151.2 (C), 149.6 (2×C), 134.5 (C), 132.3 (C), 117.6 (C), 84.7 (2×C), 82.8 (C), 77.3 (CH), 74.8 (d, J=4.1 Hz, CH), 69.7 (CH2), 68.8 (d, J=4.1 Hz, CH), 68.6 (d, J=5.9 Hz, CH), 56.0 (d, J=7.4 Hz, CH3), 51.8 (CH), 28.1 (CH3), 27.8 (CH3). MS-ESI+ for C26H40N5NaO13P+ (M+Na)+, calcd. 684.2252. found 684.2251.
E. Preparation of Compound 6a, 6b or 6c
Compound 5a, 5b or 5c (223 mg, 0.34 mmol) was dissolved in anhydrous CH2Cl2 (7 mL) under a nitrogen atmosphere. Anhydrous DMSO (104 μL, 1.46 mmol) was added and the solution was cooled to −78° C. Trifluoroacetic anhydride (104 μL, 0.74 mmol) was added dropwise and the mixture was stirred for 40 min. N,N-diisopropylethylamine (513 μL, 2.94 mmol) was added and the stirring was continued for 50 min at −78° C. Saturated NaCl solution (20 mL) was added and the mixture removed from the cold bath and diluted with CH2Cl2 (30 mL). Glacial acetic acid (170 μL, 8.75 mmol) was added and the mixture was stirred for 10 min. The layers were separated and the aqueous phase was washed with CH2Cl2 (10 mL). The combined organic phases were washed with 5% aqueous HCl, 3:1 saturated NaCl solution:10% NaHCO3 solution and saturated NaCl solution successively, dried over MgSO4, and concentrated to give compound 6a, 6b or 6c (228 mg, quant.) of suitable purity for further use. 1H NMR (500 MHz, CDCl3): δ 5.86 (m, 1 H), 5.07 (m, 1 H), 4.70-4.64 (m, 2 H), 4.49-4.40 (m, 1 H), 4.27 (m, 1 H), 3.56, m, 4 H), 1.49 (s, 9 H), 1.46 (s, 18 H) ppm. 13C NMR (500 MHz, CDCl3): δ 157.5 (C), 151.1 (C), 150.6 (2 C), 134.6 (C), 132.7 (C), 116.6 (C), 92.0 (C), 84.6 (2 C), 83.6 (C), 78.0 (CH), 76.0 (CH), 70.4 (CH2), 67.9 (CH), 56.2 (CH3) δ6.0 (CH), 28.2 (3CH3), 26.8 (6 CH3) ppm. 31P NMR (500 MHz, CDCl3): δ−6.3 ppm.
F. Preparation of compound 7: (4aR,5aR,11aR,12aS)-1,3,2-Dioxaphosphorino[4′,5′:5,6]pyrano[3,2-g]pteridin-10(4H)-one,8-amino-4-a,5a,6,9,11,11a,12,12a-octahydro-2,12,12-trihydroxy-2-oxide
Compound 6a, 6b or 6c (10 mg, 14.8 μmol was dissolved in dry acetonitrile (0.2 mL) and cooled to 0° C. Bromotrimethylsilane (19.2 μL, 148 μmol) was added dropwise and the mixture was allowed to warm to ambient temperature and stirred for 5 h during which time a precipitate formed. HCl(aq) (10 μl, 37%) was added and the mixture was stirred for a further 15 min. The mixture was centrifuged for 15 min (3000 g) and the resulting precipitate collected. Acetonitrile (0.5 mL) was added and the mixture was centrifuged for a further 15 min. The acetonitrile wash and centrifugation was repeated a further two times and the resulting solid was dried under high vacuum to give compound 7 (4 mg, 75%). 1H NMR (500 MHz, D2O): δ 5.22 (d, J=1.6 Hz, 1H), 4.34 (dt, J=13, 1.6 Hz, 1H), 4.29-4.27 (m, 1H), 4.24-4.18 (m, 1H), 3.94 (br m, 1H), 3.44 (t, J=1.4 Hz, 1H). 31P NMR (500 MHz, D2O): δ −4.8 MS-ESI+ for C10H15N5O8P+, (M+H)+calcd. 364.0653. found 364.0652.
Example 2Comparison of Precursor Z (cPMP) Prepared Synthetically to that Prepared from E. Coli in the In vitro Synthesis of Moco
In vitro synthesis of Moco was compared using samples of synthetic precursor Z (cPMP) and cPMP purified from E. coli. Moco synthesis also involved the use of the purified components E. coli MPT synthase, gephyrin, molybdate, ATP, and apo-sulfite oxidase. See U.S. Pat. No. 7,504,095 and “Biosynthesis and molecular biology of the molybdenum cofactor (Moco)” in Metal Ions in Biological Systems, Mendel, Ralf R. and Schwarz, Gunter, Informa Plc, 2002, Vol. 39, pages 317-68. The assay is based on the conversion of cPMP into MPT, the subsequent molybdate insertion using recombinant gephyrin and ATP, and finally the reconstitution of human apo-sulfite oxidase.
As shown in FIG. 1, Moco synthesis from synthetic cPMP was confirmed, and no differences in Moco conversion were found in comparison to E. coli purified cPMP.
Example 3Comparison of Precursor Z (cPMP) Prepared Synthetically to that Prepared from E. coli in the In vitro Synthesis of MPT
In vitro synthesis of MPT was compared using samples of synthetic precursor Z (cPMP) and cPMP purified from E. coli. MPT synthesis also involved the use of in vitro assembled MPT synthase from E. coli. See U.S. Pat. No. 7,504,095 and “Biosynthesis and molecular biology of the molybdenum cofactor (Moco)” in Metal Ions in Biological Systems, Mendel, Ralf R. and Schwarz, Gunter, Informa Plc, 2002, Vol. 39, pages 317-68. Three repetitions of each experiment were performed and are shown in FIGS. 2 and 3.
As shown in FIGS. 2 and 3, MPT synthesis from synthetic cPMP confirmed, and no apparent differences in MPT conversion were found when compared to E. coli purified cPMP. A linear conversion of cPMP into MPT is seen in all samples confirming the identity of synthetic cPMP (see FIG. 2). Slight differences between the repetitions are believed to be due to an inaccurate concentration determination of synthetic cPMP given the presence of interfering chromophores.
Example 4Preparation of Precursor Z (cPMP)
A. Preparation of Starting Materials

B. Introduction of the protected Phosphate

The formation of the cyclic phosphate using intermediate [10] (630 mg) gave the desired product [11] as a 1:1 mixture of diastereoisomers (494 mg, 69%).

C. Oxidation and Overall Deprotection of the Molecule
Oxidation of the secondary alcohol to the gem-diol did prove successful on intermediate [12], but the oxidized product [13] did show significant instability and could not be purified. For this reason, deprotection of the phosphate was attempted before the oxidation. However, the reaction of intermediate [11] with TMSBr led to complete deprotection of the molecule giving intermediate [14]. An attempt to oxidize the alcohol to the gem-diol using Dess-Martin periodinane gave the aromatized pteridine [15].
Oxidation of intermediate [11] with Dess-Martin periodinane gave a mixture of starting material, oxidized product and several by-products. Finally, intermediate [11] was oxidized using the method described Example 1. Upon treatment, only partial oxidation was observed, leaving a 2:1 mixture of [11]/[16]. The crude mixture was submitted to the final deprotection. An off white solid was obtained and analyzed by 1H-NMR and HPLC-MS. These analyses suggest that cPMP has been produced along with the deprotected precursor [11].
Because the analytical HPLC conditions gave a good separation of cPMP from the major impurities, this method will be repeated on a prep-HPLC in order to isolate the final material.
CLIP
BridgeBio Pharma And Affiliate Origin Biosciences Announces FDA Acceptance Of Its New Drug Application For Fosdenopterin For The Treatment Of MoCD Type A
Application accepted under Priority Review designation with Breakthrough Therapy Designation and Rare Pediatric Disease Designation previously grantedThere are currently no approved therapies for the treatment of MoCD Type A, which results in severe and irreversible neurological injury for infants and children.This is BridgeBio’s first NDA acceptanceSAN FRANCISCO, September 29, 2020 – BridgeBio Pharma, Inc. (Nasdaq: BBIO) and affiliate Origin Biosciences today announced the US Food and Drug Administration (FDA) has accepted its New Drug Application (NDA) for fosdenopterin (previously BBP-870/ORGN001), a cyclic pyranopterin monophosphate (cPMP) substrate replacement therapy, for the treatment of patients with molybdenum cofactor deficiency (MoCD) Type A.The NDA has been granted Priority Review designation. Fosdenopterin has previously been granted Breakthrough Therapy Designation and Rare Pediatric Disease Designation in the US and may be eligible for a priority review voucher if approved. It received Orphan Drug Designation in the US and Europe. This is BridgeBio’s first NDA acceptance.“We want to thank the patients, families, scientists, physicians and all others involved who helped us reach this critical milestone,” said BridgeBio CEO and founder Neil Kumar, Ph.D. “MoCD Type A is a devastating disease with a median survival of less than four years and we are eager for our investigational therapy to be available to patients, who currently have no approved treatment options. BridgeBio exists to help as many patients as possible afflicted with genetic diseases, no matter how rare. We are grateful that the FDA has accepted our first NDA for priority review and we look forward to submitting our second NDA later this year for infigratinib for second line treatment of cholangiocarcinoma.”About Fosdenopterin
Fosdenopterin is being developed for the treatment of patients with MoCD Type A. Currently, there are no approved therapies for the treatment of MoCD Type A, which results in severe and irreversible neurological injury with a median survival between 3 to 4 years. Fosdenopterin is a first-in-class cPMP hydrobromide dihydrate and is designed to treat MoCD Type A by replacing cPMP and permitting the two remaining MoCo synthesis steps to proceed, with activation of MoCo-dependent enzymes and elimination of sulfites.About Molybdenum Cofactor Deficiency (MoCD) Type A
MoCD Type A is an ultra-rare, autosomal recessive, inborn error of metabolism caused by disruption in molybdenum cofactor (MoCo) synthesis which is vital to prevent buildup of s-sulfocysteine, a neurotoxic metabolite of sulfite. Patients are often infants with severe encephalopathy and intractable seizures. Disease progression is rapid with a high infant mortality rate.Those who survive beyond the first few month’s experience profuse developmental delays and suffer the effects of irreversible neurological damage, including brain atrophy with white matter necrosis, dysmorphic facial features, and spastic paraplegia. Clinical presentation that can be similar to hypoxic-ischemic encephalopathy (HIE) or other neonatal seizure disorders may lead to misdiagnosis and underdiagnosis. Immediate testing for elevated sulfite levels and S-sulfocysteine in the urine and very low serum uric acid may help with suspicion of MoCD.About Origin Biosciences
Origin Biosciences, an affiliate of BridgeBio Pharma, is a biotechnology company focused on developing and commercializing a treatment for Molybdenum Cofactor Deficiency (MoCD) Type A. Origin is led by a team of veteran biotechnology executives. Together with patients and physicians, the company aims to bring a safe, effective treatment for MoCD Type A to market as quickly as possible. For more information on Origin Biosciences, please visit the company’s website at www.origintx.com.
About BridgeBio Pharma
BridgeBio is a team of experienced drug discoverers, developers and innovators working to create life-altering medicines that target well-characterized genetic diseases at their source. BridgeBio was founded in 2015 to identify and advance transformative medicines to treat patients who suffer from Mendelian diseases, which are diseases that arise from defects in a single gene, and cancers with clear genetic drivers. BridgeBio’s pipeline of over 20 development programs includes product candidates ranging from early discovery to late-stage development. For more information visit bridgebio.com.
| Clinical data | |
|---|---|
| Trade names | Nulibry |
| Other names | Precursor Z, ALXN1101 |
| License data | US DailyMed: Fosdenopterin |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 150829-29-1 |
| PubChem CID | 135894389 |
| DrugBank | DB16628 |
| ChemSpider | 17221217 |
| UNII | 4X7K2681Y7 |
| KEGG | D11779 |
| ChEMBL | ChEMBL2338675 |
| CompTox Dashboard (EPA) | DTXSID90934067 |
| Chemical and physical data | |
| Formula | C10H14N5O8P |
| Molar mass | 363.223 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| hideSMILESNC1=NC(=O)C2=C(N[C@@H]3O[C@@H]4COP(=O)(O)O[C@@H]4C(O)(O)[C@@H]3N2)N1 | |
| hideInChIInChI=1S/C10H14N5O8P/c11-9-14-6-3(7(16)15-9)12-4-8(13-6)22-2-1-21-24(19,20)23-5(2)10(4,17)18/h2,4-5,8,12,17-18H,1H2,(H,19,20)(H4,11,13,14,15,16)/t2-,4-,5+,8-/m1/s1Key:CZAKJJUNKNPTTO-AJFJRRQVSA-N |
//////////Fosdenopterin hydrobromide, ホスデノプテリン臭化水素酸塩水和物 , ALXN1101 HBr, UNII-X41B5W735T, X41B5W735T, D11780, BBP-870/ORGN001, Priority Review designation, Breakthrough Therapy Designation, Rare Pediatric Disease Designation, Orphan Drug Designation, molybdenum cofactor deficiency, ALXN-1101, WHO 11150, FDA 2021, APPROVALS 2021
#Fosdenopterin hydrobromide, #ホスデノプテリン臭化水素酸塩水和物 , #ALXN1101 HBr, #UNII-X41B5W735T, X41B5W735T, #D11780, #BBP-870/ORGN001, #Priority Review designation, #Breakthrough Therapy Designation, #Rare Pediatric Disease Designation, #Orphan Drug Designation, #molybdenum cofactor deficiency, #ALXN-1101, #WHO 11150, #FDA 2021, #APPROVALS 2021
C1C2C(C(C3C(O2)NC4=C(N3)C(=O)NC(=N4)N)(O)O)OP(=O)(O1)O.O.O.Br
NIROGACESTAT
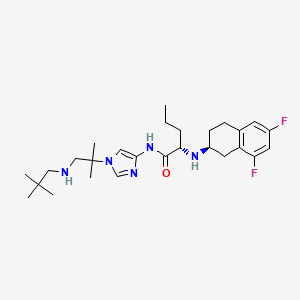

NIROGACESTAT
(2S)-2-[[(2S)-6,8-difluoro-1,2,3,4-tetrahydronaphthalen-2-yl]amino]-N-[1-[1-(2,2-dimethylpropylamino)-2-methylpropan-2-yl]imidazol-4-yl]pentanamide
489.6 g/mol, C27H41F2N5O
CAS 1290543-63-3
FDA APPROVED 11/27/2023, To treat adults with progressing desmoid tumors who require systemic treatment, Ogsiveo
PF-03084014, 1290543-63-3, PF-3084014, 865773-15-5QZ62892OFJUNII:QZ62892OFJUNII-QZ62892OFJнирогацестат [Russian] [INN]نيروغاسيستات [Arabic] [INN]尼罗司他 [Chinese] [INN]ニロガセスタット;
orphan drug designation in June 2018 for the treatment of desmoid tumors, and with a fast track designation
Nirogacestat, also known as PF-03084014, is a potent and selective gamma secretase (GS) inhibitor with potential antitumor activity. PF-03084014 binds to GS, blocking proteolytic activation of Notch receptors. Nirogacestat enhances the Antitumor Effect of Docetaxel in Prostate Cancer. Nirogacestat enhances docetaxel-mediated tumor response and provides a rationale to explore GSIs as adjunct therapy in conjunction with docetaxel for men with CRPC (castration-resistant prostate cancer).
Nirogacestat was disclosed to be a gamma-secretase inhibitor, which can inhibit Aβ-peptide production. SpringWorks Therapeutics (a spin-out of Pfizer ) is developing nirogacestat, as hydrobromide salt, a gamma-secretase inhibitor, for treating aggressive fibromatosis. In February 2021, nirogacestat was reported to be in phase 3 clinical development.
Nirogacestat is a selective gamma secretase (GS) inhibitor with potential antitumor activity. Nirogacestat binds to GS, blocking proteolytic activation of Notch receptors; Notch signaling pathway inhibition may follow, which may result in the induction of apoptosis in tumor cells that overexpress Notch. The integral membrane protein GS is a multi-subunit protease complex that cleaves single-pass transmembrane proteins, such as Notch receptors, at residues within their transmembrane domains. Overexpression of the Notch signaling pathway has been correlated with increased tumor cell growth and survival.
Nirogacestat has been used in trials studying the treatment of Breast Cancer, HIV Infection, Desmoid Tumors, Advanced Solid Tumors, and Aggressive Fibromatosis, among others.
Nirogacestat (Gamma Secretase Inhibitor)
Nirogacestat is an oral, selective, small molecule, gamma secretase inhibitor (GSI) in Phase 3 clinical development for patients with desmoid tumors. Gamma secretase is a protease complex that cleaves, or divides, multiple transmembrane protein complexes, including Notch, which, when dysregulated, can play a role in activating pathways that contribute to desmoid tumor growth.
Gamma secretase has also been shown to directly cleave BCMA, a therapeutic target that is highly expressed on multiple myeloma cells. By inhibiting gamma secretase with nirogacestat, membrane-bound BCMA can be preserved, thereby increasing target density while simultaneously reducing levels of soluble BCMA, which may serve as decoy receptors for BCMA-directed therapies. Together, these mechanisms combine to potentially enhance the activity of BCMA therapies and improve outcomes for multiple myeloma patients. SpringWorks is seeking to advance nirogacestat as a cornerstone of multiple myeloma combination therapy in collaboration with industry leaders who are advancing BCMA therapies.
SpringWorks Therapeutics Announces Clinical Collaboration with Pfizer
By Satish October 05, 2020
SpringWorks Therapeutics today announced that the company has entered into a clinical trial collaboration agreement with Pfizer to evaluate SpringWorks Therapeutics’ investigational gamma secretase inhibitor (GSI), nirogacestat, in combination with Pfizer’s anti-B-cell maturation antigen (BCMA) CD3 bispecific antibody, PF‐06863135, in patients with relapsed or refractory multiple myeloma.
Gamma secretase inhibition prevents the cleavage and shedding of BCMA from the surface of myeloma cells. In preclinical models, nirogacestat has been shown to increase the cell surface density of BCMA and reduce levels of soluble BCMA, thereby enhancing the activity of BCMA-targeted therapies, including CD3 bispecific antibodies.
Saqib Islam, Chief Executive Officer of SpringWorks Therapeutics Said: This collaboration is another important step in continuing to advance our goal of developing nirogacestat as a best-in-class BCMA potentiator, and we are pleased to work with Pfizer to study nirogacestat in combination with PF‐06863135, which has recently demonstrated promising monotherapy clinical data, We now have five collaborations with industry-leading BCMA developers to evaluate nirogacestat in combinations across modalities. We look forward to generating clinical data with our collaborators to further evaluate the ability of nirogacestat to improve outcomes for patients with multiple myeloma.

Under the terms of the agreement, Pfizer will sponsor and conduct the Phase 1b/2 study to evaluate the safety, tolerability and preliminary efficacy of the combination, and will assume all costs associated with the study, other than expenses related to the manufacturing of nirogacestat and certain expenses related to intellectual property rights. Pfizer and SpringWorks Therapeutics will also form a joint development committee to manage the clinical study, which is expected to commence in the first half of 2021.
Chris Boshoff, MD, PhD, Chief Development Officer for Pfizer Oncology at Pfizer Said: Entering into this clinical collaboration is a proud milestone in our strong relationship with SpringWorks,We believe that studying nirogacestat in combination with PF-06863135 could hold significant therapeutic promise for patients with relapsed or refractory multiple myeloma, and we look forward to working together to advance this important area of research.
In addition to its ongoing clinical collaborations with BCMA-directed therapies, SpringWorks is also currently conducting a global Phase 3, double-blind, randomized, placebo-controlled clinical trial (the DeFi Trial) to evaluate nirogacestat in adults with progressing desmoid tumors.
About Nirogacestat
Nirogacestat is an investigational, oral, selective, small molecule gamma secretase inhibitor in Phase 3 clinical development for desmoid tumors, which are rare and often debilitating and disfiguring soft-tissue tumors. Gamma secretase cleaves multiple transmembrane protein complexes, including Notch, which is believed to play a role in activating pathways that contribute to desmoid tumor growth.
In addition, gamma secretase has been shown to directly cleave membrane-bound BCMA, resulting in the release of the BCMA extracellular domain, or ECD, from the cell surface. By inhibiting gamma secretase, membrane-bound BCMA can be preserved, increasing target density while reducing levels of soluble BCMA ECD, which may serve as decoy receptors for BCMA-directed therapies. Nirogacestat’s ability to enhance the activity of BCMA-directed therapies has been observed in preclinical models of multiple myeloma. SpringWorks is evaluating nirogacestat as a BCMA potentiator and has five collaborations with industry-leading BCMA developers to evaluate nirogacestat in combinations across modalities, including with an antibody-drug conjugate, two CAR T cell therapies and two bispecific antibodies. In addition, SpringWorks and Fred Hutchinson Cancer Research Center have entered into a sponsored research agreement to further characterize the ability of nirogacestat to modulate BCMA and potentiate BCMA directed therapies using a variety of preclinical and patient-derived multiple myeloma models developed by researchers at Fred Hutch.
Nirogacestat has received Orphan Drug Designation from the U.S. Food and Drug Administration (FDA) for the treatment of desmoid tumors (June 2018) and from the European Commission for the treatment of soft tissue sarcoma (September 2019). The FDA also granted Fast Track and Breakthrough Therapy Designations for the treatment of adult patients with progressive, unresectable, recurrent or refractory desmoid tumors or deep fibromatosis (November 2018 and August 2019).
About PF‐06863135
PF‐06863135 is an anti-B-cell maturation antigen (BCMA) CD3 bispecific antibody being investigated in a Phase 1 clinical study to treat relapsed or refractory multiple myeloma. This bispecific antibody can be administered subcutaneously and has been optimized for binding affinity to both BCMA and CD3, enabling more potent T-cell-mediated tumor cell toxicity.
Source: SpringWorks Therapeutics
FDA Grants Breakthrough Designation to Nirogacestat for Desmoid Tumors
The FDA has granted nirogacestat, an investigational gamma-secretase inhibitor, with a breakthrough therapy designation for the treatment of adult patients with progressive, unresectable, recurrent or refractory desmoid tumors or deep fibromatosis.
The FDA has granted nirogacestat (PF-03084014), an investigational gamma-secretase inhibitor, with a breakthrough therapy designation for the treatment of adult patients with progressive, unresectable, recurrent or refractory desmoid tumors or deep fibromatosis.1
The breakthrough designation was granted as a result of positive findings seen in phase I and II trials of nirogacestat monotherapy in patients with desmoid tumors. A phase III trial has also been initiated investigating nirogacestat in patients with desmoid tumors or aggressive fibromatosis (NCT03785964).
“We are committed to pursuing the rapid development of nirogacestat given the important need for new therapies for patients with desmoid tumors and are pleased to receive this breakthrough therapy designation,” Saqib Islam, CEO of SpringWorks, the company developing the small molecule inhibitor, said in a statement. “We are currently enrolling adult patients in our phase III DeFi trial and will continue to work closely with the FDA with the goal of bringing nirogacestat to patients as quickly as possible.”
The open-label, single-center phase II trial of nirogacestat enrolled 17 patients with desmoid tumors who were not eligible for surgical resection or definitive radiation therapy and who had experienced disease progression after at least 1 prior treatment regimen. Patients received 150 mg twice per day of continuous, oral nirogacestat in 21-day cycles.2
The median age of patients was 34 years (range, 19-69), 82% of the patients were female, and 53% of patients had aCTNNB1T41A somatic missense mutation. The median number of prior therapies was 4 (range, 1-9), which included cytotoxic chemotherapy in 71% and a tyrosine kinase inhibitor in 59%.
Sixteen patients were evaluable for response. After a median follow-up of more than 25 months, 5 patients (29%) achieved a partial response and 11 (65%) had stable disease, for a disease control rate of 100%. Ten patients (59%) remained on treatment with nirogacestat for more than 2 years.
Grade 1/2 adverse events were observed in all patients, with diarrhea (76%) and skin disorders (71%) being the most common toxicities. The only treatment-related grade 3 event was reversible hypophosphatemia, which was reported in 8 patients (47%) and was considered to be a class effect of gamma-secretase inhibitors. Four patients met the criteria for dose reduction.
Findings from the phase I study also showed a disease control rate of 100% with nirogacestat. However, the median progression-free survival was not reached in either study due to a lack of patients progressing on treatment. Only 1 patient discontinued treatment due to an adverse event between the 2 studies.1
The FDA had previously granted nirogacestat with an orphan drug designation in June 2018 for the treatment of desmoid tumors, and with a fast track designation in November 2018 for the treatment of adult patients with progressive, unresectable, recurrent or refractory desmoid tumors or deep fibromatosis.
References
- SpringWorks Therapeutics Receives Breakthrough Therapy Designation for Nirogacestat for the Treatment of Adult Patients with Progressive, Unresectable, Recurrent or Refractory Desmoid Tumors [press release]. Stamford, CT: SpringWorks Therapeutics, Inc; August 29, 2019. https://bit.ly/30IV0Eb. Accessed September 3, 2019.
- Kummar S, O’Sullivan Coyne G, Do KT, et al. Clinical Activity of the γ-Secretase Inhibitor PF-03084014 in Adults With Desmoid Tumors (Aggressive Fibromatosis).J Clin Oncol.2017;35(14):1561-1569. doi: 10.1200/JCO.2016.71.1994.
PAPER

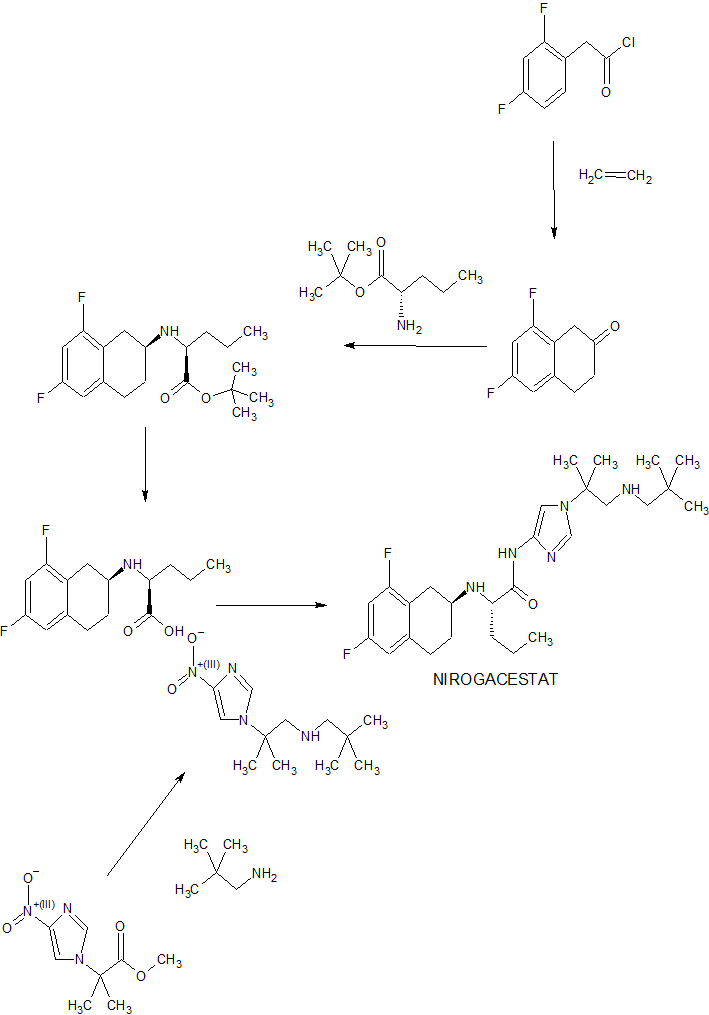
Bioorganic & medicinal chemistry letters (2011), 21(9), 2637-40.
https://www.sciencedirect.com/science/article/abs/pii/S0960894X10018822



PATENT
WO 2016089208
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016089208
PATENT
WO-2021029854
Novel, stable crystalline polymorphic (A to N) and amorphous forms of nirogacestat hydrobromide , useful for treating desmoid tumors such as multiple myeloma, a cancer having a mutation in a Notch pathway gene, adenoid cystic carcinoma and T-cell acute lymphoblastic leukemia.
(S)-2-(((S)-6,8-difluoro-l,2,3,4-tetrahydronaphthalen-2-yl)amino)-N-(l-(2- methyl- l-(neopentylamino) propan-2-yl)-lH-imidazol-4-yl)pentanamide (“Compound 1”) is a gamma-secretase inhibitor which can inhibit Ab-peptide production.
[0003] Not all compounds that are gamma-secretase inhibitors have characteristics affording the best potential to become useful therapeutics. Some of these characteristics include high affinity at the gamma-secretase, duration of gamma-secretase deactivation, oral bioavailability, tissue distribution, and stability (e.g., ability to formulate or crystallize, shelf life). Favorable characteristics can lead to improved safety, tolerability, efficacy, therapeutic index, patient compliance, cost efficiency, manufacturing ease, etc.
[0004] In addition, the isolation and commercial -scale preparation of a solid state form of hydrobromide salts of Compound 1 and corresponding pharmaceutical formulations having acceptable solid state properties (including chemical stability, thermal stability, solubility, hygroscopicity, and/or particle size), compound manufacturability (including yield, impurity rejection during crystallization, filtration properties, drying properties, and milling properties), and formulation feasibility (including stability with respect to pressure or compression forces during tableting) present a number of challenges.
[0005] Accordingly, there is a current need for one or more solid state forms of hydrobromide salts of Compound 1 that have an acceptable balance of these properties and can be used in the preparation of pharmaceutically acceptable solid dosage forms.
Crystalline Form A
[0147] In one aspect, the present disclosure relates to crystalline Form A of a hydrobromide salt of (S)-2-(((S)-6,8-difluoro-l,2,3,4-tetrahydronaphthalen-2-yl)amino)- N-(l -(2 -methyl- l-(neopentylamino) propan-2-yl)-lH-imidazol-4-yl)pentanamide having Formula (I),
[0148] In one embodiment, crystalline Form A is anhydrous.
[0149] In another embodiment, the melting point of crystalline Form A is about 254 °C.
[0150] In another embodiment, Form A is characterized by an XRPD pattern having peaks at 8.8 ± 0.2, 9.8 ± 0.2, and 23.3 ± 0.2 degrees two theta when measured by Cu Ka radiation. In another embodiment, Form A is characterized by an XRPD pattern having peaks at 8.8 ± 0.2, 9.8 ± 0.2, 23.3 ± 0.2, 25.4 ± 0.2, 28.0 ± 0.2, and 29.3 ± 0.2 degrees two theta when measured by Cu Ka radiation. In another embodiment, Form A is characterized by an XRPD pattern having peaks at 8.8 ± 0.2, 9.8 ± 0.2, 20.0 ± 0.2, 23.3 ± 0.2, 25.4 ± 0.2, 28.0 ± 0.2, 29.3 ± 0.2, and 32.5 ± 0.2 degrees two theta when measured by Cu Ka radiation.
Patent
Product case, WO2005092864 ,
hold protection in the EU states until March 2025, and expire in the US in February 2026 with US154 extension.
PATENT
WO2020208572 , co-assigned to GSK and SpringWorks, claiming a combination of nirogacestat with anti-BCMA antibody (eg belantamab mafodotin ), for treating cancer.
PATENT
US10590087 , for a prior filing from Pfizer, claiming crystalline forms of nirogacestat hydrobromide.
////////////NIROGACESTAT, orphan drug designation, esmoid tumors, fast track designation, PF-03084014, PF 03084014, QZ62892OFJ , UNII:QZ62892OFJ ,UNII-QZ62892OFJ, ,нирогацестат , نيروغاسيستات , 尼罗司他 , ニロガセスタット, phase 3
CCCC(C(=O)NC1=CN(C=N1)C(C)(C)CNCC(C)(C)C)NC2CCC3=C(C2)C(=CC(=C3)F)F
Ansuvimab-zykl

Ansuvimab-zykl
FDA APPROVED, 12/21/2020, EBANGA
To treat ebola
https://www.fda.gov/drugs/drug-safety-and-availability/fda-approves-treatment-ebola-virus
The U.S. Food and Drug Administration approved Ebanga (Ansuvimab-zykl), a human monoclonal antibody, for the treatment for Zaire ebolavirus (Ebolavirus) infection in adults and children. Ebanga blocks binding of the virus to the cell receptor, preventing its entry into the cell.
Zaire ebolavirus is one of four Ebolavirus species that can cause a potentially fatal human disease. It is transmitted through blood, body fluids, and tissues of infected people or wild animals, and through surfaces and materials, such as bedding and clothing, contaminated with these fluids. Individuals who care for people with the disease, including health care workers who do not use correct infection control precautions, are at the highest risk for infection.
During an Ebola outbreak in the Democratic Republic of the Congo (DRC) in 2018-2019, Ebanga was evaluated in a clinical trial (the PALM trial). The PALM trial was led by the U.S. National Institutes of Health and the DRC’s Institut National de Recherche Biomédicale with contributions from several other international organizations and agencies.
In the PALM trial, the safety and efficacy of Ebanga was evaluated in a multi-center, open-label, randomized controlled trial. 174 participants (120 adults and 54 pediatric patients) with confirmed Ebolavirus infection received Ebanga intravenously as a single 50 mg/kg infusion and 168 participants (135 adults and 33 pediatric patients) received an investigational control. The primary efficacy endpoint was 28-day mortality. The primary analysis population was all patients who were randomized and concurrently eligible to receive either Ebanga or the investigational control during the same time period of the trial. Of the 174 patients who received Ebanga, 35.1% died after 28 days, compared to 49.4% of the 168 patients who received a control.
The most common symptoms experienced while receiving Ebanga include: fever, tachycardia (fast heart rate), diarrhea, vomiting, hypotension (low blood pressure), tachypnea (fast breathing) and chills; however, these are also common symptoms of Ebolavirus infection. Hypersensitivity, including infusion-related events, can occur in patients taking Ebanga, and treatment should be discontinued in the event of a hypersensitivity reaction.
Patients who receive Ebanga should avoid the concurrent administration of a live virus vaccine against Ebolavirus. There is the potential for Ebanga to inhibit replication of a live vaccine virus and possibly reduce the efficacy of this vaccine.
Ebanga was granted an Orphan Drug designation, which provides incentives to assist and encourage drug development for rare diseases. Additionally, the agency granted Ebanga a Breakthrough Therapy designation.
FDA granted the approval to Ridgeback Biotherapeutics, LP.
Ansuvimab, sold under the brand name Ebanga, is a monoclonal antibody medication for the treatment of Zaire ebolavirus (Ebolavirus) infection.[1][2]
The most common symptoms include fever, tachycardia (fast heart rate), diarrhea, vomiting, hypotension (low blood pressure), tachypnea (fast breathing) and chills; however, these are also common symptoms of Ebolavirus infection.[1]
Ansuvimab was approved for medical use in the United States in December 2020.[1][2]
Chemistry
The drug is composed of a single monoclonal antibody (mAb) and was initially isolated from immortalized B-cells that were obtained from a survivor of the 1995 outbreak of Ebola virus disease in Kikwit, Democratic Republic of Congo.[3] In work supported by the United States National Institutes of Health and the Defense Advanced Projects Agency, the heavy and light chain sequences of ansuvimab mAb was cloned into CHO cell lines and initial production runs were produced by Cook Phamica d.b.a. Catalent under contract of Medimmune.[4][5]
Mechanism of action
Neutralization
Ansuvimab is a monoclonal antibody therapy that is infused intravenously into patients with Ebola virus disease. Ansuvimab is a neutralizing antibody,[3] meaning it binds to a protein on the surface of Ebola virus that is required to infect cells. Specifically, ansuvimab neutralizes infection by binding to a region of the Ebola virus envelope glycoprotein that, in the absence of ansuvimab, would interact with virus’s cell receptor protein, Niemann-Pick C1 (NPC1).[6][7][8] This “competition” by ansuvimab prevents Ebola virus from binding to NPC1 and “neutralizes” the virus’s ability to infect the targeted cell.[6]
Effector function
Antibodies have antigen-binding fragment (Fab) regions and constant fragment (Fc) regions. The Neutralization of virus infection occurs when the Fab regions of antibodies binds to virus antigen(s) in a manner that blocks infection. Antibodies are also able to “kill” virus particles directly and/or kill infected cells using antibody-mediated “effector functions” such as opsonization, complement-dependent cytotoxicity, antibody-dependent cell-mediated cytotoxicity and antibody-dependent phagocytosis. These effector functions are contained in the Fc region of antibodies, but is also dependent on binding of the Fab region to antigen. Effector functions also require the use of complement proteins in serum or Fc-receptor on cell membranes. Ansuvimab has been found to be capable of killing cells by antibody-dependent cell-mediated cytotoxicity.[3] Other functional killing tests have not been performed.
History
Ansuvimab is a monoclonal antibody that is being evaluated as a treatment for Ebola virus disease.[9] Its discovery was led by the laboratory of Nancy Sullivan at the United States National Institute of Health Vaccine Research Center and J. J. Muyembe-Tamfum from the Institut National pour la Recherche Biomedicale (INRB) in the Democratic Republic of Congo, working in collaboration with the Institute of Biomedical Research and the United States Army Medical Research Institute of Infectious Diseases.[3][10] Ansuvimab was isolated from the blood of a survivor of the 1995 outbreak of Ebola virus disease in Kikwit, Democratic Republic of Congo roughly ten years later.[3]
In 2018, a Phase 1 clinical trial of ansuvimab was conducted by Martin Gaudinski within the Vaccine Research Center Clinical Trials Program that is led by Julie E. Ledgerwood.[5][4][11] Ansuvimab is also being evaluated during the 2018 North Kivu Ebola outbreak.[12]
Ansuvimab has also shown success with lowering the mortality rate from ~70% to about 34%. In August 2019, Congolese health authorities, the World Health Organization, and the U.S. National Institutes of Health promoted the use of ansuvimab, alongside REGN-EB3, a similar Regeneron-produced monoclonal antibody treatment, over other treatments yielding higher mortality rates, after ending clinical trials during the outbreak.[13][14]
Discovery
A 2016 paper describes the efforts of how ansuvimab was originally developed as part of research efforts lead by Dr. Nancy Sullivan at the United States National Institute of Health Vaccine Research Center and Dr. J. J. Muyembe-Tamfum from the Institut National de Recherche Biomedicale (INRB) in the Democratic Republic of Congo.[3][10] This collaborative effort also involved researchers from Institute of Biomedical Research and the United States Army Medical Research Institute of Infectious Diseases.[3][10] A survivor from the 1995 outbreak of Ebola virus disease in Kikwit, Democratic Republic of Congo donated blood to the project that began roughly ten years after they had recovered.[3] Memory B cells isolated from the survivor’s blood were immortalized, cultured and screened for their ability to produce monoclonal antibodies that reacted with the glycoprotein of Ebola virus. Ansuvimab was identified from one of these cultures and the antibody heavy and light chain gene sequences were sequenced from the cells.[3] These sequences were then cloned into recombinant DNA plasmids and purified antibody protein for initial studies was produced in cells derived from HEK 293 cells.[3]
Ansuvimab and mAb100 combination
In an experiment described in the 2016 paper, rhesus macaques were infected with Ebola virus and treated with a combination of ansuvimab and another antibody isolated from the same subject, mAb100. Three doses of the combination were given once a day starting 1 day after the animals were infected. The control animal died and the treated animals all survived.[3]
Ansuvimab monotherapy
In a second experiment described in the 2016 paper, rhesus macaques were infected with Ebola virus and only treated with ansuvimab. Three doses of ansuvimab were given once a day starting 1 day or 5 days after the animals were infected. The control animals died and the treated animals all survived.[3] Unpublished data referred to in a publication of the 2018 Phase I clinical trial results of ansuvimab, reported that a single infusion of ansuvimab provided full protection of rhesus macaques and was the basis of the dosing used for human studies.[5][4]
Development
Ansuvimab was developed by the Vaccine Research Center with support of the United States National Institutes of Health and the Defense Advanced Projects Agency. The heavy and light chain sequences of ansuvimab mAb were cloned into CHO cell lines to enable large-scale production of antibody product for use in humans.[4][5]
Human safety testing
In early 2018,[9] a Phase 1 clinical trial of ansuvimab’s safety, tolerability and pharmacokinetics was conducted by Dr. Martin Gaudinski within the Vaccine Research Center Clinical Trials Program that is led by Dr. Julie E. Ledgerwood.[5][4][11] The study was performed in the United States at the NIH Clinical Center and tested single dose infusions of ansuvimab infused over 30 minutes. The study showed that ansuvimab was safe, had minimal side effects and had a half-life of 24 days.[5][4]
Ridgeback Biotherapeutics
A license for ansuvimab was obtained by Ridgeback Biotherapeutics in 2018, from the National Institutes of Health–National Institute of Allergy and Infectious Diseases.[15] Ansuvimab was given orphan drug status in May 2019 and March 2020.[16][17][18]
Experimental use in the Democratic Republic of Congo
During the 2018 Équateur province Ebola outbreak, ansuvimab was requested by the Democratic Republic of Congo (DRC) Ministry of Public Health. Ansuvimab was approved for compassionate use by the World Health Organization MEURI ethical protocol and at DRC ethics board. Ansuvimab was sent along with other therapeutic agents to the outbreak sites.[19][20][11] However, the outbreak came to a conclusion before any therapeutic agents were given to patients.[11]
Approximately one month following the conclusion of the Équateur province outbreak, a distinct outbreak was noted in Kivu in the DRC (2018–20 Kivu Ebola outbreak). Once again, ansuvimab received approval for compassionate use by WHO MEURI and DRC ethic boards and has been given to many patients under these protocols.[11] In November 2018, the Pamoja Tulinde Maisha (PALM [together save lives]) open-label randomized clinical control trial was begun at multiple treatment units testing ansuvimab, REGN-EB3 and remdesivir to ZMapp. Despite the difficulty of running a clinical trial in a conflict zone, investigators have enrolled 681 patients towards their goal of 725. An interim analysis by the Data Safety and Monitoring Board (DSMB) of the first 499 patient found that ansuvimab and REGN-EB3 were superior to the comparator ZMapp. Overall mortality of patients in the ZMapp and remdesivir groups were 49% and 53% compared to 34% and 29% for ansuvimab and REGN-EB3. When looking at patients who arrived early after disease symptoms appeared, survival was 89% for ansuvimab and 94% for REGN-EB3. While the study was not powered to determine whether there is any difference between REGN-EB3 and ansuvimab, the survival difference between those two therapies and ZMapp was significant. This led to the DSMB halting the study and PALM investigators dropping the remdesivir and ZMapp arms from the clinical trial. All patients in the outbreak who elect to participate in the trial will now be given either ansuvimab or REGN-EB3.[21][22][13][12]
In October 2020, the U.S. Food and Drug Administration (FDA) approved atoltivimab/maftivimab/odesivimab (Inmazeb, formerly REGN-EB3) with an indication for the treatment of infection caused by Zaire ebolavirus.[23]
FDA approves ansuvimab-zykl for Ebola virus infection
DECEMBER 21, 2020 BY JANICE REICHERThttps://www.antibodysociety.org/antibody-therapeutic/fda-approves-ansuvimab-zykl-for-ebola-virus-infection/embed/#?secret=zWW0Sr0BdW
On December 21, 2020, the US Food and Drug Administration approved Ebanga (ansuvimab-zykl) for the treatment for Zaire ebolavirus (Ebolavirus) infection in adults and children. Ebanga had been granted US Orphan Drug designation and Breakthrough Therapy designations. Ansuvimab is a human IgG1 monoclonal antibody that binds and neutralizes the virus.
The safety and efficacy of Ebanga were evaluated in the multi-center, open-label, randomized controlled PALM trial. In this study, 174 participants (120 adults and 54 pediatric patients) with confirmed Ebolavirus infection received Ebanga intravenously as a single 50 mg/kg infusion and 168 participants (135 adults and 33 pediatric patients) received an investigational control. The primary efficacy endpoint was 28-day mortality. Of the 174 patients who received Ebanga, 35.1% died after 28 days, compared to 49.4% of the 168 patients who received a control.
Ebanga is the 12th antibody therapeutic to be granted a first approval in the US or EU during 2020.
The Antibody Society maintains a comprehensive table of approved monoclonal antibody therapeutics and those in regulatory review in the EU or US. The table, which is located in the Web Resources section of the Society’s website, can be downloaded in Excel format.
References
- ^ Jump up to:a b c d “FDA Approves Treatment for Ebola Virus”. U.S. Food and Drug Administration. 21 December 2020. Retrieved 23 December 2020. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b “Ridgeback Biotherapeutics LP Announces the Approval of Ebanga for Ebola” (Press release). Ridgeback Biotherapeutics LP. 22 December 2020. Retrieved 23 December 2020– via Business Wire.
- ^ Jump up to:a b c d e f g h i j k l Corti D, Misasi J, Mulangu S, Stanley DA, Kanekiyo M, Wollen S, et al. (March 2016). “Protective monotherapy against lethal Ebola virus infection by a potently neutralizing antibody”. Science. 351 (6279): 1339–42. Bibcode:2016Sci…351.1339C. doi:10.1126/science.aad5224. PMID 26917593.
- ^ Jump up to:a b c d e f Clinical trial number NCT03478891 for “Safety and Pharmacokinetics of a Human Monoclonal Antibody, VRC-EBOMAB092-00-AB (MAb114), Administered Intravenously to Healthy Adults” at ClinicalTrials.gov
- ^ Jump up to:a b c d e f Gaudinski MR, Coates EE, Novik L, Widge A, Houser KV, Burch E, et al. (March 2019). “Safety, tolerability, pharmacokinetics, and immunogenicity of the therapeutic monoclonal antibody ansuvimab targeting Ebola virus glycoprotein (VRC 608): an open-label phase 1 study”. Lancet. 393 (10174): 889–898. doi:10.1016/S0140-6736(19)30036-4. PMC 6436835. PMID 30686586.
- ^ Jump up to:a b Misasi J, Gilman MS, Kanekiyo M, Gui M, Cagigi A, Mulangu S, et al. (March 2016). “Structural and molecular basis for Ebola virus neutralization by protective human antibodies”. Science. 351 (6279): 1343–6. Bibcode:2016Sci…351.1343M. doi:10.1126/science.aad6117. PMC 5241105. PMID 26917592.
- ^ Côté M, Misasi J, Ren T, Bruchez A, Lee K, Filone CM, et al. (August 2011). “Small molecule inhibitors reveal Niemann-Pick C1 is essential for Ebola virus infection”. Nature. 477 (7364): 344–8. Bibcode:2011Natur.477..344C. doi:10.1038/nature10380. PMC 3230319. PMID 21866101.
- ^ Carette JE, Raaben M, Wong AC, Herbert AS, Obernosterer G, Mulherkar N, et al. (August 2011). “Ebola virus entry requires the cholesterol transporter Niemann-Pick C1”. Nature. 477 (7364): 340–3. Bibcode:2011Natur.477..340C. doi:10.1038/nature10348. PMC 3175325. PMID 21866103.
- ^ Jump up to:a b “NIH begins testing Ebola treatment in early-stage trial”. National Institutes of Health (NIH). 2018-05-23. Retrieved 2018-10-15.
- ^ Jump up to:a b c Hayden EC (2016-02-26). “Ebola survivor’s blood holds promise of new treatment”. Nature. doi:10.1038/nature.2016.19440. ISSN 1476-4687.
- ^ Jump up to:a b c d e “NIH VideoCast – CC Grand Rounds: Response to an Outbreak: Ebola Virus Monoclonal Antibody (mAb114) Rapid Clinical Development”. videocast.nih.gov. Retrieved 2019-08-09.
- ^ Jump up to:a b Kingsley-Hall A. “Congo’s experimental mAb114 Ebola treatment appears successful: authorities | Central Africa”. http://www.theafricareport.com. Retrieved 2018-10-15.
- ^ Jump up to:a b McNeil DG (12 August 2019). “A Cure for Ebola? Two New Treatments Prove Highly Effective in Congo”. The New York Times. Retrieved 13 August 2019.
- ^ Molteni M (12 August 2019). “Ebola is Now Curable. Here’s How The New Treatments Work”. Wired. Retrieved 13 August 2019.
- ^ “Ridgeback Biotherapeutics LP announces licensing of mAb114, an experimental Ebola treatment, from the National Institute of Allergy and Infectious Diseases” (Press release). Ridgeback Biotherapeutics LP. Retrieved 2019-08-17 – via PR Newswire.
- ^ “Ansuvimab Orphan Drug Designations and Approvals”. accessdata.fda.gov. 8 May 2019. Retrieved 24 December 2020.
- ^ “Ansuvimab Orphan Drug Designations and Approvals”. accessdata.fda.gov. 30 March 2020. Retrieved 24 December 2020.
- ^ “Ridgeback Biotherapeutics LP Announces Orphan Drug Designation for mAb114”(Press release). Ridgeback Biotherapeutics LP. Retrieved 2019-08-17 – via PR Newswire.
- ^ Check Hayden, Erika (May 2018). “Experimental drugs poised for use in Ebola outbreak”. Nature. 557 (7706): 475–476. Bibcode:2018Natur.557..475C. doi:10.1038/d41586-018-05205-x. ISSN 0028-0836. PMID 29789732.
- ^ WHO: Consultation on Monitored Emergency Use of Unregistered and Investigational Interventions for Ebola virus Disease. https://www.who.int/emergencies/ebola/MEURI-Ebola.pdf
- ^ Mole B (2019-08-13). “Two Ebola drugs boost survival rates, according to early trial data”. Ars Technica. Retrieved 2019-08-17.
- ^ “Independent monitoring board recommends early termination of Ebola therapeutics trial in DRC because of favorable results with two of four candidates”. National Institutes of Health (NIH). 2019-08-12. Retrieved 2019-08-17.
- ^ “FDA Approves First Treatment for Ebola Virus”. U.S. Food and Drug Administration(FDA) (Press release). 14 October 2020. Retrieved 14 October 2020. This article incorporates text from this source, which is in the public domain.
External links
- “Ansuvimab”. Drug Information Portal. U.S. National Library of Medicine.
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | Zaire ebolavirus |
| Clinical data | |
| Trade names | Ebanga |
| Other names | Ansuvimab-zykl, mAb114 |
| License data | US DailyMed: Ansuvimab |
| Routes of administration | Intravenous |
| Drug class | Monoclonal antibody |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1] |
| Identifiers | |
| CAS Number | 2375952-29-5 |
| DrugBank | DB16385 |
| UNII | TG8IQ19NG2 |
| KEGG | D11875 |
| Chemical and physical data | |
| Formula | C6368H9924N1724O1994S44 |
| Molar mass | 143950.15 g·mol−1 |
//////////Ansuvimab-zykl , EBANGA, FDA 2020, 2020 APPROVALS, MONOCLONAL ANTIBODY, Orphan Drug designation, , Breakthrough Therapy designation , Ridgeback Biotherapeutics,
CK-101
![N-[3-[2-[2,3-Difluoro-4-[4-(2-hydroxyethyl)piperazin-1-yl]anilino]quinazolin-8-yl]phenyl]prop-2-enamide.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=117909640&t=l)

CK-101, RX-518
CAS 1660963-42-7
N-[3-[2-[[2,3-Difluoro-4-[4-(2-hydroxyethyl)piperazin-1-yl]phenyl]amino]quinazolin-8-yl]phenyl]acrylamide
N-(3-(2-((2,3-Difluoro-4-(4-(2-hydroxyethyl)piperazin-1-yl)phenyl)amino)quinazolin-8-yl)phenyl)acrylamide
EGFR-IN-3
UNII-708TLB8J3Y
Suzhou NeuPharma (Originator)
Checkpoint Therapeutics
Non-Small Cell Lung Cancer Therapy
Solid Tumors Therapy
PHASE 2 Checkpoint Therapeutics, Cancer, lung (non-small cell) (NSCLC), solid tumour
RX518(CK-101) is an orally available third-generation and selective inhibitor of certain epidermal growth factor receptor (EGFR) activating mutations, including the resistance mutation T790M, and the L858R and exon 19 deletion (del 19) mutations, with potential antineoplastic activity.
In August 2019, Suzhou Neupharma and its licensee Checkpoint Therapeutics are developing CK-101 (phase II clinical trial), a novel third-generation, covalent, EGFR inhibitor, as a capsule formulation, for the treatment of cancers including NSCLC and other advanced solid tumors. In September 2017, the FDA granted Orphan Drug designation to this compound, for the treatment of EGFR mutation-positive NSCLC; in January 2018, the capsule was being developed as a class 1 chemical drug in China.
CK-101 (RX-518), a small-molecule inhibitor of epidermal growth factor receptor (EGFR), is in early clinical development at Checkpoint Therapeutics and Suzhou NeuPharma for the potential treatment of EGFR-mutated non-small cell lung cancer (NSCLC) and other advanced solid malignancies.
In 2015, Suzhou NeuPharma granted a global development and commercialization license to its EGFR inhibitor program, excluding certain Asian countries, to Coronado Biosciences (now Fortress Biotech). Subsequently, Coronado assigned the newly acquired program to its subsidiary Checkpoint Therapeutics.
In 2017, the product was granted orphan drug designation in the U.S. for the treatment of EGFR mutation-positive NSCLC.
There are at least 400 enzymes identified as protein kinases. These enzymes catalyze the phosphorylation of target protein substrates. The phosphorylation is usually a transfer reaction of a phosphate group from ATP to the protein substrate. The specific structure in the target substrate to which the phosphate is transferred is a tyrosine, serine or threonine residue. Since these amino acid residues are the target structures for the phosphoryl transfer, these protein kinase enzymes are commonly referred to as tyrosine kinases or serine/threonine kinases.
[0003] The phosphorylation reactions, and counteracting phosphatase reactions, at the tyrosine, serine and threonine residues are involved in countless cellular processes that underlie responses to diverse intracellular signals (typically mediated through cellular receptors), regulation of cellular functions, and activation or deactivation of cellular processes. A cascade of protein kinases often participate in intracellular signal transduction and are necessary for the realization of these cellular processes. Because of their ubiquity in these processes, the protein kinases can be found as an integral part of the plasma membrane or as cytoplasmic enzymes or localized in the nucleus, often as components of enzyme complexes. In many instances, these protein kinases are an essential element of enzyme and structural protein complexes that determine where and when a cellular process occurs within a cell.
[0004] The identification of effective small compounds which specifically inhibit signal transduction and cellular proliferation by modulating the activity of tyrosine and serine/threonine kinases to regulate and modulate abnormal or inappropriate cell proliferation, differentiation, or metabolism is therefore desirable. In particular, the identification of compounds that specifically inhibit the function of a kinase which is essential for processes leading to cancer would be beneficial.
[0005] While such compounds are often initially evaluated for their activity when dissolved in solution, solid state characteristics such as polymorphism are also important. Polymorphic forms of a drug substance, such as a kinase inhibitor, can have different physical properties, including melting point, apparent solubility, dissolution rate, optical and mechanical properties, vapor pressure, and density. These properties can have a direct effect on the ability to process or manufacture a drug substance and the drug product. Moreover, differences in these properties
can and often lead to different pharmacokinetics profiles for different polymorphic forms of a drug. Therefore, polymorphism is often an important factor under regulatory review of the ‘sameness’ of drug products from various manufacturers. For example, polymorphism has been evaluated in many multi-million dollar and even multi-billion dollar drugs, such as warfarin sodium, famotidine, and ranitidine. Polymorphism can affect the quality, safety, and/or efficacy of a drug product, such as a kinase inhibitor. Thus, there still remains a need for polymorphs of kinase inhibitors. The present disclosure addresses this need and provides related advantages as well.
PATENT
WO2015027222
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015027222


PATENT
WO-2019157225
Crystalline form II-VIII of the compound presumed to be CK-101 (first disclosed in WO2015027222 ), for treating a disorder mediated by epidermal growth factor receptor (EGFR) eg cancer.
SCHEME A
Scheme B
General Procedures
Example 1: Preparation of the compound of Formula I (N-(3-(2-((2,3-difluoro-4-(4-(2-hydroxyethyl)piperazin-l-yl)phenyl)amino)quinazolin-8-yl)phenyl)acrylamide)
[0253] To a solution of l,2,3-trifluoro-4-nitrobenzene (2.5 g, 14 mmol, 1.0 eq.) in DMF (20 mL) was added K2C03 (3.8 g, 28 mmol, 2.0 eq.) followed by 2-(piperazin-l-yl)ethanol (1.8 g, 14 mmol, 1.0 eq.) at 0 °C and the mixture was stirred at r.t. overnight. The mixture was poured into ice-water (200 mL), filtered and dried in vacuo to afford 2-(4-(2,3-difluoro-4-nitrophenyl)piperazin-l-yl)ethanol (2.7 g, 67.5%).
[0254] To a solution of 2-(4-(2,3-difluoro-4-nitrophenyl)piperazin-l-yl)ethanol (2.7 g, 9.0 mmol) in MeOH (30 mL) was added Pd/C (270 mg) and the resulting mixture was stirred at r.t.
overnight. The Pd/C was removed by filtration and the filtrate was concentrated to afford 2-(4-(4-amino-2,3-difluorophenyl)piperazin-l-yl)ethanol (2.39 g, 99% yield) as off-white solid.
[0255] To a solution of 8-bromo-2-chloroquinazoline (15.4 g, 63.6 mmol, 1 eq. ) and (3-aminophenyl)boronic acid (8.7 g, 63.6 mmol, 1 eq.) in dioxane/H20 (200 mL/20 mL) was added Na2C03 (13.5 g, 127.2 mmol, 2 eq.), followed by Pd(dppf)Cl2 (2.6 g, 3.2 mmol, 0.05 eq.) under N2, then the mixture was stirred at 80 °C for 12 h. Then the solution was cooled to r.t.,
concentrated and the residue was purified via column chromatography (PE/EA=3 :2, v/v) to afford 3-(2-chloroquinazolin-8-yl)aniline as yellow solid (8.7 g, 53.7% yield).
[0256] To a solution of 3-(2-chloroquinazolin-8-yl)aniline (8.7 g, 34 mmol, 1 eq.) in DCM ( 200 mL ) cooled in ice-bath was added TEA (9.5 mL, 68 mmol, 2 eq. ), followed by acryloyl chloride (4.1 mL, 51 mmol, 1.5 eq.) dropwise. The resulting mixture was stirred at r.t. for 1 h, then washed with brine, dried over anhydrous N2S04 concentrated and the residue was purified via column chromatography (PE/EA=l : 1, v:v) to afford N-(3-(2-chloroquinazolin-8-yl)phenyl)acryl amide as yellow solid(6.6 g, 65% yield).
[0257] To a suspension of 2-(4-(4-amino-2,3-difluorophenyl)piperazin-l-yl)ethanol (83 mg,
0.32 mmol, 1 eq.) and N-(3-(2-chloroquinazolin-8-yl)phenyl)acrylamide (100 mg, 0.32 mmol, 1 eq.) in n-BuOH (5 mL) was added TFA (68 mg, 0.64 mmol, 2 eq.) and the resulting mixture was stirred at 90 °C overnight. The mixture was concentrated, diluted with DCM (20 mL) , washed with Na2C03 solution (20 mL), dried over anhydrous Na2S04, concentrated and the residue was purified via column chromatography (MeOH/DCM=l/30, v:v) to afford N-(3-(2-((2,3-difluoro-4-(4-(2-hydroxyethyl)piperazin-l-yl)phenyl)amino)quinazolin-8-yl)phenyl)acrylamide as a yellow solid(l6.3 mg, 9.5% yield). LRMS (M+H+) m/z calculated 531.2, found 531.2. 1H NMR
(CD3OD, 400 MHz) d 9.21 (s, 1 H), 7.19-8.01 (m, 10 H), 8.90 (s, 1 H), 6.41-6.49 (m, 3 H), 5.86 (m, 1 H), 3.98-4.01 (m, 3 H), 3.70-3.76 (m, 3 H), 3.40-3.49 (m, 2 H), 3.37-3.39 (m, 4 H), 3.18 (m, 2H).
Example 2. Preparation of Form I of the compound of Formula I
[0258] Crude compound of Formula I (~30 g, 75% of weight based assay) was dissolved in ethyl acetate (3 L) at 55-65 °C under nitrogen. The resulting solution was filtered via silica gel pad and washed with ethyl acetate (3 L><2) at 55-65 °C. The filtrate was concentrated via vacuum at 30-40 °C to ~2.4 L. The mixture was heated up to 75-85 °C and maintained about 1 hour.
Then cooled down to 50-60 °C and maintained about 2 hours. The heat-cooling operation was repeated again and the mixture was then cooled down to 20-30 °C and stirred for 3 hours. The resulting mixture was filtered and washed with ethyl acetate (60 mL><2). The wet cake was dried via vacuum at 30-40 °C to get (about 16 g) of the purified Form I of the compound of Formula I.
Example 3. Preparation of Form III of the compound of Formula I
[0259] The compound of Formula I (2 g) was dissolved in EtOH (40 mL) at 75-85 °C under nitrogen. n-Heptane (40 mL) was added dropwise into reaction at 75-85 °C. The mixture was stirred at 75-85 °C for 1 hour. Then cooled down to 50-60 °C and maintained about 2 hours. The heat-cooling operation was repeated again and continued to cool the mixture down to 20-30 °C and stirred for 3 hours. The resulting mixture was filtered and washed with EtOH/n-Heptane (1/1, 5 mL><2). The wet cake was dried via vacuum at 30-40 °C to get the purified Form III of the compound of Formula I (1.7 g).
Example 4. Preparation of Form IV of the compound of Formula I The crude compound of Formula I (15 g) was dissolved in ethyl acetate (600 mL) at 75-85 °C under nitrogen and treated with anhydrous Na2S04, activated carbon, silica metal scavenger for 1 hour. The resulting mixture was filtered via neutral Al203 and washed with ethyl acetate (300 mL><2) at 75-85 °C. The filtrate was concentrated under vacuum at 30-40 °C and swapped with DCM (150 mL). n-Heptane (75 mL) was added into this DCM solution at 35-45 °C, and then the mixture was cooled down to 20-30 °C slowly. The resulting mixture was filtered and washed with DCM/n-Heptane (2/1, 10 mL><3). The wet cake was dried via vacuum at 35-40 °C to get the purified Form IV of the compound of Formula I (9.6 g).
Example 5. Preparation of Form V of the compound of Formula I
[0260] Polymorph Form III of the compound of Formula I was dried in oven at 80 °C for 2 days to obtain the polymorph Form V.
Example 6. Preparation of Form VI of the compound of Formula I
[0261] The compound of Formula I (1 g) was dissolved in IPA (20 mL) at 75-85 °C under nitrogen. n-Heptane (20 mL) was added dropwise into reaction at 75-85 °C. The mixture was stirred at 45-55 °C for 16 hours. Then heated up to 75-85 °C and maintained about 0.5 hour.
Then cooled down to 45-55 °C for 0.5 hour and continued to cool the mixture down to 20-30 °C and stirred for 3 hours. Filtered and washed with IPA/n-Heptane (1/1, 3 mL><2). The wet cake was dried via vacuum at 75-80 °C for 2 hours to get the purified Form VI of the compound of Formula I.
Example 7. Preparation of Form VIII of the compound of Formula I
[0262] The polymorph Form VI of the compound of Formula I was dried in oven at 80 °C for 2 days to obtain the polymorph Form VIII.
Example 8. X-ray powder diffraction (XRD)
[0263] X-ray powder diffraction (XRD) patterns were obtained on a Bruker D8 Advance. A CuK source (=1.54056 angstrom) operating minimally at 40 kV and 40 mA scans each sample between 4 and 40 degrees 2-theta. The step size is 0.05°C and scan speed is 0.5 second per step.
Example 9. Thermogravimetric Analyses (TGA)
[0264] Thermogravimetric analyses were carried out on a TA Instrument TGA unit (Model TGA 500). Samples were heated in platinum pans from ambient to 300 °C at 10 °C/min with a nitrogen purge of 60mL/min (sample purge) and 40mL/min (balance purge). The TGA temperature was calibrated with nickel standard, MP=354.4 °C. The weight calibration was performed with manufacturer-supplied standards and verified against sodium citrate dihydrate desolvation.
Example 10. Differential scanning calorimetry (DSC)
[0265] Differential scanning calorimetry analyses were carried out on a TA Instrument DSC unit (Model DSC 1000 or 2000). Samples were heated in non-hermetic aluminum pans from ambient to 300 °C at 10 °C/min with a nitrogen purge of 50mL/min. The DSC temperature was calibrated with indium standard, onset of l56-l58°C, enthalpy of 25-29J/g.
Example 11. Hygroscopicity (DVS)
[0266] The moisture sorption profile was generated at 25°C using a DVS Moisture Balance Flow System (Model Advantage) with the following conditions: sample size approximately 5 to 10 mg, drying 25°C for 60 minutes, adsorption range 0% to 95% RH, desorption range 95% to 0% RH, and step interval 5%. The equilibrium criterion was <0.01% weight change in 5 minutes for a maximum of 120 minutes.
Example 12: Microscopy
[0267] Microscopy was performed using a Leica DMLP polarized light microscope equipped with 2.5X, 10X and 20X objectives and a digital camera to capture images showing particle shape, size, and crystallinity. Crossed polars were used to show birefringence and crystal habit for the samples dispersed in immersion oil.
Example 13: HPLC
[0256] HPLCs were preformed using the following instrument and/or conditions.
///////////////CK-101 , CK 101 , CK101 , phase II , Suzhou Neupharma, Checkpoint Therapeutics , Orphan Drug designation, EGFR mutation-positive NSCLC, NSCLC, CANCER, SOLID TUMOUR, China, RX-518, AK543910
OCCN1CCN(CC1)c5ccc(Nc2nc3c(cccc3cn2)c4cccc(NC(=O)C=C)c4)c(F)c5F

{kind=link}
{kind=link}