
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
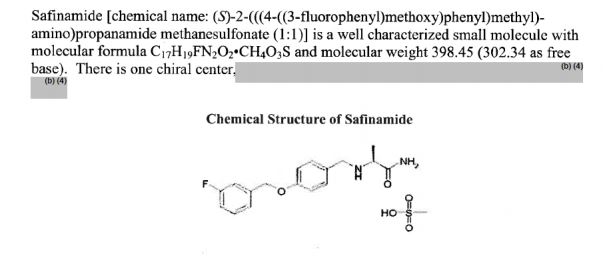
Safinamide
- Molecular Formula C17H19FN2O2
- Average mass 302.343 Da
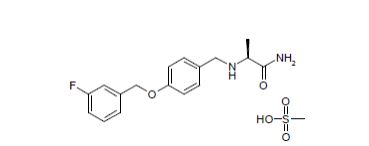
(+)-(S)-2-[[p-[(m-fluorobenzyl)oxy]benzyl]amino]propionamide monomethanesulfonate
Propanamide, 2-[[[4-[(3-fluorophenyl)methoxy]phenyl]methyl]amino]-, (2S)-, methanesulfonate
| Molecular Weight | 398.45 |
| Formula | C17H19FN2O2 ● CH4O3S |
CAS 202825-46-5 (Safinamide Mesylate)
Safinamide is a white to off-white, non-hygroscopic crystalline solid. It shows pH dependent solubility in aqueous buffers due to the secondary amine moiety, being soluble at acidic pH and practically insoluble at neutral pH.
It is freely soluble in de-ionized water, methanol and DMSO but practically insoluble in non-polar organic solvents.
Safinamide is chiral and possesses a single stereogenic centre.
Three crystalline forms are known. The anhydrous form selected for commercialisation is the most thermodynamically stable form, whilst the others are either not physiologically relevant or have very similar dissolution profiles. SOURCE EMA
Safinamide methanesulfonate was approved by European Medicine Agency (EMA) on Feb 22, 2015. It was developed by Newron and Zambon, then marketed as Xadago® by Zambon in EU.
FDA approved March 21, 2017,
- Chemistry Review(s) (PDF) for correct structure
- Chemistry Review(s) (PDF) for correct structure
Safinamide is a unique molecule with a novel dual mechanism of action based on the enhancement of the dopaminergic function (through potent reversible inhibition of MAO-B and of dopamine uptake) and inhibition of the excessive release of glutamate. It is indicated for the treatment of Parkinson’s disease (PD).
Xadago® is available as film-coated tablet for oral use, containing Eq. 50 mg/100 mg of free Safinamide. The recommended dose is 50 mg or 100 mg once daily.
March 21, 2017, Release
The U.S. Food and Drug Administration today approved Xadago (safinamide) tablets as an add-on treatment for patients with Parkinson’s disease who are currently taking levodopa/carbidopa and experiencing “off” episodes. An “off” episode is a time when a patient’s medications are not working well, causing an increase in Parkinson’s symptoms, such as tremor and difficulty walking.
“Parkinson’s is a relentless disease without a cure,” said Eric Bastings, M.D., deputy director of the Division of Neurology Products in the FDA’s Center for Drug Evaluation and Research. “We are committed to helping make additional treatments for Parkinson’s disease available to patients.”
An estimated 50,000 Americans are diagnosed with Parkinson’s disease each year, according to the National Institutes of Health, and about one million Americans have the condition. The neurological disorder typically occurs in people over age 60, though it can occur earlier, when cells in the brain that produce a chemical called dopamine become impaired or die. Dopamine helps transmit signals between the areas of the brain that produce smooth, purposeful movement – such as eating, writing, and shaving. Early symptoms of the disease are subtle and occur gradually. In some people, Parkinson’s disease progresses more quickly than in others.
The efficacy of Xadago in treating Parkinson’s disease was shown in a clinical trial of 645 participants who were also taking levodopa and were experiencing “off” time. Those receiving Xadago experienced more beneficial “on” time, a time when Parkinson’s symptoms are reduced, without troublesome uncontrolled involuntary movement (dyskinesia), compared to those receiving a placebo. The increase in “on” time was accompanied by a reduction in “off” time and better scores on a measure of motor function assessed during “on” time than before treatment.
In another clinical trial of 549 participants, the participants adding Xadago to their levodopa treatment had more “on” time without troublesome uncontrolled involuntary movement compared to those taking a placebo, and also had better scores on a measure of motor function assessed during “on” time than before treatment.
Certain patients should not take Xadago. These include patients who have severe liver problems, or who take a medicine used to treat a cough or cold called dextromethorphan. It also should not be taken by patients who take another medicine called a monoamine oxidase inhibitor (MAOI) because it may cause a sudden severe increase in blood pressure, or by those who take an opioid drug, St. John’s wort, certain antidepressants (such as serotonin-norepinephrine reuptake inhibitors, tricyclics, tetracyclics, and triazolopyridines), or cyclobenzaprine, because it may cause a life-threatening reaction called serotonin syndrome.
The most common adverse reactions observed in patients taking Xadago were uncontrolled involuntary movement, falls, nausea, and trouble sleeping or falling asleep (insomnia).
Serious, but less common, risks include the following: exacerbated high blood pressure (hypertension); serotonin syndrome when used with MAOIs, antidepressants, or opioid drugs; falling asleep during activities of daily living; hallucinations and psychotic behavior; problems with impulse control/compulsive behaviors; withdrawal-emergent hyperpyrexia (fever) and confusion; and retinal pathology.
The FDA granted approval of Xadago to Newron Pharmaceuticals.
Safinamide (INN; brand name Xadago) is a drug indicated for the treatment of Parkinson’s disease with monoamine oxidase B inhibiting and other methods of action.[2] It was approved in Europe in February 2015,[3] and in the United States on March 21, 2017[4]. It has also been tested for the use in patients with restless legs syndrome (RLS), but no study results have been published.

Medical uses
Safinamide has been approved by the European Medicines Agency for the treatment of adult patients with idiopathic Parkinson’s disease as add-on therapy to a stable dose of levodopa (L-dopa) alone or in combination with other Parkinson drugs in patients with mid-to-late-stage fluctuating disease.[5]
Contraindications
Safinamide is contraindicated in patients with severe liver impairment, with albinism, retinitis pigmentosa, severe diabetic neuropathy, uveitis and other disorders of the retina. Combination with other monoamine oxidase (MAO) inhibitors and pethidine is also contraindicated.[6]
Adverse effects
Common adverse events in clinical trials (in more than 1% of patients) included nausea, dizziness, tiredness, sleeplessness, orthostatic hypotension (low blood pressure), and headache. There was no significant difference in the occurrence of these effects between safinamide and placebo treated patients.[6][7]
In experiments with rats (but not in those with monkeys), retinopathies have been observed.[1][8]
Overdose
Expected overdose effects are hypertension (high blood pressure), orthostatic hypotension, hallucinations, psychomotor agitation, nausea, vomiting, and dyskinesia. In studies, a singe patient was suspected to have overdosed for a month; symptoms were confusion, drowsiness and mydriasis (dilation of the pupils) and subsided completely after the drug was discontinued. No specific antidote is available.[6]
Interactions
As a MAO inhibitor, safinamide can theoretically cause hypertensive crises, serotonin syndrome and other severe side effects when combined with other MAO inhibitors or with drugs that are known to interact with MAO inhibitors, such as pethidine, dextromethorphan, selective serotonin reuptake inhibitors (SSRIs), serotonin–noradrenaline reuptake inhibitors (SNRIs), tricyclic and tetracyclic antidepressants. An interaction with tyramine, a substance found in various foods, could be expected by the same reasoning but has been excluded in studies.[6]
Another theoretical interaction is with drugs with affinity to the transporter protein ABCG2 (also known as BCRP), such as pitavastatin, pravastatin, ciprofloxacin, methotrexat, and diclofenac; a study with the latter has shown no clinical relevance.[9] A study testing possible interactions with amidase inhibitors is part of the post-authorisation development plan.[1] There are no relevant interactions related to cytochrome P450 (CYP) liver enzymes, although one inactivation pathway of safinamide seems to be mediated by CYP3A4.[6]
Pharmacology
Mechanisms of action
Like the older antiparkinson drugs selegiline and rasagiline, safinamide is a selective monoamine oxidase B inhibitor, reducing degradation of dopamine; in contrast to the other two, its action is reversible. Safinamide also inhibits glutamate release[7][10] and dopamine reuptake.[11] Additionally, it blocks sodium and calcium channels,[10][12] the relevance of which for its antiparkinson action is however unknown.[6]
Pharmacokinetics
Safinamide is absorbed quickly and nearly completely from the gut and reaches highest blood plasma concentrations after 1.8 to 2.8 hours. There is no relevant first-pass metabolism; total bioavailability is 95%. The substance is bound to plasma proteins to 88–90%.[6]
The metabolism is not well understood. The principal step is mediated by amidases which have not been identified, and produces safinamide acid (NW-1153). Other relevant metabolites are O-debenzylated safinamide (NW-1199),[9] the N-dealkylated amine which is then oxidized to a carboxylic acid (NW-1689), and the glucuronide of the latter.[6][13] In tests with liver microsomes, dealkylation seemed to be mediated by CYP3A4, but other CYP enzymes appear to be involved as well. Safinamide acid binds to the organic anion transporter 3 (OAT3), but this has probably no clinical relevance. Safinamide itself transiently binds to ABCG2. No other transporter affinities have been found in preliminary studies.[6]
Safinamide is eliminated, mainly (>90%) in form of its metabolites, via the kidney, with an elimination half-life of 20 to 30 hours. Only 1.5% are found in the stool.[6]

Metabolism pathways of safinamide.[9][13] Enzymes: CYP = cytochrome P450, MAO-A = monoamine oxidase A, ALDH = aldehyde dehydrogenases, UGT = UDP-glucuronosyltransferases. Gluc = acyl glucuronide.
History
The compound was originally discovered at Farmitalia-Carlo Erba, which was acquired by Pharmacia in 1993. In 1995, Pharmacia merged with Upjohn. Safinamide was first disclosed in 1998.[14] In the course of a major restructuring in the same year, all rights for safinamide were transferred to the newly formed company Newron Pharmaceuticals, which developed the drug until it was sold to Merck KGaA in 2006.[15]
In 2007, a Phase III clinical trial was started, scheduled to run until 2011.[16] In October 2011 Merck, now Merck-Serono, announced that they would give all rights to develop the compound back to Newron because they wanted to prioritise other projects and had corrected their estimates for safinamide’s market potential downwards.[17]
The US Food and Drug Administration (FDA) refused to file Newron’s application in 2014 on formal grounds.[18] Newron re-applied in December 2014.[19] In spring 2015, the European Medicines Agency (EMA) approved the drug. Safinamide is the first antiparkinson medication to be approved for ten years.[8]
Research
Potential additional uses might be restless legs syndrome (RLS) and epilepsy.[20] They were being tested in Phase II trials in 2008, but no results are available.

(+)-(S)-2-[[p-[(m-fluorobenzyl)oxy]benzyl]amino]propionamide monomethanesulfonate
Propanamide, 2-[[[4-[(3-fluorophenyl)methoxy]phenyl]methyl]amino]-, (2S)-, methanesulfonate
| Molecular Weight | 398.45 |
| Formula | C17H19FN2O2 ● CH4O3S |
CAS 202825-46-5 (Safinamide Mesylate)
Safinamide is a white to off-white, non-hygroscopic crystalline solid. It shows pH dependent solubility in aqueous buffers due to the secondary amine moiety, being soluble at acidic pH and practically insoluble at neutral pH.
It is freely soluble in de-ionized water, methanol and DMSO but practically insoluble in non-polar organic solvents.
Safinamide is chiral and possesses a single stereogenic centre.
Three crystalline forms are known. The anhydrous form selected for commercialisation is the most thermodynamically stable form, whilst the others are either not physiologically relevant or have very similar dissolution profiles.SOURCE EMA
Safinamide methanesulfonate was approved by European Medicine Agency (EMA) on Feb 22, 2015. It was developed by Newron and Zambon, then marketed as Xadago® by Zambon in EU.
FDA approved March 21, 2017
Safinamide is a unique molecule with a novel dual mechanism of action based on the enhancement of the dopaminergic function (through potent reversible inhibition of MAO-B and of dopamine uptake) and inhibition of the excessive release of glutamate. It is indicated for the treatment of Parkinson’s disease (PD).
Xadago® is available as film-coated tablet for oral use, containing Eq. 50 mg/100 mg of free Safinamide. The recommended dose is 50 mg or 100 mg once daily.
SYNTHESIS
 |
|
| Safinamide has been obtained by reductocondensation of 4-(3-fluorobenzyloxy)benzaldehyde (I) with L-alaninamide (II) by means of sodium cyanoborohydride in methanol.EP 0400495; EP 0426816; JP 1992500215; US 5236957; US 5391577; US 5502079; WO 9014334 |
CLIP
http://pubs.rsc.org/en/content/articlehtml/2016/sc/c6sc00197a

Scheme 2 Synthesis and isolation of [18F]safinamide, [18F]FMT, and [18F]mFBG.
PATENT
Safinamide (NW- 1015, FCE-26743A, PNU- 151774E) is a sodium channel blocker, a calcium channel modulator, a monoamino oxidase B (MAO-B) inhibitor, a glutamate release inhibitor and a dopamine metabolism modulator. Safinamide is useful in the treatment of CNS disorders, in particular of epilepsy, Parkinson’s disease, Alzheimer’s disease, depression, restless legs syndrome and migraine (WO 90/ 14334, WO 2004/089353, WO 2005/ 102300 and WO 2004/062655). Ralfinamide (NW- 1029, FCE-26742A, PNU-0154339E) is a sodium channel blocker useful in the treatment of pain conditions, including chronic pain and neuropathic pain, migraine, bipolar disorders, depressions, cardiovascular, inflammatory, urogenital, metabolic and gastrointestinal disorders (WO 99/35125, WO 03/020273, WO 2004/062655, WO 2005/018627, WO 2005/070405, WO 2005/ 102300).
In particular, safinamide is specifically described in WO 90/ 14334. Safinamide, its R-enantiomer, their racemic mixture and their salts with pharmaceutically acceptable acids and the use thereof for the preparation of pharmaceutical compositions active as anti-epileptic, anti-Parkinson, neuroprotective, antidepressant, antispastic and/or hypnotic agents are specifically claimed in WO 90/ 14334. Ralfinamide is specifically described in WO 90/ 14334. Ralfinamide, its R- enantiomer, their racemic mixture and their salts with pharmaceutically acceptable acids and their use thereof for the preparation of pharmaceutical compositions active as anti-epileptic, anti-Parkinson, neuroprotective, antidepressant, antispastic and/or hypnotic agent are comprised by the claims of WO 90/ 14334.
Moreover, the use as analgesics of safinamide, ralfinamide, the respective R-enantiomers, the respective racemic mixtures and their salts with pharmaceutically acceptable acids is claimed in WO 99/035125. WO 2006/027052 A2 specifically discloses and claims the use of the single R-enantiomer of ralfinamide i.e., (R)-2-[4-(2- fluorobenzyloxy)benzylamino]propanamide (I’b), and its salts with pharmaceutically acceptable acids as a selective sodium and calcium channel modulator for the selective treatment of pathological affections wherein sodium or calcium channel mechanism(s) play(s) a pathological role, including pain, migraine, inflammatory processes affecting all body systems, disorders affecting skin and related tissue, disorders of the respiratory system, disorders of the immune and endocrinological systems, gastrointestinal, and urogenital disorders, wherein the therapeutical activity of said compound is substantially free from any MAO inhibitory side effect or exhibits significantly reduced MAO inhibitory side effect.
It has now been discovered that the large scale preparations of safinamide and ralfinamide according to the methods described in the prior art, contain two undesired impurities, i.e., respectively, (S)-2-[3-(3- fluorobenzyl)-4-(3-fluorobenzyloxy)-benzylamino]propanamide (Ha) and (S)- 2-[3-(2-fluorobenzyl)-4-(2-fluorobenzyloxy)-benzylamino]propanamide (lib), and their salt, in particular the respective methanesulfonates (lie) and (Hd)
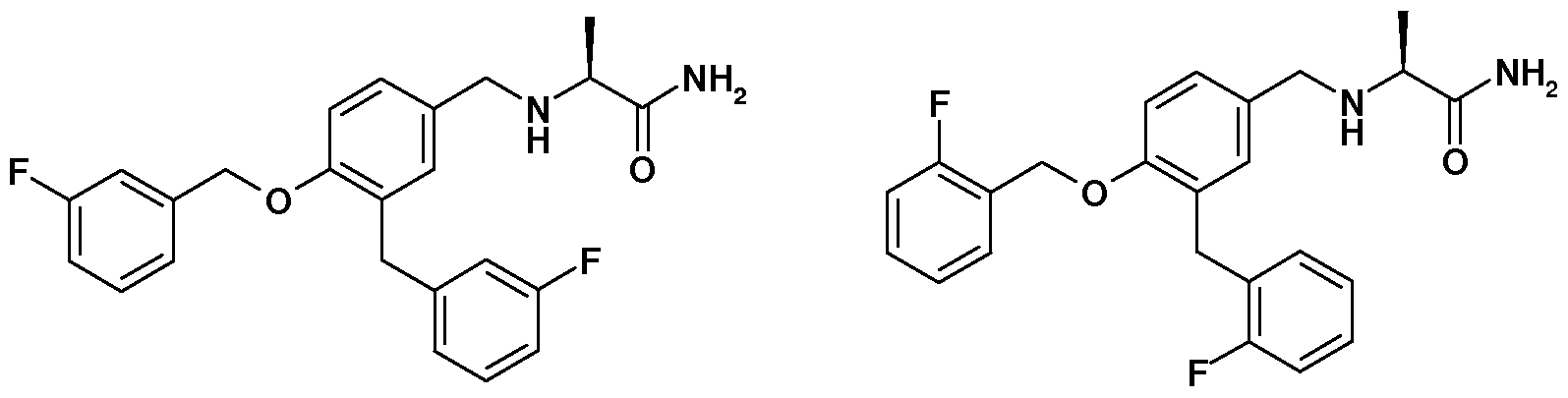
(Ha) (lib)
The same situation occurs with the preparation according the prior art methods for the R-enantiomers (I’a) and (I’b) of, respectively, safinamide and ralfinamide, the respective racemic mixtures (Ia, I’a) and (Ib, I’b), and the salts thereof with pharmaceutically acceptable acids, (I’c), (I’d) and the respective racemic mixtures (Ic, I’c) and (Id, I’d) in particular the methanesulfonates, which result to be contaminated by the respective R isomers (Il’a), (Il’b), (II’c), and (Il’d) of the above identified impurities (Ha), (lib), (lie) and (Hd) or the respective racemic mixtures (Ha, Il’a), (lib, Il’b), (Hc, II’c) and (Hd, Il’d).

PATENT
Parkinson’s disease (PD) is a progressive neurodegenerative disease characterized by bradykinesia, rigidity, resting tremor, and ataxia. These symptoms are caused by decreased dopamine release in the striatum. Clinically, PD is defined by presence of Lewy bodies, intracellular neuronal inclusions in the substantia nigra and at other sites in the brain. Estimated prevalence of this disease is 100 to 200 per 100,000 population including males and females across the entire age group. Current treatment for PD comprises dopaminergic medications that include levodopa, dopamine agonists (DAs), monoamine oxidase-B (MAO-B) inhibitors. Figure 1 provides few examples of pharmaceutically important benzyloxy-benzylamine derivatives. Many of these benzyl oxy-benzylamines with various amine functions were studied and has been patented as sodium channel blockers. Among them, safinamide ((5)-N2– {4-[3- fluorobenzyl)oxy] benzyl}- alaninamide methanesulfonate) is a noted example which is under phase III clinical trials for treatment of Parkinson’s disease. Its mechanism of action is manifold which comprise MAO-B and dopamine uptake inhibition. Further, safinamide is believed to block voltage-dependent sodium channels, modulates calcium channels and reduction of glutamate release in the central nervous system. WOl 998003472 discloses serinamide, glycinamide, alaninamide and phenylalaninamide derivatives of a compound (I). These compounds (I) are useful for the treatment of neurological diseases.
EP2474521 discloses high purity degree (S)-2-[4-(3-fluorobenzyloxy)- benzylamino]propanamide (safinamide) or (S)-2-[4-(2-fluorobenzyloxy)- benzylamino]propanamide (ralfinamide) or a salt thereof with a pharmaceutically acceptable acid with a content of the respective impurity (S)-2-[3-(3-fluorobenzyl)-4-(3- fluorobenzyloxy)-benzylamino]propanamide or (S)-2-[3-(2-fluorobenzyl)-4-(2- fluorobenzyloxy)-benzylamino]propanamide.
US2009149544 relates to novel alpha- aminoamide derivatives, their pharmaceutically acceptable salts, solvates, and hydrates thereof. The application also provides compositions comprising a compound and the use of such compositions in methods of treating diseases and conditions that are beneficially treated by administering an inhibitor of monoamine oxidase type B (MAO-B) and/or a sodium (Na.sup.+) channel blocker, and/or a calcium (Ca.sup.2+) channel modulator.
The strategy employed in the art to prepare benzyloxy-benzylamine derivatives including safinamide or its analogue ralfinamide is chiral pool approach starting from L-alaniriamide and reductively aminating with 4-(3-fluorobenzyloxy) benzaldehyde. Although this method is very simple and straightforward, it suffers from several serious drawbacks, such as need to use toxic reagents such as sodium cyanoborohydride and further formation of toxic by-products such as hydrogen cyanide and sodium cyanide and other toxic impurities in large-scale production Importantly, the possibility of generating a range of safinamide analogues by means of the chiral-pool approach is limited in terms of the structure and stereochemistry of the products because of inadequacies in the availability of D-alaninamide and its analogues
Hence, the developments of newer methods for the preparation of compounds of formula (I) comprising safinamide and related analogues are highly desirable
Example 2: Synthesis of (R)-l-(benzyIoxy)propan-2-ol [(R)-compound 3]
To a solution of (7? benzyl glycidyl ether [fR)-compound 2] (4 g, 24.4 mmol) in dry THF (10 mL) at 0 °C, a pre-cooled solution of lithium aluminium hydride (1.4 g, 36.6 mmol) in anhydrous THF (10 mL) was added slowly with stirring under nitrogen. After 60 min, the reaction mixture was quenched with 1 ml of water and 1 ml of 15 % NaOH solution and the content was stirred for 15 min. The inorganic precipitate was filtered, washed with ethyl acetate and the solvent evaporated under reduced pressure. The residue was purified by a short filtration column to afford (-fl)-compound 3 as a colorless oil (3.8 g, 95%); [a]22D = -14.5 (c 2, CHC13); IR (CHC13): vmax3418, 3087, 3063, 3030, 2963, 2924, 1952, 1873, 1600, 1495, 1454, 1363, 1244, 1099, 1028, 918, 808, 698 cm“1; Ή NMR (200 MHz, CDC13): δΗ 1.13 (d, J = 6.3 Hz, 3H), 2.5 (bs, 1H), 3.23-3.32 (dd, J = 9.8, 1.3 Hz, 1H), 3.43-3.49 (dd, J = 9.45, 3.2 Hz, 1H), 3.91-4.03 (m, 1H), 4.55 (s, 2H), 7.25-7.37 (m, 5H); I3C NMR (50 MHz, CDC13): 5C 137.8 (C), 128.3 (CH, 2 carbons), 127.7 (CH, 3 carbons), 75.7 (CH2), 73.2 (CH2), 66.4 (CH), 18.6 (CH3); MS: m/z 189 [M+Na]+.
Example 3: Synthesis of (S)-((2-azidopropoxy)methyl)benzene [(S)- compound 4]
To a stirred solution of secondary alcohol ( )-compound 3 (3 g, 18.1 mmol) in dry dichloromethane (25 mL), Et3N (3.1 mL, 21.7 mmol) at 0 °C was added, followed by drop wise addition of mesyl chloride (1.8 mL, 21.7 mmol). The reaction mixture was stirred at 0°C for 2 hours, subsequently at room temperature for 3 hours under a nitrogen atmosphere. After completion of the reaction (indicated by TLC), the reaction mixture was diluted with dichloromethane and washed with a saturated solution of sodium bicarbonate (30 mL) and water (2 x 10 mL). The organic layer was separated, dried over anhydrous Na2S04, filtered, and concentrated under reduced pressure to give the O-mesyl compound (4.3 g; crude).
To a solution of the crude 0-mesyl compound (4 g, 16.37 mmol) in dry DMF (10 mL), sodium azide (1.6 g, 24.55 mmol) was added and the reaction mixture was heated at 60°C for 6 hours under nitrogen atmosphere. After completion of the reaction (indicated by TLC), water (10 mL) was added to the reaction mixture, then extracted with ethyl acetate (2 x 15 mL). The combined organic layers were washed with brine solution, dried over anhydrous Na2S04, filtered, and concentrated under reduced pressure. Purification of the crude residue was done by column chromatography (silica gel, petroleum ether/EtOAc, 95:5) to yield (¾)-compound 4 as a colorless oil. (2.8 g; 89%); [a]22D = +6.1 (c 1.3, CHC13); IR (CHC13): vmax 3394, 3032, 2977, 2864, 2500, 2104, 1724, 1641 , 1496, 1454, 1363, 1269, 1 101 , 913, 698 αη ‘,Ή NMR (200 MHz, CDC13): δΗ 1.20 (d, J = 6.7 Hz, 3H), 3.39-3.54 (m, 2H), 3.61-3.77 (m, 1H), 4.57 (s, 2H), 7.25-7.39 (m, 5H); 13C NMR (50 MHz, CDC13): 5C 137.8 (C), 128.4 (CH, 2 carbons), 127.7 (CH), 127.5 (CH, 2 carbons), 73.7 (CH2), 73.2 (CH2), 56.9 (CH), 16.1 (CH3);MS: m/z 214 [M+Na]+.
Example 4: Synthesis of (S)-N-(l-hydroxypropan-2-yl)-2-nitrobenzenesulfonamide [(S)- compound 5]
To a solution of ^-compound 4 (2.5 g, 13.1 mmol) in methanol (15 mL), trifluoroacetic acid (2 mL) and palladium hydroxide on activated carbon (0.05 g, 10-20 wt %) were added and the reaction mixture was stirred under hydrogen (60 psi) for 8 hours. After completion of the reaction (indicated by TLC), the catalyst was filtered over a plug of celite and the solvent was evaporated under reduced pressure to half of its volume which was basified with 2.5 M methanolic NaOH. Evaporation of the remaining solvent under reduced pressure was done followed by filtration of the residue through a short bed of basic alumina (eluent; MeOH) to obtain the amino alcohol as a pale brown oil (0.94 g, crude) which was subjected to the next reaction without further purification.
To a solution of amino alcohol (0.9 g, 1 1.98 mmol) in dry dichloromethane (5 mL), 2-nitrobenzenesulfonylchloride (3.2 g, 14.37 mmol) in dichloromethane (8 mL) and triethylamine (2.6 mL, 17.97 mmol) at 0 °C were slowly added under nitrogen atmosphere. The solution was stirred for 2 hours. After completion of the reaction (indicated by TLC), water (10 mL) was added to the reaction mixture, then extracted with dichloromethane (2 x 15 mL). The combined organic layers were washed with brine solution, dried over anhydrous Na2S04, filtered, and concentrated under reduced pressure. Purification of the crude residue was done by column chromatography (silica gel, petroleum ether/EtOAc, 60:40) to yield (S)- compound 5 as a pale yellow oil (2.33 g, 75% ); [a]22D = +80.2 (c 2.1, CHClj); IR (CHC13): vmax 3546, 3367, 3022, 2883, 2401, 1594, 1542, 1412, 1362, 1216, 1170, 1 125, 1059, 971, 854, 668 cm“1; ]H NMR (200 MHz, CDC13): δΗ 1.13 (d, J = 6.5 Hz, 3H), 2.16 (bs, 1H), 3.45-3.70 (m, 3H), 5.61 (d, J = 6.6 Hz, 1H), 7.73-7.80 (m, 2H), 7.86-7.91 (m, 1H), 8.13-8.22 (m, 1H); 13C NMR (50 MHz, CDC13): 5C 147.8 (C), 134.4 (C), 133.7 (CH), 133.0 (CH), 130.9 (CH), 125.5 (CH), 66.2 (CH2), 52.5 (CH), 17.8 (CH3); MS: m/z 283 [M+Na]+.
Example 5: Synthesis of l-fluoro-3-(iodomethyl)benzene ( compound 7)
To a stirred solution of triphenyl phosphine (4.15 g, 15.85 mmol), imidazole (1.1 g, 15.85 mmol) in dry dichloromethane (20 mL), iodine (4.8 g, 19.02 mmol) at 0°C was added and the solution was stirred for 5 min. To this, 3-fluoro benzyl alcohol (compound 6) (2 g, 15.85 mmol) dissolved in dichloromethane (5 mL) was added drop wise over 10 min and the stirring was continued for 1 hour with exclusion of light. After completion of the reaction (indicated by TLC), the reaction mixture was quenched by addition of an aqueous Na2S203 solution (15 mL), then extracted with dichloromethane (2 x 20 mL). The combined organic layers were washed with brine solution, dried over anhydrous Na2S04, filtered, and concentrated under reduced pressure. Purification of the crude residue was done by column chromatography (silica gel, petroleum ether/EtOAc, 95:5) to yield compound 7 as a colorless oil (3.5 g, 95% ); (IR (CHC13): vmax 3460, 3060, 2965, 1695, 1613, 1593, 1482, 1446, 1259, 1 156, 1068, 944, 871, 782, 736, 686 cm“1 ; Ή NMR (200 MHz, CDC13): δΗ 4.42 (s, 2H), 6.89-6.99 (m, 1H), 7.05-7.17 (m, 2H), 7.21-7,29 (m, 1H); 13C NMR (50 MHz, CDC13): 6C 165.0 (C), 141.6 (C), 130.2 (CH), 124.4 (CH), 1 15.9 (CH), 1 14.7 (CH), 3.9 (C¾).
Example 6: Synthesis of (4-((3-flurobenzyl)oxy)phenyl)methanol (compound 8)
To a stirred solution of 4-(hydroxymethyl)phenol (1.57 g, 12.7 mmol) and K2C03 (8.8 g, 63.55 mmol) in dry acetonitrile (25 mL), compound 7 (3 g, 12.7 mmol) in acetonitrile was slowly added and the reaction mixture was heated at 70°C for 6 hours. After completion of the reaction (indicated by TLC), water (20 mL) was added to the reaction mixture, then extracted with ethylacetate (3 x 20 mL). The combined organic layers were washed with brine solution, dried over anhydrous Na2S04, filtered, and concentrated under reduced pressure. Purification of the crude residue was done by column chromatography (silica gel, petroleum ether/EtOAc, 70:30) to yield compound 8 as a colorless solid (2.7 g, 91% ); mp 63-65 °C; IR (CHC13): vmax 3422, 3017, 1612, 1512, 1489, 1381, 1216, 1 174, 1020, 829, 668 cm“1; Ή NMR (200 MHz, CDC13): δΗ 4.61 (s, 2H), 5.06 (s, 2H), 6.91-6.98 (m, 2H), 7.00-7.06 (m, 1H), 7.12-7.20 (m, 2H), 7.25-7.37 (m, 3H); 13C NMR (50 MHz, CDC13): 5C 165.4 (C), 160.5 (C), 158.0 (C), 139.6 (C), 133.5 (CH), 130.2 (CH), 128.7 (CH, 2 carbons), 122.7 (CH), 1 14.8 (CH, 2 carbons), 1 13.9 (CH), 69.1 (CH2), 64.9 (CH2); MS: m/z 255 [M+Na]+.
Example 7: Synthesis of l-fluoro-3-((4-(iodomethyl)phenoxy)methyI)benzene (compound 9)
To a stirred solution of triphenyl phosphine (2.82 g, 10.8 mmol), imidazole (0.73 g, 10.76 mmol) in dry dichloromethane (20 mL), iodine (3.27 g, 12.9 mmol) at 0 °C was added and the solution was stirred for 5 min. To this, compound 8 (2.5 g, 10.8 mmol) dissolved in dichloromethane (5 mL) was added drop wise over 10 min and the stirring was continued for 1 hour with exclusion of light. After completion of the reaction (indicated by TLC), the reaction mixture was quenched by addition of an aqueous Na2S203 solution (15 mL), then extracted with dichloromethane (2 x 20 mL). The combined organic layers were washed with brine solution, dried over anhydrous Na2S04, filtered, and concentrated under reduced pressure. Purification of the crude residue was done by column chromatography (silica gel, petroleum ether/EtOAc, 95:5) to yield compound 9 as a colorless oil (3.4 g, 93%); IR (CHC13): vmax 3503, 3033, 2925, 2089, 1607, 1509, 1488, 1381, 1301, 1250, 1 155, 1079, 944, 869, 776, 684 cm“1; 1H NMR (200 MHz, CDC13): δΗ 4.47 (s, 2H), 5.04 (s, 2H), 6.85-6.91 (m, 2H), 6.96-7.02 (m, 1H), 7.05-7.12 (m, 1H), 7.16-7.20 (m, 1H), 7.29-7.40 (m, 3H).
,3C NMR (50 MHz, CDC13): 6C 165.4 (C), 160.5 (C), 158.1 (C), 131.9 (C), 130.2 (CH), 130.1 (CH, 2 carbons), 122.7 (CH), 1 15.1 (CH, 2 carbons), 1 14.7 (CH), 1 13.9 (CH), 69.2 (CH2), 6.33 (CH2).
Example 8: Synthesis of (S)-N-(4-((3-flurobenzyl)oxy)benzyl)-N-(l-hydroxypropan-2-yl)-2-nitrobenzenesulfonamide [(S)-compound 10]
To a stirred solution of (^-compound 5 (1 g, 3.8 mmol) and K2C03 (2.65 g, 19.2 mmol) in dry acetonitrile (25 mL), compound 9 (1.84 g, 5.4 mmol) in acetonitrile was slowly added and the reaction mixture was heated at 70°C for 72 hours. After completion of the reaction (indicated by TLC), water (20 mL) was added to the reaction mixture, then extracted with ethylacetate (3 15 mL). The combined organic layers were washed with brine solution, dried over anhydrous Na2S04, filtered, and concentrated under reduced pressure. Purification of the crude residue was done by column chromatography (silica gel, petroleum ether/EtOAc, 80:20) to yield (¾)-compound 10 as a colorless oil (1.46 g, 80% ); [a]22D = +5.4 (c 1.5, CHC13); IR (CHC13): vmax 3445, 3020, 2928, 2400, 1613, 1544, 1512, 1453, 1371, 1216, 1 162, 1029, 852, 668 cm“1; 1H NMR (200 MHz, CDC13): δΗ 1.07 (d, J = 6.9 Hz, 3H), 1.91 (t, J = 5.2 Hz, 1H), 3.41-3.53 (m, 2H), 4.05-4.22 (m, 1H), 4.37-4.57 (m, 2H), 5.02 (m, 2H), 6.87 (d, J = 8.53 Hz, 2H), 6.97-7.12 (m, 2H), 7.20 (d, J = 7.2 Hz, 2H), 7.32 (d, J = 8.7 Hz, 2H), 7.47-7.67 (m, 3H), 7.89 (d, J = 8.09 Hz, 1H); 13C NMR (50 MHz, CDC13): 6C 165.5 (C), 160.6 (C), 158.4 (C), 147.7 (C), 139.6 (C), 134.1 (C), 133.4 (CH), 131.6 (CH), 131.4 (CH), 130.3 (CH), 129.7 (CH, 2 carbons), 124.1 (CH), 122.8 (CH), 115.1 (CH), 114. 9 (CH, 2 carbons), 114.0 (CH), 69.2 (CH2), 64.3 (CH2), 56.2 (CH), 46.9 (CH2), 15.4 (CH3); MS: m/z 497 [M+Na]+.
Example 9: Synthesis of (S)-2-(N-(4-((3-fluorobenzyl)oxy)benzyl)-2-nitrophenylsulfonamido) propanoic acid [(S)-compound 11]
A mixture of (S compound 10 (1.25 g, 2.6 mmol), TEMPO (0.028 g, 0.18 mmol), acetonitrile (20 mL), and sodium phosphate buffer (16 mL, 0.67 M, pH 6.7) was heated to 35°C. Next, sodium chlorite (0.47 g dissolved in 2 mL water, 7.9 mmol) and diluted bleach (4-6%, 0.09 mL diluted in 1 mL water) were added simultaneously over 1 hour. The reaction mixture was stirred at 35°C until the reaction was complete (3 hours, TLC), then cooled to room temperature. Water (30 mL) was added and the pH adjusted to 8 with 2 M NaOH. The reaction was quenched by pouring it into ice cold Na2S03 solution maintained at <20°C. After stirring for 30 min at room temperature, ethyl acetate (20 mL) was added and the stirring was continued for an additional 15 min. The organic layer was separated and discarded. More ethyl acetate (20 mL) was added, and the aqueous layer was acidified with 1 M HC1 to pH 3-4. The organic layer was separated, washed with water (2 x 15 mL), brine and concentrated under reduced pressure to afford the carboxylic acid (S -compound 1 1 (1.1 g, 85%); [ ]22ο = -20.4 (c 1.1, CHC13); IR (CHC13): vmax 3398, 3095, 1718, 1612, 1591, 1543, 1512, 1489, 1457, 1371, 1303, 1251, 1163, 1059, 900, 852, 831 , 778, 684 cm“1; 1H NMR (200 MHz, CDC13): 8H 1.44 (d, J = 7.3 Hz, 3H), 4.23 (d, J = 15.6 Hz, 1H), 4.64 (d, J = 15.6 Hz, 1H), 4.82-4.90 (q, J = 7.4 Hz, 1H), 4.92 (s, 2H), 6.68 (d, J = 8.6 Hz, 2H), 6.89-7.01 (m, 2H), 7.07-7.13 (m, 3H), 7.18-7.33 (m, 2H), 7.43-7.55 (m, 3H), 8.81 (bs, 1H); 13C NMR (50 MHz, CDC13): 5C 176.5 (CO), 165. 0 (C), 158.0 (C), 147.4 (C), 139.4 (C), 134.1 (C), 133.2 (CH), 131.4 (CH), 130.3 (CH), 129.9 (CH, 2 carbons), 128.4 (C), 124.1
(CH), 122.6 (CH), 1 15.0 (CH), 114.6 (CH, 2 carbons), 1 14.3 (CH), 1 13.8 (CH) 69.1 (CH2), 56.1 (CH), 49.0 (CH2), 16.8 (CH3); MS: m/z 51 1 [M+Na .
Example 10: Synthesis of (S)-2-(N-(4-((3-fluorobenzyI)oxy)benzyl)-2-nitrophenylsulfonamido) propanamide [(S)- compound 12]
To a solution of carboxylic acid (¾)-compound 1 1 (1 g, 2.04 mmol) and triethyl amine (0.34 mL, 2.4 mmol) in dry THF (20 mL), ethyl chloroformate (0.21 mL, 2.2 mmol) at 0 °C was added under nitrogen atmosphere. After 1 hour, ammonium hydroxide (25% w/v aqueous solution, 1.4 mL, 10.2 mmol) was added and the resulting reaction mixture was stirred at room temperature for 16 hours. After completion of the reaction, potassium carbonate (0.29 g, 2.1 mmol) was added and the reaction mixture was filtered, and washed with ethylacetate. The solvent was removed under reduced pressure and the crude product was subjected to column chromatography (silica gel, petroleum ether/EtOAc, 50:50) to obtain sulfonamide (Sj-compound 12 as a colorless oil (0.9 g, 91%); [a]22D = -32.1 (c 1.2, CHC13); IR (CHC13): vmax 3472, 1961 , 161 1, 1592, 1542, 1511, 1449, 1371, 1304, 1243, 1 163, 1060, 1029, 895, 852, 684 cm“1; Ή NMR (200 MHz, CDC13): δΗ 1.43 (d, J = 7.1 Hz, 3H), 4.44 (d, J = 15.4 Hz, 1H), 4.59 (d, J = 15.5 Hz, 1H), 4.60-4.71 (q, J= 7.0 Hz, 1 H), 5.01 (s, 2H), 5.50 (bs, 1H), 6.31 (bs, 1H), 6.78 (d, J = 8.71 Hz, 2H), 6.98-7.1 1 (m, 2H), 7.15-7.22 (m, 3H), 7.31-7.45 (m, 2H), 7.59-7.64 (m, 3H);13C NMR (50 MHz, CDC13): 5C 172.3 (CO), 165.5 (C), 158.2 (C), 147.5 (C), 139.6 (C), 139.4 (C), 133.6 (CH), 131.7 (CH), 130.5 (CH, 2 carbons),130.3 (CH), 128.1 (C), 124.2 (CH), 122.7 (CH), 1 15.1 (CH), 1 14.7 (CH, 2 carbons),1 14.4 (CH), 1 13.9 (CH), 69.0 (CH2), 55.7 (CH), 48.3 (CH2), 14.9 (CH3); MS: m/z 510 [M+Na]+.
Example 11: Synthesis of (S)-2-((4-((3-fluorobenzyl)oxy) benzyl) amino) propanamide [(S)-compound of formula I]
To a solution of sulfonamide (S)- compound 12 (0.8 g, 1.64 mmol), potassium carbonate (0.56 g, 4.9 mmol) in dry DMF (10 mL), thiophenol (0.2 mL, 1.9 mmol) was added. The reaction mixture was vigorously stirred for 6 hours. After completion of the reaction (indicated by TLC), water (10 mL) was added to the reaction mixture, then extracted with ethylacetate (2 x 20 mL). The combined organic layers were washed with brine solution, dried over anhydrous Na2S04, filtered, and concentrated under reduced pressure. Purification of the crude residue was done by column chromatography (silica gel, petroleum ether/EtOAc, 60:40) to yield (S) -compound of formula I as a colorless solid (0.43 g, 86% ); mp 207-09 °C; [a]22D = +3.89 (c 1.55, CHC13); IR (CHC13): vmax 3341, 2970, 2927, 2853, 1648, 1592, 1512, 1489, 1445, 1406, 1384, 1254, 1176, 1 137, 1030, 953, 928, 829, 680 cm“1; Ή NMR (200 MHz, CDC13): δΗ 1.34 (d, J = 6.9 Hz, 3H), 2.49 (bs, 2H), 3.19-3.30 (q, J = 6.8 Hz, 1H), 3.63-3.78 (dd, J = 19.4, 3.9 Hz, 2H), 5.05 (s, 2H), 5.85 (bs, 1H), 6.95 (d, J = 8.7 Hz, 2H), 7.00-7.06 (m, 1H), 7.13-7.24 (m, 4H), 7.29-7.40 (m, 1H). 13C NMR (50 MHz, CDC13): 8C 178.3 (CO), 165.4 (C), 157.7 (C), 139.6 (C), 132.1 (C), 130.2 (CH), 129.3 (CH, 2 carbons), 122.7 (CH), 1 14.9 (CH, 2 carbons), 1 14.6 (CH), 1 13.9 (CH), 69.2 (CH2), 57.5 (CH), 51.9 (CH2), 19.6 (CH3); MS: m/z 302 [M]+, 325 [M+Na]+.
Example 12: Synthesis of (S)-Safinamide mesylate
To a stirred solution of (^-compound of formula I (0.1 g, 0.33 mmol) in ethylacetate (3 mL) at 70°C, methanesulfonic acid (0.02 mL, 0.33 mmol) was added and the reaction mixture was stirred for 2 hours. Subsequently, the temperature was lowered to 35°C and the stirring was continued for additional 1 hour. The solvent was evaporated under reduced pressure and the residue was filtered through a short bed of basic alumina [eluent: EtOAc/MeOH; (95:5)] to obtain safinamide mesylate as a white solid (0.11 g, 90%); mp 209-10 °C [lit.7mp 210]; [a]22D = +9.6 (c 1.1, AcOH); {lit.7 [a] D = +12.9 (c 1.1, AcOH)} ee >98% [The ee of safinamide mesylate was determined by chiral HPLC analysis; Chiralcel OD-RH (150 x 4.6 mm) column; eluent:
Methanol/ Acetonitrile/Buffer-TEAP, pH 3 (20: 10:70); flow rate 0.5 mL/min (780 psi); detector: 224 nm] [f¾)-isomer tR = 1 1.55 min, (SJ-isomer tR = 12.94 min].

PAPERS
Synthesis2014, 46, 1751-1756.

N2-{4-[(3-Fluorobenzyl)oxy]benzyl}-L-alaninamide [(S)-14] BASE FORM
PhSH (0.2 mL, 1.9 mmol) was added to a solution of sulfonamide (S)-13 (0.8 g, 1.64 mmol) and K2CO3 (0.56 g, 4.9 mmol) in anhyd DMF (10 mL), and the mixture was vigorously stirred for 6 h. When the reaction was complete (TLC), H2O (10 mL) was added and the mixture was extracted with EtOAc (2 × 20 mL). The organic layers were combined, washed with brine (2 × 10), dried (Na2SO4), filtered, and concentrated under reduced pressure. The crude residue was purified by column chromatography [silica gel, PE–EtOAc(60:40)] to give a colorless solid; yield: 0.43 g (86%); mp 207–09 °C;
[α]D22 +3.89 (c 1.55, CHCl3).
IR (CHCl3): 3341, 2970, 2927, 2853, 1648, 1592, 1512, 1489, 1445,1406, 1384, 1254, 1176, 1137, 1030, 953, 928, 829, 680 cm–1.
1H NMR (200 MHz, CDCl3): δH = 1.34 (d, J = 6.9 Hz, 3 H), 2.49 (brs, 2 H), 3.19–3.30 (q, J = 6.8 Hz, 1 H), 3.71 (dd, J = 19.4, 3.9 Hz, 2H), 5.05 (s, 2 H), 5.85 (br s, 1 H), 6.95 (d, J = 8.7 Hz, 2 H), 7.00–7.06 (m, 1 H), 7.13–7.24 (m, 4 H), 7.29–7.40 (m, 1 H).
13C NMR (50 MHz, CDCl3): δC = 178.3 (CO), 165.4 (C), 157.7 (C),139.6 (C), 132.1 (C), 130.2 (CH), 129.3 (CH, 2 C), 122.7 (CH), 114.9 (CH, 2 C), 114.6 (CH), 113.9 (CH), 69.2 (CH2), 57.5 (CH),51.9 (CH2), 19.6 (CH3).
MS: m/z = 302 [M]+, 325 [M + Na]+.
(S)-Safinamide Mesylate (1)
MsOH (0.02 mL, 0.33 mmol) was added to a stirred solution of sulfonamide (S)-14 (0.1 g, 0.33 mmol) in EtOAc (3 mL) at 70 °C, and the mixture was stirred for 2 h. The temperature was then lowered to 35 °C, and the mixture was stirred for an additional 1 h. The solvent was evaporated under reduced pressure and the residue was filtered
through a short bed of basic alumina with elution by EtOAc–MeOH; (95:5) to give a white solid; yield: 0.11 g (90%);
mp 209–210 °C [Lit.7a 210 °C];
[α]D22 +9.6 (c 1.1, AcOH); {Lit.7 [α]D22+12.9 (c 1.1, AcOH)}.
Chiral HPLC: column: Chiralcel OD-RH (150 × 4.6 mm); eluent:MeOH–MeCN–buffer-TEAP (pH 3) (20:10:70); flow rate: 0.5mL/min (780 psi); detector: 224 nm [(R)-isomer: tR = 11.55 min;
(S)-isomer: tR = 12.94 min]; ee >98%.
7a) Pevarello, P.; Bonsignori, A.; Dostert, P.;
Heidempergher, F.; Pinciroli, V.; Colombo, M.; McArthur,
R. A.; Salvati, P.; Post, C.; Fariello, R. G.; Varasi, M. J. Med.
Chem. 1998, 41, 579.
PAPER

Chin. J. Pharmas.2012, 43, 161-163.

 …………….BASE
…………….BASE
 …………MESYLATE
…………MESYLATE
PAPER
J. Med. Chem. 2007, 50, 4909-4916.

(S)-2-[6-(3-Fluorobenzyloxy)-3,4-dihydro-1H-isoquinolin-2-yl]-propionamide (21). The title compound was obtained using the same procedure described for the synthesis of (R)-2-[6-(3-fluorobenzyloxy)-3,4-dihydro-1H-isoquinolin-2-yl]propionamide, starting from 6-(3-fluorobenzyloxy)-1,2,3,4-tetrahydroisoquinoline (0.24 g, 0.95 mmol) and (R)-2-amino-1-methyl-2-oxoethyl-2-nitrobenzenesulfonate (0.52 g, 1.9 mmol). After column chromatography
purification using 99:1 DCM/MeOH as eluent, 0.075 g (24% yield) of the title compound was obtained as a pure white solid. Mp 153- 154 °C. 1H NMR (CDCl3) ä 1.35 (d, 3H, J ) 7.0), 2.67-2.97 (m, 4H), 3.28 (q, 1H, J ) 7.0), 3.64 (d, 1H, J ) 14.2), 3.77 (d, 1H, J ) 14.2), 5.05 (s, 2H), 5.36 (br, 1H), 6.74 (d, 1H, J ) 2.5), 6.79 (dd, 1H, J ) 8.5, 2.5), 6.97 (d, 1H, J ) 8.5), 6.99-7.06 (m, 1H), 7.06-7.24 (m, 3H), 7.30-7.40 (m, 1H).
J. Med. Chem.1998, 41, 579-590.

References
- “Summary of the risk management plan (RMP) for Xadago (safinamide)” (PDF). European Medicines Agency. January 2015.
- Fariello, RG (2007). “Safinamide”. Neurotherapeutics. 4 (1): 110–116. doi:10.1016/j.nurt.2006.11.011. PMID 17199024.
- “EPAR Summary for the Public for Xadago” (PDF). European Medicines Agency. February 2015.
- “After an odyssey of setbacks, FDA finally green-lights Newron’s Parkinson’s drug Xadago”. endpts.com. Retrieved 2017-03-21.
- Lawrence, Janna (2015-01-19). “Safinamide recommended for approval as Parkinson’s disease therapy”. The Pharmaceutical Journal. Royal Pharmaceutical Society. Retrieved 2015-01-19.
- Haberfeld, H, ed. (2015). Austria-Codex (in German). Vienna: Österreichischer Apothekerverlag.
- H. Spreitzer (14 April 2014). “Neue Wirkstoffe – Safinamid”. Österreichische Apothekerzeitung (in German) (8/2014): 30.
- Klement, A (18 July 2016). “Xadago”. Österreichische Apothekerzeitung (in German) (15/2016): 10.
- “Summary of Product Characteristics for Xadago” (PDF). European Medicines Agency. 24 February 2015.
- ^ Jump up to:a b Caccia, C; Maj, R; Calabresi, M; Maestroni, S; Faravelli, L; Curatolo, L; Salvati, P; Fariello, RG (2006). “Safinamide: From molecular targets to a new anti-Parkinson drug”. Neurology. 67 (7 Suppl 2): S18–23. doi:10.1212/wnl.67.7_suppl_2.s18. PMID 17030736.
- Merck Serono: Vielversprechende Daten zur kognitiven Wirkung von Safinamid bei Parkinson im Frühstadium. (German) 8 June 2007.
- Pevarello, P; Bonsignori, A; Caccia, C; Amici, R; Salvati, P; Fariello, RG; McArthur, RA; Varasi, M (1999). “Sodium channel activity and sigma binding of 2-aminopropanamide anticonvulsants”. Bioorganic & Medicinal Chemistry Letters. 9 (17): 2521–2524. doi:10.1016/s0960-894x(99)00415-1.
- ^ Jump up to:a b Krösser, Sonja; Marquet, Anne; Gallemann, Dieter; Wolna, Peter; Fauchoux, Nicolas; Hermann, Robert; Johne, Andreas (2012). “Effects of ketoconazole treatment on the pharmacokinetics of safinamide and its plasma metabolites in healthy adult subjects”. Biopharmaceutics & Drug Disposition. 33 (9): 550. doi:10.1002/bdd.1822. PMID 23097240.
- Jump up^ Pevarello, P; Bonsignori, A; Dostert, P; Heidempergher, F; Pinciroli, V; Colombo, M; McArthur, RA; Varasi, M (1998). “Synthesis and Anticonvulsant Activity of a New Class of 2-[(Arylalkyl)amino]alkanamide Derivatives”. Journal of Medicinal Chemistry. 41 (4): 579–590. doi:10.1021/jm970599m. PMID 9484507.
- Jump up^ “Wichtigste Ergebnisse der Langzeitstudie mit Safinamid als Begleittherapie zu Levodopa bei Parkinson im fortgeschrittenen Stadium” [Major results from the long-term study of safinamide as add-on to levodopa for late-stage Parkinson] (in German). Merck KGaA. 4 November 2010.
- Jump up^ Study of Safinamide in Early Parkinson’s Disease as Add-on to Dopamine Agonist (MOTION)
- Jump up^ Merck Returns Rights for Safinamide to Newron, 21 October 2011.
- Jump up^ “Information about FDA Refusal to File” (PDF). Newron. 29 July 2014.
- “Information about FDA re-application” (PDF). Newron. 29 December 2014.
- Chazot, PL (2007). “Drug evaluation: Safinamide for the treatment of Parkinson’s disease, epilepsy and restless legs syndrome”. Current Opinion in Investigational Drugs. 8 (7): 570–579. PMID 17659477.
 |
|
| Clinical data | |
|---|---|
| Trade names | Xadago |
| AHFS/Drugs.com | UK Drug Information |
| Pregnancy category |
|
| Routes of administration |
Oral |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | 95% |
| Protein binding | 88–90% |
| Metabolism | Amidases, glucuronidation |
| Biological half-life | 20–30 hrs |
| Excretion | 76% renal, 1.5% faeces |
| Identifiers | |
| Synonyms | EMD-1195686, PNU-15774E; (2S)-2-[[4-[(3-fluorophenyl)methoxy]phenyl] methylamino]propanamide |
| CAS Number | |
| PubChemCID | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| ECHA InfoCard | 100.120.167 |
| Chemical and physical data | |
| Formula | C17H19FN2O2 |
| Molar mass | 302.34 g/mol |
| 3D model (Jmol) | |
//////////Xadago, safinamide, Newron Pharmaceuticals, FDA 2017, Parkinson’s disease, 133865-89-1 , сафинамид , سافيناميد, 沙非胺, EMD-1195686, ZP-034, FCE-28073(R-isomer), PNU-151774E, NW-1015, FCE-26743
C[C@H](NCC1=CC=C(OCC2=CC=CC(F)=C2)C=C1)C(N)=O


[…] SAFINAMIDE […]
LikeLike
[…] SAFINAMIDE […]
LikeLike