PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
Tegoprazan, a reversible H+/K+-ATPase inhibitor developed by CJ Healthcare (now inno.N), was first approved and launched in South Korea in 2019 for the treatment of gastroesophageal reflux disease (GERD).
DeveloperCJ Cheiljedang Corp.; HK inno.N; RaQualia Pharma; Shandong Luoxin Pharmaceutical
ClassAmides; Anti-inflammatories; Antibacterials; Antiulcers; Benzimidazoles; Benzopyrans; Fluorobenzenes; Small molecules
Mechanism of ActionH(+) K(+)-exchanging ATPase inhibitors; Potassium-competitive acid blockers
[0371] To a solution of 4-bromo-2-nitro-6-[(phenylmethyl)oxy]aniline (33.0 g, 102 mmol, WO 2004054984) and acetic anhydride (14.5 mL, 153 mmol) in acetic acid (90 mL) was added concentrated sulfuric acid (2 drops) at 70° C. The mixture was stirred at 70° C. for 20 minutes. After cooling to room temperature, water (800 mL) was added, and the formed precipitate was collected by filtration, and washed with diisopropyl ether to give the title compound as a brown solid (30.9 g, 83%).
[0374] A mixture of N-{4-bromo-2-nitro-6-[(phenylmethyl)oxy]phenyl}acetamide (6.5 g, 17.8 mmol, STEP 1), zinc cyanide (4.18 g, 35.6 mmol), and tetrakis(triphenylphosphine)palladium (2.06 g, 1.78 mmol) in N,N-dimethylformamide (100 mL) was heated to 170° C. for 20 minutes in the microwave synthesizer (Biotage, Emrys Optimizer). After cooling to room temperature, the suspension was filtered, and washed with ethyl acetate. The organic layers were combined, washed with water, dried over magnesium sulfate, and concentrated in vacuum. The residual solid was purified by column chromatography on silica gel eluting with hexane/ethyl acetate (3:1) to afford the title compound as a white solid (5.5 g, 99%).
[0377] A mixture of N-{4-cyano-2-nitro-6-[(phenylmethyl)oxy]phenyl}acetamide (5.5 g, 17.7 mmol, STEP 2) and iron powder (2.96 g, 53.0 mmol) in acetic acid (90 mL) was refluxed with stirring for 2 hours. After cooling to room temperature, the mixture was filtered through a pad of Celite, and the filtrate was concentrated in vacuum. The residue was poured into water, and the aqueous layer was extracted with ethyl acetate/methanol (20:1). The organic layers were combined, washed with brine, dried over magnesium sulfate, and concentrated in vacuum to afford the title compound as a brown solid (3.82 g, 82%).
[0380] A solution of 2-methyl-4-[(phenylmethyl)oxy]-1H-benzimidazole-6-carbonitrile (3.82 g, 14.5 mmol, STEP 3) and potassium hydroxide (85%, 10.2 g, 15.4 mmol) in ethylene glycol (50 mL) was heated to 170° C. for 20 minutes in the microwave synthesizer (Biotage, Emrys Optimizer). After cooling to room temperature, the mixture was acidified with 2M hydrochloric acid aqueous solution (pH=3). The precipitated solid was collected by filtration to afford the title compound as a white solid (3.83 g, 93%).
[0383] A mixture of 2-methyl-4-[(phenylmethyl)oxy-1H-benzimidazole-6-carboxylic acid (5.0 g, 17.7 mmol, STEP 4), dimethylamine hydrochloride (4.33 g, 53.1 mmol), 2-[1H-benzotriazole-1-yl]-1,1,3,3-tetramethyluronium hexafluorophosphate (10.1 g, 26.6 mmol), and triethylamine (10.7 g, 106 mmol) in N,N-dimethylformamide (80 mL) was stirred at room temperature for 1 hour. The mixture was diluted with ethyl acetate/methanol (20:1), and washed with saturated ammonium chloride aqueous solution. The organic layer was dried over magnesium sulfate, and concentrated in vacuum. The residue was purified by column chromatography on silica gel (gradient elution from ethyl acetate only to ethyl acetate methanol 5:1) to afford the title compound as a white solid (4.90 g, 89%).
[0386] To a suspension of N,N,2-trimethyl-4-[(phenylmethyl)oxy]-1H-benzimidazole-6-carboxamide (928 mg, 3.0 mmol, STEP 5) in N,N-dimethylformamide (20 mL) was added sodium hydride (60% in mineral oil, 180 mg, 4.50 mmol) at 0° C. After stirring at room temperature for 30 minutes, the reaction mixture was cooled to 0° C. To the mixture was added 4-methylbenzenesulfonyl chloride (572 mg, 3.00 mmol) at 0° C., and the reaction mixture was stirred at room temperature for 2 hours. The mixture was poured into water, and the aqueous layer was extracted with ethyl acetate. The organic layers were combined, washed with water, dried over magnesium sulfate and concentrated in vacuum. The residue was purified by column chromatography on silica gel (gradient elution from dichloromethane only to ethyl acetate only) to afford the title compound as a white solid (1.00 g, 72%).
[0389] A mixture of N,N,2-trimethyl-1-(4-methylphenyl)sulfonyl]-4-[(phenylmethyl)oxy]-1H-benzimidazole-6-carboxamide (350 mg, 0.756 mmol, STEP 6) and 20% palladium hydroxide (1.20 g) in acetic acid (20 mL) was stirred under hydrogen gas (4 atmospheres) for 4 hours. The resulted mixture was filtered through a pad of Celite, and the filtrate was concentrated in vacuum. The residue was purified by column chromatography on silica gel (gradient elution from ethyl acetate only to ethyl acetate:methanol 5:1) to afford the title compound as a white solid (131 mg, 36%).
[0392] To a solution of 5,7-difluoro-2,3-dihydro-4H-chromen-4-one (14.2 g, 77.0 mmol, US 20050038032) in methanol (200 mL) was added sodium borohydride (3.50 g, 92.5 mmol) at 0° C. The reaction mixture was stirred at the same temperature for 1 hour, and evaporated to remove methanol. The residue was quenched with water, and extracted with ethyl acetate. The extract was washed with brine, dried over magnesium sulfate, and concentrated in vacuum. The residue was purified by column chromatography on silica gel (hexane:ethyl acetate=3:1 as an eluent) to afford the title compound as a pale gray solid (9.64 g, 67%).
[0394] To a stirred mixture of 4-hydroxy-N,N,2-trimethyl-1-[(4-methylphenyl)sulfonyl]-1H-benzimidazole-6-carboxamide (110 mg, 0.294 mmol, STEP 7), 5,7-difluoro-3,4-dihydro-2H-chromen-4-ol (164 mg, 0.884 mmol, STEP 8-1) and triphenylphosphine (232 mg, 0.884 mmol) in toluene (5 mL) was added diisopropyl azodicarboxylate (DIAD) (179 mg, 0.884 mmol) at room temperature. The reaction mixture was stirred at room temperature for 6 hours and concentrated in vacuum. The residue was purified by column chromatography on silica gel (ethyl acetate:hexane gradient elution from 1:20 to 10:1) to afford a mixture of the title compound and triphenylphosphine oxide (280 mg, crude) as white solids, which was used in the next step without further purification.
[0397] To a solution of 4-[(5,7-difluoro-3,4-dihydro-2H-chromen-4-yl)oxy-N,N,2-trimethyl-1-[(4-methylphenyl)-sulfonyl]-1H-benzi midazole-6-carboxamide (280 mg, crude, STEP 8) in tetrahydrofuran (8 mL) and methanol (4 mL) was added sodium hydroxide (165 mg, 4.1 mmol) at room temperature. After stirring at room temperature for 1 hour, the mixture was quenched with saturated sodium dihydrogenphosphate aqueous solution, and extracted with ethyl acetate. The organic layers were combined, dried over magnesium sulfate and concentrated in vacuum. The residue was purified by column chromatography on silica gel (gradient elution from dichloromethane only to ethyl acetate:methanol 10:1) to afford the title compound as a white solid (74 mg, 65% for 2 steps).
Example 2(−)-4-[((4S)-5,7-Difluoro-3,4-dihydro-2H-chromen-4-yl)oxy]-N,N,2-trimethyl-1 fl-benzimidazole-6-carb oxamide andExample 3(−)-4-]((4R)-5,7-Difluoro-3,4-dihydro-2H-chromen-4-yl)oxy]-N,N,2-trimethyl-1H-benximidazole-6-carboxamide
[0400]
[0401] The fraction-1 (582 mg) and fraction-2 (562 mg) were prepared from racemic 4-[(5,7-difluoro-3,4-dihydro-2H-chromen-4-yl)oxy]-N, 1,2-trimethyl-1H-benzimidazole-6-carboxamide (1.63 g, STEP 9 in Example 1) by HPLC as follows.
Isolation Condition
[0402] Column: CHIRALCEL OJ-H (20 mm×250 mm, DAICEL)
[0403] Mobile phase: n-Hexane/Ethanol/Diethylamine (95/5/0.1)
The following is the alternative method for synthesizing (−)-4-[((4S)-5,7-difluoro-3,4-dihydro-2H-chromen-4-yl)oxy]-N,N,2-trimethyl-1H-benzimidazole-6-carboxamide.
[0411] A mixture of N-{4-bromo-2-nitro-6-[(phenylmethyl)oxy]phenyl}acetamide (120 g, 329 mmol, STEP 1 in Example 1) and iron powder (55.1 g, 986 mmol) in acetic acid (500 mL) was refluxed with stirring for 6 hours. After cooling to room temperature, the mixture was filtered through a pad of Celite, and the filtrate was concentrated in vacuum. The residue was diluted with ethyl acetate (1.5 L). The resulted precipitates were filtered through a pad of Celite, and washed with ethyl acetate (500 mL). The filterate was concentrated in vacuum, and the residue was diluted with ethyl acetate (200 mL). The brine (800 mL) was added to the organic mixture, the resulted white precipitates were collected by filtration, and washed with water (200 mL) and diethyl ether (200 mL). The white solid was dissolved with dichloromethane/methanol (10:1, 1.0 L), dried over magnesium sulfate, and concentrated. The solid was triturated with diethyl ether (300 mL), collected by filtration, and dried in vacuum to afford the title compound as a white solid (54.7 g, 53%).
[0414] To a suspension of 6-bromo-2-methyl-4-[(phenylmethyl)oxy]-1H-benzimidazole (79.2 g, 250 mmol, STEP 1) in N,N-dimethylformamide (500 mL) was added sodium hydride (60% in mineral oil, 12.0 g, 300 mmol) at 0° C. After stirring at room temperature for 20 minutes, the reaction mixture was cooled to 0° C. To the mixture was added 4-methylbenzenesulfonyl chloride (47.6 g, 250 mmol) at 0° C., and the reaction mixture was stirred at room temperature for 30 minutes. The mixture was quenched with water (800 mL), and the white precipitates were collected by filtration, washed with diisopropyl ether (500 mL), and dried in vacuum at 70° C. for 7 hours to afford the title compound as a white solid (116 g, 98%).
[0417] A mixture of 6-bromo-2-methyl-1-[(4-methylphenyl)sulfonyl]-4-[(phenylmethyl)oxy]-1H-benzimidazole (53.0 g, 112 mmol, STEP 2) and tetrakis(triphenylphosphine)palladium(0) (25.9 g, 22.4 mmol) in 2M dimethylamine tetrahydrofuran solution (580 mL) was stirred at 65° C. under carbon mono-oxide gas (1 atmosphere) for 32 hours. The mixture was cooled to room temperature, and diluted with ethyl acetate (600 mL). The organic mixture was washed with saturated ammonium chloride aqueous solution (800 mL) and brine (500 mL), dried over magnesium sulfate and concentrated in vacuum. The residue was purified by column chromatography on silica gel (hexane:ethyl acetate gradient elution from 1:2 to 1:3) to afford the title compound as a white solid (21.8 g, 42%).
[0418] 1H NMR: spectrum data were identical with STEP 6 in Example 1.
[0419] A mixture of N,N,2-trimethyl-1-[(4-methylphenyl)sulfonyl]-4-[(phenylmethyl)oxy]-1H-benzimidazole-6-carboxamide (29.0 g, 62.6 mmol, STEP 3) and 10% palladium on carbon (6.0 g) in tetrahydrofuran (200 mL) was stirred under hydrogen gas (1 atmosphere) at room temperature for 24 hours. Another 4.0 g of 10% palladium on carbon was added, and the mixture was stirred under hydrogen gas (1 atmosphere) at room temperature for additional 6 hours. The resulted mixture was filtered through a pad of Celite, and the filtrate was concentrated in vacuum to afford the title compound as a white solid (23.0 g, 98%).
[0420] 1H NMR: spectrum data were identical with STEP 7 in Example 1.
STEP 5: Methyl 3-(3,5-difluorophenoxy)acrylate
[0421] A solution of 3,5-difluorophenol (35.5 g, 273 mmol) and methyl propiolate (25.0 mL, 300 mmol) in acetonitrile (109 mL) was added to a solution of tetrabutylammonium fluoride in tetrahydrofuran (1.0 M commercial solution, 109 mL, 109 mmol) at room temperature over a period of 2 hours. After complete addition of the solution, the mixture was stirred for 1 hour. The reaction mixture was diluted with toluene (350 mL) and the organic mixture was washed twice with water (250 mL×2), dried over magnesium sulfate, and concentrated in vacuum. The residue was purified by column chromatography on amino gel (hexane:ethyl acetate=3:2 as an eluent) to afford the title compound as a yellow solid (60.0 g, quant, 1:1 mixture of cis- and trans-isomers).
[0423] A mixture of methyl 3-(3,5-difluorophenoxy)acrylate (60.0 g, 280 mmol, STEP 5), and 10% palladium on carbon (1.0 g) in methanol (300 mL) was stirred under hydrogen gas (1 atmosphere) at room temperature for 18 hours. The reaction mixture was filtered through a pad of Celite, and washed with toluene (100 mL). The filtrate was concentrated in vacuum to afford the title compound (61.0 g, quant) as a colorless oil, which was used in the next step without further purification.
[0425] A mixture of methyl 3-(3,5-difluorophenoxy)propanoate (11.6 g, 53.7 mmol, STEP 6) and trifluoromethanesulfonic acid (23.2 mL, 2.0 mL/g of substrate) was stirred at 80° C. for 2 hours. After cooling to room temperature, the reaction mixture was diluted with water (120 mL), and extracted with toluene (120 mL). The organic layer was washed successively with aqueous solution of potassium carbonate (50 mL), water (50 mL), and dried over magnesium sulfate. The organic mixture was concentrated in vacuum to afford the title compound (8.75 g, 88%) as a white solid, which was used in the next step without further purification.
[0427] To a mixture of 1 M (S)-tetrahydro-1-methyl-3,3-diphenyl-1H,3H-pyrrolo[1,2-c][1,3,2]oxazaborole toluene solution (5.43 mL, 5.43 mmol) and tetrahydrofuran (40 mL) was added 2M borane-methyl sulfide complex tetrahydrofuran solution (29.8 mL, 59.7 mmol) at 0° C. and the mixture was stirred for 20 minutes. To the mixture was added a solution of 5,7-difluoro-2,3-dihydro-4H-chromen-4-one (10.0 g, 54.3 mmol, STEP 7) in tetrahydrofuran (70 mL) at 0° C. over a period of 1 hour, and the mixture was stirred at the same temperature for 1 hour. The reaction mixture was quenched with methanol (50 mL) and stirred for 30 minutes at room temperature. The mixture was concentrated in vacuum and the residue was purified by column chromatography on silica gel (hexane:ethyl acetate=4:1 as an eluent) to afford crude white solids (8.85 g, 86% ee). The solids were recrystallized from hexane (300 mL) to give the title compound as a colorless needle crystal (5.90 g, 58%, >99% ee).
[0428] 1H NMR: spectrum data were identical with those of the racemate (STEP 8-1 in Example 1).
[0430] To a stirred mixture of 4-hydroxy-N,N,2-trimethyl-1-[(4-methylphenyl)sulfonyl]-1H-benzimidazole-6-carboxamide (21.2 g, 56.8 mmol, STEP 4), (+)-5,7-difluoro-3,4-dihydro-2H-chromen-4-ol (15.86 g, 85.1 mmol, STEP 8) and tributylphosphine (22.9 g, 113 mmol) in toluene (840 mL) was added 1,1′-(azodicarbonyl)dipiperidine (ADDP) (19.3 g, 76.5 mmol) at room temperature. After stirring at room temperature for 2 hours, the reaction mixture was filtered through a pad of Celite and washed with ethyl acetate (300 mL). The filtrate was concentrated in vacuum. The residue was purified by column chromatography on silica get (ethyl acetate:hexane gradient elution from 1:20 to 20:1) to afford crude solids (27.0 g). The solids were recrystallized from 2-propanol (130 mL) to give the title compound as a colorless crystal (23.2 g, 75%, >99% ee)
[0431] 1H NMR: spectrum data were identical with those of the racemate (STEP 8-2 in Example 1).
[0433] To a solution of (−)-4-[((4S)-5,7-difluoro-3,4-dihydro-2H-chromen-4-yl)oxy]-N,N,2-trimethyl-1-[(4-methylphenyl)-sulfonyl]-1H-benzimidazole-6-carboxamide (24.2 g, 44.7 mmol, STEP 9) in tetrahydrofuran (65 mL) and 2-propanol (220 mL) was added 2M sodium hydroxide aqueous solution (220 mL, 440 mmol) at room temperature. After stirring at room temperature for 4 hours, the mixture was diluted with ethyl acetate (1.20 L) and washed with saturated ammonium chloride aqueous solution (500 mL). The organic solution was dried over magnesium sulfate and concentrated in vacuum. The residue was purified by column chromatography on amino gel (ethyl acetate:methanol gradient elution from 50:1 to 20:1) to afford the title compound as a white solid (15.2 g, 87%, >99% ee).
[0434] 1H NMR: spectrum data were identical with those of the racemate (STEP 9 in Example 1).
[0435] Optical rotation and retention time were identical with the above.
Tegoprazan is the world’s first potassium-competitive acid blocker (P-CAB), has a mechanism similar to that of an acid pump antagonist (APA), and blocks gastric acid secretion by competing with potassium ions for binding to the enzyme H+/K+– ATPase (proton pump) that secretes H+ ions, which are a component of gastric acid, from the gastric parietal cells into the gastric lumen. Since tegoprazan is not a prodrug such as a proton pump inhibitor (PPI), it does not require an activation process, and thus acts not only on an active proton pump but also on an inactive proton pump. Thus, tegoprazan has the advantages of exhibiting its effect rapidly and reaching the maximum effect within one hour.
Meanwhile, in general, in order for a drug to exhibit an expected effect, the blood concentration of the drug needs to be maintained at a certain level or higher. To maintain the blood concentration of the drug, a patient is required to take the prescribed drug repeatedly according to a certain schedule.
In this case, taking the drug frequently decreases the patient’s medication compliance, and as a result, there are many cases where the expected therapeutic effect is not obtained. Thus, in a disease for which a drug needs to be taken for a long period of time or the blood concentration of the drug at a time when the patient cannot take the drug needs to be maintained at a certain level or higher, the frequency and method of taking the drug is also an important factor to be considered for increasing the therapeutic effect of the drug.
Accordingly, there is a need to develop a formulation capable of maintaining a therapeutically effective blood concentration of a drug because there is no problem in the absorption rate of the drug while modifying the release of the drug.
Gastric acid-related gastrointestinal diseases, such as gastroesophageal reflux disease, non-erosive reflux disease, gastric ulcers, and ulcers caused by non-steroidal anti-inflammatory drugs are the most common diseases of the gastrointestinal tract. Histamine 2 receptor blockers and proton pump inhibitors (PPIs) are used in the treatment of the above symptoms, showing good curative effects and greatly improving the quality of life of patients. However, the degree of satisfaction with existing drugs for the treatment of gastrointestinal diseases related to gastric acid is still not high. For example, during the process of taking proton pump inhibitors, the symptoms of heartburn and esophageal reflux at night are still difficult to overcome, and the related symptoms cannot be effectively relieved 3 days before taking the medicine.
Potassium ion competitive acid blocker (P-CAB) is a new mechanism of H + -K + -ATPase inhibitor, which is a reversible proton pump inhibitor. Currently on the market are Revaprazan, Vonoprazan and Tegoprazan.
Tegoprazan’s chemical name is (S)-4-((5,7-difluorochroman-4-yl)oxy)-N,N,2-trimethyl-1H-benzo [d] Imidazole-6-carboxamide, the structure is shown in formula (1):
Both WO2007072146 and CN101341149B disclose two synthetic methods of Tegoprazan:
Method one (milligram preparation method):
WO2007072146 and CN101341149B quote the synthesis method of WO2004054984 to prepare A-3 compound, then acetylate under concentrated sulfuric acid/acetic anhydride, introduce cyano group through microwave reaction to obtain A-5 compound, and then undergo reduction, ring closure, hydrolysis, condensation, and Toluenesulfonyl protection, ether hydrogenolysis, Mitsunobu reaction (Mitsunobu reaction) to obtain A-11 compound, after hydrolysis to remove the p-toluenesulfonyl protecting group to obtain A-12 compound, namely Tegorazan racemate, and finally through a chiral column Split to obtain Tegorazan with optical activity.
This synthetic route requires 12 steps of reactions (not including the preparation of 5,7-difluorochroman-4-ol), and the synthesis yield is only 2.0%; zinc cyanide is used in the reaction, which requires special treatment of wastewater; In the reaction, the protecting group (benzyl protection, p-toluenesulfonyl protection) and the removal of the protecting group need to be carried out twice. Suitable for industrial production.
Method two (ten-gram preparation method):
The obtained A-4 compound is reduced and fused under the condition of iron powder/acetic acid to obtain A-13 compound, which is protected by p-toluenesulfonyl, amidation, and debenzyl protection to obtain A-10 compound, and finally combined with a chiral alcohol The Tegorazan precursor is obtained by the Mitsunobu reaction, and then the Tegorazan is obtained by hydrolysis to protect it.
Although method 2 has been shortened compared with method 1, the synthetic route still requires 9-step reaction (excluding the preparation of chiral alcohol), the route is still longer, and the total yield is 6.8%; carbon monoxide gas is used in the reaction to pass through the coupling Co-preparation of amides requires special equipment to carry out the reaction, which poses a safety hazard; two protective groups (benzyl protection, p-toluenesulfonyl protection) and two removal of protective groups are still required in the reaction, and the reaction steps are also added. This results in low synthesis efficiency, which is not conducive to industrial production.
The comparative document CN101341149B discloses the preparation method of compound 5, that is, the tetrahydrofuran solution of 5,7-difluorochroman-4-one is added to the chiral reagent (S)-1-methyl-3,3- Diphenyl-1H,3H-pyrrolo[1,2-c][1,3,2]oxazolborane, borane-dimethyl sulfide complex and tetrahydrofuran in a mixed solution, wait until the reaction is complete After purification by column chromatography, the chiral purity was 86% ee, and then recrystallized with hexane to obtain compound 5, the optical purity of which was >99% ee, and the yield was 58%.
The comparative document CN107849003A discloses the preparation methods of compounds 3 and 5, that is, 5,7-difluorochroman-4-one is used as a raw material for reduction with a chiral ruthenium catalyst, and the yield of compound 3 is 85%. The purity is 100% ee, the yield of the obtained compound 5 is 91%, and the chiral purity is 100% ee. This method involves ruthenium reagents that are difficult to purchase commercially and are expensive.
Patent EP2390254A1 discloses the preparation method of compound 2, which uses 3-fluoro-4nitrobenzoic acid in dichloromethane with oxalyl chloride and N,N-dimethylformamide to obtain acid chloride after concentration, and then the obtained The acid chloride is dissolved in dichloromethane, and then added dropwise to a mixed solution containing dimethylamine hydrochloride and triethylamine for preparation, and the purification method adopts column chromatography for purification.
Example 1
Preparation of (S)-5,7-difluorochroman-4-ol (3)
Take a 2L three-necked flask, add anhydrous THF (400mL) and R-Me-CBS (1mol/L toluene solution, 53mL, 53mmol), protect with argon, and inject borane dimethyl sulfide complex at room temperature (10mol/L, 58.6mL, 586mmol). 5,7-Difluorochroman-4-one (98g, 533mmol) was dissolved in anhydrous tetrahydrofuran (600mL), and slowly dripped into the above system. The entire dripping process lasted 9 hours. After dripping, let it stand overnight. The reaction solution was slowly poured into methanol cooled in an ice-water bath to generate a large number of bubbles, stirred until no obvious bubbles were generated, and concentrated to remove the solvent. Add 350 mL of ethyl acetate to dissolve, wash the organic phase with water (200 mL, 200 mL) and brine (100 mL) successively, dry over anhydrous sodium sulfate, filter, and concentrate to obtain a pale yellow solid. The chiral purity measured by chiral HPLC was 94.18%ee (OZ-H column, n-hexane/isopropanol=95/5, flow rate=1 mL/min, detection wavelength 220nm).
The above solid was heated and dissolved in a mixed solvent composed of n-hexane and ethyl acetate (n-hexane/ethyl acetate = 17:1), decolorized with activated carbon and then cooled and crystallized to obtain 77.8 g of off-white solid with a yield of 78.5% . [α] D 23 = -141.4 (c = 1, MeOH). The chiral purity measured by chiral HPLC is >99.9%ee (OZ-H column, n-hexane/isopropanol=95/5, flow rate=1mL/min, detection wavelength 220nm).
Take a 1L three-necked flask, add anhydrous THF (66mL) and S-Me-CBS (1mol/L toluene solution, 9mL, 9mmol), protect with argon, and inject borane dimethyl sulfide complex at room temperature (10mol/L, 9.9mL, 99mmol). Dissolve 5,7-difluorochroman-4-one (16.6 g, 90 mmol) in anhydrous tetrahydrofuran (166 mL) and slowly drip into the above system. The entire dripping process lasted 5.5 hours. After dripping, let it stand overnight. The reaction solution was slowly poured into methanol cooled in an ice-water bath to generate a large number of bubbles, stirred until no obvious bubbles were generated, and concentrated to remove the solvent. Add 100 mL of ethyl acetate to dissolve, wash the organic phase with water (50 mL, 30 mL) and brine (20 mL) successively, dry over anhydrous sodium sulfate, filter, and concentrate to obtain an oil, which is placed at room temperature as a yellow solid. The chiral purity measured by chiral HPLC was 93.6%ee (OZ-H column, n-hexane/isopropanol=95/5, flow rate=1mL/min, detection wavelength 220nm).
The above solid was heated and dissolved in a mixed solvent consisting of n-hexane and ethyl acetate (n-hexane/ethyl acetate=17:1), and 11.1 g of needle crystals were obtained by recrystallization, with a yield of 66.5%. [α] D 20 = +141.9 (c=1, MeOH). The chiral purity measured by chiral HPLC is >99.9%ee (OZ-H column, n-hexane/isopropanol=95/5, flow rate=1 mL/min, detection wavelength 220nm).
Preparation of 3-fluoro-N,N-dimethyl-4-nitrobenzamide (2)
Suspend 3-fluoro-4-nitrobenzoic acid (60g, 324mmol) in dichloromethane (400mL), add DMF (1mL), cool in an ice water bath, add oxalyl chloride (33mL, 389mmol) dropwise, after the addition is complete Incubate and stir for 2.5h. Dimethylamine hydrochloride (26.4g, 324mmol) was added to it, the temperature was lowered to -10°C, and a mixed solution composed of triethylamine (118mL, 842mmol) and dichloromethane (120mL) was added dropwise. After the addition was completed, the temperature was kept and stirred for 20 minute. Wash with 1 mol/L hydrochloric acid (100 mL), water (50 mL, 100 mL, 100 mL), half-saturated sodium bicarbonate solution (100 mL), and brine (100 mL) in sequence. It was dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated to remove most of the solvent. About 100 mL was left. 300 mL of n-hexane was added to make slurry, filtered, and washed twice with 100 mL of n-hexane, and dried to obtain 62.5 g of light yellow solid with a yield of 91.0%.
Preparation of 3-hydroxy-N,N-dimethyl-4-nitrobenzamide (4)
Put 3-hydroxy-4-nitrobenzoic acid (20.58g, 112mmol), dimethylamine hydrochloride (9.2g, 112mmol), EDCI (23.6g, 123mmol), HOBt (15.1g, 112mmol) in 1L In the reaction flask, acetonitrile (250 mL) was added, followed by triethylamine (31.2 mL, 224 mmol), and the mixture was stirred at room temperature overnight. Concentrate to remove acetonitrile, add water (250mL), extract 8 times with dichloromethane, 150mL each time, combine the organic phases and wash with saturated sodium bicarbonate solution twice, 200mL each time, and then wash once with saturated brine (100mL) , Dried with anhydrous sodium sulfate, filtered, and concentrated to obtain 19.7 g of yellow solid with a yield of 83.9%.
Preparation of 3-hydroxy-N,N-dimethyl-4-nitrobenzamide (4)
Place 3-hydroxy-4-nitrobenzoic acid (9.15g, 50mmol) in a 500mL reaction flask, add dichloromethane (100mL), and then add 1 drop of DMF. After cooling in an ice water bath, add dropwise oxalyl chloride (5.1mL, 60mmol). Heat to reflux for 1 hour, and concentrate to remove the solvent. Add dichloromethane (100 mL) to dissolve into a solution for later use. Take another reaction flask, add 50 mL of dichloromethane and 20 mL of 33% dimethylamine aqueous solution, and cool in an ice-water bath. Add the dichloromethane solution of acid chloride dropwise to the above system while stirring, and stir for 10 minutes after dropping. The dichloromethane layer was separated, and the aqueous phase was extracted with dichloromethane 6 times, 100 mL each time. The organic phases were combined and washed with saturated brine (60 mL), dried over anhydrous sodium sulfate, filtered, and concentrated to obtain 10 g of yellow solid. The yield was 94.9%.
Compound potassium tert-butoxide (0.44g, 3.9mmol) was dissolved in anhydrous tetrahydrofuran (9mL), protected by argon, cooled in an ice water bath, and compound 3 (0.61g, 3.3mmol) in anhydrous tetrahydrofuran solution (3 mL) was added dropwise ), keep and stir for 10 minutes after the addition is complete, add dropwise an anhydrous tetrahydrofuran solution (3 mL) of compound 2 (636 mg, 3 mmol), and after the addition is complete, keep and keep stirring for 10 minutes. Add 10mL of water, extract twice with ethyl acetate, 20mL each time, combine the organic phases, wash with brine, dry with anhydrous sodium sulfate, filter, and concentrate to obtain a yellow oil, add n-hexane to make a slurry, filter, and dry to obtain 1.0g off-white Solid, the yield is 90.9%.
Compound potassium tert-butoxide (41g, 368mmol) was dissolved in anhydrous tetrahydrofuran (500mL), protected by argon, cooled in an ice water bath, compound 3 (57.9g, 311mmol) in anhydrous tetrahydrofuran solution (250 mL) was added dropwise. After the addition was completed, the mixture was kept and stirred for 10 minutes, and an anhydrous tetrahydrofuran solution (250 mL) of compound 2 (60 g, 283 mmol) was added dropwise. After the addition, the mixture was kept and stirred for 10 minutes. Add 200 mL of ice water, concentrate to remove the organic solvent, add 800 mL of water, and extract four times with ethyl acetate, 500 mL each time. Combine the obtained organic phases, wash with half-saturated brine (1L), saturated brine (500mL), dry with anhydrous sodium sulfate, filter, and concentrate to obtain a brown oil. Pour 50mL of isopropanol while hot, and add petroleum ether (500mL). ) Be beaten, filter, wash twice with a mixture of isopropanol/petroleum ether=10/100, 100mL each time, and then wash twice with a mixture of isopropanol/petroleum ether=5/100, 100mL each time Finally, it was washed with petroleum ether (100 mL) once, and left to dry at room temperature to obtain compound 6, 97.3 g of pale yellow solid, with a yield of 91.0%.
Dissolve compound 4 (1g, 4.76mmol), compound 5 (0.93g, 5mmol), and triphenylphosphine (1.5g, 5.71mmol) in anhydrous ethyl acetate (25mL), protected by argon, and cooled in an ice water bath. A mixed solution consisting of DIAD (1.1 mL, 5.71 mmol) and anhydrous ethyl acetate (1.5 mL) was added dropwise, and the mixture was stirred for 2 hours after dropping. Anhydrous zinc chloride (0.86 g, 6.3 mmol) was added, and after stirring for 1 hour, the insoluble matter was removed by filtration, and the filter cake was washed twice with 10 mL of ethyl acetate. The filtrate was washed once with a mixed solution of ammonia water (2.5 mL) and water (20 mL), then washed with water (30 mL) once, washed with brine, dried over anhydrous sodium sulfate, filtered, and concentrated to obtain an oily substance. Add isopropanol (2.4 mL) to dissolve, slowly add n-hexane (24 mL) dropwise, and stir at room temperature for 1 hour, stir and heat to 80 degrees for 30 minutes, cool down and stir overnight. Filtered to obtain 1.86g of white solid (containing hydrazine-1,2-dicarboxylic acid diisopropyl ester), chiral purity>99%ee (OZ-H chiral column, flow rate 1mL/min, detection wavelength 254nm, normal hexane Alkyl-isopropanol=80mL-20mL, temperature 28°C) was used directly in the next step without further purification.
A small amount of crude product was purified by silica gel column chromatography (0~2% ethyl acetate in dichloromethane solution), the nuclear magnetic data is: 1 HNMR (400MHz, CDCl 3 )δ7.82(d,J=8.0Hz,1H),7.40(d,J=1.6Hz,1H),7.12 (dd,J=1.6Hz,8.4Hz,1H),6.52-6.29(m,2H) , 5.64(brs,1H), 4.47-4.30(m,2H), 3.13(s,3H), 3.00(s,3H), 2.34-2.26(m,1H), 2.14-2.23(m,1H).
Compound 4 (210mg, 1mmol), compound 5 (186mg, 1mmol), triphenylphosphine (314 mg, 1.2mmol) were dissolved in anhydrous THF (5mL), protected by argon, cooled in an ice water bath, and then DIAD ( A mixed solution consisting of 236 μL, 1.2 mmol) and anhydrous THF (0.3 mL) was dripped and stirred for 5 hours. Concentrate and purify by silica gel column chromatography (0-2% ethyl acetate in dichloromethane solution). Compound 6 was obtained, with a total of 339 mg of off-white solid, with a yield of 89.7%.
Example 10
Preparation of (S)-4-amino-3-((5,7-difluorochroman-4-yl)oxy)-N,N-dimethylformamide (7)
The compound 6 (1.86 g) obtained in Example 8 was dissolved in methanol (60 mL), and dry palladium on carbon (10% palladium on carbon, 194 mg) was added. The mixture was stirred at room temperature under normal pressure for 12 hours in a hydrogen atmosphere, filtered, washed with methanol, and the filtrate was concentrated. A purple solid was obtained, and 25 mL of isopropyl ether was added for beating to obtain 1.3 g of a slightly pink solid. The yield of the two steps was 78.3%.
Preparation of (S)-4-amino-3-((5,7-difluorochroman-4-yl)oxy)-N,N-dimethylformamide (7)
The compound 6 (96 g, 254 mmol) obtained in Example 7 was dissolved in a mixed solution (500 mL) composed of methanol/tetrahydrofuran = 1/4, and 50% water content wet palladium on carbon (10% supported on carbon, 19.2 g) was added. Shake hydrogenation at ~25psi pressure. After 3 hours, it was filtered, the filtrate was concentrated to a slurry, 300 mL of isopropyl ether was added to make a slurry, and dried to obtain compound 7, 78 g of an off-white solid, with a yield of 88.6%.
Preparation of (S)-4-iminoacetamido-3-((5,7-difluorochroman-4-yl)oxy)-N,N-dimethylbenzamide (8)
Compound 7 (174mg, 0.5mmol), potassium phosphate (127mg, 0.6mmol) were suspended in dichloromethane (5mL), and 2,2,2-trichloroethylacetimide hydrochloride (9-1, 135mg , 0.6mmol), stirred at room temperature for 24h. Add 5 mL of saturated potassium carbonate solution and 15 mL of ethyl acetate and stir for 5 minutes, separate the organic phase, and extract the aqueous phase twice with ethyl acetate, each time 10 mL. The organic phases were combined, washed with brine, dried with anhydrous sodium sulfate, filtered, concentrated, and purified by silica gel column chromatography (methanol/ammonia/dichloromethane=1/1/100~3/1/100) to obtain compound 8. 60mg pale yellow foamy solid, yield 30.0%.
HR-MS: [M+H] + : Measured value 390.1601
Example 13 to Example 21
Compound 7 (174mg, 0.5mmol) was used for feeding, referring to Example 12. The specific compound 9-1, base, solvent (5mL), ratio and yield of compound 8 used are shown in the following table:
Preparation of (S)-4-iminoacetamido-3-((5,7-difluorochroman-4-yl)oxy)-N,N-dimethylbenzamide (8)
Compound 7 (1.2g, 3.4mmol) was suspended in dichloromethane (14mL), sodium acetate (367mg, 4.5mmol) and 2,2,2-trichloroethylacetimide hydrochloride ( 500mg, 2.3mmol), add three batches, and stir for 5 hours after the addition. Extract 4 times with water, 15 mL each time, combine the water phases, and backwash the water phase with isopropyl ether (25 mL) once. The resulting aqueous phase was adjusted to alkaline with potassium carbonate (2g), extracted with ethyl acetate (20mL, 15mL, 10mL), the organic phases were combined and washed once with brine, dried over anhydrous sodium sulfate, filtered, and concentrated to obtain compound 8, 1.3 g White foamy solid, yield 98.5%.
Preparation of (S)-4-iminoacetamido-3-((5,7-difluorochroman-4-yl)oxy)-N,N-dimethylbenzamide (8)
Compound 7 (1.66g, 4.76mmol) was dissolved in dichloromethane (14mL), sodium acetate (390mg, 4.76mmol) and ethylacetimide hydrochloride (9-2, 440mg, 3.57mmol) were added every 1 hour ), add a total of four batches, and stir for 1 hour after the addition. Concentrate to remove dichloromethane, add 35 mL of water, extract 3 times with ethyl acetate, 15 mL each time, and discard. The aqueous phase was adjusted to alkaline with potassium carbonate (1.3g), extracted with ethyl acetate (30 mL, 20 mL, 20 mL, 10 mL), the organic phases were combined and washed with brine once, dried over anhydrous sodium sulfate, filtered, and concentrated to obtain the compound 8. 1.37g white foam, yield 74.0%.
Compound 8 (1.3 g, 3.4 mmol) was dissolved in acetonitrile (13 mL), cooled to 5° C. in an ice-water bath, N-chlorosuccinimide (454 mg, 3.4 mmol) was added in batches, and the mixture was kept warm and stirred for 35 minutes. A solution containing sodium hydroxide (0.68 g, 17 mmol) and water (4 mL) was added, and the mixture was stirred at room temperature for 2 hours. Concentrate to remove acetonitrile, add 25mL of water, adjust the pH to about 3-4 with 1mol/L hydrochloric acid solution (17mL), extract the resulting aqueous solution with ethyl acetate (25mL, 25mL, 20mL), and then further distill the organic solvent from the aqueous phase. Adjust the pH to 8 with saturated sodium bicarbonate solution, and a white solid can be precipitated. After suction filtration, washing with water, and drying, 0.94 g of off-white solid was obtained with a yield of 72.3%. [α] D 24 = -97.8 (c = 1, MeOH).
HR-MS: [M+H] + C 20 H 20 F 2 N 3 O 3 The calculated value is 388.1467, and the measured value is 388.1470.
Compound 8 (1.5g, 3.9mmol) was dissolved in 2,2,2-trifluoroethanol (19mL), cesium carbonate (1.38g, 4.25mmol) was added, cooled in an ice water bath, and diacetyl iodobenzene (1.37g, 4.25mmol), keep stirring for 40 minutes, add water, extract twice with ethyl acetate, wash with brine, dry with anhydrous sodium sulfate, filter, and concentrate to obtain an oily substance, which is subjected to silica gel column chromatography (3-4% methanol in dichloromethane solution ) 0.6 g of off-white foamy solid was obtained, with a yield of 40.3%.
Tegorazan, also known as Tegoprazan, Tegoprazan, CJ-12420, was approved by the Korean Ministry of Food and Drug Safety (MFDS) in July 2018 for the treatment of gastroesophageal reflux disease and erosive esophagitis .
Tegoprazan was originally developed by Pfizer. In 2008, it was licensed to RaQualia Pharma (from Pfizer) for cooperative development. In 2014, it was licensed by RaQualia Pharma to CJ Health Care. Finally, CJ Health Care was successfully developed and marketed in Korea. Tegoprazan is a competitive potassium ion acid blocker (P-CAB) and hydrogen ion/potassium ion exchange ATPase (H + /K + ATPase) inhibitor. It has a fast onset and can control the pH of gastric juice for a long time. The drug was first launched in South Korea and is a brand new drug for the treatment of gastroesophageal reflux disease and erosive esophagitis.
Gastric proton pump hydrogen ion/potassium ion exchange ATPase is the main pharmacological target for the treatment of gastric acid-related diseases. Potassium Competitive Acid Blocker (P-CAB) can inhibit gastric acid secretion by competitively binding to K + H + /K + -ATPase. Studies have found that Tegoprazan is such a potassium-competitive acid blocker, which is considered to be the most advanced drug for the treatment of gastroesophageal reflux disease, because proton pump inhibitors are the most commonly used drugs for the treatment of gastroesophageal reflux disease, and Tegoprazan It just can overcome the shortcomings of proton pump inhibitors. The effectiveness and safety of Tegoprazan are mainly based on two phase III clinical trials. One of them is a double-blind, actively controlled phase III study (NCT02456935), which was conducted in South Korea, with 280 patients with erosive esophagitis as the research object, and the cumulative healing rate of erosive esophagitis at the 8th week as the primary endpoint. To compare the safety and effectiveness of Tegoprazan and the proton pump inhibitor esomeprazole. Another phase III clinical trial is a double-blind, randomized, placebo-controlled trial (NCT02556021). The trial was conducted in 324 patients in South Korea. The primary endpoint was the percentage of patients whose main symptoms (heartburn and reflux) completely resolved at 4 weeks using the reflux disease questionnaire (RDQ) to evaluate the once-daily Tegoprazan tablet ( 50mg and 100mg) in the safety and effectiveness of patients with non-erosive reflux disease. The approval of the drug on the market provides a new option for the treatment of this type of disease, and to a certain extent makes up for the shortcomings of other drugs, so that this type of disease can be better treated.
Tegoprazan chemical name is (S)-4-((5,7-difluorochroman-4-yl)oxy)-N,N,2-trimethyl-1H-benzo(d)imidazole-6-methan Amide, the chemical structure contains a benzimidazole structure and a chiral 5,7-difluorochroman-4-oxyl structure, the specific chemical structure is as follows:
Patent CN101341149B discloses the preparation method of Tegoprazan, specifically 4-hydroxy-N,N,2-trimethyl-1-[(4-tolyl)sulfonyl]-1H-benzo[d]imidazole-6-methan Amide and (S)-5,7-difluoro-3,4-dihydro-2H-chromenen-4-ol undergo condensation reaction under the action of tributylphosphine/ADDP to prepare (-)-4- [((4S)-5,7-Difluoro-3,4-2H-chromogen-4-yl)oxy]-N,N,2-trimethyl-1-[(4-tolyl) Sulfonyl]-1H-benzo[d]imidazole-6-carboxamide intermediate, the latter removes the protective group under the action of a base to complete the preparation of Tegoprazan. The specific synthesis route is as follows:
Based on the description of the above patent, the preparation of Tegoprazan mainly involves 4-hydroxy-N,N,2-trimethyl-1-[(4-tolyl)sulfonyl]-1H-benzo[d]imidazole-6- The condensation reaction of formamide and (S)-5,7-difluoro-3,4-dihydro-2H-chromenen-4-ol, this condensation reaction not only involves the use of dangerous reagents tributylphosphine and coupling Nitrogen compounds with low yield and high cost.
Therefore, the development of a new synthetic method suitable for industrialization and cost-effective synthesis of Tegoprazan and its analogs can not only reduce the risk of industrial production of Tegoprazan, but also provide more analogs for potential drugs with higher activity. Research.
The synthetic route of the present invention is as follows:
Example 1: 4-[((4S)-5,7-difluoro-3,4-2H-chromogen-4-yl)oxy]-2-methyl-1-p-toluenesulfonyl-1H -Preparation of benzo[d]imidazole-6-carboxylic acid tert-butyl ester
The 4-chloro-2-methyl-1-p-toluenesulfonyl-1H-benzo[d]imidazole-6-carboxylic acid tert-butyl ester (42.10g, 0.10mol), (S)-5,7-two Fluoro-3,4-dihydro-2H-chromenen-4-ol (28.0g, 0.15mol), copper acetate (1.0g, 5.0mmol), potassium tert-butoxide (17.0g, 0.152mol) and N 1 ,N 2 -Bis (naphthalene-1-ylmethyl)oxalamide (3.7g, 10.05mmol) was added to the reaction flask, followed by nitrogen replacement three times, and then anhydrous 1,4-dioxide was added to the reaction flask Six rings (150 mL), the reaction system was replaced with nitrogen again three times. Subsequently, the reaction system was heated to 100°C for 24 hours with stirring. After the reaction, the system naturally dropped to room temperature. The reaction system was diluted with ethyl acetate (500 mL), stirred vigorously for 0.5 hours, and filtered through Celite. The filtrate was desolventized under reduced pressure to remove the organic solvent. Add dichloromethane (1.0L) and H to the residue 2 O (400 mL), the system was stirred for 15 minutes, the organic phase was separated, the aqueous phase was extracted 3 times with dichloromethane (3×400 mL), the organic phases were combined, the solvent was removed from the organic phase under reduced pressure, and the residue was added to heptane (500 mL) Stir vigorously overnight and filter. The obtained solid compound is dried and recrystallized from ethyl acetate/heptane to obtain an off-white solid (42.83 g, 75.1%).
Example 2: (S)-4-((5,7-difluorochroman-4-yl)oxy)-N,N,2-trimethyl-1H-benzo(d)imidazole-6-methan Preparation of Tegoprazan
Add 4-bromo-N,N,2-trimethyl-1H-benzo[d]imidazole-6-carboxamide (2.82g, 10.0mmol), (S)-5,7-bis Fluoro-3,4-dihydro-2H-chromenen-4-ol (2.80g, 15mmol), cuprous iodide (100mg, 0.53mmol), sodium tert-butoxide (1.45g, 15.1mmol) and N 1 ,N 2 -Bis (phenylethyl)oxalamide (150mg, 0.51mmol) was added to the reaction flask, followed by nitrogen replacement three times, then anhydrous DMF (15mL) was added to the reaction flask, and the reaction system was replaced with nitrogen again three times. Subsequently, the reaction system was heated to 85°C for 24 hours with stirring. After the reaction, the system naturally dropped to room temperature. The reaction system was diluted with ethyl acetate (200 mL), stirred vigorously for 0.5 hours, and filtered through Celite. The filtrate was desolventized under reduced pressure to remove the organic solvent. The residue was purified by column chromatography (ethyl acetate/heptane) to obtain a white solid (3.32 g, 85.7%).
Example 3: (S)-4-((5,7-difluorochroman-4-yl)oxy)-N,1,2-trimethyl-1H-benzo(d)imidazole-6-methan Amide
Add 4-iodo-N,1,2-trimethyl-1H-benzo[d]imidazole-6-carboxamide (3.30g, 10.0mmol), (S)-5,7-difluoro to the reaction flask successively -3,4-Dihydro-2H-chromenen-4-ol (2.80g, 15mmol), cuprous iodide (60mg, 0.32mmol), sodium tert-butoxide (1.15g, 11.97mmol) and N 1 , N 2 -bis(benzyl)oxalyl diamide (135 mg, 0.50 mmol) was added to the reaction flask, followed by nitrogen replacement three times, then anhydrous DMF (15 mL) was added to the reaction flask, and the reaction system was again nitrogen replaced three times. Subsequently, the reaction system was heated to 75°C for 24 hours with stirring. After the reaction, the system naturally dropped to room temperature. The reaction system was diluted with ethyl acetate (200 mL), stirred vigorously for 1 hour, and filtered through Celite. The filtrate was desolventized under reduced pressure to remove the organic solvent. The residue was purified by column chromatography (ethyl acetate/heptane) to obtain an off-white solid (2.77 g, 71.5%).
Tegoprazan was approved by the Ministry of Food and Drug Safety (MFDS) for marketing in July 2018 for the treatment of gastroesophageal reflux disease and erosive esophagitis. Tegoprazan was originally developed by Pfizer. In 2008, it was licensed to RaQualiaPharma (separated from Pfizer) for joint development. In 2014, Tegoprazan was licensed to CJHealthCare by RaQualiaPharma. Finally, CJHealthCare was successfully developed and marketed in Korea. Tegoprazan is a competitive potassium ion acid blocker (P-CAB) and hydrogen ion/potassium ion exchange ATPase (H+/K+ATPase) inhibitor. The drug was first marketed in South Korea. Medicines for treating gastroesophageal reflux disease and erosive esophagitis. Proton pump hydrogen ion/potassium ion exchange ATPase is the main pharmacological target for the treatment of gastric acid-related diseases. Potassium-competitive acid blocker (P-CAB) can inhibit gastric acid secretion by competitively binding to K+ with H+/K+-ATPase. Research finds that Tegoprazan is such a potassium-competitive acid blocker and is considered to be the most advanced drug for treating gastroesophageal reflux disease, because proton pump inhibitors are the most commonly used drugs for treating gastroesophageal reflux disease. Tegoprazan The shortcomings of proton pump inhibitors can be just overcome. Tegoprazan’s effectiveness and safety are mainly based on two phase III clinical trials. One of them is a double-blind, active-controlled phase III study. This study was conducted in South Korea. The study used 280 patients with erosive esophagitis as the primary endpoint[1].
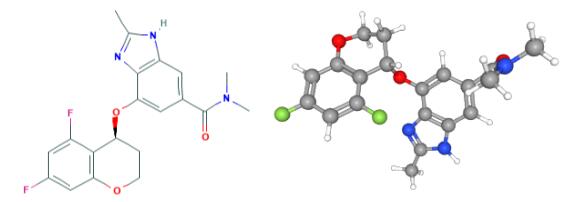
Fig 1. Chemical structure formula and three-dimensional structure of Tegoprazan
Tegoprazan, a potassium-competitive acid blocker, is a potent, oral active and highly selective inhibitor of gastric H+/K+-ATPase that could control gastric acid secretion and motility, with IC50 values ranging from 0.29-0.52 μM for porcine, canine, and human H+/K+-ATPases in vitro.
Tegoprazan inhibits porcine, canine, and human H+/K+-ATPase activity. Tegoprazan inhibits gastric H+/K+-ATPase in a potassium-competitive and reversible manner. Tegoprazan (3 μM) inhibits 86% of H+/K+-ATPase activity, whereas the inhibition is decreased to 34% after the dilution of Tegoprazan concentration to 0.15 μM[2].
Tegoprazan (1.0 mg/kg, p.o.) potently and completely inhibits histamine-induced gastric acid secretion in dogs. Tegoprazan (1.0-3.0 mg/kg, p.o.) reverses the pentagastrin-induced acidified gastric pH to the neutral range. Tegoprazan (3 mg/kg, p.o.) immediately evokes a gastric phase III contraction of the migrating motor complex in pentagastrin-treated dogs[3].
The invention relates to a method for preparing Tegoprazan chiral alcohols, in particular to the preparation method of (S) 5,7 difluoro 3,4 dihydro 2H chromogenic ene 4 alcohol. Using 5,7-difluoro-4H-benzopyran-4-ketone as starting material, the method realizes the preparation of (S)5,7-difluoro-3,4-dihydro-2H-chromogenic enone-4-alcohol by asymmetric reduction of ketone carbonyl with chiral reagent and subsequent conventional hydrogenation reaction[4].
Tegoprazan, a reversible H+/K+-ATPase inhibitor developed by CJ Healthcare (now inno.N), was first approved and launched in South Korea in 2019 for the treatment of gastroesophageal reflux disease (GERD). In 2020, the product attained supplemental approval for the treatment of gastric ulcers and Helicobacter pylori infection. Additional phase III clinical trials are being conducted by Shandong Luoxin Pharmacy Group, CJ Healthcare’s Chinese licensee. Tegoprazan was originally developed by RaQualia and licensed to CJ CheilJedang (the parent company of CJ Healthcare) in 2010 in Southeastern Asian markets; this agreement was later extended to Europe and North America in 2019. In 2015, a Chinese sublicense was granted to Shandong Luoxin Pharmacy Group. CJ Healthcare was acquired by Kolmar Korea in 2018, and renamed as inno.N in 2020.
NEW DRUG APPROVALS
one time
$10.00
References
[1] Takahashi N, et al. Tegoprazan, a Novel Potassium-Competitive Acid Blocker to Control Gastric Acid Secretion and Motility. J Pharmacol Exp Ther. 2018 Feb;364(2):275-286.
[2] Nobuyuki Takahashi and Yukinori Take.Journal of Pharmacology and Experimental Therapeutics February 2018, 364 (2) 275-286.
[3] Kim HK, Park SH, Cheung DY, Cho YS, Kim JI, Kim SS, Chae HS, Kim JK, and Chung IS (2010) Clinical trial: inhibitory effect of revaprazan on gastric acid secretion in healthy male subjects. J Gastroenterol Hepatol 25:1618–1625.
Mikami T, Ochi Y, Suzuki K, Saito T, Sugie Y, and Sakakibara M (2008) 5-Amino-6-chloro-N-[(1-isobutylpiperidin-4-yl)methyl]-2-methylimidazo[1,2-α]pyridine-8-carboxamide (CJ-033,466), a novel and selective 5-hydroxytryptamine4 receptor partial agonist: pharmacological profile in vitro and gastroprokinetic effect in conscious dogs. J Pharmacol Exp Ther 325:190–199.
/////// tegoprazan, Тегопразан , تيغوبرازان , 替戈拉生 , CJ-12420, IN-A001, K-CAB, LXI-15028, RQ-00000004, RQ-4, CJ 12420, IN A001, K CAB, LXI 15028, RQ 00000004, RQ 4, korea 2019
Peturadol CAS 686301-48-4 MFC12H20N6O MW264.33 g/mol 5-{[2-(6-amino-9H-purin-9-yl)ethyl]amino}pentan-1-olcentral analgesic, NB001, NB 001, HTS 09836, J89QT81NBQ NB-001 has been investigated for the treatment of Recurrent Herpes Labialis. NB001…
Petadeferitrin CAS911714-45-9 MFC16H21NO6S MW355.4 g/mol (4S)-2-[2-hydroxy-4-[2-(2-methoxyethoxy)ethoxy]phenyl]-4-methyl-5H-1,3-thiazole-4-carboxylic acid (4S)-2-{2-hydroxy-4-[2-(2-methoxyethoxy)ethoxy]phenyl}-4-methyl4,5-dihydro-1,3-thiazole-4-carboxylic acidiron chelating agent, SP 420, WBX54NZ436 Petadeferitrin is an orally bioavailable iron-chelating agent and derivative of desferrithiocin, with iron chelating and protective…
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
Follow my blog with Bloglovin
The title of your home page
You could put your verification ID in a
comment
Or, in its own meta tag
Or, as one of your keywords
Your content is here. The verification ID will NOT be detected if you put it here.

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....



{kind=link}
{kind=link}