
Home » Posts tagged 'PHASE 1'
Tag Archives: PHASE 1
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Varegacestat

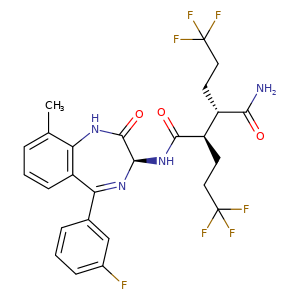
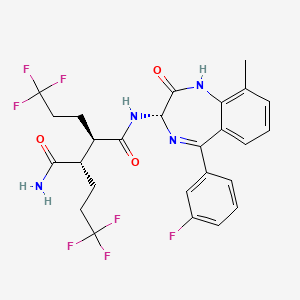
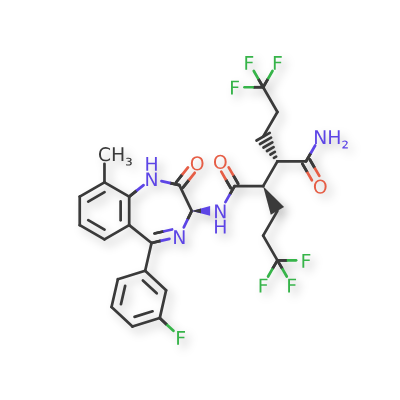
Varegacestat
CAS 1584647-27-7
MF C26H25F7N4O3 MW574.5
(2R,3S)-N1-[(3S)-5-(3-fluorophenyl)-9-methyl-2-oxo-2,3-dihydro-1H1,4-benzodiazepin-3-yl]-2,3-bis(3,3,3-trifluoropropyl)butanediamide
(2R,3S)-N1-((3S)-5-(3-FLUOROPHENYL)-2,3-DIHYDRO-9-METHYL-2-OXO-1H-1,4-BENZODIAZEPIN-3-YL)-2,3-BIS(3,3,3-TRIFLUOROPHENYL)BUTANEDIAMIDE
(2R,3S)-N1-((3S)-5-(3-FLUOROPHENYL)-9-METHYL-2-OXO-2,3-DIHYDRO-1H-1,4-BENZODIAZEPIN-3-YL)-2,3-BIS(3,3,3-TRIFLUOROPHENYL)BUTANEDIAMIDE
gamma-secretase inhibitor, antineoplastic, AL102, BMS 986115, LSK1L593UU, AL 102
BMS-986115 has been used in trials studying the treatment of Various Advanced Cancer.
Varegacestat is an orally bioavailable, gamma secretase (GS) and pan-Notch inhibitor, with potential antineoplastic activity. Upon administration, varegacestat binds to GS and blocks the proteolytic cleavage and release of the Notch intracellular domain (NICD), which would normally follow ligand binding to the extracellular domain of the Notch receptor. This prevents both the subsequent translocation of NICD to the nucleus to form a transcription factor complex and the expression of Notch-regulated genes. This results in the induction of apoptosis and the inhibition of growth of tumor cells that overexpress Notch. Overexpression of the Notch signaling pathway plays an important role in tumor cell proliferation and survival. The integral membrane protein GS is a multi-subunit protease complex that cleaves single-pass transmembrane proteins, such as Notch receptors, at residues within their transmembrane domains and leads to their activation
AL 102 (previously known as BMS 986115), was developed as an orally active a gamma-secretase and pan-Notch inhibitor. The drug participated in phase I clinical trials in solid tumor patients. The drug was safe and well-tolerated and stabilized disease for more than six months in 14% of patients, however, Bristol-Myers Squibb terminated the study because of the changes in the business objectives. Ayala, an Israeli biotech company, licensed rights for the development of AL 102 from Bristol-Myers Squibb. In December 2018, Ayala in collaborating with Novartis decided to investigate AL102 for treatment of multiple myeloma. Ayala studied AL102, an inhibitor of the Notch pathway, in blood cancers. It is known that the pathway regulates cell-fate determination during development and maintains adult tissue balance. Cumulative evidence indicates that Notch is overactive in multiple myeloma and participates in its onset and progression.
SYN
PATENTS
PATENT
https://www.google.com/patents/US20140087992
Example 1(2R,3S)—N-((3S)-5-(3-Fluorophenyl)-9-methyl-2-oxo-2,3-dihydro-1H-1,4-benzodiazepin-3-yl)-2,3-bis(3,3,3-trifluoropropyl)succinamide
Intermediate 1A: (2S,3R)-tert-Butyl 6,6,6-trifluoro-3-(((S)-5-(3-fluorophenyl)-9-methyl-2-oxo-2,3-dihydro-1H-benzo[e][1,4]diazepin-3-yl)carbamoyl)-2-(3,3,3-trifluoropropyl)hexanoate

In a 100 mL round-bottomed flask, a solution of Intermediate B-1 (1683 mg, 5.94 mmol), Et3N (1.656 mL, 11.88 mmol), and Intermediate S-1 in DMF (20 mL) was treated with o-benzotriazol-1-yl-N,N,N′,N′-tetramethyluronium tetrafluoroborate (3815 mg, 11.88 mmol) and stirred at room temperature for 1 hour. The reaction mixture was diluted with water and saturated aqueous NaHCO3. An off white precipitate formed and was filtered and washed with water. The resulting solid was dried on the filter under a stream of nitrogen to give Intermediate 1A (3.7 g, 99% yield). MS (ES): m/z=632.4[M+H+]; HPLC: RT=3.635 min Purity=98%. (H2O/MeOH with TFA, CHROMOLITH® ODS S5 4.6×50 mm, gradient=4 min, wavelength=220 nm). 1H NMR (400 MHz, methanol-d4) δ 7.53 (t, J=4.5 Hz, 1H), 7.46-7.30 (m, 3H), 7.28-7.23 (m, 1H), 7.23-7.18 (m, 2H), 5.37 (s, 1H), 2.88 (td, J=10.4, 3.4Hz, 1H), 2.60 (td, J=10.2, 4.1 Hz, 1H), 2.54-2.40 (m, 1H), 2.47 (s, 3H), 2.33-2.12 (m, 3H), 1.98-1.69 (m, 4H), 1.51 (s, 9H).
Intermediate 1B: (2S,3R)-6,6,6-Trifluoro-3-(((S)-5-(3-fluorophenyl)-9-methyl-2-oxo-2,3-dihydro-1H-benzo[e][1,4]diazepin-3-yl)carbamoyl)-2-(3,3,3-trifluoropropyl)hexanoic acid

In a 250 mL round-bottomed flask, a solution of Intermediate 1A (3.7 g, 5.86 mmol) in DCM (25 mL) was treated with TFA (25 mL) and the resulting pale orange solution was stirred at room temperature for 1.5 hours. The reaction mixture was then concentrated to give Intermediate 1B. HPLC: RT=3.12 min (H2O/MeOH with TFA, CHROMOLITH® ODS S5 4.6×50 mm, gradient=4 min, wavelength=220 nm). MS (ES): m/z=576.3 (M+H)+. 1H NMR (400 MHz, methanol-d4) δ 7.54 (t, J=4.5 Hz, 1H), 7.49-7.29 (m, 3H), 7.28-7.15 (m, 3H), 5.38 (br. s., 1H), 2.89 (td, J=10.3, 3.7 Hz, 1H), 2.67 (td, J=9.9, 4.2Hz, 1H), 2.56-2.38 (m, 1H), 2.48 (s, 3H), 2.34-2.13 (m, 3H), 2.00-1.71 (m, 4H).
Example 1
In a 250 mL round-bottomed flask, a solution of Intermediate 1B (4.04 g, 5.86 mmol) in THF (50 mL) was treated with ammonia (2M in iPrOH) (26.4 mL, 52.7 mmol), followed by HOBT (1.795 g, 11.72 mmol) and EDC (2.246 g, 11.72 mmol). The resulting white suspension was stirred at room temperature overnight. The reaction mixture was diluted with water and saturated aqueous NaHCO3. The resulting solid was filtered, rinsed with water and then dried on the filter under a stream of nitrogen. The crude product was suspended in 20 mL of iPrOH and stirred at room temperature for 20 min and then filtered and washed with iPrOH and dried under vacuum to give 2.83 g of solid. The solid was dissolved in refluxing EtOH (100 mL) and slowly treated with 200 mg activated charcoal added in small portions. The hot mixture was filtered through CELITE® and rinsed with hot EtOH. The filtrate was reduced to half volume, allowed to cool and the white precipitate formed was filtered and rinsed with EtOH to give 2.57 g of white solid. A second recrystallization from EtOH (70 mL) afforded Example 1 (2.39 g, 70% yield) as a white solid. HPLC: RT=10.859 min (H2O/CH3CN with TFA, Sunfire C18 3.5 μm, 3.0×150 mm, gradient=15 min, wavelength=220 and 254 nm); MS (ES): m/z=575.3 [M+H+]; 1H NMR (400 MHz, methanol-d4) δ 7.57-7.50 (m, 1H), 7.47-7.30 (m, 3H), 7.29-7.15 (m, 3H), 5.38 (s, 1H), 2.85-2.75 (m, 1H), 2.59 (td, J=10.5, 4.0 Hz, 1H), 2.53-2.41 (m, 4H), 2.31-2.10 (m, 3H), 1.96-1.70 (m, 4H).
PATENT
https://www.google.com/patents/WO2014047372A1?cl=en


Scheme 3


XII XI
Scheme 4

Intermediate S-l : (2R,3S)-3-(fert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid

Intermediate S-IA: 3,3,3-Trifluoro ropyl trifluoromethanesulfonate

[00180] To a cold (-25 °C) stirred solution of 2,6-lutidine (18.38 mL, 158 mmol) in DCM (120 mL) was added Tf20 (24.88 mL, 147 mmol) over 3 min, and the mixture was stirred for 5 min. To the reaction mixture was added 3,3,3-trifluoropropan-l-ol (12 g, 105 mmol) over an interval of 3 min. After 2 hr, the reaction mixture was warmed to room temperature and stirred for 1 hr. The reaction mixture was concentrated to half its volume, then purified by loading directly on a silica gel column (330g ISCO) and the product was eluted with DCM to afford Intermediate S-IA (13.74 g, 53%) as a colorless oil. 1H NMR (400 MHz, CDC13) δ ppm 4.71 (2 H, t, J= 6.15 Hz), 2.49-2.86 (2 H, m).
Intermediate S-1B: (4S)-4-Benzyl-3-(5,5,5-trifluoropentanoyl)-l,3-oxazolidin-2-one

[00181] To a stirring solution of 5,5,5-trifluoropentanoic acid (14.76 g, 95 mmol) and DMF (0.146 rriL) in DCM (50 mL) was slowly added oxalyl chloride (8.27 mL, 95 mmol). After 2h, the mixture was concentrated to dryness. A separate flask was changed with (S)-4-benzyloxazolidin-2-one (16.75 g, 95 mmol) in THF (100 mL) and then cooled to -78 °C. To the solution was slowly added n-BuLi (2.5M, 37.8 mL, 95 mmol) over 10 min, stirred for 10 min, and then a solution of the above acid chloride in THF (50 mL) was slowly added over 5 min. The mixture was stirred for 30 min, and then warmed to room temperature. The reaction was quenched with sat aq NH4C1. Next, 10% aq LiCl was then added to the mixture, and the mixture was extracted with Et20. The organic layer was washed with sat aq NaHC03 then with brine, dried (MgSC^), filtered and concentrated to dryness. The residue was purified by Si02 chromatography (ISCO, 330 g column, eluting with a gradient from 100% hexane to 100% EtOAc) to afford the product Intermediate S-IB; (25.25 g, 85%): 1H NMR (400 MHz, CDC13) δ ppm 7.32-7.39 (2 H, m), 7.30 (1 H, d, J= 7.05 Hz), 7.18-7.25 (2 H, m), 4.64-4.74 (1 H, m), 4.17-4.27 (2 H, m), 3.31 (1 H, dd, J= 13.35, 3.27 Hz), 3.00-3.11 (2 H, m), 2.79 (1 H, dd, J= 13.35, 9.57 Hz), 2.16-2.28 (2 H, m), 1.93-2.04 (2 H, m).
Intermediate S-IC: tert- utyl (3R)-3-(((4S)-4-benzyl-2-oxo-l,3-oxazolidin-3- yl)carbonyl)-6,6,6-trifluoroh xanoate

[00182] To a cold (-78 °C), stirred solution of Intermediate S-IB (3.03 g, 9.61 mmol) in THF (20 mL) was added NaHMDS (1.0M in THF) (10.6 mL, 10.60 mmol) under a nitrogen atmosphere. After 2 hours, tert-butyl 2-bromoacetate (5.62 g, 28.8 mmol) was added neat via syringe at -78 °C and stirring was maintained at the same temperature. After 6 hours, the reaction mixture was warmed to room temperature. The reaction mixture was partitioned between saturated NH4C1 and EtOAc. The organic phase was separated, and the aqueous phase was extracted with EtOAc (3x). The combined organics were washed with brine, dried (Na2s04), filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (Teledyne ISCO
CombiFlash Rf, 5% to 100% solvent A/B = hexanes/EtOAc, REDISEP® Si02 120g). Concentration of the appropriate fractions provided Intermediate S-1C (2.79 g, 67.6%) as a colorless viscous oil: 1H NMR (400 MHz, CDC13) δ ppm 7.34 (2 H, d, J= 7.30 Hz), 7.24-7.32 (3 H, m), 4.62-4.75 (1 H, m, J= 10.17, 6.89, 3.43, 3.43 Hz), 4.15-4.25 (3 H, m), 3.35 (1 H, dd, J= 13.60, 3.27 Hz), 2.84 (1 H, dd, J= 16.62, 9.57 Hz), 2.75 (1 H, dd, J = 13.35, 10.07 Hz), 2.47 (1 H, dd, J= 16.62, 4.78 Hz), 2.11-2.23 (2 H, m), 1.90-2.02 (1 H, m), 1.72-1.84 (1 H, m), 1.44 (9 H, s).
Intermediate S-ID: (2R)-2-( -tert-Butoxy-2-oxoethyl)-5,5,5-trifluoropentanoic acid

[00183] To a cool (0 °C), stirred solution of Intermediate S-1C (2.17 g, 5.05 mmol) in THF (50 mL) and water (15 mL) was added a solution of LiOH (0.242 g, 10.11 mmol) and H202 (2.065 mL, 20.21 mmol) in H20 (2 mL). After 10 min, the reaction mixture was removed from the ice bath, stirred for lh, and then cooled to 0 °C. Saturated aqueous NaHCC”3 (25 mL) and saturated aqueous Na2s03 (25 mL) were added to the reaction mixture, and the mixture was stirred for 10 min, and then partially concentrated. The resulting mixture was extracted with DCM (2x), cooled with ice and made acidic with cone. HC1 to pH 3. The mixture was saturated with solid NaCl, extracted with EtOAc (3x), and then dried over MgS04, filtered and concentrated to a colorless oil to afford Intermediate S-ID, 1.2514g, 92%): 1H NMR (400 MHz, CDCI3) δ ppm 2.83-2.95 (1 H, m), 2.62-2.74 (1 H, m), 2.45 (1 H, dd, J= 16.62, 5.79 Hz), 2.15-2.27 (2 H, m), 1.88-2.00 (1 H, m), 1.75-1.88 (1 H, m), 1.45 (9 H, s). Intermediate S-l : (2R,3S)-3-(fert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid, and Intermediate S-1E: (2R,3R)-3-(tert-butoxycarbonyl)- 6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid
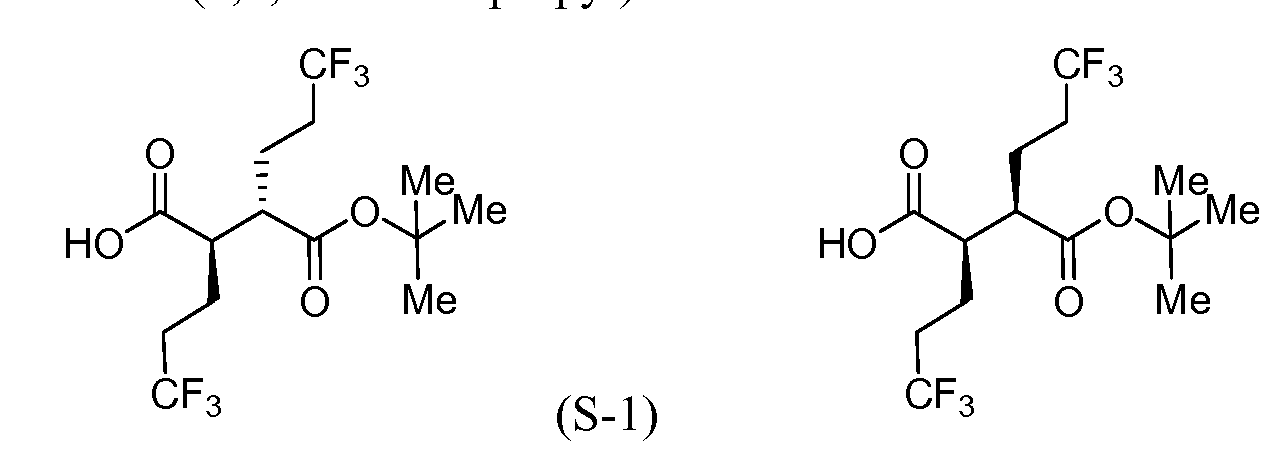
(S-1E)
[00184] To a cold (-78 °C) stirred solution of Intermediate S-1D (5 g, 18.50 mmol) in THF (60 mL) was slowly added LDA (22.2 mL, 44.4 mmol, 2.0M) over 7 min. After stirring for 2 hr, Intermediate S- 1 A (6.38 g, 25.9 mmol) was added to the reaction mixture over 3 min. After 60 min, the reaction mixture was warmed to -25 °C
(ice/MeOH/dry ice) and stirred for an additional 60 min at which time sat aq NH4C1 was added. The separated aqueous phase was acidified with IN HC1 to pH 3, and then extracted with Et20. The combined organic layers were washed with brine (2x), dried over MgS04, filtered and concentrated to provide a 1 :4 (II :I1E) mixture (as determined by 1H NMR) of Intermediate S-l and Intermediate S-1E (6.00 g, 89%) as a pale yellow solid. 1H NMR (500 MHz, CDC13) δ ppm 2.81 (1 H, ddd, J = 10.17, 6.32, 3.85 Hz), 2.63- 2.76 (1 H, m), 2.02-2.33 (4 H, m), 1.86-1.99 (2 H, m), 1.68-1.85 (2 H, m), 1.47 (9 H, s).
[00185] To a cold (-78 °C), stirred solution of a mixture of Intermediate S-l and Intermediate S-1E (5.97 g, 16.30 mmol) in THF (91 mL) was added LDA (19 mL, 38.0 mmol, 2.0M in THF/hexane/ethyl benzene) dropwise via syringe over 10 min (internal temperature never exceeded -65 °C, J-KEM® probe in reaction solution). The mixture was stirred for 15 min, and then warmed to room temperature (24 °C water bath), stirred for 15 min, and then cooled to -78 °C for 15 min. To the reaction mixture was added Et2AlCl (41 mL, 41.0 mmol, 1M in hexane) via syringe (internal temperature never exceeded -55 °C), and the mixture was stirred for 10 min, and then warmed to room temperature (24 °C bath) for 15 min and then back to -78 °C for 15 min. Meanwhile, a 1000 mL round bottom flask was charged with MeOH (145 mL) and precooled to -78 °C. With vigorous stirring the reaction mixture was transferred via cannula over 5 min to the MeOH. The flask was removed from the bath, ice was added followed by the slow addition of IN HC1 (147 mL, 147 mmol). Gas evolution was observed as the HC1 was added. The reaction mixture was allowed to warm to room temperature during which the gas evolution subsided. The reaction mixture was diluted with EtOAc (750 mL), saturated with NaCl, and the organic phase was separated, washed with a solution of potassium fluoride (8.52 g, 147 mmol) and IN HC1 (41 mL, 41.0 mmol) in water (291 mL), brine (100 mL), and then dried (Na2s04), filtered and concentrated under vacuum. 1H NMR showed the product was a 9: 1 mixture of Intermediate S-l and Intermediate S- 1E. The enriched mixture of Intermediate S-l and Intermediate S-1E (6.12 g, >99% yield) was obtained as a dark amber solid: 1H NMR (400 MHz, CDC13) δ ppm 2.64-2.76 (2 H, m), 2.04-2.35 (4 H, m), 1.88-2.00 (2 H, m), 1.71-1.83 (2 H, m), 1.48 (9 H, s).
Alternate procedure to make Intermediate S-l :
Intermediate S-IF: (2R,3 -1 -Benzyl 4-tert-butyl 2,3-bis(3,3,3-trifluoropropyl)succinate

[00186] To a stirred solution of a 9: 1 enriched mixture of Intermediate S-l and Intermediate S-1E (5.98 g, 16.33 mmol) in DMF (63 mL) were added potassium carbonate (4.06 g, 29.4 mmol) and benzyl bromide (2.9 mL, 24.38 mmol), the mixture was then stirred overnight at room temperature. The reaction mixture was diluted with EtOAc (1000 mL), washed with 10% LiCl (3×200 mL), brine (200 mL), dried (Na2S04), filtered, concentrated, and then dried under vacuum. The residue was purified by Si02 chromatography using a toluene:hexane gradient. Diastereomerically purified
Intermediate S-IF (4.81g, 65%) was obtained as a colorless solid: 1H NMR (400 MHz, chloroform-d) δ 7.32-7.43 (m, 5H), 5.19 (d, J= 12.10 Hz, 1H), 5.15 (d, J= 12.10 Hz, 1H), 2.71 (dt, J= 3.52, 9.20 Hz, 1H), 2.61 (dt, J= 3.63, 9.63 Hz, 1H), 1.96-2.21 (m, 4H), 1.69-1.96 (m, 3H), 1.56-1.67 (m, 1H), 1.45 (s, 9H).
Intermediate S-l : (2R,3S)-3-(fert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid

[00187] To a solution of Intermediate S-1F (4.81 g, 10.54 mmol) in MeOH (100 mL) was added 10% palladium on carbon (wet, Degussa type, 568.0 mg, 0.534 mmol) in a H2– pressure flask. The vessel was purged with N2 (4x), then purged with H2 (2x), and finally, pressurized to 50 psi and shaken overnight. The reaction vessel was
depressurized and purged with nitrogen. The mixture was filtered through CELITE®, washed with MeOH and then concentrated and dried under vacuum. Intermediate S-1 (3.81 g, 99% yield)) was obtained as a colorless solid: 1H NMR (400 MHz, chloroform-d) δ 2.62-2.79 (m, 2H), 2.02-2.40 (m, 4H), 1.87-2.00 (m, 2H), 1.67-1.84 (m, 2H), 1.48 (s, 9H).
Alternate procedure to make Intermediate S-1 :
Intermediate S-1 : (2R,3S)-3-(fert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid

[00188] Intermediate S-1 as a mixture with Intermediate S-IE was prepared in a similar procedure as above from Intermediate S-1D to afford a 1 :2.2 mixture of
Intermediate S-1 and Intermediate S-IE (8.60 g, 23.48 mmol), which was enriched using LDA (2.0 M solution in THF, ethyl benzene and heptane, 28.2 mL, 56.4 mmol) and diethyl aluminum chloride (1.0 M solution in hexane, 59 mL, 59.0 mmol) in THF (91 mL). After workup as described above, the resulting residue was found to be a 13.2: 1 (by 1H NMR) mixture of Intermediate S-1 and Intermediate S-IE, which was treated as follows: The crude material was dissolved in MTBE (43 mL). Hexanes (26 mL) were slowly charged to the reaction mixture while maintaining a temperature below 30 °C. The reaction mixture was stirred for 10 min. Next, tert-butylamine (2.7 mL, 1.1 eq) was charged slowly over a period of 20 minutes while maintaining a temperature below 30 °C. This addition was observed to be exothermic. The reaction mixture was stirred for 2 hrs below 30 °C and then filtered. The solid material was washed with 5:3 MTBE: hexane (80 mL), and the filtrate was concentrated and set aside. The filtered solid was dissolved in dichloromethane (300 mL), washed with IN HC1 (lOOmL), and the organic layer was washed with brine (100 mL x 2), and then concentrated under reduced pressure below 45 °C to afford Intermediate S-l (5.46 g, 64%).
A second alternate procedure for preparing Intermediate S-l :
Intermediate S-1G: tert- utyl 5,5,5-trifluoropentanoate

[00189] To a stirred solution of 5,5,5-trifluoropentanoic acid (5 g, 32.0 mmol) in THF (30 mL) and hexane (30 mL) at 0 °C, was added tert-butyl 2,2,2-trichloroacetimidate (11.46 mL, 64.1 mmol). The mixture was stirred for 15 min at 0 °C. Boron trifluoride etherate (0.406 mL, 3.20 mmol) was added and the reaction mixture was allowed to warm to room temperature overnight. To the clear reaction mixture was added solid NaHC03 (5 g) and stirred for 30 min. The mixture was filtered through MgSC^ and washed with hexanes (200 mL). The solution was allowed to rest for 45 min, and the resulting solid material was removed by filtering on the same MgSC^ filter again, washed with hexanes (100 mL) and concentrated under reduced pressure without heat. The volume was reduced to about 30 mL, filtered through a clean fritted funnel, washed with hexane (5 mL), and then concentrated under reduced pressure without heat. The resulting neat oil was filtered through a 0.45μιη nylon membrane filter disk to provide Intermediate S-1G (6.6 g, 31.4 mmol 98% yield) as a colorless oil: 1H NMR (400 MHz, CDC13) δ ppm 1.38 (s, 9 H) 1.74-1.83 (m, 2 H) 2.00-2.13 (m, 2 H) 2.24 (t, J= 7.28 Hz, 2 H). Intermediate S-1H: (4S)-4-(Propan-2-yl)-3-(5,5,5-trifluoropentanoyl)-l,3-oxazolidin-2- one

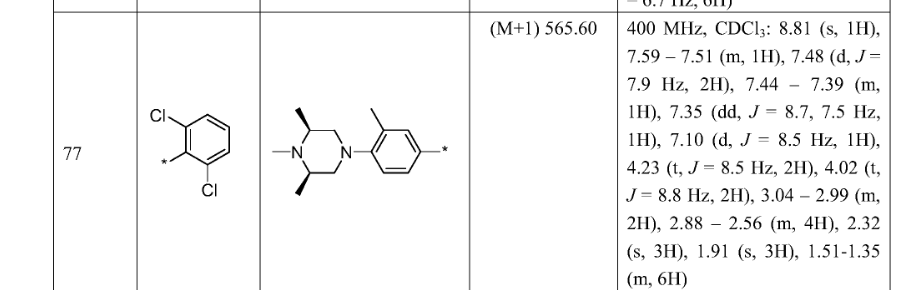
[00190] To a stirred solution of 5,5,5-trifluoropentanoic acid (5.04 g, 32.3 mmol) in DCM (50 mL) and DMF (3 drops) was added oxalyl chloride (3.4 mL, 38.8 mmol) dropwise over 5 min. The solution was stirred until all bubbling subsided. The reaction mixture was concentrated under reduced pressure to give pale yellow oil. To a separate flask charged with a solution of (4S)-4-(propan-2-yl)-l,3-oxazolidin-2-one (4.18 g, 32.4 mmol) in THF (100 mL) at -78 °C was added n-BuLi (2.5M in hexane) (13.0 mL, 32.5 mmol) dropwise via syringe over 5 min. After stirring for 10 min, the above acid chloride, dissolved in THF (20 mL), was added via cannula over 15 min. The reaction mixture was warmed to 0 °C, and was allowed to warm to room temperature as the bath warmed and stirred overnight. To the reaction mixture was added saturated NH4C1, and the mixture was extracted with EtOAc (2x). The combined organics were washed with brine, dried (Na2s04), filtered and concentrated under reduced pressure. The crude material was purified by flash chromatography (Teledyne ISCO CombiFlash Rf, 5% to 60% solvent A/B = hexanes/EtOAc, REDISEP® Si02 120g). Concentration of the appropriate fractions provided Intermediate S-1H (7.39 g, 86%) as a colorless oil: 1H NMR (400 MHz, CDC13) δ ppm 4.44 (1 H, dt, J= 8.31, 3.53 Hz), 4.30 (1 H, t, J= 8.69 Hz), 4.23 (1 H, dd, J= 9.06, 3.02 Hz), 2.98-3.08 (2 H, m), 2.32-2.44 (1 H, m, J= 13.91, 7.02, 7.02, 4.03 Hz), 2.13-2.25 (2 H, m), 1.88-2.00 (2 H, m), 0.93 (3 H, d, J= 7.05 Hz), 0.88 (3 H, d, J= 6.80 Hz).
Intermediate S-1I: (2S,3R)-tert-Butyl 6,6,6-trifluoro-3-((S)-4-isopropyl-2- oxooxazolidine-3-carbonyl)-2-(3,3,3-trifluoropropyl)hexanoate, and Intermediate S-U: (2R,3R)-tert-Butyl 6,6,6-trifluoro-3-((S)-4-isopropyl-2-oxooxazolidine-3-carbonyl)-2- (3 ,3 ,3 -trifluoropropyl)hexanoate

[00191] To a cold (-78 °C), stirred solution of diisopropylamine (5.3 mL, 37.2 mmol) in THF (59 mL) under a nitrogen atmosphere was added n-BuLi (2.5M in hexane) (14.7 mL, 36.8 mmol). The mixture was then warmed to 0 °C to give a 0.5M solution of LDA. A separate vessel was charged with Intermediate S-1H (2.45 g, 9.17 mmol). The material was azeotroped twice with benzene (the RotoVap air inlet was fitted with a nitrogen inlet to completely exclude humidity), and then toluene (15.3 mL) was added. This solution was added to a flask containing dry lithium chloride (1.96 g, 46.2 mmol). To the resultant mixture, cooled to -78 °C, was added the LDA solution (21.0 mL, 10.5 mmol) and the mixture was stirred at -78 °C for 10 min, then warmed to 0 °C for 10 min., and then cooled to -78 °C. To a separate reaction vessel containing Intermediate S-1G (3.41 g, 16.07 mmol), also azeotroped twice with benzene, was added toluene (15.3 mL), cooled to -78 °C and LDA (37.0 mL, 18.5 mmol) was added. The resulting solution was stirred at -78 °C for 25 min. At this time the enolate derived from the ester was transferred via cannula into the solution of the oxazolidinone enolate and stirred at -78 °C for an additional 5 min, at which time the septum was removed and solid powdered bis(2- ethylhexanoyloxy)copper (9.02 g, 25.8 mmol) was rapidly added to the reaction vessel and the septum was replaced. The vessel was immediately removed from the cold bath and immersed into a warm water bath (40 °C) with rapid swirling and with a concomitant color change from the initial turquoise to brown. The reaction mixture was stirred for 20 min, was then poured into 5% aqueous NH4OH (360 mL) and extracted with EtOAc (2x). The combined organics were washed with brine, dried (Na2s04), filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (Teledyne ISCO CombiFlash Rf, 0% to 60% solvent A/B = hexanes/EtOAc, REDISEP® Si02 120g). Concentration of the appropriate fractions provided a mixture of Intermediate S- II and Intermediate S-1J (2.87 g, 66%) as a pale yellow viscous oil. 1H NMR showed the product was a 1.6: 1 mixture of diastereomers S-1LS-1J as determined by the integration of the multiplets at 2.74 and 2.84 ppm: 1H NMR (400 MHz, CDC13) δ ppm 4.43-4.54 (2 H, m), 4.23-4.35 (5 H, m), 4.01 (1 H, ddd, J= 9.54, 6.27, 3.51 Hz), 2.84 (1 H, ddd, J = 9.41, 7.28, 3.64 Hz), 2.74 (1 H, ddd, J= 10.29, 6.27, 4.02 Hz), 2.37-2.48 (2 H, m, J = 10.38, 6.98, 6.98, 3.51, 3.51 Hz), 2.20-2.37 (3 H, m), 1.92-2.20 (8 H, m), 1.64-1.91 (5 H, m), 1.47 (18 H, s), 0.88-0.98 (12 H, m). Intermediate S-1 : (2R,3S)-3-(fert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid, and Intermediate S-IE: (2R,3R)-3-(tert-Butoxycarbonyl)- 6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid

(S-IE)
[00192] To a cool (0 °C), stirred solution of Intermediate S-1I and Intermediate S-1 J (4.54 g, 9.51 mmol) in THF (140 mL) and water (42 mL) were sequentially added hydrogen peroxide (30% in water) (10.3 g, 91 mmol) and LiOH (685.3 mg, 28.6 mmol). The mixture was stirred for 1 hr. At this time the reaction vessel was removed from the cold bath and then stirred for 1.5 hr. To the reaction mixture were added saturated NaHC03 (45 mL) and saturated Na2s03 (15 mL), and then the mixture was partially concentrated under reduced pressure. The resulting crude solution was extracted with DCM (3x). The aqueous phase was acidified to pH~l-2 with IN HC1, extracted with DCM (3x) and then EtOAc (lx). The combined organics were washed with brine, dried (Na2s04), filtered and concentrated under reduced pressure to provide a mixture of Intermediates S-1 and S-IE (3.00 g, 86%) as a colorless oil: 1H NMR (400 MHz, CDC13) δ ppm 2.76-2.84 (1 H, m, diastereomer 2), 2.64-2.76 (3 H, m), 2.04-2.35 (8 H, m), 1.88- 2.00 (4 H, m), 1.71-1.83 (4 H, m), 1.48 (9 H, s, diastereomer 1), 1.46 (9 H, s,
diastereomer 2); 1H NMR showed a 1.7: 1 mixture of S-1E:S-1F by integration of the peaks for the t-butyl groups. Intermediate S-1 : (2R,3S)-3-(fert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid, and Intermediate S-IF: (2R,3R)-3-(fert-Butoxycarbonyl)- 6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid

[00193] To a cold (-78 °C) stirred solution of diisopropylamine (1.7 mL, 11.93 mmol) in THF (19 mL) under a nitrogen atmosphere was added n-BuLi (2.5M in hexanes) (4.8 mL, 12.00 mmol). The mixture was stirred for 5 min and then warmed to 0 °C. In a separate vessel, to a cold (-78 °C) stirred solution of the mixture of Intermediates S-1 and S-1E (1.99 g, 5.43 mmol) in THF (18 mL) was added the LDA solution prepared above via cannula slowly over 25 min. The mixture was stirred for 15 min, then warmed to room temperature (placed in a 24 °C water bath) for 15 min, and then again cooled to -78 °C for 15 min. To the reaction mixture was added Et2AlCl (1M in hexane) (11.4 mL, 11.40 mmol) via syringe. The mixture was stirred for 10 min, warmed to room
temperature for 15 min and then cooled back to -78 °C for 15 min. Methanol (25 mL) was rapidly added, swirled vigorously while warming to room temperature, and then concentrated to ~l/4 the original volume. The mixture was dissolved in EtOAc and washed with IN HC1 (50 mL) and ice (75 g). The aqueous phase was separated and extracted with EtOAc (2x). The combined organics were washed with a mixture of KF (2.85g in 75 mL water) and IN HC1 (13 mL) [resulting solution pH 3-4], then with brine, dried (Na2s04), filtered and concentrated under reduced pressure to give a 9: 1 (S-LS-1E) enriched diastereomeric mixture (as determined by 1H NMR) of Intermediate S-1 and Intermediate S-1E (2.13 g, >99%) as a pale yellow viscous oil: 1H NMR (400 MHz, CDC13) δ ppm 2.64-2.76 (2 H, m), 2.04-2.35 (4 H, m), 1.88-2.00 (2 H, m), 1.71-1.83 (2 H, m), 1.48 (9 H, s).
Intermediate S-2: (2R,3S)-3-(fert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3- fluoropropyl)hexanoic acid

Intermediate S-2: (2R,3S)-3-(tert-Butoxycarbonyl)-7,7,7-trifluoro-2-(3,3,3- trifluoropropyl)heptanoic acid, and Intermediate S-2A: (2R,3R)-3-(tert-Butoxycarbonyl)- 7,7,7-trifluoro-2-(3,3,3-trifluoropropyl)heptanoic acid

(S-2A)
[00194] To a cold (-78 °C), stirred solution of Intermediate S-1D (1.72 g, 6.36 mmol) in THF (30 mL) was slowly added LDA (7.32 mL, 14.6 mmol) over 7 min. After stirring for 1 h, 4,4,4-trifluorobutyltrifluoromethanesulfonate (2.11 g, 8.11 mmol) was added to the reaction mixture over 2 min. After 15 min, the reaction mixture was warmed to -25 °C (ice/MeOH/dry ice) for lh, and then cooled to -78 °C. After 80 min, the reaction was quenched with a saturated aqueous NH4C1 solution (10 mL). The reaction mixture was further diluted with brine and the solution was adjusted to pH 3 with IN HC1. The aqueous layer was extracted with ether. The combined organics were washed with brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure to provide a mixture of Intermediates S-2 and S-2A (2.29 g, 95%) as a colorless oil. 1H NMR (400MHz, chloroform-d) δ 2.83-2.75 (m, 1H), 2.64 (ddd, J = 9.9, 6.7, 3.6 Hz, 1H), 2.32-2.03 (m, 5H), 1.98-1.70 (m, 3H), 1.69-1.52 (m, 3H), 1.50-1.42 (m, 9H). 1H NMR showed a 1 :4.5 mixture (S-2:S-2A) of diastereomers by integration of the peaks for the t- Bu groups.
Intermediate S-2: (2R,3S)-3-(fert-Butoxycarbonyl)-7,7,7-trifluoro-2-(3,3,3- trifluoropropyl)heptanoic acid, and Intermediate S-2A: (2R,3R)-3-(tert-Butoxycarbonyl)- 7,7,7-trifluoro-2-(3,3,3-trifluoropropyl)heptanoic acid

[00195] A mixture of Intermediate S-2 and Intermediate S-2A (2.29 g, 6.02 mmol) was dissolved in THF (38 mL) to give a colorless solution which was cooled to -78 °C. Then, LDA (7.23 mL, 14.5 mmol) (2.0M in heptane/THF/ethylbenzene) was slowly added to the reaction mixture over 3 min. After stirring for 15 min, the reaction mixture was placed in a room temperature water bath. After 15 min the reaction mixture was placed back in a -78 °C bath and then diethylaluminum chloride (14.5 mL, 14.5 mmol) (1M in hexane) was added slowly over 5 min. The reaction mixture was stirred at -78 °C. After 15 min, the reaction mixture was placed in a room temperature water bath for 10 min, and then cooled back to -78 °C. After 15 min, the reaction was quenched with MeOH (30.0 mL, 741 mmol), removed from the -78 °C bath and concentrated. To the reaction mixture was added ice and HC1 (60.8 mL, 60.8 mmol) and the resulting mixture was extracted with EtOAc (2x 200 mL). The organic layer was washed with potassium fluoride (3.50g, 60.3 mmol) in 55 mL H20 and 17.0 mL of IN HC1. The organics were dried over anhydrous magnesium sulfate and concentrated under reduced pressure to provide an enriched mixture of Intermediate S-2 and Intermediate S-2A (2.25g, 98% yield) as a light yellow oil. 1H NMR (400MHz, chloroform-d) δ 2.83-2.75 (m, 1H), 2.64 (ddd, J= 9.9, 6.7, 3.6 Hz, 1H), 2.32-2.03 (m, 5H), 1.98-1.70 (m, 3H), 1.69-1.52 (m, 3H), 1.50-1.42 (m, 9H). 1H NMR showed a 9: 1 ratio in favor of the desired diastereomer Intermediate S-2.
Intermediate S-2B: (2R,3S)-1 -Benzyl 4-tert-butyl 2,3-bis(4,4,4-trifluorobutyl)succinate

[00196] To a stirred 9: 1 mixture of Intermediate S-2 and Intermediate S-2A (2.24 g, 5.89 mmoL) and potassium carbonate (1.60 g, 11.58 mmoL) in DMF (30 mL) was added benzyl bromide (1.20 mL, 10.1 mmoL)). The reaction mixture was stirred at room temperature for 19 h. The reaction mixture was diluted with ethyl acetate (400 mL) and washed with 10% LiCl solution (3 x 100 mL), brine (50 mL), and then dried over anhydrous magnesium sulfate, filtered and concentrated to dryness under vacuum. The residue was purified by flash chromatography (Teledyne ISCO CombiFlash 0%> to 100% solvent A/B = hexane/EtOAc, REDISEP® Si02 220 g, detecting at 254 nm, and monitoring at 220 nm). Concentration of the appropriate fractions provided Intermediate S-2B (1.59 g, 57.5%). HPLC: RT = 3.863 min (CHROMOLITH® SpeedROD column 4.6 x 50 mm, 10-90% aqueous methanol over 4 minutes containing 0.1% TFA, 4 mL/min, monitoring at 220 nm), 1H NMR (400MHz, chloroform-d) δ 7.40-7.34 (m, 5H), 5.17 (d, J= 1.8 Hz, 2H), 2.73-2.64 (m, 1H), 2.55 (td, J= 10.0, 3.9 Hz, 1H), 2.16-1.82 (m, 5H), 1.79-1.57 (m, 3H), 1.53-1.49 (m, 1H), 1.45 (s, 9H), 1.37-1.24 (m, 1H).
Intermediate S-2: (2R,3S)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(4,4,4- trifluorobutyl)hexanoic acid

[00197] To a stirred solution of Intermediate S-2B (1.59 g, 3.37 mmoL) in MeOH (10 mL) and EtOAc (10 mL) under nitrogen was added 10%> Pd/C (510 mg). The atmosphere was replaced with hydrogen and the reaction mixture was stirred at room temperature for 2.5 h. The palladium catalyst was filtered off through a 4 μΜ polycarbonate film and rinsed with MeOH. The filtrate was concentrated under reduced pressure to give intermediate S-2 (1.28 g, 99%). 1H NMR (400MHz, chloroform-d) δ 2.76-2.67 (m, 1H), 2.65-2.56 (m, 1H), 2.33-2.21 (m, 1H), 2.17-2.08 (m, 3H), 1.93 (dtd, J= 14.5, 9.9, 5.2 Hz, 1H), 1.84-1.74 (m, 2H), 1.70-1.52 (m, 3H), 1.48 (s, 9H).
Intermediate A- 1 : (2-Amino-3 -methylphenyl)(3 -fluorophenyl)methanone

Intermediate A-1 A: 2-Amino- -methoxy-N,3-dimethylbenzamide

[00198] In a 1 L round-bottomed flask was added 2-amino-3-methylbenzoic acid (11.2 g, 74.1 mmol) and Ν,Ο-dimethylhydroxylamine hydrochloride (14.45 g, 148 mmol) in DCM (500 mL) to give a pale brown suspension. The reaction mixture was treated with Et3N (35 mL), HOBT (11.35 g, 74.1 mmol) and EDC (14.20 g, 74.1 mmol) and then stirred at room temperature for 24 hours. The mixture was then washed with 10% LiCl, and then acidified with IN HCl. The organic layer was washed successively with 10%> LiCl and aq NaHC03. The organic layer was decolorized with charcoal, filtered, and the filtrate was dried over MgSC^. The mixture was filtered and concentrated to give 13.22 g (92% yield) of Intermediate A-1A. MS(ES): m/z = 195.1 [M+H+]; HPLC: RT = 1.118 min. (H20/MeOH with TFA, CHROMOLITH® ODS S5 4.6 x 50 mm, gradient = 4 min, wavelength = 220 nm); 1H NMR (500MHz, chloroform-d) δ 7.22 (dd, J= 7.8, 0.8 Hz, 1H), 7.12-7.06 (m, 1H), 6.63 (t, J= 7.5 Hz, 1H), 4.63 (br. s., 2H), 3.61 (s, 3H), 3.34 (s, 3H), 2.17 (s, 3H).
Intermediate A- 1 : (2-Amino-3 -methylphenyl)(3 -fluorophenyl)methanone

[00199] In a 500 mL round-bottomed flask, a solution of l-fluoro-3-iodobenzene (13.61 mL, 116 mmol) in THF (120 mL) was cooled in a -78 °C bath. A solution of n- BuLi, (2.5M in hexane, 46.3 mL, 116 mmol) was added dropwise over 10 minutes. The solution was stirred at -78 °C for 30 minutes and then treated with a solution of
Intermediate A-1 A (6.43 g, 33.1 mmol) in THF (30 mL). After 1.5 hours, the reaction mixture was added to a mixture of ice and IN HCl (149 mL, 149 mmol) and the reaction flask was rinsed with THF (5 ml) and combined with the aqueous mixture. The resulting mixture was diluted with 10% aq LiCl and the pH was adjusted to 4 with IN NaOH. The mixture was then extracted with Et20, washed with brine, dried over MgS04, filtered and concentrated. The resulting residue was purified by silica gel chromatography (220g ISCO) eluting with a gradient from 10% EtOAc/hexane to 30% EtOAc/hexane to afford Intermediate A-l (7.11 g, 94% yield) as an oil. MS(ES): m/z = 230.1 [M+H+]; HPLC: RT = 2.820 min Purity = 99%. (H20/MeOH with TFA, CHROMOLITH® ODS S5 4.6 x 50 mm, gradient = 4 min, wavelength = 220 nm).
Intermediate B-1 : (S)-3-Amino-5-(3-fluorophenyl)-9-methyl-lH-benzo[e][l,4]diazepin- 2(3H)-one

Intermediate B-1 A: (S)-Benzyl (5-(3-fluorophenyl)-9-methyl-2-oxo-2,3-dihydro benzo[e] [ 1 ,4]diazepin-3-yl)carbamate

(B-1A)
[00225] In a 1 L round-bottomed flask, a solution of 2-(lH-benzo[d][l,2,3]triazol-l- yl)-2-((phenoxycarbonyl)amino)acetic acid (J. Org. Chem., 55:2206-2214 (1990)) (19.37 g, 62.0 mmol) in THF (135 mL) was cooled in an ice/water bath and treated with oxalyl chloride (5.43 mL, 62.0 mmol) and 4 drops of DMF. The reaction mixture was stirred for 4 hours. Next, a solution of Intermediate A- 1 (7.11 g, 31.0 mmol) in THF (35 mL) was added and the resulting solution was removed from the ice/water bath and stirred at room temperature for 1.5 hours. The mixture was then treated with a solution of ammonia, (7M in MeOH) (19.94 mL, 140 mmol). After 15 mins, another portion of ammonia, (7M in MeOH) (19.94 mL, 140 mmol) was added and the resulting mixture was sealed under N2 and stirred overnight at room temperature. The reaction mixture was then concentrated to ~l/2 volume and then diluted with AcOH (63 mL) and stir at room temperature for 4 hours. The reaction mixture was then concentrated, and the residue was diluted with 500 mL water to give a precipitate. Hexane and Et20 were added and the mixture was stirred at room temperature for 1 hour to form an orange solid. Et20 was removed under a stream of nitrogen and the aqueous layer was decanted. The residue was triturated with 40 mL of iPrOH and stirred at room temperature to give a white precipitate. The solid was filtered and washed with iPrOH, then dried on a filter under a stream of nitrogen to give racemic Intermediate B-1A (5.4 g, 41.7%yield).
[00226] Racemic Intermediate B-1A (5.9 g, 14.3 mmol) was resolved using the Chiral SFC conditions described below. The desired stereoisomer was collected as the second peak in the elution order: Instrument: Berger SFC MGIII, Column: CHIRALPAK® IC 25 x 3 cm, 5 cm; column temp: 45 °C; Mobile Phase: C02/MeOH (45/55); Flow rate: 160 mL/min; Detection at 220 nm.
[00227] After evaporation of the solvent, Intermediate B-1A (2.73 g, 46% yield) was obtained as a white solid. HPLC: RT = 3.075 min. (H20/MeOH with TFA,
CHROMOLITH® ODS S5 4.6 x 50 mm, gradient = 4 min, wavelength = 220 nm).
Chiral HPLC RT: 8.661 min (AD, 60% (EtOH/MeOH)/heptane) > 99%ee. MS(ES): m/z = 418.3 [M+H+];1H NMR (500MHz, DMSO-d6) δ 10.21 (s, 1H), 8.38 (d, J= 8.3 Hz, 1H), 7.57-7.47 (m, 2H), 7.41-7.29 (m, 8H), 7.25-7.17 (m, 2H), 5.10-5.04 (m, 3H), 2.42 (s, 3H).
Intermediate B-l : (S)-3-Amino-5-(3-fluorophenyl)-9-methyl-lH-benzo[e][l,4]diazepin- 2(3H)-one.
[00228] In a 100 mL round-bottomed flask, a solution of Intermediate B-1A (2.73 g, 6.54 mmol) in acetic acid (12 mL) was treated with HBr, 33% in HOAc (10.76 mL, 65.4 mmol) and the mixture was stirred at room temperature for 1 hour. The solution was diluted with Et20 to give a yellow precipitate. The yellow solid was filtered and rinsed with Et20 under nitrogen. The solid was transferred to 100 mL round bottom flask and water was added (white precipitate formed). The slurry was slowly made basic with saturated NaHC03. The resulting tacky precipitate was extracted with EtOAc. The organic layer was washed with water, dried over MgS04, and then filtered and
concentrated to dryness to give Intermediate B-l (1.68 g, 91% yield) as a white foam solid. MS(ES): m/z = 284.2 [M+H+]; HPLC: RT = 1.72 min (H20/MeOH with TFA, CHROMOLITH® ODS S5 4.6 x 50 mm, gradient = 4 min, wavelength = 220 nm). 1H NMR (400MHz, DMSO-d6) δ 10.01 (br. s., 1H), 7.56-7.44 (m, 2H), 7.41-7.26 (m, 3H), 7.22-7.11 (m, 2H), 4.24 (s, 1H), 2.55 (br. s., 2H), 2.41 (s, 3H). [00229] The compounds listed below in Table 6 (Intermediates B-2 to B-3) were prepared according to the general synthetic procedure described for Intermediate B-l , using the starting materials Intermediate A- 10 and Intermediate A-4, respectively.
Example 1
(2R,3S)-N-((3S)-5-(3-Fluorophenyl)-9-methyl-2-oxo-2,3-dihydro-lH-l,4-benzodiazepin- 3-yl)-2, -bis(3,3,3-trifluoropropyl)succinamide

Intermediate 1A: (2S,3R)-tert-Butyl 6,6,6-trifluoro-3-(((S)-5-(3-fluorophenyl)-9-methyl- 2-0X0-2, 3-dihydro-lH-benzo[e][l,4]diazepin-3-yl)carbamoyl)-2-(3,3 ,3- trifluoropropyl)hexanoat

[00240] In a 100 mL round-bottomed flask, a solution of Intermediate B-l (1683 mg, 5.94 mmol), Et3N (1.656 mL, 11.88 mmol), and Intermediate S-l in DMF (20 mL) was treated with o-benzotriazol-l-yl-A .A .N’.N’-tetramethyluronium tetrafluoroborate (3815 mg, 11.88 mmol) and stirred at room temperature for 1 hour. The reaction mixture was diluted with water and saturated aqueous NaHC03. An off white precipitate formed and was filtered and washed with water. The resulting solid was dried on the filter under a stream of nitrogen to give Intermediate 1A (3.7 g, 99% yield). MS(ES): m/z =
632.4[M+H+]; HPLC: RT = 3.635 min Purity = 98%. (H20/MeOH with TFA,
CHROMOLITH® ODS S5 4.6 x 50 mm, gradient = 4 min, wavelength = 220 nm). 1H NMR (400MHz, methanol-d4) δ 7.53 (t, J = 4.5 Hz, 1H), 7.46-7.30 (m, 3H), 7.28-7.23 (m, 1H), 7.23-7.18 (m, 2H), 5.37 (s, 1H), 2.88 (td, J = 10.4, 3.4 Hz, 1H), 2.60 (td, J =
10.2, 4.1 Hz, 1H), 2.54-2.40 (m, 1H), 2.47 (s, 3 H), 2.33-2.12 (m, 3H), 1.98-1.69 (m, 4H), 1.51 (s, 9H). Intermediate IB: (2S,3R)-6,6,6-Trifluoro-3-(((S)-5-(3-fluorophenyl)-9-methyl-2-oxo-
2,3-dihydro-lH-benzo[e][l,4]diazepin-3-yl)carbamoyl)-2-(3,3,3-trifluoropropyl)hexanoic acid

[00241] In a 250 mL round-bottomed flask, a solution of Intermediate 1A (3.7 g, 5.86 mmol) in DCM (25 mL) was treated with TFA (25 mL) and the resulting pale orange solution was stirred at room temperature for 1.5 hours. The reaction mixture was then concentrated to give Intermediate IB. HPLC: RT = 3.12 min (H20/MeOH with TFA, CHROMOLITH® ODS S5 4.6 x 50 mm, gradient = 4 min, wavelength = 220 nm).
MS(ES): m/z = 576.3 (M+H)+. 1H NMR (400MHz, methanol-d4) δ 7.54 (t, J= 4.5 Hz, 1H), 7.49-7.29 (m, 3H), 7.28-7.15 (m, 3H), 5.38 (br. s., 1H), 2.89 (td, J= 10.3, 3.7 Hz, 1H), 2.67 (td, J= 9.9, 4.2 Hz, 1H), 2.56-2.38 (m, 1H), 2.48 (s, 3 H), 2.34-2.13 (m, 3H), 2.00-1.71 (m, 4H).
Example 1 :
[00242] In a 250 mL round-bottomed flask, a solution of Intermediate IB (4.04 g, 5.86 mmol) in THF (50 mL) was treated with ammonia (2M in iPrOH) (26.4 mL, 52.7 mmol), followed by HOBT (1.795 g, 11.72 mmol) and EDC (2.246 g, 11.72 mmol). The resulting white suspension was stirred at room temperature overnight. The reaction mixture was diluted with water and saturated aqueous NaHC03. The resulting solid was filtered, rinsed with water and then dried on the filter under a stream of nitrogen. The crude product was suspended in 20 mL of iPrOH and stirred at room temperature for 20 min and then filtered and washed with iPrOH and dried under vacuum to give 2.83 g of solid. The solid was dissolved in re fluxing EtOH(100 mL) and slowly treated with 200 mg activated charcoal added in small portions. The hot mixture was filtered through CELITE® and rinsed with hot EtOH. The filtrate was reduced to half volume, allowed to cool and the white precipitate formed was filtered and rinsed with EtOH to give 2.57 g of white solid. A second recrystallization from EtOH (70 mL) afforded Example 1 (2.39 g, 70% yield) as a white solid. HPLC: RT = 10.859 min (H20/CH3CN with TFA, Sunfire C18 3.5μπι, 3.0x150mm, gradient = 15 min, wavelength = 220 and 254 nm); MS(ES): m/z = 575.3 [M+H+]; 1H NMR (400MHz, methanol-d4) δ 7.57-7.50 (m, 1H), 7.47-7.30 (m, 3H), 7.29-7.15 (m, 3H), 5.38 (s, 1H), 2.85-2.75 (m, 1H), 2.59 (td, J= 10.5, 4.0 Hz, 1H), 2.53-2.41 (m, 4H), 2.31-2.10 (m, 3H), 1.96-1.70 (m, 4H).
SEE
WO2012129353A1 *Mar 22, 2012Sep 27, 2012Bristol-Myers Squibb CompanyBis(fluoroalkyl)-1,4-benzodiazepinone compounds
PAPER RELATED
Structure–activity relationships in a series of (2-oxo-1,4-benzodiazepin-3-yl)-succinamides identified highly potent inhibitors of γ-secretase mediated signaling of Notch1/2/3/4 receptors. On the basis of its robust in vivo efficacy at tolerated doses in Notch driven leukemia and solid tumor xenograft models, 12 (BMS-906024) was selected as a candidate for clinical evaluation.
Discovery of Clinical Candidate BMS-906024: A Potent Pan-Notch Inhibitor for the Treatment of Leukemia and Solid Tumors
Ashvinikumar V. Gavai*†, Claude Quesnelle†, Derek Norris†, Wen-Ching Han†, Patrice Gill†, Weifang Shan†, Aaron Balog†, Ke Chen§, Andrew Tebben†, Richard Rampulla†, Dauh-Rurng Wu†, Yingru Zhang†, Arvind Mathur†,Ronald White†, Anne Rose†, Haiqing Wang†, Zheng Yang†, Asoka Ranasinghe†, Celia D’Arienzo†, Victor Guarino†, Lan Xiao†, Ching Su†, Gerry Everlof†, Vinod Arora‡, Ding Ren Shen†, Mary Ellen Cvijic†, Krista Menard†, Mei-Li Wen†, Jere Meredith‡, George Trainor†, Louis J. Lombardo†, Richard Olson‡, Phil S. Baran§,John T. Hunt†, Gregory D. Vite†, Bruce S. Fischer†, Richard A. Westhouse†, and Francis Y. Lee†
†Bristol-Myers Squibb Research and Development, Princeton, New Jersey 08543, United States
‡Bristol-Myers Squibb Research and Development, 5 Research Parkway, Wallingford, Connecticut 06492, United States
§ Department of Chemistry, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037,United StatesACS Med. Chem. Lett.
, 2015, 6 (5), pp 523–527
DOI: 10.1021/acsmedchemlett.5b00001, http://pubs.acs.org/doi/abs/10.1021/acsmedchemlett.5b00001
*Phone: 609-252-5091. E-mail: ashvinikumar.gavai@bms.com.



AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
PAPER RELATED

An enantioselective synthesis of (S)-7-amino-5H,7H-dibenzo[b,d]azepin-6-one (S–1) is described. The key step in the sequence involved crystallization-induced dynamic resolution (CIDR) of compound 7 using Boc-d-phenylalanine as a chiral resolving agent and 3,5-dichlorosalicylaldehyde as a racemization catalyst to afford S–1 in 81% overall yield with 98.5% enantiomeric excess.
Crystallization-Induced Dynamic Resolution toward the Synthesis of (S)-7-Amino-5H,7H-dibenzo[b,d]-azepin-6-one: An Important Scaffold for γ-Secretase Inhibitors
Sukhen Karmakar†, Vijay Byri†, Ashvinikumar V. Gavai‡, Richard Rampulla‡, Arvind Mathur‡, and Anuradha Gupta*†
† Department of Discovery Synthesis, Biocon Bristol-Myers Squibb Research Centre, Biocon Park, Bommasandra IV Phase, Jigani Link Road, Bengaluru 560099, India
‡Bristol-Myers Squibb Company, P.O Box 4000, Princeton, New Jersey 08543-4000, United StatesOrg. Process Res. Dev.
, Article ASAP
DOI: 10.1021/acs.oprd.6b00207, http://pubs.acs.org/doi/suppl/10.1021/acs.oprd.6b00207
*E-mail: anuradha.gupta@syngeneintl.com.
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2000007995A1 * | Aug 7, 1999 | Feb 17, 2000 | Du Pont Pharmaceuticals Company | SUCCINOYLAMINO LACTAMS AS INHIBITORS OF Aβ PROTEIN PRODUCTION |
| WO2000038618A2 * | Dec 23, 1999 | Jul 6, 2000 | Du Pont Pharmaceuticals Company | SUCCINOYLAMINO BENZODIAZEPINES AS INHIBITORS OF Aβ PROTEIN PRODUCTION |
| WO2001060826A2 * | Feb 16, 2001 | Aug 23, 2001 | Bristol-Myers Squibb Pharma Company | SUCCINOYLAMINO CARBOCYCLES AND HETEROCYCLES AS INHIBITORS OF Aβ PROTEIN PRODUCTION |
| US6737038 * | May 17, 2000 | May 18, 2004 | Bristol-Myers Squibb Company | Use of small molecule radioligands to discover inhibitors of amyloid-beta peptide production and for diagnostic imaging |
| US7053084 | Feb 17, 2000 | May 30, 2006 | Bristol-Myers Squibb Company | Succinoylamino benzodiazepines as inhibitors of Aβ protein production |
| US7456172 | Jan 13, 2006 | Nov 25, 2008 | Bristol-Myers Squibb Pharma Company | Succinoylamino benzodiazepines as inhibitors of Aβ protein production |
| US20030134841 * | Nov 1, 2002 | Jul 17, 2003 | Olson Richard E. | Succinoylamino lactams as inhibitors of A-beta protein production |
| US20120245151 * | Mar 22, 2012 | Sep 27, 2012 | Bristol-Myers Squibb Company | Bisfluoroalkyl-1,4-benzodiazepinone compounds |
//////////varegacestat, BMS-986115, BMS 986115, 3,5-dichlorosalicylaldehyde, Alzheimer’s disease, Boc-D-phenylalanine, CIDR;dibenzoazepenone, DKR; Notch inhibitors, Notch inhibitor, SAR, T-acute lymphoblastic leukemia, triple-negative breast cancer, γ-secretase inhibitor, PHASE 1, BMS, Bristol-Myers Squibb, Ashvinikumar Gavai, 1584647-27-7, LSK1L593UU, AL 102
Potrasertib

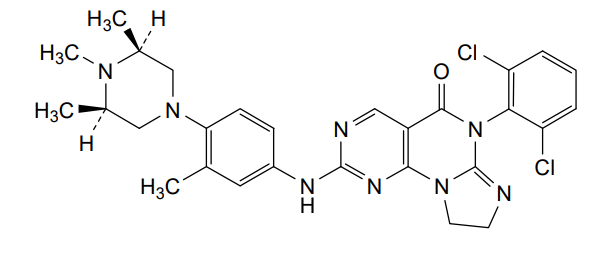
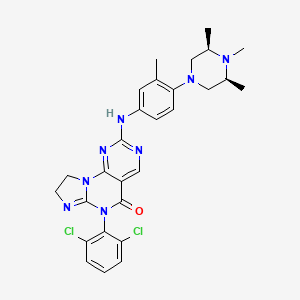
Potrasertib
CAS 2226938-19-6
MFC28H30Cl2N8O MW 565.5 g/mol
6-(2,6-dichlorophenyl)-2-{3-methyl-4-[(3R,5S)-3,4,5-trimethylpiperazin-1-yl]anilino}-8,9-dihydroimidazo[1,2-a]pyrimido[5,4-e]pyrimidin-5(6H)-one
7-(2,6-dichlorophenyl)-12-[3-methyl-4-[(3S,5R)-3,4,5-trimethylpiperazin-1-yl]anilino]-2,5,7,11,13-pentazatricyclo[7.4.0.02,6]trideca-1(13),5,9,11-tetraen-8-one
serine/ threonine kinase inhibitor, antineoplastic, IMP 7068, WEE1-IN-10, orb2664172, 621K13UG4B, Phase 1, Solid tumours
- OriginatorIMPACT Therapeutics
- ClassAntineoplastics; Small molecules
- Mechanism of ActionWEE1 protein inhibitors
- Phase ISolid tumours
- 28 Mar 2024No recent reports of development identified for phase-I development in Solid-tumours(Late-stage disease, Monotherapy) in Taiwan (PO)
- 28 Mar 2024No recent reports of development identified for phase-I development in Solid-tumours(Late-stage disease, Monotherapy) in USA (PO)
- 20 Oct 2023Efficacy, adverse events, pharmacodynamics and pharmacokinetics data from the phase I WEE1 trial in Solid tumours presented at the 48th European Society for Medical Oncology Congress (ESMO-2023)
Potrasertib is an investigational drug that is a selective inhibitor of WEE1 kinase, a protein crucial for the cell cycle. It is being studied for the treatment of various advanced solid tumors, including small cell lung cancer, ovarian, and colorectal cancers. By blocking the WEE1 kinase, potrasertib causes cancer cells with DNA damage to undergo premature, error-prone mitosis, which leads to cell death.
How it works
- Potrasertib is a serine/threonine kinase inhibitor.
- It works by targeting WEE1 kinase, which regulates the cell’s response to DNA damage.
- By inhibiting WEE1, it prevents cancer cells from repairing DNA damage before dividing, forcing them into a state that leads to cell death.
- This mechanism is particularly effective in tumors with a defective p53 gene, as these tumors rely more heavily on the WEE1 checkpoint for survival.
Potential uses
- Combination therapy: It is being explored in combination with chemotherapy (like gemcitabine and cisplatin) or radiotherapy to enhance their effectiveness against cancer.
- Monotherapy: It is also being studied as a standalone treatment for certain cancers, including ovarian, colorectal, and non-small cell lung cancer, especially those with high replication stress or WEE1 dependency.
Current status
- Potrasertib is still an investigational drug and is not yet approved for widespread clinical use.
- It is undergoing clinical trials to evaluate its safety and effectiveness in treating advanced cancers.
Potrasertib is an investigational new drug that is being evaluated by IMPACT Therapeutics for the treatment of advanced solid tumors. It is oral inhibitor of WEE1 kinase, a key regulator of cell cycle checkpoints.[1][2]
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018090939&_cid=P21-MI6TEY-70275-1

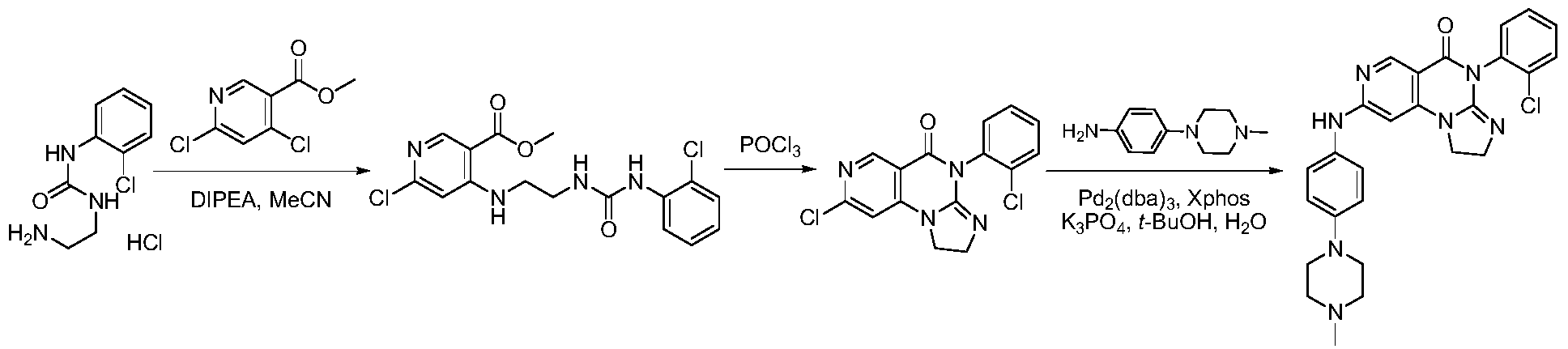
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021073491&_cid=P21-MI6TF3-70349-1
Example 1
SIMILAR NOT SAME
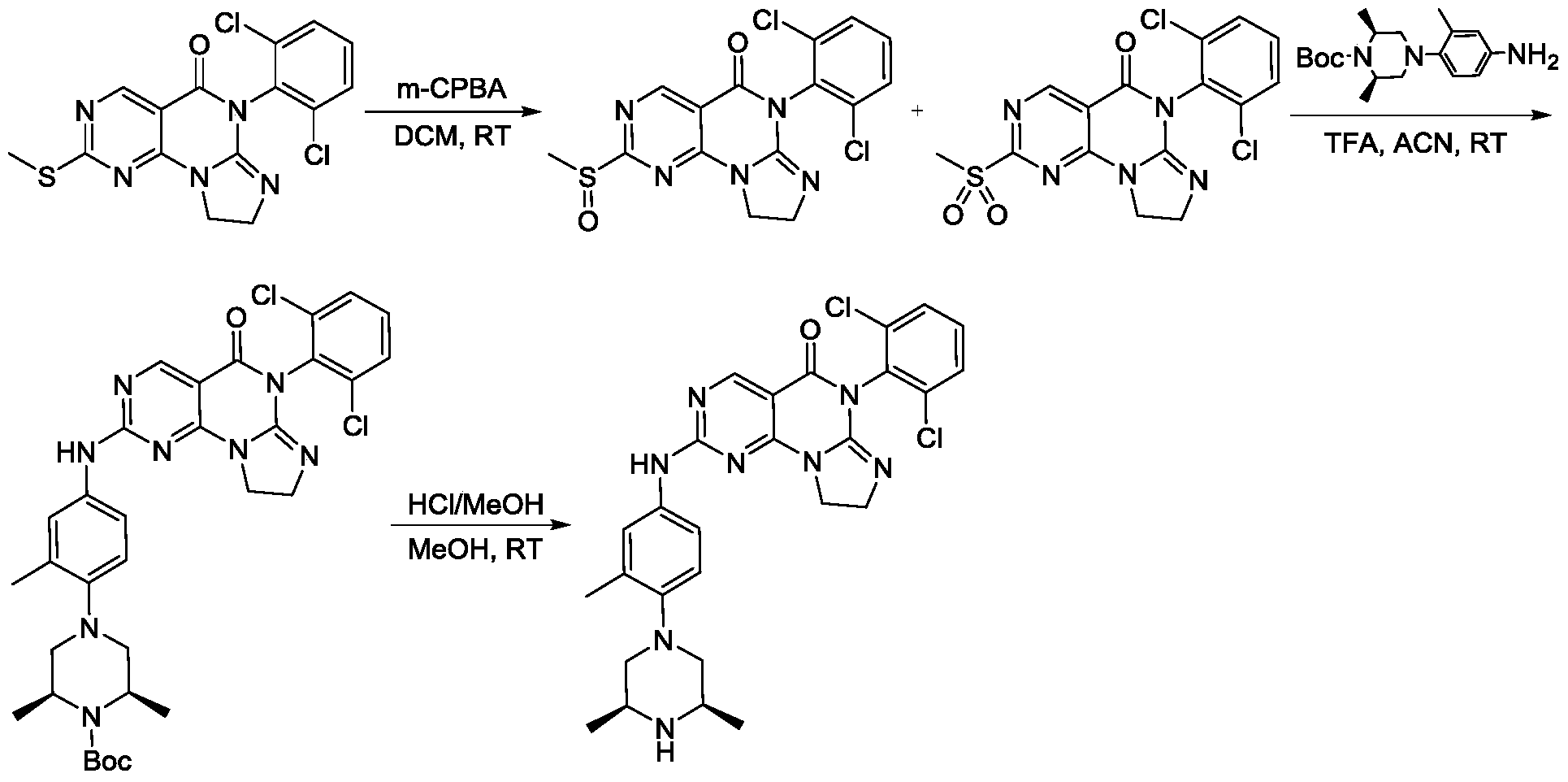
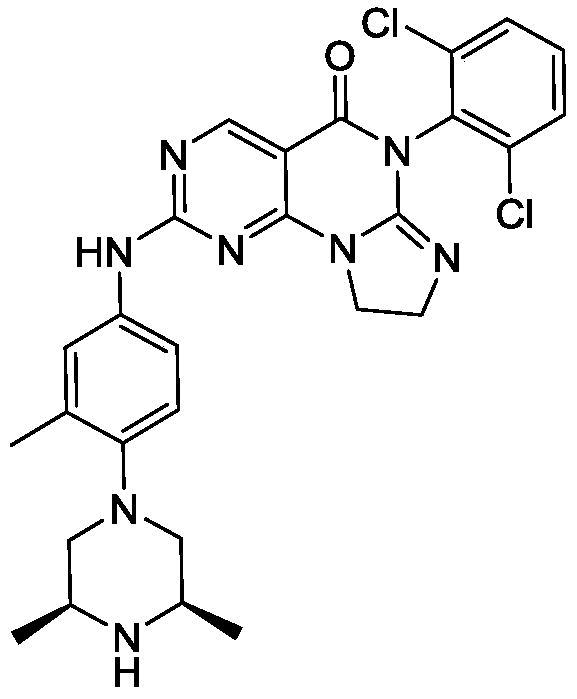
[0117]6-(2,6-dichlorophenyl)-2-((4-((3S,5R)-3,5-dimethylpiperazin-1-yl)-3-methylphenyl)amino)-8,9-dihydroimidazo[1,2-a]pyrimidino[5,4-e]pyrimidin-5(6H)-one
SIMILAR NOT SAME
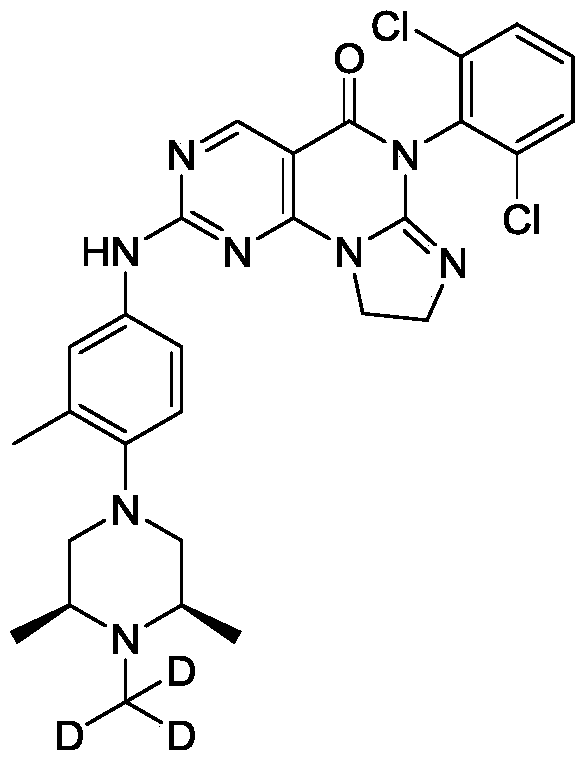
[0128]6-(2,6-dichlorophenyl)-2-((4-((3S,5R)-3,5-dimethyl-4-(methyl-d3)piperazin-1-yl)-3-methylphenyl)amino)-8,9-dihydroimidazo[1,2-a]pyrimidino[5,4-e]pyrimidin-5(6H)-one
[0130]a) Preparation of (2S,6R)-2,6-dimethyl-1-(methyl-d3)-4-(2-methyl-4-nitro)piperazine: Sodium hydride (385.03 mg, 9.63 mmol, 60% purity) was added to a solution of (3S,5R)-3,5-dimethyl-1-(2-methyl-4-nitro)piperazine (2 g, 8.02 mmol) in N,N-dimethylformamide (15 mL). The mixture was stirred at 0 °C for 25 hours, then trideuterated iodomethane (1.16 g, 8.02 mmol, 499.09 μL) was added, and the mixture was stirred at 0 °C for 2 hours. The reaction was quenched by adding an aqueous sodium bicarbonate solution (30 mL) at 0 °C, extracted with ethyl acetate (50 mL × 3), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure to obtain the target crude product (1.5 g, yellow-green solid). LC-MS(ESI): m/z(M+1) + 267.1. 1 H NMR (400MHz, CDCl
3 ): δ8.04-8.01 (m, 2H), 6.96 (d, J = 12.0Hz, 1H), 3.10 (d, J = 12Hz, 2H), 2.65 (t , J=12Hz, 2H), 2.45-2.43 (m, 2H), 2.36 (s, 3H), 1.16-1.15 (d, J=4.0Hz, 6H).
[0131]b) Preparation of 4-((3S,5R)-3,5-dimethyl-4-(methyl-d3)piperazin-1-yl)-3-methylaniline: Under nitrogen protection, palladium on carbon (281.58 μmol, 10% purity) was added to a methanol (5 mL) solution of (2S,6R)-2,6-dimethyl-1-(methyl-d3)-4-(2-methyl-4-nitro)piperazine (1.5 g, 5.63 mmol). The resulting suspension was purified multiple times under vacuum with hydrogen. The mixture was stirred at 25 °C for 12 hours under a hydrogen atmosphere (15 psi). The reaction mixture was filtered, and the filtrate was concentrated under reduced pressure to give the target crude product (1.3 g, black solid). LC-MS (ESI): m/z (M+1) + 237.1.
[0132]c) Preparation of 6-(2,6-dichlorophenyl)-2-((4-(((3S,5R)-3,5-dimethyl-4-(methyl-d3)piperazin-1-yl)-3-methylphenyl)amino)-8,9-dihydroimidazo[1,2-a]pyrimidino[5,4-e]pyrimidin-5(6H)-one: 4-((3S,5R)-3,5-dimethyl-4-(methyl-d3)piperazin-1-yl)-3-methylaniline (459.32 mg, 1.94 mmol) and the prepared 6-(2,6-dichlorophenyl)-2- A mixture (700 mg, crude) of crude (methanesulfonyl)-8,9-dihydroimidazo[1,2-a]pyrimido[5,4-e]pyrimidin-5(6H)-one and 6-(2,6-dichlorophenyl)-2-(methanesulfonyl)-8,9-dihydroimidazo[1,2-a]pyrimido[5,4-e]pyrimidin-5(6H)-one was dissolved in acetonitrile (5 mL) and trifluoroacetic acid (20.14 mg, 0.177 mmol, 13.08 μL) was added. The mixture was stirred at 20–25 °C for 2 hours, filtered, and the filtrate was concentrated under reduced pressure to give the crude product. The crude product was purified by reversed-phase HPLC to give the target compound (56.89 mg, 100.00 μmol, yellow solid, 5.66% yield). LC-MS (ESI): m/z (M+1) + 568.0.
1 H NMR (400MHz, CDCl 3 ): δ8.81 (s, 1H), 7.49 (d, J=3.8Hz, 3H), 7.41-7.34 (m, 3H), 7.02 (d, J=4.2Hz, 1H), 4.25-4.21 (m, 2H), 4.02 (t, J=8.0Hz, 2H), 2.95 (d, J=6.0Hz 2H), 2.62 (t, J=6.0Hz, 2H), 2.46-2.41 (m, 2H), 2.34 (s, 6H), 1.15 (d, J=6.4Hz, 6H).
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2022188802&_cid=P21-MI6TVM-79837-1
PAT
- 8,9-dihydroimidazo[1,2-a]pyrimido[5,4-e]pyrimidin-5(6H)-onesPublication Number: US-11345711-B2Priority Date: 2016-11-16Grant Date: 2022-05-31
- 8,9-dihydroimidazole[1,2-a]pyrimido[5,4-e]pyrimidine-5(6h)-ketone compoundPublication Number: EP-3543242-B1Priority Date: 2016-11-16Grant Date: 2024-01-03
- Compound 8,9-dihydroimidazole[1,2-a]pyrimido[5,4-e]pyrimidin-5(6H)-ketonePublication Number: ES-2968252-T3Priority Date: 2016-11-16Grant Date: 2024-05-08
- 8,9-dihydroimidazole[1,2-a]pyrimido[5,4-e]pyrimidine-5(6h)-ketone compoundPublication Number: EP-3543242-A1Priority Date: 2016-11-16
- 8,9-dihydroimidazo[1,2-a]pyrimido[5,4-e]pyrimidin-5(6H)-onesPublication Number: US-10703759-B2Priority Date: 2016-11-16Grant Date: 2020-07-07
- 8,9-DIHYDROIMIDAZO[1,2-a]PYRIMIDO[5,4-e]PYRIMIDIN-5(6H)-ONESPublication Number: US-2019308984-A1Priority Date: 2016-11-16
- 8,9-DIHYDROIMIDAZO[1,2-a]PYRIMIDO[5,4-e]PYRIMIDIN-5(6H)-ONESPublication Number: US-2020385394-A1Priority Date: 2016-11-16
- 8,9-Dihydroimidazo[1,2-a]pyrimido[5,4-e]pyrimidin-5(6H)-onesPublication Number: CN-109906227-BPriority Date: 2016-11-16Grant Date: 2022-03-11
- Dihydroimidazo pyrimido pyrimidinone compoundPublication Number: WO-2021073491-A1Priority Date: 2019-10-16
- DihydroimidazopyrimidopyrimidinonesPublication Number: CN-114502559-APriority Date: 2019-10-16
- Dihydroimidazopyrimidopyrimidinone compoundsPublication Number: CN-114502559-BPriority Date: 2019-10-16Grant Date: 2024-02-02
- Dihydroimidazo pyrimido pyrimidinone compoundPublication Number: US-2024010655-A1Priority Date: 2019-10-16
- 8,9-dihydroimidazo[1,2-a]pyrimido[5,4-e]pyrimidin-5(6h)-onesPublication Number: CA-3043945-A1Priority Date: 2016-11-16
- Use of Wee1 kinase inhibitors in the treatment of cancerPublication Number: CN-118338905-APriority Date: 2021-11-26
- Use of wee1 kinase inhibitors in the treatment of cancerPublication Number: WO-2023093840-A1Priority Date: 2021-11-26
- Use of wee1 kinase inhibitors in the treatment of cancerPublication Number: WO-2022188802-A1Priority Date: 2021-03-10
- The use of Wee1 kinase inhibitors in the treatment of cancer diseasesPublication Number: CN-117202908-APriority Date: 2021-03-10
- Use of wee1 kinase inhibitors in the treatment of cancerPublication Number: US-2024091233-A1Priority Date: 2021-03-10
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
| Clinical data | |
|---|---|
| Other names | IMP7068 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2226938-19-6 |
| PubChem CID | 139503236 |
| UNII | 621K13UG4B |
| Chemical and physical data | |
| Formula | C28H30Cl2N8O |
| Molar mass | 565.50 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- “IMP 7068”. AdisInsight. Springer Nature Switzerland AG.
- Wang Z, Li W, Li F, Xiao R (January 2024). “An update of predictive biomarkers related to WEE1 inhibition in cancer therapy”. Journal of Cancer Research and Clinical Oncology. 150 (1): 13. doi:10.1007/s00432-023-05527-y. PMC 10794259. PMID 38231277.
///////potrasertib, antineoplastic, IMP 7068, WEE1-IN-10, orb2664172, 621K13UG4B, Phase 1, Solid tumours
Edelinontrine



Edelinontrine
CRD740, PF04447943, cas 1082744-20-4
| Molecular Weight | 395.46 |
|---|---|
| Formula | C20H25N7O2 |
6-[(3S,4S)-4-methyl-1-(pyrimidin-2-ylmethyl)pyrrolidin-3-yl]-1-(oxan-4-yl)-5H-pyrazolo[3,4-d]pyrimidin-4-one
- 7N969W8Y4O
- 6-((3S,4S)-4-Methyl-1-(pyrimidin-2-ylmethyl)pyrrolidin-3-yl)-1-(tetrahydro-2H-pyran-4-yl)-1,5-dihydro-4H-pyrazolo(3,4-d)pyrimidin-4-one
- 6-[(3S,4S)-4-METHYL-1-(PYRIMIDIN-2-YLMETHYL)PYRROLIDIN-3-YL]-1-(OXAN-4-YL)-5H-PYRAZOLO[3,4-D]PYRIMIDIN-4-ONE
- 6-((3S,4S)-4-Methyl-1-(pyrimidin-2-ylmethyl)pyrrolidin-3-yl)-1-(tetrahydro-2H-pyran-4-yl)-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one
Edelinontrine (PF-04447943) is a potent inhibitor of human recombinant PDE9A (IC50=12 nM) with >78-fold selectivity, respectively, over other PDE family members (IC50>1000 nM).

PF-04447943 is a potent, selective brain penetrant PDE9 inhibitor (Ki of 2.8, 4.5 and 18 nM) for human, rhesus and rat recombinant PDE9 respectively and high selectivity for PDE9 versus PDEs1-8 and 10-11. PF-04447943 was being developed by Pfizer for the treatment of cognitive disorders. PF-04447943 attenuates a scopolamine-induced deficit in a novel rodent attention task. PF-04447943 enhances synaptic plasticity and cognitive function in rodents. PF-04447943 has completed Phase II clinical trials in subjects with mild to moderate AD in 2013 but this research was discontinued. Pfizer completes a phase I trial in Sickle cell anaemia.
SCHEME
SUDECHAIN

MAIN

Patent
WO2023114995 PFIZER
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023114995&_cid=P20-MB07JE-44537-1
PAPER
Journal of Medicinal Chemistry (2012), 55(21), 9045-9054
PATENT
WO2008139293
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2008139293&_cid=P20-MB07LY-46583-1
EXAMPLE 111
6-[(3S,4S)-4-methyl-1-(pyrimidin-2-ylmethyl)pyiτolidin-3-yl]-1-αetrahydro-2H- pyran-4-yl)-1,5-dihydro-4H-pyrazolo[3,4-d]pyrimidin-4-one

///////Edelinontrine, CRD740, PF04447943, CRD 740, PF 04447943, PHASE 1
Cinsebrutinib



Cinsebrutinib
CAS 2724962-58-5
2-fluoro-1-[(3S)-1-prop-2-enoylpiperidin-3-yl]-5,6,7,8,9,10-hexahydrocyclohepta[b]indole-4-carboxamide
- 2-fluoro-1-[(3S)-1-(prop-2-enoyl)piperidin-3-yl]-5,6,7,8,9,10-hexahydrocyclohepta[b]indole-4-carboxamide
- 2-fluoro-1-[(3S)-1-prop-2-enoylpiperidin-3-yl]-5,6,7,8,9,10-hexahydrocyclohepta[b]indole-4-carboxamide
| Molecular Weight | 383.46 |
|---|---|
| Formula | C22H26FN3O2 |
CINSEBRUTINIB is a small molecule drug with a maximum clinical trial phase of II and has 1 investigational indication.
Cinsebrutinib is a Bruton’s tyrosine kinase inhibitor, extracted from patent WO2021207549 (compound 5-6). Cinsebrutinib has the potential for cancer study.
SCHEME
INTERMEDIATE

MAIN

SYN
5-6 enantiomer A [WO2021207549A1]
GB005, Inc. WO2021207549
WO2021207549
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021207549&_cid=P22-MAAYAJ-91905-1
EXAMPLES 5-5, 5-6, 5-7
Preparation of rac-1-(1-acryloylpiperidin-3-yl)-2-fluoro-5,6,7,8,9,10-hexahydrocyclo- hepta[b]indole-4-carboxamide (Compound 5-5), (S)-1-(1-acryloylpiperidin-3-yl)-2-fluoro-5,6,7,8,9,10-hexahydrocyclohepta[b]indole-4-carboxamide (Compound 5-6) and (R)-1-(1-acryloylpiperidin-3-yl)-2-fluoro-5,6,7,8,9,10-hexahydrocyclohepta[b]indole-4- carboxamide (Compound 5-7)

STEP 1: 5-bromo-4-fluoro-2-iodoaniline
To a solution of 3-bromo-4-fluoroaniline (100.0 g, 526.3 mmol) in acetic acid (500 mL) was added N-iodosuccinimide (124.3 g, 552.5 mmol) in portions at 25 °C.
The reaction mixture was stirred for 2 hours at 25 °C. The mixture was concentrated under vacuum. The residue was diluted with saturated aqueous sodium carbonate (500 mL) and extracted with ethyl acetate (500 mL x 3). The combined organic layers were washed with water (500 mL) and brine (500 mL), dried over anhydrous sodium sulfate and concentrated under vacuum. The residue was triturated with mixed solvents of ethyl acetate and petroleum ether (300 mL, 1:4, v/v) and filtered. The solid was washed with mixed solvents of ethyl acetate and petroleum ether (50 mL x 2, 1:4, v/v) and dried under reduced pressure to give 5-bromo-4-fluoro-2-iodoaniline (88.6 g, 53%) as a light blue solid.1H NMR (300 MHz, DMSO-d6) δ 7.55 (d, J = 8.1 Hz, 1H), 6.98 (d, J = 6.3 Hz, 1H), 5.27 (brs, 2H).
STEP 2: (5-bromo-4-fluoro-2-iodophenyl)hydrazine hydrochloride
To a stirred suspension of 5-bromo-4-fluoro-2-iodoaniline (88.6 g, 280.5 mmol) in concentrated hydrochloric acid (443 mL) was added dropwise a solution of sodium nitrite (23.22 g, 337.0 mmol) in water (90 mL) at 0 °C. After stirring for 1 hour at 0 °C, the resulting mixture was added dropwise to a solution of stannous chloride dihydrate (126.61 g, 561.1 mmol) in concentrated hydrochloric acid (295 mL) at 0 °C and stirred for 1 hour at this temperature. The precipitate was collected by filtration, washed with concentrated hydrochloric acid (150 mL x 5) and ethyl acetate (300 mL), dried under reduced pressure to give (5-bromo-4-fluoro-2-iodophenyl)hydrazine hydrochloride (100.3 g, crude) as a light yellow solid.1H NMR (400 MHz, DMSO-d6) δ 10.23 (brs, 3H), 7.89 (d, J = 8.0 Hz, 1H), 7.82 (brs, 1H), 7.31-7.22 (m, 1H).
STEP 3: 1-(5-bromo-4-fluoro-2-iodophenyl)-2-cycloheptylidenehydrazine To a solution of (5-bromo-4-fluoro-2-iodophenyl)hydrazine hydrochloride (80.0 g, 217.6 mmol) in methanol (400 mL) was added cycloheptanone (24.40 g, 217.6 mmol) at 20 °C. The reaction mixture was stirred for 1 hour at 20 °C. The precipitate was collected by filtration and dried under reduced pressure to give 1-(5-bromo-4-fluoro-2-iodophenyl)-2-cycloheptylidenehydrazine (72.0 g, 78%) as an off-white solid.
1H NMR (400 MHz, DMSO-d6) δ 7.77 (d, J = 8.0 Hz, 1H), 7.44 (d, J = 6.8 Hz, 1H), 7.39 (brs, 1H), 2.50-2.44 (m, 4H), 1.80-1.67 (m, 2H), 1.64-1.48 (m, 6H).
STEP 4: 1-bromo-2-fluoro-4-iodo-5,6,7,8,9,10-hexahydrocyclohepta[b]indole A mixture of 1-(5-bromo-4-fluoro-2-iodophenyl)-2-cycloheptylidenehydrazine (72.0 g, 169.4 mmol) and concentrated sulfuric acid (18 mL) in methanol (360 mL) was stirred for 16 hours at 80 °C. The methanol was removed under reduced pressure. The residue was basified with saturated aqueous sodium carbonate until pH = 10 and extracted with ethyl acetate (600 mL x 3). The combined organic layers were washed with water (500 mL x 2) and brine (500 mL), dried over anhydrous sodium sulfate and
filtered. The filtrate was concentrated under vacuum to give 1-bromo-2-fluoro-4-iodo-5,6,7,8,9,10-hexahydrocyclohepta[b]indole (43.0 g, 80% purity, 50%) as a brown solid.
1H NMR (300 MHz, DMSO-d6) δ 10.95 (s, 1H), 7.37 (d, J = 8.7 Hz, 1H), 3.23-3.15 (m, 2H), 2.94-2.85 (m, 2H), 1.89-1.76 (m, 2H), 1.72-1.58 (m, 4H).
STEP 5: 1-bromo-2-fluoro-5,6,7,8,9,10-hexahydrocyclohepta[b]indole-4- carbonitrile
A mixture of 1-bromo-2-fluoro-4-iodo-5,6,7,8,9,10-hexahydrocyclohepta[b]indole (43.0 g, 80% purity, 84.3 mmol), zinc cyanide (4.95 g, 42.2 mmol) and tetrakis(triphenylphosphine)palladium (9.74 g, 8.4 mmol) in N,N-dimethylformamide (215 mL) was degassed and backfilled with nitrogen for three times. The reaction mixture was stirred under nitrogen at 90 °C for 2 hours. The cooled reaction mixture was diluted with water (1 L) and extracted with ethyl acetate (800 mL x 3). The combined organic layers were washed with water (500 mL x 3) and brine (800 mL), dried over anhydrous sodium sulfate, filtered and concentrated under vacuum. The residue was triturated with acetonitrile (100 mL) and filtered. The solid was washed with acetonitrile (30 mL x 2) and dried under reduced pressure to give 1-bromo-2-fluoro-5,6,7,8,9,10-hexahydrocyclohepta[b]indole-4-carbonitrile (25.5 g, 94%) as a light yellow solid. ESI-MS [M-H]- calculated for (C14H12BrFN2) 305.02, 307.02, found: 304.95, 306.95.1H NMR (300 MHz, DMSO-d6) δ 11.99 (s, 1H), 7.58 (d, J = 9.0 Hz, 1H), 3.24-3.17 (m, 2H), 2.91-2.85 (m, 2H), 1.87-1.78 (m, 2H), 1.70-1.61 (m, 4H).
STEP 6: Tert-butyl 5-(4-cyano-2-fluoro-5,6,7,8,9,10- hexahydrocyclohepta[b]indol-1-yl)-3,6-dihydropyridine-1(2H)-carboxylate A mixture of 1-bromo-2-fluoro-5,6,7,8,9,10-hexahydrocyclohepta[b]indole-4-carbonitrile (25.0 g, 81.4 mmol), tert-butyl 5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3,6-dihydropyridine-1(2H)-carboxylate (30.2 g, 97.7 mmol), [1,1′-bis(diphenylphosphino)ferrocene]dichloro-palladium(II) (5.96 g, 8.1 mmol) and potassium phosphate (51.8 g, 244.2 mmol) in tetrahydrofuran (125 mL) and water (31 mL) was degassed and backfilled with nitrogen for three times and stirred for 2 hours at 60 °C under nitrogen atmosphere. The cooled mixture was diluted with water (600 mL) and extracted with ethyl acetate (500 mL x 3). The combined organic layers was washed with water (500 mL x 2) and brine (500 mL), dried over anhydrous sodium sulfate, filtered and concentrated under vacuum to give tert-butyl 5-(4-cyano-2-fluoro-5,6,7,8,9,10-hexahydrocyclohepta[b]indol-1-yl)-3,6-dihydropyridine-1(2H)-carboxylate (45 g, crude) as a brown solid, which was used directly in next step without purification. ESI-MS [M+H-tBu]+ calculated for (C24H28FN3O2) 354.22, found: 354.05.
STEP 7: Tert-butyl 5-(4-carbamoyl-2-fluoro-5,6,7,8,9,10- hexahydrocyclohepta[b]indol-1-yl)-3,6-dihydropyridine-1(2H)-carboxylate To a mixture of 5-(4-cyano-2-fluoro-5,6,7,8,9,10-hexahydrocyclohepta[b]indol-1-yl)-3,6-dihydro-pyridine-1(2H)-carboxylate (45 g, crude) in ethanol (100 mL), tetrahydrofuran (100 mL) and water (100 mL) was added Parkin’s catalyst (2.0 g, 4.68 mmol). The reaction mixture was stirred for 16 hours at 90 °C. The cooled mixture was diluted with water (500 mL) and extracted with ethyl acetate (500 mL x 3). The combined organic layers were washed with water (500 mL x 2) and brine (500 mL), dried over anhydrous sodium sulfate, filtered and concentrated under vacuum. The residue was purified by column chromatography on silica gel eluting with ethyl acetate in petroleum ether (0 to 60%) to give tert-butyl 5-(4-carbamoyl-2-fluoro-5,6,7,8,9,10-hexahydrocyclohepta[b]indol-1-yl)-3,6-dihydropyridine-1(2H)-carboxylate (20.0 g, 57% over two steps) as a light yellow solid. ESI-MS [M+H]+ calculated for (C24H30FN3O3) 428.23, found: 428.15.1H NMR (400 MHz, DMSO-d6) δ 10.77 (s, 1H), 8.02 (s, 1H), 7.46-7.38 (m, 2H), 5.79 (s, 1H), 4.10-3.97 (m, 1H), 3.95-3.83 (m, 1H), 3.80-3.57 (m, 1H), 3.51-3.23 (m, 1H), 2.99-2.85 (m, 2H), 2.82-2.69 (m, 2H), 2.30-2.21 (m, 2H), 1.86-1.72 (m, 2H), 1.70-1.50 (m, 4H), 1.41 (s, 9H).
STEP 8: Tert-butyl 3-(4-carbamoyl-2-fluoro-5,6,7,8,9,10- hexahydrocyclohepta[b]indol-1-yl)piperidine-1-carboxylate
To a solution of tert-butyl 5-(4-carbamoyl-2-fluoro-5,6,7,8,9,10-hexahydrocyclohepta[b]indol-1-yl)-3,6-dihydropyridine-1(2H)-carboxylate (20 g, 46.8 mmol) in ethanol (300 mL) and tetrahydrofuran (300 mL) was added 10% Pd/C (15.0 g) under nitrogen atmosphere. The reaction mixture was degassed and backfilled with hydrogen for three times and stirred for 4 days at 50 °C under hydrogen (2 atm). The cooled mixture was filtered. The filtrate was concentrated under vacuum. The residue was recrystallized with tetrahydrofuran (100 mL) and petroleum ether (100 mL) to give tert-butyl 3-(4-carbamoyl-2-fluoro-5,6,7,8,9,10-hexahydrocyclohepta[b]indol-1-yl)piperidine-1-carboxylate (12.1 g, 60%) as an off-white solid. ESI-MS [M+H]+ calculated for (C24H32FN3O3) 430.24, found: 430.25.1H NMR (400 MHz, DMSO-d6) δ 10.75 (s, 1H), 8.00 (s, 1H), 7.46-7.35 (m, 2H), 4.17-3.86 (m, 2H), 3.55-3.43 (m, 1H), 3.31-3.10 (m, 1H), 3.08-2.63 (m, 5H), 2.14-1.96 (m, 1H), 1.93-1.60 (m, 9H), 1.39 (s, 9H).
STEP 9: 2-fluoro-1-(piperidin-3-yl)-5,6,7,8,9,10-hexahydrocyclohepta[b]indole- 4-carboxamide hydrochloride
Tert-butyl 3-(4-carbamoyl-2-fluoro-5,6,7,8,9,10-hexahydrocyclohepta[b]indol-1-yl)piperidine-1-carboxylate (12.1 g, 28.2 mmol) was dissolved in hydrogen chloride (150 mL, 4 M in 1,4-dioxane) and the solution was stirred for 2 hours at 25 °C. The mixture was concentrated under vacuum to give 2-fluoro-1-(piperidin-3-yl)-5,6,7,8,9,10-hexahydrocyclohepta[b]indole-4-carboxamide hydrochloride (13.4 g, crude) as a yellow solid. ESI-MS [M+H]+ calculated for (C19H24FN3O) 330.19, found: 330.10.
STEP 10: 1-(1-acryloylpiperidin-3-yl)-2-fluoro-5,6,7,8,9,10- hexahydrocyclohepta[b]indole-4-carboxamide
To a mixture of 2-fluoro-1-(piperidin-3-yl)-5,6,7,8,9,10-hexahydrocyclohepta[b]indole-4-carboxamide hydrochloride (13.4 g, crude) and sodium bicarbonate (23.7 g, 282.0 mmol) in tetrahydrofuran (300 mL) and water (150 mL) was added acryloyl chloride (2.81 g, 31.0 mmol) at 0 °C. After stirring for 1 hour at 0 °C, the mixture was diluted with water (500 mL) and extracted with ethyl acetate (400 mL x 3). The combined organic layers were washed with water (500 mL x 2) and brine (500 mL), dried over anhydrous sodium sulfate, filtered and concentrated under vacuum. The residue was recrystallized with tetrahydrofuran (290 mL), methanol (48 mL) and petroleum ether (330 mL) to give 1-(1-acryloylpiperidin-3-yl)-2-fluoro-
5,6,7,8,9,10-hexahydrocyclohepta[b]indole-4-carboxamide (6.0 g, 56% over two steps) as a white solid. ESI-MS [M+H]+ calculated for (C22H26FN3O2) 384.20, found: 384.15.
STEP 11: (S)-1-(1-acryloylpiperidin-3-yl)-2-fluoro-5,6,7,8,9,10- hexahydrocyclohepta[b]indole-4-carboxamide and (R)-1-(1-acryloylpiperidin-3- yl)-2-fluoro-5,6,7,8,9,10-hexahydrocyclohepta[b]-indole-4-carboxamide
1-(1-acryloylpiperidin-3-yl)-2-fluoro-5,6,7,8,9,10-hexahydrocyclohepta[b]indole-4-carboxamide (6.0 g) was separated by Prep-SFC with the following conditions: Column: (R,R)-Whelk-01, 2.12 x 25 cm, 5 um; Mobile Phase A: CO2, Mobile Phase B: IPA/DCM = 5:1; Flow rate: 200 mL/min; Gradient: 50% B; 220 nm; Injection Volume: 19 mL; Number Of Runs: 29; RT1: 4.97 min to afford assumed (S)-1-(1-acryloylpiperidin-3-yl)-2-fluoro-5,6,7,8,9,10-hexahydrocyclohepta[b]indole-4-carboxamide (2.55 g, 43%) as an off-white solid and RT2: 8.2 min to afford assumed (R)-1-(1-acryloylpiperidin-3-yl)-2-fluoro-5,6,7,8,9,10-hexahydrocyclohepta[b]indole-4-carboxamide (2.63 g, 44%) as an off-white solid.
Compound 5-6
ESI-MS [M+H]+ calculated for (C22H26FN3O2) 384.20, found: 384.20.1H NMR (300 MHz, DMSO-d6) δ 10.75 (s, 1H), 8.00 (s, 1H), 7.49-7.31 (m, 2H), 6.93-6.72 (m, 1H), 6.18-6.02 (m, 1H), 5.73-5.56 (m, 1H), 4.67-4.42 (m, 1H), 4.27-4.05 (m, 1H), 3.63-3.41 (m, 1.5H), 3.19-3.02 (m, 1H), 3.00-2.79 (m, 4H), 2.70-2.62 (m, 0.5H), 2.21-2.02 (m, 1H), 2.01-1.87 (m, 1H), 1.86-1.61 (m, 7H), 1.57-1.37 (m, 1H).
Protein kinases are a large group of intracellular and transmembrane signaling proteins in eukaryotic cells. These enzymes are responsible for transfer of the terminal (gamma) phosphate from ATP to specific amino acid residues of target proteins.
Phosphorylation of specific amino acid residues in target proteins can modulate their activity leading to profound changes in cellular signaling and metabolism. Protein kinases can be found in the cell membrane, cytosol and organelles such as the nucleus and are responsible for mediating multiple cellular functions including metabolism, cellular growth and differentiation, cellular signaling, modulation of immune responses, and cell death. Serine kinases specifically phosphorylate serine or threonine residues in target proteins. Similarly, tyrosine kinases, including tyrosine receptor kinases, phosphorylate tyrosine residues in target proteins. Tyrosine kinase families include: TEC, SRC, ABL, JAK, CSK, FAK, SYK, FER, ACK and the receptor tyrosine kinase subfamilies including ERBB, FGFR, VEGFR, RET and EPH. Subclass I of the receptor tyrosine kinase superfamily includes the ERBB receptors and comprises four members: ErbB1 (also called epidermal growth factor receptor (EGFR)), ErbB2, ErbB3 and ErbB4.
Kinases exert control on key biological processes related to health and disease. Furthermore, aberrant activation or excessive expression of various protein kinases are implicated in the mechanism of multiple diseases and disorders characterized by benign and malignant proliferation, as well as diseases resulting from inappropriate activation of the immune system. Thus, inhibitors of select kinases or kinase families are considered useful in the treatment of cancer, vascular disease, autoimmune diseases, and inflammatory conditions including, but not limited to: solid tumors, hematological malignancies, thrombus, arthritis, graft versus host disease, lupus erythematosus, psoriasis, colitis, illeitis, multiple sclerosis, uveitis, coronary artery vasculopathy, systemic sclerosis, atherosclerosis, asthma, transplant rejection, allergy, ischemia, dermatomyositis, pemphigus, and the like.
Tec kinases are a family of non-receptor tyrosine kinases predominantly, but not exclusively, expressed in cells of hematopoietic origin. The Tec family includes TEC, Bruton’s tyrosine kinase (BTK), inducible T-cell kinase (ITK), resting lymphocyte kinase (RLK/TXK for Tyrosine Protein Kinase), and bone marrow-expressed kinase (BMX/ETK).
BTK is important in B-cell receptor signaling and regulation of B-cell development and activation. Mutation of the gene encoding BTK in humans leads to X-linked agammaglobulinemia which is characterized by reduced immune function, including impaired maturation of B-cells, decreased levels of immunoglobulin and peripheral B cells, and diminished T-cell independent immune response. BTK is activated by Src-family kinases and phosphorylates PLC gamma leading to effects on B-cell function and survival. Additionally, BTK is important for cellular function of mast cells, macrophage and neutrophils indicating that BTK inhibition is effective in treatment of diseases mediated by these and related cells including inflammation, bone disorders, and allergic disease. BTK inhibition is also important in survival of lymphoma cells indicating that inhibition of BTK is useful in the treatment of lymphomas and other cancers. As such, inhibitors of BTK and related kinases are of great interest as anti-inflammatory, as well as anti-cancer, agents. BTK is also important for platelet function and thrombus formation indicating that BTK-selective inhibitors are also useful as antithrombotic agents. Furthermore, BTK is required for inflammasome activation, and inhibition of BTK may be used in treatment of inflammasome-related disorders, including; stroke, gout, type 2 diabetes, obesity-induced insulin resistance, atherosclerosis and Muckle-Wells syndrome. In addition, BTK is expressed in HIV infected T-cells and treatment with BTK inhibitors sensitizes infected cells to apoptotic death and results in decreased virus production. Accordingly, BTK inhibitors are considered useful in the treatment of HIV-AIDS and other viral infections.
Further, BTK is important in neurological function. Specifically targeting BTK in the brain and CNS has the potential to significantly advance the treatment of neurological diseases such as progressive and relapsing forms of MS and primary CNS lymphoma (PCNSL).
PCNSL is a rare brain tumor with an annual incidence in the United States of approximately 1900 new cases each year and constitutes approximately 3% of all newly diagnosed brain tumors.
PCNSL is highly aggressive and unlike other lymphomas outside the CNS, prognosis remains poor despite improvements in treatments in the front-line setting. High dose methotrexate remains the backbone of treatment and is used in combination with other cytotoxic agents, and more recently the addition of rituximab. From initial diagnosis, 5-year survival has improved from 19% to 30% between 1990 and 2000 but has not improved in the elderly population (>70 years), due to 20% or more of these patients being considered unfit for chemotherapy. Tumor regression is observed in ~85% of patients regardless of the treatment modality in the front-line setting, however, approximately half of these patients will experience recurrent disease within 10 -18 months after initial treatment and most relapses occur within the first 2 years of diagnosis.
Thus, the prognosis for patients with relapsed/refractory PCNSL (R/R PCNSL) remains poor with a median survival of ~ 2 months without further treatment. As there is no uniform standard of care for the treatment of R/R PCNSL, participation in clinical trials is encouraged. New safe and effective treatments are urgently needed.
BTK is involved in the signal transduction in the B cell antigen receptor (BCR) signaling pathway and integrates BCR and Toll-like receptor (TLR) signaling. Genes in these pathways frequently harbor mutations in diffuse large B-cell lymphoma (DLBCL), including CD79B and myeloid differentiation primary response 88 (MyD88). Ibrutinib, a first-generation irreversible selective inhibitor of BTK, has been approved for chronic lymphocytic leukemia/small cell lymphocytic lymphoma (CLL/SLL), previously treated Mantle Cell lymphoma (MCL) and Marginal Zone
Lymphoma (MZL), Waldenström’s macroglobulin, and previously treated chronic Graft Versus Host Disease. In clinical studies the recommended dose of Ibrutinib (480 mg/d in CLL or 560 mg/d in MCL) was escalated to 840 mg to achieve adequate brain exposure in primary CNS lymphoma.
Aberrant activation of the NF-κB pathway in PCNSL is emerging as a potential mechanism for more targeted therapy. In particular, activating mutations of CARD11 as well as of MyD88 (Toll-like receptor pathway) have been implicated. The activating exchange of leucine to proline at position 265 of MyD88, noted to occur in between 38% (11/29) and50% (7/14) of patients, is the most frequent mutation identified thus far in PCNSL. In addition, the coding region of CD79B, a component of the B-cell receptor signaling pathway, appears to contain mutations in 20% of cases, suggesting that dysregulation of the B-cell receptor and NF-κB pathways contribute to the pathogenesis of PCNSL. These data suggest that BCR pathway mutations and BTK dependence are of particular relevance to PCNSL.
Recently, several clinical studies have reported substantial single-agent clinical activity in the treatment of PCNSL with response rates of 70-77%. The majority of patients, however, discontinued therapy by 9 months. Although Ibrutinib therapy has been reported to be generally well tolerated with manageable adverse events, there are reports of sometimes fatal fungal infections. Of note, escalating doses beyond 560 mg to 840mg/day have been used to achieve higher brain exposure and these higher doses may be associated with off-target effects mediated by Ibrutinib’s kinase selectivity profile. Finally, the combination of high dose Ibrutinib in conjunction with high-dose steroids may contribute to exacerbate the increased fungal infections. Therefore, there remains a need for BTK inhibitors with an improved efficacy and safety profile due to greater brain penetration and BTK inactivation rate with greater kinase selectivity.
There remains a need for compounds that modulate protein kinases generally, as well as compounds that modulate specific protein kinases, such as BTK, as well as compounds that modulate specific protein kinases and selectively cross the blood/brain barrier for related compositions and methods for treating diseases, disorders and conditions that would benefit from such modulation and selectivity.
/////////////Cinsebrutinib, 7BS8743F3E, PHASE 1
Afimetoran
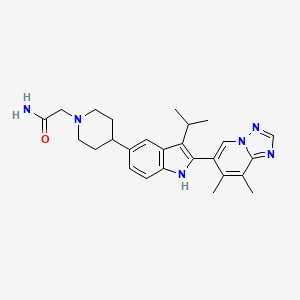

Afimetoran BMS-986256, WHO 11516
cas 2171019-55-7
2-[4-[2-(7,8-dimethyl-[1,2,4]triazolo[1,5-a]pyridin-6-yl)-3-propan-2-yl-1H-indol-5-yl]piperidin-1-yl]acetamide
C26H32N6O,444.583, phase 1
Afimetoran is an immunomodulator and an antagonist of toll-like receptors 7 and 8.1,2 It is also is under investigation in clinical trial NCT04269356 (Study to Assess the Way the Body Absorbs, Distributes, Breaks Down and Eliminates Radioactive BMS-986256 in Healthy Male Participants).

Ref
WO2018005586
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018005586&_cid=P20-M0RQ0D-09010-1
The invention further pertains to pharmaceutical compositions containing at least one compound according to the invention that are useful for the treatment of conditions related to TLR modulation, such as inflammatory and autoimmune diseases, and methods of inhibiting the activity of TLRs in a mammal.
Toll/IL-1 receptor family members are important regulators of inflammation and host resistance. The Toll-like receptor family recognizes molecular patterns derived from infectious organisms including bacteria, fungi, parasites, and viruses (reviewed in Kawai, T. et al., Nature Immunol., 11:373-384 (2010)). Ligand binding to the receptor induces dimerization and recruitment of adaptor molecules to a conserved cytoplasmic motif in the receptor termed the Toll/IL-1 receptor (TIR) domain. With the exception of TLR3, all TLRs recruit the adaptor molecule MyD88. The IL-1 receptor family also contains a cytoplasmic TIR motif and recruits MyD88 upon ligand binding (reviewed in Sims, J.E. et al., Nature Rev. Immunol., 10:89-102 (2010)).
Toll-like receptors (TLRs) are a family of evolutionarily conserved, transmembrane innate immune receptors that participate in the first-line defense. As pattern recognition receptors, the TLRs protect against foreign molecules, activated by pathogen associated molecular patterns (PAMPs), or from damaged tissue, activated by danger associated molecular patterns (DAMPs). A total of 13 TLR family members have been identified, 10 in human, that span either the cell surface or the endosomal compartment. TLR7-9 are among the set that are endosomally located and respond to single-stranded RNA (TLR7and TLR8) or unmethylated single-stranded DNA containing cytosine-phosphate-guanine (CpG) motifs (TLR9).
Activation of TLR7/8/9 can initiate a variety of inflammatory responses (cytokine production, B cell activation and IgG production, Type I interferon response). In the case of autoimmune disorders, the aberrant sustained activation of TLR7/8/9 leads to worsening of disease states. Whereas overexpression of TLR7 in mice has been shown to exacerbate autoimmune disease, knockout of TLR7 in mice was found to be protective against disease in lupus-prone MRL/lpr mice. Dual knockout of TLR7 and 9 showed further enhanced protection.
As numerous conditions may benefit by treatment involving modulation of cytokines, IFN production and B cell activity, it is immediately apparent that new compounds capable of modulating TLR7 and/or TLR8 and/or TLR9 and methods of using these compounds could provide substantial therapeutic benefits to a wide variety of patients.
The present invention relates to a new class of [1,2,4]triazolo[1,5-a]pyridinyl substituted indole compounds found to be effective inhibitors of signaling through TLR7/8/9. These compounds are provided to be useful as pharmaceuticals with desirable stability, bioavailability, therapeutic index, and toxicity values that are important to their drugability.
EXAMPLE 15
2-(4-(2-(7,8-dimethyl-[1,2,4]triazolo[1,5-a]pyridin-6-yl)-3-isopropyl-1H-indol-5-yl) piperidin-1-yl)acetamide
To a reaction flask were added
6-(3-isopropyl-5-(piperidin-4-yl)-1H-indol-2-yl)-7,8-dimethyl-[1,2,4]triazolo[1,5-a]pyrid ine, 2 HCl (47.66 g, 104 mmol), DCE (220 mL), DBU (62.4 mL, 414 mmol), and 2-bromoacetamide (17.14 g, 124 mmol). The reaction flask was capped. The reaction mixture was stirred overnight at room temperature. The reaction mixture was concentrated, diluted with water, and stirred for 30 minutes then filtered. The solid was recrystallized using ethanol to afford 2-(4-(2-(7,8-dimethyl-[1,2,4]triazolo[1,5-a] pyridin-6-yl)-3-isopropyl-1H-indol-5-yl)piperidin-1-yl)acetamide (42.3 g, 93 mmol,
90% yield) as a white solid. LCMS MH+: 445. HPLC Ret. Time 1.20 min. Method QC-ACN-TFA-XB. 1HNMR (400 MHz, DMSO-d6) δ 10.97-10.86 (m, 1H), 8.78-8.69 (m, 1H), 8.54-8.40 (m, 1H), 7.64-7.49 (m, 1H), 7.30-7.21 (m, 2H), 7.17-7.09 (m, 1H), 7.06-6.93 (m, 1H), 2.99-2.82 (m, 5H), 2.62-2.54 (m, 4H), 2.24-2.12 (m, 5H), 1.92-1.72 (m, 4H), 1.37-1.29 (m, 6H).
ACS Medicinal Chemistry Letters (2022), 13(5), 812-818 83%
References
- Bristol-Myers Squibb: Investor Series [Link]
- Bristol-Myers Squibb: Investor Series [Link]
- MedKoo Biosciences: Afimetoran [Link]
//////////////Afimetoran, BMS-986256, BMS 986256, WHO 11516, phase 1
LORPUCITINIB

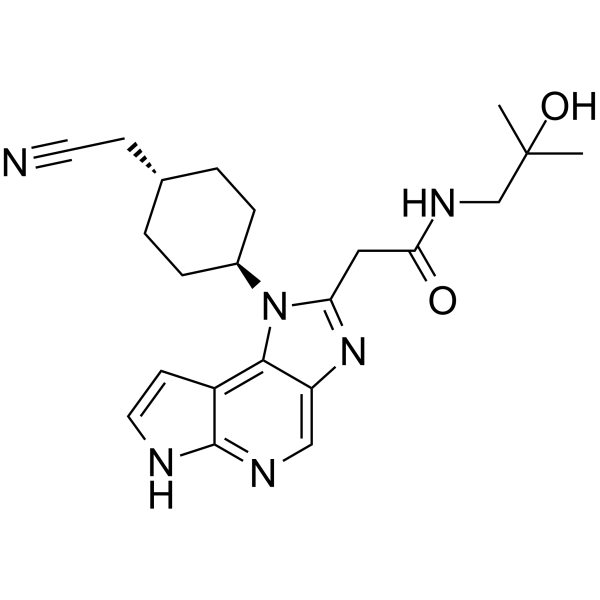
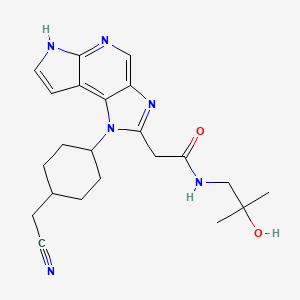
LORPUCITINIB
JNJ 64251330
2230282-02-5
UNII-OE1QTY7C25
| Molecular Weight | 408.50 |
|---|---|
| Formula | C22H28N6O2 |
| 1-(TRANS-4-(CYANOMETHYL)CYCLOHEXYL)-1,6-DIHYDRO-N-(2-HYDROXY-2-METHYLPROPYL)IMIDAZO(4,5-D)PYRROLO(2,3-B)PYRIDINE-2-ACETAMIDE |
2-[3-[4-(cyanomethyl)cyclohexyl]-3,5,8,10-tetrazatricyclo[7.3.0.02,6]dodeca-1,4,6,8,11-pentaen-4-yl]-N-(2-hydroxy-2-methylpropyl)acetamide
is a Gut-Restricted JAK Inhibitor for the research of Inflammatory Bowel Disease.
Lorpucitinib is an orally bioavailable pan-inhibitor of the Janus associated-kinases (JAKs), with potential immunomodulatory and anti-inflammatory activities. Upon oral administration, lorpucitinib works in the gastrointestinal (GI) tract where it targets, binds to and inhibits the activity of the JAKs, thereby disrupting JAK-signal transducer and activator of transcription (STAT) signaling pathways and the phosphorylation of STAT proteins. This may inhibit the release of pro-inflammatory cytokines and chemokines, reducing inflammatory responses and preventing inflammation-induced damage. The Janus kinase family of non-receptor tyrosine kinases, which includes tyrosine-protein kinase JAK1 (Janus kinase 1; JAK1), tyrosine-protein kinase JAK2 (Janus kinase 2; JAK2), tyrosine-protein kinase JAK3 (Janus kinase 3; JAK3) and non-receptor tyrosine-protein kinase TYK2 (tyrosine kinase 2), plays a key role in cytokine signaling and inflammaton.
PATENT
WO2019239387
WO2018112379
WO2018112382
PATENT
WO/2022/189496LORPUCITINIB FOR USE IN THE TREATMENT OF JAK MEDIATED DISORDERS
Example 1
[0117] 2-(1-((1r,4r)-4-(Cyanomethyl)cyclohexyl)-1,6-dihydroimidazo[4,5-d]pyrrolo[2,3-b]pyridin-2-yl)-N-(2-hydroxy-2-methylpropyl)acetamide
Step A: 2-(1-((1r,4r)-4-(Cyanomethyl)cyclohexyl)-6-(phenylsulfonyl)-1,6-dihydroimidazo[4,5-d]pyrrolo[2,3-b]pyridin-2-yl)-N-(2-hydroxy-2-methylpropyl)acetamide. To ensure dry starting material, ethyl 2-(1-((1r,4r)-4-(cyanomethyl)cyclohexyl)-6-(phenylsulfonyl)-1,6-dihydroimidazo[4,5-d]pyrrolo[2,3-b]pyridin-2-yl)acetate (Intermediate 3) was heated under vacuum at 50 °C for 18 h prior to the reaction. In a 1 L flask, ethyl 2-(1-((1r,4r)-4-(cyanomethyl)cyclohexyl)-6-(phenylsulfonyl)-1,6-dihydroimidazo[4,5-d]pyrrolo[2,3-b]pyridin-2-yl)acetate (Intermediate 3, 52.585 g, 104.01 mmol) was suspended in DMA (50 mL). 1-Amino-2-methylpropan-2-ol (50 mL) was added and the reaction was heated to 110 °C for 45 minutes, then to 125 °C for 5 hours. The reaction was cooled to room temperature and diluted with EtOAc (800 mL). The organic layer was extracted three times with a solution of water/ brine wherein the solution was made up of 1 L water plus 50 mL brine. The aqueous layers were back extracted with EtOAc (2 × 600 mL). The combined organic layers were dried over anhydrous MgSO4,
concentrated to dryness, and then dried for 3 days under vacuum to provide the title compound (65.9 g, 98% yield) as a yellow foam. The product was taken to the next step with no further purification. MS (ESI): mass calcd. for C28H32N6O4S, 548.22; m/z found, 549.2 [M+H]+.1H NMR (400 MHz, CDCl3): δ 8.76 (s, 1H), 8.26 – 8.19 (m, 2H), 7.84 (d, J = 4.1 Hz, 1H), 7.60 – 7.53 (m, 1H), 7.50 – 7.44 (m, 2H), 6.84 (d, J = 4.2 Hz, 1H), 4.76 – 4.61 (m, 1H), 3.97 (s, 2H), 3.45 (s, 1H), 3.27 (d, J = 5.9 Hz, 2H), 2.41 (d, J = 6.5 Hz, 2H), 2.38 – 2.25 (m, 2H), 2.23 – 2.12 (m, 2H), 2.09 -1.94 (m, 4H), 1.48 (qd, J = 13.6, 4.0 Hz, 2H), 1.21 (s, 6H).
[0118] Step B: 2-(1-((1r,4r)-4-(Cyanomethyl)cyclohexyl)-1,6-dihydroimidazo[4,5-d]pyrrolo[2,3-b]pyridin-2-yl)-N-(2-hydroxy-2-methylpropyl)acetamide. 2-(1-((1r,4r)-4-(Cyanomethyl)cyclohexyl)-6-(phenylsulfonyl)-1,6-dihydroimidazo[4,5-d]pyrrolo[2,3-b]pyridin-2-yl)-N-(2-hydroxy-2-methylpropyl)acetamide (65.90 g, 102.1 mmol) was added to a 1 L flask containing a stir bar. 1,4-dioxane (300 mL) was added, followed by aq KOH (3 M, 150 mL). The reaction was heated at 80 °C for 2 h. The reaction was cooled to room temperature and the solvent volume was reduced to about 200 mL on a rotovap. The residue was treated with a solution of water/brine (100 mL/100mL), then extracted with 10% MeOH in CH2Cl2 (2 x 1L). The organic layers were combined, dried over anhydrous MgSO4, and concentrated to dryness to provide a yellow solid. The solid was suspended in CH2Cl2 (200 mL), stirred vigorously for 30 minutes, and then collected by filtration. The solid was rinsed with CH2Cl2 (100 mL), dried by pulling air through the filter, and then further dried under vacuum at room temperature for 16 h to provide the title compound (41.59 g, 89% yield) as a white solid. MS (ESI): mass calcd. for C22H28N6O2, 408.23; m/z found, 409.2 [M+H]+. 1H NMR (600 MHz, DMSO-d6): δ 11.85 (s, 1H), 8.50 (s, 1H), 8.21 – 8.10 (m, 1H), 7.49 – 7.43 (m, 1H), 6.74 – 6.65 (m, 1H), 4.53 – 4.42 (m, 2H), 4.07 (s, 2H), 3.08 (d, J = 6.0 Hz, 2H), 2.58 (d, J = 6.1 Hz, 2H), 2.41 – 2.28 (m, 2H), 2.09 – 1.92 (m, 5H), 1.42 – 1.31 (m, 2H), 1.09 (s, 6H). The synthesis and active compound characterization of each of the aspects of this invention are provided herein in the form of examples. Due to the crystal structure of some of the aspects of this invention, polymorph screening may be pursued to further characterize specific forms of any such compound. This is illustrated in a non-limiting manner for compound of Formula I by the example under the heading polymorph screening.
[0119] The following compounds were prepared in reference to the foregoing synthesis:
Intermediate 1
[0120] 2-((1r,4r)-4-((5-Nitro-1-(phenylsulfonyl)-1H-pyrrolo[2,3-b]pyridin-4-yl)amino)cyclohexyl)acetonitrile
[0121] Step A: tert-butyl N-[(1r,4r)-4-(Hydroxymethyl)cyclohexyl]carbamate. To a 20-L 4-necked round-bottom flask purged and maintained with an inert atmosphere of nitrogen was placed (1r,4r)-4-[[(tert-butoxy)carbonyl]amino]cyclohexane-1-carboxylic acid (1066 g, 4.38 mol, 1.00 equiv) and THF (10 L). This was followed by the dropwise addition of BH3-Me2S (10 M, 660 mL) at -10 °C over 1 h. The resulting solution was stirred for 3 h at 15 °C. This reaction was performed three times in parallel and the reaction mixtures were combined. The reaction was then quenched by the addition of methanol (2 L). The resulting mixture was concentrated under vacuum. This resulted in of tert-butyl N-[(1r,4r)-4-(hydroxymethyl)cyclohexyl]carbamate (3000 g, 99.6%) as a white solid. MS (ESI): mass calcd. for C12H23NO3, 229.32; m/z found, 215.2 [M-tBu+MeCN+H]+; 1H NMR: (300 MHz, CDCl3): δ 4.40 (s, 1H), 3.45 (d, J = 6.3 Hz, 2H), 3.38 (s, 1H), 2.05-2.02 (m, 2H), 1.84-1.81 (m, 2H), 1.44 (s, 11H), 1.17-1.01 (m, 4H).
[0122] Step B: tert-butyl N-[(1r,4r)-4-[(Methanesulfonyloxy)methyl]cyclohexyl]carbamate. To a 20 L 4-necked round-bottom flask purged and maintained with an inert atmosphere of nitrogen, was placed tert-butyl N-[(1r,4r)-4-(hydroxymethyl)cyclohexyl]carbamate (1000 g, 4.36 mol, 1.00 equiv.), dichloromethane (10 L), pyridine (1380 g, 17.5 mol, 4.00 equiv.). This was followed by the dropwise addition of MsCl (1000 g, 8.73 mol, 2.00 equiv.) at -15 °C. The resulting solution was stirred overnight at 25 °C. This reaction was performed in parallel for 3 times and the reaction mixtures were combined. The reaction was then quenched by the addition of 2 L of water. The
water phase was extracted with ethyl acetate (1 x 9 L). The organic layer was separated and washed with 1 M HCl (3 x 10 L), NaHCO3 (saturated aq.) (2 x 10 L), water (1 x 10 L) and brine (1 x 10 L). The mixture was dried over anhydrous sodium sulfate, filtered and concentrated under vacuum. This resulted in of tert-butyl N-[(1r,4r)-4-[(methanesulfonyloxy)methyl]cyclohexyl]carbamate (3300 g, 82%) as a white solid. LC-MS: MS (ESI): mass calcd. for C13H25NO5S, 307.15; m/z found 292.1, [M-tBu+MeCN+H]+; 1H NMR: (300 MHz, CDCl3): δ 4.03 (d, J = 6.6 Hz, 2H), 3.38 (s, 1H), 3.00 (s, 3H), 2.07-2.05 (m, 2H), 1.87-1.84 (m, 2H), 1.72-1.69 (m, 1H), 1.44 (s, 9H), 1.19-1.04 (m, 4H).
[0123] Step C: tert-butyl N-[(1r,4r)-4-(Cyanomethyl)cyclohexyl]carbamate. To a 10 L 4-necked round-bottom flask, was placed tert-butyl N-[(1r,4r)-4-[(methanesulfonyloxy)methyl]cyclohexyl]carbamate (1100 g, 3.58 mol, 1.00 equiv.), DMSO (5500 mL) and NaCN (406 g, 8.29 mol, 2.30 equiv.). The resulting mixture was stirred for 5 h at 90 °C. This reaction was performed in parallel 3 times and the reaction mixtures were combined. The reaction was then quenched by the addition of 15 L of water/ice. The solids were collected by filtration. The solids were washed with water (3 x 10 L). This resulted in tert-butyl N-[(1r,4r)-4-(cyanomethyl)cyclohexyl]carbamate (2480 g, 97%) as a white solid. MS (ESI): mass calcd. for C13H22N2O2, 238.17; m/z found 224 [M-tBu+MeCN+H]+; 1H NMR: (300 MHz, CDCl3): δ 4.39 (s, 1H), 3.38 (s, 1H), 2.26 (d, J = 6.9 Hz, 2H), 2.08-2.04 (m, 2H), 1.92-1.88 (m, 2H), 1.67-1.61 (m, 1H), 1.44 (s, 9H), 1.26-1.06 (m, 4H).
[0124] Step D: 2-[(1r,4r)-4-Aminocyclohexyl]acetonitrile hydrochloride. To a 10-L round-bottom flask was placed tert-butyl N-[(1r,4r)-4-(cyanomethyl)cyclohexyl]carbamate (620 g, 2.60 mol, 1.00 equiv.), and 1,4-dioxane (2 L). This was followed by the addition of a solution of HCl in 1,4-dioxane (5 L, 4 M) dropwise with stirring at 10 °C. The resulting solution was stirred overnight at 25 °C. This reaction was performed for 4 times and the reaction mixtures were combined. The solids were collected by filtration. The solids were washed with 1,4-dioxane (3 x 3 L), ethyl acetate (3 x 3 L) and hexane (3 x 3 L). This resulted in 2-[(1r,4r)-4-aminocyclohexyl]acetonitrile hydrochloride (1753 g, 96%) as a white solid. MS (ESI): mass calcd. for C8H14N2, 138.12; m/z found 139.25, [M+H]+; 1H NMR: (300 MHz, DMSO-d6): δ 8.14 (s, 3H), 2.96-2.84 (m, 1H), 2.46 (d, J = 6.3 Hz, 2H), 1.98 (d, J = 11.1 Hz, 2H), 1.79 (d, J = 12.0 Hz, 2H), 1.64-1.49 (m, 1H), 1.42-1.29 (m, 2H), 1.18-1.04 (m, 2H).
[0125] Step E: 2-((1r,4r)-4-((5-Nitro-1-(phenylsulfonyl)-1H-pyrrolo[2,3-b]pyridin-4-yl)amino)cyclohexyl)acetonitrile. To a 1000 mL round bottom flask containing 2-[(1r,4r)-4-aminocyclohexyl]acetonitrile hydrochloride (29.10 g, 166.6 mmol) was added DMA (400 mL). The resulting suspension was treated with 4-chloro-5-nitro-1-(phenylsulfonyl)-1H-pyrrolo[2,3-b]pyridine (51.53 g, 152.6 mmol), followed by DIPEA (63.0 mL, 366 mmol). The reaction mixture was placed under N2 and heated at 80 °C for 4 h. The crude reaction mixture was cooled to room temperature and slowly poured into a vigorously stirred 2 L flask containing 1.6 L water. The resulting suspension was stirred for 15 minutes at room temperature, then filtered and dried for 16 h in a vacuum oven with heating at 70 °C to provide the title compound (63.37 g, 95%) as a yellow solid. MS (ESI): mass calcd. for C21H21N5O4S, 439.1; m/z found, 440.1 [M+H]+. 1H NMR (500 MHz, CDCl3): δ 9.10 (s, 1H), 8.99 (d, J = 7.8 Hz, 1H), 8.23 – 8.15 (m, 2H), 7.66 – 7.59 (m, 2H), 7.56 – 7.49 (m, 2H), 6.67 (d, J = 4.2 Hz, 1H), 3.95 – 3.79 (m, 1H), 2.38 (d, J = 6.2 Hz, 2H), 2.32 -2.21 (m, 2H), 2.08 – 1.98 (m, 2H), 1.88 – 1.76 (m, 1H), 1.60 – 1.32 (m, 4H).
Intermediate 2
[0126] 2-((1r,4r)-4-((5-Amino-1-(phenylsulfonyl)-1H-pyrrolo[2,3-b]pyridin-4-yl)amino)cyclohexyl)acetonitrile
[0127] 2-((1r,4r)-4-((5-Nitro-1-(phenylsulfonyl)-1H-pyrrolo[2,3-b]pyridin-4-yl)amino)cyclohexyl)acetonitrile (Intermediate 1, 58.60 g, 133.3 mmol) was dissolved in THF/MeOH (1:1, 4800 mL). The mixture was passed through a continuous-flow hydrogenation reactor (10% Pd/C), such as a Thales Nano H-Cube®, at 10 mL/min with 100 % hydrogen (atmospheric pressure, 80 °C), then the solution was concentrated to provide the product as a purple solid. The solid was triturated with EtOAc (400 mL) and then triturated again with MeOH (200 mL) then filtered and dried under vacuum to provide the title compound (50.2 g, 91.9% yield).
MS (ESI): mass calcd. for C21H23N5O2S, 409.2; m/z found, 410.2 [M+H]+. 1H NMR (400 MHz, CDCl3) δ 8.10 – 8.03 (m, 2H), 7.76 (s, 1H), 7.51 – 7.43 (m, 1H), 7.43 – 7.34 (m, 3H), 6.44 (d, J = 4.2 Hz, 1H), 4.61 (d, J = 8.5 Hz, 1H), 3.65 – 3.51 (m, 1H), 2.74 (s, 2H), 2.26 (d, J = 6.4 Hz, 2H), 2.19 – 2.05 (m, 2H), 1.97 – 1.86 (m, 2H), 1.76 – 1.59 (m, 1H), 1.33 – 1.12 (m, 4H).
Intermediate 3
[0128] Ethyl 2-(1-((1r,4r)-4-(cyanomethyl)cyclohexyl)-6-(phenylsulfonyl)-1,6-dihydroimidazo[4,5-d]pyrrolo[2,3-b]pyridin-2-yl)acetate
[0129] To a 1L round bottom flask containing a stir bar and 2-((1r,4r)-4-((5-amino-1-(phenylsulfonyl)-1H-pyrrolo[2,3-b]pyridin-4-yl)amino)cyclohexyl)acetonitrile (Intermediate 2, 58.31 g, 142.4 mmol) was added ethyl 3-ethoxy-3-iminopropanoate (60.51 g, 309.3 mmol), followed by EtOH (600 mL, dried over 3Å molecular sieves for 48 h). A reflux condenser was attached to the reaction flask, the reaction was purged with N2, and was heated at 90 °C for 9 h. The reaction mixture was cooled to room temperature and left to stand for 30 h where the product crystallized out as brown needles. The solids were broken up with a spatula and the reaction mixture was transferred to a 2 L flask. Water (1.4 L) was added slowly via separatory funnel with vigorous stirring. After addition of the water was complete, the suspension was stirred for 30 minutes. The brown needles were isolated by filtration and then dried by pulling air through the filter for 1 h. The product was transferred to a 500 mL flask and treated with EtOAc (200 mL). A small quantity of seed crystals were added, which induced the formation of a white solid precipitate. The suspension was stirred for 30 minutes at room temperature, filtered, rinsed with EtOAc (25 mL), and dried under vacuum to provide the product as a white solid (48.65 g, 68% yield). MS (ESI): mass calcd. for C26H27N5O4S, 505.2; m/z found, 506.2 [M+H]+. 1H NMR (400
MHz, CDCl3) δ 8.85 (s, 1H), 8.28 – 8.19 (m, 2H), 7.84 (d, J = 4.0 Hz, 1H), 7.61 – 7.53 (m, 1H), 7.52 – 7.43 (m, 2H), 6.84 (d, J = 4.1 Hz, 1H), 4.32 (s, 1H), 4.20 (q, J = 7.1 Hz, 2H), 4.09 (s, 2H), 2.44 (d, J = 6.2 Hz, 2H), 2.40 – 2.27 (m, 2H), 2.16 (d, J = 13.3 Hz, 2H), 2.12 – 1.96 (m, 3H), 1.54 – 1.38 (m, 2H), 1.27 (t, J = 7.1 Hz, 3H).
Polymorph screening example
[0130] Some embodiments of compound of Formula I as free bases present multiple crystalline configurations that have a complex solid-state behavior, some of which in turn can present distinguishing features among themselves due to different amounts of incorporated solvent. Some embodiments of compound of Formula I are in the form of pseudopolymorphs, which are embodiments of the same compound that present crystal lattice compositional differences due to different amounts of solvent in the crystal lattice itself. In addition, channel solvation can also be present in some crystalline embodiments of compound of Formula I, in which solvent is incorporated within channels or voids that are present in the crystal lattice. For example, the various crystalline configurations given in Table 2 were found for compound of Formula I. Because of these features, non-stoichiometric solvates were often observed, as illustrated in Table 2. Furthermore, the presence of such channels or voids in the crystal structure of some embodiments according to this invention enables the presence of water and/or solvent molecules that are held within the crystal structure with varying degrees of bonding strength. Consequently, changes in the specific ambient conditions can readily lead to some loss or gain of water molecules and/or solvent molecules in some embodiments according to this invention. It is understood that “solvation” (third column in Table 2) for each of the embodiments listed in Table 2 is the formula solvation, and that the actual determination of the same as a stoichiometry number (fourth column in Table 2) can slightly vary from the formula solvation depending on the actual ambient conditions when it is experimentally determined. For example, if about half of the water molecules in an embodiment may be present as hydrogen-bonded to the active compound in the crystal lattice, while about the other half of water molecules may be in channels or voids in the crystal lattice, then changes in ambient conditions may alter the amount of such loosely contained water molecules in voids or channels, and hence lead to a slight difference between the formula solvation that is assigned according to, for example, single crystal diffraction, and the
stoichiometry that is determined by, for example, thermogravimetric analysis coupled with mass spectroscopy.
Table 2. Embodiments of crystalline forms of compound of Formula I
[0131] The compound that was obtained as described in Example 1 was further crystallized by preparing a slurry in DCM (1:3, for example 10 g of compound in 30 ml DCM) that was stirred at 40oC for 4 hours, and further stirred for 14 hours at 25oC, then heptane was slowly added (1:2, for example 20 ml of heptane into the compound/DCM slurry/solution) at 25oC, stirred at 40oC for 4 hours, cooled to 25oC and stirred for further 14 hours at 25oC. Subsequent filtration led to compound of Formula I in the form of an off-white solid, that was identified as a monohydrate, a 1s embodiment.
CLIP
Journal of Medicinal Chemistry (2020), 63(6), 2915-2929

/////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Clip
https://clinicaltrials.gov/ct2/show/NCT04552197
The purpose of this study is to evaluate: systemic and local gut (rectum and sigmoid colon) exposure to JNJ-64251330, local tissue Pharmacodynamics (PD) using gut (rectum and sigmoid colon) biopsies (Part 1) and the effect of food on the rate and extent of absorption of JNJ-64251330 from oral tablet dosed with or without food (Part 2).
Familial adenomatous polyposis (FAP) is the most common polyposis syndrome. It is an autosomal dominant inherited disorder characterized by the early onset of hundreds to thousands of adenomatous polyps throughout the colon. JNJ-64251330 (lorpucitinib) is an oral, small molecule, potent pan-janus kinase (JAK) inhibitor that blocks phosphorylation of Signal Transducer and Activator of Transcription (STAT) proteins. pSTAT induces transcription of multiple genes involved in the progression of inflammatory disease. JNJ-64251330 has chemical properties that limits the amount of drug in the blood while delivering the drug to the tissues of the gut. Local inhibition of JAK in the gut may present a promising method to treat inflammatory diseases of the intestinal tract, such as FAP. The study consists of 3 phases: screening phase (30 days) a treatment phase (24 weeks), and follow-up visit (up to 30 days after last dose of study drug). The total duration of the study will be up to 32 weeks. Study evaluations will include efficacy via endoscopies, safety (monitoring of adverse events (AE), serious adverse events (SAEs), events of infections including tuberculosis (TB), clinical laboratory blood tests (complete blood count and serum chemistries), vital signs, and concomitant medication review), pharmacokinetics, pharmacodynamic and biomarkers evaluations.
Adenomatous polyposis coli (APC) also known as deleted in polyposis 2.5 (DP2.5) is a protein that in humans is encoded by the APC gene.[4] The APC protein is a negative regulator that controls beta-catenin concentrations and interacts with E-cadherin, which are involved in cell adhesion. Mutations in the APC gene may result in colorectal cancer.[5]
APC is classified as a tumor suppressor gene. Tumor suppressor genes prevent the uncontrolled growth of cells that may result in cancerous tumors. The protein made by the APC gene plays a critical role in several cellular processes that determine whether a cell may develop into a tumor. The APC protein helps control how often a cell divides, how it attaches to other cells within a tissue, how the cell polarizes and the morphogenesis of the 3D structures,[6] or whether a cell moves within or away from tissue. This protein also helps ensure that the chromosome number in cells produced through cell division is correct. The APC protein accomplishes these tasks mainly through association with other proteins, especially those that are involved in cell attachment and signaling. The activity of one protein in particular, beta-catenin, is controlled by the APC protein (see: Wnt signaling pathway). Regulation of beta-catenin prevents genes that stimulate cell division from being turned on too often and prevents cell overgrowth.
The human APC gene is located on the long (q) arm of chromosome 5 in band q22.2 (5q22.2). The APC gene has been shown to contain an internal ribosome entry site. APC orthologs[7] have also been identified in all mammals for which complete genome data are available.
////////////////JNJ-64251330, JNJ 64251330, LORPUCITINIB, PHASE 1, CANCER, Adenomatous Polyposis Coli
O=C(NCC(C)(O)C)CC1=NC2=CN=C(NC=C3)C3=C2N1[C@H]4CC[C@H](CC#N)CC4

NEW DRUG APPROVALS
ONE TIME
$10.00
ZY 19489, MMV 253
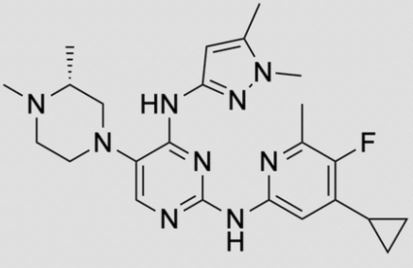

![2-N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-5-[(3R)-3,4-dimethylpiperazin-1-yl]-4-N-(1,5-dimethylpyrazol-3-yl)pyrimidine-2,4-diamine.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=92045019&t=l)
ZY 19489, MMV 253
C24 H32 FN9, 465.5
CAS 1821293-40-6
MMV253, GTPL10024, MMV674253
N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-5-((3R)-2-((1,5-dimethyl-1H-pyrazol-3-yl)amino)-3,4-dimethylpiperazin-1-yl)pyrimidin-2-amine
2-N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-5-[(3R)-3,4-dimethylpiperazin-1-yl]-4-N-(1,5-dimethylpyrazol-3-yl)pyrimidine-2,4-diamine
- N2-(4-Cyclopropyl-5-fluoro-6-methyl-2-pyridinyl)-5-[(3R)-3,4-dimethyl-1-piperazinyl]-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-2,4-pyrimidinediamine
- (R)-N2-(4-Cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-1-yl)pyrimidine-2,4-diamine

SYN
IN 201721031453
The invention relates to triaminopyrimidine compd. of formula I, pharmaceutically acceptable salts thereof, hydrates, solvates, polymorphs, optically active forms thereof, in solid state forms useful for preventing or treating malaria. The invention also relates to a process for prepn. of triaminopyrimidine compd. and intermediates thereof. Compd. I was prepd. by condensation of 5-bromouracil with tert-Bu (R)-2-methylpiperazine-1-carboxylate to give tert-Bu (R)-4-(2,4-dichloropyrimidin-5-yl)-2-methylpiperazine-1-carboxylate, which underwent chlorination followed by condensation with 1,5-dimethyl-1H-pyrazol-3-amine followed by condensation with 4-cyclopropyl-5-fluoro-6-methylpyridin-2-amine hydrochloride to give (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-5-(3-methylpiperazin-1-yl)pyrimidine-2,4-diamine, which underwent Boc-deprotection followed by methylation to give I.
SYN
WO 2019049021
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019049021
Malaria is caused by protozoan parasites of the genus Plasmodium that infect and destroy red blood cells, leading to fever, severe anemia, cerebral malaria and, if untreated, death.
International (PCT) Publication No. WO 2015/165660 (the WO ‘660) discloses triaminopyrimidine compounds, intermediates, pharmaceutical compositions and methods for use for preventing or treating malaria. The WO ‘660 discloses a process for preparation of 4-cyclopropyl-5-fluoro-6-methylpyridin-2-amine (compound 5) as depicted in scheme-1.
Scheme 1
WO ‘660 discloses a process for preparation of triaminopyrimidine compounds depicted in scheme-2.

WO ‘660 discloses the preparation of compounds 8 and 4 by using microwave technique using Biotage microwave vial. WO ‘660 in example- 13, discloses the isolation of compound 1 by concentration of reaction mixture to obtain crude product, which was purified through reverse phase HPLC GILSON instrument to obtain pure solid compound 1 in 40.8% yield, without providing the purity of the solid compound 1. The process disclosed in WO ‘660 is not industrially advantageous as it requires microwave conditions as well as chromatographic purification and provides compound 1 with lower yields. The compound 1 prepared may not be suitable for pharmaceutical preparations based on various regulatory requirements.
Polymorphism, the occurrence of different crystalline forms, is a property of some molecules. A single molecule can exist in different crystalline forms having distinct physical properties like melting point, thermal behaviors (e.g. measured by thermogravimetric analysis – TGA, or different scanning calorimetry – DSC, Powder x-ray diffraction pattern – PXRD, infrared absorption – IR). One or more these techniques may be used to distinguish different polymorphic forms of a compound.
Different salts and solid states (e.g. solvates, hydrates) of an active pharmaceutical ingredient may possess different physio-chemical properties. Such variation in the properties of different salts and solid states forms may provide a basis for improving formulation, for example, by facilitating better processing or handling characteristics, changing the dissolution profile in a favorable direction, or improving stability (both chemical and polymorph) and shelf-life. These variations in the properties of different salts and solid states forms may offer improvements to the final dosage form for example, to improve bioavailability. Different salts and solid state forms of an active pharmaceutical ingredient may also give rise to a variety of polymorphs or crystalline forms or amorphous form, which may in turn provide additional opportunities to assess variations in the properties and characteristics of an active pharmaceutical ingredient.
In view of the above, the present invention provides a process for the preparation of triaminopyrimidine compound 1 or pharmaceutically acceptable salts thereof or hydrates or solvates or polymorphs or optically active forms thereof, which is industrially scalable, environment friendly and efficient so as to obtain compounds of the invention in higher yields and purity.
The process for the preparation of triaminopyrimidine compound 1 or intermediates thereof of the present invention, takes the advantage by using appropriate solvent systems and isolation techniques as well as purification techniques, thereby to overcome problems of lower yields, chromatography purifications and microwave reactions of the prior art.
SUMMARY OF THE INVENTION
The present invention provides solid state forms of triaminopyrimidine compound
1,
1
Examples: Preparation of Intermediates
Example-1: Preparation of 6-chloro-4-cyclopropyl-3-fluoro-2-methylpyridine
In a 250 mL 4N round bottom flask, process water (30 ml) and cyclopropanecarboxylic acid (14.19 g, 164.88 mmol) were added at 25 to 35°C and started stirring. Sulphuric acid (4.4 ml, 82.44 mmol) was charged to the reaction mixture. Silver nitrate (4.18 g, 24.73 mmol), 6-Chloro-3-fluoro-2-methylpyridine (6 g, 41.22 mmol) were charged to the reaction mixture. Aqueous solution of ammonium persulphate (65.85 g, 288.54 mmol in 90 mL water) was added to the reaction mixture in 30 to 60 min at temperature NMT 60 °C. After the completion of the reaction as monitored by HPLC, toluene (30 ml) was added to the reaction mixture and stirred for 15 min. The reaction mixture filtered, separated layers from filtrate and extracted aqueous layer using toluene (30 mL). The organic layer was washed with aqueous sodium carbonate solution (30 mL) and water. The organic layer was distilled completely under vacuum at 60 °C to obtain 3.37 g syrupy mass as titled compound.
Example-2: Preparation of 6-chloro-4-cyclopropyl-3-fluoro-2-methylpyridine
In a suitable glass assembly, process water (7.5 L) and cyclopropanecarboxylic acid (3.55 Kg, 41.24 mol) were added at 25 to 35 °C and stirred. Sulphuric acid (2.02 Kg, 20.59 mol), silver nitrate (1.05 Kg, 6.21 mol), 6-chloro-3-fluoro-2-methylpyridine (1.5 Kg, 10.3 mol) were added to the reaction mixture. Aqueous solution of ammonium persulphate (16.46 g, 72.13 mmol in 22.5 L water) was added to the reaction mixture at 55 to 60 °C and maintained. After the completion of the reaction as monitored by HPLC, toluene (7.5 L) was added to the reaction mixture and stirred for 15 min. The reaction mixture was filtered, organic layer was separated and aqueous layer was extracted using toluene (6 L), filtered the reaction mixture and washed the solid with toluene (1.5 L). The combined organic layer was washed with 20% sodium carbonate solution (9 L) and water. The organic layer was concentrated completely under vacuum at 60 °C to obtain 880 g (86.50%) syrupy mass of titled compound.
Example-3: Preparation of N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-l,l-diphenyl-methanimine
In a 100 mL 3N round bottom flask, 6-chloro-4-cyclopropyl-3-fluoro-2-methylpyridine (2.69 g, 14.48 mmol) and toluene (30 mL) were added at 25 to 35 °C. Diphenylmethanimine (3.15 g, 17.38 mmol) was charged to the reaction mixture and stirred for 5-10 min under nitrogen purging. Racemic BINAP (270 mg, 0.43 mmol) and palladium acetate (98 mg, 0.43 mmol) were added to the reaction mixture. Sodium-ie/ -butoxide (2.78 g, 28.96 mmol) was added to the reaction mixture and heated to 100 to 110° C under nitrogen. After the completion of the reaction as monitored by HPLC, the reaction mixture was cooled to 25 to 35 °C and filtered over hyflo bed and washed with toluene. The filtrate containing titled compound was preserved for next stage of reaction.
Example-4: Preparation of N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-l,l-diphenyl-methanimine
In a suitable assembly, 6-chloro-4-cyclopropyl-3-fluoro-2-methylpyridine (880) and toluene (7.5 L) were added at 25 to 35 °C. Diphenylmethanimine (787 g, 4.34 mmol) and BOC anhydride (237 g, 1.086 mol) was added to the reaction mixture and stirred for 5-10 min under nitrogen purging. Racemic BINAP (67.6 g, 0.108 mmol) and palladium acetate (24.4 g, 0.108 mol) were added to the reaction mixture. S odium- ieri-butoxide (870 g, 9.05 mol) was added to the reaction mixture and heated to 100 to 110 °C under nitrogen. After the completion of the reaction as monitored by HPLC, the reaction mixture was cooled to 25 to 35 °C, water (6 L) was added. The reaction mixture was filtered over hyflo bed and washed with toluene. The filtrate containing titled compound was preserved for next stage of reaction.
Example-5: Preparation of 4-cyclopropyl-5-fluoro-6-methylpyridin-2-amine hydrochloride monohydrate
In a 100 mL 3N round bottom flask, N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-l,l-diphenylmethanimine in toluene as obtained in example-3 was added water (25 mL) at 25 to 35° C. The cone. HCl (3 mL) was charged to the reaction mixture and heated to 40 to 50 °C. After the completion of the reaction as monitored by HPLC, the reaction mixture was cooled to 25 to 35 °C. Layers were separated. The aqueous layer was treated with methylene dichloride and pH was adjusted to 7.5 to 8.5 using sodium carbonate solution, stirred for 15 min and layers were separated. Aqueous layer was extracted with methylene dichloride, charcoaled and acidified to pH 3 to 4 with aqueous HCl. The solvent was distilled completely and acetonitrile (9 mL) and ethyl acetate (9 mL) was added. The reaction mixture was stirred for 1 hour at 25 to 35° C. The product was filtered and washed with ethyl acetate. The product was dried at 50° C for 4 hours under vacuum to obtain 1.62 g title compound as monohydrate yellow crystalline solid having 99.51% HPLC purity.
Example-6: Preparation of 4-cyclopropyl-5-fluoro-6-methylpyridin-2-amine hydrochloride monohydrate
In a suitable glass assembly, N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-l,l-diphenylmethanimine in toluene as obtained in example-4 was added water (6 L) at 25 to 35° C. The cone. HCl (750 mL) was charged to the reaction mixture and heated to 40 to 50 °C. After the completion of the reaction as monitored by HPLC, the reaction mixture was cooled to 25 to 35 °C. Layers were separated. The aqueous layer was treated with methylene dichloride (3 L) and pH was adjusted to 7.5 to 8.5 using sodium carbonate solution, stirred for 15 min and layers were separated. Aqueous layer was extracted with methylene dichloride (3 L), charcoaled and acidified to pH 3 to 4 with aqueous HCl. The solvent was distilled completely and acetonitrile (1.5 L) and ethyl acetate (1.5 L) were added. The reaction mixture was stirred for 1 hour at 25 to 35° C. The product was filtered and washed with ethyl acetate. The product was dried at 50° C for 4 hours under vacuum to obtain 489 g (96.80%) title compound as monohydrate yellow crystalline solid having 99.51% HPLC purity. The crystalline compound is characterized by Powder x-ray diffraction pattern (FIG.5), Differential scanning calorimetry (FIG.6) and Thermogravimetric analysis (FIG.7).
Example 7: Preparation of 2,3-dibromobutanenitrile
In a 2 L round bottom flask, dichloromethane (550 mL) and 2-butenenitrile 110 g
(1.64 mol) were cooled to 20 to 25 °C. A solution of bromine 275 g (1.72 mol) in dichloromethane (220 mL) was dropwise added at 20 to 25 °C. Hydrobromic acid 1.43 ml (0.0082 mol) in acetic acid (33%) solution was added into the reaction mixture and stirred for 4 hours. After the completion of reaction, Na2S203 (550 mL) 4% aqueous solution was added and the reaction mixture was stirred for 15 min. The separated organic layer was distilled under vacuum completely to obtain 364.2 g (97.9%) of title compound as an oil.
Example 8: Preparation of l,5-dimethyl-lH-pyrazol-3-amine
In a 5 L round bottom flask, water (1. 36 L), sodium hydroxide 340 g (8.99 mol) were added and the reaction mixture was cooled to 0 to 5°C. A solution of methyl hydrazine sulphate 237.8 g (1.65 mol) in 680 mL water was added dropwise to the reaction mixture and stirred below 10 °C. 2,3-dibromobutanenitrile 340 g (1.5 mol) prepared in example-7 was added and the reaction mixture was stirred below 10 °C for 2 hours. After the completion of reaction, toluene (630 mL) was added and the reaction mixture was stirred for 15 min. The aqueous layer was separated and the organic layer was removed. The aqueous layer was extracted with dichloromethane (5.1 L). The combined organic layer was distilled completely under vacuum to obtain residue. Diisopropyl ether (680 mL) was added and the reaction mixture was stirred at 0 to 5 °C for 1 hour. The reaction mixture was filtered, washed with diisopropyl ether and dried to obtained 121.5 g (72.93%) of title compound having 95.63% purity.
Examples: Preparation of triaminopyrimidine compounds
Example-9: Preparation of tert-butyl (R)-4-(2,4-dioxo-l,2,3,4-tetrahydro- pyrimidin-5-yl)-2-methylpiperazine-l-carboxylate
In 2 L four neck round bottom flask, 1.25 Kg (6.545 mol) 5-bromouracil, 1.87 Kg (9.360 mol) tert-butyl (R)-2-methylpiperazine-l-carboxylate and 5L pyridine were added at 25 to 35° C. The reaction mass was stirred for 15 hours at 115 to 120°C. After completion, the reaction mass was cooled to 25 to 35°C. 12.5 L water was added and stirred for 1 hour. The reaction mass was filtered, washed with 2.5 L water and dried to obtain 1.37 Kg (67.4%) of title compound.
Example-10: Preparation of tert-butyl (R)-4-(2,4-dichloropyrimidin-5-yl)-2-methylpiperazine- 1 -carboxylate
In 20 L four neck round bottom flask, 1.36 Kg (4.382 mmol) tert-butyl (R)-4-(2,4-dioxo-1, 2,3, 4-tetrahydropyrimidin-5-yl)-2-methylpiperazine-l -carboxylate and 6.8 L phosphorus oxychloride were added at 25 to 35° C. 26.5 mL pyridine (0.329 mol) was added and the reaction mass was heated to 105 to 110 °C and stirred for 4 hours. After the completion of the reaction, phosphorus oxychloride was distilled completely at atmospheric pressure. 2.72 L acetone was added and the reaction mixture was quenched into 4.08 L water. Acetone was removed by distillation under vacuum. 20% sodium carbonate solution was added to adjust pH 7.5-8.5 of the reaction mixture. 1.14 Kg (5.258 mol) di-tert-butyl dicarbonate and 9.52 L ethyl acetate were added and stirred for 2 hours at 25 to 35 °C. After the completion of the reaction, the organic layer was separated and aqueous layer was extracted with 6.8 L ethyl acetate. The combined ethyl layers were distilled to remove ethyl acetate completely under vacuum to obtain residue. 1.36 L isopropyl alcohol was added to the residue and isopropyl alcohol was removed completely. 4.08 L isopropyl alcohol and 6.8 L water were added to the residue and stirred for 1 hour. The reaction mass was filtered, washed with water and dried to obtain 1.25 Kg of title compound.
Example-11: Preparation of tert-butyl (R)-4-(2-chloro-4-[(l,5-dimethyl-lH-pyrazol-3-yl)amino)pyrimidin-5-yl]-2-methylpiperazine-l-carboxylate
In 20 L round bottom flask, 640 g (1.843 mol) tert-butyl (R)-4-(2, 4-dichloropyrimidin-5-yl)-2-methylpiperazine-l -carboxylate, 225.3 g (2.027 g) 1,5-dimethyl-lH-pyrazol-3-amine and 9.6L toluene were added at 25 to 35°C. 1.2 Kg (3.686 mol) cesium carbonate was added. The reaction mixture was purged for 15 min under nitrogen. 12.41 g (0.0553 mol) palladium acetate and 34.43 g (0.0553 mol) racemic 2,2′-bis(diphenylphosphino)-l,l’-binaphthyl were added and the reaction mass was maintained for 16 hours at 110 to 115 °C under nitrogen. After the completion of the reaction, the reaction mixture was filtered through a celite bed and washed the bed with 1.28 L toluene. Toluene was distilled completely and 2.56 L dichlromethane was added. The compound was adsorbed by 1.92 Kg silica gel (60-120 mesh). The dichloromethane was distilled completely under vacuum and 12.8 L mixture of ethyl acetate and hexane was added to the residue and stirred for 2 hours. The silica gel was filtered and the filtrate was distilled completely under vacuum to obtain 595 g title compound.
Example-12: Preparation of tert-butyl (R)-4-(2-((4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)amino)-4-((l,5-dimethyl-lH-pyrazol-3-yl)amino) pyrimidin-5-yl)-2-methylpiperazine-l-carboxylate
In 20 L round bottom flask, 595 g (1.40 mol) tert-butyl (R)- 4-(2-chloro-4-[(l,5-dimethyl-lH-pyrazol-3-yl)amino)pyrimidin-5-yl]-2-methylpiperazine-l-carboxylate, 305 g (1.38 mol) 4-cyclopropyl-5-fluoro-6-methylpyridin-2-amine hydrochloride and 11.5 L toluene were added at 25 to 35°C. 1.08 Kg (3.32 mol) cesium carbonate was added. The reaction mixture was purged for 15 min under nitrogen. 17.21 g (27.6 mmol) palladium acetate and 6.21 g (27.6 mmol) racemic 2,2′-bis(diphenylphosphino)-l, -binaphthyl were added. The reaction mass was stirred for 6 hours at 110 tol l5 °C under nitrogen. After the completion of the reaction, the reaction mixture was filtered through a celite bed and washed with toluene. The filtrate was used as such in the next step without further treatment.
Example-13: Preparation of tert-butyl (R)-4-(2-((4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)amino)-4-((l,5-dimethyl-lH-pyrazol-3-yl)amino) pyrimidin-5-yl)-2-methylpiperazine-l-carboxylate
In 500 mL four neck round bottom flask, 7.5 g (17.77 mmol) (R)-tert-butyl 4-(2-chloro-4-[(l,5-dimethyl-lH-pyrazol-3-yl)amino)pyrimidin-5-yl]-2-methylpiperazine-l-carboxylate, 3.92 g (17.77 mmol) 4-cyclopropyl-5-fluoro-6-methylpyridin-2-amine hydrochloride compound and 150 mL toluene were added at 25 to 35 °C. 20 g (61.3 mmol) cesium carbonate was added. The reaction mixture was purged for 15 min under nitrogen. Then, 130 mg (0.58 mmol) palladium acetate and 360 mg (0.58 mmol) racemic 2,2′-bis(diphenylphosphino)-l,l’-binaphthyl were added. The reaction mass was stirred for 18 hours at 110 to 115° C under nitrogen. After completion, the reaction mixture was filtered through a celite bed and washed with toluene. The filtrate was used as such in the next step without further treatment.
2 4
Example-14: (R)-N -(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N -(1, 5-dimethyl-lH-pyrazol-3-yl)-5-(3-methylpiperazin-l-yl)pyrimidine-2,4-diamine
In 50 L glass assembly, the filtrate containing tert-butyl (R)-4-(2-((4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)amino)-4-((l,5-dimethyl-lH-pyrazol-3-yl)amino) pyrimidin-5-yl)-2-methylpiperazine-l-carboxylate from example 13 was taken. 11.5 L water and 1.28 L Cone. HC1 were added at 25 to 35 °C. The reaction mass was stirred for 2 hours at 50 to 55 °C. After the completion of the reaction, reaction mixture was cooled to room temperature and filtered over celite bed and washed with water. The separated the aqueous layer from filtrate was basified by using 20% sodium carbonate solution and extracted with 12.8 L methylene dichloride. The organic layer was distilled completely under vacuum to obtain residue. 9.6 L acetonitrile was added to the residue and heated to reflux for 30 min. The reaction mixture was cooled and stirred at 25 to 35 °C for 1 hour. The reaction mixture was filtered, washed with 640 mL acetonitrile and dried to obtain 360 g titled compound.
2 4
Example-15: (R)-N -(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N -(1,5-dimethyl-lH-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-l-yl)pyrimidine-2,4-diamine
In 250 mL four neck round bottom flask, 4.7 g (10.4 mmol) (R)-N -(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(l,5-dimethyl-lH-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-l-yl)pyrimidine-2,4-diamine was dissolved in 56 mL ethanol. 1.89 g (23.32 mmol) formaldehyde and 1.44 g (22.90 mmol) sodium cyanoborohydride were added. Adjusted pH 5-6 using acetic acid and stirred the reaction mass at 25 to 35 °C for 2 hours. After completion, ethanol was distilled completely under vacuum. 47 mL water was added to the residue. The reaction mass was basified by 20% sodium carbonate solution and extracted with methylene dichloride. Both the organic layers were combined and distilled completely under vacuum. 94 mL acetonitrile was added to the residue and heated to reflux for 15 min. The reaction mass was cooled to 25 to 35° C and stirred for 1 hour. The reaction mass was filtered, washed with 5 mL acetonitrile and dried to obtain 3.7 g title compound as crystalline solid, having HPLC purity of about 99.61%.
2 4
Example-16: (R)-N -(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N -(1,5-dimethyl-lH-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-l-yl)pyrimidine-2,4-diamine
In 20 L round bottom flask, 725 g (1.60 mol) (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(l,5-dimethyl-lH-pyrazol-3-yl)-5-(3,4-dimethylpiperazine-l-yl)pyrimidine-2,4-diamine was dissolved in 6.52 L dichloromethane. 261.5 g (3.2 mol) formaldehyde and 510.4 g (2.4 mol) sodium triacetoxyborohydride were added and stirred the reaction mixture at 25 to 35 °C for 2 hours. After the completion of the reaction, 3.63 L water was added into the reaction mixture. The reaction mixture was basified by 20% sodium carbonate solution and the organic layer was separated. The aqueous layer was extracted with 1.45 L methylene dichloride. The combined organic layers were distilled completely under vacuum. 14.5 L acetonitrile was added to the residue and heated to reflux for 15 min. The reaction mixture was cooled to 25 to 35° C and stirred for 1 hour. The reaction mass was filtered, washed with 1.45 L acetonitrile and dried to obtain 632 g of title compound as crystalline solid having 99.01% HPLC purity. The crystalline compound is characterized by Powder x-ray diffraction pattern (FIG.l) and Differential Scanning Calorimetry (FIG.2).
2 4
Example-17: Preparation of (R)-N -(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N -(l,5-dimethyl-lH-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-l-yl)pyrimidine-2,4-diamine In a 10 mL round bottom flask, 300 mg (0.644 mmol) (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(l,5-dimethyl-lH-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-l-yl)pyrimidine-2,4-diamine, 2.7 mL acetonitrile and 0.3 mL water were added and the reaction mixture was heated to reflux for 15 min. The reaction mixture was cooled to 25 to 35 °C and stirred for 1 hour. The reaction mass was filtered, washed with acetonitrile and dried to obtain 201 mg (67%) title compound as crystalline solid. The crystalline compound is characterized by Powder x-ray diffraction pattern (FIG.3) and Differential Scanning Calorimetry (FIG.4).
SYN
WO 2015165660
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015165660
Example 13
Synthetic scheme 1
Synthetic scheme 2
(R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-1-yl)pyrimidine-2,4-diamine
In a 50 mL round-bottomed flask (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-5-(3-methylpiperazin-1-yl)pyrimidine-2,4-diamine hydrochloride (190 mg, 0.42 mmol, Example 2) was taken in DCM (2 mL) to give a yellow suspension. To this Hunig’s Base (0.184 mL, 1.05 mmol) was added and the suspension turned clear. After 10 minutes, it turned into a white suspension. After another 10 minutes, the mixture was concentrated to dryness. Resultant residue was dissolved in ethanol (absolute, 99.5%) (3 mL) and formaldehyde (0.042 mL, 0.63 mmol) was added and stirred for 10 minutes. White suspension slowly cleared to yellow solution. To this clear solution sodium cyanoborohydride (26.4 mg, 0.42 mmol) was added in one portion to get white suspension. After 30 minutes LCMS showed completion of reaction. The reaction mixture was concentrated and the crude was purified through reverse phase HPLC GILSON instrument to get the pure solid of (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-1-yl)pyrimidine-2,4-diamine (80 mg, 40.8 %).1H NMR (300
MHz, DMSO-d6) δ ppm 0.67 – 0.78 (m, 2 H) 1.00 (d, J=6.22 Hz, 3 H) 1.02 – 1.08 (m, 2 H) 1.96 – 2.10 (m, 1 H) 2.23 (s, 7 H) 2.30 – 2.38 (m, 4 H) 2.73 – 2.96 (m, 4 H) 3.33 (s, 3 H) 6.83 (s, 1 H) 7.67 (d, J=5.09 Hz, 1 H) 8.00 (s, 1 H) 8.03 (s, 1 H) 9.26 (s,1 H) MS (ES+), (M+H)+ = 466.45 for C21H32FN9.
SYN
Nature Communications (2015), 6, 6715.
https://www.nature.com/articles/ncomms7715
Hameed P., S., Solapure, S., Patil, V. et al. Triaminopyrimidine is a fast-killing and long-acting antimalarial clinical candidate. Nat Commun 6, 6715 (2015). https://doi.org/10.1038/ncomms7715
The widespread emergence of Plasmodium falciparum (Pf) strains resistant to frontline agents has fuelled the search for fast-acting agents with novel mechanism of action. Here, we report the discovery and optimization of novel antimalarial compounds, the triaminopyrimidines (TAPs), which emerged from a phenotypic screen against the blood stages of Pf. The clinical candidate (compound 12) is efficacious in a mouse model of Pf malaria with an ED99 <30 mg kg−1 and displays good in vivo safety margins in guinea pigs and rats. With a predicted half-life of 36 h in humans, a single dose of 260 mg might be sufficient to maintain therapeutic blood concentration for 4–5 days. Whole-genome sequencing of resistant mutants implicates the vacuolar ATP synthase as a genetic determinant of resistance to TAPs. Our studies highlight the potential of TAPs for single-dose treatment of Pf malaria in combination with other agents in clinical development.

(A) Pyridine, microwave, 150 °C, 45 min. (B) (i) POCl3, reflux, 6 h (ii) sodium carbonate, di-tert-butyl dicarbonate, room temperature, 16 h. (C) N,N-Diisopropylethylamine (DIPEA), ethanol, microwave, 110 °C, 1 h. (D) (i) Potassium tert-butoxide, 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl (BINAP), pd2(dba)3, toluene, reflux, 12 h. (E) HCl (4 N) in dioxane, 15–30 min. (F) Compound 9, DIPEA, dichloromethane, formaldehyde (HCHO), sodium cyanoborohydride, 15 min.
Synthesis of (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1, 5-dimethyl-1H-pyrazol-3-yl)-5-(3, 4-dimethylpiperazin-1-yl)pyrimidine-2,4-diamine (12). (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-5-(3-methylpiperazin-1-yl)pyrimidine-2,4-diamine hydrochloride (compound 9, 190 mg, 0.42 mmol) was taken in dichloromethane (2 ml) to give a yellow suspension. To this Hunig’s Base (0.184 ml, 1.05 mmol) was added and the suspension turned clear. After 10 min of stirring, reaction mixture turned into a white suspension and then it was concentrated to dryness. Resultant residue was dissolved in ethanol (absolute, 99.5%) (3 ml), and formaldehyde (0.042 ml, 0.63 mmol) was added and stirred for 10 min. To this clear solution, sodium cyanoborohydride (26.4 mg, 0.42 mmol) was added in one portion to get a white suspension. The reaction mixture was concentrated and the crude product was purified through reverse-phase chromatography to get the pure off-white solid of (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1, 5-dimethyl-1H-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-1-yl)pyrimidine-2,4-diamine (80 mg, 40.8%). Yield: 40.8%, purity: >95% by HPLC (ultraviolet at 220 and 254 nm). 1H NMR (300 MHz, DMSO-d6) δ 9.26 (s,1H), 8.03 (s, 1H) 8.00 (s, 1H) 7.67 (d, J=5.1 Hz, 1H) 6.83 (s, 1H) 3.33 (s, 3H) 2.96–2.73 (m, 4H) 2.75–2.50 (m, 1H) 2.38–2.30 (m, 4H) 2.23 (s, 7H) 2.10–1.96 (m, 1H),1.08–1.02 (m, 2H) 1.00 (d, J=6.2 Hz, 3H) 0.78–0.67 (m, 2H). 13C-NMR (126 MHz, DMO-d6) δ 155.30, 154.67, 152.10, 150.93, 148.98, 146.81. 145.29, 141.95, 140.31, 138.81, 124.91, 106.20, 97.07, 58.78, 51.87, 42.16, 35.28, 17.23. 10.99 and 8.77, HRMS (ESI): m/z calculated for C24H32FN9+H [M+H]: 466.2765. Found: 466. 2838. Traces of LC-MS, HRMS, 1H NMR and 13C-NMR of compound 12 are shown in Supplementary Figs 1–3.


| Product vision |
|
| MoA |
|
| Key features |
|
| Challenges |
|
| Status |
|
| Next milestone |
|
| Previously |
|
Zydus receives Orphan Drug Designation from USFDA for ZY-19489, a novel compound to treat malaria;
ZY19489 is a novel antimalarial compound active against all current clinical strains of P. falciparum and P. vivax, including drug-resistant strains.
Zydus Cadila listed as Cadila Healthcare Limited announced that its antimalarial compound ZY19489 (MMV253), currently in development together with Medicines for Malaria Venture (MMV), a leading product development partnership (PDP) in antimalarial drug research, has received Orphan Drug Designation from the USFDA.
Orphan drug designation provides eligibility for certain development incentives, including tax credits for qualified clinical testing, prescription drug user fee exemptions, and seven-year marketing exclusivity upon FDA approval.
The company said that the Phase I study of ZY19489 has demonstrated a long half-life and potential for a single-dose cure for malaria. In a separate malaria challenge trial, potent antimalarial activity has been demonstrated following single-dose oral administration of ZY19489.
“As a global community facing threats from rapidly mutating malaria strains and the rise in artemisinin resistance cases, we have to be prepared with novel therapeutic drugs. ZY-19489 is a potential single dose radical cure for P. falciparum and P. vivax malaria which is a major global health risk today,” Pankaj R. Patel, Chairman, Zydus Group, said.
“ZY19489 is a potent, first in class molecule, originally discovered and elaborated in India” said Dr. Timothy Wells, Chief Scientific Officer, MMV. “It has tremendous potential as part of a new generation of treatments and is fully active against drug resistant strains of malaria which are increasingly a concern.”
Artemisinin resistance is seen as a mounting challenge to the global fight against malaria. ZY19489 is being developed to provide an effective alternative to the current front-line antimalarial drugs for the treatment of P. falciparum and P. vivax malaria, as artemisinin-based combination therapies (ACTs) are under threat of resistance.
As per the World Malaria Report 2021, there were an estimated 241 million cases of malaria worldwide and the estimated number of malaria deaths stood at 627,000 in 2020. A major health concern, it is estimated that a child dies from malaria every minute. About 96% of malaria deaths globally were in 29 countries. India accounted for about 82% of all malaria deaths in the WHO South-East Asia Region.
////////////ZY 19489, MMV 253, Orphan Drug Designation, PHASE 1, ZYDUS CADILA, ANTIMALARIAL
Cn1nc(Nc2nc(Nc3cc(C4CC4)c(F)c(C)n3)ncc2N2C[C@@H](C)N(C)CC2)cc1C
CC1CN(CCN1C)C2=CN=C(N=C2NC3=NN(C(=C3)C)C)NC4=NC(=C(C(=C4)C5CC5)F)C
XL 114, AUR 104 and XL 102, AUR 102 (NO CONCLUSIONS, ONLY PREDICTIONS)

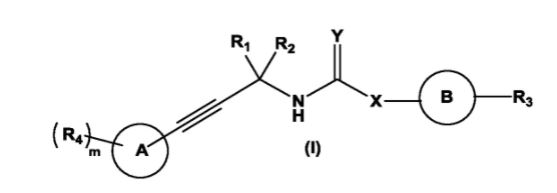
XL 114
FOR BOTH, JUST PREDICTION
PREDICTIONS
or


N[C@@H](CO)c1nc(on1)[C@@H](NC(=O)N[C@H](C(=O)O)C(C)O)CC(N)=O
![(2S)-2-[[(1S)-3-Amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]carbamoylamino]-3-hydroxybutanoic acid.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=117943691&t=l)

(2S)-2-[[(1S)-3-amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]carbamoylamino]-3-hydroxybutanoic acid
CAS 2305027-62-5
C12 H20 N6 O7, 360.32Threonine, N-[[[(1S)-3-amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]amino]carbonyl]-, (2S,3ξ)-N[C@@H](CO)c1nc(on1)[C@@H](NC(=O)N[C@H](C(=O)O)C(C)O)CC(N)=O
ALSO SEE


![(2S,3R)-2-[[(1S)-3-Amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]carbamoylamino]-3-hydroxybutanoic acid.png](http://drugapprovalsint.com/wp-content/uploads/2021/10/str1-10.jpg)
1673534-76-3C12 H20 N6 O7, 360.32
L-Threonine, N-[[[(1S)-3-amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]amino]
(2S,3R)-2-[[(1S)-3-amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]carbamoylamino]-3-hydroxybutanoic acidN-[[[(1S)-3-Amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]amino]carbonyl]-L-threonine
CAS 1673534-76-3
PD-1-IN-1 free base, EX-A1918, CS-6240, NSC-799645, CA-170 (AUPM-170)|PDL1 inhibitor, HY-101093, PD-1-IN-1
N[C@@H](CO)c1nc(on1)[C@@H](NC(=O)N[C@H](C(=O)O)[C@@H](C)O)CC(N)=O
XL 114, AUR 104
A novel covalent inhibitor of FABP5 for cancer therapy
XL 102, AUR 102
A potent, selective and orally bioavailable inhibitor of cyclin-dependent kinase 7 (CDK7)
NO CONCLUSIONS, ONLY PREDICTIONS
PREDICTIONS MORE
![(2R,3R)-2-[[(1S)-3-Amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]carbamoylamino]-3-hydroxybutanoic acid.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=155529077&t=l)

(2R,3R)-2-[[(1S)-3-amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]carbamoylamino]-3-hydroxybutanoic acid
C12H20N6O7, 360.32
![(2S,3S)-2-[[(1S)-3-Amino-1-[3-[(1S)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]carbamoylamino]-3-hydroxybutanoic acid.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=155339943&t=l)

(2S,3S)-2-[[(1S)-3-amino-1-[3-[(1S)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]carbamoylamino]-3-hydroxybutanoic acid
XL102, AUR 102
XL102 is a potent, selective and orally bioavailable covalent inhibitor of CDK7, which is an important regulator of the cellular transcriptional and cell cycle machinery. CDK7 helps regulate cell cycle progression, with overexpression observed in multiple cancers, such as breast, prostate and ovarian cancers. In preclinical studies, XL102 revealed potent anti-proliferative activity, induced cell death in a large panel of cancer cell lines and caused tumor growth inhibition and regression in xenograft models, demonstrating its potential as a targeted antitumor agent.
In late 2020, Exelixis exercised its option to in-license XL102 (formerly AUR102) from Aurigene per the companies’ July 2019 collaboration, option and license agreement. Exelixis has assumed responsibility for the future clinical development, manufacturing and commercialization of XL102. Aurigene retains limited development and commercial rights for India and Russia.
SYN

ABOUT Fatty acid-binding proteins (FABPs)
Fatty acid-binding proteins (FABPs) are involved in binding and storing hydrophobic ligands such as long-chain fatty acids, as well as transporting them to the appropriate compartments in the cell. Epidermal fatty acid-binding protein (FABP5) is an intracellular lipid-binding protein that is abundantly expressed in adipocytes and macrophages. Previous studies have revealed that the FABP5 expression level is closely related to malignancy in various types of cancer. However, its precise functions in the metabolisms of cancer cells remain unclear. Here, we revealed that FABP5 knockdown significantly induced downregulation of the genes expression, such as hormone-sensitive lipase (HSL), monoacylglycerol lipase (MAGL), elongation of long-chain fatty acid member 6 (Elovl6), and acyl-CoA synthetase long-chain family member 1 (ACSL1), which are involved in altered lipid metabolism, lipolysis, and de novo FA synthesis in highly aggressive prostate and breast cancer cells. Moreover, we demonstrated that FABP5 induced inflammation and cytokine production through the nuclear factor-kappa B signaling pathway activated by reactive oxygen species and protein kinase C in PC-3 and MDA-MB-231 cells. Thus, FABP5 might regulate lipid quality and/or quantity to promote aggressiveness such as cell growth, invasiveness, survival, and inflammation in prostate and breast cancer cells. In the present study, we have revealed for the first time that high expression of FABP5 plays a critical role in alterations of lipid metabolism, leading to cancer development and metastasis in highly aggressive prostate and breast cancer cells.
Fatty acid-binding protein, epidermal is a protein that in humans is encoded by the FABP5 gene
Function
This gene encodes the fatty acid binding protein found in epidermal cells, and was first identified as being upregulated in psoriasis tissue. Fatty acid binding proteins are a family of small, highly conserved, cytoplasmic proteins that bind long-chain fatty acids and other hydrophobic ligands. It is thought that FABPs roles include fatty acid uptake, transport, and metabolism.[6]
The phytocannabinoids (THC and CBD) inhibit endocannabinoid anandamide (AEA) uptake by targeting FABP5, and competition for FABPs may in part or wholly explain the increased circulating levels of endocannabinoids reported after consumption of cannabinoids.[7] Results show that cannabinoids inhibit keratinocyte proliferation, and therefore support a potential role for cannabinoids in the treatment of psoriasis.[8]
Interactions
FABP5 has been shown to interact with S100A7.[
ABOUT CD47/SIRPa axis
CD47/SIRPa axis is established as a critical regulator of myeloid cell activation and serves as an immune checkpoint for macrophage mediated phagocytosis. Because of its frequent upregulation in several cancers, CD47 contributes to immune evasion and cancer progression. CD47 regulates phagocytosis primarily through interactions with SIRPla expressed on macrophages. Blockade of SIRPla/CD47 has been shown to dramatically enhance tumor cell phagocytosis and dendritic cells maturation for better antigen presentation leading to substantially improved antitumor responses in preclinical models of cancer (M. P. Chao et al. Curr Opin Immunol. 2012 (2): 225-232). Disruption of CD47-SIRPa interaction is now being evaluated as a therapeutic strategy for cancer with the use of monoclonal antibodies targeting CD47 or SIRPa and engineered receptor decoys.
CD47 is expressed on virtually all non-malignant cells, and blocking the CD47 or the loss of CD47 expression or changes in membrane distribution can serve as markers of aged or damaged cells, particularly on red blood cells (RBC). Alternatively, blocking SIRPa also allows engulfment of targets that are not normally phagocytosed, for those cells where pre-phagocytic signals are also present. CD47 is a broadly expressed transmembrane glycoprotein with a single Ig-like domain and five membrane- spanning regions, which functions as a cellular ligand for SIRPa with binding mediated through the NH2-terminal V-like domain of SIRPa. SIRPa is expressed primarily on myeloid cells, including macrophages, granulocytes, myeloid dendritic cells (DCs), mast cells, and their precursors, including hematopoietic stem cells.
CD47 is also constitutively upregulated on a number of cancers such as Non-Hodgkin Lymphoma (NHL), Acute myeloid leukemia (AML), breast, colon, glioblastoma, glioma, ovarian, bladder and prostate cancers, etc. Overexpression of CD47 by tumor cells, which efficiently helps them to escape immune surveillance and killing by innate immune cells. However, in most of the tumor types, blockade of the CD47-SIRPa interaction as a single agent may not be capable of inducing significant phagocytosis and antitumor immunity, necessitating the need to combine with other therapeutic agents. The concomitant engagement of activating receptors such as Fc-receptors (FcRs) or other prophagocytic receptors (collectively known as “eat-me” signals) may be necessary for exploiting the maximum potential of the CD-47-SIPRa pathway blockade.
The role of engagement of prophagocytic receptors is proved by inefficiency to trigger phagocytosis either by anti-CD47 F(ab) fragments, single chain variable fragments of CD-47 or non-Fc portion- containing SIRPa proteins in blocking of the CD47-SIRPa interaction. When activating prophagocytic receptors are engaged, as evident in the case of using Fc portion-containing blocking anti-CD47 antibodies, CD47- SIRPa blockade is able to trigger more efficient phagocytosis. Combining CD47-SIRPa blocking agents with therapeutic antibodies (Fc-containing) targeting tumor antigens stimulate activating Fc receptors (FcRs) leading to efficient phagocytosis. The Fc portion of therapeutic antibody targeting tumor antigen also induces antibody-dependent cellular cytotoxicity (ADCC), which also adds to the therapeutic efficacy. Hence antibodies selected from the group consisting of rituximab, herceptin, trastuzumab, alemtuzumab, bevacizumab, cetuximab and panitumumab, daratumumab due to its tumor targeting nature and ADCC, can trigger more efficient phagocytosis.
Earlier approaches to disrupt CD47- SIRPa interaction utilized monoclonal antibodies targeting CD47 or SIRPa and engineered receptor decoys fused to Fc fragment. However, a concern with this approach is that CD47 is highly expressed on both hematopoietic and non-hematopoietic normal cells. Hence along with tumor cells CD47-SIRPa blocking agents containing Fc-portion may also target many normal cells potentially leading to their elimination by macrophages. The interaction of blocking antibodies with normal cells is considered as a major safety issue resulting in anemia, thrombocytopenia, and leukopenia. These agents may also affect solid tissues rich in macrophages such as liver, lung, and brain. Hence it may be ideal to block the CD47- SIRPa interaction by agents devoid of Fc portion, such as small
molecules, peptides, Fab fragments etc. while activating prophagocytic receptors in tumor cells by appropriate combinations to induce efficient phagocytosis of tumor cells.
Apart from Fc Receptors, a number of other prophagocytic receptors are also reported to promote engulfment of tumor cells in response to CD47-SIRPa blockade by triggering the phagocytosis. These include receptors for SLAMF7, Mac-l, calreticulin and possibly yet to identified receptors. B cell tumor lines such as Raji and other diffuse large B cell lymphoma express SLAMF7 and are implicated in triggering prophagocytic signals during CD47-SIRPa blockade.
Therapeutic agents known to activate prophagocytic receptors are also therefore ideal partners for use in combination with CD47-SIRPa blocking agents to achieve efficient phagocytosis. These agents include proteasome inhibitors (bortezomib, ixazomib and carfilzomib), Anthracyclines (Doxorubicin, Epirubicin, Daunorubicin, Idarubicin, Mitoxantrone) Oxaliplatin, Cyclophosphamide, Bleomycin, Vorinostat, Paclitaxel, 5-Fluorouracil, Cytarabine, BRAF inhibitory drugs (Dabrafenib, Vemurafenib), PI3K inhibitor, Docetaxel, Mitomycin C, Sorafenib, Tamoxifen and oncolytic viruses.
Apart from the specific agents known to have effect on‘eat me’ signals other agents including Abiraterone acetate, Afatinib, Aldesleukin, Aldesleukin, Alemtuzumab, Anastrozole, Axitinib, Belinostat, Bendamustine, Bicalutamide, Blinatumomab, Bosutinib, Brentuximab, Busulfan, Cabazitaxel, Capecitabine, Carboplatin, Carfilzomib, Carmustine, Ceritinib, Clofarabine, Crizotinib, Dacarbazine, Dactinomycin, Dasatinib, Degarelix, Denileukin, Denosumab, Enzalutamide, Eribulin, Erlotinib, Everolimus, Exemestane, Exemestane, Fludarabine, Fulvestrant, Gefitinib, Goserelin, Ibritumomab, Imatinib, Ipilimumab, Irinotecan, Ixabepilone, Lapatinib, Lenalidomide, Letrozole, Leucovorin, Leuprolide, Lomustine, Mechlorethamine, Megestrol, Nelarabine, Nilotinib, Nivolumab, Olaparib, Omacetaxine, Palbociclib, Pamidronate, Panitumumab, Panobinostat, Pazopanib, Pegaspargase, Pembrolizumab, Pemetrexed Disodium, Pertuzumab, Plerixafor, Pomalidomide, Ponatinib, Pralatrexate, Procarbazine, Radium 223, Ramucirumab, Regorafenib, rIFNa-2b, Romidepsin, Sunitinib, Temozolomide, Temsirolimus, Thiotepa, Tositumomab, Trametinib, Vinorelbine, Methotrexate, Ibrutinib, Aflibercept, Toremifene, Vinblastine, Vincristine, Idelalisib, Mercaptopurine and Thalidomide could potentially have effect on‘eat me’ signal pathway on combining with CD-47-SIRPa blocking agents.
In addition to the therapeutic agents mentioned above, other treatment modalities that are in use in cancer therapy also activate prophagocytic receptors, and thus can be combined with CD47-SIRPa blocking agents to achieve efficient phagocytosis. These include Hypericin-based photodynamic therapy (Hyp-PDT), radiotherapy, High-hydrostatic pressure, Photofrin-based PDT and Rose Bengal acetate -based PDT.
However, there is an unmet need for combining small molecule CD-47-SIRPa pathway inhibitors with agents capable of stimulating activating receptors such as Fc-receptors (FcRs) or other prophagocytic receptors, or combining with other treatment modalities that are in use in cancer therapy to activate prophagocytic receptors for exploiting the maximum potential of the CD-47- SIRPa pathway blockade.
CLIP
Exelixis In-Licenses Second Anti-Cancer Compound from Aurigene Following FDA Acceptance of Investigational New Drug Application for Phase 1 Clinical Trial in Non-Hodgkin’s Lymphoma
– Robust preclinical data support Exelixis’ clinical development of XL114, with phase 1 trial in Non-Hodgkin’s lymphoma expected to begin in the coming months –
– Exelixis will make an option exercise payment of $10 million to Aurigene –
https://www.businesswire.com/news/home/20211014005549/en/Exelixis-In-Licenses-Second-Anti-Cancer-Compound-from-Aurigene-Following-FDA-Acceptance-of-Investigational-New-Drug-Application-for-Phase-1-Clinical-Trial-in-Non-Hodgkin%E2%80%99s-LymphomaOctober 14, 2021 08:00 AM Eastern Daylight Time
ALAMEDA, Calif.–(BUSINESS WIRE)–Exelixis, Inc. (Nasdaq: EXEL) and Aurigene Discovery Technologies Limited (Aurigene) today announced that Exelixis has exercised its exclusive option under the companies’ July 2019 agreement to in-license XL114 (formerly AUR104), a novel anti-cancer compound that inhibits the CARD11-BCL10-MALT1 (CBM) signaling pathway, which promotes lymphocyte survival and proliferation. Exelixis has now assumed responsibility for the future clinical development, commercialization and global manufacturing of XL114. Following the U.S. Food and Drug Administration’s (FDA) recent acceptance of its Investigational New Drug (IND) application, Exelixis will soon initiate a phase 1 clinical trial evaluating XL114 monotherapy in patients with Non-Hodgkin’s lymphoma (NHL). At the American Association of Cancer Research Annual Meeting in April of this year, Aurigene presented preclinical data (Abstract 1266) demonstrating that XL114 exhibited potent anti-proliferative activity in a large panel of cancer cell lines ranging from hematological cancers to solid tumors with excellent selectivity over normal cells. In addition, oral dosing of XL114 resulted in significant dose-dependent tumor growth inhibition in diffuse large B-cell lymphoma (DLBCL) and colon carcinoma models.
“We are pleased that our agreement with Aurigene has generated a second promising compound that warrants advancement into clinical development and believe the collaboration will continue to play an important role in expanding our pipeline”
XL114 is the second molecule that Exelixis in-licensed from Aurigene under the companies’ July 2019 collaboration, option and license agreement. Exelixis previously exercised its option to in-license XL102, a potent, selective and orally bioavailable inhibitor of cyclin-dependent kinase 7 (CDK7), from Aurigene in December 2020 and initiated a phase 1 trial of XL102 as a single agent and in combination with other anti-cancer agents in patients with advanced or metastatic solid tumors in January 2021.
“We are pleased that our agreement with Aurigene has generated a second promising compound that warrants advancement into clinical development and believe the collaboration will continue to play an important role in expanding our pipeline,” said Peter Lamb, Ph.D., Executive Vice President, Scientific Strategy and Chief Scientific Officer, Exelixis. “XL114 has shown potent anti-proliferative activity in lymphoma cell lines that have aberrant activation of the CBM signaling pathway and may have a differentiated profile and potential as a best-in-class molecule that could improve outcomes for patients with Non-Hodgkin’s lymphoma and other hematologic cancers.”
XL114 was identified to have anti-proliferative activity in cell lines with constitutive activation of CBM signaling, including activated B-cell-like DLBCL (ABC-DLBCL), mantle cell lymphoma and follicular lymphoma cell lines. Further characterization of XL114 in cell-based assays demonstrated a functional role in B-cell (BCR) signaling pathways. Additionally, XL114 showed dose-dependent tumor growth inhibition in an ABC-DLBCL mouse xenograft tumor model. In preclinical development, XL114 also demonstrated a high degree of selectivity against a broad safety pharmacology panel of enzymes and receptors. While the precise molecular mechanism underlying XL114’s function in repressing BCR signaling and MALT1 activation has yet to be characterized, the fatty acid-binding protein 5 (FABP5) has been identified as a prominent XL114-binding target.
“XL114 is the second molecule that Exelixis has opted to in-license under our July 2019 agreement, underscoring the significant potential of our approach to the discovery and preclinical development of innovative cancer therapies that target novel mechanisms of action,” said Murali Ramachandra, Ph.D., Chief Executive Officer, Aurigene. “Exelixis has a track record of success in the clinical development and commercialization of anti-cancer therapies that provide patients with important new treatment options, and we are pleased that the continued advancement of XL114 will be supported by the company’s extensive clinical, regulatory and commercialization infrastructure.”
Under the terms of the July 2019 agreement, Exelixis made an upfront payment of $10 million for exclusive options to obtain an exclusive license from Aurigene to three preexisting programs, including the compounds now known as XL102 and XL114. In addition, Exelixis and Aurigene initiated three Aurigene-led drug discovery programs on mutually agreed upon targets, in exchange for an additional upfront payment of $2.5 million per program. The collaboration was expanded in 2021 to include three additional early discovery programs. Exelixis is also contributing research funding to Aurigene to facilitate discovery and preclinical development work on all nine programs. Exelixis may exercise its option for a program at any time up until the first IND for the program becomes effective. Having exercised options on two programs thus far (XL102 and XL114), if and when Exelixis exercises a future option, it will make an option exercise payment to Aurigene and assume responsibility for that program’s future clinical development and commercialization including global manufacturing. To exercise its option for XL114, Exelixis will make an option exercise payment to Aurigene of $10 million. Once Exelixis exercises its option for a program, Aurigene will be eligible for clinical development, regulatory and sales milestones, as well as royalties on future potential sales of the compound. Under the terms of the agreement, Aurigene retains limited development and commercial rights for India and Russia.
About Aurigene
Aurigene Discovery Technologies Limited is a development stage biotech company engaged in discovery and clinical development of novel and best-in-class therapies to treat cancer and inflammatory diseases and a wholly owned subsidiary of Dr. Reddy’s Laboratories Ltd. (BSE: 500124, NSE: DRREDDY, NYSE: RDY, NSEIFSC: DRREDDY). Aurigene is focused on precision-oncology, oral immune checkpoint inhibitors, and the Th-17 pathway. Aurigene’s programs currently in clinical development include an oral ROR-gamma inhibitor AUR101 for moderate to severe psoriasis in phase 2 under a U.S. FDA IND and a PD-L1/VISTA antagonist CA-170 for non-squamous non-small cell lung cancer in phase 2b/3 in India. Additionally, Aurigene has multiple compounds at different stages of pre-clinical development. Aurigene has also partnered with several large and mid-pharma companies in the U.S. and Europe and has multiple programs in clinical development. For more information, please visit Aurigene’s website at www.aurigene.com.
About Exelixis
Founded in 1994, Exelixis, Inc. (Nasdaq: EXEL) is a commercially successful, oncology-focused biotechnology company that strives to accelerate the discovery, development and commercialization of new medicines for difficult-to-treat cancers. Following early work in model system genetics, we established a broad drug discovery and development platform that has served as the foundation for our continued efforts to bring new cancer therapies to patients in need. Our discovery efforts have resulted in four commercially available products, CABOMETYX® (cabozantinib), COMETRIQ® (cabozantinib), COTELLIC® (cobimetinib) and MINNEBRO® (esaxerenone), and we have entered into partnerships with leading pharmaceutical companies to bring these important medicines to patients worldwide. Supported by revenues from our marketed products and collaborations, we are committed to prudently reinvesting in our business to maximize the potential of our pipeline. We are supplementing our existing therapeutic assets with targeted business development activities and internal drug discovery – all to deliver the next generation of Exelixis medicines and help patients recover stronger and live longer. Exelixis is a member of the Standard & Poor’s (S&P) MidCap 400 index, which measures the performance of profitable mid-sized companies. In November 2020, the company was named to Fortune’s 100 Fastest-Growing Companies list for the first time, ranking 17th overall and the third-highest biopharmaceutical company. For more information about Exelixis, please visit www.exelixis.com, follow @ExelixisInc on Twitter or like Exelixis, Inc. on Facebook.

Dinesh Chikkanna
Director, Medicinal Chemistry Aurigene Discovery Technologies

Murali Ramachandra
CEO at Aurigene Discovery Technologies

CLIP
https://cancerres.aacrjournals.org/content/81/13_Supplement/1266
Abstract 1266: Discovery and preclinical evaluation of a novel covalent inhibitor of FABP5 for cancer therapyDinesh Chikkanna, Leena Khare Satyam, Sunil Kumar Pnaigrahi, Vinayak Khairnar, Manoj Pothuganti, Lakshmi Narayan Kaza, Narasimha Raju Kalidindi, Vijaya Shankar Nataraj, Aditya Kiran Gatta, Narasimha Rao Krishnamurthy, Sandeep Patil, DS Samiulla, Kiran Aithal, Vijay Kamal Ahuja, Nirbhay Kumar Tiwari, KB Charamannna, Pravin Pise, Thomas Anthony, Kavitha Nellore, Sanjeev Giri, Shekar Chelur, Susanta Samajdar and Murali Ramachandra
DOI: 10.1158/1538-7445.AM2021-1266 Published July 2021
Proceedings: AACR Annual Meeting 2021; April 10-15, 2021 and May 17-21, 2021; Philadelphia, PA
Abstract
Dysregulated fatty acid metabolism is thought to be a hallmark of cancer, wherein fatty acids function both as an energy source and as signals for enzymatic and transcriptional networks contributing to malignancy. Fatty acid-binding protein 5 (FABP5) is an intracellular protein that facilitates transport of fatty acids and plays a role in regulating the expression of genes associated with cancer progression such as cell growth, survival, and metastasis. Overexpression of FABP5 has been reported to contribute to an aggressive phenotype and a poor survival correlation in several cancers. Therefore, inhibition of FABP5 is considered as a therapeutic approach for cancers. Phenotypic screening of a library of covalent compounds for selective sensitivity of cancer cells followed by medicinal chemistry optimization resulted in the identification of AUR104 with desirable properties. Chemoproteomic-based target deconvolution revealed FABP5 as the cellular target of AUR104. Covalent adduct formation with Cys43 of FABP5 by AUR104 was confirmed by mass spectrometry. Target occupancy studies using a biotin-tagged AUR104 demonstrated potent covalent binding to FABP5 in both cell-free and cellular conditions. Ligand displacement assay with a fluorescent fatty acid probe confirmed the competitive binding mode of AUR104 with fatty acids. Binding at the fatty acid site and covalent bond formation with Cys43 were also demonstrated by crystallography. Furthermore, AUR104 showed a high degree of selectivity against a broad safety pharmacology panel of enzymes and receptors. AUR104 exhibited potent anti-proliferative activity in a large panel of cell lines derived from both hematological and solid cancers with a high degree of selectivity over normal cells. Anti-proliferative activity in lymphoma cell lines correlated with inhibition of MALT1 pathway activity, cleavage of RelB/Bcl10 and secretion of cytokines, IL-10 and IL-6. AUR104 displayed desirable drug-like properties and dose-dependent oral exposure in pharmacokinetic studies. Oral dosing with AUR104 resulted in dose-dependent anti-tumor activity in DLBCL (OCI-LY10) and NSCLC (NCI-H1975) xenograft models. In a repeated dose MTD studies in rodents and non-rodents, AUR104 showed good tolerability with an exposure multiple of >500 over cellular EC50 for up to 8 hours. In summary, we have identified a novel covalent FABP5 inhibitor with optimized properties that showed anti-tumor activity in in vitro and in vivo models with acceptable safety profile. The data presented here strongly support clinical development of AUR104.
Citation Format: Dinesh Chikkanna, Leena Khare Satyam, Sunil Kumar Pnaigrahi, Vinayak Khairnar, Manoj Pothuganti, Lakshmi Narayan Kaza, Narasimha Raju Kalidindi, Vijaya Shankar Nataraj, Aditya Kiran Gatta, Narasimha Rao Krishnamurthy, Sandeep Patil, DS Samiulla, Kiran Aithal, Vijay Kamal Ahuja, Nirbhay Kumar Tiwari, KB Charamannna, Pravin Pise, Thomas Anthony, Kavitha Nellore, Sanjeev Giri, Shekar Chelur, Susanta Samajdar, Murali Ramachandra. Discovery and preclinical evaluation of a novel covalent inhibitor of FABP5 for cancer therapy [abstract]. In: Proceedings of the American Association for Cancer Research Annual Meeting 2021; 2021 Apr 10-15 and May 17-21. Philadelphia (PA): AACR; Cancer Res 2021;81(13_Suppl):Abstract nr 1266.
Patent
US20200147054 – COMBINATION OF SMALL MOLECULE CD-47 INHIBITORS WITH OTHER ANTI-CANCER AGENTS
Muralidhara Ramachandra
Pottayil Govindan Nair Sasikumar
Girish Chandrappa Daginakatte
Kiran Aithal Balkudru
PATENT
WO 2020095256
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020095256
Example- 1: The synthetic procedures for the preparation of compounds described in the present invention were described in co-pending Indian provisional patent application 201841001438 dated 12* Jan 2018, which is converted as PCT application
PCT/IB2019/050219, the contents of which are hereby incorporated by reference in their entirety.
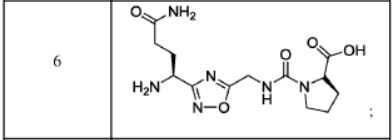
PATENT
WO 2018178947https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018178947&tab=PCTDESCRIPTION
PATENT
WO 2019138367
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019138367
PATENT
WO 2019073399
https://patents.google.com/patent/WO2019073399A1/en
Priority to IN201741036169
Example 4 of WO 2015/033299
PATENT
https://patents.google.com/patent/BR112020014202A2/en
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
PATENT
The present invention relates to substituted alkynylene compounds represented by compound of formula (I) pharmaceutically acceptable salts and stereoisomers thereof. The present invention further provides the methods of preparation of compound of formula (I) and therapeutic uses thereof as anti-cancer agents.
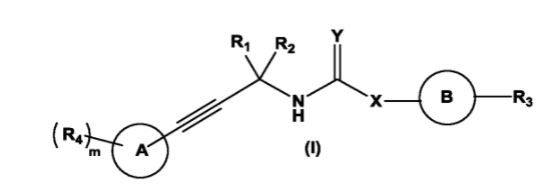
Patent
Example 1
(((S)-4-amino-1-(3-((S)-1,5-diaminopentyl)-1,2,4-oxadiazol-5-yl)-4-oxobutyl)carbamoyl)-L-proline (Compound 1)
Synthesis of Compound 1 b
Synthesis of Compound 1C
Synthesis of Compound 1d
Synthesis of Compound 1f
Synthesis of Compound 1g
Synthesis of Compound 1h
Synthesis of Compound 1i
Synthesis of compound 1j
Synthesis of Compound 1
Example 2
(S)-4-(3-((S)-1-amino-4-guanidinobutyl)-1,2,4-oxadiazol-5-yl)-4-(3-((S)-1-carboxy-2-phenylethyl) ureido)butanoic acid (Compound 7)
Synthesis of Compound 2b
Synthesis of Compound 2c
Synthesis of Compound 2d
Synthesis of Compound 2f
Synthesis of Compound 2g
Synthesis of Compound 2h
Synthesis of Compound 2i
Synthesis of Compound 2j
Synthesis of Compound 7
PATENT
WO 2015/033299
https://patents.google.com/patent/WO2015033299A1/en?oq=WO+2015%2f033299
Pottayil Govindan Nair SasikumarMuralidhara RamachandraSeetharamaiah Setty Sudarshan Naremaddepalli
Example 1: Synthesis of Compound 1

Step la:

Ethylchloroformate (1.5 g, 13.78 mniol) and N-Methylmorpholine ( 1.4 g, 13.78 mmol) were added to a solution of compound la (3 g, 11.48 mmol) in THE (30 mL) arid stirred at -20 °C. After 20 min. Liquid ammonia (0.77 g, 45.92 mmol) was added to the active mixed anhydride formed in- situ and stirred at 0-5 °C for 20 min. The completeness of the reaction was confirmed by TLC analysis. The reaction mixture was evaporated under reduced pressure and partitioned between water and ethyl acetate. Organic layer was washed with NaHCOs, citric acid, brine solution, dried over Na2S04 and evaporated under reduced pressure to get 2.9 g of compound lb (Yield: 96.3%). LCMS: 261.0 ( Vi+H ; .
Step lb:

1 b 1cTrifluroacetic anhydride (9.7 g, 46.0 mmol) was added to a solution of compound lb (8 g, 30.7 mmol) in pyridine (24.3 g, 307.0 mmol) and stirred at room temperature for 3 h. The completeness of the reaction was confirmed by TLC analysis. The reaction mixture was evaporated under reduced pressure and partitioned between water and ethyl acetate. Organic layer was washed with NaHCO?,, citric acid, brine solution, dried over Na2-S04 and evaporated under reduced pressure to afford 7 g of compound lc (Yield: 94.0%). LCMS: 187.2 (M-¾u )+.
Step lc:

1 c 1dHydroxylamine hydrochloride (3 g, 43.37 mmol) and potassium carbonate (6 g, 43.37 mmol) were added to a solution of compound lc (7 g, 28.91 mmol) in EtOH (70 m L) and stirred at 90 °C for 2 h. The completeness of the reaction was confirmed by TLC analysis. The reaction mixture was evaporated under reduced pressure and partitioned between water and ethyl acetate. Organic layer was washed with brine solution, dried over Na2S04 and evaporated under reduced pressure. The crude compound was purified by silica gel column chromatography (Eluent: 0-5% ethyl acetate in hexane) to get 4.2 g of compound Id (Yield: 52.8%). LCMS: 276.4 (M+H)+.Step Id:

Deoxo-Fluor® (1.83 g, 8.3 mmol) was added to a solution of Fmoc-Asn(Trt)-OH (4.5 g, 7.5 mmol) in CH2Q2 (50 mL) and stirred at 0 °C for 3 h. Then CH2CI2 was evaporated and triturated with hexane, decanted and evaporated under vacuum to get the corresponding acid fluoride. NMM (1.17 g, 1 1.6 mmol) and compound Id (1.6 g, 5.8 mmol) in THF were added to the acid fluoride and stirred at room temperature for 12 h. Then THF was evaporated and sodium acetate (0.72 g, 8.7 mmol) was added followed by EtOH (50 mL). The reaction mixture was stirred at 90 °C for 2 h. The completeness of the reaction was confirmed by TLC analysis. The reaction mixture was evaporated under reduced pressure and partitioned between water and ethyl acetate. Organic layer was washed with NaHCOa, citric acid, brine solution, dried over Na2S04 and evaporated under reduced pressure, which was further purified by silica gel column chromatography (Eluent: 0-5% ethyl acetate in hexane) to afford 2.8 g of compound le (Yield: 44.4%). LCMS: 836.4 (M+Hf .Step le:
Ph3

To compound le (2.3 g, 2.7 mmol) in CH2CI2 (10 mL) diethyiarnine (10 mL) was added and the reaction mixture was stirred at room temperature for 30 min. The resulting solution was concentrated in vacuum to get gummy residue. The crude compound was purified by neutral alumina column chromatography (Eluent: 0-50% ethyl acetate in hexane then 0-5% methanol in chloroform) to get 1.4 g of If (Yield: 90 %). LCMS: 636.5 (M+Na)+.

1f 1To a solution of compound If (0.45 g) in CH2CI2 (5 mL), trifluoroacetic acid (5 mL) and catalytic amount of triisopropylsilane were added and stirred for 3 h at room temperature to remove the acid sensitive protecting groups. The resulting solution was concentrated in vacuum to afford 0.29 g of crude compound 1 which was purified using prep-HPLC method described under experimental conditions. \H NMR (DMSQ-d6, 400 MHz): δ 2.58 (m, 2H), 3.53 (m, 3H), 3.91 (t, 1H), 4.36 (t, 1H), 6.91 (s, 1H), 7.45 (s, 1H); 1 C NMR (DMSO-de, 400 MHz): δ 20.85, 45.71 , 50.23, 65.55, 171.03, 171 .41, 181.66. LCMS: 216.2 (Μ+ΗΓ; HPLC: tR = 13.1 min.Example 2: Synthesis of Co

Step 2a:

1f2a
The urea linkage was carried out by the coupling compound If (2.7 g, 4.39 mmoi) in THF (30 mL) at room temperature with compound 2b (1.67 g, 4.39 mmoi). The coupling was initiated by the addition of TEA (0.9 g, 8.78 mmoi) in THF (10 m L) and the resultant mixture was stirred at room temperature. After completion of 20 h, THF was evaporated from the reaction mass, and partitioned between water and ethyl acetate. Organic layer was washed with water, brine, dried over Na2S04 and evaporated under reduced pressure to get compound 2a, which was further purified by silica gel column chromatography (Fluent: 0-50% ethyl acetate in hexane) to afford 3.46 g of compound 2a (Yield: 92.10%). LCMS 857.4 (M+H)+.

2aTo a solution of compound 2a (0.22 g, 0.25 mmol) in 0¾ί¾ (5 m L), trifluoroaeetic acid (5 mL) and catalytic amount of triisopropyisilane were added and stirred for 3h at room, temperature. The resulting solution was concentrated under reduced pressure to obtain 0.35 g of crude compound. The crude solid material was purified using preparative- HPLC method described under experimental conditions. LCMS: 347.1 (M+H)+; HPLC: tR = 12.9 min.
Synthesis of

2bTo the compound H-Ser(tBu)-OiBu (2 g, 9.2 mmol) in C I I■■(.‘{■ (20 mL), triethylamine (1.39 g, 13.8 mmol) was added and the solution was stirred at room temperature for 5-10 min. To this mixture, solution of 4-Nitrophenyl chioro formate (2.22 g, 11.04 mmol) in CH2CI2 was added and the resultant mixture was stirred at room temperature for 30 min. The completion of the reaction was confirmed by TLC analysis. After completion of reaction, reaction mixture was diluted with CH2CI2 and washed with water and 5.0 M citric acid solution, dried over Na2SC>4 and evaporated under reduced pressure to get crude compound 2b, which was further purified by silica gel column chromatography (Eiuent: 0-20% ethyl acetate in hexane) to yield 2.1 g (58.9%) of 2b.Example 3: Synthesis of Compound 3

The compound was synthesised using similar procedure as depicted in Example 1 (compound 1) and D-amino acids are linked up in reverse order. Boc-D-Thr(‘Bu)-OH was used in place of Boc-Ser(‘Bu)-OH (compound la, Example 1) and Fmoc-D- Asn(trt)-OH in place of Fmoc-Asn(trt)-OH to yield 0.15 g crude material of the title compound 3. LCMS: 230.1 (M+H)+.Example 4: Synthesis of Co
The compound was synthesised using similar procedure as depicted in Example 2 for synthesising compound 2 using

instead of H-Ser(‘Bu)-0’Bu (in synthesis of compound 2b) to yield 0.35 g crude material of the title compound. The crude solid material was purified using preparative HPLC described under experimental conditions. LCMS: 361.2 (M+H)+, HPLC: tR = 12.19 min.Example 5: Synthesis of

The compound was synthesised using similar procedure as depicted in Example 4 (compound 4) using D-amino acids are linked up in reverse order. Boc-D-Thr(‘Bu)-OH was used in place of Boc-Ser(‘Bu)-OH, Fmoc-D-Asn(trt)-OH in place of Fmoc-Asn(trt)- OH and H-D-Ser(‘Bu)-0’Bu was used in place of H-Thr^Bu^O’Bu to yield 0.3 g crude material of the title compound. The cmde solid material was purified using preparative HPLC described under experimental conditions. LCMS: 361.3 (M+H)+. HPLC: tR = 13.58 min.Example 6: Synthesis of Compound 6

The compound was synthesised using similar procedure as depicted in Example 2 by using H-Thr(‘Bu)-OMe instead of H-Ser(‘Bu)-0’Bu (in synthesis of compound 2b) to yield 0.2 g crude material of the title compound. The crude solid material was purified using preparative HPLC described under experimental conditions. LCMS: 375.1 (M+H)+, HPLC: tR = 1.84 min.Example 7: Synthesis of Compound 7

Step 7a:

1f7aThe compound 7a was synthesised using similar procedure as for compound 2a (Example 2, step 2a) using H-Thr(‘Bu)-OMe instead of H-Ser(‘Bu)-OtBu to get crude material which was further purified by silica gel column chromatography (Eluent: 0-50% ethyl acetate in he ane) to get 2.0 g of compound 7a (Yield: 74 %). LCMS: 829.2 (M+H)+.Step 7b:

7a 7bTo a solution of compound 7a (0.35 g, 4.0 mmol) in THF (5 mL) was added lithium hydroxide (0.026 g, 0.63 mmol) at 0 °C and the mixture was stirred for 2 h at room temperature. The completion of the reaction was confirmed by TLC analysis. THF was evaporated from the reaction mass, and partitioned between water and ethyl acetate. Organic layer was washed with citric acid, brine solution, dried over Na2S04 and evaporated under reduced pressure to afford 7b, which was further purified by silica gel column chromatography (Eluent: 0-5% methanol in DCM) to get 0.3 g of product 7b (Yield: 86.7%). LCMS 815.2 (M+H)+.
Step 7c:

7b 7Compound 7b (0.295 g, 0.39 mmol) was anchored to Rink amide resin (0.7 g, 0.55 mmol/g) using HOBT (0.072 g, 0.54 mmol) and DIC (0.068 g, 0.54 mmol) method in DMF (10 mL). The resin was stirred for 12 h at room temperature. The resin was washed with DCM, DMF and DCM and dried. The target compound was cleaved from the rink amide resin using TFA (5 mL) and catalytic amount of TIPS. The resin was allowed to remain at room temperature for 2 h with occasional stirring. After 2 h, TFA and TIPS were evaporated under nitrogen atmosphere and the resulting residue was washed with diethyl ether to yield 0.1 g crude material of the title compound 7. The crude solid material was purified using preparative HPLC described under experimental conditions. LCMS: 360.0 (M+H)+, HPLC: tR = 13.88 min.Example 8: Synthesis of

The compound was synthesised using similar procedure as depicted in Example 2 (compound 2) using Fmoc-Glu(0’Bu)-OH instead of Fmoc-Asn(Trt)-OH to get 0.4 g crude material of the title compound. The crude solid material was purified using preparative HPLC described under experimental conditions. LCMS: 362.1 (M+H)+. HPLC: tR = 13.27 min.
PATENThttps://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019061324&tab=FULLTEXT
Patenthttps://patents.google.com/patent/WO2019067678A1/enPATENThttps://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019061324
PATENThttps://patents.google.com/patent/WO2018073754A1/en
PATENThttps://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019087087
PAPERSScientific Reports (2019), 9(1), 1-19. https://www.nature.com/articles/s41598-019-48826-6
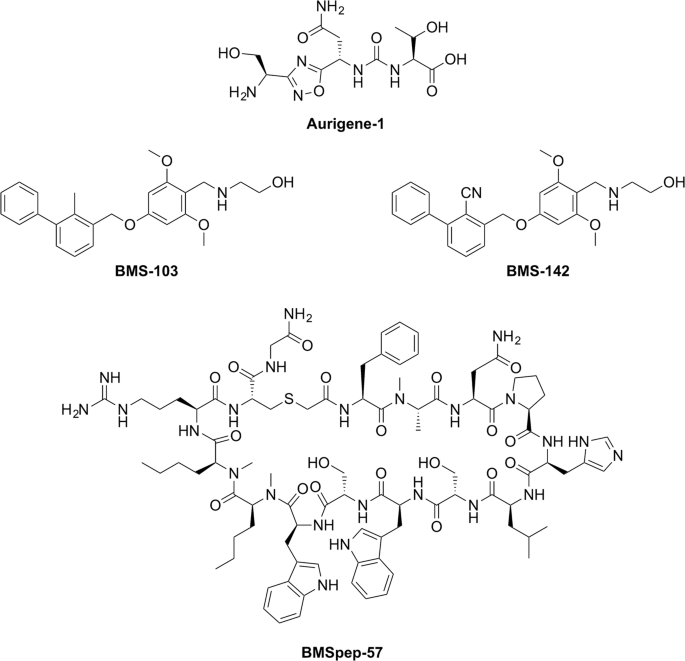
Chemical structures of PD-L1 inhibitors developed by Aurigene (Aurigene-1) and Bristol-Meyers Squibb (BMSpep-57, BMS-103, and BMS-142). Chemical structures were generated using ChemDraw Professional 15. PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019087087
L-threonine’ mentioned in compound of formula (I) thereof can be represented by any one of the following formulae:
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| US-2020289477-A1 | Conjoint therapies for immunomodulation | 2017-11-06 | |
| WO-2019073399-A1 | CRYSTALLINE FORMS OF 1,2,4-OXADIAZOLE SUBSTITUTED IN POSITION 3 | 2017-10-11 | |
| AU-2018341583-A1 | Crystal forms of immunomodulators | 2017-09-29 | |
| WO-2019061324-A1 | CRYSTALLINE FORMS OF IMMUNOMODULATORS | 2017-09-29 | |
| WO-2019067678-A1 | CRYSTALLINE FORMS OF IMMUNOMODULATORS | 2017-09-29 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| US-2020247766-A1 | Crystal forms of immunomodulators | 2017-09-29 | |
| US-2020061030-A1 | Dual inhibitors of vista and pd-1 pathways | 2016-10-20 | |
| WO-2018073754-A1 | Dual inhibitors of vista and pd-1 pathways | 2016-10-20 | |
| US-2020361880-A1 | 1,2,4-Oxadiazole and Thiadiazole Compounds as Immunomodulators | 2015-03-10 | |
| EP-3041827-B1 | 1,2,4-oxadiazole derivatives as immunomodulators | 2013-09-06 | 2018-04-18 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| EP-3363790-B1 | 1,2,4-oxadiazole derivatives as immunomodulators | 2013-09-06 | 2020-02-19 |
| US-10173989-B2 | 1,2,4-oxadiazole derivatives as immunomodulators | 2013-09-06 | 2019-01-08 |
| US-10590093-B2 | 1,2,4-oxadiazole derivatives as immunomodulators | 2013-09-06 | 2020-03-17 |
| US-2015073024-A1 | 1,2,4-Oxadiazole Derivatives as Immunomodulators | 2013-09-06 | |
| US-2017101386-A1 | 1,2,4-Oxadiazole Derivatives as Immunomodulators | 2013-09-06 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| US-2018072689-A1 | 1,2,4-Oxadiazole Derivatives as Immunomodulators | 2013-09-06 | |
| US-2019144402-A1 | 1,2,4-Oxadiazole Derivatives as Immunomodulators | 2013-09-06 | |
| US-2020199086-A1 | 1,2,4-Oxadiazole Derivatives as Immunomodulators | 2013-09-06 | |
| US-9771338-B2 | 1,2,4-oxadiazole derivatives as immunomodulators | 2013-09-06 | 2017-09-26 |
| WO-2015033299-A1 | 1,2,4-oxadiazole derivatives as immunomodulators | 2013-09-06 |
////////////Investigational New Drug Application, Phase 1, Clinical Trial, Non-Hodgkin’s Lymphoma, XL 114, AUR 104, aurigene, Exelixis
N[C@@H](CO)c1nc(on1)[C@@H](NC(=O)N[C@H](C(=O)O)C(C)O)CC(N)=O
https://patentscope.wipo.int/search/en/result.jsf?inchikey=HFOBENSCBRZVSP-WHFCDURNSA-N

NEW DRUG APPROVALS
ONE TIME
$10.00
PATENT


The present invention relates to substituted alkynylene compounds represented by compound of formula (I) pharmaceutically acceptable salts and stereoisomers thereof. The present invention further provides the methods of preparation of compound of formula (I) and therapeutic uses thereof as anti-cancer agents.
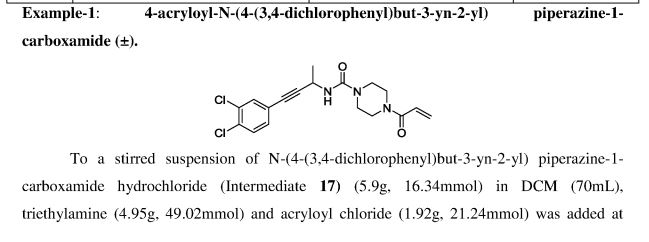
XL 102
EXELIXIS AND AURIGENE ANNOUNCE THAT PROMISING PRECLINICAL DATA TO BE PRESENTED AT THE ENA SYMPOSIUM SUPPORT THE CLINICAL DEVELOPMENT OF A NOVEL CDK7 INHIBITOR
Exelixis and Aurigene Announce That Promising Preclinical Data to Be Presented at the ENA Symposium Support the Clinical Development of a Novel CDK7 Inhibitor
– Detailed characterization of an oral inhibitor of CDK7 demonstrates potent activity against multiple hematologic and solid tumor cell lines, as monotherapy and in combination with chemotherapies –
October 09, 2020 03:02 AM Eastern Daylight Time
ALAMEDA, Calif.–(BUSINESS WIRE)–Exelixis, Inc. (Nasdaq: EXEL) and Aurigene Discovery Technologies Limited (Aurigene) today disclosed new preclinical data showing that AUR102 has potent anti-tumor activity in a large panel of cancer cell lines. AUR102 is a potent, selective, and orally bioavailable covalent inhibitor of cyclin-dependent kinase 7 (CDK7), which is an important regulator of the cellular transcriptional and cell cycle machinery. Exelixis has an exclusive option for AUR102 under its July 2019 exclusive collaboration, option and license agreement with Aurigene. The new data will be presented in a poster (Abstract 170) at the 32nd EORTC-NCI-AACR (ENA) Symposium, which is being held virtually on October 24-25, 2020.
“CDK7 plays a critical role in regulating cellular transcription and cell cycle machinery, making it an exciting target for cancer therapy”
“CDK7 plays a critical role in regulating cellular transcription and cell cycle machinery, making it an exciting target for cancer therapy,” said Murali Ramachandra, Ph.D., Chief Executive Officer of Aurigene. “The data to be presented at ENA 2020 demonstrate that AUR102 effectively engages CDK7 and inhibits a key mediator of the cell cycle and transcription. The ability to inhibit CDK7 activity with an orally available therapeutic such as AUR102 holds great potential to improve care and outcomes for patients with diverse cancer indications, including breast cancer, prostate cancer, leukemia and lymphoma.”
The abstract provides a summary of results from a detailed characterization of AUR102 in cancer cell lines and animal tumor models. Additional data will be presented in the poster. Key findings included in the abstract are:
• AUR102 exhibited potent anti-proliferative activity in a large panel of cell lines with induction of cell death in cell lines derived from multiple cancer types.
• The observed anti-proliferative activity correlated with cellular CDK7 target engagement and decreased levels of P-Ser5 RNAPII, a key mediator of transcription.
• AUR102 studies showed synergy when used in combination with multiple chemotherapies.
• Oral dosing with AUR102 resulted in dose-dependent anti-tumor activity, including complete tumor regression in diffuse large B-cell lymphoma, acute myeloid leukemia, and triple-negative breast cancer xenograft models.
• Inhibition of tumor growth was accompanied by complete target engagement as demonstrated in a parallel PK-PD study.
• AUR102 significantly impacts several pathways and key cancer driver and immune-response genes.
The study authors conclude that the data support clinical evaluation of AUR102 as a single agent and in combination with chemotherapies for the treatment of cancer.
“The exciting AUR102 data to be presented at ENA 2020 provide further validation of our partnering strategy, which gives us multiple opportunities to build a pipeline of best-in-class cancer therapies,” said Peter Lamb, Ph.D., Executive Vice President of Scientific Strategy and Chief Scientific Officer of Exelixis. “AUR102 could be the subject of an Investigational New Drug filing later this year, which would be an important value driver for the program itself and for our collaboration with Aurigene. We commend the Aurigene team on their ongoing success in building a robust body of data supporting the broad clinical potential of AUR102.”
Under the terms of the July 2019 agreement, Exelixis made an upfront payment of $10 million for exclusive options to license three preexisting programs from Aurigene. In addition, Exelixis and Aurigene initiated three Aurigene-led drug discovery programs on mutually agreed upon targets, in exchange for additional upfront option payments of $2.5 million per program. Exelixis is also contributing research funding to Aurigene to facilitate discovery and preclinical development work on all six programs. As the programs mature, Exelixis will have the opportunity to exercise an exclusive option for each program up until the time of Investigational New Drug (IND) filing acceptance. If Exelixis decides to exercise an option, it will make an option exercise payment to Aurigene and assume responsibility for that program’s future clinical development and commercialization including global manufacturing. Aurigene will be eligible for clinical development, regulatory, and sales milestones, as well as royalties on sales. Under the terms of the agreement, Aurigene retains limited development and commercial rights for India and Russia.
About Aurigene
Aurigene is a development stage biotech company engaged in discovery and clinical development of novel and best-in-class therapies to treat cancer and inflammatory diseases and a wholly owned subsidiary of Dr. Reddy’s Laboratories Ltd. (BSE: 500124, NSE: DRREDDY, NYSE: RDY). Aurigene is focused on precision-oncology, oral immune checkpoint inhibitors, and the Th-17 pathway. Aurigene’s programs currently in clinical development include an oral ROR-gamma inhibitor AUR101 for moderate to severe psoriasis in phase 2 under a U.S. FDA IND and a PD-L1/ VISTA antagonist CA-170 for non-squamous non-small cell lung cancer in phase 2b/3 in India. Additionally, Aurigene has multiple compounds at different stages of pre-clinical development. Aurigene has also partnered with several large and mid-pharma companies in the United States and Europe and has multiple programs in clinical development. For more information, please visit Aurigene’s website at http://www.aurigene.com.
About Exelixis
Founded in 1994, Exelixis, Inc. (Nasdaq: EXEL) is a commercially successful, oncology-focused biotechnology company that strives to accelerate the discovery, development and commercialization of new medicines for difficult-to-treat cancers. Following early work in model system genetics, we established a broad drug discovery and development platform that has served as the foundation for our continued efforts to bring new cancer therapies to patients in need. Our discovery efforts have resulted in four commercially available products, CABOMETYX® (cabozantinib), COMETRIQ® (cabozantinib), COTELLIC® (cobimetinib) and MINNEBRO® (esaxerenone), and we have entered into partnerships with leading pharmaceutical companies to bring these important medicines to patients worldwide. Supported by revenues from our marketed products and collaborations, we are committed to prudently reinvesting in our business to maximize the potential of our pipeline. We are supplementing our existing therapeutic assets with targeted business development activities and internal drug discovery – all to deliver the next generation of Exelixis medicines and help patients recover stronger and live longer. Exelixis is a member of Standard & Poor’s (S&P) MidCap 400 index, which measures the performance of profitable mid-sized companies. For more information about Exelixis, please visit http://www.exelixis.com, follow @ExelixisInc on Twitter or like Exelixis, Inc. on Facebook.
EXELIXIS AND AURIGENE ANNOUNCE THAT PROMISING PRECLINICAL DATA TO BE PRESENTED AT THE ENA SYMPOSIUM SUPPORT THE CLINICAL DEVELOPMENT OF A NOVEL CDK7 INHIBITOR
Exelixis and Aurigene Announce That Promising Preclinical Data to Be Presented at the ENA Symposium Support the Clinical Development of a Novel CDK7 Inhibitor
– Detailed characterization of an oral inhibitor of CDK7 demonstrates potent activity against multiple hematologic and solid tumor cell lines, as monotherapy and in combination with chemotherapies –
October 09, 2020 03:02 AM Eastern Daylight Time
ALAMEDA, Calif.–(BUSINESS WIRE)–Exelixis, Inc. (Nasdaq: EXEL) and Aurigene Discovery Technologies Limited (Aurigene) today disclosed new preclinical data showing that AUR102 has potent anti-tumor activity in a large panel of cancer cell lines. AUR102 is a potent, selective, and orally bioavailable covalent inhibitor of cyclin-dependent kinase 7 (CDK7), which is an important regulator of the cellular transcriptional and cell cycle machinery. Exelixis has an exclusive option for AUR102 under its July 2019 exclusive collaboration, option and license agreement with Aurigene. The new data will be presented in a poster (Abstract 170) at the 32nd EORTC-NCI-AACR (ENA) Symposium, which is being held virtually on October 24-25, 2020.
“CDK7 plays a critical role in regulating cellular transcription and cell cycle machinery, making it an exciting target for cancer therapy”
“CDK7 plays a critical role in regulating cellular transcription and cell cycle machinery, making it an exciting target for cancer therapy,” said Murali Ramachandra, Ph.D., Chief Executive Officer of Aurigene. “The data to be presented at ENA 2020 demonstrate that AUR102 effectively engages CDK7 and inhibits a key mediator of the cell cycle and transcription. The ability to inhibit CDK7 activity with an orally available therapeutic such as AUR102 holds great potential to improve care and outcomes for patients with diverse cancer indications, including breast cancer, prostate cancer, leukemia and lymphoma.”
The abstract provides a summary of results from a detailed characterization of AUR102 in cancer cell lines and animal tumor models. Additional data will be presented in the poster. Key findings included in the abstract are:
• AUR102 exhibited potent anti-proliferative activity in a large panel of cell lines with induction of cell death in cell lines derived from multiple cancer types.
• The observed anti-proliferative activity correlated with cellular CDK7 target engagement and decreased levels of P-Ser5 RNAPII, a key mediator of transcription.
• AUR102 studies showed synergy when used in combination with multiple chemotherapies.
• Oral dosing with AUR102 resulted in dose-dependent anti-tumor activity, including complete tumor regression in diffuse large B-cell lymphoma, acute myeloid leukemia, and triple-negative breast cancer xenograft models.
• Inhibition of tumor growth was accompanied by complete target engagement as demonstrated in a parallel PK-PD study.
• AUR102 significantly impacts several pathways and key cancer driver and immune-response genes.
The study authors conclude that the data support clinical evaluation of AUR102 as a single agent and in combination with chemotherapies for the treatment of cancer.
“The exciting AUR102 data to be presented at ENA 2020 provide further validation of our partnering strategy, which gives us multiple opportunities to build a pipeline of best-in-class cancer therapies,” said Peter Lamb, Ph.D., Executive Vice President of Scientific Strategy and Chief Scientific Officer of Exelixis. “AUR102 could be the subject of an Investigational New Drug filing later this year, which would be an important value driver for the program itself and for our collaboration with Aurigene. We commend the Aurigene team on their ongoing success in building a robust body of data supporting the broad clinical potential of AUR102.”
Under the terms of the July 2019 agreement, Exelixis made an upfront payment of $10 million for exclusive options to license three preexisting programs from Aurigene. In addition, Exelixis and Aurigene initiated three Aurigene-led drug discovery programs on mutually agreed upon targets, in exchange for additional upfront option payments of $2.5 million per program. Exelixis is also contributing research funding to Aurigene to facilitate discovery and preclinical development work on all six programs. As the programs mature, Exelixis will have the opportunity to exercise an exclusive option for each program up until the time of Investigational New Drug (IND) filing acceptance. If Exelixis decides to exercise an option, it will make an option exercise payment to Aurigene and assume responsibility for that program’s future clinical development and commercialization including global manufacturing. Aurigene will be eligible for clinical development, regulatory, and sales milestones, as well as royalties on sales. Under the terms of the agreement, Aurigene retains limited development and commercial rights for India and Russia.
About Aurigene
Aurigene is a development stage biotech company engaged in discovery and clinical development of novel and best-in-class therapies to treat cancer and inflammatory diseases and a wholly owned subsidiary of Dr. Reddy’s Laboratories Ltd. (BSE: 500124, NSE: DRREDDY, NYSE: RDY). Aurigene is focused on precision-oncology, oral immune checkpoint inhibitors, and the Th-17 pathway. Aurigene’s programs currently in clinical development include an oral ROR-gamma inhibitor AUR101 for moderate to severe psoriasis in phase 2 under a U.S. FDA IND and a PD-L1/ VISTA antagonist CA-170 for non-squamous non-small cell lung cancer in phase 2b/3 in India. Additionally, Aurigene has multiple compounds at different stages of pre-clinical development. Aurigene has also partnered with several large and mid-pharma companies in the United States and Europe and has multiple programs in clinical development. For more information, please visit Aurigene’s website at http://www.aurigene.com.
About Exelixis
Founded in 1994, Exelixis, Inc. (Nasdaq: EXEL) is a commercially successful, oncology-focused biotechnology company that strives to accelerate the discovery, development and commercialization of new medicines for difficult-to-treat cancers. Following early work in model system genetics, we established a broad drug discovery and development platform that has served as the foundation for our continued efforts to bring new cancer therapies to patients in need. Our discovery efforts have resulted in four commercially available products, CABOMETYX® (cabozantinib), COMETRIQ® (cabozantinib), COTELLIC® (cobimetinib) and MINNEBRO® (esaxerenone), and we have entered into partnerships with leading pharmaceutical companies to bring these important medicines to patients worldwide. Supported by revenues from our marketed products and collaborations, we are committed to prudently reinvesting in our business to maximize the potential of our pipeline. We are supplementing our existing therapeutic assets with targeted business development activities and internal drug discovery – all to deliver the next generation of Exelixis medicines and help patients recover stronger and live longer. Exelixis is a member of Standard & Poor’s (S&P) MidCap 400 index, which measures the performance of profitable mid-sized companies. For more information about Exelixis, please visit http://www.exelixis.com, follow @ExelixisInc on Twitter or like Exelixis, Inc. on Facebook.
Exelixis Forward-Looking Statements
This press release contains forward-looking statements, including, without limitation, statements related to: Exelixis’ and Aurigene’s plans to present preclinical data in support of the continued development of AUR102 in a poster as part of the 32nd ENA Symposium; the potential for AUR102 to improve care and outcomes for patients with diverse cancer indications, including breast cancer, prostate cancer, leukemia and lymphoma; the potential for AUR102 to be the subject of an Investigational New Drug filing later in 2020; Exelixis’ potential future financial and other obligations under the exclusive collaboration, option and license agreement with Aurigene; and Exelixis’ plans to reinvest in its business to maximize the potential of the company’s pipeline, including through targeted business development activities and internal drug discovery. Any statements that refer to expectations, projections or other characterizations of future events or circumstances are forward-looking statements and are based upon Exelixis’ current plans, assumptions, beliefs, expectations, estimates and projections. Forward-looking statements involve risks and uncertainties. Actual results and the timing of events could differ materially from those anticipated in the forward-looking statements as a result of these risks and uncertainties, which include, without limitation: the availability of data at the referenced times; the level of costs associated with Exelixis’ commercialization, research and development, in-licensing or acquisition of product candidates, and other activities; uncertainties inherent in the drug discovery and product development process; Exelixis’ dependence on its relationship with Aurigene, including Aurigene’s adherence to its obligations under the exclusive collaboration, option and license agreement and the level of Aurigene’s assistance to Exelixis in completing clinical trials, pursuing regulatory approvals or successfully commercializing partnered compounds in the territories where they may be approved; the continuing COVID-19 pandemic and its impact on Exelixis’ research and development operations; complexities and the unpredictability of the regulatory review and approval processes in the U.S. and elsewhere; Exelixis’ and Aurigene’s continuing compliance with applicable legal and regulatory requirements; Exelixis’ and Aurigene’s ability to protect their respective intellectual property rights; market competition; changes in economic and business conditions; and other factors affecting Exelixis and its product pipeline discussed under the caption “Risk Factors” in Exelixis’ Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission (SEC) on August 6, 2020, and in Exelixis’ future filings with the SEC. All forward-looking statements in this press release are based on information available to Exelixis as of the date of this press release, and Exelixis undertakes no obligation to update or revise any forward-looking statements contained herein, except as required by law.
Exelixis, the Exelixis logo, CABOMETYX, COMETRIQ and COTELLIC are registered U.S. trademarks. MINNEBRO is a registered Japanese trademark.
SY 5609
[ Fig. 0001] [ Fig. 0002]
[ Fig. 0003]
[ Fig. 0004]
SY 5609
CAS 2519828-12-5
Cancer, solid tumor
PHASE 1
A highly selective and potent oral inhibitor of cyclin-dependent kinase 7 (CDK7) for potential treatment of advanced solid tumors that harbor the Rb pa thway alterations (Syros Pharmaceuticals, Inc., Cambridge, Massachusetts, USA)
SY-5609 is an oral non-covalent CDK7 inhibitor in early clinical development at Syros Pharmaceuticals for the treatment of patients with advanced breast, colorectal, lung or ovarian cancer, or with solid tumors of any histology that harbor Rb pathway alterations.
- OriginatorSyros Pharmaceuticals
- ClassAntineoplastics; Small molecules
- Mechanism of ActionCyclin-dependent kinase-activating kinase inhibitors
- Phase IBreast cancer; Solid tumours
- 05 Aug 2021Roche plans the phase I/Ib INTRINSIC trial in Colorectal cancer (Combination therapy, Metastatic disease) in USA, Canada, Italy, South Korea, Spain and United Kingdom (NCT04929223)
- 05 Aug 2021Roche and Syros Pharmaceuticals enters into a clinical trial collaboration to evaluate atezolizumab in combination with SY 5609 in a clinical trial
- 05 Aug 2021Syros Pharmaceuticals plans a phase I trial in Cancer in second half of 2021
- NCT04247126
- https://clinicaltrials.gov/ct2/show/NCT04247126
At #ESMO21, we will be presenting new preclinical and clinical data on SY-5609, our highly selective and potent oral CDK7 inhibitor. #oncology #biotech Learn more: https://lnkd.in/gqYmWYhb
A Promising Approach for Difficult-to-Treat Cancers
SY-5609 is a highly selective and potent oral inhibitor of the cyclin-dependent kinase 7 (CDK7) in a Phase 1 dose-escalation trial in patients with advanced breast, colorectal, lung, ovarian or pancreatic cancer, or with solid tumors of any histology that harbor Rb pathway alterations.
SY-5609 represents a new approach to treating cancer that we believe has potential in a range of difficult-to-treat cancers. It has shown robust anti-tumor activity, including complete regressions, in preclinical models of breast, colorectal, lung and ovarian cancers at doses below the maximum tolerated dose. In preclinical studies of breast, lung and ovarian cancers, deeper and more sustained responses were associated with the presence of Rb pathway alterations. SY-5609 has also shown substantial anti-tumor activity in combination with fulvestrant in treatment-resistant models of estrogen receptor-positive breast cancer, including those resistant to both fulvestrant and a CDK4/6 inhibitor. Early dose-escalation data demonstrated proof-of-mechanism at tolerable doses.
Syros to Present New Data from Phase 1 Clinical Trial of SY-5609 in Oral Presentation at ESMO Congress 2021SEPTEMBER 13, 2021
Management to Host Conference Call on Monday, September 20, 2021 at 4:00 p.m. ET
CAMBRIDGE, Mass.–(BUSINESS WIRE)– Syros Pharmaceuticals (NASDAQ:SYRS), a leader in the development of medicines that control the expression of genes, today announced that it will present new data from the dose-escalation portion of the Phase 1 clinical trial of SY-5609, its highly selective and potent oral cyclin-dependent kinase 7 (CDK7) inhibitor, at the ESMO Congress 2021, taking place virtually September 16-21, 2021. The oral presentation will include safety, tolerability, and initial clinical activity data for SY-5609 in patients with breast, colorectal, lung, ovarian and pancreatic cancers, as well as in patients with solid tumors of any histology harboring Rb pathway alterations.
In separate poster presentations, Syros will present new preclinical data evaluating the antitumor and pharmacodynamic activity of intermittent dosing regimens for SY-5609 in ovarian cancer models, as well as new preclinical data evaluating antitumor activity of SY-5609 as a single agent and in combination with chemotherapy in KRAS-mutant models.
The abstracts for the two poster presentations are now available online on the ESMO conference website at: https://www.esmo.org/meetings/esmo-congress-2021/abstracts, and the presentations will become available for on-demand viewing starting September 16 at 08:30 CEST (September 16 at 2:30 a.m. ET). The abstract for the oral presentation on the Phase 1 dose-escalation data will remain embargoed until September 17 at 00:05 CEST (September 16 at 6:05 p.m. ET).
Details of the oral presentation are as follows:
Presentation Title: Tolerability and Preliminary Clinical Activity of SY-5609, a Highly Potent and Selective Oral CDK7 Inhibitor, in Patients with Advanced Solid Tumors
Session Date & Time: Monday, September 20, 17:30-18:30 CEST (11:30-12:30 p.m. ET)
Presentation Time: 17:55-18:00 CEST (11:55-12:00 p.m. ET)
Session Title: Mini Oral Session: Developmental Therapeutics
Presenter: Manish Sharma, M.D., START Midwest
Abstract Number: 518MO
Details of the poster presentations are as follows:
Presentation Title: Preclinical Evaluation of Intermittent Dosing Regimens on Antitumor and PD Activity of SY-5609, a Potent and Selective Oral CDK7 Inhibitor, in Ovarian Cancer Xenografts
Abstract Number: 14P
Presentation Title: SY-5609, a Highly Potent and Selective Oral CDK7 inhibitor, Exhibits Robust Antitumor Activity in Preclinical Models of KRAS Mutant Cancers as a Single Agent and in Combination with Chemotherapy
Abstract Number: 13P
Conference Call Information
Syros will host a conference call on Monday, September 20, 2021 at 4:00 p.m. ET to discuss the new clinical and preclinical data for SY-5609, which will be presented at the ESMO Congress 2021.
To access the live conference call, please dial 866-595-4538 (domestic) or 636-812-6496 (international) and refer to conference ID 4648345. A webcast of the call will also be available on the Investors & Media section of the Syros website at www.syros.com. An archived replay of the webcast will be available for approximately 30 days following the conference call.
About Syros Pharmaceuticals
Syros is redefining the power of small molecules to control the expression of genes. Based on its unique ability to elucidate regulatory regions of the genome, Syros aims to develop medicines that provide a profound benefit for patients with diseases that have eluded other genomics-based approaches. Syros is advancing a robust clinical-stage pipeline, including: tamibarotene, a first-in-class oral selective RARα agonist in RARA-positive patients with higher-risk myelodysplastic syndrome and acute myeloid leukemia; SY-2101, a novel oral form of arsenic trioxide in patients with acute promyelocytic leukemia; and SY-5609, a highly selective and potent oral CDK7 inhibitor in patients with select solid tumors. Syros also has multiple preclinical and discovery programs in oncology and monogenic diseases.
View source version on businesswire.com: https://www.businesswire.com/news/home/20210913005090/en/
PATENT
CN(C)C\C=C\C(=O)Nc1ccc(cc1)C(=O)Nc1cccc(c1)Nc1ncc(Cl)c(n1)c1c[NH]c2ccccc21
THZ1; 1604810-83-4; THZ-1; HY-80013
CLIP
SY 1365 MEVOCICLIB, CAS 1816989-16-8
CN(C)C\C=C\C(=O)Nc1ccc(nc1)C(=O)N[C@]1(C)C[C@@H](CCC1)Nc1ncc(Cl)c(n1)c1c[NH]c2ccccc21
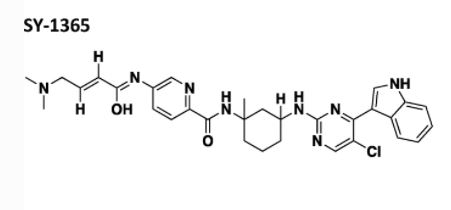
PATENT
PATENT
3-fluoro-4-(methylamino)-N-[(1S,3R)-1-methyl-3-[[4-(7-methyl-1H-indol-3-yl)-5-(trifluoromethyl)pyrimidin-2-yl]amino]cyclohexyl]benzamide (Compound 130)
3-chloro-4-[[4-(dimethylamino)-3-hydroxy-butanoyl]amino]-N-[(1S,3R)-3-[[4-(1H-indazol-3-yl)-5-(trifluoromethyl)pyrimidin-2-yl]amino]-1-methyl-cyclohexyl]benzamide (Compound 129)
4-amino-N-((1S,3R)-3-((5-chloro-4-(1H-indol-3-yl)pyrimidin-2-yl)amino)-1-methylcyclohexyl)benzamide (Compound 128)
4-amino-3-fluoro-N-[(1S,3R)-3-[[4-(1H-indazol-3-yl)-5-(trifluoromethyl)pyrimidin-2-yl]amino]-1-methyl-cyclohexyl]benzamide (Compound 127)
4-amino-N-((1S,3R)-3-((5-chloro-4-(2-methyl-1H-indol-3-yl)pyrimidin-2-yl)amino)cyclohexyl)benzamide (Compound 126)
4-amino-N-((1S,3R)-3-((5-chloro-4-(1H-indazol-3-yl)pyrimidin-2-yl)amino)cyclohexyl)benzamide (Compound 124)
Example 25 Synthesis of N1-(4-(((1S,3R)-3-((5-chloro-4-(1H-indol-3-yl)pyrimidin-2-yl)amino)cyclohexyl)carbamoyl)phenyl)oxalamide (Compound 113)
Example 24 Synthesis of N-((1S,3R)-3-((5-chloro-4-(1H-indol-3-yl)pyrimidin-2-yl)amino)cyclohexyl)-4-(4-(dimethylamino)butanamido)benzamide (Compound 105)
PATENT
4-amino-N-(3-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)tricyclo[3.3.1.13,7]decanyl)benzamide (Compound 100).
+/−)-4-amino-N-(3-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)-5 hydroxycyclohexyl)benzamide (Compound 101)
4-amino-N-((1S,3R)-3-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)cyclohexyl)benzamide (Compound 102)
(1S,3R)-N-(4-aminophenyl)-3-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)cyclohexanecarboxamide (Compound 106)
4-amino-N-((1S,3R)-3-(5-cyclopropyl-4-(1H-indol-3-yl)pyrimidin-2-ylamino)cyclohexyl)benzamide.HCl (Compound 103)
4-amino-N-((1S,3R)-3-(5-chloro-4-(pyridin-3-yl)pyrimidin-2-ylamino)cyclohexyl)benzamide (Compound 108)
4-amino-N-((1S,3R)-3-(5-cyano-4-(1H-indol-3-yl)pyrimidin-2-ylamino)cyclohexyl)benzamide (Compound 107)
(+/−)-4-amino-N-(3-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)-5-fluorocyclohexyl)benzamide (Compound 110)
4-amino-N-(5-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)bicyclo[3.1.1]heptan-1-yl)benzamide (Compound 104)
4-amino-N4(1R,5S)-5-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)-3,3-difluorocyclohexyl)benzamide (Compound 115)
4-amino-N-((1S,3R)-3-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)cyclohexyl)benzenesulfonamide (Compound 109).
4-amino-N-((1S,3R)-3-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)cyclohexyl)-2-fluorobenzamide (Compound 112)
4-amino-N-((1S,3R)-3-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)cyclohexyl)-3-fluorobenzamide (Compound 111).
(+/−)-4-amino-N-(3-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)-1-methylcyclohexyl)benzamide (Compound 116).
N-((1S,3R)-3-(4-(1H-indol-3-yl)pyrimidin-2-ylamino)cyclohexyl)-4-aminobenzamide (Compound 114).
4-amino-N-((1S,3R)-3-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)cyclohexyl)-2-morpholinobenzamide(Compound 117).
4-amino-N-((1S,3R)-3-(5-chloro-4-(1H-indol-3-yl)pyridin-2-ylamino)cyclohexyl)benzamide (Compound 118).
3-amino-N-(trans-4-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)cyclohexyl)benzamide.HCl (Compound 119).
(1S,3R)-N1-(R)-1-(4-aminophenyl)-2,2,2-trifluoroethyl)-N3-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-yl)cyclohexane-1,3-diamine (Compound 120).
(1S,3R)-N1-(4-aminobenzyl)-N3-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-yl)-N1-methylcyclohexane-1,3-diamine.HCl (Compound 122).
4-amino-N-((1S,3R)-3-(5-chloro-4-(pyrazolo[1,5-a]pyridin-3-yl)pyrimidin-2-ylamino)cyclohexyl)benzamide.HCl (Compound 123).
Synthesis of 5-amino-N-((1S,3R)-3-(5-chloro-4-(1-methyl-1H-indol-3-yl)pyrimidin-2-ylamino)cyclohexyl)picolinamide (Compound 125)
Synthesis of N-((1S,3R)-3-((5-chloro-4-(1H-indol-3-yl)pyrimidin-2-yl)amino)cyclohexyl)-4-(4-(dimethylamino)butanamido)benzamide (Compound 105)
Synthesis of N1-(4-(((1S,3R)-3-)(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-yl)amino)cyclohexyl)carbamoyl)phenyl)oxalamide (Compound 113)
Synthesis of 4-amino-N-((1S,3R)-3-((5-chloro-4-(1H-indazol-3-yl)pyrimidin-2-yl)amino)cyclohexyl)benzamide (Compound 124)
Synthesis of 4-amino-N-((1S,3R)-3-((5-chloro-4-(2-methyl-1H-indol-3-yl)pyrimidin-2-yl)amino)cyclohexyl)benzamide (Compound 126)
Synthesis of 4-amino-3-fluoro-N-[(1S,3R)-3-[[4-(1H-indazol-3-yl)-5-(trifluoromethyl)pyrimidin-2-yl]amino]-1-methyl-cyclohexyl]benzamide (Compound 127).
Synthesis of 4-amino-N-((1S,3R)-3-((5-chloro-4-(1H-indol-3-yl) pyrimidin-2-yl)amino)-1-methylcyclohexyl)benzamide (Compound 128)
Synthesis of 3-chloro-4-[[4-(dimethylamino)-3 hydroxy-butanoyl]amino]-N-[(1S,3R)-3-[[4-(1H-indazol-3-yl)-5-(trifluoromethyl)pyrimidin-2-yl]amino]-1-methyl-cyclohexyl]benzamide (Compound 129).
Synthesis of 3-fluoro-4-(methylamino)-N-[(1S,3R)-1-methyl-3-[[4-(7-methyl-1H-indol-3-yl)-5-(trifluoromethyl)pyrimidin-2-yl]amino]cyclohexyl]benzamide (Compound 130)
//////////////SY 5609, 2519828-12-5, Cancer, solid tumor, PHASE 1, SYROS

NEW DRUG APPROVALS
ONE TIME TO MAINTAIN THIS BLOG
$10.00
MAX 40279
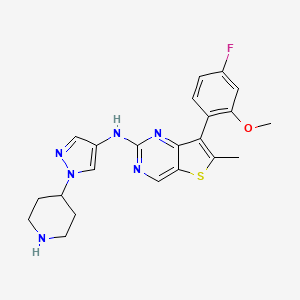
MAX 40279, EX-A4057
Max 4; MAX-40279; MAX-40279-001; MAX-40279-01
UNII-DL772G3NN7
2070931-57-4
C22H23FN6OS, 438.5
7-(4-fluoro-2-methoxyphenyl)-6-methyl-N-(1-piperidin-4-ylpyrazol-4-yl)thieno[3,2-d]pyrimidin-2-amine
Thieno[3,2-d]pyrimidin-2-amine, 7-(4-fluoro-2-methoxyphenyl)-6-methyl-N-[1-(4-piperidinyl)-1H-pyrazol-4-yl]-

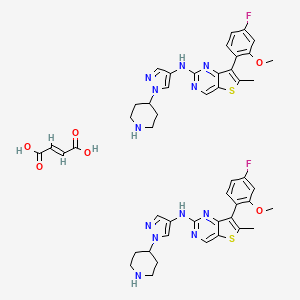
7-(4-FLUORO-2-METHOXYPHENYL)-6-METHYL-N-(1-(PIPERIDIN-4-YL)-1H-PYRAZOL-4-YL) THIENO (3,2-D)PYRIMIDIN-2-AMINE SEMI-FUMARATE CAS 2388506-43-0
- 7-(4-Fluoro-2-methoxyphenyl)-6-methyl-N-[1-(4-piperidinyl)-1H-pyrazol-4-yl]thieno[3,2-d]pyrimidin-2-amine
- Originator Maxinovel Pharmaceuticals
- ClassAntineoplastics
- Mechanism of ActionFibroblast growth factor receptor antagonists; Fms-like tyrosine kinase 3 inhibitors
- Orphan Drug StatusYes – Acute myeloid leukaemia
- Phase IAcute myeloid leukaemia; Solid tumours
Most Recent Events
- 28 Nov 2019Phase-I clinical trials in Solid tumours (Late-stage disease, Metastatic disease) in China (PO) (NCT04183764)
- 16 Apr 2019Phase-I clinical trials in Acute myeloid leukaemia (Second-line therapy or greater) in China (PO) (NCT04187495)
- 23 Jan 2019Guangzhou Maxinovel Pharmaceuticals plans a phase I trial in China (ChiCTR1900020971)
- MaxiNovel Pharmaceuticals, Inc. Announces FDA Orphan Drug Designation for MAX-40279 for the Treatment of Acute Myeloid Leukemia (AML)
March 29, 2018 11:24 AM Eastern Daylight Timehttps://www.businesswire.com/news/home/20180329005826/en/MaxiNovel-Pharmaceuticals-Inc.-Announces-FDA-Orphan-Drug-Designation-for-MAX-40279-for-the-Treatment-of-Acute-Myeloid-Leukemia-AML
GUANGZHOU, China–(BUSINESS WIRE)–MaxiNovel Pharmaceuticals, Inc. announced today that the U.S. Food and Drug Administration (“FDA”) has granted MaxiNovel Orphan Drug Designation for MAX-40279 in the treatment of Acute Myeloid Leukemia (AML).
AML is the most common acute leukemia which accounts for approximately 25% of all adult leukemias worldwide. Approximately one-third of AML patients have a FLT3 gene mutation. Such mutation can result in faster disease progression, higher relapse rates and lower rates of survival than other forms of AML. Inhibition of FLT3 mutation is of high importance in combating AML.
In the preclinical testing, MAX-40279 demonstrated potent inhibition of both FLT3 and FGFR with excellent drug concentration in the bone marrow. It is designed to overcome the observed drug resistance of the current FLT3 inhibitors due to the bone marrow FGF/FGFR pathway activation.
“We are very pleased to receive the ODD,” commented MaxiNovel’s Vice President Dr. Elizabeth Ashraf. “Our objective is to bring the best in class medicine to the patients worldwide.”
The FDA Office of Orphan Products Development grants orphan drug designation to novel drugs and biologics that are intended for the safe and effective treatment, diagnosis or prevention of rare diseases or disorders that affect fewer than 200,000 people in the United States. The designation allows manufacturers to qualify for various incentives including federal grants, tax credits for qualified clinical trials, a waiver of PDUFA filing fees and 7 years of market exclusivity upon regulatory approval.
About MaxiNovel Pharmaceuticals, Inc:
Maxinovel Pharmaceuticals, Inc. is a biotech company focusing on the discovery and development of Immuno-oncology therapy and targeted therapy. It will use its orally active Immuno-oncology product platform to bring effective combo product of multi-components in a single oral pill to the patients worldwide. For more info: www.maxinovel.com
The JAK-STAT (Janus kinase-signal transducer and activator of transcription) signal pathway is a signal transduction pathway stimulated by cytokines discovered in recent years, and it participates in many important biology such as cell proliferation, differentiation, apoptosis and immune regulation. Process (Aaronson, D Set al. Science 2002, 296, 1653-1655; O’Shea, J Jet al. Nat. Rev. Drug Discovery 2004, 3, 555-564). Compared with other signal pathways, the transmission process of this signal pathway is relatively simple. It mainly consists of three components, namely tyrosine kinase-related receptor, tyrosine kinase JAK and transcription factor STAT. JAK (Janus Kinase), a type of molecule in the cell, is rapidly recruited and activated on the receptor after receiving the signal from the upstream receptor molecule. The activated JAK catalyzes the receptor tyrosine phosphorylation, and the phosphorylation of tyrosine on the receptor molecule Amino acid is the recognition and binding site of a kind of signal molecule STAT SH2. Tyrosine phosphorylation occurs after STAT binds to the receptor. Tyrosine phosphorylated STAT forms a dimer and enters the nucleus. As an active transcription factor, dimeric STAT molecules directly affect the expression of related genes, thereby changing the proliferation or differentiation status of target cells.
The JAK-STAT pathway is widely present in various tissues and cells in the body, and has an important role in the differentiation, proliferation, and anti-infection of lymphocytes, and participates in the interaction of various inflammatory factors and signal transduction (Kiesseleva T. et al. . J. Gene, 2002, 285, 1-24). The abnormal activation of this pathway is closely related to a variety of diseases. Finding and screening JAK inhibitors can help in-depth study of the regulatory mechanism of JAK-STAT, thereby providing new drugs and methods for the prevention and treatment of related diseases
The occurrence, growth, invasion and metastasis of tumors are related to the JAK-STAT signal transduction pathway. In normal signal transduction, the activation of STATs is rapid and transient. The continuous activation of STATs is closely related to the process of malignant transformation of cells (Buettner R. et al. Clin. Cancer Res. 2002, 8(4), 945-954). STAT3 is the focus of multiple oncogenic tyrosine kinase signal channels such as EGFR, IL-6/JAK, Src, etc. It is activated in a variety of tumor cells and tissues, such as breast cancer, ovarian cancer, and head and neck squamous cells. Like cell carcinoma, prostate cancer, malignant melanoma, multiple myeloma, lymphoma, brain tumor, non-small cell lung cancer and various leukemias, etc. (Niu G. et al. Oncogene 2002, 21(13), 2000-2008 ). JAK-STAT pathway inhibitors belong to PTK inhibitors, and this enzyme is a member of the oncogene protein and proto-oncoprotein family, and plays an important role in the normal and abnormal cell proliferation. The occurrence and growth of tumors are inseparable from PTK. Therefore, JAK-STAT pathway inhibitors inhibit tumor growth by antagonizing PTK, and have obvious anti-tumor effects (Mora LBet al.J.Cancer Res.2002,62(22) , 6659-6666).
In addition, the latest research shows that: organ transplant rejection, psoriasis, tissue and organ fibrosis, bronchial asthma, ischemic cardiomyopathy, heart failure, myocardial infarction, blood system diseases, and immune system diseases are all related to JAK-STAT signaling. The pathway is closely related. This signaling pathway is not only important for maintaining the normal physiological functions of cells, but also has an important regulatory role for the occurrence and development of diseases.
The Fibroblast Growth Factor Receptor family belongs to a new type of receptor kinase family, which includes four receptor subtypes (FGFR-1,2,3) encoded by four closely related genes. And 4) and some heterogeneous molecules, which form a ternary complex with fibroblast growth factor (FGF) and heparan sulfate, and then trigger a series of signal transduction pathways to participate in the regulation of physiological processes in the organism. FGFR has a wide range of physiological and pathological effects in the body: (1) Embryo development. Studies have shown that during embryonic development, FGFR signal transduction is essential for most organ development and the formation of embryonic patterns. (2) Cell division, migration and differentiation. FGFR stimulates cell proliferation and participates in the regulation of cell transformation in the pathological process. There are many parallel pathways to achieve FGFR-mediated cell division signal transduction, which has been confirmed by many studies (JKWang et al., Oncogene 1997, 14, 1767 -1778.). (3) Bone diseases. The growth and differentiation of bones are also regulated by the FGF family, and mutations in FGFR can cause bone deformities (R. Shang et al., Cell 1994, 78, 335-342.). (4) The occurrence of tumors. FGFR can promote the migration, proliferation and differentiation of endothelial cells, and plays an important role in the regulation of angiogenesis and angiogenesis. Uncontrolled angiogenesis can lead to the occurrence of tumors and the growth of metastases (J.Folkman.Nat.Med.1995) ,1,27-31.).
FMS-like tyrosine kinase 3 (FMS-like tyrosine kinase 3, FLT3) belongs to the type III receptor tyrosine kinase (receptor tyrosine kinase III, RTK III) family member, it is composed of extracellular domain, intracellular domain and The transmembrane region is composed of 3 parts, which are first expressed in human hematopoietic stem cells. FLT3 interacts with its ligand FL to stimulate or act on stem cells, which is of great significance to the growth and differentiation of stem cells. FLT3 kinase has wild-type FLT3-WT and its main activation mutant FLT3-ITD and FLT3-D835Y. FLT3 is mainly expressed in the precursors of normal myeloid cells, but its abnormal expression is also found in a large part of acute myeloid leukemia (AML) cells.
In recent years, many large-scale studies have confirmed that activating mutations of FLT3 play a very important pathological role in the occurrence and progression of acute myeloid leukemia. FLT3 has become an important target for the treatment of acute myeloid leukemia.
rc family kinase (SFK) is a family of non-receptor tyrosine kinases, including c-Src, LYN, FYN, LCK, HCK, FGR, BLK, YES and YRK, among which LYN kinase has LYNα and LYNβ Both subtypes, LYN kinase and its two subtypes can cause similar intracellular tyrosine phosphorylation. According to the amino acid sequence, SFK can be divided into two sub-families: one family is c-Src, FYN, YES and FGR, which are widely expressed in different tissues; the other family is LCK, BLK, LYN and HCK, which are closely related to hematopoietic cells. SFK is connected to multiple signal transduction pathways in the body, and can be activated by growth factors, cytokines and immune cell receptors, G protein-coupled receptors, integrins and other cell adhesion molecules, and then activate the corresponding signal transduction pathways , Causing a variety of physiological effects of cells. The activity of SFK mainly includes the regulation of cell morphology, cell movement, cell proliferation and survival. The abnormal activation and expression of these kinases leads to the occurrence and development of a wide range of diseases, such as a large number of solid tumors, various hematological malignancies and some neuronal pathologies. Therefore, looking for SFK inhibitors is a promising research topic in the field of medicinal chemistry.

NEW DRUG APPROVALS
ONE TIME
$10.00
Patent
CN106366093A
PATENT
WO 2017012559
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017012559Example 31
N-[7-(4-Fluoro-2-methoxyphenyl)-6-methylthieno[3,2-d]pyrimidin-2-yl]-1-(piperidin-4-yl)- 1H-pyrazole-4-amine (Compound 31)
Synthesis of compound 31-e
2,4-Dichloro-6-methylthiophene [3,2-d] pyrimidine (10g, 45.6mmol) was dissolved in tetrahydrofuran (100mL) and ethanol (100mL), and the reaction solution was cooled to 0°C and divided Sodium borohydride (12.5 g, 198 mmol) was added in batches. The reaction solution was raised to room temperature and continued to stir for 16 hours, diluted with water (500 mL), and then adjusted to pH=7 with 1N aqueous hydrochloric acid. The aqueous phase was extracted with ethyl acetate (150 mL×3). The organic phase was washed sequentially with water (100mL×3) and saturated brine (100mL), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure to obtain a white solid 31-e (7.5g, yield: 88%). The product does not require further purification. LC-MS(ESI): m/z=187[M+H] + .[0492]Synthesis of compound 31-d[0493]Compound 31-e (7.5 g, 40 mmol) was dissolved in chloroform (300 mL) at 0°C, active manganese dioxide (35 g, 400 mmol) was added, the reaction solution was raised to room temperature and stirring was continued for 16 hours. The reaction solution was filtered through Celite, and the filter cake was washed with chloroform (100 mL×3). The combined filtrates were concentrated under reduced pressure to obtain white solid 31-d (6.6 g, yield: 89%), which did not require further purification. LC-MS(ESI): m/z=185[M+H]+.[0494]Synthesis of compound 31-c[0495]Compound 31-d (3.1g, 16.8mmol) was dissolved in trifluoroacetic acid (30mL) at 0℃, N-iodosuccinimide (5.7g, 25.3mmol) was added in batches, and the reaction solution was raised to Keep stirring at room temperature for 1 hour. Water (50 mL) was added to the reaction solution to quench the reaction, and it was extracted with dichloromethane (50 mL×3). The organic phase was washed successively with water (50mL×3) and saturated brine (50mL), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure to obtain a white solid 31-c (4.9g, yield: 94%). The product does not require further purification. LC-MS(ESI): m/z=311[M+H] + .[0496]Synthesis of compound 31-b[0497]Compound 31-c (615mg, 1.98mmol), 2-methoxy-4-fluorophenylboronic acid (405mg, 2.38mmol) and sodium carbonate (630mg, 5.94mmol) were suspended in dioxane (5mL) water (5mL) ), add [1,1′-bis(diphenylphosphorus)ferrocene]dichloropalladium dichloromethane complex (163mg, 0.2mmol). Replace with nitrogen 3 times, and heat to 80°C to react for 16 hours. After cooling to room temperature, the reaction solution was concentrated under reduced pressure. The residue was partitioned with dichloromethane (50mL) and water (50mL). The organic phase was dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography (petroleum Ether: dichloromethane=1:1) to obtain a white solid 31-b (240 mg, yield: 39%). LC-MS(ESI): m/z=309[M+H] + .[0498]Synthesis of compound 31-a[0499]Compound 31-b (240mg, 0.78mmol) and compound 32-c (208mg, 0.78mmol) were dissolved in N,N-dimethylformamide (3mL), potassium carbonate (323mg, 2.34mmol) was added, 2- Dicyclohexylphosphine-2′,6′-diisopropoxy-1,1′-biphenyl (112 mg, 0.24 mmol) and tris(dibenzylideneacetone) dipalladium (134 mg, 0.24 mmol). Under the protection of nitrogen, heat to 110°C to react for 16 hours. After cooling to room temperature, the reaction solution was partitioned with dichloromethane (50 mL) and water (50 mL). The organic phase was dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel thin layer chromatography preparation plate (petroleum Ether: ethyl acetate = 1:1) to obtain a yellow viscous oil 31-a (190 mg, yield: 45%). LC-MS(ESI): m/z=539[M+H] + .[0500]Synthesis of compound 31[0501]31-a (190 mg, 0.35 mmol) was dissolved in dichloromethane (3 mL), trifluoroacetic acid (3 mL) was added, and the mixture was stirred at room temperature for 3 hours. The reaction solution was concentrated under reduced pressure. The residue was layered with ethyl acetate (50mL) and 1N aqueous hydrochloric acid (50mL). The aqueous phase was adjusted to pH=10 with saturated aqueous potassium carbonate solution. 3) Washing and vacuum drying the solid to obtain a light yellow solid 31 (22 mg, yield: 14%). LC-MS(ESI): m/z=439[M+H] + .[0502]1 H-NMR (400MHz, MeOD) δ: 8.78 (d, J = 5Hz, 1H), 7.87 (s, 1H), 7.48 (s, 1H), 7.35 (m, 1H), 7.05 (dd, J = 11Hz) ,J = 2Hz, 1H), 6.91 (m, 1H), 4.10 (m, 1H), 3.79 (s, 3H), 3.22 (m, 2H), 2.77 (m, 2H), 2.47 (s, 3H), 2.03(m,2H),1.73(m,2H)ppm
PATENT
WO 2019228171
Example 1 Preparation of fumarate of fused ring pyrimidine compound as shown in formula 2
Weigh the compound N-[7-(4-fluoro-2-methoxyphenyl)-6-methylthieno[3,2-d]pyrimidin-2-yl]-1-(piperidine-4- Base)-1H-pyrazol-4-amine (synthesized according to Example 31 of patent CN106366093A) 100mg (0.228mmol, 1eq) into the vial, add 10mL 88% acetone-water solution, add the vial at about 50°C and stir until dissolved clear. 1.1 mL of fumaric acid with a concentration of 0.25 mol/L in ethanol (0.275 mmol, 1.2 eq) was slowly added dropwise to the free base solution of fused ring pyrimidine compounds, and stirred at 50 ℃ for 1 hour, and then the solution was The rate of 5°C/h was slowly reduced to room temperature, and the solid was collected and dried under vacuum at 40°C overnight.
1 H-NMR (400MHz, DMSO-d 6 ) δ: 9.45 (s, 1H), 8.94 (s, 1H), 7.75 (s, 1H), 7.78-7.33 (m, 2H), 7.15 (d, J = 6.4Hz, 1H), 6.99 (dd, J = 7.6 Hz, J = 7.2 Hz, 1H), 6.42 (s, 1H), 4.10 (m, 1H), 3.73 (s, 3H), 3.17 (d, J = 12.4 Hz, 2H), 2.77 (dd, J = 12.4 Hz, J = 11.6 Hz, 2H), 2.40 (s, 3H), 1.94 (d, J = 11.6 Hz, 2H), 1.73 (m, 2H) ppm.
PATENT
7-(4-Fluoro-2-methoxyphenyl)-6-methyl-N-(1-piperidin-4-yl)-1hydro-pyrazol-4-yl)thieno[3,2 -D]pyrimidine-2-amino is a strong JAK, FGFR, FLT3 kinase inhibitor, and has a good application prospect in the treatment of tumors, immune system diseases, allergic diseases and cardiovascular diseases. This compound is described in patent CN106366093A and has the following chemical structure:
CN106366093A discloses the preparation method of the compound:
In the above synthetic route, NaBH 4 is sodium borohydride, MnO 2 is manganese dioxide, NIS is N-iodosuccinimide, TFA is trifluoroacetic acid, and Pd(dppf)Cl 2 is [1,1′- Bis(diphenylphosphino)ferrocene]palladium dichloride, DIAD is diisopropyl azodicarboxylate, PPh 3 is triphenylphosphine, Pd/C is palladium on carbon, Pd 2 (dba) 3 is Tris(dibenzylideneacetone)dipalladium, RuPhos is 2-bicyclohexylphosphine-2′,6′-diisopropoxybiphenyl.
However, the above method has the problems of a large number of reaction steps, low yield, and requires column chromatography for separation and purification, and is not suitable for industrial scale-up production. Therefore, it is necessary to improve its preparation method.
The present invention provides a method for preparing a compound represented by formula B, which comprises the following steps: under a protective gas atmosphere, in a solvent, in the presence of a catalyst and a base, a compound represented by formula C is combined with a compound represented by formula K The compound can be subjected to the coupling reaction shown below; the catalyst includes a palladium compound and a phosphine ligand;
The preparation method of the compound represented by formula B may further include the following steps: in an organic solvent, in the presence of a base, the compound represented by formula E and the compound represented by formula D are subjected to the substitution reaction shown below, To obtain the compound represented by formula C;
The present invention provides a method for preparing a compound represented by formula C, which comprises the following steps: in an organic solvent, in the presence of a base, a compound represented by formula E and a compound represented by formula D are subjected to the following steps: Substitution reaction is enough;
Example 1: 2-Chloro-6-methylthieno[3,2-D]pyrimidine (Compound I)
Into a 500L reactor, add 10% palladium on carbon (4.6Kg), 2,4-dichloro-6-methylthieno[3,2-D]pyrimidine (24.2Kg, 109.5mol), and tetrahydrofuran (150Kg) in sequence And N,N-diisopropylethylamine (17.0Kg, 131.5mol). Fill the kettle with hydrogen, and control the hydrogen pressure at 0.5 MPa. Turn on the stirring and keep the temperature at 25±5°C to react for 120 hours. Filter, collect the filtrate, concentrate the filtrate under reduced pressure, add ethanol (58Kg) to the concentrate, and concentrate again to bring out residual tetrahydrofuran. Add ethanol (60Kg) and stir at 70±5°C until all solids are dissolved. Cool down, control the temperature at 25±5°C, add 360Kg of purified water dropwise to the kettle, control the dropping rate, and keep the temperature at 25±5°C. The solid product was separated out, centrifuged, and the filter cake was vacuum dried to obtain the product 2-chloro-6-methylthieno[3,2-D]pyrimidine 18.94Kg, yield: 93.2%. LC-MS(ESI): m/z=185.1[M+H] + .
1 H NMR (400MHz, d 6 -DMSO): δ9.30 (s, 1H), 7.34 (s, 1H), 2.73 (s, 3H).
Example 2: 2-Chloro-6-methylthieno[3,2-D]pyrimidine (Compound I)
To a 100mL reaction flask, add 10% palladium on carbon (0.17g), 2,4-dichloro-6-methylthieno[3,2-D]pyrimidine (2g, 9.2mmol), tetrahydrofuran (40mL) and N,N-Diisopropylethylamine (1.412 g, 10.9 mmol). Fill the bottle with hydrogen and control the hydrogen pressure at 0.5MPa. Turn on the stirring and keep the temperature at 25±5°C to react for 20 hours. Filter, collect the filtrate, concentrate the filtrate under reduced pressure, add ethanol (2.1 g) to the concentrate, and concentrate again to bring out residual tetrahydrofuran. Add ethanol (2.2g) and stir at 70±5°C until all solids are dissolved. Cool down, control the temperature at 25±5°C, add 13.3g of purified water dropwise to the kettle, control the dropping rate, and keep the temperature at 25±5°C. The solid product was precipitated, centrifuged, and the filter cake was vacuum dried to obtain 2.4 g of 2-chloro-6-methylthieno[3,2-D]pyrimidine as a product, with a yield of 82%. The LC-MS and 1 H NMR are the same as in Example 1.
Example 3: 7-Bromo 2-chloro-6-methylthieno[3,2-D]pyrimidine (Compound E)
Add trifluoroacetic acid (150Kg) and 2-chloro-6-methylthieno[3,2-D]pyrimidine (18.90Kg, 102.4mol) into a 500L enamel reactor. Add N-bromosuccinimide (18.33Kg, 103.0mol) under temperature control at 15±5℃. After the addition, the temperature is controlled at 25±5℃ to react for 2 hours. Sampling to monitor the reaction, there is still a small amount of raw materials remaining. Additional N-bromosuccinimide (1.0 Kg, 5.6 mol) was added, stirring was continued for 1 hour, sampling and monitoring showed that the reaction was complete. Control the temperature at 10±5°C, and add 274Kg of water dropwise. After the addition, stir at 10±5°C for 2 hours. After centrifugation, the solid was vacuum-dried to obtain the product, 7-bromo-2-chloro-6-methylthieno[3,2-D]pyrimidine, 24.68Kg, yield: 91.4%. LC-MS(ESI): m/z=265.0[M+H] + .
1 H NMR (400MHz, d 6 -DMSO): δ9.33 (s, 1H), 2.64 (s, 3H).
Example 4: 4-(p-toluenesulfonyl)-piperidine-1-tert-butyl carbonate (Compound G)
Add pyridine (176Kg) and N-BOC-4-hydroxypiperidine (36.00Kg, 178.9mol) to a 500L enamel reactor. Add p-toluenesulfonyl chloride (50.5Kg, 264.9mol) in batches under temperature control at 10±10°C. After the addition, the temperature is controlled at 25±5°C to react for 18 hours. The reaction solution was transferred to a 1000L reactor, the temperature was controlled at 15±5°C, and 710Kg of water was added dropwise. After the addition, stir at 15±5°C for 2 hours. After filtration, the solid was washed with water and dried in vacuum to obtain the product 4-(p-methylbenzenesulfonyl)-piperidine-1-carbonate tert-butyl ester, 59.3Kg, yield: 93.3%. LC-MS(ESI): m/z=378.0[M+Na] + .
Example 5: 4-(4-Nitro-1hydro-pyrazol-1-yl)piperidine-1-tert-butyl carbonate (Compound F)
Add N,N-dimethylformamide (252Kg), 4-(p-methylbenzenesulfonyl)-piperidine-1-carbonate tert-butyl ester (59.3Kg, 166.8mol), 4-nitro to the reaction kettle Pyrazole (21.5Kg, 190.1mol), and anhydrous potassium carbonate (34.3Kg, 248.2mol). The temperature was controlled at 80±5°C and the reaction was stirred for 18 hours. Cool down to 15±5°C, add 900Kg of water dropwise, control the dropping rate, and keep the temperature at 15±5°C. After the addition, stir at 5±5°C for 2 hours. After filtering, the solid was washed twice with water and dried in vacuum to obtain the product 4-(4-nitro-1hydro-pyrazol-1-yl)piperidine-1-tert-butyl carbonate 39.92Kg, yield: 80.8%. LC-MS (ESI): m/z=319.1 [M+Na] + .
1 H NMR (400MHz, d 6 -DMSO): δ8.96(s,1H), 8.27(s,1H), 4.44-4.51(m,1H), 4.06-4.08(m,2H), 2.75-2.91( m, 2H), 2.04-2.07 (m, 2H), 1.80-1.89 (m, 2H), 1.41 (s, 9H).
Example 6: 4-(4-Amino-1hydro-pyrazol-1-yl)piperidine-1-tert-butyl carbonate (Compound D)
Add 10% palladium-carbon (2.00Kg), 4-(4-nitro-1hydro-pyrazol-1-yl)piperidine-1-tert-butyl carbonate (39.94Kg, 134.09mol) to the reaction kettle, nothing Water ethanol (314Kg) and ammonia (20.0Kg, 134.09mol). Fill the kettle with hydrogen, and control the hydrogen pressure at 0.2MPa. Turn on the stirring and keep the temperature at 45±5°C to react for 4 hours. Filter, collect the filtrate, and concentrate the filtrate under reduced pressure. Add ethyl acetate (40Kg) and n-heptane (142Kg) to the concentrate, stir at 25±5°C for 1 hour, and then lower the temperature to 5±5°C and stir for 2 hours. After filtration, the solid was vacuum dried to obtain the product 4-(4-amino-1hydro-pyrazol-1-yl)piperidine-1-tert-butyl carbonate 31.85Kg, yield: 88.6%. LC-MS(ESI): m/z=267.2[M+H] + .
1 H NMR (400MHz, d 6 -DMSO): δ7.06 (s, 1H), 6.91 (s, 1H), 4.08-4.15 (m, 1H), 3.98-4.01 (m, 2H), 3.81 (brs, 2H), 2.83-2.87 (m, 2H), 1.88-1.91 (m, 2H), 1.63-1.72 (m, 2H), 1.41 (s, 9H).
Example 7: 4-(4-(7-Bromo-6-methylthieno[3,2-D]pyrimidin-2-yl)amino)-1hydro-pyrazol-1-yl)piperidine-1 -Tert-butyl carbonate (compound C)
Add n-butanol (117Kg), N,N-diisopropylethylamine (15.00Kg, 116.06mol), 4-(4-amino-1hydro-pyrazol-1-yl)piperidine to the reaction kettle 1-tert-butyl carbonate (32.02Kg, 120.22mol) and 7-bromo-2-chloro-6-methylthieno[3,2-D]pyrimidine (24.68Kg, 93.65mol). Turn on the stirring and keep the temperature at 100±5°C to react for 42 hours. Concentrate under reduced pressure. Methanol was added to the concentrate to be beaten. The solid was filtered and dried under vacuum to obtain the product 4-(4-(7-bromo-6-methylthieno[3,2-D]pyrimidin-2-yl)amino)-1hydro-pyrazol-1-yl ) Piperidine-1-tert-butyl carbonate 37.26Kg, yield: 80.6%. LC-MS(ESI): m/z=493.1[M+H] + .
1 H NMR (400MHz, d 6 -DMSO): δ9.73 (s, 1H), 8.97 (s, 1H), 8.18 (s, 1H), 7.68 (s, 1H), 4.30-4.36 (m, 1H) ,4.01-4.04(m,2H),2.87-2.93(m,2H),2.53(s,3H),2.00-2.03(m,2H),1.70-1.80(m,2H),1.41(s,9H) .
Example 8: 4-(4-((7-(4-fluoro-2-methoxyphenyl)-6-methylthieno[3,2-D]pyrimidin-2-yl)amino)-1 Hydro-pyrazol-1-yl)piperidine-1-tert-butyl carbonate (Compound B)
Add purified water (113Kg), dioxane (390Kg), 4-(4-(7-bromo-6-methylthieno[3,2-D]pyrimidin-2-yl)amino) into the reactor -1H-pyrazol-1-yl)piperidine-1-tert-butyl carbonate (37.26Kg, 93.65mol), 2-methoxy-4-fluorophenylboronic acid pinacol ester (23.05Kg, 120.22mol) , Anhydrous potassium carbonate (20.95Kg, 151.8mol), palladium acetate (0.18Kg, 0.80mol) and 2-dicyclohexylphosphine-2,4,6-triisopropylbiphenyl (0.90Kg, 1.89mol). Under the protection of nitrogen, the temperature is controlled at 70±5℃ to react for 4 hours. Cool down to 40±5°C, add ammonia water (68Kg), and stir for 8 hours. Cool down to 20±5°C and dilute with water (1110Kg). Dichloromethane extraction twice (244Kg, 170Kg). Combine the organic phases, wash sequentially with water and then with saturated brine. Add 3-mercaptopropyl ethyl sulfide-based silica (4.0Kg, used to remove heavy metal palladium) into the organic phase, and stir at 40±5°C for 20 hours. After filtration, the filtrate was concentrated under reduced pressure. The remainder was slurried sequentially with methyl tert-butyl ether and ethanol. Filter and dry in vacuo to obtain 4-(4-((7-(4-fluoro-2-methoxyphenyl)-6-methylthieno[3,2-D]pyrimidin-2-yl)amino) -1H-pyrazol-1-yl)piperidine-1-tert-butyl carbonate 34.6Kg, yield: 68.6%. LC-MS(ESI): m/z=539.3[M+H] + .
1 H NMR (400MHz, d 6 -DMSO): δ9.46 (s, 1H), 8.94 (s, 1H), 7.76 (s, 1H), 7.38 (s, 1H), 7.33 to 7.35 (m, 1H) ,7.08-7.11(m,1H),6.91-6.95(m,1H),4.03-4.12(m,3H),3.73(s,3H),2.85-2.89(m,2H),2.39(s,3H) ,1.90-1.93(m,2H),1.55-1.60(m,2H),1.41(s,9H).
Comparative Example 1: 2-Chloro-6-methylthieno[3,2-D]pyrimidine (Compound I)
Into a 100mL reaction flask, add 10% palladium on carbon (0.1g), 2,4-dichloro-6-methylthieno[3,2-D]pyrimidine (2g, 9.2mmol), methanol (40mL) and N,N-Diisopropylethylamine (1.412 g, 10.9 mmol). Fill the bottle with hydrogen and control the hydrogen pressure at 0.5MPa. Turn on the stirring and keep the temperature at 25±5°C to react for 21 hours. Filter, collect the filtrate, concentrate the filtrate under reduced pressure, add ethanol (2.1 g) to the concentrate, and concentrate again to bring out residual tetrahydrofuran. Add ethanol (2.2g) and stir at 70±5°C until all solids are dissolved. Cool down, control the temperature at 25±5°C, add 13.3g of purified water dropwise to the kettle, control the dropping rate, and keep the temperature at 25±5°C. The solid product was precipitated, centrifuged, and the filter cake was vacuum dried to obtain 1.6 g of 2-chloro-6-methylthieno[3,2-D]pyrimidine as a product, with a yield of 54%. Methoxy substituted impurities in 20% yield.
Comparative Example 2: 2-Chloro-6-methylthieno[3,2-D]pyrimidine (Compound I)
After replacing the solvent tetrahydrofuran in Example 2 with ethyl acetate, the solubility of 2-chloro-6-methylthieno[3,2-D]pyrimidine in ethyl acetate was poor, and only a small amount of product was formed, which was not calculated Specific yield.
Comparative example 3: 4-(p-toluenesulfonyl)-piperidine-1-tert-butyl carbonate (Compound G)
Triethylamine (25mL), N-BOC-4-hydroxypiperidine (5g) were added to a 100mL reaction flask. P-toluenesulfonyl chloride (7.1g) was added in batches while controlling the temperature at 10±10°C. After the addition, the temperature is controlled at 25±5℃ to react for 25 hours. Monitoring by LC-MS showed a large amount of unreacted raw materials and the reaction liquid was black and red.
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| WO-2019228171-A1 | Salt of fused ring pyrimidine compound, crystal form thereof and preparation method therefor and use thereof | 2018-05-31 | |
| AU-2016295594-A1 | Fused ring pyrimidine compound, intermediate, and preparation method, composition and use thereof | 2015-07-21 | |
| AU-2016295594-B2 | Fused ring pyrimidine compound, intermediate, and preparation method, composition and use thereof | 2015-07-21 | 2020-04-16 |
| EP-3354653-A1 | Fused ring pyrimidine compound, intermediate, and preparation method, composition and use thereof | 2015-07-21 | |
| EP-3354653-B1 | Fused ring pyrimidine compound, intermediate, and preparation method, composition and use thereof | 2015-07-21 | 2019-09-04 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| JP-2018520202-A | Fused ring pyrimidine compounds, intermediates, production methods, compositions and applications thereof | 2015-07-21 | |
| KR-20180028521-A | Condensed ring pyrimidine-based compounds, intermediates, methods for their preparation, compositions and applications | 2015-07-21 | |
| US-10494378-B2 | Fused ring pyrimidine compound, intermediate, and preparation method, composition and use thereof | 2015-07-21 | 2019-12-03 |
| US-2018208604-A1 | Fused ring pyrimidine compound, intermediate, and preparation method, composition and use thereof | 2015-07-21 | |
| WO-2017012559-A1 | Fused ring pyrimidine compound, intermediate, and preparation method, composition and use thereof | 2015-07-21 |
| CTID | Title | Phase | Status | Date |
|---|---|---|---|---|
| NCT03412292 | MAX-40279 in Subjects With Acute Myelogenous Leukemia (AML) | Phase 1 | Recruiting | 2021-05-21 |
///////////////Orphan Drug, Acute myeloid leukaemia, MAX 40279, EX-A4057, Max 4, MAX-40279, MAX-40279-001, MAX-40279-01, PHASE 1, Maxinovel Pharmaceuticals
CC1=C(C2=NC(=NC=C2S1)NC3=CN(N=C3)C4CCNCC4)C5=C(C=C(C=C5)F)OC

{kind=link}
{kind=link}