
Home » Posts tagged 'PHASE1'
Tag Archives: PHASE1
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
AD 35
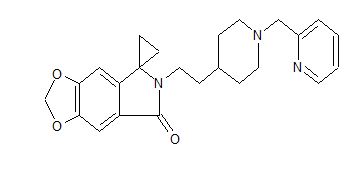
AD 35
IND-120499
- Molecular Weight, 405.49
- Spiro[cyclopropane-1,5′-[5H-1,3]dioxolo[4,5-f]isoindol]-7′(6′H)-one, 6′-[2-[1-(2-pyridinylmethyl)-4-piperidinyl]ethyl]-
6′-[2-[1-(2-Pyridinylmethyl)-4-piperidinyl]ethyl]spiro[cyclopropane-1,5′-[5H-1,3]dioxolo[4,5-f]isoindol]-7′(6’H)-one
1531586-58-9 CAS FREE FORM
1531586-64-7 PHOSPHATE
1531586-62-5 HYDROCHLORIDE
Zhejiang Hisun Pharmaceutical Co Ltd

AD-35 is known to be a neuroprotectant, useful for treating Alzheimer’s diseases.
Zhejiang Hisun Pharmaceutical is developing an oral tablet formulation of AD-35, for treating Alzheimers disease . By August 2017, the phase I multiple doses trial had been completed in the US and would be completed in China soon
CAS 1531586-64-7 PHOSPHATE
6′-[2-[1-(Pyridin-2-ylmethyl)piperidin-4-yl]ethyl]spiro[cyclopropane-1,5′-[1,3]dioxolo[4,5-f]isoindol]-7′(6’H)-one phosphate
| Molecular Formula | C24 H27 N3 O3 . H3 O4 P |
| Molecular Weight | 503.4847 |

With the rapid growth of the elderly population, the number of people suffering from Alzheimer’s disease (Alzheimer’s disease) also will be increased dramatically.Alzheimer’s disease is also known as Alzheimer-type dementia (Alzheimer type dementia), or the Alzheimer type senile dementia (senile dementia of the Alzheimer type). At present, although the prevalence of this disease on a global scale is still unknown, but according to the latest report from the US Alzheimer’s Association (the Alzheimer’s Association), and in 2011 the United States there are about 540 million people suffer from Alcatel the number of Alzheimer’s disease, and in 2050, in the United States suffering from the disease will increase to about 13.5 million. Therefore, the development of better efficacy and fewer side effects of new drugs to treat the disease it is a priority.
Alzheimer’s disease is the most common form of senile dementia, it has become the sixth leading cause of death of Americans, and 65 years and the fifth leading cause of death in Americans over 65 years. Although scientists have this disease carried out extensive and in-depth research, but so far, the exact cause of the disease remains unclear. Alzheimer’s disease is a progressive disease that continues to kill nerve cells, destroying nerve connections in the brain, resulting in brain tissue is damaged, leading to patients gradually lose memory, consciousness and judgment, and cause mood disorders and behavioral disorders in patients.
Alzheimer’s is an irreversible disease, and now there is no any drug can prevent the disease, and no drugs can cure the disease or slow the disease process. Drugs currently used to treat the disease can only alleviate or ameliorate symptoms of the disease. These drugs are FDA approved for use in the United States a total of five, four of which are acetylcholinesterase (acetylcholinesterase) inhibitors. Acetylcholine (acetylcholine) is a neurotransmitter, a chemical released by nerves, if produced in the brain acetylcholine system, i.e. damaged cholinergic system, it can result in associated with Alzheimer’s disease memory disorders; and acetylcholinesterase function is to catalyze the hydrolysis of acetylcholine, acetylcholine is decomposed. Because Alzheimer’s disease is accompanied
Attenuation of acetylcholine activity, thus inhibiting acetylcholinesterase is one way to treat this disease. As described above, in the present 5 treatment of Alzheimer’s disease drugs in clinical use, there are four acetylcholinesterase inhibitors, including acetylcholinesterase inhibitors such as donepezil (donepezil), tacrine (tacrine ), rivastigmine (rivastigmine), and galantamine (galantamine), wherein donepezil (Sugimoto et al US4895841 and 5100901;.. Pathi et al WO 2007077443;. Parthasaradhi et al WO 2005003092;. Dubey et al WO 2005076749; Gutman . et al WO 200009483;… Sugimoto et al J. Med Chem 1995, 38, 481) is a first-line treatment of Alzheimer’s disease drugs. However, donepezil and the other four drugs can only improve the patient’s symptoms, and this is the only improvement of symptoms is short, only lasting about 6-12 months, and the patient response rates to these drugs only about 50% (Alzheimer’s Association, 201 1 Alzheimer ‘Disease Facts and Figures, Alzheimer’s & Dementia, 201 1, 7 (2), 208). The present invention provides a new class of inhibitors of acetylcholinesterase, which is dioxole between a new class of derivatives of benzo, is more effective than donepezil and fewer side effects in the treatment of Alzheimer’s disease drug.
PATENT
WO 2014005421
Example 42: 6- [2- [l- (2-Pyridylmethyl) -4-piperidinyl] ethyl] spiro [[1,3] dioxolo [4,5-f ] Isoindole-7, Γ-cyclopropane-5-one (Compound No. 1-29)

To the reaction flask was added 24.3 g (0.069 mol) of compound 11-5, 36.5 g (0.26 mol) of potassium carbonate, 243 ml of ethanol, 6.1 ml (0.044 mole) of triethylamine, heated to about 50 ° C, 0.049 mol) of 2-chloromethylpyridine hydrochloride was maintained at about 50 ° C for 5 hours. The reaction was complete and 750 ml of water was added. The solid was precipitated, filtered and the cake was washed with water and dried to give 17.8 g of compound 1-29. Rate: 63.4%. ‘HNMR (CDC13 . 3 ): [delta] 1.26 (dd, 2H, J = 6.1, 7.6 Hz), 1.35 (brs,. 3 H), 1.49-1.57 (m, 4H), 1.72 (D, 2H, J = 8.6Hz) (T, 2H, J = 7.9 Hz), 3.64 (s, 2H), 6.03 (s, 2H), 2.09 (t, 2H, J = 10.4 Hz), 2.89 (d, 2H, J = 10.7 Hz) , 7.42 (s, 1 H), 7.15 (dd, 1 H, J = 5.2, 6.7 Hz), 7.24 (s, 1 H), 7.41 (d, 1 H, J = 7.7 Hz), 7.64 (td, H, J = 7.6, 1.8 Hz), 8.55 (D,. 1 H, J = 4.2 Hz); the MS (ESI): m / Z 406 [m + H] + .
Example 46: 6- [2- [l- (2-Pyridylmethyl) -4-piperidinyl] ethyl] spiro [[1,3] dioxolo [4,5-f ] Isoindole-7, Γ-cyclopropane] -5-one hydrochloride (Compound No. 1-33)

To the reaction flask was added 5 g (0.012 mol) of compound 1-29 and 25 ml of ethanol, heated at 50 ° C
(0.012 mol) of concentrated hydrochloric acid was added, and 1 g of activated charcoal was added to decolorize for 20 minutes. The filtrate was cooled to room temperature and 50 ml of isopropyl ether was added dropwise. The solid was precipitated, stirred for 1 hour, The ether cake was washed with ether and dried to give 5 g of compound 1-33 in a yield of 91.7%. Ethanol / isopropyl ether can be re-refined, the yield of about 90%. 1H-NMR is (D 2 0): 51.14 (T, 2 H, J-7.0 Hz), 1.38-1.70 (m,. 7 H), 1.96 (D, 2H, J = 13.3 Hz), 2.99-3.14 (m, H. 4 ), 3.50 (d, 2 H, J = 11.0 Hz), 4.37 (s, 2H), 5.93 (s, 2H), 6.28 (s, 1 H), 6.75 (s, 1 H), 7.47 (dd, J = 7.8, 1.7 Hz), 8.58 (d, 1 H, J = 4.4 Hz), 7.55 (d, 1 H, J = 7.8 Hz), 7.91 (td, ; MS (ESI): m / z 406 [M-Cl] & lt; + & gt ; .
Example 48: 6- [2- [l- (2-Pyridylmethyl) -4-piperidinyl] ethyl] spiro [[1,3] dioxolo [4,5-f ] Isoindole-7, Γ-cyclopropan-5-one phosphate (Compound I-3S)

To the reaction flask was added 2 g (0.0049 mole) of compound 1-29 and 40 ml of ethanol, stirred at 60 ° C until all dissolved, 0.57 g (0.0049 mole) of 85% phosphoric acid was added, stirred and solidified,
Liter of ethyl acetate, cooled to room temperature, stirred for 1 hour, filtered, and a small amount of ethyl acetate was used to wash the filter cake and dried to give 2.1 g of compound 1-35 in a yield of 84.7%. 1H-NMR (D 2 0): δ 1.10 (t, 2 H, J = 7.2 Hz), 1.33-1.64 (m, 7 H), 1.92 (d, 2 H, J = 13.4 Hz), 2.95-3.09 (m, (S, 1 H), 6.69 (s, 1 H), 7.45 (s, 2 H), 4.34 (s, (d, 1 H, J-7.8 Hz), 7.88 (td, 1 H, J = 7.7, 1.2 Hz), 8.54 (d, 1 H, J = 4.6 Hz).
PATENT
CN 103524515
https://encrypted.google.com/patents/CN103524515B?cl=en
PATENT
CN 105859732
https://www.google.com/patents/CN105859732A?cl=en

Example 14: 6- [2- [l_ (2- pyridylmethyl) -4-piperidinyl] ethyl] spiro [[1,3] dioxolo [4,5 -f] isoindole-7, prepared Γ- cyclopropane] phosphate 5-one (compound I) is

[0146] Compound was added 2g (4.9 mmol) of formula XI to the reaction flask 50mL, 40mL of ethanol, 60 ~ 70 ° C dissolved by heating, added with stirring square. 57g 85% (4.9mmol) phosphoric acid, and the precipitated solid was added dropwise 40mL of acetic acid ethyl cooled to room temperature, stirred for 1 hour, filtered, the filter cake washed with a small amount of ethyl acetate, dried to give 2.3g white solid (compound I, HPLC purity: 99.8%). Yield: 92.7%, H bandit R (D2O): δ1 · l〇 (t, 2H, J = 7.2Hz), 1.33-1.64 (m, 7H), 1.92 (d, 2H, J = 13.4Hz), 2.95 -3.09 (m, 4H), 3.46 (d, 2H, J = 10.7Hz), 4.34 (s, 2H), 5.89 (s, 2H), 6.20 (s, 1H), 6.69 (s, 1H), 7.45 ( , 7.53 (d, lH, J 7.8Hz dd, lH, J = 5.2,7.4Hz) =), 7.88 (td, lH, J =
PATENT
WO 2017177816
Process for preparing AD-35 and its intermediates – comprising the reaction of a cyano ester with a Grignard reagent, followed by condensation and further manipulative steps.
A novel intermediate of AD-35 is claimed. Also claimed is a processes for preparing 6,7-dihydro-[1,3]dioxolo[4,5-f]isoindol-5-one comprising the reaction of a cyano ester compound in an isopropyl ester (Ti(i-Pr)4)) with a Grignard reagent in the presence of an ethyl magnesium halide. Further claimed are processes for preparing synthon of intermediates. A process for preparing a benzodioxole derivative, particularly AD-35 from intermediates is also claimed.
Specific implementation plan
J Alzheimer’s Dis 2017, 56(4): 1403
| CN101626688A * | Dec 11, 2007 | Jan 13, 2010 | 雷维瓦药品公司 | Compositions, synthesis, and methods of using indanone based cholinesterase inhibitors |
| WO2014005421A1 * | Jul 3, 2013 | Jan 9, 2014 | Zhejiang Hisun Pharmaceutical Co., Ltd. | Benzodioxole derivative and preparation method and use thereof |
HAO 472
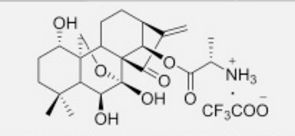
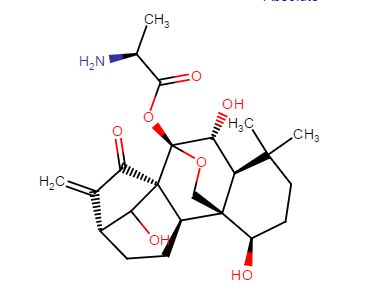 .CF3COOH
.CF3COOH
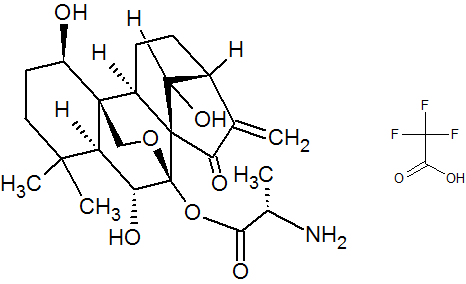
HAO 472
PHASE 1 CHINA



PRoject Name: HAO472 treatment Phase I clinical trial in relapsed / refractory AML, M2b type of AML
The main purpose: to determine HAO472 treatment of relapsed / refractory C the maximum tolerated dose (MTD). Secondary objectives: 1) evaluation of drug safety and tolerability; 2) study HAO472 in pharmacokinetic characteristics of the human body; 3) the effectiveness of HAO472 treatment of relapsed / refractory M2b type of AML.
Introduction Test
Acute myelogenous leukemia
HAO472
Phase I
Test Number: CTR20150246
Sponsor Name:
Jiangsu Hengrui Medicine Co., Ltd. 1/
2 Ruijin Hospital, Shanghai Jiaotong University School of Medicine /
3 Jiangsu Hengrui Medicine Co., Ltd. /
4 Shanghai Hengrui Medicine Co., Ltd. /

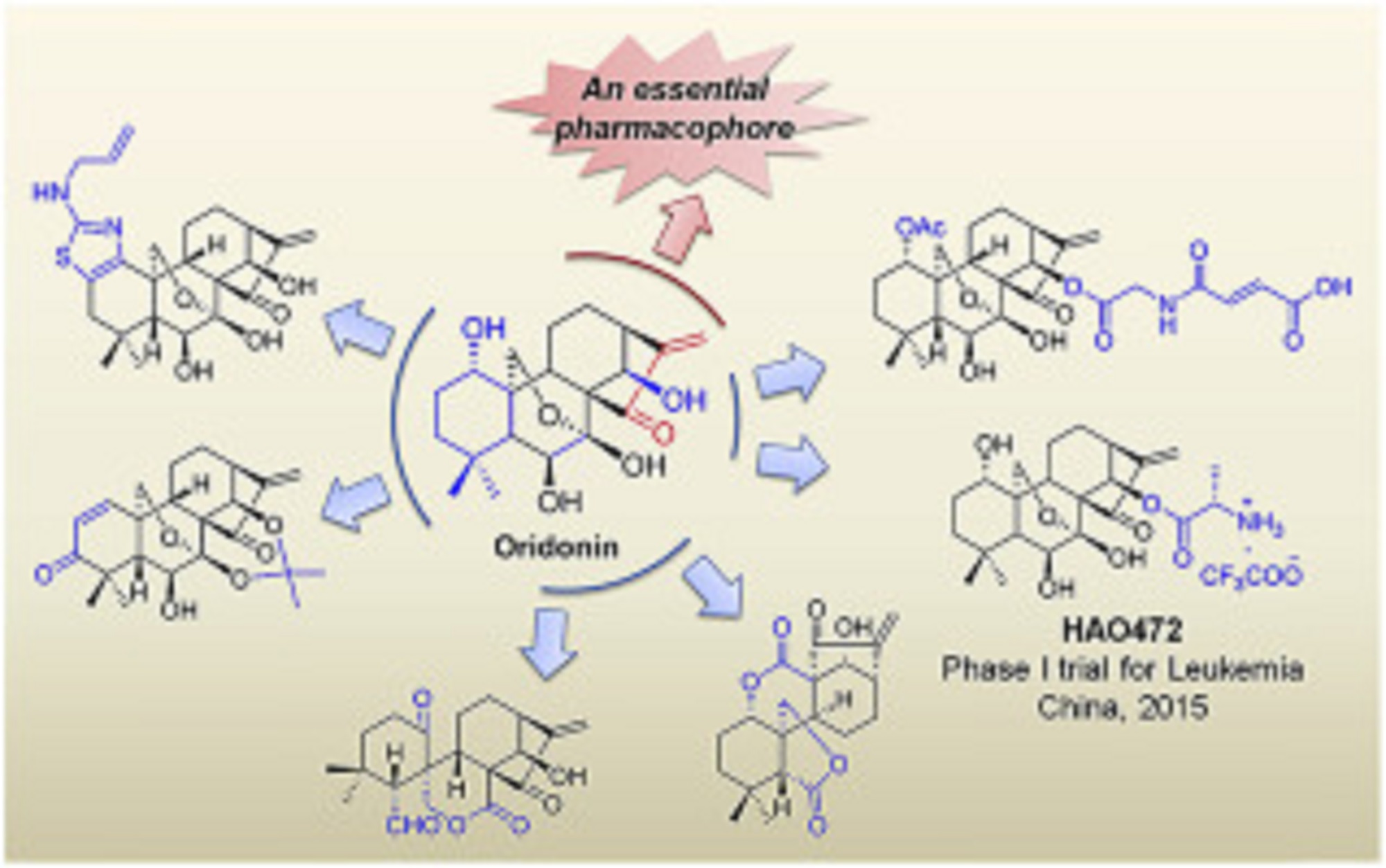
Natural products have historically been, and continue to be, an invaluable source for the discovery of various therapeutic agents. Oridonin, a natural diterpenoid widely applied in traditional Chinese medicines, exhibits a broad range of biological effects including anticancer and anti-inflammatory activities. To further improve its potency, aqueous solubility and bioavailability, the oridonin template serves as an exciting platform for drug discovery to yield better candidates with unique targets and enhanced drug properties. A number of oridonin derivatives (e.g. HAO472) have been designed and synthesized, and have contributed to substantial progress in the identification of new agents and relevant molecular mechanistic studies toward the treatment of human cancers and other diseases. This review summarizes the recent advances in medicinal chemistry on the explorations of novel oridonin analogues as potential anticancer therapeutics, and provides a detailed discussion of future directions for the development and progression of this class of molecules into the clinic.
Highlights
Oridonin displays significant anticancer activities via multi-signaling pathways.
Recent advances in medicinal chemistry of oridonin-like compounds are presented.
The article summarizes the SAR and mechanism studies of relevant drug candidates.
The milestones and future direction of oridonin-based drug discovery are discussed.
Volume 122, 21 October 2016, Pages 102–117

Discovery and development of natural product oridonin-inspired anticancer agents
- a Chemical Biology Program, Department of Pharmacology and Toxicology, University of Texas Medical Branch, Galveston, TX, 77555, United States
- b Department of Clinical Cancer Prevention, Division of Cancer Prevention and Population Sciences, The University of Texas MD Anderson Cancer Center, Houston, TX, 77030, United States

////////Natural product, Oridonin, Diterpenoids, Anticancer agents, Drug discovery, Chemical biology, AML, HAO 472, relapsed / refractory AML. Jiangsu Hengrui Medicine Co., Ltd, PHASE1, LEUKEMIA
C[C@H](N)C(=O)O[C@]15OC[C@@]2([C@H](O)CCC(C)(C)[C@@H]2[C@H]1O)[C@H]3CC[C@@H]4C(=C)C(=O)[C@@]35C4O
GSK 1070916 For Advanced solid tumor

GSK 1070916
NMI-900 , GSK-1070916, GSK-1070916A
4-[3-(4-N,N-Dimethylcarbamylaminophenyl)-1-ethyl-1H-pyrazol-4-yl]-2-[3-(dimethylaminomethyl)phenyl]-1H-pyrrolo[2,3-b]pyridine
N’-[4-[4-[2-[3-[(Dimethylamino)methyl]phenyl]-1H-pyrrolo[2,3-b]pyridin-4-yl]-1-ethyl-1H-pyrazol-3-yl]phenyl]-N,N-dimethylurea
CAS 942918-07-2,
MFC30H33N7O,
MW507.63
PHASE 1/II , Advanced solid tumor, Cancer Research Technology,
off-white solid.
1H NMR (400 MHz, DMSO-d6) δ ppm 12.14 (d, J = 1.8 Hz, 1H), 8.31 (s, 1H), 8.27 (s, 1 H), 8.07 (d, J = 4.8 Hz, 1H), 7.78 (d, J = 8.1 Hz, 1H), 7.77 (s, 1H), 7.43 (d, J = 8.6 Hz, 2H), 7.39 (d, J = 8.1 Hz, 1H), 7.27 (d, J = 8.6 Hz, 2H), 7.27 (dd, 1H), 6.79 (d, J = 5.1 Hz, 1H), 6.76 (d, J = 2.0 Hz, 1H), 4.27 (q, J = 7.3 Hz, 2H), 3.43 (s, 2H), 2.91 (s, 6H), 2.18 (s, 6H), 1.51 (t, J = 7.2 Hz, 3H).
MS m/z 508.4 [M + H]+. Anal. (C30H33N7O·1.0H2O) C, H, N.
GSK1070916 is a reversible and ATP-competitive inhibitor of Aurora B/C with IC50 of 3.5 nM/6.5 nM; displays >100-fold selectivity against the closely related Aurora A-TPX2 complex(IC50=490 nM).
NMI-900, an Aurora B/C kinase inhibitor, is under development at Cancer Research Technology in phase I/II clinical studies for the treatment of advanced and/or metastatic solid tumors. Other phase I clinical trials for the treatment of solid tumors had been previously completed, in a collaboration between GlaxoSmithKline and Cancer Research Technology, under the Cancer Research UK’s Clinical Development Partnerships (CDP) program.
The drug was originated by GlaxoSmithKline. The rights of the product were acquired by Cancer Research Technology from GlaxoSmithKline after the company elected not to take the program forward. In December 2015, the product was licensed by Cancer Research Technology to Nemucore Medical Innovations for the exclusive worldwide development and commercialization for the treatment of difficult-to-treat cancers.

PATENT
US 20070149561
https://www.google.com/patents/US20070149561
PAPER
Journal of Medicinal Chemistry (2010), 53 (10), 3973-4001
http://pubs.acs.org/doi/abs/10.1021/jm901870q
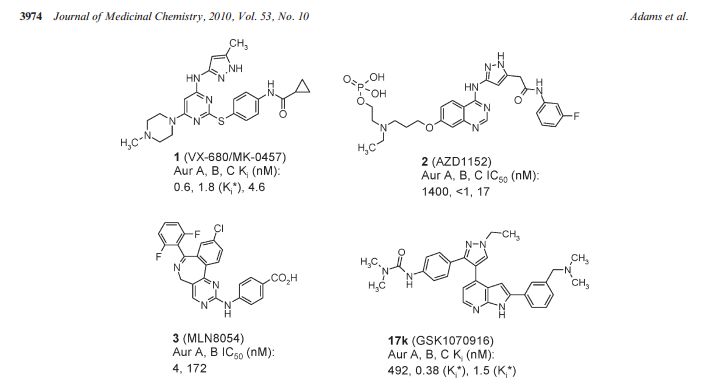
Discovery of GSK1070916, a Potent and Selective Inhibitor of Aurora B/C Kinase

The Aurora kinases play critical roles in the regulation of mitosis and are frequently overexpressed or amplified in human tumors. Selective inhibitors may provide a new therapy for the treatment of tumors with Aurora kinase amplification. Herein we describe our lead optimization efforts within a 7-azaindole-based series culminating in the identification of GSK1070916 (17k). Key to the advancement of the series was the introduction of a 2-aryl group containing a basic amine onto the azaindole leading to significantly improved cellular activity. Compound 17k is a potent and selective ATP-competitive inhibitor of Aurora B and C with Ki* values of 0.38 ± 0.29 and 1.5 ± 0.4 nM, respectively, and is >250-fold selective over Aurora A. Biochemical characterization revealed that compound 17k has an extremely slow dissociation half-life from Aurora B (>480 min), distinguishing it from clinical compounds 1 and 2. In vitro treatment of A549 human lung cancer cells with compound 17k results in a potent antiproliferative effect (EC50 = 7 nM). Intraperitoneal administration of 17k in mice bearing human tumor xenografts leads to inhibition of histone H3 phosphorylation at serine 10 in human colon cancer (Colo205) and tumor regression in human leukemia (HL-60). Compound 17k is being progressed to human clinical trials.
http://pubs.acs.org/doi/pdf/10.1021/jm901870q………..PDF FILE
PAPER
Molecules 2014, 19(12), 19935-19979; doi:10.3390/molecules191219935
http://www.mdpi.com/1420-3049/19/12/19935/htm

http://www.mdpi.com/1420-3049/19/12/19935/htm
Biological Activity of GSK-1070916
GSK1070916 is a reversible and ATP-competitive inhibitor of Aurora B/C with IC50 of 3.5 nM/6.5 nM; displays >100-fold selectivity against the closely related Aurora A-TPX2 complex(IC50=490 nM).
IC50 Value: 3.5 nM(Aurora B); 6.5 nM(Aurora C)
Target: Aurora B/C
in vitro: GSK1070916 selectively inhibits Aurora B and Aurora C with Ki of 0.38 nM and 1.5 nM over Aurora A with Ki of 490 nM. Inhibition of Aurora B and Aurora C is time-dependent, with an enzyme-inhibitor dissociation half-life of >480 min and 270 min respectively. In addition, GSK1070916 is also a competitive inhibitor with respect to ATP. Human tumor cells treated with GSK1070916 shows dose-dependent inhibition of phosphorylation on serine 10 of Histone H3, a substrate specific for Aurora B. Moreover, GSK1070916 inhibits the proliferation of tumor cells with EC50 values of <10 nM in over 100 cell lines spanning a broad range of tumor types, with a median EC50 of 8 nM. Although GSK1070916 has potent activity against proliferating cells, a dramatic shift in potency is observed in primary, nondividing, normal human vein endothelial cells. Furthermore, GSK1070916-treated cells do not arrest in mitosis but instead fails to divide and become polyploid, ultimately leading to apoptosis. In another study, it is also reported high chromosome number associated with resistance to the inhibition of Aurora B and C suggests cells with a mechanism to bypass the high ploidy checkpoint are resistant to GSK1070916.
in vivo: GSK1070916 (25, 50, or 100 mg/kg) shows dose-dependent inhibition of phosphorylation of an Aurora B–specific substrate in mice and consistent with its broad cellular activity, has antitumor effects in 10 human tumor xenograft models including breast, colon, lung, and two leukemia models.
Clinical Information of GSK-1070916
| Product Name | Sponsor Only | Condition | Start Date | End Date | Phase | Last Change Date |
|---|---|---|---|---|---|---|
| GSK-1070916 | Cancer Research UK | Advanced solid tumor | 31-MAR-10 | 31-MAR-13 | Phase 1 | 17-JUN-13 |
References on GSK-1070916
/////////////GSK1070916, GSK-1070916, 942918-07-2 GSK, phase1, Advanced solid tumor, NMI-900 , GSK-1070916, GSK-1070916A
Temanogrel


Temanogrel
APD 791
| TEMANOGREL; APD791; CHEMBL1084617; UNII-F42Z27575A; 887936-68-7; 3-Methoxy-N-[3-(2-methyl-2H-pyrazol-3-yl)-4-(2-morpholin-4-yl-ethoxy)-phenyl]-benzamide; | |
| Molecular Formula: | C24H28N4O4 |
|---|---|
| Molecular Weight: | 436.50352 g/mol |
- Originator Arena Pharmaceuticals
- Developer Arena Pharmaceuticals; Ildong Pharmaceutical
- Class Antithrombotics; Small molecules
- Mechanism of Action Serotonin 2A receptor inverse agonists
Phase I Arterial thrombosis 
Most Recent Events
- 30 Mar 2016 Arena Pharmaceuticals has patents pending for Temanogrel in 12 regions, including Brazil (Arena Pharmaceuticals 10-K; march 2016)
- 30 Mar 2016 Arena Pharmaceuticals has patent protection for Temanogrel in 87 regions, including USA, Japan, China, Germany, France, Italy, the United Kingdom, Spain, Canada, Russia, India, Australia and South Korea
- 01 Mar 2015 Ildong Pharmaceutical initiates enrolment in a phase I trial for Arterial thrombosis in South Korea (NCT02419820)
A 5-HT2A inverse agonist potentially for the reduction of the risk of arterial thrombosis.
![]()
APD-791
CAS No. 887936-68-7
Temanogrel hydrochloride
- Molecular FormulaC24H29ClN4O4
- Average mass472.965
Temanogrel, also known as APD791, is a highly selective 5-hydroxytryptamine2A receptor inverse agonist under development for the treatment of arterial thrombosis. APD791 displayed high-affinity binding to membranes (K(i) = 4.9 nM) and functional inverse agonism of inositol phosphate accumulation (IC(50) = 5.2 nM) in human embryonic kidney cells stably expressing the human 5-HT(2A) receptor. APD791 was greater than 2000-fold selective for the 5-HT(2A) receptor versus 5-HT(2C) and 5-HT(2B) receptors. APD791 inhibited 5-HT-mediated amplification of ADP-stimulated human and dog platelet aggregation (IC(50) = 8.7 and 23.1 nM, respectively)
Arterial thrombosis is the formation of a blood clot or thrombus inside an artery or arteriole that restricts or blocks the flow of blood and, depending upon location, can result in acute coronary syndrome or stroke. The formation of a thrombus is usually initiated by blood vessel injury, which triggers platelet aggregation and adhesion of platelets to the vessel wall. Treatments aimed at inhibiting platelet aggregation have demonstrated clear clinical benefits in the setting of acute coronary syndrome and stroke. Current antiplatelet therapies include aspirin, which irreversibly inhibits cyclooxygenase (COXa
Abbreviations: COX, cyclooxygenase; ADP, adenosine diphosphate; SAR, structure−activity relationship; hERG, human ether-a-go-go-related gene; CNS, central nervous system; 5-HT, serotonin; AUC, area under the plasma concentration time curve, iv, intravenous; IP, inositol phosphate.
) and results in reduced thromboxane production, clopidogrel and prasugrel, which inhibit platelet adenosine diphosphate (ADP) P2Y12 receptors, and platelet glycoprotein IIb/IIIa receptor antagonists. Another class of antiplatelet drugs, protease-activated thrombin receptor (PAR-1) antagonists, are also being evaluated in the clinic for the treatment of acute coronary syndrome. The most advanced candidate in this class, N-[(1R,3aR,4aR,6R,8aR,9S,9aS)-9-{2-[5-(3-fluorophenyl)pyridin-2-yl]vinyl}-1-methyl-3-oxoperhydro-naphtho[2,3-c]furan-6-yl]-carbamic acid ethyl ester (SCH-530348), is currently in phase 3 trials for the prevention of arterial thrombosis.

Figure 1. Serotonin and known 5-HT2A receptor antagonists.
SYNTHESIS
PAPER
Journal of Medicinal Chemistry (2010), 53(11), 4412-4421.
http://pubs.acs.org/doi/abs/10.1021/jm100044a

Serotonin, which is stored in platelets and is released during thrombosis, activates platelets via the 5-HT2A receptor. 5-HT2A receptor inverse agonists thus represent a potential new class of antithrombotic agents. Our medicinal program began with known compounds that displayed binding affinity for the recombinant 5-HT2A receptor, but which had poor activity when tested in human plasma platelet inhibition assays. We herein describe a series of phenyl pyrazole inverse agonists optimized for selectivity, aqueous solubility, antiplatelet activity, low hERG activity, and good pharmacokinetic properties, resulting in the discovery of 10k (APD791). 10k inhibited serotonin-amplified human platelet aggregation with an IC50 = 8.7 nM and had negligible binding affinity for the closely related 5-HT2B and 5-HT2C receptors. 10k was orally bioavailable in rats, dogs, and monkeys and had an acceptable safety profile. As a result, 10k was selected further evaluation and advanced into clinical development as a potential treatment for arterial
Discovery and Structure−Activity Relationship of 3-Methoxy-N-(3-(1-methyl-1H-pyrazol-5-yl)-4-(2-morpholinoethoxy)phenyl)benzamide (APD791): A Highly Selective 5-Hydroxytryptamine2A Receptor Inverse Agonist for the Treatment of Arterial Thrombosis
3-Methoxy-N-[3-(2-methyl-2H-pyrazol-3-yl)-4-(2-morpholin-4-yl-ethoxy)-phenyl]-benzamide (10k)
Additional Information
Oral administration of APD791 to dogs resulted in acute (1-h) and subchronic (10-day) inhibition of 5-HT-mediated amplification of collagen-stimulated platelet aggregation in whole blood. Two active metabolites, APD791-M1 and APD791-M2, were generated upon incubation of APD791 with human liver microsomes and were also indentified in dogs after oral administration of APD791. The affinity and selectivity profiles of both metabolites were similar to APD791. These results demonstrate that APD791 is an orally available, high-affinity 5-HT(2A) receptor antagonist with potent activity on platelets and vascular smooth muscle.(http://www.ncbi.nlm.nih.gov/pubmed/19628629).
PATENT
WO 2006055734
https://google.com/patents/WO2006055734A2?cl=en
Example 1.88: Preparation of 3-methoxy-N-[3-(2-methyl-2H-pyrazol-3-yl)-4-(2-morpholin~
4-yl-ethoxy)-phenyl]-benzamide (Compound 733).

A mixture of 3-(2-methyl-2H-pyrazol-3-yl)-4-(2-morpholin-4-yl-ethoxy)-phenylamine (120 mg, 0.40 mmole), 3-methoxy-benzoyl chloride (81 mg, 0.48 mmole), and triethylamine (0.1 mL, 0.79 mmole) in 5 mL THF was stirred at room temperature for 10 minutes. The mixture was purified by HPLC to give the title compound as a white solid (TFA salt, 88 mg, 51 %). 1H NMR ( Acetone-^, 400 MHz) 2.99-3.21 (m, 2H), 3.22-3.45 (m, 2H), 3.66 (t, J= 4.80 Hz, 2H), 3.75 (s, 3H), 3.85 (s, 3H), 3.79-3.89 (m, 4H), 4.58 (t, J= 4.80 Hz, 2H), 6.29 (d, J= 2.02 Hz IH), 7.13 (dd, J= 8.34, 2.53 Hz, IH), 7.22 (d, J= 8.84 Hz, IH), 7.42 (t, J= 7.83 Hz, IH), 7.47 (d, J= 1.77 Hz, IH), 7.52 (t, J= 1.77 Hz, IH), 7.56 (d, J= 7.07 Hz, IH), 7.80-7.83 (m, IH), 7.91-7.96 (m, IH), 9.54 (s, NH). Exact mass calculated for C24H28N4O4 436.2, found 437.5 (MH+).
References
1: Xiong Y, Teegarden BR, Choi JS, Strah-Pleynet S, Decaire M, Jayakumar H, Dosa
PI, Casper MD, Pham L, Feichtinger K, Ullman B, Adams J, Yuskin D, Frazer J,
Morgan M, Sadeque A, Chen W, Webb RR, Connolly DT, Semple G, Al-Shamma H.
Discovery and structure-activity relationship of
3-methoxy-N-(3-(1-methyl-1H-pyrazol-5-yl)-4-(2-morpholinoethoxy)phenyl)benzamide
(APD791): a highly selective 5-hydroxytryptamine2A receptor inverse agonist for
the treatment of arterial thrombosis. J Med Chem. 2010 Jun 10;53(11):4412-21.
doi: 10.1021/jm100044a. PubMed PMID: 20455563.
2: Przyklenk K, Frelinger AL 3rd, Linden MD, Whittaker P, Li Y, Barnard MR, Adams
J, Morgan M, Al-Shamma H, Michelson AD. Targeted inhibition of the serotonin
5HT2A receptor improves coronary patency in an in vivo model of recurrent
thrombosis. J Thromb Haemost. 2010 Feb;8(2):331-40. doi:
10.1111/j.1538-7836.2009.03693.x. Epub 2009 Nov 17. PubMed PMID: 19922435; PubMed
Central PMCID: PMC2916638.
3: Adams JW, Ramirez J, Shi Y, Thomsen W, Frazer J, Morgan M, Edwards JE, Chen W,
Teegarden BR, Xiong Y, Al-Shamma H, Behan DP, Connolly DT. APD791,
3-methoxy-n-(3-(1-methyl-1h-pyrazol-5-yl)-4-(2-morpholinoethoxy)phenyl)benzamide,
a novel 5-hydroxytryptamine 2A receptor antagonist: pharmacological profile,
pharmacokinetics, platelet activity and vascular biology. J Pharmacol Exp Ther.
2009 Oct;331(1):96-103. doi: 10.1124/jpet.109.153189. Epub 2009 Jul 23. PubMed
PMID: 19628629.
| Patent ID | Date | Patent Title |
|---|---|---|
| US2015361031 | 2015-12-17 | STAT3 INHIBITOR |
| US8785441 | 2014-07-22 | 3-phenyl-pyrazole derivatives as modulators of the 5-HT2A serotonin receptor useful for the treatment of disorders related thereto |
| US2013296321 | 2013-11-07 | CRYSTALLINE FORMS AND PROCESSES FOR THE PREPARATION OF PHENYL-PYRAZOLES USEFUL AS MODULATORS OF THE 5-HT2A SEROTONIN RECEPTOR |
| US2012252813 | 2012-10-04 | CRYSTALLINE FORMS OF CERTAIN 3-PHENYL-PYRAZOLE DERIVATIVES AS MODULATORS OF THE 5-HT2A SEROTONIN RECEPTOR USEFUL FOR THE TREATMENT OF DISORDERS RELATED THERETO |
| US8148417 | 2012-04-03 | PRIMARY AMINES AND DERIVATIVES THEREOF AS MODULATORS OF THE 5-HT2A SEROTONIN RECEPTOR USEFUL FOR THE TREATMENT OF DISORDERS RELATED THERETO |
| US8148418 | 2012-04-03 | ETHERS, SECONDARY AMINES AND DERIVATIVES THEREOF AS MODULATORS OF THE 5-HT2A SEROTONIN RECEPTOR USEFUL FOR THE TREATMENT OF DISORDERS RELATED THERETO |
| US2011105456 | 2011-05-05 | 3-PHENYL-PYRAZOLE DERIVATIVES AS MODULATORS OF THE 5-HT2A SEROTONIN RECEPTOR USEFUL FOR THE TREATMENT OF DISORDERS RELATED THERETO |
| US7884101 | 2011-02-08 | 3-Phenyl-pyrazole derivatives as modulators of the 5-HT2a serotonin receptor useful for the treatment of disorders related thereto |
| US2010234380 | 2010-09-16 | CRYSTALLINE FORMS AND PROCESSES FOR THE PREPARATION OF PHENYL-PYRAZOLES USEFUL AS MODULATORS OF THE 5-HT2A SEROTONIN RECEPTOR |
| US2007244086 | 2007-10-18 | 3-Phenyl-Pyrazole Derivatives as Modulators of the 5-Ht2A Serotonin Receptor Useful for the Treatment of Disorders Related Thereto |
///////////APD-791 , 887936-68-7, Temanogrel , PHASE 1, ARENA,
CN1C(=CC=N1)C2=C(C=CC(=C2)NC(=O)C3=CC(=CC=C3)OC)OCCN4CCOCC4
C(=O)(c1cc(ccc1)OC)Nc1ccc(c(c1)c1n(ncc1)C)OCCN1CCOCC1
GSK 2126458, Omipalisib, PI3K/mTOR inhibitor

GSK 2126458
CAS 1086062-66-9
OMipalisib;GSK2126458;GSK-2126458;GSK2126458 (GSK458);GSK212;
2,4-Difluoro-N-[2-methoxy-5-[4-(4-pyridazinyl)-6-quinolinyl]-3-pyridinyl]benzenesulfonamide;
2,4-Difluoro-N-[2-Methoxy-5-[4-(pyridazin-4-yl)quinolin-6-yl]pyridin-3-yl]benzenesulfonaMide
2,4-Difluoro-N-[2-methoxy-5-[4-(4-pyridazinyl)quinolin-6-yl]pyridin-3-yl]benzenesulfonamide
phosphoinositide 3 kinase inhibitor
idiopathic pulmonary fibrosis
PHASE 1
MW 505.49598
MF C25H17F2N5O3S
GSK…….http://www.gsk.com/media/280387/product-pipeline-2014.pdf
![]()
Omipalisib (GSK2126458): Omipalisib, also known as GSK2126458, is a small-molecule pyridylsulfonamide inhibitor of phosphatidylinositol 3-kinase (PI3K) with potential antineoplastic activity. PI3K inhibitor GSK2126458 binds to and inhibits PI3K in the PI3K/mTOR signaling pathway, which may trigger the translocation of cytosolic Bax to the mitochondrial outer membrane, increasing mitochondrial membrane permeability and inducing apoptotic cell death. Bax is a member of the proapoptotic Bcl2 family of proteins. PI3K, often overexpressed in cancer cells, plays a crucial role in tumor cell regulation and survival.
GlaxoSmithKline (GSK) is developing omipalisib (GSK-2126458), a phosphoinositide 3-kinase/mammalian target of rapamycin (PI3K/mTOR) inhibitor as well as mTOR complex 1 and 2 inhibitor, for the potential oral treatment of cancer and idiopathic pulmonary fibrosis
MEDKOO

![]()
![]()
![]()
![]()
|
Certificate of Analysis: |
|
|
QC data: |
GSK2126458 is a highly potent PI3K and mTOR inhibitor. In vivo, GSK2126458 showed anti-tumor activity in both pharmacodynamic and tumor growth efficacy models. GSK2126458 reduced the phosphorylated AKT, p70S6K contents in a dose and time dependent way. The IC50 of GSK2126458 is 2 nM for pAKT in the HCC1954 breast carcinoma cell line. In various human tumor cells, GSK2126458 had a width of inhibitory activity for potent cell growth and induced cell death. Notably, GSK2126458 acted mainly by not induction of apoptosis but cell cycle arrest, particularly in G1-phase
GlaxoSmithKline (GSK) is developing omipalisib (GSK-2126458), a phosphoinositide 3-kinase/mammalian target of rapamycin (PI3K/mTOR) inhibitor as well as mTOR complex 1 and 2 inhibitor, for the potential oral treatment of cancer and idiopathic pulmonary fibrosis
GSK-2126458 is a phosphatidylinositol 3-Kinase (PI3K) inhibitor in early clinical development for the oral treatment of solid tumors and for the oral treatment of lymphoma. Early clinical studies are ongoing for the treatment of idiopathic pulmonary fibrosis. The compound is being developed b GlaxoSmithKline.
In August 2009, a phase I trial began for solid tumors and lymphoma . In April 2012, phase Ib co-clinical trials in advanced prostate cancer (PC) were underway . In March 2013, a phase I trial was initiated in the UK in patients with idiopathic pulmonary fibrosis
In April 2014, a phase I, open-label, multicenter, dose-escalation study (study number P3K113794) and safety data were presented at the 105th AACR meeting in San Diego, CA. Advanced solid tumor patients (n = 69) received oral continuous GSK-2126458 or intermittent GSK-2126458 bid + trametinib. For GSK-2126458 and trametinib, the MTD in QD cohort was 2 and 1 mg, respectively, and also 1 and 1.5 mg, respectively
PAPER ![]()
![]()
![]()
![]()
![]()
![]()
![]()
Discovery of GSK2126458, a highly potent inhibitor of PI3K and the mammalian target of rampamycin
ACS Med Chem Lett 2010, 1(1): 39

Phosphoinositide 3-kinase α (PI3Kα) is a critical regulator of cell growth and transformation, and its signaling pathway is the most commonly mutated pathway in human cancers. The mammalian target of rapamycin (mTOR), a class IV PI3K protein kinase, is also a central regulator of cell growth, and mTOR inhibitors are believed to augment the antiproliferative efficacy of PI3K/AKT pathway inhibition. 2,4-Difluoro-N-{2-(methyloxy)-5-[4-(4-pyridazinyl)-6-quinolinyl]-3-pyridinyl}benzenesulfonamide (GSK2126458, 1) has been identified as a highly potent, orally bioavailable inhibitor of PI3Kα and mTOR with in vivo activity in both pharmacodynamic and tumor growth efficacy models. Compound 1 is currently being evaluated in human clinical trials for the treatment of cancer.

synthesis![]()
![]()
![]()
![]()
![]()


………………..
PATENT
WO 2008144463
http://www.google.co.in/patents/WO2008144463A1?cl=en
Example 345
2,4-difluoro-N-{2-(methyloxy)-5-[4-(4-pyridazinyl)-6-quinolinyl]-3- pyridinyl } benzenesulf onamide
a) 6-bromo-4-(4-pyridazinyl)quinoline
Dissolved 6-bromo-4-iodoquinoline (17.43 g, 52.2 mmol), 4- (tributylstannanyl)pyridazine (19.27 g, 52.2 mmol), and PdC12(dppf)-CH2C12 (2.132 g, 2.61 mmol) in 1,4-dioxane (200 mL) and heated to 105 °C. After 3 h, added more palladium catalyst and heated for 6 h. Concentrated and dissolved in methylene chloride/methanol. Purified by column chromatography (combiflash) with 2% MeOH/EtOAc to 5% MeOH/EtOAc to give the crude title compound. Trituration with EtOAc furnished 6-bromo-4-(4-pyridazinyl)quinoline (5.8 g, 20.27 mmol, 38.8 % yield). MS(ES)+ m/e 285.9, 287.9 [M+H]+.
b) 2,4-difluoro-N-{2-(methyloxy)-5-[4-(4-pyridazinyl)-6-quinolinyl]-3- pyridinyl } benzenesulf onamide A slurry of 6-bromo-4-(4-pyridazinyl)quinoline (4.8 g, 16.78 mmol), bis(pinacolato)diboron (4.69 g, 18.45 mmol) , PdC12(dppf)-CH2C12 (530 mg, 0.649 mmol) and potassium acetate (3.29 g, 33.6 mmol) in anhydrous 1,4-dioxane (120 ml) was heated at 100 °C for 3 h. The complete disappearance of the starting bromide was observed by LCMS. The reaction was then treated with N-[5-bromo-2- (methyloxy)-3-pyridinyl]-2,4-difluorobenzenesulfonamide (6.68 g, 17.61 mmol) and another portion of PdC12(dppf)-CH2C12 (550 mg, 0.673 mmol), then heated at 110 °C for 16 h. The reaction was allowed to cool to room temperature, filtered, and concentrated. Purification of the residue by chromatography (Analogix; 5% MeOH / 5% CH2C12 / 90% EtOAC) gave 6.5 g (76%) desired product. MS(ES)+ m/e 505.9 [M+H]+.
INTERMEDIATES:
Intermediate 1 Similar but not same
Scheme A:

Conditions: a) Tributyl(vinyl)tin, Pd(PPh3)4, dioxane, reflux; b) OsO4, NaIO4, 2,6- lutidine, r-BuOH, dioxane, H2O, rt; c) (4-pyridyl)boronic acid, Pd(PPh3)4, 2 M K2CO35 DMF, 100 DC.
4-(4-pyridinyl)-6-quinolinecarbaldehydeSimilar but not same

a) 4-chloro-6-ethenylquinoline
A mixture of 6-bromo-4-chloroquinoline (6.52 g, 26.88 mmol; see J. Med. Chem., H 268 (1978) ), tributyl(vinyl)tin (8.95 g, 28.22 mmol), and tetrakistriphenylphospbine palladium (0) (0.62 g, 0.54 mmol) in 1,4-dioxane (150 mL) was refluxed for 2.0 h, cooled to room temperature, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (0-4% MeOH:CH2Cl2) to give the title compound (5.1 g) as a pale yellow solid. MS (ES)+ m/e 190 [M+H]+. This material was used directly in the next step.
b) 4-chloro-6-quinolinecarbaldehyde
A mixture of 4-chloro-6-ethenylquinoline (5.1 g, 26.88 mmol), 2,6-lutidine
(5.76 g, 53.75 mmol), sodium (meta) periodate (22.99 g, 107.51 mmol), and osmium tetroxide (5.48 g of a 2.5% solution in tert-butanol, 0.538 mmol) in l,4-dioxane:H2θ (350 mL of 3: 1 mixture) was stirred for 3.5 h at room temperature and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (CH2Cb) to give the title compound (4.26 g, 83% for 2 steps) as a pale yellow solid. MS (ES)+ m/e 192 [M+H]+.
c) 4-(4-pyridmyl)-6-qumolinecarbaldehyde
A mixture of 4-chloro-6-quinolinecarbaldehyde (3.24 g, 16.92 mmol), A- pyridylboronic acid (3.12 g, 25.38 mmol), tetrakistriphenylphosphine palladium (0) (0.978 g, 0.846 mmol), and 2M aqueous K2CO3 (7.02 g, 50.76 mmol, 25.4 mis of 2M solution) in DMF (100 mL) was heated at 100 °C for 3.0 h and cooled to room temperature. The mixture was filtered through Celite and the Celite was washed with EtOAc. The filtrate was transferred to a separatory funnel, washed with water and saturated NaCl, dried (Na2SO4), filtered and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (5% MeOH:CH2Cl2) to give the title compound (2.03 g, 51%) as a tan solid. MS (ES)+ m/e 235 [M+H]+.
Intermediate 2
Preparation of 2-amino-5 -bromo-N,N-dimethyl-3 -pyridinesulfonamideSimilar but not same

a) 2-ammo-5-bromo-3-pyridinesulfonyl chloride
To a cooled (0 °C) solution of chlorosulfonic acid (58 mL) under vigorous stirring was added 5-bromo-2-pyridinamine (86.7 mmol) portionwise. The reaction mixture was then heated at reflux for 3 hrs. Upon cooling to room temperature, the reaction mixture was poured over ice (-100 g) with vigorous stirring. The resulting yellow precipitate was collected by suction filtration, washing with cold water and petroleum ether to provide the title compound as an orange-yellow solid (18.1 g, 77% yield). MS(ES)+ m/e 272.8 [M+H]+.
* Other sulfonyl chlorides can be prepared using this procedure by varying the choice of substituted aryl or heteroaryl.
b) 2-amino-5-bromo-N,N-dimethyl-3-pyridinesulfonamide
To a cold (0 DC) suspension of 2-amino-5-bromo-3-pyridinesulfonyl chloride (92.1 mmol) in dry 1,4-dioxane (92 mL) was added pyridine (101.3 mmol) followed by a 2M solution of dimethylamine in THF (101.3 mmol). The reaction was allowed to warm to rt for 2 h, heated to 50 DC for 1 h, then cooled to rt. After standing for 2 h, the precipitate was collected by filtration and rinsed with a minimal amount of cold water. Drying the precipitate to constant weight under high vacuum provided 14.1 g (55%) of the title compound as a white solid. MS(ES)+ m/e 279.8, 282.0 [M+H]+.
Intermediate 3
Preparation of 2-amino-N,N-dimethyl-5-(4,4,5,5-tetramethyl-l,3.2-dioxaborolan-2- yl)-3 -pyridinesulfonamideSimilar but not same

c) To a solution of 2-amino-5-bromo-N,N-dimethyl-3 -pyridinesulfonamide (7.14 mmol) in 1,4-dioxane (35 mL) was added 4,4,4′,4′,5,5,5′,5′-octamethyl-2,2′-bi-l,3,2- dioxaborolane (7.86 mmol), potassium acetate (28.56 mmol) and [1,1 ‘- bis(diphenylphosphmo)-ferrocene] dichloropalladium(II) dichloromethane complex (1 :1) (0.571 mmol). The reaction mixture was stirred at 100 °C for 18 h. The reaction was concentrated in vacuo, re-dissolved in ethyl acetate (50 mL) and purified on silica using 60% ethyl acetate/hexanes to yield the title compound as a tan solid (86 %). IH ΝMR (400 MHz, DMSOd6) δ ppm 8.41 (d, 1 H, J =1.52), 7.92 (d, 1 H, J = 1.77), 2.68 (s, 6 H), 1.28 (s, 12 H).
* Other boronate or boronic acids can be prepared using this procedure by varying the choice of aryl or heteroaryl bromide. Scheme 17:

Conditions: a) NaO(Rl), (Rl)OH, O 0C to room temperature; b) SnCl2-2H2O, ethyl acetate, reflux; c) (R2)SO2C1, pyridine, O 0C to room temperature.
Intermediate 4
Preparation of N-r5-bromo-2-(methyloxy)-3-pyridinyll-2,4- difluorobenzenesulfonamide
N-[5-bromo-2-(methyloxy)-3-pyridinyl]-2,4- difluorobenzenesulfonamide a) 5-bromo-2-(methyloxy)-3-nitropyridine

To a cooled (0 °C) solution of 5-bromo-2-chloro-3-nitropyridine (50 g, 211 mmol) in methanol (200 mL) was added dropwise over 10 minutes 20% sodium methoxide (50 mL, 211 mmol) solution. The reaction, which quickly became heterogeneous, was allowed to warm to ambient temperature and stirred for 16 h. The reaction was filtered and the precipitate diluted with water (200 mL) and stirred for 1 h. The solids were filtered, washed with water (3 x 100 mL) and dried in a vac oven (40 °C) to give 5-bromo-2-(methyloxy)-3-nitropyridine (36 g, 154 mmol, 73.4 % yield) as a pale yellow powder. The original filtrate was concentrated in vacuo and diluted with water (150 mL). Saturated ammonium chloride (25 mL) was added and the mixture stirred for 1 h. The solids were filtered, washed with water, and dried in a vac oven (40 °C) to give a second crop of 5-bromo-2-(methyloxy)-3- nitropyridine (9 g, 38.6 mmol, 18.34 % yield). Total yield = 90%. MS(ES)+ m/e 232.8, 234.7 [M+H]+.
b) 5-bromo-2-(methyloxy)-3-pyridinamine

To a solution of 5-bromo-2-(methyloxy)-3-nitropyridine (45 g, 193 mmol) in ethyl acetate (1 L) was added tin(II) chloride dihydrate (174 g, 772 mmol). The reaction mixture was heated at reflux for 4 h. LC/MS indicated some starting material remained, so added 20 mol% tin (II) chloride dihydrate and continued to heat at reflux. After 2 h, the reaction was allowed to cool to ambient temperature and concentrated in vacuo. The residue was treated with 2 N sodium hydroxide and the mixture stirred for 1 h. The mixture was then with methylene chloride (1 L), filtered through Celite, and washed with methylene chloride (500 mL). The layers were separated and the organics dried over magnesium sulfate and concentrated to give 5-bromo-2-(methyloxy)-3-pyridinamine (23 g, 113 mmol, 58.7 % yield). The product was used crude in subsequent reactions. MS(ES)+ m/e 201.9, 203.9 [M+H]+.
c) N-[5-bromo-2-(methyloxy)-3-pyridinyl]-2,4-difluorobenzenesulfonamide
To a cooled (0 °C) solution of 5-bromo-2-(methyloxy)-3-pyridinamine (20.3 g, 100 mmol) in pyridine (200 mL) was added slowly 2,4-difluorobenzenesulfonyl chloride (21.3 g, 100 mmol) over 15 min (reaction became heterogeneous). The ice bath was removed and the reaction was stirred at ambient temperature for 16 h, at which time the reaction was diluted with water (500 mL) and the solids filtered off and washed with copious amounts of water. The precipitate was dried in a vacuum oven at 50 °C to give N-[5-bromo-2-(methyloxy)-3-pyridinyl]-2,4- difluorobenzenesulfonamide (12 g, 31.6 mmol, 31.7 % yield) MS(ES)+ m/e 379.0, 380.9 [M+H]+.
References
|
1: Zhang Y, Xue D, Wang X, Lu M, Gao B, Qiao X. Screening of kinase inhibitors targeting BRAF for regulating autophagy based on kinase pathways. Mol Med Rep. 2014 Jan;9(1):83-90. doi: 10.3892/mmr.2013.1781. Epub 2013 Nov 7. PubMed PMID: 24213221.
2: Villanueva J, Infante JR, Krepler C, Reyes-Uribe P, Samanta M, Chen HY, Li B, Swoboda RK, Wilson M, Vultur A, Fukunaba-Kalabis M, Wubbenhorst B, Chen TY, Liu Q, Sproesser K, DeMarini DJ, Gilmer TM, Martin AM, Marmorstein R, Schultz DC, Speicher DW, Karakousis GC, Xu W, Amaravadi RK, Xu X, Schuchter LM, Herlyn M, Nathanson KL. Concurrent MEK2 mutation and BRAF amplification confer resistance to BRAF and MEK inhibitors in melanoma. Cell Rep. 2013 Sep 26;4(6):1090-9. doi: 10.1016/j.celrep.2013.08.023. Epub 2013 Sep 19. PubMed PMID: 24055054; PubMed Central PMCID: PMC3956616.
3: Kim HG, Tan L, Weisberg EL, Liu F, Canning P, Choi HG, Ezell SA, Wu H, Zhao Z, Wang J, Mandinova A, Griffin JD, Bullock AN, Liu Q, Lee SW, Gray NS. Discovery of a potent and selective DDR1 receptor tyrosine kinase inhibitor. ACS Chem Biol. 2013 Oct 18;8(10):2145-50. doi: 10.1021/cb400430t. Epub 2013 Aug 13. PubMed PMID: 23899692; PubMed Central PMCID: PMC3800496.
4: Khalili JS, Yu X, Wang J, Hayes BC, Davies MA, Lizee G, Esmaeli B, Woodman SE. Combination small molecule MEK and PI3K inhibition enhances uveal melanoma cell death in a mutant GNAQ- and GNA11-dependent manner. Clin Cancer Res. 2012 Aug 15;18(16):4345-55. doi: 10.1158/1078-0432.CCR-11-3227. Epub 2012 Jun 25. PubMed PMID: 22733540; PubMed Central PMCID: PMC3935730.
5: Greger JG, Eastman SD, Zhang V, Bleam MR, Hughes AM, Smitheman KN, Dickerson SH, Laquerre SG, Liu L, Gilmer TM. Combinations of BRAF, MEK, and PI3K/mTOR inhibitors overcome acquired resistance to the BRAF inhibitor GSK2118436 dabrafenib, mediated by NRAS or MEK mutations. Mol Cancer Ther. 2012 Apr;11(4):909-20. doi: 10.1158/1535-7163.MCT-11-0989. Epub 2012 Mar 2. PubMed PMID: 22389471.
6: Wang M, Gao M, Miller KD, Sledge GW, Zheng QH. [11C]GSK2126458 and [18F]GSK2126458, the first radiosynthesis of new potential PET agents for imaging of PI3K and mTOR in cancers. Bioorg Med Chem Lett. 2012 Feb 15;22(4):1569-74. doi: 10.1016/j.bmcl.2011.12.136. Epub 2012 Jan 10. PubMed PMID: 22297110.
7: Schenone S, Brullo C, Musumeci F, Radi M, Botta M. ATP-competitive inhibitors of mTOR: an update. Curr Med Chem. 2011;18(20):2995-3014. Review. PubMed PMID: 21651476.
8: Leung E, Kim JE, Rewcastle GW, Finlay GJ, Baguley BC. Comparison of the effects of the PI3K/mTOR inhibitors NVP-BEZ235 and GSK2126458 on tamoxifen-resistant breast cancer cells. Cancer Biol Ther. 2011 Jun 1;11(11):938-46. Epub 2011 Jun 1. PubMed PMID: 21464613; PubMed Central PMCID: PMC3127046.
ICOTINIB
ICOTINIB
4-((3-ethynylphenyl)amino)-6,7-benzo-12-crown-4-quinazoline
N-(3-Ethynylphenyl)-7,8,10,11,13,14-hexahydro[1,4,7,10]tetraoxacyclododecino[2,3-g]quinazolin-4-amine
[1,4,7,10]Tetraoxacyclododecino[2,3-g]quinazolin-4-amine, N-(3-ethynylphenyl)-7,8,10,11,13,14-hexahydro-
BPI 2009H, UNII-JTD32I0J83
610798-31-7 CAS BASE

Icotinib Hydrochloride, 1204313-51-8, CS-0918, HY-15164, Conmana Zhejiang Beta Pharma Ltd.
CLINICALS………http://clinicaltrials.gov/search/intervention=Icotinib
Icotinib Hydrochloride (BPI-2009H), or Icotinib, is a highly selective, first generation epidermal growth factor receptor tyrosine kinase inhibitor (EGFR-TKI). EGFR is an oncogenic driver and patients with somatic mutations, particularly an exon 19 deletion or exon 21 L858R mutation, within the tyrosine kinase domain have activating mutations that lead to unchecked cell proliferation. Overexpression of EGFR causes inappropriate activation of the anti-apoptotic Ras signaling pathway, found in many different types of cancer. Icotinib is a quinazoline derivative that binds reversibly to the ATP binding site of the EGFR protein, preventing completion of the signal transduction cascade.[1]
Clinical Evaluation
Icotinib is indicated for the treatment for EGFR mutation-positive, advanced or metastatic non-small cell lung cancer (NSCLC) as a second-line or third-line treatment, for patients who have failed at least one prior treatment with platinum-based chemotherapy. The ICOGEN trial was a double-blind, head-to-head phase III study comparing icotinib with gefitinib in all-comers. From 27 centers in China, 399 patients were randomized between the two treatments testing for a primary objective of progression-free survival and secondary objectives of overall survival, time to progression, quality of life, percentage of patients who achieved an objective response, and toxic effects. The ICOGEN results showed icotinib to have a median PFS of 4.6 months (95% CI 3.5 – 6.3) as compared to gefitinib which has a PFS of 3.4 months (95% CI 2.3 – 3.8). After the study was completed, post-hoc analysis revealed that in the icotinib treatment group, patients with activating EGFR mutations showed improved PFS as compared to patients with wild-type EGFR. Icotinib also was associated with fewer adverse events than gefitinib when considering all grades of reactions together (61% versus 70% respectively, p = 0.046).[2] The phase IV ISAFE trial evaluated 5,549 patients and showed icotinib to have an overall response rate of 30% and a low adverse event rate of 31.5%.[3]
Regulatory Approvals
Icotinib was approved in China by the SFDA in June, 2011.[4] Since approval, Icotinib has treated over 40,000 patients in China successfully and is now undergoing global development.
January 2014, Beta Pharma, Inc. was given a “May Proceed” from the US FDA to conduct a Phase I study for the evaluation of icotinib as a treatment of EGFR+ Non-Small Cell Lung Cancer (NSCLC).
Icotinib is a potent and specific EGFR inhibitor with IC50 of 5 nM, including the EGFR, EGFR(L858R), EGFR(L861Q), EGFR(T790M) and EGFR(T790M, L858R). Phase 4.Icotinib hydrochloride is the epidermal growth factor receptor kinase targeting a new generation of targeted anti-cancer drugs, completely independent from the original tumor clinical practitioners and experts of science, through eight years of the development, its first adaptation disease is advanced non-small cell lung cancer. Icotinib is an orally available quinazoline-based inhibitor of epidermal growth factor receptor (EGFR), with potential antineoplastic activity. Icotinib selectively inhibits the wild-type and several mutated forms of EGFR tyrosine kinase. This may lead to an inhibition of EGFR-mediated signal transduction and may inhibit cancer cell proliferation. EGFR, a receptor tyrosine kinase, is upregulated in a variety of cancer cell types. Icotinib was approved in China in 2011
Icotinib has been found to be noninferior to gefitinib in patients with non-small-cell lung cancer (NSCLC), according to reports from the phase III Chinese double-blind ICOGEN study.
“[I]cotinib is a valid therapeutic option for patients with non-small-cell lung cancer as a second-line or third-line treatment, although patients might find taking icotinib three times a day an inconvenience,” write Yan Sun (Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China) and colleagues.
Icotinib is an oral epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI) that has exhibited good antitumor activity in phase II studies. However, it has a shorter half-life than gefitinib, another TKI, which means that it needs to be taken more often.
…
-
Tyrosine kinase receptors are trans-membrane proteins that, in response to an extracellular stimulus, propagate a signaling cascade to control cell proliferation, angiogenesis, apoptosis and other important features of cell growth. One class of such receptors, epidermal growth factor receptor (EGFR) tyrosine kinases, are over-expressed in many human cancers, including brain, lung, liver, bladder, breast, head and neck, esophagus, gastrointestinal, breast, ovary, cervix or thyroid cancer.
-
EGFR is expressed in many types of tumor cells. Binding of cognate ligands (including EGF, TGFα (i.e., Transforming Growth Factor-α) and neuregulins) to the extracellular domain causes homo- or heterodimerization between family members; the juxtaposition of cytoplasmic tyrosine kinase domains results in transphosphorylation of specific tyrosine, serine and threonine residues within each cytoplasmic domain. The formed phosphotyrosines act as docking sites for various adaptor molecules and subsequent activation of signal transduction cascades (Ras/mitogen-activated, PI3K/Akt and Jak/STAT) that trigger proliferative cellular responses.
-
Various molecular and cellular biology and clinical studies have demonstrated that EGFR tyrosine kinase inhibitors can block cancer cell proliferation, metastasis and other EGFR-related signal transduction responses to achieve clinical anti-tumor therapeutic effects. Two oral EGFR kinase inhibitors with similar chemical structures are Gefitinib (Iressa; AstraZeneca), approved by the U.S. FDA for advanced non-small cell lung cancer in 2003 (and later withdrawn), and Erlotinib Hydrochloride (Tarceva; Roche and OSI), approved by the U.S. FDA for advanced non-small cell lung cancer and pancreatic cancer treatment in 2004.
-
Chinese Patent Publication No. CN1305860C discloses the structure of 4-[(3-ethynyl-phenyl)amino]-6,7-benzo-12-crown-quinoline (free base) on page 29, Example 15, Compound 23.
Icotinib was launched in China in August 2011, after approval by the State Food and Drug Administration. It is a targeted EGFR tyrosine kinase inhibitor that, like erlotinib (Tarceva) and gefitinib (Iressa), shows benefit in patients with EGFR m+ NSCLC.
……………………………………..
http://www.google.com/patents/EP2392576A1
-
Formula I (Icotinib hydrochloride):

Method 1:

Method 2:

Method 3:
-

-
BPI-02 is obtained by recrystallization.

http://www.google.com/patents/EP2392576A1 Example 1Step 1
-

-
Preparation: 16 kg (400 mol) of sodium hydroxide was dissolved in 80 L of water in a 400 L reactor, and then 18.8 L (140 mol) of triethylene glycol, 32 L of THF were added into the reactor. After cooling below 5 °C, a solution of 47.84 kg (260 mol) of tosyl chloride and 50 L of THF was added dropwise. Following the addition, the reaction mixture was kept at this temperature for 2 hours, and it was then poured into 240 L of ice water. The precipitate was formed and filtered, washed with a small amount of water, and dried. 58.64 kg of BPI-01 as a white crystalline powder was yielded at 91.4%. mp: 77-80 °C, HPLC: 97%. TLC (petroleum ether: ethyl acetate = 1:1) Rf = 0.87.
-
NMR data: 1H-NMR (CDCl3): δ ppm: 7.78 (d, 4H, J = 10.4 Hz, benzene protons by sulfonyl group); 7.34 (d, 4H, J = 11.6 Hz, benzene protons by methyl group); 4.129 (dd, 4H, J = 5.6 Hz, ethylene protons by the sulfonyl group); 3.64 (dd, 4H, J = 5.6 Hz, ethylene protons away from the sulfonyl group); 3.517 (s, 4H, ethylene protons in the middle); 2.438 (s, 6H, methyl protons on the benzene).

Step 2
-

-
Preparation: A solution containing 3.64 kg (20 mol) of ethyl 3,4-dihydroxybenzoate and 12.4 kg (89.6 mol) of potassium carbonate in 300 L of N,N-dimethylformamide was stirred and heated to 85-90 °C for about 30 minutes. A solution of 9.17 kg (20 mol) of BPI-01 in 40 L of N,N-dimethylformamide was added dropwise over 1.5-2 hours. After the addition, the reaction was kept for 30 minutes; the reaction completion was confirmed by TLC (developing solvent: petroleum ether:ethyl acetate = 1:1, Rf = 0.58). The reaction mixture was removed from the reactor and filtered. Then, the filtrate was evaporated to remove N,N-dimethylformamide; 240 L of ethyl acetate was added to dissolve the residue. After filtration and vacuum evaporation, the residual solution was extracted with 300 L of petroleum ether. After evaporation of the petroleum ether, the residual solids were re-crystallized with isopropanol in a ratio of 1:2.5 (W/V); 1.68 kg of BPI-02 as a white powder was obtained in a yield of 28%. mp: 73-76 °C, HPLC: 96.4%. NMR data: 1H-NMR (CDCl3): δ ppm: 7.701 (d, 1H, J = 2.4 Hz, benzene proton at position 6); 7.68 (s, 1 H, benzene proton at position 2); 6.966 (d, 1H, J = 10.8 Hz, benzene proton at position 5); 4.374-3.81 (q, 2H, J = 9.6 Hz, methylene protons of the ethyl); 3.78-4.23 (dd, 12H, J = 4.8 Hz, crown ether protons); 1.394 (t, 3H, J = 9.6 Hz, methyl protons of the ethyl). MS: m/z 296.

Step 3
-

-
Preparation: A solution of 592 g (2 mol) of BPI-02 and 600 mL of acetic acid in a 5 L reaction flask was cooled to 0°C; 1640 mL (25.4 mol) of concentrated nitric acid was slowly added. The internal temperature should not exceed 10 °C. While cooled below 0°C, 1 L of concentrated sulfuric acid was added dropwise. The internal temperature should not be higher than 5°C. After the addition, the reaction was kept at 0-5 °C for 1-2 hours. After completion of the reaction, the reaction solution was poured into 15 L of ice water in a plastic bucket. After mixing, filtration, and re-crystallization in ethanol, 449 g of BPI-03 as a light yellow to yellow crystalline powder was obtained in 65.7% yield. mp: 92-95 °C, HPLC: 98.2%. TLC (petroleum ether: ethyl acetate =1:1) Rf = 0.52. NMR data: 1H-NMR (CDCl3): δ ppm: 7.56 (s, 1H, benzene proton at position 5); 7.20 (s, 1H, benzene proton at position 2); 4.402 (q, 2H, J = 9.2 Hz, methylene protons of the ethyl); 4.294 (dd, 12H, J = 4.8 Hz, crown ether protons); 1.368 (t, 3H, J = 9.2 Hz, methyl protons of the ethyl).

Step 4
-

-
Preparation: In a 3 L hydrogenation reactor, 2 L of methanol and 195 g (0.57 mol) of BPI-03 were added, and then 63 mL of acetyl chloride was slowly added. After a short stir, 33 g of Pd/C containing 40% water was added. The reaction was conducted under 4 ATM hydrogen until hydrogen absorption stopped, and then the reaction was kept for 1-2 hours. After completion of the reaction, the reaction mixture was transferred into a 5 L reactor. After filtration, crystallization, and filtration, the product was obtained. The mother liquor was concentrated under vacuum, and more product was obtained. The combined crops were 168 g of BPI-04 as a white to pink crystalline powder in a yield of 85%. mp: 198-201 °C, HPLC: 99.1 %. TLC (petroleum ether: ethyl acetate = 1:1) Rf = 0.33. NMR data: 1H-NMR (DMSO-d6): δ ppm: 8-9 (br., 3H, 2 protons of the amino group and a proton of the hydrochloric acid); 7.37 (s, 1H, benzene proton at position 5); 6.55 (s, 1H , benzene proton at position 2); 4.25 (q, 2H, J = 7.06 Hz, methylene protons of the ethyl); 4.05 (dd, 12H, J = 4.04 Hz, crown ether protons); 1.31 (t, 3H, J = 7.06 Hz, methyl protons of the ethyl).

Step 5
-

-
Preparation: 1105 g (3.175 mol)of BPI-04, 4810 g (106.9 mol) of formamide, and 540 g (8.55 mol) of ammonium formate were added to a 10 L 3-neck bottle. The reaction mixture was heated to 165 °C under reflux for 4 hours. After cooling to room temperature, 3 L of water was added, and then the mixture was stirred for 10 minutes. After filtration, washing, and drying, 742 g of BPI-05 as a white crystalline powder was obtained in a yield of 80%. mp: 248-251 °C, HPLC: 99.78%. TLC (chloroform: methanol = 8:1) Rf = 0.55. NMR data: 1H-NMR (DMSO-d6): δ ppm: 12.06 (s, 1H, NH of the quinazoline); 8.0 (d, 1H, J = 3.28 Hz, proton of the quinazoline position 3); 7.62 (s, 1H, proton of the quinazoline position 6); 7.22 (s, 1H, proton of the quinazoline position 9); 4.25 (dd, 12H, J = 4.08 Hz, crown ether protons).

Step 6
-

-
Preparation: 337 g (1.13 mol) of BPI-05, 7.1 L of chloroform, 1.83 L (19.58mol) of POCI3 and 132 ml of N,N-dimethylformamide were added to a 10 L 3-neck bottle. The reaction mixture was stirred at reflux temperature. After dissolution, reaction completion was checked by TLC (developing solvent: chloroform: methanol = 15:1, Rf = 0.56); the reaction took approximately 8 hours to complete. Then, the reaction solution was cooled and evaporated under vacuum to dryness. The residue was dissolved in 4 L of chloroform; 4 kg of crushed ice was poured into the solution and the mixture was stirred for 0.5 hours. After separation, the aqueous phase was extracted twice with 2 L of chloroform. The organic phases were combined, 4 L of ice water was added and the pH was adjusted with 6 N NaOH to pH 8-9 while the temperature was maintained below 30 °C. After separation, the organic phase was washed with saturated NaCl, dried over anhydrous sodium sulfate and the solvents removed by vacuum evaporation. The residual solids were washed with acetone and filtered; 268 g of BPI-06 as a white crystalline powder was obtained in a yield of 77% with mp: 164-167°C and HPLC purity of 99%. NMR data: 1H-NMR (CDCl3): δ ppm: 8.89 (s, 1H, proton of the quinazoline position 2); 7.68 (s, 1H, proton of the quinazoline position 9); 7.42 (s, 1H, proton of the quinazoline position 6); 4.38-3.81 (dd, 12H, J = 3.88 Hz, crown ether protons).

Step 7
-
-
Preparation of the compound of the present invention: To a suspension of 20.8 g of BPI-06 in 500 mL of ethanol was added 25 mL of N,N-dimethylformamide and a solution of 8.98 g m-acetylene aniline in 200 mL of isopropanol. The reaction mixture was stirred at room temperature for 5 minutes until dissolved completely, and then the reaction solution was heated at reflux for 3 hours. After concentration and drying, the residual solids were dissolved in ethyl acetate, washed with water, and dried over anhydrous sodium sulfate. Thus, 27.1 g of the compound of Formula I was obtained as a white crystalline powder. NMR data: 1H-NMR (Bruker APX-400, solvent: DMSO-d6, TMS as internal standard): δ ppm: 3.58 (dd, 2H, two protons of the crown position 12); 3.60 (dd, 2H, two protons of the crown position 13); 3.73 (dd, 2H, two protons of the crown position 10); 3.80 (dd, 2H, two protons of the crown position 15); 4.30 (s, 1H, proton of the alkynyl); 4.34 (dd, 2H, two protons of the crown position 16); 4.40 (dd, 2H, two protons of the crown position 9); 7.39 (d, 1H, benzene proton at position 25); 7.46 (dd, 1H, benzene proton at position 26); 7.49 (s, 1H, proton of the quinazoline position 6); 7.82 (d, 1H, benzene proton at position 27); 7.94 (t due dd, 1H, proton of the quinazoline position 19); 8.85 (s, 1H, benzene proton at the position 23); 8.87 (s, 1H, proton of the quinazoline position 2); 11.70 (s, 1H, proton of the aromatic amine as salt); 14-16 (bs, 1H, hydrochloride), see Figure 5. NMR data: 13C-NMR (DMSO-d6), see Figure 6. Mass spectrometry (MS): Instrument: ZAB-HS, testing conditions: EI, 200°C, 700ev, MS measured molecular weight: m/z 427.

…………………………………..

Synthesis of compound 1 A
1 Synthesis of Compound 2

2
79.5g 3,4 – dihydroxybenzene nitrile, 272g of potassium carbonate, acetonitrile (6L) was added to a 10L three-necked reaction flask, and dissolved with stirring, heated to reflux and reflux was added dropwise an acetonitrile solution of the compound 1 (compound 1, 200 g; acetonitrile , 2L), and completion of the dropping, the HPLC monitoring of the completion of the reaction, the mixture was cooled to room temperature, filtered, and the solvent was removed, and the resulting solid was washed with ethyl acetate was dissolved, filtered, and the filtrate was concentrated, the resulting residue was dissolved in petroleum ether by rotary evaporation, the resulting solid was purified to give 18.9g of the compound 2.
1 LAI MR (CDC1 3-Sppm): 7.30 ~ 7.33 (m, 1H); 7.25 (s, 1H); 6.97-6.99 (d, 1H); 4.19 – 4.23 (m, 4H); 3.83 ~ 3.91 (m, 4H); 3.77 (s, 4H). MS: (M + H) +250 2 Synthesis of compound A

2 A
41.6g of compound 2 was dissolved in 580ml of acetic acid, dropwise addition of 83ml of fuming nitric acid at 30 ° C under completion of the dropping, the dropwise addition of 42ml of concentrated sulfuric acid at 30 ° C under the reaction at room temperature overnight, TLC monitoring completion of the reaction, the reaction solution was poured into ice water 4L , the precipitated solid was filtered, washed with cold water (500 mL X 2), vacuum 35 ° C and dried crude A compound 46g, isopropanol recrystallization was purified to give 33g of compound A.
1 LAI MR (CDC1 3-Sppm): 7.90 (s, 1H); 7.36 (s, 1H); 4.33 ~ 4.36 (m, 4H); 3.87 ~ 3.89 (m, 4H); 3.737 (s, 4H). Embodiment of Example 2 Synthesis of Compound B

AB
32g of compound A, 30.5g of iron powder, 5% acetic acid solution in methanol 1070ml 2L reaction flask was heated to reflux
TLC monitoring of the end of the reaction cooled and concentrated, dissolved in ethyl acetate, filtered, dried over anhydrous NaS0 4 23g of compound B. The solvent was removed.
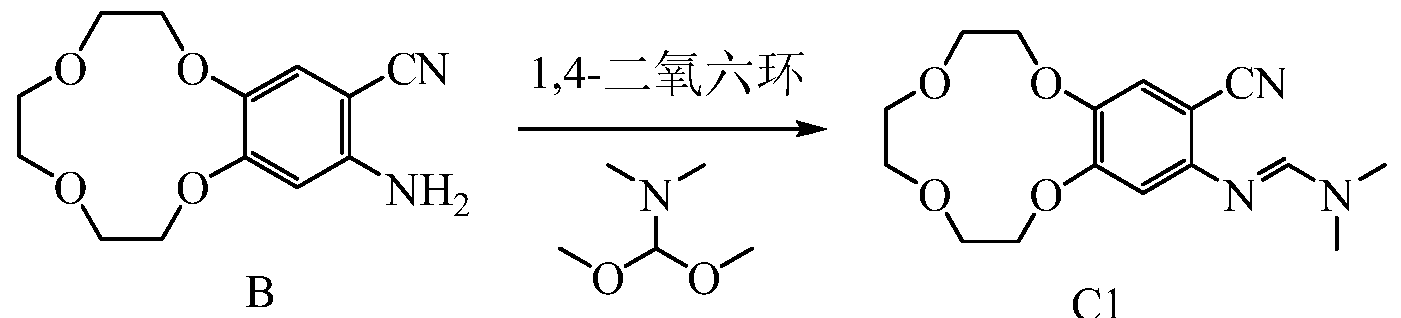
1HNMR (d 6-DMSO-Sppm): 7.07 (s, 1H); 6.36 (s, 1H); 5.73 (s, 2H); 3.95 ~ 4.22 (m, 4H); 3.77-3.78 (m, 2H); 3.34 3.62 (m, 6H).Embodiment of Example 3 Synthesis of Compound CI

B CI
500mL three-necked flask, the Add 5g compound B, 5g v, v-dimethyl formamide dimethyl acetal and 160ml of dioxane was heated to reflux the TLC monitoring progress of the reaction, the reaction time is about 12 hours, after the end of the reaction The reaction solution was cooled to room temperature, spin-dry to give 5.8g of compound Cl.
1 LAI MR (CDCl 3-Sppm): 7.56 (s, 1H); 7.15 (s, 1H); 6.51 (s, 1H); 4.12-4.18 (m, 4H); 3.89-3.91 (m, 2H); 3.78 -3.80 (m, 6H); 3.07 (s, 6H); Example 4 Icotinib Synthesis

5 g of the compound Cl, 2.2 g inter-aminophenyl acetylene, 230ml of acetic acid was added to a 500 ml reaction flask was heated to 100 ° c,
TLC monitoring of the reaction. The end of the reaction, the reaction system spin dry methanol was added, and shock dispersion, filtration, wash with methanol, 5g Icotinib.
^ M (d 6-DMSO-5ppm): 11.98 (s, IH); 9.50 (s, IH); 8.53 (s 1H); 8.14 (s, IH); 8.04-8.05 (m, IH); 7.90-7.92 (m, IH); 7.38-7.42 (m, IH); 7.31 (s IH); 7.20-7.22 (m, IH); 4.29-4.30 (m, 4H); 4.21 (s, IH); 3.74-3.81 ( m, 4H); 3.64 (s, 4H); 1.91 (s, 3H); Synthesis Example 5 Exe hydrochloride erlotinib

Exeter for Nick for; s
700mg Icotinib Add to a 100 ml reaction flask, add 40 ml of methanol, stirred pass into the hydrogen chloride gas or concentrated hydrochloric acid, and filtered to give crude hydrochloric acid Icotinib after, and purified by recrystallization from isopropanol to give 760mg hydrochloride Icotinib.
1HNMR (d 6-DMSO-Sppm): 11.37 (s, IH); 8.87 (s, IH); 8.63 (s, IH); 7.90 (s, IH); 7.78-7.80 (d, IH); 7.48-7.52 (m, IH); 7.40-7.41 (m, 2H); 4.36-4.38 (d, 4H); 4.30 (s, IH); 3.75-3.81 (d, 4H); 3.61 (s, 4H); Example 6 Synthesis of Compound B

AB
25g of compound A, 25 g of iron powder, 3% acetic acid in methanol solution 900ml with Example 2 are the same, to give 16.6g of compound B.
Embodiment of Example 7 Synthesis of Compound B

AB
40 g of compound A, 40 g of iron powder and 7% acetic acid in methanol solution was 1200ml, in Example 2, to give 28.4g of compound B.
Example 8 Compound B Synthesis

AB
25 g of compound A, 5 g of Pd / C in 3% acetic acid in methanol solution 900ml Add 2L reaction flask, of the hydrogen, TLC monitoring of the end of the reaction, filtered, and the solvent was removed to give 17g of compound B.
Example 9 Compound B Synthesis

AB
40g of compound A, 17 g of magnesium and 5% acetic acid in methanol solution 1200ml, in Example 2, to give 25.2g of compound B. Example 10 Compound B Synthesis

AB
25 g of compound A, 32.5g of zinc powder and 5% acetic acid in methanol solution 900ml with Example 2 are the same, to give 17.1g of compound B.
Example Synthesis of compound 11 B

AB
25g of compound A, 28 g of iron powder, 5% trifluoroacetic acid in methanol solution 700ml, in Example 2, 16g of compound B.
Embodiment Example 12 Synthesis of Compound C1

3g compound B, 3G v, v-dimethyl formamide dimethyl acetal and 140ml of dioxane, reflux the reaction time is 10-11 hours, the other in the same manner as in Example 3 to give 3.2g of the compound Cl.
Example 13 Synthesis of Compound C1
8g compound B, 8G N, v-dimethyl formamide dimethyl acetal and 180ml of dioxane under reflux for a reaction time of approximately 12-13 hours, with the same manner as in Example 3 to give 8.7g of compound C. Embodiment Example 14 Synthesis of Compound CI

3g compound B, 3 g of N, N-dimethyl formamide dimethyl acetal and 140ml of toluene, the reaction time is 13-15 hours under reflux, with the same manner as in Example 3 to give 2.9g of the compound Cl.
Example 15 Synthesis of Compound C1

The same as in Example 14, except that reaction time is 10 hours, to obtain 2.6g compound Cl t
Embodiment Example 16 Synthesis of Compound C1

500mL three-necked flask, add 3 g of compound B, 3.7 g v, v-dimethylformamide, diethyl acetal and 140ml of dioxane was heated to reflux, TLC monitoring the progress of the reaction, the reaction time of approximately 11-12 hours, After completion of the reaction, the mixture was cooled to room temperature, spin-dry the reaction solution to give 2.5g of the compound Cl.
Example 17 Synthesis of Compound C1

G of compound B, 5.1 g of the N, N-dimethyl formamide di-t-butyl acetal was dissolved in 140ml dioxane was heated to reflux the TLC monitoring progress of the reaction, the reaction time of approximately 11-12 hours after the completion of the reaction, was cooled to room temperature, the reaction solution was spin-dry to give 2.6g of the compound Cl.
Embodiment Example 18 Synthesis of Compound CI

3g compound B, 4.4g N, N-dimethyl formamide diisopropyl acetal was dissolved in 140ml dioxane was heated to reflux, tlc monitoring the progress of the reaction, the reaction time of approximately 11-12 hours after the completion of the reaction, was cooled to room temperature, the reaction solution was spin-dry to give 2.4g of the compound Cl.
The implementation of the synthesis of Example 19 Icotinib

3g compound Cl, 1.3 g inter-aminophenyl acetylene, 130 ml of acetic acid was added 250 ml reaction flask and heated to 70-80
V, TLC monitoring of the reaction. Spin dry the reaction system, methanol was added, and shock dispersion, filtered, and the methanol wash was 2.8g Icotinib. Implementation of Example 20 Icotinib synthesis

C1 Icotinib
. Example 25 Icotinib Hydrochloride synthesis

Icotinib Hydrochloride
The 500mg Icotinib Add to a 100 ml reaction flask, add 30ml of ethanol was stirred under hydrogen chloride gas was passed into the after, filtered crude hydrochloride Icotinib recrystallized from isopropanol to give 515mg hydrochlorideIcotinib. Example 26 Icotinib Hydrochloride Synthesis
500mg Icotinib Add 100 ml reaction flask, add 40 ml of tetrahydrofuran was stirred under hydrogen chloride gas was passed into the after, filtered crude hydrochloride Icotinib recrystallized from isopropanol to give 500mg hydrochlorideIcotinib. EXAMPLE 27 Icotinib Hydrochloride Synthesis

500mg Icotinib Add 100 ml reaction flask, add 50 ml of isopropanol and stirred under hydrogen chloride gas was passed into the after, filtered crude hydrochloride Icotinib recrystallized from isopropanol to give 500mg hydrochloride Icotinib.
………………………………………………………………….
http://www.google.com/patents/EP2392576A1 NMR data: 1H-NMR (Bruker APX-400, solvent: DMSO-d6, TMS as internal standard): δ ppm: 3.58 (dd, 2H, two protons of the crown position 12); 3.60 (dd, 2H, two protons of the crown position 13); 3.73 (dd, 2H, two protons of the crown position 10); 3.80 (dd, 2H, two protons of the crown position 15); 4.30 (s, 1H, proton of the alkynyl); 4.34 (dd, 2H, two protons of the crown position 16); 4.40 (dd, 2H, two protons of the crown position 9); 7.39 (d, 1H, benzene proton at position 25); 7.46 (dd, 1H, benzene proton at position 26); 7.49 (s, 1H, proton of the quinazoline position 6); 7.82 (d, 1H, benzene proton at position 27); 7.94 (t due dd, 1H, proton of the quinazoline position 19); 8.85 (s, 1H, benzene proton at the position 23); 8.87 (s, 1H, proton of the quinazoline position 2); 11.70 (s, 1H, proton of the aromatic amine as salt); 14-16 (bs, 1H, hydrochloride), see Figure 5. NMR data: 13C-NMR (DMSO-d6), see Figure 6. Mass spectrometry (MS): Instrument: ZAB-HS, testing conditions: EI, 200°C, 700ev, MS measured molecular weight: m/z 427.
………………………..
NEW PATENT
Zhejiang Beta Pharma Incorporation, 浙江贝达药业有限公司
http://www.google.co.in/patents/WO2013064128A1?cl=en
General synthetic route
Compound A, the present invention is provided for availability, but are not limited to, the following synthetic route to achieve:

The present invention is to provide beta available but are not limited to, the following synthetic route is now:

A BETA
The present invention is to provide a compound C, can be used, but are not limited to, the following synthetic route to achieve:

Wherein
And are independently selected from the group consisting of methyl, ethyl, propyl or isopropyl, or
, And they are connected in common to the N atom form a 3-7 membered ring. R 3 and R4 are independently selected from the group consisting of methyl, ethyl, propyl, isopropyl, n-butyl, sec-butyl, tert-butyl, iso-butyl or benzyl group, or,
R 3 and R4 to form a 3-7 membered ring.
The present C can be used for the direct preparation of Icotinib:

Wherein
And are independently selected from the group consisting of methyl, ethyl, propyl or isopropyl, or
, And they are connected in common to the N atom form a 3-7 membered ring.

Icotinib
Icotinib Hydrochloride
Example Synthesis of compound 1 A
1 Synthesis of Compound 2

2
79.5g 3,4 – dihydroxybenzene nitrile, 272g of potassium carbonate, acetonitrile (6L) was added to a 10L three-necked reaction flask, and dissolved with stirring, heated to reflux and reflux was added dropwise an acetonitrile solution of the compound 1 (compound 1, 200 g; acetonitrile , 2L), and completion of the dropping, the HPLC monitoring of the completion of the reaction, the mixture was cooled to room temperature, filtered, and the solvent was removed, and the resulting solid was washed with ethyl acetate was dissolved, filtered, and the filtrate was concentrated, the resulting residue was dissolved in petroleum ether by rotary evaporation, the resulting solid was purified to give 18.9g of the compound 2.
1 LAI MR (CDC1 3-Sppm): 7.30 ~ 7.33 (m, 1H); 7.25 (s, 1H); 6.97-6.99 (d, 1H); 4.19 – 4.23 (m, 4H); 3.83 ~ 3.91 (m, 4H); 3.77 (s, 4H). MS: (M + H) +250 2 Synthesis of compound A

2 A
41.6g of compound 2 was dissolved in 580ml of acetic acid, dropwise addition of 83ml of fuming nitric acid at 30 ° C under completion of the dropping, the dropwise addition of 42ml of concentrated sulfuric acid at 30 ° C under the reaction at room temperature overnight, TLC monitoring completion of the reaction, the reaction solution was poured into ice water 4L , the precipitated solid was filtered, washed with cold water (500 mL X 2), vacuum 35 ° C and dried crude A compound 46g, isopropanol recrystallization was purified to give 33g of compound A.
1 LAI MR (CDC1 3-Sppm): 7.90 (s, 1H); 7.36 (s, 1H); 4.33 ~ 4.36 (m, 4H); 3.87 ~ 3.89 (m, 4H); 3.737 (s, 4H). Embodiment of Example 2 Synthesis of Compound B

AB
32g of compound A, 30.5g of iron powder, 5% acetic acid solution in methanol 1070ml 2L reaction flask was heated to reflux
TLC monitoring of the end of the reaction cooled and concentrated, dissolved in ethyl acetate, filtered, dried over anhydrous NaS0 4 23g of compound B. The solvent was removed.
1HNMR (d 6-DMSO-Sppm): 7.07 (s, 1H); 6.36 (s, 1H); 5.73 (s, 2H); 3.95 ~ 4.22 (m, 4H); 3.77-3.78 (m, 2H); 3.34 3.62 (m, 6H). Embodiment of Example 3 Synthesis of Compound CI

B CI
500mL three-necked flask, the Add 5g compound B, 5g v, v-dimethyl formamide dimethyl acetal and 160ml of dioxane was heated to reflux the TLC monitoring progress of the reaction, the reaction time is about 12 hours, after the end of the reaction The reaction solution was cooled to room temperature, spin-dry to give 5.8g of compound Cl.
1 LAI MR (CDCl 3-Sppm): 7.56 (s, 1H); 7.15 (s, 1H); 6.51 (s, 1H); 4.12-4.18 (m, 4H); 3.89-3.91 (m, 2H); 3.78 -3.80 (m, 6H); 3.07 (s, 6H); Example 4 Icotinib Synthesis

5 g of the compound Cl, 2.2 g inter-aminophenyl acetylene, 230ml of acetic acid was added to a 500 ml reaction flask was heated to 100 ° c,
TLC monitoring of the reaction. The end of the reaction, the reaction system spin dry methanol was added, and shock dispersion, filtration, wash with methanol, 5g Icotinib.
^ M (d 6-DMSO-5ppm): 11.98 (s, IH); 9.50 (s, IH); 8.53 (s 1H); 8.14 (s, IH); 8.04-8.05 (m, IH); 7.90-7.92 (m, IH); 7.38-7.42 (m, IH); 7.31 (s IH); 7.20-7.22 (m, IH); 4.29-4.30 (m, 4H); 4.21 (s, IH); 3.74-3.81 ( m, 4H); 3.64 (s, 4H); 1.91 (s, 3H);
Synthesis Example 5 Exe hydrochloride erlotinib

Exeter for Nick for; s
700mg Icotinib Add to a 100 ml reaction flask, add 40 ml of methanol, stirred pass into the hydrogen chloride gas or concentrated hydrochloric acid, and filtered to give crude hydrochloric acid Icotinib after, and purified by recrystallization from isopropanol to give 760mg hydrochloride Icotinib.
1HNMR (d 6-DMSO-Sppm): 11.37 (s, IH); 8.87 (s, IH); 8.63 (s, IH); 7.90 (s, IH); 7.78-7.80 (d, IH); 7.48-7.52 (m, IH); 7.40-7.41 (m, 2H); 4.36-4.38 (d, 4H); 4.30 (s, IH); 3.75-3.81 (d, 4H); 3.61 (s, 4H);
Example 18 Synthesis of Compound CI

3g compound B, 4.4g N, N-dimethyl formamide diisopropyl acetal was dissolved in 140ml dioxane was heated to reflux, tlc monitoring the progress of the reaction, the reaction time of approximately 11-12 hours after the completion of the reaction, was cooled to room temperature, the reaction solution was spin-dry to give 2.4g of the compound Cl.
The implementation of the synthesis of Example 19 Icotinib

3g compound Cl, 1.3 g inter-aminophenyl acetylene, 130 ml of acetic acid was added 250 ml reaction flask and heated to 70-80
V, TLC monitoring of the reaction. Spin dry the reaction system, methanol was added, and shock dispersion, filtered, and the methanol wash was 2.8g Icotinib. Implementation of Example 20 Icotinib synthesis

C1 Icotinib
8g compound Cl, 3.5g inter-aminophenyl acetylene, dissolved in 380ml of acetic acid, heated to 100-120 ° C, TLC monitoring of the reaction. Spin dry the reaction system, by adding ethanol shock dispersion, filter, the ethanol wash 7.2g Icotinib. Implementation of Example 21 Icotinib Synthesis

The C1 Exeter erlotinib reaction temperature of 120-15CTC Example 4 was 2.2 g Icotinib.
Example 22 Icotinib Synthesis
3g compound Cl, 1.8 g inter-aminophenyl acetylene and 130 ml of acetic acid was added 250 ml reaction flask and heated to 90-100C, TLC monitoring of the reaction. Spin dry the reaction system, isopropanol shock dispersion, filtration, isopropyl alcohol wash was 2.9g Icotinib.
The implementation of the synthesis of Example 23 Icotinib

3G compound CI and 1.3 g of m-aminophenyl acetylene dissolved in 130ml of formic acid was heated to 80-90 ° C, TLC monitoring of the reaction. Spin dry the reaction system, methanol was added, and shock dispersion, filtered, and the methanol wash was 2.7g Icotinib.
Example 24 Icotinib synthesis

3g of compound C1 and 1.3g aminophenyl acetylene dissolved in 130ml of trifluoroacetic acid was heated to 70-80 ° C, TLC monitoring of the reaction. Spin dry the reaction system, methanol was added, and shock dispersion, filtered, and the methanol wash was 2.7g Icotinib. Example 25 Icotinib Hydrochloride synthesis

Icotinib Hydrochloride
The 500mg Icotinib Add to a 100 ml reaction flask, add 30ml of ethanol was stirred under hydrogen chloride gas was passed into the after, filtered crude hydrochloride Icotinib recrystallized from isopropanol to give 515mg hydrochloride Icotinib. Example 26 Icotinib Hydrochloride Synthesis
500mg Icotinib Add 100 ml reaction flask, add 40 ml of tetrahydrofuran was stirred under hydrogen chloride gas was passed into the after, filtered crude hydrochloride Icotinib recrystallized from isopropanol to give 500mg hydrochloride Icotinib. EXAMPLE 27 Icotinib Hydrochloride Synthesis

Exeter erlotinib erlotinib hydrochloride Exeter
500mg Icotinib Add 100 ml reaction flask, add 50 ml of isopropanol and stirred under hydrogen chloride gas was passed into the after, filtered crude hydrochloride Icotinib recrystallized from isopropanol to give 500mg hydrochloride Icotinib. Example 28 Icotinib Hydrochloride synthesis

Icotinib
Icotinib Hydrochloride
 |
|
| Clinical data | |
|---|---|
| Trade names | Conmana, Icotinib |
| Legal status |
?
|
| Routes | Oral tablets |
| Pharmacokinetic data | |
| Bioavailability | 52% |
| Metabolism | Hepatic (mainly CYP3A4, lessCYP1A2) |
| Half-life | 5.5 hrs (median) |
| Excretion | >98% as metabolites, of which >90% via faeces, 9% via urine |
| Identifiers | |
| CAS number | 1204313-51-8 |
| ATC code | ? |
| PubChem | CID 22024915 |
| DrugBank | DB00530 |
| ChemSpider | 10762174 |
| UNII | 9G6U5L461Q |
| Chemical data | |
| Formula | C22H21N3O4 |
| Mol. mass | 391.420 g/mol |
References
- Sordella, R. (20 August 2004). “Gefitinib-Sensitizing EGFR Mutations in Lung Cancer Activate Anti-Apoptotic Pathways”. Science 305(5687): 1163–1167. doi:10.1126/science.1101637. PMID 15284455.
- Shi, Yuankai; Zhang, Li; Liu, Xiaoqing; Zhou, Caicun; Zhang, Li; Zhang, Shucai; Wang, Dong; Li, Qiang; Qin, Shukui; Hu, Chunhong; Zhang, Yiping; Chen, Jianhua; Cheng, Ying; Feng, Jifeng; Zhang, Helong; Song, Yong; Wu, Yi-Long; Xu, Nong; Zhou, Jianying; Luo, Rongcheng; Bai, Chunxue; Jin, Yening; Liu, Wenchao; Wei, Zhaohui; Tan, Fenlai; Wang, Yinxiang; Ding, Lieming; Dai, Hong; Jiao, Shunchang; Wang, Jie; Liang, Li; Zhang, Weimin; Sun, Yan. “Icotinib versus gefitinib in previously treated advanced non-small-cell lung cancer (ICOGEN): a randomised, double-blind phase 3 non-inferiority trial”. The Lancet Oncology 14 (10): 953–961. doi:10.1016/s1470-2045(13)70355-3.
- Tan, Fenlai; Gu, Aiqin; Zhang, Yiping; Jiao, Shun Chang; Wang, Chang-li; He, Jintao; Jia, Xueke; Zhang, Li; Peng, Jiewen; Wu, Meina; Ying, Kejing; Wang, Junye; Ma, Kewei; Zhang, Shucai; You, Changxuan; Ding, Lieming; Wang, Yinxiang; Shen, Haijiao; Wan, Jiang; Sun, Yan (2013). “Safety and efficacy results of a phase IV, open-label, multicenter, safety-monitoring study of icotinib in treating advanced non-small cell lung cancer (NSCLC): ISAFE study”. ASCO 2013 Meeting: e19161.
- Chen, Xiaofeng; Zhu, Quan; Liu, Yiqian; Liu, Ping; Yin, Yongmei; Guo, Renhua; Lu, Kaihua; Gu, Yanhong; Liu, Lianke; Wang, Jinghua; Wang, Zhaoxia; Røe, Oluf Dimitri; Shu, Yongqian; Zhu, Lingjun; Chellappan, Srikumar P. (16 May 2014). “Icotinib Is an Active Treatment of Non-Small-Cell Lung Cancer: A Retrospective Study”. PLoS ONE 9 (5): e95897.doi:10.1371/journal.pone.0095897.
| WO2007138613A2 * | 12 Mar 2007 | 6 Dec 2007 | Venkateshappa Chandregowda | A process for synthesis of [6,7-bis-(2-methoxyethoxy)-quinazolin-4-yl]-(3-ethynylphenyl)amine hydrochloride |
| WO2010003313A1 | 7 Jul 2009 | 14 Jan 2010 | Zhejiang Beta Pharma Inc. | Icotinib hydrochloride, synthesis, crystallographic form, medical combination, and uses thereof |
| CN1305468C | 29 May 2003 | 21 Mar 2007 | 中国人民解放军第三○二医院 | Bolengsu compound and its preparation, medicine composition and use |
| US7078409 | 26 Mar 2003 | 18 Jul 2006 | Beta Pharma, Inc. | Fused quinazoline derivatives useful as tyrosine kinase inhibitors |
| Patent | Submitted | Granted |
|---|---|---|
| Icotinib Hydrochloride, Synthesis, Crystalline Forms, Pharmaceutical Compositions, and Uses Thereof [US2011182882] | 2011-07-28 | |
| Fused quinazoline derivatives useful as tyrosine kinase inhibitors [US7078409] | 2004-03-11 | 2006-07-18 |
Curis phase 1 Cancer Trial for CUDC-427 Begins

CUDC-427, GDC-0917; RG-7459
Genentech Inc (Roche Holding AG)
Curis licenses GDC-0917 from Genentech
Curis Cancer Trial Begins
Curis Inc. has initiated patient dosing in a second Phase 1 dose-escalation study of CUDC-427 that is being conducted using a continuous, twice-daily oral dosing regimen in patients with advanced and refractory solid tumors or lymphoma.
FULL STORY
About CUDC-427 (GDC-0917)
CUDC-427 is an orally bioavailable small molecule that is designed to promote cancer cell death by antagonizing IAP proteins. IAP proteins are a family of functionally and structurally related proteins that promote cancer cell survival by inhibiting programmed cell death, also known as apoptosis, which is a normal process inherent in every cell. Using IAP proteins and other anti-apoptotic factors, cancer cells evade apoptosis in response to a variety of signals, including those provided by anti-cancer agents such as chemotherapy, or naturally occurring inflammatory and immune signals transmitted through members of the tumor necrosis factor, or TNF, family of factors. Evasion from apoptosis is a fundamental mechanism whereby human cancers develop resistance to standard anti-cancer treatments. IAP inhibitors such as CUDC-427 are designed to counteract the effects of IAP proteins, thus shifting the balance away from cancer cell survival and allowing apoptosis to proceed.
CUDC-427 was designed to mimic the endogenous IAP antagonist mitochondrial protein second mitochondria-derived activator of caspases/direct IAP-binding protein (Smac/DIABLO) that is released into the cytoplasm in response to pro-apoptotic stimuli. CUDC-427 has demonstrated single-agent and combination anti-tumor activity in mouse xenograft tumor models when administered orally on a daily schedule, and IND-enabling safety studies have shown it to be well tolerated when dosed daily by oral administration, potentially enabling sustained target inhibition.
In October 2010, an open-labeled, uncontrolled, dose-escalation, Phase I clinical trial of CUDC-427 (NCT01226277; IAM4914g) began in patients with refractory solid tumors or lymphoma. Genentech recently completed this Phase I clinical trial in which 42 people received daily oral doses of CUDC-427 for two weeks, followed by a one week rest period. This 21-day cycle is repeated until disease progression or study discontinuation for any other reason. The primary endpoints of the study include evaluating the safety and tolerability and the pharmacokinetics of CUDC-427 in people with solid tumors or lymphoma and determining the maximum-tolerated-dose and a potential recommended dose for further clinical studies. Secondary endpoints include a preliminary assessment of anti-tumor activity of CUDC-427 and evaluating pharmacodynamic markers. Genentech plans to present full study results at a medical conference in mid-2013. Please refer to http://www.clinicaltrials.gov for additional study details.
About Inhibitor of Apoptosis Proteins
Impairment of programmed cell death or apoptosis often contributes to the formation and progression of cancer, and evasion of apoptosis is one of the primary strategies by which cancer cells develop resistance to anticancer therapies. Inhibitor of apoptosis (IAP) proteins are a family of functionally and structurally related proteins which include X-linked IAP (XIAP), cellular IAPs (cIAP1 and cIAP2), and melanoma IAP (ML-IAP). They confer protection from death-inducing stimuli by exerting a range of biological activities that promote cancer cell survival and proliferation. Some even directly inhibit caspases, critical players in the execution of apoptosis.
Mutations, amplifications and chromosomal translocations of IAP genes are associated with various solid and hematologic cancer types, and increased IAP expression has been associated with an unfavorable prognosis and poor outcome for patients. As a consequence, IAP proteins are considered promising molecular targets for anticancer therapy.
Amgen In Focus

Amgen In Focus
Seeking Alpha
According to Amgen, they have 45 drugs in development from Phase 1 to Phase 3. Conversely, Gilead has 32 drugs in development and Pfizer has 64. Meanwhile, Gilead only has 8 drugs in Phase 3, Pfizer has 25, and Amgen has 14. 7 of those Phase 3 …
http://seekingalpha.com/article/1510002-amgen-in-focus?source=google_news
Amgen has the second deepest pipeline of drugs of the three large cap biotechs. According to Amgen, they have 45 drugs in development from Phase 1 to Phase 3. Conversely, Gilead has 32 drugs in development and Pfizer has 64. Meanwhile, Gilead only has 8 drugs in Phase 3, Pfizer has 25, and Amgen has 14. 7 of those Phase 3 drugs are focused on cancer treatments for Amgen, more than either Pfizer or Gilead. Keep in mind that 12.4 million people learn they have cancer each year, while 7.6 million people lose that battle each year. The CDC predicts that the global number of cancer related deaths will increase by 80% by 2030. It doesn’t take a rocket scientist to know that cancer treating drugs presents the largest opportunity for any drug maker considering those statistics. Amgen has the inside track versus Gilead and Pfizer as far as quantity of drugs in late stage development.
Modality
TLC388 (Lipotecan®) Taiwan Liposome Company Hepatic cancer drug candidate gets fast track approval status from SFDA
TLC388 (Lipotecan®) structure can be figured out from a link below of a poster
http://www.tlcbio.com/files/news/2011111701580783.pdf
IT IS A CAMPOTHECIN ANALOGUE
The str can be concluded from above picture from a poster by TLC BIO
TLC388 (Lipotecan) is a potent Topoisomerase-1 inhibitor and it can disrupt both Sonic Hedgehog and HIF1-α pathways to overcome cancer drug resistance and inhibit angiogenesis induced by tumor hypoxia. This phase I first-in-human study of Lipotecan examined the MTD, safety, anti-tumor activity and pharmacokinetic profiles of TLC388 in patients with advanced incurable solid tumors.
Methods: Lipotecan was administered intravenously on day 1, 8 and 15 of a 28-day cycle. Patients underwent safety assessments regularly and tumor assessments every other cycle. Pharmacokinetic samples were drawn on days 1, 8 and 15 of cycles 1 and 2 for all treated patients.
http://mct.aacrjournals.org/cgi/content/meeting_abstract/10/11_MeetingAbstracts/A89
http://clinicaltrials.gov/show/NCT00747474
MAR19 2013
China SFDA has granted fast track approval status to Taiwan Liposome company hepatic cancer drug Lipotecan, shortening the review period. The drug will enter Phase 2 clinical trials in China in the second half of this year. Lipotecan has been granted orphan drug status by US FDA and EU EMEA for the treatment of hepatocellular carcinoma (HCC)
Nexavar is the standard of care in first line advanced liver cancer patients. Lipotecan as a second-line treatment allows patients who have failed prior treatment with Nexavar to maintain a six month course of the disease without progressing
The FDA has opened the inside track to Novartis’ experimental lung cancer drug, LDK378, which gained “Breakthrough Therapy” designation

The FDA has opened the inside track to Novartis’ experimental lung cancer drug, which gained “Breakthrough Therapy” designation that speeds the development and review schedules for new treatments. The Swiss drug giant plans to file for approval the drug, now in mid-stage clinical trials, in early 2014. Since clinical development began in 2011, the program has advanced with lightning speed compared with those that take 10 years or so to trial before submitted for approval.
While there are no guarantees of an FDA approval for Novartis’ compound, code-named LDK378, the “breakthrough” tag provides an early nod to the potential of the candidate to improve treatment for patients with metastatic non-small cell lung cancer with anaplastic lymphoma kinase (ALK) mutations.
The “breakthrough” designation is also important because Novartis’ compound and others with the coveted status have a shot to be approved by the FDA without completing all three phases of clinical trials typically required before an approval decision.
Novartis’ LDK378 joined the “breakthrough” club after showing an 80% response rate in patients studied in Phase I trial of 88 subjects with advanced cases of ALK-positive NSCLC. The company has already begun a pair of Phase II studies of the compound for patients with the same kind of ALK-positive cancers, which account for about 3% to 8% of cases of NSCLC. And plans call for kicking off Phase III development of the new drug later this year.
“LDK378 is a strong example of our research approach, which focuses on identifying the underlying cause of disease pathways,” said Alessandro Riva, Novartis’ global head of oncology development, in a statement. “This Breakthrough Therapy designation will allow us to collaborate more closely with the FDA and potentially to expedite the availability of an important new treatment option for patients with ALK+ NSCLC.”
