

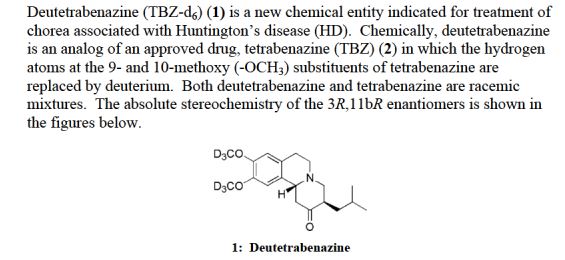
SD-809, Deutetrabenazine
- Tetrabenazine-d6
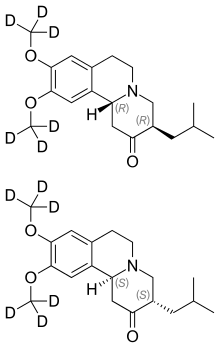
(3RS,11Brs)-9,10-di((2H3)methoxy)-3-(2-methylpropyl)-1,3,4,6,7,11b-hexahydro-2H-benzo(a)quinolizin-2-one
2H-Benzo[a]quinolizin-2-one, 1,3,4,6,7,11b-hexahydro-9,10-di(methoxy-d3)-3-(2-methylpropyl)-, (3R,11bR)-rel–
2H-Benzo(a)quinolizin-2-one, 1,3,4,6,7,11b-hexahydro-9,10-di(methoxy-d3)-3-(2-methylpropyl)-, (3R,11bR)-rel-
2H-Benzo(a)quinolizin-2-one, 1,3,4,6,7,11b-hexahydro-9,10-di(methoxy-d3)-3-(2-methylpropyl)-, (3R,11bR)-rel-
(RR,SS)-1,3,4,6,7,11b-Hexahydro-9,10-di(methoxy-d3)-3-(2-methylpropyl)-2H-benzo[a]quinolizin-2-one
(3RS,11Brs)-9,10-di((2H3)methoxy)-3-(2-methylpropyl)-1,3,4,6,7,11b-hexahydro-2H-benzo(a)quinolizin-2-one
Treatment of Chorea Associated with Huntington Disease
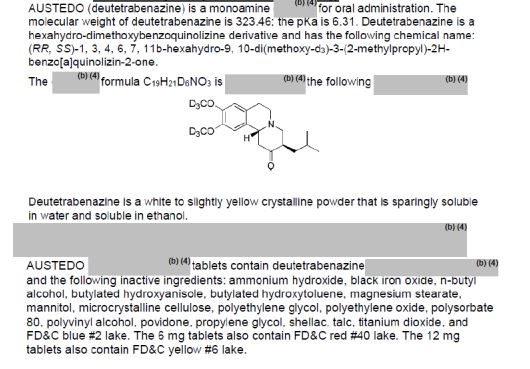
MF C19-H21-D6-N-O3
-
C19-H27-N-O3
Deutetrabenazine
TEVA

LINK……………https://newdrugapprovals.org/2015/08/15/sd-809-deutetrabenazine-nda-submitted-by-teva/
| Austedo | FDA 4/3/2017 | For the treatment of chorea associated with Huntington’s disease Drug Trials Snapshot Chemistry Review(s) (PDF) |



SD-809 was granted Orphan Drug Designation for the treatment of HD by the FDA in November 2014 and became part of Teva’s CNS portfolio with the acquisition of Auspex Pharmaceuticals in May 2015.
Teva announced that the New Drug Application (NDA) for SD-809 (deutetrabenazine) has been accepted by the U.S. Food and Drug Administration (FDA) for the treatment of chorea associated with Huntington disease (HD), a rare and fatal neurodegenerative disorder caused by the progressive breakdown of nerve cells in the brain that affects about five to seven people per 100,000 in western countries, according to the World Health Organization.
-9,10-di((2H3)methoxy)-3-(2-methylpropyl)-1,3,4,6,7,11b-hexahydro-2H-benzo(a)quinolizin-2-one")

…………………….
Patent for preparing tetrabenazine
http://www.google.com/patents/WO2012081031A1?cl=en
Chemically tetrabenazine is cis rac -1, 3, 4, 6, 7, 1 lb-hexahydro-9, 10-dimethoxy-3-(2- methylpropyl)-2Hbenzo[a]quinolizin-2-one and it is represented by compound of structural formula I.

Formula 1
The proprietary name of tetrabenazine is Xenazine and is marketed by Biovail Americas. Xenazine is indicated for the treatment of chorea associated with Huntington’s disease. U.S. patent no. 2,830,993 discloses a process for the preparation of tetrabenazine compound of structural formula I wherein 1 -carbethoxymethyl-6, 7-dimethoxy-l , 2, 3, 4- tetrahydroisoquinoline compound of structural formula IV is being reacted with mono- isobutylmalonic acid dimethyl ester compound of structural formula V and paraformaldehyde in methanol solvent to get l-carbethoxymethyl-2 (2, 2-dicarbomethoxy-4-methyl-n-pentyl)-6, 7- dimethoxy-1, 2, 3, 4-tetrahydroisoquinoline compound of structural formula VI. The 1- carbethoxymethyl-2(2,2-dicarbomethoxy-4-methyl-n-pentyl)-6,7-dimethoxy-l ,2,3,4- tetrahydroisoquinoline compound of structural formula VI is subjected to Dieckmann cyclization , hydrolysis and decarboxylation to get tetrabenazine compound of structural formula I, which is recrystallized from di-isopropyl ether solvent.

Formula I
Scheme I
U. S. patent no. 4,678,792 discloses a process for the preparation of 6, 7-dimethoxy-3, 4- dihydroisoquinoline compound of structural formula VII wherein 2-(3, 4-dimethoxyphenyl)- ethylamine compound of structural formula II is being reacted with chloral hydrate at 120°C to get N-formyl-2-(3, 4-dimethoxyphenyl)-ethylamine compound of structural formula III. The N- formyl-2-(3, 4-dimethoxyphenyl)-ethylamine compound of structural formula III is further reacted with polyphosphoric acid to get 6, 7-dimethoxy-3, 4-dihydroisoquinoline compound of structural formula VII. The 6, 7-dimethoxy-3, 4-dihydroisoquinoline compound of structural formula VII is being used as an intermediate for the preparation of tetrabenazine compound of structural formula I.

Formula III
Formula II
Polyphosphoric acid

Formula VII
Scheme II
Bull. Korean Chem. Soc. 2002 Volume (23). No. l , page no. 149 discloses N-formylation of various amines and alcohols with formic acid in toluene.
U.S. patent publication no. 2010/0130480 discloses a process for the preparation of 6, 7- dimethoxy-3, 4-dihydroisoquinoline compound of structural formula VII by reacting 2-(3, 4- dimethoxyphenyl)-ethylamine compound of structural formula II with hexamethylenetetramine in presence of acetic acid or trifluoroacetic acid.

Hexamethylenetetramine
Formula II Formula VII
U.S. patent publication no. 2008/0167337 discloses a process for the preparation of tetrabenazine compound of structural formula I wherein 6, 7-dimethoxy-3, 4-dihydroisoquinoline compound of structural formula VII is reacted with 3-dimethylaminomethyl-5-methyl-hexan-2-one methiodide compound of structural formula VIII to get crude tetrabenazine compound. The crude tetrabenazine compound was purified by employing flash column chromatography technique and

Formula VIII Formula I
The prior-art processes for preparing N-formyl-2-(3, 4-dimethoxyphenyl)-ethylamine compound of structural formula III produces below mentioned compound of structural formula XVII, XVIII, XIX, XX, XXI and XXII as a by-product of the reaction due to the demethylation and formylation of resulting hydroxy compounds.

Formula XX Formula XXI Formula XXII
The compounds of structural formula XVII, XVIII, XIX, XX, XXI and XXII are being carry- forwarded into the further steps of reactions of preparing tetrabenazine compound of structural formula I and therefore there is a need in the art to develop an improved process of preparing 6, 7-dimethoxy-3, 4-dihydroisoquinoline compound of structural formula VII, which obviates the prior-art problems. Accordingly there is provided a process of preparing tetrabenazine compound of structural formula I wherein 6, 7-dimethoxy-3, 4-dihydroisoquinoline compound of structural formula VII is being formed without the formation of above mentioned compounds of structural formula XVII, XVIII, XIX, XX, XXI and XXII.
EXAMPLE: PROCESS FOR THE PREPARATION OF SUBSTANTIAL PURE CRYSTALLINE FORM A OF TETRABENAZINE
Stage A: Process for the preparation of 6, 7-dimethoxy-3, 4-dihydroisoquinoIine
Step 1 : Process for the preparation of N-formyl-2-(3, 4-dimethoxyphenyl)-ethylamine
A solution of 2-(3, 4-dimethoxyphenyl)-ethylamine (500gm) in toluene (2000ml) was added formic acid (150gm) at 25°C, the resulting reaction mixture was diluted with toluene (500ml) and heated up to 45°C. The reaction mixture was maintained at 40-45°C for 5 hours and then the resulting reaction mixture was concentrated under reduced pressure at 50°C to get the title compound
Yield: 570gm
Purity: 99.98% (By HPLC)
Step 2: Process for the preparation of 6, 7-dimethoxy-3, 4-dihydroisoquinoline
A solution of N-formyl-2-(3, 4-dimethoxyphenyl)-ethylamine (250gm) obtained from step 1 in toluene (500ml) and polyphosphoric acid (50gm) was heated at 110°C for 5 hours. The resulting reaction mixture was cooled to 50°C, quenched with water (500ml) and pH of the resulting solution was adjusted to about 8.3 with aqueous solution of sodium hydroxide [sodium hydroxide (690gm) + water (690ml)]. The resulting reaction mass was extracted by ethyl acetate (2 1250ml), dried over anhydrous sodium sulfate (50gm) and concentrated under reduced pressure to get 6, 7-dimethoxy-3, 4-dihydroisoquinoline (190gm).
Yield: 215gm
Purity: 99.67% (By HPLC)
Stage B: Process for the preparation of 3-((dimethylamino) methyi)-5-methylhexan-2-one methiodide
Step 1 : Process for the preparation of 3-((dimethylamino) methyl)-5-methylhexan-2-one Dimethylamine hydrochloride (180gm) and paraformaldehyde (lOOgm) were added to a solution of 5-methylhexan-2-one (900ml) in methanol (1600ml). The resulting reaction mass was heated at reflux for 12 hours, and then the pH was adjusted to about 8.75 with aqueous solution of sodium hydroxide [sodium hydroxide(90gm) + water (900ml)] at 25 °C. The resulting reaction solution was extracted by toluene (2x1234ml). The organic layer was dried over anhydrous sodium sulfate (50gm) and concentrated under reduced pressure to get title compound.
Yield: 900gm
Purity: 99.80% (By HPLC)
Step 2: Process for the preparation of 3-((dimethylamino) methyl)-5-methylhexan-2-one methiodide
Methyl iodide (323gm) was added dropwise to a solution of 3-((dimethylamino) methyl)-5- methylhexan-2-one (195gm) obtained from step 1 , in ethyl acetate (1650ml) at 25-30°C in 30 minutes. The resulting reaction mixture was stirred at 25 °C for 12 hours and then the resulting solids were filtered, washed with water (200ml) and suck-dried to get wet compound (400gm). The wet compound was slurried with water (1000ml) at 25°C for 1 hour and then it was again filtered, washed with water (200ml) and dried at 45-50°C to get title compound
Yield: 300gm
Purity: 99.86% (By HPLC)
Stage C: Preparation of substantial pure crystalline form A of Tetrabenazine
3-((Dimethylamino) methyl)-5-methylhexan-2-one methiodide (80gm) was added to the solution of 6, 7-dimethoxy-3, 4-dihydroisoquinoline (40gm) in isopropanol (288ml) at 25°C and the resulting reaction mass was heated at 40-45°C for 15 hours. The resulting insoluble material was filtered, washed with isopropanol (80ml) and filtrate was concentrated under reduced pressure up to the 150ml reaction volume. The reaction solution was diluted with methylene dichloride (1200ml) and water (1000ml) and pH was adjusted to 8.5 with sodium hydroxide solution [10%, 100ml]. The organic layer was separated, washed with water (3 x 1000ml) and concentrated under reduced pressure to obtain residue. The residue was dissolved in methanol (300ml) at 50°C, and resulting solution was treated with an activated carbon (20gm) at 50-60°C for 30minutes and then it was filtered and filtrate was further stirred at 20-25°C for 2 hours. The resulting solids were filtered, washed with methanol (150ml), dried at 50-55°C for 8 hours. The resulting solids were milled, sifted through 40 mesh sieve and micronized.
Yield: 65gm
Purity: 99.96% (By HPLC)
………………………
PAPER

A concise synthesis of tetrabenazine and dihydrotetrabenazine is described. The key feature of this synthesis is the intramolecular aza-Prins-type cyclization of an amino allylsilane via oxidative C–H activation.




http://www.hgxb.com.cn/EN/abstract/abstract12047.shtml
……………
PAPER
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3126153/

The TBZ (4) for these reactions was prepared by reacting 3,4-dihydro-6,7-dimethoxyisoquinoline (3) and the Mannich base (2) as shown in Scheme 1.14 The α,β-unsaturated TBZ (5), which was the original substrate, was obtained by further treatment with chloranil in refluxing benzene.
Tetrabenazine (4a)
TLC: Rf = 0.62; silica gel; 4% MeOH/96% CH2Cl2.MS: (DCl-NH3) m/z 318 (M+H).UV: (EtOH) λmax 282.0 nm (ε4431).1H NMR: (300 MHz, CDCl3) δ 6.61 (s, 1H), 6.55 (s, 1H), 3.85 (s, 3H), 3.82 (s, 3H), 3.51 (br dd, 1H), 3.29 (dd, 1H), 3.13 (m, 2H), 2.90 (dd, 1H), 2.75 (m, 2H), 2.57 (m, 2H), 2.35 (t, 1H), 1.81 (ddd, 1H), 1.65 (m, 1H), 1.04 (ddd, 1H), 0.92 (d, 3H), 0.89 (d, 3H) ppm.13C NMR: (75 MHz, CDCl3) δ 210.00, 147.86, 147.54, 128.60, 126.11, 111.53, 107.94, 62.48, 61.52, 56.01, 55.92, 50.58, 47.62, 47.57, 36.09, 29.38, 25.44, 23.21, 22.11 ppm.EA: Anal. Calc for C19H17NO3: C, 71.89; H, 8.57; N, 4.41. Found C, 72.15; H, 8.69; N, 4.47.HPLC: Brownlee 25 cm × 4.6 mm silica gel column; 30% isopropanol/70% hexane; 1 mL/min; ret. time 5.94 min; purity >99.5%.
…………….
http://www.google.ga/patents/WO2008154243A1?cl=en
Example 10 Removal The Boc Protecting Group From First Intermediate 12 And Amino Cyclization Provide (+)-Tetrabenazine XVII

[0063] First intermediate 12 (1.0 eq) was dissolved in 10% Me2S- dichloromethane to provide an 82 mM solution. The solution was cooled to 0 0C and triisopropylsilane (1.1 eq.) followed by TFA (precooled to 0 0C) was added to the reaction mixture to provide a final concentration of 41 mM. The reaction mixture was permitted to stir at 0 0C for 1 h. Following the allotted time the reaction mixture was quenched at 0 0C by the addition of saturated aqueous potassium carbonate solution and concentrated under reduced pressure to remove the majority of the dimethylsulfide. The mixture was extracted with five portions of dichloromethane, and the combined organic extracts were washed with brine, dried (MgSO4), filtered and concentrated under reduced pressure to provide the crude product as a yellow solid. The crude product was recrystallized from 3.5% dimethoxyethane in hexanes. The resulting colorless crystals were washed with hexanes to provide pure (+)- tetrabenazine (XVII) 46%: mp 126.0 0C (3.5% DME-hexanes) (a crystal polymorph was observed at 116 0C); [α]26 D +37.2 (c 0.41, CH2Cl2); 1H NMR (CD2Cl2) δ 0.89 (apparent t, J = 7.2 Hz, 6H), 0.98 (ddd, J = 12, 6.0, 4.0 Hz, IH), 1.59-1.68 (m, IH), 1.74 (ddd, J = 12, 5.9, 5.7 Hz, IH), 2.32 (apparent t, J = 11.7 Hz, IH), 2.46 (apparent t, J = 12.3 Hz, IH), 2.55 (ddd, J = 12, 10.0, 3.8 Hz, IH), 2.65-2.73 (m, 2H), 2.83 (dd, J = 5.5, 2.8Hz, IH), 2.97-3.07 (m, IH), 3.07-3.14 (m, IH), 3.25 (dd, J =9.7, 6.3 Hz, IH), 3.47 (apparent d, J = 12Hz, IH), 3.75 (s, 3H), 3.77 (s, 3H), 6.55 (s, IH), 6.60 (s, IH) 13C NMR (CD2Cl2) δ 21.98, 23.02, 25.51, 29.46, 35.16, 47.47, 47.63, 50.47, 55.87, 56.01, 61.47, 62.46, 108.46, 111.72, 126.37, 128.96, 147.65, 147.98, 209.72; HRMS-(ESI+) calcd for (C19H27NO3 + H) ([M+H]+ 318.2069, found 318.2082.
…………….
US 20150152099



………….
NBI-98854 (CAS # 1025504-59-9), (S)-(2R,3R,l lbR)-3-isobutyl-9,10-dimethoxy-2,3,4,6,7,1 lb-hexahydro-lH-pyrido[2,l-a]isoquinolin-2-yl 2-amino-3-methylbutanoate, is a VMAT2 inhibitor. NBI-98854 is currently under investigation for the treatment of movement disorders including tardive dyskinesia. WO 2008058261; WO 2011153157; and US 8,039,627. NBI-98854, a valine ester of (+)-a-dihydrotetrabenazine, in humans is slowly hydrolyzed to (+)-a-dihydrotetrabenazine which is an active metabolite of tetrabenazine.

NBI-98854
EXAMPLE 1
D6-(±)-3-Isobutyl-9,10-dimethoxy-3,4,6,7-tetrahydro-lH-pyrido[2,l-a]isoquinolin-2(l lbH)-one ((±)-Tetrabenazine-<d6)
Step 1

[0193] Jgrt-butyl 3,4-dihydroxyphenethylcarbamate : A solution of dopamine
hydrochloride (209 g, 1.11 mol, 1.00 equiv), sodium carbonate (231 g, 2.75 mol, 2.50 equiv) and di-tert-butyl dicarbonate (263 g, 1.21 mol, 1.10) in 2.4 L tetrahydrofuran / water (5: 1) was stirred at 20°C for 2.5 h. After the starting material was consumed completedly, the reaction was diluted with ethyl acetate (2 L) and washed with water (2×600 mL). The organic phase was dried over sodium sulfate, filtered and concentrated under reduced pressure until two volumes of solvent was left. The precipitated solid was isolated by filtration and dried under vacuum to give 254 g (91%) of ieri-butyl 3,4-dihydroxyphenethylcarbamate as white solid. Ή-ΝΜΪ (300 MHz, CDC13) 8.72 (s, 1H), 8.62 (s, 1H), 6.79 (m, 1H), 6.62 (m, 1H), 6.51 (m, 1H), 6.40 (m, 1H), 3.03 (m, 2H), 2.50 (m, 2H), 1.37 (s, 1H). LC-MS: m /z = 254 (MH) +.
Step 2

[0194] D6-fert-butyl 3,4-dimethoxyphenethylcarbamate: A solution of ieri-butyl 3,4-dihydroxyphenethylcarbamate (127 g, 397 mmol, 1.00 equiv), potassium carbonate (359.3 g, 2.604 mmol, 3.00 equiv) and 18-crown-6 (1,4,7,10,13,16-hexaoxacyclooctadecane ) (68.64 g, 0.26 mmol, 0.03 equiv) in acetone (800 mL) was stirred at 38°C. After 30 min., CD3I (362 g, 2.604 mmol, 3.00 equiv) was added to the reaction, and the mixture was stirred at 38°C for 12 h. Then an additional CD3I (120 g, 0.868 mmol, 1.00 equiv) was added to the solution and the solution was stirred for 5 h. Then the mixture was cooled to room temperature and the solid was filtered. The filtrate was concentrated under vacuum. The resultant solid was dissolved in H2O (300 mL) and extracted with EA (3×300 mL), the organic layers was combined and concentrated under vacuum to give 114 g (79%) of de-tert-butyl 3,4-dimethoxyphenethylcarbamate as white
solid. ^-NMR (300 MHz, CDC13) <Π.39 (m, 5H), 6.82 (m, 1H), 6.73 (m, 2H), 5.12 (s, 1H), 3.45 (m, 2H), 2.77 (m, 2H). LC-MS: m /z = 288 (MH) +.
Step 3

[0195] D6-2-(3,4-dimethoxyphenyl)ethanamine: A solution of de-tert-butyl 3,4-dimethoxyphenethylcarbamate (128 g, 455.26 mmol, 1.00 equiv) in ethyl acetate (1.5 L) was stirred at room temperature. Then HC1 gas was introduced into the reaction mixture for 2h. The precipitated solid was isolated by filtration. The solid was dissolved in 300 mL of water. The pH value of the solution was adjusted to 12 with sodium hydroxide (solid). The resulting solution was stirred for 1 h at 5-10°C. The resulting solution was extracted with 6×800 mL of ethyl acetate and the organic layers combined, dried over sodium sulfate, and concentrated under vacuum to give 64 g (78%) of d6-2-(3,4-dimethoxyphenyl)ethanamine as yellow oil.
^-NMR (300 MHz, CDC ) 6.77 (m, 3H), 3.89 (s, 3H), 3.87 (s, 3H), 2.96 (m, 2H), 2.71 (m, 2H), 1.29 (s, 2H). LC-MS: m /z = 182 (MH) +.
Step 4

[0196] D6-N-r2-(3,4-dimethoxy-phenyl)ethyllformamide: A solution of d6-2-(3,4-dimethoxyphenyl)ethanamine (69 g, 368 mmol, 1.00 equiv) in ethyl formate(250 mL) was heated under reflux overnight. The solution was concentrated under vacuum to give 71 g (91%) of d6-N-[2-(3,4-dimethoxy-phenyl)ethyl]formamide as yellow solid. The crude solid was used in next step without purification. ^-NMR (300 MHz, CDCb) £8.17 (s, 1H), 6.81 (m, 3H), 5.53 (br, 1H).3.59 (m, 2H), 2.81 (t, 2H, / = 6.9 Hz). LC-MS: m /z = 216 (MH) +.
Step 5

[0197] D6-6,7-dimethoxy-3,4-dihvdroisoguinoline: A solution of d6-N-[2-(3,4-dimethoxy-phenyl)ethyl]formamide (71 g, 329 mmol, 1.00 equiv) in phosphorus oxychloride (100 mL) was stirred at 105°C for 1 h. Then the solution was concentrated under vacuum to remove
phosphorus oxychloride. The residual oil was dissolved in ice / water. The solution was made basic with potassium carbonate with cooling. The basic aqueous solution was extracted with dichloromethane. The collected organic phase was dried using sodium sulfate and then filtered. The dichloromethane was removed by concentration under vacuum to give an orange oil.
Purification by silica gel (ethyl acetate:petroleum ether = 1: 1 ~ ethyl acetate) to give 43 g (66%) of d6-6,7-dimethoxy-3,4-dihydroisoquinoline as orange solid (yield 66%). Ή-ΝΜΡ (300 MHz, CDC13) 8.24 (s, 1H), 6.82 (s, 1H), 6.68 (s, 1H), 3.74 (m, 2H), 2.69 (t, 2H, J = 12 Hz). LC-MS: m /z = 198 (MH) +.
Step 6

[0198] Trimethyl(5-methylhex-2-en-2-yloxy)silane: To a cold (-78°C), stirred solution of j-PrMgBr (500 mL of 2 M solution in tetrahydrofuran, 1 mol, 1.00 equiv) in anhydrous tetrahydrofuran (1 L) was added Cul (19.02 g, 0.1 mol, 0.10 equiv) and the resultant mixture was stirred for 15 min at -78°C. Anhydrous hexamethylphosphorous triamide (358.4 g, 2 mmol, 2 equiv) was added and after 20 min, a solution of methyl vinyl ketone (70 g, 0.1 mol, 1.00 equiv), trimethylsilyl chloride (217 g, 0.2 mol, 2.00 equiv), in tetrahydrofuran (200 mL) was added dropwise over 30 min. After the reaction mixture was stirred at -78 °C for lh, triethylamine (20.2g, 200 mmol, 2.00 equiv) was added and the resulting mixture stirred for 10 min at 0 °C. To this was added ie/ -butyl methyl ether (2 L), and the solution was washed with 5% ammonia solution (6×300 mL). Then the organic phase was dried over sodium sulfate and concentrated under vacuum at 25°C to give 155 g crude product as yellow liquid. The liquid was purified by distilling (64-68°C/40 mmHg) to provide 118 g (63.3%) of trimethyl(5-methylhex-2-en-2-
yloxy)silane (E:Z = 56 : 44) as a colorless oil. XH-NMR (300 MHz, J6-DMSO) 4.58 (m, 0.56H), 4.43 (m, 0.44H), 1.73 (s, 1.69H), 1.66 (s, 1.32H), 1.53 (m, 1H), 0.84 (m, 6 H), 0.15(m, 9H).
Step 7

[0199] 3-r(Dimethylamino)methyl1-5-methylhexan-2-one: To a stirred solution of trimethyl(5-methylhex-2-en-2-yloxy)silane (118 g, 633 mmol, 1.00 equiv) in anhydrous acetonitrile (800 mL) was added N-methyl-N-methylenemethanaminium iodide (128.8 g, 696.3 mmol, 1.10 equiv) in several batches and the resultant mixture was stirred at 20°C overnight. Then the solution was concentrated under vacuum to remove the solvent. The residue was dissolved in 400 mL 1 N HC1 (aq.) and extracted with ieri-butyl methyl ether. Then the water phase was basiced with 2 N aq. NaOH and extracted with ie/ -butyl methyl ether. The organic phase was dried and concentrated under vacuum. The liquid was purified by distilling (80°C/0.5 mmHg) to provide 50 g (46%) of 3-[(dimethylamino)methyl]-5-methylhexan-2-one as a colorless oil. XH-NMR (300 MHz, J6-DMSO) £0.92 (d, 3H), 0.98 (d, 3H), 1.11-1.23 (m, 1H), 1.23-1.38 (m, 1H), 1.54-1.70 (m, 1H), 2.30 (s, 3H), 3.01 (s, 9H), 3.10-3.32 (m, 2H), 3.81-3.88 (m, 1H).
Step 8

[0200] 2-Acetyl-N,N V,4-tetramethylpentan-l-aminium iodide: A solution of 3-[(dimethylamino)methyl]-5-methylhexan-2-one (50 g, 15.00 mmol, 1.00 equiv) and methyl iodide (4.26 g, 30.00 mmol, 2.00 equiv) in 50 mL diethyl ether was stirred overnight at room temperature. The precipitated solid was isolated by filtration and dried under vacuum to give 79 g (86%) of 2-acetyl-N,N,N,4-tetramethylpentan-l-aminium iodide as white solid. XH-NMR (300 MHz, Je-DMSO) 0.89-0.98 (m, 6H), 1.11-1.20 (m, 1H), 1.40 (m, 1H), 1.66 (m, 1H), 2.30 (s, 3H), 3.01(s, 9H), 3.21 (m, 2H), 3.85 (m, 1H).
Step 9

[0201] Ρό- (±) -tetrabenazine : A solution of d6-6,7-dimethoxy-3,4-dihydroisoquinoline (33.4 g, 169 mmol, 1.10 equiv) and 2-acetyl-N,N,N,4-tetramethylpentan-l-aminium iodide (48 g, 153 mmol, 1.00 equiv) in 300ml of methanol was heated under reflux for 48 h. Then 150 mL water was added. The solution was cooled to room temperature. The precipitated solid was isolated by filtration and dried under vacuum to give 38 g of crude d6-tetrabenazine as yellow solid. The crude tetrabenazine was dissolved in ieri-butyl methyl ether (15 volumes), the mixture was heated until the solid was almost dissolved. The yellow solid which was unsolvable was filtered. The filtrate was concentrated under vacuum until 2 volumes ieri-butyl methyl ether was left. The solid was filtered and collected. The above solid was dissolved in ethanol (4 volumes), then the mixture was heated until the solid was dissolved. The solution was stirred and cooled to room temperature at the rate of 20°C/h. Then the mixture was stirred at 0°C for lh. The precipitated solid was isolated by filtration and dried under vacuum to give 25 g (50.4%) of tetrabenazine-<d6 as white solid.
^-NMR (300 MHz, CD2C12) £6.61 (s, 1H), 6.55 (s, 1H), 3.84 (s, 3H), 3.82 (s, 3H), 3.50 (d, 1H, / = 12 Hz), 3.27 (dd, 1H, / = 11.4Hz, / = 6.3 Hz), 3.11 (m, 2H), 2.84 (dd, 1H, / = 10.5 Hz, / = 3 Hz), 2.74 (m, 2H), 2.56 (m, 2H), 2.31 (t, 1H, J = 12 Hz), 1.76 (m, 1H), 1.63 (m, 1H), 0.98 (m, 1H), 0.89 (m, 6H).
LC-MS: m /z = 324 (MH) +.
………………
NMR PREDICT
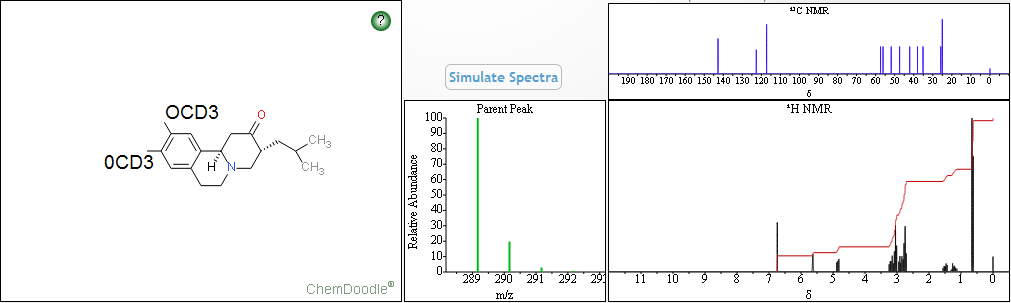

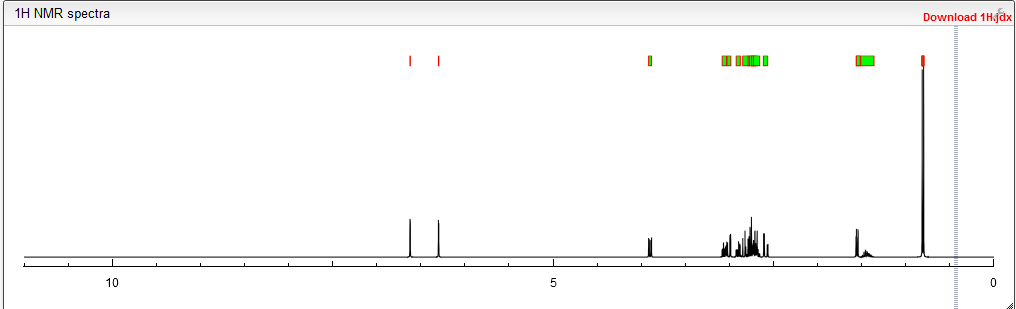

Watch out will be updated……………….


Rob Koremans, MD, President and CEO of Global Specialty Medicines at Teva.

Michael Hayden, M.D., Ph.D., President of Global R&D and Chief Scientific Officer at Teva
| WO2009070552A1 * | 25 nov. 2008 | 4 juin 2009 | Gen Electric | Alpha-fluoroalkyl tetrabenazine and dihydrotetrabenazine imaging agents and probes |
| WO2012000308A1 * | 27 juin 2011 | 5 janv. 2012 | China Pharmaceutical University | A method for resolution of tetrabenazine |
| WO2012081031A1 * | 11 avr. 2011 | 21 juin 2012 | Enaltec Labs Pvt. Ltd. | Process for preparing tetrabenazine |
| WO2013041621A1 * | 20 sept. 2012 | 28 mars 2013 | Basf Se | Low molecular weight modulators of the cold-menthol receptor trpm8 and use thereof |
| WO2015048370A1 * | 26 sept. 2014 | 2 avr. 2015 | Auspex Pharmaceuticals, Inc. | Benzoquinolone inhibitors of vmat2 |
| US7897769 | 25 oct. 2007 | 1 mars 2011 | General Electric Company | Intermediates for fluorinated dihydrotetrabenazine ether imaging agents and probes |
| US7897770 | 25 oct. 2007 | 1 mars 2011 | General Electric Company | Fluorinated dihydrotetrabenazine ether imaging agents and probes |
| US7902364 | 29 nov. 2007 | 8 mars 2011 | General Electric Company | Alpha-fluoroalkyl tetrabenazine and dihydrotetrabenazine imaging agents and probes |
| US7910738 | 29 nov. 2007 | 22 mars 2011 | General Electric Company | Intermediates for alpha-fluoroalkyl tetrabenazine and dihydrotetrabenazine imaging agents and probes |
| US7919622 | 7 déc. 2007 | 5 avr. 2011 | Kande Kankanamalage Dayarathna Amarasinghe | Intermediates for fluorinated tetrabenazine carbinol compounds imaging agents and probes |
| US8013161 | 7 déc. 2007 | 6 sept. 2011 | General Electric Company | Fluoroalkyl tetrabenazine carbinol compounds as imaging agents and probes |
| US8053578 | 16 juil. 2008 | 8 nov. 2011 | General Electric Company | Alpha-fluoroalkyl dihydrotetrabenazine imaging agents and probe |
| WO2007017654A1 | 4 août 2006 | 15 févr. 2007 | Cambridge Lab Ireland Ltd | 3, hb cis dihydrotetrabanezine for the treatment of schizophrenia and other psychoses |
| US3132147 * | 15 juin 1962 | 5 mai 1964 | Titre non disponible | |
| US4193998 * | 14 juin 1978 | 18 mars 1980 | Chinoin Gyogyszer Es Vegyeszeti Termekek Gyara Rt | 1,2,3,4,6,7-Hexahydro-11BαH-benzo[a]quinolizine-derivatives |
| US4686226 * | 3 sept. 1985 | 11 août 1987 | Merck & Co., Inc. | Substituted benzo[b]furo- and benzo[b]thieno quinolizines |
| US5118690 * | 21 oct. 1991 | 2 juin 1992 | John Wyeth & Brother Limited | Pharmaceutical tetrahydroisoquinolines |
| US5272270 * | 12 août 1991 | 21 déc. 1993 | Consortium Fur Elektrochemische Industrie Gmbh | Process for the preparation of 1-alkylisoquinoline derivatives |
| US5278308 * | 28 févr. 1992 | 11 janv. 1994 | The Trustees Of The University Of Pennsylvania | Iodine derivatives of tetrabenazine |
| US20020055637 * | 21 déc. 2001 | 9 mai 2002 | Song Liu | Methods for synthesis of amino-tetrahydroisoquinoline-carboxylic acids |
| US20040082647 * | 21 avr. 2003 | 29 avr. 2004 | G.D. Searle, Llc | Method for the preparation of tetrahydrobenzothiepines |
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO1994000460A1 * | Jun 23, 1993 | Jan 6, 1994 | Univ California | SYNTHESIS OF N-FORMYL-3,4-DI-t-BUTOXYCARBONYLOXY-6-(TRIMETHYLSTANNYL)-L-PHENYLALANINE ETHYL ESTER AND ITS REGIOSELECTIVE RADIOFLUORODESTANNYLATION TO 6-[18F]FLUORO-L-DOPA |
| WO2008058261A1 * | Nov 8, 2007 | May 15, 2008 | Neurocrine Biosciences Inc | Substituted 3-isobutyl-9, 10-dimethoxy-1,3,4,6,7,11b-hexahydro-2h-pyrido[2,1-a] isoquinolin-2-ol compounds and methods relating thereto |
| WO2008154243A1 * | Jun 4, 2008 | Dec 18, 2008 | Gen Electric | Method for making tetrabenazine compounds |
| WO2010044981A2 * | Sep 18, 2009 | Apr 22, 2010 | Auspex Pharmaceutical ,Inc. | Benzoquinoline inhibitors of vesicular monoamine transporter 2 |
| EP0154842A2 * | Feb 16, 1985 | Sep 18, 1985 | Dr. Karl Thomae GmbH | Medicament containing quaternary 3,4-dihydroisoquinoline salts |
| US2830993 | May 18, 1956 | Apr 15, 1958 | Quinolizine derivatives | |
| US4678792 | Feb 28, 1985 | Jul 7, 1987 | Dr. Karl Thomae Gmbh | Quaternary 3,4-dihydro-isoquinolinium salts |
| US20080167337 | Nov 8, 2007 | Jul 10, 2008 | Gano Kyle W | Substituted 3-isobutyl-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2h-pyrido[2,1-a]isoquinolin-2-ol compounds and methods relating thereto |
| US20100130480 | Sep 18, 2009 | May 27, 2010 | Auspex Pharmaceuticals, Inc. | Benzoquinoline inhibitors of vesicular monoamine transporter 2 |
| Reference | ||||
| 1 | * | BROSSI, A. ET AL: “Synthesis in the emetine series. I. 2-Oxohydrobenzo[a]quinolizines“, HELVETICA CHIMICA ACTA, vol. 41, 1958, pages 119-139, XP002659731, | ||
| 2 | BULL. KOREAN CHEM. SOC. vol. 23, no. 1, 2002, page 149 | |||
| 3 | * | DATABASE CA [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; AL-HIARI, YUSUF M. ET AL: “Synthesis of 1-benzyl-1,2,3,4-tetrahydroisoquinoline, Part I: Grignard synthesis of 1-(substituted benzyl)-1,2,3,4-tetrahydroisoquinoline models with potential antibacterial activity“, XP002659739, retrieved from STN Database accession no. 2009:467462 | ||
| 4 | * | DATABASE CA [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; DE LUCA, LIDIA ET AL: “A new, simple procedure for the synthesis of formyl amides“, XP002659734, retrieved from STN Database accession no. 2004:1062632 | ||
| 5 | * | DATABASE CA [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; DOMINGUEZ, ESTHER ET AL: “Solvent effect on the Bischler-Napieralski reaction. Synthesis of 3-aryl-3,4-dihydroisoquinolines“, XP002659736, retrieved from STN Database accession no. 99:158206 | ||
| 6 | * | DATABASE CA [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; FALCK, J. R. ET AL: “Oxazoline chemistry. Preparation of isoquinolines and 2,2′-bisoxazolines“, XP002659744, retrieved from STN Database accession no. 1981:497646 | ||
| 7 | * | DATABASE CA [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; FUKUDA, TSUTOMU ET AL: “Synthesis of both enantiomers of protoberberines via laterally lithiated (S)-4-isopropyl-2-(o-tolyl)oxazolines“, XP002659742, retrieved from STN Database accession no. 2008:192807 | ||
| 8 | * | DATABASE CA [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; JAHANGIR ET AL: “Aza analogs of protoberberine and phthalideisoquinoline alkaloids“, XP002659741, retrieved from STN Database accession no. 1986:572799 | ||
| 9 | * | DATABASE CA [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; MENENDEZ, J. C. ET AL: “Synthesis and antibacterial activity of some 1-thia-4,8-diazaspiro[4.5]decan-3-ones, thiazolo[2,3-a]isoquinolin-3-ones and 1,3-thiazino[2,3-a]isoquinolin-4-ones“, XP002659740, retrieved from STN Database accession no. 1989:114772 | ||
| 10 | * | DATABASE CA [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; NARASIMHAN, N. S. ET AL: “Unusual products in Bischler-Napieralski reaction“, XP002659743, retrieved from STN Database accession no. 1981:46871 | ||
| 11 | * | DATABASE CA [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; REIMANN, EBERHARD ET AL: “Protoberberines from Reissert-Compounds. Part IX [1]. An Alternative Approach to Dibenzoquinolizine- and Isoquinonaphthyridin-13a-carboxylic Acids, a Novel Synthesis of Alangimarine“, XP002659738, retrieved from STN Database accession no. 143:267131 | ||
| 12 | * | DATABASE CA [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; SHAFIK, RAGAB M. ET AL: “.alpha.-Phenyl-.beta.-(3,4-dimethoxy)phen ethylamines: novel inhibitors of choline acetyltransferase from Torpedo electric organ“, XP002659735, retrieved from STN Database accession no. 1985:61873 | ||
| 13 | * | DATABASE CA [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; WANG, CHENG-XUE ET AL: “Synthesis of rutaecarpine and quinazolone compounds“, XP002659737, retrieved from STN Database accession no. 2009:92700 | ||
| 14 | * | RISHEL, MICHAEL J. ET AL: “Asymmetric Synthesis of Tetrabenazine and Dihydrotetrabenazine“, JOURNAL OF ORGANIC CHEMISTRY, vol. 74, no. 10, 2009, pages 4001-4004, XP002659732, | ||
| 15 | * | SCHWARTZ, D. E. ET AL: “Metabolic studies of tetrabenazine, a psychotropic drug in animals and man“, BIOCHEMICAL PHARMACOLOGY, vol. 15, no. 5, 1966, pages 645-655, XP002659733, | ||
update on 2018
Novel Process for Preparation of Tetrabenazine and Deutetrabenazine
 , Yogesh Dadaji Pawar , Dnyaneshwar Tukaram Singare, Tushar Nandkumar Deshpande, and Girij Pal Singh
, Yogesh Dadaji Pawar , Dnyaneshwar Tukaram Singare, Tushar Nandkumar Deshpande, and Girij Pal SinghAbstract

A novel process for the synthesis of tetrabenazine (1) and deutetrabenazine (2), two well-known drugs used for the treatment of chorea associated with Huntington’s disease, has been developed. All of the reaction parameters were optimized through a series of reactions and by using Design of Experiment techniques. The newly developed methods are industrially scalable and employ cheap, commercially available raw materials and hence are highly efficient. The added advantage is that the developed processes evade the use of genotoxic alkylating agents and therefore could be considered as safe and viable alternatives to the existing methods.

Chemical structures of 1 and 2.
-
(a) Brossi, A.; Lindlar, H.; Walter, M.; Schnider, O. Helv. Chim. Acta 1958, 41, 119– 139, DOI: 10.1002/hlca.660410117
.
(b) Brossi, A.; Schnider, O.; Walter, M. Quinolizine derivatives.U.S. Patent 2,830,993, 1958.
-
Pletscher, A.; Brossi, A.; Gey, K. F. Int. Rev. Neurobiol. 1962, 4, 275– 306, DOI: 10.1016/S0074-7742(08)60024-0
-
Mestre, T.; Ferreira, J.; Coelho, M. M.; Rosa, M.; Sampaio, C. Cochrane Database Syst. Rev. 2009, 3,CD006456, DOI: 10.1002/14651858.CD006456.pub2
-
U.S. Food and Drug Administration. Novel Drug Approvals for 2017.https://www.fda.gov/Drugs/DevelopmentApprovalProcess/DrugInnovation/ucm537040 (accessed Jan 15, 2018).SYNTHESIS
-
Brossi, A. Preparation of substituted 2-oxobenzoquinolizines. U.S. Patent 3,045,021, 1962.
-
Gant, T. G.; Shahbaz, M. M. Benzoquinoline inhibitors of vesicular monoamine transporter-2. U.S. Patent 8,524,733, 2013.
Large-Scale Preparation of Tetrabenazine (1)
Purification of 1
1 (9.6 g, 96%). HPLC purity: 99.75%.
FTIR (in KBr): 2942, 2919, 1701, 1516, 1465, 1370, 1263, 1159, 1010, 860, 749 cm–1.
1H NMR (500 MHz, DMSO-d6): δ 6.70 (s, 1H), 6.69 (s, 1H), 3.72 (s, 6H), 3.46 (d, 1H, J = 10.0 Hz), 3.24 (dd, 1H, J = 11.5, 6.0 Hz), 3.15–3.11 (m, 1H), 2.95–2.89 (m, 1H), 2.85 (dd, 1H, J = 13.0, 3.0 Hz), 2.69–2.65 (m, 2H), 2.52–2.46 (m, 2H), 2.28 (t, 1H, J= 12.0 Hz), 1.66–1.63 (m, 2H), 0.94–0.85 (m, 7H).
ESI-MS: m/z 318.3 [M + H]+.



Large-Scale Preparation of Deutetrabenazine (2)FTIR (in KBr): 2942, 2920, 2246, 2067, 1700, 1513, 1269, 1113, 990, 747 cm–1. 1H NMR (500 MHz, CDCl3): δ 6.63 (s, 1H), 6.56 (s, 1H), 3.53 (d, 1H, J = 10.5 Hz), 3.32–3.30 (m, 1H), 3.17–3.13 (m, 2H), 2.92 (dd, 1H, J = 13.5,3.0 Hz), 2.77–2.73 (m, 2H), 2.64–2.53 (m, 2H), 2.37 (t, 1H, J = 11.5 Hz), 1.84–1.79 (m, 1H), 1.69–1.67 (m, 1H), 1.08–1.04 (m, 1H), 0.94–0.91 (m, 6H). ESI-MS: m/z 324.4 [M + H]+
Large-Scale Purification of 2
Mp: 128.75–129.42 °C.
FTIR (in KBr): 2942, 2920, 2246, 2067, 1700, 1513, 1269, 1113, 990, 747 cm–1.
1H NMR (500 MHz, DMSO-d6): δ 6.69 (s, 2H), 3.46 (d, 1H, J = 10.0 Hz), 3.25 (dd, 1H, J = 11.5, 6.0 Hz), 3.15–3.11 (m, 1H), 2.95–2.89 (m, 1H), 2.85 (dd, 1H, J = 13.5, 3.0 Hz), 2.70–2.64 (m, 2H), 2.52–2.44 (m, 2H), 2.28 (t, 1H, J = 11.5 Hz), 1.66–1.63 (m, 2H), 0.93–0.85 (m, 7H).
ESI-MS: m/z 324.4 [M + H]+.
[α]D −0.3 [c 0.3, DCM at 25 °C].



सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये। औकात बस इतनी देना, कि औरों का भला हो जाये।
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE




 LIONEL MY SON
LIONEL MY SON
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
///////


[…] Deutetrabenazine […]
LikeLike