
Home » Posts tagged 'GENERIC DRUG'
Tag Archives: GENERIC DRUG
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
SPIRONOLACTONE, спиронолактон , سبيرونولاكتون , 螺内酯 ,

Spironolactone
Spironolactone, Supra-puren, Suracton, спиронолактон, سبيرونولاكتون ,
螺内酯 , Abbolactone, Aldactide, SNL, Spiroctanie, Sprioderm, Verospirone, Opianin
7α-Acetylthio-17α-hydroxy-3-oxopregn-4-ene-21-carboxylic acid γ-lactone
(1’S,2R,2’R,9’R,10’R,11’S,15’S)-9′-(acetylsulfanyl)-2′,15′-dimethylspiro[oxolane-2,14′-tetracyclo[8.7.0.02,7.011,15]heptadecan]-6′-ene-5,5′-dione
| CAS 52-01-7 |
MF C24H32O4S, MW 416.573 Da
Spironolactone, marketed under the brand name Aldactone among others, is a medication primarily used to treatfluid build-up due to heart failure, liver scarring, or kidney disease.[1] Other uses include high blood pressure, low blood potassium that does not improve with supplementation, early puberty, excessive hair growth in women,[1] and as a component of hormone replacement therapy for transgender women.[6] It is taken by mouth.[1]
Common side effects include electrolyte abnormalities particularly high blood potassium, nausea, vomiting, headache, a rash, and a decreased desire for sex. In those with liver or kidney problems extra care should be taken.[1]Spironolactone has not been well studied in pregnancy and should not be used to treat high blood pressure of pregnancy.[7] It is a steroid that blocks mineralocorticoid receptors. It also blocks androgen, and blocks progesterone. It belongs to a class of medications known as potassium-sparing diuretics.[1]
Spironolactone was introduced in 1959.[8][9] It is on the World Health Organization’s List of Essential Medicines, the most important medications needed in a basic health system.[10] It is available as a generic medication.[1] The wholesale cost in the developing world as of 2014 is between 0.02 and 0.12 USD per day.[11] In the United States it costs about 0.50 USD per day.[1]
|
Title: Spironolactone
CAS Registry Number: 52-01-7
CAS Name: (7a,17a)-7-(Acetylthio)-17-hydroxy-3-oxopregn-4-ene-21-carboxylic acid g-lactone
Additional Names: 17-hydroxy-7a-mercapto-3-oxo-17a-pregn-4-ene-21-carboxylic acid g-lactone, acetate; 3-(3-oxo-7a-acetylthio-17b-hydroxy-4-androsten-17a-yl)propionic acid g-lactone
Manufacturers’ Codes: SC-9420
Trademarks: Aldactone (Pharmacia & Upjohn); Aquareduct (Azupharma); Practon (Pfizer); Osyrol (Aventis); Sincomen (Schering AG); Spirobeta (Betapharm); Spiroctan (Ferlux); Spirolone (APS); Spironone (Dexo); Verospiron (Richter Gedeon); Xenalon (Mepha)
Molecular Formula: C24H32O4S
Molecular Weight: 416.57
Percent Composition: C 69.20%, H 7.74%, O 15.36%, S 7.70%
Literature References: Aldosterone antagonist. Prepn: Cella, Tweit, J. Org. Chem. 24, 1109 (1959); US 3013012 (1961 to Searle); Tweit et al., J. Org. Chem. 27, 3325 (1962). Activity and metabolic studies: Gerhards, Engelhardt, Arzneim.-Forsch. 13, 972 (1963). Crystal and molecular structure: Dideberg, Dupont, Acta Crystallogr. B28, 3014 (1972). Comprehensive description: J. L. Sutter, E. P. K. Lau, Anal. Profiles Drug Subs. 4, 431-451 (1975). Review of carcinogenetic risk: IARC Monographs 24, 259-273 (1980). Review of antiandrogen effects and clinical use in hirsutism: R. R. Tremblay, Clin. Endocrinol. Metab. 15, 363-371 (1986); of clinical efficacy in hypertension: A. N. Brest, Clin. Ther. 8, 568-585 (1986). Review of pharmacology: H. A. Skluth, J. G. Gums,DICP Ann. Pharmacother. 24, 52-59 (1990). Clinical trial in congestive heart failure: B. Pitt et al., N. Engl. J. Med. 341, 709 (1999).
Properties: Crystals from methanol, mp 134-135° (resolidifies and dec 201-202°). [a]D20 -33.5° (chloroform). uv max: 238 nm (e20200). Practically insol in water. Sol in alcohol; freely sol in benzene, chloroform. LD50 in rats, mice, rabbits (mg/kg): 790, 360, 870 i.p. (IARC, 1980).
Melting point: mp 134-135° (resolidifies and dec 201-202°)
Optical Rotation: [a]D20 -33.5° (chloroform)
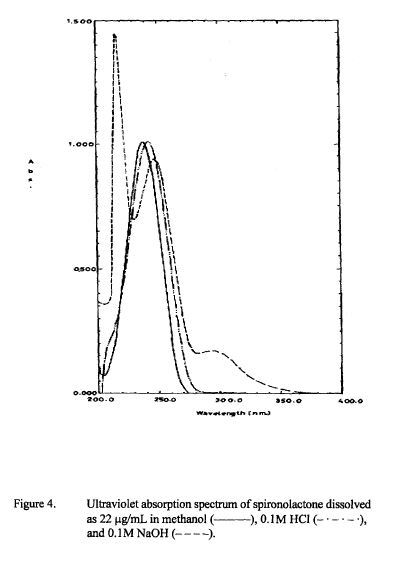
Absorption maximum: uv max: 238 nm (e 20200)
Toxicity data: LD50 in rats, mice, rabbits (mg/kg): 790, 360, 870 i.p. (IARC, 1980)
Therap-Cat: Diuretic.
Therap-Cat-Vet: Diuretic.
Keywords: Aldosterone Antagonist; Diuretic; Steroids
|
Medical uses
Spironolactone is used primarily to treat heart failure, edematous conditions such as nephrotic syndrome or ascites in people with liver disease, essential hypertension, hypokalemia, secondary hyperaldosteronism (such as occurs with hepatic cirrhosis), and Conn’s syndrome (primary hyperaldosteronism). On its own, spironolactone is only a weak diuretic because it primarily targets the distal nephron (collecting tubule), where only small amounts of sodium are reabsorbed, but it can be combined with other diuretics to increase efficacy.
Spironolactone is an antagonist of the androgen receptor (AR) as well as an inhibitor of androgen production. Due to the antiandrogenic effects that result from these actions, it is frequently used off-label to treat a variety of dermatological conditions in which androgens, such as testosterone and dihydrotestosterone (DHT), play a role. Some of these uses include androgenic alopecia in men (either at low doses or as a topical formulation) and women, and hirsutism, acne, and seborrhea in women.[12] Spironolactone is the most commonly used drug in the treatment of hirsutism in the United States.[13] Higher doses of spironolactone are not recommended in males due to the high risk of feminization and other side effects. Similarly, it is also commonly used to treat symptoms of hyperandrogenism in polycystic ovary syndrome.[14]
Spironolactone (SL) is known to be a potent aldosterone antagonist at mineralocorticoid steroid hormone receptors, and it is widely used in humans for the treatment of essential hypertension, congestive heat failure and refractory edema or hyperaldosteronism. However, the prolonged use of SL is associated with undesirable endocrine side effects such as gynecomastia and lose of libido in men and menstrual irregularities in women due to interaction of SL with gonadal steroid hormone biosynthesis and target cell gonadal steroid receptors.
The nature and prevalence of the undesirable side effects limit the usefulness of spironolactone as a therapeutic agent. Gynecomastia or tender breast enlargement has been found to occur in 10% of hypertensive patients using spironolactone for therapy as compared to 1% of men in the placebo group. Recent studies by Pitt, et al. with spironolactone have shown that in patients with congestive heart failure (CHF) taking digoxin and a loop diuretic—spironolactone therapy in conjunction with digitalis and ACE inhibitor—reduces mortality by 30%. See Pitt, B., et al., The Effect of Spironolactone on Morbidity and Mortality in Patients with Severe Heart Failure, Randomized Aldactone Evaluation Study Investigors; N. Engl. J. Med., 1999, 341:709-717. These authors stated that the 30% reduction in the risk of death among patients in the group receiving spironolactone could be attributed to a lower risk of both death from progressive heart failure and sudden death from cardiac arrhythmic causes. In addition, they found that the frequency of hospitalization for worsening heart failure is 35% lower in the spironolacotone treated group than in the placebo group. These authors concluded that patients who received spironolactone had a significant improvement in the symptoms of severe heart failure caused by systolic left ventricular dysfunction. Overall, 8% of the patients in the spironolactone group discontinued treatment because of adverse events. The purpose of the present invention is to make available the individual chiral isomers of spironolactone that would be effective in treating CHF and in reducing hypertension, and at the same time would be devoid of undesirable side effects such as gynecomastia, lose of libido in men, and menstrual irregularities in women.
Spironolactone is the name commonly used for a specific spirolactone that has the full chemical name 17-hydroxy-7-alpha-mercapto-3-oxo-17-alpha-pregn-4-ene-21-carboxylic acid gamma-lactone acetate. The term “spirolactone” denotes that a lactone 10 ring (i.e., a cyclic ester) is attached to another ring structure in a spiro configuration (i.e., the lactone ring shares a single carbon atom with the other ring). Spirolactones that are coupled to steroids are the most important class of spirolactones from a pharmaceutical perspective, so they are widely referred to in the pharmaceutical arts simply as spirolactones. As used herein, “spironolactone” refers to a molecule comprising a lactone structure coupled via a spiro configuration to a steroid structure or steroid derivative.
Spironolactone, its activities, and modes of synthesis and purification are described in a number of U.S. patents, notably U.S. Pat. Nos. 3,013,012, 4,529,811 and 4,603,128.
Intracellular receptors (IRs) form a class of structurally-related genetic regulators that act as ligand-dependent transcription factors. See Evans, R. M., “The Steroid and Thyroid Hormone Receptor Superfamily”, Science, May 13, 1988; 240(4854):889-95. Steroid receptors are a recognized subset of the IRs, including the progesterone receptor (PR), androgen receptor (AR), estrogen receptor (ER), which can be referred to collectively as the gonadal steroid receptors, glucocorticoid receptor (GR), and mineralocorticoid receptor (MR). Regulation of a gene by such factors requires both the IR itself and a corresponding ligand that has the ability to selectively bind to the IR in a way that affects gene transcription.
Ligands for the IRs can include low molecular weight native molecules, such as the hormones aldosterone, progesterone, estrogen and testosterone, as well as synthetic derivative compounds such as medroxyprogesterone acetate, diethylstilbesterol and 19-nortestosterone. These ligands, when present the fluid surrounding a cell, pass through the outer cell membrane by passive diffusion and bind to specific IR proteins to create a ligand/receptor complex. This complex then translocates to the cell’s nucleus, where it binds to a specific gene or genes present in the cell’s DNA. Once bound to DNA, the complex modulates the production of the protein encoded by that gene. In this regard, a compound that binds to an IR and mimics the effect of the native ligand is referred to as an “agonist”, while a compound that binds to an IR and inhibits the effect of the native ligand is called an “antagonist”.
The therapeutic mechanism of action of spironolactone involves binding to intracellular mineralocorticoid receptors (MRs) in kidney epithelial cells, thereby inhibiting the binding of aldosterone. Spironolactone has been found to counteract the sodium reabsorption and potassium excretion effects of aldosterone and other mineralocorticoids. Spironolactone has also been shown to interfere with testosterone biosynthesis, has anti-androgen action and inhibits adrenal aldosterone biosynthesis. Large doses of spironolactone in children appear to decrease the testosterone production rate.
Spironolactone is found to exhibit intra-individual variability of pharmacokinetic parameters and it presumably belongs to the group of drugs with high inter-subject variability. Spironolactone has poor water solubility and dissolution rate.
In order to prolong the half-life and decrease the side effects associated with spironolactone, syntheses of spironolactone derivatives have been developed (e.g. synthesis of mexrenone, prorenone, spirorenone). Slight modifications of the spironolactone steroid skeleton, e.g. such as formation of 11β-allenic and epoxy compounds, have been shown to effect important variations in the affinity and specificity for the mineralocorticoid receptor. These results suggest that it is possible to develop spironolactone analogues that do not interact with the androgen receptor or cytochrome P-450 and are therefore free of spironolactone undesirable side-effects.
METABOLISM

SYNTHESIS
METHOD 1 REF 150

REF 130, 150
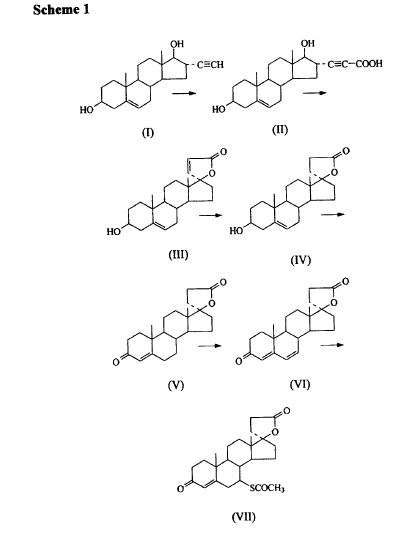

METHOD 2 REF 140
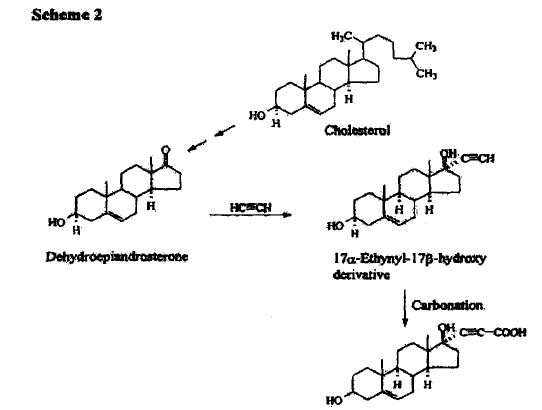
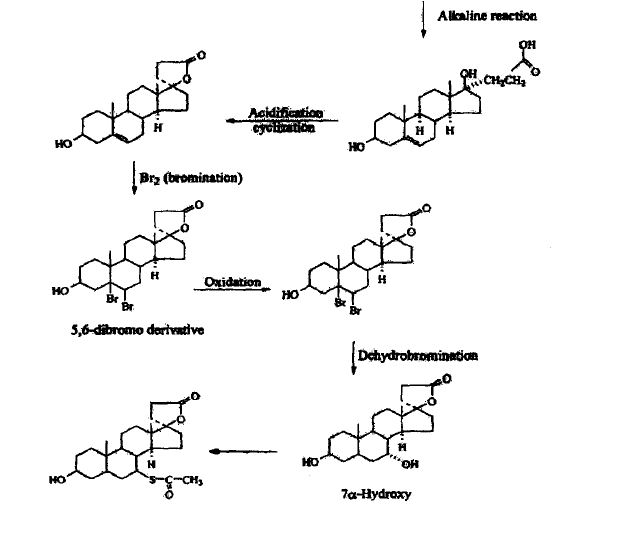

METHOD 3 REF 150
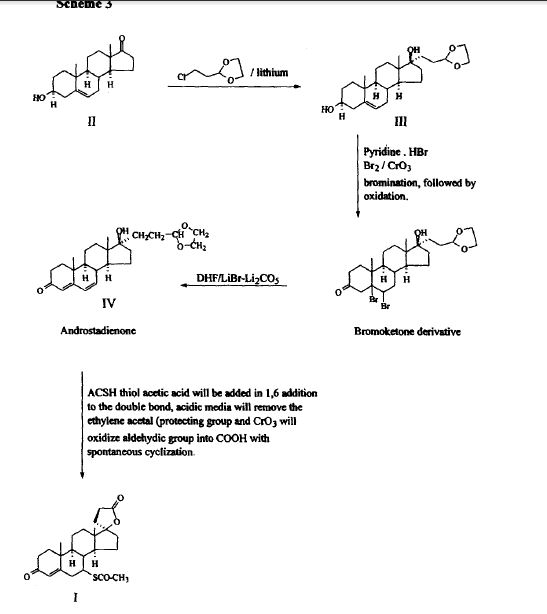
Synthesis
Cella, John A.; Tweit, Robert C. (1959). Journal of Organic Chemistry 24: 1109. doi:.
(See also part 1 and part 3)

SPECTROSCOPY UV
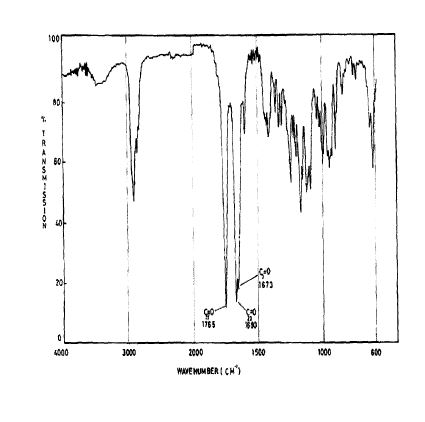
SPECTROSCOPY IR
KBR
The principal absorption peaks of the spectrum shown in Figure 5 were noted at 1765,
1693, 1673, 1240, 1178, 1135, 1123 and 1193 cm -1.
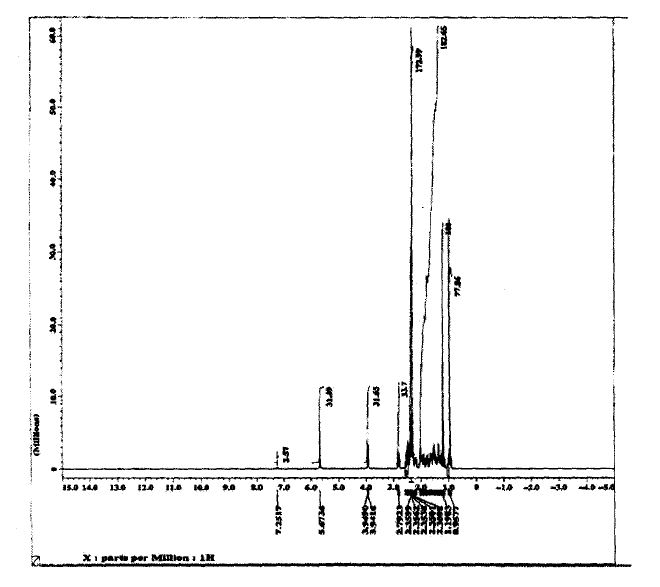
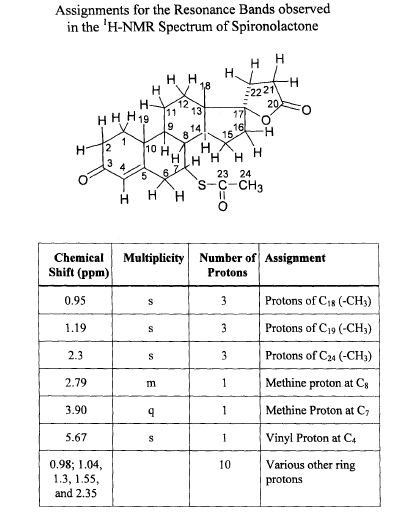
SPECTROSCOPY 1H NMR
SPECTROSCOPY 13C NMR
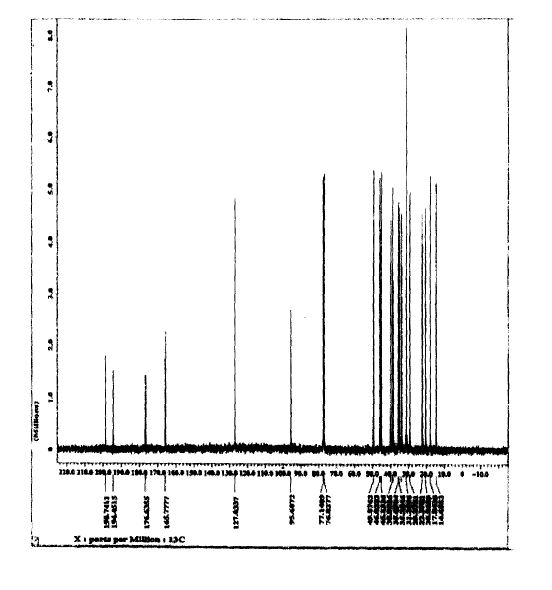
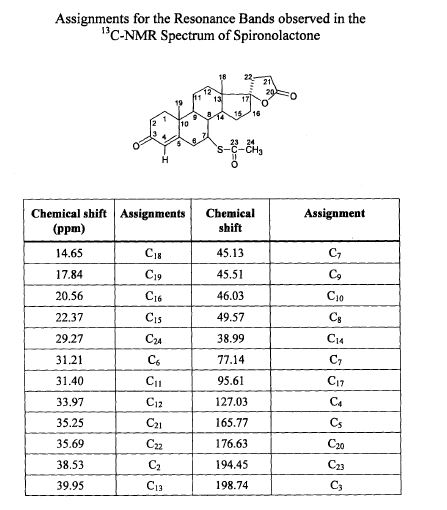
SPECTROSCOPY MASS SPECTRUM
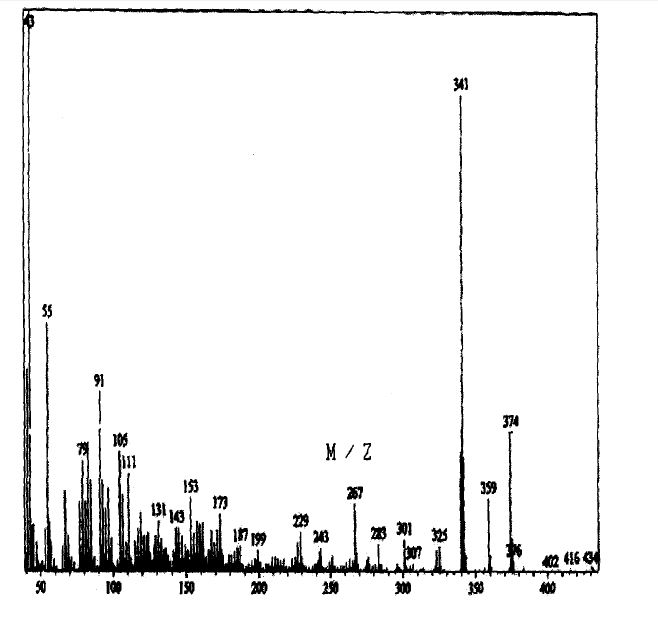
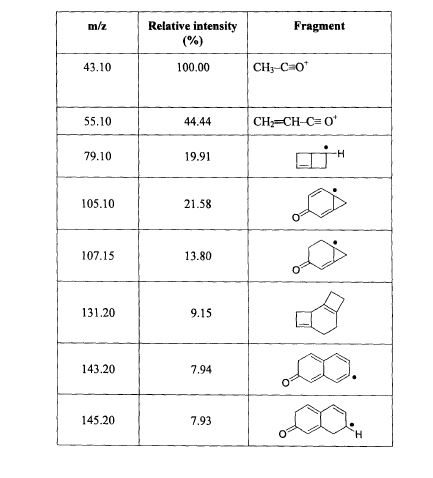
130 J.A. Cola, E.A. Brown, and R.R. Burtner, 3. Org. Chem., 24, 1109(1959).
140 Remington’s: The Science and Practice of Pharmacy, 19 t~ edn.Volume II, K.G. Alfonso, ed.; Mack Publishing Co., Pennsylvania (1995) p.1048.
150. G. Anner and H. Wehrli (Ciba-Geigy, A.-G.), German Often 2,625,723 (cl.C07J21/00), Dec,1976; Swiss Appl. 75/7, 696, 13Jun. 1975; pp. 37.
ANALYTICAL
-
High-Performance Liquid Chromatographic Conditions Column LiChrosorb RP-8, 5 μm. 150 × 4.6 mm I.D. Eluent Acetonitrile-0.05 M phosphate buffer, pH 4 (45:55) Flow-rate 1 ml/min Temperature 25° C. Detector UV detector, wavelength 286 nm or 271 nm Recorder Chart speed 0.5 cm/min Sample loop 10 μl -
The concentration of canrenone is determined in plasma and urine samples by high-performance liquid chromatography (HPLC) with UV-detection. An aliquot of 300 ng of spironolactone derivative is added to the samples as internal standard, which are then extracted twice with 1 ml n-hexane-toluene (1:1, v/v). The organic phase is taken to dryness and re-dissolved in 250 μl HPLC eluent (methanol-water, 60:40, v/v). (25×4.6 mm; 5 μm). Detection is performed with the UV detector set at λ=285 nm.
Flurometric Method
- Five ml of water is a reagent blank and 5 ml of working standards containing 0.05 μg and 0.20 μg of SC-9376 are carried through the entire procedure. Lower sales are read vs. the 0.05 μg standard at full scale, and higher samples vs. the 0.20 μg standard. Fluorescence readings are proportional to the concentrations of the standards in this range.
- Pipette 0.2 ml of heparinized plasma into a 50-ml polyethylene-stoppered centrifuge tube, dilute to 5 ml with water and add 15 ml of methylene chloride (Du Pont refrigeration grade, redistilled). Shake for 30 seconds, centrifuge and discard the aqueous supernatant. Add 1 ml 0.1 N NaOH, shake 15 seconds, centrifuge and discard the supernatant. Transfer a 10-ml aliquot of the methylene chloride phase to another tube containing 2 ml of 65% aqueous sulfuric acid, shake 30 seconds, centrifuge and remove organic phase by aspiration. The material is allowed to stand at room temperature for about 1 hour and then about 1 ml of the sulfuric acid phase in transferred to a quartz cuvette. Fluorescence intensity is determined in an Aminco-Bowman spectrophotofluorometer (activation maximum, 465 nm).
- Gas Liquid Chromatography
- The GLC estimation is carried out on a Fractovap Model 251 series 2150 (Carlo Erba) instrument equipped with a Nickel-63 electron capture detector. A 6-foot, 0.4 mm internal diameter, U-shaped glass column, packed with OV-17 2% or XE-60 1% on gas chrom A, 100-120 mesh (Applied Science Lab) is conditioned for 3 days before use. Argon with 10% methane which passed through a molecular sieve before entering the column is used as the carrier gas. The conditions of analysis are: column 255° C., detector 275° C., carrier gas flow 30 ml/min. Samples are injected on the column with a 10 μl Hamilton syringe. The injector in not heated.
PATENT
https://www.google.com/patents/US20090325918
EXAMPLE 1Chiral Separation
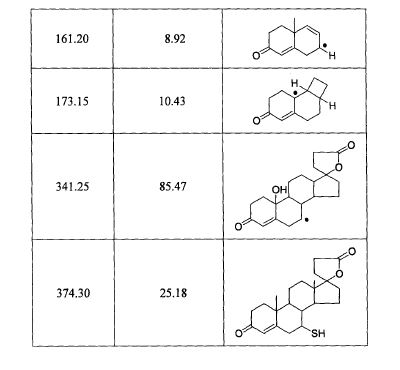
The separation of 7 beta isomer of SL is schematically described below.
-
 Chromatographic Method for Isolation of SL IsomersThe basic method is described in Chan, Ky, et al., J. Chromatog, Nov. 15, 1991:571 (1-2) 291-297. The separation is performed using spectra-physics HPLC instrument and UV variable wavelength detector set at 254 nm. For chiral separation, the chromatographic column is either a pre-packed 25 mm×4.6 mm ID Cyclobond 1 (5 μm particle size), or a pre-packed 150 mm×4 mm ID Resolvosil BSA-7 column (5 μm) operated using the conditions described herein.Analysis of the isomers present in the peaks in the chromatograms and their chiral extract purity analysis can be determined in each case by high resolution NMR spectroscopy using a chiral shift reagent. Based on this information and the determination of molecular weight by mass spectrometry and/or optical activity, structural configuration is assigned to each isomer. Eluted samples of isomers may be re-chromatographed in order to obtain adequate quantities of isomers having desired optical purity for study. For future use, reference standards that are optically pure will be compared for confirmation of purity and identity to the isolated isomers that are obtained after their chromatographic separation.
Chromatographic Method for Isolation of SL IsomersThe basic method is described in Chan, Ky, et al., J. Chromatog, Nov. 15, 1991:571 (1-2) 291-297. The separation is performed using spectra-physics HPLC instrument and UV variable wavelength detector set at 254 nm. For chiral separation, the chromatographic column is either a pre-packed 25 mm×4.6 mm ID Cyclobond 1 (5 μm particle size), or a pre-packed 150 mm×4 mm ID Resolvosil BSA-7 column (5 μm) operated using the conditions described herein.Analysis of the isomers present in the peaks in the chromatograms and their chiral extract purity analysis can be determined in each case by high resolution NMR spectroscopy using a chiral shift reagent. Based on this information and the determination of molecular weight by mass spectrometry and/or optical activity, structural configuration is assigned to each isomer. Eluted samples of isomers may be re-chromatographed in order to obtain adequate quantities of isomers having desired optical purity for study. For future use, reference standards that are optically pure will be compared for confirmation of purity and identity to the isolated isomers that are obtained after their chromatographic separation.
EXAMPLE 2Chemical Synthesis of Optical Isomers
- As an example, the desire spironolactone 7-beta-isomer is synthesized following the scheme that is described below:
-
 Diene (i) is prepared from commercially available starting materials using methods well known in the art of chemical synthesis.Diene (i) is treated with acetic acid and the mixture is heated to reflux to yield 7-alpha-acetate ester (ii). The 7-alpha-ester (ii) is further subjected to nucleophilic substitution, followed by hydrolysis to obtain the 7-beta-isomer (iii). The 7-beta-isomer (iii) is then esterified with an acyl halide in the presence of a base to generate the desired spironolactone 7-beta-isomer (iv).
Diene (i) is prepared from commercially available starting materials using methods well known in the art of chemical synthesis.Diene (i) is treated with acetic acid and the mixture is heated to reflux to yield 7-alpha-acetate ester (ii). The 7-alpha-ester (ii) is further subjected to nucleophilic substitution, followed by hydrolysis to obtain the 7-beta-isomer (iii). The 7-beta-isomer (iii) is then esterified with an acyl halide in the presence of a base to generate the desired spironolactone 7-beta-isomer (iv).
EXAMPLE 3Preparation of Radiolabeled Probe Compounds of the Invention
- Using known methods, the compounds of the invention may be prepared as radiolabeled probes by carrying out their synthesis using precursors comprising at least one atom that is a radioisotope. The radioisotope is preferably selected from at least one of carbon (preferably
14
- C), hydrogen (preferably
3
- H), sulfur (preferably
35
- S), or iodine (preferably I). Such radiolabeled probes are conveniently synthesized by a radioisotope supplier specializing in customer synthesis of radiolabeled probe compounds. Such suppliers include Amersham Corporation, Arlington Heights, Ill.; Cambridge Isotope Laboratories, Inc., Andover, Mass.; SRI International, Menlo Park, Calif.; Wizard Laboratories, West Sacramento, Calif.; ChemSyn Laboratories, Lexena, Kans.; American Radiolabeled Chemicals, Inc., St. Louis, Mo.; and Moravek Biochemicals Inc., Brea, Calif.
- Tritium labeled probe compounds are also conveniently prepared catalytically via platinum-catalyzed exchange in tritiated acetic acid, acid-catalyzed exchange in tritiated trifluoroacetic acid, or heterogeneous-catalyzed exchange with tritium gas. Tritium labeled probe compounds can also be prepared, when appropriate, by sodium borotritide reduction. Such preparations are also conveniently carried out as a custom radiolabeling by any of the suppliers listed in the preceding paragraph using the compound of the invention as substrate.
- EXAMPLE 4Isolation and Purification Procedure
- The optical isomers of spironolactones may be isolated from fluid sample such as urine or blood as follows:
- Extraction from Urine
- The urine sample is extracted with dichloromethane and the extract washed with NaOH (0.1 N) and then with water to neutrality. The residue obtained after evaporation of the dichloromethane extract is purified on TLC in three different systems: benzene-acetone-water, (150:100:0.4); chloroform-ethanol, (90:10); ethyl acetate-cyclohexane-ethanol, (45:25:10), using aldosterone as reference standard.
- The extract is then purified by high performance liquid chromatography (HPLC) on a Waters 6000 A, 480 U.V. detector instrument with radial pressure. The extract is first run through a C
18
- 10μ column using methanol-water (70:30) as the eluent, followed by a silica 5μ column using dichloromethane-methanol (95:5). In both cases, the rate of the eluent is 1.5 ml/min. A small part of the extract is subjected to heptafluorobutyrylation for GLC investigation.
References
- “Spironolactone”. The American Society of Health-System Pharmacists. Retrieved Oct 24, 2015.
- “Spironolactone: MedlinePlus Drug Information”. Retrieved 2016-01-20.
- “Spironolactone”. Merriam-Webster Dictionary.
- “Spironolactone”. Dictionary.com Unabridged. Random House.
- Harry G. Brittain (26 November 2002). Analytical Profiles of Drug Substances and Excipients. Academic Press. p. 309. ISBN 978-0-12-260829-2. Retrieved 27 May 2012.
- Maizes, Victoria (2015). Integrative Women’s Health (2 ed.). p. 746.ISBN 9780190214807.
- “Spironolactone Pregnancy and Breastfeeding Warnings”. Retrieved 29 November2015.
- Camille Georges Wermuth (24 July 2008). The Practice of Medicinal Chemistry. Academic Press. p. 34. ISBN 978-0-12-374194-3. Retrieved 27 May 2012.
- Marshall Sittig (1988). Pharmaceutical Manufacturing Encyclopedia. William Andrew. p. 1385. ISBN 978-0-8155-1144-1. Retrieved 27 May 2012.
- “WHO Model List of EssentialMedicines” (PDF). World Health Organization. October 2013. Retrieved 22 April 2014.
- “Spironolactone”. International Drug Price Indicator Guide. Retrieved 29 November2015.
- Hughes BR, Cunliffe WJ (May 1988). “Tolerance of spironolactone”. The British Journal of Dermatology 118 (5): 687–91. doi:10.1111/j.1365-2133.1988.tb02571.x.PMID 2969259.
- Victor R. Preedy (1 January 2012). Handbook of Hair in Health and Disease. Springer Science & Business Media. pp. 132–. ISBN 978-90-8686-728-8.
- Loy R, Seibel MM (December 1988). “Evaluation and therapy of polycystic ovarian syndrome”. Endocrinology and Metabolism Clinics of North America 17 (4): 785–813.PMID 3143568.
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
7α-Acetylthio-17α-hydroxy-3-oxopregn-4-ene-21-carboxylic acid γ-lactone
|
|
| Clinical data | |
| Pronunciation | /spɪˌroʊnəˈlæktoʊn, spaɪ–, spə–, –ˈrɒ–, –noʊ–/or /ˌspaɪrənoʊˈlæktoʊn/[2][3][4] |
| Trade names | Aldactone |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a682627 |
| Pregnancy category |
|
| Routes of administration |
Oral[1] |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Protein binding | 90%+[5] |
| Metabolism | Hepatic CYP450 |
| Biological half-life | 1.3-2 hours |
| Excretion | Urine, bile |
| Identifiers | |
| CAS Number | 52-01-7 |
| ATC code | C03DA01 (WHO) |
| PubChem | CID 5833 |
| IUPHAR/BPS | 2875 |
| DrugBank | DB00421 |
| ChemSpider | 5628 |
| UNII | 27O7W4T232 |
| KEGG | D00443 |
| ChEBI | CHEBI:9241 |
| ChEMBL | CHEMBL1393 |
| Chemical data | |
| Formula | C24H32O4S |
| Molar mass | 416.574 g/mol |
///////Spironolactone, Supra-puren, Suracton, спиронолактон, سبيرونولاكتون ,
螺内酯 , Abbolactone, Aldactide, SNL, Spiroctanie, Sprioderm, Verospirone, Opianin
O=C5O[C@@]4([C@@]3([C@H]([C@@H]2[C@H](SC(=O)C)C/C1=C/C(=O)CC[C@]1(C)[C@H]2CC3)CC4)C)CC5
Rapamycin (Sirolimus) For the prophylaxis of organ rejection in patients receiving renal transplants.

Rapamycin (Sirolimus)
(3S,6R,7E,9R,10R,12R,14S,15E,17E,19E,21S,23S,26R,27R,34aS)-9,10,12,13,14,21,22,23,24,25, 26,27,32,33,34,34a-Hexadecahydro-9,27-dihydroxy-3-[(1R)-2-[(1S,3R,4R)-4-hydroxy-3-methoxycyclohexyl]-1-methylethyl]-10,21-dimethoxy-6,8,12,14,20,26-hexamethyl-23,27-epoxy-3H-pyrido[2,1-c][1,4]oxaazacyclohentriacontine-1,5,11,28,29(4H,6H,31H)-pentone
Wyeth Pharmaceuticals (Originator)
M.Wt:914.18
Formula:C51H79NO13
53123-88-9 cas no
Antifungal and immunosuppressant. Specific inhibitor of mTOR (mammalian target of Rapamycin). Complexes with FKBP-12 and binds mTOR inhibiting its activity. Inhibits interleukin-2-induced phosphorylation and activation of p70 S6 kinase. Induces autophagy in yeast and mammalian cell lines.
Rapamycin is a triene macrolide antibiotic, which demonstrates anti-fungal, anti-inflammatory, anti-tumor and immunosuppressive properties. Rapamycin has been shown to block T-cell activation and proliferation, as well as, the activation of p70 S6 kinase and exhibits strong binding to FK-506 binding proteins. Rapamycin also inhibits the activity of the protein, mTOR, (mammalian target of rapamycin) which functions in a signaling pathway to promote tumor growth. Rapamycin binds to a receptor protein (FKBP12) and the rapamycin/FKB12 complex then binds to mTOR and prevents interaction of mTOR with target proteins in this signaling pathway. Rapamycin name is derived from the native word for Easter Island, Rapi Nui.
- (-)-Rapamycin
- Antibiotic AY 22989
- AY 22989
- AY-22989
- CCRIS 9024
- HSDB 7284
- NSC 226080
- Rapammune
- Rapamune
- Rapamycin
- SILA 9268A
- Sirolimus
- UNII-W36ZG6FT64
- WY-090217
- A 8167
A macrolide compound obtained from Streptomyces hygroscopicus that acts by selectively blocking the transcriptional activation of cytokines thereby inhibiting cytokine production. It is bioactive only when bound to IMMUNOPHILINS. Sirolimus is a potent immunosuppressant and possesses both antifungal and antineoplastic properties.
Sirolimus (INN/USAN), also known as rapamycin, is an immunosuppressant drug used to prevent rejection in organ transplantation; it is especially useful in kidney transplants. It prevents activation of T cells and B cells by inhibiting their response to interleukin-2 (IL-2). Sirolimus is also used as a coronary stent coating. Sirolimus works, in part, by eliminating old and abnormal white blood cells.[citation needed] Sirolimus is effective in mice with autoimmunity and in children with a rare condition called autoimmune lymphoproliferative syndrome (ALPS).

A macrolide, sirolimus was discovered by Brazilian researchers as a product of the bacterium Streptomyces hygroscopicus in a soil sample fromEaster Island[1] — an island also known as Rapa Nui.[2] It was approved by the FDA in September 1999 and is marketed under the trade nameRapamune by Pfizer (formerly by Wyeth).
Sirolimus was originally developed as an antifungal agent. However, this use was abandoned when it was discovered to have potent immunosuppressive and antiproliferative properties. It has since been shown to prolong the life of mice and might also be useful in the treatment of certain cancers.
Unlike the similarly named tacrolimus, sirolimus is not a calcineurin inhibitor, but it has a similar suppressive effect on the immune system. Sirolimus inhibits the response tointerleukin-2 (IL-2), and thereby blocks activation of T and B cells. In contrast, tacrolimus inhibits the secretion of IL-2.
The mode of action of sirolimus is to bind the cytosolic protein FK-binding protein 12(FKBP12) in a manner similar to tacrolimus. Unlike the tacrolimus-FKBP12 complex which inhibits calcineurin (PP2B), the sirolimus-FKBP12 complex inhibits themammalian target of rapamycin (mTOR, rapamycin being an older name for sirolimus) pathway by directly binding the mTOR Complex1 (mTORC1).
mTOR has also been called FRAP (FKBP-rapamycin associated protein), RAFT (rapamycin and FKBP target), RAPT1, or SEP. The earlier names FRAP and RAFT were coined to reflect the fact that sirolimus must bind FKBP12 first, and only the FKBP12-sirolimus complex can bind mTOR. However, mTOR is now the widely accepted name, since Tor was first discovered via genetic and molecular studies of sirolimus-resistant mutants of Saccharomyces cerevisiae that identified FKBP12, Tor1, and Tor2 as the targets of sirolimus and provided robust support that the FKBP12-sirolimus complex binds to and inhibits Tor1 and Tor2.
 rapamycin
rapamycin
Unlike the similarly named tacrolimus, sirolimus is not a calcineurin inhibitor, but it has a similar suppressive effect on the immune system. Sirolimus inhibits the response to interleukin-2 (IL-2), and thereby blocks activation of T and B cells. In contrast, tacrolimus inhibits the secretion of IL-2.
The mode of action of sirolimus is to bind the cytosolic protein FK-binding protein 12 (FKBP12) in a manner similar to tacrolimus. Unlike the tacrolimus-FKBP12 complex which inhibits calcineurin (PP2B), the sirolimus-FKBP12 complex inhibits the mammalian target of rapamycin(mTOR, rapamycin being an older name for sirolimus) pathway by directly binding the mTOR Complex1 (mTORC1).
mTOR has also been called FRAP (FKBP-rapamycin associated protein), RAFT (rapamycin and FKBP target), RAPT1, or SEP. The earlier names FRAP and RAFT were coined to reflect the fact that sirolimus must bind FKBP12 first, and only the FKBP12-sirolimus complex can bind mTOR. However, mTOR is now the widely accepted name, since Tor was first discovered via genetic and molecular studies of sirolimus-resistant mutants of Saccharomyces cerevisiae that identified FKBP12, Tor1, and Tor2 as the targets of sirolimus and provided robust support that the FKBP12-sirolimus complex binds to and inhibits Tor1 and Tor2.

Rapamycin and its preparation are described in US Patent No. 3,929,992, issued December 30, 1975. Alternatively, rapamycin may be purchased commercially [Rapamune®, Wyeth].
Rapamycin (Sirolimus) is a 31-member natural macrocyclic lactone [C51H79N1O13; MWt=914.2] produced by Streptomyces hygroscopicus and found in the 1970s (U.S. Pat. No. 3,929,992; 3,993,749). Rapamycin (structure shown below) was approved by the Food and Drug Administration (FDA) for the prophylaxis of renal transplant rejection in 1999.

Rapamycin resembles tacrolimus (binds to the same intracellular binding protein or immunophilin known as FKBP-12) but differs in its mechanism of action. Whereas tacrolimus and cyclosporine inhibit T-cell activation by blocking lymphokine (e.g., IL2) gene transcription, sirolimus inhibits T-cell activation and T lymphocyte proliferation by binding to mammalian target of rapamycin (mTOR). Rapamycin can act in synergy with cyclosporine or tacrolimus in suppressing the immune system.
Rapamycin is also useful in preventing or treating systemic lupus erythematosus [U.S. Pat. No. 5,078,999], pulmonary inflammation [U.S. Pat. No. 5,080,899], insulin dependent diabetes mellitus [U.S. Pat. No. 5,321,009], skin disorders, such as psoriasis [U.S. Pat. No. 5,286,730], bowel disorders [U.S. Pat. No. 5,286,731], smooth muscle cell proliferation and intimal thickening following vascular injury [U.S. Pat. Nos. 5,288,711 and 5,516,781], adult T-cell leukemia/lymphoma [European Patent Application 525,960 A1], ocular inflammation [U.S. Pat. No. 5,387,589], malignant carcinomas [U.S. Pat. No. 5,206,018], cardiac inflammatory disease [U.S. Pat. No. 5,496,832], anemia [U.S. Pat. No. 5,561,138] and increase neurite outgrowth [Parker, E. M. et al, Neuropharmacology 39, 1913-1919, 2000].
Although rapamycin can be used to treat various disease conditions, the utility of the compound as a pharmaceutical drug has been limited by its very low and variable bioavailability and its high immunosuppressive potency and potential high toxicity. Also, rapamycin is only very slightly soluble in water. To overcome these problems, prodrugs and analogues of the compound have been synthesized. Water soluble prodrugs prepared by derivatizing rapamycin positions 31 and 42 (formerly positions 28 and 40) of the rapamycin structure to form glycinate, propionate, and pyrrolidino butyrate prodrugs have been described (U.S. Pat. No. 4,650,803). Some of the analogues of rapamycin described in the art include monoacyl and diacyl analogues (U.S. Pat. No. 4,316,885), acetal analogues (U.S. Pat. No. 5,151,413), silyl ethers (U.S. Pat. No. 5,120,842), hydroxyesters (U.S. Pat. No. 5,362,718), as well as alkyl, aryl, alkenyl, and alkynyl analogues (U.S. Pat. Nos. 5,665,772; 5,258,389; 6,384,046; WO 97/35575).
………………………………………..
Synthesis
ref are independent of body…see below for this clip
Several total synthese of rapamycin have been reported3,4as well as many fragments and part-syntheses. Rapamycin is a complicated molecule comprising a 31-membered ring including a pipecolinyl group and pyranose ring, a conjugated triene system and a tri-carbonyl region. It also has 15 chiral centres, meaning the number of possible stereoisomers is enormous. The synthesis of rapamycin therefore presents a huge challenge to synthetic chemists.
In the following synthesis, published in three separate papers5,6,7two fragments of C10-C21 and C22-C42 are prepared separately, before being combined to give the total synthesis of rapamycin. Only the main outline of the synthesis will be shown as it is too long and complicated to show in great detail. For the full experimental details of the synthesis see the literature (ref. nos. given above).

In the retro-synthesis shown the molecule is disconnected at the ester group next to carbon 1 and the C21-C22 double bond of the triene to give the synthetic precursors 2 and 3. Further disconnections of 3 will be shown later. First the C10-C21 fragment is synthesised.
Synthesis of C10-C21 fragment
The synthesis uses (R)-methyl 3-hydroxy-2-methylpropionate (8) as a starting material.

The starting material 8 is converted to an alcohol by a four-step process; protection of the alcohol as aTHP ether followed by reduction, ether formation and deprotection steps. Substitution of the hydroxyl group in the product for a bromine leads to the formation of the bromide 9. Reaction of 9 with methyl acetoacetate gave ester 10.

Catalytic reduction of 10 using the conditions of Noyori produced ester 11, which was then converted to its Weinreb amide 12. Overall, compound 12 was produced in 54% yield from an inexpensive starting material. Vinyl bromide 13 was metalated with t-BuLi and the resulting vinyllithium was combined with 12 and the PMB-protecting group was removed to give 14. The remaining carbonyl group in 14 was selectively reduced to a hyrdoxy group. In order to differentiate the 1,3-diol a lactol was formed, where one hydroxy group ended up in the ring. To acheive this an oxidation was performed using RuCl2(PPh3)3 resulting in formation of a lactol. The two remaining alcohol groups could then be methylated using MeI forming 15.

The lactol ring opening was achieved using TiCl4 and thiol HS(CH2)2SH to form a dithiolane. The freed alcohol was then protected as its TBS ether and the same protecting group selectively removed from the primary alcohol to form 16. To avoid removing the dithiolane group at a later stage in the synthesis the thio-acetal was converted to the dimethyl acetal 17 using PhI(OCOCF3)2 and methanol.

The next stage in the synthesis was to extend 17 for the building of the triene region. The terminal alcohol was oxidised to its aldehyde using BaMnO4 , then a Wittig reaction was carried out using Ph3P=CHCO2Et and CH2Cl2 to form the second double bond. Reduction of the ester group to an alcohol was carried out using DIBAL-H, then treatment with PPh3 and exposure to the air gave rapamycin fragment 2.
Synthesis of C22-C42 fragment
Here the retro-synthesis of 3 is shown, giving the three synthetic precursors 5, 6 and 7

It was thought 4 could be obtained by alkylative coupling of a vinyllithium species generated from 7 to the Weinreb amide 6. The nucleophilic opening of epoxide 5 by the lithiated sulfone from phenyl sulfone 4 would then produce the desired fragment.
The ester 18 was used as a starting material to make fragment 6.

A Wittig reaction followed by reduction and protection steps produced 19. This was hydrogenated using a rhodium catalyst to give syn-dimethyl product 20. The minor anti diastereomer was successfully separated off. 20 was oxidised then underwent an aldol condensation to give adduct 21.

Transamination of 21 and protection of the alcohol with PMB resulted in amide 6, corresponding to the C22-C28 segment of rapamycin.
The vinyl bromide 7 was prepared using ester 22 as a starting material.

Reduction of 22 followed by dibromoolefination resulted in product 23. Acetylene 24 was prepared using n-BuLi, THF and MeI, then sulfenylation with Ph2S2 and bromination gave fragment 7.

Iodination and alkylation of starting material 25 with the lithiated allylic sulfide shown followed by a number of further steps resulted in its conversion to fragment 5.

Fragments 7 was first converted to its vinyllithium using t-BuLi then combined with 6 forming an enone in 78% yield. Stereoselective reduction of the carbonyl group using Zn(BH4)2 gave an alcohol which was protected with DEIPS giving 28. The phenyl sulfide was oxidised to a sulfone using m-CPBA in excess pyridine.

Lithiation and addition of the epoxide 5 resulted in the hydroxy sulfone in a 4:1 ratio of two diastereomers which were separated by HPLC. Metalation using n-BuLi followed by oxidation formed the total C22-C42 fragment.
Total synthesis of rapamycin through the combination of C10-C21 and C22-C42 fragments.
Fragment 3 (C22-C42) was treated with (S)-Boc-pipecolinal, followed by a Swern oxidation resulted in the aldehyde 29.

Condensation with the lithium salt of phosphine oxide 2 (C10-C21) produced the triene shown below.

The triene was hydrolysed with pyridinium p-toluenesulfonic acid and an aldol reaction was performed. Treatment with triethylsilyl triflate produced an amino acid which was subjected to Mukaiyama macrocyclization conditions to form the 31-membered ring. Finally, deprotection steps were performed to give synthetic rapamyin (1). This was judged to be identical to natural rapamycin by comparison of physical properties, 1H-NMR, 13C-NMR, IR and UV spectral data.
3. K. C. Nicolaou, T. K. Chakraborty, A. D. Piscopio, N. Minowa, P. Bertinato; J. Am. Chem. Soc.; 115; 1993; 4419
4. C. M. Hayward, D. Yohannes, S. J. Danishefsky; J. Am. Chem. Soc.; 115; 1993; 9345
5. S. D. Meyer, T. Miwa, M. Nakatsuka, S. L. Schreiber; J. Org. Chem.; 57; 1992; 5058-5060
6. D. Romo, D. D. Johnson, L. Plamondon, T. Miwa, S. L. Schreiber; J. Org. Chem.; 57; 1992; 5060-5063
7. S. D. Meyer, D. Romo, D. D. Johnson, S. L. Schreiber; J. Am. Chem. Soc.; 115; 1993; 7906-7907
………………………………………….
Synthesis
PREPARATION
CUT PASTE FROM TEXT
In one embodiment of this invention rapamycin is prepared in the followingmanner: 4
A suitable fermenter is charged with production meis reached in the fermentation mixture after 2-8 days,
usually after about 5 days, as determined by the cup plate method and Candida albicans as the test organism. The mycelium is harvested by filtration with diatomaceous earth. Rapamycin is then extracted from the mycelium with a water-miscible solvent, for example a lower alkanol, preferably methanol or ethanol. The latter extract is then concentrated, preferably under reduced pressure, and the resulting aqueous phase is extracted with a water-immiscible solvent. A preferred water-immiscible solvent for this purpose is methylene dichloride although chloroform, carbon tetrachloride, benzene, n-butanol and the like may also be used. The latter extract is concentrated, preferably under reduced pressure, to afford the crude product as an oil.
The product may be purified further by a variety of methods. Among the preferred methods of purification is to dissolve the crude product in a substantially nonpolar, first solvent, for example petroleum ether or hexane, and to treat the resulting solution with a suit able absorbent, for example charcoal or silica gel, so that the antibiotic becomes absorbed on the absorbant. The absorbant is then separated and washed or eluted with a second solvent more polar than the first solvent, for example ethyl acetate, methylene dichloride, or a mixture of methylene dichloride and ether (preferred). Thereafter, concentration of the wash solution or eluate affords substantially pure rapamycin. Further purification is obtained by partial precipitation with a nonpolar solvent, for example, petroleum ether, hexane, pentane and the like, from a solution of the rapamycin in a more polar solvent, for example, ether, ethyl acetate, benzene and the like. Still-further purification is obtained by column chromatography, preferably employing silica gel, and by crystallization of the rapamycin from ether.
In another preferred embodiment of this invention a first stage inoculum of S treptomyces hygroscopicus NRRL 5491 is prepared in small batches in a medium containing soybean flour, glucose, ammonium sulfate, and calcium carbonate incubated at about 25C at pH 7.l-7.3 for 24 hrs. with agitation, preferably on a gyrotary shaker. The growth thus obtained is used to inoculate a number of somewhat larger batches of the same medium as described above which are incubated at about 25C and pH 7.1-7.3 for 18 hrs. with agitation, preferably on a reciprocating’shaker, to obtain a sec- “ond stagc inoculum which is used to inoculate the production stage fermenters.
6 5.86′.2.-The fermenters are inoculated with the second stage inoculum described above and incubated at about 25C with’ agitationand aeration while controlling and ‘mai’ntaining the mixture at approximately pH 6.0 by
addition offa base, for example, sodium hydroxide, potassium hydroxide or preferably ammonium hydroxide, as required from time to time. Addition of a source -of assimilable carbon, preferably glucose, is started when theconcentrationof the latter in the broth has dropped to about 0.5% wt/vol, normally about 48 hrs after. the start of fermentation, and is maintained until the end ofthe particular run. In this manner a fermentation broth containing about 60 ug/ml of rapamycin as determined by the assay method described above is obtained in 45 days, when fermentation is stopped.
‘ Filtration of the’mycelium, mixing the latter with a watef-miscible ‘lower’ alkanol, preferably methanol, followed by extraction with a halogenated aliphatic hydrocarbon, preferably trichloroethane, and evaporation of the solvents yields a first oily residue. This first oily residue is dissolved in a lower aliphatic ketone, preferably acetone, filtered from insoluble impurities, the filtrate evaporated to yield a second oily residue which is extractedjwith a water-miscible lower alkanol,
preferably methanol, and the latter extract is evaporated to yield crude rapamycin as a third oily residue. This third oily residue is dissolved in a mixture of a lower aliphatic ketone and a lower aliphatic hydrocarbon, preferably acetone-hexane, an absorbent such as charcoal or preferably silica gel is added to adsorb the rapamycin, the latter is eluted from the adsorbate with a similar but more polar solvent mixture, for example a mixture as above but containing a higher proportion of the aliphatic ketone, the eluates are evaporated and the residue is crystallized from diethyl ether, to yield pure crystalline rapamycin. In this manner a total of 45-5 8% of the rapamycin initially present in the fermentation mixture is recovered as pure crystalline rapamycin.
CHARACTERIZATION solvent systems; for example, ether-hexane 40:60 (Rf 0.42), ‘isopropyl alcoholvbenzene 15:85 (Rf= 0.5) and ethanol-benzene 20:80 (Rf f 0.43);
d. rapamycin obtained from four successive fermentation batchesgave the following values on repeated The production stage fermenters are equipped with 7 devices for controlling and maintaining pH at a predetermined level and for continuous metered addition of elemental analyses:
AVER- e. rapamycin exhibits the following characteristic absorption maxima in its ultraviolet absorption spectrum ethanol):
f. the infrared absorption spectrum of rapamycin in chloroform is reproduced in FIG. 1 and shows characteristic absorption bands at 3560, 3430, 1730, 1705 and 1630-1610 cm;
Further infrared absorption bands are characterized by the following data given in reciprocal centimeters with (s) denoting a strong, (m) denoting a medium, and ![]() denoting a weak intensity band. This classification is arbitrarily selected in such a manner that a band is denoted as strong (s) if its peak absorption is more than two-thirds of the background in the same region; medium (m) if its peak is between one-third and twothirds of the background in the same region; and weak
denoting a weak intensity band. This classification is arbitrarily selected in such a manner that a band is denoted as strong (s) if its peak absorption is more than two-thirds of the background in the same region; medium (m) if its peak is between one-third and twothirds of the background in the same region; and weak ![]() if its peak is less than one-third of the background in the same region.
if its peak is less than one-third of the background in the same region.
2990 cm (m) 1158 cm” (m) 2955 cm (s) 1129 cm (s) 2919 cm (s) 1080 cm (s) 2858 cm (s) 1060 cm (s) 2815 cm (m) 1040 cm (m) 1440 cm (s) 1020 crn’ (m) 1365 cm (m) 978 cm” (s) 1316 cm (in) 905 cm (m) 1272 cm (m) 888 cm” ![]() 1178 cm (s) 866 cm-
1178 cm (s) 866 cm- ![]() g. the nuclear magnetic resonance spectrum of rapamycinin deuterochloroform is reproduced in FIG. 2; SEE PATENT
g. the nuclear magnetic resonance spectrum of rapamycinin deuterochloroform is reproduced in FIG. 2; SEE PATENT
CLAIMS
l. Rapamycin, an antibiotic which a. is a colourless, crystalline compound with a melting point of 183 to l8SC, after recrystallization from ether;
b. is soluble in ether, chloroform, acetone, methanol and dimethylformamide, very sparingly soluble in hexane and petroleum ether and substantially insoluble in water;
c. shows a uniform spot on thin layer plates of silica gel”,
d. has a characteristic elemental analysis of about C,
e. exhibits the following characteristic absorption maxima in its ultraviolet absorption spectrum (95% ff has ‘a characteristic infrared absorption spectrum shown in accompanying FIG. 1; SEE PATENT
……………………………………………..
Rapamycin synthetic studies. 1. Construction of the C(27)-C(42) subunit. Tetrahedron Lett 1994, 35, 28, 4907

A partial synthesis of rapamycin has been reported: The condensation of sulfone (I) with epoxide (II) by means of butyllithium followed by desulfonation with Na/Hg gives the partially protected diol (III), which is treated with methanesulfonyl chloride and NaH to afford the epoxide (IV). Ring opening of epoxide (IV) with LiI and BF3.Et2O followed by protection of the resulting alcohol with PMBOC(NH)CCl3 yields the primary iodo compound (V). The condensation of (V) with the fully protected dihydroxyaldehyde (VI) (see later) by means of butyllithium in THF/HMPT gives the fully protected trihydroxyketone (VII), which is hydrolyzed with camphorsulfonic acid (CSA) to the corresponding gemdiol and reprotected with pivaloyl chloride (the primary alcohol) and tert-butyldimethylsilyl trifluoromethanesulfonate (the secondary alcohol), yielding a new fully protected trihydroxyketone (VIII). Elimination of the pivaloyl group with DIBAL and the dithiane group with MeI/CaCO3 affords the hydroxyketone (IX), which is finally oxidized with oxalyl chloride to the ketoaldehyde (X), the C(27)-C(42) fragment [the C(12)-C(15) fragment with the C(12)-substituent based on the IUPAC nomenclature recommendations]. The fully protected dihydroxyaldehyde (VI) is obtained as follows: The reaction of methyl 3-hydroxy-2(R)-methylpropionate (XI) with BPSCl followed by reduction with LiBH4 to the corresponding alcohol and oxidation with oxalyl chloride gives the aldehyde (XII), which is protected with propane-1,3-dithiol and BF3.Et2O to afford the dithiane compound (XIII). Elimination of the silyl group with TBAF followed by esterification with tosyl chloride, reaction with NaI and, finally, with sodium phenylsulfinate gives the sulfone (XIV), which is condensed with the partially protected dihydroxyaldehyde (XV), oxidized with oxalyl chloride and desulfonated with Al/Hg to afford the dithianyl ketone (XVI). The reaction of (XVI) with lithium hexamethyldisilylazane gives the corresponding enolate, which is treated with dimethyllithium cuprate to yield the fully protected unsaturated dihydroxyaldehyde (VI).
……………………………………………
……………………………
The Ley Synthesis of RapamycinRapamycin (3) is used clinically as an immunosuppressive agent. The synthesis of 3 (Angew. Chem. Int. Ed. 2007, 46, 591. DOI: 10.1002/anie.200604053) by Steven V. Ley of the University of Cambridge was based on the assembly and subsequent coupling of the iododiene 1 and the stannyl alkene 2.
The lactone of 1 was prepared by Fe-mediated cyclocarbonylation of the alkenyl epoxide 5, following the protocol developed in the Ley group.
The cyclohexane of 2 was constructed by SnCl4-mediated cyclization of the allyl stannane 9, again employing a procedure developed in the Ley group. Hydroboration delivered the aldehyde 11, which was crotylated with 12, following the H. C. Brown method. The alcohol so produced (not illustrated) was used to direct the diastereoselectivity of epoxidation, then removed, to give 13. Coupling with 14 then led to 2.
Combination of 1 with 2 led to 15, which was condensed with catechol to give the macrocycle 16. Exposure of 16 to base effected Dieckmann cyclization, to deliver the ring-contracted macrolactone 17, which was carried on to (-)-rapamycin (3).
|




……………………………….
Total Synthesis of Rapamycin
Angewandte Chemie International Edition
Volume 46, Issue 4, pages 591–597, January 15, 2007

PREVIEW THIS ARTICLE WITH READCUBE
![]()
……………………..

Ley, Maddess, Tackett, Watanabe, Brennan, Spilling, Scott and Osborn. ACIEE, 2006, EarlyView. DOI:10.1002/anie.200604053.
It’s been in the works for quite a while, but Steve Ley’s synthesis of Rapamycin has just been published. This complex beast has a multitude of biological activities, including an interesting immunosuppressive profile, resulting in clinical usage following organ transplantation. So, unsurprisingly, it’s been the target of many projects, with complete total syntheses published by Smith, Danishefsky, Schreiber and KCN.
So what makes this one different? Well, it does have one of the most interesting macrocyclisations I’ve seen since Jamison’s paper, and a very nice demonstration of the BDA-aldol methodology. The overall strategy is also impressive, so on with the retro:

First stop is the BDA-aldol; this type of chemistry is interesting, because the protecting group for the diol is also the stereo-directing group. The stereochemistry for this comes from a glycolic acid, and has been usedin this manner by the group before. The result is as impressive as ever, with a high yield, and presumably a very high d.r. (no mention of actual numbers).

The rest of the fragment synthesis was completed in a succinct and competent manner, but using relatively well known chemistry. However, I was especially impressed with the macrocyclisation I mentioned:

Tethering the free ends of the linear precursor with a simple etherification/esterification onto catechol gave then a macrocycle holding the desired reaction centres together. Treatment of this with base then induces a Dieckmann-condensation type cyclisation to deliver the desired macrocycle. Of course, at this stage, only a few more steps were required to complete the molecule, and end an era of the Wiffen Lab.
………………………………
Drugs Fut 1999, 24(1): 22
DOI: 10.1358/dof.1999.024.01.474036

In CDCl3 rapamycin exists as a mixture of conformers in a 3:1 ratio, which complicates the NMR spectrum. In the table below the chemical shifts of the carbons and hydrogens of the major isomer only are given.
| Carbon No. | Carbon Type | Major carbon | Major proton | Carbon No. | Carbon Type | Major carbon | Major proton |
|
1
|
C=O | 169.2 |
–
|
28
|
CH-OH | 77.3 | 4.17 |
|
2
|
CH | 51.3 | 5.29 |
29
|
C=C | 136.1 |
–
|
|
3
|
CH2 | 27.0 | 2.34, 1.76 |
30
|
CH=C | 126.8 | 5.42 |
|
4
|
CH2 | 20.6 | 1.78, 1.47 |
31
|
CH | 46.6 | 3.33 |
|
5
|
CH2 | 25.3 | 1.75, 1.48 |
32
|
C=O | 208.2 |
–
|
|
6
|
CH2 | 44.2 | 3.59, 3.44 |
33
|
CH2 | 40.7 | 2.74, 2.60 |
|
8
|
C=O | 166.8 |
–
|
34
|
CH-OCO | 75.7 | 5.17 |
|
9
|
C=O | 192.5 |
–
|
35
|
CH | 33.1 | 1.98 |
|
10
|
O-C-OH | 98.5 |
–
|
36
|
CH2 | 38.4 | 1.22, 1.12 |
|
11
|
CH | 33.7 | 1.98 |
37
|
CH | 33.2 | 1.39 |
|
12
|
CH2 | 27.3 | 1.60, 1.60 |
38
|
CH2 | 34.2 | 2.10, 0.68 |
|
13
|
CH2 | 31.3 | 1.62, 1.33 |
39
|
CH-OCH3 | 84.4 | 2.93 |
|
14
|
67.2 | 3.86 |
40
|
CH-OH | 73.9 | 3.37 | |
|
15
|
CH2 | 38.8 | 1.85, 1.52 |
41
|
CH2 | 31.3 | 1.99, 1.33 |
|
16
|
CH-OCH3 | 84.4 | 3.67 |
42
|
CH2 | 31.7 | 1.70, 1.00 |
|
17
|
C=C | 135.5 |
–
|
43
|
11-CH3 | 16.2 | 0.95 |
|
18
|
CH=C | 129.6 | 5.97 |
44
|
17-CH3 | 10.2 | 1.65 |
|
19
|
CH=C | 126.4 | 6.39 |
45
|
23-CH3 | 21.5 | 1.05 |
|
20
|
CH=C | 133.6 | 6.32 |
46
|
25-CH3 | 13.8 | 1.00 |
|
21
|
CH=C | 130.1 | 6.15 |
47
|
29-CH3 | 13.0 | 1.74 |
|
22
|
CH=C | 140.2 | 5.54 |
48
|
31-CH3 | 16.0 | 1.11 |
|
23
|
CH | 35.2 | 2.32 |
49
|
35-CH3 | 15.9 | 0.92 |
|
24
|
CH2 | 40.2 | 1.50, 1.20 |
50
|
16-OCH3 | 55.8 | 3.13 |
|
25
|
CH | 41.4 | 2.74 |
51
|
27-OCH3 | 59.5 | 3.34 |
|
26
|
C=O | 215.6 |
–
|
52
|
39-OCH3 | 56.5 | 3.41 |
|
27
|
CH-OCH3 | 84.9 | 3.71 |
REFERENCES
- Vézina C, Kudelski A, Sehgal SN (October 1975). “Rapamycin (AY-22,989), a new antifungal antibiotic”. J. Antibiot. 28 (10): 721–6. doi:10.7164/antibiotics.28.721. PMID 1102508.
- Pritchard DI (2005). “Sourcing a chemical succession for cyclosporin from parasites and human pathogens”. Drug Discovery Today 10 (10): 688–691. doi:10.1016/S1359-6446(05)03395-7. PMID 15896681.
Wu X, Wang L, Han Y, Regan N, Li PK, Villalona MA, Hu X, Briesewitz R, Pei D.
ACS Comb Sci. 2011 Sep 12;13(5):486-95. doi: 10.1021/co200057n. Epub 2011 Jul 28.
Gibbons JJ, Abraham RT, Yu K.
Semin Oncol. 2009 Dec;36 Suppl 3:S3-S17. doi: 10.1053/j.seminoncol.2009.10.011. Review.
Ayral-Kaloustian S, Gu J, Lucas J, Cinque M, Gaydos C, Zask A, Chaudhary I, Wang J, Di L, Young M, Ruppen M, Mansour TS, Gibbons JJ, Yu K.
J Med Chem. 2010 Jan 14;53(1):452-9. doi: 10.1021/jm901427g.
6. Fluorescent probes to characterise FK506-binding proteins.
Kozany C, März A, Kress C, Hausch F.
Chembiochem. 2009 May 25;10(8):1402-10. doi: 10.1002/cbic.200800806.
7. Recent advances in the chemistry, biosynthesis and pharmacology of rapamycin analogs.
Graziani EI.
Nat Prod Rep. 2009 May;26(5):602-9. doi: 10.1039/b804602f. Epub 2009 Mar 5. Review.
8 Total synthesis of rapamycin.
Ley SV, Tackett MN, Maddess ML, Anderson JC, Brennan PE, Cappi MW, Heer JP, Helgen C, Kori M, Kouklovsky C, Marsden SP, Norman J, Osborn DP, Palomero MA, Pavey JB, Pinel C, Robinson LA, Schnaubelt J, Scott JS, Spilling CD, Watanabe H, Wesson KE, Willis MC.
Chemistry. 2009;15(12):2874-914. doi: 10.1002/chem.200801656.
Evans AC, Longbottom DA, Matsuoka M, Davies JE, Turner R, Franckevicius V, Ley SV.
Org Biomol Chem. 2009 Feb 21;7(4):747-60. doi: 10.1039/b813494d. Epub 2009 Jan 6.
Maddess ML, Tackett MN, Ley SV.
Prog Drug Res. 2008;66:13, 15-186. Review.
Zhang J, Rodila R, Watson P, Ji Q, El-Shourbagy TA.
Biomed Chromatogr. 2007 Oct;21(10):1036-44.
Sormani R, Yao L, Menand B, Ennar N, Lecampion C, Meyer C, Robaglia C.
BMC Plant Biol. 2007 Jun 1;7:26.
13 Total synthesis of rapamycin.
Maddess ML, Tackett MN, Watanabe H, Brennan PE, Spilling CD, Scott JS, Osborn DP, Ley SV.
Angew Chem Int Ed Engl. 2007;46(4):591-7. No abstract available.
14 Drug evaluation: AP-23573–an mTOR inhibitor for the treatment of cancer.
Elit L.
IDrugs. 2006 Sep;9(9):636-44.
15 lipase-catalyzed regioselective esterification of rapamycin: synthesis of temsirolimus (CCI-779).
Gu J, Ruppen ME, Cai P.
Org Lett. 2005 Sep 1;7(18):3945-8.
Elit L.
Curr Opin Investig Drugs. 2002 Aug;3(8):1249-53. Review.
Dumont FJ.
Curr Opin Investig Drugs. 2001 Sep;2(9):1220-34. Review.
18 Kuo et al (1992) Rapamycin selectively inhibits interleukin-2 activation of p70 S6 kinase. Nature 358 70. PMID:1614535.
19 Huang et al (2003) Rapamycins: mechanism of action and cellular resistance. Cancer Biol.Ther. 2 221. PMID:12878853.
20 Kobayashi et al (2007) Rapamycin, a specific inhibitor of the mammalian target of rapamycin, suppresses lymphangiogenesis and lymphatic metastasis. Cancer Sci. 98 726. PMID: 17425689.
21 Fleming et al (2011) Chemical modulators of autophagy as biological probes and potential therapeutics. 7 9. PMID:21164513.
22 J Am Chem Soc1993,115,(10):4419
23 Tetrahedron Lett1994,35,(28):4911
24 Chemistry (Weinheim)1995,1,(5):318
24
 SIROLIMUS
SIROLIMUS
FEMALE FERTILITY
PATENTS
| Canada | 2293793 | APPROVED2006-07-11 | EXP 2018-06-11 |
| Canada | 2103571 | 2003-04-29 | 2012-02-21 |
| United States | 5989591 | 1998-09-11 | 2018-09-11 |
| United States | 5212155 | 1993-05-18 | 2010-05-18 |
| WO1998054308A2 * | May 28, 1998 | Dec 3, 1998 | Biotica Tech Ltd | Polyketides and their synthesis and use |
| EP0589703A1 * | Sep 23, 1993 | Mar 30, 1994 | American Home Products Corporation | Proline derivative of rapamycin, production and application thereof |
| US20010039338 * | Jun 7, 2001 | Nov 8, 2001 | American Home Products Corporation | Regioselective synthesis of rapamycin derivatives |
| WO2007067560A2 * | Dec 6, 2006 | Jun 14, 2007 | Clifford William Coughlin | Scalable process for the preparation of a rapamycin 42-ester from a rapamycin 42-ester boronate |
| WO2012131019A1 | Mar 30, 2012 | Oct 4, 2012 | Sandoz Ag | Regioselective acylation of rapamycin at the c-42 position |
| US7622578 | Dec 6, 2006 | Nov 24, 2009 | Wyeth | Scalable process for the preparation of a rapamycin 42-ester from a rapamycin 42-ester boronate |
| US3929992 | Apr 12, 1974 | Dec 30, 1975 | Ayerst Mckenna & Harrison | Rapamycin and process of preparation |
| US5646160 | May 26, 1995 | Jul 8, 1997 | American Home Products Corporation | Method of treating hyperproliferative vascular disease with rapamycin and mycophenolic acid |
| US5665772 | Sep 24, 1993 | Sep 9, 1997 | Sandoz Ltd. | O-alkylated rapamycin derivatives and their use, particularly as immunosuppressants |
| US5728710 | Jul 16, 1993 | Mar 17, 1998 | Smithkline Beecham Corporation | Rapamycin derivatives |
| US5957975 | Dec 15, 1997 | Sep 28, 1999 | The Centre National De La Recherche Scientifique | Stent having a programmed pattern of in vivo degradation |
| US5985890 | Jun 5, 1996 | Nov 16, 1999 | Novartis Ag | Rapamycin derivatives |
| US6001998 | Oct 13, 1995 | Dec 14, 1999 | Pfizer Inc | Macrocyclic lactone compounds and their production process |
| US6015815 | Sep 24, 1998 | Jan 18, 2000 | Abbott Laboratories | Tetrazole-containing rapamycin analogs with shortened half-lives |
| US6187568 | Aug 20, 1999 | Feb 13, 2001 | Pfizer Inc | Macrocyclic lactone compounds and their production process |
| US6273913 | Apr 16, 1998 | Aug 14, 2001 | Cordis Corporation | Modified stent useful for delivery of drugs along stent strut |
| US6585764 | Jun 4, 2001 | Jul 1, 2003 | Cordis Corporation | Stent with therapeutically active dosage of rapamycin coated thereon |
| US6641611 | Nov 26, 2001 | Nov 4, 2003 | Swaminathan Jayaraman | Therapeutic coating for an intravascular implant |
| US6805703 | Sep 18, 2001 | Oct 19, 2004 | Scimed Life Systems, Inc. | Protective membrane for reconfiguring a workpiece |
| US7025734 | Sep 28, 2001 | Apr 11, 2006 | Advanced Cardiovascular Systmes, Inc. | Guidewire with chemical sensing capabilities |
| US7056942 | Jan 16, 2004 | Jun 6, 2006 | Teva Pharmaceutical Industries Ltd. | Carvedilol |
| US7820812 * | Jul 23, 2007 | Oct 26, 2010 | Abbott Laboratories | Methods of manufacturing crystalline forms of rapamycin analogs |
| US20010027340 | Jun 4, 2001 | Oct 4, 2001 | Carol Wright | Stent with therapeutically active dosage of rapamycin coated thereon |
| US20010029351 | May 7, 2001 | Oct 11, 2001 | Robert Falotico | Drug combinations and delivery devices for the prevention and treatment of vascular disease |
| US20020005206 | May 7, 2001 | Jan 17, 2002 | Robert Falotico | Antiproliferative drug and delivery device |
| US20020007213 | May 7, 2001 | Jan 17, 2002 | Robert Falotico | Drug/drug delivery systems for the prevention and treatment of vascular disease |
| US20020082680 | Sep 7, 2001 | Jun 27, 2002 | Shanley John F. | Expandable medical device for delivery of beneficial agent |
| US20020123505 | Sep 10, 2001 | Sep 5, 2002 | Mollison Karl W. | Medical devices containing rapamycin analogs |
| US20030129215 | Sep 6, 2002 | Jul 10, 2003 | T-Ram, Inc. | Medical devices containing rapamycin analogs |
| US20040072857 | Jul 2, 2003 | Apr 15, 2004 | Jacob Waugh | Polymerized and modified rapamycins and their use in coating medical prostheses |
| US20050033417 | Jul 1, 2004 | Feb 10, 2005 | John Borges | Coating for controlled release of a therapeutic agent |
| US20050101624 | Nov 12, 2003 | May 12, 2005 | Betts Ronald E. | 42-O-alkoxyalkyl rapamycin derivatives and compositions comprising same |
| US20050152842 | Dec 22, 2004 | Jul 14, 2005 | Chun Li | Poly (L-glutamic acid) paramagnetic material complex and use as a biodegradable MRI contrast agent |
| US20050175660 | Oct 29, 2004 | Aug 11, 2005 | Mollison Karl W. | Medical devices containing rapamycin analogs |
| US20050208095 | Nov 22, 2004 | Sep 22, 2005 | Angiotech International Ag | Polymer compositions and methods for their use |
| US20050209244 | Feb 27, 2003 | Sep 22, 2005 | Prescott Margaret F | N{5-[4-(4-methyl-piperazino-methyl)-benzoylamido]-2-methylphenyl}-4-(3-pyridyl)-2-pyrimidine-amine coated stents |
| US20050239178 | Apr 25, 2005 | Oct 27, 2005 | Wyeth | Labeling of rapamycin using rapamycin-specific methylases |
| US20060094744 | Sep 28, 2005 | May 4, 2006 | Maryanoff Cynthia A | Pharmaceutical dosage forms of stable amorphous rapamycin like compounds |
| US20060229711 | Apr 4, 2006 | Oct 12, 2006 | Elixir Medical Corporation | Degradable implantable medical devices |
| US20070015697 | Nov 1, 2005 | Jan 18, 2007 | Peyman Gholam A | Enhanced ocular neuroprotection and neurostimulation |
| US20070059336 | Feb 27, 2006 | Mar 15, 2007 | Allergan, Inc. | Anti-angiogenic sustained release intraocular implants and related methods |
| US20070207186 | Mar 3, 2007 | Sep 6, 2007 | Scanlon John J | Tear and abrasion resistant expanded material and reinforcement |
| US20080086198 | May 24, 2007 | Apr 10, 2008 | Gary Owens | Nanoporous stents with enhanced cellular adhesion and reduced neointimal formation |
| EP1236478A1 | Feb 27, 2002 | Sep 4, 2002 | Medtronic Ave, Inc. | Peroxisome proliferator-activated receptor gamma ligand eluting medical device |
| EP1588727A1 | Apr 20, 2005 | Oct 26, 2005 | Cordis Corporation | Drug/drug delivery systems for the prevention and treatment of vascular disease |
| WO1993016189A1 | Feb 11, 1993 | Aug 19, 1993 | Pfizer | Novel macrocyclic lactones and a productive strain thereof |
| WO1994009010A1 | Sep 24, 1993 | Apr 28, 1994 | Sandoz Ag | O-alkylated rapamycin derivatives and their use, particularly as immunosuppressants |
| WO1996041807A1 | Jun 5, 1996 | Dec 27, 1996 | Sylvain Cottens | Rapamycin derivatives |
| WO1998007415A2 | Aug 18, 1997 | Feb 26, 1998 | Ciba Geigy Ag | Methods for prevention of cellular proliferation and restenosis |
| WO2001087263A2 | May 14, 2001 | Nov 22, 2001 | Cordis Corp | Delivery systems for treatment of vascular disease |
| WO2001087342A2 | May 14, 2001 | Nov 22, 2001 | Cordis Corp | Delivery devices for treatment of vascular disease |
| WO2001087372A1 | Apr 25, 2001 | Nov 22, 2001 | Cordis Corp | Drug combinations useful for prevention of restenosis |
| WO2001087373A1 | May 14, 2001 | Nov 22, 2001 | Cordis Corp | Delivery devices for treatment of vascular disease |
| WO2001087374A1 | May 14, 2001 | Nov 22, 2001 | Cordis Corp | Delivery systems for treatment of vascular disease |
| WO2001087375A1 | May 14, 2001 | Nov 22, 2001 | Cordis Corp | Delivery devices for treatment of vascular disease |
| WO2001087376A1 | May 14, 2001 | Nov 22, 2001 | Cordis Corp | Drug/drug delivery systems for the prevention and treatment of vascular disease |
| WO2002056790A2 | Dec 18, 2001 | Jul 25, 2002 | Avantec Vascular Corp | Delivery of therapeutic capable agents |
| WO2002065947A2 | Feb 18, 2002 | Aug 29, 2002 | Jomed Gmbh | Implants with fk506 for prophylaxis and treatment of restonoses |
| WO2003064383A2 | Feb 3, 2003 | Aug 7, 2003 | Ariad Gene Therapeutics Inc | Phosphorus-containing compounds & uses thereof |
| WO2006116716A2 | Apr 27, 2006 | Nov 2, 2006 | William A Dunn | Materials and methods for enhanced degradation of mutant proteins associated with human disease |

![]()
A plaque, written in Brazilian Portuguese, commemorating the discovery of sirolimus on Easter Island, near Rano Kau
mTOR inhibitor
temsirolimus (CCI-779), everolimus (RAD001), deforolimus (AP23573), AP21967, biolimus, AP23102, zotarolimus (ABT 578), sirolimus (Rapamune), and tacrolimus (Prograf).\
SIROLIMUS
1H NMR

13 C NMR

HPLC

MIDAZOLAM


MIDAZOLAM
8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine
59467-70-8 CAS NO OF FREE BASE
59467-94-6 MALEATE, Launched – 1982, Roche (Originator)
59467-96-8 (HCl)
A short-acting hypnotic-sedative drug with anxiolytic and amnestic properties. It is used in dentistry, cardiac surgery, endoscopic procedures, as preanesthetic medication, and as an adjunct to local anesthesia. The short duration and cardiorespiratory stability makes it useful in poor-risk, elderly, and cardiac patients. It is water-soluble at pH less than 4 and lipid-soluble at physiological pH.
Midazolam (/mɪˈdæzəlæm/, marketed in English-speaking countries and Mexico under the trade names Dormicum, Hypnovel, andVersed,) is a short-acting drug in the benzodiazepine class developed by Hoffmann-La Roche in the 1970s. The drug is used for treatment of acute seizures, moderate to severe insomnia, and for inducing sedation and amnesia before medical procedures. It possesses profoundly potentanxiolytic, amnestic, hypnotic, anticonvulsant, skeletal muscle relaxant, and sedative properties.[6][7][8] Midazolam has a fast recovery time and is the most commonly used benzodiazepine as a premedication for sedation; less commonly it is used for induction and maintenance of anesthesia.Flumazenil, a benzodiazepine antagonist drug, can be used to treat an overdose of midazolam, as well as to reverse sedation.[7] However, flumazenil can trigger seizures in mixed overdoses and in benzodiazepine-dependent individuals, so is not used in most cases.[9][10]
midazolam
Administration of midazolam by the intranasal or the buccal route (absorption via the gums and cheek) as an alternative to rectally administereddiazepam is becoming increasingly popular for the emergency treatment of seizures in children. Midazolam is also used for endoscopyprocedural sedation and sedation in intensive care. The anterograde amnesia property of midazolam is useful for premedication before surgery to inhibit unpleasant memories. Midazolam, like many other benzodiazepines, has a rapid onset of action, high effectiveness and low toxicity level. Drawbacks of midazolam include drug interactions, tolerance, and withdrawal syndrome, as well as adverse events including cognitive impairment and sedation. Paradoxical effects occasionally occur, most commonly in children and the elderly, particularly after intravenous administration. The drug has also recently been hastily introduced for use in executions in the USA in combination with other drugs.
Midazolam is a short-acting benzodiazepine in adults with an elimination half-life of one to four hours; however, in the elderly, as well as young children and adolescents, the elimination half-life is longer. Midazolam is metabolised into an active metabolite alpha1-hydroxymidazolam. Age related deficits, renal and liver status affect the pharmacokinetic factors of midazolam as well as its active metabolite. However, the active metabolite of midazolam is minor and contributes to only 10 percent of biological activity of midazolam. Midazolam is poorly absorbed orally with only 50 percent of the drug reaching the bloodstream. Midazolam is metabolised by cytochrome P450 (CYP) enzymes and by glucuronide conjugation. The therapeutic as well as adverse effects of midazolam are due to its effects on the GABAA receptors; midazolam does not activate GABAA receptors directly but, as with other benzodiazepines, it enhances the effect of the neurotransmitter GABA on the GABAA receptors (↑ frequency of Cl− channel opening) resulting in neural inhibition. Almost all of the properties can be explained by the actions of benzodiazepines on GABAA receptors. This results in the following pharmacological properties being produced: sedation, hypnotic, anxiolytic, anterograde amnesia, muscle relaxation and anti-convulsant.Midazolam maleate is a benzodiazepine that is commercialized by Astellas Pharma and Roche as an intravenous or intramuscular injection for the long-term sedation of mechanically ventilated patients under intensive care. The drug is also available in a tablet formulation, and is currently distributed in various markets, including Germany, Japan, Switzerland and the U.K. In March 2002, two lots of a syrup formulation were recalled in the U.S. due to the potential presence of a crystalline precipitate of an insoluble complex of midazolam and saccharin. Subsequently, the injection and syrup formulations of the product were both withdrawn from the U.S. market. In 2010, a Pediatric Use Marketing Authorization (PUMA) was filed for approval in the E.U. by ViroPharma for the treatment of prolonged, acute, convulsive seizures in infants, toddlers, children and adolescents, from 3 months to less than 18 years. In 2011, a positive opinion was assigned to the PUMA and final approval was assigned in June 2011. The product was launched in the U.S. in November 2011. This product was filed for approval in Japan in 2013 by Astellas Pharma for the conscious sedation in dentistry and dental surgery. In the same year the product was approved for this indication.
In terms of clinical development, a nasal formulation of the drug is in phase III clinical trials at Upsher-Smith for rescue treatment of seizures in patients on stable anti-epileptic drug regimens who require control of intermittent bouts of increased seizure activity (seizure clusters). The Hopitaux de Paris had been developing a sublingual tablet formulation of midazolam to be used in combination with morphine for the treatment of pain in children following bone fractures; however, no recent development has been reported for this indication. NovaDel Pharma had been developing the compound preclinically for the treatment of generalized anxiety, however no recent developments have been reported.
Midazolam achieves its therapeutic effect through interaction with the gamma-aminobutyric acid benzodiazepine (GABA-BZ) receptor complex. Subunit modulation of the GABA-BZ receptor chloride channel macromolecular complex is hypothesized to be responsible for some of the pharmacological properties of benzodiazepines, which include sedative, anxiolytic, muscle relaxant, and anticonvulsive effects in animal models. GABA acts at inhibitory synapses in the brain by binding to specific transmembrane receptors in the plasma membrane of both pre- and post-synaptic neurons, opening ion channels and bringing about a hyperpolarization via either chloride or potassium ion flow.
In 2008, fast track designation was assigned to midazolam maleate in the U.S. for the treatment of seizure disorders.
In 2009, Orphan Drug Designation was received in the U.S. by for the treatment of seizure disorders in patients who require control of intermittent bouts of increased seizure activity (e.g. acute repetitive seizures, seizure clusters). This designation was assigned in the U.S. for the treatment of nerve agent-induced seizures.
In 2010, midazolam maleate was licensed to Upsher-Smith by Ikano Therapeutics for the treatment of acute repetitive seizure in patients with epilepsy. However, in 2010, Ikano closed and dissolved its business. Previously, Ikano had transferred to Upsher-Smith ownership of it nasal midazolam maleate program.
Midazolam is among about 35 benzodiazepines which are currently used medically, and was synthesised in 1975 by Walser and Fryer at Hoffmann-LaRoche, Inc in the United States.Owing to its water solubility, it was found to be less likely to cause thrombophlebitis than similar drugs.The anticonvulsant properties of midazolam were studied in the late 1970s, but not until the 1990s did it emerge as an effective treatment for convulsive status epilepticus. As of 2010, it is the most commonly used benzodiazepine in anesthetic medicine. In acute medicine, midazolam has become more popular than other benzodiazepines, such as lorazepam and diazepam, because it is shorter lasting, is more potent, and causes less pain at the injection site.Midazolam is also becoming increasingly popular in veterinary medicine due to its water solubility.
Midazolam is a water-soluble benzodiazepine available as a sterile, nonpyrogenic parenteral dosage form for intravenous or intramuscular injection. Each mL contains midazolam hydrochloride equivalent to 1 mg or 5 mg midazolam compounded with 0.8% sodium chloride and 0.01% edetate disodium with 1% benzyl alcohol as preservative, and sodium hydroxide and/or hydrochloric acid for pH adjustment. pH 2.9-3.7.
Midazolam is a white to light yellow crystalline compound, insoluble in water. The hydrochloride salt of midazolam, which is formed in situ, is soluble in aqueous solutions. Chemically, midazolam HCl is 8-chloro-6-(2-fluorophenyl)-1-methyl-4H– imidazo[1,5-a] [1,4] benzodiazepine hydrochloride. Midazolam hydrochloride has the molecular formula C18H13ClFN3•HCl, a calculated molecular weight of 362.25 and the following structural formula:
 |
In the Netherlands, midazolam is a List II drug of the Opium Law. Midazolam is a Schedule IV drug under the Convention on Psychotropic Substances. In the United Kingdom, midazolam is a Class C controlled drug. In the United States, midazolam (DEA number 2884) is on the Schedule IV list of the Controlled Substances Act as a non-narcotic agent with low potential for abuse.
midaolam hydrochloride NDA 018654, 075154
REF
U.S. Pat. No. 4,280,957
U.S. Pat. No. 5,693,795
U.S. Pat. No. 6,512,114
Midazolam Maleate
Drugs Fut 1978, 3(11): 822
Bioorganic and Medicinal Chemistry, 2012 , vol. 20, 18 pg. 5658 – 5667
Journal of Heterocyclic Chemistry, 1983 , vol. 20, 3 pg. 551 – 558.. 32 maleate
WO 2001070744
WO 2001002402
WO 2012075286
US2011/275799 A1… no 5
Journal of Organic Chemistry, 1978 , vol. 43, p. 936,942, mp free base, nmr
| US4280957 | May 15, 1978 | Jul 28, 1981 | Hoffmann-La Roche Inc. | Imidazodiazepines and processes therefor |
| US6262260 * | Mar 23, 2000 | Jul 17, 2001 | Abbott Laboratories | Process for the preparation of midazolam |
| US6512114 | Jun 30, 1999 | Jan 28, 2003 | Abbott Laboratories | Process for the preparation of Midazolam |
……………………….
introduction
4H-imidazo[1,5-a][1,4]benzodiazepines or, more simply, imidazobenzodiazepines, are a class of benzodiazepines having the general formula (I),

wherein the 1,4-diazepine ring is fused with a 1,3-imidazole ring. The main compounds part of the 4H-imidazo[1,5-a][1,4]benzodiazepines are Midazolam of formula (IV):

an active ingredient currently commercially available as a hydrochloride salt under the name of Versed or Hypnovel for anaesthetic and sedative use and the maleate salt currently commercially available under the name Dormicum or Flormidal.
Other important compounds are Climazolam of formula (VII):

Imidazenil of formula (VIII):

1-Hydroxymidazolam of formula (IX):

and Desmethyl midazolam of formula (X):

all these being biologically active substances and having psychotropic and sedative action.
The synthesis of the Midazolam as described in U.S. Pat. No. 4,280,957 of Hoffmann-La Roche provides for the decarboxylation reaction of the 8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine-3-carboxylic acid of formula (VI) according to the following scheme:

The process for preparing the intermediate (VI) via basic hydrolysis of the corresponding ester is described in such patent publication and it is well known in the art.
The thermal decarboxylation reaction in high boiling solvent such as mineral oil at 230° C. for 5 min results in a mixture of products of Midazolam of formula (IV) and of Isomidazolam of formula (IV-bis), a non-pharmacologically active isomer, at a 80:20 ratio. The two products are separated by chromatography.
At industrial level, the formation of the Isomidazolam isomer impurity requires a further isomerisation reaction performed on the mixture of the two compounds to convert the isomer into the active product. The reaction mixture obtained from the thermal decarboxylation is thus subjected to basic treatment under the action of KOH in EtOH followed by an acid treatment which thus provides a mixture of Midazolam-Isomidazolam at a 95:5 ratio. The final removal of the Isomidazolam impurity from the product occurs through crystallisation of the product from AcOEt and EtOH. In order to limit this isomerisation treatment, in the subsequent U.S. Pat. No. 5,693,795 of Hoffmann-La Roche dated 1999, there is described a process for performing the decarboxylation of the compound of formula (VI) in n-butanol in a continuous tubular reactor with a 4 minutes permanence period with a yield between 47-77%. However, the reaction, performed at high temperature and pressure (280° C., 100 bars) results in the formation of a considerable percentage of Isomidazolam (85:15 Midazolam/Isomidazolam ratio) which still requires the basic isomerisation step.
Lastly, in U.S. Pat. No. 6,512,114 of Abbott Laboratories there is described the decarboxylation of the compound of formula (VI) in mineral oil or in N,N-Dimethylacetamide (DMA) at 160-230° C. for at least 3 hours obtaining a 3/1 to 6/1 Midazolam/Isomidazolam ratio with a yield of isolated product equal to just 54%.
Though performed using dedicated apparatus and in extreme conditions, the prior art processes do not allow selectively performing the decarboxylation reaction of the intermediate (VI) to Midazolam thus requiring a further synthetic passage followed by crystallisation with ensuing reduction of the overall yield.
Midazolam (8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine) is represented by the following structural formula (I):

Midazolam is a central nervous system (CNS) depressant, used for short term treatment of insomnia. Like other benzodiazepines, midazolam binds to benzodiazepine receptors in the brain and spinal cord and is thus used as a short-acting hypnotic-sedative drug with anxiolytic and amnestic properties. It is currently used in dentistry, cardiac surgery, endoscopic procedures, as a preanesthetic medication, as an adjunct to local anesthesia and as a skeletal muscle relaxant. Depending on the pH value, midazolam can exist in solution as a closed ring form (I) as well as an open ring form (IA), which are in equilibrium, as shown in Scheme 1:

The amount of the open ring form (IA) is dependent upon the pH value of the solution. At a pH value of about 3, the content of the open ring form (IA) can be 40%, while at pH value of 7.5, the closed ring form (I) can be more than 90%.
Clinical studies have demonstrated that there are no significant differences in the clinical activity between midazolam hydrochloride and midazolam maleate, however the use of intravenous midazolam hydrochloride has been associated, in some cases, with respiratory depression and arrest.
U.S Pat. No. 4,280,957 (hereinafter the ‘957 patent) describes a synthetic process for preparing midazolam, which is depicted in Scheme 2 below. This process includes reacting 2-aminomethyl-7-chloro-2,3-dihydro-5-(2-fluorophenyl)-1H-1,4-bezodiazepine (II) with acetic anhydride in dichloromethane to produce 2-acetamido-methyl-7-chloro-2,3-dihydro-5-(2-fluorophenyl)-1H-1,4-bezodiazepine (III). The latter is heated with polyphosphoric acid at 150° C. to produce 8-chloro-6-(2-fluorophenyl)-3a,4-dihydro-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine of formula (IV), which is purified by column chromatography. Compound IV is then mixed with toluene and manganese dioxide and heated to reflux to afford midazolam base, which is crystallized from ether to yield a product with mp of 152-154° C.

The ‘957 patent further describes an alternative process which includes reacting 2-aminomethyl-7-chloro-2,3-dihydro-5-(2-fluorophenyl)-1H-1,4-bezodiazepine (II) (optionally as a dimaleate salt) with triethylorthoacetate in ethanol and in the presence of p-toluenesulfonic acid to afford 8-chloro-6-(2-fluorophenyl)-3a,4-dihydro-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine (IV). This product is dissolved in xylene and treated with activated manganese dioxide to afford the crude base, which is reacted in situ with maleic acid in ethanol and crystallized by addition of ether to produce the midazolam maleate having melting point of 148-151° C. The process is depicted in Scheme 3 below.

The preparation of midazolam maleate from the isolated midazolam base is also described in a further example of the ‘957 Patent, wherein a warm solution of midazolam base in ethanol is combined with a warm solution of maleic acid in ethanol. The mixture is diluted with ether and at least part of the solvents is evaporated using a steam bath to obtain crystalline midazolam maleate having melting point of 148-151° C. The yield and the purity of the obtained midazolam maleate are not disclosed.
U.S. Pat. No. 6,512,114 (hereinafter the ‘114 patent) describes another synthetic process for preparing midazolam, which is depicted in Scheme 4 below. According to this Process, the starting material 8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine-3-carboxylic acid (V) is heated in mineral oil for 3 hours at 230° C. until it is decarboxylated, followed by treatment with potassium tert-butoxide, to afford midazolm (I), isomidazolam (VI) and a midazolam dimmer (VII). Midazolam base is obtained in 54.5% yield after two re-crystallizations from ethyl acetate and heptane; however, the purity of the product is not disclosed.

The preparation of midazolam by conventional routes is liable to produce impurities such as isomidazolam (VI) and a midazolam dimmer (VII), and possibly other impurities. There is, therefore, a need in the art for a midazolam purification process that will provide highly pure midazolam containing minimal amounts of impurities produced. The present invention provides such a process.
This example describes the preparation of midazolam base as taught in the ‘957 patent.
16 g (0.03 mol) of 2-aminomethyl-7-chloro-5-(2-fluorophenyl)-2,3-dihydro-1H-1,4-bezodiazepine dimaleate was dissolved in 200 ml of toluene and 10 ml of 25% ammonium hydroxide solution was added and mixing was maintained for an hour. Then, the phases were separated and the toluene phase was dried by azeotropic distillation using a Dean Stark apparatus. 7 ml (0.038 mol) of triethylorthoacetate was added and the solution was heated to reflux for 4 hours, after which time the solution was left to cool to ambient temperature. 25 ml of methyl tert-butyl ether was added and the mixture was cooled overnight to produce 8-chloro-6-(2-fluorophenyl)-3a,4-dihydro-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine, which was isolated by filtration. The product was mixed with 200 ml of toluene and dried by azeotropic distillation using a Dean Stark apparatus. Then, 30 g of manganese dioxide was added and the mixture was heated to reflux for two hours. The excess manganese dioxide was filtered off to afford a solution of midazolam base in toluene, which was evaporated to obtain a product having 97.9% purity and containing 0.44% of impurity VIII and 1.14% of impurity IX (according to HPLC).
…………………………
EXAMPLE 28
2-Aminomethyl-7-chloro-2,3-dihydro-5-(2-fluorophenyl)-1H-1,4-benzodiazepine dimaleate
A suspension of 17 g (0.05 m) of 7-chloro-1,3-dihydro-5-(2-fluorophenyl)-2-nitromethylene-2H-1,4-benzodiazepine-4-oxide in 200 ml of tetrahydrofuran and 100 ml of methanol was hydrogenated in presence of 17 g of Raney nickel at an initial pressure of 155 psi for 24 hrs. The catalyst was removed by filtration and the filtrate was evaporated. The residue was dissolved in 50 ml of 2-propanol and warmed on the steambath. A warm solution of 17 g of maleic acid in 60 ml of ethanol was added and the salt was allowed to crystallize by cooling in the ice bath. The final product consisted of yellow crystals with mp 196
EXAMPLE 14
8-Chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine
Acetic anhydride, 7 ml., was added to a solution of 6.16 g. of crude 2-aminomethyl-7-chloro-2,3-dihydro-5-(2-fluorophenyl)-1H-1,4-benzodiazepine in 200 ml. of methylene chloride. The solution was layered with 200 ml. of saturated aqueous sodium bicarbonate and the mixture was stirred for 20 minutes. The organic layer was separated, washed with sodium bicarbonate, dried over sodium sulfate and evaporated to leave 6.2 g. resinous 2-acetaminomethyl-7-chloro-2,3-dihydro-5-(2-fluorophenyl)-1H-1,4-benzodiazepine. This material was heated with 40 g. of polyphosphoric acid at 150 water, made alkaline with ammonia and ice and extracted with methylene chloride. The extracts were dried and evaporated and the residue (5.7 g.) was chromatographed over 120 g. of silica gel using 20% methanol in methylene chloride. The clean fractions were combined and evaporated to yield resinous 8-chloro-3a,4-dihydro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[ 1,5-a][1,4]benzodiazepine. A mixture of this material with 500 ml. of toluene and 30 g. of manganese dioxide was heated to reflux for 11/2 hours. The manganese dioxide was separated by filtration over celite. The filtrate was evaporated and the residue was crystallized from ether to yield a product with m.p. 152 was recrystallized from methylene chloride/hexane
EXAMPLE 49
8-Chloro-6-(2-fluorophenyl)-1-methyl-6H-imidazo[1,5-a][1,4]benzodiazepine
Potassium t-butoxide, 0.625 g. (5.5 mmol), was added to a solution of 1.625 g. (5 mmol) of 8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine in 20 ml. of dimethylformamide cooled to -30 nitrogen for 10 min. at -30 ml. of glacial acetic acid and was then partitioned between aqueous bicarbonate and toluene/methylene chloride (3:1 v/v). The organic layer was separated, dried and evaporated. The residue was chromatographed over 60 g. of silica gel using 25% (v/v) methylene chloride in ethyl acetate. The less polar product was eluted first and was crystallized from ethylacetate/hexane to yield product with m.p. 180
EXAMPLE 50
8-Chloro-6-(2-fluorphenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine
Potassium t-butoxide, 0.125 g. (1.1 mmol) was added to a solution of 0.325 g. (1 mmol) of 8-chloro-6-(2-fluorophenyl)-1-methyl-6H-imidazo[1,5-a][1,4]benzodiazepine in 20 ml. of dimethylformamide cooled to -30 -30 by addition of 0.2 ml. of glacial acetic acid and was partitioned between aqueous sodium bicarbonate and methylene chloridetoluene (1:3). The organic phase was washed with water, dried and evaporated. The residue was chromatographed over 20 g. of silica gel using ethyl acetate for elution. After elution of starting material, product was collected and crystallized from ether/hexane, m.p. 156
hyd and dihydrochloride
EXAMPLE 24
8-Chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine dihydrochloride
A solution of 0.32 g (1 mmol) of 8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine in 5 ml of ethanol was treated with excess ethanolic hydrogen chloride. The salt was crystallized by addition of 2-propanol and ether. The colorless crystals were collected, washed with ether and dried to leave a final product with mp 290
EXAMPLE 258-Chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine hydrochloride
A solution of 0.325 g (1 mmol) of 8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine in 3 ml of ethanol was combined with a suspension of 0.4 g (1 mmol) of the dihydrochloride of this compound in 5 ml of ethanol. After filtration, the solution was treated with ether and heated on the steambath for 5 min to crystallize. The crystals were collected, washed with ether and dried to leave the monohydrochloride with mp 295
maleate
EXAMPLE 22
8-Chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine maleate
A warm solution of 6.5 g (0.02 m) of 8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine in 30 ml of ethanol was combined with a warm solution of 2.6 g (0.022 m) of maleic acid in 20 ml of ethanol. The mixture was diluted with 150 ml of ether and heated on the steam bath for 3 min. After cooling, the crystals were collected, washed with ether and dried in vacuo to yield a final product with mp 148
…
Synthesis
Midazolam, can be described according to scheme 4 indicated below:

 was prepared according to processes known in the art (e.g. U.S. Pat. No. 4,280,957) which comprise the basic hydrolysis of the corresponding ester.
was prepared according to processes known in the art (e.g. U.S. Pat. No. 4,280,957) which comprise the basic hydrolysis of the corresponding ester.For the reactions performed in the microreactor, the solutions containing the substrates to be decarboxylated were loaded into 5 and 10 mL gastight glass syringes (Hamilton, item n. 81527, 81627) mounted on syringe pumps (KD Scientifics, model KDS100). A pipe made of PTFE® (OD=1.58 mm, ID=0.8 mm, Supelco, item n. 58696-U) was used for making the reaction channel.A counterpressure valve sold by Swagelok (item n. SS-SS1-VH) was used for regulating the flow within the channel.Example 1Synthesis of the Compound of Formula (V)—Example of the Invention

50 g (0.135 mol) of 8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepin-3-carboxylic acid of formula (VI) and 250 mL of ethanol were loaded into a two-neck 500 mL flask, equipped with a magnetic stirrer. 40 mL of an aqueous solution of 1 M HCl are dripped in about 10 minutes. The open di-hydrochloride intermediate of formula (V) starts precipitating into the reaction environment already after 3 minutes from the beginning of the addition of the acid solution. The mixture is maintained stirred at RT for 3 hrs and then it is filtered on buckner washing the solid with ethanol. The moist product is dried in an oven under vacuum at 60° C. up to reaching a constant weight. A light yellow crystalline product is obtained (51.5 g, 83% yield). The crude product was used for the decarboxylation without further purifications.
ESI-MS [MeCN+0.1% HCOOH]: m/z 388 (V); 370 (VI).
1H-NMR (250 MHz, CD3OD): 2.52 (s, 3H); 4.27-4.41 (m, 2H); 7.22-8.1 (m, 7H). M.p.: 217° C.
Example 2
Synthesis of Midazolam of Formula (IV)—Performed in Batch—Example of the Invention

30 g (0.065 mol) of 5-(aminomethyl)-1-{(4-chloro-2-[(2-fluorophenyl)carbonyl]phenyl}-2-methyl-1H-imidazole-4-carboxylic acid dihydrochloride of formula (V) and 90 mL of NMP are loaded into a three-neck 250 mL flask, equipped with a magnetic stirrer and coolant. The mass is heated using an oil bath at T=195-203° C. for one hour. Thus, 1 mL of solution is collected for performing HPLC analysis. The reaction product is Midazolam having 82% titre (w/w) (determined via HPLC titre correcting it using the solvent) and it contains 1% of Isomidazolam. The product is extracted using Isopropyl acetate after raising the pH to 10 by adding aqueous Na2CO3.
Example 3
Synthesis of Midazolam of Formula (IV)—Performed in a Micro-Reactor—Example of the Invention

3.22 g (7 mmol) of 5-(aminomethyl)-1-{4-chloro-2-[(2-fluorophenyl)carbonyl]phenyl}-2-methyl-1H-imidazole-4-carboxylic acid dihydrochloride of formula (V) and 10 mL of NMP are loaded into a 10 mL flask equipped with a magnetic stirrer. In order to facilitate the complete solubilisation of the substrate, it is necessary to slightly heat the reaction mixture (about 40° C.) for a few minutes. The solution thus obtained is transferred into a 10 mL gastight glass syringe mounted on a KDS100 syringe pump (FIG. 1) and the flow is regulated at 1.0 mL/h so as to set a residence period of 30 minutes at 200° C. The reaction product is Midazolam having an 89% titre (w/w) (determined via HPLC titre correcting it using the solvent) and containing 3% (w/w) of Isomidazolam.
Example 4Synthesis of Midazolam of formula (IV)—Comparison of the InventionA table is reported which summarises the results of the decarboxylation of the compound of formula (V) and (V-bis) (for the latter see Examples 6 and 7) obtained according to some embodiments of the invention and those obtained by way of experiment through the decarboxylation of the intermediate of formula (VI) (process of the prior art) both performed in 3 volumes of NMP at 200° C., both in batch method (Example 4) and in continuous method with the microreactor (MR) made of PTFE of FIG. 1. (Examples 4-1, 4-2, 4-3).
| Example | substrate | Mode | Solv. | T° C. | t min. | Midazolam (p/p) | Isomidaz. (P/P) |
| 2 | (V) | Batch | NMP | 200 | 60 | 82 | 1 |
| 3 | (V) | MR | NMP | 200 | 30 | 89 | 3 |
| 7 | (V-bis) | Batch | NMP | 200 | 60 | 68 | 3 |
| 4 | (VI) | Batch | NMP | 200 | 60 | 78 | 18 |
| 4-1 | (VI) | MR | NMP | 200 | 38 | 81 | 17 |
| 4-2 | (VI) | MR | NMP | 200 | 20 | 77 | 18 |
| 4-3 | (VI) | MR | NMP | 200 | 15 | 58 | 22 |
| U.S. Pat. No. | (VI) | Tubular | n-BuOH | 290 | 4 | 85 * | 15 * |
| 5,693,795 | reactor | ||||||
| U.S. Pat. No. | (VI) | Batch | Olio | 230 | 180 | 75 * | 25 * |
| 6,512,114 | min. | 87.5 * | 12.5 * | ||||
| or DMA | |||||||
| * = Midazolam/Isomidazolam ratio only (other impurities not considered). | |||||||
The product of the comparative experiments 4, 4-1, 4-2, 4-3 and of the two USA patents should be subjected to a further isomerisation process to reduce the high amount of Isomidazolam so as to be able to obtain Midazolam free of Isomidazolam after further crystallization, which would not be required for the product obtained according to the invention (examples 2 and 3).

A 4-neck RBF was charged under nitrogen flow with: 10 g of Midazolam (IV) (prepared according to example 2) and 40 mL of Ethanol. The slurry was stirred until complete dissolution at 25/30° C. In an other flask was prepared the following solution: 3.72 g of maleic acid are dissolved in 15 mL of Ethanol. The slurry was stirred until complete dissolution at 25/30° C. The maleic acid solution is dropped in 30/40 minutes and keeping T=25/30° C. into the solution containing Midazolam. The slurry was cooled down at −15° C. in one hour and kept at that temperature for at least 2 hours. The slurry was then filtered and the cake was washed with 40 mL of cool Ethanol. The filter was discharged and the product was dried at 40° C. under vacuum for 2 hours and then at 60° C. for 8 hours. 12.8 g of Midazolam Maleate as white solid were collected (Molar yield=94.5%). m.p.=149-152° C. (by DSC).

A 4-neck RBF was charged under nitrogen flow with: 1 g of Midazolam (IV) (prepared according to example 2) and 15 mL of Ethanol. The slurry was stirred until complete dissolution at 25/30° C. 5 mL of a ethanolic solution of Hydrochloric acid 2N were slowly added. 20 mL of Isopropanol were added over 30 minutes at RT. The slurry was cooled down at −15° C. in one hour and kept at that temperature for at least 2 hours. The slurry was then filtered and the cake was washed with 10 mL of cool isopropanol. The filter was discharged and the product was dried at 40° C. under vacuum for 2 hours and then at 60° C. for 8 hours. Midazolam dihydrochloride as white solid was collected.
MIDAZOLAM HYDROCHLORIDE
Example 10
Preparation of 8-Chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine hydrochloride (Midazolam hydrochloride)

A 4-neck RBF was charged under nitrogen flow with: 1 g of Midazolam (IV) (prepared according to example 2) and 10 mL of Ethanol. The slurry was stirred until complete dissolution at 25/30° C. In an other flask was prepared the following suspension: 1.22 g of Midazolam dihydrochloride (prepared according to example 9) and 15 mL of Ethanol. The Midazolam ethanolic solution was added to the Midazolam dihydrochloride suspension. After filtration, the solution was treated with MTBE and heated at 60° C. until crystallization. After cooling to RT, the slurry was filtered, the cake washed with MTBE and the product was dried to provide Midazolam (mono)hydrochloride as a white solid.
…..


NEW DRUG APPROVALS
ONE TIME
$10.00
TELMISARTAN ..Actavis’ Generic Version of Micardis Receives FDA Approval
DUBLIN, Jan. 8, 2014 /PRNewswire/ — Actavis plc today announced that it has received approval from the U.S. Food and Drug Administration (FDA) on its Abbreviated New Drug Application (ANDA) for Telmisartan Immediate-Release Tablets, 20 mg, 40 mg and 80 mg, a generic equivalent to Boehringer Ingelheim’s Micardis. Actavis intends to launch the product immediately.
IRBESARTAN

IRBESARTAN, SR 47436, BMS-186295
Avapro® (Bristol-Myers Squibb) and Karvea®
(Sanofi-Winthrop)
2-butyl-3-({4-[2-(2H-1,2,3,4-tetrazol-5-yl)phenyl]phenyl}methyl)-1,3-diazaspiro[4.4]non-1-en-4-one
138402-11-6 CAS NO
U.S. Patents 5,270,317 and 5,352,788, 6,162,922
The compound prepared according to US 5270317 is polymorph A
-
Irbesartan is known by following chemical names:
- (a) 2-Butyl-3-[[2′-(1H-tetrazol-5-yl)[1,1′-biphenyl]-4-yl]methyl]-1,3-diazaspiro[4,4]non-1-en-4-one
- (b) 2-Butyl-3-[p-(o-1H-tetrazol-5-ylphenyl)benzyl]-1,3-diazaspiro[4,4]non-1-en-4-one
- (c) 2-n-butyl-4-spirocyclopentane-1-[(2′-(tetrazol-5-yl)biphenyl-4-yl) methyl]-2-imidazolin-5-one.
-
The structural formula of Irbesartan is represented below.

Irbesartan
-
The synthesis of irbesartan is first disclosed in US5270317 (equivalentEP0454511 ) and subsequently, several other patents disclose the synthesis of irbesartan by different methods. Basically the synthesis of this molecule involves two common intermediates namely spiroimidazole and substituted 4′-bromomethylbiphenyl.
-
US 5270317 describes preparation of irbesartan wherein 1-[(2′-cyanobiphenyl-4-yl)methyl]-2-n-butyl-4-spirocyclopentane-2-imidazolin -5-one which is reacted with tributyltin azide in xylene at reflux temperature for 66 hours to give a product which is isolated from the reaction mass as trityl irbesartan and then deprotected in methanol/THF mixture using 4N hydrochloric acid to get irbesartan.
-
US5629331 describes a process for the preparation of irbesartan from 1-[(2′-cyanobiphenyl)4-yl)methyl]-2-n-butyl-4-spirocyclopentane-2-imidazolin-5-one using sodium azide, TEA.HCl in N-methylpyrrolidone. The product is isolated from the alkaline reaction mass after acidification to pH 4.7 to 5.8 and the crude product is recrystallised from IPA/water to get Form A and ethanol/water to get Form B.
Irbesartan (INN) /ɜrbəˈsɑrtən/ is an angiotensin II receptor antagonist used mainly for the treatment of hypertension. Irbesartan was developed by Sanofi Research (now part ofsanofi-aventis). It is jointly marketed by sanofi-aventis and Bristol-Myers Squibb under thetrade names Aprovel, Karvea, and Avapro.
It is marketed in Brazil by Sanofi-Aventis under the trade name Aprovel .

As with all angiotensin II receptor antagonists, irbesartan is indicated for the treatment ofhypertension. Irbesartan may also delay progression of diabetic nephropathy and is also indicated for the reduction of renal disease progression in patients with type 2 diabetes,[1]hypertension and microalbuminuria (>30 mg/24 hours) or proteinuria (>900 mg/24 hours).[2]
Irbesartan is also available in a combination formulation with a low dose thiazide diuretic, invariably hydrochlorothiazide, to achieve an additive antihypertensive effect. Irbesartan/hydrochlorothiazide combination preparations are marketed under similar trade names to irbesartan preparations, including Irda, CoIrda, CoAprovel, Karvezide,Avalide and Avapro HCT.
A large randomized trial following 4100+ men and women with heart failure and normal ejection fraction (>=45%) over 4+ years found no improvement in study outcomes or survival with irbesartan as compared to placebo.[3]

BMS annual sales approx $1.3bn. Sanofi-aventis annual sales approx $2.1bn. In the United States, a generic version is available. Patent expired March 2012.
- Lewis EJ, Hunsicker LG, Clarke WR, Berl T, Pohl MA, Lewis JB, Ritz E, Atkins RC, Rohde R, Raz I; Collaborative Study Group. (2001). “Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes”. N Engl J Med 345 (12): 851–60. doi:10.1056/NEJMoa011303.PMID 11565517.
- Rossi S, editor. Australian Medicines Handbook 2006. Adelaide: Australian Medicines Handbook; 2006. ISBN 0-9757919-2-3
- Massie BM, Carson PE, McMurray JJ, Komajda M, McKelvie R, Zile MR, Anderson S, Donovan M, Iverson E, Staiger C, Ptaszynska A (December 2008). “Irbesartan in patients with heart failure and preserved ejection fraction”. N. Engl. J. Med. 359 (23): 2456–67.doi:10.1056/NEJMoa0805450. PMID 19001508.
4……….C. A. Bernhart, P. M. Perreaut, B. P. Ferrari, Y. A. Muneaux,
J.-L. A. Assens, J. Clement, F. Haudricourt, C. F. Muneaux,
J. E. Taillades, M.-A. Vignal, J. Gougat, P. R. Guiraudou, C.
A. Lacour, A. Roccon, C. F. Cazaubon, J.-C. Brelihre, G. Le
Fur, D. Nisato, J. Med. Chem. 1993, 36, 3371–3380.
5…. K. F. Croom, M. P. Curran, K. L. Goa, Drugs 2004 64,
999–1028.
6… C. Bernhard, J.-C. Breliere, J. Clement, D. Nisato, P. M. Perreaut, C. F. Muneaux, (Elf Sanofi) US 5 270 317; Chem. Abstr. 1993, 119, 95560.
7. S. Chava, M. Bandari, K. S. Mathuresh, (Matrix Laboratories) WO 2005/122699; Chem. Abstr. 2005, 144, 88292.
5. S. Zupan~i~, A. Pe~avar, R. Zupet, (Krka) WO 2006/073376;
Chem. Abstr. 2006, 145, 124576.
8. C. V. Kavitha, S. L. Gaonkar, J. N. Chandra, S. Narendra, C.
T. Sadashiva, K. S. Rangappa, Bioorg. Med. Chem. 2007, 15,
7391–7398.
9. S. Rádl, J. Stach, O. Klecán, (Zentiva) WO 2005/021535;
Chem. Abstr. 2005, 142, 298118.
10. B. Satyanarayana, Y. Anjaneyulu, P. Veerasomaiah, P. P.
Reddy, Heterocycl. Commun. 2007, 13, 223–228.
11. V. V. Korrapati, P. Rao, R. Dandala, V. K. Handa, I. V. S. Rao,
A. Rani, A. Naidu, Synth. Commun. 2007, 37, 2897–2905.
12. J. Havlí~ek, Z. Mandelová, R. Weisemann, I. Strˇelec, S.
Rádl, Collect. Czech. Chem. Commun. 2009, 77, 347.
Irbesartan of formula (I).

The chemical name of Irbesartan is 2-Butyl-3-[[2′-(lH-tetrazol-5-yl)[l,l’-biphenyl]-4- yl]methyl]-l,3-diazaspiro[4,4]non-l-en-4-one and formula is C2SH2SN6O and molecular weight is 428.53. The current pharmaceutical product containing this drug is being sold by Sanofi Synthelabo using the tradename AVAPRO, in the form of tablets. Irbesartan is useful in the treatment of diabetic neuropathy, heart failure therapy and hypertension. Irbesartan is angiotension II type I (AΙIi)-receptor antagonist. Angiotension II is the principal pressor agent of the rennin-angiotension system and also stimulates aldosterone synthesis and secretion by adrenal cortex, cardiac contraction, renal resorption of sodium, activity of the sympathetic nervous system and smooth muscle cell growth. Irbesartan blocks the vasoconstrictor and aldosterone- secreting effects of angiotension II by selectively binding to the ATi angiotension II receptor. U.S. Pat. Nos. 5,270,317 and 5,559,233 describes a process for the preparation of N- substituted heterocyclic derivatives which involves reacting a heterocyclic compound of the formula

with a (biphenyl-4-yl)methyl derivative of the formula

wherein R1, R2, R3, R4, R5, and t, z and Hal have the meanings given in said U.S. Pat. No.
5,270,317, in the presence of an inert solvent such as DMF, DMSO or THF, with a basic reagent, for example KOH, a metal alcoholate, a metal hydride, calcium carbonate or triethylamine. The products of the reaction were purified by chromatography.
U.S. Pat. Nos. 5,352,788, and 5,559,233, and WO 91/14679 also describe identical alkylation of the nitrogen atom of the heterocyclic compound with the halo-biphenyl compound using the same inert solvent and the same basic reagents.
-
US5629331 describes a process for the preparation of irbesartan from 1-[(2′-cyanobiphenyl)4-yl)methyl]-2-n-butyl-4-spirocyclopentane-2-imidazolin-5-one using sodium azide, TEA.HCl in N-methylpyrrolidone. The product is isolated from the alkaline reaction mass after acidification to pH 4.7 to 5.8 and the crude product is recrystallised from IPA/water to get Form A and ethanol/water to get Form B.
-
WO 2005/051943 A1 describes a process for the preparing irbesartan wherein 1-[(2′-cyanobiphenyl-4-yl)methyl]-2-n-butyl-4-spirocyclopentane-2-imidazolin-5-one is reacted with tributyltin chloride, sodium azide and TBAB in toluene at reflux temperature for 20 hours. Product is isolated from the reaction mass as trityl irbesartan and then deprotected in methanol and formic acid to get irbesartan.
-
WO 2006/023889 describes a method for preparing irbesartan, wherein 1-(2′-cyanobiphenyl-4-yl)methyl)-2-n-butyl-4-spirocyclopentane-2-imidazolin-5-one is reacted with sodium azide and triethylamine hydrochloride in N-methyl-2-pyrrolidone to give irbesartan.
-
WO 2005/113518 describes a process for preparing irbesartan wherein cyano irbesartan in xylene, is reacted with tributyltin chloride and sodium azide at reflux temperature till reaction is completed followed by aqueous work-up and recrystallization to give irbesartaN
-
The process involving use of zinc salt for the transformation of nitrile to tetrazole is a safe and efficient process as reported in JOC (2001) 66, 7945-50. The use of zinc salt for transforming nitrile to tetrazole has also been published in WO9637481 and US5502191
Also Canadian Patent No. 2050769 describes the alkylation of the nitrogen atom of the heterocycle of the formula
with a compound of the formula

wherein X, R1, Z1 and Z6 have the meanings given therein, in the presence of N,N- dimethylformamide and a basic reagent, such as alkali metal hydrides for example sodium or potassium hydride.
All of the above identified patents describe alkylation in solvents, such as N5N- dimethylformamide or DMSO, etc. in the presence of a basic reagent, for example, a metal hydride or a metal alcoholate etc. The strong bases, such as metal hydride or a metal alcoholate require anhydrous reaction conditions. Since N,N-dimethylformamide is used as a solvent, its removal requires high temperature concentration by distillation, which can result in degradation of the final product. The product intermediate is also purified by chromatography which is commercially not feasible and cumbersome on large scale. Another process given in Canadian Patent No. 2050769 provides synthetic scheme as herein given below.

This process comprises the steps of protecting carboxylic group present on cyclopentane ring which is deprotected in consecutive step by vigourous hydrogenation condition in autoclave which is operationally difficult at a large scale.
US Patent No. 2004242894 also discloses the process of preparation of lrbesartan from 4- bromomethyl biphenyl 2′-(lH-tetrazol (2-triphenylmethyl) 5-yl) and Ethyl ester of 1- Valeramido cyclopentanecarboxylic acid in toluene in presence of base and PTC, and then hydrolyzing the protecting group. However this requires chromatographic purification.
This patent also discloses the process of preparation of tetrazolyl protected lrbesartan using 2,6 lutidine and oxalylchloride in toluene. However in this process the yield is as low as 30%.
US Patent No. 2004192713 discloses the process of preparation of lrbesartan by condensing the two intermediates via Suzuki coupling reaction. The reaction scheme is as given herein below.

However, this process has several disadvantages such as use of the reagents like butyl lithium and triisobutyl borate at low temp such as -20 to -30°C under Argon atmosphere condition which is difficult to maintain at commercial scale.
WO2005113518 discloses the process of preparation of Irbesartan by condensing n- pentanoyl cycloleucine (V) with 2-(4-aminomethyl phenyl) benzonitrile (VI) using dicyclocarbodiimide (DCC) and 1 -hydroxy benzotriazole as catalyst to give an open chain intermediate of formula (VIII) which is then cyclized in the presence of an acid, preferably trifluoro acetic acid to give cyano derivative of formula (VII) and which in turn is converted to Irbesartan by treating it with tributyl tin chloride and sodium azide.

In this application further describes another process comprising the steps of reacting 2- butyl-l,3-diazasρiro[4,4]non-l-en-4-one monohydrochloride (A) with 4-bromobenzyl bromide (B) in presence of base and solvent to give 3-[4-bromobenzyl]-2-butyl-l,3- diazaspiro[4,4]non-l-en-4-one (C) which is condensed with 2-[2′-(triphenylmethyl-2’H- tetrazol-5′-yl)phenyl boronic acid in the presence of tetrakis triphenyl phosphine palladium and base to give lrbesartan (I). However these processes suffer with several disadvantages such as it uses trifluoroacetic acid for the cyclization step which is highly corrosive material. The process requires an additional step of activation by DCC. This step not only increases number of steps but also create problem in handling DCC at an industrial scale as it is highly prone to hazard which makes the process least preferred on a large scale production of lrbesartan. Further it uses phenyl boronic acid derivative and triphenyl phosphine complex which are harmful for the skin and eye tissue and also harmful for respiratory system. Tetrakis triphenyl phosphine palladium is also a costly material which increases overall cost for the production of lrbesartan. Moreover the yield is as low as 22%. All the above patents/applications are incorporated herein as reference. In summary, prior art relating to the process for the preparation of lrbesartan suffers with several drawbacks such as i) It requires chromatographic purification of intermediates at various stages. ii) It requires specific autoclave conditions for a deprotection of protecting group. iii) It requires maintaining low temperature conditions such as -300C and requires special handling care and air and moisture tight condition with the reagents such as butyl lithium and triisobutyl borate. iv) It uses hazardous and highly corrosive reagents, v) It suffers low yield problem. vi) All the process is having more number of reaction steps.
- Irbesartan is described in Bernhart et al., U.S. Patent No. 5,270,317
-
Irbesartan, is a potent, long-acting angiotensin II receptor antagonist which is particularly useful in the treatment of cardiovascular ailments such as hypertension and heart failure. Its chemical name is2-n-butyl-4-spirocyclopentane-1-[(2′-(tetrazol-5-yl)biphenyl-4-yl)methyl]-2-imidazolin-5-one.
Irbesartan is an antihypertensive agent known from EP 454511. From EP 708103, which discloses their X-ray spectra, two polymorphs are known where form A can be produced form a solvent system containing less than 10% of water, while Form B from a system with more than 10% of water. The specific morphological variant of form A can be prepared having properties as disclosed in EP 1089994. Additional form has been disclosed in WO 04089938. Amorphous irbesartan is known from WO 03050110. It is said that Irbesartan produced as taught in EP 454511 is a fluffy material with relatively low bulk and tap densities and undesirable flow characteristics, which consequently has unadvantageous electrostatic properties, among them a high chargeability as measured by tribugeneration between -30 and -40 nanocoulomb/g (10‘9As/g). Alternativelyirbesartan could be prepared by complex process using sonifications and/or temperature oscillations according to EP 1089994 to exhibit a chargeability as measured by tribugeneration between -0 and -10 nanocoulomb/g.
According to EP 454511 a solid composition in form of tablets is prepared by mixing the active ingredient with a vehicle such as gelatine, starch, lactose, magnesium stearate, talc, gum Arabic or the like and can be optionally coated. The compositions containing from 20% to 70% by weight of irbesartan are known from EP 747050.
WO 04/007482 teaches the acidification to pH 2 – 3,5 of trityl irbesartan, which is sufficient to remove the protecting group, but not to convert into an acid addition salt; WO 04/065383 is likewise silent on hydrohalide acid addition salts. WO
06/011859 relates to the preparation of a hydrochloride salt of irbesartan in order to incorporate it into a pharmaceutical formulation. W099/38847 mentions optional conversion of irbesartan into hydrochloride, hydrobromide or hydrogen sulfate salts
……………………………………………
…………………

Example 1Preparation of Compounds of formula IVa and IVb:
-

-
A jacketed 1,000 mL 3-neck flask was charged with 4′-methylbiphenyl-2-carbonitrile (Compound 1, 100.0 g) and CH2CI2 (500 mL) under nitrogen. To a 500 mL Erlenmeyer flask with magnetic stirrer, sodium bromate (NaBrO3; 31.2 g) was dissolved in water (170 mL). The NaBrO3 solution was transferred to the 1,000 mL flask and the reaction mixture was cooled to about 5 °C or less. Aqueous HBr solution (48 %, 105.0 g) was added to the 1,000 mL flask and the resulting reaction mixture was recycled though a UV lamp reactor. The reaction mixture was kept at 0-20 °C and the recycling was continued until the reaction was deemed complete by HPLC. Optionally, additional sodium bromate and hydrogen bromide may be added. The relative amounts of Compound 2 and Compound 3 were about 80-90% and about 10-20% respectively. Aqueous sodium metabisulfite solution (2.0 g of in 10 mL water) was added to the reaction mixture. Allow the phases to settle and the methylene chloride phase was washed with water and used in the next step without further purification.
Example 2Preparation of Compound II:
-

-
A 1L 3-neck flask was charged with Compound V (134.0 g), MTBAC (5.0 g) and CH2Cl2 (170 mL) and cool to -5 to 5 °C. An aqueous solution of KOH (182.6 g in 212 mL water) was added slowly to the 1L flask and the reaction temperature was kept at ≤ 5 °C. The methylene chloride solution of Compound IVa and Compound IVb from Example 1 was added to the reaction mixture slowly, while maintaining the temperature at 0-10 °C. Diethyl phosphite (39.66g) was added drop wise at 0-10 °C. Check the reaction mixture for completion of the reduction reaction, and additional diethyl phosphite may be added.
-
The reaction mixture was allowed to warm to ambient (20-30 °C) and agitated until the reaction was deemed complete by HPLC. Water (150 mL) was added and the phases were separated. The organic layer was extracted with water (230 mL) and polish filtered.
-
The methylene chloride (which contained the crude Compound II) was distilled off and exchanged with about 400 mL of methyl tert-butyl ether (MTBE) (optionally, the MTBE recycled from washing below can be used here). Upon cooling, crystallization occurred (optionally seeds were added) and after further cooling to below 25°C, crystals of Compound II were isolated, washed with MTBE and dried in vacuum at a temperature of less than 60°C. HPLC retention time: 18.126 min. Typically, the yield was about 85 to about 88%. Alternatively, IPA could be used as the crystallization and washing solvent
-
Optionally, the solvent (i.e., MTBE or IPA) used to wash the crystals of Compound II above can be recycled and used to crystallize the crude Compound II in the next batch. Since the washed solvent contains Compound II as well as impurities, it was surprisingly found that the washed solvent can be recovered and used again in crystallizing the crude compound of formula II in the next batch without sacrificing its purity while increasing its yield.
Example 3Preparation of Compound I:
-

-
A reactor was charged with Compound II (1 kg), triethylamine chlorhydrate (0.713 kg), sodium azide (0.337 kg) and N-methyl pyrrolidinone (2.07 kg), and the reaction mixture was heated to about 122°C under stirring. After completion of the reaction as determined by HPLC, the reaction mixture was cooled to about 45°C, and an aqueous solution of sodium hydroxide (35%, 5.99 kg) and water (3.0 kg) were added, the resulting mixture was stirred at a temperature between about 20 and about 40°C for about 0.5 hours. The aqueous phase was discarded and the organic phase was treated with toluene (1.73 kg) and water (5.0 kg), and stirred for about 0.5 hours at about 20 – about 30°C. The toluene phase was discarded and the aqueous phase was washed with ethyl acetate (1.8 kg) and treated with aqueous HCl until pH was adjusted to about 4.8 – about 5.2. Precipitation occurred and the resulting suspension was stirred for about 1 hour at about 20 – about 25°C. The precipitation was collected and washed with water three times (1.0 kg x 3). The crude wet product was recrystallized using a mixture of iso-propanol (0.393 kg) and water (4.5 kg). HPLC retention time: 11.725 min. The yield for Compound I was about 87%.
…………………………………………….
SPECTRAL DATA
The ESI mass spectrum of irbesartan showed a protonated molecular ion peak at m/z 429.3 confirming the molecular weight 428. The fragmentation pattern of parent ion 429.3 showed the fragment ions at m/z 385.9, 235.1, 207, 195.4, 192.1, 180.2 and 84
The FT-IR spectrum exhibited a characteristic stretching absorption band at 1732 cm-1 for the carbonyl group of amide functionality. The presence of this band at higher frequency was due to the ring stretching due to five member ring system. Another band at 1614cm-1 was due to C=N stretching vibrations
1H and 13C- NMR were recorded using DMSO-d6 as a solvent. In 1H-NMR the signal due to tetrazole NH proton was not detected may probably due to the tautomerism.
SEE
http://orgspectroscopyint.blogspot.in/2013/12/irbesartan-spectral-data.html
DP 1 IS IMPURITY
………………………………………….
NMR
1H-NMR (DMSO d6): δppm 0.78 (t, 3H); 1.17-1.30 (sex, 2H); 1.40-1.50 (quent, 2H); 1.64-1.66 (m, 2H); 1.80-1.82 (m, 6H); 2.22-2.29 (t, 2H); 4.67 (s, 2H); 7.07 (s, 4H); 7.50- 7.68 (m, 4H) M+: 429.6
,…………………..
m.p:181-182oC,
IR (KBr, cm-1) 1732 (C=O), 1616 (C=N); 1H NMR (DMSO-d6): δ 7.95–7.32 (m, 8 H), 4.80 –4.60 (s, 2 H), 3.60– 3.00 (br s, 1 H), 2.40– 2.20 (t, 2 H , J = 6.04 Hz), 2.00– 1.60 (m, 8 H),1.60–1.45 (quint, 2 H), 1.40– 1.20 (sext, 2 H), 0.91–0.70 (t, 3H, J = 7.41 Hz);
13C-NMR (DMSOd6): δ 186.5, 162.0,155.9, 141.9, 139.2, 137.2. 131.9, 131.4, 130.1, 128.7, 127.1, 124.3, 76.7, 43.1,
37.7, 28.3, 27.4, 26.3, 22.4, 14.5;
MS: m/z= 429 [M+1];
Anal. Calcd for C25H28N6O : C, 70.07; H,
6.59; N, 19.61. Found: C, 70.04; H, 6.57; N, 19.58.
http://www.acgpubs.org/OC/2011/Volume%204/Issue%201/13-OC-1106-199.pdf
………………………………………………..
1H NMR in DMSO-D6 : 7.68 (d. 2H, Ar-H), 7.52 (d, 2 H, Ar-H), 7.08 (s, 4 H, Ar-H), 4.68(s, 2H, -CH2), 2.69(t,2H,-CH2),2.18(m,2H,-CH2),1.83(m,2H,-CH2),1.81 (t, 2H, -CH2), 1.65 (t, 2H, -CH2), 1.45 (m, 2 H, -CH2), 1.24(m , 2H, -CH2), 0.77 (t, 3H, -CH3),
IR (KBR): 3061 (Aromatic C-H stretching), 2960 (Aliphatic C-H stretching), 3443 (N-H stretching), 1733 (C=0 stretching), 1617(CN stretching), 1337.99(CN stretching), 1407(N=N stretching) cm“1.
……………………….
HPLC condition:
Column: Alltima C18 (Alltech 88050) 15.0cm in length x 4.6mm in internal diameter and 5 micron particle size;
Column temperature: 40 C;
Solvent A: Buffer solution A 1.1 g of heptanesulfonic acid in 1 liter of water and adjust the pH to 2.5;
Solvent B: Methanol Flow rate: 1.2mL/min;
Gradient Elution Condition:
Time% A % %B
0 min 50 50
35 min 15 85
Detector: 240 nm;
Injection volume: 10 uL.
The chromatographic purity of
the compounds was analyzed using Agilent 1200 series HPLC instrument under the following conditions:
Column : Symmetry C18, 4.6 × 75 mm, 3.5 µm
Mobile phase : Eluent A: Deionized water, Eluent B: HPLC grade Methanol
Chromatographic Conditions
a. Column temperature : Ambient
b. Sample compartment : Ambient
c. Detector : 225 nm
d. Injection volume : 10 µL
e. Run time : 45 minutes
f. Flow rate :1.0 mL/min
g. Injector :Auto sampler with variable volume injector
h. Diluent : HPLC grade Acetonitrile
Argatroban

Argatroban
Molecular Formula: C23H36N6O5S
Formula Weight: 508.63
CAS No.: 74863-84-6
(2R,4R)-1-[(2S)-5-(diaminomethylideneamino)-2-
[[(3R)-3-methyl-1,2,3,4-tetrahydroquinolin-8-yl]
sulfonylamino]pentanoyl]-4-methyl-piperidine-2-
carboxylic acid
PATENT
US 7,589,106, 7,687,516, EP 0008746; US 4258192, US 4201863
Argatroban is an anticoagulant that is a small molecule direct thrombin inhibitor.[1] In 2000, argatroban was licensed by the Food and Drug Administration (FDA) for prophylaxis or treatment of thrombosis in patients with heparin-induced thrombocytopenia (HIT). In 2002, it was approved for use during percutaneous coronary interventions in patients who have HIT or are at risk for developing it. In 2012, it was approved by the MHRA in the UK for anticoagulation in patients with Heparin-Induced Thrombocytopenia Type II (HIT) who require parenteral antithrombotic therapy.[2]
Argatroban is given intravenously and drug plasma concentrations reach steady state in 1-3 hours.[3] Argatroban is metabolized in the liver and has a half-life of about 50 minutes. It is monitored by PTT. Because of its hepatic metabolism, it may be used in patients with renal dysfunction. (This is in contrast to lepirudin, a direct thrombin inhibitor that is primarily renally cleared).

Argatroban is used as an anticoagulant in individuals with thrombosis and heparin induced thrombocytopenia. Often these individuals require long term anticoagulation. If warfarin is chosen as the long term anticoagulant, this poses particular challenges due to the falsely elevated prothrombin time and INR caused by argatroban. The combination of argatroban and warfarin may raise the INR to greater than 5.0 without a significant increased risk of bleeding complications.[4] One solution to this problem is to measure the chromogenic factor X level. A level < 40-45% typically indicates that the INR will be therapeutic (2-3) when the argatroban is discontinued.
……………………………………………………….
Argatroban monohydrate

Argatroban is a synthetic direct thrombin inhibitor and the chemical name is 1-[5-[(aminoiminomethyl) amino]-1-oxo2[[(1,2,3,4-tetrahydro-3-methyl-8-quinolinyl)sulfonyl]amino]pentyl]-4-methyl-2- piperidinecarboxylic acid, monohydrate. Argatroban has 4 asymmetric carbons. One of the asymmetric carbons has an R configuration (stereoisomer Type I) and an S configuration (stereoisomer Type II). Argatroban consists of a mixture of R and S stereoisomers at a ratio of approximately 65:35.
The molecular formula of argatroban is C23H36N6O5S•H2O. Its molecular weight is 526.66 g/mol. cas 141396-28-3
Argipidine, Argatroban monohydrate, GN1600, DK-7419, MDI-805 Acova, Slonnon, Novastan
Mitsubishi Chemical (Originator), Encysive Pharmaceuticals (Licensee), Mitsubishi Pharma (Distributor), Daiichi Pharmaceutical (Codevelopment), GlaxoSmithKline (Codevelopment), Mitsubishi Pharma (Codevelopment), Sanofi-SynthLabo (Codevelopment)
Antithrombocytopenic, CARDIOVASCULAR DRUGS, Cerebrovascular Diseases, Treatment of, HEMATOLOGIC DRUGS, Hematopoiesis Disorders Therapy, Ischemic Stroke, Treatment of, NEUROLOGIC DRUGS, Peripheral Vascular Disease, Treatment of, Stroke, Treatment of, Treatment of Peripheral Obstructive Vascular Disease, Thrombin Inhibitors
Synthesis of argatroban on the method reported in the literature there are two synthetic routes, patent EP8746, US4258192, US4201863, JP8115267 relates to a route is: with 4 – methyl-piperidine as a starting material was prepared first intermediate body (2R, 4R) -4 – methyl-2 – ethyl-piperidine, and the first and a t-BOC protected amino nitro-L-arginine condensation, and then the 3 – methyl – 8 – quinoline sulfonyl chloride condensation after hydrolysis, hydrogenation, hydration be argatroban. This entry route synthesis process complicated procedure to be carried out under the protection of nitrogen, the raw material is highly toxic gas phosgene, the operation more difficult.
US4117127, JP02-212473, EP823430, EP8746, JC S Perk Transl 1981 (5), JP02-212473 relates to an alternative route is: nitro L-arginine prior to the 3 – methyl-8 – subsequent condensation quinoline sulfonyl chloride Intermediate (2R, 4R) -4 – methyl-2 – piperidinecarboxylate condensation, and then after hydrolysis, hydrogenation, hydration be argatroban. This synthetic route despite the relatively simple process method, to obtain raw materials, but this method using reagents such as phosphorus oxychloride, phosphorus trichloride has a pungent odor, easy to absorb moisture in humid air, intense smoke, environmental pollution, greater stimulation of the body’s respiratory tract, can cause eye and skin irritation and burning, and the use of this method, complex operation, low yield, high cost.
Patent CN100586946C Argatroban discloses a method for separating optically active isomeric compounds, the feedstock argatroban mixed solvent of alcohol and water was heated to reflux 5-10 hours, cooled and allowed to stand, and filtered to give White crystalline product, dried, repeated 2-6 times. [0008] Patent CN101033223A discloses a Argatroban is the main by-product (2R, 4R)-l_ [N2-(3_ methyl-8 – quinolinesulfonyl)-L-arginyl] -4 – methyl-2 – carboxylic acid, argatroban, and the byproducts are difficult to isolate, argatroban two diastereomeric isomer 21 (S) and 21 (R) separation of work attracted a lot of research persons. Because both physical and chemical properties are very similar, so separation is very difficult. 1993 Rawson, Thomas E.; VanGorp, Kimmie A.; Yang, Janet so first by high pressure liquid chromatography and column chromatography separation to obtain a single 21 (S) and 21 (R) argatroban [ Journal of Pharmaceutical Sciences vol. 82, No. 6,672]; Thibaudeau Karen et al. reported Protein A chromatographic separation [US6440417]. However, due to the separation of these methods a small amount of low efficiency, so there is no practical value industrialization. 2006 China Tianjin Weijie Technology Co., Ltd. Song Honghai et al. Reported using recrystallization Separation 21 (S) and 21 (R) argatroban way [0 With 951,936 it], so that the mass 21 (5) Aga music classes as possible, but the law of low yield, complicated operation, high cost, and a large amount of a small amount of 21 (S) of 21 (R)-product argatroban, from the viewpoint of industrial production, is still a ideal method. [0009] These methods can be effectively prepared argatroban, but the purity of the desired product is not high, poor color, content is low, affecting the quality of the results of its preparation.
U.S. Pat. No. 4,201,863 (6 May 1980) and EP 8746 (filed on 22 Aug. 1979 with priority based on the application for the cited US patent) describe a class of N2-arylsulphonyl-L-argininamide drugs, with anti-thrombotic activity, and the processes for obtaining them. Of these, the compound 4-methyl-1-[N2-(3-methyl-1,2,3,4-tetrahydro-8-quinolinesulphonyl)-L-arginyl]-2-piperidine carboxylic acid (argatroban, isomers mixture) is described. The described process comprises the synthesis of an intermediate NG-substituted-N2-quinolinesulphonyl-L-argininamide from which the desired compound is obtained by catalyzed hydrogenolysis or acidolysis and catalyzed hydrogenation. The general conditions provided for the hydrogenolysis and hydrogenation reaction are: i) inert solvents (methanol, ethanol, tetrahydrofuran or dioxane); ii) presence of a catalyst (Raney nickel, palladium, platinum, ruthenium, rhodium); iii) hydrogen atmosphere at a pressure between 1 and 100 kg/cm2 and preferably between 5 and 50 kg/cm2; iv) temperature between 0° C. and 200° C. and preferably between 50° C. and 150° C.; v) reaction temperature from 2 hours to 120 hours. The crude product obtained is then purified by trituration or by re-crystallization from diethyl ether-tetrahydrofuran, diethyl ether-methanol or from water-methanol or by chromatography. No example is given of this purification step. In particular, both U.S. Pat. No. 4,210,863 and EP 8746 in example 1(E) describe the preparation of argatroban, isomers mixture. This compound is obtained in amorphous form by hydrogenation of [NG-nitro-N2-(3-methyl-8-quinolinesulphonyl)-L-arginyl]-4-methyl-2-piperidine carboxylic acid in ethanol in the presence of Pd/C with hydrogen pressure of 10 kg/cm2 at 100° C. for 8 hours. The catalyst is removed by filtration of the ethanol solution which is then evaporated without further purification and/or re-crystallization steps. In the US patent at issue as indeed in patent application EP 8746, no mention is made of polymorphic forms of the compounds and, for the obtained compound, the following characteristics are reported: Amorphous solid, I.R. (KBr) (cm−1) 3400; 1620; 1460; 1380; Molecular composition (%): theoretical C 54.31; H 7.13; N 16.52; found (%) C 54.01; H 6.98; N 16.61.
U.S. Pat. No. 4,258,192 (24 Mar. 1981) (continuation-in-part of the aforesaid patent application U.S. Pat. No. 4,201,863) and the same patent application EP 8746 describe the stereoisomers and the preparation thereof, including argatroban used as an active principle in medicaments, i.e. the stereoisomer (2R,4R)-4methyl-[4N2-(3-methyl-1,2,3,4-tetrahydro-8-quinolinesulphonyl)-L-arginyl]-2-piperidine carboxylic acid, with the following characteristics: melting point (m.p.). 188-191° C.; I.R. (KBr) (cm−1) 3400, 1620, 1460, 1380; Molecular composition (%): theoretical C 54.31; H 7.13; N 16.52; found (%) C 54.05; H 6.94; N 16.65. The compound is prepared according to the description given in examples 1(E) in U.S. Pat. No. 4,258,192 and 2(E) and 3 in EP 8746 respectively by hydrogenation of (2R,4R) 1-[NG-nitro-N2-(3-methyl-1,2,3,4-tetrahydro-8-quinolinesulphonyl)-L-arginyl]-2-piperidine carboxylic acid in ethanol in presence of acetic acid catalyzed by Pd/C. After filtering the mass to remove the catalyst, the solvent is evaporated and the residue suspended in chloroform, the solution treated with a saturated sodium bicarbonate solution or 1N sodium hydroxide solution and after washing, the solvent is evaporated. The compound is then re-crystallized from ethanol. Again in this case, no reference is made to the obtainment of monohydrate polymorphic forms.
Said polymorphic forms are described instead in the publication Biochem. Biophys. Res. Comm. 1981, 101, 440-446 in the context of stereoisomer preparation. The monohydrate polymorph of the (2R,4R) stereoisomer is prepared by re-crystallization from ethanol/water and the reported characteristics are: m.p. 176-180° C.; [α]D 27 +76.1° (c 1, 0.2N HCl).
U.S. Pat. No. 5,925,760 (20 Jul. 1999) and EP 0823430 (filed 4 Aug. 1997) subsequently describe a new method for preparing argatroban by means of a new intermediate N2-(3-methyl-8-quinolinesulphonyl)-NG-nitro-L-arginine. In particular the patent makes reference to the preparation of a crystalline monohydrate form of argatroban, referring back to examples (D) and (E) of Japanese patent publication No. (Hei)-2-31055/1990 and generically to an I.R. spectrum identical to that of the commercially available argatroban compound. The relevant example in the cited patent publication is example (E), while example (D) concerns the preparation of (2R,4R)-1-[NG-nitro-N2-(3-methyl-8-quinolinesulphonyl)-L-arginyl]-4-methyl-2-piperidine carboxylic acid. This compound represents the starting compound for argatroban preparation by catalytic reduction in the presence of Pd/C. The crude argatroban obtained is then purified by extraction with chloroform, treatment with a saturated sodium bicarbonate solution and, after solvent evaporation, re-crystallization from ethanol or from 15% alcohol in water. It should be noted however that the Japanese patent makes no mention of the monohydrate form of argatroban being obtained and that for the compound the following characteristics are reported: m.p. 188-191° C.; molecular composition (theoretical/found) (%): C 54.31/54.01; H 7.13/6.98; N 16.52/16.61; I.R. (KBr) (cm−1) 3400; 1620; 1460; 1380. These analytical data, with the exception of the unreported melting point, are the same as those indicated in the cited patent documents describing a mixture of (2R,4R)-4methyl-[4N2-(3S-methyl-1,2,3,4-tetrahydro-8-quinolinesulphonyl)-L-arginyl]-2-piperidine carboxylic acid and (2R,4R)-4methyl-1-[N2-(3R-methyl-1,2,3,4-tetrahydro-8-quinolinesulphonyl)-L-arginyl]-2-piperidine carboxylic acid isomers of argatroban, but do not correspond to the melting point given in the publication, being the only document that identifies the monohydrate form of argatroban.
More recently, patent application CN 1,951,937 (filing date 10 Nov. 2006) described a method for preparing hydrated argatroban by treating argatroban with large quantities of water (more than 60 and up to 80 volumes of distilled water per gram of argatroban) at a temperature of 80-100° C. for a time of 0.5-1 hour and crystallization by cooling. The water content reported is comprised between 3.3 and 3.8% and the ratio of dextroisomer R to levoisomer S is R:S=63-67: 37-33.
Argatroban is a compound of wide therapeutic use, for which reason the need still exists to provide a compound of pharmaceutically acceptable quality obtained by easily industrialized and economically convenient methods. With regard to the monohydrate, this form is preferable for the applicative purpose since the anhydrous form is unstable and tends to become hydrated and/or wet. Moreover it crystallizes only with difficulty at the correct ratio between the diastereoisomers.

…..

the protection of 4-methylpiperidine (I) with (Boc)2O gives the carbamate (II), which is condensed with benzyl chloroformate by means of sec-butyl lithium and TMEDA in ethyl ether to yield (?-trans-1-(tert-butoxycarbonyl)-4-methylpiperidine-2-carboxylic acid benzyl ester (III). Deprotection of the NH group of (III) with HCl in ethyl acetate affords (?-trans-4-methylpiperidine-2-carboxylic acid benzyl ester (IV), which is condensed with the protected arginine derivative (V) by means of isobutyl chloroformate and TEA to provide the corresponding amide as a diastereomeric mixture. Resolution of this mixture by flash chromatography furnishes the desired diastereomer (VI), which is treated with HCl in ethyl acetate in order to remove the Boc-protecting group to yield compound (VII). Condensation of compound (VII) with 3-methylquinoline-8-sulfonyl chloride (VIII) by means of TEA in dichloromethane affords the expected sulfonamide (IX). Finally, this compound is submitted to hydrogenation with H2 over Pd/C in AcOH/ethanol in order to produce debenzylation, cleavage of the NO2 group and hydrogenation of the pyridine ring to yield argatroban.
………….
Argatroban, i.e., (2R,4R)-1-((2S)-5-((Aminoiminomethyl)amino)-1-oxo-2-((1,2,3,4-tetrahydro-3-methyl-8-quinolinyl)sulfonyl)amino) pentyl)-4-methyl-2-piperidine carboxylic acid, has two diastereoisomers: 21(R) and 21(S). Usually the ratio of 21(R) to 21(S) is 64-65: 36-35 (U.S. Pat. No. 6,440,417, Cossy. J., et al, Bioorganic & Medicine Chemistry Letters, 11 (2001), 1989-1992, Journal of pharmaceutical Sciences, Vol. 82, No. 6, 672 (1993)).
The structure formula of Argatroban is reported below:

-
- 21(S) Argatroban, X=CH3, Y═H;
- 21(R) Argatroban, X=H, Y=CH3;
- Argatroban, 21(S): 21 (R)=35:65.
The chemical names of the two diastereoisomers mentioned above are:
- 21(S) Argatroban: (2R,4R)-1-((2S)-5-((Aminoiminomethyl)amino)-1-oxo-2-((((3S)-1,2,3,4-tetrahydro-3-methyl-8-quinolinyl)sulfonyl)amino)pentyl)-4-methyl-2-piperidine carboxylic acid (121785-72-6); and
- 21(R) Argatroban: (2R,4R)-1-((2S)-5-((Aminoiminomethyl)amino)-1-oxo-2-((((3R)-1,2,3,4-tetrahydro-3-methyl-8-quinolinyl)sulfonyl)amino)pentyl)-4-methyl-2-piperidine carboxylic acid (121785-71-5).
In 1978, S. Akamoto et al from Japanese Mitsubishi Chemical Corporation first disclosed the anti-thrombin activity of Argatroban monohydrate (U.S. Pat. No. 4,101,653). In the next 20 years, numerous researchers had in-depth studies on Argatroban about its biological activity and medicine values. In 1981, S. Akamoto compared Argatroban with heparin in vivo (Okamoto, S. et al., Biochem. Biophys. Res. Commun. 101, 440 (1981)); T. Kumoto disclosed its three-dimensional selective activity (Kumada, T. et al., Thromb. Res. 24, 285 (1981)). In 1984, R. Kumato made a clinical evaluation of hemodialysis of Argatroban (Kikumoto, R. et al., Biochemistry 23, 85 (1984)), and in 1986, he further disclosed that Argatroban can inhibit the thrombin activity of mammals, and can be used as active ingredient to treat and prevent thrombosis and as an inhibitor of platelet aggregation. Argatroban monohydrate can be used as a selective anti-thrombosis agent for treatment of chronic arterial blockage and cerebral thrombosis, etc (JP 61-48829). In 1992 and 1993, Taparelli and Jakubowski separately disclosed the reversibility of Argatroban in anti-thrombin (Taparelli, C., Trends Pharmacol. Sci., 1993, 14, 366, Jakubowski, J. A. et al, Rep. Med. Chem., 1992, 27, 99). In 1990s, many researchers such as L. R. Buch reported other related research (Buch, L. R., Cadiosvasc. Drug Rev., 1991, 9, 247, Strupcnewski, J. D. et al., Academic: San Diego, 1991; Vol. 26, p 299, Brundish, D. et al., J. Med. Chem. 1999; 42, 4584, Shebuski, R. J., Academic: San Diego, 1999; Vol. 26, p 98). In 1992, Argatroban monohydrate was first approved as an anti-thrombin medicine in Japan (Hijikata-Okunomiya, A., et al, Thromb. Hemostasis, 1992, 18, 135).
Updated in sept 2015, reader there may be duplication
| Argatroban |
| AC1L99H9; AC-15185; TL8005144; C04931; | |
| Molecular Formula: | C23H36N6O5S |
|---|---|
| Molecular Weight: | 508.63414 g/mol |
(2R,4R)-1-[5-(diaminomethylideneamino)-2-[(3-methyl-1,2,3,4-tetrahydroquinolin-8-yl)sulfonylamino]pentanoyl]-4-methylpiperidine-2-carboxylic acid, cas 74863-84-6
| Patent | Submitted | Granted |
|---|---|---|
| Prodrugs of (2R)-2-Propyloctanoic Acid For the Treatment of Stroke [US7495029] | 2008-06-05 | 2009-02-24 |
Tomiya Mano, Jin Shiomura, “Argatroban preparations for ophthalmic use.” U.S. Patent US5506241, issued October, 1986.
Argatroban is an anticoagulant that is a small molecule direct thrombin inhibitor.[1] In 2000, argatroban was licensed by the Food and Drug Administration (FDA) for prophylaxis or treatment of thrombosis in patients with heparin-induced thrombocytopenia (HIT). In 2002, it was approved for use during percutaneous coronary interventions in patients who have HIT or are at risk for developing it. In 2012, it was approved by the MHRA in the UK for anticoagulation in patients with Heparin-Induced Thrombocytopenia Type II (HIT) who require parenteral antithrombotic therapy.[2]
Argatroban is given intravenously and drug plasma concentrations reach steady state in 1-3 hours.[3] Argatroban is metabolized in the liver and has a half-life of about 50 minutes. It is monitored by PTT. Because of its hepatic metabolism, it may be used in patients with renal dysfunction. (This is in contrast to lepirudin, a direct thrombin inhibitor that is primarily renally cleared).
Argatroban is a direct, selective thrombin inhibitor. The American College of Cardiologists (ACC) recommend using bivalirudin or argatroban in patients who have had, or at risk for, heparin induced thrombocytopenia (HIT) and are undergoing percutaneous coronary intervention. Argatroban is a non-heparin anticoagulant shown to both normalize platelet count in patients with HIT and prevent the formation of thrombi. Parental anticoagulants must be stopped and a baseline activated partial thromboplastin time must be obtained prior to administering argatroban.

Transitioning to warfarin in individuals with heparin induced thrombocytopenia
Argatroban is used as an anticoagulant in individuals with thrombosis and heparin induced thrombocytopenia. Often these individuals require long term anticoagulation. If warfarin is chosen as the long term anticoagulant, this poses particular challenges due to the falsely elevated prothrombin time and INR caused by argatroban. The combination of argatroban and warfarin may raise the INR to greater than 5.0 without a significant increased risk of bleeding complications.[4] One solution to this problem is to measure the chromogenic factor X level. A level < 40-45% typically indicates that the INR will be therapeutic (2-3) when the argatroban is discontinued.

http://www.google.com/patents/WO2009124906A2?cl=en
………….
NMR paper
Complete 1H and 13C assignments of (21R) and (21S) diastereomers of argatroban
Article first published online: 20 DEC 2007
DOI: 10.1002/mrc.2122, http://onlinelibrary.wiley.com/doi/10.1002/mrc.2122/abstract



click on image for clear view
1H NMR PREDICT


13C NMR PREDICT
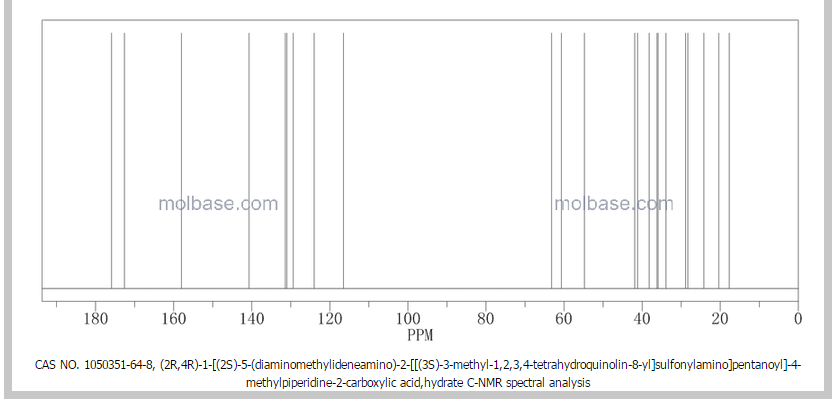
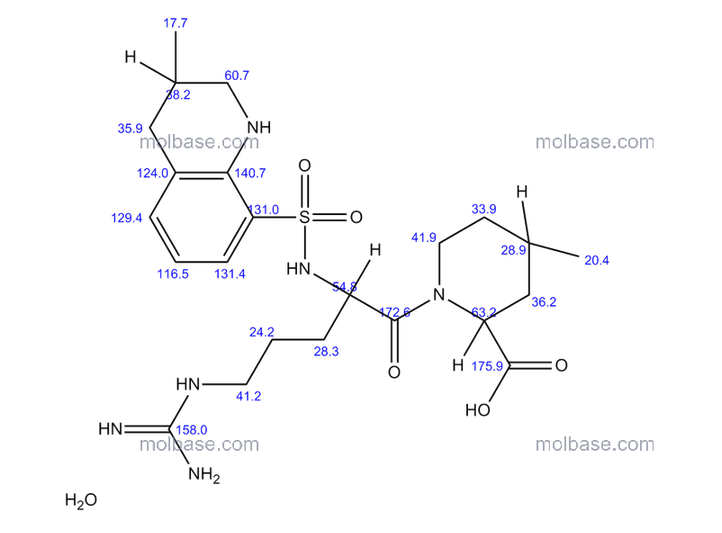
References
1 Di Nisio M, Middeldorp S, Buller HR. Direct thrombin inhibitors. N Engl J Med 2005;353:1028-40. PMID 16148288
2http://www.pharmatimes.com/Article/12-07-03/UK_launch_for_Mitsubishi_s_blood_thinner_Exembol.aspx
3Dhillon S. Argatroban: A Review of its Use in the Management of Heparin-Induced Thrombocytopenia. Am J Cardiovasc Drugs 2009; 9 (4): 261-82. Link text
- Hursting MJ, Lewis BE, Macfarlane DE. (2005). “Transitioning from argatroban to warfarin therapy in patients with heparin-induced thrombocytopenia.”. Clin Appl Thromb Hemost 11 (3): 279–87. doi:10.1177/107602960501100306. PMID 16015413.
External links
| EP0823430A1 * | Aug 4, 1997 | Feb 11, 1998 | Mitsubishi Chemical Corporation | Method for preparing n2-arylsulfonyl-l-argininamides |
| US4201863 * | Aug 31, 1978 | May 6, 1980 | Mitsubishi Chemical Industries, Limited | N2 -Arylsulfonyl-L-argininamides and the pharmaceutically acceptable salts thereof |
| Reference | ||||
|---|---|---|---|---|
| 1 | None | |||
| 2 | * | OKAMOTO S ET AL: “Potent inhibition of thrombin by the newly synthesized arginine derivative No. 805. The importance of stereo-structure of its hydrophobic carboxamide portion” BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS, ACADEMIC PRESS INC. ORLANDO, FL, US, vol. 101, no. 2, 30 July 1981 (1981-07-30), pages 440-446, XP024844713 ISSN: 0006-291X [retrieved on 1981-07-30] cited in the application | ||
| 3 | * | SONG H: “Method for preparing argatroban monohydrate in pure water” CASREACT,, 25 April 2007 (2007-04-25), XP002493272 & CN 1 951 937 A (TIANJIN WEIJIE TECHNOLOGY CO L [CN]) 25 April 2007 (2007-04-25) | ||
| Citing Patent | Filing date | Publication date | Applicant | Title |
| WO2012136504A1 | Mar 26, 2012 | Oct 11, 2012 | Lundbeck Pharmaceuticals Italy S.P.A. | Method for the preparation of process intermediates for the synthesis of argatroban monohydrate |
| CN102408468A * | Sep 20, 2011 | Apr 11, 2012 | 海南灵康制药有限公司 | Argatroban compound and preparation method thereof |
| EP2752412A1 | Mar 26, 2012 | Jul 9, 2014 | Lundbeck Pharmaceuticals Italy S.p.A. | Intermediates for the synthesis of Argatroban monohydrate |
Argatroban is a synthetic direct thrombin inhibitor and the chemical name is 1-[5[(aminoiminomethyl)amino]1-oxo-2-[[(1,2,3,4-tetrahydro-3-methyl-8-quinolinyl)sulfonyl]amino]pentyl]-4methyl-2-piperidinecarboxylic acid, monohydrate. Argatroban has 4 asymmetric carbons. One of the asymmetric carbons has an R configuration (stereoisomer Type I) and an S configuration (stereoisomer Type II). Argatroban consists of a mixture of R and S stereoisomers at a ratio of approximately 65:35.
The molecular formula of argatroban is C23H36N6O5S•H2O. Its molecular weight is 526.66 g/mol. The structural formula is:
|
Argatroban Injection is a sterile, non-pyrogenic, clear, colorless to pale yellow isotonic solution. It is supplied in a single use polyolefin bag containing 250 mg of argatroban in 250 mL sodium chloride solution (1 mg/mL). Each mL contains 1 mg argatroban, 9 mg sodium chloride, USP, and 3 mg sorbitol, NF in water for injection, USP. The pH of the solution is between 3.2 to 7.5.
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(2R,4R)-1-[(2S)-5-(diaminomethylideneamino)-2-
[[(3R)-3-methyl-1,2,3,4-tetrahydroquinolin-8-yl] sulfonylamino]pentanoyl]-4-methyl-piperidine-2- carboxylic acid |
|
| Clinical data | |
| Trade names | Argatroban |
| AHFS/Drugs.com | monograph |
| Routes of administration |
intravenous |
| Pharmacokinetic data | |
| Bioavailability | 100% (intravenous) |
| Protein binding | 54% |
| Metabolism | hepatic |
| Biological half-life | 39 and 51 minutes |
| Identifiers | |
| CAS Registry Number | 74863-84-6 |
| ATC code | B01AE03 |
| PubChem | CID: 440542 |
| DrugBank | DB00278 |
| ChemSpider | 389444 |
| UNII | OCY3U280Y3 |
| KEGG | C04931 |
| ChEMBL | CHEMBL1166 |
| Chemical data | |
| Formula | C23H36N6O5S |
| Molecular mass | 508.635 g/mol |
//////
Drug spotlight- Zafirlukast

ZAFIRLIKAST 
cyclopentyl 3-{2-methoxy-4-[(o-tolylsulfonyl)carbamoyl]benzyl}-1-methyl-1H-indol-5-ylcarbamate 107753-78-6
Matassa, V.G. et al, J. Med. Chem., v. 33, 1781 (1990);
U. S. Patent No. 4,859,692;
U. S. Patent No. 5,993,859;
http://www.accessdata.fda.gov/drugsatfda_docs/label/2011/020547s031lbl.pdf
Zafirlukast is an oral leukotriene receptor antagonist (LTRA) for the maintenance treatment of asthma, often used in conjunction with an inhaled steroid and/or long-acting bronchodilator. It is available as a tablet and is usually dosed twice daily. Another leukotriene receptor antagonist is montelukast (Singulair), taken once daily. Zileuton (Zyflo), also used in the treatment of asthma via its inhibition of 5-lipoxygenase, is taken four times per day.
Zafirlukast blocks the action of the cysteinyl leukotrienes on the CysLT1 receptors, thus reducing constriction of the airways, build-up of mucus in the lungs andinflammation of the breathing passages.
Zafirlukast is marketed by Astra Zeneca with the brand names Accolate, Accoleit, and Vanticon. It was the first LTRA to be marketed in the USA and is now approved in over 60 countries, including the UK, Japan, Taiwan, Italy, Spain, Canada, Brazil, China and Turkey
Healthy young men who received a single oral 40 mg dose attained peak plasma zafirlukast concentrations that averaged 607 μg/L at 3.4 hours. The elimination half-life ranged from 12 to 20 hours. In another study involving a 20 mg single oral dose in healthy men, the elimination half-life averaged 5.6 hours.[1][2]
A letter was submitted to the FDA by Zeneca Pharmaceuticals on July 22, 1997, notifying them of a change in product labeling that includes the following potential reaction in patients undergoing a dosage reduction of oral steroids who are currently taking zafirlukast:
PRECAUTIONS-Eosinophilic Conditions: The reduction of the oral steroid dose, in some patients on ACCOLATE therapy, has been followed in rare cases by the occurrence of eosinophilia, vasculitic rash, worsening pulmonary symptoms, cardiac complications, and/or neuropathy sometimes presenting as Churg–Strauss syndrome, a systemic eosinophilic vasculitis. Although a causal relationship with ACCOLATE has not been established, caution is required when oral steroid reduction is being considered.1
NDA..020547 26/09/1996, ACCOLATE, ASTRAZENECA, 20MG TABLET
| US Patent No | Expirey Date | patent use code |
|---|---|---|
| 5482963 | Jan 9, 2013 | |
| 5612367 | Mar 18, 2014 | U-189 |
Brief background information
| Salt | ATC | Formula | MM | CAS |
|---|---|---|---|---|
| – | R03DC01 | C 31 H 33 N 3 O 6 S | 575.69 g / mol | 107753-78-6 |
| monohydrate | R03DC01 | C 31 H 33 N 3 O 6 S · H 2 O | 593.70 g / mol | 143052-93-1 |
| calcium (2: 1) | R03DC01 | C 62 H 64 CaN 6 O 12 S 2 | 1189.43 g / mol | 107753-86-6 |
Application
-
antihistamine effect
-
LTD4-antagonist
Classes of substances
-
Benzenesulfonamide (s -imidy), as well as their derivatives
-
Esters of carbamic acid
-
Cyclopentanes
-
Hydroxybenzoic acid amides, and hydroxy acids alkoksibenzoynyh
-
Indoles
-
-
-
-
Zafirlukast is a synthetic, selective peptide leukotriene receptor antagonist (LTRA), with the chemical name 4(5-cyclopentyloxy-carbonylamino-1-methyl-indol-3ylmethyl)-3-methoxy-N-o-tolylsulfonylbenzamide. The molecular weight of zafirlukast is 575.7 and the structural formula is:

Zafirlukast, a fine white to pale yellow amorphous powder, is practically insoluble in water. It is slightly soluble in methanol and freely soluble in tetrahydrofuran, dimethylsulfoxide, and acetone.The empirical formula is: C31H33N3O6S
- Fischer JD, Song MH, Suttle AB, Heizer WD, Burns CB, Vargo DL, Brouwer KL. Comparison of zafirlukast (Accolate) absorption after oral and colonic administration in humans. Pharmaceut. Res. 17: 154-159, 2000.
- Bharathi DV, Naidu A, Jagadeesh B, Laxmi KN, Laxmi PR, Reddy PR, Mullangi R. Development and validation of a sensitive LC-MS/MS method with electrospray ionization for quantitation of zafirlukast, a selective leukotriene antagonist in human plasma: application to a clinical pharmacokinetic study. Biomed. Chromatogr. 22: 645-653, 2008.
- Zafirlukast (U.S. National Library of Medicine)
- Zafirlukast (patient information)

 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
| cyclopentyl 3-{2-methoxy-4-[(o-tolylsulfonyl)carbamoyl]benzyl}-1-methyl-1H-indol-5-ylcarbamate | |
| Clinical data | |
| Trade names | Accolate |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a697007 |
| Pregnancy cat. | B1 (Australia), B (United States) |
| Legal status | POM (UK) |
| Routes | Oral |
| Pharmacokinetic data | |
| Bioavailability | Unknown |
| Protein binding | 99% |
| Metabolism | Hepatic (CYP2C9-mediated) |
| Half-life | 10 hours |
| Excretion | Biliary |
| Identifiers | |
| CAS number | 107753-78-6 |
| ATC code | R03DC01 |
| PubChem | CID 5717 |
| IUPHAR ligand | 3322 |
| DrugBank | DB00549 |
| ChemSpider | 5515 |
| UNII | XZ629S5L50 |
| KEGG | D00411 |
| ChEBI | CHEBI:10100 |
| ChEMBL | CHEMBL603 |
| Chemical data | |
| Formula | C31H33N3O6S |
| Mol. mass | 575.676 g/mol |
Trade Names
| Country | Trade name | Manufacturer |
|---|---|---|
| United Kingdom | Akkolat | AstraZeneca |
| Italy | Akkoleit | – “- |
| Zafirst | Chiesi | |
| Japan | Akkolat | AstraZeneca |
| USA | – “- | Zeneca |
| Ukraine | No | No |
Formulations
-
Tablets of 20 mg, 40 mg

is a first anti-asthmatic leukotriene antagonist (Matassa, V.G. et al, J. Med. Chem., v. 33, 1781 ‘(1990); U. S. Patent No. 4,859,692 and The Merck Index, 12th Edition, 10241). Methods for the preparation of Zafirlukast are described in J. Med. Chem., v. 33, 1781 (1990), U. S. Patent 4,859,692 and U.S. Patent 5,993,859 starting from methyl 3-methoxy-4-(l-methyl-5-nitroindol-3-ylmethyl)benzoate [la]



in the presence of an equivalent quantity of silver(I) oxide,

The above process has serious disadvantages in the isolation of the product [4] in step (b) which is due to the fact that alkylation of indole, that is unsubstituted at positions 1-, 2- and 3-, at the 3-position, is accompanied by the undesired process of poly alkylation, to form polysubstituted indoles of formula [7] and/or formula [8] :

while at the same time some quantity of the starting unreacted indole remains in the reaction mixture. Most common methods for the separation of alkyl (indol-3-ylmethyl)benzoate of formula [4] from by-products of polyalkylation and starting unreacted indole, which are all covalent compounds with similar physical properties, include column chromatography that is an unpractical method for industrial scale applications.
Formula (I) compound for the synthesis of an important intermediate of zafirlukast.Reported in the patent EP199543 synthesized compound (I) of the conventional method, the following formula:

(A) (I)
In this method, Intermediate A and 5 – nitro-indole silver oxide in the presence of a catalyst, for docking composite formula (I) compound. Reported only 45% of the reaction yield, the reaction is difficult to complete the reaction and post-treatment using chromatographic methods, resulting in product purification more difficult. And the use of more expensive silver oxide catalysts, high cost.
W00246153 reported a catalyst for the above reaction to zinc bromide, Compound (I), after treatment of the compound (I) with sodium hydroxide hydrolysis of the intermediate (B), separating the product and raw materials purification products.

The method reported in the literature a yield of 60%, but the actual operation is repeated only about 30% yield, and the operation is complicated, cumbersome and costly.
zaafirlukast is a selective and competitive receptor antagonist of leukotriene D4 and E4 (LTD4 and LTE4), components of slow-reacting substance of anaphylaxis (SRSA). Cysteinyl leukotriene production and receptor occupation have been correlated with the pathophysiology of asthma, including airway edema, smooth muscle constriction, and altered cellular activity associated with the inflammatory process, which contribute to the signs and symptoms of asthma.
The cysteinyl leukotrienes (LTC4 LTD4, LTE4) are the products of arachidonic acid metabolism and are various cells, including mast cells and eosinophills, these eicosinoids bind to cysteinyl leukotriene (CysLT) receptors. The CysLT type-1 (CysLT1) receptor is found in human airway and other pro-inflammatory cells. CysLTs have been correlated with the pathophysiology of asthma.
Zafirlukast is a synthetic, selective peptide leukotriene receptor antagonist (LTRA), useful for the treatment of asthma and is commercially available in products sold under the brand name ACCOLATE™ as 10 and 20 mg tablets for oral administration. ACCOLATE™ is indicated for the prophylaxis and treatment of asthma in adults and children 5 years of age and older.
ACCOLATE™ film coated tablets contain amorphous zafirlukast as the active ingredient and the excipients croscarmellose sodium, lactose, magnesium stearate, microcrystalline cellulose, povidone, hypromellose, and titanium dioxide.
The greatest prevalence of asthma is in preschool children; however, the clinical utility of asthma therapy for this age group is limited by a narrow therapeutic index, long-term tolerability, and frequency and/or difficulty of administration. Asthma treatment requires an immediate perceivable effect. Inhalation therapy is a very common therapy prescribed for young children; inhalation therapy has the disadvantage of high dose variability.
 Process for the preparation of zafirlukast
Process for the preparation of zafirlukastUS 20040186300 A1



An Improved and Scalable Process for Zafirlukast: An Asthma Drug
Melting range: 142−145 °C; MS (m/z): 576 (M+ + H); IR (KBr, cm−1): 3326 (NH), 1679 (−C═O), 1H NMR (CDCl3) δ 7.0−8.0 (m, 11H), 3.7 (s, 3H), 4.0 (s, 2H), 3.9 (s, 3H), 2.6 (s, 3H), 1.45−1.8 (s, 9H). ……………………………………………………………….. US 20040186300 A1 http://www.google.com/patents/US20040186300 zafirlukast ethanolate as white powder with mp 132-133° C. (dec.) and 99.8% purity by HPLC. 1H NMR (CDCl3, δ, ppm): 1.22 (t, J 7.05 Hz, 3H), 1.45-1.87 (m, 8H), 2.66 (s, 3H), 3.67 (s, 3H), 3.73 (q, J 7.05 Hz, 4H), 3.79 (s, 3H), 3.98 (s, 2H), 5.08-5.23 (m, 1H), 6.58 (s, 1H), 6.73 (s, 1H), 7.01-7.51 (m, 9H), 8.23 (d, J 7.52 Hz, 1H), 9.67 (s, 1H).
Synthesis pathway
| Synthesis a) |
|---|
      |
| Synthesis of b) |
   |
-
Synthesis a)
-
US 4,859,692 (ICI; 08/22/1989; GB -prior. 4/17/1985; 17.10.1985).
-
EP 199 543 (ICI, Zeneca; appl. 16.4.1986; GB -prior. 4/17/1985).
-
-
Synthesis of b)
-
EP 490 649 (ICI, Zeneca; 11.12.1991; GB -prior. 12.12.1990).
-
Matassa, G. et al .: J. Med. Chem. (JMCMAR) 33, 1781 (1990).
-
Srinivas, K. et al .: Org. Process Res. Dev. (OPRDFK) 8 (6), 952 (2004).
-
added info Asthma is a disease that causes swelling and narrowing the airways of the lungs. Airways are air carriers to and from lungs. Swollen and narrower airways affect the air flow to and from the lungs and this lead to tightness of chest, wheezing, shortness of breath and cough. These symptoms are often occurs in early morning and in night. Asthma is caused by genetic and environmental factors, it was not curable completely but this can be controlled with good medical care. Leukotriene antagonists also known as leukast are the medicaments that are used to reduce leukotrienes, which are produced by several types of cells and causes inflammation in asthma and bronchitis. Leukotriene antagonists that are available in market are Montelukast, Zafirlukast and Pranlukast. Zafirlukast is the first leukast compound approved for management of Asthma. US FDA approved zafirlukast in the form of 10 mg and 20 mg tablet with the brand name of Accolate®.1 Subsequently this was approved and launched by innovator in few other countries. There are many synthetic routes for the preparation of Zafirlukast 4 is well documented in literature. Some of the key approaches are discussed here under. Scientists from ICI Americas Inc2 have reported process for the synthesis of 4, which starts with esterification of 3-methoxy-4-methyl benzoic acid 53 using methanol in presence of acetyl chloride PRODUCT PATENT ROUTE Allylic bromination of methyl ester 54 using bromine in presence of CCl4 resulted bromo compound 55, which was reacted with 5-nitro indole 124 using silver oxide as catalyst to obtain condensed compound 125. N-methylation of 125 utilizing methyl iodide in presence of NaH afforded N-methyl indole derivative 57. Thus obtained 57 was subjected to reduction using palladium carbon (Pd/C) in methanol followed by reacted with cyclopentyl chloroformate to obtain compound 59. Hydrolysis of 59 using LiOH.H2O subsequently reaction with o-toluene sulfonamide (OTSA) in presence of 1-[3-(dimethylamino)propyl]-3-ethyl carbodiimide hydrochloride (DMAPEC) and DMAP furnished zafirlukast 4. Matassa et al3 also reported similar procedure for the synthesis of Zafirlukast 4. 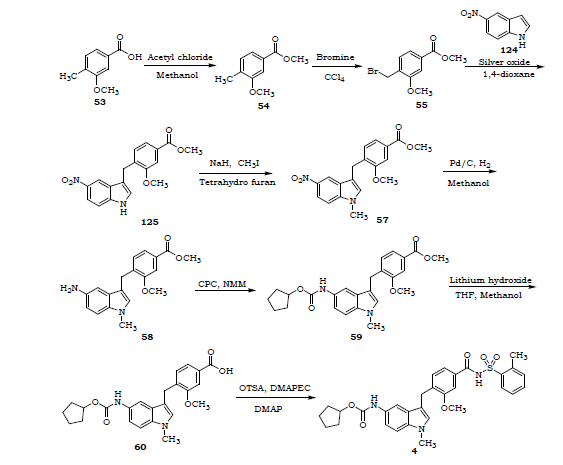

Teva Gets Orphan Drug Designation for Treanda
Teva Announces Additional Regulatory Exclusivity for TREANDA® (Bendamustine HCI) for Injection
Orphan Designation combined with pediatric extension provides regulatory exclusivity through April 2016 for indolent B-cell non-Hodgkin lymphoma indication
JERUSALEM, November 27, 2013 –(BUSINESS WIRE)–Teva Pharmaceutical Industries Ltd. (NYSE: TEVA) today announced that the U.S. Food and Drug Administration (FDA) has granted orphan drug exclusivity for TREANDA through October 2015 for indolent B-cell non-Hodgkin lymphoma (iNHL) that has progressed during or within six months of treatment with rituximab or a rituximab-containing regimen.http://www.pharmalive.com/teva-announces-additional-regulatory-exclusivity-for-treanda
read my old post, contains synthesis
https://newdrugapprovals.wordpress.com/2013/09/19/fda-oks-tevas-injectable-treanda/
SCRIP Awards 2013 -Best Company in an Emerging Market – Dr Reddy’s Laboratories – India, Novartis’s Bexsero, Best New Drug

The SCRIP Awards 2013 celebrated achievements in the global biopharma industry last night at the Lancaster, London.
Hosted by Justin Webb, the evening was a fantastic mix of dining, entertainment and awards.
Among the winners were:
- Novartis’s Bexsero, Best New Drug
- Genmab, Biotech Company of the Year
- Regeneron Pharmaceuticals and Sanofi’s Phase IIa study dupilumab in asthma, Clinical Advance of the Year
You can view the full roll of honour by clicking on the button below.
It was a great night and we would like to thank all those who entered and attended this year’s awards.
Finally congratulations to our winners and a huge thanks to our sponsors for helping us make it such a fantastic success.
Don’t forget to check our website in the next couple of days for all the pictures from the night.
2013 Winners
Best Company in an Emerging Market – Sponsored by Clinigen Group
- Dr Reddy’s Laboratories – India
Best Technological Development in Clinical Trials
- Quintiles’s Infosario Safety
Best Partnership Alliance
- AstraZeneca with Bristol-Myers Squibb and Amylin in diabetes
Financing Deal of the Year
- Mesoblast’s equity financing of Aus$170m
Best Advance in an Emerging Market
- Novartis’s Jian Kang Kuai Che Healthcare Project in China
Clinical Advance of the Year – Sponsored by Quintiles
- Regeneron Pharmaceuticals and Sanofi’s Phase IIa study dupilumab in in asthma
Licensing Deal of the Year – Sponsored by Hume Brophy
- AstraZeneca and Horizon Discovery for the development and commercialization of the HD-001 kinase target program for multiple cancer types
Executive of the Year
- Roch Doliveux, chairman and chief executive officer of UCB
Biotech Company of the Year
- Genmab
Best Contract Research Organization
- Quintiles
Management Team of the Year
- Regeneron Pharmaceuticals’ CEO Leonard S Schleifer and CSO George D Yancopoulos
Best New Drug – Sponsored by INC Research
- Novartis’ Bexsero (meningococcal group B vaccine)
Pharma Company of the Year – Sponsored by ICON
- Astellas
Lifetime Achievement Award
- Prof Dr Désiré Collen
…….read about bexero at

DR ANTHONY MELVIN CRASTO Ph.D

Ziprasidone


Ziprasidone
Ziprasidone (marketed as Geodon, Zeldox by Pfizer) was the fifth atypical antipsychotic to gain approval (February 2001) in the United States. It is approved by the U.S. Food and Drug Administration (FDA) for the treatment of schizophrenia, and acute mania and mixed states associated withbipolar disorder. Its intramuscular injection form is approved for acute agitation in schizophrenic patients for whom treatment with just ziprasidone is appropriate.
Ziprasidone is also used off-label for depression, bipolar maintenance, mood disorders, anxiety, aggression, dementia, attention deficit hyperactivity disorder, obsessive compulsive disorder, autism, and post-traumatic stress disorder.

Ziprasidone synthesis: John A. Lowe, Arthur A. Nagel. Pfizer Inc. U.S. Patent 4,831,031 (1989).
The oral form of ziprasidone is the hydrochloride salt, ziprasidone hydrochloride. The intramuscular form, on the other hand, is the mesylate salt, ziprasidone mesylate trihydrate, and is provided as a lyophilized powder.
Ziprasidone, chemically named (5-[2-{4-(l,2-benzisothiazol-3-yl)piperizin- 0 1 -yl } ethyl] -6-chlorooxindole)hydrochloride hydrate, is a substituted benzisothiazolylpiperazine. The free base of ziprasidone has the following structure:

Ziprasidone and some of its uses are described by U.S. Patent Nos. 4,831,031 and 0 5,312,925.
Like clozapine and risperidone, ziprasidone is a highly potent and selective 5-HT2receptor and dopamine D2 receptor antagonist. Seeger, T.F. et al, J. Pharmacol. Exp. Ther.. 275(1): 101-1 13 (1995). Ziprasidone is characterized as an antipsychotic, but may also have anxiolytic and antidepressant effects due to its ability to inhibit serotonin and 5 noradrenaline reuptake. Davis, R. and Markham, A., CNS Drugs, 8(2):154-159 (1997). The therapeutic potential of ziprasidone may also be enhanced by its high affinity for the 5-HT1A, 5-HT1D, 5-HT2Creceptor subtypes. Seeger, T.F. et al, J. Pharmacol. Exp. Ther.. 275(1):101-113 (1995).
The metabolism of ziprasidone is complex. When administered orally to
30 healthy humans, the drug is extensively metabolized by at least four major pathways: 1) N- dealkylation of the ethyl side chain attached to the piperazinyl nitrogen; 2) oxidation at sulfur resulting in the formation of sulfoxide or sulfone; 3) reductive cleavage of the bensisothiazole moiety; and 4) hydration of the C=N bond and subsequent sulfur oxidation or N-dearylation of the benzisothiazole moiety. Prakash, C. et al, Drug Metab. Dispos.,
35 25(7):863-872 (1997). At least 12 human metabolites have been identified: ziprasidone sulfoxide (ZIP-SO); ziprasidone sulfone (ZIP-SO2); 3-(piperazine-l -yl)-l,2-benzisothiazole (BITP); BITP sulfoxide; BITP sulfone; 6-chloro-5-(2-piperazinJ-yl-ethyl)JJ-dihydro- indol-2-one; 6-chloro-5-(2- {4-[imino-(2-mercapto-phenyl)methyl]-piperazin- 1 -yl} ethyl)- 1 ,3-dihydro-indol-2-one; 6-chloro-5-(2- {4-[imino-(2-methylsulfanyl-phenyl)methyI]- piperazin-1-yl} ethyl)- l,3-dihydro-indol-2-one; S-methyl-dihydro-ziprasidone; S-methyl- dihydro-ziprasidone sulfoxide; dihydro-ziprasidone sulfoxide; and (6-chloro-2-oxo-2,3- dihydro-lH-indol-5-yl)acetic acid. Two metabolites, ZIP-SO and ZIP-SO2, both of which are formed by oxidation of the ziprasidone sulfur atom are discussed herein. These metabolites have the following structures:

Ziprasidone Sulfoxide (ZIP-SO)

Ziprasidone Sulfone (ZIP-SO2)
Both ZIP-SO and ZIP-SO2 are minor metabolites, and account for less than about 10% and less than about 3% of ziprasidone metabolites found in human urine, respectively. Prakash, C. et al, Drug Metab. Dispos.. 25(7):863-872 (1997). It has been reported that neither metabolite likely contributes to the antipsychotic activity of ziprasidone. Prakash, C. et al, Drug Metab. Dispos., 25(7):863-872 (1997). Indeed, it has been reported that ziprasidone metabolites in general are not active at the D2 and 5-HT2A receptor sites. Ereshefsky, L., JL Clin. Psvch.. 57(suppl. l l):12-25 (1996).
Ziprasidone offers a number of benefits, but unfortunately many adverse effects are associated with its administration. Examples of adverse affects of ziprasidone include, but are not limited to, nausea, somnolence, asthenia, dizziness, extra-pyramidal symptoms, akathisia, cardiovascular disturbances, male sexual dysfunction, and elevated serum liver enzyme levels. Davis, R. and Markham, A., CNS Drugs, 8(2): 154-159 (1997). These adverse effects can significantly limit the dose level, frequency, and duration of drug therapy. It is thus desirable to find a compound which possesses advantages of ziprasidone but fewer of its disadvantages.
3. SUMMARY OF THE INVENTION
This invention relates to novel methods using, and compositions comprising, ziprasidone metabolites, preferably, ziprasidone sulfoxide and ziprasidone sulfone. These metabolites, prior to the present invention, have been reported to have little or no in vivo activity. The present invention encompasses the in vivo use of these metabolites, and their incorporation into pharmaceutical compositions and single unit dosage forms useful in the treatment and prevention of disorders that are ameliorated by the inhibition of serotonin reuptake at 5-HT2receptors and/or the inhibition of dopamine reuptake at dopamine D2 receptors. Such disorders include psychotic and neuroleptic disorders. In a preferred embodiment, ziprasidone metabolites are used in the treatment or prevention of neuroleptic and related disorders in mammals, including humans.

-
Ziprasidone (5-(2-(4-(1,2-benzisothiazol-3-yl-1-piperazinyl)-ethyl)-6-chloro-1,3-dihydro-2-(1H)-indol-2-one) is a potent antipsychotic agent and is useful for treating various disorders including schizophrenia, anxiety and migraine pain. Ziprasidone has been approved by the FDA for treatment of schizophrenia and goes by the brand name Geodon™ in the United States. Ziprasidone has also been indicated as useful for treating Tourette’s Syndrome (United States Patent 6,127,373), glaucoma and ischemic retinopathy (EP 985414 A2), and psychiatric conditions including dementia of the Alzheimer’s type, bipolar disorders, mood disorders, panic disorders, agoraphobia, social phobia, panic disorder, post-traumatic stress disorder, acute stress disorder, substance-induced anxiety disorder, anxiety disorders not otherwise specified, dyskinesias and behavioral manifestations of mental retardation, conduct disorder, and autistic disorder (United States Patent 6,245,766).
-
United States Patent 4,831,031 describes a genus of compounds encompassing ziprasidone and the synthesis of such compounds. Another method for synthesizing ziprasidone is described in United States Patent 5,206,366. A method for specifically synthesizingziprasidone hydrochloride monohydrate is described in United States Patent 5,312,925. A method for synthesizing ziprasidone mesylate dihydrate is described in United States Patent 6,245,765; and a method for synthesizing ziprasidone mesylate trihydrate is described in United States Patent 6,110,918. United States Patents 5,338,846; 5,359,068; and 6,111,105 also describe methods for synthesizing ziprasidoneand/or intermediates therefore.
-
The structure of ziprasidone can be depicted as:

(H. Howard, et al., “Ziprasidone Hydrochloride”, Drugs of the Future1994, 19(6): 560-563. As can be seen from the structure above, the compound ziprasidone comprises a chlorine atom.
-
Methods of introducing halogens into organic compounds are summarized in many organic text books. For example, J. March,Advanced Organic Chemistry, 4th Edition, pp. 587-591, and references cited therein, has a discussion of halogenation chemistry. More specifically, formation of chloro-aromatic compounds are frequently formed by a variety of methods also well known to those skilled in the art, and again summarized in J. March, Advanced Organic Chemistry, 4th Edition, Chapter 11, “Aromatic Electrophilic Substitution”. The chemistry to add a halogen, or more specifically a chlorine, to an aromatic group is thus well known to those skilled in the art. It is also known that such chemistry usually results in some mixtures of molecules, one of which is commonly the unreacted starting material not containing the chlorine atom. Further, over-chlorination is a problem well known to those skilled in the art; it is common to form some dichloro-compound impurities when the mono-chloro is desired and some trichloro-compound impurities when the dichloro- is desired. Over-chlorination is typically controlled by limiting the amount of the chlorinating reagent used. Unfortunately, control of over-chlorinated analogs in the drug substance by limiting the amount of chlorinating reagent utilized in the introduction of the aromatic chlorine substituent would be expected to result in more of a des-chloro impurity (unreacted starting material not containing the chlorine atom).
-
-
6-chlorooxindole (6-chloro-1,3-dihydro-2H-indol-2-one).
-
Although there are many known routes to 6-chlorooxindole, starting materials therefore are typically a substituted 4-chlorotoluene or 1,4-dichloro-nitrobenzene (see, G. J. Quallich and P. M. Morrissey,Synthesis, 1993, 51-53; and references cited therein; and F. R. Busch and R. J. Shine, “Development of an Efficient Process to 6-Chlorooxindole”, presented at the 208th ACS National Meeting in Washington D.C. in the Symposium on Technical Achievements in Organic Chemistry, 1994, (talk #126).). However, the concept of controlling chlorinated isomers, over-chlorination, or des-chloro impurities for the synthesis of 6-chlorooxindole is not described in the prior art. Other methods of synthesizing 6-chlorooxindole can be determined by a person of ordinary skill in the art, and such methods are included in the step of obtaining a batch of 6-chlorooxindole for the above-described method of this invention. Furthermore, a batch of 6-chlorooxindole can be obtained by purchase from manufacturers of organic chemicals, for example Plaistow, Ltd., Little Island, County Cork, Ireland or Finorga, Route de Givors, 38670 Chasse-Sur-Rhone, France.
-
Ziprasidone has two major fragments, benzisothiazol and substituted oxindole. In from 2 – mercapto acid methyl ester ( 1 ), the alkaline conditions with hydroxylamine-O-sulfonic acid reaction ring closure under alkaline conditions to obtain 5 . 5 3 can also be prepared from the disulfide, disulfides 3 by three methods (anthranilic acid by diazotization pass sulfur dioxide gas, o-fluorinated thiol acid and two xenon reaction, or dibromoethoxychlorophosphonazo acid and sulfur in copper iodide reaction), 3 and chlorinated sulfoxide and sulfone chlorination reaction of 4 , 4and ammonia reaction again 5 . 5 by chlorination with phosphorus oxychloride, the reaction of piperazine 7 . 7 may be made of the compound 8 ( 8 can be from 2 – cyano bromobenzene After the i-PrMgCl, ZnBr 2 , S 2 Cl 2 prepared in one-pot reaction) was prepared in DMSO and directly in the hot reaction piperazine.
Oxindole fragment from 6 – chloro-indol-2 – one ( 10 ) starts, the FC acylation later reduction with triethylsilane 12 , 12 and 7 occurs in alkaline aqueous solution S N 2 reaction with hydrochloric acid salt to obtain ziprasidone hydrochloride.
United States Patent 5,206,366,



MORE INFO UPDATED
Ziprasidone is an antipsychotic agent with the following chemical name: 5-[2-[4-(1,2-benzisothiazol-3-yl)-1-piperazinyl]ethyl]-6-chloro-1,3-dihydro-2H-indol-2-one of formula (I)
Ziprasidone is disclosed in U.S. Pat. Nos. 4,831,031 and 5,312,925 (assigned to Pfizer). Ziprasidone inhibits synaptic reuptake of serotonin and norepinephrine. No appreciable affinity was exhibited for other receptor/binding sites tested, including the cholinergic muscarinic receptor. The mechanism of action of ziprasidone, as with other drugs having efficacy in schizophrenia, is unknown. However, it has been proposed that this drug’s efficacy in schizophrenia is mediated through a combination of dopamine type 2 (D 2) and serotonin type 2 (5HT 2) antagonism.Ziprasidone’s antagonism of histamine H receptors may explain the somnolence observed with this drug.
U.S. Pat. No. 5,312,925 (Pfizer Inc.) describes a process for the synthesis of monohydrate of 5-(2-(4-(1,2-benzisothiazol-3-yl)piperazinyl)ethyl)-6-chloro-1,3-dihydro-2H-indol-2-one hydrochloride and its characterization based on IR, XRD and moisture content. The ‘925 patent also discloses that the hemihydrate may be obtained by the process described in Example 16 of U.S. Pat. No. 4,831,031 and its characterization by IR, XRD and moisture content. It also discloses the IR, XRD and moisture content of anhydrous Ziprasidone hydrochloride. According to the invention in the ‘925 patent, Ziprasidone of water content of 3.97, 2.55 and 0.37% were used for the IR and XRD study of Ziprasidone hydrochloride monohydrate, hemihydrate and anhydrous. In this invention, the monohydrate ofZiprasidone hydrochloride was prepared by reacting anhydrous 5-(2-(4-(1,2-benzisothiazol-3-yl)piperazinyl)ethyl)-6-chloro-1,3-dihydro-2H-indol-2-one with aqueous hydrochloric acid. The temperature range of the reaction was maintained between 60 to 65° C. and aqueous hydrochloride used for salt formation was around 0.7 M. Depending on the reaction temperature and other conditions, the reaction times were set around 3 to 24 hours. The final product thus obtained was dried carefully in monitored conditions to make certain that water content was from about 3.8% to about 4.5% to obtain the stable monohydrate.
U.S. Pat. No. 6,150,366, discloses a manufacturing process of ziprasidonehydrochloride monohydrate, comprises: 1) dissolving, ziprasidone free base in a solvent comprising THF and water, in a volume ratio of about 22-35 unit volumes of THF to about 1.5-8 volumes of water; 2) heating the solution resulting from step (1); 3) adding HCl to the solution resulting from step (2); and 4) cooling the solution resulting from step (3) and crystals collected by filtration and drying.
U.S. Pat. No. 5,206,366 and U.S. Pat. No. 5,338,846 describe a process for preparing ziprasidone by reacting 1-(1,2-benzisothiazol-3-yl) piperazine with 5-(2-chloroethyl)-6-chloro-oxindole in water with a neutralizing agent such as sodium carbonate under reflux.
J. Med. Chem. 1996, 39, 143-148 discloses preparation of ziprasidone by reacting 1-(1,2-benzisothiazol-3-yl)piperazine with 5-(2-bromoethyl)-6-chloro-oxindole in isoamyl alcohol solvent in the presence of sodium carbonate.
Some salts of ziprasidone, and in particular, its hydrochloride salt is a potent commercial antipsychotic agent useful in the treatment of various disorders, including schizophrenia and anxiety diseases. Ziprasidone hydrochloride is currently marketed under the proprietary name of Geodon. Other salts ofziprasidone are also reported to be effective for the treatment of the same type of diseases.
Some of the processes described in the aforementioned patents necessitate the use of ion-exchange catalyst (i.e. sodium iodide) and/or phase transfer catalysts (for example tetra butyl ammonium bromide or tetra butyl phosphoriium bromide) in order for the coupling reaction producing ziprasidone to take place. For example, U.S. Pat. No. 4,831,031 indicates that arylpiperazinyl-ethyl (or butyl)-heterocydic compounds may be prepared by reacting piperazines of the formula II with compounds of the formula III as follows in [Scheme 1]:

Wherein Hal is fluoro, chloro, bromo or iodo; and Ar, n, X and Y are as defined therein with reference to formula I. According to the ‘031 patent the coupling reaction is generally conducted in a polar solvent, such as a lower alcohol, dimethylformamide or methylisobutylketone, and in the presence of a weak base and that, preferably, the reaction is carried out in the presence of a catalytic amount of sodium iodide, hydrogen chloride and neutralizing agent such as sodium carbonate.
In some instances, the ziprasidone obtained was purified by column chromatography, thus making the process impractical for large-scale preparations. Another process uses potentially explosive gases such as hydrogen in the presence of catalysts, for example zinc, palladium on carbon, followed by acid treatment to carry out a reduction and cyclization of an intermediate, in order to obtain ziprasidone.
Despite various processes disclosed in the prior art for the preparation of ziprasidone and salts thereof, still there is a need for a good process for producing ziprasidone and pharmaceutically acceptable acid addition salts of ziprasidone thereof, in high purity. One of the major problems faced in the prior art is formation of sticky material and difficult stirrability of the reaction mass. This problem is especially acute in large scale manufacturing.
picked up from polish site…translation is machine, please bear for errors
The first stages are two simple reactions: reduction of Wolf-Kiżnera and Friedel-Crafts

The next step is to reduce the use of triethylsilane and trifluoroacetic acid [2], and then the coupling with a compound 5 to obtain the final product.

a) method 1

b) method 2

In the second method, we have marked with an interesting transition.As you zoom scale but found that method 2 is the only possible one. Just destroy the product hydrochloride and get a clean API ready for tableting. [1] Bhugra D. The global prevalence of schizophrenia .. “PLoS Medicine”. 5 (2), pp. E151, 175 [2] Tetrahedron Letters “Selectivities in Ionic Reductions of Alcohols and Ketones with Triethyisilane / Trifluoroacetic Acid” 38, (6), 1997, pp. 1013-1016
1H NMR PREDICT

13C NMR PREDICT

http://www.google.com/patents/EP1476162A1?cl=en
Scheme 1
Stepl

MW = 167.59 MW = 244.08 Step 2

MW = 244.08 MW = 230.09 Step 3

MW = 230.09 MW = 255.76

MW = 412.94 Step 4

ziprasidone hydrochloride
MW = 412.94 monohydrate MW = 467.42
Scheme 2

Example 1 : Synthesis of Ziprasidone
Step 1 : Friedel-Crafts Acylation of 6-chloro-1 ,3-dihydro-2H-indol-2-one
Methylene chloride (310 L) and aluminum chloride (172.3 kg) were combined. Chloroacetyl chloride (66.7 kg) was added, and the resulting mixture was stirred for
45 minutes. 6-Chloro-1 ,3-dihydro-2/-/-indol-2-one (61.8 kg) was added. The reaction mixture was stirred at 28 to 32° C for 19.5 hours and then cooled to 15 to 20 C. Water (805 L) was cooled to 5 to 10 C. The reaction was quenched by the slow addition of the reaction mixture to the cold water. After the quench was complete, the mixture was heated to reflux, and the methylene chloride was removed by atmospheric distillation at 43 to 57° C. The resulting mixture was cooled to 15 to 20° C and stirred for 1 hour. The solids were isolated by filtration and washed with water (114 L) followed by methanol (114 L).- The solids were dried in a suitable dryer.
6-Chloro-5-(chloroacetyl)-1,3-dihydro-2/-/-indol-2-one, yield: 91.3 kg (101.4%). Note: A weight yield in excess of 100% resulted due to small amounts of residual salts which were removed in the following step.
The resulting 6-chloro-5-(chloroacetyl)-1,3-dihydro-2H-indol-2-one was carried through the following step in portions, one of which is detailed below.
Step 2: Trifluoroacetic Acid/Silane Reduction of 6-Chloro-5-(chloroacetyl)-1 ,3- dihydro-2H-indol-2-one
Trifluoroacetic acid (278 kg) and (74.2 kg) were combined and stirred slowly at 24 to 28° C. Triethylsilane (77.9 kg) was charged to the stirring mixture. The reaction temperature was allowed to exotherm slightly during this addition and was maintained between 50 to 62° C during the reaction period. , The reaction mixture was stirred for 8 hours, cooled to 38 C, and sampled for reaction completion. The reaction mixture was stirred at 50 to 54° C for an additional 3 hours. After the reaction was determined to be complete, the reaction mixture was cooled to 18° C, and quenched with water (594 L). The resulting slurry was stirred for 30 minutes at 10 to 15° C, and the solids were isolated by filtration. The product was rinsed from the tank and the product cake was washed with water (83 L) followed by methanol (76 L).
In each of two batches of equal size, tetrahydrofuran (742 L), Darco KB-B (1.9 kg), and the wet product cake were combined and heated to reflux. The resulting mixture was stirred at reflux for 30 minutes and filtered through a sparkler filter (pre- coated with filteraid) at 50 to 60° C to remove the carbon. The tank and sparkler were rinsed with hot tetrahydrofuran (38 L). Following the filtration the two batches were combined. The solution was concentrated in vacuo and stirred at 4 to 5° C for 1 hour. The solids were isolated by filtration and washed with cold tetrahydrofuran (38
L). The solids were dried in vacuo at 45 to 73° C until a loss on drying of 0.45% was achieved, giving 6-Chloro-5-(2-chloroethyl)-1 ,3-dihydro-2H-indol-2-one, yield: 60.1 kg (85.9%).
The resulting 6-chloro-5-(2-chloroethyl)-1 ,3-dihydro-2H-indol-2-one was combined with material of comparable quality and carried through the following step.
Step 3: Coupling of 6-Chloro-5-(2-chloroethyl)-1 ,3-dihydro-2H-indol-2-one and 3-(1-Piperazinyl)-1 ,2-benzisothiazole Monohydrochloride
Water (780 L) and sodium carbonate (126.0 kg) were combined and the mixture was stirred to dissolve. 3-(1-Piperazinyl)-1 ,2-benzisothiazole monohydrochloride (155.0 kg) and 6-chloro-5-(2-chloroethyl)-1 ,3-dihydro-2W-indol-2- one (150.4 kg) were added, and the reaction mixture was heated to reflux (-100° C). After 24 and 28 hours, the reaction slurry was sampled for reaction completion assay. The reaction was determined to be complete after the assay of the second sample. Water (1251 L) was added and the slurry was cooled to temperatures between 18 to
22° C. The solids were isolated by filtration and washed with water (302 L). The water wet solids were combined with isopropanol (940 L) and the resulting mixture was stirred for approximately 2 hours at ambient temperature. The solids were isolated by filtration, washed with isopropanol (89 L), and dried in vacuo at less than 43° C, giving 5-[2-[4-(2,3-Benzisothiazol-3-yl)-1-piperazinyl]ethyl]-6-chloro-1 ,3- dihydro-2H-indol-2-one, yield: 202.8 kg (80.8%).
The resulting 5-[2-[4-(2,3-benzisothiazol-3-yl)-1 -piperazinyl]ethyl]-6-chloro- 1 ,3-dihydro-2/- -indol-2-one was divided into two portions. These batches were carried separately through the following additional purification and resulted in material of comparable quality. The processing of one of these batches is detailed below.
Step 3R: Purification of 5-[2-[4-(2,3-Benzisothiazol-3-yl)-1-piperazinyl]ethyl]- 6-chloro-1 ,3-dihydro-2H-indol-2-one 5-[2-[4-(2,3-Benzisothiazol-3-yl)-1-piperazinyl]ethyl]-6-chloro-1 ,3-dihydro-2H- indol-2-one (51 kg), filteraid (4 kg) and tetrahydrofuran (2678 L) were combined. The mixture was heated to reflux (-65° C) for -1 hour, filtered while maintaining the temperature above 55° C, and rinsed with tetrahydrofuran (570 L). The product rich filtrate was partially concentrated in vacuo. 5-[2-[4-(2,3-Benzisothiazol-3-yl)-1- piperazinyl]ethyl]-6-chloro-1 ,3-dihydro-2H-indol-2-one (51 kg), filteraid (4 kg) and tetrahydrofuran (2675 L) were combined. The mixture was heated to reflux (-65° C) for -1 hour, filtered while maintaining the temperature above 55° C, and rinsed with tetrahydrofuran (560 L). The product rich filtrate was combined with the partially concentrated mixture above and concentrated in vacuo. The resulting mixture was cooled to 0 to 5° C. The solids were isolated by filtration, washed with filtered tetrahydrofuran (113 L), and dried in vacuo at less than 41° C, giving ziprasidone, free base, yield: 79.3 kg (77.7 %).
A portion of the batch was combined with material of comparable quality which had been recrystallized separately and the batch was carried through the following step.
Example 2: Crystallization Salt Formation of Ziprasidone Hydrochloride Monohydrate Tetrahydrofuran (2715 L), water (307 L), and 5-[2-[4-(2,3-benzisothiazol-3-yl)-
1-piperazinyl]ethyl]-6-chloro-1 ,3-dihydro-2 – -indol-2-one (100.0 kg) were combined, heated to reflux (- 64° C), and stirred for -30 minutes. The solution was filtered and rinsed with tetrahydrofuran (358 L).
Water (203 L) and concentrated hydrochloric acid (29 L) were combined and stirred at ambient temperature. The resulting aqueous hydrochloric acid solution was charged to the 5-[2-[4-(2,3-benzisothiazpl-3-yl)-1-piperazinyl]ethyl]-6-chloro-1 ,3- dihydro-2 – -indol-2-one solution over a period of 27 minutes. The reaction mixture was cooled .to temperatures between 1 and 5° C over a period of -2 hours. The mixture was stirred between 1 and 5 C for -10 hours. The solids were isolated by filtration, washed with cold tetrahydrofuran (358 L), and dried until a water content of
4.1% was obtained.
Ziprasidone Hydrochloride Monohydrate, yield: 108.6 kg (96.0 % weight yiplrl)
The solids were milled on a Bauermeister mill. Example 3: Purification of 6-Chloro-5-(2-chloroethyl)-1,3-dihydro-2H-indol-2- one To Remove 5-(2-Chloroethyl)-1,3-dihydro-2H-indol-2-one
A 100 mL round bottom flask equipped with a magnet stirrer and reflux condenser was charged with 4.0 g (17.4 mmoles) of 6-chloro-5-(2-chloroethyl)-1 ,3- dihydro-2H-indol-2-one (Compound 3) and 36 mL of acetonitriie and 4.0 mL of water were added. The slurry was gently heated and stirred overnight (-18 hrs at -78° C). The heating was then removed and the slurry cooled to 0 to 5° C, and stirred for an additional hour. The product was collected by filtration, washed with a small portion of acetonitriie and the product dried under vacuum at 50° C, to give 3.77 g (94.3% yield) of 6-chloro-5-(2-chloroethyl)-1,3-dihydro-2H-ιndol-2-one. The level of the des- chloro impurity had been reduced from 1280 ppm to 230 ppm.
Example 4: Experimental Determination of Purge Factor for Compound 6 (1,3-Dihydro-2/Y-indol-2one)
A batch of 6-chloro-1 ,3-dihydro-2W-indol-2-one which contained a very high content of 1 ,3-dihydro-2λ7-indol-2-one was selected. This was intentionally selected so that higher levels of the impurity would be easier to measure, and to determine the purge factor for this impurity. An additional reason for this strategy of starting with material which was very high in the impurity for purposes of determining the purge factor of the impurity was to avoid having the material purge to less than the limit of analytical detection during the synthesis; thus resulting in a zero value in the final product. Since the purge factor is a ratio, it is not meaningful to divide by a zero result. (The material with the high level of impurity was used for this experiment but was NOT subsequently used in any studies with human subjects.) A batch of 6- chloro-1 ,3-dihydro-2H-indol-2-one which contained 4000 ppm of jl ,3-dihydro-2 – – ιndol-2-one was processed through the standard synthetic process according to Examples 1 and 2 above.
Following the first two steps of the synthesis, the level of the corresponding des-chloro impurity was measured, using the method described. It was found that
1700 ppm of 5-(2-chloroethyl)-1 ,3-dihydro-2H-indol-2-one (Compound 8 of Scheme 2, above) was present in 6-chloro-5-(2-chloroethyl)-1,3-dihydro-2 – -indol-2-one (Compound 3 of Scheme 1 , above). The processing was continued to 5-[2-[4-(1 ,2)- benzisothiazol-3-yl)-1-piperazinyl]ethyl]-6-chloro-1 ,3-dihydro-2H-indol-2-one hydrochloride monohydrate, where it was determined that 600 ppm of 5-[2-[4-(1,2)- benzisothiazol-3-yl)-1-piperazinyl]ethyl]-1 ,3-dihydro-2H-indol-2-one (Compound 9 of Scheme 2, above) was present.
Thus the purge factor through the entire synthesis for the des-chloro analogs was from 4000 ppm to 600 ppm, or approximately a 6-fold decrease. Minor run to run variations in processing can lead to small differences in the yield and quality of the materials produced. A 20% error in the reproducibility of the impurity formation, that is if 500 ppm in one run expecting between 400 and 600 ppm in other experiments, is then allowed for. In the case of the synthesis described in Examples 1 and 2, with 5 processing steps, the additive experimental error could result in as much as a 2-fold difference in the level of the impurity. Thus, for the purpose of setting the upper limit, where the drug is going to be used by human subjects a conservative 3-fold purge factor was utilized. Therefore, to insure that the product produced would not contain over 100 ppm of 5-[2-[4-(1 ,2)-benzisothiazol-3-yl)-1-piperazinyl]ethyl]-1,3-dihydro-2H- indol-2-one (Compound 9), a limit of 300 ppm of 1 ,3-dihydro-2H-indol-2-one
(Compound 6) in 6-chloro-1 ,3-dihydro-2H-indol-2-one (Compound 1) was determined.
…………………………………………..

The condensation of 1-(1,2-benzoisothiazol-3-yl)piperazine (I) with 6-chloro-5-(2-chloroethyl)-2-indolinone (II) in refluxing water or refluxing methyl isobutyl ketone gives the target indolinone derivative.
AU 8812537; EP 0281309; JP 1988301861

Wolff-Kishner reduction of 6-chloroisatin (I) gives 6-chlorooxindole (II), which is treated with chloroacetyl chloride under Friedel-Crafts conditions to yield 5-chloroacetyl-6-chlorooxindole (III). The ketone (III) is reduced using triethylsilane in trifluoroacetic acid to produce 6-chloro-5-(2-chloroethyl)oxindole (IV). 1,2-Benzisothiazolin-3-one (V) is converted to 3-chloro-1,2-benzisothiazole (VI) using phosphorus oxychloride and is then condensed with piperazine to provide 1-(1,2-benzisothiazol-3-yl)piperazine (VII). Finally, intermediate (VII) is alkylated by compound (IV) in the presence of sodium carbonate in water and is converted to the salt with aqueous hydrochloric acid.
US 4831031

WO 9500510
The nitration of 2,5-dichlorotoluene (I) with HNO3 in H2SO4/AcOH gives 2,5-dichloro-4-methylnitrobenzene (II), which is treated with t-butoxybis(dimethylamino)methane (III) in refluxing THF to yield 2,5-dichloro-4-[2-(dimethylamino)vinyl]nitrobenzene (IV). The condensation of (IV) with 1-(1,2-benzoisothiazol-3-yl)piperazine (V) in AcOH affords the disubstituted piperazine (VI), whose double bond is reduced by means of NaBH(OAc)3 in dichloroethane/AcOH to provide the saturated compound (VII). The condensation of (VII) with dimethyl malonate (VIII) by means of KOH in NMP gives the alkylated malonic ester (IX), which is hydrolyzed and monodecarboxylated with refluxing 3N HCl to yield the phenylacetic acid (X). The esterification of (X) with SOCl2 and methanol affords the methyl ester (XI), which is finally cyclized to the target indolone by reduction of its nitro group with sodium hydrosulfite in refluxing THF/ethanol. Alternatively, compound (VII) can be condensed with methyl cyanacetate (XII) by means of KOH in NMP to give the alkylated cyanacetic ester (XIII), which is hydrolyzed with refluxing 3N HCl to afford the already reported phenylacetic acid (X).
……………………
J Label Compd Radiopharm 1994,34(2),117
The Friedel Crafts condensation of 6-chloroindolin-2-one (I) with 14C labeled 2-chloroacetyl chloride (II) by means of AlCl3 in CS2 gives 6-chloro-5-(2-chloroacetyl)indolin-2-one (III), which is reduced with trimethylsilane in TFA to yield the labeled chloroethyl derivative (IV). Finally, this compound is condensed with 3-(1-piperazinyl)-1,2-benzoisothiazole (V) by means of Na2CO3 in refluxing water to provide the target radiolabeled compound.

………………….

The bromination of 3-chloro-1,2-benzoisothiazole (I) with Br2 in AcOH using FeCl3 as catalyst gives a mixture of 3,5-dibromo-1,2-benzoisothiazole (II) and 3,7-dibromo-1,2-benzoisothiazole (III) that are separated by flash chromatography. The desired isomer (III) is condensed with piperazine (IV) in refluxing diglyme to yield 7-bromo-3-(1-piperazinyl)-1,2-benzoisothiazole (V), which is condensed with 6-chloro-5-(2-chloroethyl)indolin-2-one (VI) by means of Na2CO3 in refluxing water to afford the brominated adduct (VII). Finally, this compound is debrominated with tritium gas over a Pd/BaSO4 catalyst in THF to provide the target radiolabeled compoun
……………….

The current process for ziprasidone involves preparation and isolation of the key intermediate 6-chloro-5-(2-chloroethyl)oxindole. An improved process for the synthesis of this intermediate is reported here. The new process involves use of a novel Lewis acid-mediated selective deoxygenation of the precursor ketone with tetramethyldisiloxane. The new method affords the desired compound in a one-pot process obviating the need for isolation of the potentially hazardous precursor ketone. This process was successfully scaled up to multikilo scale.

A new, one-step commercial process for the preparation of 3-(1-piperazinyl)-1,2-benzisothiazole, a key intermediate in the synthesis of ziprasidone has been developed: The reaction of 2-cyanophenyl disulfide (I) with piperazine (II) by means of DMSO and isopropanol at 120-5 C.
…………………………..

J Med Chem 1996,39(1),143
A new synthesis for ziprasidone hydrochloride has been reported: The condensation of 6-chloroindolin-2-one (I) with bromoacetic acid (II) by means of polyphosphoric acid (PPA) gives 5-(bromoacetyl)-6-chloroindolin-2-one (III), which is reduced with triethylsilane and trifluoroacetic acid to the corresponding 2-bromoethyl derivative (IV). Finally, this compound is condensed with 4-(3-benzisothiazolyl)piperazine (V) by means of Na2CO3 in DMF or isobutyl methyl ketone.
……………………….
http://www.google.com/patents/WO2004050655A1?cl=en
REFERENCE EXAMPLE 1 Preparation of 5-(2-Chloro ethyl) -6-chloro oxindole
Triethylsilane (57.2 gm) was added slowly to the reaction mixture of 5-(2-
Chloro acetyl) -6-chloro oxindole (50.0 gm) and trifluoroacetic acid (175 mL) below the temperature of 45°C. The reaction was maintained at 40-45°C for 6 hours. The reaction mass was cooled to 0°C to -5°C and maintained stirring for 90 minutes. The separated solid was filtered and washed with water (50 mL). Then the wet compound was further slurred in water (250 mL) for 90 minutes. The resultant solid was filtered, washed with water (50 mL) and dried at a temperature of 70-75°C to afford 5-(2-chloroethyl) -6-chloro oxindole (43.5 gm).
REFERENCE EXAMPLE 2 Preparation of Ziprasidone base
Refluxed the reaction mixture of 5-(2-chloroethyl) -6-chloro oxindole (100 gm), 3-(l-piperazinyl)-l,2-benzisothiazole (104.7 gm), sodium carbonate (92.2 gm), sodium iodide (6.4 gm), terra butyl ammonium bromide (28 gm) and cyclohexane (1000 mL) till the reaction was completed. The reaction mass was cooled to a temperature of 30°C and the solid was filtered. To the wet compound was added water (1000 mL) and continued stirring for 45 minutes. The solid was filtered and washed with water (100 mL).
To the water wet compound was added acetone (500 mL) and there was stirring for 2 hours at room temperature. The compound was filtered and washed with acetone (200 mL) and dried at a temperature of 70-75°C to afford the Cmde Ziprasidone base (156.9 g) REFERENCE EXAMPLE 3
Preparation of Ziprasidone base
Charged 5-(2-chloroethyl) -6-chloro oxindole (50 gm), 3-(l-piperazinyl)-
1,2-benzisothiazole (47.5 gm) and cyclohexane (500 mL) into an autoclave. To this sodium carbonate (46 gm), sodium iodide (3.2 gm), terra butyl phosphonium bromide (14.8 gm) was added and the reaction was maintained at a temperature of 95-102°C and the pressure was kept at 2.5 kg/cm till the reaction was completed. The reaction mass was cooled to 30°C and water (250 mL) was added. The resulting compound was filtered and washed with water (100 mL). The wet compound was further slurred in water (500 mL), filtered and washed with water (100 mL). To the water wet compound was added acetone (500 mL) and was stirred at room temperature for 2 hours and 30 minutes. The solid was filtered, washed with acetone (100 L) and dried at a temperature of 60-65°C to afford the Ziprasidone base (65.7 gm).
EXAMPLE 1
Preparation of amorphous form of Ziprasidone hydrochloride Ziprasidone (5g) and 50 mL of acetic acid were placed into a round bottom flask and heated to 45 – 50°C. Added was 25 mL of aqueous hydrochloric acid slowly to the mixture over 20 min. Then the reaction mixture was refluxed. Water (10 mL) was added, followed by addition of 50 mL of Isopropanol. The reaction mass was cooled to 50°C and distilled off the solvent completely under vacuum. The material formed was scratched from the flask.
EXAMPLE 2
Preparation of crystalline form of Ziprasidone
Sodium carbonate (56.3 g) and 500 mL of water were placed into a round bottom flask. Added was 50 g of 3-(l-piperazinyl)-l,2-benzisothiazole hydrochloride and 50 g of 6-Chloro-5-(2-Chloroethyl) oxindole. The reaction mixture was then refluxed for
15 hours. The reaction completion was monitored by TLC. The reaction mass was cooled to room temperature. The resulting compound was filtered and washed with 50 mL of water. The wet compound and 250 mL of acetone were placed into a flask and the reaction mixture was sthred at room temperature for 2 hours. The reaction mixture was filtered to give a solid cake, which was washed with 50 mL of acetone. The wet cake and
750 mL of methanol were placed into a flask, which was heated to 50°C, and 14 mL of methane sulfonic acid was added to the solution over 20 minutes. The resulting reaction mass was cooled to room temperature and was subjected to a filtration to give a solid compound, which was washed with methanol. The wet compound and 750 mL of water were placed into a flask, and then pH of the solution was adjusted to pH 9 with caustic lye.
The reaction mixture was then stirred at room temperature for 1 hour and filtered. The filtered compound was washed with water and dried at 70°C to give 65 g of crystalline form Ziprasidone base.
……………………..
http://www.google.com/patents/US7087611
Ziprasidone hydrochloride (5-[2-[4-(1,2-benzisothiazol-3-yl)-1-piperazinyl]ethyl]-6-chloro-1,3-dihydro-2H-indol-2-one hydrochloride), I is a potent neuroleptic agent useful in the treatment of psychotic disorders, schizophrenia, and anxiety diseases. It is currently marketed under the proprietary name of Geodon.

Ziprasidone hydrochloride is known to exist in three crystalline forms; namely, the monohydrate, hemihydrate and anhydrous form as disclosed in U.S. Pat. Nos. 4,831,031 and 5,312,925, both of which are herein incorporated by reference. U.S. Pat. No. 5,312,925 states that ziprasidone hydrochloride monohydrate is substantially hygroscopically stable, thus alleviating potential problems due to weight changes of the active pharmaceutical ingredient during the final formulation process. Nevertheless a very low aqueous solubility is observed for this crystalline form.
U.S. Pat. No. 4,831,031 discloses that arylpiperazinyl-ethyl (or butyl)-heterocyclic compounds II may be prepared by reacting piperazines of the formula III with compounds of the formula IV as follows:

The ‘031 patent indicates that this coupling reaction is generally conducted in a polar solvent, such as a lower alcohol, dimethylformamide or methyl isobutyl ketone, and in the presence of a weak base and that, preferably, the reaction is in the further presence of a catalytic amount of sodium iodide, and a neutralizing agent for hydrochloride such as sodium carbonate. At example 16, the ‘031 patent discloses a process in which a solution of ziprasidone free base is taken up in dichloromethane and then reacted with ether saturated with HCl to afford a precipitate which is subsequently filtered, slurried with acetone and filtered again to give ziprasidone hydrochoride hemihydrate.
Additionally, the methods described in the prior art for the preparation of some of these crystalline forms, for instance ziprasidone hydrochloride anhydrate provide inconsistent reproducibility. For example, ziprasidone hydrochloride anhydrate has been prepared by prolonged drying in air at 50° C. of the corresponding monohydrate form, as disclosed in U.S. Pat. No. 5,312,925. However, repeated attempts to prepare the anhydrous form of ziprasidone hydrochloride in our laboratory by using the above mentioned conditions failed to produce the expected anhydrate and instead ziprasidone hydrochloride having variable contents of water, including that corresponding to the hemihydrate form, were obtained. Furthermore, even when more drastic conditions were used, (i.e., higher temperatures, longer drying times, under vacuum) anhydrous product was still not obtained.
EXAMPLE 3
Preparation of 5-[2-[4-(1,2-benzisothiazol-3-yl)-1-piperazinyl]ethyl]-6-chloro-1, 3-dihydro-2H-indol-2-one hydrochloride anhydrate.
To a 3-necked flask equipped with mechanical stirrer, thermometer and nitrogen inlet was added 5-(2-(4-(1,2-benzisothiazol-3-yl)-1-piperazinyl)ethyl)-6-chloro-1,3-dihydro-2H-indol-2-one free base (5.0 g) and 1-methyl-2-pyrrolidinone (60 mL) under nitrogen and the suspension was warmed up to 35–40° C. to dissolution. The flask was cooled to about 25° C. A 20.5% anhydrous solution of hydrogen chloride in isopropanol (6.45 g) was added and the mixture was stirred at about 25° C. for about 3 h. The product was collected by filtration on a Buchner funnel. The filter cake is rinsed twice with 10 mL of isopropanol at 20–25° C. and the damp cake transferred to a flask equipped with magnetic stirrer and nitrogen inlet. Isopropanol was added (30 mL) and the suspension stirred at 20–25° C. for about 2 h. The product was collected by filtration on a Buchner funnel. The filter cake is rinsed with 3×10 mL of isopropanol at 20–25° C. and transferred to a drying oven and dried in vacuo at 70–75° C. for 43 h. This afforded 4.52 g of anhydrous ziprasidone hydrochloride. The material contained 3.1% and 0.26% of residual NMP and IPA, respectively as determined by NMR.
………………………..
http://www.google.com/patents/WO2000059489A2?cl=en
5. EXAMPLES 5.1. EXAMPLE 1: SYNTHESIS OF ZIPRASIDONE
To a 125 mL round bottom flask equipped with an N2 inlet and condenser are added 0.73 g (3.2 mmol) 5-(2-chloroethyl)-6-chloro-oxindole, 0.70 g (3.2 mmol) N-(l,2- benzisothiazol-3-yl)piperazine, 0.68 g (6.4 mmol) sodium carbonate, 2 mg sodium iodide, and 30 mL methylisobutyl ketone. The reaction is refluxed for 40 hours, cooled, filtered, and evaporated. The residue is chromatographed on silica gel, eluting the by-products with ethyl acetate (1 L) and the product with 4 % methanol in ethyl acetate (1.5 L). The product fractions (Ry = 0.2 in 5 % methanol in ethyl acetate) are evaporated, taken up in methylene chloride, and precipitated by addition of ether saturated with HC1; the solid is filtered and washed with ether, dried, and washed with acetone. The latter is done by slurrying the solid with acetone and filtering. Ziprasidone is obtained as a high melting, non-hygroscopic solid product having an expected melting point of 288°C to 288.5°C.
……………………
Combined serotonin (5HT2) and dopamine (D2) receptor antagonist. Prepn: J. A. Lowe III, A. A. Nagel, EP281309; eidem, US 4831031 (1988, 1989 both to Pfizer). Clinical pharmacology: C. J. Bench et al., Psychopharmacology 112, 308 (1993). HPLC determn in serum: J. S. Janiszewski et al., J. Chromatogr. B 668, 133 (1995). Receptor binding profile: A. W. Schmidt et al., Eur. J. Pharmacol. 425, 197 (2001). Review of pharmacology and clinical experience: G. L. Stimmel et al., Clin. Ther. 24, 21-37 (2002); P. D. Harvey, C. R. Bowie, Expert Opin. Pharmacother. 6, 337-346 (2005).
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US4831031 * | Jan 22, 1988 | May 16, 1989 | Pfizer Inc. | Aryl piperazinyl-(C2 or C4) alkylene heterocyclic compounds having neuroleptic activity |
| US5206366 | Aug 26, 1992 | Apr 27, 1993 | Pfizer Inc. | Process for preparing aryl piperazinyl-heterocyclic compounds |
| US5312925 | Sep 1, 1992 | May 17, 1994 | Pfizer Inc. | Neuroleptic agents |
| US5338846 | Apr 20, 1993 | Aug 16, 1994 | Pfizer Inc. | Process for preparing aryl piperazinyl-heterocyclic compounds with a piperazine salt |
| US5935960 | Feb 7, 1997 | Aug 10, 1999 | Pfizer Inc. | Treatment of schizophrenia |
| US6150366 | May 27, 1999 | Nov 21, 2000 | Pfizer Inc. | Crystalline ziprasidone free base or crystalline ziprasidone hydrochloride particles with specific particle size are useful as antipsychosis agent |
| US20040152711 | Dec 4, 2003 | Aug 5, 2004 | Dr. Reddy’s Laboratories Limited | Crystal structure and amorphous form; psychological disorders |
| CA2166203A1 | Apr 6, 1994 | Jan 5, 1995 | Processes and intermediates for the preparation of 5-[2-(4-(benzoisothiazol-3-yl)-piperazin-1-yl)ethyl]- 6-chloro-1,3-dihydro-indol-2-one | |
| CA2245269A1 | Aug 7, 1998 | Feb 11, 1999 | Pfizer Prod Inc | Solid pharmaceutical dispersions with enhanced bioavailability |
| CA2252898A1 | Apr 10, 1997 | Nov 13, 1997 | Pfizer | Mesylate dihydrate salts of 5-(2-(4-(1,2-benzisothiazol-3-yl)-1-piperazinyl)-ethyl)-6-chloro-1,3-dihydro-2(1h)-indol-2-one(=ziprasidone), its preparation and its use as dopamine d2 antagonist |
| WO1995000510A1 | Apr 6, 1994 | Jan 5, 1995 | Pfizer | Processes and intermediates for the preparation of 5-[2-(4-(benzoisothiazol-3-yl)-piperazin-1-yl)ethyl]-6-chloro-1,3-dihydro-indol-2-one |
| WO2003070246A1 | Feb 17, 2003 | Aug 28, 2003 | Pfizer Prod Inc | Controlled synthesis of ziprasidone and compositions thereof |
| WO2004050655A1 | Dec 4, 2003 | Jun 17, 2004 | Akundi Surya Prabhakar | Polymorphic forms of ziprasidone and its hydrochloride |
| US7939662 | Dec 6, 2007 | May 10, 2011 | Apotex Pharmachem Inc. | Amorphous ziprasidone hydrochloride (5-[2-[4-(1,2-benzisothiazol-3-yl)-1-piperazinyl]ethyl]-6-chloro-1,3-dihydro-2H-indol-2-one hydrochloride) and processes to produce the same |
| US4831031 * | Jan 22, 1988 | May 16, 1989 | Pfizer Inc. | Aryl piperazinyl-(C2 or C4) alkylene heterocyclic compounds having neuroleptic activity |
| US5206366 * | Aug 26, 1992 | Apr 27, 1993 | Pfizer Inc. | Process for preparing aryl piperazinyl-heterocyclic compounds |
| WO1997042190A1 * | Mar 26, 1997 | Nov 13, 1997 | Frank R Busch | Mesylate trihydrate salt of 5-(2-(4-(1,2-benzisothiazol-3-yl)-1-piperazinyl)ethyl)-6-chloro-1,3-dihydro-2(1h)-indol-2-one (=ziprasidone), its preparation and its use as dopamine d2 antagonist |
| WO1997042191A1 * | Apr 10, 1997 | Nov 13, 1997 | Pfizer | Mesylate dihydrate salts of 5-(2-(4-(1,2-benzisothiazol-3-yl)-1-piperazinyl)-ethyl)-6-chloro-1,3-dihydro-2(1h)-indol-2-one (=ziprasidone), its preparation and its use as dopamine d2 antagonist |
| WO2003099198A2 * | May 26, 2003 | Dec 4, 2003 | Srinivasu Kilaru | A process for the preparation of oxindole derivatives |
| WO2004050655A1 * | Dec 4, 2003 | Jun 17, 2004 | Akundi Surya Prabhakar | Polymorphic forms of ziprasidone and its hydrochloride |
| US6110918 * | Mar 26, 1997 | Aug 29, 2000 | Pfizer Inc | Mesylate trihydrate salt of 5-(2-(4-(1,2-benzisothiazol-3-yl)-1-piperazinyl)ethyl)-6-chloro-1,3-dihy dro-2(1H)-indol-2-one (=ziprasidone), its preparation and its use as dopamine D2 antagonist |
| US6245765 | Apr 10, 1997 | Jun 12, 2001 | Pfizer Inc | Psychological disorders |
| US7087611 | Aug 30, 2004 | Aug 8, 2006 | Apotex Pharmachem Inc. | Reacting ziprasidone with hydrochloric acid in solvent; salt formation; drying |
| US7488729 | Dec 4, 2003 | Feb 10, 2009 | Dr. Reddy’s Laboratories Limited | Polymorphic forms of ziprasidone and its hydrochloride salt and process for preparation thereof |
| US7790886 | Jan 5, 2009 | Sep 7, 2010 | Dr. Reddy’s Laboratories, Inc. | providing alcoholic solvent, desolventizing to form solid mass, isolating amorphous form; psychosis |
| US7939662 | Dec 6, 2007 | May 10, 2011 | Apotex Pharmachem Inc. | Amorphous ziprasidone hydrochloride (5-[2-[4-(1,2-benzisothiazol-3-yl)-1-piperazinyl]ethyl]-6-chloro-1,3-dihydro-2H-indol-2-one hydrochloride) and processes to produce the same |
| US8097610 | Aug 24, 2006 | Jan 17, 2012 | Shionogi & Co., Ltd. | Derivative having PPAR agonistic activity |
| US4831031 † | Jan 22, 1988 | May 16, 1989 | Pfizer Inc. | Aryl piperazinyl-(C2 or C4) alkylene heterocyclic compounds having neuroleptic activity |
| US5206366 † | Aug 26, 1992 | Apr 27, 1993 | Pfizer Inc. | Process for preparing aryl piperazinyl-heterocyclic compounds |
| US5312925 † | Sep 1, 1992 | May 17, 1994 | Pfizer Inc. | Neuroleptic agents |
| US6111105 † | Oct 11, 1996 | Aug 29, 2000 | Pfizer, Inc. | Chemical intermediates for ziprasidone, a neuroleptic agent; reacting a 2-(piperidino) or (1,2-benzisoxazol-3-yl) or (2-cyanophenyl)thio),3-cyanobenzene with piperazine at a temperature from 80-170 degrees c. |
| US6245765 † | Apr 10, 1997 | Jun 12, 2001 | Pfizer Inc | Psychological disorders |
