
Home » Posts tagged '2020 APPROVALS'
Tag Archives: 2020 APPROVALS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
DOFETILIDE

Dofetilide
115256-11-6[RN]
6756
b-((p-Methanesulfonamidophenethyl)methylamino)methanesulfono-p-phenetidide
Methanesulfonamide, N-[4-[2-[methyl[2-[4-[(methylsulfonyl)amino]phenoxy]ethyl]amino]ethyl]phenyl]-
MFCD00869707 [MDL number]
- Molecular FormulaC19H27N3O5S2
- Average mass441.565 Da
- UK68798UNII:R4Z9X1N2NDUNII-R4Z9X1N2NDXelideβ-((p-Methanesulfonamidophenethyl)methylamino)methanesulfono-p-phenetidideдофетилидدوفيتيليد多非利特
N-[4-[2-[methyl[2-[4-[(methylsulfonyl)amino]phenoxy]ethyl]amino]ethyl]phenyl]-methanesulfonamideNCGC00164549-01PB0478000SMR000466333Tikosyn (TN)Research Code:UK-68798Trade Name:Tikosyn®MOA:Atrial potassium channel blockerIndication:Atrial flutter; Atrial fibrillationStatus:ApprovedCompany:Pfizer (Originator)Sales:ATC Code:C01BD04
INDIA 31/7/2020 APPROVED CDSCO
Dofetilide was first approved by the U.S. Food and Drug Administration (FDA) on Oct 1, 1999, then approved by European Medicine Agency (EMA) on Nov 29, 1999. It was developed and marketed as Tikosyn® by Pfizer.
Dofetilide is a selective blocker of delayed rectifier outward potassium current (IKr). It is indicated for the maintenance of normal sinus rhythm (delay in time to recurrence of atrial fibrillation/atrial flutter [AF/AFl]) in patients with atrial fibrillation/atrial flutter of greater than one week duration who have been converted to normal sinus rhythm.
Tikosyn® is available capsule for oral use, containing 0.125, 0.25 or 0.5 mg of free Dofetilide. The recommended dose is 500 µg orally twice daily.
Dofetilide is a class III antiarrhythmic agent.[1] It is marketed under the trade name Tikosyn by Pfizer, and is available in the United States in capsules containing 125, 250, and 500 µg of dofetilide. It is not available in Europe[2] or Australia.[3] In the United States it is only available by mail order or through specially trained local pharmacies.[4]
Medical uses
Dofetilide is used for the maintenance of sinus rhythm in individuals prone to the occurrence of atrial fibrillation and flutter arrhythmias, and for chemical cardioversion to sinus rhythm from atrial fibrillation and flutter.[5][6]
Based on the results of the Danish Investigations of Arrhythmias and Mortality on Dofetilide (“DIAMOND”) study,[7] dofetilide does not affect mortality in the treatment of patients post-myocardial infarction with left ventricular dysfunction, however it was shown to decrease all-cause readmissions as well as CHF-related readmissions.[8] Because of the results of the DIAMOND study, some physicians use dofetilide in the suppression of atrial fibrillation in individuals with LV dysfunction, however use appears limited: After initially receiving marketing approval in Europe in 1999, Pfizer voluntarily withdrew this approval in 2004 for commercial reasons[2] and it is not registered in other first world countries.
It has clinical advantages over other class III antiarrhythmics in chemical cardioversion of atrial fibrillation, and maintenance of sinus rhythm, and does not have the pulmonary or hepatotoxicity of amiodarone, however atrial fibrillation is not generally considered life-threatening, and dofetilide causes an increased rate of potentially life-threatening arrhythmias in comparison to other therapies.[9]
Contraindications
Prior to administration of the first dose, the corrected QT (QTc) must be determined. If the QTc is greater than 440 msec (or 500 msec in patients with ventricular conduction abnormalities), dofetilide is contraindicated. If heart rate is less than 60 bpm, the uncorrected QT interval should be used. After each subsequent dose of dofetilide, QTc should be determined and dosing should be adjusted. If at any time after the second dose of dofetilide the QTc is greater than 500 msec (550 msec in patients with ventricular conduction abnormalities), dofetilide should be discontinued. [4]
Adverse effects
Torsades de pointes is the most serious side effect of dofetilide therapy. The incidence of torsades de pointes is 0.3-10.5% and is dose-related, with increased incidence associated with higher doses. The majority of episodes of torsades de pointes have occurred within the first three days of initial dosing. Patients should be hospitalized and monitored for the first three days after starting dofetilide.[10]
The risk of inducing torsades de pointes can be decreased by taking precautions when initiating therapy, such as hospitalizing individuals for a minimum of three days for serial creatinine measurement, continuous telemetry monitoring and availability of cardiac resuscitation.
Pharmacology
Mechanism of action
Dofetilide works by selectively blocking the rapid component of the delayed rectifier outward potassium current (IKr).[11]
This causes the refractory period of atrial tissue to increase, hence its effectiveness in the treatment of atrial fibrillation and atrial flutter.
Dofetilide does not affect dV/dTmax (the slope of the upstroke of phase 0 depolarization), conduction velocity, or the resting membrane potential.

Dofetilide synthesis
There is a dose-dependent increase in the QT interval and the corrected QT interval (QTc). Because of this, many practitioners will initiate dofetilide therapy only on individuals under telemetry monitoring or if serial EKG measurements of QT and QTc can be performed.
Pharmacokinetics
Peak plasma concentrations are seen two to three hours after oral dosing when fasting. Dofetilide is well absorbed in its oral form, with a bioavailability of >90%. Intravenous administration of dofetilide is not available in the United States. [12]
The elimination half-life of dofetilide is roughly 10 hours; however, this varies based on many physiologic factors (most significantly creatinine clearance), and ranges from 4.8 to 13.5 hours. Due to the significant level of renal elimination (80% unchanged, 20% metabolites), the dose of dofetilide must be adjusted to prevent toxicity due to impaired renal function.[13]
Dofetilide is metabolized predominantly by CYP3A4 enzymes predominantly in the liver and GI tract. This means that it is likely to interact with drugs that inhibit CYP3A4, such as erythromycin, clarithromycin, or ketoconazole, resulting in higher and potentially toxic levels of dofetilide. [14]
Metabolism
A steady-state plasma level of dofetilide is achieved in 2–3 days.
80% of dofetilide is excreted by the kidneys, so the dose of dofetilide should be adjusted in individuals with chronic kidney disease, based on creatinine clearance.
In the kidneys, dofetilide is eliminated via cation exchange (secretion). Agents that interfere with the renal cation exchange system, such as verapamil, cimetidine, hydrochlorothiazide, itraconazole, ketoconazole, prochlorperazine, and trimethoprim should not be administered to individuals taking dofetilide.
About 20 percent of dofetilide is metabolized in the liver via the CYP3A4 isoenzyme of the cytochrome P450 enzyme system. Drugs that interfere with the activity of the CYP3A4 isoenzyme can increase serum dofetilide levels. If the renal cation exchange system is interfered with (as with the medications listed above), a larger percentage of dofetilide is cleared via the CYP3A4 isoenzyme system.
History
After its initial US FDA approval, due to the pro-arrhythmic potential it was only made available to hospitals and prescribers that had received education and undergone specific training in the risks of treatment with dofetilide; however, this restriction was subsequently removed in 2016. [15
SYN


REF
Reference:1. US5079248A / US4959366A.
2. J. Med. Chem. 1990, 33, 1151-1155.
SYN
SYN
SYN
SYN
EP 0245997; JP 1987267250; US 4959366; US 5079248
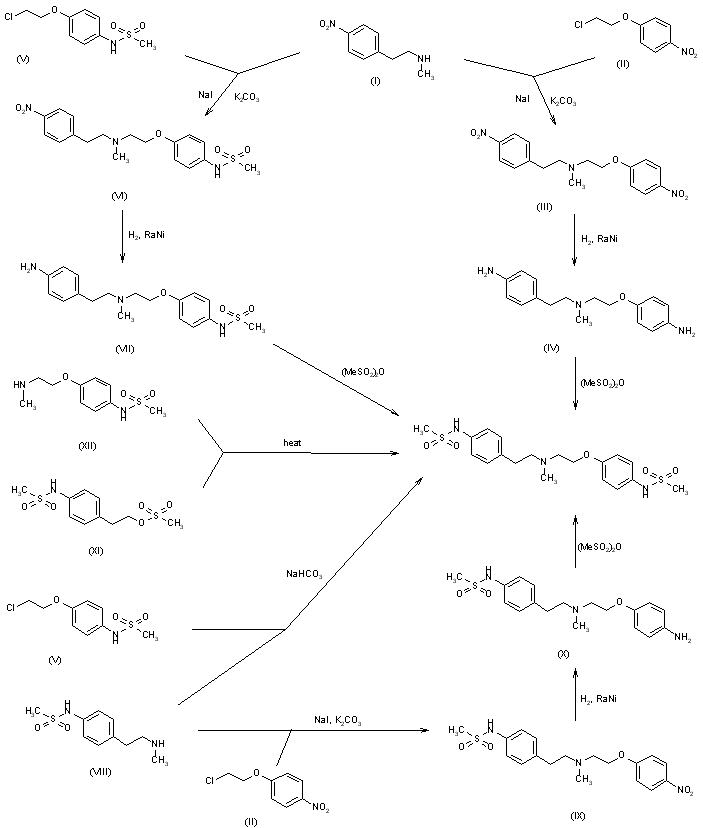
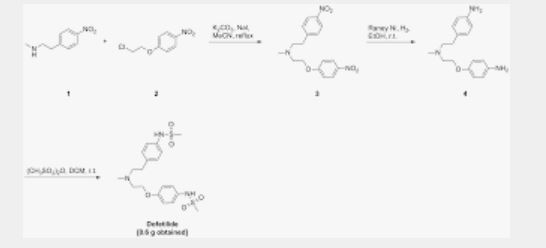
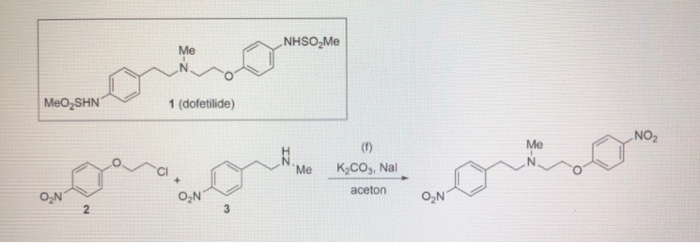
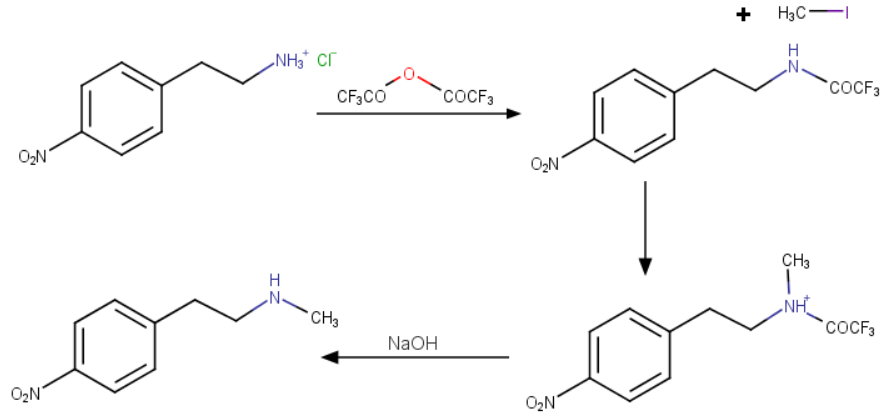
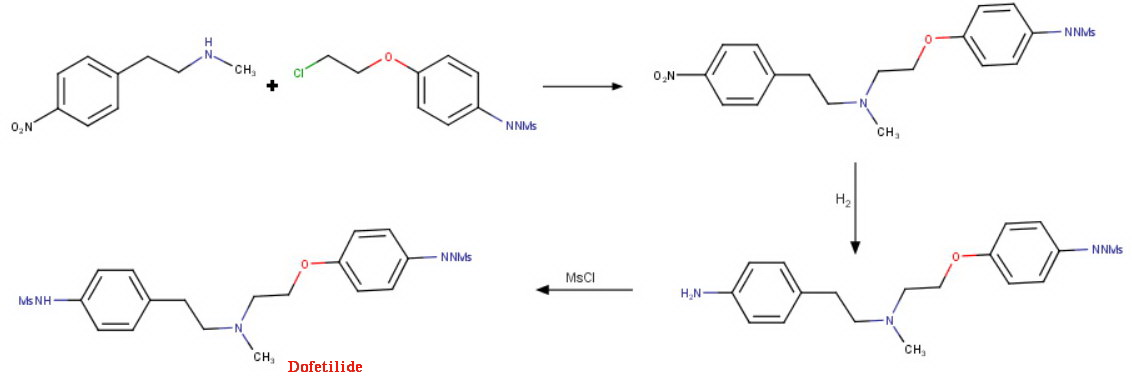
This compound can be prepared by several related ways: 1) The condensation of N-methyl-2-(4-nitrophenyl)ethylamine (I) with 4-(2-chloroethoxy)nitrobenzene (II) by means of NaI and K2CO3 in refluxing acetonitrile gives 1-(4-nitrophenoxy)-5-(4-nitrophenyl)-3-methyl-3-azapentane (III), which is reduced with H2 over Pd/C in ethanol, yielding the corresponding diamino derivative (IV). Finally, this compound is acylated with methanesulfonyl anhydride in dichloromethane. 2) The condensation of (I) with N-[4-(2-chloroethoxy)phenyl]methanesulfonamide (V) with NaI and K2CO3 as before gives 1-[4-(methanesulfonamide)phenoxy]-3-methyl-5-(4-nitrophenyl)-3-azapentane (VI), which is reduced with H2 over Pd/C as before, yielding the corresponding amino derivative (VII). Finally, this compound is acylated with methanesulfonyl anhydride as usual. 3) The condensation of (II) with N-[4-[2-(methylamino)ethyl]phenyl]methanesulfonamide (VIII) with NaI and K2CO3 as usual gives 1-[4-(methanesulfonamido)phenyl]-3-methyl-5-(4-nitrophenoxy)-3-azapentane (IX), which is reduced with H2 and RaNi to the corresponding amino derivative (X). Finally, this compound is acylated with methanesulfonyl chloride and pyridine. 4) By condensation of N-[4-[2-(methanesulfonyloxy)ethyl]phenyl]methanesulfonamide (XI) with N-[4-[2-(methylamino)ethoxy]phenyl]methanesulfonamide (XII) in refluxing ethanol. 5) By condensation of (V) with (VIII) by means of NaHCO3.
References
- ^ Lenz TL; Hilleman DE (July 2000). “Dofetilide, a new class III antiarrhythmic agent”. Pharmacotherapy. 20 (7): 776–86. doi:10.1592/phco.20.9.776.35208. PMID 10907968.
- ^ Jump up to:a b Wathion, Noel (2004-04-13). “Public Statement on Tikosyn (dofetilide): Voluntary Withdrawal of the Marketing Authorisation in the European Union” (PDF). European Agency for the Evaluation of Medicinal Products.
- ^ Australian Medicines Handbook 2014
- ^ Jump up to:a b TIKOSYN® (dofetilide). Pfizer. <http://www.tikosyn.com/>.
- ^ Banchs JE; Wolbrette DL; Samii SM; et al. (November 2008). “Efficacy and safety of dofetilide in patients with atrial fibrillation and atrial flutter”. J Interv Card Electrophysiol. 23(2): 111–5. doi:10.1007/s10840-008-9290-6. PMID 18688699. S2CID 25162347.
- ^ Lenz TL; Hilleman DE (November 2000). “Dofetilide: A new antiarrhythmic agent approved for conversion and/or maintenance of atrial fibrillation/atrial flutter”. Drugs Today. 36 (11): 759–71. doi:10.1358/dot.2000.36.11.601530. PMID 12845335.
- ^ Torp-Pedersen C, Møller M, Bloch-Thomsen PE, et al. (September 1999). “Dofetilide in patients with congestive heart failure and left ventricular dysfunction. Danish Investigations of Arrhythmia and Mortality on Dofetilide Study Group”. The New England Journal of Medicine. 341 (12): 857–65. doi:10.1056/NEJM199909163411201. PMID 10486417.
- ^ Torp-Pedersen C; ller M; Mø Bloch-Thomsen PE; et al. (September 1999). “Dofetilide in patients with congestive heart failure and left ventricular dysfunction. Danish Investigations of Arrhythmia and Mortality on Dofetilide Study Group”. N. Engl. J. Med. 341 (12): 857–65. doi:10.1056/NEJM199909163411201. PMID 10486417.
- ^ Micromedex Drugdex drug evaluations micromedex.com
- ^ Torp-Pedersen C, Møller M, Bloch-Thomsen PE, et al. Dofetilide in patients with congestive heart failure and left ventricular dysfunction. Danish Investigations of Arrhythmia and Mortality on Dofetilide Study Group. N Engl J Med 1999; 341:857.
- ^ Roukoz H; Saliba W (January 2007). “Dofetilide: a new class III antiarrhythmic agent”. Expert Rev Cardiovasc Ther. 5 (1): 9–19. doi:10.1586/14779072.5.1.9. PMID 17187453. S2CID 11255636.
- ^ 1Rasmussen HS, Allen MJ, Blackburn KJ, et al. Dofetilide, a novel class III antiarrhythmic agent. J Cardiovasc Pharmacol 1992; 20 Suppl 2:S96.
- ^ “Dofetilide.” Lexicomp. Wulters Kluwer Health, n.d. Web. <online.lexi.com>.
- ^ Walker DK, Alabaster CT, Congrave GS, et al. Significance of metabolism in the disposition and action of the antidysrhythmic drug, dofetilide. In vitro studies and correlation with in vivo data. Drug Metab Dispos 1996; 24:447.
- ^ “Information for Tikosyn (dofetilide)”. US Food and Drug Administration. 2016-03-09.
DofetilideCAS Registry Number: 115256-11-6CAS Name:N-[4-[2-[Methyl[2-[4-[(methylsulfonyl)amino]phenoxy]ethyl]amino]ethyl]phenyl]methanesulfonamideAdditional Names: 1-(4-methanesulfonamidophenoxy)-2-[N-(4-methanesulfonamidophenethyl)-N-methylamino]ethaneManufacturers’ Codes: UK-68798Trademarks: Tikosyn (Pfizer)Molecular Formula: C19H27N3O5S2Molecular Weight: 441.56Percent Composition: C 51.68%, H 6.16%, N 9.52%, O 18.12%, S 14.52%Literature References: Potassium channel blocker. Prepn: J. E. Arrowsmith et al.,EP245997; P. E. Cross et al.,US4959366 (1987, 1990 both to Pfizer); idemet al.,J. Med. Chem.33, 1151 (1990). HPLC determn in urine: D. K. Walker et al.,J. Chromatogr.568, 475 (1991). Mechanism of action study: D. Carmeliet, J. Pharmacol. Exp. Ther.262, 809 (1992). Review of pharmacology and pharmacokinetics: H. S. Rasmussen et al.,ibid.20, Suppl. 2, S96-S105 (1992). Clinical trial in atrial fibrillation and flutter: B. L. Norgaard et al.,Am. Heart J.137, 1062 (1999); in congestive heart failure: C. Torp-Pedersen et al.,N. Engl. J. Med.341, 857 (1999).Properties: Crystals from ethyl acetate/methanol (10:1), mp 147-149° (Cross); from hexane/ethyl acetate, mp 151-152° (Arrowsmith). Also reported as white crystalline solid, mp 161° (Rasmussen). pKa 7.0, 9.0, 9.6. Distribution coefficient (pH 7.4): 0.96. Sol in 0.1M NaOH, acetone, 0.1M HCl; very slightly sol in water, propan-2-ol.Melting point: mp 147-149° (Cross); mp 151-152° (Arrowsmith); mp 161° (Rasmussen)pKa: pKa 7.0, 9.0, 9.6Therap-Cat: Antiarrhythmic (class III).Keywords: Antiarrhythmic; Potassium Channel Blocker.
| Clinical data | |
|---|---|
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a601235 |
| ATC code | C01BD04 (WHO) |
| Pharmacokinetic data | |
| Bioavailability | 96% (oral) |
| Protein binding | 60% -70% |
| Elimination half-life | 10 hours |
| Identifiers | |
| IUPAC name[show] | |
| CAS Number | 115256-11-6 |
| PubChem CID | 71329 |
| IUPHAR/BPS | 2604 |
| DrugBank | DB00204 |
| ChemSpider | 64435 |
| UNII | R4Z9X1N2ND |
| KEGG | D00647 |
| ChEBI | CHEBI:4681 |
| ChEMBL | ChEMBL473 |
| CompTox Dashboard (EPA) | DTXSID5046433 |
| ECHA InfoCard | 100.166.441 |
| Chemical and physical data | |
| Formula | C19H27N3O5S2 |
| Molar mass | 441.56 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES[hide]O=S(=O)(Nc1ccc(cc1)CCN(CCOc2ccc(cc2)NS(=O)(=O)C)C)C | |
| InChI[hide]InChI=1S/C19H27N3O5S2/c1-22(13-12-16-4-6-17(7-5-16)20-28(2,23)24)14-15-27-19-10-8-18(9-11-19)21-29(3,25)26/h4-11,20-21H,12-15H2,1-3H3 Key:IXTMWRCNAAVVAI-UHFFFAOYSA-N |
////////////DOFETILIDE, 2020 APPROVALS, INDIA 2020, UK 68798, UNII:R4Z9X1N2ND, дофетилид , دوفيتيليد ,多非利特 , TIKOSYN
Lurbinectedin

Lurbinectedin
(1’R,6R,6aR,7R,13S,14S,16R)-5-(Acetyloxy)-2′,3′,4′,6,6a,7,9′-decahydro-8,14-dihydroxy-6′,9-dimethoxy-4,10,23-trimethyl-spiro(6,16-(epithiopropaneoxymethano)-7.13-imino-12H-1,3-dioxolo[7,8]soquino[3,2-b][3]benzazocine-20,1′-[1H]pyrido[3,4-b]indol]-19-one
| Molecular Weight | 784.87 |
| Formula | C41H44N4O10S |
| CAS No. | 497871-47-3 (Lurbinectedin); |
| Chemical Name | Spiro[6,16-(epithiopropanoxymethano)-7,13-imino-12H-1,3-dioxolo[7,8]isoquino[3,2-b][3]benzazocine-20,1′-[1H]pyrido[3,4-b]indol]-19-one, 5-(acetyloxy)-2′,3′,4′,6,6a,7,9′,13,14,16-decahydro-8,14-dihydroxy-6′,9-dimethoxy-4,10,23-trimethyl-, (1’R,6R,6aR,7R,13S,14S,16R)- (9CI) |
fda approved , 6/15/2020 , ZEPZELCA, Pharma Mar S.A.
To treat metastatic small cell lung cancer
Drug Trials Snapshot
Research Code:PM-01183; PM-1183
MOA:RNA polymerase inhibitor
Indication:Ovarian cancer; Breast cancer; Non small cell lung cancer (NSCLC)лурбинектединلوربينيكتيدين芦比替定(1R,1’R,2’R,3’R,11’S,12’S,14’R)-5′,12′-Dihydroxy-6,6′-dimethoxy-7′,21′,30′-trimethyl-27′-oxo-2,3,4,9-tetrahydrospiro[β-carboline-1,26′-[17,19,28]trioxa[24]thia[13,30]diazaheptacyclo[12.9.6.13,11. 02,13.04,9.015,23.016,20]triaconta[4,6,8,15,20,22]hexaen]-22′-yl acetate [ACD/IUPAC Name]2CN60TN6ZS497871-47-3[RN]9397
Lurbinectedin is in phase III clinical development for the treatment of platinum refractory/resistant ovarian cancer.
Phase II clinical trials are also ongoing for several oncology indications: non-small cell lung cancer, breast cancer, small cell lung cancer, head and neck carcinoma, neuroendocrine tumors, biliary tract carcinoma, endometrial carcinoma, germ cell tumors and Ewing’s family of tumors.
Lurbinectedin, sold under the brand name Zepzelca, is a medication for the treatment of adults with metastatic small cell lung cancer (SCLC) with disease progression on or after platinum-based chemotherapy.[1][2][3]
The most common side effects include leukopenia, lymphopenia, fatigue, anemia, neutropenia, increased creatinine, increased alanine aminotransferase, increased glucose, thrombocytopenia, nausea, decreased appetite, musculoskeletal pain, decreased albumin, constipation, dyspnea, decreased sodium, increased aspartate aminotransferase, vomiting, cough, decreased magnesium and diarrhea.[1][2][3]
Lurbinectedin is a synthetic tetrahydropyrrolo [4, 3, 2-de]quinolin-8(1H)-one alkaloid analogue with potential antineoplastic activity.[4] Lurbinectedin covalently binds to residues lying in the minor groove of DNA, which may result in delayed progression through S phase, cell cycle arrest in the G2/M phase and cell death.[4]
Lurbinectedin was approved for medical use in the United States in June 2020.[5][1][2][3][6]
Structure
Lurbinectedin is structurally similar to trabectedin, although the tetrahydroisoquinoline present in trabectedin is replaced with a tetrahydro β-carboline which enables lurbinectedin to exhibit increased antitumor activity compared with trabectedin.[7]
Biosynthesis
Lurbinectedin a marine agent isolated from the sea squirt species Ecteinascidia turbinata. Synthetic production is necessary because very small amounts can be obtained from sea organisms. For example, one ton (1000 kg) of sea squirts are required to produce one gram of trabectedin, which is analogue of lurbinectedin. Complex synthesis of lurbinectedin starts from small, common starting materials that require twenty-six individual steps to produce the drug with overall yield of 1.6%.[8][9]
Mechanism of action
According to PharmaMar,[10] lurbinectedin inhibits the active transcription of the encoding genes. This has two consequences. On one hand, it promotes tumor cell death, and on the other it normalizes tumor microenvironment. Active transcription is the process by which there are specific signal where information contained in the DNA sequence is transferred to an RNA molecule. This activity depends on the activity of an enzyme called RNA polymerase II. Lurbinectedin inhibits transcription through a very precise mechanism. Firstly, lurbinectedin binds to specific DNA sequences. It is at these precise spots that slides down the DNA to produce RNA polymerase II that is blocked and degraded by lurbinectedin. Lurbinectedin also has important role in tumor microenvironment. The tumor cells act upon macrophages to avoid them from behaving like an activator of the immune system. Literally, macrophages work in any tumor’s favor. Macrophages can contribute to tumor growth and progression by promoting tumor cell proliferation and invasion, fostering tumor angiogenesis and suppressing antitumor immune cells.[11][12] Attracted to oxygen-starved (hypoxic) and necrotic tumor cells they promote chronic inflammation. So, not only that macrophages inhibit immune system avoiding the destruction of tumor cells, but they also create tumor tissue that allows tumor growth. However, macrophages associated with tumors are cells that are addicted to the transcription process. Lurbinectedin acts specifically on the macrophages associated with tumors in two ways: firstly, by inhibiting the transcription of macrophages that leads to cell death and secondly, inhibiting the production of tumor growth factors. In this way, lurbinectedin normalizes the tumor microenvironment.
History
Lurbinectedin was approved for medical use in the United States in June 2020.[5][1][2][3][6]
Efficacy was demonstrated in the PM1183-B-005-14 trial (Study B-005; NCT02454972), a multicenter open-label, multi-cohort study enrolling 105 participants with metastatic SCLC who had disease progression on or after platinum-based chemotherapy.[3][6] Participants received lurbinectedin 3.2 mg/m2 by intravenous infusion every 21 days until disease progression or unacceptable toxicity.[3] The trial was conducted at 26 sites in the United States, Great Britain, Belgium, France, Italy, Spain and Czech Republic.[6]
The U.S. Food and Drug Administration (FDA) granted the application for lurbinectedin priority review and orphan drug designations and granted the approval of Zepzelca to Pharma Mar S.A.[3][13]
Research
Clinical Trials
Lurbinectedin can be used as monotherapy in the treatment of SCLC. Lurbinectedin monotherapy demonstrated the following clinical results in relapsed extensive stage SCLC:
- For sensitive disease (chemotherapy-free interval of ≥ 90 days) overall response rate (ORR) was 46.6% with 79.3% disease control rate and median overall survival (OS) being increased to 15.2 months.[14]
- For resistant disease (chemotherapy-free interval of < 90 days) overall response rate (ORR) was 21.3% with 46.8% disease control rate and 5.1 months median overall survival (OS).[14]
Lurbinectedin is also being investigated in combination with doxorubicin as second-line therapy in a randomized Phase III trial.[medical citation needed] While overall survival in this trial is not yet known, response rates at second line were
- 91.7% in sensitive disease with median progression-free survival of 5.8 months, and
- 33.3% in resistant disease with median progression-free of 3.5 months.[15]
Lurbinectedin is available in the U.S. under Expanded Access Program (EAP).[15][16]
SYN
SYN
WO2011/147828
Ecteinascidins is a group of naturally occurring marine compounds and analogs thereof, which are well identified and structurally characterized, and are disclosed to have antibacterial and cytotoxic properties. See for example, European Patent 309.477; WO 03/66638; WO 03/08423; WO 01 /771 15; WO 03/014127; R. Sakai et al., 1992, Proc. Natl. Acad. Sci. USA 89, pages 1 1456- 1 1460; R. Menchaca et al., 2003, J. Org. Chem. 68(23), pages 8859-8866; and I. Manzanares et al., 2001 , Curr. Med. Chem. Anti-Cancer Agents, 1 , pages 257-276; and references therein. Examples of ecteinascidins are provided by ET-743, ET-729, ET-745, ET-759A, ET-759B, ET-759C, ET-770, ET-815, ET-731 , ET-745B, ET-722, ET-736, ET-738, ET-808, ET-752, ET-594, ET-552, ET-637, ET-652, ET-583, ET-597, ET-596, ET-639, ET-641 , and derivatives thereof, such as acetylated forms, formylated forms, methylated forms, and oxide forms.
The structural characterizations of such ecteinascidins are not given again explicitly herein because from the detailed description provided in such references and citations any person of ordinary skill in this technology is capable of obtaining such information directly from the sources cited here and related sources.
At least one of the ecteinascidin compounds, ecteinascidin 743 (ET-743), has been extensively studied, and it will be referred to
specifically herein to illustrate features of this invention. ET-743 is being employed as an anticancer medicament, under the international nonproprietary name (INN) trabectedin, for the treatment of patients with advanced and metastatic soft tissue sarcoma (STS), after failure of anthracyclines and ifosfamide, or who are unsuited to receive such agents, and for the treatment of relapsed platinum- sensitive ovarian cancer in combination with pegylated liposomal doxorubicin.
ET-743 has a complex tris(tetrahydroisoquinoline) structure of formula
It was originally prepared by isolation from extracts of the marine tunicate Ecteinascidia turbinata. The yield was low, and alternative preparative processes had been sought.
The first synthetic process for producing ecteinascidin compounds was described in US Patent 5,721 ,362. This process employed sesamol as starting material and yielded ET-743 after a long and complicated sequence of 38 examples each describing one or more steps in the synthetic sequence.
An improvement in the preparation of one intermediate used in such process was disclosed in US Patent 6,815,544. Even with this improvement, the total synthesis was not suitable for manufacturing ET-743 at an industrial scale.
A hemisynthetic process for producing ecteinascidin compounds was described in EP 1.185.536. This process employs cyanosafracin B as starting material to provide ET-743. Cyanosafracin B is a pentacyclic antibiotic obtained by fermentation from the bacteria Pseudomonas fluorescens.
Cyanosafracin B
An improvement in such hemisynthetic process was disclosed in
EP 1.287.004.
To date four additional synthetic process (2 total and 2 formal synthesis) have been disclosed in patent applications JP 2003221395, WO 2007/045686, and WO 2007/087220 and in J. Org. Chem. 2008, 73, pages 9594-9600.
WO 2007/045686 also relates to the synthesis of Ecteinascidins-583 and 597 using intermediate compounds of formula:
Total synthesis strategies for the synthesis of the pentacyclic core -743 are overviewed in Figure I.
X = OH or CI
R = Protecting Group
WO2007087220 JOC 2008, 73, 9594-9600
EXAMPLE 3: SYNTHESIS OF COMPOUND 17.
Scheme X above provides an example of the synthesis of compound 17 from intermediate 10.
Compounds 16 and 17 are obtainable from intermediate 15 using the same procedures than those previously described in WO03/014127.
SYN

Reference:
1. WO2003014127A1.
https://patents.google.com/patent/WO2003014127A1/en
The ecteinascidins are exceedingly potent antitumour agents isolated from the marine tunicate Ecteinascidia turbinata. Several ecteinascidins have been reported previously in the patent and scientific literature. See, for example:
U.S. Patent No 5.256.663, which describes pharmaceutical compositions comprising matter extracted from the tropical marine invertebrate, Ecteinascidia turbinata, and designated therein as ecteinascidins, and the use of such compositions as antibacterial, antiviral, and/ or antitumour agents in mammals.
U.S. Patent No 5.089.273, which describes novel compositions of matter extracted from the tropical marine invertebrate, Ecteinascidia turbinata, and designated therein as ecteinascidins 729, 743, 745, 759A, 759B and 770. These compounds are useful as antibacterial and/or antitumour agents in mammals.
U.S. Patent No 5.149.804 which describes Ecteinascidins 722 and 736 (Et’s 722 and 736) isolated from the Caribbean tunicate Ecteinascidia turbinata and their structures. Et’s 722 and 736 protect mice in vivo at very low concentrations against P388 lymphoma, B 16 melanoma, and Lewis lung carcinoma.
U.S. Patent No 5.478.932, which describes ecteinascidins isolated from the Caribbean tunicate Ecteinascidia turbinata, which provide in vivo protection against P388 lymphoma, B 16 melanoma, M5076 ovarian sarcoma, Lewis lung carcinoma, and the LX- 1 human lung and MX- 1 human mammary carcinoma xenografts.
U.S. Patent No 5.654.426, which describes several ecteinascidins isolated from the Caribbean tunicate Ecteinascidia turbinata, which provide in vivo protection against P388 lymphoma, B 16 melanoma, M5076 ovarian sarcoma, Lewis lung carcinoma, and the LX-1 human lung and MX- 1 human mammary carcinoma xenografts.
U.S. Patent No 5.721.362 which describes a synthetic process for the formation of ecteinascidin compounds and related structures.
U.S. Patent No 6.124.292 which describes a series of new ecteinascidin- like compounds.
WO 0177115, WO 0187894 and WO 0187895, which describe new synthetic compounds of the ecteinascidin series, their synthesis and biological properties.
See also: Corey, E.J., J. Am. Chem. Soc, 1996, 118 pp. 9202-9203; Rinehart, et al., Journal of Natural Products, 1990, “Bioactive Compounds from Aquatic and Terrestrial Sources”, vol. 53, pp. 771- 792; Rinehart et al., Pure and Appl. Chem., 1990, “Biologically active natural products”, vol 62, pp. 1277- 1280; Rinehart, et al., J. Org. Chem., 1990, “Ecteinascidins 729, 743, 745, 759A, 759B, and 770: potent Antitumour Agents from the Caribbean Tunicate Ecteinascidia tuminata”, vol. 55, pp. 4512-4515; Wright et al., J. Org. Chem., 1990, “Antitumour Tetrahydroisoquinoline Alkaloids from the Colonial ascidian Ecteinascidia turbinata”, vol. 55, pp. 4508-4512; Sakai et al., Proc. Natl. Acad. Sci. USA 1992, “Additional anitumor ecteinascidins from a Caribbean tunicate: Crystal structures and activities in vivo”, vol. 89, 1 1456- 1 1460; Science 1994, “Chemical Prospectors Scour the Seas for Promising Drugs”, vol. 266, pp.1324; Koenig, K.E., “Asymmetric Synthesis”, ed. Morrison, Academic Press, Inc., Orlando, FL, vol. 5, 1985, p. 71; Barton, et al., J. Chem Soc. Perkin Trans., 1 , 1982, “Synthesis and Properties of a Series of Sterically Hindered Guanidine bases”, pp. 2085; Fukuyama et al., J. Am. Chem. Soc, 1982, “Stereocontrolled Total Synthesis of (+)-Saframycin B”, vol. 104, pp. 4957; Fukuyama et al., J. Am. Chem. Soc, 1990, “Total Synthesis of (+) – Saframycin A”, vol. 112, p. 3712; Saito, et al., J. Org. Chem., 1989, “Synthesis of Saframycins. Preparation of a Key tricyclic Lactam Intermediate to Saframycin A”, vol. 54, 5391; Still, et al., J Org. Chem., 1978, “Rapid Chromatographic Technique for Preparative Separations with Moderate Resolution”, vol. 43, p. 2923; Kofron, W.G.; Baclawski, L.M., J. Org. Chem., 1976, vol. 41, 1879; Guan et al., J. Biomolec Struc & Dynam., vol. 10, pp. 793-817 (1993); Shamma et al., “Carbon- 13 NMR Shift Assignments of Amines and Alkaloids”, p. 206 (1979); Lown et al., Biochemistry, 21, 419-428 (1982); Zmijewski et al., Chem. Biol. Interactions, 52, 361-375 (1985); Ito, CRC Crit. Rev. Anal. Chem., 17, 65- 143 (1986); Rinehart et al., “Topics in Pharmaceutical Sciences 1989”, pp. 613-626, D. D. Breimer, D. J. A. Cromwelin, K. K. Midha, Eds., Amsterdam Medical Press B. V., Noordwijk, The Netherlands (1989); Rinehart et al., “Biological Mass Spectrometry”, 233-258 eds. Burlingame et al., Elsevier Amsterdam (1990); Guan et al., Jour. Biomolec. Struct. & Dynam., vol. 10 pp. 793-817 (1993); Nakagawa et al., J. Amer. Chem. Soc, 11 1 : 2721-2722 (1989);; Lichter et al., “Food and Drugs from the Sea Proceedings” (1972), Marine Technology Society, Washington, D.C. 1973, 117- 127; Sakai et al., J. Amer. Chem. Soc, 1996, 1 18, 9017; Garcϊa-Rocha et al., Brit. J. Cancer, 1996, 73: 875-883; and pommier et al., Biochemistry, 1996, 35: 13303- 13309;
In 2000, a hemisynthetic process for the formation of ecteinascidin compounds and related structures such as phthalascidin starting from natural bis(tetrahydroisoquinoline) alkaloids such as the saframycin and safracin antibiotics available from different culture broths was reported; See Manzanares et al., Org. Lett., 2000, “Synthesis of Ecteinascidin ET-743 and Phthalascidin Pt-650 from Cyanosafracin B”, Vol. 2, No 16, pp. 2545-2548; and International Patent Application WO 00 69862.
Ecteinascidin 736 was first discovered by Rinehart and features a tetrahydro-β-carboline unit in place of the tetrahydroisoquinoline unit more usually found in the ecteinascidin compounds isolated from natural sources; See for example Sakai et al., Proc. Natl. Acad. Sci. USA 1992, “Additional antitumor ecteinascidins from a Caribbean tunicate: Crystal structures and activities in vivo”, vol. 89, 11456-11460.
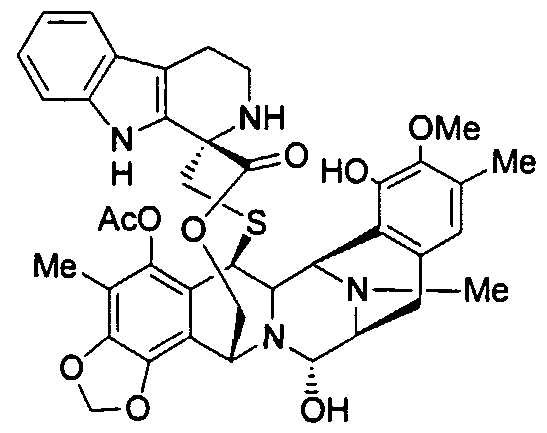
Et-736
WO 9209607 claims ecteinascidin 736, as well as ecteinascidin 722 with hydrogen in place of methyl on the nitrogen common to rings C and D of ecteinascidin 736 and O-methylecteinascidin 736 with methoxy in place of hydroxy on ring C of ecteinascidin 736.
Despite the positive results obtained in clinical applications in chemotherapy, the search in the field of ecteinascidin compounds is still open to the identification of new compounds with optimal features of cytotoxicity and selectivity toward the tumour and with a reduced systemic toxicity and improved pharmacokinetic properties.
PATENT
WO2001087894A1.
PATENT
US 20130066067
https://patents.google.com/patent/US20130066067A1/en
- Ecteinascidins is a group of naturally occurring marine compounds and analogs thereof, which are well identified and structurally characterized, and are disclosed to have antibacterial and cytotoxic properties. See for example, European Patent 309.477; WO 03/66638; WO 03/08423; WO 01/77115; WO 03/014127; R. Sakai et al., 1992, Proc. Natl. Acad. Sci. USA 89, pages 11456-11460; R. Menchaca et al., 2003, J. Org. Chem. 68(23), pages 8859-8866; and I. Manzanares et al., 2001, Curr. Med. Chem. Anti–Cancer Agents, 1, pages 257-276; and references therein. Examples of ecteinascidins are provided by ET-743, ET-729, ET-745, ET-759A, ET-759B, ET-759C, ET-770, ET-815, ET-731, ET-745B, ET-722, ET-736, ET-738, ET-808, ET-752, ET-594, ET-552, ET-637, ET-652, ET-583, ET-597, ET-596, ET-639, ET-641, and derivatives thereof, such as acetylated forms, formylated forms, methylated forms, and oxide forms.
- [0003]
The structural characterizations of such ecteinascidins are not given again explicitly herein because from the detailed description provided in such references and citations any person of ordinary skill in this technology is capable of obtaining such information directly from the sources cited here and related sources. - [0004]
At least one of the ecteinascidin compounds, ecteinascidin 743 (ET-743), has been extensively studied, and it will be referred to specifically herein to illustrate features of this invention. ET-743 is being employed as an anticancer medicament, under the international nonproprietary name (INN) trabectedin, for the treatment of patients with advanced and metastatic soft tissue sarcoma (STS), after failure of anthracyclines and ifosfamide, or who are unsuited to receive such agents, and for the treatment of relapsed platinum-sensitive ovarian cancer in combination with pegylated liposomal doxorubicin. - [0005]
ET-743 has a complex tris(tetrahydroisoquinoline) structure of formula - [0006]
It was originally prepared by isolation from extracts of the marine tunicate Ecteinascidia turbinata. The yield was low, and alternative preparative processes had been sought. - [0007]
The first synthetic process for producing ecteinascidin compounds was described in U.S. Pat. No. 5,721,362. This process employed sesamol as starting material and yielded ET-743 after a long and complicated sequence of 38 examples each describing one or more steps in the synthetic sequence. - [0008]
An improvement in the preparation of one intermediate used in such process was disclosed in U.S. Pat. No. 6,815,544. Even with this improvement, the total synthesis was not suitable for manufacturing ET-743 at an industrial scale. - [0009]
A hemisynthetic process for producing ecteinascidin compounds was described in EP 1.185.536. This process employs cyanosafracin B as starting material to provide ET-743. Cyanosafracin B is a pentacyclic antibiotic obtained by fermentation from the bacteria Pseudomonas fluorescens. - [0010]
An improvement in such hemisynthetic process was disclosed in EP 1.287.004. - [0011]
To date four additional synthetic process (2 total and 2 formal synthesis) have been disclosed in patent applications JP 2003221395, WO 2007/045686, and WO 2007/087220 and in J. Org. Chem. 2008, 73, pages 9594-9600. - [0012]
WO 2007/045686 also relates to the synthesis of Ecteinascidins-583 and 597 using intermediate compounds of formula: - [0013]
Total synthesis strategies for the synthesis of the pentacyclic core of ET-743 are overviewed in FIG. 1.
PAPER
Angewandte Chemie, International Edition (2019), 58(12), 3972-3975.
https://onlinelibrary.wiley.com/doi/abs/10.1002/anie.201900035
An efficient and scalable approach is described for the total synthesis of the marine natural product Et‐743 and its derivative lubinectedin, which are valuable antitumor compounds. The method delivers 1.6 % overall yield in 26 total steps from Cbz‐protected (S)‐tyrosine. It features the use of a common advanced intermediate to create the right and left parts of these compounds, and a light‐mediated remote C−H bond activation to assemble a benzo[1,3]dioxole‐containing intermediate.

Synthesis of lactone SI-5. A mixture of 19 (98.0 mg, 0.16 mmol, 1.0 equiv), 2-(5-methoxy-1H-indol-3-yl) ethanamine hydrochloride salt (357.8 mg, 1.58 mmol, 10.0 equiv) and NaOAc (144 mg, 1.74 mmol, 11.0 equiv) in anhydrous EtOH (5.0 mL) was stirred at 60 oC for 5 h. The cooled mixture was extracted with ethyl acetate, and the organic layer was dried over sodium sulfate and concentrated. The residue was purified by flash column chromatography (eluting with DCM/MeOH = 20:1) to afford compound SI-5 (109 mg, 87%). [α]𝐷 20 = -27.7 (c = 1.0, CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.61 (s, 1H), 7.13 (d, J = 8.8 Hz, 1H), 6.82 (d, J = 2.2 Hz, 1H), 6.75 (dd, J = 8.8, 2.4 Hz, 1H), 6.66 (s, 1H), 6.22 (d, J = 1.0 Hz, 1H), 6.02 (d, J = 1.0 Hz, 1H), 5.78 (s, 1H), 5.08 (d, J = 11.7 Hz, 1H), 4.55 (s, 1H), 4.32 (s, 1H), 4.27 (d, J = 3.8 Hz, 1H), 4.23–4.15 (m, 2H), 3.81 (s, 3H), 3.79 (s, 3H), 3.47–3.39 (m, 2H), 3.20–3.10 (m, 1H), 3.06 (d, J = 18.1 Hz, 1H), 2.93 (dd, J = 18.2, 9.1 Hz, 1H), 2.86–2.76 (m, 1H), 2.62 (dt, J = 14.9, 4.8 Hz, 1H), 2.56–2.47 (m, 2H), 2.37 (s, 3H), 2.30–2.27 (m, 1H), 2.26 (s, 3H), 2.22 (s, 3H), 2.06 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 171.6, 168.8, 154.0, 148.2, 145.8, 143.1, 141.3, 140.5, 131.4, 130.8, 130.7, 129.4, 127.3, 120.9, 120.8, 118.4, 118.4, 113.9, 113.8, 112.2, 111.8, 110.2, 102.2, 100.5, 62.6, 61.4, 60.7, 60.5, 59.6, 59.6, 55.9, 54.9, 54.8, 42.1, 41.6, 39.9, 39.5, 29.5, 24.0, 20.8, 16.0, 9.9; HRMS (ESI) m/z calcd. for C42H43N5O9S [M + H]+ 794.2860, found 794.2858

Lurbinectedin: To a solution of SI-5 (80 mg, 0.1 mmol, 1.0 equiv) in acetonitrile and water (3:2, v/v, 10 mL) was added silver nitrate (514 mg, 3 mmol, 30.0 equiv). The suspension was stirred at 25 oC for 24 h before a mixture of saturated brine (5.0 mL) and saturated sodium hydrogen carbonate (5 mL) were added. The resultant mixture was stirred at 25 oC for 15 min before it was filtered through celite and extracted with ethyl acetate (3 × 20 mL). The combined organic layers were dried over sodium sulfate and concentrated, and the residue was purified by flash column chromatography (eluting with DCM/MeOH = 20:1) to afford Lurbinectedin (71 mg, 89%). [α]𝐷 20 = -45.0 (c = 1.0, CHCl3) 1H NMR (400 MHz, CDCl3) δ 7.61 (s, 1H), 7.13 (d, J = 8.8 Hz, 1H), 6.82 (d, J = 2.2 Hz, 1H), 6.74 (dd, J = 8.8, 2.4 Hz, 1H), 6.67 (s, 1H), 6.19 (d, J = 1.1 Hz, 1H), 5.99 (d, J = 1.1 Hz, 1H), 5.77 (br s, 1H), 5.20 (d, J = 11.3 Hz, 1H), 4.82 (s, 1H), 4.53–4.40 (m, 2H), 4.18–4.08 (m, 2H), 3.81 (s, 3H), 3.79 (s, 3H), 3.49 (d, J = 4.2 Hz, 1H), 3.24–3.13 (m, 2H), 3.01 (d, J = 17.9 Hz, 1H), 2.88–2.79 (m, 2H), 2.63 (dt, J = 15.0, 4.9 Hz, 1H), 2.56–2.47 (m, 2H), 2.37 (s, 3H), 2.32–2.27 (m, 1H), 2.26 (s, 3H), 2.19 (s, 3H), 2.05 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 171.4, 168.8, 153.8, 147.9, 145.5, 142.9, 141.1, 140.7, 131.8, 131.3, 130.7, 129.1, 127.3, 121.4, 121.0, 118.2, 115.6, 112.9, 111.9, 111.7, 110.0, 101.8, 100.4, 82.0, 62.4, 61.9, 60.4, 57.8, 57.5, 56.0, 55.8, 55.0, 42.2, 41.3, 39.8, 39.3, 29.3, 23.6, 20.6, 15.9, 9.7; HRMS (ESI) m/z calcd. for C41H44N4O10S [M – OH]+ 767.2745, found 767.2742.
References
- ^ Jump up to:a b c d e “Zepzelca- lurbinectedin injection, powder, lyophilized, for solution”. DailyMed. 15 June 2020. Retrieved 24 September 2020.
- ^ Jump up to:a b c d “Jazz Pharmaceuticals Announces U.S. FDA Accelerated Approval of Zepzelca (lurbinectedin) for the Treatment of Metastatic Small Cell Lung Cancer” (Press release). Jazz Pharmaceuticals. 15 June 2020. Retrieved 15 June 2020 – via PR Newswire.
- ^ Jump up to:a b c d e f g “FDA grants accelerated approval to lurbinectedin for metastatic small”. U.S. Food and Drug Administration (FDA). 15 June 2020. Retrieved 16 June 2020.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b “Lurbinectedin”. National Cancer Institute. Retrieved 15 June 2020. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b “Zepzelca: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 15 June 2020.
- ^ Jump up to:a b c d “Drug Trials Snapshots: Zepzelca”. U.S. Food and Drug Administration (FDA). 15 June 2020. Retrieved 28 June 2020. This article incorporates text from this source, which is in the public domain.
- ^ Takahashi, Ryoko; Mabuchi, Seiji; Kawano, Mahiru; Sasano, Tomoyuki; Matsumoto, Yuri; Kuroda, Hiromasa; Kozasa, Katsumi; Hashimoto, Kae; Sawada, Kenjiro; Kimura, Tadashi (17 March 2016). “Preclinical Investigations of PM01183 (Lurbinectedin) as a Single Agent or in Combination with Other Anticancer Agents for Clear Cell Carcinoma of the Ovary”. PLOS ONE. 11 (3): e0151050. Bibcode:2016PLoSO..1151050T. doi:10.1371/journal.pone.0151050. PMC 4795692. PMID 26986199.
- ^ Total synthesis of marine antitumor agents trabectedin and lurbinectedin | https://www.sciencedaily.com/releases/2019/02/190219111659.htm
- ^ A Scalable Total Synthesis of the Antitumor Agents Et‐743 and Lurbinectedin | https://onlinelibrary.wiley.com/doi/full/10.1002/anie.201900035
- ^ PharmaMar presentation of Lurbinectedin’s Mechanism of Action Lurbinectedin Mechanisim of Action | https://www.youtube.com/watch?v=8daELhxAXcQ
- ^ Qian BZ, Pollard JW (April 2010). “Macrophage diversity enhances tumor progression and metastasis”. Cell. 141 (1): 39–51. doi:10.1016/j.cell.2010.03.014. PMC 4994190. PMID 20371344.
- ^ Engblom C, Pfirschke C, Pittet MJ (July 2016). “The role of myeloid cells in cancer therapies”. Nature Reviews. Cancer. 16 (7): 447–62. doi:10.1038/nrc.2016.54. PMID 27339708. S2CID 21924175.
- ^ “Lurbinectedin Orphan Drug Designation and Approval”. U.S. Food and Drug Administration (FDA). 1 August 2018. Retrieved 16 June 2020.
- ^ Jump up to:a b Paz-Ares, Luis G.; Trigo Perez, Jose Manuel; Besse, Benjamin; Moreno, Victor; Lopez, Rafael; Sala, Maria Angeles; Ponce Aix, Santiago; Fernandez, Cristian Marcelo; Siguero, Mariano; Kahatt, Carmen Maria; Zeaiter, Ali Hassan; Zaman, Khalil; Boni, Valentina; Arrondeau, Jennifer; Martinez Aguillo, Maite; Delord, Jean-Pierre; Awada, Ahmad; Kristeleit, Rebecca Sophie; Olmedo Garcia, Maria Eugenia; Subbiah, Vivek (20 May 2019). “Efficacy and safety profile of lurbinectedin in second-line SCLC patients: Results from a phase II single-agent trial”. Journal of Clinical Oncology. 37 (15_suppl): 8506. doi:10.1200/JCO.2019.37.15_suppl.8506.
- ^ Jump up to:a b Calvo, E.; Moreno, V.; Flynn, M.; Holgado, E.; Olmedo, M.E.; Lopez Criado, M.P.; Kahatt, C.; Lopez-Vilariño, J.A.; Siguero, M.; Fernandez-Teruel, C.; Cullell-Young, M.; Soto Matos-Pita, A.; Forster, M. (October 2017). “Antitumor activity of lurbinectedin (PM01183) and doxorubicin in relapsed small-cell lung cancer: results from a phase I study”. Annals of Oncology. 28 (10): 2559–2566. doi:10.1093/annonc/mdx357. PMC 5834091. PMID 28961837. Lay summary.
- ^ Farago, Anna F; Drapkin, Benjamin J; Lopez-Vilarino de Ramos, Jose Antonio; Galmarini, Carlos M; Núñez, Rafael; Kahatt, Carmen; Paz-Ares, Luis (January 2019). “ATLANTIS: a Phase III study of lurbinectedin/doxorubicin versus topotecan or cyclophosphamide/doxorubicin/vincristine in patients with small-cell lung cancer who have failed one prior platinum-containing line”. Future Oncology. 15 (3): 231–239. doi:10.2217/fon-2018-0597. PMC 6331752. PMID 30362375.
External links
- “Lurbinectedin”. Drug Information Portal. U.S. National Library of Medicine.
- “Lurbinectedin”. NCI Dictionary of Cancer Terms. National Cancer Institute.
- Clinical trial number NCT02454972 for “Clinical Trial of Lurbinectedin (PM01183) in Selected Advanced Solid Tumors” at ClinicalTrials.gov
FDA grants accelerated approval to lurbinectedin for metastatic small cell lung cancer
On June 15, 2020, the Food and Drug Administration granted accelerated approval to lurbinectedin(ZEPZELCA, Pharma Mar S.A.) for adult patients with metastatic small cell lung cancer (SCLC) with disease progression on or after platinum-based chemotherapy.
Efficacy was demonstrated in the PM1183-B-005-14 trial (Study B-005; NCT02454972), a multicenter open-label, multi-cohort study enrolling 105 patients with metastatic SCLC who had disease progression on or after platinum-based chemotherapy. Patients received lurbinectedin 3.2 mg/m2 by intravenous infusion every 21 days until disease progression or unacceptable toxicity.
The main efficacy outcome measures were confirmed overall response rate (ORR) determined by investigator assessment using RECIST 1.1 and response duration. Among the 105 patients, the ORR was 35% (95% CI: 26%, 45%), with a median response duration of 5.3 months (95% CI: 4.1, 6.4). The ORR as per independent review committee was 30% (95% CI: 22%, 40%) with a median response duration of 5.1 months (95% CI: 4.9, 6.4).
The most common adverse reactions (≥20%), including laboratory abnormalities, were myelosuppression, fatigue, increased creatinine, increased alanine aminotransferase, increased glucose, nausea, decreased appetite, musculoskeletal pain, decreased albumin, constipation, dyspnea, decreased sodium, increased aspartate aminotransferase, vomiting, cough, decreased magnesium and diarrhea.
The recommended lurbinectedin dose is 3.2 mg/m2 every 21 days.
View full prescribing information for ZEPZELCA.
This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
This review was conducted under Project Orbis, an initiative of the FDA Oncology Center of Excellence. Project Orbis provides a framework for concurrent submission and review of oncology drugs among international partners. For this application, a modified Project Orbis was undertaken because of the timing of submission to other regulatory agencies. FDA is collaborating with the Australian Therapeutic Goods Administration (TGA). FDA approved this application 2 months ahead of the goal date. The review is ongoing for the Australian TGA.
FDA granted lurbinectedin orphan drug designation for the treatment of SCLC and priority review to this application. A description of FDA expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.
REFERENCES
1: Calvo E, Moreno V, Flynn M, Holgado E, Olmedo ME, Lopez Criado MP, Kahatt C, Lopez-Vilariño JA, Siguero M, Fernandez-Teruel C, Cullell-Young M, Soto Matos-Pita A, Forster M. Antitumor activity of lurbinectedin (PM01183) and doxorubicin in relapsed small-cell lung cancer: results from a phase I study. Ann Oncol. 2017 Oct 1;28(10):2559-2566. doi: 10.1093/annonc/mdx357. PubMed PMID: 28961837.
2: Erba E, Romano M, Gobbi M, Zucchetti M, Ferrari M, Matteo C, Panini N, Colmegna B, Caratti G, Porcu L, Fruscio R, Perlangeli MV, Mezzanzanica D, Lorusso D, Raspagliesi F, D’Incalci M. Ascites interferes with the activity of lurbinectedin and trabectedin: Potential role of their binding to alpha 1-acid glycoprotein. Biochem Pharmacol. 2017 Nov 15;144:52-62. doi: 10.1016/j.bcp.2017.08.001. Epub 2017 Aug 4. PubMed PMID: 28782526.
3: Belgiovine C, Bello E, Liguori M, Craparotta I, Mannarino L, Paracchini L, Beltrame L, Marchini S, Galmarini CM, Mantovani A, Frapolli R, Allavena P, D’Incalci M. Lurbinectedin reduces tumour-associated macrophages and the inflammatory tumour microenvironment in preclinical models. Br J Cancer. 2017 Aug 22;117(5):628-638. doi: 10.1038/bjc.2017.205. Epub 2017 Jul 6. PubMed PMID: 28683469; PubMed Central PMCID: PMC5572168.
4: Jimeno A, Sharma MR, Szyldergemajn S, Gore L, Geary D, Diamond JR, Fernandez Teruel C, Soto Matos-Pita A, Iglesias JL, Cullell-Young M, Ratain MJ. Phase I study of lurbinectedin, a synthetic tetrahydroisoquinoline that inhibits activated transcription, induces DNA single- and double-strand breaks, on a weekly × 2 every-3-week schedule. Invest New Drugs. 2017 Aug;35(4):471-477. doi: 10.1007/s10637-017-0427-2. Epub 2017 Jan 20. PubMed PMID: 28105566.
5: Paz-Ares L, Forster M, Boni V, Szyldergemajn S, Corral J, Turnbull S, Cubillo A, Teruel CF, Calderero IL, Siguero M, Bohan P, Calvo E. Phase I clinical and pharmacokinetic study of PM01183 (a tetrahydroisoquinoline, Lurbinectedin) in combination with gemcitabine in patients with advanced solid tumors. Invest New Drugs. 2017 Apr;35(2):198-206. doi: 10.1007/s10637-016-0410-3. Epub 2016 Nov 21. PubMed PMID: 27873130.
6: Harlow ML, Maloney N, Roland J, Guillen Navarro MJ, Easton MK, Kitchen-Goosen SM, Boguslawski EA, Madaj ZB, Johnson BK, Bowman MJ, D’Incalci M, Winn ME, Turner L, Hostetter G, Galmarini CM, Aviles PM, Grohar PJ. Lurbinectedin Inactivates the Ewing Sarcoma Oncoprotein EWS-FLI1 by Redistributing It within the Nucleus. Cancer Res. 2016 Nov 15;76(22):6657-6668. doi: 10.1158/0008-5472.CAN-16-0568. Epub 2016 Oct 3. PubMed PMID: 27697767; PubMed Central PMCID: PMC5567825.
7: Céspedes MV, Guillén MJ, López-Casas PP, Sarno F, Gallardo A, Álamo P, Cuevas C, Hidalgo M, Galmarini CM, Allavena P, Avilés P, Mangues R. Lurbinectedin induces depletion of tumor-associated macrophages, an essential component of its in vivo synergism with gemcitabine, in pancreatic adenocarcinoma mouse models. Dis Model Mech. 2016 Dec 1;9(12):1461-1471. Epub 2016 Oct 20. PubMed PMID: 27780828; PubMed Central PMCID: PMC5200894.
8: Santamaría Nuñez G, Robles CM, Giraudon C, Martínez-Leal JF, Compe E, Coin F, Aviles P, Galmarini CM, Egly JM. Lurbinectedin Specifically Triggers the Degradation of Phosphorylated RNA Polymerase II and the Formation of DNA Breaks in Cancer Cells. Mol Cancer Ther. 2016 Oct;15(10):2399-2412. Epub 2016 Sep 14. PubMed PMID: 27630271.
9: Metaxas Y, Cathomas R, Mark M, von Moos R. Combination of cisplatin and lurbinectedin as palliative chemotherapy in progressive malignant pleural mesothelioma: Report of two cases. Lung Cancer. 2016 Dec;102:136-138. doi: 10.1016/j.lungcan.2016.07.012. Epub 2016 Jul 14. PubMed PMID: 27440191.
10: Lima M, Bouzid H, Soares DG, Selle F, Morel C, Galmarini CM, Henriques JA, Larsen AK, Escargueil AE. Dual inhibition of ATR and ATM potentiates the activity of trabectedin and lurbinectedin by perturbing the DNA damage response and homologous recombination repair. Oncotarget. 2016 May 3;7(18):25885-901. doi: 10.18632/oncotarget.8292. PubMed PMID: 27029031; PubMed Central PMCID: PMC5041952.
11: Takahashi R, Mabuchi S, Kawano M, Sasano T, Matsumoto Y, Kuroda H, Kozasa K, Hashimoto K, Sawada K, Kimura T. Preclinical Investigations of PM01183 (Lurbinectedin) as a Single Agent or in Combination with Other Anticancer Agents for Clear Cell Carcinoma of the Ovary. PLoS One. 2016 Mar 17;11(3):e0151050. doi: 10.1371/journal.pone.0151050. eCollection 2016. PubMed PMID: 26986199; PubMed Central PMCID: PMC4795692.
12: Pernice T, Bishop AG, Guillen MJ, Cuevas C, Aviles P. Development of a liquid chromatography/tandem mass spectrometry assay for the quantification of PM01183 (lurbinectedin), a novel antineoplastic agent, in mouse, rat, dog, Cynomolgus monkey and mini-pig plasma. J Pharm Biomed Anal. 2016 May 10;123:37-41. doi: 10.1016/j.jpba.2016.01.043. Epub 2016 Jan 21. PubMed PMID: 26871278.
13: Elez ME, Tabernero J, Geary D, Macarulla T, Kang SP, Kahatt C, Pita AS, Teruel CF, Siguero M, Cullell-Young M, Szyldergemajn S, Ratain MJ. First-in-human phase I study of Lurbinectedin (PM01183) in patients with advanced solid tumors. Clin Cancer Res. 2014 Apr 15;20(8):2205-14. doi: 10.1158/1078-0432.CCR-13-1880. Epub 2014 Feb 21. PubMed PMID: 24563480.
14: Romano M, Frapolli R, Zangarini M, Bello E, Porcu L, Galmarini CM, García-Fernández LF, Cuevas C, Allavena P, Erba E, D’Incalci M. Comparison of in vitro and in vivo biological effects of trabectedin, lurbinectedin (PM01183) and Zalypsis® (PM00104). Int J Cancer. 2013 Nov;133(9):2024-33. doi: 10.1002/ijc.28213. Epub 2013 May 25. PubMed PMID: 23588839.
15: Vidal A, Muñoz C, Guillén MJ, Moretó J, Puertas S, Martínez-Iniesta M, Figueras A, Padullés L, García-Rodriguez FJ, Berdiel-Acer M, Pujana MA, Salazar R, Gil-Martin M, Martí L, Ponce J, Molleví DG, Capella G, Condom E, Viñals F, Huertas D, Cuevas C, Esteller M, Avilés P, Villanueva A. Lurbinectedin (PM01183), a new DNA minor groove binder, inhibits growth of orthotopic primary graft of cisplatin-resistant epithelial ovarian cancer. Clin Cancer Res. 2012 Oct 1;18(19):5399-411. doi: 10.1158/1078-0432.CCR-12-1513. Epub 2012 Aug 15. PubMed PMID: 22896654.
| Clinical data | |
|---|---|
| Pronunciation | LOOR-bih-NEK-teh-din |
| Trade names | Zepzelca |
| Other names | PM-01183 |
| AHFS/Drugs.com | Professional Drug Facts |
| MedlinePlus | a620049 |
| License data | US DailyMed: Lurbinectedin |
| Pregnancy category | US: N (Not classified yet) |
| Routes of administration | Intravenous |
| Drug class | Antineoplastic agent |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1] |
| Identifiers | |
| IUPAC name[show] | |
| CAS Number | 497871-47-3 |
| PubChem CID | 57327016 |
| DrugBank | 12674 |
| ChemSpider | 32701856 |
| UNII | 2CN60TN6ZS |
| KEGG | D11644 |
| ChEMBL | ChEMBL4297516 |
| CompTox Dashboard (EPA) | DTXSID30198065 |
| Chemical and physical data | |
| Formula | C41H44N4O10S |
| Molar mass | 784.88 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES[hide]CC1=CC2=C([C@@H]3[C@@H]4[C@H]5C6=C(C(=C7C(=C6[C@@H](N4[C@H]([C@H](C2)N3C)O)COC(=O)[C@@]8(CS5)C9=C(CCN8)C2=C(N9)C=CC(=C2)OC)OCO7)C)OC(=O)C)C(=C1OC)O | |
| InChI[hide]InChI=1S/C41H44N4O10S/c1-17-11-20-12-25-39(48)45-26-14-52-40(49)41(38-22(9-10-42-41)23-13-21(50-5)7-8-24(23)43-38)15-56-37(31(45)30(44(25)4)27(20)32(47)33(17)51-6)29-28(26)36-35(53-16-54-36)18(2)34(29)55-19(3)46/h7-8,11,13,25-26,30-31,37,39,42-43,47-48H,9-10,12,14-16H2,1-6H3/t25-,26-,30+,31+,37+,39-,41+/m0/s1Key:YDDMIZRDDREKEP-HWTBNCOESA-N |
//////////lurbinectedin, FDA 2020, 2020 APPROVALS, ORPHAN, priority review , ZEPZELCA, Pharma Mar, PM-1183, PM 1183, PM 01183, лурбинектедин , لوربينيكتيدين , 芦比替定
Cc1cc2c(c(c1OC)O)[C@@H]3[C@@H]4[C@H]5c6c(c7c(c(c6OC(=O)C)C)OCO7)[C@@H](N4[C@H]([C@H](C2)N3C)O)COC(=O)[C@@]8(CS5)c9c(c1cc(ccc1[nH]9)OC)CCN8
Naxitamab

(Heavy chain)
QVQLVESGPG VVQPGRSLRI SCAVSGFSVT NYGVHWVRQP PGKGLEWLGV IWAGGITNYN
SAFMSRLTIS KDNSKNTVYL QMNSLRAEDT AMYYCASRGG HYGYALDYWG QGTLVTVSSA
STKGPSVFPL APSSKSTSGG TAALGCLVKD YFPEPVTVSW NSGALTSGVH TFPAVLQSSG
LYSLSSVVTV PSSSLGTQTY ICNVNHKPSN TKVDKRVEPK SCDKTHTCPP CPAPELLGGP
SVFLFPPKPK DTLMISRTPE VTCVVVDVSH EDPEVKFNWY VDGVEVHNAK TKPREEQYNS
TYRVVSVLTV LHQDWLNGKE YKCKVSNKAL PAPIEKTISK AKGQPREPQV YTLPPSRDEL
TKNQVSLTCL VKGFYPSDIA VEWESNGQPE NNYKTTPPVL DSDGSFFLYS KLTVDKSRWQ
QGNVFSCSVM HEALHNHYTQ KSLSLSPGK
(Light chain)
EIVMTQTPAT LSVSAGERVT ITCKASQSVS NDVTWYQQKP GQAPRLLIYS ASNRYSGVPA
RFSGSGYGTE FTFTISSVQS EDFAVYFCQQ DYSSFGQGTK LEIKRTVAAP SVFIFPPSDE
QLKSGTASVV CLLNNFYPRE AKVQWKVDNA LQSGNSQESV TEQDSKDSTY SLSSTLTLSK
ADYEKHKVYA CEVTHQGLSS PVTKSFNRGE C
(Disulfide bridge: H22-H95, H146-H202, H222-L211, H228-H’228, H231-H’231, H263-H323, H369-H427, H’22-H’95, H’146-H’202, H’222-L’211, H’263-H’323, H’369-H’427, L23-L88, L131-L191, L’23-L’88, L’131-L’191)
Naxitamab
ナキシタマブ;
Antineoplastic, Anti-GD2 antibody
| Formula | C6414H9910N1718O1996S44 |
|---|---|
| CAS | 1879925-92-4 |
| Mol weight | 144434.4882 |
FDA APPROVED 2020/11/25, Danyelza
FDA grants accelerated approval to naxitamab for high-risk neuroblastoma in bone or bone marrow
On November 25, 2020, the Food and Drug Administration granted accelerated approval to naxitamab (DANYELZA, Y-mAbs Therapeutics, Inc.) in combination with granulocyte-macrophage colony-stimulating factor (GM-CSF) for pediatric patients one year of age and older and adult patients with relapsed or refractory high-risk neuroblastoma in the bone or bone marrow demonstrating a partial response, minor response, or stable disease to prior therapy.
Efficacy was evaluated in patients with relapsed or refractory neuroblastoma in the bone or bone marrow enrolled in two single-arm, open-label trials: Study 201 (NCT 03363373) and Study 12-230 (NCT 01757626). Patients with progressive disease following their most recent therapy were excluded. Patients received 3 mg/kg naxitamab administered as an intravenous infusion on days 1, 3, and 5 of each 4-week cycle in combination with GM-CSF subcutaneously at 250 µg/m2/day on days -4 to 0 and at 500 µg/m2/day on days 1 to 5. At the investigator’s discretion, patients were permitted to receive pre-planned radiation to the primary disease site in Study 201 and radiation therapy to non-target bony lesions or soft tissue disease in Study 12-230.
The main efficacy outcome measures were confirmed overall response rate (ORR) per the revised International Neuroblastoma Response Criteria (INRC) and duration of response (DOR). Among 22 patients treated in the multicenter Study 201, the ORR was 45% (95% CI: 24%, 68%) and 30% of responders had a DOR greater or equal to 6 months. Among 38 patients treated in the single-center Study 12-230, the ORR was 34% (95% CI: 20%, 51%) with 23% of patients having a DOR greater or equal to 6 months. For both trials, responses were observed in either the bone, bone marrow or both.
The prescribing information contains a Boxed Warning stating that naxitamab can cause serious infusion-related reactions and neurotoxicity, including severe neuropathic pain, transverse myelitis and reversible posterior leukoencephalopathy syndrome (RPLS). To mitigate these risks, patients should receive premedication prior to each naxitamab infusion and be closely monitored during and for at least two hours following completion of each infusion.
The most common adverse reactions (incidence ≥25% in either trial) in patients receiving naxitamab were infusion-related reactions, pain, tachycardia, vomiting, cough, nausea, diarrhea, decreased appetite, hypertension, fatigue, erythema multiforme, peripheral neuropathy, urticaria, pyrexia, headache, injection site reaction, edema, anxiety, localized edema, and irritability. The most common Grade 3 or 4 laboratory abnormalities (≥5% in either trial) were decreased lymphocytes, decreased neutrophils, decreased hemoglobin, decreased platelet count, decreased potassium, increased alanine aminotransferase, decreased glucose, decreased calcium, decreased albumin, decreased sodium and decreased phosphate.
The recommended naxitamab dose is 3 mg/kg/day (up to 150 mg/day) on days 1, 3, and 5 of each treatment cycle, administered after dilution as an intravenous infusion in combination with GM-CSF, subcutaneously at 250 µg/m2/day on days -4 to 0 and at 500 µg/m2/day on days 1 to 5. Treatment cycles are repeated every 4 to 8 weeks.
View full prescribing information for DANYELZA. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761171lbl.pdf
This review used the Real-Time Oncology Review (RTOR) pilot program and the Assessment Aid, a voluntary submission from the applicant to facilitate the FDA’s assessment.
This application was granted accelerated approval based on overall response rate and duration of response. Continued approval may be contingent upon verification and description of clinical benefit in confirmatory trials.
This application was granted priority review, breakthrough therapy, and orphan drug designation. A priority review voucher was issued for this rare pediatric disease product application. A description of FDA expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.
////////////Naxitamab, priority review, breakthrough therapy, orphan drug, FDA 2020, 2020 APPROVALS, Danyelza, MONOCLONAL ANTIBODY, PEPTIDE, ナキシタマブ,
Ansuvimab-zykl

Ansuvimab-zykl
FDA APPROVED, 12/21/2020, EBANGA
To treat ebola
https://www.fda.gov/drugs/drug-safety-and-availability/fda-approves-treatment-ebola-virus
The U.S. Food and Drug Administration approved Ebanga (Ansuvimab-zykl), a human monoclonal antibody, for the treatment for Zaire ebolavirus (Ebolavirus) infection in adults and children. Ebanga blocks binding of the virus to the cell receptor, preventing its entry into the cell.
Zaire ebolavirus is one of four Ebolavirus species that can cause a potentially fatal human disease. It is transmitted through blood, body fluids, and tissues of infected people or wild animals, and through surfaces and materials, such as bedding and clothing, contaminated with these fluids. Individuals who care for people with the disease, including health care workers who do not use correct infection control precautions, are at the highest risk for infection.
During an Ebola outbreak in the Democratic Republic of the Congo (DRC) in 2018-2019, Ebanga was evaluated in a clinical trial (the PALM trial). The PALM trial was led by the U.S. National Institutes of Health and the DRC’s Institut National de Recherche Biomédicale with contributions from several other international organizations and agencies.
In the PALM trial, the safety and efficacy of Ebanga was evaluated in a multi-center, open-label, randomized controlled trial. 174 participants (120 adults and 54 pediatric patients) with confirmed Ebolavirus infection received Ebanga intravenously as a single 50 mg/kg infusion and 168 participants (135 adults and 33 pediatric patients) received an investigational control. The primary efficacy endpoint was 28-day mortality. The primary analysis population was all patients who were randomized and concurrently eligible to receive either Ebanga or the investigational control during the same time period of the trial. Of the 174 patients who received Ebanga, 35.1% died after 28 days, compared to 49.4% of the 168 patients who received a control.
The most common symptoms experienced while receiving Ebanga include: fever, tachycardia (fast heart rate), diarrhea, vomiting, hypotension (low blood pressure), tachypnea (fast breathing) and chills; however, these are also common symptoms of Ebolavirus infection. Hypersensitivity, including infusion-related events, can occur in patients taking Ebanga, and treatment should be discontinued in the event of a hypersensitivity reaction.
Patients who receive Ebanga should avoid the concurrent administration of a live virus vaccine against Ebolavirus. There is the potential for Ebanga to inhibit replication of a live vaccine virus and possibly reduce the efficacy of this vaccine.
Ebanga was granted an Orphan Drug designation, which provides incentives to assist and encourage drug development for rare diseases. Additionally, the agency granted Ebanga a Breakthrough Therapy designation.
FDA granted the approval to Ridgeback Biotherapeutics, LP.
Ansuvimab, sold under the brand name Ebanga, is a monoclonal antibody medication for the treatment of Zaire ebolavirus (Ebolavirus) infection.[1][2]
The most common symptoms include fever, tachycardia (fast heart rate), diarrhea, vomiting, hypotension (low blood pressure), tachypnea (fast breathing) and chills; however, these are also common symptoms of Ebolavirus infection.[1]
Ansuvimab was approved for medical use in the United States in December 2020.[1][2]
Chemistry
The drug is composed of a single monoclonal antibody (mAb) and was initially isolated from immortalized B-cells that were obtained from a survivor of the 1995 outbreak of Ebola virus disease in Kikwit, Democratic Republic of Congo.[3] In work supported by the United States National Institutes of Health and the Defense Advanced Projects Agency, the heavy and light chain sequences of ansuvimab mAb was cloned into CHO cell lines and initial production runs were produced by Cook Phamica d.b.a. Catalent under contract of Medimmune.[4][5]
Mechanism of action
Neutralization
Ansuvimab is a monoclonal antibody therapy that is infused intravenously into patients with Ebola virus disease. Ansuvimab is a neutralizing antibody,[3] meaning it binds to a protein on the surface of Ebola virus that is required to infect cells. Specifically, ansuvimab neutralizes infection by binding to a region of the Ebola virus envelope glycoprotein that, in the absence of ansuvimab, would interact with virus’s cell receptor protein, Niemann-Pick C1 (NPC1).[6][7][8] This “competition” by ansuvimab prevents Ebola virus from binding to NPC1 and “neutralizes” the virus’s ability to infect the targeted cell.[6]
Effector function
Antibodies have antigen-binding fragment (Fab) regions and constant fragment (Fc) regions. The Neutralization of virus infection occurs when the Fab regions of antibodies binds to virus antigen(s) in a manner that blocks infection. Antibodies are also able to “kill” virus particles directly and/or kill infected cells using antibody-mediated “effector functions” such as opsonization, complement-dependent cytotoxicity, antibody-dependent cell-mediated cytotoxicity and antibody-dependent phagocytosis. These effector functions are contained in the Fc region of antibodies, but is also dependent on binding of the Fab region to antigen. Effector functions also require the use of complement proteins in serum or Fc-receptor on cell membranes. Ansuvimab has been found to be capable of killing cells by antibody-dependent cell-mediated cytotoxicity.[3] Other functional killing tests have not been performed.
History
Ansuvimab is a monoclonal antibody that is being evaluated as a treatment for Ebola virus disease.[9] Its discovery was led by the laboratory of Nancy Sullivan at the United States National Institute of Health Vaccine Research Center and J. J. Muyembe-Tamfum from the Institut National pour la Recherche Biomedicale (INRB) in the Democratic Republic of Congo, working in collaboration with the Institute of Biomedical Research and the United States Army Medical Research Institute of Infectious Diseases.[3][10] Ansuvimab was isolated from the blood of a survivor of the 1995 outbreak of Ebola virus disease in Kikwit, Democratic Republic of Congo roughly ten years later.[3]
In 2018, a Phase 1 clinical trial of ansuvimab was conducted by Martin Gaudinski within the Vaccine Research Center Clinical Trials Program that is led by Julie E. Ledgerwood.[5][4][11] Ansuvimab is also being evaluated during the 2018 North Kivu Ebola outbreak.[12]
Ansuvimab has also shown success with lowering the mortality rate from ~70% to about 34%. In August 2019, Congolese health authorities, the World Health Organization, and the U.S. National Institutes of Health promoted the use of ansuvimab, alongside REGN-EB3, a similar Regeneron-produced monoclonal antibody treatment, over other treatments yielding higher mortality rates, after ending clinical trials during the outbreak.[13][14]
Discovery
A 2016 paper describes the efforts of how ansuvimab was originally developed as part of research efforts lead by Dr. Nancy Sullivan at the United States National Institute of Health Vaccine Research Center and Dr. J. J. Muyembe-Tamfum from the Institut National de Recherche Biomedicale (INRB) in the Democratic Republic of Congo.[3][10] This collaborative effort also involved researchers from Institute of Biomedical Research and the United States Army Medical Research Institute of Infectious Diseases.[3][10] A survivor from the 1995 outbreak of Ebola virus disease in Kikwit, Democratic Republic of Congo donated blood to the project that began roughly ten years after they had recovered.[3] Memory B cells isolated from the survivor’s blood were immortalized, cultured and screened for their ability to produce monoclonal antibodies that reacted with the glycoprotein of Ebola virus. Ansuvimab was identified from one of these cultures and the antibody heavy and light chain gene sequences were sequenced from the cells.[3] These sequences were then cloned into recombinant DNA plasmids and purified antibody protein for initial studies was produced in cells derived from HEK 293 cells.[3]
Ansuvimab and mAb100 combination
In an experiment described in the 2016 paper, rhesus macaques were infected with Ebola virus and treated with a combination of ansuvimab and another antibody isolated from the same subject, mAb100. Three doses of the combination were given once a day starting 1 day after the animals were infected. The control animal died and the treated animals all survived.[3]
Ansuvimab monotherapy
In a second experiment described in the 2016 paper, rhesus macaques were infected with Ebola virus and only treated with ansuvimab. Three doses of ansuvimab were given once a day starting 1 day or 5 days after the animals were infected. The control animals died and the treated animals all survived.[3] Unpublished data referred to in a publication of the 2018 Phase I clinical trial results of ansuvimab, reported that a single infusion of ansuvimab provided full protection of rhesus macaques and was the basis of the dosing used for human studies.[5][4]
Development
Ansuvimab was developed by the Vaccine Research Center with support of the United States National Institutes of Health and the Defense Advanced Projects Agency. The heavy and light chain sequences of ansuvimab mAb were cloned into CHO cell lines to enable large-scale production of antibody product for use in humans.[4][5]
Human safety testing
In early 2018,[9] a Phase 1 clinical trial of ansuvimab’s safety, tolerability and pharmacokinetics was conducted by Dr. Martin Gaudinski within the Vaccine Research Center Clinical Trials Program that is led by Dr. Julie E. Ledgerwood.[5][4][11] The study was performed in the United States at the NIH Clinical Center and tested single dose infusions of ansuvimab infused over 30 minutes. The study showed that ansuvimab was safe, had minimal side effects and had a half-life of 24 days.[5][4]
Ridgeback Biotherapeutics
A license for ansuvimab was obtained by Ridgeback Biotherapeutics in 2018, from the National Institutes of Health–National Institute of Allergy and Infectious Diseases.[15] Ansuvimab was given orphan drug status in May 2019 and March 2020.[16][17][18]
Experimental use in the Democratic Republic of Congo
During the 2018 Équateur province Ebola outbreak, ansuvimab was requested by the Democratic Republic of Congo (DRC) Ministry of Public Health. Ansuvimab was approved for compassionate use by the World Health Organization MEURI ethical protocol and at DRC ethics board. Ansuvimab was sent along with other therapeutic agents to the outbreak sites.[19][20][11] However, the outbreak came to a conclusion before any therapeutic agents were given to patients.[11]
Approximately one month following the conclusion of the Équateur province outbreak, a distinct outbreak was noted in Kivu in the DRC (2018–20 Kivu Ebola outbreak). Once again, ansuvimab received approval for compassionate use by WHO MEURI and DRC ethic boards and has been given to many patients under these protocols.[11] In November 2018, the Pamoja Tulinde Maisha (PALM [together save lives]) open-label randomized clinical control trial was begun at multiple treatment units testing ansuvimab, REGN-EB3 and remdesivir to ZMapp. Despite the difficulty of running a clinical trial in a conflict zone, investigators have enrolled 681 patients towards their goal of 725. An interim analysis by the Data Safety and Monitoring Board (DSMB) of the first 499 patient found that ansuvimab and REGN-EB3 were superior to the comparator ZMapp. Overall mortality of patients in the ZMapp and remdesivir groups were 49% and 53% compared to 34% and 29% for ansuvimab and REGN-EB3. When looking at patients who arrived early after disease symptoms appeared, survival was 89% for ansuvimab and 94% for REGN-EB3. While the study was not powered to determine whether there is any difference between REGN-EB3 and ansuvimab, the survival difference between those two therapies and ZMapp was significant. This led to the DSMB halting the study and PALM investigators dropping the remdesivir and ZMapp arms from the clinical trial. All patients in the outbreak who elect to participate in the trial will now be given either ansuvimab or REGN-EB3.[21][22][13][12]
In October 2020, the U.S. Food and Drug Administration (FDA) approved atoltivimab/maftivimab/odesivimab (Inmazeb, formerly REGN-EB3) with an indication for the treatment of infection caused by Zaire ebolavirus.[23]
FDA approves ansuvimab-zykl for Ebola virus infection
DECEMBER 21, 2020 BY JANICE REICHERThttps://www.antibodysociety.org/antibody-therapeutic/fda-approves-ansuvimab-zykl-for-ebola-virus-infection/embed/#?secret=zWW0Sr0BdW
On December 21, 2020, the US Food and Drug Administration approved Ebanga (ansuvimab-zykl) for the treatment for Zaire ebolavirus (Ebolavirus) infection in adults and children. Ebanga had been granted US Orphan Drug designation and Breakthrough Therapy designations. Ansuvimab is a human IgG1 monoclonal antibody that binds and neutralizes the virus.
The safety and efficacy of Ebanga were evaluated in the multi-center, open-label, randomized controlled PALM trial. In this study, 174 participants (120 adults and 54 pediatric patients) with confirmed Ebolavirus infection received Ebanga intravenously as a single 50 mg/kg infusion and 168 participants (135 adults and 33 pediatric patients) received an investigational control. The primary efficacy endpoint was 28-day mortality. Of the 174 patients who received Ebanga, 35.1% died after 28 days, compared to 49.4% of the 168 patients who received a control.
Ebanga is the 12th antibody therapeutic to be granted a first approval in the US or EU during 2020.
The Antibody Society maintains a comprehensive table of approved monoclonal antibody therapeutics and those in regulatory review in the EU or US. The table, which is located in the Web Resources section of the Society’s website, can be downloaded in Excel format.
References
- ^ Jump up to:a b c d “FDA Approves Treatment for Ebola Virus”. U.S. Food and Drug Administration. 21 December 2020. Retrieved 23 December 2020. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b “Ridgeback Biotherapeutics LP Announces the Approval of Ebanga for Ebola” (Press release). Ridgeback Biotherapeutics LP. 22 December 2020. Retrieved 23 December 2020– via Business Wire.
- ^ Jump up to:a b c d e f g h i j k l Corti D, Misasi J, Mulangu S, Stanley DA, Kanekiyo M, Wollen S, et al. (March 2016). “Protective monotherapy against lethal Ebola virus infection by a potently neutralizing antibody”. Science. 351 (6279): 1339–42. Bibcode:2016Sci…351.1339C. doi:10.1126/science.aad5224. PMID 26917593.
- ^ Jump up to:a b c d e f Clinical trial number NCT03478891 for “Safety and Pharmacokinetics of a Human Monoclonal Antibody, VRC-EBOMAB092-00-AB (MAb114), Administered Intravenously to Healthy Adults” at ClinicalTrials.gov
- ^ Jump up to:a b c d e f Gaudinski MR, Coates EE, Novik L, Widge A, Houser KV, Burch E, et al. (March 2019). “Safety, tolerability, pharmacokinetics, and immunogenicity of the therapeutic monoclonal antibody ansuvimab targeting Ebola virus glycoprotein (VRC 608): an open-label phase 1 study”. Lancet. 393 (10174): 889–898. doi:10.1016/S0140-6736(19)30036-4. PMC 6436835. PMID 30686586.
- ^ Jump up to:a b Misasi J, Gilman MS, Kanekiyo M, Gui M, Cagigi A, Mulangu S, et al. (March 2016). “Structural and molecular basis for Ebola virus neutralization by protective human antibodies”. Science. 351 (6279): 1343–6. Bibcode:2016Sci…351.1343M. doi:10.1126/science.aad6117. PMC 5241105. PMID 26917592.
- ^ Côté M, Misasi J, Ren T, Bruchez A, Lee K, Filone CM, et al. (August 2011). “Small molecule inhibitors reveal Niemann-Pick C1 is essential for Ebola virus infection”. Nature. 477 (7364): 344–8. Bibcode:2011Natur.477..344C. doi:10.1038/nature10380. PMC 3230319. PMID 21866101.
- ^ Carette JE, Raaben M, Wong AC, Herbert AS, Obernosterer G, Mulherkar N, et al. (August 2011). “Ebola virus entry requires the cholesterol transporter Niemann-Pick C1”. Nature. 477 (7364): 340–3. Bibcode:2011Natur.477..340C. doi:10.1038/nature10348. PMC 3175325. PMID 21866103.
- ^ Jump up to:a b “NIH begins testing Ebola treatment in early-stage trial”. National Institutes of Health (NIH). 2018-05-23. Retrieved 2018-10-15.
- ^ Jump up to:a b c Hayden EC (2016-02-26). “Ebola survivor’s blood holds promise of new treatment”. Nature. doi:10.1038/nature.2016.19440. ISSN 1476-4687.
- ^ Jump up to:a b c d e “NIH VideoCast – CC Grand Rounds: Response to an Outbreak: Ebola Virus Monoclonal Antibody (mAb114) Rapid Clinical Development”. videocast.nih.gov. Retrieved 2019-08-09.
- ^ Jump up to:a b Kingsley-Hall A. “Congo’s experimental mAb114 Ebola treatment appears successful: authorities | Central Africa”. http://www.theafricareport.com. Retrieved 2018-10-15.
- ^ Jump up to:a b McNeil DG (12 August 2019). “A Cure for Ebola? Two New Treatments Prove Highly Effective in Congo”. The New York Times. Retrieved 13 August 2019.
- ^ Molteni M (12 August 2019). “Ebola is Now Curable. Here’s How The New Treatments Work”. Wired. Retrieved 13 August 2019.
- ^ “Ridgeback Biotherapeutics LP announces licensing of mAb114, an experimental Ebola treatment, from the National Institute of Allergy and Infectious Diseases” (Press release). Ridgeback Biotherapeutics LP. Retrieved 2019-08-17 – via PR Newswire.
- ^ “Ansuvimab Orphan Drug Designations and Approvals”. accessdata.fda.gov. 8 May 2019. Retrieved 24 December 2020.
- ^ “Ansuvimab Orphan Drug Designations and Approvals”. accessdata.fda.gov. 30 March 2020. Retrieved 24 December 2020.
- ^ “Ridgeback Biotherapeutics LP Announces Orphan Drug Designation for mAb114”(Press release). Ridgeback Biotherapeutics LP. Retrieved 2019-08-17 – via PR Newswire.
- ^ Check Hayden, Erika (May 2018). “Experimental drugs poised for use in Ebola outbreak”. Nature. 557 (7706): 475–476. Bibcode:2018Natur.557..475C. doi:10.1038/d41586-018-05205-x. ISSN 0028-0836. PMID 29789732.
- ^ WHO: Consultation on Monitored Emergency Use of Unregistered and Investigational Interventions for Ebola virus Disease. https://www.who.int/emergencies/ebola/MEURI-Ebola.pdf
- ^ Mole B (2019-08-13). “Two Ebola drugs boost survival rates, according to early trial data”. Ars Technica. Retrieved 2019-08-17.
- ^ “Independent monitoring board recommends early termination of Ebola therapeutics trial in DRC because of favorable results with two of four candidates”. National Institutes of Health (NIH). 2019-08-12. Retrieved 2019-08-17.
- ^ “FDA Approves First Treatment for Ebola Virus”. U.S. Food and Drug Administration(FDA) (Press release). 14 October 2020. Retrieved 14 October 2020. This article incorporates text from this source, which is in the public domain.
External links
- “Ansuvimab”. Drug Information Portal. U.S. National Library of Medicine.
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | Zaire ebolavirus |
| Clinical data | |
| Trade names | Ebanga |
| Other names | Ansuvimab-zykl, mAb114 |
| License data | US DailyMed: Ansuvimab |
| Routes of administration | Intravenous |
| Drug class | Monoclonal antibody |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1] |
| Identifiers | |
| CAS Number | 2375952-29-5 |
| DrugBank | DB16385 |
| UNII | TG8IQ19NG2 |
| KEGG | D11875 |
| Chemical and physical data | |
| Formula | C6368H9924N1724O1994S44 |
| Molar mass | 143950.15 g·mol−1 |
//////////Ansuvimab-zykl , EBANGA, FDA 2020, 2020 APPROVALS, MONOCLONAL ANTIBODY, Orphan Drug designation, , Breakthrough Therapy designation , Ridgeback Biotherapeutics,
Amikacin sulfate
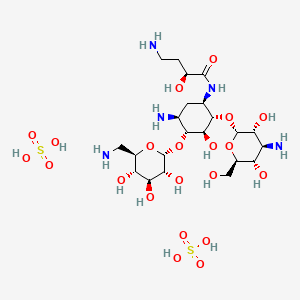

Amikacin sulfate
アミカシン硫酸塩 , BB K 8
| Formula | C22H43N5O13. 2H2SO4 |
|---|---|
| CAS | 39831-55-5FREE 37517-28-5 |
| Mol weight | 781.7595 |
EU APPROVED, 2020/10/27, Arikayce liposomal
Antibacterial, Protein biosynthesis inhibitor
(2S)-4-amino-N-[(1R,2S,3S,4R,5S)-5-amino-2-[(2S,3R,4S,5S,6R)-4-amino-3,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-4-[(2R,3R,4S,5S,6R)-6-(aminomethyl)-3,4,5-trihydroxyoxan-2-yl]oxy-3-hydroxycyclohexyl]-2-hydroxybutanamide;sulfuric acid AmikacinCAS Registry Number: 37517-28-5
CAS Name:O-3-Amino-3-deoxy-a-D-glucopyranosyl-(1®6)-O-[6-amino-6-deoxy-a-D-glucopyranosyl-(1®4)]-N1-[(2S)-4-amino-2-hydroxy-1-oxobutyl]-2-deoxy-D-streptamine
Additional Names: 1-N-[L(-)-4-amino-2-hydroxybutyryl]kanamycin AMolecular Formula: C22H43N5O13Molecular Weight: 585.60Percent Composition: C 45.12%, H 7.40%, N 11.96%, O 35.52%
Literature References: Semisynthetic aminoglycoside antibiotic derived from kanamycin A. Prepn: Kawaguchi et al.,J. Antibiot.25, 695 (1972); H. Kawaguchi, T. Naito, DE2234315; H. Kawaguchi et al.,US3781268 (both 1973 to Bristol-Myers). Biological formation from kanamycin A: L. M. Cappelletti, R. Spagnoli, J. Antibiot.36, 328 (1983). Microbiological evaluation: Price et al.,ibid.25, 709 (1972). Pharmacokinetics: Cabana, Taggart, Antimicrob. Agents Chemother.3, 478 (1973). In vitro studies: Yu, Washington, ibid.4, 133 (1973); Bodey, Stewart, ibid. 186. Pharmacology in humans: Bodey et al.,ibid.5, 508 (1974). Toxicity studies: Fujisawa et al.,J. Antibiot.27, 677 (1974). Review: K. A. Kerridge in Pharmacological and Biochemical Properties of Drug Substancesvol. 1, M. E. Goldberg, Ed. (Am. Pharm. Assoc., Washington, DC, 1977) pp 125-153. Comprehensive description: P. M. Monteleone et al.,Anal. Profiles Drug Subs.12, 37-71 (1983).Properties: White crystalline powder from methanol-isopropanol, mp 203-204° (sesquihydrate). [a]D23 +99° (c = 1.0 in water). LD50 in mice of solns pH 6.6, pH 7.4 (mg/kg): 340, 560 i.v. (Kawaguchi).Melting point: mp 203-204° (sesquihydrate)Optical Rotation: [a]D23 +99° (c = 1.0 in water)Toxicity data: LD50 in mice of solns pH 6.6, pH 7.4 (mg/kg): 340, 560 i.v. (Kawaguchi)
Derivative Type: SulfateCAS Registry Number: 39831-55-5Trademarks: Amiglyde-V (Fort Dodge); Amikin (BMS); Amiklin (BMS); BB-K8 (BMS); Biklin (BMS); Lukadin (San Carlo); Mikavir (Salus); Novamin (BMS); Pierami (Fournier)Molecular Formula: C22H43N5O13.2H2SO4Molecular Weight: 781.76Percent Composition: C 33.80%, H 6.06%, N 8.96%, O 42.98%, S 8.20%Properties: Amorphous form, dec 220-230°. [a]D22 +74.75° (water).Optical Rotation: [a]D22 +74.75° (water)
Therap-Cat: Antibacterial.Therap-Cat-Vet: Antibacterial.Keywords: Antibacterial (Antibiotics); Aminoglycosides.
Amikacin Sulfate is the sulfate salt of amikacin, a broad-spectrum semi-synthetic aminoglycoside antibiotic, derived from kanamycin with antimicrobial property. Amikacin irreversibly binds to the bacterial 30S ribosomal subunit, specifically in contact with 16S rRNA and S12 protein within the 30S subunit. This leads to interference with translational initiation complex and misreading of mRNA, thereby hampering protein synthesis and resulting in bactericidal effect. This agent is usually used in short-term treatment of serious infections due to susceptible strains of Gram-negative bacteria.Amikacin disulfate is an aminoglycoside sulfate salt obtained by combining amikacin with two molar equivalents of sulfuric acid. It has a role as an antibacterial drug, an antimicrobial agent and a nephrotoxin. It contains an amikacin(4+).
Amikacin sulfate is semi-synthetic aminoglycoside antibiotic derived from kanamycin. It is C22H43N5O13•2H2SO4•O-3-amino-3-deoxy-α-D-glucopyranosyl-(1→4)-O-[6-amino-6-deoxy-α-Dglucopyranosyl-( 1→6)]-N3-(4-amino-L-2-hydroxybutyryl)-2-deoxy-L-streptamine sulfate (1:2)
|
M.W. 585.61The dosage form is supplied as a sterile, colorless to light straw colored solution for intramuscular or intravenous use. Each mL contains 250 mg amikacin (as the sulfate), 0.66% sodium metabisulfite, 2.5% sodium citrate dihydrate with pH adjusted to 4.5 with sulfuric acid.
Amikacin is an antibiotic medication used for a number of bacterial infections.[4] This includes joint infections, intra-abdominal infections, meningitis, pneumonia, sepsis, and urinary tract infections.[4] It is also used for the treatment of multidrug-resistant tuberculosis.[5] It is used by injection into a vein using an IV or into a muscle.[4]
Amikacin, like other aminoglycoside antibiotics, can cause hearing loss, balance problems, and kidney problems.[4] Other side effects include paralysis, resulting in the inability to breathe.[4] If used during pregnancy it may cause permanent deafness in the baby.[4][1] Amikacin works by blocking the function of the bacteria’s 30S ribosomal subunit, making it unable to produce proteins.[4]
Amikacin was patented in 1971, and came into commercial use in 1976.[6][7] It is on the World Health Organization’s List of Essential Medicines.[8] It is derived from kanamycin.[4]
Medical uses
Amikacin is most often used for treating severe infections with multidrug-resistant, aerobic Gram-negative bacteria, especially Pseudomonas, Acinetobacter, Enterobacter, E. coli, Proteus, Klebsiella, and Serratia.[9] The only Gram-positive bacteria that amikacin strongly affects are Staphylococcus[9] and Nocardia.[10] Amikacin can also be used to treat non-tubercular mycobacterial infections and tuberculosis (if caused by sensitive strains) when first-line drugs fail to control the infection.[4] It is rarely used alone.[11]
It is often used in the following situations:[4]
- Bronchiectasis[12]
- Bone and joint infections
- Granulocytopenia, when combined with ticarcillin, in people with cancer[13]
- Intra-abdominal infections (such as peritonitis) as an adjunct to other medicines, like clindamycin, metronidazole, piperacillin/tazobactam, or ampicillin/sulbactam
- Meningitis:
- for meningitis by E. coli, as an adjunct to imipenem
- for meningitis caused by Pseudomonas, as an adjunct to meropenem
- for meningitis caused by Acetobacter, as an adjunct to imipenem or colistin
- for neonatal meningitis caused by Streptococcus agalactiae or Listeria monocytogenes, as an adjunct to ampicillin
- for neonatal meningitis caused by Gram negative bacteria such as E. coli, as adjunct to a 3rd-generation cephalosporin
- Mycobacterial infections, including as a second-line agent for active tuberculosis.[14] It is also used for infections by Mycobacterium avium, M. abcessus, M. chelonae, and M. fortuitum.
- Rhodococcus equi, which causes an infection resembling tuberculosis
- Respiratory tract infections, including as an adjunct to beta-lactams or carbapenem for hospital-acquired pneumonia
- Sepsis, including that in neonates,[9] as an adjunct to beta-lactams or carbapenem
- Skin and suture-site infections[9]
- Urinary tract infections that are caused by bacteria resistant to less toxic drugs (often by Enterobacteriaceae or P. aeruginosa)
Amikacin may be combined with a beta-lactam antibiotic for empiric therapy for people with neutropenia and fever.[4]
Available forms[
Amikacin may be administered once or twice a day and is usually given by the intravenous or intramuscular route, though it can be given via nebulization. There is no oral form available, as amikacin is not absorbed orally. In people with kidney failure, dosage must be adjusted according to the creatinine clearance, usually by reducing the dosing frequency.[9] In people with a CNS infection such as meningitis, amikacin can be given intrathecally (by direct injection into the spine) or intraventricularly (by injection into the ventricles of brain).[4]
An liposome inhalation suspension is also available and approved to treat Mycobacterium avium complex (MAC) in the United States.[15][16] The application for Arikayce was withdrawn in the European Union because the Committee for Medicinal Products for Human Use (CHMP) was of the opinion that the benefits of Arikayce did not outweigh its risks.[17]
Special populations
Amikacin should be used in smaller doses in the elderly, who often have age-related decreases in kidney function, and children, whose kidneys are not fully developed yet. It is considered pregnancy category D in both the United States and Australia, meaning they have a probability of harming the fetus.[4] Around 16% of amikacin crosses the placenta; while the half-life of amikacin in the mother is 2 hours, it is 3.7 hours in the fetus.[9] A pregnant woman taking amikacin with another aminoglycoside has a possibility of causing congenital deafness in her child. While it is known to cross the placenta, amikacin is only partially secreted in breast milk.[4]
In general, amikacin should be avoided in infants.[18] Infants also tend to have a larger volume of distribution due to their higher concentration of extracellular fluid, where aminoglycosides reside.[3]
The elderly tend to have amikacin stay longer in their system; while the average clearance of amikacin in a 20-year-old is 6 L/hr, it is 3 L/hr in an 80-year-old.[19]
Clearance is even higher in people with cystic fibrosis.[20]
In people with muscular disorders such as myasthenia gravis or Parkinson’s disease, amikacin’s paralytic effect on neuromuscular junctions can worsen muscle weakness.[4]
Adverse effects
Side-effects of amikacin are similar to those of other aminoglycosides. Kidney damage and ototoxicity (which can lead to hearing loss) are the most important effects, occurring in 1–10% of users.[12] The nephro- and ototoxicity are thought to be due to aminoglycosides’ tendency to accumulate in the kidneys and inner ear.[3]

Diagram of the inner ear. Amikacin causes damage to the cochlea and vestibules.
Amikacin can cause neurotoxicity if used at a higher dose or for longer than recommended. The resulting effects of neurotoxicity include vertigo, numbness, tingling of the skin (paresthesia), muscle twitching, and seizures.[4] Its toxic effect on the 8th cranial nerve causes ototoxicity, resulting in loss of balance and, more commonly, hearing loss.[3] Damage to the cochlea, caused by the forced apoptosis of the hair cells, leads to the loss of high-frequency hearing and happens before any clinical hearing loss can be detected.[9][21] Damage to the ear vestibules, most likely by creating excessive oxidative free radicals. It does so in a time-dependent rather than dose-dependent manner, meaning that risk can be minimized by reducing the duration of use.[22]
Amikacin causes nephrotoxicity (damage to the kidneys), by acting on the proximal renal tubules. It easily ionizes to a cation and binds to the anionic sites of the epithelial cells of the proximal tubule as part of receptor-mediated pinocytosis. The concentration of amikacin in the renal cortex becomes ten times that of amikacin in the plasma;[18] it then most likely interferes with the metabolism of phospholipids in the lysosomes, which causes lytic enzymes to leak into the cytoplasm.[3] Nephrotoxicity results in increased serum creatinine, blood urea nitrogen, red blood cells, and white blood cells, as well as albuminuria (increased output of albumin in the urine), glycosuria (excretion of glucose into the urine), decreased urine specific gravity, and oliguria (decrease in overall urine output).[9][21] It can also cause urinary casts to appear.[3] The changes in renal tubular function also change the electrolyte levels and acid-base balance in the body, which can lead to hypokalemia and acidosis or alkalosis.[22] Nephrotoxicity is more common in those with pre-existing hypokalemia, hypocalcemia, hypomagnesemia, acidosis, low glomerular filtration rate, diabetes mellitus, dehydration, fever, and sepsis, as well as those taking antiprostaglandins.[4][18][3][22] The toxicity usually reverts once the antibiotic course has been completed,[3] and can be avoided altogether by less frequent dosing (such as once every 24 hours rather than once every 8 hours).[18]
Amikacin can cause neuromuscular blockade (including acute muscular paralysis) and respiratory paralysis (including apnea).[4]
Rare side effects (occurring in fewer than 1% of users) include allergic reactions, skin rash, fever, headaches, tremor, nausea and vomiting, eosinophilia, arthralgia, anemia, hypotension, and hypomagnesemia. In intravitreous injections (where amikacin is injected into the eye), macular infarction can cause permanent vision loss.[9][12]
The amikacin liposome inhalation suspension prescribing information includes a boxed warning regarding the increased risk of respiratory conditions including hypersensitivity pneumonitis (inflamed lungs), bronchospasm (tightening of the airway), exacerbation of underlying lung disease and hemoptysis (spitting up blood) that have led to hospitalizations in some cases.[15][16] Other common side effects in patients taking amikacin liposome inhalation suspension are dysphonia (difficulty speaking), cough, ototoxicity (damaged hearing), upper airway irritation, musculoskeletal pain, fatigue, diarrhea and nausea.[15][16]
Contraindications
Amikacin should be avoided in those who are sensitive to any aminoglycoside, as they are cross-allergenic (that is, an allergy to one aminoglycoside also confers hypersensitivity to other aminoglycosides). It should also be avoided in those sensitive to sulfite (seen more among people with asthma),[9] since most amikacin usually comes with sodium metabisulfite, which can cause an allergic reaction.[4]
In general, amikacin should not be used with or just before/after another drug that can cause neurotoxicity, ototoxicity, or nephrotoxicity. Such drugs include other aminoglycosides; the antiviral acyclovir; the antifungal amphotericin B; the antibiotics bacitracin, capreomycin, colistin, polymyxin B, and vancomycin; and cisplatin, which is used in chemotherapy.[4]
Amikacin should not be used with neuromuscular blocking agents, as they can increase muscle weakness and paralysis.[4]
Interactions
Amikacin can be inactivated by other beta-lactams, though not to the extent as other aminoglycosides, and is still often used with penicillins (a type of beta-lactam) to create an additive effect against certain bacteria, and carbapenems, which can have a synergistic against some Gram-positive bacteria. Another group of beta-lactams, the cephalosporins, can increase the nephrotoxicity of aminoglycoside as well as randomly elevating creatinine levels. The antibiotics chloramphenicol, clindamycin, and tetracycline have been known to inactivate aminoglycosides in general by pharmacological antagonism.[4]
The effect of amikacin is increased when used with drugs derived from the botulinum toxin,[12] anesthetics, neuromuscular blocking agents, or large doses of blood that contains citrate as an anticoagulant.[4]
Potent diuretics not only cause ototoxicity themselves, but they can also increase the concentration of amikacin in the serum and tissue, making the ototoxicity even more likely.[4] Quinidine also increases levels of amikacin in the body.[12] The NSAID indomethacin can increase serum aminoglycoside levels in premature infants.[4] Contrast mediums such as ioversol increases the nephrotoxicity and otoxicity caused by amikacin.[12]
Amikacin can decrease the effect certain vaccines, such as the live BCG vaccine (used for tuberculosis), the cholera vaccine, and the live typhoid vaccine by acting as a pharmacological antagonist.[12]
Pharmacology
Mechanism of action

The 30S subunit of the prokaryotic ribosome. The orange represents the 16S rRNA, and the blue represents the various proteins attached.
Amikacin irreversibly binds to 16S rRNA and the RNA-binding S12 protein of the 30S subunit of prokaryotic ribosome and inhibits protein synthesis by changing the ribosome’s shape so that it cannot read the mRNA codons correctly.[9][23] It also interferes with the region that interacts with the wobble base of the tRNA anticodon.[24] It works in a concentration-dependent manner, and has better action in an alkaline environment.[3]
At normal doses, amikacin-sensitive bacteria respond within 24–48 hours.[9]
Resistance
Amikacin evades attacks by all antibiotic-inactivating enzymes that are responsible for antibiotic resistance in bacteria, except for aminoacetyltransferase and nucleotidyltransferase.[25] This is accomplished by the L-hydroxyaminobuteroyl amide (L-HABA) moiety attached to N-1 (compare to kanamycin, which simply has a hydrogen), which blocks the access and decreases the affinity of aminoglycoside-inactivating enzymes.[25][26][27] Amikacin ends up with only one site where these enzymes can attack, while gentamicin and tobramycin have six.[11]
Bacteria that are resistant to streptomycin and capreomycin are still susceptible to amikacin; bacteria that are resistant to kanamycin have varying susceptibility to amikacin. Resistance to amikacin also confers resistance to kanamycin and capreomycin.[28]
Resistance to amikacin and kanamycin in Mycobacterium, the causative agent of tuberculosis, is due to a mutation in the rrs gene, which codes for the 16S rRNA. Mutations such as these reduce the binding affinity of amikacin to the bacteria’s ribosome.[29] Variations of aminoglycoside acetyltransferase (AAC) and aminoglycoside adenylyltransferase (AAD) also confer resistance: resistance in Pseudomonas aeruginosa is caused by AAC(6′)-IV, which also confers resistance to kanamycin, gentamicin, and tobramycin, and resistance in Staphylococcus aureus and S. epidermidis is caused by AAD(4′,4), which also confers resistance to kanamycin, tobramycin, and apramycin.[26] Some strains of S. aureus can also inactivate amikacin by phosphorylating it.[13]
Pharmacokinetics
Amikacin is not absorbed orally and thus must be administered parenterally. It reaches peak serum concentrations in 0.5–2 hours when administered intramuscularly. Less than 11% of the amikacin actually binds to plasma proteins. It is distributed into the heart, gallbladder, lungs, and bones, as well as in bile, sputum, interstitial fluid, pleural fluid, and synovial fluids. It is usually found at low concentrations in the cerebrospinal fluid, except when administered intraventricularly.[4] In infants, amikacin is normally found at 10–20% of plasma levels in the spinal fluid, but the amount reaches 50% in cases of meningitis.[9] It does not easily cross the blood-brain barrier or enter ocular tissue.[3]
While the half-life of amikacin is normally two hours, it is 50 hours in those with end-stage renal disease.[11]
The vast majority (95%) of amikacin from an IM or IV dose is secreted unchanged via glomerular filtration and into the urine within 24 hours.[4][11] Factors that cause amikacin to be excreted via urine include its relatively low molecular weight, high water solubility, and unmetabolized state.[18]
Chemistry
Amikacin is derived from kanamycin A:[30][31]

Veterinary use
While amikacin is only FDA-approved for use in dogs and for intrauterine infection in horses, it is one of the most common aminoglycosides used in veterinary medicine,[32] and has been used in dogs, cats, guinea pigs, chinchillas, hamsters, rats, mice, prairie dogs, cattle, birds, snakes, turtles and tortoises, crocodilians, bullfrogs, and fish.[3][33][34] It is often used for respiratory infections in snakes, bacterial shell disease in turtles, and sinusitis in macaws. It is generally contraindicated in rabbits and hares (though it has still been used) because it harms the balance of intestinal microflora.[3]
In dogs and cats, amikacin is commonly used as a topical antibiotic for ear infections and for corneal ulcers, especially those that are caused by Pseudomonas aeruginosa. The ears are often cleaned before administering the medication, since pus and cellular debris lessen the activity of amikacin.[32] Amikacin is administered to the eye when prepared as an ophthalmic ointment or solution, or when injected subconjunctivally.[35] Amikacin in the eye can be accompanied by cephazolin. Despite its use there amikacin (and all aminoglycosides) are toxic to intraocular structures.[36]
In horses, amikacin is FDA-approved for uterine infections (such as endometriosis and pyometra) when caused by susceptible bacteria.[37] It is also used in topical medication for the eyes and arthroscopic lavage; when combined with a cephalosporin, is used to treat subcutaneous infections that are caused by Staphylococcus. For infections in the limbs or joints, it is often administered with a cephalosporin via limb perfusion directly into the limb or injected into the joint.[32][38] Amikacin is also injected into the joints with the anti-arthritic medication Adequan in order to prevent infection.[39]
Side effects in animals include nephrotoxicity, ototoxicity, and allergic reactions at IM injection sites. Cats tend to be more sensitive to the vestibular damage caused by ototoxicity. Less frequent side effects include neuromuscular blockade, facial edema, and peripheral neuropathy.[3][32]
The half-life in most animals is one to two hours.[40]
Treating overdoses of amikacin requires kidney dialysis or peritoneal dialysis, which reduce serum concentrations of amikacin, and/or penicillins, some of which can form complexes with amikacin that deactivate it.[3]
Liposome inhalation suspension
Amikacin liposome inhalation suspension was the first drug approved under the US limited population pathway for antibacterial and antifungal drugs (LPAD pathway).[15] It also was approved under the accelerated approval pathway.[15] The U.S. Food and Drug Administration (FDA) granted the application for amikacin liposome inhalation suspension fast track, breakthrough therapy, priority review, and qualified infectious disease product (QIDP) designations.[15] The FDA granted approval of Arikayce to Insmed, Inc.[15]
The safety and efficacy of amikacin liposome inhalation suspension, an inhaled treatment taken through a nebulizer, was demonstrated in a randomized, controlled clinical trial where patients were assigned to one of two treatment groups.[15] One group of patients received amikacin liposome inhalation suspension plus a background multi-drug antibacterial regimen, while the other treatment group received a background multi-drug antibacterial regimen alone.[15] By the sixth month of treatment, 29 percent of patients treated with amikacin liposome inhalation suspension had no growth of mycobacteria in their sputum cultures for three consecutive months compared to 9 percent of patients who were not treated with amikacin liposome inhalation suspension.[15]

SYN

SYN
https://www.mdpi.com/1420-3049/22/12/2267/htm
Scheme 1. Original chemical reactions sequence to obtain amikacin by modification of kanamycin A.PATENThttps://patents.google.com/patent/CN105440090A/zh
Amikacin is a semi-synthetic aminoglycoside antibiotic with a broad antibacterial spectrum and strong antibacterial activity against a variety of bacteria; its sulfate has become a clinically commonly used first-line anti-infective drug in the world and continues to Develop new dosage forms and uses.
[0003] Amikacin sulfate is suitable for Pseudomonas aeruginosa and other Pseudomonas, Escherichia coli, Proteus, Klebsiella, Enterobacter, Serratia, Acinetobacter Severe infections caused by other sensitive gram-negative bacilli and Staphylococcus (methicillin-sensitive strains), such as bacteremia or sepsis, bacterial endocarditis, lower respiratory tract infections, bone and joint infections, biliary tract infections, abdominal infections, Complex urinary tract infections, skin and soft tissue infections, etc. Because it is stable to most aminoglycoside inactivating enzymes, it is especially suitable for the treatment of serious infections caused by gram-negative bacilli against kanamycin, gentamicin or tobramycin-resistant strains.
[0004] Amikacin, also known as amikacin, has a molecular weight of 585. The most commonly used synthetic route is a silyl protecting routes, such as the document “amikacin by New Method” (Author: Jiangzhong Liang, Wang Yu; Fine & Specialty Chemicals, 2004, 12 (10), 26- 28) The main process recorded is: (1) Using kanamycin A (KMA) as a raw material to protect the 11 amino groups and hydroxyl groups of kanamycin to obtain methylsilyl kanamycin; (2) ) Using YN-phthalimido-α-hydroxybutyric acid (PHBA) and N-hydroxy-phthalimide (NOP) as raw materials in dicyclohexylcarbodiimide (DCC) The active ester compound is prepared in the presence; (3) acylation (transesterification reaction) with methylsilyl kanamycin and active ester, and then acidolysis and hydrazinolysis reactions to obtain amikacin. As shown in the following route:
[0005] 1. Silanization protection reaction:
[0006]
[0007] 2. Preparation of Living King®:

[0008]

[0009] 3. Acylation reaction:
U
[0011] 4. Acidolysis reaction:
[0012]

[0013] 5. Hydrazine reaction:
[0014]

[0015] The acylation reaction in the above route adopts a transesterification reaction between a silyl group protection reactant and an independently prepared active ester. Due to the active transesterification reaction, a large excess of reactant active ester is needed to improve the reaction yield, and there is an independent unit reaction for preparing active ester, and the raw material N-hydroxy-phthalimide is used. (NOP), increasing the usage amount of reaction solvent, the solvent in the process is volatile, the loss is large, the environment is affected, and the production cost is increased.
[0016] How to find a direct one-step acylation reaction between the silyl group protection and YN-phthalimido-α-hydroxybutyric acid (PHBA), which can not only ensure the synthesis yield, but also reduce the synthesis The steps are easy to operate, and the N-hydroxy-phthalimide (NOP), the raw material for preparing active esters, is no longer used, and the acylation reaction conditions that reduce solvent consumption are a very beneficial synthetic process line.
Example 1
[0046] 600mL of acetonitrile was put into the silanization reaction flask, and 0.1 billion kanamycin A (KMA) was added. After the feeding port was closed and stirred for 10 minutes, hexamethyldisilazane (HMDS) was added. 400mL, heated to reflux, refluxed at 75~80°C for 7hr. Use drinking water to cool the outside of the reaction flask to lower the temperature to below 35°C, and let it stand for natural layering. Separate and collect the lower layer to obtain a silyl group protected product.
[0047] Add 1000mL acetone to the silyl group protection product, start stirring, add 60g γ-N-phthalimido-α-hydroxybutyric acid (PHBA), and then add 2.5g catalyst 4-N, N -Lutidine (DMAP), cooled to -15~-1 (TC〇
[0048] Dissolve 60gN, N-bicyclohexylcarbodiimide with 300mL of acetone, add its flow to the above-mentioned reactant, control the flow rate of 5mL/min, and control the temperature of the reactant to -15~-10°C; the flow is completed Continue the reaction for 1 hour.
[0049] After the completion of the acylation reaction, the material was transferred to the acidolysis bottle, the stirring was turned on, and 400mL of 4.0mol/L hydrochloric acid was added for acidolysis, and the feed solution was pH 3.0 and allowed to stand for 60 minutes. The lower acid hydrolysis solution was collected by suction filtration, and the filter cake (DCU) was washed three times with 150 mL of deionized water, and the washing water was incorporated into the acid hydrolysis solution.
[0050] The acid hydrolysate was transferred to a distillation flask. Turn on the vacuum, the degree of vacuum: <0.07Mpa, the distillation temperature is controlled at 40~68°C, the distillation time: 2.5 hours after the distillation is complete; transfer the PKS concentrate in the distillation flask into the hydrazinolysis flask, and add 7.Omol/ L ammonia water 200mL, so that the pH of the material solution reaches 8.0; add 180mL hydrazine hydrate, increase the temperature, the temperature is 85~95°C, hydrazinolysis 3.5 hours, use drinking water to cool outside the hydrazinolysis bottle, and cool to 40 °C.
[0051] Add 4.0111〇1/1 hydrochloric acid 12001^ to the hydrazinolysis bottle, adjust? !1 is 4.0. Turn on the vacuum filtration. With 5001 ^ deionized water top washing filter, 1510mL of amikacin synthetic solution, amikacin content 5.8% (g/mL), relative to the synthetic yield of kanamycin A is 72.5 %.
[0052] Example 2
[0053] 600mL of acetonitrile was put into the silanization reaction flask, 0.1 billion kanamycin A (KMA) was added, the feeding port was closed and stirred for 10 minutes, and hexamethyldisilazane (HMDS) was added 500mL, heated to reflux, refluxed at 75~80°C for 8hr. After the reaction is completed, cool down to 40°C with drinking water and let stand for natural layering. Separate and collect the lower layer to obtain a silyl group protected product.
[0054] Add 1000mL acetone to the silyl group protection product, start stirring, add 70g Y-N-phthalimido-α-hydroxybutyric acid (PHBA), and add 3.0g catalyst 1-hydroxybenzo Triazole (HOBT), after the material is dissolved, the temperature is reduced to -15~-10°C.
[0055] Dissolve 70g of N,N-bicyclohexylcarbodiimide with 300mL of acetone, add its flow to the above-mentioned reactants, control the flow rate of 6mL/min, and control the temperature of the reactants from -15 to -10°C; the flow is completed Continue the reaction for 1.5 hours.
[0056] After the acylation reaction is completed, the material is transferred to the acidolysis bottle, the stirring is turned on, and 6.0m〇l/L hydrochloric acid 300mL is added for acidolysis, the feed solution is pH 2.0, and the acidolysis is completed, and it is allowed to stand for 50 minutes. The lower acid hydrolysis solution was collected by suction filtration, the filter cake (DCU) was washed three times with 200 mL of deionized water, and the washing water was incorporated into the acid hydrolysis solution.
[0057] Transfer the acid hydrolysate into a distillation flask. Turn on the vacuum, vacuum degree: <-0.07Mpa, the distillation temperature is controlled at 40~68°C, the distillation time is 3.0 hours, except for acetone. After the distillation is completed, transfer the PKS concentrate in the distillation flask into the hydrazinolysis flask, add 150 mL of 10.0 mol/L ammonia water, the pH of the feed solution is 8.5; add 200 mL of hydrazine hydrate, increase the temperature at 85~95 °C, hydrazinolysis 4 After hours, use drinking water to cool down outside the hydrazinolysis bottle to 45°C.
[0058] Add 6.0111〇1/1 hydrochloric acid 10001^ to the hydrazinolysis bottle, adjust? !1 is 3.0. Turn on the vacuum filtration, use 8001^ deionized water to wash and filter the fish, to obtain 1620 mL of amikacin synthetic solution, and the amikacin content is 5.5% (g/mL). The synthetic yield relative to kanamycin A was 73.7%.
References
- ^ Jump up to:a b c “Amikacin Use During Pregnancy”. Drugs.com. 2 December 2019. Retrieved 13 March 2020.
- ^ “Amikacin 250 mg/ml Injection – Summary of Product Characteristics (SmPC)”. (emc). 16 September 2015. Retrieved 13 March 2020.
- ^ Jump up to:a b c d e f g h i j k l m n Plumb, Donald C. (2011). “Amikacin Sulfate”. Plumb’s Veterinary Drug Handbook (7th ed.). Stockholm, Wisconsin; Ames, Iowa: Wiley. pp. 39–43. ISBN 978-0-470-95964-0.
- ^ Jump up to:a b c d e f g h i j k l m n o p q r s t u v w x y z aa “Amikacin Sulfate”. The American Society of Health-System Pharmacists. Archived from the original on 20 December 2016. Retrieved 8 December 2016.
- ^ World Health Organization (2009). Stuart MC, Kouimtzi M, Hill SR (eds.). WHO Model Formulary 2008. World Health Organization. p. 137. hdl:10665/44053. ISBN 9789241547659.
- ^ Fischer, Janos; Ganellin, C. Robin (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 507. ISBN 9783527607495. Archived from the original on 20 December 2016.
- ^ Oxford Handbook of Infectious Diseases and Microbiology. OUP Oxford. 2009. p. 56. ISBN 9780191039621. Archived from the original on 24 November 2015.
- ^ World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
- ^ Jump up to:a b c d e f g h i j k l m “Amikacin sulfate injection, solution”. DailyMed. 10 April 2019. Retrieved 13 March 2020.
- ^ Scholar, Eric M.; Pratt, William B. (22 May 2000). The Antimicrobial Drugs (2nd ed.). Oxford University Press, USA. pp. 15–19. ISBN 978-0-19-975971-2. Archived from the original on 10 September 2017.
- ^ Jump up to:a b c d Cunha, Burke A. (1 November 2006). “New Uses for Older Antibiotics: Nitrofurantoin, Amikacin, Colistin, Polymyxin B, Doxycycline, and Minocycline Revisited”. Medical Clinics of North America. Antimicrobial Therapy. 90 (6): 1089–1107. doi:10.1016/j.mcna.2006.07.006. ISSN 0025-7125. PMID 17116438.
- ^ Jump up to:a b c d e f g “amikacin (Rx)”. Medscape. WebMD. Archived from the original on 9 August 2017. Retrieved 9 August 2017.
- ^ Jump up to:a b Aronson J. K., ed. (2016). “Amikacin”. Meyler’s Side Effects of Drugs (16th ed.). Oxford: Elsevier. pp. 207–209. ISBN 978-0-444-53716-4.
- ^ Vardanyan, Ruben; Hruby, Victor (2016). “Chapter 32: Antimicobacterial Drugs”. Synthesis of Best-Seller Drugs. Boston: Academic Press. pp. 669–675. ISBN 978-0-12-411492-0.
- ^ Jump up to:a b c d e f g h i j “FDA approves a new antibacterial drug to treat a serious lung disease using a novel pathway to spur innovation”. U.S. Food and Drug Administration (FDA)(Press release). Retrieved 12 November 2018. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c “Arikayce- amikacin suspension”. DailyMed. 30 September 2018. Retrieved 13 March 2020.
- ^ “Arikayce: Withdrawn application”. European Medicines Agency (EMA). 14 October 2016. Retrieved 13 March 2020.
- ^ Jump up to:a b c d e Ettinger, Stephen J.; Feldman, Edward C. (24 December 2009). Textbook of Veterinary Internal Medicine. Elsevier Health Sciences. pp. 1976, 523. ISBN 978-1-4377-0282-8. Archived from the original on 10 September 2017.
- ^ Maire, P.; Bourguignon, L.; Goutelle, S.; Ducher, M.; Jelliffe, R. (2017). “Chapter 20 – Individualizing Drug Therapy in the Elderly”. Individualized Drug Therapy for Patients. Boston: Academic Press. pp. 373–382. ISBN 978-0-12-803348-7.
- ^ Eghianruwa, Kingsley (2014). Essential Drug Data for Rational Therapy in Veterinary Practice. Author House. p. 16. ISBN 978-1-4918-0000-3. Archived from the original on 10 September 2017.
- ^ Jump up to:a b Morris, Daniel O.; Kennis, Robert A. (11 October 2012). Clinical Dermatology, An Issue of Veterinary Clinics: Small Animal Practice, E-Book. Elsevier Health Sciences. p. 29. ISBN 978-1-4557-7377-0. Archived from the original on 10 September 2017.
- ^ Jump up to:a b c Corti, Natascia; Taegtmeyer, Anne; Imhof, Alexander (1 January 2011). “Miscellaneous antibacterial drugs”. Side Effects of Drugs Annual. A worldwide yearly survey of new data in adverse drug reactions. 33: 509–540. doi:10.1016/B978-0-444-53741-6.00026-X. ISBN 9780444537416. ISSN 0378-6080.
- ^ Bauman, Robert W. (2015). Microbiology: with diseases by body system (4th ed.). Boston: Pearson. ISBN 978-0-321-91855-0.
- ^ “Amikacin”. DrugBank. 2 August 2017. Archived from the original on 16 August 2017. Retrieved 10 August 2017.
- ^ Jump up to:a b Mudd, Efrain (7 August 2017). “O Aminoglycosides”. Pharmacological Sciences. Archived from the original on 16 August 2017. Retrieved 14 August 2017.
- ^ Jump up to:a b Kondo, Shinichi; Hotta, Kunimoto (1 January 1999). “Semisynthetic aminoglycoside antibiotics: Development and enzymatic modifications”. Journal of Infection and Chemotherapy. 5 (1): 1–9. doi:10.1007/s101560050001. ISSN 1341-321X. PMID 11810483. S2CID 38981498.
- ^ Park, Je Won; Ban, Yeon Hee; Nam, Sang-Jip; Cha, Sun-Shin; Yoon, Yeo Joon (1 December 2017). “Biosynthetic pathways of aminoglycosides and their engineering”. Current Opinion in Biotechnology. Chemical biotechnology: Pharmaceutical biotechnology. 48: 33–41. doi:10.1016/j.copbio.2017.03.019. ISSN 0958-1669. PMID 28365471.
- ^ Caminero, José A; Sotgiu, Giovanni; Zumla, Alimuddin; Migliori, Giovanni Battista (1 September 2010). “Best drug treatment for multidrug-resistant and extensively drug-resistant tuberculosis”. The Lancet Infectious Diseases. 10 (9): 621–629. doi:10.1016/S1473-3099(10)70139-0. ISSN 1473-3099. PMID 20797644.
- ^ Ahmad, Suhail; Mokaddas, Eiman (1 March 2014). “Current status and future trends in the diagnosis and treatment of drug-susceptible and multidrug-resistant tuberculosis”. Journal of Infection and Public Health. 7 (2): 75–91. doi:10.1016/j.jiph.2013.09.001. ISSN 1876-0341. PMID 24216518.
- ^ Kawaguchi, H.; Naito, T.; Nakagawa, S.; Fujisawa, K. I. (December 1972). “BB-K 8, a new semisynthetic aminoglycoside antibiotic”. The Journal of Antibiotics. 25 (12): 695–708. doi:10.7164/antibiotics.25.695. ISSN 0021-8820. PMID 4568692. Archived from the original on 16 August 2017.
- ^ Monteleone, Peter M.; Muhammad, Naseem; Brown, Robert D.; McGrory, John P.; Hanna, Samir A. (1 January 1983). Amikacin Sulfate. Analytical Profiles of Drug Substances. 12. pp. 37–71. doi:10.1016/S0099-5428(08)60163-X. ISBN 9780122608124. ISSN 0099-5428.
- ^ Jump up to:a b c d Forney, Barbara. “Amikacin for Veterinary Use”. Wedgewood Pharmacy. Archived from the original on 16 August 2017. Retrieved 9 August 2017.
- ^ Riviere, Jim E.; Papich, Mark G. (13 May 2013). Veterinary Pharmacology and Therapeutics. John Wiley & Sons. p. 931. ISBN 978-1-118-68590-7. Archived from the original on 10 September 2017.
- ^ Mader, Douglas R.; Divers, Stephen J. (12 December 2013). Current Therapy in Reptile Medicine and Surgery – E-Book. Elsevier Health Sciences. p. 382. ISBN 978-0-323-24293-6. Archived from the original on 10 September 2017.
- ^ Maggs, David; Miller, Paul; Ofri, Ron (7 August 2013). Slatter’s Fundamentals of Veterinary Ophthalmology – E-Book. Elsevier Health Sciences. p. 37. ISBN 978-0-323-24196-0. Archived from the original on 10 September 2017.
- ^ Hsu, Walter H. (25 April 2013). Handbook of Veterinary Pharmacology. John Wiley & Sons. p. 486. ISBN 978-1-118-71416-4.
- ^ US National Library of Medicine (9 March 2017). “Amiglyde-V- amikacin sulfate injection”. DailyMed. Archived from the original on 16 August 2017. Retrieved 8 August2017.
- ^ Orsini, James A. (1 August 2017). “Update on Managing Serious Wound Infections in Horses: Wounds Involving Joints and Other Synovial Structures”. Journal of Equine Veterinary Science. 55: 115–122. doi:10.1016/j.jevs.2017.01.016. ISSN 0737-0806.
- ^ Wanamaker, Boyce P.; Massey, Kathy (25 March 2014). Applied Pharmacology for Veterinary Technicians – E-Book. Elsevier Health Sciences. p. 392. ISBN 978-0-323-29170-5.
- ^ Papich, Mark G. (October 2015). “Amikacin”. Saunders Handbook of Veterinary Drugs: Small and Large Animal (4th ed.). Elsevier Health Sciences. pp. 25–27. ISBN 978-0-323-24485-5. Archived from the original on 10 September 2017.
External links
- “Amikacin”. Drug Information Portal. U.S. National Library of Medicine.
- “Amikacin sulfate”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Amikin, Amiglyde-V, Arikayce, others |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a682661 |
| License data | US DailyMed: Amikacin |
| Pregnancy category | AU: D[1]US: D (Evidence of risk)[1] |
| Routes of administration | intramuscular, intravenous |
| Drug class | Aminoglycoside |
| ATC code | D06AX12 (WHO) J01GB06 (WHO), S01AA21 (WHO), J01RA06 (WHO), QD06AX12 (WHO), QJ01GB06 (WHO), QS01AA21 (WHO), QJ01RA06 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only)UK: POM (Prescription only) [2]US: ℞-onlyEU: Rx-only |
| Pharmacokinetic data | |
| Bioavailability | >90%[3] |
| Protein binding | 0–11% |
| Metabolism | Mostly unmetabolized |
| Elimination half-life | 2–3 hours |
| Excretion | Kidney |
| Identifiers | |
| IUPAC name[show] | |
| CAS Number | 37517-28-5 |
| PubChem CID | 37768 |
| DrugBank | DB00479 |
| ChemSpider | 34635 |
| UNII | 84319SGC3C |
| KEGG | D02543 as salt: D00865 |
| ChEBI | CHEBI:2637 |
| ChEMBL | ChEMBL177 |
| CompTox Dashboard (EPA) | DTXSID3022586 |
| ECHA InfoCard | 100.048.653 |
| Chemical and physical data | |
| Formula | C22H43N5O13 |
| Molar mass | 585.608 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES[hide]O=C(N[C@H]3[C@H](O[C@H]1O[C@@H]([C@@H](O)[C@H](N)[C@H]1O)CO)[C@@H](O)[C@H](O[C@H]2O[C@H](CN)[C@@H](O)[C@H](O)[C@H]2O)[C@@H](N)C3)[C@@H](O)CCN | |
| InChI[hide]InChI=1S/C22H43N5O13/c23-2-1-8(29)20(36)27-7-3-6(25)18(39-22-16(34)15(33)13(31)9(4-24)37-22)17(35)19(7)40-21-14(32)11(26)12(30)10(5-28)38-21/h6-19,21-22,28-35H,1-5,23-26H2,(H,27,36)/t6-,7+,8-,9+,10+,11-,12+,13+,14+,15-,16+,17-,18+,19-,21+,22+/m0/s1 Key:LKCWBDHBTVXHDL-RMDFUYIESA-N |
/////////Amikacin sulfate, Arikayce liposomal, EU 2020, 2020 APPROVALS, Antibacterial, Protein biosynthesis inhibitor, アミカシン硫酸塩 , BB K 8, AMIKACIN
C1C(C(C(C(C1NC(=O)C(CCN)O)OC2C(C(C(C(O2)CO)O)N)O)O)OC3C(C(C(C(O3)CN)O)O)O)N.OS(=O)(=O)O.OS(=O)(=O)O
Tirbanibulin
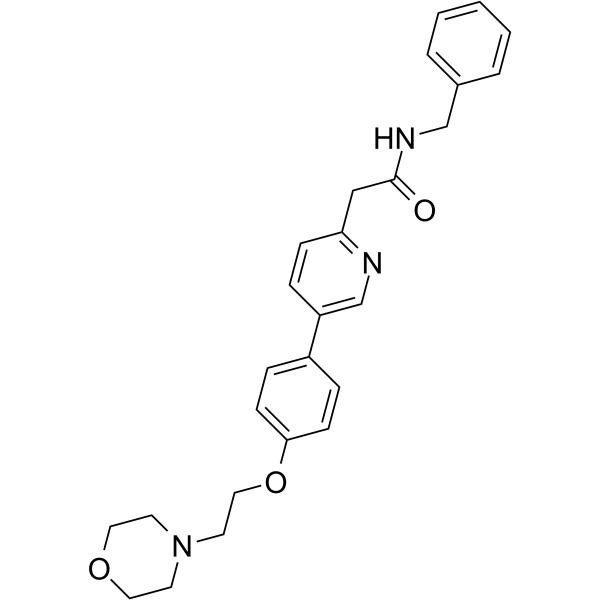
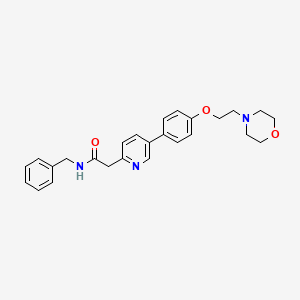
Tirbanibulin
CAS 897016-82-9, 1038395-65-1 DI HCL
1080645-95-9 MESYLATE
N-benzyl-2-[5-[4-(2-morpholin-4-ylethoxy)phenyl]pyridin-2-yl]acetamide
| Molecular Weight | 431.53 |
|---|---|
| Formula | C₂₆H₂₉N₃O₃ |
FDA APPROVED 12/14/2020, Klisyri
To treat actinic Keratosis of the face or scalp
Tirbanibulin (KX2-391) is an inhibitor of Src that targets the peptide substrate site of Src, with GI50 of 9-60 nM in cancer cell lines.
- Originator Kinex Pharmaceuticals
- Developer Almirall S.A.; Athenex; Hanmi Pharmaceutical; Kinex Pharmaceuticals; PharmaEssentia Corporation
- ClassAcetamides; Amides; Antineoplastics; Antipsoriatics; Morpholines; Phenyl ethers; Pyridines; Skin disorder therapies; Small molecules
- Mechanism of ActionAngiogenesis inhibitors; Src-Family kinase inhibitors; Tubulin polymerisation inhibitors
- PreregistrationActinic keratosis
- Phase IIPsoriasis
- Phase I/IISolid tumours
- Phase IPhotodamage
- PreclinicalSkin cancer
- 09 Mar 2020FDA assigns PDUFA action date of 30/12/2020 for tirbanibulin for Actinic keratosis
- 09 Mar 2020US FDA accepts NDA for tirbanibulin for Actinic keratosis for review
- 02 Mar 2020European Medicines Agency accepts Marketing Authorization Application for tirbanibulin for Actinic keratosis for review
KX-01 is a dual inhibitor of Src kinase and tubulin polymerization. KX01 promotes the induction of p53, G2/M arrest of proliferating cell populations and subsequent apoptosis via the stimulation of Caspase-3 and PARP cleavage. The drug was developed by Kinex Pharmaceuticals and reached phase II of clinical trials for the treatment of Castration-Resistant Prostate Cancer and Actinic Keratosis. KX-01 demonstrated good in vitro pofile against different cancer cell lines with IC50 in nanomolar range.
Tirbanibulin (Mesylate) (KX2-391 (Mesylate)) is an inhibitor of Src that targets the peptide substrate site of Src, with GI50 of 9-60 nM in cancer cell lines.
Tirbanibulin (KX2-391) is a Src inhibitor that is directed to the Src substrate pocket. Tirbanibulin (KX2-391) shows steep dose-response curves against Huh7 (GI50=9 nM), PLC/PRF/5 (GI50=13 nM), Hep3B (GI50=26 nM), and HepG2 (GI50=60 nM), four hepatic cell cancer (HCC) cell lines[1]. Tirbanibulin (KX2-391) is found to inhibit certain leukemia cells that are resistant to current commercially available drugs, such as those derived from chronic leukemia cells with the T3151 mutation. Tirbanibulin (KX2-391) is evaluated in engineered Src driven cell growth assays inNIH3T3/c-Src527F and SYF/c-Src527F cells and exhibits GI50 with 23 nM and 39 nM, respectively[2].
Orally administered Tirbanibulin (KX2-391) is shown to inhibit primary tumor growth and to suppress metastasis, in pre-clinical animal models of cancer[2].
[1]. Lau GM, et al. Expression of Src and FAK in hepatocellular carcinoma and the effect of Src inhibitors on hepatocellular carcinoma in vitro. Dig Dis Sci, 2009, 54(7), 1465-1474. [2]. Fallah-Tafti A, et al. Thiazolyl N-benzyl-substituted acetamide derivatives: synthesis, Src kinase inhibitory and anticancer activities. Eur J Med Chem, 2011, 46(10), 4853-4858.

Approval allows Almirall to move forward with the topical ointment for individuals with AK on the face or scalp.

The US Food and Drug Administration (FDA) has approved tirbanibulin (Klisyri) as a topical treatment for actinic keratosis (AK).
The approval, awarded to Almirall, S.A., will allow the novel, topical first-in-class microtubule inhibitor for treatment of the disease on the face or scalp, representing a significant breakthrough in treatment of AK because of its short treatment protocol of once daily application for 5 days.
Actinic keratosis represents the second most common diagnosis in dermatology in the US, with a reported prevalence between 11-25%.
“Early diagnosis and treatment of actinic keratosis (AK) is critical, since those who already have an AK are likely to develop more actinic keratoses (plural) in the future,” said Deborah S. Sarnoff, MD, President of the Skin Cancer Foundation, said in a statement. “Patients with AK are at higher risk for skin cancer, since AKs can progress into squamous cell carcinoma (SCC), a common and sometimes invasive form of skin cancer.”
The approval is based on recent data from a large phase 3 clinical study, as well as 2 randomized, double-blind, vehicle-controlled phase 3 studies evaluating the efficacy and safety of tirbanibulin ointment 1% in adults with AK on the face or scalp.
“These studies enrolled a total of 702 patients across 62 sites in the United States, providing robust data,” Andrew Blauvelt, MD, MBA, President of Oregon Medical Research Center, and one of the lead investigators of the studies, said in a statement. “Tirbanibulin achieved a significantly higher number of patients with complete (100%) clearance of AK lesions in the treated area compared to vehicle (44% vs. 5% in study 1 and 54% vs. 13% in study 2), as well as reaching the secondary endpoint of partial (≥75%) clearance of lesions.”
PATENT
WO 2006071960
US 20070015752
US 20080287436
WO 2008082637
WO 2008002676
US 20090318450
https://patents.google.com/patent/US20090318450A1/en
- .
- [0374]A 1 L single-necked round-bottomed flask was charged with 7 (61.4 g, 0.172 mol), benzyl amine (55.6 g, 0.519 mol, 3 eq), and anhydrous anisole (300 g) and then stirred at reflux until reaction was essentially complete (23 h, 165° C. oil bath temperature; internal temperature was 147° C.) and then allowed to cool to near room temperature. A portion (1 mL) of the reaction mixture was diluted with toluene (1 mL) resulting in the complete crystallization of that portion. This seed was then added to the reaction mixture and allowed to stand until the whole reaction mixture had crystallized to a single block. Toluene (150 mL) was added and the mixture swirled to break up the solid. Heptane/toluene (1:1, 100 mL) was added and the solid mixture broken up further. Finally, heptane (50 mL, then 25 mL) was added and the mixture broken up even further, allowing to stand an additional 30 min before filtering the solid. Filtration of the solid, washing with 2:1 toluene/heptane (300 mL), 1:2 toluene/heptane (300 mL), and then heptane (2×300 mL), and then drying (air, then high vac) gave 60.16 g (yield of 81%) of title product as a white solid (>98.9% AUC). Another 2.5 g of less pure (97.4%) material was obtained from the mother liquors.
- [0375]1H NMR (CDCl3) δ 2.60 (t, 4 H), 2.83 (t, 2 H), 3.74 (t, 4 H), 3.82 (s, 2 H), 4.18 (t, 2 H), 4.49 (d, 2 H), 7.01 (d, 2 H), 7.2-7.35 (m, 6 H), 7.49 (d, 2 H), 7.64 (br t, 1 H), 7.81 (dd, 1 H), 8.69 (fine d, 1 H). MS (from LC/MS): m/z 432.5 [M+1].
- [0376]To a stirred suspension of KX2-391 (free base, 60.00 g) in absolute EtOH (600 mL) was added 170 mL of 2.5 M HCl (in ethanol), 25 mL EtOH being added to wash down the sides of the flask. The resulting homogeneous solution was stirred at room temperature (20 min) and then evaporated to near dryness (to frothing). After chasing with EtOH (2×150 mL), the residue was taken up again in EtOH (150 mL) and then was followed by the slow addition of heptane until the mixture appeared saturated (33 mL required for cloudiness to remain). After sitting overnight, two layers had formed. After adding additional heptane (250 mL) crystallization still could not be induced and so the reaction mixture was concentrated to a volume of ˜200 mL at which time the mixture was homogeneous. This thick homogeneous solution was added dropwise to very rapidly stirred (mechanical) EtOAc (2 L). After the addition was complete, a 25 mL EtOH rinse of the original flask and addition funnel was added to the rapidly stirred mixture. The rapid stirring was continued for another ˜1 h and then the mixture was filtered and the solid (partly gummy) was washed with EtOAc (300 mL) and then heptane. As soon as the heptane wash began, the solid got much gummier. The fritted Buchner funnel and its contents were covered (paper towel/rubber band) and immediately placed in the vacuum oven. After overnight vacuum at ˜45° C., the vacuum was released under nitrogen, and the Buchner funnel containing the product (foamy solid) was immediately placed in a zip-lock back and then, under nitrogen (glove bag), transferred to a bottle and the foamy solid broken up (spatula) to a powder. A second night under high vacuum (˜45° C.) resulted in only 1.3 g of additional weight loss. Constant weight was essentially attained with the third night of high vacuum (˜45° C.) where only 0.2 g of weight was lost. The final weight of material was 68.05 g (yield of 97%), containing 0.29 eq (4.8% w/w) of EtOAc, 0.035 eq (0.3% w/w) EtOH, and 0.03 eq (0.6% w/w) heptane. The purity was 99.6%.
- [0377]1H NMR (DMSO-d6) δ 3.1-3.3 (m, 2 H), 3.45-3.65 (m, 4 H), 3.8-4.0 (m, 4 H), 4.11 (s, 2 H), 4.32 (d, 2 H), 4.57 (t, 2 H), 7.19 (d, 2 H), 7.2-7.4 (m, 5 H), 7.88 (d, 2 H), 7.93 (d, 1 H), 8.68 (dd, 1 H), 8.99 (br t, 1 H), 9.10 (fine d, 1 H), 11.8 (br s, 1 H). MS (from LC/MS): m/z 432.5 [M+1 of free base].
- [0378]Elemental analysis (for C26H29N3O3.2 HCl.0.035 EtOH.0.29.EtOAc.0.03 heptane.0.8 H2O):
- [0379]Calculated (%): C, 60.03; H, 6.54; N, 7.65; Cl, 12.91
- [0380]Observed (%):C, 59.85/59.97; H, 6.54/6.47; N, 7.67/7.67; Cl, 13.10/13.24
- [0381]Calculated FW: 534.63 (does not take into account the 0.8 H2O which probably arose during handling of this very hygroscopic powder, since 1H NMR shows no evidence for H2O).
- [0382]The ethyl chloride level in this material was measured and found to be 98 ppm. The sample was also analyzed and found to contain 5,800 ppm of heptane.
- [0383]Analysis of another portion of this sample yielded the following results: 99.6% AUC, 1640 ppm ethanol, 41,480 ppm ethyl acetate, 5600 ppm heptane, no anisole detected, and 120 ppm ethyl chloride.
- [0384]A procedure for recrystallizing the salt was also developed using the above dried salt. This procedure would work just was well on the highly pure crude salt (containing residual EtOH) obtained from concentrating the HCl salt-forming reaction mixture:
- [0385]The salt (575 mg) was dissolved in twice the mass of absolute EtOH (1.157 g) and then heated under nitrogen. To this hot solution (stirred) was added 1.6 g of 25% EtOH (in EtOAc) followed by the addition of EtOAc (0.25 mL) resulting in a cloudiness that remained. The cloudy hot solution was allowed to cool to room temperature during which time crystallization occurred. After crystallization was complete (2 h), the crystalline solid was filtered, washed with anhydrous EtOAc (˜40 mL), and vacuum dried to give 424 mg of the dihydrochloride salt of KX2-391 as a free-flowing solid (tiny beads, 99.8% AUC) containing only 0.05 eq (0.45% w/w) of EtOH and 0.015 eq (0.26% w/w) of EtOAc. Slightly better recovery (460 mg from 586 mg) was attained using isopropanol/EtOAc but the level of solvent entrapment was higher [0.085 eq (1.0% w/w) of isopropanol and 0.023 eq (0.4% w/w) of EtOAc].
PATENT
WO 2009051848
https://patents.google.com/patent/WO2009051848A1/en
].

Preparation of 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)-N-benzylacetamide (Compound (I) free base).
[ 000242 ] A l L single-necked round-bottomed flask was charged with 7 (61.4 g, 0.172 mol), benzyl amine (55.6 g, 0.519 mol, 3 eq), and anhydrous anisole (300 g) and then stirred at reflux until reaction was essentially complete (23 h, 165 0C oil bath temperature; internal temperature was 147 0C) and then allowed to cool to near room temperature. A portion (1 mL) of the reaction mixture was diluted with toluene (1 mL) resulting in the complete crystallization of that portion. This seed was then added to the reaction mixture and allowed to stand until the whole reaction mixture had crystallized to a single block. Toluene (150 mL) was added and the mixture swirled to break up the solid. Heptane/toluene (1:1, 100 mL) was added and the solid mixture broken up further. Finally, heptane (50 mL, then 25 mL) was added and the mixture broken up even further, allowing to stand an additional 30 min before filtering the solid. Filtration of the solid, washing with 2:1 toluene/heptane (300 mL), 1:2 toluene/heptane (300 mL), and then heptane (2 x 300 mL), and then drying (air, then high vac) gave 60.16 g (yield of 81%) of title product as a white solid (>98.9% AUC). Another 2.5 g of less pure (97.4%) material was obtained from the mother liquors.
[000243 ] 1H NMR (CDCl3) δ 2.60 (t, 4 H), 2.83 (t, 2 H), 3.74 (t, 4 H), 3.82 (s, 2 H), 4.18 (t, 2 H), 4.49 (d, 2 H), 7.01 (d, 2 H), 7.2-7.35 (m, 6 H), 7.49 (d, 2 H), 7.64 (br t, 1 H), 7.81 (dd, 1 H), 8.69 (fine d, 1 H). MS (from LC/MS): m/z 432.5 [M + I].

Preparation of 4-(2-(4-(6-(2-(benzylamino)-2-oxoethyl)pyridinium-3-yl)phenoxy)ethyl)- morpholin-4-ium chloride (Compound (I), diHCI salt).
[000244 ] To a stirred suspension of compound (I) (free base, 60.00 g) in absolute EtOH (600 mL) was added 170 mL of 2.5 M HCl (in ethanol), 25 mL EtOH being added to wash down the sides of the flask. The resulting homogeneous solution was stirred at room temperature (20 min) and then evaporated to near dryness (to frothing). After chasing with EtOH (2 x 150 mL), the residue was taken up again in EtOH (150 mL) and then was followed by the slow addition of heptane until the mixture appeared saturated (33 mL required for cloudiness to remain). After sitting overnight, two layers had formed. After adding additional heptane (250 mL) crystallization still could not be induced and so the reaction mixture was concentrated to a volume of -200 mL at which time the mixture was homogeneous. This thick homogeneous solution was added dropwise to very rapidly stirred (mechanical) EtOAc (2 L). After the addition was complete, a 25 mL EtOH rinse of the original flask and addition funnel was added to the rapidly stirred mixture. The rapid stirring was continued for another ~1 h and then the mixture was filtered and the solid (partly gummy) was washed with EtOAc (300 mL) and then heptane. As soon as the heptane wash began, the solid got much gummier. The fritted Buchner funnel and its contents were covered (paper towel/rubber band) and immediately placed in the vacuum oven. After overnight vacuum at -45 0C, the vacuum was released under nitrogen, and the Buchner funnel containing the product (foamy solid) was immediately placed in a zip-lock back and then, under nitrogen (glove bag), transferred to a bottle and the foamy solid broken up (spatula) to a powder. A second night under high vacuum (-45 0C) resulted in only 1.3 g of additional weight loss. Constant weight was essentially attained with the third night of high vacuum (-45 0C) where only 0.2 g of weight was lost. The final weight of material was 68.05 g (yield of 97%), containing 0.29 eq (4.8% w/w) of EtOAc, 0.035 eq (0.3% w/w) EtOH, and 0.03 eq (0.6% w/w) heptane. The purity was 99.6%.
[000245] 1H NMR (DMSO-Cl6) δ 3.1-3.3 (m, 2 H), 3.45-3.65 (m, 4 H), 3.8-4.0 (m, 4 H), 4.11 (s, 2 H), 4.32 (d, 2 H), 4.57 (t, 2 H), 7.19 (d, 2 H), 7.2-7.4 (m, 5 H), 7.88 (d, 2 H), 7.93 (d, 1 H), 8.68 (dd, 1 H), 8.99 (br t, 1 H), 9.10 (fine d, 1 H), 11.8 (br s, 1 H). MS (from LC/MS): m/z 432.5 [M + 1 of free base].
[000246] Elemental analysis (for C26H29N3O3 • 2 HCl • 0.035 EtOH • 0.29 EtOAc • 0.03 heptane • 0.8 H2O): a. Calculated (%): C, 60.03; H, 6.54; N, 7.65; Cl, 12.91 b. Observed (%):C, 59.85/59.97; H, 6.54/6.47; N, 7.67/7.67; Cl, 13.10/13.24
[ 000247] Calculated FW: 534.63 (does not take into account the 0.8 H2O which probably arose during handling of this very hygroscopic powder, since 1H NMR shows no evidence for H2O).
[ 000248] The ethyl chloride level in this material was measured and found to be 98 ppm. The sample was also analyzed and found to contain 5,800 ppm of heptane.
[000249] Analysis of another portion of this sample yielded the following results: 99.6% AUC, 1640 ppm ethanol, 41,480 ppm ethyl acetate, 5600 ppm heptane, no anisole detected, and 120 ppm ethyl chloride.
[000250] A procedure for recrystallizing the salt was also developed using the above dried salt. This procedure would work just was well on the highly pure crude salt (containing residual EtOH) obtained from concentrating the HCl salt-forming reaction mixture:
[000251] The salt (575 mg) was dissolved in twice the mass of absolute EtOH (1.157 g) and then heated under nitrogen. To this hot solution (stirred) was added 1.6 g of 25% EtOH (in EtOAc) followed by the addition of EtOAc (0.25 mL) resulting in a cloudiness that remained. The cloudy hot solution was allowed to cool to room temperature during which time crystallization occurred. After crystallization was complete (2 h), the crystalline solid was filtered, washed with anhydrous EtOAc (~40 mL), and vacuum dried to give 424 mg of the dihydrochloride salt of compound (I) as a free-flowing solid (tiny beads, 99.8% AUC) containing only 0.05 eq (0.45% w/w) of EtOH and 0.015 eq (0.26% w/w) of EtOAc. Slightly better recovery (460 mg from 586 mg) was attained using isopropanol/EtOAc but the level of solvent entrapment was higher [0.085 eq (1.0% w/w) of isopropanol and 0.023 eq (0.4% w/w) ofEtOAc].
Example 3: Large Scale Synthesis of Compound (I) di-HCl
[000252 ] Reagents and solvents were used as received from commercial suppliers. Progress of the reactions was monitored by HPLC, GC/MS, or 1H NMR. Thin-layer chromatography (TLC) was performed using Analtech silica gel plates and visualized by UV light (254 nm). High pressure liquid chromatography (HPLC) was performed on an Agilent 1100 Series instruments. Proton and carbon nuclear magnetic resonance spectra were obtained using a Bruker AV 300 at 300 MHz for proton and 75 MHz for carbon. The solvent peak was used as the reference peak for proton and carbon spectra. Preparation of 4-(2-(4-Bromophenoxy)ethyl)morpholine (2)
[000253 ] A 50 L jacketed reactor equipped with a reflux condenser and temperature probe was charged with 4-(3-chloropropyl)morpholine (2.44 kg, 0.54 mol), 4-bromophenol (2.27 kg, 0.54 mol, 1.0 equiv.), powdered potassium carbonate (6.331 kg, 1.88 mol, 3.50 equiv.), and DMF (12.2 L) and stirred. The reaction mixture was then heated to 60-65 0C and stirred overnight. After 17.5 h, the reaction mixture was cooled to 20-25 °C. The reaction mixture was charged to a different reactor equipped with bottom valve for the work-up. While maintaining a temperature between 20-30 0C, DI water (48.7 L) was charged to the reactor. The phases were separated. The aqueous layer was extracted with MTBE (3 x 24.4 L). To the combined organics, DI water (18.3 L) and then 6M sodium hydroxide (18.2 L) were added. The mixture was stirred for 2-5 minutes and the phases were separated. The organic phase was washed with water (24.4 L) and brine (24.4 L), dried over magnesium sulfate, filtered, and concentrated to give 337Og of a yellow oil (89% crude yield, 99.4% AUC by HPLC).
Preparation of 6-fluoropyridin-3-ylboronic acid (4)
[000254] A 72 L reactor equipped with reflux condenser, and temperature probe. To the reactor 5-bromo-2-fluoropyridine (1.17 L, 0.568 mol), toluene (18.2 L), and triisopropyl borate (3.13 L, 0.68 mol, 1.2 equiv.) were charged and stirred. Tetrahydrofuran (4.4 L) was added to the reactor and the reaction mixture was cooled to between —35 to -50 0C. While maintaining a temperature between -35 to —45 0C, n-butyl lithium (2.5 M solution of hexanes, 5.44 L, 0.68 mol, 1.2 equiv.) was cautiously added to the reactor. After 5 h, the reaction was deemed complete and the reaction mixture was warmed to between -15 to -20 0C. To the reaction was added 2M HCl (11.80L) to the reactor while maintaining a temperature between -15 0C and 0 0C. The reaction mixture was stirred at 18 to 23 0C for (16 h) and the phases were separated. The organics were then extracted with 6 M sodium hydroxide (6.0 L). The acidic anbasic aqueous phases were mixed in the reactor and 6 M HCl (2.5 L) was added until pH 7.5 was achieved. Sodium chloride (6.0 kg) was then added to the aqueous phase. The aqueous phase was then extracted with THF (3 * 20 L). The combined organics were dried with magnesium sulfate and concentrated to give 1300 g of a tan solid (81% crude yield).
Preparation of 4-(2-(4-(6-fluoropyridin-3-yl)phenoxy)ethyl)morpholine (5) [000255] A 72 L reactor equipped with reflux condenser, sparging tube, bubbler, and temperature probe was charged with 6-fluoropyridin-3-ylboric acid (2.84 kg, 1.24 equiv.), 4- (2-(4-bromophenoxy)ethyl)morpholine (4.27 kg, 1.0 equiv.), and DME (27 L). Agitation was started and sodium carbonate (4.74 kg, 3.0 equiv.) as a solution in DI water (17.1 L) was then charged to the reaction mixture. Argon was bubbled through the reaction mixture for 50 minutes. Under an argon atmosphere, tetrakis(triphenylphosphine)palladium (750 g, 0.04 equiv.) was added to the reaction mixture as a slurry in DME (1.0 L). The reaction mixture was heated to 75 – 85 0C and stirred overnight (17 h). The reaction mixture was cooled to between 18 – 22°C. DI water (26.681kg) and MTBE (26.681 L) were charged to the reactor and stirred for 5 minutes. The phases were separated and the aqueous phase was extracted with MTBE (2 x 26.7 L). The combined organics were extracted with 2M HCl (1 x 15.0 L, 3 x 21.8 L). The aqueous phase was then charged back to the reactor and ethyl acetate was added (26.7 L). The pH was adjusted to 6.2 using 6 M sodium hydroxide (26.7 L) while maintaining a temperature between 15 – 25 0C. The phases were separated and the aqueous phase was extracted with ethyl acetate (2 x 26.7 L). The combined organics were dried with magnesium sulfate and concentrated to give 4555 g of a residue (101% crude yield, 67.1% AUC by HPLC).
Purification of 4-(2-(4-(6-fluoropyridin-3-yl)phenoxy)ethyl)morpholine (5)
[000256] The crude product (575 g) was purified by silica gel chromatography by eluting with methanol/ethyl acetate/heptane (30% ethyl acetate/heptane, 50% ethyl acetate/heptane, 75% ethyl acetate/heptane, 100% ethyl acetate, and 5% methanol/ethyl acetate). Concentration of the pure fractions by TLC (10% methanol/dichloromethane, Rf = 0.3) provided 420 g of a light brown solid (73% recovery, >99.9% AUC by HPLC).
Preparation of 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)acetonitrile (6)
[ 000257] A 1 M solution of NaHMDS (2.0 L, 5.0 equiv.) in THF was charged to a 5-L flask and cooled to -20 to -15 0C. While maintaining a temperature below -10 0C, fluoride (119.7g, 1.0 equiv.) in THF (500 mL) was charged to the flask over 20 minutes. Acetonitrile (82.5 mL, 4.0 equiv.) in THF (170 mL) was added to the flask over 20 minutes, while maintaining a temperature below —100C. The reaction mixture was then stirred for 1 h. To the reaction was added brine (1.5 L, 12.6 vol.) at a rate as to maintain a temperature below 10 0C. The solution was then warmed to room temperature and the layers were allowed to separate. The mixture was filtered over Celite and washed with THF (I x 200 mL, 1 x 100 mL). The aqueous phase was extracted with toluene (750 mL). The combined organics were dried with magnesium sulfate, filtered, washed with toluene (2 * 25OmL), and concentrated to dryness. Toluene (IL) was added and the solution was concentrated to dryness again to give 169.8 g of an oil. MTBE (1190 mL, 7 vol.) was added to the oil at 50 0C and stirred for 15 minutes. Heptane (850 mL, 5vol.) was added over ten minutes at 50 0C. The mixture was then cooled to room temperature over 1.5 h and stirred for 2 h. The slurry was filtered, washed with 1 :4 MBTE/heptane (2 x 100 mL), and dried in an oven overnight at 45 0C to give 102.3 g of an off-white solid (80% yield, 98.8% AUC by HPLC).
Preparation of methyl 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)acetate (7)
[000258] Nitrile 6 (101 g) and methanol (1.01 L, 10 vol.) were charged to a 3-L flask equipped with stir bar and thermocouple. Concentrated H2SO4 (175 mL, 10.0 equiv.) was added drop wise to the solution over 15 minutes while maintaining a temperature below 60 0C. Followed by 30% fuming sulfuric acid (124 mL) was added drop wise to the solution while maintaining a temperature below 60 0C. The solution was then heated to reflux with a heating mantle and stirred overnight. When the reaction was deemed complete, it was cooled to 20 0C. In a second flask (22 L), saturated sodium bicarbonate (10.7 L) and dichloromethane (1.1 L) were charged and cooled to 15 0C. While maintaining a temperature below 20 0C, the reaction mixture was added to the sodium bicarbonate/dichloromethane mixture. The quench was stirred for 15 minutes and the phases were separated. The aqueous phase was extracted with dichloromethane (I x 55OmL, 1 x 30OmL). The combined organics were dried with magnesium sulfate and concentrated to dryness to give 105 g of an orange solid (94% crude yield, 97.7% AUC by HPLC).
Preparation of 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)-N-benzylacetamide (Compound (I))
[ 000259] Ester 7 (103 g), anisole (513 mL, 5 vol.), and benzylamine (94 mL, 3.0 equiv.) were charged to a 3 L flask equipped with thermocouple and overhead stirrer. The reaction mixture was then heated to 142 0C and stirred for two days. The reaction mixture was cooled to 45-50 0C and stirred for 2 hours. To the mixture was added n-heptane (1.5 L) dropwise over an hour. The solution was cooled to room temperature over three hours and then stirred overnight. The resulting slurry was filtered, washed with 4: 1 Anisole/n-heptane (200 mL) and n-heptane (3 x 100 mL). Drying in the oven overnight, the resulting product was 112. Ig of a tan solid (90% yield, 99.6% AUC by HPLC). The use of a single isomer of heptane was essential to adequately quantitate the residual solvent.
Preparation of 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)-N-benzylacetamide dihydrochloride salt (Compound (I) 2HC1)
[000260 ] EtOH (1.0 L) was charged to a 2-L flask and acetyl chloride (62.5 raL, 3.0 equiv.) was added slowly to the flask and stirred for 40 minutes. The resulting solution was added to compound (I) (100 g) over 30 minutes while maintaining a temperature of 30 0C. The solution was concentrated to a mass of 270 g. The concentrated solution was added to ethyl acetate (2 L) over 20 minutes with rapid stirring. The mixture was stirred overnight and then filtered under nitrogen to give two distinct solid products, tan solids (73.5 g) and darker solids (42.2 g). The solids were dry blended to give a combined yield of 99%. The HPLC analysis indicated 99.0% purity (AUC).
Analysis indicated that ethanol was present at 2530 ppm, ethyl acetate at 48,110 ppm, ethyl chloride at 170 ppm, and no heptane and anisole were detected. Palladium content was assayed three times and measured to be 29 ppm, 2 ppm, and less than 1 ppm.
Crystallization Study of Compound (I) 2HCl
Preparation of 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)-N-benzylacetamide (Compound (I))
[000268] To a 22-L reactor was charged compound 7 (650 g, 1.82 mol), anisole (3.25
L, 5 vol, anhydrous) and benzylamine (600 mL, 0.92 vol, 3 equiv). The batch (approximately 18 °C) was heated to 142 ± 5 °C over 1 hour 44 minutes, with dissolution occurring at 30 0C. The batch was maintained at 142 ± 5 0C for 69 hours 30 minutes at which point HPLC analysis indicated that compound 7 was 0.9% by conversion (specification <1.7% by conversion). The batch was cooled to 45-50 0C over 5 hours 12 minutes (to aid cooling the nitrogen flow was increased once the batch was approximately 72 0C). At that temperature range, the batch was poorly stirring and on mixing, the batch temperature increased to 52 0C. It was >50 °C for <15 minutes. The batch was aged for 2 hours 2 minutes once initially <50 0C, then n-heptane (9.75 L, 15 vol, 99%) was added to the batch over 1 hour 56 minutes, maintaining the batch temperature at 45-50 °C. The heating was then discontinued and the batch cooled to 25 0C over 10 hours 32 minutes and then to approximately 20 °C over 20 minutes. The total time the batch was maintained <25 0C was 4 hours 50 minutes (2 hours 47 minutes at approximately 20 0C). The batch was filtered under suction via a 24-inch polypropylene filter funnel (fitted with a PTFE cloth) and the reactor rinsed with anisole/n- heptane (1.3 L, 4: 1) and the rinse transferred to the cake. The cake was then washed successively with two portions of /i-heptane (1.3 L, 0.65 L). The total filtration time was 39 minutes. The batch (net wet weight 1004 g of KX2391) was transferred to three glass trays and placed into a vacuum oven set at 50 0C and dried to constant weight over 96 hours 26 minutes.
Preparation of 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)-N-benzylacetamide mesylate (Compound (I)-MSA)
[000269] Compound (I) (520 g, 1.21 mol) was transferred to reactor 1 using acetone (41.6 vol, 80 vol, ACS) to facilitate the transfer. The batch was heated to 50 ± 5 0C over 33 minutes with dissolution occurring at 30 0C . The batch was clarified into a second reactor via a transfer pump fitted with an inline filter (Pall P/N 12077, 10 micron) and reheated from 46 0C to 50 ± 5 0C. Methanesulfonic acid (121.4 g, 1.05 equiv, 99% extra pure) was added to the pale yellow batch over 12 minutes and the heating then discontinued. After fourteen minutes, white solids were observed, which increased in number to give after 59 minutes a white suspension. The batch was in the range of 25 ± 5 0C after 7 hours 51 minutes and aged for a further 19 hours 21 minutes (10 hours 30 minutes at <27 0C). The batch was filtered under suction via a 24-inch polypropylene filter (PTFE cloth) and the reactor rinsed with acetone (2.0 L, clarified, ACS) and the rinse transferred to the cake. The cake was covered with a stainless steel cover and sucked dry under a flow of nitrogen. The total filtration time was 21 minutes. The batch (net wet weight 764 g) was transferred to three glass drying trays and dried in a vacuum oven to constant weight at 25 ± 5 °C over 21 hours 54 minutes (565 g, 89% of theory). A sample was removed for analysis and the batch maintained in vacuo at 25 ± 5 °C. The batch was then transferred to two 80-oz amber glass bottles (Teflon lined polypropylene closure), blanketed with argon and stored at -10 to -20 °C.
PATENT
WO 2010135429
https://patents.google.com/patent/WO2010135429A2/en
Preparation of KX2-391 and its salts
[00045] The synthesis of 4-(2-(4-(6-fluoropyridin-3-yl)phenoxy)ethyl)morpholine is shown in the scheme below:

[00046] 4-(2-(4-(6-fluoropyridin-3-yl)phenoxy)ethyl)morpholine (5) was synthesized in 3 steps. Intermediate 2 was synthesized using an ether coupling reaction e.g., using Williamson ether synthesis. Ether formation between 4-(2-chloroethyl)morpholine (1) and A- bromophenol was carried out in the presence of potassium carbonate and DMF to afford 4-(2- (4-bromophenoxy)ethyl)morpholine (2). Rigorously dry conditions were not essential for this reaction and a basic wash with sodium hydroxide was used to remove any remaining A- bromophenol. In another aspect of the invention, intermediate 2 is synthesized using any ether formation reaction. Intermediate 2 is synthesized starting from compound 1 containing any leaving group. For example, the skilled chemist would start with compounds of the
general formula

wherein the leaving group “LG” includes but is not limited to halogen, tosylate, mesylate, trifluate, etc.
[00047] Compound 5 was formed using a Suzuki reaction. Formation of the aryl borate, 6-fluoropyridin-3-yl-3-boronic acid (4), was carried out by forming the aryl anion using n-BuLi followed by in situ quenching with triisopropylborate (Li, et ah, J. Org. Chem. 2002, 67, 5394-5397). The resulting 6-fluoropyridin-3-yl-3-boronic acid (4) was coupled to 4-(2-(4-bromophenoxy)ethyl)morpholine (2) in a solution of DME and aqueous sodium carbonate using tetrakis(triphenylphosphine)palladium to afford 4-(2-(4-(6-fluoropyridin-3- yl)phenoxy)ethyl)morpholine (5), which was purified using silica gel chromatography. The skilled chemist would know that other transition metal coupling reaction are used to prepare compound 5.
[00048] The synthesis of 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)-JV- benzylacetamide dihydro chloride is shown below:

[00049] 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)-N-benzylacetamide dihydrochloride (KX2-391 HCl) was synthesized in four linear steps. The fluoride of 4-(2-(4- (6-fluoropyridin-3-yl)phenoxy)ethyl)morpholine (5) was displaced by the anion of acetonitrile formed using commercially available NaHMDS. Acetonitrile was added slowly to a cooled mixture of compound 5 and base to form 2-(5-(4-(2- morpholinoethoxy)phenyl)pyridin-2-yl)acetonitrile (6). In another aspect of the invention, intermediate 5 may have a leaving group other than fluorine. Thus, compounds of the general formula:

would be pursued where LG includes other leaving groups known to the skilled chemist.
[00050] Acid catalyzed methanolysis of 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-
2-yl)acetonitrile (6) was carried out using a mixture of concentrated sulfuric and fuming sulfuric acid. The use of fuming sulfuric acid removed residual water from the reaction mixture and reduced the amount of carboxylic acid by-product formed. The reaction mixture was quenched by adding the reaction mixture to a solution of saturated sodium bicarbonate and dichloromethane while maintaining the temperature below 20 ºC. Any carboxylic acid contaminant was readily removed with aqueous work-up. In another aspect of the invention, other acid catalyzed conditions are used by the skilled artisan for alcoho lysis of the nitrile of compound 6 to produce compound 7.
[00051] The resulting methyl 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2- yl)acetate (7) and benzyl amine were coupled in anisole at high temperature to afford 2-(5-(4- (2-morpholinoethoxy)phenyl)pyridin-2-yl)-N-benzylacetamide (KX2-391). An HCl solution formed by adding acetyl chloride to absolute ethanol was added to KX2-391 to form the bis- HCl salt, 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)-N-benzylacetamide dihydrochloride, (KX2-di-HCl).
[00052] The synthesis of the mesylate salt of KX2-391 (KX2-391 -MSA) is depicted in the scheme below:

[00053] 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)-N-benzylacetamide mesylate (KX2-391 MSA) was synthesized in four linear steps starting from compound 5.
The first 3 steps were carried out similar to the procedure discussed above for KX2-391 2HCl to afford methyl 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)acetate (KX2-391). KX2-
391 was converted to the methanesulfonate salt by treatment with methanesulfonic acid
(MSA) in acetone at 50 ºC to afford 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)-JV- benzylacetamide mesylate (KX2-391 MSA).
EXAMPLES Example 1: Small Scale Synthesis of KX2-391

[000343] The preliminary synthesis described below was illustrated in
US20060160800A1. This procedure is useful for small scale reactions, for example, reactions that produce up to 50 g of product.
[000344] For the following synthesis, unless otherwise noted, reagents and solvents were used as received from commercial suppliers. Proton and carbon nuclear magnetic resonance spectra were obtained on a Bruker AC 300 or a Bruker AV 300 spectrometer at 300 MHz for proton and 75 MHz for carbon. Spectra are given in ppm (δ) and coupling constants, J, are reported in Hertz. Tetramethylsilane was used as an internal standard for proton spectra and the solvent peak was used as the reference peak for carbon spectra. Mass spectra and LC-MS mass data were obtained on a Perkin Elmer Sciex 100 atmospheric pressure ionization (APCI) mass spectrometer. LC-MS analyses were obtained using a Luna C8(2) Column (100 x 4.6 mm, Phenomenex) with UV detection at 254 nm using a standard solvent gradient program (Method B). Thin-layer chromatography (TLC) was performed using Analtech silica gel plates and visualized by ultraviolet (UV) light, iodine, or 20 wt % phosphomolybdic acid in ethanol. HPLC analyses were obtained using a Prevail Cl 8 column (53 x 7 mm, Alltech) with UV detection at 254 nm using a standard solvent gradient program (Method A or B). Method A:
A = Water with 0.1 v/v Trifluoroacetic Acid
B = Acetonitrile with 0.1 v/v Trifluoroacetic Acid

Method B:
A = Water with 0.02 v/v Trifluoroacetic Acid
B = Acetonitrile with 0.02 v/v Trifluoroacetic Acid

Synthesis of Η-benzyl-2- (5-bromopyridin-2-yl)acetamide :

[000345] A flask was charged with 5-(5-bromopyridin-2(lH)-ylidene)-2,2-dimethyl- l,3-dioxane-4,6-dione (1.039 g, 3.46 mmol), benzylamine (0.50 mL, 4.58 mmol), and toluene (20 mL). The reaction was brought to reflux under nitrogen for 18 hours, then cooled and placed in a freezer until cold. The product was collected by filtration and washed with hexanes to yield a mass of bright white crystals (1.018 g, 96%).
Synthesis of 4- (2- (4- (4, 4, 5, 5-tetramethylfl, 3, 2] dioxaborolan-2-yl)- phenoxy) ethyl)morpholine :

[000346] To a stirring solution of 4-(4,4,5,5-tetramethyl[l,3,2]dioxaborolan-2-yl)- phenol (2.55 g, 11.58 mmol), 2-morpholin-4-ylethanol (1.60 mL, 1.73 g, 13.2 mmol) and triphenyl phosphine (3.64 g, 13.9 mmol) in methylene chloride (60 mL) at 0 ºC was added dropwise DIAD (2.82 g, 13.9 mmol). The reaction was allowed to warm to room temperature and stir overnight. After 18 hours, additional portions of triphenyl phosphine (1.51 g, 5.8 mmol), 2-morpholin-4-ylethanol (0.70 mL, 5.8 mmol), and DIAD (1.17 g, 5.8 mmol) were added. After stirring an additional 2 hours at room temperature the reaction was concentrated and the residue purified by flash chromatography (5% to 25% EtOAc in CHCI3) to provide the product as a white solid (2.855 g, 74%).
Synthesis of 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)-N-benzylacetamide KX2-391

[000347] A lO rnL reaction tube with a septum closure and stir bar was charged with N- benzyl-2-(5-bromopyridin-2-yl)acetamide (123 mg, 0.403 mmol), 4-(2-(4-(4,4,5,5- tetramethyl[l,3,2]dioxaborolan-2-yl)-phenoxy)ethyl)morpholine (171 mg, 0.513 mmol), and FibreCat 1007 (30 mg, 0.015 mmol). Ethanol (3 mL) was added, followed by aqueous potassium carbonate solution (0.60 mL, 1.0 M, 0.60 mmol). The tube was sealed and heated under microwave conditions at 150 ºC for 10 minutes. The reaction was cooled and concentrated to remove the majority of the ethanol, and then taken up in 10 mL of ethyl acetate and washed successively with water and saturated sodium chloride solution. The organic layer was dried with MgSO4, filtered and concentrated to a white solid. This white solid was triturated with ethyl ether to give KX2-391 as a white solid (137 mg, 79%): mp 135-137 ºC; 1H NMR (300 MHz,CDCl3) δ 8.70 (d, IH, J=2.0 Hz), 7.81 (dd, IH, J=2.4 Hz, J=8.0Hz), 7.65 (br s, IH), 7.49 (d, 2H, J=8.8 Hz), 7.37-7.20 (m, 6H), 7.01 (d, 2H, J=8.8 Hz), 4.49 (d, 2H, J=5.8 Hz), 4.16 (t, 2H, J=5.7 Hz, 3.82 (s, 2H), 3.78-3.72 (m, 4H), 2.84 (t, 2H, J=5.7 Hz), 2.62-2.58 (m, 4H); HPLC (Method B) 98.0% (AUC), tR = 1.834 min.; APCI MS m/z 432 [M+H]+.
Example 2: Intermediate Scale Synthesis of KX2-391 di-hydrochloride
[000348] The synthesis outlined in this example can be used on intermediate-scale reactions. The preparation of batches of at least 50 g of the dihydrochloride salt of KX2-391 is shown in Scheme 1. The linear synthesis consisted of 6 steps, a seventh step being the preparation of one of the reagents, 6-fluoropyridin-3-ylboronic acid (which is also available commercially). The overall yield of the sequence was 35% with an average yield of 83%, with the lowest yielding step being that of 68%. Of the seven steps only one required chromatography. The procedure listed below was performed on a 70 g scale.
[000349] The first step is a Williamson ether synthesis between 4-bromophenol (131 g) and N-chloroethylmorpholine (1 as the HCl salt; 141 g) using K2CO3 powder (3 to 3.5 equivalents) as the base and having acetonitrile as the solvent. The ingredients were mixed and stirred at reflux overnight with high conversion (96.3-99.1%). After dilution with dichloromethane and heptane, the reaction mixture was filtered and evaporated to give the desired product 2 in essentially a quantitative yield (216 g). Note that with similar substrates (e.g., 4-bromo-3-fluorophenol), conversions (even with extensive heating) were not always so high (e.g., 59.9-98.3%). Both the alkyl chloride and the K2CO3 are preferably purchased from Aldrich. If continued heating does not drive reaction to completion, unreacted bromophenol can readily be removed by dissolving the crude reaction mixture in 4 parts toluene and washing out the phenol with 4 parts 15% aqueous NaOH. [000350] One of the reagents required for the second step (Suzuki coupling) was 6- fluoropyridin-3-ylboronic acid (4). Although available commercially, this reagent was readily prepared by lithium-bromide exchange of 5-bromo-2-fluoropyridine (3, 102 g) with n- butyllithium (1.2 eq) at low temperatures (<-60 ºC) in TBME followed by the addition of triisopropylborate (1.65 eq). Both stages of the reaction are brief, with an overall reaction time (including addition times) of ~3 h. Quenching is achieved with aqueous 24% NaOH, which also extracts the product leaving impurities in the organic layer. Once the aqueous layer is removed, it is then neutralized with HCl and extracted with EtOAc. After drying the organics and diluting with some heptane, concentration leads to precipitation/ crystallization of the product. Filtration gave the boronic acid 4 in relatively high purity (96.4% AUC) and good yield (69 g, 79-90%; see note on estimation of yield in the experimental section), which can be used without further purification.
[000351] The second reaction step in the linear sequence (a Suzuki coupling) is a simple reaction to set up; all the reagents [2 (111 g), aqueous Na2CO3, DME, and Pd(PPh3)4 (0.04 eq)] were charged to the reaction flask and the mixture heated at reflux; note that the reaction mixture was degassed to remove oxygen. Once the reaction is complete (within 7 h), the work-up involved decanting (or siphoning off) of reaction solution from the organic salts on the side of the flask (there was no visible aqueous layer), the flask was rinsed, and dried, and the solvent was removed from the combined organics. Crystallization of crude 5 from isopropanol/heptane provided material of improved purity compared to the crude, but still required chromatography (ratio of silica gel to crude was -8.5:1) to obtain material of adequate purity (>98%); the yield was 68% (79.5 g). Use of clean 5 prevented the need for chromatography in the next step, acetonitrile displacement of the fluorine atom. [000352] The replacement of fluoride with acetonitrile was also a simple reaction, and a simple room temperature crystallization of the crude product provided clean 6 in high yield and purity. The reaction involved initial formation of the “enolate” from acetonitrile (6.5 eq) using potassium hexamethyldisilane KHMDS (8 eq)/THF at -10 ºC followed immediately by the addition of fluoride 5 (79 g). The reaction was quick and after one hour quenching was achieved with saturated brine. After drying and evaporation of solvent of the organics, the resulting crude mixture consisted of only two components, the desired product and a much less polar product from apparent self-condensation of acetonitrile. The crude mixture was swirled in isopropanol/heptane and allowed to sit overnight, which resulted in complete crystallization of the product, which was filtered off and washed to provide high purity 6 (99.3% AUC) in good yield (64 g, 76%).
[000353] Methanolysis of 6 (64 g) was accomplished by heating in 40% H2SO4 (in
MeOH) until the reaction was complete (25 h). The reaction was then cooled, stirred with MgSO4 to convert traces of hydro lyzed product (ArCH2-CO2Me) back to product, and then added to cooled, aqueous K2CO3, with simultaneous extraction into dichloromethane. Drying and evaporation of most of the DCM followed by addition of 5% EtOAc (in heptane) and further concentration resulted in the crystallization of the product. Filtration of the solid and washing gave high purity (98.9% AUC) 7 in good yield (82%), additional high purity product (4 g) being obtained from the mother liquors for a total yield of 61.7 g (87%). [000354] The amidation step also involved charging of the reaction vessel with the ingredients (7 (61 g), benzyl amine (3 eq), and high boiling anisole) and then heating at reflux until the reaction was complete. Cooling of the reaction mixture resulted in complete crystallization of the target compound with high purity (98.9%) and good yield (81%). [000355] The final step was the formation of the dihydro chloric salt of the target compound. In order to ensure complete protonation at both basic sites, the reaction was conducted in absolute ethanol, which freely dissolved the dihydrochloride salt. After evaporation to near dryness, the reaction mixture was “chased” with ethanol twice to remove excess hydrogen chloride. The resulting viscous oil was dissolved in ethanol (2 parts) and then added, with rapid stirring, to a large volume (20 parts) EtOAc (ethyl acetate). Filtration, washing with ethyl acetate (no heptane) and vacuum drying provided the dihydrochloride salt of KX2-391 as a creamy-white powder. A total of 68 g (yield of 97%) was obtained of the final salt in high purity (99.6% AUC), which contained traces of EtOAc (4.8% w/w), EtOH (0.3% w/w), and heptane (0.6% w/w; from a final wash with heptane prior to vacuum drying). This salt was also crystallized (instead of the precipitation method described above) from hot EtOH/EtOAc to afford crystalline beads that had much lower entrapped solvent levels (only 0.26% w/w of EtOAc and 0.45% w/w of EtOH) and was free-flowing.

Preparation of 4-(2-(4-bromophenoxy)ethyl)morpholine (2):
[000356] A 5 L three-necked round-bottomed flask, equipped with mechanical stirrer, thermometer with adapter, condenser, and nitrogen inlet (on top of condenser), was charged with 1 (140.7 g, 0.756 mol), 4-bromophenol (130.6 g, 0.755 mol), anhydrous K2CO3 powder (367.6 g, 2.66 mol, 3.5 eq), and acetonitrile (1.3 L). The mixture was vigorously stirred (blade touching bottom of flask) at 80 ºC (overnight), followed by dilution with DCM (500 mL) and heptane (200 mL) and filtration through Celite. Evaporation to dryness (rotovap, then high vac) gave 2 as a light yellow oil (216.00 g, yield of 100%, 96.3% AUC, contains 3.7% unreacted bromophenol). This material was used successfully without further purification.
[000357] 1H NMR (CDCl3) δ 2.57 (t, 4 H), 2.79 (t, 2 H), 3.73 (t, 4 H), 4.08 (t, 2 H), 6.78
(d, 2 H), 7.37 (d, 2 H). MS (from LC/MS): m/z 287.1 [M + I].
[000358] That the bromophenol can be readily removed was demonstrated on a 2 g sample by first dissolving the sample in toluene (8 g) and washing with 8 g of 15% aqueous NaOH; liquid chromatography showed no trace of unreacted bromophenol in the recovered product (1.97 g; 98.5% recovery).

Preparation of 6-fluoropyridin-3-ylboronic acid (4):
[000359] To stirred and cooled (dry ice-acetone bath) anhydrous [TBME] (620 mL; in a
3 L three-necked round-bottomed flask equipped with mechanical stirrer, temperature probe with adapter, and nitrogen inlet) was added (via syringe) 2 M BuLi (352 mL, 0.704 mol, 1.2 eq). To this rapidly stirred and cooled (< -75 ºC) mixture was added a solution of 3 (102.2 g, 0.581 mol) in anhydrous TBME (100 mL) over a period of 13 min during which time the internal temperature rose to -62 ºC. The reaction was stirred for another 45 min (the temperature was maintained between -62 ºC and -80 ºC), followed by the rapid and sequential addition of four portions of triisopropylborate (total of 180 g, 0.957 mol, 1.65 eq). At the end of the addition the internal temperature had risen to -33 ºC. After stirring an additional 45 min over the cold bath (internal temperature lowered from -33 ºC to -65 ºC), the cold bath was removed and the stirred mixture on its own rose to -22 ºC over a period of 50 min. After warming (via water bath) to 6 ºC over a period of 15 min, the stirred reaction mixture was placed in an ice-water bath and then quenched under nitrogen with a cooled solution of NaOH (160 g) in water (500 mL). Once the addition was complete, the internal temperature was 20 ºC. This mixture was stirred at room temperature for 1.5 h. The aqueous layer was removed, neutralized to pH 7 with -350 mL concentrated HCl, and then extracted with EtOAc (3 x 1 L). Because the pH was now 8-9, the aqueous layer was adjusted to pH 7 using ~15 mL concentrated HCl and extracted further (2 x 1 L) with ethyl acetate. The combined EtOAc extracts were dried (Na2SO4), filtered, and concentrated to a volume of -150 mL. With swirling of the concentrate, heptane was added in portions (total volume of 300 mL) resulting in the precipitation/crystallization of the product. Filtration, washing of the solid with heptane (100 mL, 300 mL, then another 300 mL), and air drying gave the title product as an off-white solid (68.6 g, yield of 79-90%*; LC purity of 96.4%, NMR showed an estimated 5.5% w/w of heptane), which was used successfully without further purification. LC/MS showed it to be a mixture of the two following entities, the intensity of the higher molecular weight entity being major (*Note: yield of reaction is 79% if the boronic acid is assumed to be the only constituent and is 90% if it is assumed that the cyclic borate is the only constituent):

1H NMR (CDCl3) δ 7.14 (dd, 1 H), 8.27 (ddd, 1 H), 8.39 (br s, 2 H, 2 OH), 8.54 (fine d, 1 H). MS (from LC/MS): m/z 143.0 [M + 1; for boronic acid] and 370.0 [M + 1; for cyclic borate above].

[000360] A 2 L three-necked round-bottomed flask equipped with mechanical stirrer, thermometer and adapter, condenser, and nitrogen inlet (at top of condenser) was charged with 2 (110.7 g, 0.387 mol), 4 (71.05 g, 0.477 mol, 1.23 eq) and DME (700 mL). The resulting stirred solution was degassed by passing a rapid stream of nitrogen through the stirred solution over a period of 5 min followed by the addition of a degassed solution of Na2CO3 (121.06 g, 1.142 mol, 3 eq) in H2O (250 mL) and also solid Pd(PPh3)4 (19.8 g, 0.044 eq). Immediately after the last addition, the head space above the reaction mixture was purged with nitrogen and the mixture then stirred at 80-85 ºC (internal temperature) for 7 h, followed by cooling to room temperature. Because of the lack of an aqueous layer, the supernatant was decanted, leaving behind the inorganic salts (with adsorbed water). The reaction flask with the inorganic salts was washed with 50% dichloromethane/ethyl acetate (2 x 250 mL), the washes being added to the decanted supernatant. These combined organics were dried (Na2SO4), filtered, and evaporated to dryness to a dark brown oil (148 g). To this oil was added 15O g of 50% heptane/isopropyl alcohol (IPA) and after swirling and cooling (via ice water bath), crystallization began. Additional heptane (50 g) was added and the resulting solid was filtered, washed, and air dried to give 48 g of a light brown solid. After evaporating the filtrate to dryness, the resulting mixture was swirled in 100 mL of 50% heptane/IPA followed by the addition of more heptane (-100 mL), stoppering and placing in the freezer for crystallization. The resulting solid was filtered, washed with heptane, and air dried to give 61 g of a gummy solid. Evaporation of the resulting filtrate gave an oil (34 g) which contained significant less polar impurities including Ph3P=O and so it was partitioned between 2 N HCl (240 mL) and EtOAc (220 mL). The bottom aqueous layer was removed and then stirred with EtOAc while neutralizing with K2CO3 to a pH of 7-8. The EtOAc layer was dried, filtered, and evaporated to dryness (22 g). The 48 g, 61 g, and 22 g portions were chromato graphed over silica gel (1.1 Kg) packed in DCM. Elution with DCM (400 mL), 50% DCM/EtOAc (5 L), and then 50% DCM/EtOAc (8 L) containing increasing amounts of MeOH/Et3N (beginning with 1.5% MeOH/1% Et3N and ending with 5% MeOH/3% Et3N) gave 77.68 g of a viscous oil (purity 98.0%) which immediately crystallized upon swirling in heptane (300 mL). Filtration, washing with heptane and air drying gave 75.55 g (98.7% AUC) of solid 5. Additional pure 5 (total of 3.9 g, 98.6-99.3% AUC) was obtained from earlier chromatographic fractions containing Ph3P=O by cleaning them up as done for the above 34 g sample, followed by evaporative crystallization. The total yield of 5 was 79.5 g (68%). 1H NMR (CDCl3) δ 2.59 (t, 4 H), 2.84 (t, 2 H), 3.75 (t, 4 H), 4.16 (t, 2 H), 6.97 (dd, 1 H), 7.01 (d, 2 H), 7.46 (d, 2 H), 7.92 (ddd, 1 H), 8.37 (fine d, 1 H). MS (from LC/MS): m/z 303.2 [M + I].

Preparation of 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)acetonitrile (6):
[000361] A 3 L three-necked round-bottomed flask was equipped with mechanical stirrer, thermometer and adapter, additional funnel, and nitrogen inlet (on top of addition funnel, positive pressure through a bubbler). With a rapid stream of nitrogen going through the bubbler, the stopper was removed and the flask was charged with KHMDS (415.8 g, 2.08 mol) and then anhydrous THF (1 L). To the stirred and cooled (ice/methanol bath, internal temperature of solution was -8 ºC) KHMDS/THF solution was added dropwise a solution of MeCN (70 g) in THF (110 mL) over a period of 22 min followed immediately by the relatively rapid (4 min) addition of a solution of 5 (79.06 g, 0.262 mol) in THF (400 mL), after which time the internal temperature of the reaction mixture had reached 10 ºC. With continued cooling (1 h) the internal temperature was -6 ºC and by TLC the reaction appeared complete. After an additional 30 min (internal temperature of -3 ºC), the reaction mixture was quenched with saturated brine (1 L) and diluted with EtOAc (500 mL). After removing the aqueous layer, the organic solution was dried (Na2SO4), filtered, and evaporated to dryness (to an oil) followed by completely dissolving in IPA (150 mL), diluting with heptane (300 mL), adding seed crystals (prepared by dissolving -100 mg of crude oil in IPA (-150 mg) and diluting with heptane (-2.5 mL)), and allowing to stand overnight. After stirring to break up the crystalline solid, the solid was filtered, washed with 250 mL 2:1 heptane/IP A and then multiple washes with heptane and air dried to give 64.38 g (yield of 76%) of title product 6 as a crystalline tan solid (LC purity of 99.3%). Another 5.88 g of less pure material was obtained from the filtrate.
[000362] 1H NMR (CDCl3) δ 2.59 (t, 4 H), 2.84 (t, 2 H), 3.74 (t, 4 H), 3.97 (s, 2 H),
4.17 (t, 2 H), 7.02 (d, 2 H), 7.46 (d, 1 H), 7.51 (d, 2 H), 7.87 (dd, 1 H), 8.77 (fine d, 1 H). MS (from LC/MS): m/z 324 A [M + I].

Preparation of methyl 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)acetate (7): [000363] A 2 L single-necked round-bottomed flask was charged with 6 (64.00 g, 0.198 mol) and MeOH (360 g) followed by the slow, careful, and dropwise addition OfH2SO4 (240 g) and the resulting homogeneous solution stirred at reflux (115 ºC oil bath) until the reaction was complete (25 h with 0.8% unreacted starting material) with 3.5% ArCH2CO2H. After brief cooling, MgSO4 (75 g) was added and the mixture swirled and allowed to stand an additional 45 min (composition now 96.3% product, 0.8% unreacted starting material, and 2.5% ArCH2CO2H). The reaction mixture was then added slowly to a rapidly stirred and cooled (ice-water bath) mixture of DCM (2 L) and a solution OfK2CO3 (450 g) in H2O (600 mL). The resulting emulsion was allowed to stand overnight. The clear portions of organic solution were siphoned off and the remainder portions were treated iteratively with water and DCM, the clear organics being combined with the original portion that was siphoned off. The combined organics were dried (Na2SO4), filtered, and concentrated to a volume of ~1.2 L followed by the addition of 300 mL of 5% EtOAc (in heptane) and then heptane (300 mL) and the mixture concentrated (rotovap with heat) again to remove the DCM. At this point 15 mL EtOAc was added and the hot mixture swirled until crystallization had begun, swirling continued until crystallization was near complete, and then allowed to stand and cool to room temperature for complete crystallization. The solid was then filtered, washed with 300 mL 5% EtOAc (in heptane) and heptane (100 mL) and then fully air dried to give 57.74 g (yield of 82%) of 7 as a light yellow solid (98.9% AUC). Another 3.94 g of clean product (97.9% AUC) was obtained from the filtrate (total yield of 87%).
[000364] 1H NMR (CDCl3) δ 2.60 (t, 4 H), 2.84 (t, 2 H), 3.74 (overlapping t and s, 6 H),
3.89 (s, 2 H), 4.17 (t, 2 H), 7.01 (d, 2 H), 7.34 (d, 1 H), 7.49 (d, 2 H), 7.80 (dd, 1 H), 8.74 (fine d, 1 H). MS (from LC/MS): m/z 357.4 [M + I].

Preparation of 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)-N-benzylacetamide (KX2-391 free base).
[000365] A l L single-necked round-bottomed flask was charged with 7 (61.4 g, 0.172 mol), benzyl amine (55.6 g, 0.519 mol, 3 eq), and anhydrous anisole (300 g) and then stirred at reflux until reaction was essentially complete (23 h, 165 ºC oil bath temperature; internal temperature was 147 ºC) and then allowed to cool to near room temperature. A portion (1 mL) of the reaction mixture was diluted with toluene (1 mL) resulting in the complete crystallization of that portion. This seed was then added to the reaction mixture and allowed to stand until the whole reaction mixture had crystallized to a single block. Toluene (150 mL) was added and the mixture swirled to break up the solid. Heptane/toluene (1 :1, 100 mL) was added and the solid mixture broken up further. Finally, heptane (50 mL, then 25 mL) was added and the mixture broken up even further, allowing to stand an additional 30 min before filtering the solid. Filtration of the solid, washing with 2:1 toluene/heptane (300 mL), 1 :2 toluene/heptane (300 mL), and then heptane (2 x 300 mL), and then drying (air, then high vac) gave 60.16 g (yield of 81%) of title product as a white solid (≥98.9% AUC). Another 2.5 g of less pure (97.4%) material was obtained from the mother liquors. 1H NMR (CDCl3) δ 2.60 (t, 4 H), 2.83 (t, 2 H), 3.74 (t, 4 H), 3.82 (s, 2 H), 4.18 (t, 2 H), 4.49 (d, 2 H), 7.01 (d, 2 H), 7.2-7.35 (m, 6 H), 7.49 (d, 2 H), 7.64 (br t, 1 H), 7.81 (dd, 1 H), 8.69 (fine d, 1 H). MS (from LC/MS): m/z 432.5 [M + I].
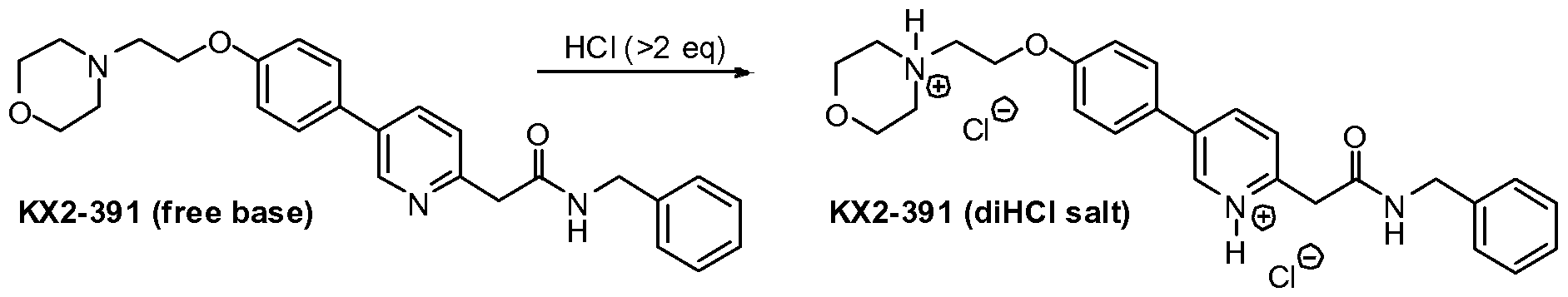
Preparation of 4-(2-(4-(6-(2-(benzylamino)-2-oxoethyl)pyridinium-3-yl)phenoxy)ethyl)- morpholin-4-ium chloride (KX2-391, diHCl salt).
[000366] To a stirred suspension of KX2-391 (free base, 60.00 g) in absolute EtOH (600 niL) was added 170 niL of 2.5 M HCl (in ethanol), 25 niL EtOH being added to wash down the sides of the flask. The resulting homogeneous solution was stirred at room temperature (20 min) and then evaporated to near dryness (to frothing). After chasing with EtOH (2 x 150 mL), the residue was taken up again in EtOH (150 mL) and then was followed by the slow addition of heptane until the mixture appeared saturated (33 mL required for cloudiness to remain). After sitting overnight, two layers had formed. After adding additional heptane (250 mL) crystallization still could not be induced and so the reaction mixture was concentrated to a volume of -200 mL at which time the mixture was homogeneous. This thick homogeneous solution was added dropwise to very rapidly stirred (mechanical) EtOAc (2 L). After the addition was complete, a 25 mL EtOH rinse of the original flask and addition funnel was added to the rapidly stirred mixture. The rapid stirring was continued for another ~1 h and then the mixture was filtered and the solid (partly gummy) was washed with EtOAc (300 mL) and then heptane. As soon as the heptane wash began, the solid got much gummier. The fritted Buchner funnel and its contents were covered (paper towel/rubber band) and immediately placed in the vacuum oven. After overnight vacuum at ~45 ºC, the vacuum was released under nitrogen, and the Buchner funnel containing the product (foamy solid) was immediately placed in a zip-lock back and then, under nitrogen (glove bag), transferred to a bottle and the foamy solid broken up (spatula) to a powder. A second night under high vacuum (-45 ºC) resulted in only 1.3 g of additional weight loss. Constant weight was essentially attained with the third night of high vacuum (~45 ºC) where only 0.2 g of weight was lost. The final weight of material was 68.05 g (yield of 97%), containing 0.29 eq (4.8% w/w) of EtOAc, 0.035 eq (0.3% w/w) EtOH, and 0.03 eq (0.6% w/w) heptane. The purity was 99.6%.
1H NMR (DMSO-d6) δ 3.1-3.3 (m, 2 H), 3.45-3.65 (m, 4 H), 3.8-4.0 (m, 4 H), 4.11 (s, 2 H), 4.32 (d, 2 H), 4.57 (t, 2 H), 7.19 (d, 2 H), 7.2-7.4 (m, 5 H), 7.88 (d, 2 H), 7.93 (d, 1 H), 8.68 (dd, 1 H), 8.99 (br t, 1 H), 9.10 (fine d, 1 H), 11.8 (br s, 1 H). MS (from LC/MS): m/z 432.5 [M + 1 of free base].
Elemental analysis (for C26H29N3O3 • 2 HCl • 0.035 EtOH • 0.29 EtOAc • 0.03 heptane • 0.8 H2O): Calculated (%): C, 60.03; H, 6.54; N, 7.65; Cl, 12.91 Observed (%):C, 59.85/59.97; H, 6.54/6.47; N, 7.67/7.67; Cl, 13.10/13.24 Calculated FW: 534.63 (does not take into account the 0.8 H2O which probably arose during handling of this very hygroscopic powder, since 1H NMR shows no evidence for H2O). [000367] The ethyl chloride level in this material was measured and found to be 98 ppm. The sample was also analyzed and found to contain 5,800 ppm of heptane. [000368] Analysis of another portion of this sample yielded the following results: 99.6% AUC, 1640 ppm ethanol, 41,480 ppm ethyl acetate, 5600 ppm heptane, no anisole detected, and 120 ppm ethyl chloride.
[000369] A procedure for recrystallizing the salt was also developed using the above dried salt. This procedure would work just was well on the highly pure crude salt (containing residual EtOH) obtained from concentrating the HCl salt-forming reaction mixture: [000370] The salt (575 mg) was dissolved in twice the mass of absolute EtOH (1.157 g) and then heated under nitrogen. To this hot solution (stirred) was added 1.6 g of 25% EtOH (in EtOAc) followed by the addition of EtOAc (0.25 mL) resulting in a cloudiness that remained. The cloudy hot solution was allowed to cool to room temperature during which time crystallization occurred. After crystallization was complete (2 h), the crystalline solid was filtered, washed with anhydrous EtOAc (~40 mL), and vacuum dried to give 424 mg of the dihydrochloride salt of KX2-391 as a free-flowing solid (tiny beads, 99.8% AUC) containing only 0.05 eq (0.45% w/w) of EtOH and 0.015 eq (0.26% w/w) of EtOAc. Slightly better recovery (460 mg from 586 mg) was attained using isopropanol/EtOAc but the level of solvent entrapment was higher [0.085 eq (1.0% w/w) of isopropanol and 0.023 eq (0.4% w/w) OfEtOAc].
Example 3: Large Scale Synthesis of KX2-391 di-HCl
[000371] Reagents and solvents were used as received from commercial suppliers.
Progress of the reactions was monitored by HPLC, GC/MS, or 1H NMR. Thin-layer chromatography (TLC) was performed using Analtech silica gel plates and visualized by UV light (254 nm). High pressure liquid chromatography (HPLC) was performed on an Agilent 1100 Series instruments. Proton and carbon nuclear magnetic resonance spectra were obtained using a Bruker AV 300 at 300 MHz for proton and 75 MHz for carbon. The solvent peak was used as the reference peak for proton and carbon spectra.
Preparation of 4-(2-(4-Bromophenoxy)ethyl)morpholine (2) [000372] A 50 L jacketed reactor equipped with a reflux condenser and temperature probe was charged with 4-(3-chloropropyl)morpholine (2.44 kg, 0.54 mol), 4-bromophenol (2.27 kg, 0.54 mol, 1.0 equiv.), powdered potassium carbonate (6.331 kg, 1.88 mol, 3.50 equiv.), and DMF (12.2 L) and stirred. The reaction mixture was then heated to 60-65 ºC and stirred overnight. After 17.5 h, the reaction mixture was cooled to 20-25 ºC. The reaction mixture was charged to a different reactor equipped with bottom valve for the work-up. While maintaining a temperature between 20-30 ºC, DI water (48.7 L) was charged to the reactor. The phases were separated. The aqueous layer was extracted with MTBE (3 x 24.4 L). To the combined organics, DI water (18.3 L) and then 6M sodium hydroxide (18.2 L) were added. The mixture was stirred for 2-5 minutes and the phases were separated. The organic phase was washed with water (24.4 L) and brine (24.4 L), dried over magnesium sulfate, filtered, and concentrated to give 337Og of a yellow oil (89% crude yield, 99.4% AUC by HPLC).
Preparation of 6-fluoropyridin-3-ylboronic acid (4)
[000373] A 72 L reactor equipped with reflux condenser, and temperature probe. To the reactor 5-bromo-2-fluoropyridine (1.17 L, 0.568 mol), toluene (18.2 L), and triisopropyl borate (3.13 L, 0.68 mol, 1.2 equiv.) were charged and stirred. Tetrahydrofuran (4.4 L) was added to the reactor and the reaction mixture was cooled to between -35 to -50 ºC. While maintaining a temperature between -35 to -45 ºC, n-butyl lithium (2.5 M solution of hexanes, 5.44 L, 0.68 mol, 1.2 equiv.) was cautiously added to the reactor. After 5 h, the reaction was deemed complete and the reaction mixture was warmed to between -15 to -20 ºC. To the reaction was added 2M HCl (11.80L) to the reactor while maintaining a temperature between -15 ºC and 0 ºC. The reaction mixture was stirred at 18 to 23 ºC for (16 h) and the phases were separated. The organics were then extracted with 6 M sodium hydroxide (6.0 L). The acidic anbasic aqueous phases were mixed in the reactor and 6 M HCl (2.5 L) was added until pH 7.5 was achieved. Sodium chloride (6.0 kg) was then added to the aqueous phase. The aqueous phase was then extracted with THF (3 x 20 L). The combined organics were dried with magnesium sulfate and concentrated to give 1300 g of a tan solid (81% crude yield).
Preparation of 4-(2-(4-(6-fluoropyridin-3-yl)phenoxy)ethyl)morpholine (5)
[000374] A 72 L reactor equipped with reflux condenser, sparging tube, bubbler, and temperature probe was charged with 6-fluoropyridin-3-ylboric acid (2.84 kg, 1.24 equiv.), A- (2-(4-bromophenoxy)ethyl)morpholine (4.27 kg, 1.0 equiv.), and DME (27 L). Agitation was started and sodium carbonate (4.74 kg, 3.0 equiv.) as a solution in DI water (17.1 L) was then charged to the reaction mixture. Argon was bubbled through the reaction mixture for 50 minutes. Under an argon atmosphere, tetrakis(triphenylphosphine)palladium (750 g, 0.04 equiv.) was added to the reaction mixture as a slurry in DME (1.0 L). The reaction mixture was heated to 75 – 85 ºC and stirred overnight (17 h). The reaction mixture was cooled to between 18 – 22ºC. DI water (26.681kg) and MTBE (26.681 L) were charged to the reactor and stirred for 5 minutes. The phases were separated and the aqueous phase was extracted with MTBE (2 x 26.7 L). The combined organics were extracted with 2M HCl (1 x 15.0 L, 3 x 21.8 L). The aqueous phase was then charged back to the reactor and ethyl acetate was added (26.7 L). The pH was adjusted to 6.2 using 6 M sodium hydroxide (26.7 L) while maintaining a temperature between 15 – 25 ºC. The phases were separated and the aqueous phase was extracted with ethyl acetate (2 x 26.7 L). The combined organics were dried with magnesium sulfate and concentrated to give 4555 g of a residue (101% crude yield, 67.1% AUC by HPLC).
Purification of 4-(2-(4-(6-fluoropyridin-3-yl)phenoxy)ethyl)morpholine (5)
[000375] The crude product (575 g) was purified by silica gel chromatography by eluting with methanol/ethyl acetate/heptane (30% ethyl acetate/heptane, 50% ethyl acetate/heptane, 75% ethyl acetate/heptane, 100% ethyl acetate, and 5% methanol/ethyl acetate). Concentration of the pure fractions by TLC (10% methanol/dichloromethane, Rf = 0.3) provided 420 g of a light brown solid (73% recovery, >99.9% AUC by HPLC).
Preparation of 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)acetonitrile (6)
[000376] A 1 M solution of NaHMDS (2.0 L, 5.0 equiv.) in THF was charged to a 5-L flask and cooled to -20 to -15 ºC. While maintaining a temperature below -10 ºC, fluoride (119.7g, 1.0 equiv.) in THF (500 mL) was charged to the flask over 20 minutes. Acetonitrile (82.5 mL, 4.0 equiv.) in THF (170 mL) was added to the flask over 20 minutes, while maintaining a temperature below -10 ºC. The reaction mixture was then stirred for 1 h. To the reaction was added brine (1.5 L, 12.6 vol.) at a rate as to maintain a temperature below 10 ºC. The solution was then warmed to room temperature and the layers were allowed to separate. The mixture was filtered over Celite and washed with THF (1 x 200 mL, 1 x 100 mL). The aqueous phase was extracted with toluene (750 mL). The combined organics were dried with magnesium sulfate, filtered, washed with toluene (2 x 25OmL), and concentrated to dryness. Toluene (IL) was added and the solution was concentrated to dryness again to give 169.8 g of an oil. MTBE (1190 niL, 7 vol.) was added to the oil at 50 ºC and stirred for 15 minutes. Heptane (850 rnL, 5vol.) was added over ten minutes at 50 ºC. The mixture was then cooled to room temperature over 1.5 h and stirred for 2 h. The slurry was filtered, washed with 1 :4 MBTE/heptane (2 x 100 mL), and dried in an oven overnight at 45 ºC to give 102.3 g of an off-white solid (80% yield, 98.8% AUC by HPLC).
Preparation of methyl 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)acetate (7)
[000377] Nitrile 6 (101 g) and methanol (1.01 L, 10 vol.) were charged to a 3-L flask equipped with stir bar and thermocouple. Concentrated H2SO4 (175 mL, 10.0 equiv.) was added drop wise to the solution over 15 minutes while maintaining a temperature below 60 ºC. Followed by 30% fuming sulfuric acid (124 mL) was added drop wise to the solution while maintaining a temperature below 60 ºC. The solution was then heated to reflux with a heating mantle and stirred overnight. When the reaction was deemed complete, it was cooled to 20 ºC. In a second flask (22 L), saturated sodium bicarbonate (10.7 L) and dichloromethane (1.1 L) were charged and cooled to 15 ºC. While maintaining a temperature below 20 ºC, the reaction mixture was added to the sodium bicarbonate/dichloromethane mixture. The quench was stirred for 15 minutes and the phases were separated. The aqueous phase was extracted with dichloromethane (I x 55OmL, 1 x 30OmL). The combined organics were dried with magnesium sulfate and concentrated to dryness to give 105 g of an orange solid (94% crude yield, 97.7% AUC by HPLC).
Preparation of 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)-N-benzylacetamide (KX2-391)
[000378] Ester 7 (103 g), anisole (513 mL, 5 vol.), and benzylamine (94 mL, 3.0 equiv.) were charged to a 3 L flask equipped with thermocouple and overhead stirrer. The reaction mixture was then heated to 142 ºC and stirred for two days. The reaction mixture was cooled to 45-50 ºC and stirred for 2 hours. To the mixture was added n-heptane (1.5 L) dropwise over an hour. The solution was cooled to room temperature over three hours and then stirred overnight. The resulting slurry was filtered, washed with 4:1 Anisole/n-heptane (200 mL) and n-heptane (3 χ100 mL). Drying in the oven overnight, the resulting product was 112. Ig of a tan solid (90% yield, 99.6% AUC by HPLC). The use of a single isomer of heptane was essential to adequately quantitate the residual solvent. See Figure 5 for 1H NMR of KX2- 391. Preparation of 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)-N-benzylacetamide dihydrochloride salt (KX2-391 2HC1)
[000379] EtOH (1.0 L) was charged to a 2-L flask and acetyl chloride (62.5 niL, 3.0 equiv.) was added slowly to the flask and stirred for 40 minutes. The resulting solution was added to KX2-391 (100 g) over 30 minutes while maintaining a temperature of 30 ºC. The solution was concentrated to a mass of 270 g. The concentrated solution was added to ethyl acetate (2 L) over 20 minutes with rapid stirring. The mixture was stirred overnight and then filtered under nitrogen to give two distinct solid products, tan solids (73.5 g) and darker solids (42.2 g). The solids were dry blended to give a combined yield of 99%. The HPLC analysis indicated 99.0% purity (AUC). Analysis indicated that ethanol was present at 2530 ppm, ethyl acetate at 48,110 ppm, ethyl chloride at 170 ppm, and no heptane and anisole were detected. Palladium content was assayed three times and measured to be 29 ppm, 2 ppm, and less than 1 ppm.
PATENT
CN 106810490
| 2-(5-(4-(2-morpholinylethoxy)phenyl)pyridine-2-yl)-N-benzyl-acetamide, development code KX -01, KX2-391, have the structure shown in formula I. |
| |
| Patent CN10118473B and US7300931B disclose compound KX2-391, and disclose its application in the treatment of cell proliferative disorders. KX2-391 and its pharmaceutically acceptable salts are effective Src tyrosine kinase inhibitors, which can effectively treat diseases and disorders regulated by Src kinase. KX2-391 has a GI50 of 9-60 nM in cancer cell lines and is currently in clinical phase II. |
| KX2-391 has polymorphism. Polymorphism refers to the phenomenon that the same compound can form two or more molecular spatial arrangements by controlling its different production conditions to produce different solid crystals. Different crystal forms of the same compound have the same chemical composition. , But the microscopic crystal structure is different, which leads to differences in their appearance, physical and chemical properties and biological activity. The phenomenon of polymorphism directly affects the processing performance of the drug formulation, and affects the stability, solubility, and bioavailability of the drug, and further affects the quality, safety, effectiveness and application of the drug. Therefore, in drug research and development, the polymorphism of drugs should be fully considered. At present, KX2-391 is still in the research and development stage, and a comprehensive study of its solid form is of great significance to the research and development of KX2-391 and the approval of the market. |
| Example 1 |
| 2-(5-(4-(2-morpholinylethoxy)phenyl)pyridin-2-yl)-N-benzyl-acetamide (KX2-391) crystal form (i.e. having formula (I) The structure of the crystalline diaryl compound, the subsequent examples are referred to as the preparation of KX2-391 crystal form B) |
| Put KX2-391 (5.0g) in a 500ml round bottom flask, add 150ml methanol to dissolve KX2-391 completely, and place it at 50°C and stir. 300ml of purified water was gradually added dropwise. After the addition, the resulting slurry was stirred at room temperature for 1 hour to crystallize, filtered with suction, and dried under vacuum at 50°C. The resulting solid was KX2-391 crystal form B. The purity detected by HPLC is ≥99.83%. |
| Example 2 Preparation of KX2-391 crystal form B |
| Put KX2-391 (5.0g) in a 100ml round bottom flask, add 25ml of DMSO to dissolve KX2-391 completely, and stir at room temperature. Gradually add 50ml of purified water dropwise. After the dropwise addition, the resulting slurry was stirred at 0°C for 1h to crystallize, filtered with suction, and dried under vacuum at 50°C. The resulting solid was KX2-391 crystal form B. HPLC detection purity ≥99.81%. |
| Example 3 Preparation of KX2-391 crystal form B |
| Put KX2-391 (5.0g) in a 250ml round bottom flask, add 15ml of dichloromethane to dissolve KX2-391 completely, and stir at 30°C. Gradually add 100ml of n-heptane dropwise. After the dropwise addition, the resulting slurry was stirred at room temperature for 0.5h to crystallize, filtered with suction, and dried under vacuum at 50°C. The resulting solid was KX2-391 crystal form B. HPLC detection purity ≥99.80%. |
| Example 4 Preparation of KX2-391 crystal form B |
| Put KX2-391 (2.0g) in a 500ml round bottom flask, add 100ml of acetone to completely dissolve KX2-391, and stir at room temperature. Gradually add 150 ml of n-hexane, and after the addition is complete, the resulting slurry is stirred at 0°C for 1 h to crystallize, filtered with suction, and dried in vacuum at 50°C. The obtained solid is KX2-391 crystal form B. HPLC detection purity ≥99.79%. |
| Example 5 Preparation of KX2-391 crystal form B |
| Put KX2-391 (2.0g) in a 250ml round bottom flask, add 50ml of THF to dissolve KX2-391 completely, and place it at 40°C and stir. Gradually add 100 ml of methyl tert-butyl ether dropwise. After the dropwise addition, the resulting slurry was stirred at room temperature for 2 hours to crystallize, filtered with suction, and dried under vacuum at 50°C. The resulting solid was KX2-391 crystal form B. HPLC detection purity ≥99.81%. |
| Example 6 Detection of KX2-391 crystal form B |
| The KX2-391 crystal form B prepared in Example 1 was tested by XRPD method. The equipment used is RIGAKU TTR III X-ray powder diffractometer, measurement conditions and methods: Cu (target), 40KV-30mA (working voltage and current), 2θ=2~50 degrees (scanning range), 4.0deg /min. (scanning speed), the obtained spectrum is shown in Figure 1. It can be seen from Figure 1 that the XRPD spectrum of KX2-391 crystal form B provided in Example 1 is 2.10, 3.68, 4.16, 6.24, 8.33, There are peaks at 12.53, 16.26, 16.75, 18.33, 19.05, 19.85, 21.00, 21.50, 21.92, 22.50, 23.16, 25.08, 25.35, 25.70, 27.49, 29.67, 33.97, and 38.43. |
| The invention also adopts the DSC-TGA method to detect the crystal form B of KX2-391 provided by the invention. The equipment used is METTLER TOLEDO’s TGA-DSC, testing environment conditions 22℃, relative humidity RH68%, temperature range 0-400℃, heating rate 12℃/min, protective gas N 2 , The resulting maps are shown in Figure 2 and Figure 3. It can be seen from Figure 2 that the DSC spectrum of KX2-391 crystal form B provided in Example 1 has endothermic peaks at 126.9°C and 137.4°C. It can be seen from Figure 3 that the TGA pattern of KX2-391 crystal form B provided in Example 1 has no significant weight loss before 200°C. |
PAPER
Journal of Medicinal Chemistry (2018), 61(11), 4704-4719.
[1]. Lau GM, et al. Expression of Src and FAK in hepatocellul
https://pubs.acs.org/doi/10.1021/acs.jmedchem.8b00164
Abstract

The discovery of potent, peptide site directed, tyrosine kinase inhibitors has remained an elusive goal. Herein we describe the discovery of two such clinical candidates that inhibit the tyrosine kinase Src. Compound 1 is a phase 3 clinical trial candidate that is likely to provide a first in class topical treatment for actinic keratosis (AK) with good efficacy and dramatically less toxicity compared to existing standard therapy. Compound 2 is a phase 1 clinical trial candidate that is likely to provide a first in class treatment of malignant glioblastoma and induces 30% long-term complete tumor remission in animal models. The discovery strategy for these compounds iteratively utilized molecular modeling, along with the synthesis and testing of increasingly elaborated proof of concept compounds, until the final clinical candidates were arrived at. This was followed with mechanism of action (MOA) studies that revealed tubulin polymerization inhibition as the second MOA.

[1]. Lau GM, et al. Expression of Src and FAK in hepatocellular carcinoma and the effect of Src inhibitors on hepatocellular carcinoma in vitro. Dig Dis Sci, 2009, 54(7), 1465-1474.
//////////Tirbanibulin, Klisyri, FDA 2020, 2020 APPROVALS, KX2 391, KX 2391, KX-01, actinic Keratosis
O=C(CC1=NC=C(C2=CC=C(OCCN3CCOCC3)C=C2)C=C1)NCC4=CC=CC=C4
Lonafarnib

Lonafarnib
- Molecular FormulaC27H31Br2ClN4O2
- Average mass638.822 Da
193275-84-2[RN]
1-Piperidinecarboxamide, 4-[2-[4-[(11R)-3,10-dibromo-8-chloro-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridin-11-yl]-1-piperidinyl]-2-oxoethyl]-
4-[2-[4-[(11R)-3,10-Dibromo-8-chloro-6,11-dihydro-5Hbenzo[5,6]cyclohepta[1,2-b]pyridin-11-yl]-1-piperidinyl]-2-oxoethyl]-1-piperidinecarboxamide
8191(+)-4[2-[4-(8-Chloro-3,10-dibromo-6,11-dihydro-5H-benzo[5,6] cyclohepta[1,2-b]-pyridin-11(R)-yl-1-piperidinyl]-2-oxo-ethyl]-1-piperidinecarboxamide
(R)-4-(2-(4-(3,10-dibromo-8-chloro-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridin-11-yl)piperidin-1-yl)-2-oxoethyl)piperidine-1-carboxamide
4-{2-[4-(3,10-dibromo-8-chloro-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridin-11-yl)piperidin-1-yl]-2-oxoethyl}piperidine-1-carboxamide
D04768
lonafarnibum TM5989100 UNII:IOW153004F
FDA APPROVED 11/20/2020, Zokinvy
To treat rare conditions related to premature aging
Press Release
Drug Trials Snapshotлонафарниб [Russian] [INN]لونافارنيب [Arabic] [INN]氯那法尼 [Chinese] [INN]
Lonafarnib, sold under the brand name Zokinvy, is a medication used to reduce the risk of death due to Hutchinson-Gilford progeria syndrome and for the treatment of certain processing-deficient progeroid laminopathies in people one year of age and older.[1][2]
The most common side effects included nausea vomiting, diarrhea, infection, decreased appetite and fatigue.[1]
Medical uses
Lonafarnib is indicated to be used to reduce the risk of death due to Hutchinson-Gilford progeria syndrome and for the treatment of certain processing-deficient progeroid laminopathies in people one year of age and older.[1][2]
Contraindications
Lonafarnib is contraindicated for co-administration with strong or moderate CYP3A inhibitors and inducers, as well as midazolam and certain cholesterol-lowering medications.[1]
History
Lonafarnib, a farnesyltransferase inhibitor, is an oral medication that helps prevent the buildup of defective progerin or progerin-like protein.[1] The effectiveness of lonafarnib for the treatment of Hutchinson-Gilford progeria syndrome was demonstrated in 62 patients from two single-arm trials (Trial 1/NCT00425607 and Trial 2/NCT00916747) that were compared to matched, untreated patients from a separate natural history study.[1][2] Compared to untreated patients, the lifespan of Hutchinson-Gilford progeria syndrome patients treated with lonafarnib increased by an average of three months through the first three years of treatment and by an average of 2.5 years through the maximum follow-up time of 11 years.[1] Lonafarnib’s approval for the treatment of certain processing-deficient progeroid laminopathies that are very rare took into account similarities in the underlying genetic mechanism of disease and other available data.[1] The participants were from 34 countries around the world, including the United States.[2]
The U.S. Food and Drug Administration (FDA) granted the application for lonafarnib priority review, orphan drug, and breakthrough therapy designations.[1] In addition, the manufacturer received a rare pediatric disease priority review voucher.[1] The FDA granted the approval of Zokinvy to Eiger BioPharmaceuticals, Inc.[1]

Research
Lonafarnib is a farnesyltransferase inhibitor (FTI) that has been investigated in a human clinical trial as a treatment for progeria, which is an extremely rare genetic disorder in which symptoms resembling aspects of aging are manifested at a very early age.[3][4]
Lonafarnib is a synthetic tricyclic halogenated carboxamide with antineoplastic properties.[5] As such, it is used primarily for cancer treatment. For those with progeria, research has shown that the drug reduces the prevalence of stroke and transient ischemic attack, and the prevalence and frequency of headaches while taking the medication.[6] A phase II clinical trial was completed in 2012, which showed that a cocktail of drugs that included lonafarnib and two other drugs met clinical efficacy endpoints that improved the height and diminished the rigidity of the bones of progeria patients.
SYN
| EP 1019392; EP 1380581; JP 1999501671; WO 9723478 |
Introduction of a bromine atom at the 10-position of the benzocycloheptapyridine (I) was achieved by the following sequence. Nitration of (I) using NaNO3-H2SO4 afforded a mixture of nitro compounds (II) and (III), from which the major 9-nitro isomer (III) was separated by silica gel chromatography. Reduction of the nitro group of (III) with iron filings and CaCl2 in refluxing aqueous ethanol gave amine (IV), which was brominated at position 10 with Br2 in AcOH. The brominated aniline (VI) was then deaminated by diazotization, followed by reduction of the resulting diazonium salt with hypophosphorous acid to give trihalo compound (VI). Hydrolysis of carbamate group of (VI) in boiling concentrated HCl afforded piperidine (VII). Subsequent reduction of the C-11 double bond of (VII) was carried out using DIBAL-H in refluxing toluene to afford the corresponding racemic piperidine. Separation of enantiomers was achieved by HPLC on a ChiralPak AD column or by chemical resolution using N-acetyl-L-phenylalanine as the resolving agent. The appropriate R-(+) enantiomer (VIII) was coupled with N-Boc-piperidylacetic acid (IX) in the presence of EDC and HOBt to yield protected amide (X). Hydrolysis of the Boc protecting group was performed with trifluoroacetic acid, and the resulting piperidine (XI) was finally treated with trimethylsilyl isocyanate to give the desired carboxamide (3-5).
SYN2
EP 1091954; JP 2002519419; WO 0001689
J Org Chem 2000,65(18),5451
The starting product is the benzocyclohetapyridine (VII), already reported as intermediate (VII) in the synthesis of 25468001a. Compound (VII) is resolved into its atropaisomers by digestion with Toyobo LIP-300 enzyme in the presence of trifluroethyl isobutyrate (XII) to give a mixture of unreacted (-)-(XIII) and acylated compound (+)-(XIV) that are separated by acid extraction. The undesired atropaisomer (-)-(XIII) can be recovered by thermal razemization in diethyleneglycol dibutyl ether at 210 C and new enzymatic separation. The acid hydrolysis of the separated amide (+)-(XIV) produces the desired atropaisomer (+)-(XIII), which is reduced to the (R)-(+)-(VIII), intermediate already reported with no. (VIII) in the synthesis of 25468001a. (6,7)
SYN 3
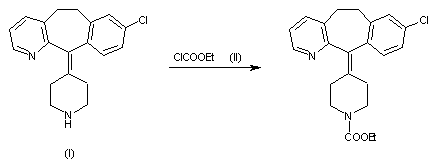
1) By carboxylation of 8-chloro-6,11-dihydro-11-(4-piperidylidene)-5H-benzo[5,6]cyctohepta[1,2-b]pyridine (I) with ethyl chloroformate (II) in refluxing benzene.
SYN 4
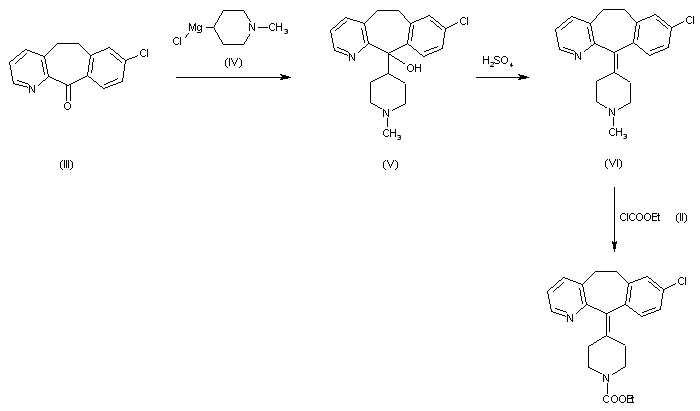
2) By reaction of 8-chloro-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridin-11-one (III) with the Grignard reagent (IV) to give the tertiary carbinol (V), which is dehydrated with 85% H2SO4 affording 8-chloro-11-piperidinylidene derivative (VI). Finally, cornpound (VI) is treated with ethyl chloroformate (II) in toluene.
SYN 5
J Med Chem 1997,40(26),4290
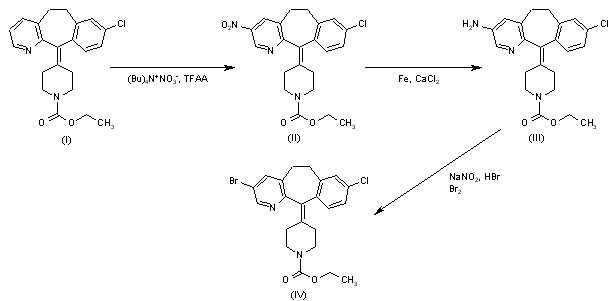
The nitration of loratadine (I) (1) by means of tetrabutylammonium nitrate and trifluoroacetic anhydride (TFAA) in dichloromethane gives the 3-nitro derivative (II), which is reduced with iron filings and CaCl2 in refluxing ethanol/water to yield the 3-amino derivative (III). Treatment of compound (III) with NaNO2, HBr and Br2 provides 4-(3-bromo-8-chloro-5,6-dihydro-1H-benzo[5,6]-cyclohepta[1,2-b]pyridin-11-ylidene)piperidine-1-carboxylic acid ethyl ester (IV) (see scheme 25468001a, intermediate (I).(2)
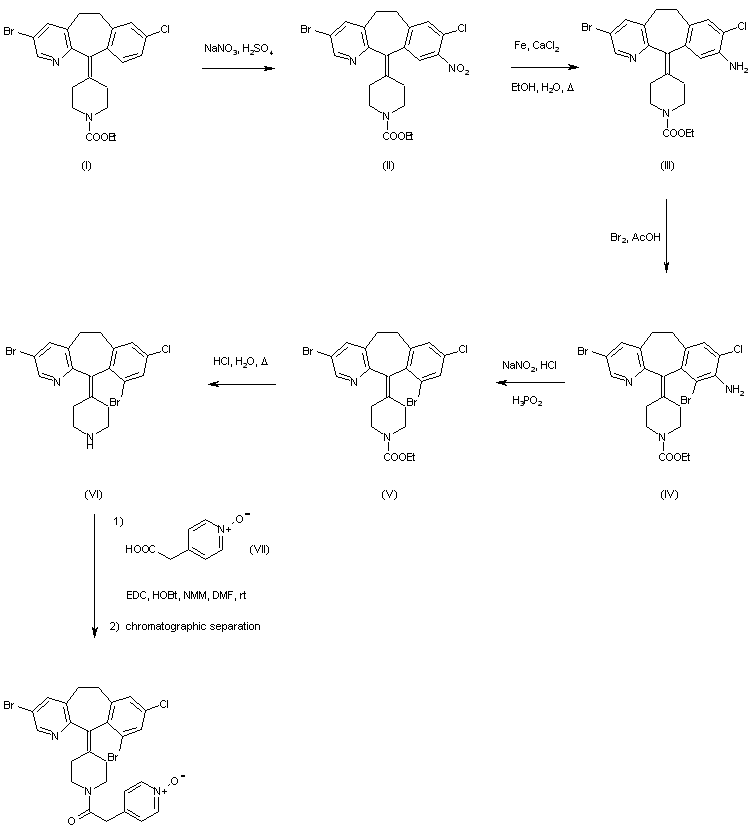
Benzocycloheptapyridine (I) was nitrated with NaNO3 and H2SO4 to afford (II) as the major isomer. Reduction of (III) with iron and CaCl2 gave amine (III), which was brominated to provide (IV). Removal of the amino group of (IV) was accomplished by diazotization, followed by reduction with hypophosphorous acid to give (V). Then, hydrolysis of the carbamate group of (V) in refluxing hydrochloric acid furnished piperidine (VI). Subsequent coupling of (VI) with pyridineacetic acid N-oxide (VII) using EDC and HOBt yielded the corresponding amide. Finally, separation of the target (+)-atropoisomer was achieved by chiral chromatography.
References
- ^ Jump up to:a b c d e f g h i j k “FDA Approves First Treatment for Hutchinson-Gilford Progeria Syndrome and Some Progeroid Laminopathies”. U.S. Food and Drug Administration (FDA) (Press release). 20 November 2020. Retrieved 20 November 2020. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c d “Drug Trials Snapshots: Zokinvy”. U.S. Food and Drug Administration. 20 November 2020. Retrieved 11 December 2020. This article incorporates text from this source, which is in the public domain.
- ^ Liu G, Marrinan CH, Taylor SA, Black S, Basso AD, Kirschmeier P, et al. (September 2007). “Enhancement of the antitumor activity of tamoxifen and anastrozole by the farnesyltransferase inhibitor lonafarnib (SCH66336)”. Anti-Cancer Drugs. 18 (8): 923–31. doi:10.1097/CAD.0b013e3280c1416e (inactive 2020-09-10). PMID 17667598.
- ^ “The FTI Drug Lonafarnib”, Progeria Research Foundation. Accessed October 3, 2017.
- ^ “Lonafarnib”. NCI Drug Dictionary. National Cancer Institute. 2011-02-02.
- ^ Ullrich NJ, Kieran MW, Miller DT, Gordon LB, Cho YJ, Silvera VM, et al. (July 2013). “Neurologic features of Hutchinson-Gilford progeria syndrome after lonafarnib treatment”. Neurology. 81 (5): 427–30. doi:10.1212/WNL.0b013e31829d85c0. PMC 3776537. PMID 23897869.
External links
- “Lonafarnib”. Drug Information Portal. U.S. National Library of Medicine.
- “Experimental Drug Is First To Help Kids With Premature-Aging Disease”, NPR, September 24, 2012
- Clinical trial number NCT00425607 for “Phase II Trial of Lonafarnib (a Farnesyltransferase Inhibitor) for Progeria” at ClinicalTrials.gov
- Clinical trial number NCT00916747 for “Study of Zoledronic Acid, Pravastatin, and Lonafarnib for Patients With Progeria” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Zokinvy |
| Other names | SCH 66336 |
| License data | US DailyMed: Lonafarnib |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only |
| Identifiers | |
| IUPAC name[show] | |
| CAS Number | 193275-84-2 |
| PubChem CID | 148195 |
| IUPHAR/BPS | 8024 |
| DrugBank | DB06448 |
| ChemSpider | 130645 |
| UNII | IOW153004F |
| KEGG | D04768 |
| ChEBI | CHEBI:47097 |
| ChEMBL | ChEMBL298734 |
| PDB ligand | 336 (PDBe, RCSB PDB) |
| CompTox Dashboard (EPA) | DTXSID90172927 |
| ECHA InfoCard | 100.204.509 |
| Chemical and physical data | |
| Formula | C27H31Br2ClN4O2 |
| Molar mass | 638.83 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES[hide]C=12CCC=3C=C(C=C(C3[C@H](C1N=CC(=C2)Br)C4CCN(CC4)C(=O)CC5CCN(CC5)C(N)=O)Br)Cl | |
| InChI[hide]InChI=1S/C27H31Br2ClN4O2/c28-20-12-19-2-1-18-13-21(30)14-22(29)24(18)25(26(19)32-15-20)17-5-9-33(10-6-17)23(35)11-16-3-7-34(8-4-16)27(31)36/h12-17,25H,1-11H2,(H2,31,36)/t25-/m1/s1 Key:DHMTURDWPRKSOA-RUZDIDTESA-N |
/////////lonafarnib, Zokinvy, FDA 2020, 2020 APPROVALS, лонафарниб , لونافارنيب , 氯那法尼 , D 04768, lonafarnibum, TM 5989100
Gallium 68 PSMA-11
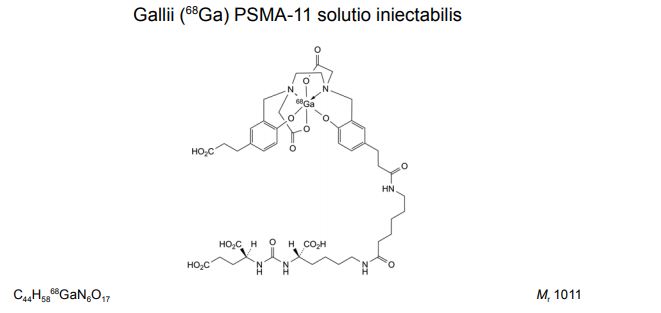

Gallium 68 PSMA-11
FDA APPROVED, 12/1/2020, Gallium 68 PSMA-11
For detection and localization of prostate cancer
Press Release
Drug Trials Snapshot

Chemical structure of 18F-labeled radiotracers. [18F]DCFPyL (A), [18F]PSMA-1007 (B), [18F]CTT1057 (C), (D) [18F]JK-PSMA-7 and (E) [18F]AIF-PSMA-11. The urea backbone of (A), (B), (D) and (E) is marked in blue, while the phosphoramidate of [18F]CTT1057 in (C) is highlighted in orange. Modified from Behr et al. [32], © by the Society of Nuclear Medicine and Molecular Imaging, Inc.
PSMA-11, also known as HBED-CC-PSMA or Psma-hbed-CC, is used to make gallium Ga 68-labeled PSMA-11, which has potential use as a tracer for PSMA-expressing tumors during positron emission tomography (PET). Upon intravenous administration of gallium Ga 68-labeled PSMA-11, the Glu-urea-Lys(Ahx) moiety targets and binds to PSMA-expressing tumor cells. Upon internalization, PSMA-expressing tumor cells can be detected during PET imaging. PSMA, a tumor-associated antigen and type II transmembrane protein, is expressed on the membrane of prostatic epithelial cells and overexpressed on prostate tumor cells

Name: PSMA-11
CAS#: 1366302-52-4
Chemical Formula: C44H62N6O17
Exact Mass: 946.4171
(3S,7S)-22-(3-(((2-((5-(2-Carboxyethyl)-2-hydroxybenzyl)(carboxymethyl)amino)ethyl)(carboxymethyl)amino)methyl)-4-hydroxyphenyl)-5,13,20-trioxo-4,6,12,19-tetraazadocosane-1,3,7-tricarboxylic acid
The Food and Drug Administration (FDA) has approved Gallium 68 PSMA-11 (Ga 68 PSMA-11), the first drug for positron emission tomography (PET) imaging of prostate-specific membrane antigen (PSMA) positive lesions in men with prostate cancer.
Ga 68 PSMA-11, a radioactive diagnostic agent, is indicated for patients with suspected prostate cancer metastasis who are potentially curable by surgery or radiation therapy. It is also indicated for patients with suspected prostate cancer recurrence based on elevated serum prostate-specific antigen (PSA) levels.
The approval was based on efficacy and safety data from 2 prospective clinical trials (Trial 1 and 2) with a total of 960 men with prostate cancer who each received 1 injection of Ga 68 PSMA-11. Trial 1 included 325 patients with biopsy-proven prostate cancer who underwent PET/CT or PET/MRI scans performed with Ga 68 PSMA-11. Results from the study showed that positive readings in the pelvic lymph nodes on Ga 68 PSMA-11 PET were associated with a clinically important rate of metastatic cancer confirmed by surgical pathology in those who proceeded to surgery.
In Trial 2, 635 patients with rising serum PSA levels after prostate surgery or radiotherapy received a single Ga 68 PSMA-11 PET/CT scan or PET/MR scan. Findings demonstrated that 74% of patients had at least 1 positive lesion detected by Ga 68 PSMA-11 PET, and local recurrence or metastasis of prostate cancer was confirmed in 91% of cases.
 This is the first drug approved for PET imaging of prostate-specific membrane antigen positive lesions in men with prostate cancer.
This is the first drug approved for PET imaging of prostate-specific membrane antigen positive lesions in men with prostate cancer.
REF
REFERENCES
1: Meißner S, Janssen JC, Prasad V, Brenner W, Diederichs G, Hamm B, Hofheinz F, Makowski MR. Potential of asphericity as a novel diagnostic parameter in the evaluation of patients with (68)Ga-PSMA-HBED-CC PET-positive prostate cancer lesions. EJNMMI Res. 2017 Oct 23;7(1):85. doi: 10.1186/s13550-017-0333-9. PubMed PMID: 29058157; PubMed Central PMCID: PMC5651532.
2: Verburg FA, Pfister D, Drude NI, Mottaghy FM, Behrendt F. PSA levels, PSA doubling time, Gleason score and prior therapy cannot predict measured uptake of [(68)Ga]PSMA-HBED-CC lesion uptake in recurrent/metastatic prostate cancer. Nuklearmedizin. 2017 Oct 18;56(6). doi: 10.3413/Nukmed-0917-17-07. [Epub ahead of print] PubMed PMID: 29044297.
3: Amor-Coarasa A, Kelly JM, Gruca M, Nikolopoulou A, Vallabhajosula S, Babich JW. Continuation of comprehensive quality control of the itG (68)Ge/(68)Ga generator and production of (68)Ga-DOTATOC and (68)Ga-PSMA-HBED-CC for clinical research studies. Nucl Med Biol. 2017 Oct;53:37-39. doi: 10.1016/j.nucmedbio.2017.07.006. Epub 2017 Jul 14. PubMed PMID: 28803001.
4: Janssen JC, Woythal N, Meißner S, Prasad V, Brenner W, Diederichs G, Hamm B, Makowski MR. [(68)Ga]PSMA-HBED-CC Uptake in Osteolytic, Osteoblastic, and Bone Marrow Metastases of Prostate Cancer Patients. Mol Imaging Biol. 2017 Dec;19(6):933-943. doi: 10.1007/s11307-017-1101-y. PubMed PMID: 28707038.
5: Damle NA, Tripathi M, Chakraborty PS, Sahoo MK, Bal C, Aggarwal S, Arora G, Kumar P, Kumar R, Gupta R. Unusual Uptake of Prostate Specific Tracer (68)Ga-PSMA-HBED-CC in a Benign Thyroid Nodule. Nucl Med Mol Imaging. 2016 Dec;50(4):344-347. Epub 2016 Mar 22. PubMed PMID: 27994690; PubMed Central PMCID: PMC5135692.
6: Behrendt F, Krohn T, Mottaghy F, Verburg FA. [(68)Ga]PSMA-HBED-CC PET/CT to differentiate between diffuse bone metastases of prostate cancer and osteopoikilosis. Nuklearmedizin. 2016 Dec 6;55(6):N64-N65. PubMed PMID: 27922151.
7: Krohn T, Birmes A, Winz OH, Drude NI, Mottaghy FM, Behrendt FF, Verburg FA. The reconstruction algorithm used for [(68)Ga]PSMA-HBED-CC PET/CT reconstruction significantly influences the number of detected lymph node metastases and coeliac ganglia. Eur J Nucl Med Mol Imaging. 2017 Apr;44(4):662-669. doi: 10.1007/s00259-016-3571-6. Epub 2016 Nov 29. PubMed PMID: 27900518.
8: Berliner C, Tienken M, Frenzel T, Kobayashi Y, Helberg A, Kirchner U, Klutmann S, Beyersdorff D, Budäus L, Wester HJ, Mester J, Bannas P. Detection rate of PET/CT in patients with biochemical relapse of prostate cancer using [(68)Ga]PSMA I&T and comparison with published data of [(68)Ga]PSMA HBED-CC. Eur J Nucl Med Mol Imaging. 2017 Apr;44(4):670-677. doi: 10.1007/s00259-016-3572-5. Epub 2016 Nov 28. PubMed PMID: 27896369.
9: Sathekge M, Lengana T, Modiselle M, Vorster M, Zeevaart J, Maes A, Ebenhan T, Van de Wiele C. (68)Ga-PSMA-HBED-CC PET imaging in breast carcinoma patients. Eur J Nucl Med Mol Imaging. 2017 Apr;44(4):689-694. doi: 10.1007/s00259-016-3563-6. Epub 2016 Nov 8. PubMed PMID: 27822700; PubMed Central PMCID: PMC5323468.
10: Rauscher I, Maurer T, Beer AJ, Graner FP, Haller B, Weirich G, Doherty A, Gschwend JE, Schwaiger M, Eiber M. Value of 68Ga-PSMA HBED-CC PET for the Assessment of Lymph Node Metastases in Prostate Cancer Patients with Biochemical Recurrence: Comparison with Histopathology After Salvage Lymphadenectomy. J Nucl Med. 2016 Nov;57(11):1713-1719. Epub 2016 Jun 3. PubMed PMID: 27261524.
11: Verburg FA, Behrendt FF, Mottaghy FM, Pfister D, Steib F, Knuechel R. Strong [(68)Ga]PSMA-HBED-CC accumulation in non-cancerous prostate tissue surrounding a PSMA-negative prostate carcinoma recurrence. Nuklearmedizin. 2016 Sep 26;55(5):N44-5. PubMed PMID: 27668299.
12: Kanthan GL, Izard MA, Emmett L, Hsiao E, Schembri GP. Schwannoma Showing Avid Uptake on 68Ga-PSMA-HBED-CC PET/CT. Clin Nucl Med. 2016 Sep;41(9):703-4. doi: 10.1097/RLU.0000000000001281. PubMed PMID: 27405039.
13: Noto B, Vrachimis A, Schäfers M, Stegger L, Rahbar K. Subacute Stroke Mimicking Cerebral Metastasis in 68Ga-PSMA-HBED-CC PET/CT. Clin Nucl Med. 2016 Oct;41(10):e449-51. doi: 10.1097/RLU.0000000000001291. PubMed PMID: 27355852.
14: Pfob CH, Ziegler S, Graner FP, Köhner M, Schachoff S, Blechert B, Wester HJ, Scheidhauer K, Schwaiger M, Maurer T, Eiber M. Biodistribution and radiation dosimetry of (68)Ga-PSMA HBED CC-a PSMA specific probe for PET imaging of prostate cancer. Eur J Nucl Med Mol Imaging. 2016 Oct;43(11):1962-70. doi: 10.1007/s00259-016-3424-3. Epub 2016 May 20. PubMed PMID: 27207281.
15: Amor-Coarasa A, Schoendorf M, Meckel M, Vallabhajosula S, Babich JW. Comprehensive Quality Control of the ITG 68Ge/68Ga Generator and Synthesis of 68Ga-DOTATOC and 68Ga-PSMA-HBED-CC for Clinical Imaging. J Nucl Med. 2016 Sep;57(9):1402-5. doi: 10.2967/jnumed.115.171249. Epub 2016 Apr 21. PubMed PMID: 27103024.
16: Prasad V, Steffen IG, Diederichs G, Makowski MR, Wust P, Brenner W. Biodistribution of [(68)Ga]PSMA-HBED-CC in Patients with Prostate Cancer: Characterization of Uptake in Normal Organs and Tumour Lesions. Mol Imaging Biol. 2016 Jun;18(3):428-36. doi: 10.1007/s11307-016-0945-x. PubMed PMID: 27038316.
17: Pfister D, Porres D, Heidenreich A, Heidegger I, Knuechel R, Steib F, Behrendt FF, Verburg FA. Detection of recurrent prostate cancer lesions before salvage lymphadenectomy is more accurate with (68)Ga-PSMA-HBED-CC than with (18)F-Fluoroethylcholine PET/CT. Eur J Nucl Med Mol Imaging. 2016 Jul;43(8):1410-7. doi: 10.1007/s00259-016-3366-9. Epub 2016 Mar 19. PubMed PMID: 26993315.
18: Kanthan GL, Coyle L, Kneebone A, Schembri GP, Hsiao E. Follicular Lymphoma Showing Avid Uptake on 68Ga PSMA-HBED-CC PET/CT. Clin Nucl Med. 2016 Jun;41(6):500-1. doi: 10.1097/RLU.0000000000001169. PubMed PMID: 26914565.
19: Kanthan GL, Hsiao E, Kneebone A, Eade T, Schembri GP. Desmoid Tumor Showing Intense Uptake on 68Ga PSMA-HBED-CC PET/CT. Clin Nucl Med. 2016 Jun;41(6):508-9. doi: 10.1097/RLU.0000000000001192. PubMed PMID: 26909712.
20: Eiber M, Weirich G, Holzapfel K, Souvatzoglou M, Haller B, Rauscher I, Beer AJ, Wester HJ, Gschwend J, Schwaiger M, Maurer T. Simultaneous (68)Ga-PSMA HBED-CC PET/MRI Improves the Localization of Primary Prostate Cancer. Eur Urol. 2016 Nov;70(5):829-836. doi: 10.1016/j.eururo.2015.12.053. Epub 2016 Jan 18. PubMed PMID: 26795686.
//////////Gallium 68 PSMA-11, FDA 2020, 2020 APPROVALS, RADIO ACTIVE
Lumasiran
 |
The molecular formula of lumasiran sodium is C530H669F10N173O320P43S6Na43 and the molecular weight is 17,286 Da.
lumasiran
CAS 1834610-13-7
FDA APPROVED, 11/23/2020, Oxlumo
To treat hyperoxaluria type 1
Press Release
Drug Trials Snapshot
RNA, (Gm-sp-Am-sp-Cm-Um-Um-Um-(2′-deoxy-2′-fluoro)C-Am-(2′-deoxy-2′-fluoro)U-(2′-deoxy-2′-fluoro)C-(2′-deoxy-2′-fluoro)C-Um-Gm-Gm-Am-Am-Am-Um-Am-Um-Am), 3′-[[(2S,4R)-1-[29-[[2-(acetylamino)-2-deoxy-β-D-galactopyranosyl]oxy]-14,14-bis[[3-[[3-[[5-[[2-(acetylamino)-2-deoxy-β-D-galactopyranosyl]oxy]-1-oxopentyl]amino]propyl]amino]-3-oxopropoxy]methyl]-1,12,19,25-tetraoxo-16-oxa-13,20,24-triazanonacos-1-yl]-4-hydroxy-2-pyrrolidinyl]methyl hydrogen phosphate], complex with RNA (Um-sp-(2′-deoxy-2′-fluoro)A-sp-Um-Am-Um-(2′-deoxy-2′-fluoro)U-Um-(2′-deoxy-2′-fluoro)C-(2′-deoxy-2′-fluoro)C-Am-Gm-Gm-Am-(2′-deoxy-2′-fluoro)U-Gm-(2′-deoxy-2′-fluoro)A-Am-Am-Gm-Um-Cm-sp-Cm-sp-Am) (1:1)
Nucleic Acid Sequence
Sequence Length: 44, 23, 2115 a 8 c 7 g 14 umultistranded (2); modified
OXLUMO is supplied as a sterile, preservative-free, clear, colorless-to-yellow solution for subcutaneous administration containing the equivalent of 94.5 mg of lumasiran (provided as lumasiran sodium) in 0.5 Ml of water for injection and sodium hydroxide and/or phosphoric acid to adjust the pH to ~7.0.
Lumasiran An investigational RNAi Therapeutic for Primary Hyperoxaluria Type 1 (PH1)
Overview • Lumasiran (ALN-GO1) is an investigational, subcutaneously administered (under the skin) RNA interference (RNAi) therapeutic targeting glycolate oxidase (GO) in development for the treatment of primary hyperoxaluria type 1 (PH1).
• PH1 is a rare, life-threatening disease that can cause serious damage to kidneys and progressively to other organs.1
• PH1 is characterized by the pathologic overproduction of oxalate by the liver. Oxalate is an end product of metabolism that, when in excess, is toxic and accumulates in the kidneys forming calcium oxalate crystals.1,2
• Symptoms of PH1 are often associated with recurrent kidney stones and include flank pain, urinary tract infections, painful urination, and blood in the urine.2,3
• Currently, the only curative treatment is a liver transplant, to correct the metabolic defect, combined with a kidney transplant, to replace the terminally damaged kidneys.1,3 Clinical Development
• The safety and efficacy of lumasiran are being evaluated in a randomized, double-blind, placebo-controlled, global, multicenter Phase 3 study of approximately 30 PH1 patients, called ILLUMINATE-A (NCT03681184).
• The primary endpoint is percent change in 24-hour urinary oxalate excretion from baseline to Month 6.
• Key secondary and exploratory endpoints in ILLUMINATE-A will evaluate additional measures of urinary oxalate, estimated glomerular filtration rate (eGFR), safety, and tolerability.
Regulatory Designations • Breakthrough Therapy Designation by the U.S. Food and Drug Administration (FDA) • Priority Medicines (PRIME) Designation from the European Medicines Agency (EMA) • Orphan Drug Designations in both the U.S. and the European Union
/////////lumasiran, fda 2020, 2020 approvals, Oxlumo, Breakthrough Therapy Designation, Orphan Drug, Priority Medicines (PRIME) Designation
Setmelanotide


Setmelanotide
Ac-Arg-Cys(1)-D-Ala-His-D-Phe-Arg-Trp-Cys(1)-NH2
- Molecular FormulaC49H68N18O9S2
- Average mass1117.309 Da
- N-acetyl-L-arginyl-L-cysteinyl-D-alanyl-L-histidyl-D-phenylalanyl-L-arginyl-L-tryptophyl-L-cysteinamide (2->8)-disulfide
1,2-Dithia-5,8,11,14,17,20-hexaazacyclotricosane-4-carboxamide, 22-[[(2S)-2-(acetylamino)-5-[(diaminomethylene)amino]-1-oxopentyl]amino]-10-[3-[(diaminomethylene)amino]propyl]-16-(1H-imidazol-5-ylmeth yl)-7-(1H-indol-3-ylmethyl)-19-methyl-6,9,12,15,18,21-hexaoxo-13-(phenylmethyl)-, (4R,7S,10S,13R,16S,19R,22R)- [ACD/Index Name]10011920014-72-8[RN]Imcivree [Trade name]N2-acetyl-L-arginyl-L-cysteinyl-D-alanyl-L-histidyl-D-phenylalanyl-L-arginyl-Ltryptophyl- L-cysteinamide, cyclic (2-8)-disulfideN7T15V1FUYRM-493, BIM-22493UNII-N7T15V1FUYсетмеланотид [Russian] [INN]سيتميلانوتيد [Arabic] [INN]司美诺肽 [Chinese] [INN](4R,7S,10S,13R,16S,19R,22R)-22-[[(2S)-2-acetamido-5-(diaminomethylideneamino)pentanoyl]amino]-13-benzyl-10-[3-(diaminomethylideneamino)propyl]-16-(1H-imidazol-5-ylmethyl)-7-(1H-indol-3-ylmethyl)-19-methyl-6,9,12,15,18,21-hexaoxo-1,2-dithia-5,8,11,14,17,20-hexazacyclotricosane-4-carboxamide
FDA 11/25/2020, Imcivree, To treat obesity and the control of hunger associated with pro-opiomelanocortin deficiency, a rare disorder that causes severe obesity that begins at an early age
Drug Trials Snapshot, 10MG/ML, SOLUTION;SUBCUTANEOUS, Orphan


update Imcivree EMA APPROVED 2021/7/16
DESCRIPTION
IMCIVREE contains setmelanotide acetate, a melanocortin 4 (MC4) receptor agonist. Setmelanotide is an 8 amino acid cyclic peptide analog of endogenous melanocortin peptide α-MSH (alpha-melanocyte stimulating hormone).
The chemical name for setmelanotide acetate is acetyl-L-arginyl-L-cysteinyl-D-alanyl-Lhistidinyl-D-phenylalanyl-L-arginyl-L-tryptophanyl-L-cysteinamide cyclic (2→8)-disulfide acetate. Its molecular formula is C49H68N18O9S2 (anhydrous, free-base), and molecular mass is 1117.3 Daltons (anhydrous, free-base).
The chemical structure of setmelanotide is:
 |
IMCIVREE injection is a sterile clear to slightly opalescent, colorless to slightly yellow solution. Each 1 mL of IMCIVREE contains 10 mg of setmelanotide provided as setmelanotide acetate, which is a salt with 2 to 4 molar equivalents of acetate, and the following inactive ingredients: 100 mg N-(carbonyl-methoxypolyethylene glycol 2000)-1,2-distearoyl-glycero-3phosphoethanolamine sodium salt, 8 mg carboxymethylcellulose sodium (average MWt 90,500), 11 mg mannitol, 5 mg phenol, 10 mg benzyl alcohol, 1 mg edetate disodium dihydrate, and Water for Injection. The pH of IMCIVREE is 5 to 6.
Setmelanotide is a peptide drug and investigational anti-obesity medication which acts as a selective agonist of the MC4 receptor. Setmelanotide binds to and activates MC4 receptors in the paraventricular nucleus (PVN) of the hypothalamus and in the lateral hypothalamic area (LHA), areas involved in the regulation of appetite, and this action is thought to underlie its appetite suppressant effects. Setmelanotide increases resting energy expenditure in both obese animals and humans. Setmelanotide has been reported to possess the following activity profile (cAMP, EC50): MC4 (0.27 nM) > MC3 (5.3 nM) ≈ MC1 (5.8 nM) > MC5 (1600 nM) ≟ MC2 (>1000 nM).
Setmelanotide, sold under the brand name Imcivree, is a medication for the treatment of obesity.[1]
The most common side effects include injection site reactions, skin hyperpigmentation (skin patches that are darker than surrounding skin), headache and gastrointestinal side effects (such as nausea, diarrhea, and abdominal pain), among others.[1] Spontaneous penile erections in males and adverse sexual reactions in females have occurred with treatment.[1] Depression and suicidal ideation have also occurred with setmelanotide.[1]
SYN
WO 2011060355



Medical uses
Setmelanotide is indicated for chronic weight management (weight loss and weight maintenance for at least one year) in people six years and older with obesity due to three rare genetic conditions: pro-opiomelanocortin (POMC) deficiency, proprotein subtilisin/kexin type 1 (PCSK1) deficiency, and leptin receptor (LEPR) deficiency confirmed by genetic testing demonstrating variants in POMC, PCSK1, or LEPR genes considered pathogenic (causing disease), likely pathogenic, or of uncertain significance.[1] Setmelanotide is the first FDA-approved treatment for these genetic conditions.[1]
Setmelanotide is not approved for obesity due to suspected POMC, PCSK1, or LEPR deficiency with variants classified as benign (not causing disease) or likely benign or other types of obesity, including obesity associated with other genetic syndromes and general (polygenic) obesity.[1]
Setmelanotide binds to and activates MC4 receptors in the paraventricular nucleus (PVN) of the hypothalamus and in the lateral hypothalamic area (LHA), areas involved in the regulation of appetite, and this action is thought to underlie its appetite suppressant effects.[2] In addition to reducing appetite, setmelanotide increases resting energy expenditure in both obese animals and humans.[3] Importantly, unlike certain other MC4 receptor agonists, such as LY-2112688, setmelanotide has not been found to produce increases in heart rate or blood pressure.[4]
Setmelanotide has been reported to possess the following activity profile (cAMP, EC50): MC4 (0.27 nM) > MC3 (5.3 nM) ≈ MC1 (5.8 nM) > MC5 (1600 nM) ≟ MC2 (>1000 nM).[5] (19.6-fold selectivity for MC4 over MC3, the second target of highest activity.)
History
Setmelanotide was evaluated in two one-year studies.[1] The first study enrolled participants with obesity and confirmed or suspected POMC or PCSK1 deficiency while the second study enrolled participants with obesity and confirmed or suspected LEPR deficiency; all participants were six years or older.[1] The effectiveness of setmelanotide was determined by the number of participants who lost more than ten percent of their body weight after a year of treatment.[1]
The effectiveness of setmelanotide was assessed in 21 participants, ten in the first study and eleven in the second.[1] In the first study, 80 percent of participants with POMC or PCSK1 deficiency lost ten percent or more of their body weight.[1] In the second study, 46 percent of participants with LEPR deficiency lost ten percent or more of their body weight.[1]
The study also assessed the maximal (greatest) hunger in sixteen participants over the previous 24 hours using an eleven-point scale in participants twelve years and older.[1] In both studies, some, but not all, of participants’ weekly average maximal hunger scores decreased substantially from their scores at the beginning of the study.[1] The degree of change was highly variable among participants.[1]
The U.S. Food and Drug Administration (FDA) granted the application for setmelanotide orphan disease designation, breakthrough therapy designation, and priority review.[1] The FDA granted the approval of Imcivree to Rhythm Pharmaceutical, Inc.[1]
Research
Setmelanotide is a peptide drug and investigational anti-obesity medication which acts as a selective agonist of the MC4 receptor.[6][4] Its peptide sequence is Ac-Arg-Cys(1)-D-Ala-His-D-Phe-Arg-Trp-Cys(1)-NH2. It was first discovered at Ipsen and is being developed by Rhythm Pharmaceuticals for the treatment of obesity and diabetes.[6] In addition, Rhythm Pharmaceuticals is conducting trials of setmelanotide for the treatment of Prader–Willi syndrome (PWS), a genetic disorder which includes MC4 receptor deficiency and associated symptoms such as excessive appetite and obesity.[7] As of December 2014, the drug is in phase II clinical trials for obesity and PWS.[6][8][9][needs update] So far, preliminary data has shown no benefit of Setmelanotide in Prader-Willi syndrome.[10]
PATENT
WO 2007008704
WO 2011060355
WO 2011060352
US 20120225816
PAPER
Journal of Medicinal Chemistry, 61(8), 3674-3684; 2018
PATENT
https://patents.google.com/patent/US9314509
Synthesis of Example 1i.e., Ac-Arg-cyclo(Cys-D-Ala-His-D-Phe-Arg-Trp-Cys)-NH2

The title peptide having the above structure was assembled using Fmoc chemistry on an Apex peptide synthesizer (Aapptec; Louisville, Ky., USA). 220 mg of 0.91 mmol/g (0.20 mmoles) Rink Amide MBHA resin (Polymer Laboratories; Amherst, Mass., USA) was placed in a reaction well and pre-swollen in 3.0 mL of DMF prior to synthesis. For cycle 1, the resin was treated with two 3-mL portions of 25% piperidine in DMF for 5 and 10 minutes respectively, followed by 4 washes of 3-mL DMF—each wash consisting of adding 3 mL of solvent, mixing for 1 minute, and emptying for 1 minute. Amino acids stocks were prepared in NMP as 0.45M solutions containing 0.45M HOBT. HBTU was prepared as a 0.45M solution in NMP and DIPEA was prepared as a 2.73M solution in NMP. To the resin, 2 mL of the first amino acid (0 9 mmoles, Fmoc-Cys(Trt)-OH) (Novabiochem; San Diego, Calif., USA) was added along with 2 mL (0.9 mmoles) of HBTU and 1.5 mL (4.1 mmoles) of DIPEA. After one hour of constant mixing, the coupling reagents were drained from the resin and the coupling step was repeated. Following amino acid acylation, the resin was washed with two 3-mL aliquots of DMF for 1 minute. The process of assembling the peptide (deblock/wash/acylate/wash) was repeated for cycles 2-9 identical to that as described for cycle 1. The following amino acids were used: cycle 2) Fmoc-Trp(Boc)-OH (Genzyme; Cambridge, Mass., USA); cycle 3) Fmoc-Arg(Pbf)-OH (Novabiochem); cycle 4) Fmoc-DPhe-OH (Genzyme); cycle 5) Fmoc-His(Trt)-OH (Novabiochem); cycle 6) Fmoc-D-Ala-OH (Genzyme); cycle 7) Fmoc-Cys(Trt)-OH, (Novabiochem); and cycle 8) Fmoc-Arg(Pbf)-OH (Genzyme). The N-terminal Fmoc was removed with 25% piperidine in DMF as described above, followed by four 3-mL DMF washes for 1 minute. Acetylation of the N-terminus was performed by adding 0.5 mL of 3M DIPEA in NMP to the resin along with 1.45 mL of 0.45M acetic anhydride in NMP. The resin was mixed for 30 minutes and acetylation was repeated. The resin was washed with 3 mL of DMF for a total of 5 times followed with 5 washes with 5 mL of DCM each.
To cleave and deprotect the peptide, 5mL of the following reagent was added to the resin: 2% TIS/5% water/5% (w/v) DTT/88% TFA. The solution was allowed to mix for 3.5 hours. The filtrate was collected into 40 mL of cold anhydrous ethyl ether. The precipitate was pelleted for 10 minutes at 3500 rpm in a refrigerated centrifuge. The ether was decanted and the peptide was re-suspended in fresh ether. The ether workup was performed three times. Following the last ether wash, the peptide was allowed to air dry to remove residual ether.
The peptide was dissolved in 10% acetonitrile and analyzed by mass spectrometry and reverse-phase HPLC employing a 30×4.6 cm C18 column (Vydac; Hesperia, Calif., USA) with a gradient of 2-60% acetonitrile (0.1% TFA) over 30 minutes. This analysis identified a product with ˜53% purity. Mass analysis employing electrospray ionization identified a main product containing a mass of 1118.4 corresponding to the desired linear product. The crude product (˜100 mg) was diluted to a concentration of 2 mg/mL in 5% acetic acid. To this solution, 0.5M iodine/methanol was added dropwise with vigorous stirring until a pale yellow color was achieved. The solution was vigorously stirred for another 10 minutes. Excess iodine was then quenched by adding 1.0M sodium thiosulfate under continuous mixing until the mixture was rendered colorless. The peptide was re-examined by mass spectrometry analysis and HPLC. Mass spectrometry analysis identified a main species with a mass of 1116.4 which indicated successful oxidation to form the cyclic peptide. The peptide solution was purified on a preparative HPLC equipped with a C18 column using a similar elution gradient. The purified product was re-analyzed by HPLC for purity (>95%) and mass spectrometry (1116.9 which is in agreement with the expected mass of 1117.3) and subsequently lyophilized. Following lyophilization, 28 mg of purified product was obtained representing a 24% yield.
The other exemplified peptides were synthesized substantially according to the procedure described for the above-described synthetic process. Physical data for select exemplified peptides are given in Table 1.
TABLE 1 Example Mol. Wt. Mol. Wt. Purity Number (calculated) (ES-MS) (HPLC) 1 1117.3 1116.9 95.1% 2 1117.3 1116.8 99.2% 3 1280.5 1280.6 98.0% 5 1216.37 1216.20 99.9%
Preparation of Pamoate Salt of Example 1
The acetate salt of Example 1 (200 mg, 0.18 mmole) was dissolved in 10 mL of water. Sodium pamoate (155 mg, 0.36 mmole) was dissolved in 10 mL of water. The two solutions were combined and mixed well. The precipitates were collected by centrifugation at 3000 rpm for 20 minutes, washed for three times with water, and dried by lyophilization.
References
- ^ Jump up to:a b c d e f g h i j k l m n o p q r “FDA approves first treatment for weight management for people with certain rare genetic conditions”. U.S. Food and Drug Administration (FDA) (Press release). 27 November 2020. Retrieved 27 November 2020. This article incorporates text from this source, which is in the public domain.
- ^ Kim GW, Lin JE, Blomain ES, Waldman SA (January 2014). “Antiobesity pharmacotherapy: new drugs and emerging targets”. Clinical Pharmacology and Therapeutics. 95 (1): 53–66. doi:10.1038/clpt.2013.204. PMC 4054704. PMID 24105257.
- ^ Chen KY, Muniyappa R, Abel BS, Mullins KP, Staker P, Brychta RJ, et al. (April 2015). “RM-493, a melanocortin-4 receptor (MC4R) agonist, increases resting energy expenditure in obese individuals”. The Journal of Clinical Endocrinology and Metabolism. 100 (4): 1639–45. doi:10.1210/jc.2014-4024. PMC 4399297. PMID 25675384.
- ^ Jump up to:a b Kievit P, Halem H, Marks DL, Dong JZ, Glavas MM, Sinnayah P, et al. (February 2013). “Chronic treatment with a melanocortin-4 receptor agonist causes weight loss, reduces insulin resistance, and improves cardiovascular function in diet-induced obese rhesus macaques”. Diabetes. 62 (2): 490–7. doi:10.2337/db12-0598. PMC 3554387. PMID 23048186.
- ^ Muniyappa R, Chen K, Brychta R, Abel B, Mullins K, Staker P, et al. (June 2014). “A Randomized, Double-Blind, Placebo-Controlled, Crossover Study to Evaluate the Effect of a Melanocortin Receptor 4 (MC4R) Agonist, RM-493, on Resting Energy Expenditure (REE) in Obese Subjects” (PDF). Endocrine Reviews. Rhythm Pharmaceuticals. 35 (3). Retrieved 2015-05-21.
- ^ Jump up to:a b c Lee EC, Carpino PA (2015). “Melanocortin-4 receptor modulators for the treatment of obesity: a patent analysis (2008-2014)”. Pharmaceutical Patent Analyst. 4 (2): 95–107. doi:10.4155/ppa.15.1. PMID 25853469.
- ^ “Obesity and Diabetes Caused by Genetic Deficiencies in the MC4 Pathway”. Rhythm Pharmaceuticals. Retrieved 2015-05-21.
- ^ Jackson VM, Price DA, Carpino PA (August 2014). “Investigational drugs in Phase II clinical trials for the treatment of obesity: implications for future development of novel therapies”. Expert Opinion on Investigational Drugs. 23 (8): 1055–66. doi:10.1517/13543784.2014.918952. PMID 25000213. S2CID 23198484.
- ^ “RM-493: A First-in-Class, Phase 2-Ready MC4 Agonist: A New Drug Class for the Treatment of Obesity and Diabetes”. Rhythm Pharmaceuticals. Archived from the original on 2015-06-14. Retrieved 2015-05-21.
- ^ Duis J, van Wattum PJ, Scheimann A, Salehi P, Brokamp E, Fairbrother L, et al. (March 2019). “A multidisciplinary approach to the clinical management of Prader-Willi syndrome”. Molecular Genetics & Genomic Medicine. 7 (3): e514. doi:10.1002/mgg3.514. PMC 6418440. PMID 30697974.
ADDITIONAL INFORMATION
The peptide sequence is Ac-Arg-Cys(1)-D-Ala-His-D-Phe-Arg-Trp-Cys(1)-NH2. It is being researched by Rhythm Pharmaceuticals for the treatment of obesity and diabetes. In addition, Rhythm Pharmaceuticals is conducting trials of setmelanotide for the treatment of Prader–Willi syndrome (PWS), a genetic disorder which includes MC4 receptor deficiency and associated symptoms such as excessive appetite and obesity. As of December 2014, the drug is in phase II clinical trials for obesity and PWS.
L-Cysteinamide, N2-acetyl-L-arginyl-L-cysteinyl-D-alanyl-L-histidyl-D-phenylalanyl-L-arginyl-L-tryptophyl-, cyclic (2->8)-disulfide
Ac-Arg-Cys(1)-D-Ala-His-D-Phe-Arg-Trp-Cys(1)-NH2
REFERENCES
1: Lee EC, Carpino PA. Melanocortin-4 receptor modulators for the treatment of obesity: a patent analysis (2008-2014). Pharm Pat Anal. 2015;4(2):95-107. doi: 10.4155/ppa.15.1. PubMed PMID: 25853469.
2: Chen KY, Muniyappa R, Abel BS, Mullins KP, Staker P, Brychta RJ, Zhao X, Ring M, Psota TL, Cone RD, Panaro BL, Gottesdiener KM, Van der Ploeg LH, Reitman ML, Skarulis MC. RM-493, a melanocortin-4 receptor (MC4R) agonist, increases resting energy expenditure in obese individuals. J Clin Endocrinol Metab. 2015 Apr;100(4):1639-45. doi: 10.1210/jc.2014-4024. Epub 2015 Feb 12. PubMed PMID: 25675384; PubMed Central PMCID: PMC4399297.
3: Clemmensen C, Finan B, Fischer K, Tom RZ, Legutko B, Sehrer L, Heine D, Grassl N, Meyer CW, Henderson B, Hofmann SM, Tschöp MH, Van der Ploeg LH, Müller TD. Dual melanocortin-4 receptor and GLP-1 receptor agonism amplifies metabolic benefits in diet-induced obese mice. EMBO Mol Med. 2015 Feb 4;7(3):288-98. doi: 10.15252/emmm.201404508. PubMed PMID: 25652173; PubMed Central PMCID: PMC4364946.
4: Jackson VM, Price DA, Carpino PA. Investigational drugs in Phase II clinical trials for the treatment of obesity: implications for future development of novel therapies. Expert Opin Investig Drugs. 2014 Aug;23(8):1055-66. doi: 10.1517/13543784.2014.918952. Epub 2014 Jul 7. Review. PubMed PMID: 25000213.
5: Kievit P, Halem H, Marks DL, Dong JZ, Glavas MM, Sinnayah P, Pranger L, Cowley MA, Grove KL, Culler MD. Chronic treatment with a melanocortin-4 receptor agonist causes weight loss, reduces insulin resistance, and improves cardiovascular function in diet-induced obese rhesus macaques. Diabetes. 2013 Feb;62(2):490-7. doi: 10.2337/db12-0598. Epub 2012 Oct 9. PubMed PMID: 23048186; PubMed Central PMCID: PMC3554387.
6: Kumar KG, Sutton GM, Dong JZ, Roubert P, Plas P, Halem HA, Culler MD, Yang H, Dixit VD, Butler AA. Analysis of the therapeutic functions of novel melanocortin receptor agonists in MC3R- and MC4R-deficient C57BL/6J mice. Peptides. 2009 Oct;30(10):1892-900. doi: 10.1016/j.peptides.2009.07.012. Epub 2009 Jul 29. PubMed PMID: 19646498; PubMed Central PMCID: PMC2755620.
External links
- “Setmelanotide”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Imcivree |
| Other names | RM-493; BIM-22493; IRC-022493; N2-Acetyl-L-arginyl-L-cysteinyl-D-alanyl-L-histidyl-D-phenylalanyl-L-arginyl-L-tryptophyl-L-cysteinamide, cyclic (2-8)-disulfide |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only |
| Identifiers | |
| IUPAC name[show] | |
| CAS Number | 920014-72-8 |
| PubChem CID | 11993702 |
| ChemSpider | 10166169 |
| UNII | N7T15V1FUY |
| KEGG | D11927 |
| Chemical and physical data | |
| Formula | C49H68N18O9S2 |
| Molar mass | 1117.32 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES[hide]C[C@@H]1C(=O)N[C@H](C(=O)N[C@@H](C(=O)N[C@H](C(=O)N[C@H](C(=O)N[C@@H](CSSC[C@@H](C(=O)N1)NC(=O)[C@H](CCCN=C(N)N)NC(=O)C)C(=O)N)Cc2c[nH]c3c2cccc3)CCCN=C(N)N)Cc4ccccc4)Cc5cnc[nH]5 | |
| InChI[hide]InChI=1S/C49H68N18O9S2/c1-26-41(70)63-37(20-30-22-55-25-59-30)46(75)64-35(18-28-10-4-3-5-11-28)44(73)62-34(15-9-17-57-49(53)54)43(72)65-36(19-29-21-58-32-13-7-6-12-31(29)32)45(74)66-38(40(50)69)23-77-78-24-39(47(76)60-26)67-42(71)33(61-27(2)68)14-8-16-56-48(51)52/h3-7,10-13,21-22,25-26,33-39,58H,8-9,14-20,23-24H2,1-2H3,(H2,50,69)(H,55,59)(H,60,76)(H,61,68)(H,62,73)(H,63,70)(H,64,75)(H,65,72)(H,66,74)(H,67,71)(H4,51,52,56)(H4,53,54,57)/t26-,33+,34+,35-,36+,37+,38+,39+/m1/s1Key:HDHDTKMUACZDAA-PHNIDTBTSA-N |
///////////Setmelanotide, FDA 2020, 2020 APPROVALS, Imcivree, Orphan, PEPTIDE, ANTIOBESITY, UNII-N7T15V1FUY, сетмеланотид , سيتميلانوتيد , 司美诺肽 , BIM 22493, RM 493
CC1C(=O)NC(C(=O)NC(C(=O)NC(C(=O)NC(C(=O)NC(CSSCC(C(=O)N1)NC(=O)C(CCCN=C(N)N)NC(=O)C)C(=O)N)CC2=CNC3=CC=CC=C32)CCCN=C(N)N)CC4=CC=CC=C4)CC5=CN=CN5

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}