
Home » Posts tagged 'Phase3 drugs' (Page 2)
Tag Archives: Phase3 drugs
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Cempra Provides Guidance on the Clinical Program Required for Regulatory Approval for Solithromycin for Community-Acquired Bacterial Pneumonia (CABP)

solithromycin
(3aS,4R,7S,9R,10R,11R,13R,15R,15aR)-1-[4-[4-(3-aminophenyl)-1H-1,2,3-triazol-1-yl]butyl]-4-ethyl-7-fluorooctahydro-11-methoxy-3a,7,9,11,13,15-hexamethyl-10-{[3,4,6-trideoxy-3-(dimethylamino)-β-D–xylo-hexopyranosyl]oxy}-2H-Oxacyclotetradecino[4,3-d]oxazole-2,6,8,14(1H,7H,9H)-tetrone
| Legal status | Phase III clinical trials, North America, South America, Europe |
|---|---|
| Routes | oral, intravenous |
| Identifiers | |
| CAS number | 760981-83-7 |
Cempra Provides Guidance on the Clinical Program Required for Regulatory …
The Herald | HeraldOnline.com
The Phase 3 solithromycin clinical program in CABP will be planned to consist of an oral trial and an intravenous (IV)-to-oral clinical trial. Cempra followed the CABP guidance that the FDA proposed in a November, 2011, meeting of the Anti-Infective …
READ ALL AT
http://www.heraldonline.com/2013/06/13/4944834/cempra-provides-guidance-on-the.html
Solithromycin (formerly known as CEM-101 and OP-1068) is a novel ketolide antibiotic undergoing clinical development for the treatment of community-acquired pneumonia (CAP) and other infections.It is expected to be the first macrolide antibiotic available in intravenous, oral, and pediatric suspension formulations in over 20 years.
Solithromycin exhibits excellent in vitro activity against a broad spectrum of Gram-positive respiratory tract pathogens, including macrolide-resistant strains. Solithromycin has activity against a wide variety of pathogens, and further research is being conducted for other infections.
- May 2011: Solithromycin is in a Phase 2 clinical trial for serious community-acquired bacterial pneumonia (CABP) and in a Phase 1 clinical trial with an intravenous formulation.
- September 2011 : Encouraging results from the phase 2 clinical trial versus levofloxacin were reported.
Intravenous formulation of Melphalan, which is in a Phase III trial for use as a conditioning treatment prior to autologous stem cell transplant for patients with multiple myeloma

Mephalan
15 march 2013
Spectrum Pharmaceuticals has licensed an investigational multiple myeloma drug from Ligand Pharmaceuticals in a deal that could be worth over $50 million.
The treatment in question is an intravenous formulation of melphalan, which is in a Phase III trial for use as a conditioning treatment prior to autologous stem cell transplant for patients with multiple myeloma. Spectrum is assuming the responsibility for the trial and hopes to file Captisol-enabled melphalan in the first half of 2014.
The Captisol technology used to reformulate melphalan allows for longer administration durations and slower infusion rates. It has been used with six US Food and Frug Administration-approved products, including Onyx Pharmaceuticals’ multiple myeloma drug Kyprolis (carfilzomib )and Pfizer’s antifungal Vfend (voriconazole).
Melphalan hydrochloride (trade name Alkeran) is a chemotherapy drug belonging to the class of nitrogen mustard alkylating agents.
An alkylating agent adds an alkyl group (CnH2n+1) to DNA. It attaches the alkyl group to the guanine base of DNA, at the number 7 nitrogen atom of the imidazole ring.
Otherwise known as L-Phenylalanine Mustard, or L-PAM, melphalan is a phenylalanine derivative of mechlorethamine.
Uses
It is used to treat multiple myeloma[1] and ovarian cancer, and occasionally malignant melanoma.
The agent was first investigated as a possible drug for use in melanoma. It was not found to be effective, but has been found to be effective in the treatment of myeloma.
Oral or intravenous; dosing varies by purpose and route of administration as well as patient weight.
Melphalan Prescribing Information: Alkeran[2]
Melphalan Patient Information: MedlinePlus[3]
Melphalan Material Safety Data Sheet (MSDS): Sequoia Research Products[4]
- Facon T, Mary JY, Hulin C, et al. (October 2007). “Melphalan and prednisone plus thalidomide versus melphalan and prednisone alone or reduced-intensity autologous stem cell transplantation in elderly patients with multiple myeloma (IFM 99-06): a randomised trial”. Lancet 370 (9594): 1209–18. doi:10.1016/S0140-6736(07)61537-2. PMID 17920916.
- celgene.com
- nlm.nih.gov
- seqchem.com
Sinovac Reports Preliminary Top-Line Results from Phase III Clinical Trial for EV71 Vaccine Candidate Against Hand, Foot and Mouth Disease
")
BEIJING, March 14, 2013
Sinovac Biotech Ltd. a leading provider of vaccines in China, announced today preliminary top-line data from its Phase III clinical trial assessing the efficacy, immunogenicity and safety of the Company’s proprietary Enterovirus 71 (“EV71”) vaccine against hand, foot and mouth disease (“HFMD”).
The primary objective of the study was to evaluate the efficacy of the EV71 vaccine in the prevention of HFMD caused by EV71 in infants of 6 to 35 months old. The preliminary Phase III data showed that Sinovac’s EV71 vaccine was 95.4% (95% CI: 87.5%, 98.3%) efficacious against HFMD caused by EV71.
The Phase III trial showed good immunogenicity and safety for Sinovac’s EV71 vaccine. The overall incidence of serious adverse events in this trial was 2.2% among the EV71 candidate vaccine recipients and 2.6% among those receiving a control vaccine during the fourteen months observation period. The difference in rates of serious adverse events (“SAEs”) is not statistically significant. Most of the SAEs were considered unlikely to be vaccine-related.
The double-blinded, randomized, placebo controlled Phase III clinical trial was conducted at three sites across China’s Jiangsu province. Approximately 10,000 healthy infants completed the two dose vaccination schedule (at 0 and 28 days) in the first quarter of 2012, prior to the HFMD epidemic season in China, followed by active monitoring period.
In parallel, Sinovac conducted another clinical study that was comprised of 1,400 volunteers and designed to evaluate the consistency of three consecutive lots of EV71 vaccine manufactured by the Company. The trial was conducted in children from 6 month to 5 years old. After receiving the vaccine, the ratios of neutralizing antibody GMTs on the 56th day of any two groups were calculated and the 95% confidence intervals of the ratios are all between 0.67 and1.5, which indicates the immunogenicity of the three vaccine lots is equivalent. The study results showed consistent immune response for all three lots and a good safety profile. With immunogenicity equivalent across the three consecutive lots, the results showed Sinovac’s vaccine production process and quality are stable.
In March 2008, an EV71 outbreak in Fuyang City of China’s Anhui Province caused 23 fatalities, and attracted significant attention from the government and medical communities. In May 2008, the PRC Ministry of Health identified EV71 as a Class C infectious disease according to prevention and control regulations. EV71 outbreaks have increased over the last five years, with over 1 million cases identified and 500 to 900 reported fatalities each year.
Dr. Weidong Yin , Chairman, President and CEO of Sinovac, commented, “We are excited to report an over 95% efficacy rate from the Phase III trial on our proprietary EV71 vaccine candidate. The conclusion of this trial marks an important milestone in the development of our proprietary vaccine. Hand, foot, and mouth disease continues to represent a significant unmet public health need and economic burden in China, as well as several other Asian countries. Our EV71 vaccine is poised to provide an effective solution to prevent hand, food and mouth disease caused by EV71, a much needed resource given the current limited prevention and EV71 specific treatment methods. At Sinovac, we are committed to our stated mission to develop and supply vaccines to eliminate human diseases.”
Professor Hua Wang, Lead Principal Investigator, stated, “The Phase III study for Sinovac’s EV71 vaccine candidate met its primary objective. The trial results demonstrated that the vaccine is not only safe, but shows significant efficacy in subjects.”
The Company’s next step is to finalize the clinical report, which will become an important part of documents to be filed with the PRC State Food and Drug Administration (“SFDA”) for the application of new drug certificate, GMP certification, and the production license in order to commence the commercial production of the vaccine. In parallel, Sinovac’s dedicated EV71 vaccine manufacturing facility has been completed and is ready for the GMP inspection by SFDA.
Sinovac obtained clinical research approval for its proprietary EV71 vaccine candidate from the SFDA in December 2010, and completed Phase I and II clinical trials in 2011. The preliminary results of the Phase I and Phase II studies confirmed that Sinovac’s vaccine candidate has good safety and immunogenicity profile.
About Sinovac
Sinovac Biotech Ltd. is a China-based biopharmaceutical company that focuses on research, development, manufacturing and commercialization of vaccines that protect against human infectious diseases including hepatitis A and B, seasonal influenza, H5N1 pandemic influenza and mumps, as well as animal rabies vaccine. In 2009, Sinovac was the first company worldwide to receive approval for its H1N1 influenza vaccine, Panflu.1, and has manufactured it for the Chinese Central Government, pursuant to the government-stockpiling program. The Company is also the only supplier of the H5N1 pandemic influenza vaccine to the government-stockpiling program. Sinovac is developing a number of new pipeline vaccines including vaccines for enterovirus 71 (against hand, foot, and mouth disease), pneumococcal conjugate, pneumococcal polysaccharides, varicella and rubella. Sinovac sells its vaccines mainly in China and exports selected vaccines to Mongolia, Nepal, and the Philippines.

A sensor-adaptor mechanism for enterovirus uncoating from structures of EV71
Xiangxi Wang, Wei Peng, Jingshan Ren, Zhongyu Hu, Jiwei Xu, Zhiyong Lou, Xumei Li, Weidong Yin, Xinliang Shen, Claudine Porta, Thomas S Walter, Gwyndaf Evans, Danny Axford, Robin Owen, David J Rowlands, Junzhi Wang*, David I Stuart*, Elizabeth E Fry* & Zihe Rao*
Enterovirus 71 1 (EV71) is a major agent of hand, foot and mouth disease in children that can cause severe central nervous system disease and death. No vaccine or antiviral therapy is available. High-resolution structural analysis of the mature virus and natural empty particles shows that the mature virus is structurally similar to other enteroviruses. In contrast, the empty particles are markedly expanded and resemble elusive enterovirus-uncoating intermediates not previously characterized in atomic detail. Hydrophobic pockets in the EV71 1 capsid are collapsed in this expanded particle, providing a detailed explanation of the mechanism for receptor-binding triggered virus uncoating. These structures provide a model for enterovirus uncoating in which the VP1 1 GH loop acts as an adaptor-sensor for cellular receptor attachment, converting heterologous inputs to a generic uncoating mechanism, highlighting new opportunities for therapeutic intervention. [ Nat Struct Mol Biol. 2012 Mar 4. doi: 10.1038/nsmb.2255. Epub ahead of print. PMID: 22388738 ][ PDF ]
Biogen submits haemophilia A drug to FDA

Mar 14 2013
Biogen Idec has filed the first long-lasting Factor VIII treatment for haemophilia A with the US Food and Drug Administration.
The US biotech major has submitted recombinant factor VIII Fc fusion protein (rFVIIIFc), the first haemophilia A product candidate “in a new class of long-lasting clotting factor therapies being developed with the goal of reducing the burden of treatment for this condition”. If approved, rFVIIIFc will be the first major advance in haemophilia A treatment in more than two decades, Biogen claims.
The regulatory submission is based on results from A-LONG, the largest Phase III study in haemophilia A to date. Glenn Pierce, Biogen’s head of global medical affairs, noted that in that trial, patients were able to inject rFVIIIFc once-weekly to twice-weekly, “which creates the potential for those currently on prophylactic treatment to reduce injections by 50 to 100 per year”. Moreover, patients currently treating bleeding episodes could potentially dose once per week “and maintain significant protection from bleeding with about the same total number of injections each year they use to treat bleeding episodes today”, he added.
Earlier this month, the FDA accepted for review the company’s BLA for its factor IX candidate, rFIXFc, for use in patients with haemophilia B.
Links
Phase III trial of Merck’s Vytorin passes vital safety test
mar 13 2013
Merck & Co’s stock enjoyed a boost yesterday after it revealed it has been given permission to continue a late-stage trial of its cholesterol buster Vytorin.
The Whitehouse Station, New Jersey-based firm must have a breathed a sigh of relief when the Data Safety Monitoring Board issued a green light for the Phase III IMPROVE for a second time, having found no significant safety concerns raised by the data.
After an earlier planned review of data last year, the Board, rather unusually, said it would undertake a second interim analysis at a later date, which had led to some concerns that there may be issues that could lead to the trial being halted, according to media reports.
However, it seems these fears are unfounded at this point, as the18,000-plus patient study – which is designed to determine whether Vytorin is more effective at reducing the risk of heart attack, stroke and death in patients with heart disease than simvastatin alone – has been cleared to conclude.
The drugmaker said the trial should finish in September next year, and it will no doubt be hoping for a positive outcome to prove the benefits of Vytorin – a combination of the generic simvastatin and the still-patented Zetia (ezetimibe) – and breathe a little new life into its heart franchise.
Citi Investment Research analyst Andrew Baum, however, expressed doubt in a research note the final analysis will show Merck’s drug is more effective than generic competition, according to the Associated Press.
 |
|
|---|---|
 |
|
| Combination of | |
| Ezetimibe | via Niemann-Pick C1-Like 1 protein |
| Simvastatin | Statin HMG-CoA reductase inhibitor |
Cangrelor, AR-C69931MX Shows Improvement Over Plavix in Phase III Trial

Cangrelor, AR-C69931MX
[dichloro-[[[(2R,3S,4R,5R)-3,4-dihydroxy-5-[6-(2-methylsulfanylethylamino)-2-(3,3,3-trifluoropropylsulfanyl)purin-9-yl]oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-hydroxyphosphoryl]methyl]phosphonic acid
N-[2-(Methylthio)ethyl]-2-[(3,3,3-trifluoropropyl)thio]-5¢-adenylic acid monoanhydride with (dichloromethylene)bis[phosphonic acid]
163706-06-7 cas no
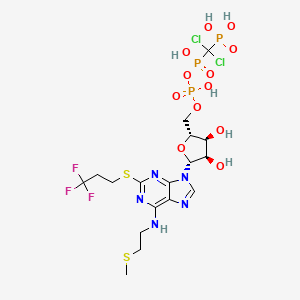 Cangrelor
CangrelorUPDATE
Company:
Approval Status:
Approved June 2015
Specific Treatments:
For reducing periprocedural thrombotic events
Therapeutic Areas
Company:
Approval Status:
Approved June 2015
Specific Treatments:
For reducing periprocedural thrombotic events
Therapeutic Areas
Kengreal (cangrelor)
MAR 09, 2013
The Medicines Company said yesterday it will pursue marketing approvals for its anti-clotting drug candidate Cangrelor after it met its primary efficacy endpoint in a Phase III clinical trial of improvement compared with Plavix (clopidogrel).
The intravenous small molecule antiplatelet agent reduced by 22% the likelihood of patients experiencing death, myocardial infarction, ischemia-driven revascularization, or stent thrombosis within 48 hours of taking it—to 4.7% from 5.9% of subjects randomized during CHAMPION PHOENIX. The Phase III trial compared Cangrelor to oral Plavix in 11,145 patients undergoing percutaneous coronary intervention.
Cangrelor also showed a 38% reduction (0.8% compared with 1.4%) over Plavix in the likelihood of patients experiencing the key secondary endpoint, incidence of stent thrombosis at 48 hours.
Cangrelor is designed to prevent platelet activation and aggregation that leads to thrombosis in acute care settings, including in patients undergoing percutaneous coronary intervention. During CHAMPION PHOENIX, Cangrelor made its best showing in patients with Q-wave myocardial infarction (QMI), lowering by 39% (to 0.2% compared with 0.3%) the incidence of QMI. Cangelor’s most disappoint showing was its inability to lower the odds of death compared with Clopidogrel; both drugs showed a likelihood of 0.3%.
“Our next step is to submit for market approvals in the U.S. and Europe. We anticipate submitting these data for a new drug application to the U.S. Food and Drug Administration in the second quarter with findings of prior trials, including the BRIDGE trial in patients awaiting open heart surgery,” Simona Skerjanec, PharmD, senior vp and innovation leader for antiplatelet therapies at The Medicines Company, said in a statement.
Cangrelor is a P2Y12 inhibitor under investigation as an antiplatelet drug[1] for intravenous application. Some P2Y12 inhibitors are used clinically as effective inhibitors of adenosine diphosphate-mediated platelet activation and aggregation.[1] Unlike clopidogrel (Plavix), which is a prodrug, cangrelor is an active drug not requiring metabolic conversion.
Poor interim results led to the abandonment of the two CHAMPION clinical trials in mid 2009.[2] The BRIDGE study, for short term use prior to surgery, continues.[3] The CHAMPION PHOENIX trial was a randomized study of over 11,000 patients published in 2013. It found usefulness of cangrelor in patients getting cardiac stents. Compared with clopidogrel given around the time of stenting, intravenous ADP-receptor blockade with cangrelor significantly reduced the rate of stent thrombosis and myocardial infarction.[4] Reviewers have questioned the methodology of the trial.[5]

One particularly preferred example of a reversible, short-acting P2Y12 inhibitor is cangrelor. Cangrelor is a potent, direct, and reversible antagonist of the platelet P2Y12 receptor. Cangrelor has a half-life of approximately less than 10 minutes, allowing for a return to normal platelet function in a very short period of time upon discontinuation of the drug. By reducing the need for a compound to be metabolized for activity, and by having a relatively short half-life, reversible, short-acting P2Y12 inhibitors are considered “reversible,” meaning that full platelet functionality may return rather quickly as compared to thienopyridines.
The binding of cangrelor to the P2Y12 receptor inhibits platelet activation as well as aggregation when mediated in whole or in part via this receptor. Cangrelor can be derived completely from synthetic materials, and is an analogue of adenosine triphosphate (ATP). ATP is a natural antagonist of the P2Y12 receptor sites and is found in humans.
The chemical structure for cangrelor is depicted below as Formula I.

Cangrelor is clinically well tolerated and safe and has no drug-drug interaction with aspirin, heparin or nitroglycerin. Unlike orally dosed thienopyridines, cangrelor can be administered intravenously and binds directly to P2Y12 receptor sites of platelets. In each of the embodiments of the present invention, the term “cangrelor” encompasses the compound of Formula I as well as tautomeric, enantiomeric and diastereomeric forms thereof, and racemic mixtures thereof, other chemically active forms thereof, and pharmaceutically acceptable salts of these compounds, including a tetrasodium salt. These alternative forms and salts, processes for their production, and pharmaceutical compositions comprising them, are well known in the art and set forth, for example, in U.S. Pat. No. 5,721,219. Additional disclosure relevant to the production and use of cangrelor may be found in U.S. Pat. Nos. 5,955,447, 6,130,208 and 6,114,313, as well as in U.S. Appln. Publication Nos. 2006/0270607 and 2011/0112030.
Invasive procedures means any technique where entry to a body cavity is required or where the normal function of the body is in some way interrupted by a medical procedure and/or treatment that invades (enters) the body, usually by cutting or puncturing the skin and/or by inserting instruments into the body. Invasive procedures can include coronary artery bypass grafting (CABG), orthopedic surgeries, urological surgeries, percutaneous coronary intervention (PCI), other general invasive procedures, such as endarterectomy, renal dialysis, cardio-pulmonary bypass, endoscopic procedures or any medical, surgical, or dental procedure that could result in excessive bleeding or hemorrhage to the patient.
Perioperative means the period of a patient’s invasive procedure which can occur in hospitals, surgical centers or health care providers’ offices. Perioperative includes admission, anesthesia, surgery, to recovery.
Thrombosis is the formation of a blood clot (thrombus) inside a blood vessel obstructing the flow of blood through the circulatory system. When a blood vessel is injured, the body uses platelets and fibrin to form a blood clot to prevent blood loss. Some examples of the types of thrombosis include venous thrombosis which includes deep vein thrombosis, portal vein thrombosis, renal vein thrombosis, jugular vein thrombosis, Budd-Chiari syndrome, Paget-Schroetter disease, cerebral venous sinus thrombosis, cerebral venous sinus thrombosis and arterial thrombosis which includes stroke and myocardial infarction.
The compound cangrelor from the Medicines Company is represented by the structure

 OR
ORRN: 163706-36-3



…………
J. Med. Chem., 1999, 42 (2), pp 213–220
http://pubs.acs.org/doi/full/10.1021/jm981072s
10l (AR-C69931MX)
N6–(2-Methylthioethyl)-2-(3,3,3-trifluoropropylthio)-5‘-adenylic Acid, Monoanhydride withDichloromethylenebis(phosphonic acid) (10l). Prepared as the triammonium salt in 4% yield from 3l: 1H NMR δ(D2O) 8.30 (1H, s, H8), 5.97 (1H, d, J = 5.5 Hz, H1‘), 4.65 (1H, m, H2‘), 4.47 (1H, m, H3‘), 4.28 (1H, m, H4‘), 4.17 (2H, m, H5‘a and H5‘b), 3.67 (br s, NHCH2), 3.21 (2H, t, J = 7.6 Hz, SCH2), 2.72 (2H, t, J = 6.6 Hz, SCH2CH2CF3), 2.58 (2H, m, NCH2CH2), 2.04 (3H, s, SCH3);31P NMR δ(D2O) 8.80 (d, 1P, J = 18.6 Hz, Pγ), 0.42 (dd, 1P, J1 = 18.9 Hz, J2 = 28.9 Hz, Pβ), −9.41 (d, 1P, J = 29.0 Hz, Pα). Anal. (C17H34Cl2F3N8O12P3S2·3H2O) H, N, S; C: calcd, 23.16; found, 23.66.
References
- Cangrelor Attenuates Coated-Platelet Formation
- CHAMPION Trials With Cangrelor Stopped for Lack of Efficacy
- What Cangrelor Failure Means to Medicines
- Effect of Platelet Inhibition with Cangrelor during PCI on Ischemic Events (2013) Bhatt, DL etal. New England Journal of Medicine March 10, 2013 DOI: 10.1056/NEJMoa1300815 (published initially online).
- The Duel between Dual Antiplatelet Therapies (2013) Lange, RA and Hillis, LD. New England Journal of Medicine March 10, 2013 DOI: 10.1056/NEJMe1302504
- 15th European Federation for Medicinal Chemistry International Symposium on Medicinal Chemistry (Sept 6 1998, Edinburgh)1998,:Abst P.281
- Specific P2Y12 purinoceptor antagonist; inhibits ADP-induced platelet aggregation. Prepn: A. H. Ingall et al., WO 9418216 (1994 to Fisons); eidem, US 5721219 (1998 to Astra); and in vivo antithrombotic activity: idem et al., J. Med. Chem. 42, 213 (1999).
- In vivo antithrombotic effects in canine arterial thrombosis: J. Huang et al., J. Pharmacol. Exp. Ther. 295, 492 (2000).
- Mechanism of action study: A. Ishii-Watabe et al., Biochem. Pharmacol. 59, 1345 (2000).
- Clinical safety assessment and evaluation in acute coronary syndromes: R. F. Storey et al., Thromb. Haemostasis 85, 401 (2001); in angina pectoris and non-Q-wave myocardial infarction: F. Jacobsson et al., Clin. Ther. 24, 752 (2002).
- Clinical pharmacodynamics compared with clopidogrel: R. F. Storey et al., Platelets 13, 407 (2002).
- Review of clinical development: S. C. Chattaraj, Curr. Opin. Invest. Drugs2, 250-255 (2001).
- WO2013/103567 A2,
- Journal of Medicinal Chemistry, 1999 , vol. 42, 2 p. 213 – 220
Phase 2 Drug: Ustekinumab A monoclonal antibody against the p40 subunit of IL-12/23 Other Name: Stelara
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | IL-12 and IL-23
|

Ustekinumab, CAS number 815610-63-0, is also known by it’s brand name Stelara, which is marketed by Janssen Biotech, Inc. Developed as a treatment for adults with moderate to severe plaque psoriasis
Rockefeller University, MAR 2013
http://clinicaltrials.gov/ct2/show/NCT01806662
Atopic dermatitis (AD) is a chronic disease associated with intense itching, which affects most aspects of everyday life in the majority of patients. Acute inflammation and extensor/facial involvement is common in infants, whereas chronic inflammation increases in prevalence with age, as do localization to flexures. AD has a complex background characterized by immune activation, increased epidermal thickness in chronic diseased skin, and defective barrier function. In normal, healthy skin, the outer layer of the epidermis, the stratum corneum is made up flattened dead cells called corneocytes held together by a mixture of lipids and proteins. The stratum corneum and, in particular, the lipid layer are vital in providing a natural barrier function that locks water inside the skin and keeps allergens and irritants out. In people with AD, the barrier function is defective, which leads to dry skin. As the skin dries out, it cracks allowing allergens and irritants to penetrate.
Mild AD can be controlled with emollients and topical medications. However, moderate to severe AD is extremely difficult to control and requires systemic treatment that is often unsatisfactory due to impracticality and lack of effectiveness. Only three therapeutic options exist for moderate to severe AD, including: 1) oral steroids 2) cyclosporine A (CsA), that is not widely used in the US as it is not FDA approved for AD and 3) ultraviolet phototherapy. Oral steroids and CsA treatments have major side effects and UV radiation therapy is highly inconvenient for patients. Several biologic medications, such as TNF-alpha inhibitors, are effective, convenient, and relatively safe therapies for psoriasis, but have thus far not shown efficacy in AD. Ustekinumab is a unique biologic medication that may specifically target AD.
The investigators study will determine whether there is a reversal of the skin thickness and the immune pathways involved in the disease during treatment with Ustekinimab and what specific immune cells are involved. The investigators are also interested to understand how the clinical reversal of the disease will correlate with tissue reversal of the disease.
Detailed Description:
In psoriasis, epidermal hyperplasia is driven by underlying immune activation, whether as a direct response to IL-20 family cytokines that induces hyperplasia and inhibits keratinocyte terminal differentiation or as an indirect response to immune-mediated injury to keratinocytes. The epidermal reaction in psoriasis is largely restored to normal with selective immune suppression. Hence, one might hypothesize that similar epidermal responses should occur in the presence of “generalized” cellular immune activation, in diseases with similar inflammatory infiltrate and epidermal hyperplasia, such as AD. In fact, psoriasis and AD share features of dense T-cells and dentritic cell infiltrates, as well as over-expression of IL-22 in skin lesions. These diseases also share similar epidermal hyperplasia in their chronic phases.
Work from the investigators group showed that IL-22 is a key cytokine in the pathogenesis of both AD and psoriasis. The investigators have demonstrated that in psoriasis, ustekinumab suppresses the production of IL-12, IL-23, and IL-22. Additionally, by RT-PCR the investigators demonstrated that the mRNA expression of p40 cytokine and the IL23R is up-regulated in AD as compared to both normal skin and psoriasis. The investigators therefore hypothesize that ustekinumab will suppress IL-22 and possibly also p40 production in AD lesions and reverse both the epidermal growth/differentiation defects and the underlying immune activation, and hence will suppress disease activity. Interestingly, p40 was also found to be significantly up-regulated in non-lesional AD skin as compared with normal skin.
Although AD is thought to be predominately a disease of Th2-type cells, in the chronic stage, there is large Th1 component. To date, the precise mechanism by which sequential activation of Th2 and Th1 cells in AD is achieved remains unknown. IL-12 induces the differentiation and maturation of human Th cells into Th1-type cells. Recent circumstantial evidence suggests that in AD patients IL-12 may facilitate a change from the Th2-type to a Th1 cytokine profile. IL-12 was recently shown to be highly elevated in pediatric AD and its levels were strongly associated with disease severity.
Expression of IL-12 p40 mRNA is significantly enhanced in lesional skin from AD, suggesting that the enhanced local production of IL-12 in dendritic cells and macrophages may be responsible for the increased production of IFN-γ in chronic lesions potentially suggesting that IL-12 may have a pivotal role in promoting inflammation in atopic dermatitis. Topical steroids which constitute a mainstay of therapy in AD are known to strongly down-regulate IL-12 expression, possibly also indicating that targeted anti IL-12 therapy might important role in treating AD.
Recently, the Th1/Th2 paradigm in autoimmunity and allergy has been revisited to include a role for a new population of IL-17-producing Th cells known as Th17. Th17 cells are characterized by the production of inflammatory cytokines such as IL-17A, IL-17F, IL-22, and IL-26. One of the key factors involved in naive Th-cell commitment to a Th17 phenotype is IL-23.
Patients with acute AD were found to have increased Th17 T-cells in peripheral blood by flow cytometry and intracellular cytokine staining 26 as well as by immunohistochemistry (IHC) in lesions. Since IL-23 is the major inducer of Th17 T-cells, as well as “T22” T-cells, neutralization of IL-23 could potentially result in both decreased Th17 signal in acute AD as well as decreased “T22/IL22″ signal. Therefore the investigators postulate that ustekinumab in AD will act both inhibiting the IL-12-dependent Th1 shift in chronic AD stage as well as the pathogenic IL-22/”T22” axis in this disease.
Ustekinumab [1] (INN, experimental name CNTO 1275, proprietary commercial name Stelara,[2] Centocor) is a human monoclonal antibody. It is directed against interleukin 12 and interleukin 23, naturally occurring proteins that regulate the immune system and immune-mediated inflammatory disorders.[3]
In two Phase III trials for moderate to severe psoriasis, the longest >76 weeks, ustekinumab was safe and effective.[4][5]
A third Phase III trial, ACCEPT, compared the efficacy and safety of ustekinumab with etanercept in the treatment of moderate to severe plaque psoriasis.[6] This trial found a significantly higher clinical response with ustekinumab over the 12-week study period compared to high-dose etanercept.[6] It also demonstrated the clinical benefit of ustekinumab among patients who failed to respond to etanercept.[6]
Ustekinumab is approved in Canada, Europe and the United States to treat moderate to severe plaque psoriasis.[7]
As of November 2009, the drug is being investigated for the treatment of psoriatic arthritis.[8][9] It has also been tested in Phase II studies for multiple sclerosis[10] and sarcoidosis, the latter versus golimumab (Simponi).[11]
- Cingoz, Oya (2009). “Ustekinumab”. MAbs 1 (3): 216–221. doi:10.4161/mabs.1.3.8593. PMC 2726595. PMID 20069753.
- ^ European Medicines Agency, 20 November 2008, http://www.emea.europa.eu/pdfs/human/opinion/Stelara_58227008en.pdf
- ^ Reddy M, Davis C, Wong J, Marsters P, Pendley C, Prabhakar U (May 2007). “Modulation of CLA, IL-12R, CD40L, and IL-2Ralpha expression and inhibition of IL-12- and IL-23-induced cytokine secretion by CNTO 1275”. Cell. Immunol. 247 (1): 1–11. doi:10.1016/j.cellimm.2007.06.006. PMID 17761156.
- ^ Leonardi CL, Kimball AB, Papp KA, et al. (May 2008). “Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 76-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 1)”. Lancet 371 (9625): 1665–74. doi:10.1016/S0140-6736(08)60725-4. PMID 18486739.
- ^ Papp KA, Langley RG, Lebwohl M, et al. (May 2008). “Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 52-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 2)”. Lancet 371 (9625): 1675–84. doi:10.1016/S0140-6736(08)60726-6. PMID 18486740.
- ^ a b c Griffiths C, Strober B, van de Kerkhof P et al. (2010). “Comparison of Ustekinumab and Etanercept for Moderate-to-Severe Psoriasis”. N Engl J Med 362 (2): 118–28. doi:10.1056/NEJMoa0810652. PMID 20071701.
- ^ Medarex to Receive Milestone Payment for Approval of STELARA(TM) (Ustekinumab) for the Treatment of Moderate to Severe Plaque Psoriasis
- ^ ClinicalTrials.gov NCT00267956 A Study of the Safety and Efficacy of CNTO 1275 in Patients With Active Psoriatic Arthritis
- ^ ClinicalTrials.gov NCT01009086 A Study of the Safety and Efficacy of Ustekinumab in Patients With Psoriatic Arthritis
- ^ ClinicalTrials.gov NCT00207727 A Safety and Efficacy Study of CNTO1275 in Patients With Multiple Sclerosis
- ^ ClinicalTrials.gov NCT00955279 A Study to Evaluate the Safety and Effectiveness of Ustekinumab or Golimumab Administered Subcutaneously (SC) in Patients With Sarcoidosis
- ^ http://www.empr.com/stelara-approved-for-moderate-to-severe-psoriasis/article/149760/
- ^ a b Centocor 12/19/08 Press Release, http://www.centocor.com/centocor/i/press_releases/FDA_ISSUES_COMPLETE_RESPONSE_LETTER_TO_CENTOCOR_FOR_USTEKINUMAB_BIOLOGIC_LICENSE_APPLICATION_
- ^ Johnson LL. “Study: Drug for serious psoriasis tops competition” The Associated Press. 18 Sept 2008.[dead link]
- ^ Wild, David (November 2011), “Novel IL-12/23 Antagonist Shows Potential in Severe Crohn’s”, Gastroenterology & Endoscopy News 62 (11), retrieved 2011-12-04
- ^ a b c Weber J, Keam SJ (2009). “Ustekinumab”. BioDrugs 23 (1): 53–61. doi:10.2165/00063030-200923010-00006. PMID 19344192.
- ^ Segal BM, Constantinescu CS, Raychaudhuri A, Kim L, Fidelus-Gort R, Kasper LH (September 2008). “Repeated subcutaneous injections of IL12/23 p40 neutralising antibody, ustekinumab, in patients with relapsing-remitting multiple sclerosis: a phase II, double-blind, placebo-controlled, randomised, dose-ranging study”. Lancet Neurol 7 (9): 796–804. doi:10.1016/S1474-4422(08)70173-X. PMID 18703004.
- ^ “Important Safety Information”. STELARA® (ustekinumab). Janssen Biotech.
External links
- Centocor Ortho Biotech official site
- CNTO 1275 research studies registered with U.S. National Institutes of Health:
- ClinicalTrials.gov NCT00207727 Phase II Study on Multiple Sclerosis
- ClinicalTrials.gov NCT00320216 Phase II Study on Psoriasis
- ClinicalTrials.gov NCT00267969 Phase III Study on Psoriasis
- ClinicalTrials.gov NCT00307437 Phase III Study on Psoriasis
- ClinicalTrials.gov NCT00267956 Phase II Study on Psoriatic Arthritis
- Sylvester, Bruce (2006-03-06). “CNTO 1275 Shows Efficacy for Psoriasis: Presented at AAD”. Doctor’s Guide Publishing. Retrieved 2007-01-25.
ViiV Healthcare presents phase III SAILING study data of dolutegravir vs raltegravir in treatment-experienced adults with HIV-1

Dolutegravir
| Identifiers | |
|---|---|
| CAS number | 1051375-16-6 |
8 TH MATCH 2013
ViiV Healthcare, a global specialist HIV company established in November 2009 by GSK and Pfizer dedicated to delivering advances in treatment and care for people living with HIV, has announced 24-week data from the phase III SAILING (ING111762) study evaluating the investigational integrase inhibitor dolutegravir in patients with HIV-1 who are failing on current therapy, but had not been treated with an integrase inhibitor.
At 24 weeks, 79% of study participants receiving the once-daily dolutegravir regimen were virologically suppressed (HIV-1 RNA <50 c/mL) vs. 70% of participants on the twice-daily raltegravir regimen. This difference in response was statistically significant with a 95% confidence interval for the difference of 3.4% to 15.9% (p=0.003).
The SAILING study was designed to demonstrate non-inferiority of a regimen containing dolutegravir versus raltegravir (both with up to two background agents) and the analysis met this criterion; statistical superiority was concluded as part of a pre-specified testing procedure. These data were presented at the 20th Conference on Retroviruses and Opportunistic Infections (CROI) in Atlanta, Georgia.
Differences in treatment outcome in favour of the dolutegravir arm were driven by greater virologic response: at Week 24, 15% of patients receiving the dolutegravir regimen had virologic non-response vs. 24% of patients receiving the raltegravir regimen. In addition, fewer subjects failed therapy with integrase inhibitor resistance on dolutegravir (n=2) than on raltegravir (n=10, p=0.016).
Overall, the tolerability of dolutegravir (DTG) was similar to that of raltegravir (RAL). At 24 weeks, 2% of subjects on the dolutegravir regimen discontinued due to adverse events (AEs) vs. 4% of subjects on the raltegravir regimen. The rate of drug-related AEs was similar for both arms (DTG 20%, RAL 23%) and commonly reported AEs (defined as events that occurred in more than 10% of subjects) were similar on both arms, namely diarrhoea (20% DTG, 17% RAL) and upper respiratory tract infection (11% DTG, 8% RAL).
“People living with HIV who have developed resistance to more than one antiretroviral drug class face increasingly narrow treatment options and clinical decisions become increasingly complex. We welcome these initial results supporting the efficacy and tolerability of dolutegravir as a potentially useful addition in the management of HIV in treatment-experienced patients.” said John Pottage, chief scientific and medical officer, ViiV Healthcare. “These encouraging data were included as part of the comprehensive clinical data package supporting recent regulatory submissions for dolutegravir and we look forward to receiving the primary analysis at 48 weeks in due course.”
The primary objective of the ongoing double-blind, double-dummy phase III SAILING study is to demonstrate the antiviral activity of once-daily dolutegravir 50mg compared to twice-daily raltegravir 400mg over 48 weeks in HIV-1 infected, antiretroviral-experienced, integrase inhibitor-naïve adults. At baseline, 715 study participants were randomised 1:1 to receive either dolutegravir or raltegravir plus investigator-selected background regimen of no more than 2 agents, one of which was fully active. All subjects had documented genotypic or phenotypic resistance to agents from at least two antiretroviral therapy drug classes, and ongoing virologic replication. Median baseline HIV-1 RNA levels were 4.18 log10 c/mL and median baseline CD4+ cell counts were 200 cells/mm3. The study population included 32% women, 42% were of African American/African heritage, and 46% of study participants were classified as CDC Class C (patients who have one or more AIDS-defining illness). The 48-week primary analysis of this study will be presented at a future scientific meeting.
S/GSK1349572 (dolutegravir, DTG) is an investigational integrase inhibitor currently in development for the treatment of HIV; it does not require an additional pharmacokinetic boosting drug to be added to the regimen. Integrase inhibitors block HIV replication by preventing the viral DNA from integrating into the genetic material of human immune cells (T-cells). This step is essential in the HIV replication cycle and is also responsible for establishing chronic infection.
SAILING is the fourth phase III dolutegravir study reporting in 2012 and 2013. Data from the two studies in treatment-naïve populations, SPRING-2 (ING113086) and SINGLE (ING114467), were announced in April and July of 2012 respectively. Data from VIKING-3 (ING112574) in integrase inhibitor-resistant patients were announced in November 2012. Dolutegravir is not yet approved as a treatment for HIV or any other indication anywhere in the world.
Dolutegravir[1] is an experimental new drug under investigation for the treatment of HIV infection. Dolutegravir is an integrase inhibitor. Also known as S/GSK1349572 or just “572”, the drug is under development by GlaxoSmithKline (GSK). Studies have shown dolutegravir to be effective in patients with resistance to the integrase inhibitor, raltegravir.[2] Clinical trials are underway to support dolutegravir in combination with abacavir and lamivudine, in a new new fixed dose combination called 572-Trii.[3] In February, 2013 the Food and Drug Administration announced that it would fast track dolutegravir’s approval process.[4]
Results from the 96-week comparison with efavirenz, SPRING-1, showed dolutegravir 50mg orally to be effective at reducing HIV viral load and raising CD4 counts in integrase-naive patients. [5]
References
- [1] American Medical Association (AMA), STATEMENT ON A NONPROPRIETARY NAME ADOPTED BY THE USAN COUNCIL (Dolutegravir) Accessed 3 December 2011.
- Dolutegravir (“572”) Holds Up in Heavily Raltegravir-Resistant Patients, Phase 2B Study Finds Nelson Vergel. The Body PRO. Accessed 23 April 2011.
- Shionogi-ViiV Healthcare Starts Phase 3 Trial for “572-Trii” Test positive airwave. The Body PRO. Accessed 23 April 2011.
- “GSK wins priority status for new HIV drug in U.S”. Reuters. 16 February 2013. Retrieved 18 February 2013.
- Horn, Tim. ViiV’s Dolutegravir Continues to Show Well After 96 Weeks, Versus Sustiva, for First-Time Treatment. AIDSmeds.com 7 Mar 2012. Accessed 14 Mar 2012.
Takeda Submits Marketing Authorisation Application for Vedolizumab in Moderately to Severely Active Ulcerative Colitis and Crohn’s Disease in the European Union

March 7, 2013
Pharmaceutical Company Limited (“Takeda”) today announced that a Marketing Authorisation Application (MAA) has been submitted to The European Medicines Agency (EMA) for vedolizumab, an investigational, gut-selective humanized monoclonal antibody for the treatment of adults with moderately to severely active ulcerative colitis (UC) and Crohn’s disease (CD), the two most common types of inflammatory bowel disease (IBD). If approved, vedolizumab would be the first and only gut-selective biologic agent for UC and CD on the market.
“Ulcerative colitis and Crohn’s disease are chronic debilitating diseases with important unmet medical needs, often affecting young people in the prime of their lives,” said Asit Parikh, M.D., Ph.D., vice president, general medicine, Takeda. “We are encouraged by the findings of GEMINI, the vedolizumab Phase 3 clinical development program, which studied approximately 3,000 patients in nearly 40 countries, making it the largest IBD clinical trial program conducted to date.”
Nearly four million people worldwide are affected by IBD, with UC affecting more than 500,000 people and CD affecting approximately 230,000 people in the EU. Crohn’s disease and ulcerative colitis are chronic diseases that cause inflammation of the lining of the digestive tract. Inflammation caused by CD can involve varying areas of the digestive tract, while UC impacts the colon only. CD and UC can be both painful and debilitating, which may sometimes lead to serious complications and can significantly impact the quality of life for patients.
The MAA submission was supported by Phase 3 clinical studies, GEMINI I, GEMINI II, GEMINI III and GEMINI LTS (Long-term Safety), which are part of the GEMINI Studies™, a four-study clinical research program to investigate the efficacy and safety of vedolizumab on clinical response and remission in moderately to severely active CD and UC patients, who had failed at least one conventional or anti-TNFα therapy.
“With a targeted mechanism of action, vedolizumab has clinical promise as a potential treatment option for people with moderate to severely active CD and UC,” said Paul Rutgeerts, M.D., Ph.D., F.R.C.P., professor of medicine, Catholic University of Leuven, Belgium. “While there is no known cure, there is a need for new CD and UC treatment options, in an effort to provide patients with additional choices for managing their disease, reducing symptoms and achieving remission.”
About Crohn’s disease and ulcerative colitis
Crohn’s disease (CD) and ulcerative colitis (UC) are the two most common forms of inflammatory bowel disease (IBD), which is marked by inflammation in the lining of the GI tract. CD can impact any part of the digestive tract, and common symptoms may include abdominal pain, diarrhea, rectal bleeding, weight loss, and/or fever. UC impacts the large intestine only, which includes the colon and the rectum. The most common symptoms of UC include abdominal discomfort and blood or pus in diarrhea. There is no known cause for CD or UC, although many researchers believe that the interaction of an outside agent, such as a virus or bacteria, with the body’s immune system may trigger them. No cure exists for CD or UC; the aim of IBD treatments is to induce and maintain remission, or achieve extended periods of time when patients do not experience symptoms.
About vedolizumab
Vedolizumab was developed for the treatment of CD and UC, as a gut-selective, humanized monoclonal antibody that specifically antagonizes the alpha4beta7 (α4β7) integrin, which is expressed on a subset of circulating white blood cells. These cells have been shown to play a role in mediating the inflammatory process in CD and UC. α4β7 binds with a specific adhesion molecule primarily expressed in the intestinal tract. Therefore, vedolizumab, by preventing this interaction, has a gut selective effect.
About Takeda Pharmaceutical Company Limited
Located in Osaka, Japan, Takeda is a research-based global company with its main focus on pharmaceuticals. As the largest pharmaceutical company in Japan and one of the global leaders of the industry, Takeda is committed to strive towards better health for patients worldwide through leading innovation in medicine. Additional information about Takeda is available through its corporate website, http://www.takeda.com.
Vedolizumab is a monoclonal antibody being developed by Millennium Pharmaceuticals, Inc. for the treatment of ulcerative colitis and Crohn’s disease.It binds to integrin α4β7(LPAM-1, lymphocyte Peyer’s patch adhesion molecule 1).[1][2]
The molecule was first identified by Dr. Andrew Lazarovits [1][2] as the murine MLN0002 homologue. His discovery of the mouse equivalent of this antibody—originally applied to anti-rejection strategies in kidney transplantation—was published in the journal Nature in 1996. The drug was then licensed to Millennium Pharmaceuticals of Boston for further development.
As of October 2009, vedolizumab is undergoing Phase III trials.[3] Clinical trials indicate that Vedolizumab was found safe and highly effective for inducing and maintaining clinical remission in patients with moderate to severe ulcerative colitis [3]. Dr. Brian Faegan, head researcher, reported an absence of any instances of progressive multifocal leukoencephalopathy (PML), which is a particularly important finding [4]. It looks like it will be an effective abiologic agent without some of the toxicity issues previously seen with anti-TNF drugs .
It is widely believed now that “vedolizumab can be used either as a first-line treatment or in case of anti-TNF failure”
- Statement On A Nonproprietary Name Adopted By The USAN Council – Vedolizumab, American Medical Association.
- Soler, D; Chapman, T; Yang, LL; Wyant, T; Egan, R; Fedyk, ER (2009). “The binding specificity and selective antagonism of vedolizumab, an anti-alpha4beta7 integrin therapeutic antibody in development for inflammatory bowel diseases”. The Journal of Pharmacology and Experimental Therapeutics 330 (3): 864–75. doi:10.1124/jpet.109.153973. PMID 19509315.
- ClinicalTrials.gov NCT00790933 Study of Vedolizumab (MLN0002) in Patients With Moderate to Severe Crohn’s Disease (GEMINI II)

Phase 3-Trius Therapeutics will soon be reporting data from its second phase III trial of Tedizolid
Tedizolid
(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)pyridin-3-yl]phenyl}-5-(hydroxymethyl)-1,3-oxazolidin-2-one
- Molecular Formula: C17H15FN6O3
- Average mass: 370.337799
856866-72-3 cas no
Torezolid (also known as TR-701 and now tedizolid[1]) is an oxazolidinone drug being developed by Trius Therapeutics (originator Dong-A Pharmaceuticals) for complicated skin and skin-structure infections (cSSSI), including those caused by Methicillin-resistantStaphylococcus aureus (MRSA).[2]
As of July 2012, tedizolid had completed one phase III trial, with another one under way. [3]Both trials compare a six-day regimen of tedizolid 200mg once-daily against a ten-day regimen of Zyvox (linezolid) 600mg twice-daily.
The prodrug of tedizolid is called “TR-701”, while the active ingredient is called “TR-700”.[4][5]

March 5 2013
Trius Therapeutics will soon be reporting data from its second phase III trial (ESTABLILSH-2) and the recently announced publication of the data from its first phase III trial (ESTABLISH-1) in the Journal of the American Medical Association (JAMA)
- “Trius grows as lead antibiotic moves forward”. 31 Oct 2011.
- “Trius Completes Enrollment In Phase 2 Clinical Trial Evaluating Torezolid (TR-701) In Patients With Complicated Skin And Skin Structure Infections”. Jan 2009.
- http://clinicaltrials.gov/ct2/results?flds=Xf&flds=a&flds=b&term=tedizolid&phase=2&fund=2&show_flds=Y
- PMID 19528279 In vitro activity of TR-700, the active ingredient of the antibacterial prodrug TR-701, a novel oxazolidinone antibacterial agent.
- PMID 19218276 TR-700 in vitro activity against and resistance mutation frequencies among Gram-positive pathogens.

{kind=link}