


| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US8383596 | ANTIBACTERIAL AMINOGLYCOSIDE ANALOGS |
2010-04-22
|
|
| US8822424 | Antibacterial aminoglycoside analogs |
2013-01-04
|
2014-09-02
|
| US2012208781 | AMINOGLYCOSIDE DOSING REGIMENS |
2011-11-11
|
2012-08-16
|
| US2012214759 | TREATMENT OF KLEBSIELLA PNEUMONIAE INFECTIONS WITH ANTIBACTERIAL AMINOGLYCOSIDE COMPOUNDS |
2011-11-11
|
2012-08-23
|
| US2012214760 | TREATMENT OF URINARY TRACT INFECTIONS WITH ANTIBACTERIAL AMINOGLYCOSIDE COMPOUNDS |
2011-11-11
|
2012-08-23
|
Home » Posts tagged 'Fast Track Designation'
Tag Archives: Fast Track Designation
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Birelentinib
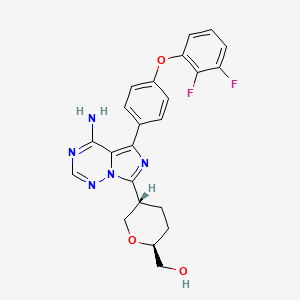
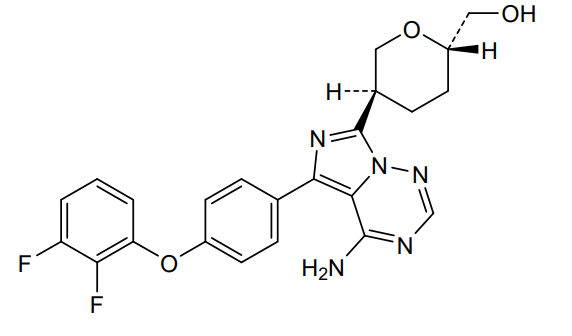
Birelentinib
CAS 2662512-15-2
MF C23H21F2N5O3 MW453.4 g/mol
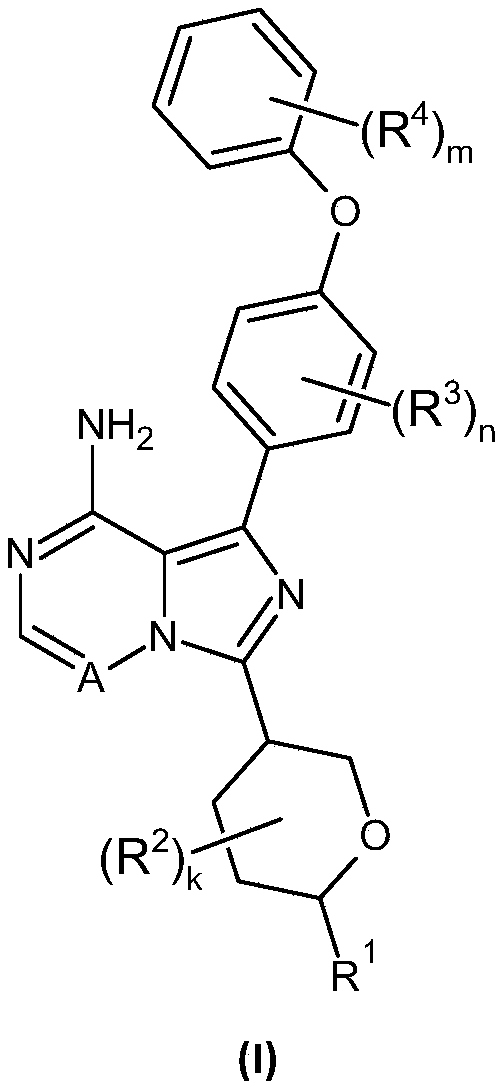
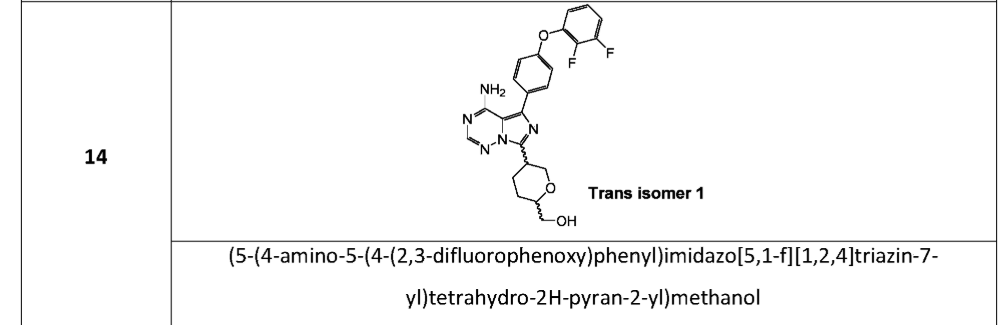
[(2S,5S)-5-[4-amino-5-[4-(2,3-difluorophenoxy)phenyl]imidazo[5,1-f][1,2,4]triazin-7-yl]oxan-2-yl]methanol
[(2S,5S)-5-{4-amino-5-[4-(2,3-difluorophenoxy)phenyl]imidazo[5,1-f][1,2,4]triazin-7-yl}oxan-2-yl]methanol
tyrosine kinase inhibitor, antineoplastic, DZD8586, DZD 8586, Fast Track designation, BTK-IN-30, Z2F599L9GD
Birelentinib (also known as DZD8586) is a first-in-class, non-covalent dual inhibitor of LYN (lymphocyte-specific protein tyrosine kinase) and BTK (Bruton’s tyrosine kinase).
It is currently being developed by Dizal Pharmaceutical as an oral therapy for various B-cell malignancies.
Clinical Status and FDA Designations
As of late 2025, birelentinib has received significant attention for its potential in treating resistant blood cancers:
- Fast Track Designation: In August 2025, the U.S. FDA granted Fast Track designation to birelentinib for adult patients with relapsed or refractory (R/R) chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL).
- Target Population: It is specifically intended for those who have failed at least two prior therapies, including a covalent BTK inhibitor and a BCL-2 inhibitor.
- Key Trials: It is being evaluated in multiple studies, including the Phase 3 Tai-Shan6 trial comparing it against standard treatments like bendamustine and rituximab.
Unique Therapeutic Properties
Birelentinib is designed to overcome common drug resistance mechanisms found in existing treatments:
- Overcoming Resistance: It targets both BTK-dependent pathways (including the common C481X mutation) and BTK-independent B-cell receptor (BCR) signaling pathways.
- Blood-Brain Barrier (BBB) Penetration: A notable feature is its ability to fully penetrate the blood-brain barrier, which may offer therapeutic benefits for patients with central nervous system (CNS) involvement.
- Efficacy: Early Phase 1/2 data presented at the ASH Annual Meeting and EHA Congress in 2025 showed an Objective Response Rate (ORR) of 84.2% in heavily pretreated patients
Birelentinib is an orally bioavailable non-covalent dual inhibitor of tyrosine-protein kinases Lyn (LYN) and BTK (Bruton’s tyrosine kinase; Bruton agammaglobulinemia tyrosine kinase), with potential antineoplastic activity. Upon oral administration, birelentinib targets and inhibits both LYN and BTK, thereby blocking both BTK-dependent and BTK-independent B-cell antigen receptor (BCR) signaling pathways. This prevents the proliferation of malignant B-cells in which the BCR signaling pathway is overactivated. Birelentinib is able to cross the blood-brain barrier (BBB) and thus potentially useful in the treatment of central nervous system (CNS) metastases
SYN’
SYN



AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

References
Publication Number: WO-2021136219-A1
Priority Date: 2020-01-02
/////////birelentinib, tyrosine kinase inhibitor, antineoplastic, DZD8586, DZD 8586, Fast Track designation, BTK-IN-30, Z2F599L9GD
Atebimetinib
Atebimetinib
CAS 2669009-92-9
MF C23H27FN4O6S MW506.5 g/mol
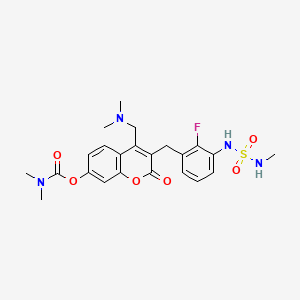
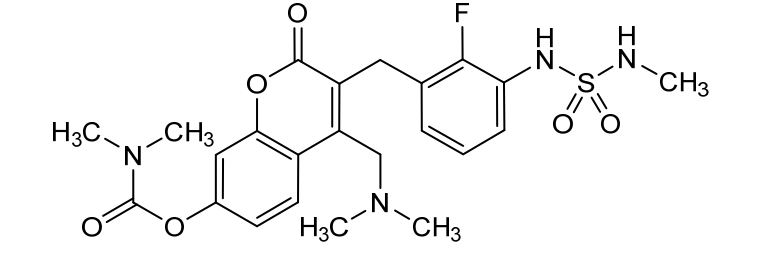
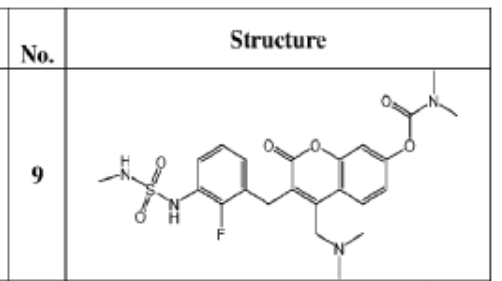
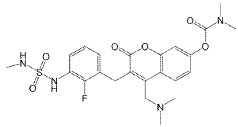
[4-[(dimethylamino)methyl]-3-[[2-fluoro-3-(methylsulfamoylamino)phenyl]methyl]-2-oxochromen-7-yl] N,N-dimethylcarbamate
4-[(dimethylamino)methyl]-3-({2-fluoro-3-[(methylsulfamoyl)amino]phenyl}methyl)-2-oxo-2H-1-benzopyran-7-yl
dimethylcarbamate
MEK tyrosine kinase inhibitor, antineoplastic, IMM-104, IMM 104, Fast Track designation, TEL9243A3N
Atebimetinib (IMM-104) is an investigational oral, deep cyclic inhibitor (DCI) that targets the MAP kinase (MAPK) pathway in solid tumors, particularly RAS-mutant pancreatic cancer. Designed for rapid, pulsatile inhibition to minimize resistance and side effects, it is currently in Phase 2a trials, having shown promising, durable tumor shrinkage and high 1-year survival rates.
Key Aspects of Atebimetinib:
- Mechanism of Action: As a DCI, it works differently from standard inhibitors by targeting MAPK with a short half-life, allowing for rapid “pulsing” that suppresses tumor growth while permitting healthy cells to recover, thus improving tolerability.
- Targeted Cancers: Primarily aimed at RAS-mutant advanced or metastatic solid tumors, including pancreatic ductal adenocarcinoma (PDAC).
- Clinical Trial Results: In a Phase 2a study (NCT05585320), the combination of atebimetinib with modified chemotherapy showed a 64% overall survival (OS) rate at 12 months for first-line pancreatic cancer patients.
- Fast Track Designation: In 2024, the FDA granted fast track designation for atebimetinib to treat patients with pancreatic adenocarcinoma who have progressed after one line of therapy.
- Advantage over Traditional Inhibitors: It is designed to avoid typical MAP kinase inhibitor adverse events like pyrexia (fever) while overcoming the rapid resistance often seen in other therapies.
Atebimetinib is being developed by Immuneering Corporation.
Development Status
- FDA Designations: In 2024, the FDA granted atebimetinib Fast Track designation for patients with pancreatic adenocarcinoma (PDAC) who have progressed after one line of treatment.
- Future Plans: A global registrational Phase 3 trial, named MAPKeeper 301, is planned to begin dosing patients in mid-2026.
Clinical Trial Results (Phase 2a)
Recent data from the Phase 2a trial (as of early 2026) showed significant survival benefits when combined with modified chemotherapy (gemcitabine and nab-paclitaxel) for first-line pancreatic cancer:
- Overall Survival (OS): Reported at 94% at 6 months, 86% at 9 months, and 64% at 12 months. This is roughly double the 1-year survival rate typically seen with standard chemotherapy alone (~35%).
- Progression-Free Survival (PFS): Median PFS reached 8.5 months.
- Disease Control Rate: Approximately 81% of patients achieved disease control.
SYN
WO2023076991 COMBINATION THERAPY FOR TREATING ABNORMAL CELL GROWTH
SYN
WO2025010293 MEK IMMUNE ONCOLOGY INHIBITOR PHARMACEUTICAL COMPOSITIONS
EXAMPLE 1A
Synthesis of Compound A
[0198] Compound A was prepared in 1 step:
[0199] 4-(bromomethyl)-3-(2-fluoro-3-((N-methylsulfamoyl)amino)benzyl)-2-oxo-2H-chromen-7-yl dimethylcarbamate (22.22 g, 34.79 mmol) was suspended in methanol. Dimethylamine 2M was added and the formed reaction mixture was stirred until full conversion was observed. After full conversion the reaction was concentrated under reduced pressure. IM HC1 was added to the residue and the water layer was extracted with CH2CI2. The water layer was made basic with solid Na CCE. The basic water layer was extracted with CH2CI2. The organic layer from the basic extraction was washed with brine, dried over Na2SO4, filtered and concentrated under reduced pressure to obtain the title compound (13.23 g, 25.7 mmol, yield: 74%) as a light yellow amorphous solid.
[0200] Yield: Compound A was isolated as a light yellow solid (74% over 1 step). Analysis: LCMS (Method T): tR = 1.53 min; m/z calculated for [M+H]+ = 507.2, found = 507.2; 1H NMR (400 MHz, DMSO) d 9.38 (s, 1H), 8.08 (d, J = 8.8 Hz, 1H), 7.28 (td, J = 8.0, 1.6 Hz, 1H), 7.25 – 7.18 (m, 2H), 7.15 (dd, J = 8.8, 2.4 Hz, 1H), 7.00 (t, J = 7.9 Hz, 1H), 6.90 – 6.77 (m, 1H), 4.04 (s, 2H), 3.64 (s, 2H), 3.06 (s, 3H), 2.93 (s, 3H), 2.52 (d, J = 4.9 Hz, 3H), 2.19 (s, 6H).
ADVT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- Mek immune oncology inhibitor pharmaceutical compositionsPublication Number: WO-2025010293-A2Priority Date: 2023-07-03
- Inhibiting mitogen-activated protein (map)/erk kinase (mek)1 and mek2 and related methods of treatmentPublication Number: WO-2024220440-A1Priority Date: 2023-04-17
- Methods of treating cancer with a ras mutationPublication Number: WO-2024186693-A1Priority Date: 2023-03-03
- Combination therapy for treating abnormal cell growthPublication Number: WO-2023235356-A1Priority Date: 2022-06-03
- Combination therapy for treating abnormal cell growthPublication Number: WO-2023147297-A2Priority Date: 2022-01-25
- Methods of treating abnormal cell growthPublication Number: WO-2023081676-A1Priority Date: 2021-11-02
- Combination therapy for treating abnormal cell growthPublication Number: WO-2023076991-A1Priority Date: 2021-10-28
- Mek inhibitors and therapeutic uses thereofPublication Number: US-2023119327-A1Priority Date: 2020-01-10
//////atebimetinib, FLAX LAB, antineoplastic, IMM-104, IMM 104, Fast Track designation, TEL9243A3N
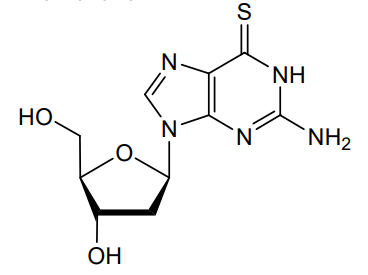
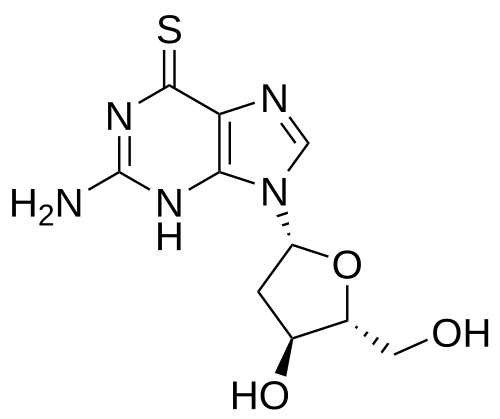
Ateganosine
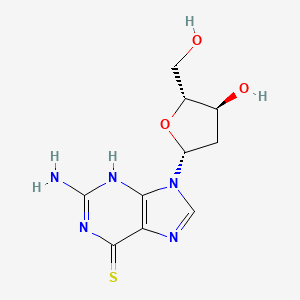
Ateganosine
CAS 789-61-7
MF C10H13N5O3S MW 283.31 g/mol
2′-deoxy-6-thioguanosine
nucleoside analogue, antineoplastic
- 6-THIO-2′-DEOXYGUANOSINE
- 2′-Deoxythioguanosine
- TGdR
- Thioguanine deoxyriboside
- KR0RFB46DF
- NSC-71261
Ateganosine is a telomerase inhibitor[1] and apoptosis inducer currently under investigation for the treatment of various cancers, including non-small cell lung cancer (NSCLC).[2]
Beta-Thioguanine Deoxyriboside is a thiopurine nucleoside derivative with antineoplastic activity. After conversion to the triphosphate, beta-thioguanine deoxyriboside is incorporated into DNA, resulting in inhibition of DNA replication. This agent is cytotoxic against leukemia cell lines and has demonstrated some activity against leukemia cells in vivo. Beta-thioguanine deoxyriboside demonstrates antineoplastic activity against 6-thioguanine-resistant tumor cells. (NCI04)
- THIO Sequenced With Cemiplimab in Advanced NSCLCCTID: NCT05208944Phase: Phase 2Status: RecruitingDate: 2025-05-31
- A Phase III Study With THIO + Cemiplimab vs Chemotherapy as 3rd Line Treatment in Advanced/Metastatic NSCLCCTID: NCT06908304Phase: Phase 3Status: Not yet recruitingDate: 2025-04-08

AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- Eglenen-Polat B, Kowash RR, Huang HC, Siteni S, Zhu M, Chen K, et al. (January 2024). “A telomere-targeting drug depletes cancer initiating cells and promotes anti-tumor immunity in small cell lung cancer”. Nature Communications. 15 (1) 672. Bibcode:2024NatCo..15..672E. doi:10.1038/s41467-024-44861-8. PMC 10803750. PMID 38253555.
- “Ateganosine”. PatSnap.
| Clinical data | |
|---|---|
| Other names | 2′-Deoxythioguanosine |
| Identifiers | |
| IUPAC name | |
| CAS Number | 789-61-7 |
| PubChem CID | 3000603 |
| DrugBank | DB18117 |
| ChemSpider | 2272164 |
| UNII | KR0RFB46DF |
| KEGG | D13071 |
| ChEMBL | ChEMBL3250476 |
| CompTox Dashboard (EPA) | DTXSID4021345 |
| Chemical and physical data | |
| Formula | C10H13N5O3S |
| Molar mass | 283.31 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
////////Ateganosine, nucleoside analogue, antineoplastic, 6-THIO-2′-DEOXYGUANOSINE, 2′-Deoxythioguanosine, TGdR, Thioguanine deoxyriboside, KR0RFB46DF, fast track designation, NSC-71261, NSC 71261
IMIPRIDONE

![7-Benzyl-4-(2-methylbenzyl)-1,2,6,7,8,9-hexahydroimidazo[1,2-A]pyrido[3,4-E]pyrimidin-5(4H)-one.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=73777259&t=l)
IMIPRIDONE
CAS No. : 1616632-77-9
Molecular Weight, 386.4964
Related CAS #: 41276-02-2 (TIC10 isomer) 1616632-77-9 (free base) 1638178-82-1 (HCl) 1777785-71-3 (HBr) 2007141-57-1 (2HBr)
TIC 10, 0NC 201, OP 10
Synonym: ONC201; ONC 201; ONC-201; NSC350625; NSC-350625; NSC 350625; TIC10; TIC 10; TIC-10; TRAIL inducing compound 10; imipridone
7-benzyl-4-(2-methylbenzyl)-1,2,6,7,8,9-hexahydroimidazo[1,2-a]pyrido[3,4-e]pyrimidin-5(4H)-one
2,4,6,7,8,9-Hexahydro-4-((2-methylphenyl)methyl)-7-phenylmethyl)imidazo)(1,2-a)pyrido(3,4-e)pyrimidin-5(1H)-one
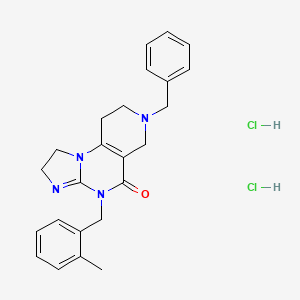
ONC-201 Dihydrochloride
459.4
UNII-53VG71J90J
53VG71J90J
Q27896336
1638178-82-1
- A TRAIL-dependent antitumor agent.
TIC10 (ONC-201) is a potent, orally active, and stable tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) inducer which acts by inhibiting Akt and ERK, consequently activating Foxo3a and significantly inducing cell surface TRAIL. TIC10 can cross the blood-brain barrier.
ONC-201, also known as TIC10, is a potent, orally active, and stable small molecule that transcriptionally induces TRAIL in a p53-independent manner and crosses the blood-brain barrier. TIC10 induces a sustained up-regulation of TRAIL in tumors and normal cells that may contribute to the demonstrable antitumor activity of TIC10. TIC10 inactivates kinases Akt and extracellular signal-regulated kinase (ERK), leading to the translocation of Foxo3a into the nucleus, where it binds to the TRAIL promoter to up-regulate gene transcription. TIC10 is an efficacious antitumor therapeutic agent that acts on tumor cells and their microenvironment to enhance the concentrations of the endogenous tumor suppressor TRAIL.
Akt/ERK Inhibitor ONC201 is a water soluble, orally bioavailable inhibitor of the serine/threonine protein kinase Akt (protein kinase B) and extracellular signal-regulated kinase (ERK), with potential antineoplastic activity. Upon administration, Akt/ERK inhibitor ONC201 binds to and inhibits the activity of Akt and ERK, which may result in inhibition of the phosphatidylinositol 3-kinase (PI3K)/Akt signal transduction pathway as well as the mitogen-activated protein kinase (MAPK)/ERK-mediated pathway. This may lead to the induction of tumor cell apoptosis mediated by tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL)/TRAIL death receptor type 5 (DR5) signaling in AKT/ERK-overexpressing tumor cells. The PI3K/Akt signaling pathway and MAPK/ERK pathway are upregulated in a variety of tumor cell types and play a key role in tumor cell proliferation, differentiation and survival by inhibiting apoptosis. In addition, ONC201 is able to cross the blood-brain barrier.
SYN
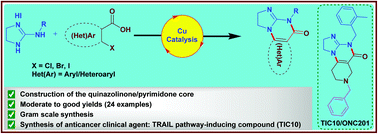
Organic & Biomolecular Chemistry, 19(39), 8497-8501; 2021
Herein, we present a copper-catalyzed tandem reaction of 2-aminoimidazolines and ortho-halo(hetero)aryl carboxylic acids that causes the regioselective formation of angularly fused tricyclic 1,2-dihydroimidazo[1,2-a]quinazolin-5(4H)-one derivatives. The reaction involved in the construction of the core six-membered pyrimidone moiety proceeded via regioselective N-arylation–condensation. The presented protocol been successfully applied to accomplish the total synthesis of TIC10/ONC201, which is an active angular isomer acting as a tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL): a sought after anticancer clinical agent.

7-Benzyl-4-(2-methylbenzyl)-1,2,6,7,8,9-hexahydroimidazo[1,2-a]pyrido[3,4-e]pyrimidin-5(4H)-one (6): Pale orange semi-solid, 202 mg (0.521 mmol), 52 % Rf = 0.25 (CH3OH/CHCl3 5:95); IR 1490, 1610, 1644, 2882, 2922 cm-1 ; 1H-NMR (500 MHz, CDCl3) δ = 2.39 (s, 3H), 2.54 (t, J = 5.5 Hz, 2H), 2.72 (t, J = 5.7 Hz, 2H), 3.31 (s, 2H), 3.67 (s, 2H), 3.84-3.91 (m, 4H), 5.04 (s, 2H), 7.02-7.04 (m, 1H), 7.08-7.12 (m, 3H), 7.26- 7.34 (m, 5H). 13C{1H}-NMR (101 MHz, CDCl3) δ = 19.3, 26.8, 43.4, 46.9, 48.2, 49.6, 50.45, 62.3, 102.1, 125.2, 125.9, 126.8, 127.4, 128.45, 129.2, 130.2, 134.2, 135.6, 137.9, 145.7, 153.3, 161.4; MS (ESI, m/z): [M+H]+ 387; HRMS (ESI, m/z): calcd for C24H27N4O [M+H]+ found 387.2183.
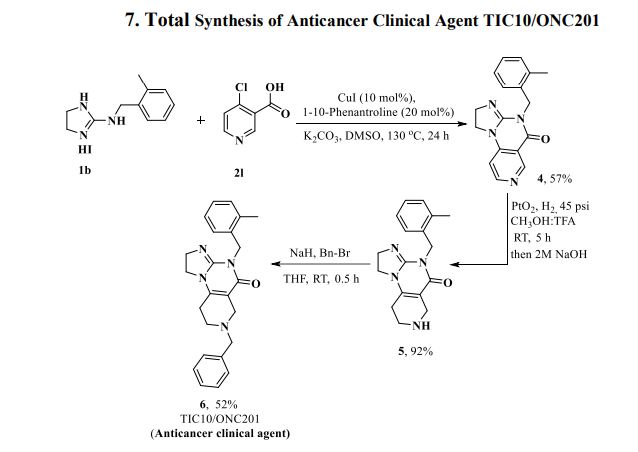


PATENT
https://patents.google.com/patent/WO2017132661A2/en
Scheme 1.


Scheme 2.



NEW DRUG APPROVALS
ONE TIME
$10.00
//////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
CLIP
https://mdanderson.elsevierpure.com/en/publications/discovery-and-clinical-introduction-of-first-in-class-imipridone-Discovery and clinical introduction of first-in-class imipridone ONC201
Abstract
ONC201 is the founding member of a novel class of anti-cancer compounds called imipridones that is currently in Phase II clinical trials in multiple advanced cancers. Since the discovery of ONC201 as a p53-independent inducer of TRAIL gene transcription, preclinical studies have determined that ONC201 has anti-proliferative and pro-apoptotic effects against a broad range of tumor cells but not normal cells. The mechanism of action of ONC201 involves engagement of PERK-independent activation of the integrated stress response, leading to tumor upregulation of DR5 and dual Akt/ERK inactivation, and consequent Foxo3a activation leading to upregulation of the death ligand TRAIL. ONC201 is orally active with infrequent dosing in animals models, causes sustained pharmacodynamic effects, and is not genotoxic. The first-in-human clinical trial of ONC201 in advanced aggressive refractory solid tumors confirmed that ONC201 is exceptionally well-tolerated and established the recommended phase II dose of 625 mg administered orally every three weeks defined by drug exposure comparable to efficacious levels in preclinical models. Clinical trials are evaluating the single agent efficacy of ONC201 in multiple solid tumors and hematological malignancies and exploring alternative dosing regimens. In addition, chemical analogs that have shown promise in other oncology indications are in pre-clinical development. In summary, the imipridone family that comprises ONC201 and its chemical analogs represent a new class of anti-cancer therapy with a unique mechanism of action being translated in ongoing clinical trials.
////////////IMIPRIDONE, TIC 10, ONC 201, NSC 350625, OP 10, Fast Track Designation, Orphan Drug Designation, Rare Pediatric Disease Designation, PHASE 3, GLIOMA, CHIMERIX
O=C1N(CC2=CC=CC=C2C)C3=NCCN3C4=C1CN(CC5=CC=CC=C5)CC4
NIROGACESTAT
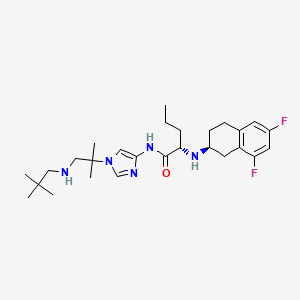

NIROGACESTAT
(2S)-2-[[(2S)-6,8-difluoro-1,2,3,4-tetrahydronaphthalen-2-yl]amino]-N-[1-[1-(2,2-dimethylpropylamino)-2-methylpropan-2-yl]imidazol-4-yl]pentanamide
489.6 g/mol, C27H41F2N5O
CAS 1290543-63-3
FDA APPROVED 11/27/2023, To treat adults with progressing desmoid tumors who require systemic treatment, Ogsiveo
PF-03084014, 1290543-63-3, PF-3084014, 865773-15-5QZ62892OFJUNII:QZ62892OFJUNII-QZ62892OFJнирогацестат [Russian] [INN]نيروغاسيستات [Arabic] [INN]尼罗司他 [Chinese] [INN]ニロガセスタット;
orphan drug designation in June 2018 for the treatment of desmoid tumors, and with a fast track designation
Nirogacestat, also known as PF-03084014, is a potent and selective gamma secretase (GS) inhibitor with potential antitumor activity. PF-03084014 binds to GS, blocking proteolytic activation of Notch receptors. Nirogacestat enhances the Antitumor Effect of Docetaxel in Prostate Cancer. Nirogacestat enhances docetaxel-mediated tumor response and provides a rationale to explore GSIs as adjunct therapy in conjunction with docetaxel for men with CRPC (castration-resistant prostate cancer).
Nirogacestat was disclosed to be a gamma-secretase inhibitor, which can inhibit Aβ-peptide production. SpringWorks Therapeutics (a spin-out of Pfizer ) is developing nirogacestat, as hydrobromide salt, a gamma-secretase inhibitor, for treating aggressive fibromatosis. In February 2021, nirogacestat was reported to be in phase 3 clinical development.
Nirogacestat is a selective gamma secretase (GS) inhibitor with potential antitumor activity. Nirogacestat binds to GS, blocking proteolytic activation of Notch receptors; Notch signaling pathway inhibition may follow, which may result in the induction of apoptosis in tumor cells that overexpress Notch. The integral membrane protein GS is a multi-subunit protease complex that cleaves single-pass transmembrane proteins, such as Notch receptors, at residues within their transmembrane domains. Overexpression of the Notch signaling pathway has been correlated with increased tumor cell growth and survival.
Nirogacestat has been used in trials studying the treatment of Breast Cancer, HIV Infection, Desmoid Tumors, Advanced Solid Tumors, and Aggressive Fibromatosis, among others.
Nirogacestat (Gamma Secretase Inhibitor)
Nirogacestat is an oral, selective, small molecule, gamma secretase inhibitor (GSI) in Phase 3 clinical development for patients with desmoid tumors. Gamma secretase is a protease complex that cleaves, or divides, multiple transmembrane protein complexes, including Notch, which, when dysregulated, can play a role in activating pathways that contribute to desmoid tumor growth.
Gamma secretase has also been shown to directly cleave BCMA, a therapeutic target that is highly expressed on multiple myeloma cells. By inhibiting gamma secretase with nirogacestat, membrane-bound BCMA can be preserved, thereby increasing target density while simultaneously reducing levels of soluble BCMA, which may serve as decoy receptors for BCMA-directed therapies. Together, these mechanisms combine to potentially enhance the activity of BCMA therapies and improve outcomes for multiple myeloma patients. SpringWorks is seeking to advance nirogacestat as a cornerstone of multiple myeloma combination therapy in collaboration with industry leaders who are advancing BCMA therapies.
SpringWorks Therapeutics Announces Clinical Collaboration with Pfizer
By Satish October 05, 2020
SpringWorks Therapeutics today announced that the company has entered into a clinical trial collaboration agreement with Pfizer to evaluate SpringWorks Therapeutics’ investigational gamma secretase inhibitor (GSI), nirogacestat, in combination with Pfizer’s anti-B-cell maturation antigen (BCMA) CD3 bispecific antibody, PF‐06863135, in patients with relapsed or refractory multiple myeloma.
Gamma secretase inhibition prevents the cleavage and shedding of BCMA from the surface of myeloma cells. In preclinical models, nirogacestat has been shown to increase the cell surface density of BCMA and reduce levels of soluble BCMA, thereby enhancing the activity of BCMA-targeted therapies, including CD3 bispecific antibodies.
Saqib Islam, Chief Executive Officer of SpringWorks Therapeutics Said: This collaboration is another important step in continuing to advance our goal of developing nirogacestat as a best-in-class BCMA potentiator, and we are pleased to work with Pfizer to study nirogacestat in combination with PF‐06863135, which has recently demonstrated promising monotherapy clinical data, We now have five collaborations with industry-leading BCMA developers to evaluate nirogacestat in combinations across modalities. We look forward to generating clinical data with our collaborators to further evaluate the ability of nirogacestat to improve outcomes for patients with multiple myeloma.

Under the terms of the agreement, Pfizer will sponsor and conduct the Phase 1b/2 study to evaluate the safety, tolerability and preliminary efficacy of the combination, and will assume all costs associated with the study, other than expenses related to the manufacturing of nirogacestat and certain expenses related to intellectual property rights. Pfizer and SpringWorks Therapeutics will also form a joint development committee to manage the clinical study, which is expected to commence in the first half of 2021.
Chris Boshoff, MD, PhD, Chief Development Officer for Pfizer Oncology at Pfizer Said: Entering into this clinical collaboration is a proud milestone in our strong relationship with SpringWorks,We believe that studying nirogacestat in combination with PF-06863135 could hold significant therapeutic promise for patients with relapsed or refractory multiple myeloma, and we look forward to working together to advance this important area of research.
In addition to its ongoing clinical collaborations with BCMA-directed therapies, SpringWorks is also currently conducting a global Phase 3, double-blind, randomized, placebo-controlled clinical trial (the DeFi Trial) to evaluate nirogacestat in adults with progressing desmoid tumors.
About Nirogacestat
Nirogacestat is an investigational, oral, selective, small molecule gamma secretase inhibitor in Phase 3 clinical development for desmoid tumors, which are rare and often debilitating and disfiguring soft-tissue tumors. Gamma secretase cleaves multiple transmembrane protein complexes, including Notch, which is believed to play a role in activating pathways that contribute to desmoid tumor growth.
In addition, gamma secretase has been shown to directly cleave membrane-bound BCMA, resulting in the release of the BCMA extracellular domain, or ECD, from the cell surface. By inhibiting gamma secretase, membrane-bound BCMA can be preserved, increasing target density while reducing levels of soluble BCMA ECD, which may serve as decoy receptors for BCMA-directed therapies. Nirogacestat’s ability to enhance the activity of BCMA-directed therapies has been observed in preclinical models of multiple myeloma. SpringWorks is evaluating nirogacestat as a BCMA potentiator and has five collaborations with industry-leading BCMA developers to evaluate nirogacestat in combinations across modalities, including with an antibody-drug conjugate, two CAR T cell therapies and two bispecific antibodies. In addition, SpringWorks and Fred Hutchinson Cancer Research Center have entered into a sponsored research agreement to further characterize the ability of nirogacestat to modulate BCMA and potentiate BCMA directed therapies using a variety of preclinical and patient-derived multiple myeloma models developed by researchers at Fred Hutch.
Nirogacestat has received Orphan Drug Designation from the U.S. Food and Drug Administration (FDA) for the treatment of desmoid tumors (June 2018) and from the European Commission for the treatment of soft tissue sarcoma (September 2019). The FDA also granted Fast Track and Breakthrough Therapy Designations for the treatment of adult patients with progressive, unresectable, recurrent or refractory desmoid tumors or deep fibromatosis (November 2018 and August 2019).
About PF‐06863135
PF‐06863135 is an anti-B-cell maturation antigen (BCMA) CD3 bispecific antibody being investigated in a Phase 1 clinical study to treat relapsed or refractory multiple myeloma. This bispecific antibody can be administered subcutaneously and has been optimized for binding affinity to both BCMA and CD3, enabling more potent T-cell-mediated tumor cell toxicity.
Source: SpringWorks Therapeutics
FDA Grants Breakthrough Designation to Nirogacestat for Desmoid Tumors
The FDA has granted nirogacestat, an investigational gamma-secretase inhibitor, with a breakthrough therapy designation for the treatment of adult patients with progressive, unresectable, recurrent or refractory desmoid tumors or deep fibromatosis.
The FDA has granted nirogacestat (PF-03084014), an investigational gamma-secretase inhibitor, with a breakthrough therapy designation for the treatment of adult patients with progressive, unresectable, recurrent or refractory desmoid tumors or deep fibromatosis.1
The breakthrough designation was granted as a result of positive findings seen in phase I and II trials of nirogacestat monotherapy in patients with desmoid tumors. A phase III trial has also been initiated investigating nirogacestat in patients with desmoid tumors or aggressive fibromatosis (NCT03785964).
“We are committed to pursuing the rapid development of nirogacestat given the important need for new therapies for patients with desmoid tumors and are pleased to receive this breakthrough therapy designation,” Saqib Islam, CEO of SpringWorks, the company developing the small molecule inhibitor, said in a statement. “We are currently enrolling adult patients in our phase III DeFi trial and will continue to work closely with the FDA with the goal of bringing nirogacestat to patients as quickly as possible.”
The open-label, single-center phase II trial of nirogacestat enrolled 17 patients with desmoid tumors who were not eligible for surgical resection or definitive radiation therapy and who had experienced disease progression after at least 1 prior treatment regimen. Patients received 150 mg twice per day of continuous, oral nirogacestat in 21-day cycles.2
The median age of patients was 34 years (range, 19-69), 82% of the patients were female, and 53% of patients had aCTNNB1T41A somatic missense mutation. The median number of prior therapies was 4 (range, 1-9), which included cytotoxic chemotherapy in 71% and a tyrosine kinase inhibitor in 59%.
Sixteen patients were evaluable for response. After a median follow-up of more than 25 months, 5 patients (29%) achieved a partial response and 11 (65%) had stable disease, for a disease control rate of 100%. Ten patients (59%) remained on treatment with nirogacestat for more than 2 years.
Grade 1/2 adverse events were observed in all patients, with diarrhea (76%) and skin disorders (71%) being the most common toxicities. The only treatment-related grade 3 event was reversible hypophosphatemia, which was reported in 8 patients (47%) and was considered to be a class effect of gamma-secretase inhibitors. Four patients met the criteria for dose reduction.
Findings from the phase I study also showed a disease control rate of 100% with nirogacestat. However, the median progression-free survival was not reached in either study due to a lack of patients progressing on treatment. Only 1 patient discontinued treatment due to an adverse event between the 2 studies.1
The FDA had previously granted nirogacestat with an orphan drug designation in June 2018 for the treatment of desmoid tumors, and with a fast track designation in November 2018 for the treatment of adult patients with progressive, unresectable, recurrent or refractory desmoid tumors or deep fibromatosis.
References
- SpringWorks Therapeutics Receives Breakthrough Therapy Designation for Nirogacestat for the Treatment of Adult Patients with Progressive, Unresectable, Recurrent or Refractory Desmoid Tumors [press release]. Stamford, CT: SpringWorks Therapeutics, Inc; August 29, 2019. https://bit.ly/30IV0Eb. Accessed September 3, 2019.
- Kummar S, O’Sullivan Coyne G, Do KT, et al. Clinical Activity of the γ-Secretase Inhibitor PF-03084014 in Adults With Desmoid Tumors (Aggressive Fibromatosis).J Clin Oncol.2017;35(14):1561-1569. doi: 10.1200/JCO.2016.71.1994.
PAPER

Bioorganic & medicinal chemistry letters (2011), 21(9), 2637-40.
https://www.sciencedirect.com/science/article/abs/pii/S0960894X10018822



PATENT
WO 2016089208
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016089208
PATENT
WO-2021029854
Novel, stable crystalline polymorphic (A to N) and amorphous forms of nirogacestat hydrobromide , useful for treating desmoid tumors such as multiple myeloma, a cancer having a mutation in a Notch pathway gene, adenoid cystic carcinoma and T-cell acute lymphoblastic leukemia.
(S)-2-(((S)-6,8-difluoro-l,2,3,4-tetrahydronaphthalen-2-yl)amino)-N-(l-(2- methyl- l-(neopentylamino) propan-2-yl)-lH-imidazol-4-yl)pentanamide (“Compound 1”) is a gamma-secretase inhibitor which can inhibit Ab-peptide production.
[0003] Not all compounds that are gamma-secretase inhibitors have characteristics affording the best potential to become useful therapeutics. Some of these characteristics include high affinity at the gamma-secretase, duration of gamma-secretase deactivation, oral bioavailability, tissue distribution, and stability (e.g., ability to formulate or crystallize, shelf life). Favorable characteristics can lead to improved safety, tolerability, efficacy, therapeutic index, patient compliance, cost efficiency, manufacturing ease, etc.
[0004] In addition, the isolation and commercial -scale preparation of a solid state form of hydrobromide salts of Compound 1 and corresponding pharmaceutical formulations having acceptable solid state properties (including chemical stability, thermal stability, solubility, hygroscopicity, and/or particle size), compound manufacturability (including yield, impurity rejection during crystallization, filtration properties, drying properties, and milling properties), and formulation feasibility (including stability with respect to pressure or compression forces during tableting) present a number of challenges.
[0005] Accordingly, there is a current need for one or more solid state forms of hydrobromide salts of Compound 1 that have an acceptable balance of these properties and can be used in the preparation of pharmaceutically acceptable solid dosage forms.
Crystalline Form A
[0147] In one aspect, the present disclosure relates to crystalline Form A of a hydrobromide salt of (S)-2-(((S)-6,8-difluoro-l,2,3,4-tetrahydronaphthalen-2-yl)amino)- N-(l -(2 -methyl- l-(neopentylamino) propan-2-yl)-lH-imidazol-4-yl)pentanamide having Formula (I),
[0148] In one embodiment, crystalline Form A is anhydrous.
[0149] In another embodiment, the melting point of crystalline Form A is about 254 °C.
[0150] In another embodiment, Form A is characterized by an XRPD pattern having peaks at 8.8 ± 0.2, 9.8 ± 0.2, and 23.3 ± 0.2 degrees two theta when measured by Cu Ka radiation. In another embodiment, Form A is characterized by an XRPD pattern having peaks at 8.8 ± 0.2, 9.8 ± 0.2, 23.3 ± 0.2, 25.4 ± 0.2, 28.0 ± 0.2, and 29.3 ± 0.2 degrees two theta when measured by Cu Ka radiation. In another embodiment, Form A is characterized by an XRPD pattern having peaks at 8.8 ± 0.2, 9.8 ± 0.2, 20.0 ± 0.2, 23.3 ± 0.2, 25.4 ± 0.2, 28.0 ± 0.2, 29.3 ± 0.2, and 32.5 ± 0.2 degrees two theta when measured by Cu Ka radiation.
Patent
Product case, WO2005092864 ,
hold protection in the EU states until March 2025, and expire in the US in February 2026 with US154 extension.
PATENT
WO2020208572 , co-assigned to GSK and SpringWorks, claiming a combination of nirogacestat with anti-BCMA antibody (eg belantamab mafodotin ), for treating cancer.
PATENT
US10590087 , for a prior filing from Pfizer, claiming crystalline forms of nirogacestat hydrobromide.
////////////NIROGACESTAT, orphan drug designation, esmoid tumors, fast track designation, PF-03084014, PF 03084014, QZ62892OFJ , UNII:QZ62892OFJ ,UNII-QZ62892OFJ, ,нирогацестат , نيروغاسيستات , 尼罗司他 , ニロガセスタット, phase 3
CCCC(C(=O)NC1=CN(C=N1)C(C)(C)CNCC(C)(C)C)NC2CCC3=C(C2)C(=CC(=C3)F)F
Teprotumumab-trbw
Tepezza (teprotumumab-trbw)
Company: Horizon Therapeutics plc
Date of Approval: January 21, 2020
Treatment for: Thyroid Eye Disease
UNIIY64GQ0KC0A
CAS number1036734-93-6
R-1507 / R1507 / RG-1507 / RG1507 / RO-4858696 / RO-4858696-000 / RO-4858696000 / RO4858696 / RO4858696-000 / RV-001 / RV001
Tepezza (teprotumumab-trbw) is a fully human monoclonal antibody (mAb) and a targeted inhibitor of the insulin-like growth factor 1 receptor (IGF-1R) for the treatment of active thyroid eye disease (TED).
FDA Approves Tepezza (teprotumumab-trbw) for the Treatment of Thyroid Eye Disease (TED) – January 21, 2020
Today, the U.S. Food and Drug Administration (FDA) approved Tepezza (teprotumumab-trbw) for the treatment of adults with thyroid eye disease, a rare condition where the muscles and fatty tissues behind the eye become inflamed, causing the eyes to be pushed forward and bulge outwards (proptosis). Today’s approval represents the first drug approved for the treatment of thyroid eye disease.
“Today’s approval marks an important milestone for the treatment of thyroid eye disease. Currently, there are very limited treatment options for this potentially debilitating disease. This treatment has the potential to alter the course of the disease, potentially sparing patients from needing multiple invasive surgeries by providing an alternative, non surgical treatment option,” said Wiley Chambers, M.D., deputy director of the Division of Transplant and Ophthalmology Products in the FDA’s Center for Drug Evaluation and Research. “Additionally, thyroid eye disease is a rare disease that impacts a small percentage of the population, and for a variety of reasons, treatments for rare diseases are often unavailable. This approval represents important progress in the approval of effective treatments for rare diseases, such as thyroid eye disease.”
Thyroid eye disease is associated with the outward bulging of the eye that can cause a variety of symptoms such as eye pain, double vision, light sensitivity or difficulty closing the eye. This disease impacts a relatively small number of Americans, with more women than men affected. Although this condition impacts relatively few individuals, thyroid eye disease can be incapacitating. For example, the troubling ocular symptoms can lead to the progressive inability of people with thyroid eye disease to perform important daily activities, such as driving or working.
Tepezza was approved based on the results of two studies (Study 1 and 2) consisting of a total of 170 patients with active thyroid eye disease who were randomized to either receive Tepezza or a placebo. Of the patients who were administered Tepezza, 71% in Study 1 and 83% in Study 2 demonstrated a greater than 2 millimeter reduction in proptosis (eye protrusion) as compared to 20% and 10% of subjects who received placebo, respectively.
The most common adverse reactions observed in patients treated with Tepezza are muscle spasm, nausea, alopecia (hair loss), diarrhea, fatigue, hyperglycemia (high blood sugar), hearing loss, dry skin, dysgeusia (altered sense of taste) and headache. Tepezza should not be used if pregnant, and women of child-bearing potential should have their pregnancy status verified prior to beginning treatment and should be counseled on pregnancy prevention during treatment and for 6 months following the last dose of Tepezza.
The FDA granted this application Priority Review, in addition to Fast Track and Breakthrough Therapy Designation. Additionally, Tepezza received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases or conditions. Development of this product was also in part supported by the FDA Orphan Products Grants Program, which provides grants for clinical studies on safety and efficacy of products for use in rare diseases or conditions.
The FDA granted the approval of Tepezza to Horizon Therapeutics Ireland DAC.
Teprotumumab (RG-1507), sold under the brand name Tepezza, is a medication used for the treatment of adults with thyroid eye disease, a rare condition where the muscles and fatty tissues behind the eye become inflamed, causing the eyes to be pushed forward and bulge outwards (proptosis).[1]
The most common adverse reactions observed in people treated with teprotumumab-trbw are muscle spasm, nausea, alopecia (hair loss), diarrhea, fatigue, hyperglycemia (high blood sugar), hearing loss, dry skin, dysgeusia (altered sense of taste) and headache.[1] Teprotumumab-trbw should not be used if pregnant, and women of child-bearing potential should have their pregnancy status verified prior to beginning treatment and should be counseled on pregnancy prevention during treatment and for six months following the last dose of teprotumumab-trbw.[1]
It is a human monoclonal antibody developed by Genmab and Roche. It binds to IGF-1R.
Teprotumumab was first investigated for the treatment of solid and hematologic tumors, including breast cancer, Hodgkin’s and non-Hodgkin’s lymphoma, non-small cell lung cancer and sarcoma.[2][3] Although results of phase I and early phase II trials showed promise, research for these indications were discontinued in 2009 by Roche. Phase II trials still in progress were allowed to complete, as the development was halted due to business prioritization rather than safety concerns.
Teprotumumab was subsequently licensed to River Vision Development Corporation in 2012 for research in the treatment of ophthalmic conditions. Horizon Pharma (now Horizon Therapeutics, from hereon Horizon) acquired RVDC in 2017, and will continue clinical trials.[4] It is in phase III trials for Graves’ ophthalmopathy (also known as thyroid eye disease (TED)) and phase I for diabetic macular edema.[5] It was granted Breakthrough Therapy, Orphan Drug Status and Fast Track designations by the FDA for Graves’ ophthalmopathy.[6]
In a multicenter randomized trial in patients with active Graves’ ophthalmopathy Teprotumumab was more effective than placebo in reducing the clinical activity score and proptosis.[7] In February 2019 Horizon announced results from a phase 3 confirmatory trial evaluating teprotumumab for the treatment of active thyroid eye disease (TED). The study met its primary endpoint, showing more patients treated with teprotumumab compared with placebo had a meaningful improvement in proptosis, or bulging of the eye: 82.9 percent of teprotumumab patients compared to 9.5 percent of placebo patients achieved the primary endpoint of a 2 mm or more reduction in proptosis (p<0.001). Proptosis is the main cause of morbidity in TED. All secondary endpoints were also met and the safety profile was consistent with the phase 2 study of teprotumumab in TED.[8] On 10th of July 2019 Horizon submitted a Biologics License Application (BLA) to the FDA for teprotumumab for the Treatment of Active Thyroid Eye Disease (TED). Horizon requested priority review for the application – if so granted (FDA has a 60-day review period to decide) it would result in a max. 6 month review process.[9]
History[edit]
Teprotumumab-trbw was approved for use in the United States in January 2020, for the treatment of adults with thyroid eye disease.[1]
Teprotumumab-trbw was approved based on the results of two studies (Study 1 and 2) consisting of a total of 170 patients with active thyroid eye disease who were randomized to either receive teprotumumab-trbw or a placebo.[1] Of the subjects who were administered Tepezza, 71% in Study 1 and 83% in Study 2 demonstrated a greater than two millimeter reduction in proptosis (eye protrusion) as compared to 20% and 10% of subjects who received placebo, respectively.[1]
The U.S. Food and Drug Administration (FDA) granted the application for teprotumumab-trbw fast track designation, breakthrough therapy designation, priority review designation, and orphan drug designation.[1] The FDA granted the approval of Tepezza to Horizon Therapeutics Ireland DAC.[1]
References
- ^ Jump up to:a b c d e f g h “FDA approves first treatment for thyroid eye disease”. U.S. Food and Drug Administration (FDA) (Press release). 21 January 2020. Retrieved 21 January 2020.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ https://clinicaltrials.gov/ct2/show/NCT01868997
- ^ http://adisinsight.springer.com/drugs/800015801
- ^ http://www.genmab.com/product-pipeline/products-in-development/teprotumumab
- ^ http://adisinsight.springer.com/drugs/800015801
- ^ http://www.genmab.com/product-pipeline/products-in-development/teprotumumab
- ^ Smith, TJ; Kahaly, GJ; Ezra, DG; Fleming, JC; Dailey, RA; Tang, RA; Harris, GJ; Antonelli, A; Salvi, M; Goldberg, RA; Gigantelli, JW; Couch, SM; Shriver, EM; Hayek, BR; Hink, EM; Woodward, RM; Gabriel, K; Magni, G; Douglas, RS (4 May 2017). “Teprotumumab for Thyroid-Associated Ophthalmopathy”. The New England Journal of Medicine. 376 (18): 1748–1761. doi:10.1056/NEJMoa1614949. PMC 5718164. PMID 28467880.
- ^ “Horizon Pharma plc Announces Phase 3 Confirmatory Trial Evaluating Teprotumumab (OPTIC) for the Treatment of Active Thyroid Eye Disease (TED) Met Primary and All Secondary Endpoints”. Horizon Pharma plc. Retrieved 22 March 2019.
- ^ “Horizon Therapeutics plc Submits Teprotumumab Biologics License Application (BLA) for the Treatment of Active Thyroid Eye Disease (TED)”. Horizon Therapeutics plc. Retrieved 27 August 2019.
External links
- “Teprotumumab”. Drug Information Portal. U.S. National Library of Medicine.
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | IGF-1R |
| Clinical data | |
| Other names | teprotumumab-trbw, RG-1507 |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| DrugBank | |
| ChemSpider |
|
| UNII | |
| KEGG | |
| ChEMBL | |
| ECHA InfoCard | 100.081.384 |
| Chemical and physical data | |
| Formula | C6476H10012N1748O2000S40 |
| Molar mass | 145.6 kg/mol g·mol−1 |
/////////Teprotumumab-trbw, APPROVALS 2020, FDA 2020, ORPHAN, BLA, fast track designation, breakthrough therapy designation, priority review designation, and orphan drug designation, Tepezza, Horizon Therapeutics, MONOCLONAL ANTIBODY, 2020 APPROVALS, active thyroid eye disease, Teprotumumab
https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-thyroid-eye-disease
Beperminogene perplasmid, ベペルミノゲンペルプラスミド
1gctgcttcgc gatgtacggg ccagatatac gcgttgacat tgattattga
51ctagttatta atagtaatca attacggggt cattagttca tagcccatat
101atggagttcc gcgttacata acttacggta aatggcccgc ctggctgacc
151gcccaacgac ccccgcccat tgacgtcaat aatgacgtat gttcccatag
201taacgccaat agggactttc cattgacgtc aatgggtgga gtatttacgg
251taaactgccc acttggcagt acatcaagtg tatcatatgc caagtacgcc
301ccctattgac gtcaatgacg gtaaatggcc cgcctggcat tatgcccagt
351acatgacctt atgggacttt cctacttggc agtacatcta cgtattagtc
401atcgctatta ccatggtgat gcggttttgg cagtacatca atgggcgtgg
451atagcggttt gactcacggg gatttccaag tctccacccc attgacgtca
501atgggagttt gttttggcac caaaatcaac gggactttcc aaaatgtcgt
551aacaactccg ccccattgac gcaaatgggc ggtaggcgtg tacggtggga
601ggtctatata agcagagctc tctggctaac tagagaaccc actgcttact
651ggcttatcga aattaatacg actcactata gggagaccca agctggctag
701cgtttaaact taagcttggt accgagctcg gatccgccag cccgtccagc
751agcaccatgt gggtgaccaa actcctgcca gccctgctgc tgcagcatgt
801cctcctgcat ctcctcctgc tccccatcgc catcccctat gcagagggac
851aaaggaaaag aagaaataca attcatgaat tcaaaaaatc agcaaagact
901accctaatca aaatagatcc agcactgaag ataaaaacca aaaaagtgaa
951tactgcagac caatgtgcta atagatgtac taggaataaa ggacttccat
1001tcacttgcaa ggcttttgtt tttgataaag caagaaaaca atgcctctgg
1051ttccccttca atagcatgtc aagtggagtg aaaaaagaat ttggccatga
1101atttgacctc tatgaaaaca aagactacat tagaaactgc atcattggta
1151aaggacgcag ctacaaggga acagtatcta tcactaagag tggcatcaaa
1201tgtcagccct ggagttccat gataccacac gaacacagct ttttgccttc
1251gagctatcgg ggtaaagacc tacaggaaaa ctactgtcga aatcctcgag
1301gggaagaagg gggaccctgg tgtttcacaa gcaatccaga ggtacgctac
1351gaagtctgtg acattcctca gtgttcagaa gttgaatgca tgacctgcaa
1401tggggagagt tatcgaggtc tcatggatca tacagaatca ggcaagattt
1451gtcagcgctg ggatcatcag acaccacacc ggcacaaatt cttgcctgaa
1501agatatcccg acaagggctt tgatgataat tattgccgca atcccgatgg
1551ccagccgagg ccatggtgct atactcttga ccctcacacc cgctgggagt
1601actgtgcaat taaaacatgc gctgacaata ctatgaatga cactgatgtt
1651cctttggaaa caactgaatg catccaaggt caaggagaag gctacagggg
1701cactgtcaat accatttgga atggaattcc atgtcagcgt tgggattctc
1751agtatcctca cgagcatgac atgactcctg aaaatttcaa gtgcaaggac
1801ctacgagaaa attactgccg aaatccagat gggtctgaat caccctggtg
1851ttttaccact gatccaaaca tccgagttgg ctactgctcc caaattccaa
1901actgtgatat gtcacatgga caagattgtt atcgtgggaa tggcaaaaat
1951tatatgggca acttatccca aacaagatct ggactaacat gttcaatgtg
2001ggacaagaac atggaagact tacatcgtca tatcttctgg gaaccagatg
2051caagtaagct gaatgagaat tactgccgaa atccagatga tgatgctcat
2101ggaccctggt gctacacggg aaatccactc attccttggg attattgccc
2151tatttctcgt tgtgaaggtg ataccacacc tacaatagtc aatttagacc
2201atcccgtaat atcttgtgcc aaaacgaaac aattgcgagt tgtaaatggg
2251attccaacac gaacaaacat aggatggatg gttagtttga gatacagaaa
2301taaacatatc tgcggaggat cattgataaa ggagagttgg gttcttactg
2351cacgacagtg tttcccttct cgagacttga aagattatga agcttggctt
2401ggaattcatg atgtccacgg aagaggagat gagaaatgca aacaggttct
2451caatgtttcc cagctggtat atggccctga aggatcagat ctggttttaa
2501tgaagcttgc caggcctgct gtcctggatg attttgttag tacgattgat
2551ttacctaatt atggatgcac aattcctgaa aagaccagtt gcagtgttta
2601tggctggggc tacactggat tgatcaacta tgatggccta ttacgagtgg
2651cacatctcta tataatggga aatgagaaat gcagccagca tcatcgaggg
2701aaggtgactc tgaatgagtc tgaaatatgt gctggggctg aaaagattgg
2751atcaggacca tgtgaggggg attatggtgg cccacttgtt tgtgagcaac
2801ataaaatgag aatggttctt ggtgtcattg ttcctggtcg tggatgtgcc
2851attccaaatc gtcctggtat ttttgtccga gtagcatatt atgcaaaatg
2901gatacacaaa attattttaa catataaggt accacagtca tagctgttaa
2951cccgggtcga agcggccgct cgagtctaga gggcccgttt aaacccgctg
3001atcagcctcg actgtgcctt ctagttgcca gccatctgtt gtttgcccct
3051cccccgtgcc ttccttgacc ctggaaggtg ccactcccac tgtcctttcc
3101taataaaatg aggaaattgc atcgcattgt ctgagtaggt gtcattctat
3151tctggggggt ggggtggggc aggacagcaa gggggaggat tgggaagaca
3201atagcaggca tgctggggat gcggtgggct ctatggcttc tactgggcgg
3251ttttatggac agcaagcgaa ccggaattgc cagctggggc gccctctggt
3301aaggttggga agccctgcaa agtaaactgg atggctttct tgccgccaag
3351gatctgatgg cgcaggggat caagctctga tcaagagaca ggatgaggat
3401cgtttcgcat gattgaacaa gatggattgc acgcaggttc tccggccgct
3451tgggtggaga ggctattcgg ctatgactgg gcacaacaga caatcggctg
3501ctctgatgcc gccgtgttcc ggctgtcagc gcaggggcgc ccggttcttt
3551ttgtcaagac cgacctgtcc ggtgccctga atgaactgca agacgaggca
3601gcgcggctat cgtggctggc cacgacgggc gttccttgcg cagctgtgct
3651cgacgttgtc actgaagcgg gaagggactg gctgctattg ggcgaagtgc
3701cggggcagga tctcctgtca tctcaccttg ctcctgccga gaaagtatcc
3751atcatggctg atgcaatgcg gcggctgcat acgcttgatc cggctacctg
3801cccattcgac caccaagcga aacatcgcat cgagcgagca cgtactcgga
3851tggaagccgg tcttgtcgat caggatgatc tggacgaaga gcatcagggg
3901ctcgcgccag ccgaactgtt cgccaggctc aaggcgagca tgcccgacgg
3951cgaggatctc gtcgtgaccc atggcgatgc ctgcttgccg aatatcatgg
4001tggaaaatgg ccgcttttct ggattcatcg actgtggccg gctgggtgtg
4051gcggaccgct atcaggacat agcgttggct acccgtgata ttgctgaaga
4101gcttggcggc gaatgggctg accgcttcct cgtgctttac ggtatcgccg
4151ctcccgattc gcagcgcatc gccttctatc gccttcttga cgagttcttc
4201tgaattatta acgcttacaa tttcctgatg cggtattttc tccttacgca
4251tctgtgcggt atttcacacc gcatcaggtg gcacttttcg gggaaatgtg
4301cgcggaaccc ctatttgttt atttttctaa atacattcaa atatgtatcc
4351gctcatgaga caataaccct gataaatgct tcaataatag cacgtgctaa
4401aacttcattt ttaatttaaa aggatctagg tgaagatcct ttttgataat
4451ctcatgacca aaatccctta acgtgagttt tcgttccact gagcgtcaga
4501ccccgtagaa aagatcaaag gatcttcttg agatcctttt tttctgcgcg
4551taatctgctg cttgcaaaca aaaaaaccac cgctaccagc ggtggtttgt
4601ttgccggatc aagagctacc aactcttttt ccgaaggtaa ctggcttcag
4651cagagcgcag ataccaaata ctgttcttct agtgtagccg tagttaggcc
4701accacttcaa gaactctgta gcaccgccta catacctcgc tctgctaatc
4751ctgttaccag tggctgctgc cagtggcgat aagtcgtgtc ttaccgggtt
4801ggactcaaga cgatagttac cggataaggc gcagcggtcg ggctgaacgg
4851ggggttcgtg cacacagccc agcttggagc gaacgaccta caccgaactg
4901agatacctac agcgtgagct atgagaaagc gccacgcttc ccgaagggag
4951aaaggcggac aggtatccgg taagcggcag ggtcggaaca ggagagcgca
5001cgagggagct tccaggggga aacgcctggt atctttatag tcctgtcggg
5051tttcgccacc tctgacttga gcgtcgattt ttgtgatgct cgtcaggggg
5101gcggagccta tggaaaaacg ccagcaacgc ggccttttta cggttcctgg
5151ccttttgctg gccttttgct cacatgttct t
Beperminogene perplasmid
ベペルミノゲンペルプラスミド
HGF plasmid
- DNA (human hepatocyte growth factor plasmid pVAX1 cDNA)
- DNA (plasmid pVAX1HGF/MGBI)
- AMG-0001
DS-992
Nucleic Acid Sequence
Sequence Length: 51811342 a 1223 c 1314 g 1302 t
APPROVED, japan 2019, Collategene, 2019/3/29
Antiparkinsonian, Angiogenesis inducing agent
CAS: 627861-07-8
- Originator AnGes MG
- Developer AnGes MG; Osaka University Hospital
- Class Antiparkinsonians; Gene therapies; Ischaemic heart disorder therapies; Vascular disorder therapies
- Mechanism of Action Angiogenesis inducing agents; Gene transference; Hepatocyte growth factor expression stimulants
- Available For Licensing Yes – Ischaemic heart disorders; Lymphoedema; Parkinson’s disease
- Registered Peripheral arterial disorders
- Phase I/II Lymphoedema
- No development reported Arteriosclerosis obliterans; Ischaemic heart disorders; Parkinson’s disease; Thromboangiitis obliterans
- 26 Mar 2019 Registered for Peripheral arterial disorders in Japan (IM)
- 21 Feb 2019 The Pharmaceutical Affairs and Food Sanitation Council recommends conditional and time-limited approval of beperminogene perplasmid for the improvement of ulcers associated with chronic peripheral arterial disease
- 21 Feb 2019 AnGes plans a clinical study to assess the efficacy of beperminogene perplasmid in improvement of pain at rest in chronic peripheral arterial disorders
- In 2010, the product received fast track designation in the U.S. for the treatment of critical limb ischemia
HGF Plasmid (Beperminogene Perplasmid)Critical Limb Ischemia (Arteriosclerosis Obliterans & Buerger’s Disease) AMG0001 Injection, JAPAN AND US ALLIANCE Mitsubishi Tanabe Pharma
PATENT
WO 2017126488
US 20170283446
Expert Review of Cardiovascular Therapy (2014), 12(10), 1145-1156.
////////////Beperminogene perplasmid, japan 2019, ベペルミノゲンペルプラスミド , AnGes MG, Osaka University Hospital, Critical Limb Ischemia, Arteriosclerosis Obliterans, Buerger’s Disease, AMG0001, AMG-0001, DS-992 , HGF plasmid , fast track designation
Plazomicin sulfate, プラゾマイシン硫酸塩 ,


Plazomicin
- Molecular FormulaC25H48N6O10
- Average mass592.683 Da
(2S)-4-Amino-N-[(1R,2S,3S,4R,5S)-5-amino-4-{[(2S,3R)-3-amino-6-{[(2-hydroxyéthyl)amino]méthyl}-3,4-dihydro-2H-pyran-2-yl]oxy}-2-{[3-désoxy-4-C-méthyl-3-(méthylamino)-β-L-arabinopyranosyl]oxy}-3-hyd roxycyclohexyl]-2-hydroxybutanamide [French][ACD/IUPAC Name]
1154757-24-0 [RN]
9522
ACHN-490
Plazomicin Sulfate
| Molecular Formula: | C25H50N6O14S |
|---|---|
| Molecular Weight: | 690.763 g/mol |
Plazomicin Sulfate; UNII-A78L6MT746; Plazomicin Sulfate [USAN]; A78L6MT746; 1380078-95-4; Plazomicin sulfate (USAN),
|
6′-(hydroxylethyl)-1-(haba)-sisomicin
Plazomicin is a neoglycoside antibiotic with activity against a broad range of Gram-positive and Gram-negive pathogens. Plazomicin showed potent in vitro activity against multidrug-resistant Klebsiella pneumoniae and Escherichia coli.
- Mechanism of ActionProtein synthesis inhibitors
- Orphan Drug StatusNo
- New Molecular EntityYes
Highest Development Phases
- MarketedUrinary tract infections
- RegisteredPyelonephritis
- PreregistrationBacteraemia; Nosocomial pneumonia
- PreclinicalGram-negative infections
- No development reportedRespiratory tract infections; Tularaemia; Yersinia infections
Most Recent Events
- 27 Jun 2018Registered for Pyelonephritis (Treatment-resistant) in USA (IV)- First Global Approval
- 27 Jun 2018Registered for Urinary tract infections (Treatment-resistant) in USA (IV)- First Global Approval
- 26 Jun 2018Achaogen receives complete response letter from the FDA for Plazomicin in Bloodstream infection
| Synonyms: O-2-Amino-2,3,4,6-tetradeoxy-6-[(2-hydroxyethyl)amino]-α-D-glycero-hex-4-enopyranosyl-(1→4)-O-[3-deoxy-4-C-methyl-3-(methylamino)-β-L-arabinopyranosyl-(1→6)]-N1-[(2S)-4-amino-2-hydroxy-1-oxobutyl]-2-deoxy-D-streptamine; ACHN 490; |
| CAS Number: 1154757-24-0
Sulfate 1380078-95-4, プラゾマイシン硫酸塩; |
| Achaogen (USA)Phase II completed |
| Mol. Formula: C25H48N6O10 |
| Aminoglycosides, Broad-spectrum, |
| Mol. Weight: 592.68 |
FDA
Click to access 210303Orig1s000lbl.pdf
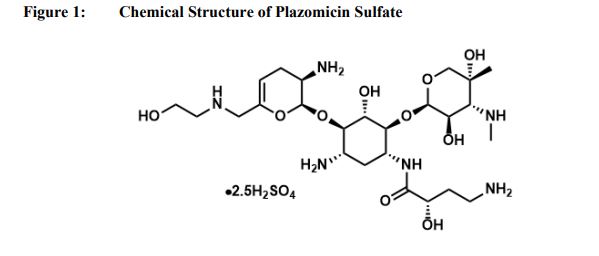
Developed by Achaogen biopharmaceuticals, plazomicin is a next-generation aminoglycoside synthetically derived from [DB12604]. The structure of plazomicin was established via appending hydroxylaminobutyric acid to [DB12604] at position 1 and 2-hydroxyethyl group at position 6′ [A33942]. It was designed to evade all clinically relevant aminoglycoside-modifying enzymes, which contribute to the main resistance mechanism for aminoglycoside therapy [A33942]. However, acquired resistance of aminoglycosides may arise through over expression of efflux pumps and ribosomal modification by bacteria, which results from amino acid or rRNA sequence mutations [A33942]. Like other aminoglycosides, plazomicin is ineffective against bacterial isolates that produce 16S rRNA methyltransferases [FDA Label]. Plazomicin mediates the antibacterial activity against pathogens including carbapenem-resistant (CRE) and extended-spectrum beta-lactamase (ESBL) producing _Enterobacteriaceae_. It mediates the antibacterial activity by binding to bacterial 30S ribosomal subunit and inhibiting protein synthesis [FDA Label]. On June 28th, 2018, plazomicin sulfate was approved by the FDA for use in adult patients for the treatment of complicated urinary tract infections (cUTI) including Pyelonephritis. It is marketed as Zemdri and is administered via once-daily intravenous infusion.
Developed by Achaogen biopharmaceuticals, plazomicin is a next-generation aminoglycoside synthetically derived from Sisomicin. The structure of plazomicin was established via appending hydroxylaminobutyric acid to Sisomicin at position 1 and 2-hydroxyethyl group at position 6′ [1]. It was designed to evade all clinically relevant aminoglycoside-modifying enzymes, which contribute to the main resistance mechanism for aminoglycoside therapy [1]. However, acquired resistance of aminoglycosides may arise through over expression of efflux pumps and ribosomal modification by bacteria, which results from amino acid or rRNA sequence mutations [1]. Like other aminoglycosides, plazomicin is ineffective against bacterial isolates that produce 16S rRNA methyltransferases [Label]. Plazomicin mediates the antibacterial activity against pathogens including carbapenem-resistant (CRE) and extended-spectrum beta-lactamase (ESBL) producing Enterobacteriaceae. It mediates the antibacterial activity by binding to bacterial 30S ribosomal subunit and inhibiting protein synthesis [Label]. On June 28th, 2018, plazomicin sulfate was approved by the FDA for use in adult patients for the treatment of complicated urinary tract infections (cUTI) including Pyelonephritis. It is marketed as Zemdri and is administered via once-daily intravenous infusion.
Plazomicin (INN,[1] ZEMDRI) is a next-generation aminoglycoside (“neoglycoside”) antibacterial derived from sisomicin by appending a hydroxy-aminobutyric acid (HABA) substituent at position 1 and a hydroxyethyl substituent at position 6′.[2][3]
Plazomicin has been reported to demonstrate in vitro synergistic activity when combined with daptomycin or ceftobiprole versus methicillin-resistant Staphylococcus aureus (MRSA), vancomycin-resistant S. aureus (VRSA) and against Pseudomonas aeruginosawhen combined with cefepime, doripenem, imipenem or piperacillin/tazobactam.[3] It also demonstrates potent in vitro activity versus carbapenem-resistant Acinetobacter baumannii.[4]
In 2012, U.S. Food and Drug Administration granted fast track designation for the development and regulatory review of plazomicin.[5]
It is being developed by Achaogen, Inc. to treat serious bacterial infections due to multidrug-resistant Enterobacteriaceae, including carbapenem-resistant Enterobacteriaceae (CRE)[6] and was in Phase III clinical trials as of April 7, 2016.[7]
In June 2018 the FDA approved plazomicin (ZEMDRI) for adults with complicated urinary tract infections (cUTI), including pyelonephritis, caused by Escherichia coli, Klebsiella pneumoniae, Proteus mirabilis, or Enterobacter cloacae, in patients who have limited or no alternative treatment options. Zemdri is an intravenous infusion, administered once daily.[8][9] The FDA declined approval for treating bloodstream infections due to lack of effectiveness.[10]
To continue the development of plazomicin, the company has received a contract option of US$ 60M from the Biomedical Advanced Research and Development Authority (BARDA) to support a global Phase III clinical study. The study will evaluate plazomicin in treating patients with serious Gram-negative bacterial infections due to carbapenem-resistant Enterobacteriaceae. The study is expected to start in the fourth quarter of 2013 [4].
PATENT
WO 2009067692
WO 2010132770
PAPER
Synthesis and spectrum of the neoglycoside ACHN-490
Antimicrobial Agents and Chemotherapy (2010), 54, (11), 4636-4642
https://aac.asm.org/content/54/11/4636



PAPER
Plazomicin Retains Antibiotic Activity against Most Aminoglycoside Modifying Enzymes
ACS Infectious Diseases (2018), 4, (6), 980-987.
https://pubs.acs.org/doi/abs/10.1021/acsinfecdis.8b00001


PAPER
Effects of the 1-N-(4-Amino-2S-hydroxybutyryl) and 6′-N-(2-Hydroxyethyl) Substituents on Ribosomal Selectivity, Cochleotoxicity, and Antibacterial Activity in the Sisomicin Class of Aminoglycoside Antibiotics
ACS Infectious Diseases (2018), 4, (7), 1114-1120.
https://pubs.acs.org/doi/abs/10.1021/acsinfecdis.8b00052

Syntheses of the 6′-N-(2-hydroxyethyl) and 1-N-(4-amino-2S-hydroxybutyryl) derivatives of the 4,6-aminoglycoside sisomicin and that of the doubly modified 1-N-(4-amino-2S-hydroxybutyryl)-6′-N-(2-hydroxyethyl) derivative known as plazomicin are reported together with their antibacterial and antiribosomal activities and selectivities. The 6′-N-(2-hydroxyethyl) modification results in a moderate increase in prokaryotic/eukaryotic ribosomal selectivity, whereas the 1-N-(4-amino-2S-hydroxybutyryl) modification has the opposite effect. When combined in plazomicin, the effects of the two groups on ribosomal selectivity cancel each other out, leading to the prediction that plazomicin will exhibit ototoxicity comparable to those of the parent and the current clinical aminoglycoside antibiotics gentamicin and tobramycin, as borne out by ex vivo studies with mouse cochlear explants. The 6′-N-(2-hydroxyethyl) modification restores antibacterial activity in the presence of the AAC(6′) aminoglycoside-modifying enzymes, while the 1-N-(4-amino-2S-hydroxybutyryl) modification overcomes resistance to the AAC(2′) class but is still affected to some extent by the AAC(3) class. Neither modification is able to circumvent the ArmA ribosomal methyltransferase-induced aminoglycoside resistance. The use of phenyltriazenyl protection for the secondary amino group of sisomicin facilitates the synthesis of each derivative and their characterization through the provision of sharp NMR spectra for all intermediates.
https://pubs.acs.org/doi/suppl/10.1021/acsinfecdis.8b00052/suppl_file/id8b00052_si_001.pdf




4 (19 mg, 40%). [α]D 25 = +46.5 (c = 0.01, H2O);
1 H NMR (600 MHz, D2O): δ 5.51 ( s, 1H, H-1ʹ), 5.16 (t, J = 3.5 Hz, H, H-4ʹ), 4.99 (d , J = 4.0 Hz, 1H, H-1ʹʹ), 4.11 (dd , J =9.4 Hz, 3.9 Hz, 1H, CH(OH)CH2CH2), 4.00 (d , J = 12.8 Hz, 1H, H-5ʹʹ), 3.99-3.93 (m, 1H, H-1), 3.84 (dd, J = 11.0 Hz, 4.0 Hz, 1H, H-2ʹʹ), 3.81 (t, J = 9.9 Hz, 1H, H-4), 3.77 (t, J = 5.3 Hz, 1H, H-2ʹ), 3.71 (t, J = 5.1 Hz, 2H, NHCH2CH2O), 3.69 – 3.65 (m, 2H, H-6, H-6ʹ), 3.64 – 3.44 (m , 2H, H-5, H-6ʹ), 3.35 – 3.26 (m , 1H, H-3), 3.24 (d, J = 12.8 Hz, 1H, H-5ʹʹ), 3.15 (d, J = 11.0 Hz, 1H, H-3ʹʹ), 3.09 – 3.06 (m, 2H, NHCH2CH2O), 3.01 (t, J = 7.2 Hz, 2H, CH(OH)CH2CH2), 2.74 (s, 3H, NCH3), 2.58 – 2.52 (m, 1H, H-3ʹ), 2.29 – 2.24 (m, 1H, H-3ʹ), 2.07 (dt, J = 13.2 Hz, 4.4 Hz, 1H, H-2), 2.04 – 1.98 (m, 1H, CH(OH)CH2CH2), 1.84 – 1.79 (m, 1H, CH(OH)CH2CH2), 1.64 (q, 1H, J = 12.5 Hz, H-2), 1.17 (s, 3H, 4ʹʹ-CH3);
13C NMR (151 MHz, D2O): δ 181.2 (s, CH3COOH), 175.4 (s, NHCO), 141.7 (s, C-5ʹ), 102.5 (s, C-4ʹ), 98.0 (s, C-1ʹʹ), 96.9 (s, C-1ʹ), 79.8 (s, C-4), 78.8 (s, C-6), 73.8 (s, C-5), 69.8 (s, C-4ʹʹ), 69.4 (s, CH(OH)CH2CH2), 66.8 (s, C-5ʹʹ), 65.9 (s, C-2ʹʹ), 64.2 (s, C-3ʹʹ), 56.4 (s, NHCH2CH2O), 48.8 (s, C-1), 48.31 (s, NHCH2CH2O), 48.26 (s, C-3), 47.9 (s, C-6ʹ), 45.9 (s, C2ʹ), 36.8 (s, CH(OH)CH2CH2), 34.9 (s, NCH3), 30.7 (s, CH(OH)CH2CH2), 30.4 (s, C-2), 23.1 (s, CH3COOH), 23.0 (s, C-3ʹ), 20.8 (s, 4ʹʹ-CH3).
ESI-HRMS: m/z calcd. for C25H49N6O10 [M+H]+ 593.3510, found: 593.3481.
PATENT
http://www.google.com/patents/US20100099661
Common Intermediates Sisomicin

Amberlite IRA-400 (OH form) (200 g) was washed with MeOH (3×200 m1). To a stirring suspension of the washed resin in MeOH (150 mL) was added sisomicin sulfate (20.0 g, 0.029 mol) and the mixture was stirred overnight. The resin was then filtered and washed with MeOH (100 mL) and the combined organic layers were concentrated to dryness to yield the desired sisomicin (11.57 g, 0.026 mol, 89.6% yield): MS m/e [M+H]+ calcd 448.3, found 448.1.
Example 1 6′-(2-Hydroxy-ethyl)-1-(4-amino-2(S)-hydroxy-butyryl)-sisomicin

6′-(2-tert-Butyldimethylsililoxy-ethyl)-2′,3,3″-triBoc-1-(N-Boc-4-amino-2(S)-hydroxy-butyryl)-sisomicin
2′,3,3″-triBoc-1-(N-Boc-4-amino-2(S)-hydroxy-butyryl)-sisomicin (0.10 g, 0.105 mmol) was treated with tert-butyldimethylsilyloxy acetaldehyde following Procedure 1-Method A to yield the desired 6′-(2-tert-butyldimethylsilyloxy-ethyl)-2′,3,3″-triBoc-1-(N-Boc-4-amino-2(S)-hydroxy-butyryl)-sisomicin (MS m/e [M+H]+ calcd 1107.6, found 1107.4), which was carried through to the next step without further purification.

6′-(2-Hydroxy-ethyl)-1-(4-amino-2(S)-hydroxy-butyryl)-sisomicin
6′ -(2-tert-butyldimethylsililoxy-ethyl)-2′,3,3″-triBoc-1-(N-Boc-4-amino-2(S)-hydroxy-butyryl)-sisomicin (0.105 mmol) was submitted to Procedure 3-Method B for Boc removal to yield a crude, which was purified by RP HPLC Method 1-Column A to yield 6′-(2-hydroxy-ethyl)-1-(4-amino-2(S)-hydroxy-butyryl)-sisomicin: MS m/e [M+H]+ calcd 593.3, found 593.2, [M+Na]+615.3 ; CLND 97.5% purity.
- Achaogen. Study for the treatment of complicated urinary tract infection and acute pyelonephritis.Available online: http://www.clinicaltrials.gov/ct2/show/NCT01096849 (accessed on 11 April 2013).
- Zhanel, G.G.; Lawson, C.D.; Zelenitsky, S.; Findlay, B.; Schweizer, F.; Adam, H.; Walkty, A.; Rubinstein, E.; Gin, A.S.; Hoban, D.J.; et al. Comparison of the next-generation aminoglycoside plazomicin to gentamicin, tobramycin and amikacin. Expert Rev. Anti-Infect. Ther. 2012, 10, 459–473, doi:10.1586/eri.12.25.
- Endimiani, A.; Hujer, K.M.; Hujer, A.M.; Armstrong, E.S.; Choudhary, Y.; Aggen, J.B.; Bonomo, R.A. ACHN-490, a neoglycoside with potent in vitro activity against multidrug-resistant Klebsiella pneumoniae isolates. Antimicrob. Agents Chemother. 2009, 53, 4504–4507.
- Achaogen. Achaogen pipeline. Available online: http://www.achaogen.com (accessed on 30 August 2012).
- Achaogen. Achaogen Awarded $60M Contract Option by BARDA for the Clinical Development of Plazomicin. Available online: http://www.achaogen.com/news/151/15 (accessed on 19 June 2013).
- Achaogen. Achaogen announces all objectives met in Phase 2 Plazomicin complicated urinary tract infections study and start of first-in-human study with ACHN-975. Available online: http://www.achaogen.com/uploads/news/id148/Achaogen_PressRelease_2012–05–15.pdf (accessed on 10 April 2013).
- Achaogen. Achaogen Announces Agreement with FDA on a Special Protocol Assessment for a Phase 3 Clinical Trial of Plazomicin to Treat Infections Caused by Carbapenem-Resistant Enterobacteriaceae (CRE); Achaogen: San Francisco, CA, USA, 2013.
- Comparison of the next-generation aminoglycoside plazomicin to gentamicin, tobramycin and amikacin
-
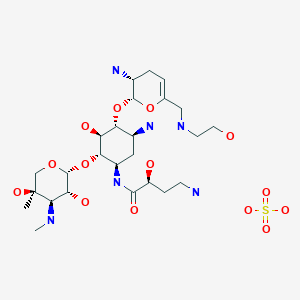
4-23-2010ANTIBACTERIAL AMINOGLYCOSIDE ANALOGS

| US8318685 | Nov 14, 2011 | Nov 27, 2012 | Achaogen, Inc. | Antibacterial aminoglycoside analogs |
| US8367625 | Apr 7, 2011 | Feb 5, 2013 | Achaogen, Inc. | Antibacterial aminoglycoside analogs |
| US8372813 | Apr 7, 2011 | Feb 12, 2013 | Achaogen, Inc. | Antibacterial aminoglycoside analogs |
| US8377896 | Mar 9, 2011 | Feb 19, 2013 | Isis Pharmaceuticals, Inc | Antibacterial 4,6-substituted 6′, 6″ and 1 modified aminoglycoside analogs |
| US8399419 | Mar 9, 2011 | Mar 19, 2013 | Achaogen, Inc. | Antibacterial aminoglycoside analogs |
| US8481502 | Apr 6, 2012 | Jul 9, 2013 | Achaogen, Inc. | Antibacterial aminoglycoside analogs |
| US8492354 | Nov 14, 2011 | Jul 23, 2013 | Achaogen, Inc. | Antibacterial aminoglycoside analogs |
| US8524675 | Nov 14, 2011 | Sep 3, 2013 | Achaogen, Inc. | Antibacterial aminoglycoside analogs |
| US8524689 | Nov 14, 2011 | Sep 3, 2013 | Achaogen, Inc. | Antibacterial aminoglycoside analogs |
| US8569264 | Jan 5, 2012 | Oct 29, 2013 | Isis Pharmaceuticals, Inc. | Antibacterial 4,5-substituted aminoglycoside analogs having multiple substituents |
| US8653041 | Oct 15, 2012 | Feb 18, 2014 | Achaogen, Inc. | Antibacterial aminoglycoside analogs |
| US8653042 | Nov 14, 2011 | Feb 18, 2014 | Achaogen, Inc. | Antibacterial aminoglycoside analogs |
| US8658606 | Nov 14, 2011 | Feb 25, 2014 | Achaogen, Inc. | Antibacterial aminoglycoside analogs |
References
- Jump up^ “WHO Drug Information, Vol. 26, No. 3, 2012. International Nonproprietary Names for Pharmaceutical Substances (INN). Recommended International Nonproprietary Names: List 68”(PDF). World Health Organization. p. 314. Retrieved 27 April 2016.
- Jump up^ Aggen, JB; Armstrong, ES; Goldblum, AA; Dozzo, P; Linsell, MS; Gliedt, MJ; Hildebrandt, DJ; Feeney, LA; Kubo, A; Matias, RD; Lopez, S; Gomez, M; Wlasichuk, KB; Diokno, R; Miller, GH; Moser, HE (30 August 2010). “Synthesis and Spectrum of the Neoglycoside ACHN-490” (PDF). Antimicrobial Agents and Chemotherapy. 54 (11): 4636–4642. doi:10.1128/AAC.00572-10. PMC 2976124
 . PMID 20805391. Retrieved 27 April2016.
. PMID 20805391. Retrieved 27 April2016. - ^ Jump up to:a b Zhanel, GG; Lawson, CD; Zelenitsky, S; Findlay, B; Schweizer, F; Adam, H; Walkty, A; Rubinstein, E; Gin, AS; Hoban, DJ; Lynch, JP; Karlowsky, JA (10 January 2014). “Comparison of the Next-Generation Aminoglycoside Plazomicin to Gentamicin, Tobramycin and Amikacin”. Expert Review of Anti-infective Therapy. 10 (4): 459–73. doi:10.1586/eri.12.25. PMID 22512755.
- Jump up^ García-Salguero, C; Rodríguez-Avial, I; Picazo, JJ; Culebras, E (October 2015). “Can Plazomicin Alone or in Combination Be a Therapeutic Option against Carbapenem-Resistant Acinetobacter baumannii?” (PDF). Antimicrobial Agents and Chemotherapy. 59 (10): 5959–66. doi:10.1128/AAC.00873-15. PMC 4576036 . Retrieved 27 April 2016.
- Jump up^ “Achaogen Announces Plazomicin Granted QIDP Designation by FDA”. GlobeNewswire, Inc. Retrieved 27 April 2016.
- Jump up^ “Achaogen — Plazomicin”. Achaogen, Inc. Retrieved 27 April2016.
- Jump up^ “Plazomicin — AdisInsight”. Springer International Publishing AG. Retrieved 27 April 2016.
- Jump up^ “Medscape Log In”. http://www.medscape.com. Retrieved 2018-07-03.
- Jump up^ “BioCentury – FDA approves plazomicin for cUTI, but not blood infections”. http://www.biocentury.com. Retrieved 2018-06-28.
- Jump up^ “Drugs@FDA: FDA Approved Drug Products”. http://www.accessdata.fda.gov. Retrieved 2018-06-28.
 |
|
| Names | |
|---|---|
| IUPAC name
(2S)-4-Amino-N-[(1R,2S,3S,4R,5S)-5-amino-4-[[(2S,3R)-3-amino-6-[(2-hydroxyethylamino)methyl]-3,4-dihydro-2H-pyran-2-yl]oxy]-2-[(2R,3R,4R,5R)-3,5-dihydroxy-5-methyl-4-(methylamino)oxan-2-yl]oxy-3-hydroxycyclohexyl]-2-hydroxybutanamide
|
|
| Other names
6′-(hydroxylethyl)-1-(HABA)-sisomicin
|
|
| Identifiers | |
|
3D model (JSmol)
|
|
| ChEMBL | |
| ChemSpider | |
| KEGG | |
|
PubChem CID
|
|
| UNII | |
| Properties | |
| C25H48N6O | |
| Molar mass | 592.683 g/mol |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F],
|
|
Achaogen is a clinical-stage biopharmaceutical company passionately committed to the discovery, development, and commercialization of novel antibacterials to treat multi-drug resistant, or MDR, gram-negative infections.

Achaogen (a-KAY-o-jen) is developing plazomicin, its lead product candidate, for the treatment of serious bacterial infections due to MDR Enterobacteriaceae, including carbapenem-resistant Enterobacteriaceae, or CRE. In 2013, the Centers for Disease Control and Prevention identified CRE as a “nightmare bacteria” and an immediate public health threat that requires “urgent and aggressive action.” We expect to initiate a Phase 3 superiority trial of plazomicin in the first quarter of 2014.
CRE are one of many types of MDR gram-negative pathogens threatening patients. Bacteria such as Pseudomonas aeruginosa, Acinetobacter baumannii, and extended-spectrum beta-lactamase producing Enterobacteriaceae each pose “serious” resistance threats, according to the CDC, and also drive a great need for new, safe, and effective antibiotics. We have assembled the chemistry and microbiology expertise and capabilities required to develop new agents for the treatment of gram-negative infections. Plazomicin was the first clinical candidate from our gram-negative antibiotic discovery engine. In addition, our research and development pipeline includes two antipseudomonal programs targeting P. aeruginosa—a program to discover and develop small molecule inhibitors of LpxC, which is an enzyme essential for the synthesis of the outer membrane of gram-negative bacteria, and a therapeutic antibody program. We are also pursuing small molecule research programs targeting other essential gram-negative enzymes.
Achaogen has built an exceptional research and development team with deep expertise in the discovery and development of new drugs from research through commercialization. Our executive team has over 60 years of combined industry experience, and a proven track record of leadership, global registration, and lifecycle management for over 20 products. Our facility is located on the shores of the San Francisco Bay, ten minutes from the San Francisco International Airport, and only fifteen minutes from downtown San Francisco.

ZEMDRITM (plazomicin) Approved by FDA for the Treatment of Adults with Complicated Urinary Tract Infections (cUTI)
― ZEMDRI is a new treatment for patients with cUTI, including pyelonephritis, due to certain Enterobacteriaceae ―
― ZEMDRI is the only once-daily aminoglycoside therapy approved for use in cUTI ―
― ZEMDRI has microbiological activity against pathogens designated by the CDC as urgent and serious public health threats, including carbapenem-resistant (CRE) and extended spectrum beta-lactamase (ESBL)- producing Enterobacteriaceae ―
SOUTH SAN FRANCISCO, Calif., June 26, 2018 (GLOBE NEWSWIRE) — Achaogen, Inc. (NASDAQ:AKAO), a biopharmaceutical company developing and commercializing innovative antibacterial agents to address multidrug resistant (MDR) gram-negative infections, today announced that the U.S. Food and Drug Administration (FDA) has approved ZEMDRI™ (plazomicin) for adults with complicated urinary tract infections (cUTI), including pyelonephritis, caused by certain Enterobacteriaceae in patients who have limited or no alternative treatment options. ZEMDRI is an intravenous infusion, administered once daily.
“The approval of ZEMDRI marks a significant milestone for Achaogen and we are excited to offer healthcare practitioners a new treatment option for patients with certain serious bacterial infections. ZEMDRI is designed to retain its potent activity in the face of certain difficult-to-treat MDR infections, including CRE and ESBL- producing Enterobacteriaceae,” said Blake Wise, Achaogen’s Chief Executive Officer. “Today’s milestone was made possible by our employees, by patients and investigators involved in our clinical trials, and by BARDA, who contributed significant funding for the development of ZEMDRI. This marks an important step in our commitment to fighting MDR bacteria and we are excited to launch ZEMDRI, a much needed once-daily antibiotic.”
“Bacteria continue to circumvent existing antibiotics, making certain infections notoriously hard to treat and putting some patients at high risk for mortality,” said James A. McKinnell, Assistant Professor of Medicine at the David Geffen School of Medicine and LA Biomed at Harbor-UCLA. “Aminoglycosides are a familiar and very effective class of antibiotics. I look forward to adding plazomicin to my short list of available treatment options and to its potential impact on patient outcomes.”
Regarding the potential indication for plazomicin for the treatment of bloodstream infection (BSI), the FDA issued a Complete Response Letter (CRL) stating that the CARE study does not provide substantial evidence of effectiveness of plazomicin for the treatment of BSI. The Company intends to meet with the FDA to determine whether there is a feasible resolution to address the CRL.
Achaogen will work with hospitals, providers, and insurers to ensure patients are able to receive this treatment. Patients, physicians, pharmacists, or other healthcare professionals with questions about ZEMDRI should contact 1.833.252.6400 or visit www.ZEMDRI.com.
ZEMDRI Phase 3 Clinical Results
The approval of ZEMDRI is supported in part by data from the EPIC (Evaluating Plazomicin In cUTI) clinical trial, which was the first randomized controlled study of once-daily aminoglycoside therapy for the treatment of cUTI, including pyelonephritis.
In the Phase 3 EPIC cUTI trial, ZEMDRI demonstrated non-inferiority to meropenem for the co-primary efficacy endpoints of composite cure (clinical cure and microbiological eradication) in the microbiological modified intent-to-treat (mMITT; N=388) population at Day 5 and test-of-cure (TOC) visit (Day 17 + 2). Composite cure rates at Day 5 were 88.0% (168/191) for ZEMDRI vs 91.4% (180/197) for meropenem (difference -3.4%, 95% CI, -10.0 to 3.1). Composite cure rates at TOC were 81.7% (156/191) for ZEMDRI vs 70.1% (138/197) for meropenem (difference 11.6%, 95% CI, 2.7 to 20.3). Composite cure at the TOC visit in patients with concomitant bacteremia at baseline was achieved in 72.0% (18/25) of patients in the ZEMDRI group and 56.5% (13/23) of patients in the meropenem group. The most common side effects (≥1% of patients treated with ZEMDRI) were decreased kidney function, diarrhea, hypertension, headache, nausea, vomiting, and hypotension.1
The FDA approved a breakpoint of <= 2 mcg/mL; greater than 99% of Escherichia coli, Klebsiella pneumoniae and Enterobacter cloacae in U.S. surveillance are susceptible to Zemdri when applying this breakpoint.2
About cUTI
cUTI is defined as a UTI occurring in a patient with an underlying complicating factor of the genitourinary tract, such as a structural or functional abnormality.3 Patients with pyelonephritis, regardless of underlying abnormalities of the urinary tract, are considered a subset of patients with cUTI.4 An estimated 3 million cases of cUTI are treated in the hospital setting in the US each year.5 Enterobacteriaceae are the most common pathogens causing cUTIs6, and resistance within this family is a global concern. High rates of resistance to previous mainstays of therapy necessitate alternative treatment options. Ineffectively managed cUTI can lead to increased treatment failure rates, recurrence of infection, increased re-hospitalization, and increased morbidity and mortality. cUTI infections place an economic burden on hospitals and payers.6,7
About ZEMDRI
ZEMDRI is an aminoglycoside with once-daily dosing that has activity against certain Enterobacteriaceae, including CRE and ESBL- producing Enterobacteriaceae. Achaogen’s EPIC clinical trial successfully evaluated the safety and efficacy of ZEMDRI in adult patients with cUTI, including pyelonephritis. ZEMDRI was engineered to overcome aminoglycoside-modifying enzymes, the most common aminoglycoside-resistance mechanism in Enterobacteriaceae, and has in vitro activity against ESBL- producing, aminoglycoside- resistant, and carbapenem- resistant isolates. The Centers for Disease Control and Prevention (CDC) has characterized ESBL- producing Enterobacteriaceae as a “serious threat” and CRE as “nightmare bacteria”, which is an immediate public health threat that requires urgent and aggressive action.
Working in the Lab

Working in the Lab
Achaogen, Inc.
Blake Wise, Chief Executive Officer at Achaogen

Blake Wise, Chief Executive Officer at Achaogen
Achaogen, Inc.
High-Resolution Achaogen company logo

High-Resolution Achaogen company logo
Achaogen, Inc.
/////////Plazomicin, ZEMDRI, FDA 2018, fast track designation, Plazomicin SULFATE, ACHN 490 sulfate, cUTI, Achaogen
CC1(COC(C(C1NC)O)OC2C(CC(C(C2O)OC3C(CC=C(O3)CNCCO)N)N)NC(=O)C(CCN)O)O
CN[C@@H]1[C@@H](O)[C@@H](O[C@H]2[C@@H](C[C@H](N)[C@@H](O[C@H]3OC(CNCCO)=CC[C@H]3N)[C@@H]2O)NC(=O)[C@@H](O)CCN)OC[C@]1(C)O
FDA approves first drug Epidiolex (cannabidiol) comprised of an active ingredient derived from marijuana to treat rare, severe forms of epilepsy
The U.S. Food and Drug Administration today approved Epidiolex (cannabidiol) [CBD] oral solution for the treatment of seizures associated with two rare and severe forms of epilepsy, Lennox-Gastaut syndrome and Dravet syndrome, in patients two years of age and older. This is the first FDA-approved drug that contains a purified drug substance derived from marijuana. It is also the first FDA approval of a drug for the treatment of patients with Dravet syndrome.
June 25, 2018
Release
The U.S. Food and Drug Administration today approved Epidiolex (cannabidiol) [CBD] oral solution for the treatment of seizures associated with two rare and severe forms of epilepsy, Lennox-Gastaut syndrome and Dravet syndrome, in patients two years of age and older. This is the first FDA-approved drug that contains a purified drug substance derived from marijuana. It is also the first FDA approval of a drug for the treatment of patients with Dravet syndrome.
CBD is a chemical component of the Cannabis sativa plant, more commonly known as marijuana. However, CBD does not cause intoxication or euphoria (the “high”) that comes from tetrahydrocannabinol (THC).
It is THC (and not CBD) that is the primary psychoactive component of marijuana.
“This approval serves as a reminder that advancing sound development programs that properly evaluate active ingredients contained in marijuana can lead to important medical therapies. And, the FDA is committed to this kind of careful scientific research and drug development,” said FDA Commissioner Scott Gottlieb, M.D. “Controlled clinical trials testing the safety and efficacy of a drug, along with careful review through the FDA’s drug approval process, is the most appropriate way to bring marijuana-derived treatments to patients. Because of the adequate and well-controlled clinical studies that supported this approval, prescribers can have confidence in the drug’s uniform strength and consistent delivery that support appropriate dosing needed for treating patients with these complex and serious epilepsy syndromes. We’ll continue to support rigorous scientific research on the potential medical uses of marijuana-derived products and work with product developers who are interested in bringing patients safe and effective, high quality products. But, at the same time, we are prepared to take action when we see the illegal marketing of CBD-containing products with serious, unproven medical claims. Marketing unapproved products, with uncertain dosages and formulations can keep patients from accessing appropriate, recognized therapies to treat serious and even fatal diseases.”
Dravet syndrome is a rare genetic condition that appears during the first year of life with frequent fever-related seizures (febrile seizures). Later, other types of seizures typically arise, including myoclonic seizures (involuntary muscle spasms). Additionally, status epilepticus, a potentially life-threatening state of continuous seizure activity requiring emergency medical care, may occur. Children with Dravet syndrome typically experience poor development of language and motor skills, hyperactivity and difficulty relating to others.
Lennox-Gastaut syndrome begins in childhood. It is characterized by multiple types of seizures. People with Lennox-Gastaut syndrome begin having frequent seizures in early childhood, usually between ages 3 and 5. More than three-quarters of affected individuals have tonic seizures, which cause the muscles to contract uncontrollably. Almost all children with Lennox-Gastaut syndrome develop learning problems and intellectual disability. Many also have delayed development of motor skills such as sitting and crawling. Most people with Lennox-Gastaut syndrome require help with usual activities of daily living.
“The difficult-to-control seizures that patients with Dravet syndrome and Lennox-Gastaut syndrome experience have a profound impact on these patients’ quality of life,” said Billy Dunn, M.D., director of the Division of Neurology Products in the FDA’s Center for Drug Evaluation and Research. “In addition to another important treatment option for Lennox-Gastaut patients, this first-ever approval of a drug specifically for Dravet patients will provide a significant and needed improvement in the therapeutic approach to caring for people with this condition.”
Epidiolex’s effectiveness was studied in three randomized, double-blind, placebo-controlled clinical trials involving 516 patients with either Lennox-Gastaut syndrome or Dravet syndrome. Epidiolex, taken along with other medications, was shown to be effective in reducing the frequency of seizures when compared with placebo.
The most common side effects that occurred in Epidiolex-treated patients in the clinical trials were: sleepiness, sedation and lethargy; elevated liver enzymes; decreased appetite; diarrhea; rash; fatigue, malaise and weakness; insomnia, sleep disorder and poor quality sleep; and infections.
Epidiolex must be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks. As is true for all drugs that treat epilepsy, the most serious risks include thoughts about suicide, attempts to commit suicide, feelings of agitation, new or worsening depression, aggression and panic attacks. Epidiolex also caused liver injury, generally mild, but raising the possibility of rare, but more severe injury. More severe liver injury can cause nausea, vomiting, abdominal pain, fatigue, anorexia, jaundice and/or dark urine.
Under the Controlled Substances Act (CSA), CBD is currently a Schedule I substance because it is a chemical component of the cannabis plant. In support of this application, the company conducted nonclinical and clinical studies to assess the abuse potential of CBD.
The FDA prepares and transmits, through the U.S. Department of Health and Human Services, a medical and scientific analysis of substances subject to scheduling, like CBD, and provides recommendations to the Drug Enforcement Administration (DEA) regarding controls under the CSA. DEA is required to make a scheduling determination.
The FDA granted Priority Review designation for this application. Fast-Track designation was granted for Dravet syndrome. Orphan Drug designation was granted for both the Dravet syndrome and Lennox-Gastaut syndrome indications.
The FDA granted approval of Epidiolex to GW Research Ltd.

/////////// Epidiolex, cannabidiol, fda 2018, Dravet syndrome, epilepsy, Priority Review , Fast-Track designation, Orphan Drug designation
VORAPAXAR SULPHATE

VORAPAXAR
Thrombosis, Antiplatelet Therapy, PAR1 Antagonists , MERCK ..ORIGINATOR
Ethyl N-[(3R,3aS,4S,4aR,7R,8aR,9aR)-4-[(E)-2-[5-(3-fluorophenyl)-2-pyridyl]vinyl]-3-methyl-1-oxo-3a,4,4a,5,6,7,8,8a,9,9a-decahydro-3H-benzo[f]isobenzofuran-7-yl]carbamate
Carbamic acid, [(1R,3aR,4aR,6R,8aR,9S,9aS)-9-[(1E)-2-[5-(3-fluorophenyl)-2- pyridinyl]ethenyl]dodecahydro-1-methyl-3-oxonaphtho[2,3-c]furan-6-yl]-, ethyl ester
Carbamic acid, N-[(1R,3aR,4aR,6R,8aR,9S,9aS)-9-[(E)-2-[5-(3-fluorophenyl)-2-pyridinyl]ethenyl]dodecahydro-1-methyl-3-oxonaphtho[2,3-c]furan-6-yl]-, ethyl ester
Ethyl [(1R,3aR,4aR,6R,8aR,9S,9aS)-9-{(E)-2-[5-(3-fluorophenyl)-2-pyridinyl]vinyl}-1-methyl-3-oxododecahydronaphtho[2,3-c]furan-6-yl]carbamate
Ethyl ((1R,3aR,4aR,6R,8aR,9S,9aS)-9-((1E)-2-(5-(3-fluorophenyl)pyridin-2-yl)ethenyl)- 1-methyl-3-oxododecahydronaphtho(2,3-c)furan-6-yl)carbamate
Carbamic acid, ((1R,3aR,4aR,6R,8aR,9S,9aS)-9-((1E)-2-(5-(3-fluorophenyl)-2- pyridinyl)ethenyl)dodecahydro-1-methyl-3-oxonaphtho(2,3-c)furan-6-yl)-, ethyl ester
618385-01-6 CAS NO FREE FORM
CAS Number: 705260-08-8 SULPHATE
Has antiplatelet activity.
Also known as: SCH-530348, MK-5348
Molecular Formula: C29H33FN2O4
Molecular Weight: 492.581723
ZCE93644N2
- UNII-ZCE93644N2
- Zontivity
Registered – 2015 MERCK Thrombosis
Vorapaxar (formerly SCH 530348) is a thrombin receptor (protease-activated receptor, PAR-1) antagonist based on the natural product himbacine. Discovered by Schering-Plough and currently being developed by Merck & Co., it is an experimental pharmaceutical treatment for acute coronary syndrome chest pain caused by coronary artery disease.[1]
In January 2011, clinical trials being conducted by Merck were halted for patients with stroke and mild heart conditions.[2] In a randomized double-blinded trial comparing vorapaxar with placebo in addition to standard therapy in 12,944 patients who had acute coronary syndromes, there was no significant reduction in a composite end point of death from cardiovascular causes, myocardial infarction, stroke, recurrent ischemia with rehospitalization, or urgent coronary revascularization. However, there was increased risk of major bleeding.[3]
A trial published in February 2012, found no change in all cause mortality while decreasing the risk of cardiac death and increasing the risk of major bleeding.[4]
SCH-530348 is a protease-activated thrombin receptor (PAR-1) antagonist developed by Schering-Plough and waiting for approval in U.S. for the oral secondary prevention of cardiovascular events in patients with a history of heart attack and no history of stroke or transient ischemic attack. The drug candidate is being investigated to determine its potential to provide clinical benefit without the liability of increased bleeding; a tendency associated with drugs that block thromboxane or ADP pathways. In April 2006, SCH-530348 was granted fast track designation in the U.S. for the secondary prevention of cardiovascular morbidity and mortality outcomes in at-risk patients.
Vorapaxar was recommended for FDA approval on January 15, 2014.[5]
Vorapaxar is a protease-activated thrombin receptor (PAR-1) antagonist developed by Schering-Plough (now, Merck & Co.) and approved in the U.S. in 2014 for the reduction of thrombotic cardiovascular events in patients with a history of myocardial infarction or with peripheral arterial disease. However, in 2018 Aralez discontinued U.S. commercial operations. In 2015, the product was approved in the E.U. for the reduction of atherothrombotic events in adult patients with a history of myocardial infarction. In April 2006, vorapaxar was granted fast track designation in the U.S. for the secondary prevention of cardiovascular morbidity and mortality outcomes in at-risk patients. In 2016, Aralez Pharmaceuticals acquired the U.S. and Canadian rights to the product pursuant to an asset purchase agreement entered into between this company and Merck & Co.
Merck & Co (following its acquisition of Schering-Plough) has developed and launched vorapaxar (Zontivity; SCH-530348; MK-5348), an oral antagonist of the thrombin receptor (protease-activated receptor-1; PAR1); the product is marketed in the US by Aralez Pharmaceuticals
WO-03089428, published in October 2003, claims naphtho[2,3-c]furan-3-one derivatives as thrombin receptor antagonists. WO-03033501 and WO-0196330, published in April 2003 and December 2001, respectively, claim himbacine analogs as thrombin receptor antagonists. WO-9926943 published in June 1999 claims tricyclic compounds as thrombin receptor antagonists
 VORAPAXAR
VORAPAXAR17 JAN 2014
FDA advisory panel votes to approve Merck & Co’s vorapaxar REF 6
https://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/204886Orig1s000ChemR.pdf
Zontivity (vorapaxar) tablets NDA 204886



VORAPAXAR SULPHATE

CAS Number: 705260-08-8 SULPHATE
Molecular Formula: C29H33FN2O4.H2O4S
Molecular Weight: 590.7
Chemical Name: Ethyl [(1R,3aR,4aR,6R,8aR,9S,9aS)-9-[(1E)-2-[5-(3-fluorophenyl)pyridin-2- yl]ethenyl]-1-methyl-3-oxododecahydronaphtho[2,3-c]furan-6-yl]carbamate sulfate
Synonyms: Carbamic acid, [(1R,3aR,4aR,6R,8aR,9S,9aS)-9-[(1E)-2-[5-(3-fluorophenyl)-2- pyridinyl]ethenyl]dodecahydro-1-methyl-3-oxonaphtho[2,3-c]furan-6-yl]-,ethyl ester,sulfate; SCH-530348
Vorapaxar Sulfate (SCH 530348) a thrombin receptor (PAR-1) antagonist for the prevention and treatment of atherothrombosis.
POLYMORPH
U.S.Pat. No. 7,304,078 discloses Vorapaxar base. U.S.Pat. No. 7,235,567 discloses Polymorph I and II of vorapaxar sulphate
CN 106478608 provides a crystalline polymorph A
EMA
Atherosclerosis and ischemic cardiovascular (CV) diseases like coronary artery disease (CAD) are progressive systemic disorders in which clinical events are precipitated by episodes of vascular thrombosis. Patients with an established history of atherothrombotic or athero-ischemic disease are at particular risk of future cardiac or cerebral events, and vascular death. Anti-thrombotic therapy options in patients with stable atherosclerosis are not well-established. Long-term therapies to effectively modulate the key components responsible for atherothrombosis in secondary prevention of ischemic CV disease are therefore required. Vorapaxar is a first – in – class selective antagonist of the protease-activated receptor 1 (PAR-1), the primary thrombin receptor on human platelets, which mediates the downstream effects of this critical coagulation factor in hemostasis and thrombosis. Thrombin-induced platelet activation has been implicated in a variety of cardiovascular disorders including thrombosis, atherosclerosis, and restenosis following percutaneous coronary intervention (PCI). As an antagonist of PAR-1, vorapaxar blocks thrombin-mediated platelet aggregation and thereby has the potential to reduce the risk of atherothrombotic complications of coronary disease. The applicant has investigated whether a new class of antiplatelet agents, PAR-1 antagonists, can further decrease the risk of cardiovascular events in a population of established atherothrombosis when added to standard of care, in secondary prevention of ischemic diseases. The following therapeutic indication has been submitted for vorapaxar: Vorapaxar is indicated for the reduction of atherothrombotic events in patients with a history of MI. Vorapaxar has been shown to reduce the rate of a combined endpoint of cardiovascular death, MI, stroke, and urgent coronary revascularization. Vorapaxar will be contraindicated in patients with a history of stroke or TIA. The indication sought in the current application is supported by the efficacy results of the TRA 2P-TIMI, which is considered the pivotal trial for this indication. During the procedure, the applicant requested the possibility of extending the indication initially sought for, to extend it to the population of PAD patients. This request was discussed at the CHMP and not accepted by the Committee.
Introduction The finished product is presented as immediate release film-coated tablets containing 2.5 mg of vorapaxar sulfate as active substance per tablet, corresponding to 2.08 mg vorapaxar. Other ingredients are: lactose monohydrate, microcrystalline cellulose (E460), croscarmellose sodium (E468), povidone (E1201) , magnesium stearate (E572), hypromellose (E464), titanium dioxide (E171), triacetin (glycerol triacetate) (E1518), iron oxide yellow (E172), as described in section 6.1 of the SmPC. The product is available in Aluminium–Aluminium blisters (Alu-Alu) as described in section 6.5 of the SmPC.
General information The chemical name of the active substance vorapaxar sulfate is ethyl[(1R,3aR,4aR,6R,8aR,9S,9aS)- -9-{(1E)-2-[5-(3-fluorophenyl)pyridin-2-yl]ethen-1-yl}-1-methyl-3-oxododecahydronaphtho[2,3-c] furan-6-yl]carbamate sulfate, corresponding to the molecular formula C29H33FN2O4 • H2SO4 and has a relative molecular mass 590.7. It has the following structure:
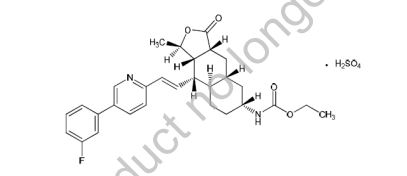
The structure of the active substance has been confirmed by mass spectrometry, infrared spectroscopy, 1H- and 13C-NMR spectroscopy and X-ray crystallography, all of which support the chemical structure elemental analysis. It appears as a white to off-white, slightly hygroscopic, crystalline powder. It is freely soluble in methanol and slightly soluble in ethanol and acetone but insoluble to practically insoluble in aqueous solutions at pH above 3.0. The highest solubility in aqueous solution can be achieved at pH 1.0 or in simulated gastric fluids at pH 1.4. The dissociation constant of vorapaxar sulfate was determined to be pKa = 4.7 and its partition coefficient LogP was determined to be 5.1. Vorapaxar sulfate contains seven chiral centers and a trans double bond. The seven chiral centres are defined by the manufacturing process of one of the intermediates in the vorapaxar synthesis and potential enantiomers are controlled by appropriate specifications. The cis-isomer of the double bond is controlled by a highly stereo-specific process reaction resulting in non-detectable levels of cis-isomer impurity. The cis-isomer impurity is controlled in one of the intermediates as an unspecified impurity. A single crystalline stable anhydrous form has been observed.
GENERAL INTRODUCTION
SIMILAR NATURAL PRODUCT
+ HIMBACINE


Himbacine is an alkaloid muscarinic receptor antagonist displaying more potent activity associated with M2 and M2 subtypes over M1 or M3. Observations show himbacine bound tightly to various chimeric receptors in COS-7 cells as well as possessed the ability to bind to cardiac muscarinic receptors allosterically. Recent studies have produced series of thrombin receptor (PAR1) antagonists derived from himbacine Himbacine is an inhibitor of mAChR M2 and mAChR M4.
Technical Information
| Physical State: | Solid |
| Derived from: | Australian pine Galbulimima baccata |
| Solubility: | Soluble in ethanol (50 mg/ml), methanol, and dichloromethane. Insoluble in water. |
| Storage: | Store at -20° C |
| Melting Point: | 132-134 °C |
| Boiling Point: | 469.65 °C at 760 mmHg |
| Density: | 1.08 g/cm3 |
| Refractive Index: | n20D 1.57 |
| Optical Activity: | α20/D +51.4º, c = 1.01 in chloroform |
| Application: | An alkaloid muscarinic receptor antagonist |
| CAS Number: | 6879-74-9 |
| Molecular Weight: | 345.5 |
| Molecular Formula: | C22H35NO2 |
General scheme:

PATENT
WO 2006076415
WO 2006076452
WO 2003089428
US 6063847
CN 107540564
WO 2008005344
CN 106749138
PATENT
Example 1:
[0027] The steel shed amide (300mg, 7. 93mmol) and 15 blood THF was added to 100 blood Ξ jar. The starting material II (2.OOg, 5. 89mmol) was dissolved in 15mL of THF dropwise via pressure-equalizing dropping funnel to the reaction system, the process temperature will produce a large number of bubbles -2 ~ 0 ° C, in the process, Lan mix of about 0.1 until no bubbles generate. THF solution containing 13 Blood Ship (0.75 Yap, 2. 95mmol) is transferred to a pressure-equalizing dropping funnel. It was slowly added dropwise to the reaction system. After the completion of dropwise continue to embrace mix ratio. After the treatment, at 0 ° C under 0.8 blood, Imol / L 1 fat slowly dropped into the embrace mixed reaction system, after adding the right amount of water, acetic acid extraction. The combined organic phase with Imol / L of 0H (17mLX3) washing the organic phase coating. Tu brine, dried over anhydrous sulfate steel, 25 ° C under reduced pressure to spin dry to give 1. 75g light yellow oil, yield 91%.
[0028] After the content was determined using the external standard method, first prepared by a qualified reference determine its content, W this as a standard substance, measuring the external standard method to get the content of 99%.
[0029] Zan NMR: (400MHz, CD3CN):… 5 46 of r, 1H), 4 70 (td, 1H), 4 03 based 2H), 3 69-3 57 (m, 2 Η).. , 3. 45-3. 32 (based, IH), 2. 77 (br, IH), 2. 61-2. 51 (m, IH), 2. 49-2. 39 (m, 1 field, 2 30 of r IH), 2 .12-1. 92 (m, IH), 1. 87 (dt, IH), 1. 81-1. 72 (m, IH), 1. 61-1. 50 ( …. m, IH), 1 48 (d, 3H), 1 23-1 09 (m, 7H), 1. 05-0 90 (m, 2H);
[0030] MS (ES +) m / z: 326. 24 [M + + field.
[Cited 00] Example 2:
[003 cited the steel shed amide (312mg, 8. 25mmol) and 16 blood THF was added to the lOOmL Ξ jar. The starting material II (2.OOg, 5. 89mmol) was dissolved in 15mL of THF dropwise via pressure-equalizing dropping funnel to the reaction system, the process temperature will produce a large number of bubbles -2 ~ -5 ° C, in the process and takes about 45min mix until no bubbles generate. The 13 ships of blood containing 60g, 2. 36mmol) in THF solution was transferred to a pressure-equalizing dropping funnel. It was slowly added dropwise to the reaction system. After the completion of dropwise continue to embrace mix ratio. After the treatment, at 0 ° C under 0.8 blood, Imol / L 1 fat slowly dropped into the embrace mixed reaction system, after adding the right amount of water, acetic acid extraction. The combined organic phase with llmol / L of 0H (17mLX3) washing the organic phase coating. Tu brine, dried over anhydrous sulfate steel, 25 ° C under reduced pressure to spin dry to give 1. 65g light yellow oil.
[0033] Determination of Reference Example 1 in an amount of 98.7%.
[0034] MS (ES +) m / z: 326. 24 [M + + field.
[003 cited Example 3:
[0036] 50 single jar of blood, condenser. Intermediate inb (l.〇〇g, 3. 07mmol) was dissolved in 10ml of dichloromethane burn during and after the blood was added to a 50-port flask, make dioxide of 32g, 3.68mmol), the reaction of reflux. After completion of the reaction by TLC, cooled to 20 ~ 25 ° C after suction filtration, the filter cake rinsed with methylene burning (the X3 3 blood), at 30 ° CW and the filtrate was concentrated to dryness. To the residue was added 5 blood acetic acid, at 20 ~ 25 ° C after mixing 0. embrace of suction, the resulting cake was vacuum dried at 30 ° C 10 ~ 12h. Give 0. 87g of white solid.
[0037] Electric NMR: (400MHz, CD3CN):. 9 74 oriented 1H), 5 40 of r, 1H), 4 77-4.66 (m, 1H), 4 09-3 98 (m, 2H…. ), 3. 49-3. 37 (m, IH), 2. 75-2. 64 (m, 2H), 2. 55-2. 48 (m, IH), 1. 95-1. 87 (m , 2H), 1. 89-1 .77 (m, 2H), 1. 61-1. 49 (m, IH), 1. 32-1. 13 (m, 9H), 1. 08-0. 82 (m, 2H);
[0038] MS (ES +) m / z: 324. 33 [M + + field.
PATENT
CN 106478608 crystal
https://patents.google.com/patent/CN106478608A/en
The present invention provides a crystalline polymorph A one kind of the compound of formula I:

In another embodiment, the present invention provides a method of preparing a crystalline polymorph of compound A I,

Which comprising, a) the compound II is dissolved in acetonitrile and stirred to form a mixture; b) heating the mixture to 50 ° C ~ 70 ° C; c) adding sulfuric acid to the heated mixture; d) evaluating the temperature was lowered to 0 ° C ~ 20 ° C, seeded and stirred to precipitate crystals.
Preparation [0042] A crystalline polymorph of the compound of Example 1 I

Compound II (1. 0g) was dissolved in 5. 0ml of acetonitrile, stirred and heated to 50 ° C ~ 70 ° C was added and this temperature was added 1.2ml 2N H2S04 / acetonitrile solution and then lowering the temperature of the system to 15 ° C ~ 20 ° C, the system was added to the appropriate amount of seed crystals and stirred for 2h, the precipitated solid was filtered and the cake washed twice with 2. 5ml of acetonitrile to give a white solid, the white solid was placed under 40 ° C desolventizing 2 hours and then dried at 80 ° C for vacuo to give a white solid 0. 83 g, 69. 3% yield, HPLC:. 99 94%. A powder X-ray diffraction spectrum shown in Figure 1, a DSC endothermic curve shown in Figure 2, which HPLC profile shown in Fig.
PATENT
https://patents.google.com/patent/CN106478608A/en
PATENT
WO 2009093972 synthesis
https://encrypted.google.com/patents/WO2009093972A1?cl=ko&hl=en&output=html_text
Clip
Vorapaxar sulfate (Zontivity)
Merck Sharp & Dohme successfully obtained approval in the EU in 2014 for vorapaxar sulfate, marketed as Zontivity. The drug is a first-in-class thrombin receptor (also referred to as a protease-activated or PAR-1) antagonist which, when used in conjunction with antiplatelet therapy, has been shown to reduce the chance of
myocardial infarction and stroke, particularly in patients with a history of cardiac events.277
Antagonism of PAR-1 allows for thrombin-mediated fibrin deposition while blocking thrombinmediated platelet activation.277 Although a variety of papers and patents describe the synthesis of vorapaxar sulfate (XXXVII),278–282 a combination of two patents describe the largest-scale synthesis reported in the literature, and this is depicted in Scheme 52.


Retrosynthetically, the drug can be divided into olefination partners 306 and 305.283,284 Lactone 305
is further derived from synthons 300 and 299, which are readily prepared from commercially available starting materials. Dienyl acid 300 was constructed in two steps starting from commercial vinyl bromide 307, which first undergoes a Heck reaction with methacrylate (308) followed by saponification of the ester to afford the desired acid 300 in 71% over two steps (Scheme 53).
The synthesis of alcohol 299 begins with tetrahydropyranyl (THP) protection of enantioenriched alcohol 295 to afford butyne 297 (Scheme 52). Lithiation of this system followed by trapping with (benzyloxy)chloroformate and Dowex work-up to remove the protective functionality provided acetyl ester 298. Hydrogenation of the alkyne with Lindlar’s catalyst delivered cis-allylic alcohol 299 in 93% yield. Acid 300 was then esterified with alcohol 299 by way of a 1,3-dicyclohexylcarbodiimide (DCC) coupling and, upon heating in refluxing xylenes, an intramolecular Diels–
Alder reaction occurred. Subsequent subjection to DBU secured the tricyclic system 301 in 38% over three steps as a single enantiomer.
Diastereoselective hydrogenation reduced the olefin with concomitant benzyl removal to give key fragment 302. Next, acidic revelation of the ketone followed by reductive amination with ammonium formate delivered primary amines 303a/303b as a mixture of diastereomers. These amines were then converted to the corresponding carbamates, and resolution by means of recrystallization yielded 50% of 304 as the desired diastereomer. Acid 304
was treated with oxalyl chloride and the resulting acid chloride was reduced to aldehyde 305 in 66% overall yield. Finally, deprotonation of phosphonate ester 306 (whose synthesis is described in Scheme 54) followed by careful addition of 305 and acidic quench delivered vorapaxar sulfate (XXXVII) in excellent yield over the
two-step protocol.
The preparation of vorapaxar phosponate ester 306 (Scheme 54)commenced from commercial sources of 5-(3-fluorophenyl)-2-methylpyridine (310). Removal of the methyl proton with LDA followed by quench with diethyl chlorophosphonate resulted in phosponate ester 306.

277. Frampton, J. E. Drugs 2015, 75, 797.
278. Chackalamannil, S.; Wang, Y.; Greenlee, W. J.; Hu, Z.; Xia, Y.; Ahn, H.; Boykow,G.; Hsieh, Y.; Palamanda, J.; Agans-Fantuzzi, J.; Kurowski, S.; Graziano, M.;Chintala, M. J. Med. Chem. 2008, 51, 3061.
279. Sudhakar, A.; Kwok, D.; Wu, G. G.; Green, M. D. WO Patent 2006076452A2,2006.
280. Wu, G. G.; Sudhakar, A.; Wang, T.; Ji, X.; Chen, F. X.; Poirier, M.; Huang, M.;Sabesan, V.; Kwok, D.; Cui, J.; Yang, X.; Thiruvengadam, T.; Liao, J.; Zavialov, I.;Nguyen, H. N.; Lim, N. K. WO Patent 2006076415A2, 2006.
281. Yong, K. H.; Zavialov, I. A.; Yin, J.; Fu, X.; Thiruvengadam, T. K. US Patent20080004449A1, 2008.
282. Chackalamannil, S.; Clasby, M.; Greenlee, W. J.; Wang, Y.; Xia, Y.; Veltri, E.;Chelliah, M. WO Patent 03089428A1, 2003.
283. Thiruven-Gadam, T. K.; Wang, T.; Liao, J.; Chiu, J. S.; Tsai, D. J. S.; Lee, H.; Wu,W.; Xiaoyong, F. WO Patent 2006076564A1, 2006.
284. Chackalamannil, S.; Asberon, T.;Xia, Y.; Doller, D.; Clasby, M. C.; Czarniecki,M. F. US Patent 6,063,847, 2000.
PRODUCT PATENT
SYNTHESIS
Inventor Samuel ChackalamannilMartin C. ClasbyWilliam J. GreenleeYuguang WangYan XiaEnrico P. VeltriMariappan ChelliahWenxue Wu
Original Assignee Schering Corporation
Priority date 2002-04-16
THE EXACT BELOW COMPD IS 14
Example 2
Step 1 :

Phosphonate 7, described in US 6,063,847, (3.27 g, 8.1 mmol) was dissolved in THF (12 ml) and C(O)Oled to 0 °C, followed by addition of 2.5 M n- BuLi (3.2 ml, 8.1 mmol). The reaction mixture was stirred at 0 °C for 10 min and warmed up to rt. A solution of aldehyde 6, described in US 6,063,847, in THF (12 ml) was added to the reaction mixture. The reaction mixture was stirred for 30 min. Standard aqueous work-up, followed by column chromatography (30-50% EtOAc in hexane) afforded product 8. 1HNMR (CDCI3): δ 0.92-1.38 (m, 31 H), 1.41 (d, J= 6 Hz, 3H), 1.40-1.55 (m, 2H), 1.70-1.80 (m, 2H), 1.81-1.90 (m, 2H), 2.36 (m, 2H), 2.69 (m, 1 H), 3.89 (m, 4H), 4.75 (m, 1 H), 6.28-6.41 (m, 2H), 7.05-7.15 (m, 2H), 8.19 (br s, 1 H). Step 2:

Compound 8 (2.64 g, 4.8 mmol) was dissolved in THF (48 ml). The reaction mixture was C(O)Oled to 0 °C followed by addition of 1 M TBAF (4.8 ml). The reaction mixture was stirred for 5 min followed by standard aqueous work-up. Column chromatography (50% EtOAc/hexane) afforded product 9 (1.9 g, 100%). 1HNMR (CDCI3): δ 1.15-1.55 (m, 6H), 1.41 (d, J= 6 Hz, 3H), 1.70-1.82 (m, 3H), 1.85-1.90 (m, 1 H), 2.36 (m, 2H), 2.69 (m, 1 H), 3.91 (m, 4H), 4.75 (m, 1 H), 6.18- 6.45 (m, 2H), 7.19 (br s, 2H), 8.19 (br s, 1 H). Step 3:

To a solution of compound 9 (250 mg, 0.65 mmol) in pyridine (5 ml) C(O)Oled to 0 °C was added Tf2O (295 μL, 2.1 mmol). The reaction mixture was stirred overnight at rt. Standard aqueous work-up followed by column chromatography afforded product 10 (270 mg, 80%). 1HNMR (CDCI3): δ 1.15-1.55 (m, 6H), 1.41 (d, J= 6 Hz, 3H), 1.70-1.82 (m, 3H), 1.85-1.90 (m, 1 H), 2.36 (m, 2H), 2.69 (m, 1 H), 3.91 (m, 4H), 4.75 (m, 1 H), 6.42-6.68 (m, 2H), 7.25 (m, 1 H), 7.55 (m, 1 H), 8.49 (d, J= 2.8 Hz, 1 H).

Compound 10 (560 mg, 1.1 mmol), 3-fluorophenyl boronic acid (180 mg, 1.3 mmol) and K2CO3 (500 mg, 3.6 mmol) were mixed with toluene (4.4 ml), H2O (1.5 ml) and EtOH (0.7 ml) in a sealed tube. Under an atmosphere of N2, Pd(Ph3P)4 (110 mg, 0.13 mmol) was added. The reaction mixture was heated at 100 °C for 2 h under N2. The reaction mixture was C(O)Oled down to rt, poured to EtOAc (30 ml) and washed with water (2X20 ml). The EtOAc solution was dried with NaHCO3 and concentrated at reduced pressure to give a residue. Preparative TLC separation of the residue (50% EtOAc in hexane) afforded product 11 (445 mg, 89%). 1HNMR (CDCI3): δ 1.15-1.59 (m, 6H), 1.43 (d, J= 6 Hz, 3H), 1.70-1.79 (m, 2H), 1.82 (m, 1H), 1.91 (m, 2H), 2.41 (m, 2H), 2.69 (m, 1 H), 3.91 (m, 4H), 4.75 (m, 1 H), 6.52-6.68 (m, 2H), 7.15 (m, 1 H), 7.22 (m, 2H), 7.35 (m, 1 H), 7.44 (m, 1 H), 7.81 (m, 1 H), 8.77 (d, J= 1.2 Hz, 1 H). Step 5:

Compound 11 (445 mg, 0.96 mmol) was dissolved in a mixture of acetone (10 ml) and 1 N HCI (10 ml). The reaction mixture was heated at 50 °C for 1 h.
Standard aqueous work-up followed by preparative TLC separation (50% EtOAc in hexane) afforded product 12 (356 mg, 89%). 1HNMR (CDCI3): δ 1.21-1.45 (m, 2H), 1.47 (d, J= 5.6 Hz, 3H), 1.58-1.65 (m, 2H), 2.15 (m, 1 H), 2.18-2.28 (m, 2H), 2.35- 2.51 (m, 5H), 2.71 (m, 1 H), 4.79 (m, 1 H), 6.52-6.68 (m, 2H), 7.15 (m, 1 H), 7.22 (m, 2H), 7.35 (m, 1 H), 7.44 (m, 1 H), 7.81 (m, 1 H), 8.77 (d, J= 1.2 Hz, 1 H). Step 6:

Compound 12 (500 mg, 4.2 mmol) was dissolved in EtOH (40 ml) and CH2CI2 (15 ml) NH3 (g) was bubbled into the solution for 5 min. The reaction mixture was C(O)Oled to 0 °C followed by addition of Ti(O/‘Pr)4 (1.89 ml, 6.3 mmol). After stirring at 0 °C for 1 h, 1 M TiCI (6.3 ml, 6.3 mmol) was added. The reaction mixture was stirred at rt for 45 min and concentrated to dryness under reduced pressure. The residue was dissolved in CH3OH (10 ml) and NaBH3CN (510 mg, 8 mmol) was added. The reaction mixture was stirred overnight at rt. The reaction mixture was poured to 1 N NaOH (100 ml) and extracted with EtOAc (3x 100 ml). The organic layer was combined and dried with NaHC03. Removal of solvent and separation by PTLC (5% 2 M NH3 in CH3OH/ CH2CI2) afforded β-13 (spot 1 , 30 mg, 6%) and α-13 (spot 2, 98 mg, 20%). β-13: 1HNMR (CDCI3): δ 1.50-1.38 (m, 5H), 1.42 (d, J= 6 Hz, 3H), 1.51-1.75 (m, 5H), 1.84 (m, 2H), 2.38 (m, 1 H), 2.45 (m, 1 H), 3.38 (br s, 1 H), 4.78 (m, 1 H), 6.59 (m, 2H), 7.15 (m, 1 H), 7.26 (m, 2H), 7.36 (m, 1 H), 7.42 (m, 1 H), 7.82 (m, 1 H), 8.77 (d, J= 2 Hz, 1 H). α-13:1HNMR (CDCI3): δ 0.95 (m, 2H), 1.02-1.35 (m, 6H), 1.41 (d, J= 6 Hz, 3H), 1.82-1.95 (m, 4H), 2.37 (m; 2H), 2.69 (m, 2H), 4.71 (m, 1 H), 6.71 (m, 2H), 7.11 (m, 1 H), 7.25 (m, 2H), 7.38 (m, 1 H), 7.42 (m, 1 H), 7.80 (m, 1 H), 8.76 (d, J= 1.6 Hz, 1 H). Step 7:
Compound α-13 (300 mg, 0.71 mmol) was dissolved in CH2CI2 (10 ml) followed by addition of Et3N (0.9 ml). The reaction mixture was C(O)Oled to 0 °C and ethyl chloroformate (0.5 ml) was added. The reaction mixture was stirred at rt for 1 h. The reaction mixture was directly separated by preparative TLC (EtOAc/ hexane, 1 :1) to give the title compound (14) VORAPAXAR (300 mg, 86%). MS m/z 493 (M+1).
HRMS Calcd for C29H34N2O4F (M+1 ): 493.2503, found 493.2509.
PATENT
SYNTHESIS 1
http://www.google.com/patents/WO2006076564A1
VORAPAXAR= COMPD A
Example 6 – Preparation of Compound A

To a three-neck flask equipped with an agitator, thermometer and nitrogen inertion was added 7A (13.0 g), THF (30 mL). The mixture was cooled to below -200C after which lithium diisopropylamide (2M, 20 mL) was slowly added. The reaction mixture was agitated for an additional hour (Solution A). To another flask was added 6 (10.0 g) and THF (75 mL) . The mixture was stirred for about 30 minutes and then slowly transferred into the solution A while maintaining the temperature below 200C. The mixture was stirred at below -200C for an additional hour before quenching the reaction by adding 20 mL of water. The reaction mixture was warmed to 00C and the pH was adjusted to about 7 by addition of 25% HaSO4 (11 mL). The mixture was further warmed to 200C and then diluted with 100 mL of ethyl acetate and 70 mL of water. The two phases that had formed were separated and the aqueous layer was extracted with 50 mL of ethyl acetate. The solvents THF and ethyl acetate were then replaced with ethanol, and the Compound A was precipitated out as a crystalline solid from ethanol with seeding at 35 to 4O0C. After cooling to O0C, the suspension was stirred for an additional hour and then the product was filtered and washed with cold ethanol. The product was dried at 50 – 600C under vacuum to provide an off-white solid. VORAPAXAR
Yield: 12.7 g, (90%). m.p. 104.90C (DSC onset point).
1H NMR (CDCl3) δ 8.88 (d, J = 2.4 Hz, IH), 8.10 (dd, J = 8.2, 2.4 Hz, IH), 7.64 (IH), 7.61 (d, J = 8.8 Hz, IH), 7.55 (m, J = 8.2, 6.2 Hz, IH), 7.51 (d, J = 8.0 Hz, IH), 7.25 (dt, J = 9.0, 2.3 Hz, IH), 7.08 (d, J = 8.0 Hz, IH), 6.68 (dd, J = 15.4, 9.4 Hz, IH), 6.58 (d, J = 9.6 Hz, IH), 4.85 (dd, J = 14.2, 7.2 Hz, IH), 3.95 (dd, J = 14.2, 7.1 Hz, 2H), 3.29 (m, IH), 2.66 (m, J = 12.0, 6.4 Hz, IH), 2.33 (m, 2H), 1.76 (m, 4H), 1.30 (d, J = 5.6 Hz, 3H), 1.19 (m, 4H), 1.14 (t, J = 7.2 Hz, 3H), 0.98 (m, IH), 0.84 (m, IH). MS (EI) m/z: calcd. 492, found 492.
BISULPHATE SALT
Example 7 – Preparation of an Acid Salt (bisulfate) of Compound A:
Compound IA (5 g) was dissolved in about 25 mL of acetonitrile.
The solution was agitated for about 10 minutes and then heated to about 50 0C. About 6 mL of 2M sulfuric acid in acetonitrile was added into the heated reaction mixture. The solid salt of Compound A precipitated out during the addition of sulfuric acid in acetonitrile. After addition of sulfuric acid solution, the reaction mixture was agitated for 1 hour before cooling to room temperature. The precipitated solid was filtered and washed with about 30 mL of acetonitrile. The wet solid was dried under vacuum at room temperature for 1 hour and at 80 0C for about 12 hours to provide about 5 g white solid (yield 85%). m.p. 217.0 0C. 1H NMR (DMSO) 9.04 (s, IH), 8.60 (d, J = 8.1 Hz, IH), 8.10 (d, J = 8.2 Hz, IH), 7.76 (d, J = 10.4, IH), 7.71 (d, J = 7.8 Hz, IH), 7.60 (dd, J = 8.4, 1.8 Hz, IH), 7.34 (dd, 8.4, 1.8 Hz, IH), 7.08 (d, J = 8.0 Hz, IH), 7.02 (m, IH), 6.69 (d, J = 15.8 Hz, IH), 4.82 (m, IH), 3.94 (dd, J = 14.0, 7.0 Hz, 2H), 3.35 (brs, IH), 2.68 (m, IH), 2.38 (m, 2H), 1.80-1.70 (m, 4H), 1.27 (d, J = 5.8 Hz, 3H), 1.21 (m, 2H), 1.13 (t, J = 7.0 Hz, 3H), 0.95 (m, IH, 0.85 (m, IH). MS (EI) m/z calcd. 590, found 492.
INTERMEDIATE 6
Example 5- Preparation of Compound 6

To a three-neck flask equipped with an agitator, thermometer and nitrogen inert were added the crude product solution of Compound 5 (containing about 31 g. of Compound 5 in 300 mL solution) and anhydrous DMF (0.05 mL). After the mixture was agitated for 5 minutes, oxalyl chloride (12.2 mL) was added slowly while maintaining the batch temperature between 15 and 25°C. The reaction mixture was agitated for about an hour after the addition and checked by NMR for completion of reaction. After the reaction was judged complete, the mixture was concentrated under vacuum to 135 mL while maintaining the temperature of the reaction mixture below 300C. The excess oxalyl chloride was removed completely by two cycles of vacuum concentration at below 500C with replenishment of toluene (315 mL) each time, resulting in a final volume of 68 mL. The reaction mixture was then cooled to 15 to 25°C, after which THF (160 mL) and 2,6-lutidine (22 mL) were added. The mixture was agitated for 16 hours at 20 to 25°C under 100 psi hydrogen in the presence of dry 5% Pd/C (9.0 g). After the reaction was judged complete, the reaction mixture was filtered through celite to remove catalyst. More THF was added to rinse the hydrogenator and catalyst, and the reaction mixture was again filtered through celite. Combined filtrates were concentrated under vacuum at below 25°C to 315 mL. MTBE (158 mL) and 10% aqueous solution of phosphoric acid (158 mL) were added for a thorough extraction at 100C to remove 2,6- lutidine. Then phosphoric acid was removed by extracting the organic layer with very dilute aqueous sodium bicarbonate solution (about 2%), which was followed by a washing with dilute brine. The organic solution was concentrated atmospherically to a volume of 90 mL for solvent replacement. IPA (315 mL) was added to the concentrated crude product solution. The remaining residual solvent was purged to <_ 0.5% of THF (by GC) by repeated concentration under vacuum to 68 mL, with replenishment of IPA (315 mL) before each concentration. The concentrated (68 mL) IPA solution was heated to 50°C, to initiate crystallization. To this mixture n-heptane (68 mL) was added very slowly while maintaining the batch temperature at 50°C. The crystallizing mixture was cooled very slowly over 2.5 hours to 25°C. Additional n- heptane (34 mL) was added very slowly into the suspension mixture at 250C. The mixture was further cooled to 200C, and aged at that temperature for about 20 hours. The solid was filtered and washed with a solvent mixture of 25% IPA in n-heptane, and then dried to provide
19.5 g of a beige colored solid of Compound 6. (Yield: 66%) m.p. 169.30C. IH NMR (CD3CN) δ 9.74 (d, J = 3.03 Hz, IH), 5.42 (br, IH), 4.69 (m, IH), 4.03 (q, J = 7.02 Hz, 2H), 3.43 (qt, J = 3.80, 7.84 Hz, IH), 2.67 (m, 2H), 2.50 (dt, J = 3.00, 8.52 Hz, IH), 1.93 (d, J = 12.0 Hz, 2H), 1.82 (dt, J = 3.28, 9.75 Hz, 2H), 1.54 (qd, J = 3.00, 10.5 Hz, IH), 1.27 (d, J = 5.97 Hz, 3H), 1.20 (m, 6H), 1.03 – 0.92 (m, 2H). MS (ESI) m/z (M++1): calcd. 324, found 324.
INTERMEDIATE 7A
Example 4 – Preparation of Compound 7A
+ 1-Pr2NLi + (EtO)2POCI – + LiCI
![]()
8

7A
To a 10 L three-necked round bottomed flask equipped with an agitator, thermometer and a nitrogen inlet tube, was added 20Og of
Compound 8 (1.07 mol, from Synergetica, Philadelphia, Pennsylvania). THF (1000 mL) was added to dissolve Compound 8. After the solution was cooled to -80 0C to -50 0C, 2.0 M LDA in hexane/THF(1175 mL, 2.2 eq) was added while maintaining the batch temperature below -50 0C. After about 15 minutes of agitation at -800C to -50 0C, diethyl chlorophosphate (185 mL, 1.2 eq) was added while maintaining the batch temperature below -50 0C. The mixture was agitated at a temperature from -800C to – 50 0C for about 15 minutes and diluted with n-heptane (1000 mL). This mixture was warmed up to about -35 0C and quenched with aqueous ammonium chloride (400 g in 1400 mL water) at a temperature below -10 0C. This mixture was agitated at -150C to -10 0C for about 15 minutes followed by agitation at 150C to 25 0C for about 15 minutes. The aqueous layer was split and extracted with toluene (400 mL). The combined organic layers were extracted with 2N hydrochloric acid (700 mL) twice. The product-containing hydrochloric acid layers were combined and added slowly to a mixture of toluene (1200 mL) and aqueous potassium carbonate (300 g in 800 mL water) at a temperature below 30 0C. The aqueous layer was extracted with toluene (1200 mL). The organic layers were combined and concentrated under vacuum to about 600 ml and filtered to remove inorganic salts. To the filtrate was added n-heptane (1000 ml) at about 55 0C. The mixture was cooled slowly to 40 0C, seeded, and cooled further slowly to -10 0C. The resulting slurry was aged at about -10 0C for 1 h, filtered, washed with n- heptane, and dried under vacuum to give a light brown solid (294 g, 85% yield), m.p. 52 0C (DSC onset point).1H NMR (CDCl3) δ 8.73 (d, J = 1.5 Hz, IH), 7.85 (dd, Ji = 8.0 Hz, J2 = 1.5 Hz, IH), 7.49 (dd, Ji = 8.0 Hz, J2 = 1.3 Hz, IH), 7.42 (m, IH), 7.32 (d, J = 7.8 Hz, IH), 7.24 (m, IH), 7.08 (dt, Ji = 8.3 Hz, J2 = 2.3 Hz, IH), 4.09 (m, 4H), 3.48 (d, J = 22.0 Hz, 2H), 1.27 (t, J = 7.0 Hz, 6H). MS (ESI) for M+H calcd. 324, found 324.
Example 3 – Preparation of Compound 5:

4 5
To a three-necked round bottomed flask equipped with an agitator, thermometer and a nitrogen inlet tube was added a solution of Compound 4 in aqueous ethanol (100 g active in 2870 ml). The solution was concentrated to about 700 ml under reduced pressure at 350C to 40°C to remove ethyl alcohol. The resultant homogeneous mixture was cooled to 200C to 300C and its pH was adjusted to range from 12 to 13 with 250 ml of 25% sodium hydroxide solution while maintaining the temperature at 20-300C. Then 82 ml of ethyl chloroformate was slowly added to the batch over a period of 1 hour while maintaining the batch temperature from 200C to 300C and aged for an additional 30 minutes. After the reaction was judged complete, the batch was acidified to pH 7 to 8 with 10 ml of concentrated hydrochloric acid (37%) and 750 ml of ethyl acetate. The pH of the reaction mixture was further adjusted to pH 2 to 3 with 35% aqueous hydrochloric acid solution. The organic layer was separated and the aqueous layer was extracted again with 750 ml of ethyl acetate. The combined organic layers were washed twice with water (200 ml) . Compound 5 was isolated from the organic layer by crystallization from ethyl acetate and heptane mixture (1: 1 mixture, 1500 ml) at about 700C to 80 0C. The solid was filtered at 500C to 60 °C, washed with heptane and then dried to provide an off-white solid (yield 50%). m.p. 197.7°C. 1HNMR (CD3CN) δ 5.31 (brs, IH), 4.67 (dt, J = 16.1, 5.9 Hz, IH), 4.03 (q, J = 7.1 Hz, 2H), 3.41 (m, IH), 2.55 – 2.70 (m, 2H), 1.87 – 1.92 (m, IH), 1.32 – 1.42 (m, IH), 1.30 (d, J = 5.92 Hz, 3H), 1.30 – 1.25 (m, 6H), 0.98 (qt, J = 15.7, 3.18 Hz, 2H). MS (ESI) M+l m/z calculated 340, found 340.
Example 2 – Preparation of Compound 4;

3 4
7.4 kg of ammonium formate was dissolved in 9L of water at 15- 250C, and then cooled to 0-100C. 8.9 kg of Compound 3 was charged at 0-150C followed by an addition of 89L of 2B ethyl alcohol. The batch was cooled to 0-50C 0.9 kg of 10% Palladium on carbon (50% wet) and 9 L of water were charged. The batch was then warmed to 18-280C and agitated for 5 hours, while maintaining the temperature between 18-28 0C. After the reaction was judged complete, 7 IL of water was charged. The batch was filtered and the wet catalyst cake was then washed with 8OL of water. The pH of the filtrate was adjusted to 1-2 with 4N aqueous hydrochloric acid solution. The solution was used in the next process step without further isolation. The yield is typically quantiative. m.p. 216.40C. IH NMR (D2O+1 drop HCl) δ 3.15 (m, IH), 2.76 (m, IH), 2.62 (m, IH), 2.48 (dd,J-5.75Hz, IH), 1.94 (m, 2H), 1.78 (m, 2H), 1.38 (m, 2H), 1.20 (m, 6H), 1.18 (m, IH), 0.98 (q,J=2.99Hz, IH).
Example 1 – Preparation of Compound 3

2B 3
To a reactor equipped with an agitator, thermometer and nitrogen, were added about 10.5 kg of 2B, 68 L of acetone and 68 L of IN aqueous hydrochloric acid solution. The mixture was heated to a temperature between 50 and 600C and agitated for about 1 hour before cooling to room temperature. After the reaction was judged complete, the solution was concentrated under reduced pressure to about 42 L and then cooled to a temperature between 0 and 50C. The cooled mixture was agitated for an additional hour. The product 3 was filtered, washed with cooled water and dried to provide an off-white solid (6.9 kg, yield 76%). m.p. 2510C. Η NMR (DMSO) δ 12.8 (s, IH), 4.72 (m, J = 5.90 Hz, IH), 2.58 (m, 2H), 2.40 (m, J = 6.03 Hz, 2H), 2.21 (dd, J = 19.0, 12.8 Hz, 3H), 2.05 (m, IH), 1.87 (q, J = 8.92 Hz, IH), 1.75 (m, IH), 1.55 (m, IH), 1.35 (q, J = 12.6 Hz, IH), 1.27 (d, J = 5.88 Hz, 3H). MS (ESI) M+l m/z calcd. 267, found 267.
NOTE
Compound 7A may be prepared from Compound 8 by treating Compound 8 with diethylchlorophosphate:

Compound 8 may be obtained by the process described by Kyoku, Kagehira et al in “Preparation of (haloaryl)pyridines,” (API Corporation, Japan). Jpn. Kokai Tokkyo Koho (2004). 13pp. CODEN: JKXXAF JP
2004182713 A2 20040702. Compound 8 is subsequently reacted with a phosphate ester, such as a dialkyl halophosphate, to yield Compound 7A. Diethylchlorophosphate is preferred. The reaction is preferably conducted in the presence of a base, such as a dialkylithium amide, for example diisopropyl lithium amide.
Paper
J Med Chem 2008, 51(11): 3061
http://pubs.acs.org/doi/abs/10.1021/jm800180e
The discovery of an exceptionally potent series of thrombin receptor (PAR-1) antagonists based on the natural product himbacine is described. Optimization of this series has led to the discovery of 4 (SCH 530348), a potent, oral antiplatelet agent that is currently undergoing Phase-III clinical trials for acute coronary syndrome (unstable angina/non-ST segment elevation myocardial infarction) and secondary prevention of cardiovascular events in high-risk patients.
Ethyl [(3aR,4aR,8aR,9aS)-9(S)-[(E)-2-[5-(3-fluorophenyl)-2-
pyridinyl]ethenyl]dodecahydro-1(R)-methyl-3-oxonaphtho[2,3-c]furan-6(R)-yl]carbamate (4).
4 (300 mg, 86%). MS m/z 493 (M+1).
HRMS Calcd for C29H34N2O4F
(M+1): 493.2503, found 493.2509; mp125 °C;
[]D20 6.6 (c 0.5, MeOH).
1HNMR (CDCl3):
http://pubs.acs.org/doi/suppl/10.1021/jm800180e/suppl_file/jm800180e-file002.pdf
0.88-1.18 (m, 5 H), 1.22-1.30 (m, 3 H), 1.43 (d, J = 5.85 Hz, 3 H), 1.88-2.10 (m, 4 H), 2.33-2.42 (m, 2 H),
2.75-2.67 (m, 1 H), 3.52-3.60 (m, 1 H), 4.06-4.14 (m, 2 H), 4.54-4.80 (m, 1 H), 4.71-4.77 (m, 1 H),
6.55-6.63 (m, 2 H), 7.07-7.12 (m, 1 H), 7.26-7.29 (m, 2 H), 7.34 (d, J = 8.05 Hz, 1 H), 7.41-7.46 (m, 1 H), 7.80-7.82 (m, 1 H), 8.76-8.71 (m, 1 H).
PATENT
IN 201621010411
An improved process for preparation of Vorapaxar intermediates and a novel polymorphic form of Vorapaxar
ALEMBIC PHARMACEUTICALS LIMITED
Vorapaxar Sulfate is indicated for the reduction of thrombotic cardiovascular events in patients with a history of myocardial infarction (MI) or with peripheral arterial disease (PAD). ZONTIVITY has been shown to reduce the rate of a combined endpoint of cardiovascular death, MI, stroke, and urgent coronary revascularization (UCR).
According to present invention Vorapaxar sulfate is synthesized from compound of formula 1.
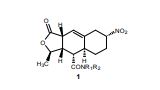
wherein R1 and R2 are each independently selected from the group consisting of H, alkyl, alkenyl, alkynyl, alkoxy, cycloalkyl, aryl, alkylaryl, arylalkyl, and heteroaryl groups. Process for the preparation of compound of formula 1 is disclosed in U.S Pat. No. 7,605,275. It disclosed preparation of compound of formula 1 via cyclization of compound 2 in presence of solvent selected from xylene, N-methylpyrrolidinone, Dimethylsulfoxide, diphenyl ether, dimethylacetamide. This cyclization step takes approximately 6-8 hrs.
There is need to develop a process which takes less time for cyclization step to prepare compound of formula 1. Therefore, our scientist works tenaciously to develop process which takes approximately 1-2 hrs for cyclization of compound 1.
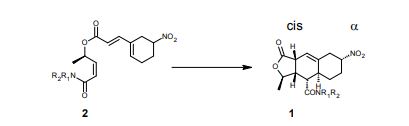
5 According to present invention Vorapaxar sulfate is synthesized from intermediate compound of formula-II.
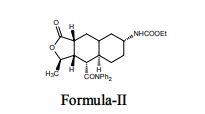
Formula-II Compound of formula-II is critical intermediate in the preparation of Vorapaxar Sulfate.
10 Patent WO2006076415 discloses the process of preparation of above Formula-II in example 7, in which purification/crystallisation step involves treating the reaction mixture having compound of Formula-II with an ethanol/water mixture followed by azeotropic distillation of the mixture. This process yielded formula-II with low yields and with low purities. WO2009055416 (page 9, second paragraph) discloses that use of various solvent systems for
15 formula-II purification such as Methyl-tert-Butyl Ether (MTBE) and various solvent/antisolvent systems, for example, ethylacetate/heptane and toluene/heptane and by using these solvent systems, compound of formula-II are obtained as oil. These oils did not yield a reduced impurity profile in synthesis of the compound of Formula II, nor provide an improvement in the quality of the product compound of Formula II.
20 The inventors surprisingly found that using the process according to the invention provides formula-II with improved yield and high purity. Further, present invention provides a process for the preparation of novel crystalline form of Vorapaxar base. The present invention also relates to novel impurity and process for its preparation.
U.S.Pat. No. 7,304,078 discloses Vorapaxar base. U.S.Pat. No. 7,235,567 discloses Polymorph I and II of vorapaxar sulphate
Example 1- Preparation of compound 1a:
Process A: 5.0 g of compound 2a was suspended in 10.0 ml silicone oil at room temperature. The reaction mixture was then heated to 125°C and stirred for 30 min. Then reaction mass was further heated up to 150°C and stirred for 30 min. After completion of reaction, the reaction mass was cooled to 50-60°C and 25 ml of cyclohexane was added to the reaction mass. The reaction mass was cooled slowly up to room temperature and stirred for 30 min.
15 The precipitated product was filtered off and washed with 5.0 ml Cyclohexane. Wet solid was suspended in mixture of 45.0 ml isopropyl alcohol and 20.0 ml denatured ethanol at 40-45°C and further epimerized with 0.17 ml DBU. The crystallized solid was filtered off with suction, washed with mixture of 1.5 ml Isopropyl alcohol and 0.67 ml denatured ethanol and dried.
20 Process B: 5.0 g of compound 2a was suspended in 10.0 ml paraffin oil at room temperature. The reaction mixture was then heated to 125°C and stirred for 30 min. Then reaction mass was further heated up to 150°C and stirred for 30 min. After completion of reaction, the reaction mass was cooled to 50-60°C and 25 ml of cyclohexane was added to the reaction mass. The reaction mass was cooled slowly up to room temperature and stirred for 30 min.
25 The precipitated product was filtered off and washed with 5.0 ml Cyclohexane. Wet solid was suspended in mixture of 45.0 ml isopropyl alcohol and 20.0 ml denatured ethanol at 40-45°C and further epimerized with 0.17 ml DBU. The crystallized solid was filtered off with suction, washed with mixture of 1.5 ml Isopropyl alcohol and 0.67 ml denatured ethanol and dried. Yield: 4.3 g
Process C: 5.0 g of compound 2a was charged in reaction vessel at room temperature. The solid was then heated to 125°C and stirred for 30 min. Then reaction mass was further heated up to 150°C and stirred for 30 min. After completion of reaction, the reaction mass was cooled to 50-60°C and was added mixture of 45.0 ml isopropyl alcohol and 20.0 ml
5 denatured ethanol at 50-60°C. This was cooled to 40-45°C and further epimerized with 0.17 ml DBU. The crystallized solid was filtered off with suction, washed with mixture of 1.5 ml Isopropyl alcohol and 0.67 ml denatured ethanol and dried. Yield: 4.5 g Example 2: Preparation of Intermediate (Formula-II) of vorapaxar
10 Example 2(a): 50.0g of 1,3,3a,4,4a,5,6,7,8,9a-Decahydro-3-methyl-7-nitro-1-oxo-N,Ndiphenylnaphtho[2,3-c]furan-4-carboxamide compound was suspended in 300.0 ml THF, 15 g 10% Pd/C (50% wet) and 200 ml Process water at room temperature. The reaction mixture was heated to 45°C and drop wise formic acid (35 ml) was added and then stirred for 15 hrs. After completion of reaction, the reaction mass was cooled to 25-30°C and 100 ml THF was
15
added and pH was made acidic with 2M sulfuric acid solution. The reaction mass was filtered and washed with 150 ml THF, 150 ml water. Organic and aqueous layer were separated and aqueous layer was extracted with THF. Organic layers were combined and washed with water. The organic layer was cooled up to 5-10°C, 20 ml of TEA and 13 ml of Ethyl chloro formate were added. The reaction mass was stirred for 30 min. After completion of reaction,
20
reaction mass was washed with 2M sulfuric acid solution and distilled out reaction mass completely under vacuum. Acetonitrile (50 ml) was added to residue and heated up to 40- 45°C. Cooled the reaction mass up to 25-30°C and filtered the solid. Purity: 94-96% Example 2(b): Crystallization with Acetonitrile Acetonitrile (50 ml) was added to above obtained solid and heated to 40-45°C. Cooled the
25 reaction mass slowly up to 25-30°C and then up to 5-10°C. The reaction mass was stirred and the solid was filtered. XRD: Fig-1 Purity: 98-99% Example 2(c): Crystallization with Ethyl acetate To the solid obtained in example-1(a) Ethyl acetate (30 ml) was added. The reaction mass was heated up to 70-75°C and stirred for 10-15 min. The reaction mass was cooled slowly up 30 to 25-30°C and then up to 5-10°C. The reaction mass was stirred for 30 min. The solid was filtered and washed with Ethyl acetate. XRD: Fig-2 Purity: 98-99%
Example 3: Preparation of Amorphous Form of Vorapaxar base Vorapaxar base (10.0 g) was dissolved in 500 ml of 40% Ethyl acetate in Cyclohexane. The solvent was then completely removed under vacuum at 45-50o C to give a solid. Yield: 9.8 g
Example 3 (a): Preparation of crystalline vorapaxar base 5 (2-{[Ethyl (ethylperoxy)phosphory]methyl}-5-(3-fluorophenyl)pyridine) (10 g) was dissolved in THF (30ml) at 25±5°C under Nitrogen. Cool the reaction mass up to -30 to – 50°C. Add drop wise LDA (2.0 M solution in THF). After 1 hr add drop wise (N- [(1R,3aR,4aR,6R,8aR,9S,9aS)-9-formyl dodecahydro-1-methyl-3-oxonaphtho[2,3-c]furan-6- yl]-ethyl ester Carbamic acid) solution (10 g dissolved in 70 ml THF). After completion of 10 reaction mass quench the reaction mass to sulphuric acid solution. Separate the layers and distilled out organic layer under vacuum get foamy residue. (purity 82%) Add MIBK (10 ml) in above residue and stir it at 40-50°C till clear solution. Add drop wise n-Heptane (10 ml) and stir the reaction mass for 30 min. Gradually cool the reaction mass up to 25-30°C. Stir the reaction mass for 24 hrs. Filter the solid and washed it with n-Heptane (5.0 ml). Dry the 15 solid. Yield: 7.0 g. XRD: Fig-3 purity 96%
Example 3(b): Preparation of crystalline vorapaxar base Vorapaxar advance intermediate (2-{[Ethyl (ethylperoxy)phosphory]methyl}-5-(3- fluorophenyl)pyridine) (10 g) was dissolved in THF (30ml) at 25±5°C under Nitrogen. Cool the reaction mass up to -30 to -50°C. Add drop wise LDA (2.0 M solution in THF). After a 1
20 hr add drop wise VORA-Aldehyde (N-[(1R,3aR,4aR,6R,8aR,9S,9aS)-9-formyl dodecahydro1-methyl-3-oxonaphtho[2,3-c]furan-6-yl]-ethyl ester Carbamic acid) solution (10 g dissolved in 70 ml THF). After completion of reaction mass quench the reaction mass to sulphuric acid solution. Separate the layers and distilled out organic layer under vacuum get foamy residue (purity 82%). Add MTBE (10 ml) in above residue and stir it at 40-50°C till clear solution.
25 Add drop wise n-Heptane (30 ml) and stir the reaction mass for 30 min. Gradually cool the reaction mass up to 25-30°C. Stir the reaction mass for 24 hrs. Filter the solid and washed it with n-Heptane (5.0 ml). Dry the solid. Yield: 8.5.0 g. XRD: Fig-4 purity 97%
References
- Samuel Chackalamannil; Wang, Yuguang; Greenlee, William J.; Hu, Zhiyong; Xia, Yan; Ahn, Ho-Sam; Boykow, George; Hsieh, Yunsheng et al. (2008). “Discovery of a Novel, Orally Active Himbacine-Based Thrombin Receptor Antagonist (SCH 530348) with Potent Antiplatelet Activity”. Journal of Medicinal Chemistry 51 (11): 3061–4.doi:10.1021/jm800180e. PMID 18447380.
- Merck Blood Thinner Studies Halted in Select Patients, Bloomberg News, January 13, 2011
- Tricoci et al. (2012). “Thrombin-Receptor Antagonist Vorapaxar in Acute Coronary Syndromes”. New England Journal of Medicine 366 (1): 20–33.doi:10.1056/NEJMoa1109719. PMID 22077816.
- Morrow, DA; Braunwald, E; Bonaca, MP; Ameriso, SF; Dalby, AJ; Fish, MP; Fox, KA; Lipka, LJ; Liu, X; Nicolau, JC; Ophuis, AJ; Paolasso, E; Scirica, BM; Spinar, J; Theroux, P; Wiviott, SD; Strony, J; Murphy, SA; TRA 2P–TIMI 50 Steering Committee and, Investigators (Apr 12, 2012). “Vorapaxar in the secondary prevention of atherothrombotic events.”. The New England Journal of Medicine 366 (15): 1404–13. doi:10.1056/NEJMoa1200933.PMID 22443427.
- “Merck Statement on FDA Advisory Committee for Vorapaxar, Merck’s Investigational Antiplatelet Medicine”. Merck. Retrieved 16 January 2014.
- http://www.forbes.com/sites/larryhusten/2014/01/15/fda-advisory-panel-votes-in-favor-of-approval-for-mercks-vorapaxar/
- SCH-530348 (Vorapaxar) is an investigational candidate for the prevention of arterial thrombosis in patients with acute coronary syndrome and peripheral arterial disease. “Convergent Synthesis of Both Enantiomers of 4-Hydroxypent-2-ynoic Acid Diphenylamide for a Thrombin Receptor Antagonist Sch530348 and Himbacine Analogues.” Alex Zaks et al.: Adv. Synth. Catal. 2009, 351: 2351-2357 Full text;
- Discovery of a novel, orally active himbacine-based thrombin receptor antagonist (SCH 530348) with potent antiplatelet activity
J Med Chem 2008, 51(11): 3061
- Stu Borman (2005). “Hopes Ride on Drug Candidates: Researchers reveal potential new medicines for thrombosis, anxiety, diabetes, and cancer”. Chemical & Engineering News 83 (16): 40–44.
PATENTS
- WO 2003089428
- WO 2006076452
- US 6063847
- WO 2006076565
- WO 2008005344
- WO2010/141525
- WO2008/5353
- US2008/26050
- WO2006/76564 mp, nmr
|
3-21-2012
|
EXO-SELECTIVE SYNTHESIS OF HIMBACINE ANALOGS
|
|
|
10-14-2011
|
EXO- AND DIASTEREO- SELECTIVE SYNTHESIS OF HIMBACINE ANALOGS
|
|
|
8-3-2011
|
Exo- and diastereo-selective syntheses of himbacine analogs
|
|
|
3-18-2011
|
COMBINATION THERAPIES COMPRISING PAR1 ANTAGONISTS WITH NAR AGONISTS
|
|
|
8-11-2010
|
Exo-selective synthesis of himbacine analogs
|
|
|
6-4-2010
|
SYNTHESIS Of DIETHYLPHOSPHONATE
|
|
|
5-12-2010
|
THROMBIN RECEPTOR ANTAGONISTS
|
|
|
3-31-2010
|
Synthesis of diethyl{[5-(3-fluorophenyl)-pyridine-2yl]methyl}phosphonate
|
|
|
12-4-2009
|
Local Delivery of PAR-1 Antagonists to Treat Vascular Complications
|
|
|
12-2-2009
|
SYNTHESIS OF HIMBACINE ANALOGS
|
|
10-21-2009
|
Exo- and diastereo- selective syntheses of himbacine analogs
|
|
|
6-31-2009
|
Synthesis of 3-(5-nitrocyclohex-1-enyl) acrylic acid and esters thereof
|
|
|
6-3-2009
|
Synthesis of himbacine analogs
|
|
|
1-23-2009
|
METHODS AND COMPOSITIONS FOR TREATING CARDIAC DYSFUNCTIONS
|
|
|
9-26-2008
|
REDUCTION OF ADVERSE EVENTS AFTER PERCUTANEOUS INTERVENTION BY USE OF A THROMBIN RECEPTOR ANTAGONIST
|
|
|
2-8-2008
|
IMMEDIATE-RELEASE TABLET FORMULATIONS OF A THROMBIN RECEPTOR ANTAGONIST
|
|
|
1-32-2008
|
SOLID DOSE FORMULATIONS OF A THROMBIN RECEPTOR ANTAGONIST
|
|
|
12-5-2007
|
Thrombin receptor antagonists
|
|
|
11-23-2007
|
THROMBIN RECEPTOR ANTAGONISTS
|
|
|
8-31-2007
|
THROMBIN RECEPTOR ANTAGONISTS AS PROPHYLAXIS TO COMPLICATIONS FROM CARDIOPULMONARY SURGERY
|
|
8-31-2007
|
CRYSTALLINE POLYMORPH OF A BISULFATE SALT OF A THROMBIN RECEPTOR ANTAGONIST
|
|
|
6-27-2007
|
Crystalline polymorph of a bisulfate salt of a thrombin receptor antagonist
|
|
|
8-4-2006
|
Preparation of chiral propargylic alcohol and ester intermediates of himbacine analogs
|
|
|
9-31-2004
|
Methods of use of thrombin receptor antagonists
|
| US6063847 * | Nov 23, 1998 | May 16, 2000 | Schering Corporation | Thrombin receptor antagonists |
| US6326380 * | Apr 7, 2000 | Dec 4, 2001 | Schering Corporation | Thrombin receptor antagonists |
| US20030216437 * | Apr 14, 2003 | Nov 20, 2003 | Schering Corporation | Thrombin receptor antagonists |
| US20040176418 * | Jan 9, 2004 | Sep 9, 2004 | Schering Corporation | Crystalline polymorph of a bisulfate salt of a thrombin receptor antagonist |
| WO2011128420A1 | Apr 14, 2011 | Oct 20, 2011 | Sanofi | Pyridyl-vinyl pyrazoloquinolines as par1 inhibitors |
//////////////fast track designation , VORAPAXAR, FDA 2014, EU 2016, Zontivity, NDA 204886, MERCK, VORAPAXAR SULPHATE
CCOC(=O)NC1CCC2C(C1)CC3C(C2C=CC4=NC=C(C=C4)C5=CC(=CC=C5)F)C(OC3=O)C

{kind=link}
{kind=link}
{kind=link}