


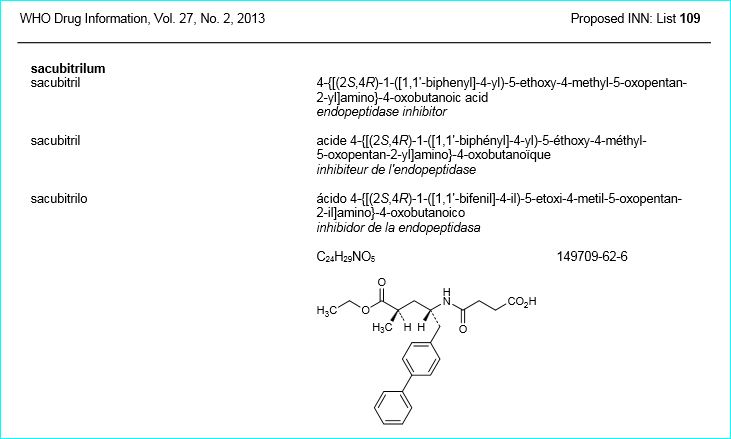
Sacubitril, AHU 377
NEPRILYSIN INHIBITOR
FOR HEART FAILURE
CAS 149709-62-6
CAS SODIUM SALT 149690-05-1
(2R,4S)-5-(biphenyl-4-yl)-4-((3-carboxypropionyl)amino)-2-methylpentanoic acid ethyl ester
5-(Biphenyl-4-yl)-4(S)-(3-carboxypropionamido)-2(R)-methylbutyric acid ethyl ester
N-(3-carboxy-1-oxopropyl)-(4S)-(p-phenylphenylmethyl)-4-amino-2R-methyl butanoic acid ethyl ester
[1,1′-Biphenyl]-4-pentanoic acid, γ-[(3-carboxy-1-oxopropyl)amino]-α-methyl-, α-ethyl ester, (αR,γS)-
- [1,1′-Biphenyl]-4-pentanoic acid, γ-[(3-carboxy-1-oxopropyl)amino]-α-methyl-, ethyl ester, [S-(R*,S*)]-
- (2R,4S)-4-[(3-Carboxy-1-oxopropyl)amino]-4-[(p-phenylphenyl)methyl]-2-methylbutanoic acid ethyl ester
- (2R,4S)-5-(Biphenyl-4-yl)-4-[(3-carboxypropionyl)amino]-2-methylpentanoic acid ethyl ester
-
Formula C24H29NO5 MW 411.49 g/mol
AHU377; AHU-377; Sacubitril; 149709-62-6; UNII-17ERJ0MKGI; Alpha-ethyl (alphaR,gammaS)-gamma-<(3-carboxy-1-oxopropyl)amino>-alpha-methyl<1,1′-biphenyl>-4-pentanoate
Sacubitril sodium
149690-05-1
Sacubitril is an antihypertensive drug used in combination with valsartan. The combination drug, valsartan/sacubitril, known during trials as LCZ696 and marketed under the brand name, Entresto, is a treatment for heart failure.[1] It was approved under the FDA’spriority review process for use in heart failure on July 7, 2015.


Mechanism of action
Sacubitril is a prodrug that is activated to LBQ657 by de-ethylation via esterases.[2] LBQ657 inhibits the enzyme neprilysin,[3] which is responsible for the degradation of atrial and brain natriuretic peptide, two blood pressure lowering peptides that work mainly by reducing blood volume.[4]


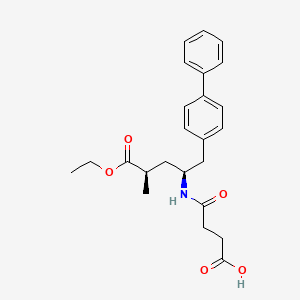
SYNTHESIS
CLICK ON IMAGE FOR CLEAR VIEW
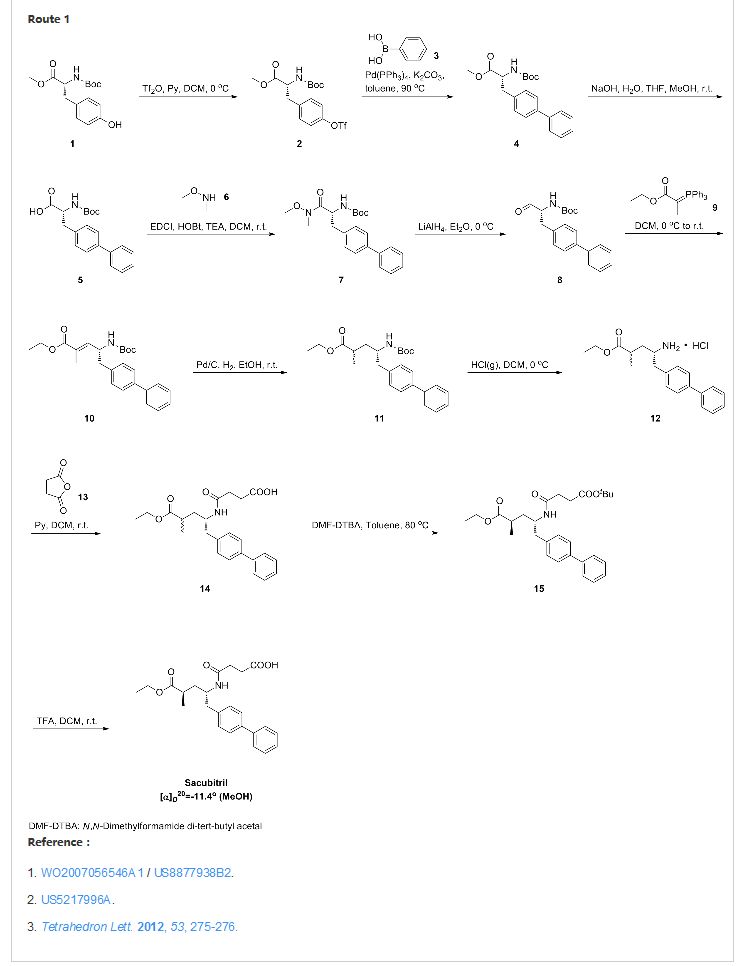
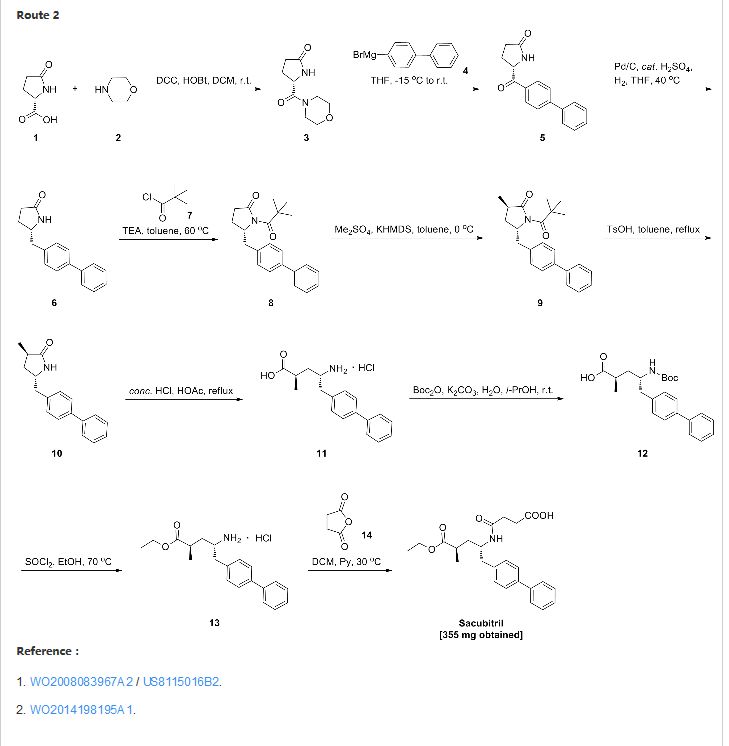
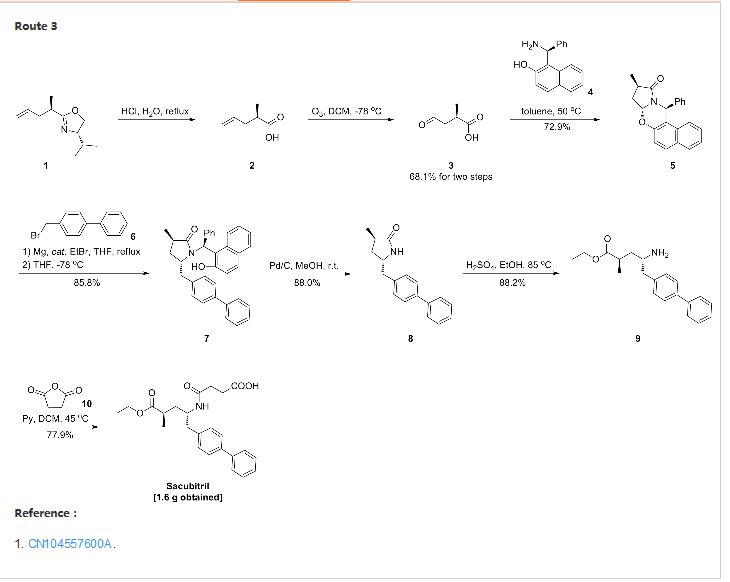
SYNTHESIS

WO-2008031567
http://www.google.com/patents/WO2008031567A1?cl=en
the following steps:


and optionally the following additional steps:


Example 1 :
(E)-(R)-5-biphenyl-4-yl-4-fert-butoxycarbonylamino-2-methylpent-2-enoic acid

(E)-(R)-5-Biphenyl-4-yl-4-tert-butoxycarbonylamino-2-methylpent-2-enoic acid ethyl ester (CAS# 149709-59-1) is hydrolysed using lithium hydroxide in ethanol to yield (£)-(f?)-5-biphenyl-4-yl-4-te/t-butoxycarbonylamino-2-methylpent-2-enoic acid as a white solid. δH (400 MHz; DMSO) 1.31 (9H1 s, (CH3J3), 1.59 (3H, s, 1- CH3), 2.68 (1H, dd, J 6.8, 13.2, 5-HA), 2.86 (1H, m, 5-HB), 4.44 (1H, m, 4-H), 6.51 (1H1 d, J 9.2, 3-H), 7.16 (1H, d, J 8.0, NH), 7.26 (2H, d, J 8.0, Ar-ortho- H(Ph)), 7.31 (1H, t, J 7.6, Ar-(Ph)-para-H), 7.40 (2H, t, J 8.0, Ar-(Ph)-metø-H), 7.54 (2H, d, J 8.0, Ar-mefa-H(Ph), 7.60 (2H, d, J 7.6, Ar-(Ph)-ort/vo-H), 12.26 (1H, s, CO2H); m/z (+ESI) 404 ([MNa]+, 17%), 382 ([MHf, 2), 326 (10), 264 (100), 167 (13).
Example 2:
(2/?,4S)-5-biphenyl-4-yl-4-fert-butoxycarbonylamino-2-methylpentanoic acid in crystalline form [2(i-a)]

2(i-a) To a suspension of (E)-(f?)-5-biphenyl-4-yl-4-te/t-butoxycarbonylamino-2- methylpent-2-enoic acid [2(ii-a)] (200 g, 524.3 mmol) in degassed ethanol (900 ml) at 40 °C a solution of diiodo(p-cymene)ruthenium(ll) dimer (0.052 g, 0.0524 mmol) and (αf?,αf?)-2,2>-bis(α-Λ/,Λ/-dimethylaminophenylmethyl)-(S,S)- 1 ,1′-bis[di(3,5-dimethyl-4-methoxyphenyl)phosphine]ferrocene (= Mandyphos SL-M004-1) (0.116 g, 0.110 mmol) is added in degassed ethanol (100 ml). The solution is degassed using vacuum and a pressure of 20 bar hydrogen applied. The mixture is stirred at 40 0C for 6 h. Vessel is then purged with nitrogen. Ethanol (700 ml) is removed by distillation, lsopropyl acetate (600 ml) is added. Solvent (600 ml) is removed by distillation, lsopropyl acetate (600 ml) is added. Solvent (600 ml) is removed by distillation, lsopropyl acetate (300 ml) is added and the solution is heated to reflux. Heptane fraction (1200 ml) is added and the mixture is cooled to room temperature. The solid is collected by filtration and washed with heptane fraction-isopropyl acetate 2 : 1 mixture (360 ml). The solid is dried overnight at 50 0C under 1-50 mbar vacuum to afford the title compound as a white/off-white solid [Ratio 2(i-a) : 2(i-b) 99 : 1, as determined by HPLC analysis]. Mpt 146-147 0C; δH (500 MHz; DMSO) 1.07 (3H1 d, J 7.0, 1-CH3), 1.34 (9H, s, (CH3)3), 1-38 (1H, m, 3-HA), 1.77 (1H, m, 3-HB), 2.43 (1H, m, 2-H), 2.70 (2H, d, J 7.0, 5-H)1 3.69 (1 H, m, 4-H), 6.74 (1 H, d, J 9.0, NH)1 7.27 (2H, d, J 8.0, Ar-ortA;o-H(Ph)), 7.36 (1H, t, J 7.0, Ar-(Ph)-para-H), 7.46 (2H, t, J 7.5, Ar-(Ph)- meta-H), 7.57 (2H, d, J 8.0, Ar-mefa-H(Ph), 7.64 (2H, d, J 7.5, Ar-(Ph)-orfΛo-H), 12.01 (1H, s, CO2H); δc (500 MHz, DMSO) 18.1 (1-CH3), 28.3 [(CH3)3], 35.9 (2- C), 37.9 (3-C), 40.7 (5-C), 50.0 (4-C), 77.4 [(C(CH3)3], 126.3, 126.5, 127.2, 128.9, 129.8 (Ar-CH), 137.7 (Ar-/pso-C(Ph)), 138.3 (Ar-para-C(Ph)), 140.1 (Ar- (Ph)-/pso-C), 155.2 (NCO), 177.2 (CO2H); mlz (+ESI) 406 ([MNa]+, 6%), 384 ([MH]+, 31 ), 328 (100), 284 (19); Found: [MH]+, 384.21691. C23H30NO4 requires MH 384.21693
PATENT
http://www.google.com/patents/EP0555175A1
Example 1….THE SODIUM SALT
To a solution of N-(3-carbo(t)butoxy-1-oxopropyl)-(4S)-(p-phenylphenylmethyl)-4-amino-2R-methylbutanoic acid ethyl ester (0.80 g) in 15 ml of CH2CI2 at room temperature are added 3 ml of trifluoroacetic acid. The mixture is stirred overnight and concentrated. The residue is dissolved in tetrahydrofuran (THF), and 6.5 ml of 1 N NaOH is added. The mixture is concentrated and triturated with ether. The solid can be recrystallized from methylene chloride-hexane to give sodium N-(3-carboxy-1-oxopropyl)-(4S)-(p-phenylphenylmethyl)-4-amino-2R-methyl butanoic acid ethyl ester melting at 159-160°C; [a]D20 = – 11.4° (methanol).
-
 SODIUM SALT
SODIUM SALT

The starting material is prepared as follows:
A solution of a-t-BOC-(R)-tyrosine methyl ester (5.9 g, 20 mmol) and pyridine (8 mL, 100 mmol) in methylene chloride (30 mL) is cooled to 0-5°C. Trifluoromethanesulfonic anhydride (4 mL, 23 mmol) is added at 0-5°C, and the resulting mixture is held for another 30 minutes. The reaction mixture is diluted with water (60 mL) and methylene chloride (100 mL), and washed sequentially with 0.5 N sodium hydroxide solution (1 x 50 mL), water (1 x 60 mL), 10% citric acid solution (2 x 75 mL) and water (1 x 60 mL). The organic phase is dried over MgS04 and concentrated to an oil. The oil is purified by column chromatography (silica gel, hexane/ethyl acetate, 2:1 to give methyl(R)-2-(t-butoxycarbonylamino)-3-[4-(trifluoromethylsulfonyloxy)phenyl]-propionate which crystallizes on standing; m.p. 46-48°C; [a]20 D-36.010 (c=1, CHCI3).
Nitrogen is passed through a suspension of (R)-2-(t-butoxycarbonylamino)-3-[4-(trifluoromethylsulfonyloxy)-phenyl]-propionate (1.75mmol), phenylboronic acid (3.5 mmol), anhydrous potassium carbonate (2.63 mmol) and toluene (17 mL) for 15 minutes. Tetrakis(triphenyiphosphine)paiiadium(0) is added, and the mixture is heated at 85-90° for 3 hours. The reaction mixture is cooled to 25°C, diluted with ethyl acetate (17 mL) and washed sequentially with saturated sodium bicarbonate (1 x 20 mL), water (1 x 20 mL), 10% citric acid (1 x 20 mL), water (1 x 20 mL) and saturated sodium chloride solution (1 x 20 mL). The organic phase is concentrated, and the residue is purified by column chromatography (silica gel, hexane/ethyl acetate 2:1) to yield methyl (R)-2-(t-butoxycarbonylamino)-3-(p-phenylphenyl)-propionate which can also be called N-(R)-t-butoxycarbonyl-(p-phenylphenyl)-alanine methyl ester.
To a solution of N-(R)-t-butoxycarbonyl-(p-phenylphenyl)-alanine methyl ester (6.8 g) in 60 ml of THF and 20 ml of methanol are added 20 ml of aqueous 1 N sodium hydroxide solution. The mixture is stirred for 1 h at room temperature and then acidified with 21 ml of 1 N hydrochloric acid. The aqueous solution is extracted 3x with ethyl acetate. The combined organic extracts are dried (MgS04), filtered and concentrated to give N-(R)-t-butoxycarbonyl-(p-phenylphenyl)-alanine, m.p. 98-99°C; [a]2°D -18.59° (c=1, methanol).
To a solution of N-(R)-t-butoxycarbonyl-(p-phenylphenyl)-alanine (4.8 g) in 70 ml of methylene chloride (CH2CI2) at 0°C with 1.65 g of N,O-dimethylhydroxylamine HCI, 1.7 g of triethylamine and 2.85 g of hydroxybenzotriazole are added 5.37 g of 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride. The mixture is stirred 17 h at room temperature. The mixture is concentrated taken up in ethyl acetate (EtOAc) and washed with saturated sodium bicarbonate, 1N HCI and brine, then dried (MgS04), filtered and concentrated to give N-(R)-t-butoxycarbonyl-(p-phenylphenyl)-alanine N,O-dimethyl hydroxylamine amide.
To a 0°C solution of N-(R)-t-butoxycarbonyl-(p-phenylphenyl)-alanine N,O-dimethyl hydroxylamine amide (5.2 g) in 250 ml of diethyl ether are added 0.64 g of lithium aluminum hydride. The reaction is stirred for 30 min. and quenched with aqueous potassium hydrogen sulfate. The mixture is stirred for additional 5 min., poured onto 1N HCI, extracted (3x) with EtOAc, dried (MgS04), filtered, and concentrated to give N-(R)-4-t-butoxycarbonyl-(p-phenylphenyl)-alanine carboxaldehyde as a colorless oil.
To a 0°C solution of N-(R)-t-butoxycarbonyl-(p-phenylphenyl)-alanine carboxaldehyde (4.4 g) in 200 ml of CH2CI2are added 10 g of carboethoxyethylidene phenyl phosphorane. The mixture is warmed to room temperature, stirred for 1 h, washed with brine, dried (MgS04), filtered and concentrated. The residue is chromatographed on silica gel eluting with (1:2) ether:hexane to give N-t-butoxycarbonyl-(4R)-(p-phenylphenylme- thyl)-4-amino-2-methyl-2-butenoic acid ethyl ester.
A solution of N-t-butoxycarbonyl-(4R)-(p-phenylphenylmethyl)-4-amino-2-methyl-2-butenoic acid ethyl ester (4.2 g) in 400 ml of ethanol is suspended with 2.0 g of 5% palladium on charcoal and then is hydrogenated at 50 psi for 6h. The catalyst is removed by filtration and the filtrate is concentrated to give N-t-butoxycarbonyl(4S)-(p-phenylphenylmethyl)-4-amino-2-methylbutanoic acid ethyl ester as a 80:20 mixture of diastereomers.
To the N-t-butoxycarbonyl(4S)-(p-phenylphenylmethyl)-4-amino-2-methylbutanoic acid ethyl ester (4.2 g) in 40 ml of CH2CI2 at 0°C is bubbled dry hydrogen chloride gas for 15 min. The mixture is stirred 2 h and concentrated to give (4S)-(p-phenylphenylmethyl)-4-amino-2-methylbutanoic acid ethyl ester hydrochloride as a 80:20 mixture of diastereomers.
To a room temperature solution of the above amine salt (3.12 g) in 15 ml of CH2CI2 and 15 ml of pyridine are added 13.5 g of succinic anhydride. The mixture is stirred for 17 h, concentrated, dissolved in ethyl acetate, washed with 1N HCI and brine, and dried (MgS04) to give N-(3-carboxy-1-oxopropyl)-(4S)-(p-phenylphenyl- methyl)-4-amino-2-methylbutanoic acid ethyl ester as a 80:20 mixture of diastereomers.
The above N-(3-carboxy-1-oxopropyl)-(4S)-(p-phenylphenylmethyl)-4-amino-2-methylbutanoic acid ethyl ester diastereomeric mixture (3.9 g) and N,N-dimethylformamide-di-t-butyl acetal (8.8 ml) are heated at 80°C in 40 ml of toluene for 2 h. The mixture is poured onto ice- 1N HCI, extracted with ether, chromatographed on silica gel eluting with (2:1) toluene:ethyl acetate to give N-(3-carbo(t)butoxy-1-oxopropyl)-(4S)-(p-phenylphe- nylmethyl)-4-amino-2R-methylbutanoic acid ethyl ester as the more polar material and the corresponding (S,S) diastereomer as the less polar material.
Example 2………THE ACID
To a solution of N-(3-carboxy-1-oxopropyl)-(4S)-(p-phenylphenylmethyl)-4-amino-(2R)-methylbutanoic acid ethyl ester (0.33 g) in 20 ml of (1:1) ethanol:tetrahydrofuran (THF) at room temperature are added 5 ml of 1 N sodium hydroxide solution (NaOH) and stirred for 17 h. The mixture is concentrated, dissolved in water and washed with ether. The aqueous layer is acidified with 1 N hydrochloric acid (HCI), extracted 3x with ethyl acetate (EtOAc), dried over magnesium sulfate (MgS04), filtered and concentrated. The residue is triturated with ether to yield N-(3-carboxy-1-oxopropyl)-(4S)-(p-phenylphenylmethyl)-4-amino-(2R)-methylbutanoic acid melting at 158-164°C, [α]D 20= -23.5° (methanol).
PATENT
CN 104557600
http://www.google.com/patents/CN104557600A?cl=zh

United States Patent US5217996 and international patent W02008031567, W02010136474 and W02012025501 reported a synthetic route follows to the chiral amino alcohols as raw materials, oxidized to the aldehyde, Victoria ladder tin reaction, chiral hydrogenation and amidation condensation reaction to obtain the objective product.

In addition, the international patent TO2008083967, TO2011088797, TO2012025502 and TO2014198195 reported that another type of preparation. The route through the 2-oxo-proline as raw material, carboxyl activating biphenyl substituted carbonyl reduction, chiral methylation, ring-opening reaction and amide condensation reaction to obtain the objective product.


Example Eight:
in the reaction flask was added (2R, 4S) -2- methyl-4-amino -5- (l, P- biphenyl-4-yl) – pentanoic acid ethyl ester (VII) (1.55g, 5mmol ), Jie of pyridine (1.2g, 15mmol) and dichloromethane burning 25mL, stirring to dissolve, butyric anhydride (1.0g, 10mmol), was heated to 4〇-45 ° C, the reaction was stirred for 6 hours. Fill Gaudin anhydride (0. 5g, 5mmol), the reaction was continued for 4 hours and the end of the reaction by TLC. Concentrated under reduced pressure, the residue was recrystallized from ethyl acetate and n-hexane to give an off-white solid sand sacubitril Kubica song (I) L 6g, a yield of 77.9%;
1H NMR (CDCl3) S 7.51 (d, 2H), 7.46 ( d, 2H), 7.36 (m, 2H), 7. 27 (m, 1H), 7. 17 (d, 2H), 5. 72 (d, 1H), 4. 19 (brs, 1H), 4. 06 (q, 2H), 2. 87-2. 72 (m, 2H), 2. 62-2. 54 (m, 2H), 2. 49 (brs, 1H), 2. 43-2. 33 ( m, 2H), I 88 (m 1H), I 54-1 43 (m, 1H), I. 18 (t, 3H), l 10 (d, 3H);…..
FAB-MSm / z : 412 [M + H] +.
PATENT
http://www.google.com/patents/WO2014198195A1?cl=en
Example 7
(2 Standby

Acetyl chloride (1 mL) 0 ° C was added ethanol (10 mL), and at room temperature for 0.5 hours, the compound (3R, 5S) -5- biphenyl-4-methyl-1- (2,2- methyl-propionyl) -3-methyl pyrrolidone (520 mg, 1.49 mmol), the reaction was refluxed for 3 days. After cooling to room temperature, and concentrated. The reaction mixture was dissolved in 8 mL of dichloromethane and pyridine 1: 1 mixed solution, and then butyryl anhydride (223 mg, 2.23 mmol). 30 ° C overnight. LC-MS detection, a small amount of starting material remaining, fill Gading anhydride (75 mg, 0.74 mmol), continue to reflect four hours. Concentrated and reverse phase column chromatography to give a white foam solid (2R, 4S) -5- biphenyl-4-yl-4- (3-carboxy – propionylamino) -2-methyl – acetic acid ester a (355 mg, 58%) and white solid (2R, 4S) -5- biphenyl-4-yl-4- (3-carboxy – propionylamino) -2-methyl – pentanoic acid b ( 13 mg, 2.3%).
a: 1H MR (400 MHz, CDCl 3 ) [delta] 7.51 (d, = 7.8 Hz, 2H), 7.46 (d, = 7.8 Hz, 2H), 7.36 (t, J = 7.6 Hz, 2H), 7.27 (t, J = 7.2 Hz, IH), 7.17 (d, J = 7.9 Hz, 2H), 5.72 (d, J = 8.1 Hz, IH), 4.19 (brs, IH), 4.06 (q, J = 7.0 Hz, 2H) , 2.87-2.72 (m, 2H), 2.62-2.54 (m, 2H), 2.49 (brs, IH), 2.43-2.33 (m, 2H), 1.88 (ddd, = 13.2, 9.5, 3.9 Hz, IH), 1.54-1.43 (m, IH), 1.18 (t, = 7.0 Hz, 3H), 1.10 (d, = 7.2 Hz, 3H).
LC-MS: t R = 3.43 min; [M + H] +: 412.0.
……………………
Paper
JOURNAL OF MEDICINAL CHEMISTRY, vol. 38, no. 10, 1995, pages 1689-1700,
http://pubs.acs.org/doi/pdf/10.1021/jm00010a014
NOTE———–DIACID
(aR,yS)-y-[ (3-Carbo-1-oxopropyl)aminol-a-methyl- [l,l’-biphenyllpentanoic Acid (21a).
To the sodium salt of 19a (0.73 g, 1.68 mM) in 20 mL of THF:EtOH was added 1 N NaOH (5.0 mL, 5.0 “01). The reaction mixture was stirred overnight and then washed with ether. The aqueous layer was acidified with 1 N HCI, re-extracted with EtOAc (3 x 10 mL), dried (MgSO& and evaporated to dryness. The solid was recrystallized from ethanol to yield 435 mg of 21a DIACID OF SACUBITRIL
melting at 165-167 “C:
[a] D25~ -28.73 (c = 10.1 in MeOH);
‘H NMR, DMSOD6
PPM 12.0, (s, 2H), 7.75 (d, 1H), 7.62 (d, 2H), 7.55 (d, 2H), 7.45 (t, 2H), 7.32 (t, lH), 7.25 (d, 2H), 4.92 (m, lH), 2.70 (d, 2H), 2.35 (t, 3H), 2.25 (m, 2H), 1.75 (m, lH), 1.32 (m, lH), 1.03 (d, 3H).
Anal. (C22H25N05) C,H,N
Note diacid is sacubitrilat (LBQ657)
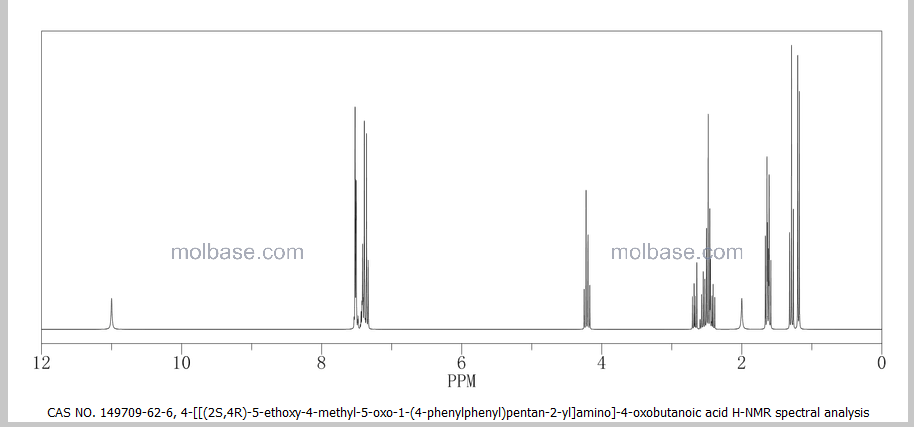
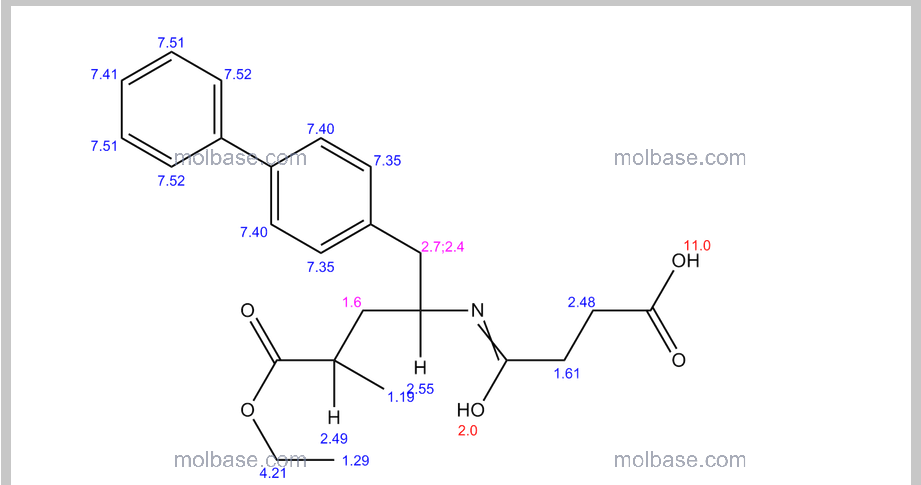
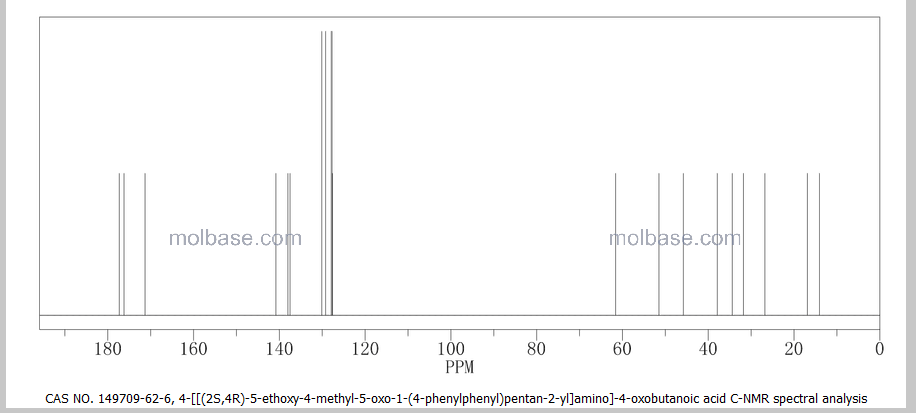
NMR PREDICT
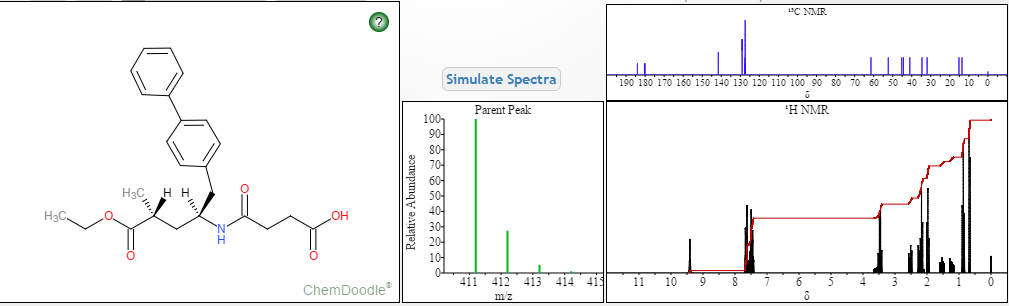
1H NMR PREDICT
13C NMR PREDICT
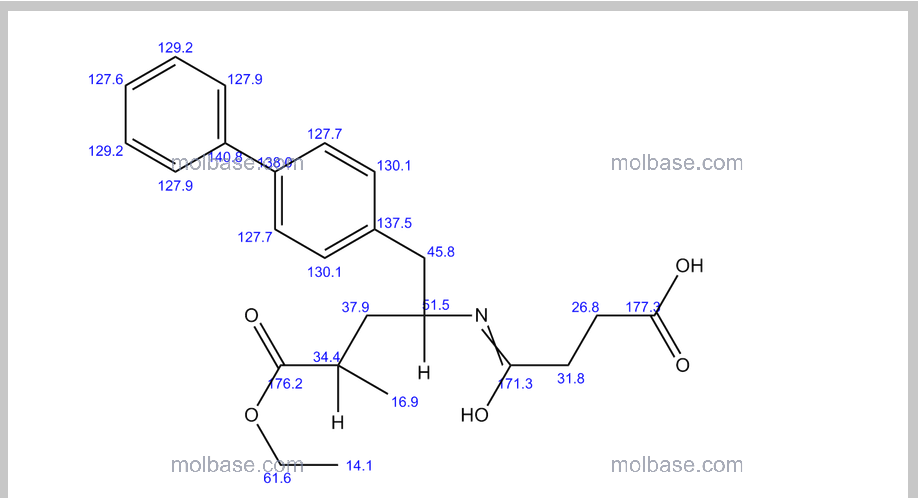
COSY PREDICT
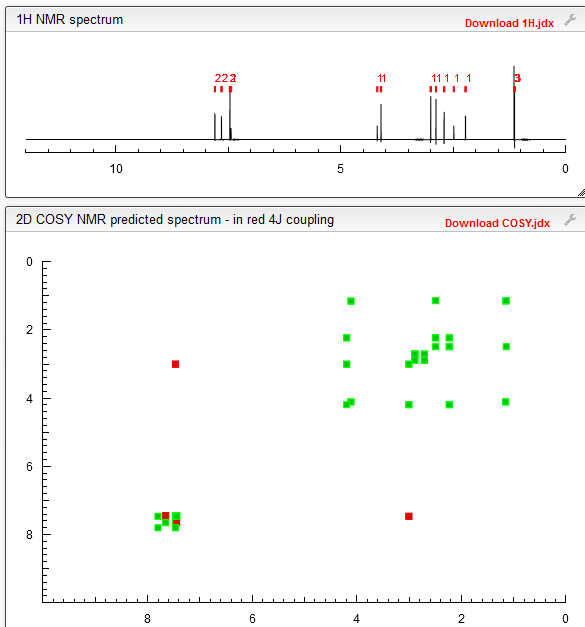
…………….

NMR…..http://www.chemietek.com/Files/Line3/CHEMIETEK,%20AHU-377%20,%20Lot%2001,%20NMR-MeOD,%201.1.pdf
- Mol. Formula:C24H29NO5 ∙ C4H11NO3
- MW:532.6
- HPLC………http://www.chemietek.com/Files/Line2/CHEMIETEK,%20AHU-377%20,%20Lot%2001,%20HPLC.pdf
update………
PATENT
WO2016180275
- John J.V. McMurray, Milton Packer, Akshay S. Desai, et al. for the PARADIGM-HF Investigators and Committees (August 30, 2014).“Angiotensin–Neprilysin Inhibition versus Enalapril in Heart Failure”. N Eng J Med 371. doi:10.1056/NEJMoa1409077.
- Solomon, SD. “HFpEF in the Future: New Diagnostic Techniques and Treatments in the Pipeline”. Boston. p. 48. Retrieved 2012-01-26.
- Gu, J.; Noe, A.; Chandra, P.; Al-Fayoumi, S.; Ligueros-Saylan, M.; Sarangapani, R.; Maahs, S.; Ksander, G.; Rigel, D. F.; Jeng, A. Y.; Lin, T. H.; Zheng, W.; Dole, W. P. (2009). “Pharmacokinetics and Pharmacodynamics of LCZ696, a Novel Dual-Acting Angiotensin Receptor-Neprilysin Inhibitor (ARNi)”. The Journal of Clinical Pharmacology 50 (4): 401–414. doi:10.1177/0091270009343932.PMID 19934029.
- Schubert-Zsilavecz, M; Wurglics, M. “Neue Arzneimittel 2010/2011.” (in German)


| WO2004085378A1 * | Mar 15, 2004 | Oct 7, 2004 | Joseph D Armstrong Iii | Process for the preparation of chiral beta amino acid derivatives by asymmetric hydrogenation |
| WO2006057904A1 * | Nov 18, 2005 | Jun 1, 2006 | Merck & Co Inc | Stereoselective preparation of 4-aryl piperidine amides by asymmetric hydrogenation of a prochiral enamide and intermediates of this process |
| WO2006069617A1 * | Dec 5, 2005 | Jul 6, 2006 | Dsm Fine Chem Austria Gmbh | Process for transition metal-catalyzed asymmetric hydrogenation of acrylic acid derivatives, and a novel catalyst system for asymmetric transition metal catalysis |
| US5217996 * | Jan 22, 1992 | Jun 8, 1993 | Ciba-Geigy Corporation | Biaryl substituted 4-amino-butyric acid amides |
| Reference | ||
|---|---|---|
| 1 | * | KSANDER, GARY M. ET AL: “Dicarboxylic Acid Dipeptide Neutral Endopeptidase Inhibitors” JOURNAL OF MEDICINAL CHEMISTRY, vol. 38, no. 10, 1995, pages 1689-1700, XP002340280 cited in the application |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
4-{[(2S,4R)-1-(4-Biphenylyl)-5-ethoxy-4-methyl-5-oxo-2-pentanyl]amino}-4-oxobutanoic acid
|
|
| Clinical data | |
| Legal status |
|
| Identifiers | |
| CAS Registry Number | 149709-62-6 |
| ATC code | None |
| PubChem | CID: 9811834 |
| ChemSpider | 7987587 |
| Synonyms | AHU-377; AHU377 |
| Chemical data | |
| Formula | C24H29NO5 |
| Molecular mass | 411.49 g/mol |
|
|
| Systematic (IUPAC) name | |
|---|---|
|
4-{[(2S,4R)-1-(4-Biphenylyl)-5-ethoxy-4-methyl-5-oxo-2-pentanyl]amino}-4-oxobutanoic acid
|
|
| Identifiers | |
| CAS Number | 149709-62-6 |
| ATC code | None |
| PubChem | CID: 9811834 |
| ChemSpider | 7987587 |
| Synonyms | AHU-377; AHU377 |
| Chemical data | |
| Formula | C24H29NO5 |
| Molecular mass | 411.49 g/mol |
Date: 1 September 2016 at 15:16
Subject: LCZ696 (SACUBITRIL+VALSARTAN)/ Changzhou Comwin Fine Chemicals Co,. Ltd
To: amcrasto <amcrasto@gmail.com>
Dear SirHow do you do! Sincerely hope my email will bring you more LCZ696 (SACUBITRIL+VALSARTAN) possibilities.I am Wang Zhuo of ComWin from China, and in charge of LCZ696 (SACUBITRIL+VALSARTAN) global marketing.
LCZ696 (sacubitril/Valsartan) is a combination drug for use in heart failure developed by Novartis. It consists of valsartan and sacubitril, in a 1:1 mixture by molecule count. It was approved by US FDA in July 2015.
Sacubitril (Hemicalcium) is a neprilysin inhibitor, We have developed this project since 2nd half of 2014. At present, some intermediates are in commercial scale, and some are in pilot production.
We will put our most focus on this project from 2nd half of this year. Certainly we will file DMF for Sacubitril and make submission to regulartory market. We have sold Sacbutitril to some EU customers for evaluation purpose, such as Teva, Chemo. Also, we are doing the development of LCZ696 (the final API). It’s co-crystallized valsartan and sacubitril, in a one-to one molar ratio. One LCZ696 complex consists of six valsartan anions, six sacubitril anions, 18 sodium cations, and 15 molecules of water. Now we have the sample of LCZ696 in kilogram grade.
Best Regards
Wang Zhuo
Sales Executive
Changzhou ComWin Fine Chemicals Co.,Ltd.
24th Floor, Jiaye International Commercial Plaza
99 Yanling West Road, Changzhou
Jiangsu 213003 China
Tel: 0086 519 8663 2882
Fax: 0086 519 8661 3190
email: wang.zhuo@comwin-china.com
www.comwin-china.com


सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये। औकात बस इतनी देना, कि औरों का भला हो जाये।
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE




 LIONEL MY SON
LIONEL MY SON
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
////// antihypertensive drug, Heart Failure, Sacubitril, AHU 377
CCOC(=O)[C@H](C)C[C@@H](Cc1ccc(cc1)c2ccccc2)NC(=O)CCC(=O)O
CCOC(=O)[C@H](C)C[C@@H](CC1=CC=C(C=C1)C2=CC=CC=C2)NC(=O)CCC(=O)O

