
Home » Posts tagged 'Phase3 drugs'
Tag Archives: Phase3 drugs
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Nemonoxacin….TaiGen’s pneumonia antibiotic Taigexyn 奈诺沙星 gets marketing approval in Taiwan

Nemonoxacin 奈诺沙星
378746-64-6 CAS
TG-873870
-
C20-H25-N3-O4
- 371.4345
WARNER CHILCOTT ORIGINATOR
CLINICAL TRIALS http://clinicaltrials.gov/search/intervention=Nemonoxacin
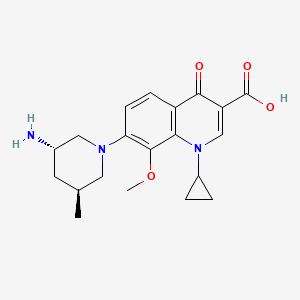
(3S,5S)-7-[3-amino-5-methyl-piperidinyl]-l-cyclopropyl-l,4- dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid
7-[3(S)-Amino-5(S)-methylpiperidin-1-yl]-1-cyclopropyl-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid
Taigexyn has been approved in Taiwan IN 2014
“TAIPEI, MARCH 13, 2014 /PRNEWSWIRE/ — TAIGEN BIOTECHNOLOGY …”
13.03.14 |
13.03.14 |
TaiGen Biotechnology Receives Marketing Approval from the Taiwan Food and Drug Administration for Taigexyn in Taiwan
TAIPEI, March 13, 2014 /PRNewswire/ — TaiGen Biotechnology Company, Limited (“TaiGen”) today announced that the Taiwan Food and Drug Administration (TFDA) has approved the new drug application (NDA) of Taigexyn® (nemonoxacin) oral formulation (500 mg) for the treatment of community-acquired bacterial pneumonia (CAP). With this NDA approval, Taiwan is the first region to grant marketing approval to Taigexyn®. An NDA for Taigexyn® was also submitted to China FDA (CFDA) in April 2013 and is currently under review.
Nemonoxacin is a novel non-fluorinated quinolone antibiotic undergoing clinical trials.
Taigexyn Granted QIDP and Fast Track Designations
TaiGen Biotechnology announced that the FDA has granted nemonoxacin (Taigexyn) Qualified Infectious Disease Product (QIDP) and Fast Track designations for community-acquired bacterial pneumonia (CAP) and acute bacterial skin and skin structure infections (ABSSSI).
Nemonoxacin is a novel non-fluorinated quinolone broad spectrum antibiotic available in both oral and intravenous formulations. Nemonoxacin demonstrates activity against gram-positive and gram-negative bacteria and atypical pathogens. Nemonoxacin also possesses activities against methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant pathogens.
Nemonoxacin is a novel non-flourinated quinolone antibiotic registered in Taiwan for the oral treatment of community-acquired pneumonia. Clinical trials are in development at TaiGen Biotechnology for the treatment of diabetic foot infections and for the treatment of moderate to severe community-acquired pneumonia with an intravenous formulation. The drug is thought to accomplish its antibacterial action through topoisomerase inhibition.
Originally developed at Procter & Gamble, nemonoxacin was the subject of a strategic alliance formed in January 2005 between P&G and TaiGen to further the development and commercialization of nemonoxacin. In 2012, the product was licensed by TaiGen Biotechnology to Zhejiang Medicine in China for manufacturing, sales and marketing. In 2014, TaiGen out-licensed the exclusive rights of the product in Russian Federation, Commonwealth Independent States and Turkey to R-Pharm.
TaiGen has completed two Phase 2 clinical studies, one in CAP and the other in diabetic foot infections with demonstrated efficacy and safety. In the clinical trials conducted to date, nemonoxacin has shown activity against drug-resistant bacteria such as MRSA, quinolone-resistant MRSA, as well as quinolone-resistant Streptococcus pneumoniae.

Malate salt
Nemonoxacin malate anhydrous
951163-60-3 CAS NO, MW: 505.5209
Nemonoxacin malate hemihydrate
951313-26-1, MW: 1029.0566
Chemical structure of nemonoxacin as a malate salt (C20H25N3O4·C4H6O5·H2O). Nemonoxacin is the free base, and its molecular mass is 371.44 g/mol. The molecular mass of the salt, nemonoxacin malate, is 514.53 g/mol.


……………………..
isomeric compounds are:

(3S,5S)-7-[3-amino-5-methyl-piperidinyl]-l-cyclopropyl-l,4-dihydro-8- methoxy-4-oxo-3 -quinolinecarboxylic acid
COMPD1…….DESIRED

(3S,5R)-7-[3-amino-5-methyl-piperidinyl]-l-cyclopropyl-l,4-dihydro-8- methoxy-4-oxo-3 -quinolinecarboxylic acid
COMPD 1’….NOT DESIRED
Example 1
Malate salts of (3S,5S)-7-[3-amino-5-methyl-piperidinyl]-l-cyclopropyl-l,4- dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid (Compound 1) and (3S,5R)-7- [3-ammo-5-methyl-piperidinyl]- 1 -cyclopropyl- 1 ,4-dihydro-8-methoxy-4-oxo-3- quinolinecarboxylic acid (Compound 1′) were synthesized as follows:
(A) Synthesis of (3S,5S)-(5-Methyl-piperidin-3-yl)-carbamic acid tert-butyl ester (Compound 9) and (3S,5R)-(5-Methyl-piperidin-3-yl)-carbamic acid tert-butyl ester (Compound 9′): Compound 9′ was synthesized as shown in Scheme 1 below:
Scheme 1

3 4 Boc

A 50-L reactor was charged with Compound 2 (5.50 kg, 42.60 mol), methanol (27 L) and cooled to 10-150C. Thionyl chloride (10.11 kg, 2.0 equiv.) was added via an addition funnel over a period of 65 min, with external cooling to keep temperature below 30°. The resulting solution was stirred at 250C for 1.0 hour, after which methanol was removed under reduced pressure. The oily residue was azeotroped with ethyl acetate (3 x 2.5 L) to remove residual methanol, dissolved in ethyl acetate (27.4 L), charged into a 50 L reactor, and neutralized by slow addition of triethylamine (3.6 kg) below 3O0C. The resulting suspension was filtered to remove triethylamine hydrochloride.
The filtrate was charged to a 50 L reactor, along with DMAP (0.53 kg). Di- fert-butyl dicarbonate (8.43 kg) was added via hot water heated addition funnel, over a period of 30 min at a temperature of 20-300C. The reaction was complete after 1 hour as determined by TLC analysis. The organic phase was washed with ice cold IN HCl (2 x 7.5 L), saturated sodium bicarbonate solution (1 x 7.5 L), dried over magnesium sulfate, and filtered. After ethyl acetate was removed under reduced pressure, crystalline slurry was obtained, triturated with MTBE (10.0 L), and filtered to afford Compound 3 as a white solid (5.45 kg, 52.4%).
Anal. Calcd for CHHI7NO5 : C, 54.3; H, 7.04; N, 5.76. Found: C, 54.5; H, 6.96; N, 5.80. HRMS (ESI+) Expected for CHHI8NO5, [M+H] 244.1185. Found
244.1174; 1H NMR (CDCl3, 500 MHz):δ=4.54 (dd, J= 3.1, 9.5 Hz, IH), 3.7 (s, 3H), 2.58-2.50 (m, IH), 2.41 (ddd, IH, J= 17.6, 9.5, 3.7), 2.30-2.23 (m, IH), 1.98-1.93 (m, IH), 1.40 (s, 9H); 13C NMR (CDCl3, 125.70 MHz) δ 173.3, 171.9, 149.2, 83.5, 58.8, 52.5, 31.1, 27.9, 21.5. Mp 70.20C.
A 50-L reactor was charged with Compound 3 (7.25 kg, 28.8 mol), DME (6.31 kg), and Bredereck’s Reagent (7.7 kg, 44.2 mole). The solution was agitated and heated to 750C + 50C for three hours. The reaction was cooled to O0C over an hour, during which time a precipitate formed. The mixture was kept at O0C for an hour, filtered, and dried in a vacuum oven for at least 30 hours at 3O0C + 50C to give compound 4 as a white crystalline solid (6.93 kg, 77.9%).
Anal. Calcd for Ci4H22N2O5: C, 56.4; H, 7.43; N, 9.39. Found C, 56.4; H, 7.32; N, 9.48; HRMS (ESI+) Expected for Ci4H22N2O5, [M+H] 299.1607. Found 299.1613; 1H NMR (CDCl3, 499.8 MHz) δ = 7.11 (s, IH), 4.54 (dd, IH, J= 10.8, 3.6), 3.74 (s, 3H), 3.28-3.19 (m, IH), 3.00 (s, 6H), 2.97-2.85 (m,lH), 1.48 (s, 9H); 13C NMR (CDCl3, 125.7 MHz) δ = 172.6, 169.5, 150.5, 146.5, 90.8, 82.2, 56.0, 52.3, 42.0, 28.1, 26.3. MP 127.90C. A 10-gallon Pfaudler reactor was charged with ESCAT 142 (Engelhard Corp.
N.J, US) 5% palladium powder on carbon (50% wet, 0.58 kg wet wt), Compound 4 (1.89 kg, 6.33 mol), and isopropanol (22.4 Kg). After agitated under a 45-psi hydrogen atmosphere at 450C for 18 hrs, the reaction mixture was cooled to room temperature and filtered though a bed of Celite (0.51 kg). The filtrate was evaporated under reduced pressure to give a thick oil, which was solidified on standing to afford Compound 5 (1.69 kg, 100%) as a 93:7 diastereomeric mixture.
A sample of product mixture was purified by preparative HPLC to give material for analytical data. Anal. Calcd for Ci2Hi9NO5: C, 56.0; H, 7.44; N, 5.44. Found C, 55.8; H, 7.31; N, 5.44; MS (ESI+) Expected for Ci2Hi9NO5, [M+H] 258.1342. Found 258.1321; 1H NMR (CDCl3, 499.8 MHz) δ = 4.44 (m, IH), 3.72 (s, 3H), 2.60-2.48 (m, 2H), 1.59-1.54 (m, IH), 1.43 (s, 9H), 1.20 (d, j = 6.8 Hz,3H); 13C NMR (CDCl3, 125.7 MHz) δ = 175.7, 172.1, 149.5, 83.6, 57.4, 52.5, 37.5, 29.8, 27.9, 16.2. Mp 89.90C.
A 50-L reactor was charged with Compound 5 (3.02 kg, 11.7 mol), absolute ethanol (8.22 kg), and MTBE (14.81 kg). Sodium borohydride (1.36 kg, 35.9 mol) was added in small portions at 00C + 50C. A small amount of effervescence was observed. The reaction mixture was warmed to 1O0C + 50C and calcium chloride dihydrate (2.65 kg) was added in portions at 1O0C + 50C over an hour. The reaction was allowed to warm to 2O0C + 50C over one hour and agitated for an additional 12 hours at 200C + 50C. After the reaction was cooled to -50C + 50C, ice-cold 2N HCl (26.9 kg) was added slowly at of O0C + 50C. Agitation was stopped. The lower aqueous phase was removed. The reactor was charged with aqueous saturated sodium bicarbonate (15.6 kg) over five minutes under agitation. Agitation was stopped again and the lower aqueous phase was removed. The reactor was charged with magnesium sulfate (2.5 kg) and agitated for at leastlO minutes. The mixture was filtered though a nutsche filter, and concentrated under reduced pressure to afford Compound 6 (1.80 kg, 66%). Anal. Calcd for CnH23NO4: C, 56.6 H, 9.94; N, 6.00. Found C, 56.0; H, 9.68;
N, 5.96; HRMS (ESI+) Expected for CnH24NO4, [M+H] 234.1705. Found 234.1703; 1H NMR (CDCl3, 500 MHz) δ = 6.34 (d, J= 8.9 Hz, IH, NH), 4.51 (t, J= 5.8, 5.3 Hz, IH, NHCHCH2OH), 4.34 (t, J= 5.3, 5.3 Hz, IH, OBCHCH2OH), 3.46-3.45, (m, IH, NHCH), 3.28 (dd, J= 10.6, 5.3 Hz, NHCHCHHOH), 3.21 (dd, J= 10.2, 5.8 Hz , IH, CH3CHCHHOH), 3.16 (dd, J = 10.2, 6.2 Hz, IH, NHCHCHHOH), 3.12 (dd, J= 10.6, 7.1 Hz , IH, CH3CHCHHOH), 1.53-1.50 (m, IH, CH3CHCHHOH), 1.35 (s, 9H, 0(CHB)3, 1.30 (ddd, J = 13.9, 10.2, 3.7 Hz, IH, NHCHCHHCH), 1.14 (ddd, J= 13.6, 10.2, 3.4 Hz, IH, NHCHCHHCH), 0.80 (d, J= 6.6 Hz, 3H, CH3); 13C NMR (CDCl3, 125.7 MHz) δ 156.1, 77.9, 50.8, 65.1, 67.6, 65.1, 35.6, 32.8, 29.0, 17.1. Mp 92.10C. A 50 L reactor was charged with a solution of Compound 6 (5.1 kg) in isopropyl acetate (19.7 kg). The reaction was cooled to 150C + 5°C and triethylamine (7.8 kg) was added at that temperature. The reactor was further cooled to O0C + 50C and methanesulfonyl chloride (MsCl) (6.6 kg) was added. The reaction was stirred for a few hours and monitored for completion by HPLC or TLC. The reaction was quenched by saturated aqueous bicarbonate solution. The organic phase was isolated and washed successively with cold 10% aqueous triethylamine solution, cold aqueous HCl solution, cold saturated aqueous bicarbonate solution, and finally saturated aqueous brine solution. The organic phase was dried, filtered, and concentrated in vacuo below 550C + 50C to afford compound 7 as a solid/liquid slurry, which was used in the subsequent reaction without further purification.
After charged with 9.1 kg of neat benzylamine, a 50 L reactor was warmed to 550C, at which temperature, a solution of compound 7 (8.2 kg) in 1,2- dimethoxyethane (14.1 kg) was added. After the addition, the reaction was stirred at 6O0C + 50C for several hours and monitored for completion by TLC or HPLC. The reaction was cooled to ambient temperature and the solvent was removed under vacuum. The residue was diluted with 11.7 kg of 15% (v/v) ethyl acetate/hexanes solution and treated, while agitating, with 18.7 kg of 20% (wt) aqueous potassium carbonate solution. A triphasic mixture was obtained upon standing. The upper organic layer was collected. The isolated middle layer was extracted twice again with 11.7 kg portions of 15% (v/v) ethyl acetate/hexanes solution. The combined organic layers were concentrated under vacuum to give an oily residue. The residue was then purified by chromatography to afford Compound 8 as an oil. A 40 L pressure vessel was charged with 0.6 kg 50% wet, solid palladium on carbon (ElOl, 10 wt. %) under flow of nitrogen. A solution of Compound 8 (3.2 kg) in 13.7 kg of absolute ethanol was then added to the reactor under nitrogen. The reactor was purged with nitrogen and then pressurized with hydrogen at 45 psi. The reaction was then heated to 45°C. It was monitored by TLC or LC. Upon completion, the reaction was cooled to ambient temperature, vented, and purged with nitrogen. The mixture was filtered through a bed of Celite and the solid was washed with 2.8 kg of absolute ethanol. The filtrate was concentrated under vacuum to afford Compound 9 as a waxy solid.
TLC R/(Silica F254, 70:30 v/v ethyl acetate-hexanes, KMnO4 stain) = 0.12; 1H NMR (300 MHz, CDCl3) δ 5.31 (br s, IH), 3.80-3.68 (m, IH), 2.92 (d, J=I 1.4 Hz,
IH), 2.77 (AB quart, JAB=12.0 Hz, v=50.2 Hz, 2H), 2.19 (t, J=10.7 Hz, IH), 1.82-1.68 (m, 2H), 1.54 (br s, IH), 1.43 (s, 9H), 1.25-1.15 (m, IH), 0.83 (d, J=6.6 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ: 155.3, 78.9, 54.3, 50.8, 45.3, 37.9, 28.4, 27.1, 19.2; MS (ESI+) m/z 215 (M+H), 429 (2M+H). Similarly, (3S,5R)-(5-Methyl-piperidin-3-yl)-carbamic acid tert-butyl ester
(Compound 9′) was synthesized as shown in Scheme 2.
Scheme 2

HN Boc HN Boc
NaBH4,EtOH w – “ MsCI1TEA . „ _. – – _. „ Benzyl Amine
THF EA1CoId

(B) Synthesis of l-Cyclopropyl-7-fluoro-8-methoxy-4-oxo-l,4-dihydro-quinoline-3- carboxylic acid (Compound 10): Compound 10 was prepared according to the method described in U.S. Patent
6,329,391.
(C) Synthesis of borone ester chelate of l-Cyclopropyl-7-fluoro-8-methoxy-4-oxo- l,4-dihydro-quinoline-3-carboxylic acid (Compound 11):
Scheme 3

Toluene, tert-Butylmethyl ether 20-500C, filter
A reactor was charged with boron oxide (2.0 kg, 29 mol), glacial acetic acid (8.1 L, 142 mol), and acetic anhydride (16.2 L, 171 mol). The resulting mixture was refluxed at least 2 hours, and then cooled to 400C, at which temperature, 7- fluoroquinolone acid compound 10 (14.2 kg, 51 mol) was added. The mixture was refluxed for at least 6 hours, and then cooled to about 900C. Toluene (45 L) was added to the reaction. At 5O0C, terϊ-butylmethyl ether (19 L) was added to introduce precipitation. The mixture was then cooled to 200C and filtered to isolate the precipitation. The isolated solid was then washed with teτt-butylmethyl ether (26 L) prior to drying in a vacuum oven at 4O0C (50 torr) to afford Compound 11 in a yield of 86.4%. Raman (cm 1): 3084.7, 3022.3, 2930.8, 1709.2, 1620.8, 1548.5, 1468.0, 1397.7, 1368.3, 1338.5, 1201.5, 955.3, 653.9, 580.7, 552.8, 384.0, 305.8. NMR (CDCl3, 300 MHz) δ (ppm): 9.22 (s, IH), 8.38-8.33 (m, IH), 7.54 (t, J=9.8 Hz, IH), 4.38-4.35 (m, IH), 4.13 (s, 3H), 2.04 (s, 6H), 1.42-1.38 (m, 2H), 1.34-1.29 (m, 2H). TLC (Whatman MKC18F Silica, 6θA, 200 μm), Mobile Phase: 1 :1 (v/v) CH3CN : 0.5N NaCl (aq), UV (254/366 nm) visualization; R^O.4-0.5. (D) Synthesis of malate salt of (3S,5S)-7-[3-amino-5-methyl-piperidmyl]-l- cyclopropyl-l,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid (Compound 1) and malate salt of (3S,5R)-7-[3-amino-5-methyl-piperidmyl]-l-cyclopropyl-l,4- dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid (Compound 1′)
Compound 1 was synthesized from compound 9 as shown in Scheme 4 below:
Scheme 4

5O0C 3 d
a 6 0 N HCI (aq) CH2CI2 35°40°C 12 h t> Extract pH ad]ust to ~7-8 50″-65″C filter



A reactor was charged with Compound 11 (4.4 kg, 10.9 mol), Compound 9 (2.1 kg, 9.8 mol), triethylamine (TEA) (2.1 L, 14.8 mol), and acetonitrile (33.5 L, 15.7 L/kg). The resulting mixture was stirred at approximately 500C till completion of the reaction, as monitored by HPLC or reverse phase TLC. It was cooled to approximately 35°C and the reaction volume was reduced to approximately half by distillation of acetonitrile under vacuum between 0-400 torr. After 28.2 kg of 3.0 N NaOH (aq) solution was added, the reaction mixture was warmed to approximately 4O0C, distilled under vacuum until no further distillates were observed, and hydro lyzed at room temperature. Upon completion of hydrolysis, which was monitored by HPLC or reverse phase TLC, 4-5 kg of glacial acetic acid was added to neutralize the reaction mixture.
The resulting solution was extracted 3 times with 12.7 kg (9.6 L) of dichloromethane. The organic layers were combined and transferred to another reactor. The reaction volume was reduced to approximately a half by evaporation at 400C. After 20.2 Kg 6.0N HCl (aq) solution was added, the reaction mixture was stirred for at least 12 hours at 35°C. After the reaction was completed as monitored by HPLC or reverse phase TLC, agitation was discontinued to allow phase separation. The organic phase was removed and the aqueous layer was extracted with 12.7 kg (9.6 L) of dichloromethane. The aqueous layer was diluted with 18.3 kg distilled water and warmed to approximately 500C. Dichloromethane was further removed by distillation under vacuum (100-400 torr).
The pH of the aqueous solution was then adjusted to 7.8-8.1 by adding about 9.42 kg of 3.0 N NaOH (aq) below 65°C. The reaction mixture was stirred at 500C for at least an hour and then cooled to room temperature. The precipitate was isolated by suction filtration, washed twice with 5.2 kg of distilled water, and dried with suction for at least 12 hours and then in a convection oven at 55°C for additional 12 hours. Compound 12 (3.2 kg, 79%) was obtained as a solid.
A reactor was charged with 3.2 kg of Compound 12 and 25.6 kg of 95% ethanol. To the reactor was added 1.1 kg of solid D,L-malic acid. The mixture was refluxed temperature (~80°C). Distilled water (-5.7 L) was added to dissolve the precipice and 0.2 kg of activated charcoal was added. The reaction mixture was passed through a filter. The clear filtrate was cooled to 45°C and allowed to sit for at least 2 hours to allow crystallization. After the reaction mixture was further cooled to 5°C, the precipitate was isolated by suction filtration, washed with 6.6 kg of 95% ethanol, and dried with suction for at least 4 hours. The solid was further dried in a convection oven at 450C for at least 12 hours to afford 3.1 kg of Compound 1 (yield: 70%). NEMONOXACIN
NMR (D2O, 300 MHz) δ (ppm): 8.54 (s, IH), 7.37 (d, J=9.0 Hz, IH), 7.05 (d, J=9.0 Hz, IH), 4.23-4.18 (m, IH), 4.10-3.89 (m, IH), 3.66 (br s, IH), 3.58 (s, 3H), 3.45 (d, J=9.0 Hz, IH), 3.34 (d, J=9.3 Hz, IH), 3.16 (d, J=12.9 Hz, IH), 2.65 (dd, J=16.1, 4.1 Hz, IH), 2.64-2.53 (m, IH), 2.46 (dd, J=16.1, 8.0 Hz, IH), 2.06 (br s, IH), 1.87 (d, J=14.4 Hz, IH), 1.58-1.45 (m, IH), 1.15-0.95 (m, 2H), 0.91 (d, J=6.3 Hz, 3H), 0.85-0.78 (m, 2H).
Similarly, Compound 1′ was synthesized from Compound 9′ as shown in Scheme 5 below:
Scheme 5

(3S,5R)-7-[3-amino-5-methyl-piperidinyl]-l-cyclopropyl-l,4-dihydro-8- methoxy-4-oxo-3 -quinolinecarboxylic acid
COMPD 1’….NOT DESIRED
…………………
US2007/232650 A1,
malate salts of

(3S,5S)-7-[3-amino-5-methyl-piperidinyl]-1-cyclopropyl-1,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid (hereinafter Compound I, see also intermediate (23) in Section D, of Detailed Description of the Invention).
EXAMPLES Example 1 Synthesis of (3S,5S)-7-[3-amino-5-methyl-piperidinyl]-1-cyclopropyl-1,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid and malate salt thereof A. Synthesis of (3S,5S)-(5-Methyl-piperidin-3-yl)-carbamic acid tert-butyl ester (8)

(2S)-1-(1,1-Dimethylethyl)-5-oxo-1,2-pyrrolidinedicarboxylic acid-2-methyl ester, (2). A 50-L reactor is charged with compound (1) (5.50 Kg, 42.60 mol), methanol (27 L) and cooled to 10-15° C. Thionyl chloride (10.11 Kg, 2.0 equiv.) is added via addition funnel over a period of 65 min, with external cooling to maintain temperature at <30°. The resulting solution is stirred at 25° C.+5° C. for 1.0 hour, after which the methanol is distilled off under reduced pressure. The resulting thick oil is azeotroped with ethyl acetate (3×2.5 L) to remove residual methanol. The residue is dissolved in ethyl acetate (27.4 L), charged into a 50 L reactor, and neutralized by the addition of triethylamine (3.6 Kg) from an addition funnel over 30 minutes. The temperature of the neutralization is maintained below 30° C. via external cooling. The resulting suspension of triethylamine hydrochloride is removed by filtration, and the clarified mother liquor solution is charged to a 50 L reactor, along with DMAP (0.53 Kg). Di-tert-butyl dicarbonate (8.43 Kg) is added via hot water heated addition funnel, over a period of 30 min with external cooling to maintain temperature at about 20-30° C. The reaction is complete after 1 hour as determined by TLC analysis. The organic phase is washed with ice cold 1N HCl (2×7.5 L), saturated sodium bicarbonate solution (1×7.5 L), and dried over magnesium sulfate. The mixture is filtered through a nutsche filter and ethyl acetate is removed under reduced pressure to yield a crystalline slurry that is triturated with MTBE (10.0 L) and filtered to afford intermediate (2) as a white solid (5.45 Kg, 52.4%). Anal. Calcd for C11H17NO5: C, 54.3; H, 7.04; N, 5.76. Found: C, 54.5; H, 6.96; N, 5.80. HRMS (ESI+) Expected for C11H18NO5, [M+H] 244.1185. Found 244.1174; 1H NMR (CDCl3, 500 MHz): δ=4.54 (dd, J=3.1, 9.5 Hz, 1H), 3.7 (s, 3H), 2.58-2.50 (m, 1H), 2.41 (ddd, 1H, J=17.6, 9.5, 3.7), 2.30-2.23 (m, 1H), 1.98-1.93 (m, 1H), 1.40 (s, 9H); 13C NMR (CDCl3, 125.70 MHz) δ 173.3, 171.9, 149.2, 83.5, 58.8, 52.5, 31.1, 27.9, 21.5; Mp 70.2° C.
(2S,4E)-1-(1,1-Dimethylethyl)-4-[(dimethylamino)methylene]-5-oxo-1,2-pyrrolidinedicarboxylic acid-2-methyl ester (3). A 50-L reactor is charged with intermediate (2) (7.25 Kg, 28.8 mol), DME (6.31 Kg), and Bredereck’s Reagent (7.7 Kg, 44.2 mole). The solution is agitated and heated to 75° C.±5° C. for at least three hours. The progress of the reaction is monitored by HPLC. The reaction is cooled to 0° C.±5° C. over on hour during which time a precipitate forms. The mixture is held at 0° C.±5° C. for one hour and filtered though a nutsche filter and the product dried in a vacuum oven for at least 30 hours at 30° C.±5° C. to give intermediate (3) as a white crystalline solid (6.93 Kg, 77.9%). Anal. Calcd for C14H22N2O5: C, 56.4; H, 7.43; N, 9.39. Found C, 56.4; H, 7.32; N, 9.48; HRMS (ESI+) Expected for C14H22N2O5, [M+H] 299.1607. Found 299.1613; 1H NMR(CDCl3, 499.8 MHz)δ=7.11 (s, 1H), 4.54 (dd, 1H, J=10.8, 3.6), 3.74 (s, 3H), 3.28-3.19 (m, 1H), 3.00 (s, 6H), 2.97-2.85 (m, 1H), 1.48 (s, 9H); 13C NMR (CDCl3, 125.7 MHz) δ=172.6, 169.5, 150.5, 146.5, 90.8, 82.2, 56.0, 52.3, 42.0, 28.1, 26.3. Mp 127.9° C.
(2S,4S)-1-(1,1-Dimethylethyl)-4-methyl-5-oxo-1,2-pyrrolidinedicarboxylic acid-2-methyl ester (4). A 10-gallon Pfaudler reactor is inerted with nitrogen and charged with ESCAT 142 5% palladium powder on carbon (50% wet, 0.58 Kg wet wt.), intermediate (3) (1.89 Kg, 6.33 mol) and isopropanol (22.4 Kg). The reaction mixture is agitated under a 45-psi hydrogen atmosphere at 45° C. for 18 hrs. The reaction mixture is then cooled to room temperature and filtered though a bed of Celite (0.51 Kg) in a nutsche filter to remove catalyst. The mother liquor is evaporated under reduced pressure to give a thick oil that crystallizes on standing to afford 4 (1.69 Kg, 100%) as a 93:7 diastereomeric mixture. A sample of product mixture is purified by preparative HPLC to give material for analytical data. Anal. Calcd for C12H19NO5: C, 56.0; H, 7.44; N, 5.44. Found C, 55.8; H, 7.31; N, 5.44; MS (ESI+) Expected for C12H19NO5, [M+H] 258.1342. Found 258.1321; 1H NMR (CDCl3, 499.8 MHz) δ=4.44 (m, 1H), 3.72 (s, 3H), 2.60-2.48 (m, 2H), 1.59-1.54 (m, 1H), 1.43 (s, 9H), 1.20 (d, j=6.8 Hz,3H); 13C NMR (CDCl3, 125.7 MHz) δ=175.7, 172.1, 149.5, 83.6, 57.4, 52.5, 37.5, 29.8, 27.9, 16.2. Mp 89.9° C.
(1S,3S)-(4-Hydroxyl-1-hydroxymethyl-3-methyl-butyl)-carbamic acid tert-butyl ester (5). A 50-L reactor is charged with intermediate (4) (3.02 Kg, 11.7 mol), absolute ethanol (8.22 Kg), and MTBE (14.81 Kg). The solution is agitated and cooled to 0° C.±5° C. and sodium borohydride (1.36 Kg, 35.9 mol) is added in small portions so as to maintain reaction temperature at 0° C.±5° C. A small amount of effervescence is observed. The reaction mixture is warmed to 10° C.±5° C. and calcium chloride dihydrate (2.65 Kg) is added portion wise at a slow rate over an hour so as to maintain a reaction temperature of 10° C.±5° C. The reaction is allowed to warm to 20° C.±5° C. over one hour and agitated for an additional 12 hours at 20° C.±5° C. The reaction is cooled to −5° C.±5° C., ice-cold 2N HCl (26.9 Kg) is added at a rate to maintain a reaction temperature of 0° C.±5° C. Agitation is stopped to allow phases to separate. The lower aqueous phase (pH=1) is removed. The reactor is charged with aqueous saturated sodium bicarbonate (15.6 Kg) over five minutes. Agitation is stopped to allow phases to separate. The lower aqueous phase (pH=8) is removed. The reactor is charged with magnesium sulfate (2.5 Kg) and agitated for at least 10 minutes. The mixture is filtered though a nutsche filter, and condensed under reduced pressure to afford intermediate (5) (1.80 Kg, 66%). Anal. Calcd for C11H23NO4: C, 56.6; H, 9.94; N, 6.00. Found C, 56.0; H, 9.68; N, 5.96; HRMS (ESI+) Expected for C11H24NO4, [M+H] 234.1705. Found 234.1703; 1H NMR (CDCl3, 500 MHz)δ=6.34(d, J=8.9 Hz, 1H, NH), 4.51 (t, J=5.8, 5.3 Hz, 1H, NHCHCH2OH), 4.34 (t, J=5.3, 5.3 Hz, 1H, CH3CHCH2OH), 3.46-3.45, (m, 1H, NHCH), 3.28 (dd, J=10.6, 5.3 Hz, NHCHCHHOH), 3.21 (dd, J=10.2, 5.8 Hz, 1H, CH3CHCHHOH), 3.16 (dd, J=10.2, 6.2 Hz, 1H, NHCHCHHOH), 3.12 (dd, J=10.6, 7.1 Hz, 1H, CH3CHCHHOH), 1.53-1.50 (m, 1H, CH3CHCHHOH), 1.35 (s, 9H, O(CH 3)3, 1.30 (ddd, J=13.9, 10.2, 3.7 Hz, 1H, NHCHCHHCH), 1.14 (ddd, J=13.6, 10.2, 3.4 Hz, 1H, NHCHCHHCH), 0.80 (d, J=6.6 Hz, 3H, CH3); 13C NMR (CDCl3, 125.7 MHz) δ 156.1, 77.9, 50.8, 65.1, 67.6, 65.1, 35.6, 32.8, 29.0, 17.1. Mp 92.1° C.
(2S,4S)-Methanesulfonic acid 2-tert-butoxycarbonylamino-5-methanesulfonyloxy-4-methyl-pentyl ester (6). A 50 L reactor is charged with a solution of intermediate (5) (5.1 Kg) in isopropyl acetate (i-PrOAc) 11.8 Kg followed by a rinse with an additional 7.9 Kg i-PrOAc. The reaction is cooled to 15° C.±5° C. and triethylamine (TEA) (7.8 Kg) is added while maintaining the set temperature. The reactor is further cooled to 0° C.±5° C. and methanesulfonyl chloride (MsCl) (6.6 Kg) is added to the reaction solution while maintaining the set temperature. The reaction is stirred for a few hours and monitored for completion by HPLC or TLC. The reaction is quenched by the addition of a saturated aqueous bicarbonate solution and the resulting isolated organic phase is washed successively with cold 10% aqueous triethylamine solution, cold aqueous HCl solution, cold saturated aqueous bicarbonate solution, and finally saturated aqueous brine solution. The organic phase is dried, filtered, and concentrated in vacuo below 55° C.±5° C. until a solid/liquid slurry containing intermediate (6) is obtained. The slurry is used crude in subsequent reaction without further characterization.
(3S,5S)-(1-Benzyl-5-methyl-piperidin-3-yl)-carbamic acid tert-butyl ester (7). A 50 L reactor is charged with 9.1 Kg of neat benzylamine. The reactor is brought to 55° C. and a solution of intermediate (6) (8.2 Kg) in 1,2-dimethoxyethane (DME) (14.1 Kg) is added to the reactor while maintaining a temperature of 60° C.±5° C. After complete addition of this solution, the reaction is stirred at 60° C.±5° C. for several hours and monitored for completion by TLC or HPLC. The reaction is cooled to ambient temperature and volatiles (DME) are removed by rotary evaporation under vacuum. The residue is diluted with 11.7 Kg of 15% (v/v) ethyl acetate/hexanes solution and treated, while agitating, with 18.7 Kg of 20% (wt) aqueous potassium carbonate solution. A triphasic mixture is obtained upon settling. The bottom aqueous phase is removed and the middle phase is set aside. The upper organic phase is collected and held for combination with extracts from additional extractions. The isolated middle phase is extracted twice again with 11.7 Kg portions of 15% (v/v) ethyl acetate/hexanes solution, each time combining the extracts with original organic phase. The combined organic extracts are transferred into a rotary evaporator and solvent is removed under vacuum until an oily residue remains. The residue is then purified via large-scale preparative chromatography to afford purified intermediate (7) as an oil.
(3S,5S)-(5-Methyl-piperidin-3-yl)-carbamic acid tert-butyl ester (8). A 40 L pressure vessel is charged with 0.6 Kg 50% wet, solid palladium on carbon (E101, 10 wt. %) under flow of nitrogen. A solution of 3.2 Kg intermediate (7) in 13.7 Kg of absolute ethanol is then charged to the reactor under nitrogen. The reactor is purged with nitrogen and is then pressurized with hydrogen at 45 psi. The reaction is then heated to 45° C. while maintaining a hydrogen pressure of 45 psi. The reaction is monitored by TLC or LC until complete. The reaction is cooled to ambient temperature, vented, and purged with nitrogen. The reactor contents are filtered through a bed of Celite and the solids are washed with 2.8 Kg of absolute ethanol. The filtrate is concentrated by rotary evaporation under vacuum until a waxy solid is obtained to afford intermediate (8): TLC Rf (Silica F254, 70:30 v/v ethyl acetate-hexanes, KMnO4 stain)=0.12; 1H NMR (300 MHz, CDCl3) δ 5.31 (br s, 1H), 3.80-3.68 (m, 1H), 2.92 (d, J=11.4 Hz, 1H), 2.77 (AB quart, JAB=12.0 Hz, Δν=50.2 Hz, 2H), 2.19 (t, J=10.7 Hz, 1H), 1.82-1.68 (m, 2H), 1.54 (br s, 1H), 1.43 (s, 9H), 1.25-1.15 (m, 1H), 0.83 (d, J=6.6 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 155.3, 78.9, 54.3, 50.8, 45.3, 37.9, 28.4, 27.1, 19.2; MS (ESI+) m/z 215 (M+H), 429 (2M+H).
B. Synthesis of 1-Cyclopropyl-7-fluoro-8-methoxy-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid (19)


Intermediate (12): A reactor is charged with a solution of intermediate (11) (1.2 Kg, 7.7 mol, 1.0 eq) in anhydrous toluene (12 L) followed by ethylene glycol (1.8 L, 15.7 mol, 4.2 eq) and solid p-toluenesulfonic acid (120 g, 10 wt. %). The reaction mixture is stirred at ambient temperature for at least 30 minutes and then heated to reflux, collecting the water/toluene azeotrope in a Dean Stark type trap apparatus until the reaction is complete as determined by TLC analysis (15% EtOAc/Hexanes v/v). Upon completion, the reaction is cooled to ambient temperature and poured into an aqueous solution of sodium bicarbonate (6 L). The organic toluene phase was removed and washed with saturated sodium bicarbonate solution (6 L), distilled water (2×6 L), and saturated aqueous brine (6 L). The organic phase was removed and dried over MgSO4, filtered, and evaporated under reduced pressure to afford intermediate (12) as an oil (1.3 Kg, 86%). The material is used without further purification in subsequent reaction steps.
Intermediate (13): A reactor is charged with a solution of intermediate (12) (1.2 Kg, 6.0 mol, 1.0 eq) in anhydrous tetrahydrofuran (12 L) and n-butyllithium (2.5M in hexanes, 2.6 L, 6.6 mol, 1.1 eq) is added at −40° C., while maintaining this temperature throughout the addition. The reaction is stirred for at least one hour at −40° C. and trimethylborate (0.9 L, 7.8 mol, 1.3 eq) is added to the mixture while maintaining the temperature at or below −40° C. The reaction mixture is stirred for at least one hour at −40° C. until complete as determined by TLC analysis (30% EtOAc/Hexanes v/v). The reaction is warmed slightly to −30° C. and acetic acid (3 L) is added slowly. Upon complete addition, water is added (0.5 L) to the reaction and the mixture is allowed to quickly warm to ambient temperature while stirring overnight. Organic solvent is removed from the reaction by distillation under reduced pressure at 45° C. To the reaction residue is added 3-4 volumes of water (6 L) and 30% hydrogen peroxide (0.7 L, 1.0 eq) slowly at ambient temperature with cooling provided to control the exotherm. The reaction is stirred for at least an hour at ambient temperature until complete as determined by TLC (15% EtOAc/Hexanes v/v). The reaction mixture is cooled to 0-5° C. and excess peroxide is quenched with the addition of 10% aqueous sodium bisulfite solution (2 L). The mixture is tested to ensure a negative peroxide result and the reaction is acidified by the addition of 6N HCl (aq) (1.2 L). The reaction is stirred until the hydrolysis reaction is complete as determined by TLC or NMR analysis. The resulting solids are collected by suction filtration to afford intermediate (13) as a yellow solid (1.0 Kg, 79%).
Intermediate (14): A reactor is charged with intermediate (13) (0.53 Kg, 3.0 mol, 1.0 eq) and dissolved in dry toluene (2.7 Kg, 3.1 L). To this solution is added dimethylsulfate (0.49 Kg, 3.9 mol, 1.30 eq) followed by solid potassium carbonate (0.58 Kg, 4.2 mol, 1.4 eq). The reaction mixture is heated to reflux and held for at least 1 hour until complete as determined by HPLC. During this time, vigorous gas evolution is observed. The reaction is then cooled to ambient temperature and diluted with distilled water (3.2 L) along with 30% NaOH (aq) (0.13 Kg, 0.33 eq). The aqueous phase is separated and the remaining toluene phase is extracted twice more with distilled water (3.2 L) combined with 30% NaOH (aq) (0.13 Kg, 0.33 eq), removing the aqueous phase each time. The organic upper phase is concentrated by distillation in vacuo (<100 mbar) at approximately 40° C. until a concentrated toluene solution is achieved. The resulting solution is cooled to ambient temperature, checked for quality and yield by HPLC, and carried forward to the next step in the synthesis without further purification (theoretical yield for intermediate (14) assumed, 0.56 Kg).
Intermediate (15a,b): A reactor is charged with 1.8 Kg (2.1 L) anhydrous toluene along with sodium hydride (0.26 Kg, 6.6 mol, 2.20 eq) as a 60 wt. % dispersion in mineral oil. To this mixture is added (0.85 Kg, 7.2 mol, 2.4 eq) diethylcarbonate as the reaction mixture is heated to 90° C. over 1 hour. A solution of intermediate (14) (˜1.0 eq) in toluene from the previous step is added to the reaction while maintaining a temperature of 90° C.±5° C. Gas evolution can be observed during this addition. After complete addition, the reaction is stirred for at least 30 minutes or until complete as determined by HPLC analysis. Upon completion, the mixture is cooled to ambient temperature and diluted with 10 wt. % aqueous sulfuric acid (3.8 Kg, 3.9 mol, 1.3 eq) with agitation. The phases are allowed to separate and the lower aqueous phase is removed. The remaining organic phase is concentrated in vacuo (<100 mbar) at approximately 40° C. until a concentrated toluene solution is achieved. The resulting solution is cooled to ambient temperature and carried forward to the next step in the synthesis without further purification (theoretical yield for intermediate (15a,b) assumed, 0.85 Kg).
Intermediate (16a,b; 17a,b): A reactor is charged with a solution of intermediate (15a,b) (0.85 Kg, ˜3.0 mol, ˜1.0 eq) in toluene from the previous step. To the reactor is then added dimethylformamide-dimethylacetal (0.54 Kg, 4.5 mol, 1.5 eq) and the resulting solution is heated to reflux temperature (˜95-105° C.). The lower boiling solvent (methanol from reaction) is allowed to distill off while the temperature is maintained at ≧90° C. Heating is continued for at least 1 hour or until complete as determined by HPLC analysis. Upon completion, the reaction containing the mixture of intermediate (16a,b), is cooled to ambient temperature and toluene (1.8 Kg, 2.1 L) along with cyclopropylamine (0.21 Kg, 3.6 mol, 1.2 eq) are added to the reaction. The reaction is stirred at ambient temperature for at least 30 minutes until complete as determined by HPLC. Upon completion, the reaction is diluted with 10 wt. % aqueous sulfuric acid (2.9 Kg, 3.0 mol, 1.0 eq) with agitation, and the phases are then allowed to separate. The aqueous phase is removed and the organic phase is concentrated under reduced pressure (<100 mbar) at approximately 40° C. by distillation. When the desired concentration is achieved, the solution is cooled to ambient temperature and the toluene solution containing the mixture of intermediate (17a,b) is carried forward to the next step in the synthesis without further purification (theoretical yield for intermediate (17a,b) assumed, ˜1.1 Kg).
Intermediate (18): A reactor is charged with a solution of the mixture of intermediate (17a,b) (˜4.7 Kg, ˜3.0 mol) at ambient temperature. To the reactor is added N,O-bis(trimethylsilyl)acetamide (0.61 Kg, 3.0 mol, 1.0 eq) and the reaction is heated to reflux temperature (˜105-115° C.) for at least 30 minutes or until complete as determined by HPLC analysis. If not complete, an additional amount of N,O-bis(trimethylsilyl)acetamide (0.18 Kg, 0.9 mol, 0.3 eq) is added to the reaction to achieve completion. Upon completion, the reaction is cooled to below 40° C. and organic solvent is removed under reduced pressure (<100 mbar) at approximately 40° C. by distillation until a precipitate is formed. The reaction is cooled to ambient temperature and the precipitated solids are isolated by suction filtration and washed with distilled water twice (1×1.8 L, 1×0.9 L). The solid is dried to afford intermediate (18) as a white solid (0.76 Kg, 82%). The material is used without further purification in the next reaction step.
Intermediate (19): A reactor is charged with solid intermediate (18) (0.76 Kg, ˜2.5 mol, ˜1.0 eq) at ambient temperature followed by ethanol (5.3 Kg, 6.8 L) and 32 wt. % aqueous hydrochloric acid (1.1 Kg, 10 mol). The reaction mixture is brought to reflux temperature (76-80° C.) during which time the mixture first becomes homogeneous and later becomes heterogeneous. The mixture is heated at reflux for at least 5 hours or until complete as determined by TLC analysis (15% EtOAc/Hexanes v/v). Upon completion, the reaction is cooled to 0° C.±5° C. and the precipitated solid is isolated by filtration and washed with distilled water (1.7 Kg) followed by ethanol (1.7 Kg). The isolated solid is dried to afford intermediate (19) as a white solid (0.65 Kg, ˜95%). 1H NMR (CDCl3, 300 MHz) δ (ppm): 14.58 (s, 1H), 8.9 (s, 1H), 8.25 (m, 1H), 7.35 (m, 1H), 4.35 (m, 1H), 4.08 (s, 3H), 1.3 (m, 2H), 1.1 (m, 2H) 19F NMR (CDCl3+CFCl3, 292 MHz) δ (ppm): −119. HPLC: 99.5% by area.
C. Synthesis of borone ester chelate of 1-Cyclopropyl-7-fluoro-8-methoxy-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid (20)

A reactor is charged with boron oxide (2.0 Kg, 29 mol) followed by dilution with glacial acetic acid (8.1 L, 142 mol) and acetic anhydride (16.2 L, 171 mol). The resulting mixture is heated to reflux temperature for at least 2 hours. The reaction contents are cooled to 40° C. and the solid 7-fluoroquinolone acid intermediate (19) (14.2 Kg, 51 mol) is added to the reaction mixture. The mixture is again heated to reflux temperature for at least 6 hours. Reaction progress is monitored by HPLC and NMR. The mixture is cooled to approximately 90° C. and toluene (45 L) is added to the reaction. The reaction is further cooled to 50° C. and tert-butylmethyl ether (19 L) is added to the reaction mixture to bring about precipitation of the product. The mixture is then cooled to 20° C. and the solid product 19 is isolated by filtration. The isolated solids are then washed with tert-butylmethyl ether (26 L) prior to drying in a vacuum oven at 40° C. (50 torr). The product yield obtained for intermediate (20) in this reaction is 86.4%. Raman (cm−1): 3084.7, 3022.3, 2930.8, 1709.2, 1620.8, 1548.5, 1468.0, 1397.7, 1368.3, 1338.5, 1201.5, 955.3, 653.9, 580.7, 552.8, 384.0, 305.8. NMR (CDCl3, 300 MHz) δ (ppm): 9.22 (s, 1H), 8.38-8.33 (m, 1H), 7.54 (t, J=9.8 Hz, 1H), 4.38-4.35 (m, 1H), 4.13 (s, 3H), 2.04 (s, 6H), 1.42-1.38 (m, 2H), 1.34-1.29 (m, 2H). TLC (Whatman MKC18F Silica, 60 Å, 200 μm), Mobile Phase: 1:1 (v/v) CH3CN:0.5N NaCl (aq), UV (254/366 nm) visualization; Rf=0.4-0.5.
D. Coupling of 1-Cyclopropyl-7-fluoro-8-methoxy-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid (20) to (3S,5S)-(5-Methyl-piperidin-3-yl)-carbamic acid tert-butyl ester (8), and synthesis of malate salt of (3S,5S)-7-[3-amino-5-methyl-piperidinyl]-1-cyclopropyl-1,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid (25)

A reactor is charged with solid intermediate (20) (4.4 Kg, 10.9 mol) followed by dilution with a solution of triethylamine (TEA) (2.1 L, 14.8 mol) and piperidine side chain intermediate (8) (2.1 Kg, 9.8 mol) in acetonitrile (33.5 L, 15.7 L/Kg) at room temperature. The resulting mixture is warmed to approximately 50° C. until reaction is judged complete. Reaction progress is monitored by HPLC or reverse phase TLC. When complete, the reaction is cooled to approximately 35° C. and reaction volume is reduced to approximately half by distillation of acetonitrile under vacuum between 0-400 torr. The reactor is then charged with 28.2 Kg of 3.0N NaOH (aq) solution and the temperature is raised to approximately 40° C. Distillation under vacuum is continued between 1-4 hours or until no further distillates are observed. The reaction is then cooled to room temperature and the hydrolysis reaction is monitored by HPLC or reverse phase TLC. Upon completion, the reaction mixture is neutralized to a pH of between 6-8 by adding ˜4-5 Kg of glacial acetic acid. The reactor is then charged with 12.7 Kg (9.6 L) of dichloromethane as an extraction solvent, the mixture is agitated, phases are allowed to separate, and the organic dichloromethane phase is removed. The extraction process is repeated two additional times using 12.7 Kg (9.6 L) of dichloromethane, collecting the lower, organic phase each time. The aqueous phase is discarded and the organic extracts are combined in a single reactor. The reactor contents are heated to 40° C. and the reaction volume is reduced to approximately one half by distillation. The reactor is then charged with 20.2 Kg 6.0N HCl (aq) solution, the temperature is adjusted to 35° C., and agitation is allowed for at least 12 hours to permit the Boc deprotection reaction to occur. The reaction is monitored by HPLC or reverse phase TLC. When complete, agitation is discontinued and the phases are allowed to separate. The lower, organic phase is removed and set aside. The reactor is then charged with 12.7 Kg (9.6 L) of dichloromethane as an extraction solvent, the mixture is agitated, phases are allowed to separate, and the organic dichloromethane phase is removed. The organic extracts are combined and discarded. The remaining aqueous phase is diluted with 18.3 Kg distilled water and the temperature is raised to approximately 50° C. Distillation under vacuum (100-400 torr) is performed to remove residual dichloromethane from the reaction. The pH of the reaction is then adjusted to between 7.8-8.1 using about 9.42 Kg of 3.0N NaOH (aq) solution while keeping the temperature of the reaction below 65° C. The reaction is cooled to 50° C. and the precipitated solids are aged for at least an hour prior to cooling the mixture to room temperature. The solids are isolated by suction filtration and washed twice with 5.2 Kg portions of distilled water. The solids are dried for at least 12 hours with suction and then for an additional 12 hours in a convection oven at 55° C. The yield achieved for intermediate (23) in this example is 3.2 Kg (79%). A reactor is charged with 3.2 Kg solid intermediate (23) and the solids are suspended in 25.6 Kg of 95% ethanol as solvent. To the reactor is then added 1.1 Kg of solid D,L-malic acid (24), and the mixture is heated to reflux temperature (˜80° C.). Distilled water (˜5.7 L) is added to the reaction until a complete solution is achieved and 0.2 Kg of activated charcoal is added. The reaction mixture is passed through a filter to achieve clarification, cooled to 45° C. and held for a period of at least 2 hours to allow crystallization to occur. The reaction mixture is further cooled to 5° C. and the suspended solids are isolated by suction filtration. The solids are then washed with 6.6 KG of 95% ethanol and dried for at least 4 hours with suction under vacuum. The solids are then further dried in a convection oven for at least 12 hours at 45° C. to afford 3.1 Kg of intermediate (24) (70%). NMR (D2O, 300 MHz) δ (ppm): 8.54 (s, 1H), 7.37 (d, J=9.0 Hz, 1H), 7.05 (d, J=9.0 Hz, 1H), 4.23-4.18 (m, 1H), 4.10-3.89 (m, 1H), 3.66 (br s, 1H), 3.58 (s, 3H), 3.45 (d, J=9.0 Hz, 1H), 3.34 (d, J=9.3 Hz, 1H), 3.16 (d, J=12.9 Hz, 1H), 2.65 (dd, J=16.1, 4.1 Hz, 1H), 2.64-2.53 (m, 1H), 2.46 (dd, J=16.1, 8.0 Hz, 1H), 2.06 (br s, 1H), 1.87 (d, J=14.4 Hz, 1H), 1.58-1.45 (m, 1H), 1.15-0.95 (m, 2H), 0.91 (d, J=6.3 Hz, 3H); 0.85-0.78 (m, 2H). TLC (Whatman MKC18F Silica, 60 Å, 200 μm), Mobile Phase: 1:1 (v/v) CH3CN:0.5N NaCl (aq), UV (254/366 nm) visualization. HPLC: Mobile Phase H2O with 0.1% formic acid/Acetonitrile with 0.1% formic acid, gradient elution with 88% H2O/formic acid to 20% H2O/formic acid, Zorbax SB-C8 4.6 mm×150 mm column, Part No. 883975.906, 1.5 ml/min rate, 20 min run time, 292 nm, Detector Model G1314A, S/N JP72003849, Quat Pump Model G1311A, S/N US72102299, Auto Sampler Model G1313A, S/N DE14918139, Degasser Model G1322A, S/N JP73007229; approximate retention time for intermediate (19): 13.0 min; approximate retention time for intermediate (20): 11.6 min; approximate retention time for intermediate (21): 16.3 min; approximate retention time for intermediate (22): 18.2 min; approximate retention time for intermediate (23): 8.6 min; approximate retention time for compound (25): 8.6 min.
………………..
REF
A. ARJONA ET AL: “Nemonoxacin“, DRUGS OF THE FUTURE, vol. 34, no. 3, 1 January 2009 (2009-01-01), page 196, XP55014485, ISSN: 0377-8282, DOI: 10.1358/dof.2009.034.03.1350294
| 2 | * | ANONYMOUS: “TaiGen Announces Positive Data From the Phase II Study of Nemonoxacin (TG-873870) in Community-Acquired Pneumonia“, INTERNET CITATION, [Online] 7 April 2008 (2008-04-07), page 1, XP007919900, Retrieved from the Internet: URL:http://www.taigenbiotech.com/news.html#16> [retrieved on 2011-12-12] |
| 3 | * | ANONYMOUS: “TaiGen Biotechnology Initiates Phase II Trial Of Nemonoxacin For Treatment Of Adult Community Acquired Pneumonia (CAP)“, 20070108, [Online] 8 January 2007 (2007-01-08), page 1, XP007919910, Retrieved from the Internet: URL:http://www.taigenbiotech.com/news.html#11> [retrieved on 2011-12-12] |
| 4 | * | ANONYMOUS: “TaiGen Initiates Phase 1B Trial of a Novel Quinolone Antibiotic“, 20050618, 18 June 2005 (2005-06-18), pages 1-2, XP007919904, |
| 5 | * | See also references of WO2010002415A1 |
| WO2007110834A2 * | Mar 26, 2007 | Oct 4, 2007 | Procter & Gamble | Malate salts, and polymorphs of (3s,5s)-7-[3-amino-5-methyl-piperidinyl]-1-cyclopropyl-1,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid |
| WO2009023473A2 * | Aug 5, 2008 | Feb 19, 2009 | Chi-Hsin Richard King | Antimicrobial parenteral formulation |
| WO2010009014A2 * | Jul 10, 2009 | Jan 21, 2010 | Taigen Biotechnology Co., Ltd. |
|
7-4-2012
|
TREATMENT OF ANTIBIOTIC-RESISTANT BACTERIA INFECTION
|
|
|
4-18-2012
|
Coupling Process For Preparing Quinolone Intermediates
|
|
|
10-19-2011
|
Malate salts, and polymorphs of (3S,5S)-7-[3-amino-5-methyl-piperidinyl]-1-cyclopropyl-1,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid
|
|
|
6-18-2010
|
STEREOSELECTIVE SYNTHESIS OF PIPERIDINE DERIVATIVES
|
|
|
2-19-2010
|
PNEUMONIA TREATMENT
|
|
|
5-6-2009
|
Hydride reduction process for preparing quinolone intermediates
|
|
|
2-13-2009
|
ANTIMICROBIAL PARENTERAL FORMULATION
|
|
|
11-26-2008
|
Coupling process for preparing quinolone intermediates
|
| US8158798 | Oct 27, 2008 | Apr 17, 2012 | Taigen Biotechnology Co., Ltd. | Coupling process for preparing quinolone intermediates |
| US8211909 | Sep 8, 2008 | Jul 3, 2012 | Taigen Biotechnology Co., Ltd. | Treatment of antibiotic-resistant bacteria infection |
| WO2010002965A2 * | Jul 1, 2009 | Jan 7, 2010 | Taigen Biotechnology Co., Ltd. | Pneumonia treatmen |
WO 2007110834
WO 2007110835
WO 2007110836
WO 1999014214
WO 2010077798

1, nemonoxacin; 2, delafloxacin; 3, finafloxacin; 4, zabofloxacin; 5, JNJ-Q2; 6, DS-8587; 7, KPI-10; 8, ozenoxacin; 9, chinfloxacin; 10, ACH-702.

Late-stage success for Sanofi/Regeneron RA drug sarilumab

SARILUMAB
PRONUNCIATION sar il’ ue mab
THERAPEUTIC CLAIM Treatment of rheumatoid arthritis and
ankylosing spondylitis
CHEMICAL NAMES
1. Immunoglobulin G1, anti-(human interleukin 6 receptor α) (human REGN88 heavy
chain), disulfide with human REGN88 light chain, dimer
2. Immunoglobulin G1, anti-(human interleukin-6 receptor subunit alpha (IL-6RA,
membrane glycoprotein 80, CD126)); human monoclonal RGN88 γ1 heavy chain (219-
214′)-disulfide with human monoclonal RGN88 κ light chain dimer (225-225”:228-
228”)-bisdisulfide
MOLECULAR FORMULA C6388H9918N1718O1998S44
MOLECULAR WEIGHT 144.13 kDa
SPONSOR Regeneron Pharmaceuticals, Inc.
CODE DESIGNATION REGN88, SAR153191
CAS REGISTRY NUMBER 1189541-98-7
sarilumab
Sarilumab (REGN88/SAR153191) is a fully-human monoclonal antibody directed against the IL-6 receptor (IL-6R). Sarilumab is a subcutaneously delivered inhibitor of IL-6 signaling, which binds with high affinity to the IL-6 receptor. It blocks the binding of IL-6 to its receptor and interrupts the resultant cytokine-mediated inflammatory signaling.
Sanofi and Regeneron’s investigational rheumatoid arthritis drug sarilumab has succeeded in a late-stage trial.
The year-long Phase III study enrolled 1,200 patients with active, moderate-to-severe RA who were inadequate responders to methotrexate. Patients were randomised to one of three subcutaneous treatment groups, all in combination with MTX and dosed every other week – sarilumab 200mg, 150mg or placebo.http://www.pharmatimes.com/Article/13-11-22/Late-stage_success_for_Sanofi_Regeneron_RA_drug.aspx
Sarilumab is a human monoclonal antibody against the interleukin-6 receptor.
Regeneron and Sanofi are currently co-developing the drug for the treatment of rheumatoid arthritis, for which it is in phase III trials. Development inankylosing spondylitis has been suspended after the drug failed to show clinical benefit over methotrexate in a phase II trial.[1][2]
On May 15th, 2013, both companies announced that 2 new trials were starting (COMPARE and ASCERTAIN) and the first patients had already been enrolled.[3]
On November 22nd, 2013, both companies On May 15th, 2013, both companies announced positive phase 3 results for the RA-MOBILITY trial
- “Statement On A Nonproprietary Name Adopted By The USAN Council: Sarilumab”. American Medical Association.
- http://investor.regeneron.com/releasedetail.cfm?releaseid=590869
- http://en.sanofi.com/Images/33027_20130515_sari_en.pdf
fully human monoclonal antibody directed against the interleukin-6 receptor (IL-6R) in combination with methotrexate (MTX) therapy improved disease signs and symptoms as well as physical functionw while inhibiting progression of joint damage in adults with RA who saw little improvement through MTX therapy alone.
Sarilumab met all three primary endpoints of the 52-week SARIL-RA-MOBILITY Phase III trial by demonstrating clinically relevant and statistically significant improvements compared to the placebo group in the two groups treated with the drug candidate. The trial enrolled about 1,200 patients with active, moderate-to-severe rheumatoid arthritis who were inadequate responders to MTX therapy.
Of patients treated with the 200 mg dose of sarilumab plus MTX, 66% saw improvement in signs and symptoms of RA at 24 weeks, as measured by the American College of Rheumatology score of at-least 20% improvement. The percentage dipped to 58% of sarilumab 150 mg dose patients, and 33% of placebo patients.
Sarilumab 200 mg patients showed the least progression of structural damage after 52 weeks, registering a 0.25 change in the modified Van der Heijde total Sharp score. That contrasts with scores of 0.90 in patients taking sarilumab 150 mg, and 2.78 in the placebo group.
In addition, sarilumab 200 mg patients showed improvement in physical function, as measured by change from baseline in the Health Assessment Question-Disability at week 16. However, the companies did not quantify those results in their announcement. Sanofi and Regeneron said additional analyses of efficacy and safety data from SARIL-RA-MOBILITY will be presented “at a future medical conference.”
“We are encouraged by these Phase III results and the impact sarilumab demonstrated on inhibition of progression of structural damage assessed radiographically in this study,” Tanya M. Momtahen, Sanofi’s sarilumab global project head, said in a statement.
Sarilumab—known as SAR153191 and REGN88—blocks the binding of IL-6 to its receptor and interrupts the resultant cytokine-mediated inflammatory signaling characteristic of RA. Sarilumab was developed using Regeneron’s VelocImmune® antibody technology.
The positive results continue what has been mostly strong success in clinical trials for the partners, whose development collaborations include alirocumab (REGN727), dupilumab (REGN668), and enoticumab (REGN421). Alirocumab is a PCSK9 antibody being evaluated for its ability to manage LDL cholesterol, including in people who do not get to their target LDL levels using statin medicines alone. Dupilumab is an antibody to the receptors for interleukin-4 and interleukin-13 under evaluation in atopic dermatitis and eosinophilic asthma. Enoticumab is a fully human monoclonal antibody to delta-like ligand-4 (Dll4) now in Phase I study for advanced malignancies.
On its own, however, Sanofi’s R&D efforts have shown more mixed results, with the pharma giant earlier this month ending development of cancer drug candidate fedratinib (SAR302503) after it was placed on clinical hold by the FDA following reports that some patients in clinical trials developed symptoms consistent with Wernicke’s encephalopathy. Another cancer compound, iniparib, had its development halted earlier this year after a disappointing Phase III trial.
Daiichi Sankyo anticoagulant edoxaban succeeds in Phase III

Edoxaban, DU-176b
Daiichi Sankyo, APPROVED IN JAPAN as tosylate monohydrate salt in 2011 for the prevention of venous embolism in patients undergoing total hip replacement surgery
for synthesis see….http://www.sciencedirect.com/science/article/pii/S0968089613002642 Bioorganic & Medicinal Chemistry 21 (2013) 2795–2825, see s[pecific page 2808 for description ie 14/31 of pdf
WO 2010071121, http://www.google.com/patents/WO2010071121A1
WO 2007032498
N’-(5-chloropyridin-2-yl)-N-[(1S,2R,4S)-4-(dimethylcarbamoyl)-2-[(5-methyl-6,7-dihydro-4H-[1,3]thiazolo[5,4-c]pyridine-2-carbonyl)amino]cyclohexyl]oxamide
NOV20, 2013
Daiichi Sankyo will file edoxaban on both sides of the Atlantic shortly after the bloodthinner proved as effective and safer than warfarin in a Phase III trial of patients with atrial fibrillation.
The company has presented data on edoxaban, a once-daily oral factor Xa inhibitor, at the American Heart Association meeting in Dallas, from a study involving 21,105 patients across 46 countries. The drug, evaluated in 60mg and 30mg doses, met its primary endpoint of non-inferiority compared to warfarin for the prevention of stroke or systemic embolic events in patients with non-valvular AF.http://www.pharmatimes.com/Article/13-11-20/Daiichi_Sankyo_anticoagulant_edoxaban_succeeds_in_Phase_III.aspx
Edoxaban (INN, codenamed DU-176b, trade name Lixiana) is an anticoagulant drug which acts as a direct factor Xa inhibitor. It is being developed by Daiichi Sankyo. It was approved in July 2011 in Japan for prevention of venous thromboembolisms (VTE) following lower-limb orthopedic surgery.[1]
In animal studies, edoxaban is potent, selective for factor Xa and has good oral bioavailability.[2]
Daichi Sankyo’s edoxaban tosilate is an orally administered
coagulation factor Xa inhibitor that was approved and launched
in Japan for the preventive treatment of venous thromboembolic
events (VTE) in patients undergoing total knee arthroplasty, total
hip arthroplasty, or hip fracture surgery. Edoxaban has been
shown to have a rapid onset of anticoagulant effect due to short
Tmax (1–2 h) after dosing and sustained for up to 24 h post-dose.
Marketed under the brand name Lixiana, it is currently in phase
III studies in the US for the prevention of stroke and systemic embolic
events in patients with atrial fibrillation (AF) and venous
thromboembolism (VTE).
Several Phase II clinical trials have been conducted, for example for thromboprophylaxis after total hip replacement[3] (phase III early results compare well to enoxaparin[4]), and for stroke prevention in patients with atrial fibrillation[5][6].Those papers follow similar recent major trials showing similar results for the other new factor Xa inhibitors, rivaroxaban and apixaban.
A large phase III trial showed that edoxaban was non inferior to warfarin in preventing recurrent venous thromboembolic events with fewer episodes of major bleeding.[7]
- “First market approval in Japan for LIXIANA (Edoxaban)”. Press Release. Daiichi Sankyo Europe GmbH. 2011-04-22.
- Furugohri T, Isobe K, Honda Y, Kamisato-Matsumoto C, Sugiyama N, Nagahara T, Morishima Y, Shibano T (September 2008). “DU-176b, a potent and orally active factor Xa inhibitor: in vitro and in vivo pharmacological profiles”. J. Thromb. Haemost. 6 (9): 1542–9. doi:10.1111/j.1538-7836.2008.03064.x. PMID 18624979.
- Raskob, G.; Cohen, A. T.; Eriksson, B. I.; Puskas, D.; Shi, M.; Bocanegra, T.; Weitz, J. I. (2010). “Oral direct factor Xa inhibition with edoxaban for thromboprophylaxis after elective total hip replacement”. Thrombosis and Haemostasis 104 (3): 642–649. doi:10.1160/TH10-02-0142.PMID 20589317. edit
- “Phase III Trial Finds Edoxaban Outclasses Enoxaparin in Preventing Venous Thromboembolic Events”. 8 Dec 2010.
- Weitz JI, Connolly SJ, Patel I, Salazar D, Rohatagi S, Mendell J, Kastrissios H, Jin J, Kunitada S (September 2010). “Randomised, parallel-group, multicentre, multinational phase 2 study comparing edoxaban, an oral factor Xa inhibitor, with warfarin for stroke prevention in patients with atrial fibrillation”. Thromb. Haemost. 104 (3): 633–41. doi:10.1160/TH10-01-0066.
- Edoxaban versus Warfarin in Patients with Atrial Fibrillation Robert P. Giugliano, M.D., Christian T. Ruff, M.D., M.P.H., Eugene Braunwald, M.D., Sabina A. Murphy, M.P.H., Stephen D. Wiviott, M.D., Jonathan L. Halperin, M.D., Albert L. Waldo, M.D., Michael D. Ezekowitz, M.D., D.Phil., Jeffrey I. Weitz, M.D., Jindřich Špinar, M.D., Witold Ruzyllo, M.D., Mikhail Ruda, M.D., Yukihiro Koretsune, M.D., Joshua Betcher, Ph.D., Minggao Shi, Ph.D., Laura T. Grip, A.B., Shirali P. Patel, B.S., Indravadan Patel, M.D., James J. Hanyok, Pharm.D., Michele Mercuri, M.D., and Elliott M. Antman, M.D. for the ENGAGE AF-TIMI 48 InvestigatorsDOI: 10.1056/NEJMoa1310907
- “Edoxaban versus Warfarin for the Treatment of Symptomatic Venous Thromboembolism”. N. Engl. J. Med. August 2013. doi:10.1056/NEJMoa1306638. PMID 23991658.
- WO 03/000657 pamphlet WO 03/000680 pamphlet WO 03/016302 pamphlet WO 04/058715 pamphlet WO 05/047296 pamphlet WO 07/032498 pamphlet WO 08/129846 pamphlet WO 08/156159 pamphlet
- J Am Chem Soc 1978, 100(16): 5199
Drug formulation , lixiana, edoxaban tosylate monohydrate, CAS 912273-65-5, C24 H30 Cl N7 O4 S . C7 H8 O3 S . H2 O, 738.274
-
N1-(5-chloropyridin-2-yl)-N2-((1S,2R,4S)-4-[(dimethylamino)carbonyl]-2-{[(5-methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-yl)carbonyl]amino}cyclohexyl)ethanediamide p-toluenesulfonic acid monohydrate represented by the following formula (A) (hereinafter, also referred to as compound A) :
-


-
is known as a compound that exhibits an inhibitory effect on activated blood coagulation factor X (FXa), and is useful as a preventive and/or therapeutic drug for thrombotic diseases (Patent Literature 1 to 8).
-
For example, a method comprising mixing the free form of compound A represented by the following formula (B) (hereinafter, also referred to as compound B):
-

-
with p-toluenesulfonic acid or p-toluenesulfonic acid monohydrate, followed by crystallization from aqueous ethanol, is known as a method for obtaining compound A (Patent Literature 1 to 8). These literature documents do not make any mention about adding p-toluenesulfonic acid or p-toluenesulfonic acid monohydrate in a stepwise manner in the step of obtaining compound A from compound B.
Citation ListPatent Literature
-
- Patent Literature 1: International Publication No. WO 03/000657
- Patent Literature 2: International Publication No. WO 03/000680
- Patent Literature 3: International Publication No. WO 03/016302
- Patent Literature 4: International Publication No. WO 04/058715
- Patent Literature 5: International Publication No. WO 05/047296
- Patent Literature 6: International Publication No. WO 07/032498
- Patent Literature 7: International Publication No. WO 08/129846
- Patent Literature 8: International Publication No. WO 08/156159
SIMILAR

OTHER SALTS

Edoxaban hydrochloride
CAS Number: 480448-29-1
Molecular Formula: C24H30ClN7O4S · HCl
Molecular Weight: 584.52 g.mol-1
Edoxaban is reported to be a member of the so-called “Xaban-group” and as such to be a low molecular inhibitor of the enzyme factor Xa, participating in the blood coagulation system. Therefore, edoxaban is classified as an antithrombotic drug and its possible medical indications are reported to be treatment of thrombosis and thrombosis prophylaxis after orthopaedic operations, such as total hip replacement, as well as for stroke prevention in patients with atrial fibrillation, the prophylaxis of the acute coronary syndrome and the prophylaxis after thrombosis and pulmonary embolism.
The IUPAC name for edoxaban is N’-(5-chloropyridin-2-yl)-N-[(15,2^,4S)-4- (dimethylcarbamoyl)-2-[(5-methyl-6,7-dihydro-4H-[l ,3]thiazolo[5,4-c]pyridine-2- carbonyl)amino]cyclohexyl]oxamide. The chemical structure of edoxaban is shown in the formula (1) below:

formula ( 1 ) While Edoxaban is reported to be soluble in strongly acidic aqueous solutions, its solubility is considered to be very low in neutral or alkaline aqueous media. EP 2 140 867 A 1 claims an edoxaban-containing pharmaceutical composition comprising a water-swelling additive and/or a sugar alcohol. Further, it is alleged that compositions comprising lactose or cornstarch do not have good dissolution properties. The claimed pharmaceutical compositions in EP 2 140 867 Al are considered to show good dissolution properties in a neutral aqueous medium as well. Tablets comprising said composition were produced by wet granulation. However, it turned out that prior art pharmaceutical formulations comprising edoxaban being suitable for oral administration are still improvable with regards to dissolution rate and bioavailability. Further, stability and content uniformity of the known formulations could be improved. Further, due to the intolerance of many people to sugar alcohol(s), such as sorbitol, the use of sugar alcohol(s) should be avoided.
Amgen: Phase III Melanoma treatment, Talimogene Laherparepvec Improves Survival
 |
|
|---|---|
| Transmission electron micrograph of an unmodified herpes simplex virus |
Talimogene Laherparepvec
Amgen Presents Interim Overall Survival Data From Phase 3 Study Of Talimogene Laherparepvec In Patients With Metastatic Melanoma
T-VEC was engineered from herpes simplex 1 (HSV-1), a relatively innocuous virus that normally causes cold sores. A number of genetic modifications were made to the virus in order to:
- Attenuate the virus (so it can no longer cause herpes)
- Increase selectivity for cancer cells (so it destroys cancer cells while leaving healthy cells unharmed)
- Secrete the cytokine GM-CSF (a protein naturally secreted in the body to initiate an immune response)
T-VEC has a dual mechanism of action, destroying cancer both by directly attacking cancer cells and also by helping the immune system to recognize and destroy cancer cells. T-VEC is injected directly into a number of a patient’s tumors. The virus invades both cancerous and healthy cells, but it is unable to replicate in healthy cells and thus they remain unharmed. Inside a cancer cell, the virus is able to replicate, secreting GM-CSF in the process. Eventually overwhelmed, the cancer cell lyses (ruptures), destroying the cell and releasing new viruses, GM-CSF, and an array of tumor-specific antigens (pieces of the cancer cell that are small enough to be recognized by the immune system).
The GM-CSF attracts dendritic cells to the site. Dendritic cells are immune cells that process and present antigens to the immune system so that the immune system can then identify and destroy whatever produced the antigen. The dendritic cells pick up the tumor antigens, process them, and then present them on their surface to cytotoxic (killer) T cells. Now the T cells are essentially “programmed” to recognize the cancer as a threat. These T cells lead an immune response that seeks and destroys cancer cells throughout the body (eg, tumors and cancer cells that were not directly injected with T-VEC).

In this way, T-VEC has both a direct effect on injected tumors and a systemic effect throughout the entire body. Because the adaptive immune system “remembers” a target once it has been identified, there is high likelihood that the effect of an oncolytic virus like T-VEC will be durable (eg, prevent relapse). And it is for this reason that T-VEC does not need to be injected into every tumor, just a few in order to start the immune process.
Clinical efficacy in unresectable melanoma has been demonstrated in Phase II and Phase III clinical trials.
The Phase II clinical trial was published in the Journal of Clinical Oncology in 2009. 50 patients with advanced melanoma (most of whom had failed previous treatment) were treated with T-VEC. The overall response rate (patients with a complete or partial response per RECIST criteria) was 26% (16% complete responses, 10% partial responses). Another 4% of patients had a surgical complete response, and another 20% had stable disease for at least 3 months. On an extension protocol, 3 more patients achieved complete responses, and overall survival was 54% at 1 year and 52% at 2 years—demonstrating that responses to T-VEC are quite durable.
Consistent with other immunotherapies, some patients exhibited initial disease progression before responding to therapy because of the time it takes to generate the full immune response. Responses were seen in both injected and uninjected tumors (including those in visceral organs), demonstrating the systemic immunotherapeutic effect of T-VEC. Treatment was extremely well tolerated, with only Grade 1 or 2 drug-related side effects, the most common being mild flu-like symptoms.

|

|
Amgen announced the initial results of the Phase III OPTiM trial on Mar. 19, 2013. This global, randomized, open-label trial compared T-VEC with subcutaneously administered GM-CSF (2:1 randomization) in 430 patients with unresectable stage IIIB, IIIC or IV melanoma. The primary endpoint was durable response rate (DRR), defined as a complete or partial tumor response lasting at least 6 months and starting within 12 months of treatment.

T-VEC was proven to offer superior benefits in metastatic melanoma. DRR was achieved in 16% of patients receiving T-VEC compared with only 2% in the GM-CSF control group (P<.0001). The greatest benefit was seen in patient with stage IIIB or IIIC melanoma, with a 33% DRR vs 0% with GM-CSF. The objective response rate (any response) with T-VEC was 26%, with an impressive 11% of patients experiencing a complete response (complete disappearance of melanoma throughout the body). This demonstrated once again that T-VEC has a systemic immune effect that destroys distant, uninjected tumors. According to Financial Times one of the investigators involved questioned the ethics of the trial design, as the control arm received subcutaneous GM-CSF instead of standard care

A trend toward improved survival with T-VEC was observed in a pre-specified interim analysis of this endpoint, with the final survival data (event-driven) expected in late 2013. At the interim analysis, T-VEC was associated with a 21% reduced risk of death. The most common side effects with T-VEC were fatigue, chills, and fever. No serious side effect occurred in more than 3% of patients in either arm of the study.
The investigators concluded that “T-VEC represents a novel potential [treatment] option for melanoma with regional or distant metastases.” The success of T-VEC in the OPTiM trial represents the first Phase III proof of efficacy for a virus-based oncolytic immunotherapy.
Gene Therapy for Melanoma: Progress and Perspectives

FIGURE 1.
Schematic representation of the wild type counterpart of the typically used recombinant viral vectors. (A) Gammaretroviruses and (B) lentiviruses share similar structures, but differ greatly in their genomes and their impact on cellular function. Gag, pro, pol and env genes encode structural proteins of the capsid, protease, reverse transcriptase and envelope proteins, respectively. The additional lentiviral genes perform regulatory functions as well as alter cellular function. (C) The serotype 5 adenovirus has a protein capsid (non-enveloped) and a large, complex genome that encodes critical genes for viral replication (E1a, E1b) as well as structural and functional genes that regulate both viral and cellular activities.
Introduction
Gene therapy, the therapeutic transfer of genetic information to a target cell, continues to be a promising alternative in the fight against cancer. In the case of melanoma, the use of an experimental treatment is justified since this disease is incurable in its advanced stages. Is gene therapy a viable option for the treatment of melanoma patients? In this chapter, we will attempt to answer this question by exploring the intersection between the technology of gene therapy and the biology of melanoma, a point at which opportunities for intervention are revealed.
Gene Therapy for Melanoma: Progress and Perspectives
Bryan E. Strauss1 and Eugenia Costanzi-Strauss2
[1] Cancer Institute of Sao Paulo, University of Sao Paulo School of Medicine, Brazil
[2] University of Sao Paulo, Biomedical Sciences Institute, Brazil

……………….
Enbrel (etanercept), Biosimilar innovator drug companies scrambling to copy

Enbrel (etanercept)

http://www.biosimilarnews.com/enbrel-patent-in-the-us
Biosimilars are protein products that are sufficiently similar to a biopharmaceutical already approved by a regulatory agency. Several biotechnology companies and generic drug manufacturers in Asia and Europe are developing biosimilars of tumor necrosis factor inhibitors and rituximab. A biosimilar etanercept is already being marketed in Colombia and China. In the US, several natural source products and recombinant proteins have been approved as generic drugs under Section 505(b)(2) of the Food, Drug, and Cosmetic Act. However, because the complexity of large biopharmaceuticals makes it difficult to demonstrate that a biosimilar is structurally identical to an already approved biopharmaceutical, this Act does not apply to biosimilars of large biopharmaceuticals. Section 7002 of the Patient Protection and Affordable Care Act of 2010, which is referred to as the Biologics Price Competition and Innovation Act of 2009, amends Section 351 of the Public Health Service Act to create an abbreviated pathway that permits a biosimilar to be evaluated by comparing it with only a single reference biological product.
Amgen announced the issuance of U.S. Patent No. 8,063,182 related to Enbrel (etanercept).owned by Hoffmann-la roche and licensed to Amgen (exp2028) VIA immunex
A biosimilar etanercept, manufactured in China by CP Guojian Pharmaceutical Co., Ltd. (Shanghai), is already being marketed in China as Yisaipu [3] and in Colombia as Etanar [4]. Several biotechnology companies in Asia are also developing biosimilar versions of tumor necrosis factor inhibitors. Protalix Biotherapeutics, Inc. (Carmiel, Israel) is developing a biosimilar etanercept that is expressed in plant cells [5]. Mycenax Biotech (Taiwan) has completed early-phase clinical trials of a biosimilar etanercept in Southeast Asia: a phase I trial among 24 healthy subjects in South Korea and a phase I/II trial that enrolled 18 patients with rheumatoid arthritis in Taiwan [6]. Avesthagen (Bangalore, India) has received a patent from the Indian patent office for a biosimilar etanercept [7]. In South Korea, both Celltrion (Yeonsu-gu Incheon City) and Aprogen (Daejeon) are developing a biosimilar of infliximab [8] and LG Life Sciences (Seoul) is developing biosimilars of both etanercept and infliximab to treat rheumatoid arthritis and other inflammatory diseases [9].

Drug developers:
- Avesthagen: Avent™ in clinical studies
![]()
read this doc
http://www.avesthagen.com/docs/020910pr.pdf
…………………………………………………………………………………..
- BioXpress Therapeutics: Biosimilar in active development

http://www.bioxpress.com/pipeline/
………………………………………………………………………………………
- Cipla:Etacept, Launches biosimilar in India on April 17, at a price of Rs. 6,150 ($113.43), 30% less than the innovator product.

- read this
………………………………………………………………………………….
- Hanwha Chemical: HD203 “scheduled for launch,” company states on its website without including a date, following submission for marketing approval to South Korea’s Korea Ministry of Food and Drug Safety following completion of Phase I and Phase III trials. Hanwha has said it will seek a partner to commercialize HD203 and a biosimilar for Herceptin (trastuzumab).
- http://www.thepharmaletter.com/file/105028/merck-co-links-with-koreas-hanwha-on-biosimilar-of-enbrel.html
………………………………………………………………………………
- LG Life Sciences: LBEC0101 completed Phase I trial in South Korea
![]()
http://www.lgls.co.kr/rd/pipeline.jsp
………………………………………………………………………………
- Mycenax Biotech: TuNEX in Phase III clinical trials in Japan and South Korea
…………………………………………………………………………..
- Protalix Biotherapeutics: PRX-106 in preclinical studies
http://www.protalix.com/product-development/prx-106.asp
![]()
…………………………………………………………………………………
- Shanghai CP Goujian Pharmaceutical: Etanar®, marketed in Colombia; Yisaipu, marketed in China
……………………………………………………………………………………………..
Recently discontinued effort: Merck & Co. and Hanwha Chemical: Hanwha disclosed December 18, 2012, that Merck terminated agreement to develop and manufacture the biosimilar MK-8953, now called HD203, as well as market it in all countries except South Korea and Turkey, an up to $720 million deal signed June 2011.1
Nature and indication: Tumor necrosis factor (TNF) blocker for rheumatoid arthritis, polyarticular Juvenile Idiopathic Arthritis (JIA) in patients aged two years or older; psoriatic arthritis; ankylosing spondylitis; and plaque psoriasis
2012 sales: $7.963 billion (includes $4.236 billion Amgen + $3.737 billion Pfizer). Amgen markets Enbrel in U.S. and Canada under an agreement with Pfizer set to expire October 31, 2013
Patent status: Patents set to expire in EU in 2015; in U.S., 2019, 2023, 2028, and 2029
Etanercept is a fusion protein produced by recombinant DNA, which fuses a soluble human TNF receptor with an IgG1 antibody. This modified protein works by blocking TNF activity, thereby reducing their ability to cause an inflammatory response as well as severe, chronic pain and discomfort to patients. The fusion protein is protected by five different molecule Key patent families (Fig 2) and are all considered to be a constraint to generic entry until expiry. Although the patent families are owned by different patentees, Amgen have entered into licensing agreements with all parties allowing them sole distributing and marketing rights of Enbrel®.
- Public Health Service Act Sec. 262. Regulation of biological products.http://www.fda.gov/RegulatoryInformation/Legislation/ucm149278.htm
- Woodcock J, Griffin J, Behrman R, Cherney B, Crescenzi T, Fraser B, Hixon D, Joneckis C, Kozlowski S, Rosenberg A, Schrager L, Shacter E, Temple R, Webber K, Winkle H. The FDA’s assessment of follow-on protein products: a historical perspective. Nat Rev Drug Discov. 2007;6:437–442. doi: 10.1038/nrd2307. [PubMed] [Cross Ref]
- Yisaipu. http://www.cpgj-pharm.com/en/product_patient.asp?proid=22&action=intro
- Rondon F, Bautista A, Salazar JC, Casas N, Santos P, Vargas F, Marquez J. Etanar therapy in real-life patients with rheumatoid arthritis [abstract]Arthritis Rheum. 2010;62(Suppl 10):1811.
- Pipeline products. http://www.protalix.com/pipeline_products.html
- Biosimilar TuNEX® completes Phase I/II clinical trial in Taiwan, Phase I in Korea. http://www.mycenax.com.tw/webe/html/02news/news_show.aspx?page=1
- Singh K. Avesthagen gets patent for Enbrel biosimilar. Economic Times. 2010.
- Korea’s Celltrion and Aprogen in race to sell biosimilars in Japan.http://sis.windhover.com/buy/abstract.php?id=28101102003
- LG Life Sciences’ Pipeline Overview. http://thinklgls.com/rnd/pipeline
- Dr. Reddy’s Marketed Pharmaceutical Products.http://www.drreddys.com/products/bio_mproducts.html#
- TL011 in severe, active rheumatoid arthritis patients.http://clinicaltrials.gov/ct2/show/NCT01123070
- GP2013 in the treatment of RA patients refractory to or intolerant of standard therapy. http://www.clinicaltrials.gov/ct2/show/NCT01274182
- View Opportunities. http://www.sourcegenerics.com/viewAllListing.asp
- Rudick RA, Simonian NA, Alam JA, Campion M, Scaramucci JO, Jones W, Coats ME, Goodkin DE, Weinstock-Guttman B, Herndon RM, Mass MK, Richert JR, Salazar AM, Munschauer FE, Cookfair DL, Simon JH, Jacobs LD. Incidence and significance of neutralizing antibodies to interferon beta-1a in multiple sclerosis. Multiple Sclerosis Collaborative Research Group (MSCRG)Neurology. 1998;50:1266–1272. [PubMed]
- Casadevall N, Nataf J, Viron B, Kolta A, Kiladjian JJ, Martin-Dupont P, Michaud P, Papo T, Ugo V, Teyssandier I, Varet B, Mayeux P. Pure red-cell aplasia and antierythropoietin antibodies in patients treated with recombinant erythropoietin. N Engl J Med. 2002;346:469–475. doi: 10.1056/NEJMoa011931. [PubMed] [Cross Ref]
- Schellekens H, Jiskoot W. Eprex-associated pure red cell aplasia and leachates. Nat Biotechnol. 2006;24:613–614. doi: 10.1038/nbt0606-613.[PubMed] [Cross Ref]
- Bennett CL, Luminari S, Nissenson AR, Tallman MS, Klinge SA, McWilliams N, McKoy JM, Kim B, Lyons EA, Trifilio SM, Raisch DW, Evens AM, Kuzel TM, Schumock GT, Belknap SM, Locatelli F, Rossert J, Casadevall N. Pure red-cell aplasia and epoetin therapy. N Engl J Med. 2004;351:1403–1408. doi: 10.1056/NEJMoa040528. [PubMed] [Cross Ref]
- Federal Food, Drug, and Cosmetic Act (FD&C Act) SEC. 505. [21 USC §355] New Drugs.http://www.fda.gov/RegulatoryInformation/Legislation/FederalFoodDrugandCosmeticActFDCAct/FDCActChapterVDrugsandDevices/ucm108125.htm
- Committee for Medicinal Products for Human Use. Guideline on similar biological medicinal products. London: European Medicines Agency; 2005.
- Committee for Medicinal Products for Human Use. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. London: European Medicines Agency; 2006.
- European Medicines Agency. Work plan for the biosimilar medicinal products working party (BMWP) 2011. London: European Medicines Agency; 2010.
- H. R. 3590–686. Patient Protection and Affordable Care Act. Title VII– Improving Access to Innovative Medical Therapies. Subtitle A–Biologics Price Competition and Innovation. Sec. 7002. Approval Pathway for Biosimilar Biological Products.http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/UCM216146.pdf
- Implementation of the Biologics Price Competition and Innovation Act of 2009.http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/ucm215089.htm
see details of etanercept

Etanercept
ATC (Anatomical Therapeutic Chemical Classification)
L04AA11,L04AB01
CAS registry number (Chemical Abstracts Service)
0185243-69-0
Chemical Formula
C2224-H3472-N618-O701-S36
Molecular Weight
51238
Therapeutic Categories
Immunosuppressant
Disease-modifying antirheumatic drug, DMARD
Biological response modifier, BRM
Anti-inflammatory agent
Tumor necrosis factor alpha (TNF-α) inhibitor
Chemical Name
Dimeric fusion protein consisting of the extracellular ligand-binding portion of the human 75 kilodalton (p75) tumor necrosis factor receptor (TNFR) linked to the Fc portion of human IgG1
is made from the combination of two naturally occurring soluble human 75-kilodalton TNF receptors linked to an Fc portion of an IgG1. The effect is an artificially engineered dimeric fusion protein.
Sandoz launches Phase III clinical trial for biosimilar etanercept
Trial expected to support registration in the U.S. and European Union
• Sandoz continues to advance biosimilar pipeline with seven Phase III trials across five molecules
• Global program underscores Sandoz’s leadership in biosimilarsHolzkirchen, Germany, June 24, 2013 – Sandoz, the global leader in biosimilars, announced it has initiated a major Phase III clinical trial with its biosimilar version of etanercept (Amgen’s Enbrel®).
• Sandoz continues to advance biosimilar pipeline with seven Phase III trials across five molecules
• Global program underscores Sandoz’s leadership in biosimilarsHolzkirchen, Germany, June 24, 2013 – Sandoz, the global leader in biosimilars, announced it has initiated a major Phase III clinical trial with its biosimilar version of etanercept (Amgen’s Enbrel®).
Etanercept was the first biologic approved in the US for the treatment of Rheumatoid arthritis (RA), it was then later approved by the FDA for other forms of arthritis and psoriasis. Patents in families 1992-10-08 and 1999-04-19 protect the aforementioned indications, as well as the use of Etanercept as adjunctive therapy with Methotrexate for RA. Patents in the family protecting the market authorised indications are considered to constrain biosimilar entry for the indicated use, however it would be possible for generic manufacturers to ‘carve out’ market authorised indications thus circumventing these constraining patents prior to expiry.
Read more at
Read more at
http://www.drugs.com/news/novartis-begins-enbrel-phase-iii-trial-45414.html
| Etanercept (trade name Enbrel) is a biopharmaceutical that treats autoimmune diseases by interfering with tumor necrosis factor (TNF; a soluble inflammatory cytokine) by acting as a TNF inhibitor. It has U.S. F.D.A. approval to treat rheumatoid, juvenile rheumatoid andpsoriatic arthritis, plaque psoriasis and ankylosing spondylitis. TNF-alpha is the “master regulator” of the inflammatory (immune) response in many organ systems. Autoimmune diseases are caused by an overactive immune response. Etanercept has the potential to treat these diseases by inhibiting TNF-alpha. Etanercept is a fusion protein produced by recombinant DNA. It fuses the TNF receptor to the constant end of the IgG1 antibody. First, the developers isolated the DNA sequence that codes the human gene for soluble TNF receptor 2, which is a receptor that binds to tumor necrosis factor-alpha. Second, they isolated the DNA sequence that codes the human gene for the Fc end of immunoglobulin G1 (IgG1). Third, they linked the DNA for TNF receptor 2 to the DNA for IgG1 Fc. Finally, they expressed the linked DNA to produce a protein that links the protein for TNF receptor 2 to the protein for IgG1 Fc.The prototypic fusion protein was first synthesized and shown to be highly active and unusually stable as a modality for blockade of TNF in vivo in the early 1990s by Bruce A. Beutler, an academic researcher then at the University of Texas Southwestern Medical Center at Dallas, and his colleagues.[2][3][4] These investigators also patented the protein, selling all rights to its use to Immunex, a biotechnology company that was acquired by Amgen in 2002.It is a large molecule, with a molecular weight of 150 kDa., that binds to TNFα and decreases its role in disorders involving excess inflammation in humans and other animals, including autoimmune diseases such as ankylosing spondylitis, juvenile rheumatoid arthritis, psoriasis, psoriatic arthritis, rheumatoid arthritis, and, potentially, in a variety of other disorders mediated by excess TNFα.In North America, etanercept is co-marketed by Amgen and Pfizer under the trade name Enbrel in two separate formulations, one in powder form, the other as a pre-mixed liquid. Wyeth is the sole marketer of Enbrel outside North America excluding Japan whereTakeda Pharmaceuticals markets the drug.Etanercept is an example of a protein-based drug created using the tools of biotechnologyand conceived through an understanding afforded by modern cell biology.  |

Figure 2: Molecule Key Patents landscape
International Market
Patents protecting the various technologies of the Etanercept molecule (Fig. 2) across all five families have now expired in Europe, Canada and Australia. In Europe, SPCs and paediatric extensions were granted based on the EP0418014 (1989-09-05) and EP0939121 (1989-09-12) however the last of the paediatric extensions expired in early August, 2015. Finland has been granted a national patent disclosing the Etanercept sequence in the family with priority US40324189A (1989-09-05), which would constrain generic entry until April, 2020. Cyprus has also received a five year patent extension on a national patent set to expire in mid-2016 and would be a constraint for biosimilars entering the market there.
Although the Etanercept molecule is no longer protected in the European, Canadian or Australian markets, no biosimilar has been approved in these major markets suggesting the difficulty of developing a biosimilar which complies with the stringent regulatory pathways in place. Having said that, Merck and Samsung Bioepis (a joint venture from electronics giant Samsung and biotech firm Biogen Idec) has submitted their Etanercept biosimilar candidate SB4 to the EMA, which is currently awaiting review. If approved, it is expected that they will obtain further approval in other territories where Etanercept is no longer protected. With the regulatory approval pathways differing from country to country, Etanercept biosimilars have been approved in smaller markets including India, China and South Korea.
US Market
In the US, the ‘molecule’ patents protecting active ingredient Etanercept have all expired aside from US8,063,182 (‘182) and US8,163,522 (‘522) members from priority CH331989 (1989-09-12) owned by Roche (exclusively licensed to Amgen), which are set to expire in 2028 and 2029, respectively. These patents members disclose a portion of the Etanercept sequence, so are considered to constrain biosimilar entry until expiry. The members are continuation patents filed from US5,610,279 (another member of the same family) and while they were both filed in May, 1995, were not issued until 2011 (‘182) and 2012 (‘522). Under the 35 U.S. Code § 154, these patents received 17 year patent term from the issuing date. Since these patents were applied for in 1995 during the transitional period of the TRIPS agreement, they were not published by the USPTO until they were issued. This situation often gives rise to the term ‘submarine patents’.
Currently there is no system to link relevant patents to biologic drugs in the US as with small molecule drugs (Orange Book) which makes filing biosimilars in the US a convoluted process. While the FDA are currently working on an equivalent to the Orange Book, the ‘Purple book’, companies wishing to develop biosimilars in the US need to do considerable patent landscape searching in order to avoid infringement of any patents potentially protecting the biologic drug. In the case of US member ‘182 and ‘522, upon inspection these patents are clearly relevant to Enbrel®, however without a registry there is no easy way of making this link. The patents have been flagged in the Key Patent module in Ark due to SPCs and paediatric extensions on the equivalent EP0939121 member and litigation in the US (see below).
Currently, biologic drugs approved in the US receive a 12 year data exclusivity period and in Europe, an 8 year data exclusivity period with additional 2 year market exclusivity, starting from the market authorisation date. Enbrel® was approved in 1998 and 2000, in the US and Europe, respectively and data exclusivity protection has therefore now expired.
Development of biosimilars takes considerably longer than generic medicine making it a costly venture for generic pharmaceutical manufacturers. According to Amgen, Enbrel® was protected by US5395760 (‘760) and US5605690 (‘690) members from priority 1989-09-05 which were set to lose patent protection in 2012 and 2014, respectively. In 2004, Sandoz began developing GP2015 a biosimilar equivalent of Etanercept, investing millions of dollars in the hope that they would be ready to launch by the time all the patent protection for Enbrel® expired. Currently, GP2015 is in Phase III study in the US and European Union for patients with moderate to severe chronic plaque-type psoriasis with respect to PASI 75 response rate at Week 12.
In June 2013, Sandoz filed a suit against Amgen and Roche in the US District Court for the Northern District of California seeking declaratory judgment of non-infringement, invalidity and unenforceability of the ‘182 and ‘522 patents. Sandoz claimed a ‘case of controversy’ regarding the patents, as their research and development was based on the understanding that ‘760 and ‘690 patents members were protecting Enbrel®. With the issuing of ‘182 and ‘522 patents this has essentially delayed the prospect of an Etanercept biosimilar from entering the US market until 2029.
Amgen and Roche sought a dismissal of the proceeding due to lack of subject matter jurisdiction, which was granted. Although Sandoz appealed the decision, the Court of Appeals affirmed the dismissal, since there was no real and immediate controversy as Sandoz had not yet filed an FDA application, and they had based their suit on future events and were not able to establish “real and immediate injury or threat of future injury.”

Soliris Gets Thumbs Up From EMA’s COMP


eculizumab
CAS number 219685-50-4
Alexion’s Soliris® (eculizumab) Receives Positive Opinion from the Committee for Orphan Medicinal Products for Treatment of Neuromyelitis Optica (NMO)
Alexion Pharmaceuticals, Inc. (Nasdaq: ALXN) today announced that Soliris® (eculizumab), the company’s first-in-class terminal complement inhibitor, has received a positive opinion for orphan medicinal product designation from the Committee for Orphan Medicinal Products (COMP) of the European Medicines Agency (EMA) for the treatment of neuromyelitis optica (NMO), a life-threatening, ultra-rare neurological disorder. The positive opinion of the COMP has now been forwarded to the European Commission for final approval and publication in the community register. Soliris is not approved in any country for the treatment of patients with NMO
http://www.pharmalive.com/soliris-gets-thumbs-up-from-emas-comp
Soliris is a formulation of eculizumab which is a recombinant humanized monoclonal IgG2/4;κ antibody produced by murine myeloma cell culture and purified by standard bioprocess technology. Eculizumab contains human constant regions from human IgG2 sequences and human IgG4 sequences and murine complementarity-determining regions grafted onto the human framework light- and heavy-chain variable regions. Eculizumab is composed of two 448 amino acid heavy chains and two 214 amino acid light chains and has a molecular weight of approximately 148 kDa.
Eculizumab (INN and USAN; trade name Soliris®) is a humanized monoclonal antibody that is a first-in-class terminal complement inhibitor and the first therapy approved for the treatment of paroxysmal nocturnal hemoglobinuria (PNH), a rare, progressive, and sometimes life-threatening disease characterized by excessive destruction of red blood cells (hemolysis).[1] It costs £400,000 ($US 600,000) per year per patient.[1]
Eculizumab also is the first agent approved for the treatment of atypical hemolytic uremic syndrome (aHUS), an ultra-rare genetic disease that causes abnormal blood clots to form in small blood vessels throughout the body, leading to kidney failure, damage to other vital organs and premature death.[2][3]
In clinical trials in patients with PNH, eculizumab was associated with reductions in chronic hemolysis, thromboembolic events, and transfusion requirements, as well as improvements in PNH symptoms, quality of life, and survival.[1][4][5][6] Clinical trials in patients with aHUS demonstrated inhibition of thrombotic microangiopathy (TMA),[7] the formation of blood clots in small blood vessels throughout the body,[1][3][4] including normalization of platelets and lactate dehydrogenase (LDH), as well as maintenance or improvement in renal function.[7]
Eculizumab was discovered and developed by Alexion Pharmaceuticals and is manufactured by Alexion. It was approved by the United States Food and Drug Administration (FDA) on March 16, 2007 for the treatment of PNH, and on September 23, 2011 for the treatment of aHUS. It was approved by the European Medicines Agency for the treatment of PNH on June 20, 2007, and on November 24, 2011 for the treatment of aHUS. Eculizumab is currently being investigated as a potential treatment for other severe, ultra-rare disorders
- Hillmen, Young, Schubert, P, N, J, et al (2006). “The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria”.N Engl J Med 355 (12): 1233–1243. doi:10.1056/NEJMMoa061648. PMID 16990386.
- Noris, Caprioli, Bresin, M, J, E, et al. (2010). “Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype”. Clin J Am Soc Nephrol 5: 1844–1859.
- Caprioli, Noris, Brioschi, J, M, S, et al (2006). “Genetics of HUS: the impact of MPC, CFH, and IF mutations on clinical presentation, response to treatment, and outcome”. Blood 108: 1267–1279.
- Hillman, Hall, Marsh, P, C, JC, et al (2004). “Effect of eculizumab on hemolysis and transfusion requirements in patients with paroxysmal nocturnal hemoglobinuria”. N Eng J Med 350: 552–559.
- Ray, Burrows, Ginsberg, Burrows, JG, RF, JS, EA (2000). “Paroxysmal nocturnal hemoglobinuria and the risk of venous thrombosis: review and recommendations for management of the pregnant and nonpregnant patient”. Haemostasis 30: 103–107.
- Kelly, Hill, Arnold, RJ, A, LM, et al (2011). “Long-term treatment with eculizumab in paroxysmal nocturnal hemoglobinuria: sustained efficacy and improved survival”. Blood 117: 6786–6792.
- .Soliris® (eculizumab) prescribing information (2011). Cheshire, CT: Alexion Pharmaceuticals.http://www.soliris.net/sites/default/files/assets/soliris)pi.pdf.
Array Starts First Phase 3 Trial, Shifts to Late-Stage Development

HY-15202
MEK162
(Synonyms ARRY-162; ARRY-438162; MEK 162; ARRY 162; ARRY 438162)
MEK162 M.Wt: 441.23
MEK162 Formula: C17H15BrF2N4O3
MEK162 Storage: at -20℃ 2 years
MEK162 CAS No.: 606143-89-9
http://clinicaltrials.gov/ct2/show/NCT00959127
|
Array Starts First Phase 3 Trial, Shifts to Late-Stage Development read all at |
Sanofi’s new insulin U300 superior to Lantus: study

Sanofi’s investigational diabetes drug U300, cas no 160337-95-1, insuline glargine, new formulation is better at controlling dangerous low blood sugar events at night than its blockbuster Lantus, according to data from a phase III clinical programme.

insulin glargine
Lantus, developed in the 1990s, is currently Sanofi’s top-selling product, generating $6.6bn last year. But the drug is expected to lose its patent in 2015.
http://www.medscape.com/viewarticle/805067 says no cancer risk
http://clinicaltrials.gov/ct2/show/NCT01689142 reports clinical trials
To compare the efficacy of a new formulation of insulin glargine and Lantus in terms of change of HbA1c from baseline to endpoint (scheduled at month 6 [week 26]) in patients with type 2 diabetes mellitus.
Secondary Objectives:
- To compare a new formulation of insulin glargine and Lantus in terms of change in fasting plasma glucose, pre-injection plasma glucose, 8-point self-measured plasma glucose profile.
- To compare a new formulation of insulin glargine and Lantus in terms of occurrence of hypoglycemia
Insulin glargine is produced by recombinant DNA technology using a non-pathogenic laboratory strain of Escherichia coli (K12) as the production organism. It is an analogue of human insulin made by replacing the asparagine residue at position A21 of the A-chain with glycine and adding two arginines to the C-terminus (positions B31 and 32) of the B-chain. The resulting protein is soluble at pH 4 and forms microprecipitates at physiological pH 7.4. Small amounts of insulin glargine are slowly released from microprecipitates giving the drug a long duration of action (up to 24 hours) and no pronounced peak concentration.

Let’s Set a Global Drug Quality Benchmark by Kiran M Shaw, Biocon

With Indian-made generics accounting for a US market share of over 25 per cent, it is not surprising that it is gaining significant mindshare of the Food and Drug Administration ( FDA). The spate of quality issues with leading Indian pharmaceutical companies in the past couple of years however should not be viewed in isolation. Big Pharma in the West, too, has been facing increasing flak from the FDA and other regulators over good manufacturing practice ( GMP) violations. High profile names like J& J, Genzyme (Sanofi), GSK, Sandoz, Watson, Teva and many others have encountered their share of quality problems and have been served with ‘warning letters’ from FDA
http://kiranmazumdarshaw.blogspot.in/2013/06/lets-set-global-drug-quality-benchmark.html
READ ALL AT THE LINK ABOVE
