2016

PNQ 370
Advinus Therapeutics Ltd
Adenosine A2a receptor antagonist
for treating disease or disorder susceptible to improvement by antagonism of A2A receptor.
Advinus Therapeutics is investigating PNQ-370, presumed to be lead from a series of small molecule therapeutics including PD-2 and PD-3, as adenosine A2a receptor antagonist, for the potential treatment of Parkinson’s disease . In November 2012, this drug was in preclinical development .

KEEP WATCHING THIS POST AS I ARRIVE AT THE STRUCTURE…………..



ONE OF THE ABOVE OR SIMILAR
INTRODUCTION
The effects of adenosine are mediated through at least four specific cell membrane receptors so far identified and classified as Ai, A2A, A2B and A3 belonging to G protein-coupled receptor family. The Ai and A3 receptors down-regulate cellular cAMP levels through their coupling to G protein, which inhibit adenylate cyclase. In contrast, A2A and A2B receptors couple to G protein that activate adenylate cyclase and increase intracellular levels of cAMP. Through these receptors, adenosine regulates the wide range of physiological functions.
Advances in understanding the role of adenosine and its receptors in physiology and pathophysiology, as well as new developments in medicinal chemistry of these receptors have identified potential therapeutic areas for drug development. With the combination of pharmacological data, using selective ligands and genetically modified mice, important progress has been made toward an understanding of the role of ARs in a variety of diseases, such as inflammatory conditions, sepsis, heart attack, ischemia-reperfusion injury, vascular injury, spinal cord injury, chronic obstructive pulmonary disease (COPD), asthma, diabetes, obesity, inflammatory bowel disease, retinopathy, and Parkinson’s Disease (PD).
Happy new year wishes 2016


Movement disorder constitutes a serious health problem, especially among the elderly. These movement disorders can often be the result of brain lesions. Disorders involving the basal ganglia which result in movement disorders include Parkinson’s disease, Huntington’s chorea and Wilson’s disease. Tremor, rigidity, akinesia and postural changes are four classic symptoms of Parkinson’s disease, it is also associated with depression, dementia and overall cognitive decline. Parkinson’s disease has a prevalence of 1 per 1000 of the total population and increases to 1 per 100 for those aged over 60 years. Degeneration of dopaminergic neurons in the substantia nigra and the subsequent reductions in the interstitial concentrations of dopamine in the striatum are critical to the development of Parkinson’s disease. About 80% of cells from the substantia nigra can be destroyed before the clinical symptoms of Parkinson’s disease become apparent
PD is a progressive, incurable disorder with no definite preventive treatment, although drugs are available to alleviate the symptoms and/or slow down the progress of the disease. Current therapy is based on dopamine replacement therapy, the most common drug treatments being dopaminomimetic agents, including L-DOPA, a dopamine precursor, as well as direct or indirect dopamine receptor agonists. L-DOPA is the mainstay in the treatment of PD, but because of tolerance problems and a wide range of adverse reactions, including involuntary movements and vomiting, a strong demand for new therapies exists. Among the various strategies, A2A AR blockers are considered a potential approach to treatment of the disease. Within the brain A2A ARs are richly expressed in the striatum, nucleus accumbens, and olfactory tubercle. A coexpression of A2A with D2 dopamine receptors has been reported in the GABAergic striatopallidal neurons where adenosine and dopamine agonists exert antagonistic effects in the regulation of locomotor activity. Activation of A2A ARs in striatopallidal neurons decreases the affinity of D2 receptors for dopamine, antagonizing the effects of D2 receptors.
The negative interaction between A2A and D2 receptors is at the basis of the use of A2A antagonists as a novel therapeutic approach in the treatment of PD. (Pharmacol. Ther. 2005, 105, 267). The recent discovery that the A2A can form functional heteromeric receptor complexes with other Gprote in-coupled receptors such as D2 and the mGlu5 receptors has also suggested new opportunities for the potential of A2A antagonists in PD. (J. Mol. Neurosci. 2005, 26, 209).
A2A knockout (KO) mice transient focal ischemia caused less neuronal damage in comparison to their wild-type (WT) littermates (J. Neurosci. 1999, 19, 9192.). Therefore, it seems that tonic activation of A2A ARs may be responsible for dangerous signal during injury, in contrast to the neuroprotective effects induced by endogenous Al activation. Recently, selective inactivation or reconstitution of A2A ARs in bone-marrow cells revealed their contribution to the development of ischemic brain injury (J.F. Nat. Med. 2004, 10, 1081) Blockade of A2A ARs has recently been implicated in the treatment of movement disorders such as Parkinson’s disease (Trends Pharmacol. Sci. 1997, 18, 338-344) and in the treatment of cerebral ischaemia (Life Sci. 1994, 55, 61-65).
The potential utility of A2A AR antagonists in the treatment of Parkinson’s disease has been reviewed (CNS drugs, 1998, 10, 31 1-320). One advantage of A2A AR antagonist therapy is that the underlying neurodegenerative disorder may also be treated ((Ann. N. Y. Acad. Sci. 1997, 825 (Neuroprotective Agents), 3048). In particular, blockade of A2A AR function confers neuroprotection against MPTP-induced neurotoxicity in mice (Neurosci. 2001, 21, RC143).
Alzheimer’s disease (AD) is a neurodegenerative disorder of the central nervous system manifested by cognitive and memory deterioration, a variety of neuropsychiatric symptoms, behavioral disturbances, and progressive impairment of daily life activities. Recent research suggests that adenosine receptors play important roles in the modulation of cognitive function. Epidemiological studies have found an association between coffee (a nonselective adenosine receptor antagonist) consumption and improved cognitive function in AD patients and in the elderly. Long-term administration of caffeine in transgenic animal models showed a reduced amyloid burden in brain with better cognitive performance.

Advinus’ Pharma Development Bangalore operation, located on a 8-acre campus with 220,000 sq ft of modern facilities, offers end-to-end pre-clinical to early clinical development platform for pharma product development
Antagonists of adenosine A2A receptors mimic these beneficial effects of caffeine on cognitive function. Neuronal cell cultures with amyloid beta in the presence of an A2A receptor antagonist completely prevented amyloid beta-induced neurotoxicity. These findings suggest that the adenosinergic system constitutes a new therapeutic target for AD, and caffeine and A2A receptor antagonists may have promise to manage cognitive dysfunction in AD (Curr Neuropharmacol. 2009 September; 7(3): 207-216).
High expression of A2A ARs has been found in platelets, leukocytes, vascular smooth muscle, and endothelial cells with important implications in the regulation of inflammatory responses. It is now well established that stimulation of the A2A AR in immune cells induces anti-inflammatory effects, mostly due to its ability to increase cAMP levels, which has strong immunosuppressive effects (Trends Immunol. 2005, 26, 299). Stimulation of A2A ARs inhibits neutrophil adherence to the endothelium, degranulation of activated neutrophils and monocytes, plus superoxide anion generation. A2A ARs have been recently defined as sensors and terminators of proinflammatory activities. The strongest evidence for the key role of A2A in inflammation is derived by the elegant study using mice deficient in A2A ARs (Nature 2001, 414, 916).

The state-of-the-art facility in Pune, Advinus Drug Discovery, develops its own drug candidates to out-license them at preclinical or clinical stages
In this model the lack of A2A subtype leads to increased tissue inflammation and damage, thus suggesting a negative and nonredundant regulatory role for the A2A AR. This model permits one to appreciate that adenosinergic regulation of immune cells is fundamental in normal physiological control of inflammation in vivo in spite of the fact that other Gs-protein-coupled receptors and cAMP elevating ligands are present, such as cathecolamines, prostaglandins, dopamine, and histamine (Trends Immunol. 2005, 26, 299). Interestingly, the A2A AR has been demonstrated to be involved in promotion of wound healing and angiogenesis in healing wounds (Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 289, R283).
Moreover, it plays an active role in the pathogenesis of dermal fibrosis, suggesting a role for antagonists as novel therapeutic approach in the treatment and prevention of dermal fibrosis in diseases such as scleroderma (Arthritis Rheum. 2006, 54, 2632) as well as hepatic fibrosis (Br. J. Pharmacol. 2006 Aug; 148(8): 1 144-55). Studies also suggest that A2A receptor antagonists may be beneficial for social memory impairment and hypertension (Behav Brain Res. 2005 Apr 30;159(2):197-205), sepsis (J Immunol. 2006 May 1 ; 176(9): 5616-26), spinal cord injury and neuroprotection (J Neuroinflammation. 201 1 Apr 12;8:31), retinopathy (IVOS, Jan. 2000, vol. 41 (1), 230-243, depression (Neurology. 2003 Dec 9;61(1 1 Suppl 6):S82-7), narcolepsy and other sleep related disorders (Prog Neurobiol. 2007 Dec;83(5):332-47), attention-deficit hyperactivity disorder (ADHD) (Behav Pharmacol. 2009 Mar;20(2): 134-45; Clinical Genetics (2000), 58(1), 31-40 and references therein),

Dr Rashmi Barbhaiya, CEO & Managing Director

… Dr Rashmi Barbhaiya, CEO & Managing Director and Dr Kasim Mookthiar, Chief Scientific Officer and SVP, Drug Discovery, Advinus Therapeutics …
Antagonists of the A2A receptor are potentially useful therapies for the treatment of addiction. Major drugs of abuse (opiates, cocaine, ethanol, and the like) either directly or indirectly modulate dopamine signaling in neurons particularly those found in the nucleus accumbens, which contain high levels OfA2A adenosine receptors. Dependence has been shown to be augmented by the adenosine signaling pathway, and it has been shown that administration of an A2A receptor antagonist redues the craving for addictive substances (“The Critical Role of Adenosine A2A Receptors and Gi βγ Subunits in Alcoholism and Addiction: From Cell Biology to Behavior”, by Ivan Diamond and Lina Yao, (The Cell Biology of Addiction, 2006, pp 291-316) and “Adaptations in Adenosine Signaling in Drug Dependence: Therapeutic Implications”, by Stephen P. Hack and Macdonald J. Christie, Critical Review in Neurobiology, Vol. 15, 235-274 (2003)). See also Alcoholism: Clinical and Experimental Research (2007), 31(8), 1302-1307.
A2A receptors may be beneficial for the treatment or prevention of disorders such as a movement disorder, for example, Parkinson’s disease or progressive supernuclear palsy, Restless leg syndrome, nocturnal myoclonus, cerebral ischaemia, Huntington’s disease, multiple system atrophy, corticobasal degeneration, Wilson’s disease or other disorders of basal ganglia which results in dyskinesias, post traumatic stress disorder. See for example WO200013682, WO200012409, WO2009156737, WO20091 1442, WO2008121748, WO2001092264, WO2007038284, WO2008002596, WO20091 1 1449, WO20091 1 1442, WO2008121748, WO2009156737, WO2003022283, WO2005044245, WO2008077557, WO20091 1 1449, WO2009705138, WO20091 1 1442, WO2007035542, WO20080870661, WO2008070529, WO20051 16026, WO2009055548, WO2007133983, WO2010045006, WO2010045015, WO2010045008 WO2009015236.
https://patentscope.wipo.int/search/en/detail.jsf;jsessionid=9B4D4A1C3A9C0C5ACBBBA119D16D32E2.wapp2nC?docId=WO2012038980&recNum=1&maxRec=&office=&prevFilter=&sortOption=&queryString=&tab=PCTDescription

centre: Mr Ratan Tata, Chairman, Tata Sons, flanked by Dr Rashmi Barbhaiya (left), Managing Director and CEO, Advinus, and Mr R. Gopalakrishnan, …
ONE EXAMPLE………..
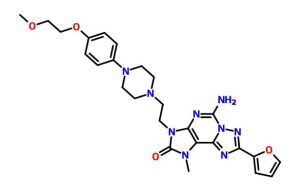
COMPD A1
MF C26 H31 N9 O4
2H-[1,2,4]Triazolo[5,1-i]purin-2-one, 5-amino-8-(2-furanyl)-1,3-dihydro-3-[2-[4-[4-(2-methoxyethoxy)phenyl]-1-piperazinyl]ethyl]-1-methyl-
mw 533.58
cas 1367365-26-1
| Molecular Formula: |
C26H31N9O4 |
| Molecular Weight: |
533.58224 g/mol |

A1
5-amino-8-(furan-2-yl)-3-[2-[4-[4-(2-methoxyethoxy)phenyl]piperazin-1-yl]ethyl]-1-methyl-[1,2,4]triazolo[5,1-f]purin-2-one
WO2012038980
Example Al :
5-amino-8-(2-furyl)-3-[2-[4-[4-(2-methoxyethoxy)phenyl]piperazin- 1 -yl]ethyl]- 1 -methyl-[ 1 ,2,4]triazolo[5, 1 -f]purin-2-one
5-Amino-8-(2-furyl)-3-[2-[4-[4-(2-methoxyethoxy)phenyl]piperazin-l-
5-amino-8-(2-furyl)-3-[2-[4-[4-(2-methoxyethoxy)phenyl]piperazin- 1 -yl]ethyl]- 1 -methyl-[ 1 ,2,4]triazolo[5, 1 -f]purin-2-one
Step-1 : 2-[(2,5-Diamino-6-chloro-pyrimidin-4-yI)amino]ethanol
A mixture of 4,6-dichloropyrimidine-2,5-diamine (28g, 156mmol), ethanolamine (18ml, 312mmol) and ethanol (250ml) were heated at 100-1 10 °C for 16 hours. The mixture was cooled and solvent was removed. To the residue methanol (100ml) was added and stirred for 20 minutes. The solid was filtered off to obtain 2-[(2,5-diamino-6-chloro-pyrimidin-4-yl)amino]ethanol (22.0g, 70%).
‘H MR(400MHz, DMSO d6): δ 3.36-3.40 (m, 2H); 3.50-3.54 (m, 2H); 3.88 (bs, 2H); 4.74 (t, J=5.6Hz, 1H); 5.63 (bs, 2H); 6.51 (t, J=5.6Hz, 1H)
Step-2: 2-Amino-6-chloro-9-(2-hydroxyethyl)-7H-purin-8-one
A mixture of 2-[(2,5-diamino-6-chloro-pyrimidin-4-yl)amino]ethanol obtained in step 1 (l O.Og, 49.26mmol) in acetonitrile (400ml) were cooled to 0 °C. To this reaction mixture K2C03 (20.39gm, 147.7mmol) and 4-nitrophenyl chloroformate (19.8g, 98.52mmol)was added and stirred at 25-27 °C for 24 hours. This reaction mixture was filtered and washed with acetonitrile (300ml) and diethyl ether (300ml) respectively. Solid obtained was dried to obtain crude 2-amino-6-chloro-9-(2-hydroxyethyl)-7H-purin-8-one as a yellow solid. Small amount of crude material was purified by column chromatography to obtain pure product. ‘HNMR(400MHz, DMSO d6): δ 3.61-3.66 (m, 2H); 3.72-3.75 (m, 2H); 4.85 (t, J=6Hz, 1H); 6.60 (s, 2H); 1 1.21 (s, 1 H)
Step-3: 2-Amino-6-chloro-9-(2-hydroxyethyl)-7-methyl-purin-8-one
A mixture of 2-amino-6-chloro-9-(2-hydroxyethyl)-7H-purin-8-one obtained in step 2 (13g, 56.7mmol) , K2C03 (1 1.5g, 84mmol), methyl iodide (12g, 85.15mmol) and DMF (130ml) were stirred at 25-30 °C for 16 hours. The reaction mixture was concentrated and purified by column chromatography using 60-120 silica gel and 4% methanol in DCM as an eluent to obtain 2-amino-6-chloro-9-(2-hydroxyethyl)-7-methyl-purin-8-one (8g, 58%) as an off white solid.
‘HNMR(400MHz, DMSO d6): δ 3.42 (s, 3H); 3.65 (t, J=5.6Hz, 2H); 3.78 (t, J=5.6Hz, 2H); 4.85 (t, J=5.6Hz, 1H); 6.69 (bs, 2H).
Step-4: 2-Amino-6-hydrazino-9-(2-hydroxyethyl)-7-methyI-purin-8-one
A mixture of 2-amino-6-chloro-9-(2-hydroxyethyl)-7-methyl-purin-8-one obtained in step 3 (8g, 32.9mmol) , Hydrazine hydrate (16ml ,32.9mmol) and ethanol (300ml) were heated at 100-1 10 °C for 16 hours. The reaction mixture was concentrated and solid obtained was filtered off and dried to obtain 2-amino-6-hydrazino-9-(2-hydroxyethyl)-7-methyl-purin-8-one (7g, 89 %) as an off white solid.
‘HNMR(400MHz, DMSO d6): δ 3.37 (s, 3H); 3.58-3.61 (m, 2H); 3.71 (t, J=6Hz, 2H); 4.29 (bs, 2H); 4.87 (t, J=5.6Hz, 1H), 6.00 (bs, 2H); 7.63 (s, 1H).
Step-5: N’-[2-Amino-9-(2-hydroxyethyl)-7-methyl-8-oxo-purin-6-yl]furan-2-carbohydrazide
2-amino-6-hydrazino-9-(2-hydroxyethyl)-7-methyl-purin-8-one (4.5g, 18.18mmol) obtained in step 4, 2-furoic acid (2.53g, 22.5mmol), HOBT (2.53g, 18.8 mmol) and N-methylmorpholine were taken in dimethylformamide (40ml). l-Ethyl-3(3′-dimethylaminopropryl)carbodiimide hydrochloride (EDCI.HCl) (5.4g, 28.2mmol) was added to the reaction mixture and stirred at 25-27 °C for 14 hours. The reaction mixture was evaporated and residue was purified by column chromatography to obtain N’-[2-amino-9-(2-hydroxyethyl)-7-methyl-8-oxo-purin-6-yl]furan-2-carbohydrazide (5.3g, 84%) as an off white solid.
‘HNMR (400MHZ, DMSO d6): δ 3.43 (s, 3H); 3.59-3.63 (m, 2H); 3.74 (t, J=6Hz, 2H); 4.88 (t, J=5.6Hz, 1H); 5.98 (bs, 2H); 6.67 (bs, 1H); 7.25 (d, J=3.2Hz, 1H); 7.90 (s, 1H); 8.35 (s, 1H); 10.28 (s, lH).
Step-6: 5-Amino-8-(2-furyl)-3-(2-hydroxyethyl)-l-methyl-[l^,4]triazolo[5,l-flpurin-2-one
A mixture of N’-[2-amino-9-(2-hydroxyethyl)-7-methyl-8-oxo-purin-6-yl]furan-2-carbohydrazide obtained in step 5 (5.3g, 15.9mmol), Ν,Ο-bistrimethylsilylacetamide (27ml, 1 1 1.4mmol) and hexamethyldisilazane (83ml, 397mmol) were heated at 1 10-120 °C for 16 hours. The reaction mixture was quenched with methanol (100ml) and water (100ml) and organic volatiles were evaporated. The solid obtained was filtered off and washed with water (30ml) followed by diethyl ether (100ml) to obtain 5-amino-8-(2-furyl)-3-(2-hydroxyethyl)-l-methyl-[l,2,4]triazolo[5,l-f]purin-2-one (3.50g, 71%) as an off white solid.
‘HNMR (400MHZ, DMSO d6): δ 3.56 (s, 3H); 3.67-3.70 (m, 2H); 3.84-3.87 (m, 2H); 4.88 (t, J=5.6Hz, 1H); 6.73 (bs, 1H); 7.20 (bs, 1H); 7.79 (bs, 2H); 7.94 (bs, 1H).
Step-7: 2-[5-Amino-8-(2-furyl)-l-methyl-2-oxo-[l,2,4]triazolo[5,l-fJpurin-3-yl]ethyl 4-methylbenzenesulfonate
A mixture of 5-amino-8-(2-furyl)-3-(2-hydroxyethyl)-l -methyl-[l,2,4]triazolo[5, l-fJpurin-2-one obtained in step 6 (3.5g, l lmmol), p-toluene sulphonylchloride (5.2 g, 27mmol) were taken in pyridine (30ml)and stirred at 25-27 °C for 16 hours. To the reaction mixture hexane (100ml) was added and solid obtained was filtered off and washed with water (100ml) followed by hexane (100ml) to obtain 2-[5-amino-8-(2-furyl)-l-methyl-2-oxo-[l,2,4]triazolo[5, l-f]purin-3-yl]ethyl 4-methylbenzenesulfonate (4.1g, 78%) as a brown solid. ‘HNMR (400MHz, DMSO d6): δ 2.02 (s, 3H); 3.49 (s, 3H); 3.99 (t, J=4.8Hz, 2H); 4.71 (t, J=4.8Hz, 2H); 6.73-6.75 (m, 1H); 7.01 (d, J=8Hz, 2H); 7.23 (d, J=3.2Hz, 1H); 7.41 (d, J=8.4Hz, 2H); 7.78 (bs, 2H); 7.96 (d, J=1.2Hz, 1H).
Step-8: : 5-Amino-8-(2-furyl)-3-[2-[4-[4-(2-methoxyethoxy)phenyl]piperazin-l-yl]ethyl]-l-methyl-[l,2,4]triazolo[5,l-f)purin-2-one
A mixture of 2-[5-amino-8-(2-furyl)-l-methyl-2-oxo-[l ,2,4]triazolo[5, l-f]purin-3-yl]ethyl 4-methylbenzenesulfonate obtained in step 7 (0.25g, 0.533mmol), l-[4-(2-Methoxy-ethoxy)-phenyl]-piperazine (0.188g, 0.799mmol) and DIPEA (0.27ml, 1.599mmol) were taken in DMF (5ml) and stirred at 80 °C for 16 hours. To the reaction mixture water (100ml) was added and solid obtained was filtered off. The crude product was purified by column chromatography to obtain 5-amino-8-(2-furyl)-3-[2-[4-[4-(2-methoxyethoxy)phenyl]piperazin- 1 -yl]ethyl]- 1 -methyl-[ 1 ,2,4]triazolo[5, 1 -f]purin-2-one (0.135g, 47%) as an off white solid
‘HNMR (400MHz, DMSO d6): δ 2.60 (bs, 4H); 2.68 (t, J=6.4Hz, 2H); 2.96 (bs, 4H); 3.29 (s, 3H); 3.56 (s, 3H); 3.59-3.62 (m, 2H); 3.94-4.00 (m, 4H); 6.71 -6.73 (m, 1H); 6.79-6.86 (m, 4H); 7.19 (dd, J=3.2Hz, 1.2Hz, 1H); 7.80 (bs, 2H); 7.94 (bs, 1H).
ANOTHER……..
Example Gl: 5-Amino-l-ethyl-8-(2-furyl)-3-[2-[4-[4-(2-methoxyethoxy)phenyl]piperazin-l-yl]ethyl]-[l,2,4]triazolo[5,l-i]purin-2-one

Step-1 : 2-Amino-6-chloro-7-ethyl-9-(2-hydroxyethyl)purin-8-one
(Procedure is same as step-3 in example Al)
‘HNMR (400MHz, DMSO d6): δ 1.21 (t, J=7.2Hz, 3H); 3.64 (s, 2H); 3.78 (t, J=6Hz, 2H);
3.92 (q, J=7.2Hz, 2H); 4.92 (bs, I H); 6.7 (bs, 2H).
Step-2 : 2-Amino-7-ethyl-6-hydrazino-9-(2-hydroxyethyl)purin-8-one
(Procedure is same as step-4 in example Al)
‘ HNMR (400MHz, DMSO d6): δ 1.07 (t, J=6.8Hz, 3H); 3.59 (q, J=6Hz, 2H); 3.72 (t, J=6Hz,
2H); 3.91 (q, J=6.8Hz, 2H); 4.32 (bs, 2H); 4.86 (t, J=5.6Hz, IH); 5.99 (bs, 2H), 7.55 (bs, IH).
Step-3: N’-[2-Amino-7-ethyl-9-(2-hydroxyethyl)-8-oxo-purin-6-yl]furan- 2carbohydrazide (Procedure is same as step-5 in example Al)
Crude product was used in next step
Step-4: 5-Amino-l-ethyI-8-(2-furyl)-3-(2-hydroxyethyl)-[l,2,4]triazolo[5,l-flpurin-2-one
(Procedure is same as step-6 in example Al)
‘H MR (400MHZ, DMSO d6): δ 1.34 (t, J=7.2Hz, 3H); 3.67 (q, J=5.6Hz, 2H); 3.84 (t, J=5.6Hz, 2H); 4.01 (q, J=7.2Hz, 2H); 4.87 (t, J=6Hz, IH); 6.70 (bs, IH); 7.17 (d, J=2.8Hz, I H); 7.18 (bs, 2H); 7.92 (bs, IH).
Step-5: 2-[5-Amino-l-ethyl-8-(2-furyl)-2-oxo-[l,2,4]triazoIo[5,l-f|purin-3-yl]ethyl 4- methylbenzenesulfonate (procedure is same as step-7 in example Al)
lHNMR (400MHz, DMSO d6): δ 1.35 (t, J=7.2Hz, 3H); 2.00 (s, 3H); 3.95-4.00 (m, 4H); 4.47 (bs, 2H); 6.74 (s, IH); 7.00 (d, J=7.6Hz, 2H); 7.22 (s, IH); 7.42 (d, J=7.6Hz, 2H); 7.78 (bs, 2H); 7.97 (bs, IH).
Step-6: 5-Amino-l-ethyl-8-(2-furyl)-3-[2-[4-[4-(2-methoxyethoxy)phenyi]piperazin-l- yl]ethyl]-[l,2,4]triazolo[5,l-f]purin-2-one (procedure is same as step-8 in example Al)
HNMR(400MHz, DMSO d6): δ 1.35 (t, J=7.2Hz, 3H); 2.60 (bs, 4H); 2.68 (t, J=6.8Hz, 2H); 2.95 (bs, 4H); 3.28(s, 3H);3.61 (t, J=4.4Hz, 2H); 3.94-4.04 (m, 6H); 6.72 (dd, J=2Hz, 3.6Hz, I H); 6.78-6.85 (m, 4H); 7.19 (d, J=3.2Hz, IH); 7.81(bs, 2H); 7.94 (s, IH).
Representative compounds of the present disclosure were tested and had micromolar to nanomolar activity.

 A1 ABOVE
A1 ABOVE
A7 ABOVE
A9 ABOVE
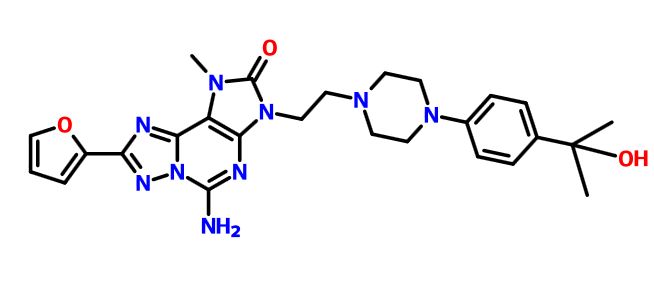
A13 ABOVE
A31 ‘HNMR (400MHz, DMSO d6): δ 2.62 (bs,4H); 2.68 (t, J=6.8Hz, 2H); 2.85 (bs, 4H); 3.28 (s, 3H); 3.57 (s, 3H); 3.59-3.62 (m, 2H); o 3.95 (t, J=6.8Hz, 2H); 4.01-4.04 (m, 2H);
5-Amino-3-[2-[4-[2-fluoro-4-(2- 6.66-6.68 (m, 1H); 6.72 (dd, J=2 Hz,3.6Hz, methoxyethoxy)phenyl]piperazin-l-yl]ethyl]-8- 1H); 6.79 (dd, J=2.8Hz, 14Hz, 1H); 6.92 (t, (2-furyl)- 1 -methyl-[ 1 ,2,4]triazolo[5, 1 -f|purin-2- J=9.6Hz, 1H); 7.19 (d, J=3.2Hz, 1 H); 7.93 one (bs, 2H); 7.93-7.94 (m, 1H).
A31 ABOVE
A32 HNM (400MHz, DMSO d6): δ 2.59 (bs,
4H); 2.68(t, J=6.4Hz, 2H); 3.27(t, J=4.8Hz, 4H); 3.56 (s, 3H); 3.96 (t, J=6.4Hz, 2H);
0 6.72(dd, J=2Hz, 3.6Hz, 1H); 6.99 (d, J=8.8Hz,
4-[4-[2-[5-Amino-8-(2-furyl)-l-methyl-2-oxo- 2H); 7.19 (d, J=3.6Hz, 1H);7.56 (d, J=8.8Hz, [ 1 ,2,4]triazolo[5, 1 -f]purin-3-yl]ethyl]piperazin- 2H); 7.80 (bs, 2H); 7.93 (bs, lH).
l-yl]benzonitrile
A32 ABOVE
A36 ‘HNMR(400MHz, CDCI3): δ θ.09 (d,
J=4.4Hz, 2H); 0.50 (d, J=6.8Hz, 2H); 0.82- 0.89 (m, 1H); 2.24 (d, J=6.0Hz, 2H): 2.52- 2.72 (m, 8H); 2.80 (t, J=6.4Hz, 2H); 3.76 (s,
5-Amino-3-[2-[4-(cyclopropylmethyl)piperazin- 3H); 4.07 (t, J=6.8Hz, 2H); 5.89 (bs, 2H); l -yl]ethyl]-8-(2-furyl)-l-methyl- 6.61 (bs, 1H); 7.22 (d, J=2.4Hz, 1H); 7.64 (s, [ 1 ,2,4]triazolo[5, 1 -f]purin-2-one 1H).
A36 ABOVE
A38 ‘HNMR(400MHz, CDCI3): δ 2.62 . (t,
J=4.4Hz, 4H); 2.79 (t, J=6.4Hz, 2H); 2.81 (s, 6H); 3.22 (t, J=4.4Hz, 4H): 3.77 (s, 3H); 4.06 (t, J=6.8Hz, 2H); 5.74 (bs, 2H); 6.60 (dd,
4-[2-[5-Amino-8-(2-fiiryl)- 1 -methyl-2-oxo- J=2.0Hz, 3.2Hz, 1H); 7.24 (d, J=3.6Hz, 1H);
[ 1 ,2,4]triazolo[5, 1 -f]purin-3-yl]ethyl]-N,N- 7.65 (s, 1H).
dimethy l-piperazine- 1 -sulfonamide
A38 ABOVE
A39 ‘HNMR(400MHZ, DMSO d6): δ 1.89-1.94
im, 1H); 2.09-2.18 .(m, 1 H); 2.60 (bs, 4H); 2.67 (t, J=6.4Hz, 2H); 2.96 (bs, 4H); 3.56 (s, 3H); 3.69-3.85 (m, 4H); 3.95 (t, J=6.4Hz, 
2H); 4.89 (bs, 1H); 6.72 (dd, J=2.0, 3.2Hz,
5-Amino-8-(2-furyl)-l -methyl-3-[2-[4-(4- 1H); 6.78 (d, J=9.2Hz, 2H); 6.85 (d, J=9.2Hz, tetrahydrofuran-3-yloxyphenyl)piperazin- 1 – 2H): 7.20 (d, J=3.2Hz, 1 H); 7.80 (bs, 2H); yl]ethyl]-[l ,2,4]triazolo[5,l-f]purin-2-one
7.93 (s, 1H).
A39 ABOVE
A42 ‘HNMR(400MHz, CDCI3): δ
2.26 (s,3H); 2.94-2.97 (m, 6H); 3.72 (s, 2H); 3.75 (s, 3H); 4.17 (t, J=6.4Hz, 2H); 5.74 (bs, 2H); 6.59 (dd, J=1.6Hz, 3.6Hz, 1H);7.13 (s, J=3.6Hz, IH); 7.21-7.24 (m, IH); 7.63 (s,
5-Amino-8-(2-furyl)-l-methyl-3-[2-(3-methyl- IH); 8.20 (bs, IH),
7,8-dihydro-5H- 1 ,6-naphthyridin-6-yl)ethyl]- [ 1 ,2,4]triazolo[5, 1 -f]purin-2-one
A42 ABOVE
A57 HNMR(400MHz, DMSO d6): δ 2.95 (t,
J=8Hz, 2H); 3.52 (s, 3H); 3.69 (s, 3H ), 3.97 (t, J=8Hz, 2H); 6.71 (dd, J=2Hz, 3.6Hz, I H );
5-Amino-8-(2-furyl)-3-[2-(4- 6.80 (dd, J=2Hz, 6.8Hz, 2H); 7.10 (d, methoxyphenyl)ethyl]- 1 -methyl- J=8.8Hz, 2H); 7.18 (dd, J=0.8Hz, 3.2Hz, I H );
[ 1 ,2,4]triazolo[5, 1 -f]purin-2-one 7.80 (bs, 2H), 7.94 (dd, J=lHz, 2Hz, I H ).
A57 ABOVE
A58 HNMR(400MHz, DMSO d6): δ 2.61 (bs,
4H); 2.68 (bs, 2H); 3.05(bs, 4H); 3.57 (s, 3H ), 3.96 (bs, 2H); 6.72 (bs, IH); 6.92 (d, J=8Hz, 2H); 7.01 (d, J=10Hz, 2H );7.03(d, J=148Hz, IH); 7.19 (bs , 1 H); 7.80 (bs, 2H); 7.94 (s,
5-amino-3-[2-[4-[4- IH).
(difluoromethoxy)phenyl]piperazin-l-yl]ethyl]- 8-(2-furyl)- 1 -methyl-[ 1 ,2,4]triazolo[5, 1 -fjpurin- 2-one
A58 ABOVE
A62 O ‘HNMR (400MHz, DMSO d6): δ 0.66-0.70
(m, 4H); 1.90-1.94 (m, lH); 2.41 (bs, 4H); 2.65 (t, J=6Hz, 2H); 3.38 (bs, 2H); 3.56 (bs, 5H); 3.93 (t, J=6.4 Hz, 2H); 6.71 (bs, 1H );
5-Amino-3-[2-[4- 7.19 (d, J=2.4Hz, 1H); 7.79 (bs, 2H); 7.93 (bs,
(cyclopropanecarbonyl)piperazin- 1 -yl]ethyl]-8- 1H).
(2-furyl)- 1 -methyl-[ 1 ,2,4]triazolo[5, 1 -fjpurin-2- one
A62 ABOVE
A63 ‘HNMR (400MHz, DMSO d6): δ 0.07-0.10
(m, 2H); 0.40-0.44 (m, 2H); 0.88-0.94 (m,lH); 2.21 (d, J=6.4Hz, 2H); 2.41-2.45 (m, 4H); 2.64 (t, J=6.4Hz, 2H); 3.38 (bs,4H); 3.56
5-Amino-3-[2-[4-(2- (s, 3H); 3.93 (t, J=6.4Hz, 2H); 6.72 (dd, cyclopropylacetyl)piperazin-l -yl]ethyl]-8-(2- J=2Hz,3.6 Hz, 1H); 7.19-7.20 (m, 1H); 7.80 fury 1)- 1 -methyl-[ 1 ,2,4]triazolo[5, 1 -fJpurin-2- (bs, 2H); 7.93 (d, J=0.8 Hz, 1H).
one
A63 ABOVE

C1 ABOVE

E1 ABOVE

D3 ABOVE
G1 ABOVE
 G2
G2
 H2
H2
 M1
M1
 M2
M2

M3

M6
ETC AS IN TABLE……………..

Dr Kasim Mookthiar, CSO & Executive VP (Drug Discovery),
Dr Nimish Vachharajani, Senior VP & Head (Pharmaceuticals & Agrochemical Development),
/////////
n21c(nc4c(c1nc(n2)c3occc3)N(C(N4CCN5CCN(CC5)c6ccc(cc6)OCCOC)=O)C)N
CN1C2=C(N=C(N3C2=NC(=N3)C4=CC=CO4)N)N(C1=O)CCN5CCN(CC5)C6=CC=C(C=C6)OCCOC

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....











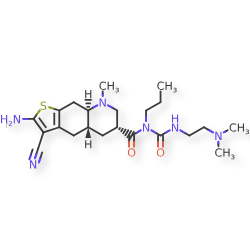
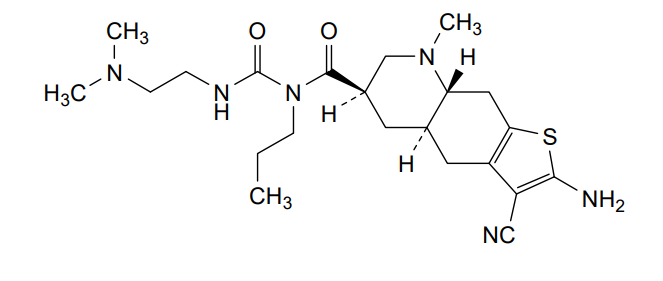
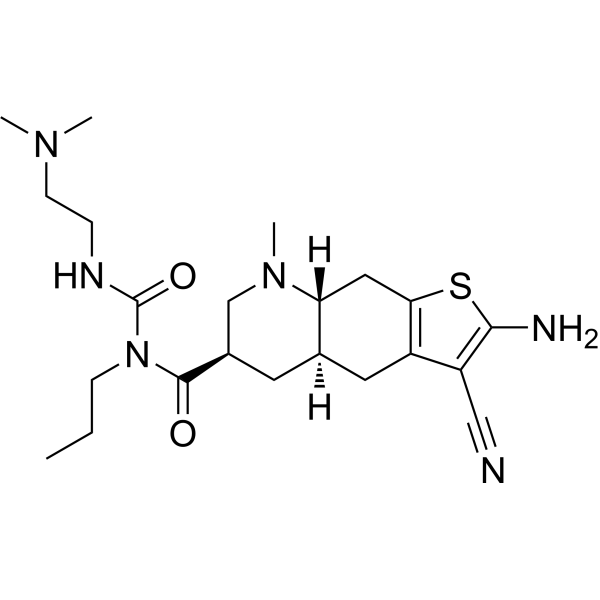

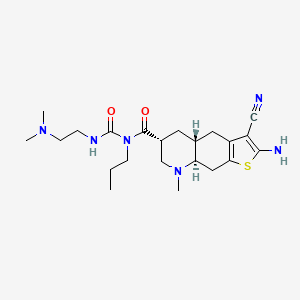
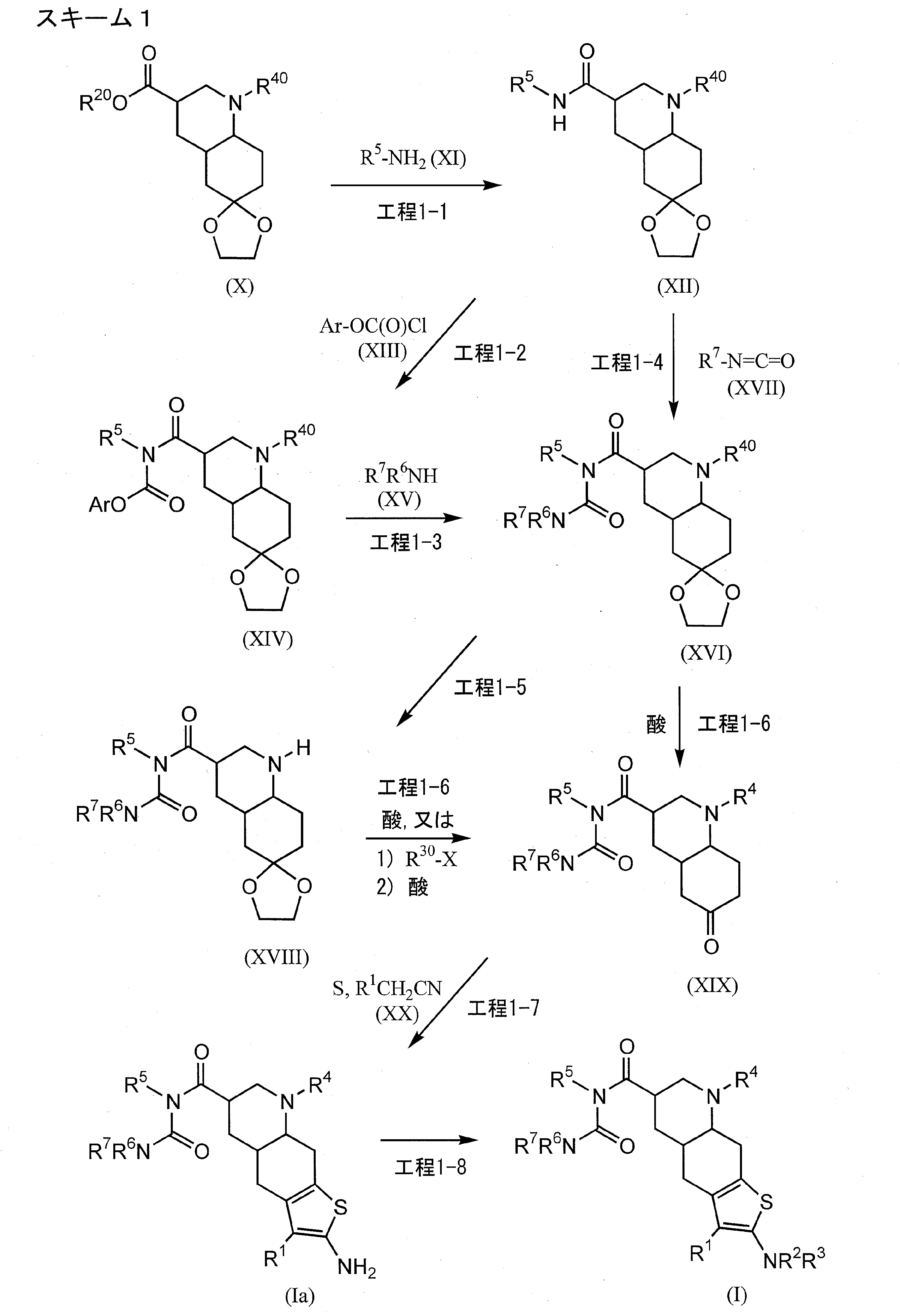
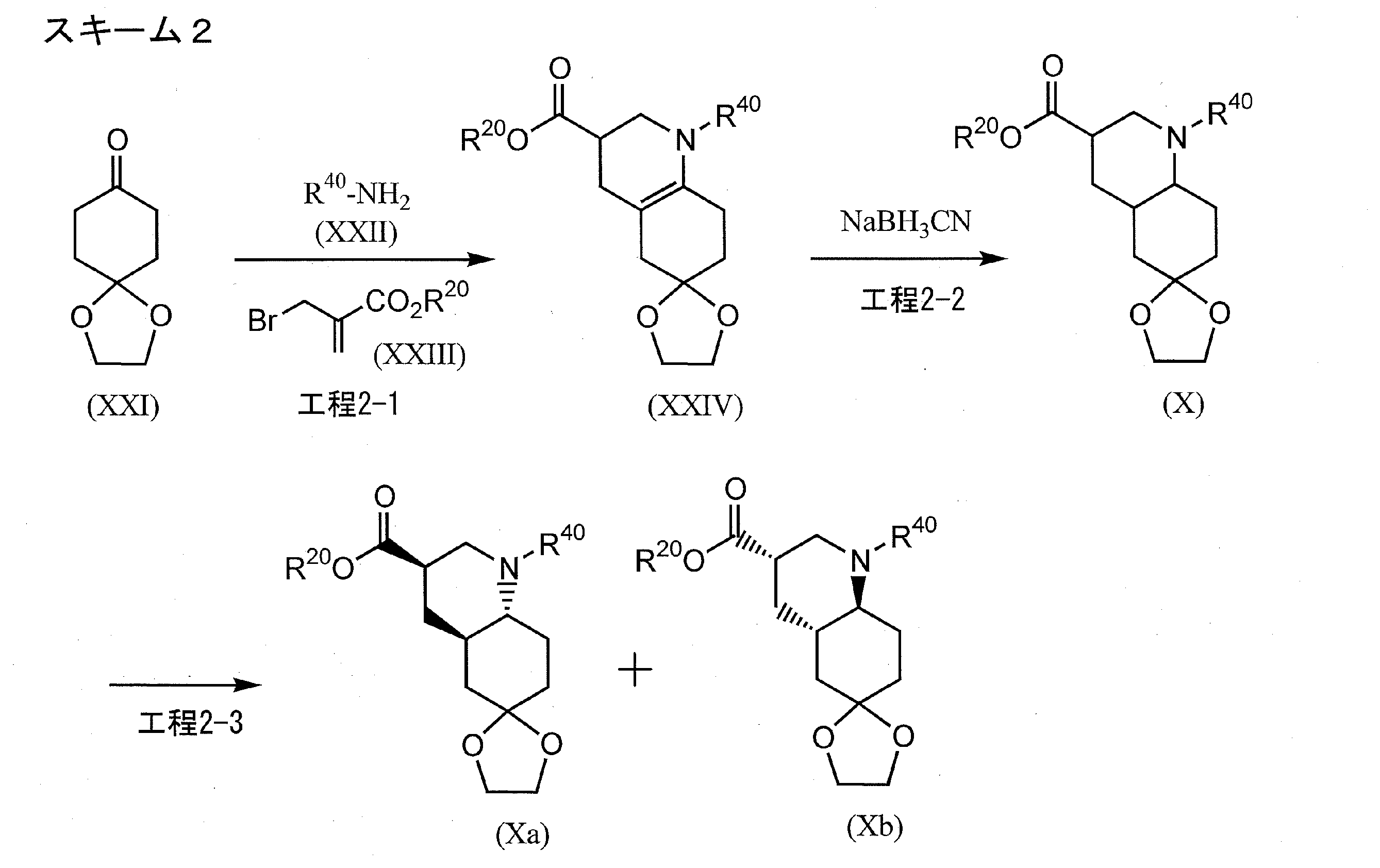
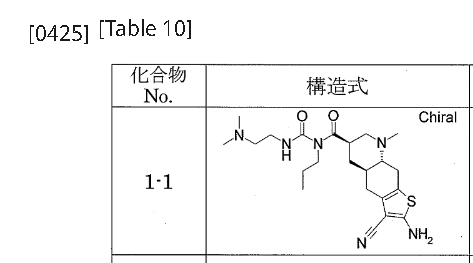
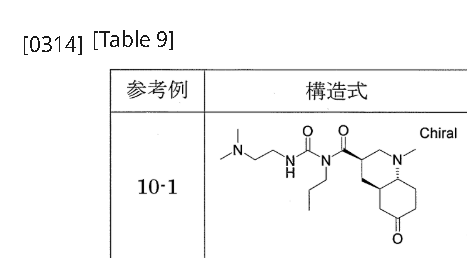
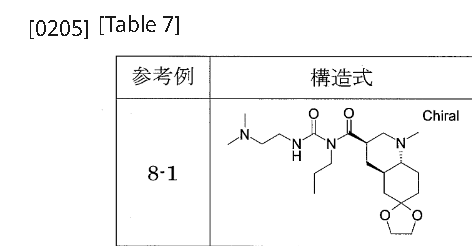















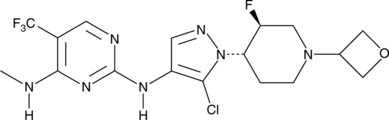

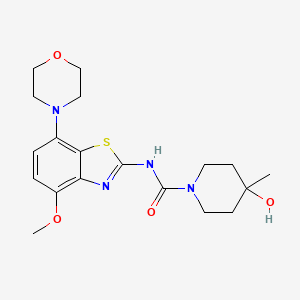











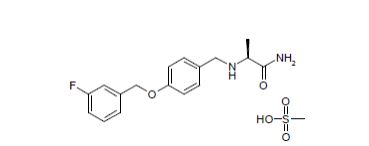
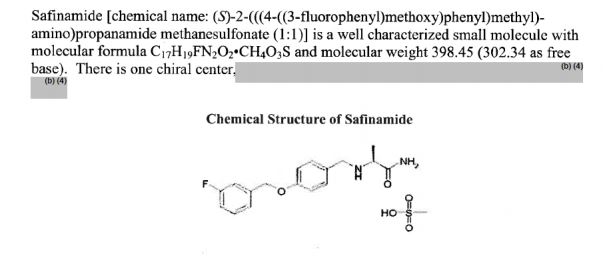











 …………….BASE
…………….BASE …………MESYLATE
…………MESYLATE


























































{kind=link}