PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
Difluprednate is a topical corticosteroid used for the symptomatic treatment of inflammation and pain associated with ocular surgery.
Difluprednate is a corticosteroid, It is chemically a butyrate ester of 6(alpha),9(alpha)-difluoro prednisolone acetate. Accordingly, difluprednate is sometimes abbreviated DFBA, for difluoroprednisolone butyrate acetate.
Difluprednate is a topical corticosteroid indicated for the treatment of infammation and pain associated with ocular surgery. It is a butyrate ester of 6(α), 9(α)-difluoro prednisolone acetate. Difluprednate is abbreviated DFBA, or difluoroprednisolone butyrate acetate. It is indicated for treatment of endogenous anterior uveiti.
Approval
On June 24, 2008, the US Food and Drug Administration (FDA) approved difluprednate for the treatment of post-operative ocular inflammation and pain.[1] It is marketed by Alcon under the tradename Durezol.
WO/2022/118271DIFLUPREDNATE FOR REDUCING THE ADVERSE EFFECTS OF OCULAR INFLAMMATION
SYN 1
Synthetic Reference
Process for preparation of Difluprednate from sterol fermentation product; Ding, Kai; Xu, Feifei; Assignee Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Peop. Rep. China; East China University of Science and Technology; 2014; Patent Information; Aug 06, 2014; CN; 103965277; A
SYN 2
Synthetic Reference
Preparation method of Difluprednate; Tian, Yuan; Zhou, Shengan; Guo, Bin; Xu, Zhiguo; Assignee Guangzhou Renheng Pharmaceutical Technology Co., Ltd., Peop. Rep. China 2017; Patent Information; May 10, 2017; CN; 106632561; A
SYN3
Synthetic Reference
Shailesh, Singh; Bharat, Suthar; Jain, Ashish; Gaikwad, Vinod; Kulkarni, Kuldip. Process for preparing difluprednate. Assignee Ajanta Pharma Ltd., India. IN 2013MU02535. (2015).
SYN4
Synthetic Reference
Sun, Hongbin; Chen, Bo. Method for preparation of Difluprednate. Assignee China Pharmaceutical University, Peop. Rep. China. CN 103509075. (2014).
Embodiment 1:4, pregnant steroid-17 α of 9 (11)-diene, 21-dihydroxyl-3,20-diketone-21-acetic ester (formula III compound)
10g hydrocortisone-21 acetic ester (formula II compound) is joined in 250mL eggplant type bottle, add 50mL N, dinethylformamide and 8.8mL pyridine, slowly heat up and make material dissolution complete, slowly cooling afterwards, slowly be added dropwise to 4.4mL methylsulfonyl chloride, add rear solution to be yellow completely.Be warming up to 85 ℃ of stirrings, the reaction solution thick one-tenth that can slowly become sticky is faint yellow, adds slightly some DMFs and makes reaction solution dilution, can normally stir, and keeps this thermotonus one hour, and reaction solution slowly becomes grey black during this period.TLC follows the tracks of (sherwood oil: ethyl acetate=1: 1) show that reaction finishes.Stop heating, treat that the backward reaction solution of slow cooling adds 200mL methyl alcohol, stir 1min, reaction flask is placed in to crystallization under ice-water bath.Suction filtration after 1h, makes water and methanol wash filter cake, crude product productive rate 100%.With methyl alcohol-methylene dichloride mixed solvent system recrystallization, obtain sterling, M.P.231-235 ℃, productive rate 90%. 1H-NMR(300MHz,CDCl 3):δ(ppm)5.75(1H,s,4-H),5.55(1H,s,11-H),5.07(1H,d,J=5Hz,21-H),4.84(1H,d,J=5Hz,21-H),2.15(3H,s,H-21-OAc),1.31(3H,s,19-CH 3),0.65(3H,s,18-CH 3),0.66-2.90(m,17H,backbone).
By 9.4g4, pregnant steroid-17 α of 9 (11)-diene, 21-dihydroxyl-3,20-diketone-21-acetic ester (formula III compound) and 10g4-Dimethylamino pyridine add in 1000mL eggplant-shape bottle, add again 50mL diethylene glycol dimethyl ether and 260mL methylene dichloride, heated and stirred makes dissolution of solid, slowly adds 32mL butyryl oxide slightly after cooling, is warming up to 80 ℃ of return stirrings.After 23h, TLC follows the tracks of, and raw material primitive reaction is complete, stops heating and stirs.Vacuum concentration is removed methylene dichloride.After being down to room temperature, add frozen water in reaction flask, white solid standing to be separated out.Suction filtration, saturated sodium bicarbonate aqueous solution washing leaching cake, dries under infrared lamp, obtain 4,9 (11)-diene-17 α, 21-dihydroxyl-3,20-ketone-21-acetic ester 17 iophenoxic acid esters (formula IV compound) sterling 10.65g, M.P220-224 ℃, productive rate 95.9%. 1H-NMR(500MHz,CDCl 3):δ(ppm)5.75(1H,s,4-H),5.54(1H,m,11-H),4.87(1H,d,J=4.8Hz,O=C-CH 2-O,21-H),4.64-4.91(2H,ABq,J=16.6Hz,21-H),2.75(2H,m,2-H),0.70(3H,s,18-CH 3),0.95(3H,t,J=4.4Hz),1.34(3H,s,18-CH 3),1.66(2H,m,-CH 2CH 3),2.17(3H,s,O=C-CH 3),2.32(2H,t,J=4.3Hz,O=C-CH 2),? 13C-NMR(75MHz,CDCl 3):δ(ppm)199.1,198.9,173.4,170.4,169.1,144.1,124.1,118.5,94.5,66.9,48.2,46.3,40.9,37.5,36.4,34.2,33.8,32.7,32.2,32.1,30.6,26.2,24.5,20.5,18.3,13.7,13.6;ESI-MS?m/z:457.2[M+H +],479.2[M+Na +];HRMS?for?C 27H 36O 6+Na +?calcd?479.2410,found479.2402.
10g4, pregnant steroid-17 α of 9 (11)-diene, 21-dihydroxyl-3,20-diketone-21-acetic ester 17 iophenoxic acid esters add in 250mL eggplant type bottle, then add 80mL methylvinyl acetate, slowly drip while stirring the 1mL vitriol oil.Be warming up to 80 ℃ of stirring reactions, solution is thin out yellow clarification slowly.(sherwood oil: ethyl acetate=3: 1), raw material reaction is complete produces new point to TLC after 30min.Stop heating, wait to be cooled to 50 ℃, add 1mL triethylamine, be stirred to and be down to room temperature.Add water in reaction solution, ethyl acetate aqueous layer extracted three times, saturated common salt water washing organic phase twice, anhydrous sodium sulfate drying.After 30min, steam organic solvent and obtain brown color oily matter.Column chromatography is purified and is obtained 3,5,9 (11) pregnant steroid-3 of triolefin, 17 α, 21 trihydroxy–3,20-diketone-3,21-diacetate esters 17 iophenoxic acid esters, productive rate 90%. 1H-NMR(300MHz,CDCl 3):δ(ppm)5.74(1H,s,4-H),5.53(1H,s,11-H),5.45(1H,s,6-H),4.64-4.91(2H,ABq,J=16.6Hz,21-H),2.17(3H,s,-COCH 3),1.17(3H,s,19-CH 3),0.96(3H,t,J=7.5Hz),0.70(3H,s,18-CH 3).
14g4, 9 (11)-diene-6 α-fluoro-17 α, 21-dihydroxyl-3, 20-diketone-21-acetic ester 17 iophenoxic acid esters (formula VII) and 9 (11)-diene-6 β-fluoro-17 α, 21-dihydroxyl-3, the mixture of 20-diketone-21-acetic ester 17 iophenoxic acid esters (formula VI) adds in dry three-necked bottle, add while stirring 400mL acetum, under room temperature, slowly pass into anhydrous hydrogen chloride gas (98% vitriol oil is added dropwise in 37% concentrated hydrochloric acid solution and makes) until saturated, be stirred to raw material and be dissolved into yellow solution completely, continue to stir 2h, TLC monitoring reacts completely, stop stirring, in reaction solution, add the aqueous solution, after separating out solid, suction filtration, saturated sodium bicarbonate aqueous solution washing, dry, be weighed as 13g, productive rate is 93%. 1H?NMR(300MHz,CDCl 3):δ(ppm)6.10(s,1H),5.61(s,1H),5.41-5.16(m,1H),4.64-4.91(2H,ABq,J=16.6Hz,21-H),2.82(dd,J=28.3,15.7Hz,3H),2.50(s,2H),2.32(t,J=7.4Hz,2H),2.17(s,3H),1.96(s,5H),1.66(d,J=7.4Hz,2H),1.46(s,2H),1.33(s,3H),0.96(s,3H),0.71(s,3H).
13g 6 α-fluoro-4; 9; (11)-diene-pregnant steroid-3,20-22 ketone-17-butyric ester-20-acetic ester is dissolved in and fills 300mL1, in the eggplant type bottle of 4 dioxane; add while stirring 40mL 0.46mol/L high chloro acid solution; under room temperature, stir after several minutes, add 14g N-succinimide in reaction system, under nitrogen protection, stir; raw material dissolves gradually, and it is faint yellow that reaction solution is.(the sherwood oil: ethyl acetate=12: 5) monitoring, raw material primitive reaction is complete, adds 10%Na of TLC after 2h 2sO 3unnecessary N-succinimide is fallen in aqueous solution cancellation, and checks (it is blue that test paper no longer becomes) with starch-kalium iodide test paper.Add water in reaction flask, ethyl acetate extraction three times, twice of saturated common salt water washing organic phase, anhydrous sodium sulfate drying organic phase, after 30min, be spin-dried for organic phase, obtain faint yellow oily matter, column chromatography purification (sherwood oil: ethyl acetate=12: 1) obtain white solid 6 α-fluoro-9 α-bromo-11 beta-hydroxies-4-alkene-pregnant steroid-3, the about 14g of 20-diketone-17-butyric ester-20-acetic ester, productive rate is 89%. 1H-NMR(300MHz,CDCl 3):δ(ppm)5.93(1H,d,J=4.5,4-H),5.06(1H,m,6-H),4.64-4.91(2H,ABq,J=16.6Hz,21-H),2.17(3H,s,-COCH 3),1.84(3H,s,18-CH 3),0.96(3H,t,J=7.5Hz),1.02(3H,s,19-CH 3),4.72(1H,s,11-H);ESI-MS?m/z:593.3,595.3[M+Na +].
100mg 6 α-fluoro-9 β, 11 beta epoxides-4-alkene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester drops in the Plastic Bottle of tetrafluoroethylene, adds 2mL methylene dichloride to dissolve, and stirs at-20 ℃.1mL Olah reagent with under 1mL methylene dichloride low temperature, mix after, be slowly added dropwise in reaction system, maintain low temperature and stir 2 hours, TLC monitoring reaction finishes.Reaction flask shifts out low-temp reaction groove, is slowly added dropwise to the 1mol/L NaOH aqueous solution by excessive HF cancellation, is adjusted to pH7~8.Add chloroform in reaction system, extraction, organic layer is used respectively aqueous hydrochloric acid and the saturated common salt water washing of 3mol/L, anhydrous sodium sulfate drying, after standing 30min, steams except organic solvent, column chromatography is further purified and is obtained white solid powder 6 α, 9 α-fluoro-11 beta-hydroxies-4-alkene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester, productive rate 90%. 1H-NMR(300MHz,CDCl 3):δ(ppm)?6.11(1H,d,J=4.5Hz,4-H),5.27(1H,m,6-H),4.64-4.91(2H,ABq,J=16.6Hz,21-H),2.17(3H,s,-COCH 3),4.40(1H,d,J=4.5Hz,11-H),1.02(3H,s,18-CH 3),0.96(3H,t,J=7.5Hz),1.52(3H,s,19-CH 3);ESI-MS?m/z:533.3[M+Na +]
40mg 6 α, 9 α-fluoro-11 beta-hydroxies-4-alkene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester is dissolved in 3mL dioxane, adds 28mgDDQ, and 100 ℃ of return stirrings heat up.TLC monitoring reaction (sherwood oil: ethyl acetate=12: 8) after 13h, generate the larger product of polarity, steam except organic solvent dioxane, obtain brown color oily matter, add a small amount of methylene dichloride lysate, suction filtration, elimination solid residue, filtrate is washed with sodium bicarbonate aqueous solution after adding a small amount of methylene dichloride again, steams except organic phase rear pillar Chromatographic purification, obtain white solid powder 6 α, 9 α-fluoro-11 beta-hydroxies-Isosorbide-5-Nitrae-diene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester, be title molecule difluprednate, productive rate 70%. 1h-NMR (300MHz, CDCl 3): δ (ppm) 7.20 (1H, d, J=4.5Hz, 1-H), 6.43 (1H, s, 4-H), 6.38 (1H, d, J=6Hz, 2-H), 5.36 (1H, m, 6-H), 4.64-4.91 (2H, ABq, J=16.6Hz, 21-H), 4.43 (1H, d, J=4.5Hz, 11-H), 2.27 (2H, m ,-CH 2-CH 3), 2.17 (3H, s, O=C-CH 3), 1.55 (3H, s, 19-CH 3), 1.02 (3H, s, 18-CH 3), 0.93 (3H, t, J=4.5Hz, 0=C-CH 2cH 2cH 3); ESI-MS m/z:509.3[M+H +]; HRMS for C 27h 35o 7f 2+ H +calcd 509.2351, found 509.2356.M.P.188-190 ℃ (literature value M.P.190-194 ℃); [α] d22=+30.1 ° of (literature values [α] d22=+31.7 °).
Claims (6)
Hide Dependent
1. a method of preparing difluprednate, as following reaction formula:
Specifically comprise the following steps:
(1) by hydrocortisone-21-acetic ester (formula II compound):
Carry out dehydration reaction, generate formula III compound:
(2) formula III compound is carried out to butyric acid esterification, obtains formula IV compound:
(3) formula IV compound is carried out to the reaction of enolization esterifying reagent, obtains formula V compound:
(4) formula V compound is reacted with fluoro reagent and obtains formula VI and formula VII compound:
(5) by formula VI compound, through configuration reversal, reaction obtains formula VII compound;
(6) formula VII compound is reacted with N-bromo-succinimide and water, obtains formula VIII compound:
(7) formula VIII compound epoxidation under alkaline condition is obtained to formula IX compound:
(8) formula IX compound is reacted with fluorination reagent and obtains formula X compound:
(9) dehydrogenation of formula X compound oxidation is obtained to formula I compound (difluprednate).
2. method as claimed in claim 1, is characterized in that, in step (2), formula III compound is obtained to formula IV compound through fourth esterification, and the fourth esterifying reagent adopting is butyryl oxide or butyryl chloride; The alkaline catalysts adopting is pyridine, triethylamine or DMAP; The solvent adopting is methylene dichloride, diethylene glycol dimethyl ether, 1, the mixture of the optional solvents in 2-ethylene dichloride, dioxane, trichloromethane, DMF, methyl-sulphoxide, N,N-dimethylacetamide or above-mentioned solvent.
3. method as claimed in claim 1, is characterized in that, in step (3), formula IV compound is obtained to formula V compound through enolization esterification, and the enolization esterifying reagent adopting is diacetyl oxide, Acetyl Chloride 98Min., methylvinyl acetate or vinyl-acetic ester; The catalyzer adopting is the vitriol oil or tosic acid; The solvent adopting is the mixture of the optional solvents in methylene dichloride, chloroform, toluene, methylvinyl acetate, vinyl-acetic ester or above-mentioned solvent.
4. method as claimed in claim 1, is characterized in that, in step (4), formula V compound is obtained to formula VI compound and formula VII compound through fluoridizing, and the fluoro reagent adopting is Selectfluor or Accufluor; The solvent adopting is the mixture of the optional solvents in methylene dichloride, chloroform, toluene, acetonitrile or above-mentioned solvent.
5. method as claimed in claim 1, it is characterized in that, in step (8), formula IX compound is obtained to formula X compound through fluoridizing open loop, the fluorination reagent adopting is aqueous hydrogen fluoride solution, hydrogen fluoride pyridine solution (Olah reagent) or hydrogen fluoride triethylamine solution; The solvent adopting is methylene dichloride, chloroform, 1, the mixture of the optional solvents in 2-ethylene dichloride, tetrahydrofuran (THF), toluene or above-mentioned solvent; Range of reaction temperature is-50~50 ℃.
6. a key intermediate compound for synthetic difluprednate, shown in IV compound:
Difluprednate ophthalmic emulsion 0.05% is also being studied in other ocular inflammatory diseases, including a phase 3 study evaluating difluprednate for the treatment of anterior uveitis[2][3]
Today, the U.S. Food and Drug Administration approved Piqray (alpelisib) tablets, to be used in combination with the FDA-approved endocrine therapy fulvestrant, to treat postmenopausal women, and men, with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative, PIK3CA-mutated, advanced or metastatic breast cancer (as detected by an FDA-approved test) following progression on or after an endocrine-based regimen.
The FDA also approved the companion diagnostic test, therascreen PIK3CA RGQ PCR Kit, to detect the PIK3CA mutation in a tissue and/or a liquid biopsy. Patients who are negative by
May 24, 2019
Today, the U.S. Food and Drug Administration approved Piqray (alpelisib) tablets, to be used in combination with the FDA-approved endocrine therapy fulvestrant, to treat postmenopausal women, and men, with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative, PIK3CA-mutated, advanced or metastatic breast cancer (as detected by an FDA-approved test) following progression on or after an endocrine-based regimen.
The FDA also approved the companion diagnostic test, therascreen PIK3CA RGQ PCR Kit, to detect the PIK3CA mutation in a tissue and/or a liquid biopsy. Patients who are negative by the therascreen test using the liquid biopsy should undergo tumor biopsy for PIK3CA mutation testing.
“Piqray is the first PI3K inhibitor to demonstrate a clinically meaningful benefit in treating patients with this type of breast cancer. The ability to target treatment to a patient’s specific genetic mutation or biomarker is becoming increasingly common in cancer treatment, and companion diagnostic tests assist oncologists in selecting patients who may benefit from these targeted treatments,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “For this approval, we employed some of our newer regulatory tools to streamline reviews without compromising the quality of our assessment. This drug is the first novel drug approved under the Real-Time Oncology Review pilot program. We also used the updated Assessment Aid, a multidisciplinary review template that helps focus our written review on critical thinking and consistency and reduces time spent on administrative tasks.”
Metastatic breast cancer is breast cancer that has spread beyond the breast to other organs in the body (most often the bones, lungs, liver or brain). When breast cancer is hormone-receptor positive, patients may be treated with anti-hormonal treatment (also called endocrine therapy), alone or in combination with other medicines, or chemotherapy.
The efficacy of Piqray was studied in the SOLAR-1 trial, a randomized trial of 572 postmenopausal women and men with HR-positive, HER2-negative, advanced or metastatic breast cancer whose cancer had progressed while on or after receiving an aromatase inhibitor. Results from the trial showed the addition of Piqray to fulvestrant significantly prolonged progression- free survival (median of 11 months vs. 5.7 months) in patients whose tumors had a PIK3CA mutation.
Common side effects of Piqray are high blood sugar levels, increase in creatinine, diarrhea, rash, decrease in lymphocyte count in the blood, elevated liver enzymes, nausea, fatigue, low red blood cell count, increase in lipase (enzymes released by the pancreas), decreased appetite, stomatitis, vomiting, weight loss, low calcium levels, aPTT prolonged (blood clotting taking longer to occur than it should), and hair loss.
Health care professionals are advised to monitor patients taking Piqray for severe hypersensitivity reactions (intolerance). Patients are warned of potentially severe skin reactions (rashes that may result in peeling and blistering of skin or mucous membranes like the lips and gums). Health care professionals are advised not to initiate treatment in patients with a history of severe skin reactions such as Stevens-Johnson Syndrome, erythema multiforme, or toxic epidermal necrolysis. Patients on Piqray have reported severe hyperglycemia (high blood sugar), and the safety of Piqray in patients with Type 1 or uncontrolled Type 2 diabetes has not been established. Before initiating treatment with Piqray, health care professionals are advised to check fasting glucose and HbA1c, and to optimize glycemic control. Patients should be monitored for pneumonitis/interstitial lung disease (inflammation of lung tissue) and diarrhea during treatment. Piqray must be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks.
Piqray is the first new drug application (NDA) for a new molecular entity approved under the Real-Time Oncology Review (RTOR) pilot program, which permits the FDA to begin analyzing key efficacy and safety datasets prior to the official submission of an application, allowing the review team to begin their review and communicate with the applicant earlier. Piqray also used the updated Assessment Aid (AAid), a multidisciplinary review template intended to focus the FDA’s written review on critical thinking and consistency and reduce time spent on administrative tasks. With these two pilot programs, today’s approval of Piqray comes approximately three months ahead of the Prescription Drug User Fee Act (PDUFA) VI deadline of August 18, 2019.
The FDA granted this application Priority Review designation. The FDA granted approval of Piqray to Novartis. The FDA granted approval of the therascreen PIK3CA RGQ PCR Kit to QIAGEN Manchester, Ltd.
Yescarta is the second gene therapy product approval in the U.S.
The U.S. Food and Drug Administration today approved Yescarta (axicabtagene ciloleucel), a cell-based gene therapy, to treat adult patients with certain types of large B-cell lymphoma who have not responded to or who have relapsed after at least two other kinds of treatment. Yescarta, a chimeric antigen receptor (CAR) T cell therapy, is the second gene therapy approved by the FDA and the first for certain types of non-Hodgkin lymphoma (NHL). Continue reading.
Axicabtagene ciloleucel is a chimeric antigen receptor (CAR) T cell therapy for the treatment of Diffuse large B-cell lymphoma (DLBCL), which is a type of a non-Hodgkin lymphoma (NHL). It is the second cell-based gene therapy that is FDA-approved but the first in the treatment of large B-cell lymphoma in adult patients. Uniquely, axicabtagene ciloleucel utilizes each patient’s own immune system where each dose of the drug consists of the patient’s genetically modified T-cells that were previously collected. The modified version of the T-cell expresses a new gene that targets and kills the lymphoma cells and is infused back into the patient.
Diffuse large B-cell lymphoma (DLBCL) is the most common type of NHL in adults that mostly originates from the lymph nodes but can initiate outside of the lymphatic system. Lymphoma cells appear to be much larger in size than normal lymphocytes. In a multicenter clinical trial, the patients who were treated with axicabtagene ciloleucel achieved the complete remission rate of 51%.
Developed by Kite Pharma, Inc., it was approved on October 18th, 2017 by the FDA as an intravenously infused anticancer therapy and is marketed under the brand name Yescarta
The person’s T-cells are engineered to target CD19.[1] The CD19 are found on the surface of certain cancerous cells.[1] The cost for treatment is 373,000 USD in the United States.[2]
Side effects
Because treatment with axicabtagene carries a risk of cytokine release syndrome and neurological toxicities, the FDA has mandated that hospitals be certified for its use.[1]
The U.S. Food and Drug Administration today approved Vabomere for adults with complicated urinary tract infections (cUTI), including a type of kidney infection, pyelonephritis, caused by specific bacteria. Vabomere is a drug containing meropenem, an antibacterial, and vaborbactam, which inhibits certain types of resistance mechanisms used by bacteria.
The U.S. Food and Drug Administration today approved Vabomere for adults with complicated urinary tract infections (cUTI), including a type of kidney infection, pyelonephritis, caused by specific bacteria. Vabomere is a drug containing meropenem, an antibacterial, and vaborbactam, which inhibits certain types of resistance mechanisms used by bacteria.
“The FDA is committed to making new safe and effective antibacterial drugs available,” said Edward Cox, M.D., director of the Office of Antimicrobial Products in the FDA’s Center for Drug Evaluation and Research. “This approval provides an additional treatment option for patients with cUTI, a type of serious bacterial infection.”
The safety and efficacy of Vabomere were evaluated in a clinical trial with 545 adults with cUTI, including those with pyelonephritis. At the end of intravenous treatment with Vabomere, approximately 98 percent of patients treated with Vabomere compared with approximately 94 percent of patients treated with piperacillin/tazobactam, another antibacterial drug, had cure/improvement in symptoms and a negative urine culture test. Approximately seven days after completing treatment, approximately 77 percent of patients treated with Vabomere compared with approximately 73 percent of patients treated with piperacillin/tazobactam had resolved symptoms and a negative urine culture.
The most common adverse reactions in patients taking Vabomere were headache, infusion site reactions and diarrhea. Vabomere is associated with serious risks including allergic reactions and seizures. Vabomere should not be used in patients with a history of anaphylaxis, a type of severe allergic reaction to products in the class of drugs called beta-lactams.
To reduce the development of drug-resistant bacteria and maintain the effectiveness of antibacterial drugs, Vabomere should be used only to treat or prevent infections that are proven or strongly suspected to be caused by susceptible bacteria.
Vabomere was designated as a qualified infectious disease product (QIDP). This designation is given to antibacterial products that treat serious or life-threatening infections under the Generating Antibiotic Incentives Now (GAIN) title of the FDA Safety and Innovation Act. As part of its QIDP designation, Vabomere received a priority review.
The FDA granted approval of Vabomere to Rempex Pharmaceuticals.
FDA approves Intrarosa for postmenopausal women experiencing pain during sex
The U.S. Food and Drug Administration approved Intrarosa (prasterone) to treat women experiencing moderate to severe pain during sexual intercourse (dyspareunia), a symptom of vulvar and vaginal atrophy (VVA), due to menopause. Intrarosa is the first FDA approved product containing the active ingredient prasterone, which is also known as dehydroepiandrosterone (DHEA).
The U.S. Food and Drug Administration approved Intrarosa (prasterone) to treat women experiencing moderate to severe pain during sexual intercourse (dyspareunia), a symptom of vulvar and vaginal atrophy (VVA), due to menopause. Intrarosa is the first FDA approved product containing the active ingredient prasterone, which is also known as dehydroepiandrosterone (DHEA).
During menopause, levels of estrogen decline in vaginal tissues, which may cause a condition known as VVA, leading to symptoms such as pain during sexual intercourse.
“Pain during sexual intercourse is one of the most frequent symptoms of VVA reported by postmenopausal women,” said Audrey Gassman, M.D., deputy director of the Division of Bone, Reproductive, and Urologic Products (DBRUP) in the Office of Drug Evaluation III in the FDA’s Center for Drug Evaluation and Research (CDER). “Intrarosa provides an additional treatment option for women seeking relief of dyspareunia caused by VVA.”
Efficacy of Intrarosa, a once-daily vaginal insert, was established in two 12-week placebo-controlled clinical trials of 406 healthy postmenopausal women, 40 to 80 years of age, who identified moderate to severe pain during sexual intercourse as their most bothersome symptom of VVA. Women were randomly assigned to receive Intrarosa or a placebo vaginal insert. Intrarosa, when compared to placebo, was shown to reduce the severity of pain experienced during sexual intercourse.
The safety of Intrarosa was established in four 12-week placebo-controlled trials and one 52-week open-label trial. The most common adverse reactions were vaginal discharge and abnormal Pap smear.
Although DHEA is included in some dietary supplements, the efficacy and safety of those products have not been established for diagnosing, curing, mitigating, treating or preventing any disease.
Intrarosa is marketed by Quebec-based Endoceutics Inc.
The FDA, an agency within the U.S. Department of Health and Human Services, protects the public health by assuring the safety, effectiveness, and security of human and veterinary drugs, vaccines and other biological products for human use, and medical devices. The agency also is responsible for the safety and security of our nation’s food supply, cosmetics, dietary supplements, products that give off electronic radiation, and for regulating tobacco products.
The GDUFA (Generic Drug User Fee Amendments) is a legislative package which came into force in 2012 and entitles the US-American FDA to collect fees from generic drug manufacturers, who strive for a marketing authorisation for the American market. An annual fee has to be paid after the successful registration.
The core of the document is the obligation to “Self-Identify” for those companies that have to submit essential site-related information to the FDA. The details of this self-identification are set in a Guidance for Industry entitled “Self-Identification of Generic Drug Facilities, Sites, and Organizations” published on 22 September 2016 by the FDA in the finalised form.
The Guidance describes the following elements:
1. Which types of generic facilities, sites, and organizations are required to self-identify?
2. What information is requested?
3. What technical standards are to be used for electronically submitting the requested information?
4. What is the penalty for failing to self-identify?
Hereinafter, you will find a short summary of these four topics:
1. Companies that manufacture finished generic medicinal products for human use or the APIs for them, or both are required to self-identify as well as companies that package the finished generic drug into the primary container and label it. Besides, sites that – pursuant to a contract with the applicant (generic drug manufacturer) – repack/redistribute the finished drug from a primary container into a different primary container are also required to submit a self-identification as well as sites that perform bioequivalence/bioavailability studies. Last but not least, the obligation to self-identify also concerns sites that are listed in the application dossier as contract laboratories for the sampling and performing of analytical testing.
2. Essential data are: the D-U-N-S number (a unique nine-digit sequence specific for each site / each distinct physical location of an entity), the “Facility Establishment Identifier, FEI” (an identifier used by the FDA for the planning and tracking of inspections) and general information with regard to the facility (company owner, type of business operation, contact data, information about the manufacture of non generic drugs).
4. Companies that fail to self-identify do not have to expect an explicit penalty. However, such a failure leads to two drawbacks: first, the likelihood of a site inspection by the FDA prior to approval is higher. The second drawback which is much more serious is that all the APIs or finished drugs from a manufacturer who hasn’t self-identified are deemed misbranded. For the FDA, such products are not allowed for importation in the USA.
To the satisfaction of the FDA, the regulations set in the GDUFA and the provisions laid down in the new Guidance represent a major contribution to an enhanced transparency in particular of complex supply chains.
//////////GDUFA, FDA, new Guidance, Self-Identification, Generic Drug Manufacturers
The U.S. Food and Drug Administration today approved Amjevita(adalimumab-atto) as a biosimilar toHumira (adalimumab) for multiple inflammatory diseases.
The U.S. Food and Drug Administration today approved Amjevita (adalimumab-atto) as a biosimilar to Humira (adalimumab) for multiple inflammatory diseases.
Amjevita is approved for the following indications in adult patients:
moderately to severely active rheumatoid arthritis;
active psoriatic arthritis;
active ankylosing spondylitis (an arthritis that affects the spine);
moderately to severely active Crohn’s disease;
moderately to severely active ulcerative colitis; and
moderate to severe plaque psoriasis.
Amjevita is also indicated for moderately to severely active polyarticular juvenile idiopathic arthritis in patients four years of age and older.
Health care professionals should review the prescribing information in the labeling for detailed information about the approved uses.
“This is the fourth FDA-approved biosimilar. The biosimilar pathway is still a new frontier and one that we expect will enhance access to treatment for patients with serious medical conditions,” said Janet Woodcock, M.D., director of the FDA’s Center for Drug Evaluation and Research.
Biological products are generally derived from a living organism and can come from many sources, including humans, animals, microorganisms or yeast. A biosimilar is a biological product that is approved based on a showing that it is highly similar to an already-approved biological product and has no clinically meaningful differences in terms of safety, purity and potency (i.e., safety and effectiveness) from the reference product, in addition to meeting other criteria specified by law.
The FDA’s approval of Amjevita is based on review of evidence that included structural and functional characterization, animal study data, human pharmacokinetic and pharmacodynamics data, clinical immunogenicity data and other clinical safety and effectiveness data that demonstrates Amjevita is biosimilar to Humira. It has been approved as a biosimilar, not as an interchangeableproduct.
The most serious known side effects with Amjevita are infections and malignancies. The most common expected adverse reactions with Amjevita are infections and injection site reactions.
Like Humira, the labeling for Amjevita contains a Boxed Warning to alert health care professionals and patients about an increased risk of serious infections leading to hospitalization or death. The Boxed Warning also notes that lymphoma and other malignancies, some fatal, have been reported in children and adolescent patients treated with tumor necrosis factor blockers, including adalimumab products. The drug must be dispensed with a patient Medication Guide that describes important information about its uses and risks.
Amjevita is manufactured by Amgen, Inc., of Thousand Oaks, California. Humira was approved in December 2002 and is manufactured by AbbVie Inc. of North Chicago, Illinois.
The U.S. Food and Drug Administration approved Adlyxin (lixisenatide), a once-daily injection to improve glycemic control (blood sugar levels), along with diet and exercise, in adults with type 2 diabetes.
The U.S. Food and Drug Administration approved Adlyxin (lixisenatide), a once-daily injection to improve glycemic control (blood sugar levels), along with diet and exercise, in adults with type 2 diabetes.
“The FDA continues to support the development of new drug therapies for diabetes management,” said Mary Thanh Hai Parks, M.D., deputy director, Office of Drug Evaluation II in the FDA’s Center for Drug Evaluation and Research. “Adlyxin will add to the available treatment options to control blood sugar levels for those with type 2.”
Type 2 diabetes affects more than 29 million people and accounts for more than 90 percent of diabetes cases diagnosed in the United States. Over time, high blood sugar levels can increase the risk for serious complications, including heart disease, blindness and nerve and kidney damage.
Adlyxin is a glucagon-like peptide-1 (GLP-1) receptor agonist, a hormone that helps normalize blood sugar levels. The drug’s safety and effectiveness were evaluated in 10 clinical trials that enrolled 5,400 patients with type 2 diabetes. In these trials, Adlyxin was evaluated both as a standalone therapy and in combination with other FDA-approved diabetic medications, including metformin, sulfonylureas, pioglitazone and basal insulin. Use of Adlyxin improved hemoglobin A1c levels (a measure of blood sugar levels) in these trials.
In addition, more than 6,000 patients with type 2 diabetes at risk for atherosclerotic cardiovascular disease were treated with either Adlyxin or a placebo in a cardiovascular outcomes trial. Use of Adlyxin did not increase the risk of cardiovascular adverse events in these patients.
Adlyxin should not be used to treat people with type 1 diabetes or patients with increased ketones in their blood or urine (diabetic ketoacidosis).
The most common side effects associated with Adlyxin are nausea, vomiting, headache, diarrhea and dizziness. Hypoglycemia in patients treated with both Adlyxin and other antidiabetic drugs such as sulfonylurea and/or basal insulin is another common side effect. In addition, severe hypersensitivity reactions, including anaphylaxis, were reported in clinical trials of Adlyxin.
The FDA is requiring the following post-marketing studies for Adlyxin:
Clinical studies to evaluate dosing, efficacy and safety in pediatric patients.
A study evaluating the immunogenicity of lixisenatide.
Adlyxin is manufactured by Sanofi-Aventis U.S. LLC, of Bridgewater, New Jersey.
Sanofi (formerly sanofi-aventis, formerly Aventis), under license from Zealand Pharma, has developed and launched lixisenatide
Lixisenatide (trade name Lyxumia) is a once-daily injectable GLP-1 receptor agonist for the treatment of diabetes, discovered by Zealand Pharma A/S of Denmark and licensed and developed by Sanofi.[1] Lixisenatide was accepted for review by the US FDA on February 19, 2013, and approved by the European Commission on February 1, 2013.[2] On September 12, 2013, Sanofi delayed the approval process in the US, citing internal data from a cardiovascular risk study. The drug will likely be resubmitted for approval in 2015.
Lixisenatide is a once-daily injectable GLP-1 receptor agonist discovered by Zealand Pharma A/S of Denmark and licensed and developed by Sanofi. As of September 2010 it is in clinical trials for diabetes. Lixisenatide was accepted for review by the US FDA on February 19, 2013, and approved by the European Commission on February 1, 2013. The drug will likely be resubmitted for approval in 2015.
Mechanism of action
GLP-1 is a naturally-occurring peptide that is released within minutes of eating a meal. It is known to suppress glucagon secretion from pancreatic alpha cells and stimulate insulin secretion by pancreatic beta cells. GLP-1 receptor agonists are used as an add-on treatment for type 2 diabetes and their use is endorsed by the European Association for the Study of Diabetes, the American Diabetes Association, the American Association of Clinical Endocrinologists and the American College of Endocrinology.
Physical and chemical properties
Lixisenatixe has been described as “des-38-proline-exendin-4 (Heloderma suspectum)-(1–39)-peptidylpenta-L-lysyl-L-lysinamide”, meaning it is derived from the first 39 amino acids in the sequence of the peptide exendin-4, found in the Gila monster (Heloderma suspectum), omitting proline at position 38 and adding six lysine residues. Its complete sequence is:[3]
The title method comprises the steps of: (1) coupling Fmoc-Lys(Boc)-OH and resin to obtain Fmoc-Lys(Boc)-resin, (2) protecting amino acid with Fmoc, conducting solid-phase synthesis to obtain lixisenatide wholly protected 20-44-peptide resin, (3) conducting solid-phase synthesis to obtain wholly protected 15-19-peptide resin, (4) coupling the wholly protected 20-44-peptide resin and wholly protected 15-19-peptide resin, (5) coupling other amino acids till solid-phase synthesis finishes, (6) cracking lixisenatide peptide resin to obtain crude peptide, and (7) purifying through RP-HPLC. The method improves crude peptide purity and purifn. yield.
MACHINE TRANSLATION FROM CHINESE, PL BEAR WITH SOME IREGULARITES IN GRAMMAR
利西拉, the English name: Lixisenatide, is a polypeptide containing 44 amino acids, the structural formula is as follows: peptide sequence as follows:
H-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Lys-Gln-Met-Glu-Glu-Glu-Al a-Val-Arg-Leu-Phe-IIe-Glu -Trp-Leu-Lys-Asn-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Pro-Pr O-Ser-Lys-Lys-Lys-Lys-Lys-Lys-NH 2 Li Xila to (Lixisenatide ) by Sanofi-Aventis developed once a day subcutaneously with glucagon-like peptide -I (GLP-I) receptor agonists, for the treatment of type II diabetes, on February 1, 2013 Sanofi Lee Division -Aventis of exenatide is approved EMEA, for the adjuvant treatment of poorly stable dose of basal insulin (or metformin) in the treatment of type II diabetes to improve HbAlc and postprandial blood glucose levels.
CN201210030151. 2 used in a pure solid phase sequential coupling method synthetic peptides.The method amino resin as the carrier, using conventional coupling sequence, the final cut to give Li Xila.
US6528486 patent for the compound, synthetic methods mentioned it to phase condensation method Fmoc / tBu strategy.
The [0005] W02005058954 synthesis method including the gradual condensation process Fmoc / tBu strategy, Boc strategy of gradual condensation methods and genetic engineering.
The W02001004156 synthesis method for the gradual condensation process Fmoc / tBu strategy.
Since Li Xila abroad mostly used to synthesize Fmoc solid phase synthesis method, a gradual shrinking gradually synthesis step more, resulting in more types of product impurities, US 20130284912 Special Report polypeptide impurity: Di-Ser33- Leisy pull and Di-Ala35- Li Xila come, Di-Ser 33- Li Xila come and Di-Ala35- Li Xila to atmosphere amino acid sequence as follows: Di-Ser33- Li Xila to the amino acid sequence: H-His -Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Lys-Gln-Met-Glu-Glu-Glu-Al a-Val-Arg-Leu-Phe-IIe-Glu-Trp- Leu-Lys-Asn-Gly-Gly-Pro-Ser-Ser-Ser-Gly-Ala-Pr 〇-Pr〇-Ser-Lys_Lys_Lys_Lys_Lys_LyS-NH2 Di-Ala35- Li Xila to the amino acid sequence: H-His-Gly- Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Lys-Gln-Met-Glu-Glu-Glu-Al a-Val-Arg-Leu-Phe-IIe-Glu-Trp-Leu-Lys -Asn-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Ala-Pr 〇-Pr〇-Ser-Lys_Lys_Lys_Lys_Lys_LyS-NH2 toxicity of these impurities are impurities larger, and very difficult to separate from the main peak , the presence of the impurities seriously affect 利西拉 to content and the use of safety.Hence the need to find an effective way to remove it and to reach the high standard level of 0.1% or less.The present inventors have found that this impurity is difficult to remove by means of the prior art, although there are ways to remove part of, but removal is not ideal, it is difficult to achieve high quality standards is likely to cause 利西拉 level while reducing their yield.
In summary, the existing Li Xila to the solid phase synthesis, low yield of the synthesis, impurities, in particular, are not well controlled impurity Di-Ser 33- Li Xila come and Di-Ala35 – Li Xila to, does not apply to industrial production
Example i ^ a: Preparation 利西拉 to fine peptide acetate Weigh 利西拉 above 44. 70g to 45L crude peptide was dissolved in water, purified by C18 column, the first purification conditions: mobile phase: A phase: 0 I% TFA; B phase: acetonitrile; gradient program was: 15% B, 60 minutes to 60% B; detection wavelength 220 nm; peak fraction collection purposes.The second purification conditions: mobile phase was: A phase: 0 3% HAC; B phase: acetonitrile; gradient program was: 10% B, 60 minutes to 60% B; detection wavelength 220 nm; peak fraction collection purposes.Desalting conditions: Mobile phase: A phase: an aqueous solution of 20 mmol / L ammonium acetate: acetonitrile = 95: 5; B phase: water: acetonitrile = 95: 5; C phase: 0.03% aqueous solution of acetic acid: acetonitrile = 95 : 5; D phase: 0.03% aqueous solution of acetic acid: acetonitrile = 50: 50; gradient program: mobile phase A isocratic for 15 minutes, convert isocratic mobile phase B for 10 minutes, is converted into the flow Phase C isocratic 10 minutes, converted into a mobile phase D isocratic 25 minutes; detection wavelength 220 nm; peak fraction collection purposes; rotary evaporation concentrated and lyophilized to give Li Xila acetate fine peptide 22. 65g which HPLC spectrum shown in Figure 5, HPLC purity of 99.75% (area normalization method), Di-Ser33- Li Xila come to 0.03% (area normalization method), Di-Ala35- Li Xila to the content of 0.05% (area normalization method).Purification total yield of 51%, total yield 41%.Its mass spectrum as shown in Figure 6, [M + H] + = 4858. 691, 利西拉 precise molecular weight to the theoretical: 4857.53, the sample mass is consistent with the theoretical molecular weight.
MACHINE TRANSLATION FROM CHINESE, PL BEAR WITH SOME IREGULARITES IN GRAMMAR
Example 2: Preparation 利西拉 to crude peptide
利西拉 [0116] Example 24 was prepared to be placed 125.4g peptide resin cleavage reaction to 10ml / g resin ratio added lysis reagent (TFA: thioanisole: EDT: TIS: water = 86: 5 : 5: 3: 1 (V / V)), stirred at room temperature 2.5h.The reaction was purified by frit funnel filtration, the filtrate was collected, the resin was washed 3 times and then a small amount of TFA, the combined filtrates concentrated under reduced pressure.Frozen precipitation in anhydrous ether was added, washed three times with anhydrous diethyl ether, and dried in vacuo to give a white solid powder, i.e. Li Xila to crude peptide 47.lg, by weight of the crude peptide yield 97.2%, HPLC purity 63.8% 0
利西拉 to crude peptide preparation: 27 patients [0117] Example
利西拉 [0118] The Example 25 was prepared to be placed 123.7g peptide resin cleavage reaction to 10ml / g resin ratio added lysis reagent (TFA: thioanisole: EDT: TIS: water = 86: 5 : 5: 3: 1 (V / V)), stirred at room temperature 2.5h.The reaction was purified by frit funnel filtration, the filtrate was collected, the resin was washed 3 times and then a small amount of TFA, the combined filtrates concentrated under reduced pressure.Frozen precipitation in anhydrous ether was added, washed three times with anhydrous diethyl ether, and dried in vacuo to give a white solid powder, i.e. Li Xila to crude peptide 46.9g, yield the crude peptide by weight 96.5%, HPLC purity 64.2% 0
28 Example 2: Preparation 利西拉 to fine peptide acetate
Example weighed 26 to 27 after 利西拉 to any 30.0g crude peptide was dissolved in 3000ml of water using Waters2545RP-HPLC system, wavelength 230nm, 50 X 250mm column of reverse phase C18 column, 0.2% TFA conventional / acetonitrile mobile phase were fractionated peaks of fractions, refined peptide purity greater than 98.5%.The fine peptide solution using Waters2545RP-HPLC system, 50 X 250mm column was C18 reverse phase column, 0.1% acetic acid / acetonitrile mobile phase transfer salt, the purpose of peak fractions were collected, concentrated by rotary evaporation and lyophilized to give Li Xila acetate fine salt peptide> 9.0g, RP-HPLC purity ≥98.5%.Purification Yield ≥30%, total yield ≥29.0%.
PATENT
CN 102875663
MACHINE TRANSLATION FROM CHINESE, PL BEAR WITH SOME IREGULARITES IN GRAMMAR
[0239] The crude peptide Li Xila to 4000g (including Li Xila to 1139g) was dissolved with purified water 100L, collected by filtration and the filtrate set aside.
[0240] purification chromatographic conditions:
[0241] HPLC Model: Novasep LC450
Column: 450X250mm, built-phenyl silane bonded silica gel as stationary phase filler, the filler particle size of 10 μ m0
flow rate: 5000ml / min.
The detection wavelength: 280nm.
Mobile phase A phase: 10% 30mM D- 30mM sodium tartrate and disodium hydrogenphosphate in methanol / 90% aqueous (v / v), adjusted to pH 2.5 with phosphoric acid.
[0246] Mobile phase A phase preparation process: Weigh 1280g 2070g D- sodium tartrate and disodium hydrogenphosphate, after an appropriate amount of purified water was dissolved through 0.45 μ m membrane filter, the filtrate collected all 300L tank, added 30L chromatographically pure After methanol was added to the 300L scale purification of water, adjusted to pH 2.5 with phosphoric acid.Repeat preparation run.
[0247] The mobile phase B phase: HPLC grade acetonitrile.
[0249] sample volume: 250.0g (6250ml).
[0250] Purification: column equilibration the sample so that after 5 minutes, run a gradient purification, monitoring and staging purposes peak fractions were collected.The collected fractions (chromatographic conditions purity testing to the same conditions as above 利西拉 determination to area normalization method measured) purity test, the purity of greater than or equal to 98% of the fractions after removing most of the acetonitrile in turn salt; purity of 70% or more less than 98% of the fraction recovered after removal of most of the acetonitrile and the purification procedure is repeated, again collected purity greater than or equal to 98% of the fraction after removal of most of the acetonitrile are also used to turn salt; purity of less than 70 % of fractions by waste disposal.
[0251] points and 16 injections, repeat the above operation.
[0252] turn salt chromatographic conditions:
[0253] HPLC Model: Novasep LC450
[0254] Column: 450 X 250mm, built-C8 reversed-phase chromatography packing, the particle size of the filler is 10 μ m.
[0255] flow rate: 5000ml / min.
[0256] The detection wavelength: 280nm.
[0257] Mobile phase A phase: 0.2% acetic acid (v / v) solution.
[0258] The mobile phase B phase: HPLC grade acetonitrile.
[0259] gradient
[0260] sample volume: 2500ml.
[0261] Purification: The column equilibration the sample for 5 minutes, run a gradient purification, monitoring and collecting the target peak fractions.The purpose of the peak fractions were concentrated by rotary evaporation under reduced pressure to 9000ml after lyophilization.
[0262] After the freeze-dried to give a white powder refined peptide 704g.Purity of 98.39%, the impurity content of less than 0.5%.Purification yield 61.8% (in crude Li Xila to content), total yield of 17.6%.
[0096] To the resulting Fmoc-Lys (Boc) -Lys (Boc) -Lys (Boc) -RinkAmide-MBHAResin mouth of a 20% strength piperidine / DMF solution for 10 minutes, the reaction was drained, washed with DMF Resin 6 (50ml * 6).Weigh Fmoc-Lys (Boc) -〇H3.52g, H0Bt1.01g, HBTU2.84g, TMP1.98ml, DMF50ml added to dissolve slowly with stirring under ice-cooling for 3 minutes, at room temperature for 2 hours, the reaction Ninhydrin detection method completed, pumping off the reaction solution, DMF the resin was washed twice (50mlX2), DCM the resin was washed twice (50mlX2), to give Fmoc-Lys (B oc) -Lys (Boc) -Lys (Boc) -Lys (Boc) -RinkAmide-MBHAResin.As used in the above operation Fmoc-Lys (Boc) -OH: HOBt: HBTU: TMP ratio is 1: 1: 1: 2, wherein Fmoc-Lys (Boc) -OH is the number of moles of Fmoc-RinkAmide-MBHAResin number of moles 3 times.
[0097] Li Xila fully protected side chain was prepared to -Rink Amide-MBHA Resin:
[0098] To the resulting Fmoc-Lys (Boc) -Lys (Boc) -Lys (Boc) -Lys (Boc) -RinkAmide-MBHA Resin added 20% piperidine / DMF solution for 10 minutes, drained reaction solution, washed 6 times with DMF.Weigh Jie 111〇 (3-1 ^ 8 billion (3) -0 13.528, 1 (»Shu 1.018,01 (:!! 1.391111 added 50,111,101 ^ dissolve slowly stirring for 3 minutes in an ice bath, poured into the solid phase resin is mixed with the reaction column, at room temperature for 2 hours, the reaction Ninhydrin detection method is completed, the reaction solution was deprived, DMF the resin was washed twice (50ml X 2), DCM the resin was washed twice (50ml X 2), to give Fmoc-Lys ( Boc) -Lys (Boc) -Lys (Boc) -Lys (Boc) -Lys (Boc) -Rink Amide-MBHAResin above operation used by the Fmoc-Lys (Boc) -〇H:. HOBt: DIC ratio is 1: 1: L2, which Fmoc-Lys (Boc) is three times the number of moles -〇H Fmoc-Rink Amide-MBHA Resin moles of repeat after the coupling step, followed by the completion of the 39 lysine to first. connecting protected amino acids histidine, followed by addition of 20% piperidine / DMF solution for 10 minutes, the reaction was drained, DMF the resin was washed six times (50ml X 6), DCM the resin was washed six times (50ml X 6 ), MeOH contraction of the resin three times with MeOH 50ml, each contraction 5min. After the resin was dried in vacuo to give a full side-chain protected peptide resin to the Li Xila 27. 5g, weight resin 17. 5g.
[0099] Li Xila to crude peptide preparation:
[0100] Weigh side chains fully protected Li Xila to -Rink Amide-MBHA Resin 27. 5 grams, into a round bottom flask.Configuration 275 ml lysis buffer, wherein trifluoroacetic acid: thioanisole: ethanedithiol: anisole, phenol = 93: 4: 1: 1.5: 2 (volume ratio).Lysate in the refrigerator after the pre-freeze 1 hour before Sheng Youli put to Silas to -Rink Amide-MBHA Resin round bottom flask, stirred at room temperature for 2 hours.The reaction mixture was filtered, the resin was washed with 20ml TFA and the combined filtrate.
[0101] The volume of the filtrate was slowly poured into 2,750 ml of diethyl ether frozen (frozen advance ether), a white precipitate appears, at 3000 rpm / centrifuged 5 minutes, the resulting solid was washed twice with ether, then the solid was dried under vacuum to give Li Xila trifluoroacetate crude peptide to 15. 3g.
[0102] Li Xila to large scale production of fine peptide:
[0103] Sample Preparation: The crude peptide was dissolved in water, the sample was completely dissolved by membrane filtration, the filtrate was collected for use.
[0104] Purification conditions: Column: octadecyl silane bonded silica gel as stationary phase column, the column diameter and length: 300_X250mm.Mobile phase: A phase: 35mm〇l / L phosphoric acid solution adjusted with triethylamine to pH 6. 7; B phase: acetonitrile, flow rate: 2200ml / min, Gradient: B%: 12% ~32%, detection wavelength: 280nm .The injection volume was 75g.Purification process: the column with 50% acetonitrile rinse clean after balance sample, sample amount is 75g.Linear gradient 120min, the purpose of collecting peaks will be collected 利西拉 solution was concentrated by rotary evaporation under reduced pressure to about 80mg / ml and reserve the water temperature exceeds 40 ° C without conditions.
[0105] turn salt: turn salt conditions: Column: octadecyl silane bonded silica gel as stationary phase column, the column diameter and length: 300mmX250mm.Mobile phase: A phase: mass concentration of 0.2% aqueous acetic acid; B phase: HPLC grade acetonitrile, flow rate: 2200ml / min, detection wavelength: 280nm.Gradient: B%: 6% ~36%.The injection volume was 48-60g.Salt transfer process: the column with 50% acetonitrile rinse clean after the sample, the sample volume is 1600ml sample solution.Linear gradient 90min, the purpose of collecting peaks collected Li Xila to solutions were concentrated by rotary evaporation to about 80ml / g after go to the appropriate size vials, then freeze-dried to obtain the purity of greater than 99.5% The Li Xila come.
Sanofi Provides Update on Lixisenatide New Drug Application in U.S.
Paris, France – September 12, 2013 – Sanofi (EURONEXT: SAN and NYSE: SNY) announced today its decision to withdraw the lixisenatide New Drug Application (NDA) in the U.S., which included early interim results from the ongoing ELIXA cardiovascular (CV) outcomes study. The company plans to resubmit the NDA in 2015, after completion of the ELIXA CV study.
The decision to withdraw the lixisenatide application follows discussions with the U.S. Food and Drug Administration (FDA) regarding its proposed process for the review of interim data. Sanofi believes that potential public disclosure of early interim data, even with safeguards, could potentially compromise the integrity of the ongoing ELIXA study. Sanofi’s decision is not related to safety issues or deficiencies in the NDA………………………read all at
Christensen, M; Knop, FK; Holst, JJ; Vilsboll, T (2009). “Lixisenatide, a novel GLP-1 receptor agonist for the treatment of type 2 diabetes mellitus”. IDrugs : the investigational drugs journal12 (8): 503–13. PMID19629885.
The U.S. Food and Drug Administration approved Xiidra (lifitegrast ophthalmic solution) for the treatment of signs and symptoms of dry eye disease, on Monday, July 11, 2016. Xiidra is the first medication in a new class of drugs, called lymphocyte function-associated antigen 1 (LFA-1) agonist, approved by the FDA for dry eye disease.
FDA approves new medication for dry eye disease
July 12, 2016
Release
The U.S. Food and Drug Administration approved Xiidra (lifitegrast ophthalmic solution) for the treatment of signs and symptoms of dry eye disease, on Monday, July 11, 2016. Xiidra is the first medication in a new class of drugs, called lymphocyte function-associated antigen 1 (LFA-1) agonist, approved by the FDA for dry eye disease.
“Normal tear production is needed for clear vision and eye health,” said Edward Cox, M.D., director of the Office of Antimicrobial Products in the FDA’s Center for Drug Evaluation and Research. “This approval will provide a new treatment option for patients with dry eye disease.”
Dry eye disease includes a group of conditions in which the eye does not produce an adequate volume of tears or when the tears are not of the correct consistency. The chance of experiencing dry eye increases with age, affecting approximately five percent of the adult population age 30-40 and 10 to 15 percent of adults over age 65, and is more common among women. When severe and left untreated, this condition can lead to pain, ulcers or scars on the part of the eye called the cornea. Dry eye can make it more difficult to perform some activities, such as using a computer or reading for an extended period of time, and it can decrease tolerance for dry environments, such as the air inside an airplane.
The safety and efficacy of Xiidra was assessed in over a thousand patients, in four separate, randomized, controlled studies. These studies included patients 19–97 years of age, of which the majority were female (76 percent). Patients were randomized equally to receive either Xiidra eyedrops or placebo eyedrops, which were used twice a day for twelve weeks. The studies found that groups treated with Xiidra demonstrated more improvement in both the signs and the symptoms of eye dryness than the groups treated with placebo.
The most common side effects of Xiidra include eye irritation, discomfort or blurred vision and an unusual taste sensation (dysgeusia).
Dry eye disease does not routinely occur in children. Safety and efficacy in pediatric patients below the age of 17 years has not been studied.
Xiidra is manufactured by Shire US Inc., of Lexington, Massachusetts.
Trademarks: Lifitegrast Molecular Formula: C29H24Cl2N2O7S CAS Registry Number: 1025967-78-5 Molecular Weight: 615.49 Activity: SAR 1118 is a potent novel small molecule lymphocyte function-associated antigen-1 (LFA-1/ICAM-1) antagonist for a broad range of ocular inflammatory conditions including dry eye and diabetic macular edema. Intermediates:
Lifitegrast Synthesis: US20110092707A1
References:
1. Burnier, J. Crystalline Pharmaceutical and Methods of Preparation and Use Thereof. US20110092707A1
2. Zhong, M.; et. al. Discovery of tetrahydroisoquinoline (THIQ) derivatives as potent and orally bioavailable LFA-1/ICAM-1 antagonists. Bioorg. Med. Chem. Lett. 20 (2010) 5269-5273.
3. Zeller, J. R.; et. al. Lfa-1 inhibitor and polymorph thereof. WO2014018748A1
4. Zhong, M.; et. al. Modulators of cellular adhesion. WO2005044817A1
///////Xiidra, Shire US Inc., Lexington, Massachusetts, fda 2016, lifitegrast ophthalmic solution, FDA, approves, new medication , dry eye disease
(4R–cis)-1-[[4-[[4-[3,3-Dibutyl-7-(dimethylamino)-2,3,4,5-tetrahydro-4-hydroxy-1,1-dioxido-1-benzothiepin-5-yl]phenoxy]methyl]phenyl]methyl]-4-aza-1-azoniabicyclo[2.2.2]octane Chloride Salt
It is well established that agents which inhibit the 20 transport of bile acids across the ileum can also cause a decrease in the level of cholesterol in blood serum. Stedronski, in “Interaction of bile acids and cholesterol with nonsystemic agents having hypocholesterolemic properties,” Biochimica et Biophysica Acta, 1210 (1994) 255- 25287, discusses biochemistry, physiology, and known active agents affecting bile acids and cholesterol.
A class of ileal bile acid transport-inhibiting compounds which was recently discovered to be useful for influencing the level of blood serum cholesterol is 30 tetrahydrobenzothiepine-l,l-dioxides (THBDO compounds). (U.S. Patent Application No. 08/816,065)
Some classes of compounds show enhanced potency as pharmaceutical therapeutics after they have been enantiomerically-enriched (see, for example, Richard B. Silverman, The Organic Chemistry of Drug Design and Drug Action, Academic Press, 1992, pp. 76-82) . Therefore, THBDO compounds that have been enantiomerically-enriched are of particular interest.
A class of chemistry useful as intermediates in the preparation of racemic THBDO compounds is tetrahydrobenzothiepine-1-oxides (THBO compounds) . THBDO compounds and THBO compounds possess chemical structures in which a phenyl ring is fused to a seven-member ring. A method of preparing enantiomerically-enriched samples of another phenyl/seven-member fused ring system, the benzothiazepines, is described by Higashikawa (JP 59144777) , where racemic benzothiazepine derivatives are optically resolved on a chromatographic column containing chiral crown ethers as a stationary phase. Although optical resolution is achieved, the Higashikawa method is limited to producing only small quantities of the enantiomerically-enriched benzothiazepine derivatives. Giordano (CA 2068231) reports the cyclization of (2S, 3S) -aminophenylthiopropionates in the presence of a phosphonic acid to produce (2S, 3S) -benzothiazepin-4-ones . However, that preparation is constrained by the need to use enantiomerically-enriched starting materials rather than racemic starting materials. In addition, the Giordano method controls the stereochemistry of the seven-member ring of the benzothiazepin-4-one only at the 2- and 3 -positions. The 4- and 5-positions of the seven-member ring of the benzothiazepin-4-one are not asymmetric centers, and the stereochemistry at these sites therefore cannot be controlled by the Giordano method. A method by which enantiomerically-enriched 1,5- benzothiazepin-3-hydroxy-4 (5H) -one compounds have been produced is through the asymmetric reduction of 1,5- benzothiazepin-3,4 (2H, 5H) -dione compounds, reported by Yamada, et al . (J“. Org. Chem. 1996, 61 (24), 8586-8590). The product is obtained by treating the racemic 1,5- benzothiazepin-3,4 (2H, 5H) -dione with the reaction product of an optically active alpha-amino acid and a reducing agent, for example sodium borohydride. Although a product with high optical purity was achieved, the method is limited by the use of a relatively expensive chemical reduction step.
The microbial reduction of racemic 1, 5-benzothiazepin- 3 , 4 (2H, 5H) -dione compounds to produce enantiomerically- enriched 1, 5-benzothiazepin-3-hydroxy-4 (5H) -one compounds is reported by Patel et al . , U.S. Patent 5,559,017. This method is limited by the inherent problems of maintaining a viable and pure bacterial culture of the appropriate species and variety. In addition, that method is limited in scale, producing only microgram quantities of the desired product. Until now, there have been no reported processes for preparing enantiomerically-enriched THBDO compounds or enantiomerically-enriched THBO compounds. Furthermore, there have been no reported processes for controlling the stereochemistry at the 4- and 5-positions of the seven- member rings of THBDO compounds or THBO compounds
FDA Grants Breakthrough Designation to Shire’s Rare GI Therapies
Tue, 06/14/2016
Shire announced that the U.S. Food and Drug Administration (FDA) has granted Breakthrough Therapy Designation for two investigational products for rare diseases: SHP621 (budesonide oral suspension, or BOS) for eosinophilic esophagitis (EoE), and SHP625 (maralixibat) for progressive familial intrahepatic cholestasis type 2 (PFIC2).
“Receiving Breakthrough Therapy Designation on two pipeline products this past week reflects the potential of our strong and innovative pipeline of more than 60 programs,” said Flemming Ornskov, M.D., MPH, and CEO, Shire. “Shire is committed to bringing innovation to the rare and specialty areas we focus on. We persevere to see compounds through the many stages of development through their challenges and successes, and always keep patients with unmet needs top of mind.”
EoE is a serious, chronic and rare disease that stems from an elevated number of eosinophils, a type of white blood cell, that infiltrate the walls of the esophagus. EoE is characterized by an inflammation of the esophagus that may lead to difficulty swallowing (dysphagia). The diagnosed prevalence of EoE ranges from approximately 15-55 cases per 100,000 persons, with high-end estimates reported by studies in Western regions.
PFIC refers to a group of autosomal-recessive liver disorders of childhood that disrupt bile formation and present with cholestasis. The symptoms of PFIC include severe itching of the skin (pruritus), and jaundice. PFIC is estimated to affect 1 in 50,000 to 1 in 100,000 births. PFIC2 is the most common type of PFIC, accounting for around half of cases.
According to the FDA, Breakthrough Therapy Designation is granted to a therapy that is intended to treat a serious or life-threatening disease or condition and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement on one or more clinically significant endpoints over current standard of care. Under the designation, the FDA provides intensive guidance, organizational commitment involving senior managers, and eligibility for rolling and priority review of the application; this process helps ensure patients have access to therapies as soon as possible, pending approval. Breakthrough Therapy Designation does not guarantee that FDA will ultimately approve BOS for EoE or maralixibat for PFIC2, and the timing of any such approval is uncertain.
“On behalf of patients in the United States with EoE and PFIC2, we are so pleased that the FDA has granted Breakthrough Therapy Designation to BOS and maralixibat,” said Philip J. Vickers, Ph.D., Head of R&D, Shire. “We look forward to working with the agency to continue their development and, pending FDA approval, deliver these therapeutic options to the patients who need them most.”
It is well established that agents which inhibit the transport of bile acids across the tissue of the ileum can also cause a decrease in the levels of cholesterol in blood serum. Stedronski, in “Interaction of bile acids and cholesterol with nonsystemic agents having hypocholesterolemic properties,” Biochimica et Biophysica Acta, 1210 (1994) 255-287 discusses biochemistry, physiology, and known active agents surrounding bile acids and cholesterol. Bile acids are actively transported across the tissue of the ileum by an apical sodium co-dependent bile acid transporter (ASBT), alternatively known as an ileal bile acid transporter (IBAT). A class of ASBT-inhibiting compounds that was recently discovered to be useful for influencing the level of blood serum cholesterol comprises tetrahydrobenzothiepine oxides (THBO compounds, PCT Patent Application No. WO 96/08484). Further THBO compounds useful as ASBT inhibitors are described in PCT Patent Application No. WO 97/33882. Additional THBO compounds useful as ASBT inhibitors are described in U.S. Patent No. 5,994,391. Still further THBO compounds useful as ASBT inhibitors are described in PCT Patent Application No. WO 99/64409. Included in the THBO class are tetrahydrobenzo-thiepine-l -oxides and tetrahydrobenzothiepine- 1,1 -dioxides. THBO compounds possess chemical structures in which a phenyl ring is fused to a seven-member ring.
Published methods for the preparation of THBO compounds include the synthesis through an aromatic sulfone aldehyde intermediate. For example l-(2,2-dibutyl-3-oxopropylsulfonyl)-2-((4-methoxyphenyl)methyl)benzene (29) was cyclized with potassium t-butoxide to form tetrahydrobenzothiepine- 1,1 -dioxide (svn-24) as shown in Eq. 1.
Compound 29 was prepared by reacting 2-chloro-5-nitrobenzoic acid chloride with anisole in the presence of aluminum trichloride to produce a chlorobenzophenone compound; the chlorobenzophenone compound was reduced in the presence of trifluoromethanesulfonic acid and triethylsilane to produce a chlorodiphenylmethane compound; the chlorodiphenylmethane compound was treated with lithium sulfide and 2,2-dibutyl-3-(methanesulfonato)propanal to produce l-(2,2-dibutyl-3-oxopropylthio)-2-((4-methoxyphenyl)methyl)-4-dimethylaminobenzene (40); and 40 was oxidized with m-chloroperbenzoic acid to produce 29. The first step of that method of preparing compound 29 requires the use of a corrosive and reactive carboxylic acid chloride that was prepared by the reaction of the corresponding carboxylic acid with phosphorus pentachloride. Phosphorus pentachloride readily hydrolyzes to produce volatile and hazardous hydrogen chloride. The reaction of 2,2-dibutyl-3-(methanesulfonato)propanal with the lithium sulfide and the chlorodiphenylmethane compound required the intermediacy of a cyclic tin compound to make the of 2,2-dibutyl-3-(methanesulfonato)propanal. The tin compound is expensive and creates a toxic waste stream. In WO 97/33882 compound syn-24 was dealkylated using boron tribromide to produce the phenol compound 28. Boron tribromide is a corrosive and hazardous material that generates hydrogen bromide gas and requires special handling. Upon hydrolysis, boron tribromide also produces borate salts that are costly and time-consuming to separate and dispose of.
An alternative method of preparing THBO compounds was described in WO 97/33882, wherein a 1,3-propanediol was reacted with thionyl chloride to form a cyclic sulfite compound. The cyclic sulfite compound was oxidized to produce a cyclic sulfate compound. The cyclic sulfate was condensed with a 2-methylthiophenol that had been deprotonated with sodium hydride. The product of the condensation was a (2-methylphenyl) (3′-hydroxypropyl)thioether compound. The thioether compound was oxidized to form an thioether aldehyde compound. The thioether aldehyde compound was further oxidized to form an aldehyde sulfone compound which in turn was cyclized in the presence of potassium t-butoxide to form a 4-hydroxytetrahydrobenzothiepine 1,1 -dioxide compound. This cyclic sulfate route to THBO compounds requires an expensive catalyst. Additionally it requires the use of SOCI2, which in turn requires special equipment to handle. PCT Patent Application No. WO 97/33882 describes a method by which the phenol compound 28 was reacted at its phenol hydroxyl group to attach a variety of functional groups to the molecule, such as a quaternary ammonium group. For example, (4R,5R)-28 was reacted with l,4-bis(chloromethyl)benzene (?,??’-dichloro-p-xylene) to produce the chloromethyl benzyl- ether (4R,5R)-27. Compound (4R,5R)-27 was treated with diazabicyclo[2.2.2]octane (DABCO) to produce (4R,5R)-l-((4-(4-(3,3-dibutyl-7-(dimemylamino)-2,3,4,5-tetrahydro-4-hydroxy-l , 1 -dioxido-1 -benzothiepin-5-yl)phenoxy)methyl)phenyl)methyl-4-aza-l-azomabicyclo[2.2.2]octane chloride (41). This method suffers from low yields because of a propensity for two molecules of compound (4R,5R)-28 to react with one molecule of l,4-bis(chloromethyl)benzene to form a bis(benzothiepine) adduct. Once the bis-adduct forms, the reactive chloromethyl group of compound (4R,5R)-27 is not available to react with an amine to form the quaternary ammonium product.
A method of preparing enantiomerically enriched tetrahydrobenzothiepine oxides is described in PCT Patent Application No. WO 99/32478. In that method, an aryl-3- hydroxypropylsulfide compound was oxidized with an asymmetric oxidizing agent, for example (lR (->(8,9-dichloro-10-camphorsulfonyl)oxaziridine, to yield a chiral aryl-3-hydroxypropylsulfoxide. Reaction of the aryl-3-hydroxypropylsulfoxide with an oxidizing agent such as sulfur trioxide pyridine complex yielded an aryl-3-propanalsulfoxide. The aryl- 3-propanalsulfoxide was cyclized with a base such as potassium t-butoxide to enantioselectively produce a tetrahydrobenzothiepine- 1 -oxide. The tetrahydrobenzothiepine- 1 -oxide was further oxidized to produce a tetrahydrobenzothiepine- 1 , 1 -dioxide. Although this method could produce tetrahydrobenzothiepine- 1,1 -dioxide compounds of high enantiomeric purity, it requires the use of an expensive asymmetric oxidizing agent. Some 5-amidobenzothiepine compounds and methods to make them are described in
PCT Patent Application Number WO 92/18462. In Svnlett. 9, 943-944(1995) 2-bromophenyl 3-benzoyloxy-l-buten-4-yl sulfone was treated with tributyl tin hydride and AIBN to produce 3-benzoyloxytetrahydrobenzothiepine-1,1 -dioxide. In addition to forming the desired ASBT inhibitors, it is also desirable to form such
ASBT inhibitors of higher purity and having lower levels of residual solvent impurities. This is especially so with respect to ASBT inhibitors having a positively charged substituent, for example, the compounds designated as 41 (supra) and 60 (infra). It is further desirable to provide methods for making such high purity ASBT inhibitors.
( 4R, 5R) -26 A 1000 mL 4 neck jacketed Ace reactor flask was fitted with a mechanical stirrer, a nitrogen inlet, an addition funnel or condenser or distilling head with receiver, a thermocouple, four internal baffles and a 28 mm Teflon turbine agitator. The flask was purged with nitrogen gas and charged with 25.0 grams of (4R,5R)-28 and 125 mL of N,N-dimethylacetamide (DMAC). To this was added 4.2 grams of 50% sodium hydroxide. The mixture was heated to 50°C and stiπed for 15 minutes. To the flask was added 8.3 grams of 55 dissolved in 10 mL of DMAC, all at once. The temperature was held at 50°C for 24 hrs. To the flask was added 250 mL of toluene followed by 125 mL of dilution water. The mixture was stiπed for 15 minutes and the layers were then allowed to separate at 50°C. The flask was then charged with 125 mL of saturated sodium chloride solution and stiπed 15 minutes. Layers separated cleanly in 30 seconds at 50°C. Approximately half of the solvent was distilled off under vacuum at 50°C. The residual reaction mixture contained (4R,5R)-26.
Step 2. Preparation of (4R.5RV27.
( 4R, 5R) -27 Toluene was charged back to the reaction mixture of Step 1 and the mixture was cooled to 35°C. To the mixture was then added 7.0 grams of thionyl chloride over 5 minutes. The reaction was exothermic and reached 39°C. The reaction turned cloudy on first addition of thionyl chloride, partially cleared then finally remained cloudy. The mixture was stirred for 0.5 hr and was then washed with 0.25N NaOH. The mixture appeared to form a small amount of solids that diminished on stirring, and the layers cleanly separated. The solvent was distilled to a minimum stir volume under vacuum at 50°C. The residual reaction mixture contained (4R,5R)-27.
Step 3. Preparation of 41. To the reaction mixture of Step 2 was charged with 350 mL of methyl ethyl ketone (MEK) followed by 10.5 mL water and 6.4 grams of diazabicyclo[2.2.2]octane (DABCO) dissolved in 10 mL of MEK. The mixture was heated to reflux, and HPLC showed <0.5% of (4R,5R)-27. The reaction remained homogenous initially then crystallized at the completion of the reaction. An additional 5.3 mL of water was charged to the flask to redissolve product. Approximately 160 mL of solvent was then distilled off at atmospheric pressure. The mixture started to form crystals after 70 mL of solvent was distilled. Water separated out of distillate indicating a ternary azeotrope between toluene, water and methyl ethyl ketone (MEK). The mixture was then cooled to 25°C. The solids were filtered and washed with 150 mL MEK, and let dry under vacuum at 60°C. Isolated 29.8.0 g of off-white crystalline 4 Example 11a. Alternate Preparation of (4R,5R)-l-((4-(4-(3,3-dibutyl-7-(dimemylamino)-2,3,4,5-tetrahydro- 4-hydroxy- 1 , 1 -dioxido- 1 -benzithiepin-5-yl)phenoxy)methyl)phenyl)methyl-4-aza- 1 – azoniabicyclo[2.2.2]octane chloride, Form II of 41
A 1000 mL 4 neck jacketed Ace reactor flask is fitted with a mechanical stiπer, a nitrogen inlet, an addition funnel or condenser or distilling head with receiver, a thermocouple, four internal baffles and a 28 mm Teflon turbine agitator. The flask is purged with nitrogen gas and charged with 25.0 grams of (4R,5R)-28 and 100 mL of N,N-dimethylacetamide (DMAC). The mixture is heated to 50°C and to it is added 4.02 grams of 50% sodium hydroxide. The mixture is stiπed for 30 minutes. To the flask is added 8.7 grams of 55 dissolved in 12.5 mL of DMAC, all at once. The charge vessel is washed with 12.5 mL DMAC and the wash is added to the reactor. The reactor is stiπed for 3 hours. To the reactor is added 0.19 mL of 49.4% aq. NaOH and the mixture is stirred for 2 hours. To the mixture is added 0.9 g DABCO dissolved in 12.5 mL DMAC. The mixture is stiπed 30 to 60 minutes at 50°C. To the flask is added 225 mL of toluene followed by 125 mL of dilution water. The mixture is stiπed for 15 minutes and the layers are then allowed to separate at 50°C. The bottom aqueous layer is removed but any rag layer is retained. The flask is then charged with 175 mL of 5% hydrochloric acid solution and stiπed 15 minutes. Layers are separated at 50°C to remove the bottom aqueous layer, discarding any rag layer with the aqueous layer. Approximately half of the solvent is distilled off under vacuum at a maximum pot temperature of 80°C. The residual reaction mixture contains (4R,5R)-26.
Step 2. Preparation of (4R.5RV27.
Toluene (225 mL) is charged back to the reaction mixture of Step 1 and the mixture is cooled to 30°C. To the mixture is then added 6.7 grams of thionyl chloride over 30 to 45 minutes. The temperature is maintained below 35°C. The reaction turns cloudy on first addition of thionyl chloride, then at about 30 minutes the layers go back together and form a clear mixture. The mixture is stiπed for 0.5 hr and is then charged with 156.6 mL of 4% NaOH wash over a 30 minute period. The addition of the wash is stopped when the pH of the mixture reaches’ 8.0 to 10.0. The bottom aqueous layer is removed at 30°C and any rag layer is retained with the organic layer. To the mixture is charged 175 mL of saturated NaCl wash with agitation. The layers are separated at 30°C and the bottom aqueous layer is removed, discarding any rag layer with the aqueous layer. The solvent is distilled to a minimum stir volume under vacuum at 80°C. The residual reaction mixture contains (4R,5R)-27.
Step 3. Preparation of 41. To the reaction mixture of Step 2 is charged 325 mL of methyl ethyl ketone (MEK) and 13 mL water. Next, the reactor is charged 6.2 grams of diazabicyclo[2.2.2]octane (DABCO) dissolved in 25 mL of MEK. The mixture is heated to reflux and held for 30 minutes. Approximately 10% of solvent volume is then distilled off. The mixture starts to form crystals during distillation. The mixture is then cooled to 20°C for 1 hour. The off-white crystalline 41 (Form U) is filtered and washed with 50 mL MEK, and let dry under vacuum at 100°C.
Example lib. Alternate Preparation of (4R,5R)-1 -((4-(4-(3,3-dibutyl-7-(dimethylamino)-2,3,4,5-tetrahydro- 4-hydroxy- 1 , 1 -dioxido- 1 -benzithiepin-5-yl)phenoxy)methyl)phenyl)methyl-4-aza- 1 – azoniabicyclo[2.2.2]octane chloride, Form II of 41
A 1000 mL 4 neck jacketed Ace reactor flask is fitted with a mechanical stiπer, a nitrogen inlet, an addition funnel or condenser or distilling head with receiver, a thermocouple, four internal baffles and a Teflon turbine agitator. The flask is purged with nitrogen gas and charged with 25.0 grams of (4R,5R)-28 and 125 mL of N,N-dimethylacetamide (DMAC). The mixture is heated to 50°C and to it is added 7.11 grams of 30% sodium hydroxide over a period of 15 to 30 minutes with agitation. The mixture is stiπed for 30 minutes. To the flask is added 9.5 grams of solid 55. The reactor is stiπed for 3 hours. To the mixture is added 1.2 g of solid DABCO. The mixture is stiπed 30 to 60 minutes at 50°C. To the flask is added 225 mL of toluene followed by 125 mL of water. The mixture is stirred for 15 minutes and the layers are then allowed to separate at 50°C. The bottom aqueous layer is removed but any rag layer is retained with the organic layer. The flask is then charged with 175 mL of 5% hydrochloric acid solution and stirred 15 minutes. Layers are separated at 50°C to remove the bottom aqueous layer, discarding any rag layer with the aqueous layer. The flask is then charged with 225 mL of water and stirred 15 minutes. The layers are allowed to separate at 50°C. The bottom aqueous layer is removed, discarding any rag layer with the aqueous layer. Approximately half of the solvent is distilled off under vacuum at a maximum pot temperature of 80°C. The residual reaction mixture contains (4R,5R)-26.
Step 2. Preparation of (4R.5RV27.
Toluene (112.5 mL) is charged back to the reaction mixture of Step 1 and the mixture is cooled to 25°C. To the mixture is then added 7.3 grams of thionyl chloride over 15 to 45 minutes. The temperature of the mixture is maintained above 20°C and below 40°C. The reaction turns cloudy on first addition of thionyl chloride, then at about 30 minutes the layers go back together and form a clear mixture. The mixture is then charged with 179.5 mL of 4% NaOH wash over a 30 minute period. The mixture is maintained above 20°C and below 40°C during this time. The addition of the wash is stopped when the pH of the mixture reaches 8.0 to 10.0. The mixture is then allowed to separate at 40°C for at least one hour.
The bottom aqueous layer is removed and any rag layer is retained with the organic layer. To the mixture is charged 200 mL of dilution water. The mixture is stiπed for 15 minutes and then allowed to separate at 40°C for at least one hour. The bottom aqueous layer is removed, discarding any rag layer with the aqueous layer. The solvent is distilled to a minimum stir volume under vacuum at 80°C. The residual reaction mixture contains (4R,5R)-2 .
Step 3. Preparation of 41. To the reaction mixture of Step 2 is charged 350 mL of methyl ethyl ketone (MEK) and 7 mL water. The mixture is stiπed for 15 minutes and the temperature of the mixture is adjusted to 25°C. Next, the reactor is charged with 6.7 grams of solid diazabicyclo[2.2.2]octane (DABCO). The mixture is maintained at 25°C for three to four hours. It is then heated to 65°C and maintained at that temperature for 30 minutes. The mixture is then cooled to 25°C for 1 hour. The off-white crystalline 41 (Form II) is filtered and washed with 50 mL MEK, and let dry under vacuum at 100°C.
Example 12. Alternate preparation of (4R,5R)-1 -((4-(4-(3,3-dibutyl-7-(dimethylamino)-2,3,4,5-tetrahydro- 4-hydroxy- 1 , 1 -dioxido- 1 -benzithiepin-5-yl)phenoxy)methyl)phenyl)methyl-4-aza- 1 – azoniabicyclo[2.2.2]octane chloride, Form I of 41
(4R,5R)-27 (2.82 kg dry basis, 4.7 mol) was dissolved in MTBE (9.4 L). The solution of (4R,5R)-22 was passed through a 0.2 mm filter cartridge into the feeding vessel. The flask and was rinsed with MTBE (2 x 2.5 L). The obtained solution as passed through the cartridge filter and added to the solution of (4R,5R)-2 in the feeding vessel. DABCO (diazabicyclo[2.2.2]octane, 0.784 kg, 7.0 mol) was dissolved in MeOH (14.2 L). The DABCO solution was passed through the filter cartridge into the 100 L nitrogen-flushed reactor. The Pyrex bottle and the cartridge filter were rinsed with MeOH (7.5 L) and the solution was added to the reactor. The (4R,5R)-22 solution was added from the feeding vessel into the reactor at 37°C over a period of 10 min, while stirring. Methanol (6.5 L) was added to the Pyrex bottle and via the cartridge filter added to the feeding vessel to rinse the remaining (4R,5R)-2 into the reactor. The reaction mixture was brought to 50-60°C over 10-20 min and stiπed at that temperature for about 1 h. The mixture was cooled to 20-25°C over a period of 1 h. To the reaction mixture, methyl t-butyl ether (MTBE) (42 L) was added over a period of 1 h and stiπed for a minimum of 1 h at 20 – 25°C. The suspension was filtered through a Buchner funnel. The reactor and the filter cake were washed with MTBE (2 x 14 L). The solids were dried on a rotary evaporator in a 20 L flask at 400 – 12 mbar, 40°C, for 22 h. A white crystalline solid was obtained. The yield of 4 . (Form I) was 3.08 kg (2.97 kg dry, 93.8 %) and the purity 99.7 area % (HPLC; Kromasil C 4, 250 x 4.6 mm column; 0.05% TFA in H2O/0.05% TFA in ACN gradient, UV detection at 215 nm).
Example 12a. Conversion of Form I of Compound 41 into Form II of Compound 41.
To 10.0 grams of Form I of 4 . in a 400 mL jacketed reactor is added 140 mL of MEK. The reactor is stirred (358 φm) for 10 minutes at 23 °C for 10 minutes and the stirring rate is then changed to 178 φm. The suspension is heated to reflux over 1 hour using a programmed temperature ramp (0.95°C/minute) using batch temperature control (cascade mode). The delta Tmaχ is set to 5°C. The mixture is held at reflux for 1 hour. The mixture is cooled to
25°C. After 3 hours at 25°C, a sample of the mixture is collected by filtration. Filtration is rapid (seconds) and the filtrate is clear and colorless. The white solid is dried in a vacuum oven (80°C, 25 in. Hg) to give a white solid. The remainder of the suspension is stirred at 25°C for 18 hours. The mixture is filtered and the cake starts to shrink as the mother liquor reaches the top of the cake. The filtration is stopped and the reactor is rinsed with 14 mL of MEK. The reactor stirrer speed is increased from 100 to 300 φm to rinse the reactor. The rinse is added to the filter and the solid is dried with a rapid air flow for 5 minutes. The solid is dried in a vacuum oven at 25 in. Hg for 84 hours to give Form II of 4
Department of Discovery Chemistry and Department of Cardiovascular Disease, Pharmacia, 700 Chesterfield Parkway W, Chesterfield, Missouri 63017, Office of Science and Technology, Chemical Science Division, Pharmacia, 800 Lindbergh Boulevard, Creve Coeur, Missouri 63167, Department of Pharmaceutical Sciences, Pharmacia, Skokie, Illinois, and Department of Chemistry, University of Missouri, St. Louis, Missouri
In the preceding paper several compounds were reported as potent apical sodium-codependent bile acid transporter (ASBT) inhibitors. Since the primary site for active bile acid reabsorption is via ASBT, which is localized on the luminal surface of the distal ileum, we reasoned that a nonsystemic inhibitor would be desirable to minimize or eliminate potential systemic side effects of an absorbed drug. To ensure bioequivalency and product stability, it was also essential that we identify a nonhygroscopic inhibitor in its most stable crystalline form. A series of benzothiepines were prepared to refine the structure−activity relationship of the substituted phenyl ring at the 5-position of benzothiepine ring and to identify potent, crystalline, nonhygroscopic, and efficacious ASBT inhibitors with low systemic exposure.
compd
R
IC50 (nM)b
hygroscp I wt gain (%)c
hygroscp II % wt gain (%)d
crystallinitye
74
OCH2C6H4(p)CH2(N+)DB
0.28
1.59
2.1
yes
(4R–cis)-1-[[4-[[4-[3,3-Dibutyl-7-(dimethylamino)-2,3,4,5-tetrahydro-4-hydroxy-1,1-dioxido-1-benzothiepin-5-yl]phenoxy]methyl]phenyl]methyl]-4-aza-1-azoniabicyclo[2.2.2]octane Chloride Salt(74). Following a similar procedure as in General Method B, the title compound 74 was prepared from the corresponding chloromethyl benzyl ether and DABCO as a white solid, mp 223−230 °C (dec); 1H NMR (CDCl3) δ 0.89 (m, 6H), 1.27−1.52 (br m, 10H), 1.63 (m, 1H), 2.20 (m, 1H), 2.81 (s, 6H), 3.06 (ABq, JAB = 15.1 Hz, J = 43.3 Hz, 2H), 3.16 (s, 6H), 3.76 (s, 6H), 4.11 (d, J = 7.7 Hz, 1H), 5.09 (s, 2H), 5.14 (s, 2H), 5.48 (s, 1H), 5.96 (s, 1H), 6.49 (d, J = 8.9 Hz, 1H), 6.99 (d, J = 8.0 Hz, 2H), 7.26 (m, 1H), 7.44 (d, J = 8.0 Hz, 2H), 7.52 (d, J = 7.4 Hz, 2H), 7.68 (d, J = 7.4 Hz, 2H), 7.87 (d, J = 8.9 Hz, 1H). HRMS calcd for C40H56N3O4S: 674.3992; found, 674.4005. Anal. Calcd for C40H56N3O4S: ‘ C, 67.62; H, 7.95; N, 5.92; S, 4.51. Found: C, 67.48; H, 8.32; N, 5.85; S, 4.60.
a All compounds were prepared using method B in Scheme 3.b Taurocholate is transported across the baby hamster kidney cell membrane.c % weight gain in a 25 °C, 57% humidity chamber for 2 weeks.d % weight gain in a 40 °C, 80% humidity chamber for 2 weeks.e Crystallinity as determined by X-ray powder diffraction analysis.f (N+)DB is a DABCO terminal group with the quaternary ammonium attached to the linke
Example 10. Preparation of enantiomerically-enriched (4R.5R)-1- r.4- r _4- .3.3 -Dibutyl-7- (dimethylamino) -2.3 ,4.5- tetrahydro-4-hydroxy-1, l-dioxido-l-benzothiepin-5- yl] henoxy] ethyl] phenyl1methyl] -4-aza-l- azoniabicyclo [2.2.2] octane chloride ( (4R,5R) -XXVII) ♦
( (4R,5R) -XXVII) * = chiral center
Step 1. Preparation of 4-flUoro-2- ( (4- methoxyphenyl) methyl) -phenol To a stirred solution of 23.66 g of 95% sodium hydride (0.94 mol) in 600 mL of dry toluene was added 100.0 g of 4- fluorophenol (0.89 mol) at 0°C. The mixture was stirred at 90°C for 1 hour until gas evolution stopped. The mixture was cooled down to room temperature and a solution of 139.71 g of 3 -methoxybenzyl chloride (0.89 mol) in 400 mL of dry toluene was added. After refluxing for 24 hours, the mixture was cooled to room temperature and quenched with 500 mL of water. The organic layer was separated, dried over MgS04, and concentrated under high vacuum. The remaining starting materials were removed by distillation. The crude dark red oil was filtered through a layer of 1 L of silica gel with neat hexane to yield 53.00 g (25.6%) of the product as a pink solid: *H NMR (CDC13) d 3.79 (s, 3H) , 3.90 (s, 2H) , 4.58 (s, IH) , 6.70-6.74 (m, IH) , 6.79-6.88 (m, 4H) , 7.11-7.16 (m, 2H) .
Step 2. Preparation of 4-fluoro-2- ( (4- methoxyphenyl) methyl) -thiophenol
Step 2a. Preparation of thiocarbamate To a stirred solution of 50.00 g (215.30 mmol) of 4- fluoro-2- ( ( -methoxyphenyl) methyl) -phenol in 500 mL of dry DMF was added 11.20 g of 60% sodium hydride dispersion in mineral oil (279.90 mmol) at 2°C. The mixture was allowed to warm to room temperature and 26.61 g of dimethylthiocarbamoyl chloride (215.30 mmol) was added. The reaction mixture was stirred at room temperature overnight. The mixture was quenched with 100 mL of water in an ice bath. The solution was extracted with 500 mL of diethyl ether. The ether solution was washed with 500 mL of water and 500 mL of brine. The ether solution was dried over MgS04 and stripped to dryness. The crude product was filtered through a plug of 500 mL silica gel using 5% ethyl acetate/hexane to yield 48.00 g (69.8%) of the product as a pale white solid: XH NMR (CDC13) d 3.21 (s, 3H) , 3.46 (s, 3H) , 3.80 (s, 3H) , 3.82 (s, 2H) , 6.78-6.86 (m, 3H) , 6.90- 7.00 (m, 2H) , 7.09 (d, J = 8.7 Hz, 2H) .
Step 2b. Rearrangement and hydrolysis of thiocarbamate to 4-fluoro-2- ( (4 -methoxyphenyl) methyl) -thiophenol A stirred solution of 48.00 g (150.29 mmol) of thiocarbamate (obtained from Step 2a) in 200 mL of diphenyl ether was refluxed at 270°C overnight. The solution was cooled down to room temperature and filtered through 1 L of silica gel with 2 L of hexane to remove phenyl ether. The rearrangement product was washed with 5% ethyl acetate/hexane to give 46.00 g (95.8%) of the product as a pale yellow solid: XH NMR (CDC13) d 3.02 (s, 3H) , 3.10 (s, 3H) , 3.80 (s, 3H) , 4.07 (s, 2H) , 6.82-6.86 (m, 3H) , 6.93 (dt, J = 8.4 Hz, 2.7 Hz, IH) , 7.08 (d, J = 8.7 Hz, 2H) , 7.49 (dd, J = 6.0 Hz, 8.7 Hz, IH) . To a solution of 46.00 g (144.02 mmol) of the rearrangement product (above) in 200 mL of methanol and 200 mL of THF was added 17.28 g of NaOH (432.06 mmol) . The mixture was refluxed under nitrogen overnight . The solvents were evaporated off and 200 mL of water was added. The aqueous solution was washed with 200 mL of diethyl ether twice and placed in an ice bath. The aqueous mixture was acidified to pH 6 with concentrated HCl solution. The solution was extracted with 300 mL of diethyl ether twice. The ether layers were combined, dried over MgS04 and stripped to dryness to afford 27.00 g (75.5%) of the product as a brown oil: XH NMR (CDC13) d 3.24 (s, IH) , 3.80 (s, 3H) , 3.99 (s, 2H) , 6.81-6.87 (m, 4H) , 7.09 (d, J = 8.7 Hz, 2H) , 7.27- 7.33 (m, IH) .
Step 3. Preparation of dibutyl cyclic sulfate
Step 3a. Preparation of 2 , 2-dibutyl-l, 3-propanediol . To a stirred solution of di-butyl-diethylmalonate (Aldrich) (150g, 0.55 mol in dry THF (700ml) in an acetone/dry ice bath was added LAH (1 M THF) 662 ml (1.2 eq. , 0.66 mol) dropwise maintaining the temperature between -20 to 0°C. The reaction was stirred at RT overnight. The reaction was cooled to -20°C and 40 ml of water, and 80 mL of 10% NaOH and 80 ml of water were added dropwise. The resulting suspension was filtered. The filtrate was dried over sodium sulphate and concentrated in vacuo to give diol 598.4 g (yield 95%) as an oil. MS spectra and proton and carbon NMR spectra were consistent with the product.
Step 3b. Preparation of dibutyl cyclic sulfite
A solution of 2 , 2-dibutyl-l, 3-propanediol (103 g, 0.548 0 mol, obtained from Step 3a) and triethylamine (221 g, 2.19 mol) in anhydrous methylene chloride (500 ml) was stirred at 0°C under nitrogen. To the mixture, thionyl chloride (97.8* g, 0.‘82 mol) was added dropwise and within 5 min the solution turned yellow and then black when the addition was 5 completed within half an hour. The reaction mixture was stirred for 3 hrs. at 0°C. GC showed that there was no starting material left. The mixture was washed with ice water twice then with brine twice . The organic phase was dried over magnesium sulfate and concentrated under vacuum 0 to give 128 g (100%) of the dibutyl cyclic sulfite as a black oil. Mass spectrum (MS) was consistent with the product .
Step 3c. Oxidation of dibutyl cyclic sulfite to 5 dibutyl cyclic sulfate
To a solution of the dibutyl cyclic sulfite (127.5 g , 0.54 mol, obtained from Step 3b) in 600 ml acetonitrile and 500 ml of water cooled in an ice bath under nitrogen was added ruthenium (III) chloride (1 g) and sodium periodate 0 (233 g, 1.08 mol) . The reaction was stirred overnight and the color of the solution turned black. GC showed that there was no starting material left. The mixture was extracted with 300 ml of ether and the ether extract was washed three times with brine. The organic phase was dried over magnesium sulfate and passed through celite. The filtrate was 5 concentrated under vacuum and to give 133 g (97.8%) of the dibutyl cyclic sulfate as an oil. Proton and carbon NMR and MS were consistent with the product.
Step 4. Preparation of aryl-3-hydroxypropylsulfide
10 To a stirred solution of 27.00 g (108.73 mmol) of 4- fluoro-2- ( (4-methoxyphenyl) methyl) thiophenol (obtained from Step 2) in 270 mL of diglyme was added 4.35 g of 60% sodium-, hydride dispersion in mineral oil (108.73 mmol) at 0°C. After gas evolution ceased, 29.94 g (119.60 mmol) of the
15 dibutyl cyclic sulfate (obtained from Step 3c) was added at 0°C and stirred for 10 minutes. The mixture was allowed to warm up to room temperature and stirred overnight. The solvent was evaporated and 200 mL of water was added. The solution was washed with 200 mL of diethyl ether and added
2025 mL of concentrated sulfuric acid to make a 2.0 M solution that was refluxed overnight. The solution was extracted with ethyl acetate and the organic solution was dried over MgS04 and concentrated in vacuo. The crude aryl-3 – hydroxypropylsulfide was purified by silica gel
25 chromatography (Waters Prep 500) using 8% ethyl acetate/hexane to yield 33.00 g (72.5%) of the product as a light brown oil: E NMR (CDC13) d 0.90 (t, J = 7.1 Hz, 6H) , 1.14-1.34 (m, 12H) , 2.82 (s, 2H) , 3.48 (s, 2H) , 3.79 (s, 3H) , 4.10 (s, 2H) , 6.77-6.92 (m, 4H) , 7.09 (d, J = 8.7 Hz,
To a stirred solution of 20.00 g (47.78 mmol) of aryl- 53 -hydroxypropylsulfide (obtained from Step 4) in 1 L of methylene chloride was added 31.50 g of 96% (12?) – ( -) – (8 , 8- dichloro-10-camphor-sulfonyl) oxaziridine (100.34 mmol, Aldrich) at 2°C. After all the oxaziridine dissolved the mixture was placed into a -30 °C freezer for 72 hours. The
10 solvent was evaporated and the crude solid was washed with 1 L of hexane. The white solid was filtered off and the hexane solution was concentrated in vacuo. The crude oil was purified on a silica gel column (Waters Prep 500) using 15% ethyl acetate/hexane to afford 19.00 g (95%) of the
207.00 (d, J = 8.1 Hz, 2H) , 7.18-7.23 (m, IH) , 7.99-8.04 (m, IH) . Enantiomeric excess was determined by chiral HPLC on a (2?,2?) -Whelk-0 column using 5% ethanol/hexane as the eluent. It showed to be 78% e.e. with the first eluting peak as the major product.
25
Step 6. Preparation of enantiomerically-enriched aryl-3- propanalsulfoxide
To a stirred solution of 13.27 g of triethylamine (131.16 mmol, Aldrich) in 200 mL dimethyl sulfoxide were
30 added 19.00 g (43.72 mmol) of enantiomerically-enriched aryl-3 -hydroxypropylsulfoxide (obtained from Step 5) and 20.96 g of sulfur trioxide-pyridine (131.16 mmol, Aldrich) at room temperature. After the mixture was stirred at room temperature for 48 hours, 500 mL of water was added to the mixture and stirred vigorously. The mixture was then 5 extracted with 500 mL of ethyl acetate twice. The ethyl acetate layer was separated, dried over MgS04, and concentrated in vacuo. The crude oil was filtered through 500 mL of silica gel using 15% ethyl acetate/hexane to give 17.30 g (91%) of the enantiomerically-enriched aryl-3-
Step 7. Preparation of the enantiomerically-enriched tetrahydrobenzothiepine-1-oxide (4R, 5R)
20 To a stirred solution of 17.30 g (39.99 mmol) of enantiomerically-enriched aryl-3 -propanalsulfoxide (obtained from Step 6) in 300 mL of dry THF at -15°C was added 48 mL of 1.0 M potassium t-butoxide in THF (1.2 equivalents) under nitrogen. The solution was stirred at -15°C for 4 hours.
25 The solution was then quenched with 100 mL of water and neutralized with 4 mL of concentrated HCl solution at 0°C. The THF layer was separated, dried over MgS04, and concentrated in vacuo. The enantiomerically-enriched tetrahydrobenzothiepine-1-oxide (4R,5R) was purified by
To a stirred solution of 13.44 g (31.07 mmol) of enantiomerically-enriched tetrahydrobenzothiepine-1-oxide (obtained from Step 7) in 150 mL of methylene chloride was added 9.46 g of 68% m-chloroperoxybenzoic acid (37.28 mmol,
15 Sigma) at 0 °C. After stirring at 0 °C for 2 hours, the mixture was allowed to warm up to room temperature and stirred for 4 hours. 50 mL of saturated Na2S03 was added into the mixture and stirred for 30 minutes. The solution was then neutralized with 50 mL of saturated NaHC03 solution.
20 The methylene chloride layer was separated, dried over MgS04, and concentrated in vacuo to give 13.00 g (97.5%) of the enantiomerically-enriched tetrahydrobenzothiepine-1, 1- dioxide (4R,5R) as a light yellow solid: ‘H NMR (CDC13) d 0.89-0.95 (m, 6H) , 1.09-1.42 (m, 12H) , 2.16-2.26 (m, IH) ,
30 Step 9. Preparation of enantiomerically-enriched 7-
(dimethylamino) tetrahydrobenzothiepine-1 , 1-dioxide (4R.5R) – To a solution of 13.00 g (28.98 mmol) of enantiomerically-enriched tetrahydrobenzothiepine-1, 1- dioxide (obtained from Step 8) in 73 mL of dimethylamine (2.0 M in THF, 146 mmol) in a Parr Reactor was added ca . 20 5 mL of neat dimethylamine . The mixture was sealed and stirred at 110 °C overnight, and cooled to ambient temperature. The excess dimethylamine was evaporated. The crude oil was dissolved in 200 mL of ethyl acetate and washed with 100 mL of water, dried over MgS04 and
10 concentrated in vacuo. Purification on a silica gel column (Waters Prep 500) using 20% ethyl acetate/hexane gave 12.43 g (90.5%) of the enantiomerically- enriched 7- (dimethylamino) tetrahydrobenzothiepine-1, 1-dioxide (4R, 5R) as a colorless solid: *H NMR (CDC13) d 0.87-0.93 (m, 6H) ,
20 IH) . The product was determined to have 78% e.e. by chiral HPLC on a Chiralpak AD column using 5% ethanol/hexane as the eluent. Recrystallization of this solid from ethyl acetate/hexane gave 1.70 g of the racemic product. The remaining solution was concentrated and recrystallized to
25 give 9.8 g of colorless solid. Enantiomeric excess of this solid was determined by chiral HPLC on a Chiralpak AD column using 5% ethanol/hexane as the eluent. It showed to have 96% e.e with the first eluting peak as the major product.
30 Step 10: Demethylation of 5- (4 ‘ -methoxyphenyl) -7-
(dimethylamino) tetrahydrobenzothiepine-1.1-dioxide (4R, 5R) To a solution of 47 g (99 mmol) of enantiomeric- enriched (dimethylamino) tetrahydrobenzothiepine-1, 1-dioxide (obtained from Step 9) in 500 mL of methylene chloride at -10 °C was added dropwise a solution of boron tribromide (297 mL, 1M in methylene chloride, 297 mmol), and the resulting solution was stirred cold (-5 °C to 0 °C) for 1 hour or until the reaction was complete. The reaction was cooled in an acetone-dry ice bath at -10 °C, and slowly quenched with 300 mL of water. The mixture was warmed to 10 °C, and further diluted with 300 mL of saturated sodium bicarbonate solution to neutralize the mixture. The aqueous layer was separated and extracted with 300 mL of methylene chloride, and the combined extracts were washed with 200 mL of water, brine, dried over MgS04 and concentrated in vacuo. The residue was dissolved in 500 mL of ethyl acetate and stirred with 50 mL of glacial acetic acid for 30 minutes at ambient temperature. The mixture was washed twice with 200 mL of water, 200 mL of brine, dried over MgS04 and concentrated in vacuo to give the crude 4-hydroxyphenyl intermediate. The solid residue was recrystallized from methylene chloride to give 37.5 g (82%) of the desired (4R, 5R) -5- (4′ – hydoxyphenyl) -7- (dimethylamino) tetrahydrobenzothiepine-1, 1- dioxide as a white solid: *H NMR (CDC13) d 0.84-0.97 (m, 6H) , 1.1-1.5 (m, 10H) , 1.57-1.72 (m, IH) , 2.14-2.28 (m, IH) , 2.83 (s, 6H) , 3.00 (d, J = 15.3 Hz, IH) , 3.16 (d, J – 15.3 Hz, IH) , 4.11 (s, 2H) , 5.48 (s, IH) , 6.02 (d, J – 2.4 Hz, IH) , 6.55 (dd, J“ = 9, 2.4 Hz, IH) , 6.88 (d, 8 , 7 Hz , 2H) , 7.38 (d, J – 8.7 Hz, 2H) , 7.91 (d, J“ = 9 Hz, 2H) .
Step 11: Preparation of enantiomerically-enriched chlorobenzyl intermediate Treat a solution of enantiomerically-enriched (4R,5R)- 5- (4′ -hydoxypheny1) -7- (dimethylamino) tetrahydrobenzothiepine-1, 1-dioxide (5.0 g, 10.9 mmol, obtained from Step 10) in acetone (100 mL) at 25 °C under N2 with powdered 5 K2C03 (2.3 g, 16.3 mmol, 1.5 eq.) and a, a’ -dichloro-p-xylene (6.7 g, 38.1 mmol, 3.5 eq.) . Stir the resulting solution at 65 °C for about 48 hours. Cool the reaction mixture to 25 °C and concentrate to 1/5 of original volume. Dissolve the residue in EtOAc (150 mL) and wash with water (2 x 150 mL) .
10 Extract the aqueous layer with EtOAc (2 x 150 mL) and wash the combined organic extracts with saturated aqueous NaCI (2 x 150 mL. Dry the combined extracts with MgS04 and concentrate in vacuo to provide the crude product . Purification by flash chromatography (5.4 x 45 cm silica,
1525-40% EtOAc/hexane) will afford the enantiomerically- enriched chlorobenzyl intermediate .
Step 12: Preparation of enantiomerically-enriched (4R.5R)- 1- r [4- [ [4- [3 , 3-Dibutyl-7- (dimethylamino) -2,3 , 4 , 5-tetrahvdro-
Treat a solution of the enantiomerically-enriched chlorobenzyl intermediate (4.6 g, 7.7 mmol, obtained from
25 above in Step 11) in acetonitrile (100 mL) at 25 °C under N2 with diazabicyclo [2.2.2] -octane (DABCO, 0.95 g, 8.5 mmol, 1.1 eq.) and stir at 35 °C for 2 hours. Collect the precipitated solid and wash with CH3CN. Recrystallization from CH3OH/Et20 will give the desired title compound (XXVII) .
ANY ERROR, EMAIL amcrasto@gmail.com, +919323115463

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
















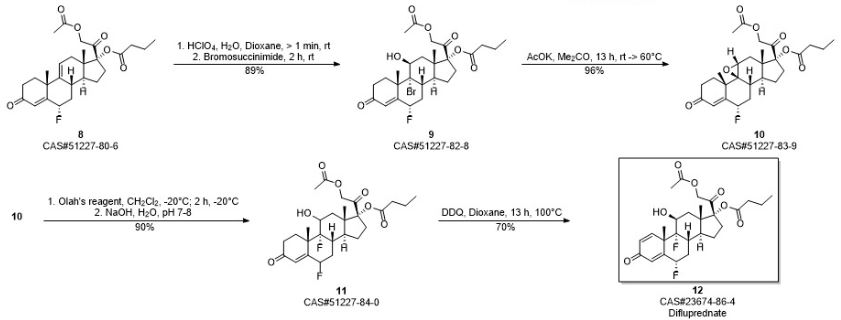




























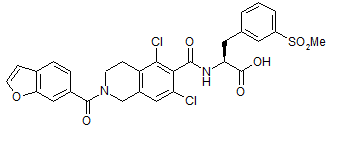

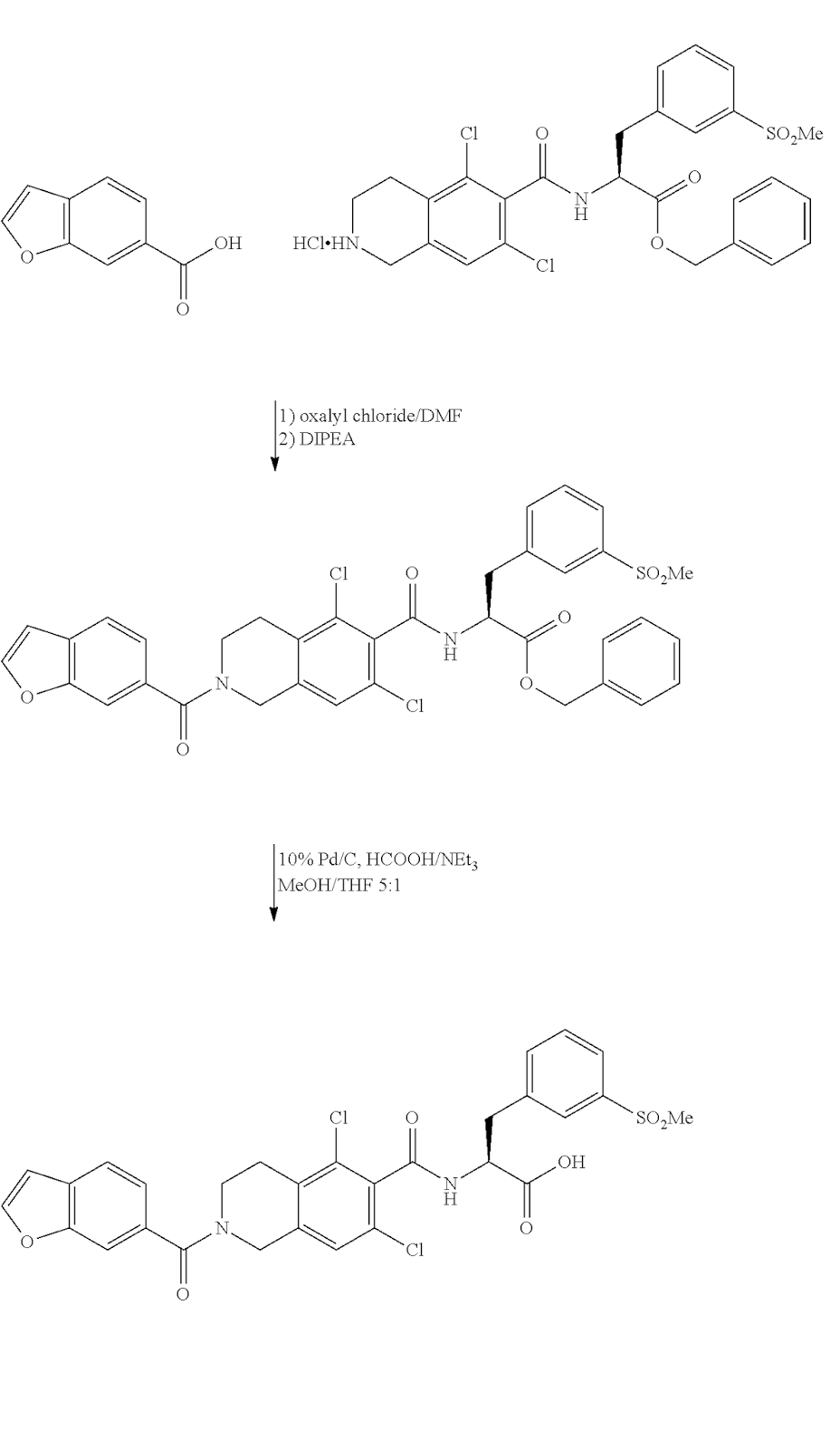
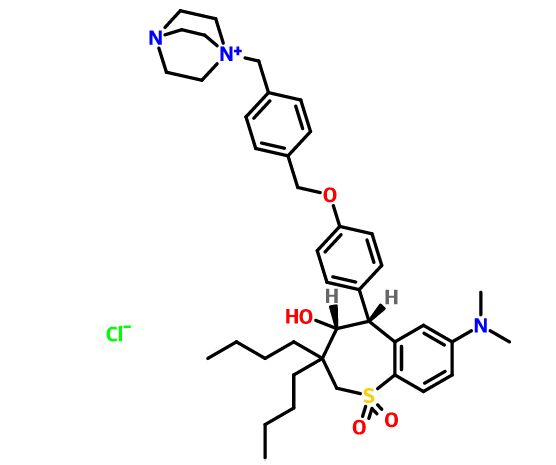


















{kind=link}
{kind=link}