



| Patent | Submitted | Granted |
|---|---|---|
| BENZIMIDAZOLE MODULATORS OF VR1 [US2011190344] | 2011-08-04 | |
| BENZIMIDAZOLE MODULATORS OF VR1 [US2011190364] | 2011-08-04 | |
| Benzimidazole Modulators of VR1 [US7951829] | 2007-11-08 | 2011-05-31 |
Home » Posts tagged 'PHASE1' (Page 2)
Tag Archives: PHASE1
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Phase 1-Sangamo Presents New Clinical Data at CROI 2013 Demonstrating Persistent Immune System Improvements After Treatment With ZFN Therapeutic(R) SB-728-T

The gene therapy diminished the levels of virus and eradicated in patients having naturally occurring mutation of gene, found a preliminary trail of HIV treatment. The first phase of very small trail tested the SB-728-T gene treatment that is intended to interrupt theCCR5 gene used by HIV to contaminate immune system cells.
The first clinical trial using zinc-finger nucleases to provide long-term resistance to HIV-1 infection has been given the go-ahead by the US Food and Drug Administration. Sangamo BioSciences of Richmond, California, and its clinical partner, the University of Pennsylvania, have begun enrolling the first 12 people in a phase 1 clinical trial to evaluate SB-728-T, a novel zinc-finger DNA-binding nuclease that permanently disrupts the CCR5 gene on CD4+ T cells (Nat. Biotechnol. 26, 808–816, 2008
Data Demonstrate that SB-728-T Possesses Necessary Immunologic Properties to Support a ‘Functional Cure’ for HIV/AIDS
RICHMOND, Calif., March 6, 2013
Sangamo BioSciences, Inc. announced new data from its program to develop a ‘functional cure’ for HIV/AIDS in two presentations at the 20th Conference on Retroviruses and Opportunistic Infections (CROI), held in Atlanta from March 3 to 6, 2013.
The first presentation described data from the SB-728-T Phase 1 study (SB-728-902, Cohorts 1-3) demonstrating that SB-728-T treatment of HIV-infected subjects leads to durable reconstitution of the immune system driven by increases in total CD4+ central memory T-cells (TCM) and CCR5-protected TCM. TCM are long-lived, self-renewing cells that have the ability to remember and react against foreign antigens including HIV. The data also showed that certain cell surface marker and gene expression profiles may predict which patients will likely respond best to SB-728-T treatment.
About Sangamo
Sangamo BioSciences, Inc. is focused on research and development of novel DNA-binding proteins for therapeutic gene regulation and genome editing. The Company has ongoing Phase 2 clinical trials to evaluate the safety and efficacy of a novel ZFP Therapeutic® for the treatment of HIV/AIDS. Sangamo’s other therapeutic programs are focused on monogenic diseases, including hemophilia, Huntington’s disease and hemoglobinopathies such as beta-thalassemia and sickle cell anemia. Sangamo’s core competencies enable the engineering of a class of DNA-binding proteins known as zinc finger DNA-binding proteins (ZFPs). Engineering of ZFPs that recognize a specific DNA sequence enables the creation of sequence-specific ZFP Nucleases (ZFNs) for gene modification and ZFP transcription factors (ZFP TFs) that can control gene expression and, consequently, cell function. Sangamo has entered into a strategic collaboration with Shire AG to develop therapeutics for hemophilia, Huntington’s disease and other monogenic diseases and has established strategic partnerships with companies in non-therapeutic applications of its technology including Dow AgroSciences and Sigma-Aldrich Corporation. For more information about Sangamo, visit the company’s website atwww.sangamo.com.
Sanofi And Regeneron Report Positive Proof-of-Concept Data For Dupilumab, An IL-4R Alpha Antibody, In Atopic Dermatitis
| Monoclonal antibody | |
|---|---|
| Source | Human |
| Target | IL4 receptor alpha |
Treatment of atopic diseases
Immunoglobulin, anti-(human interleukin 4 receptor α) (human REGN668 heavy chain),
disulfide with human REGN668 κ-chain, dimer
Immunoglobulin G4, anti-(human interleukin-4 receptor subunit alpha (IL-4R-alpha,
CD124)); human monoclonal REGN668 des-452-lysine{CH3107K>-}-[233-
proline{H10S>P}]γ4 heavy chain (139-219′)-disulfide with human monoclonal REGN668
κ light chain, dimer (231-231”:234-234”)-bisdisulfide
1190264-60-8 cas no
REGN668, SAR231893
MOLECULAR FORMULA- C6512H10066N1730O2052S46
Dupilumab is a monoclonal antibody designed for the treatment of atopic diseases.[1]
This drug was developed by Regeneron Pharmaceuticals.
- Statement On A Nonproprietary Name Adopted By The USAN Council – Dupilumab,American Medical Association.
Phase 1b Data Presented at Late Breaking Session of 71st Annual Meeting of the American Academy of Dermatology
PARIS and TARRYTOWN, N.Y., March 2, 2013 – Sanofi and Regeneron Pharmaceuticals, Inc. today announced that pooled data from two Phase 1b trials with dupilumab (REGN668/SAR231893), an investigational, high-affinity, subcutaneously administered, fully-human antibody targeting the alpha subunit of the interleukin 4 receptor (IL-4R alpha), were presented at the 71st Annual Meeting of the American Academy of Dermatology (AAD) in Miami.
The primary objective of the Phase 1b studies was to assess the safety profile of dupilumab. Other exploratory endpoints included pharmacokinetic, biomarker, and efficacy parameters. The efficacy data showed that treatment with four weekly subcutaneous injections of dupilumab at either 150 milligrams (mg) or 300mg per week, significantly improved the signs and symptoms of patients with moderate-to-severe atopic dermatitis (AD) whose disease was not adequately controlled with topical medications. Specifically, patients treated with dupilumab had significant improvements in body surface area (BSA) score, Investigator Global Assessment (IGA) score, and Eczema Area Severity Index (EASI) from baseline to week 4 compared to placebo (p<0.05 vs. placebo for all measures and doses). The significant improvements in BSA, IGA, and EASI scores were maintained at week 8 in the 300mg dose group (p<0.05 vs. placebo). A responder analysis demonstrated that at week 4, 54.5% of patients treated with the 150mg dose and 71.4% of patients treated with the 300mg dose achieved a reduction in EASI score of 50% or greater compared to 18.8% with placebo (p<0.05). The most common adverse events (AEs) were nasopharyngitis (19.6% vs 12.5% for placebo) and headache (11.8% vs 6.3% for placebo).
“Despite existing therapies, a significant proportion of patients with moderate-to-severe atopic dermatitis continue to suffer from inflamed skin and intractable itch, which significantly impacts their quality of life,” said Dr. Eric Simpson, Associate Professor, Director of Clinical Studies, Oregon Health and Science University, Portland, Oregon, USA, and Principal Investigator of the study. ”The early phase results with this biologic therapy, which has a novel mechanism of action, are encouraging to those of us who treat these patients and warrant further clinical investigation.”
“Through blockade of IL-4R alpha, dupilumab modulates signaling of both the IL-4 and IL-13 pathway, which have been implicated in the pathophysiology of allergic disease,” said George D. Yancopoulos, M.D., Ph.D., Chief Scientific Officer of Regeneron and President of Regeneron Laboratories. ”We look forward to presenting additional data from a 12-week, Phase 2a trial in atopic dermatitis, as well as starting a larger Phase 2b trial with dupilumab in patients with atopic dermatitis, later this year.”
Presented today in a late-breaking clinical trials session at the AAD meeting, the Phase 1b trials included 67 patients randomized to three different doses of dupilumab (75mg, n=8; 150mg, n=22; 300mg, n=21) and placebo (n=16). The primary objective of the Phase 1b studies was to assess the safety profile of dupilumab. Other endpoints included pharmacokinetic, biomarker, and efficacy parameters. Following the 4-week treatment period, patients in the studies were followed for an additional 4 weeks for a total of 8 weeks.
About IL-4R and the IL-4/IL-13 Pathway
Atopic dermatitis and some types of asthma are characterized by the induction of a specific type of an immune response that is driven by a subset of immune cells called Type 2 helper T cells, or Th2 cells. IL-4 and IL-13 are key cytokines that are required for the initiation and maintenance of this Th2 immune response. Both IL-4 and IL-13 signaling occurs through two different IL-4 receptors (Type I and II), which both contain a common IL-4R alpha subunit.
About Dupilumab (SAR231893/REGN668)
Dupilumab is a fully human monoclonal antibody directed against IL-4R alpha and is administered via subcutaneous injection. By blocking IL-4R alpha dupilumab modulates signaling of both IL-4 and IL-13, drivers of a Th2 immune response. Dupilumab was created using Regeneron’s pioneering VelocImmune® technology and is being co-developed with Sanofi. Dupilumab is currently being studied in both atopic dermatitis and asthma.
About Atopic Dermatitis
Atopic dermatitis (AD) is a chronic, immune-mediated, inflammation of the skin that is characterized by poorly defined erythema (redness) with edema (swelling), weeping in the acute stage, and skin thickening (lichenification) in the chronic stage. Chronic and/or relapsing lesions, along with pruritus (itching) and scratching are the hallmarks of the disease. The prevalence of AD is estimated to be between 1% and 3% of adults. For many patients, topical therapies are not effective for keeping the disease under control and the only approved systemic therapies to treat AD are prednisone and cyclosporine (in Europe). Moderate-to-severe atopic dermatitis can negatively impact patients’ lives and is associated with a high burden to society both in terms of direct costs of medical care and prescription drugs, as well as loss of productivity.
About Sanofi
Sanofi, a global and diversified healthcare leader, discovers, develops and distributes therapeutic solutions focused on patients’ needs. Sanofi has core strengths in the field of healthcare with seven growth platforms: diabetes solutions, human vaccines, innovative drugs, consumer healthcare, emerging markets, animal health and the new Genzyme. Sanofi is listed in Paris (EURONEXT: SAN) and in New York (NYSE:SNY).
About Regeneron Pharmaceuticals, Inc.
Regeneron is a leading science-based biopharmaceutical company based in Tarrytown, New York that discovers, invents, develops, manufactures, and commercializes medicines for the treatment of serious medical conditions. Regeneron markets medicines for eye diseases, colorectal cancer, and a rare inflammatory condition and has product candidates in development in other areas of high unmet medical need, including hypercholesterolemia, rheumatoid arthritis, asthma, and atopic dermatitis. For additional information about the company, please visitwww.regeneron.com.
Researchers Begin Shigella Vaccine Trial , WRSs2 and WRSs3

FEB2013
PHASE 1 Safety and Immunogenicity of Two Live, Attenuated Oral Shigella Sonnei Vaccines WRSs2 and WRSs3
Phase 1, randomized, double-blind, placebo controlled, dose-escalation, inpatient study of single doses of S. sonnei. Enroll serial groups up to 90 subjects. Evaluate safety and tolerance of WRSs2 by monitoring presence and severity of clinical signs and symptoms, evaluate the immune response in blood and stool following ingestion of WRSs2
http://clinicaltrials.gov/show/NCT01336699
Shigellosis is one of those nasty bacterial diseases that follows the cringeworthy fecal-oral route to infect humans and other primates. Mild cases bring stomachaches; the severe end includes cramping, vomiting, fever, diarrhea, and it generally only gets more disgusting from there. While the disease can occur all over the world—estimates suggest ninety million cases of Shigellosis dysentery each year—the greatest mortality occurs in the third world. Hoping to stem transmission, or, at least, minimize the damage it causes, the World Health Organization has long called for a vaccine to stop Shigella infection.
And, today, scientists are one step closer. The National Institutes of Health announced that two Shigella vaccine have entered early-stage human clinical trials:
Researchers have launched an early-stage human clinical trial of two related candidate vaccines to prevent infection with Shigella, bacteria that are a significant cause of diarrheal illness, particularly among children. The Phase 1 clinical trial, funded by the National Institute of Allergy and Infectious Diseases (NIAID), part of the National Institutes of Health, will evaluate the vaccines for safety and their ability to induce immune responses among 90 healthy adults ages 18 to 45 years. The trial is being conducted at the Cincinnati Children’s Hospital Medical Center, one of the eight NIAID-funded Vaccine and Treatment Evaluation Units in the United States.

Researchers have launched an early-stage human clinical trial of two related candidate vaccines to prevent infection with Shigella, bacteria that are a significant cause of diarrheal illness, particularly among children. The Phase I clinical trial, funded by the National Institute of Allergy and Infectious Diseases (NIAID), part of the National Institutes of Health, will evaluate the vaccines for safety and their ability to induce immune responses among 90 healthy adults ages 18 to 45 years. The trial is being conducted at the Cincinnati Children’s Hospital Medical Center, one of the eight NIAID-funded Vaccine and Treatment Evaluation Units in the United States ….
…. Led by principal investigator Robert W. Frenck, Jr., M.D., director of clinical medicine at Cincinnati Children’s, the new clinical trial will evaluate two related candidate vaccines, known as WRSs2 and WRSs3, which have been found to be safe and effective when tested in guinea pigs and nonhuman primates. Both target Shigella sonnei, one of the bacteria’s four subtypes and the cause of most shigellosis outbreaks in developed and newly industrialized countries. Though neither candidate vaccine has been tested in humans, a precursor to both, known as WRSs1, was found to be safe and generated an immune response in small human trials in the United States and Israel. This early work was supported by NIAID, the U.S. Department of Defense and the Walter Reed Army Institute of Research. All three versions of the vaccine were developed by researchers at the Walter Reed institute.
Janssen Research Development, LLC , JNJ 39439335, Mavatrep

Mavatrep; UNII-F197218T99; Mavatrep (USAN); JNJ-39439335; 956274-94-5;
2-(2-(2-(2-(4-trifluoromethylphenyl)vinyl)-1H-benzimidazol-5-yl)phenyl)propan-2-ol
(E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol
Phase I Musculoskeletal pain; Pain
- 01 Mar 2013 Janssen Research and Development completes a phase I trial in Japanese and Caucasian adult male volunteers in the US (NCT01631487)
- 01 Mar 2013 Janssen completes enrolment in its phase I trial for Pain (in volunteers) in the USA (NCT01631487)
- 05 Feb 2013 Janssen Research and Development initiates enrolment in a phase I trial for Pain (Japanese and Caucasian volunteers) in USA (NCT01631487)
- Originator Johnson & Johnson Pharmaceutical Research & Development
- Developer Janssen Research & Development
- Class Analgesics; Benzimidazoles; Small molecules
- Mechanism of Action TRPV1 receptor antagonists
| PHASE 1 Johnson & Johnson Pharmaceutical Research & Development, L.L.C. |
|
| Public title: | A Clinical Study to Investigate the Effect on Pain Relief of a Single Dose of JNJ-39439335 in Patients With Chronic Osteoarthritis Pain of the Knee |
http://clinicaltrials.gov/ct2/show/NCT01006304
http://apps.who.int/trialsearch/trial.aspx?trialid=NCT00933582
http://www.ama-assn.org/resources/doc/usan/mavatrep.pdf SEE STRUCTURE IN THIS FILE
MAVATREP IS JNJ-39439335
—
(E)-2-(2-(2-(4-(trifluoromethyl)styryl)-1H-benzo[d]imidazol-6-yl)phenyl)propan-2-ol hydrochloride
956282-89-6 CAS NO OF HCl SALT
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.5b00271, http://pubs.acs.org/doi/abs/10.1021/acs.oprd.5b00271

The process development of Mavatrep (1), a potent transient receptor potential vanilloid-1 (TRPV1) antagonist, is described. The two key synthetic transformations are the synthesis of (E)-6-bromo-2-(4-(trifluoromethyl)styryl)1H-benzo[d]imidazole (4) and the Suzuki coupling of 4 with 3,3-dimethyl-3H-benzo[c][1,2]oxaborol-1-ol (5). Compound 1a was prepared in four chemical steps in 63% overall yield.




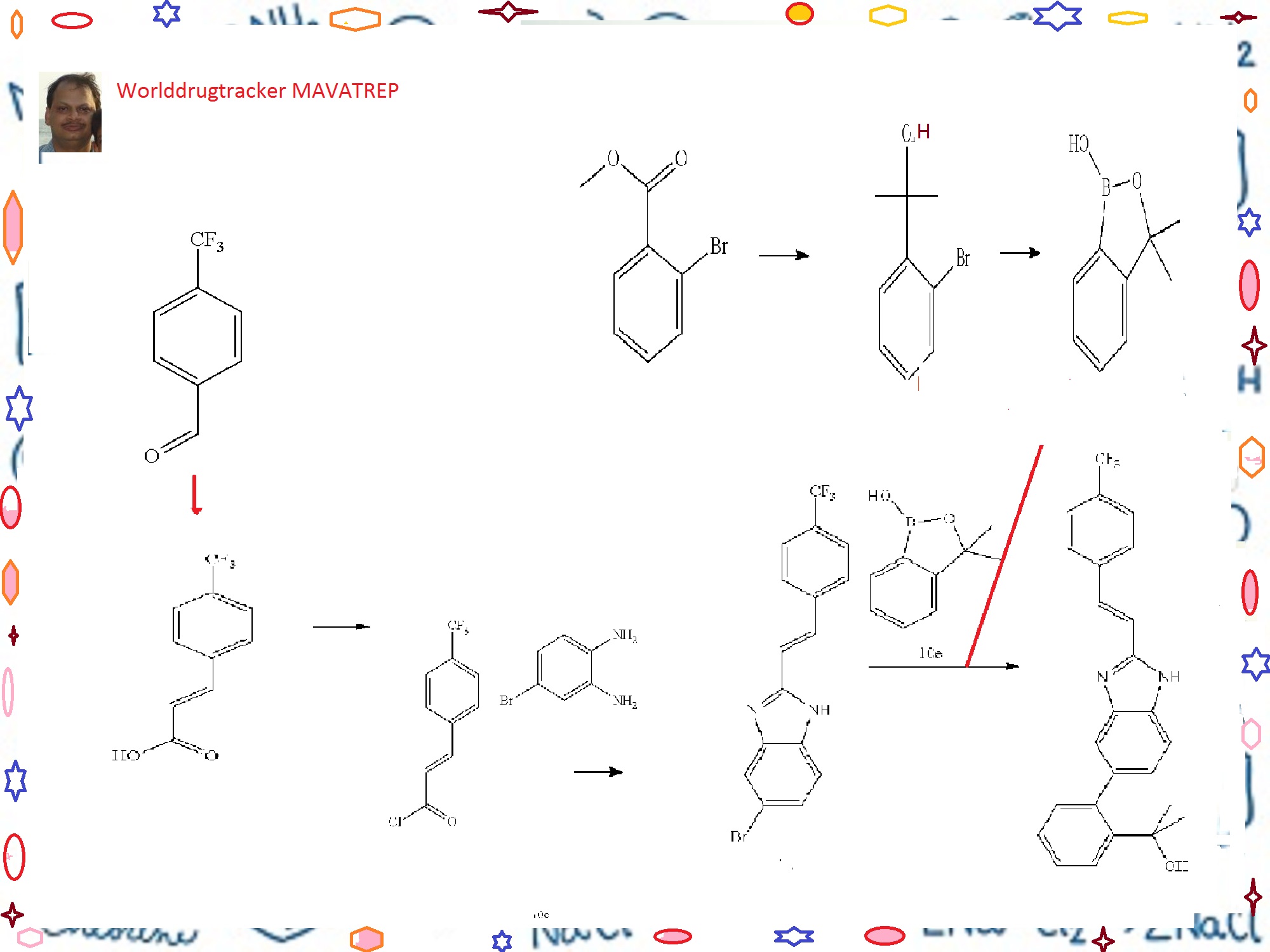
CLICK ON IMAGE FOR CLEAR VIEW
Example 10 (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol(Cpd 18)
Step A. 3-(4-trifluoromethyl-phenyl)-acrylic acid
-
[0278]A solution of 4-trifluoromethylbenzaldehyde (7.7 mL, 57.7 mmol), malonic acid (12.0 g, 115.4 mmol), 0.567 μL piperidine (5.75 mmol) in 30 mL of pyridine was stirred at 70° C. for 18 h. The reaction solution was cooled to room temperature. Water (300 mL) was added and the resulting mixture was acidified to pH 4 (litmus) using concentrated hydrochloric acid to give a precipitate. The solid was filtered, and washed with water until the filtrate was neutral. The solid product was dried in vacuo to give the title Compound 10a as a white powder (11.2 g, 90%). 1HNMR (400 MHz, DMSO-d6) δ (ppm): 12.60 (bs, 1H), 7.92 (d, 2H, J=8.2 Hz), 7.77 (d, 2H, J=8.2 Hz), 7.66 (d, 1H, J=16.0 Hz), 6.70 (d, 1H, J=16.0 Hz).
-
[0000]

Step B. (E)-5-bromo-2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazole
-
[0279]A solution of Compound 10a (20.6 g, 95.4 mmol) in anhydrous methylene chloride (200 mL) was treated with oxalyl chloride (16.6 mL, 190 mmol) and “3 drops” of anhydrous dimethylformamide. The resulting solution was stirred at room temperature under an argon atmosphere for 18 h. The solvent was concentrated to give 3-(4-trifluoromethyl-phenyl)-acryloyl chloride Compound 10b as a solid, which was used without further purification in the next step.
-
[0280]To a solution of 4-bromo-benzene-1,2-diamine (16.1 g, 86.7 mmol) in acetic acid (100 mL) was added dropwise a solution of Compound 10b (assumed 95.4 mmol) in acetic acid (100 mL). The reaction mixture was stirred at 100° C. for 18 h. The reaction mixture was cooled to room temperature, and a mixture of ethyl acetate and hexanes 3:7 (500 mL) was added. The mixture was triturated at room temperature for 3 h to give a precipitate. The solid was filtered, and dried in vacuo to give the title Compound 10c (23.2 g, 73%). 1H NMR (400 MHz, DMSO-d6/CDCl3) δ (ppm): 8.45 (d, 1H, J=16.7 Hz), 7.84-7.90 (m, 1H), 7.74 (d, 2H, J=8.3
-
[0281]Hz), 7.56-7.62 (m, 3H), 7.50-7.52 (m, 1H), 7.34 (d, 1H, 16.7 Hz).
-
[0000]

Step C. 2-(2-bromo-phenyl)-propan-2-ol
-
[0282]To a solution of methyl 2-bromobenzoate (20.76 g, 96 mmol) in 120 mL of anhydrous ether under Argon at 0° C. was slowly added methylmagnesium bromide (77 mL, 3.26 M) at a rate that the internal temperature of the mixture was below 20° C. A white suspension resulted, and the mixture was stirred at room temperature for 2 h. The mixture was cooled in an ice-water bath. To the reaction mixture was very slowly added hydrochloric acid (400 mL, 0.5 M). The pH of the final mixture was adjusted to less than about 6 with few drops of 2M hydrochloric acid. The layers were separated, and the aqueous layer was extracted twice with ether. The organic layers were combined and dried over magnesium sulfate. The organic fraction was filtered, and the filtrate was concentrated to yield the title compound as a pale yellow liquid, which was distilled under vacuum to afford the title Compound 10d as a colorless liquid (16.9 g, 82%, b.p. about 65-70° C./0.3 mmHg). 1H NMR (400 MHz, CDCl3) δ (ppm): 7.67 (dd, 1H, J=1.7, 7.9 Hz), 7.58 (dd, 1H, J=1.3, 7.9 Hz), 7.30 (ddd, 1H, J=1.4, 7.4, 7.9 Hz), 7.10 (ddd, 1H, J=1.7, 7.4, 7.8 Hz), 2.77 (br s, 1H), 1.76 (s, 6H).
-
[0000]

Step D. 3,3-dimethyl-3H-benzo[c][1,2]oxaborol-1-ol
-
[0283]To a solution of n-BuLi (166 mL, 2.6 M, 432 mmol) in 200 mL of THF at −78° C. under argon was slowly added a solution of Compound 10d (42.2 g, 196 mmol) in 60 mL of THF at a rate that the internal temperature remained below −70° C. The mixture was stirred at −75° C. for 2 h. To the reaction mixture was then added triisopropylborate (59 mL, 255 mmol) in three portions. The mixture was allowed to warm slowly to room temperature overnight. The mixture was then cooled to 0° C., and was carefully quenched with dilute hydrochloric acid (250 mL, 2N). The mixture was then stirred at room temperature for 1 h. The pH of the mixture was checked and adjusted to acidic using additional 2N HCl if prophetic. The two layers were separated, and the aqueous layer was extracted twice with ether. The organic layers were combined, and dried with magnesium sulfate and filtered. The filtrate was concentrated under reduced pressure to yield a pale yellow oil. The residue was then diluted with ethyl acetate (400 mL) and, washed with 1N sodium hydroxide solution (150 mL×3). The basic aqueous layers were combined and acidified with 2N HCl. The clear solution turned cloudy when the acid was added. The mixture was extracted with ether (150 mL×3). The organic layers were combined and dried with magnesium sulfate. The solution was filtered, and the filtrate was concentrated under reduced pressure to yield the title Compound 10e as a colorless oil (26.2 g, 82%) which was used without further purification in the next step. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 9.00 (s, 1H), 7.66 (dm, 1H, J=7.3 Hz), 7.45 (dt, 1H, J=1.1, 7.7 Hz), 7.40 (dm, 1H, J=7.6 Hz), 7.31 (dt, 1H, J=1.2, 7.1 Hz), 1.44 (s, 6H).
-
[0000]

Step E. (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol
-
[0284]To a mixture of Compound 10e (11.7 g, 71 mmol), Compound 10c (19.9 g, 54 mmol), sodium carbonate (46 g, 435 mmol) and PdCl2(dppf).CH2Cl2 (8.9 g, 11 mmol) in a 1 L round bottom flask equipped with water condenser was added 400 mL of anhydrous DME and 200 mL of water. The mixture was evacuated and filled with Argon three times. The mixture was heated to 100° C. for 20 h. The mixture was then cooled to room temperature. The biphasic system was transferred to a 1 L separatory funnel and the two layers were separated. The organic layer was washed with brine (2×300 mL). The aqueous layers were combined and extracted with ethyl acetate once (about 300 mL). The organic layers were combined, dried with sodium sulfate, and filtered. The volume of the filtrate was reduced to about 170 mL under reduced pressure. The mixture was then filtered through a pad of silica gel and the pad was washed with ethyl acetate until the filtrate did not contain any product. After concentration, a light pink/beige solid was obtained. The solid was triturated with 50 mL ethyl acetate, and the mixture was heated to 85° C. for 5 min. The mixture was slowly cooled to r.t., then cooled at 0° C. for 0.5 h. The mixture was filtered, and the solid was washed with cold ethyl acetate twice, and dried under vacuum at 40° C. to yield the title Compound 18 as a light beige solid (7.58 g, 33%). RP-HPLC 95% pure.
-
1H NMR (400 MHz, DMSO-d6) δ (ppm): 12.73 (m, 1H,), 7.90 (d, 2H, J=8.2 Hz), 7.85 (dd, 1H, J=8.0, 0.6 Hz), 7.78 (d, 2H, J=8.4 Hz), 7.74 (d, 1H, J=16.8 Hz), 7.59-7.47 (m, 1H), 7.41 (s, 1H), 7.37-7.32 (m, 2H), 7.21 (dt, 1H, J=1.2, 7.4 Hz), 7.06 (s, 1H), 7.02 (d, 1H, J=7.4 Hz), 4.85 (s, 1H), 1.21 (s, 6H).
-
Mass Spectrum (LCMS, APCI pos.) Calcd. For C25H21F3N2O: 423.2 (M+H). Found 423.3.
-
m.p. (uncorr.) 250-251° C.
Example 10.1 Scale Up Preparation of (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol (Cpd 18) Step A. 3-(4-trifluoromethyl-phenyl)-acrylic acid
-
[0286]A 2-L 4-neck round bottom flask equipped with an air condenser/argon inlet, mechanical stirrer, thermocouple and a stopper was charged with 4-(trifluoromethyl)benzaldehyde (250 g, 196.2 mL, 1.44 mol), malonic acid (302.6 g, 2.87 mol), and pyridine (750 mL). An exotherm developed (about 38-40° C.), which was maintained for 30 min. Piperidine (14.202 mL, 143.58 mmol) was then added to the reaction and a second exotherm developed (Tmax about 42° C. after about 10 min.). The reaction was stirred for 30 min and then heated to 60° C. for 18 h (overnight). The reaction appeared to be complete by TLC, and was cooled to about 40° C., diluted into water (2 L; done to prevent reaction freezing), cooled to room temperature, and further diluted with water (4 L, 6 L total). The slurry was acidified to pH=2.0-3.0 with concentrated hydrochloric acid (about 675-700 mL). The material was stirred for 30 min., and a white solid was collected by filtration. The filter cake was washed with water until the filtrate was neutral (pH about 5.5-6, 2.5 L), air-dried in a Buchner funnel for 2 h, and then further dried in a vacuum oven at 60° C. overnight to provide 300.5 g (96%) of the title Compound 10a as a white solid.
Step B. (E)-5-bromo-2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazole
-
[0287]To a 5-L 4-neck round bottom flask equipped with a magnetic stirrer, argon inlet-argon outlet to a carbonate scrub, two stoppers, and a room temperature water bath was charged with 4-(trifluoromethyl)cinnamic acid (315 g, 1.46 mol) and dichloromethane (3.15 L) to give a slurry. To the slurry was added oxalyl chloride (151.71 mL, 1.75 mol) and DMF (1.13 mL, 14.57 mmol). Upon addition of DMF, gas evolution commenced, and the reaction was continued for about 3 h during which time a solution developed. When the reaction was complete (LC-MS), it was concentrated to dryness to give 342.4 g of 3-(4-trifluoromethyl-phenyl)-acryloyl chloride Compound 10b (>100%) as a yellow oily solid.
-
[0288]A 5-L 4-neck round bottom flask equipped with mechanical stirrer, thermocouple, air condenser with argon inlet, and a stopper was charged with 4-bromo-benzene-1,2-diamine (244 g, 1.27 mol) and acetic acid (2.13 L). To this solution was added a solution of Compound 10b (327 g, 1.39 mol) in toluene (237 mL). After this addition, the temperature spiked to 45° C. in about 30 seconds and then subsided. The reaction was then heated to 90° C. for 16 h (overnight). The reaction was cooled to 40° C., and poured into a mixed solution of EtOAc and heptane (about 1:3, 5.75 L) and a precipitate occurred. The resulting slurry was stirred for 3 h, and the solid was collected by filtration, washed with EtOAc:heptane (1:3, 3 L), and then dried in a vacuum oven (60° C.) to give 324.3 g (65%) of the title Compound 10c as a partial acetate salt.
Step C. 2-(2-bromo-phenyl)-propan-2-ol
-
[0289]A 12-Liter 4-neck flask equipped with a thermocouple, condenser, septum, addition funnel and overhead mechanical stirrer under argon was charged with methyl-2-bromobenzoate (226.5 g, 1.05 mol) and THF (1.6 L, 19.66 mol). The mixture was cooled to a temperature between 2 and 5° C. with stirring and held for 30 min. To the solution was slowly added methyl magnesium bromide in diethyl ether (3M, 1.05 L; 3.15 mol) via the addition funnel at a rate to maintain the reaction temperature below 15° C. An exotherm was observed during the addition, the reaction temperature warmed from 3 to 15° C. The addition of 1.05 L Grignard was complete in 4 h (approximate feed rate was 4.17 mL/min). The reaction mixture appeared to be off-white/yellow slurry. The reaction was allowed to warm to room temperature and stirred overnight (15 h). The reaction was sampled by HPLC/TLC and showed no starting material present. The ice bath was again applied to the reaction flask and a 0.5 M HCl solution (4.5 L; 2.25 mol) was slowly added over a period of 2 h. The temperature increased dramatically from 0 to 15° C. After the quench was complete, the reaction was stirred at room temperature for 30 min. Additional 2 N HCl (500 mL; 1.00 mol) was slowly added to maintain a pH less than 6. MTBE (1 L) was added to help with the phase split. The reaction was stirred at room temperature for 1 to 2 h to dissolve the solid material into the aqueous phase (most likely Mg(OH)2 which is very basic). The pH must be checked and adjusted with additional acid when necessary. The phases were separated and the aqueous layer was washed with an additional 1 L MTBE (2×500 mL). The organic phases were combined, washed with NaHCO3 solution (2×300 mL), dried over MgSO4, filtered and the filtrate was concentrated under vacuum to yield the title Compound 10d (220.83 g, 97.48% yield) as a clear yellow oil.
Step D. 3,3-dimethyl-3H-benzo[c][1,2]oxaborol-1-ol
-
[0290]A 12-Liter 4-neck round bottom flask equipped with a thermocouple, condenser, addition funnel and overhead mechanical stirrer under dry Argon was charged with anhydrous THF, (3 L) and chilled to −70 to −78° C. via a dry ice/acetone bath. n-Butyl lithium (2.5N in hexanes, 860 mL, 2.15 mol) was slowly added via addition funnel. An exotherm was observed as the temperature rose from −78 to −70° C. To the addition funnel was added a solution of Compound 10d (220 g, 979.97 mmol) in anhydrous THF (1 L). The 2-(2-bromophenyl)propan-2-ol solution was slowly added to the n-BuLi solution. The addition took 90 min in order to maintain a reaction temperature below −70° C. After the addition was complete, the reaction mixture was stirred at −70 to −75° C. for 30 min. The triethylborate (230 mL, 1.35 mol) was quickly added in 3 portions at −70° C. An exotherm was observed, the batch temperature rose from −70 to −64° C. The reaction was stirred at −70° C. and slowly warmed to room temperature over night. After the reaction was cooled to 0-5° C., the reaction was slowly quenched with 2 M HCl (1 L, 2.00 mol) added via the addition funnel while maintaining the batch temperature 0-5° C. The reaction mixture was stirred for 1 h. The aqueous phase pH was 9-10. The pH was then adjusted to acidic (4-5) with 2 M HCl (200 mL). The two phases were separated and the aqueous layer was extracted with MTBE (2×500 mL). The combined organic phases were dried with anhydrous magnesium sulfate. The solution was filtered and concentrated to yield a yellow oil. The yellow oil was diluted with MTBE (1.5 L) and washed with 1M NaOH (3×500 mL). The product containing basic aqueous phases were combined and acidified with 2 M HCl (800 mL) (the clear solution turns turbid with the addition of acid). After stirring the turbid solution for 15 min (pH=4-5) (Note 1), it was extracted with MTBE (2×500 mL). The organic phases were combined and dried over MgSO4. The solution was filtered and the filtrate was concentrated to yield the title Compound 10e as a clear yellow oil (121.78 grams, 77% yield).
Step E. (E)-2-(2-{2-[2-(4-Trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol
-
A 5-L 4-neck flask equipped with a thermocouple controller, condenser, overhead mechanical stirrer, Firestone Valve® and a nitrogen inlet/outlet was charged with dimethoxyethane (2 L), DI water (1 L) and sodium carbonate (230.9 g, 2.18 mol). The solution was degassed and purged with N2 three times. Compound 10e (71.7 g, 0.35 mol) and Compound 10c (100.0 g, 0.27 mol) were added to the degassed solution. The solution was degassed and purged with N2 three times. PdCl2(dppf) (44.48 g, 54.4 mmol) was added to the solution, and the solution was degassed and purged with N2 three times. The resulting two-phase suspension was heated to reflux for 18 h, and then cooled to room temperature. The reaction mixture was transferred to a 12-L separatory funnel, and the layers were separated. The organic layer was washed with brine (1 L). The two aqueous layers were combined and extracted with EtOAc (1 L). The combined organic layers were dried (Na2SO4), filtered, and the filtrate was concentrated to an oil. Two separate 100 g coupling reactions were combined and purified by chromatography in 10 successive chromatography runs on an ISCO preparative chromatography system (10×1.5 Kg SiO2, 5 column volumes of EtOAc, 250 mL/min flow rate). The combined fractions were transferred to two 22 L 4-neck round bottom flasks, and Silicycle Si-thiol functionalized silica gel (2 g) was added to each solution. The solutions were warmed to 40° C. and aged for 1 h. The solutions were filtered thru a medium glass funnel and washed with EtOAc (4 L) and combined. The filtrate was evaporated to a semi solid, which was transferred to a 2 L round bottom flask, to which EtOAc (0.4 L) was added. The resulting white precipitate slurry was cooled to −5° C. and stirred for 1 h. The slurry was filtered and washed twice with cold EtOAc (100 mL). The solids were dried in a vacuum oven at 40° C. for 40 h to afford 84.0 g (36.5% yield, 98.8 area % purity) of the title Compound 18 as a white solid. Anal. Calcd for C25H21N2OF3.0.04% H2O.0.15 mol MeOH: C, 70.48; H, 5.14: N, 6.42; F, 13.06 Found: C, 70.54; H, 4.83: N, 6.18; F, 13.33
Example 10.2 (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol monosodium salt (Cpd 18)
-
A 5-L 4-neck flask equipped with a thermocouple controller, an overhead mechanical stirrer, and a nitrogen inlet/outlet was charged with (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol. Compound 18 (125.0 g, 0.510 mol) and MeOH (1.25 L). A solution of sodium methoxide in methanol (0.5 M, 592 mL, 0.3 mol) was added. The reaction was heated to 65° C. for 30 min and all solids dissolved. The solution was cooled and evaporated to dryness. The foam was collected by scraping it out of the flask. The solids were placed in vacuum oven for 24 h at 40° C. to afford 139 g (about 100% isolated yield) of the title Compound 18 monosodium salt as a yellowish solid. 1H NMR (400 MHz, DMSO-d6) δ 7.80-7.84 (m, 3H), 7.74 (d, 2H, J=8.59 Hz), 7.65 (d, 1H, J=16.4 Hz), 7.40-7.44 (m, 2H), 7.25-7.37 (m, 2H), 7.16-7.20 (m, 1H), 7.01-7.05 (m, 1H), 6.84-6.87 (m, 1H), 1.23 (s, 6H). Mass Spectrum (LCMS, APCI pos.) Calcd. For C25H21F3N2O: 423.2 (M+H). Found 423.3. m.p. (uncorr.) 258-259° C.
Example 10.3 (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol hydrochloride salt (Cpd 18)
-
A 250-mL separatory funnel was charged with (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol. Compound 18 (1.0 g, 2.4 mmol) and EtOAc (20 mL). Aqueous HCl (1M, 20 mL) was added to the white slurry, and the separatory funnel was shaken. The solid product quickly dissolved, and a white precipitate started to form. The organic layer was transferred to a 100 mL round bottom flask equipped with a magnetic stir bar, and was stirred for 2 h. The thick slurry was filtered, rinsed with EtOAc (2×5 mL), and put into a vacuum oven at 40° C. for 36 h to afford 0.95 g (87.5%) of the title Compound 18 hydrochloride salt.
////////////Phase I, Musculoskeletal pain, Pain, Mavatrep, JNJ 39439335,
FDA approves UCLA IND application to commence embryonic stem cell-based trial
12 feb 2013
The USFDA has approved the investigator investigational new drug (IND) application of University of California, Los Angeles (UCLA), the clinical partner of Advanced Cell Technology (ACT), to commence a clinical trial using the human embryonic stem cells (hESCs)-derived cells to treat severe myopia.
Embryonic stem cell-based trial was designed to assess the hESC-derived ACT’s retinal pigment epithelial (RPE) cells in patients with severe myopia (nearsightedness).
ACT chairman and CEO Gary Rabin said, “We are pleased to be on track to broaden the scope of our RPE program with the initiation of the new Investigator IND.”
Human embryonic stem cells (hESCs) are pluripotent cells derived from the inner cell mass of the blastocyst. They have the ability to renew themselves and to differentiate into a variety of different cell types that are found in the body. Unlike somatic or ‘adult’ stem cells, hESCs proliferate indefinitely. This, together with their ability to differentiate into most adult cell types, has resulted in the preferred use of these cells for research and therapeutic applications, as they represent a potentially indefinite source of therapeutic cells. Any cell therapy derived from hESCs would be allogeneic by nature. Some current studies involve the potential therapeutic application of hESCs for spinal cord injury, age-related macular degeneration (AMD), cardiovascular diseases, and diabetes. Among the start up cell therapy companies, Geron and Advanced Cell Technologies have pioneered clinical trials using cells differentiated from hESCs.
Verastem Initiates Phase 1/1b Clinical Trial of VS-6063 Plus Paclitaxel for Patients with Ovarian Cancer
February 05, 2013
Verastem, Inc., (NASDAQ: VSTM), a clinical-stage biopharmaceutical company focused on discovering and developing drugs to treat cancer by the targeted killing of cancer stem cells, announced the initiation of a Phase 1/1b trial of VS-6063 in combination with paclitaxel for the treatment of advanced ovarian cancer.
Our unique understanding of cancer stem cell biology, coupled with our focus on execution in research and development, has allowed us to build the leading portfolio of FAK inhibitors”
VS-6063 is a potent inhibitor of focal adhesion kinase (FAK) and has demonstrated signs of clinical activity in ovarian cancer in a single agent, Phase 1 clinical trial.
“In the Phase 1 study we demonstrated that VS-6063 as a single agent was generally well tolerated, giving us optimism that this novel agent can be combined with the widely used drug paclitaxel,” said Principal Investigator Howard “Skip” Burris, III, M.D., Chief Medical Officer and Executive Director, Drug Development Program, Sarah Cannon Research Institute, Nashville, TN. “Moreover, clinically meaningful disease stabilization for about 6 months was observed in 3 of 4 patients with ovarian cancer treated with a dose of VS-6063 in the range of expected activity. All of these patients had received multiple lines of prior chemotherapy, including platinum-based treatment which has been shown to preferentially select chemo-resistant cancer stem cells.”
Robert Weinberg, Ph.D., Verastem co-founder and chair of the Scientific Advisory Board, has demonstrated that the FAK pathway is a critical component for the growth and survival of cancer stem cells, which are an underlying cause of tumor metastasis and recurrence.
“We believe that cancer stem cells are ultimately responsible for disease progression with ovarian and other cancers,” said Dr. Joanna Horobin, Verastem Chief Medical Officer. “Through FAK inhibition, we have the potential to provide more durable clinical responses for these diseases. Verastem has advanced the science of targeting cancer stem cells so that we can now clinically evaluate the therapeutic benefit of this approach in the treatment of cancer.”
The Phase 1b trial is an open-label, multicenter, dose-escalation trial of VS-6063 in combination with paclitaxel. The endpoints of the study are safety, tolerability and efficacy as determined by Response Evaluation Criteria in Solid Tumors (RECIST) and proprietary cancer stem cell biomarkers. The study will enroll up to 30 patients with ovarian cancer at three U.S. locations.
