




| Patent | Submitted | Granted |
|---|---|---|
| ORGANIC COMPOUNDS [US2009156585] | 2009-06-18 | |
| METHODS OF TREATMENT AND PHARMACEUTICAL COMPOSITION [US8101659] | 2008-10-23 | 2012-01-24 |
| Substituted Aminobutyric Derivatives as Neprilysin Inhibitors [US2010305145] | 2010-12-02 | |
| PROCESS FOR PREPARING BIARYL SUBSTITUTED 4-AMINO-BUTYRIC ACID OR DERIVATIVES THEREOF AND THEIR USE IN THE PRODUCTION OF NEP INHIBITORS [US2009326066] | 2009-12-31 | |
| Process for preparing 5-biphenyl-4-amino-2-methyl pentanoic acid [US8115016] | 2010-05-06 | 2012-02-14 |
| Methods of treatment and pharmaceutical composition [US7468390] | 2003-07-31 | 2008-12-23 |
| Process for Preparing 5-biphenyl-4-amino-2-methyl Pentanoic Acid [US2014249320] | 2014-03-25 | 2014-09-04 |
| Substituted Aminobutyric Derivatives as Neprilysin Inhibitors [US2012252830] | 2012-06-07 | 2012-10-04 |
| Process for preparing 5-biphenyl-4-amino-2-methyl pentanoic acid [US8716495] | 2011-12-21 | 2014-05-06 |
Home » Posts tagged 'Heart Failure'
Tag Archives: Heart Failure
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Zamicastat
CAS 1080028-80-3 BASE
1383828-47-4 OF HCL SALT
C21 H21 F2 N3 O S BASE
2H-Imidazole-2-thione, 1-[(3R)-6,8-difluoro-3,4-dihydro-2H– 1-benzopyran-3-yl]-1,3-dihydro-5-[2-[(phenylmethyl)amino] ethyl] -(R)-5-(2-(Benzylamino)ethyl)-1-(6,8-difluorochroman-3-yl)-1H-imidazole-2(3H)-thione
(R)-5-(2-(Benzylamino)ethyl)-1-(6,8-difluorochroman-3-yl)-1H-imidazole-2(3H)-thione
Molecular Weight, 401.47 BASE

- BIAL – PORTELA & CA., S.A. [PT/PT]; À Avenida da Siderurgia Nacional P-4745-457 S. Mamede do Coronado (PT)

- Zamicastat is a dopamine beta-monooxygenase inhibitor in phase I clinical studies at BIAL for the treatment of hypertension and heart failure.
- Zamicastat is a potent and selective dopamine β-mono-oxygenase inhibitor. Zamicastat Prevents the Deterioration of Cardiometabolic and Inflammatory Biomarkers in a Genetic Model of Salt-sensitive Hypertension. Chronic high salt intake deteriorates several cardiometabolic and inflammatory biomarkers in Dahl/SS rats, which can be prevented by dopamine β-hydroxylase inhibition with zamicastat.
- crystalline forms of l-[(3R)-6,8-difluoro- 3,4-dihydro-2H-l-benzopyran-3-yl]-l,3-dihydro-5-[2-[(phenylmethyl)amino]ethyl]-2H- imidazole-2-thione, i.e. the Renantiomer of

and processes for preparing the same. Background and prior art:Interest in the development of inhibitors of dopamines-hydroxylase (ϋβΗ) has centred on the hypothesis that inhibition of this enzyme may provide significant clinical improvements in patients suffering from cardiovascular disorders such as hypertension or chronic heart failure. The rationale for the use of ϋβΗ inhibitors is based on their capacity to inhibit the biosynthesis of noradrenaline, which is achieved via enzymatic hydroxylation of dopamine. Activation of neurohumoral systems, chiefly the sympathetic nervous system, is the principal clinical manifestation of congestive heart failure (Parmley, W.W., Clinical Cardiology, 18: 440-445, 1995). Congestive heart failure patients have elevated concentrations of plasma noradrenaline (Levine, T.B. et al., Am. J. Cardiol., 49: 1659-1666, 1982), increased central sympathetic outflow (Leimbach, W.N. et al., Circulation, 73: 913- 919, 1986) and augmented cardiorenal noradrenaline spillover (Hasking, G.J. et al., Circulation, 73:615-621, 1966). Prolonged and excessive exposure of the myocardium to noradrenaline may lead to down-regulation of cardiac β] -adrenoceptors, remodelling of the left ventricle, arrhythmias and necrosis, all of which can diminish the functional integrity of the heart. Congestive heart failure patients who have high plasma concentrations of noradrenaline also have the most unfavourable long-term prognosis (Cohn, J.N. et al., N. Engl. J. Med., 311 :819-823, 1984). Of greater significance is the observation that plasma noradrenaline concentrations are already elevated in asymptomatic patients with no overt heart failure and can predict ensuing mortality and morbidity (Benedict, C.R. et al., Circulation, 94:690-697, 1996). An activated sympathetic drive is not therefore merely a clinical marker of congestive heart failure, but may contribute to progressive worsening of the disease.
Potent dopamines-hydroxylase inhibitors having high potency and significantly reduced brain access are disclosed in WO 2008/136695. WO 2008/136695 describes compounds of formula I:

I where Rls R2 and R3 are the same or different and signify hydrogens, halogens, alkyl, nitro, amino, alkylcarbonylamino, alkylamino or dialkylamino group; R4 signifies -alkylaryl or – alkylheteroaryl; X signifies CH2, oxygen atom or sulphur atom; n is 2 or 3; including the individual (R)- and (S)-enantiomers or mixtures of enantiomers thereof; and including pharmaceutically acceptable salts and esters thereof, wherein the term alkyl means hydrocarbon chains, straight or branched, containing from one to six carbon atoms, optionally substituted by aryl, alkoxy, halogen, alkoxycarbonyl or hydroxycarbonyl groups; the term aryl means a phenyl or naphthyl group, optionally substituted by alkyl, alkyloxy, halogen or nitro group; the term halogen means fluorine, chlorine, bromine or iodine; the term heteroaryl means heteroaromatic group. In particular, WO 2008/136695 describes l-[(3R)-6,8-difluoro-3,4-dihydro-2H-l-benzopyran-3-yl]-l,3-dihydro-5-[2- [(phenylmethyl)amino]ethyl]-2H-Imidazole-2-thione.
Processes for the preparation of compounds of formula I, and in particular l-[(3R)-6,8- difluoro-3,4-dihydro-2H-l-benzopyran-3-yl]-l,3-dihydro-5-[2-[(phenylmethyl)amino] ethyl] -2H-Imidazole-2-thione, are described in WO 2008/136695 and are incorporated by reference herein. It is known that polymorphic forms of the same drug may have substantially different pharmaceutically important properties such as dissolution characteristics and bioavailability as well as stability of the drug. Furthermore, different forms may have different particle size, hardness and glass transition temperature. Thus, one form may provide significant advantages over other forms of the same drug in solid dosage form manufacture processes, such as accurate measurement of the active ingredients, easier filtration, or improved stability during granulation or storage. Furthermore, a particular process suitable for one form may also provide drug manufacturers several advantages such as economically or environmentally suitable solvents or processes, or higher purity or yield of the desired product.
PATENT
http://www.google.com/patents/WO2012087174A2?cl=en
Preparation of compound 2
[0090] Six lots of compound 2 (designated as lots 1, 2, 3, 4, 5 and 6) were prepared. The starting materials were prepared according to the following experimental protocols.
Lot 1 (Form A)
To a suspension of (R)-5-(2-aminoethyl)-l-(6,8-difluorochroman-3-yl)-lH- imidazole-2(3H)-thione (6.23 g, 20 mmol) in a mixture of Dichloromethane (DCM – 40 ml) and Methanol (40.0 ml) was added BENZALDEHYDE (2.230 ml, 22.00 mmol). To the resulting clear solution SODIUM CYANOBOROHYDRIDE (1.9 g, 28.7 mmol) was added in portions at 20-25°C to avoid intensive foaming and the solution was stirred at 20- 25°C for 40 h. The solution was quenched at 20-25°C with IN HC1 (35 ml), neutralised with 3N NaOH (35 ml), the mixture was extracted with DCM (200 ml). The organic phase was washed with brine, dried (MgS04), evaporated to dryness. The oily residue crystallised from 2-propanol (40 ml) at 20-25°C over a week-end. The crystals were collected, washed with 2-propanol, dried to give 5.2 g of the crude product. Re- crystallisation from 2-propanol-DCM hasn’t removed all impurities. Everything collected, evaporated with silica, applied on a column, eluted with Ethyl Acetate (EA)->EA-MeOH 9:1->4: 1, fractions 8-25 collected to give 3.8 g. Re-crystallised from 2-propanol (45 ml) and DCM (120 ml, removed on a rotavap) to give 2.77 g => initial lot (a) (HPLC 98.3% area) and 0.3 g of undissolved filtered off, by TLC right product. Initial lot (a) re- crystallised from 2-propanol (35 ml) and DCM (95 ml, removed on a rotavap) to give 2.51 g => initial lot (b) (HPLC 98.3% area). Combined with the above undissolved, re- crystallised from acetonitrile (200 ml, reflux to ice bath) to give 2.57 g => initial lot (c) (HPLC 98.8% area). Re-crystallised from acetonitrile (180 ml, reflux to 15°C) to give 2.25 g => Lot 1 (HPLC 99.2% area), mp 190-92°C. Lot 2 (Form A)
[0092] (R)-5-(2-(benzylamino)ethyl)-l-(6,8-difluorochroman-3-yl)-lH-imidazole- 2(3H)-thione (12 g, 29.9 mmol) was dissolved with heating to reflux in Tetrahydrofuran (300 ml), the solution was cooled to 5-10°C, Water (510 ml) was added slowly (approx 10 min) with stirring. The mixture was stirred for 1 h, solid was collected, washed with water, dried to give 11.73 g of product, by HPLC 1% of (R)-5-(2-Aminoethyl)-l-(6,8- difluorochroman-3-yl)-l,3-dihydroimidazole-2-thione hydrochloride and 1% of less polar impurity. The product was dissolved in Tetrahydrofuran (300 ml) with heating to reflux, 2- Propanol (150 ml) was added, the solution was concentrated to approx 100 ml (crystallisation occured), stirred in ice for 1.5 h. Solid was collected, washed with 2- propanol, dried to give 11.2 g of product, by HPLC 0.8% of (R)-5-(2-aminoethyl)-l-(6,8- difluorochroman-3-yl)-lH-imidazole-2(3H)-thione hydrochloride and 0.5% of less polar impurity. The product was dissolved in Tetrahydrofuran (300 ml) with heating to reflux, 2- Propanol (150 ml) was added, the solution was concentrated to approx 100 ml (crystallisation occured), stirred at 20-25°C for 1 h. Solid was collected, washed with 2- propanol, dried to give (R)-5-(2-(benzylamino)ethyl)-l-(6,8-difluorochroman-3-yl)-lH- imidazole-2(3H)-thione (10.22 g, 25.5 mmol, 85 % yield).,
Lot 3 (form B)
To (R)-5-(2-aminoethyl)-l-(6,8-difluorochroman-3-yl)-lH-imidazole-2(3H)- thione (2.36 g, 7.58 mmol) in a mixture of Methanol (15.00 ml) and Dichloromethane (15 ml) was added BENZALDEHYDE (0.845 ml, 8.34 mmol). To the resulting clear solution SODIUM CYANOBOROHYDRIDE (0.702 g, 10.61 mmol) was added in portions at 20- 25°C to avoid intensive foaming and the solution was stirred at 20-25°C for 40 h. The solution was quenched at 20-25°C with IN HC1 (12 ml), neutralised with 3N NaOH (12 ml), the mixture was extracted with DCM (100 ml). The organic phase was washed with brine, dried (MgS04), evaporated to dryness. The residue was purified on a column with EA-MeOH 9: 1 as eluent, fractions collected, concentrated to approx 20 ml, cooled in ice. The precipitate collected, washed with Ethyl Acetate-Petroleum Ether 1 : 1, dried on air to give (R)-5-(2-(benzylamino)ethyl)-l-(6,8-difluorochroman-3-yl)-lH-imidazole-2(3H)- thione (1.55 g, 3.86 mmol, 50.9 % yield). Lot 4 (Form A)
To a 500 mL flask set up for atmospheric distillation was added (R)-5-(2- (benzylamino)ethyl)-l-(6,8-difluorochroman-3-yl)-lH-imidazole-2(3H)-thione (20 g, 49,8 mmol) and Tetrahydrofuran (400 ml) to afford a suspension. The suspension was heated until full dissolution was achieved (61°C) whereupon it was filtered. The resulting solution was then heated to 66°C in order to commence the distillation. A mixture of Water (125 ml) & 2-Propanol (125 ml) was added at the same rate as the distillate was collected. The distillation was continued until 400 mL of distillate was collected. Crystallisation commenced after ~320 mL of distillate was collected. The suspension was cooled to 20°C and aged for 45 min. before filtering and washing with additional 2- propanol (80 mL) and then dried under vacuum at 50°C overnight to give (R)-5-(2- (benzylamino)ethyl)-l-(6,8-difluorochroman-3-yl)-lH-imidazole-2(3H)-thione (18.79 g, 94%). Lot 5 (Form A)
To a mixture of Methanol (66 L) and Water (10 L) at 20°C was added purified (R)-5-(2-(benzylamino)ethyl)-l-(6,8-difluorochroman-3-yl)-lH-imidazole-2(3H)-thione hydrochloride (4.37 kg, 9.98 mol) to afford a suspension. The reaction mixture was then heated to 67°C to affect complete dissolution, whereupon IN Sodium hydroxide (10.48 Ls 10.48 mol, 1.05 eq) was added in a single portion. The reaction mixture was adjusted back to 67°C and held at 67°C for 30 min. The reaction mixture was then cooled to 20°C and aged at 20°C for at least 30 min. The reaction was then filtered and the filter cake washed with aqueous Methanol (1 : 1 v/v, 20 L), sucked down for 15 min. and then dried at 45°C under vacuum, to afford (R)-5-(2-(benzylamino)ethyl)-l-(6,8-difluorochroman-3-yl)-lH- imidazole-2(3H)-thione (3.855 kg, 96%) as a pale tan crystalline solid.
PATENT
WO 2015038022
http://www.google.com/patents/WO2015038022A1?cl=en
processes .
(J?) -5- (2-Aminoethyl) -1- (6, 8-difluorochroman-3-yl) -1, 3-dihydroimidazole-2 -thione hydrochloride (the compound of formula 1, below) is a potent, non-toxic and peripherally selective inhibitor of ϋβΗ, which can be used for treatment of certain cardiovascular disorders. Compound 1 is disclosed in WO2004/033447 , along with processes for its preparation.

1
The process disclosed in WO2004/033447 involves the reaction of ( R) – 6 , 8 -difluorochroman-3 -ylamine hydrochloride (the structure of ( R) -6, 8-difluorochroman-3 -ylamine is shown below as compound QA) , [4 – ( tert-butyldimethylsilanyloxy) -3 -oxobutyl] carbamic acid tert-butyl ester and potassium thiocyanate .

QA
(R) -6 , 8-difluorochroman- 3 -ylamine (compound QA) is a key intermediate in the synthesis of compound 1. The stereochemistry at the carbon atom to which the amine is attached gives rise to the stereochemistry of compound 1, so it is advantageous that compound QA is present in as pure enantiomeric form as possible. In other words, the (R) -enantiomer of compound QA should be in predominance, with little or no (S) enantiomer present. Thus, the process for preparing compound QA will advantageously produce compound QA with as high enantiomeric excess (ee) as possible.
Advantageous processes for preparing, for example, the compound of formula QA have now been found. In one aspect, the processes involve a biotransformation step. In another aspect, the processes involve chemical transformation. The processes may also be employed in the preparation of similar precursors useful in the production of other peripherally-selective inhibitors of dopamine -β -hydroxylase .
WO2008/136695 discloses a compound of formula YA, its (R) or (S) enantiomer, a mixture of its (R) and (S) enantiomers, or pharmaceutically acceptable salts thereof.

YA
The (R) -enantiomer of the compound of formula YA has been found to be a potent dopamines-hydroxylase inhibitor having high potency and significantly reduced brain access.
As disclosed in WO2008/136695 , the compound of formula YA may be prepared by reacting the compound of formula 1 with benzaldehyde under reductive alkylation conditions. In particular, (R) -5- (2 -aminoethyl ) -1- (6 , 8-difluorochroman-3 -yl) – 1 , 3 -dihydroimidazole-2 -thione and benzaldehyde may be reacted in the presence of a solvent or mixture of solvents, and a reducing agent such as sodium cyanoborohydride or sodium triacetoxyborohydride .
process comprises the following steps:

The route from 2 , 4-difluorophenol may be as described 9/064210.
Preferably, the reagents and conditions are:
(i) H2S04, acetic acid
(ii) NaOCl, MeOH/water
(iii) Ru-based catalyst, H2, 30 bars, MeOH
(iv) aqueous KOH, MeOH, L-tartaric acid
(v) KSCN, AcOH/lPA
(vi) NaBH4, BF3.THF complex, THF then IPA
n one aspect, the process comprises the following steps


i. KOH, Thioglycolic acid or cysteine
ii. MEK
According to an aspect of the present invention, there is provided the following 2 -part synthetic route from the starting material 2 , 4 -difluorophenol to (R) -5- (2 -aminoethyl ) -1- (6 , 8-difluorochroman-3 -yl) -1 , 3 -dihydroimidazole-2 – thione
hydrochloride :
Part (1)


Preferred reagents and conditions:
a) HMTA, CF3COOH, 115°C, 18 hours
b) CH2CHCN, DABCO, DMF, water, 70°C, 16 hours
c) H2S04, AcOH, 100°C, 1 hour
d) NaClO, NaOH, MeOH, 25°C, 24 hours
e) (R) -C3 -TunePhosRu (acac) 2 S/C 3000, 30 bar H2, MeOH, 80°C, 20 hours
f) Water, 2-propanol, reflux to 20°C
g) 40% KOH, MeOH, reflux, 24 hours
h) L-tartaric acid, ethanol, water, RT, 1 hour
Part (2)

![]()

Preferred reagents and conditions
a’) methyl vinyl ketone, t-BuONa, EtOAc, EtOH, 40-50°C, 2-3 hours
Br2, MeOH, 20-25°C, 5 hours
water, reflux, 1 hour
KOH, AcOH, reflux, 1 hour
HCl, water, 2-propanol, 75 °C, 4 hours
KSCN, AcOH, 100°C, 2-4 hours
NaHC03, water, EtOH
NaBH4, 2-propanol, THF, water, 20-25°C, 16 hours
HCl, 2-propanol, water, reflux, 1-2 hours
The ( R ) -5- (2-Aminoethyl) -1- (6, 8-difluorochroman-3 -yl) -1,3-dihydroimidazole-2 – thione hydrochloride may then be used to
prepare (R) -5- (2- (benzylamino) ethyl) -1- (6, 8-difluorochroman-3 -yl) -lH-imidazole-2 (3H) -thione as follows.

Preferred reaction conditions/reagents:
q) NaBH(OAc)3, PhCHO, IPA;
t) NaOH, MeOH , H20
Either r) and s) :
r) HCI aq;
s) MeOH/Toluene;
Or n) , o) and p) :
n) HCI aq;
o) MeOH, toluene;
p) IPA.
EXAMPLES
Example 1
Nitro chromene synthesis

To 3 , 5-difluoro-2-hydroxybenzaldehyde (lOg, 63mmol, leq) , di-n-butylamine (4.1g, 32mmol, 0.5eq) , phtalic anhydride (18.7g, 126mmol, 2eq) in toluene (500mL) was added nitroethanol (5.75g, 63mmol, leq) . The round bottomed flask fitted with a dean stark apparatus was refluxed for 18h. The mixture was cooled and nitroethanol (5.75g, 63mmol, leq) was added. The resulting reaction mixture was then reflux for 12h. After cooling, the solution was evaporated down to approximately 150mL and purified over silica gel (eluent ethyl acetate : hexane 1:1) this gave several fractions that contained only the product by TLC, these was evaporated under reduced pressure to yield 1.8g which was 100% pure by HPLC aera. Several more fractions were collected containing a mixture of product and starting material. These were combined and washed with 2% NaOH solution (2x50mL) to remove starting material. The organic layer was washed with water (50mL) , dried over sodium sulfate and evaporated under reduced pressure to give 2.49g of brown solid ( 100% pure by HPLC aera) . More fractions were collected. These were combined, washed with 2% NaOH solution (3xl00mL) , water (lOOmL) and dried over sodium sulfate. This was then filtered and evaporated down in vacuum to yield 6.14g of a brown solid which was 91.3% pure by HPLC aera. 6 , 8 -difluoro-3 -nitro-2H-chromene (9.90g, 73.4%) was obtained as a brown solid.
Example 2
Nitro chromene synthesis with column purification
To a solution of isobenzofuran-1 , 3 -dione (4,68 g, 31,6 mmol) , 3 , 5-difluoro-2 -hydroxybenzaldehyde (2,5 g, 15,81 mmol) in Toluene (25 ml) was added 2 -nitroethanol (2,88 g, 31,6 mmol). The resulting mixture was heated to reflux overnight (Dean stark) .
The reaction conversion was checked by TLC (eluent PE/EtOAc 9:1) . A yellow spot was observed and corresponds to the expected product .
Reaction was cooled to room temperature and a plug of silica gel was performed. A pale brown solid (3.9g) was obtained. “””H-NMR showed presence of product and starting material. The solid was dissolved in diethylether and the organic layer was washed with aqueous sodium carbonate, dried over Na2S04, filtered and concentrated under reduced pressure. A pale brown solid (1.7g,) was obtained. The 1H-NMR was indicated no starting material but still polymer from nitroethanol and residue of phtalic anhydride. A second silica plug (eluent: PE/EtOAc 95:5) was done. A pale yellow solid (1.5g) was obtained. 1H-NMR of solid showed only product and polymer. The solid was recrystallized from methanol/water . A pale yellow solid (1.05g, 31.2%) was obtained.
Example 3
Nitro chromene synthesis without column purification
To a solution of isobenzofuran- 1 , 3 -dione (18,74 g, 127 mmol) , 3 , 5-difluoro-2 -hydroxybenzaldehyde (10 g, 63,3 mmol) in Toluene (100 ml) was added 2 -nitroethanol (6,86 ml, 95 mmol) . The resulting mixture was heated to reflux for 24h (Dean stark) .
The reaction conversion was checked by HPLC and by 1H-NMR. Only 50% conversion was obtained.
The reaction mixture was cooled to room temperature and diluted with DCM (lOOmL) and 1M NaOH solution (200mL) .
The biphasic system was stirred for 30 minutes and then separated (very difficult to see phase separation) . The aqueous layer was washed with DCM (50mL) and the combined organic layers were washed twice with water (2x50ml) , dried over sodium sulfate. The filtered organic layer was concentrated under reduced pressure. To the residue was added methanol (50mL) . The methanol was then removed by distillation under reduced pressure. A brown solution precipitated when most of the methanol was removed. More methanol was added and more solid crushed out then few drops of water was added to increase the product precipitation. The brown slurry was stirred for 30 minutes and filtered. The brown solid was washed with methanol/water (1:9, 5mL) and dried in a vacuum oven at 40°C for 12h.6, 8-difluoro-3 -nitro-2H-chroraene (4,9 g, 22,99 mmol,) was obtained as brown solid in 36.3% yield.
HPLC showed a purity of 98% and 1H-NMR confirmed the structure and purity around 95%
Example 4
Reduction of nitro chromene to nitro-alkane (racemic mixture)

To a suspension of 6 , 8 -difluoro-3 -nitro-2H-chromene (213mg, 0,999 mmol) and silica (0,8 g, 0,999 mmol) in a mixture of CHC13 (10 ml) and IPA (3,4 ml) at 0°C was added portion wise sodium borohydride (95 mg, 2,498 mmol). The resulting mixture was stirred at 0°C for 45 minutes. Reaction conversion was checked by HPLC. 1 mL of acetic acid was added at 0°C and the resulting mixture was stirred for 30 minutes at room temperature. The slurry was filtered and the silica was washed with DCM. The filtrate was diluted with ethyl acetate and water and the biphasic system was separated. The aqueous layer was back extracted with ethyl acetate. The combined organic layers were washed with brine, dried over MgS04, filtered and concentrated under reduced pressure.
6 , 8-difluoro-3 -nitrochroman (196mg, 0,911 mmol, 91 % yield) was obtained as a pale yellow oil.
Example 5
Preparation of 6 , 8 -difluorochroman-3 -one from nitro chromene

A solution of 6, 8-difluoro-3 -nitro-2H-chromene (lOOmg, 0,469 mmol) in acetic acid (0.5 ml) is added slowly to a stirred slurry of iron (262 mg, 4,69 mmol) in acetic acid (1 ml) at 60.deg. C. The reaction mixture is stirred at 60. °C for 2 hour then allowed to cool to room temperature and stirred overnight. The reaction mixture is poured onto ice-water (30 ml) and filtered through Celite. The solid was wash with dichloromethane (DCM) (50 ml) . The organic portion is separated and washed with water (2 x 30 ml) and brine (30 ml) , dried over MgS04, filtered and concentrated in vacuo to give a brown oil. 6,8-difluorochroman-3 -one (75 mg, 0,407 mmol, 87 % yield) was obtained as a brown oil.
Example 6
Preparation of 6 , 8-difluorochroman-3 -one from methyl 6,8-difluoro-2H-chromen-3 -yl-carbamate

Methanol (1000m ml) was added to a slurry of methyl fluoro-2H-chromen-3 -yl -carbamate (250 g, 1.037 mol) hydrogen chloride 6N (2000 ml, 12 mol) at room temperature. The resulting mixture was reflux and stirred for 2 hours. Reaction monitored by HPLC.
Reaction was not complete but was stopped in order to avoid degradation of the product. The yellow solution was cooled to room temperature. A slurry (two type of solid) was observed and diluted with diethyl ether (300mL) . The resulting slurry was stirred at 5°C for 1 hour then filtered. The yellow solid was washed with water. The resulting wet yellow solid was suspended in diethylether (400mL) and petroleum ether (PE) (400mL) was added. Slight yellow solid was stirred at room temperature overnight, filtered and washed with PE (300mL) , dried in a vacuum oven at 30 °C for 4h. The wet sample was checked by NMR. No starting material was detected. A pale yellow solid (72.5g, solid 1) was obtained. The mother liquors were concentrated to dryness. A yellow solid was obtained, suspended in diethyl ether and PE. The slurry was then stirred for 4 hours, filtered, washed with PE . A dark yellow solid (4.5g, solid 2) was obtained. Solid 1 (2g) was diluted in DCM and washed with water (pH =6). The organic layer was then dried over Na2S04, filtered, concentrated to dryness. A crystalline pale yellow solid (1.9g, solid 3) was obtained. NMR showed the same purity for solid 3 as for solid 1. The remaining part of solid 1 was then diluted in DCM. The resulting organic layer was washed with water, dried over Na2S04, filtered and then concentrated to dryness. Slight yellow crystalline solid (68.5g, solid 4) was obtained. NMR confirmed high quality material.
Loss on Drying (LOD) : 1.03% .
Example 7
Biotransformation: Transaminases

Codexis transaminases ATA-025, ATA-251 and ATA-P2-A07 recognized 6 , 8 -difluorochroman-3 -one as the substrate and produced the corresponding 6 , 8 -difluorochroman-3 -amine .
PATENT
WO 2014077715
WO 2013002660
WO 2008136695
REFERNCES
International Journal of Pharmaceutics (Amsterdam, Netherlands) (2016), 501(1-2), 102-111.
| WO2012087174A2 | Dec 21, 2011 | Jun 28, 2012 | BIAL – PORTELA & Cª., S.A. | Crystalline forms and processes for their preparation |
| WO2012087174A3 * | Dec 21, 2011 | May 10, 2013 | BIAL – PORTELA & Cª., S.A. | Crystalline forms and processes for their preparation |
| WO2013002660A2 | Jun 29, 2012 | Jan 3, 2013 | BIAL – PORTELA & Cª, S.A. | Process |
| WO2014077715A1 * | Nov 14, 2013 | May 22, 2014 | BIAL – PORTELA & Cª, S.A. | 1,3-dihydroimidazole-2-thione derivatives for use in the treatment of pulmonary arterial hypertension and lung injury |
| US8481582 | May 6, 2008 | Jul 9, 2013 | Bial-Portela & Ca, S.A. | 1,3-dihydroimidazole-2-thione derivatives as inhibitors of dopamine-beta-hydroxylase |
| US8865913 | Jun 19, 2013 | Oct 21, 2014 | Bial-Portela & Ca, S.A. | Crystalline forms and processes for their preparation |
| WO1995007284A1 * | Aug 29, 1994 | Mar 16, 1995 | Smithkline Beecham Plc | Phosphinic acid derivatives with anti-hyper glycemic and/or anti-obesity activity |
| WO2006044293A2 * | Oct 11, 2005 | Apr 27, 2006 | Pharmacopeia Drug Discovery, Inc. | Bicyclic compounds as selective melanin concentrating hormone receptor antagonists for the treatment of obesity and related disorders |
| WO2012007548A1 * | Jul 14, 2011 | Jan 19, 2012 | Dsm Ip Assets B.V. | (r)-selective amination |
| WO2013002660A2 * | Jun 29, 2012 | Jan 3, 2013 | BIAL – PORTELA & Cª, S.A. | Process |
| GR1005093B * | Title not available |
///////Zamicastat, BIA-5-1058, dopamine beta-monooxygenase inhibitor, phase I, clinical studies, BIAL, treatment of hypertension , heart failure.
S=C4NC=C(CCNCc1ccccc1)N4[C@@H]2Cc3cc(F)cc(F)c3OC2
Sacubitril

Sacubitril, AHU 377
NEPRILYSIN INHIBITOR
FOR HEART FAILURE
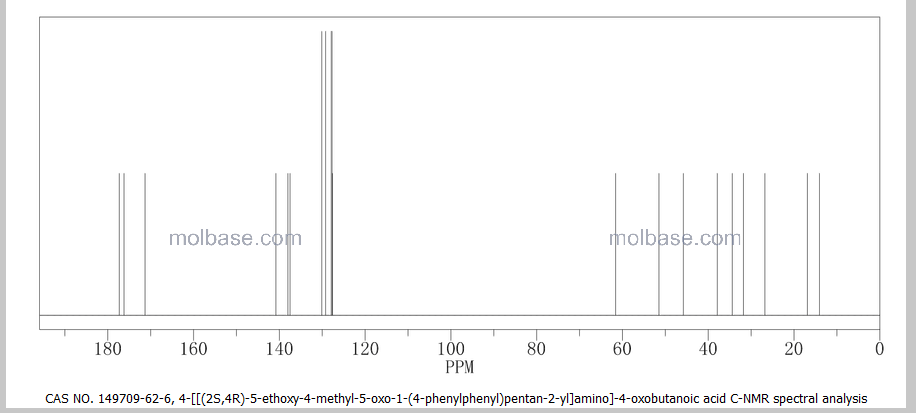
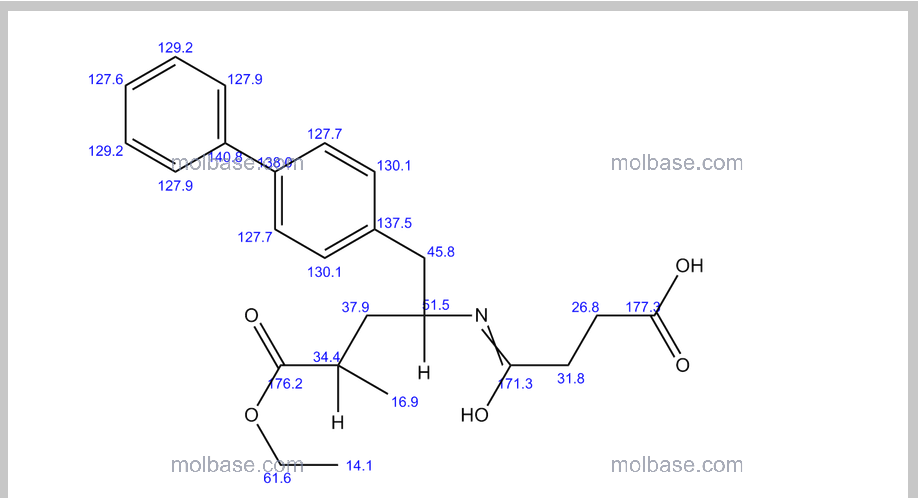
CAS 149709-62-6
CAS SODIUM SALT 149690-05-1
(2R,4S)-5-(biphenyl-4-yl)-4-((3-carboxypropionyl)amino)-2-methylpentanoic acid ethyl ester
5-(Biphenyl-4-yl)-4(S)-(3-carboxypropionamido)-2(R)-methylbutyric acid ethyl ester
N-(3-carboxy-1-oxopropyl)-(4S)-(p-phenylphenylmethyl)-4-amino-2R-methyl butanoic acid ethyl ester
[1,1′-Biphenyl]-4-pentanoic acid, γ-[(3-carboxy-1-oxopropyl)amino]-α-methyl-, α-ethyl ester, (αR,γS)-
- [1,1′-Biphenyl]-4-pentanoic acid, γ-[(3-carboxy-1-oxopropyl)amino]-α-methyl-, ethyl ester, [S-(R*,S*)]-
- (2R,4S)-4-[(3-Carboxy-1-oxopropyl)amino]-4-[(p-phenylphenyl)methyl]-2-methylbutanoic acid ethyl ester
- (2R,4S)-5-(Biphenyl-4-yl)-4-[(3-carboxypropionyl)amino]-2-methylpentanoic acid ethyl ester
-
Formula C24H29NO5 MW 411.49 g/mol
AHU377; AHU-377; Sacubitril; 149709-62-6; UNII-17ERJ0MKGI; Alpha-ethyl (alphaR,gammaS)-gamma-<(3-carboxy-1-oxopropyl)amino>-alpha-methyl<1,1′-biphenyl>-4-pentanoate
Sacubitril sodium
149690-05-1
Sacubitril is an antihypertensive drug used in combination with valsartan. The combination drug, valsartan/sacubitril, known during trials as LCZ696 and marketed under the brand name, Entresto, is a treatment for heart failure.[1] It was approved under the FDA’spriority review process for use in heart failure on July 7, 2015.


Mechanism of action
Sacubitril is a prodrug that is activated to LBQ657 by de-ethylation via esterases.[2] LBQ657 inhibits the enzyme neprilysin,[3] which is responsible for the degradation of atrial and brain natriuretic peptide, two blood pressure lowering peptides that work mainly by reducing blood volume.[4]


SYNTHESIS
CLICK ON IMAGE FOR CLEAR VIEW
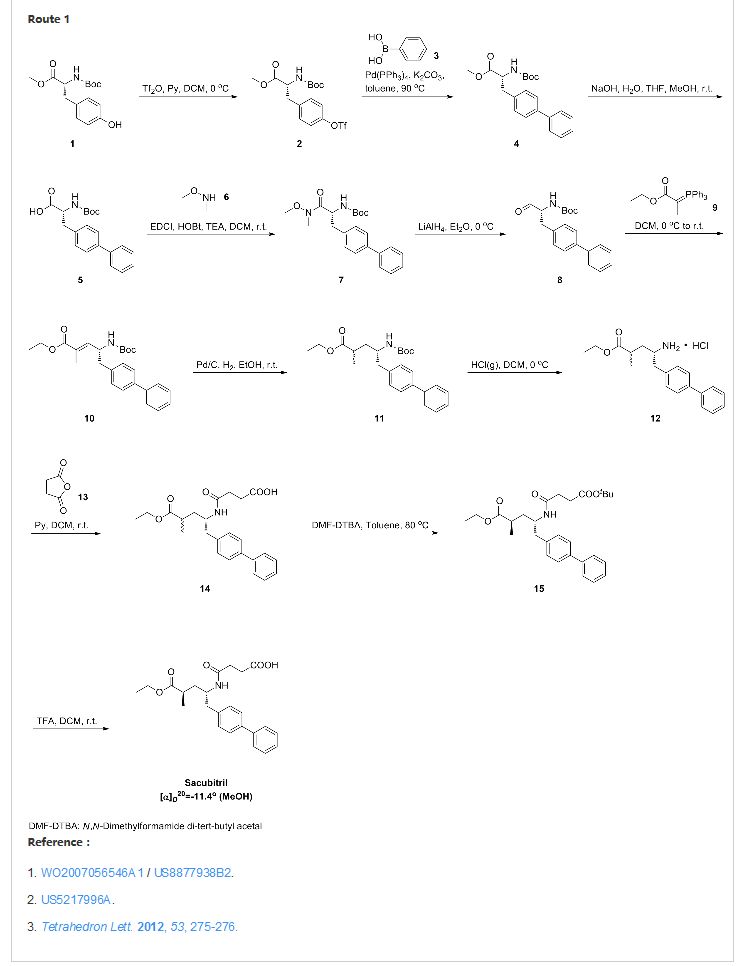
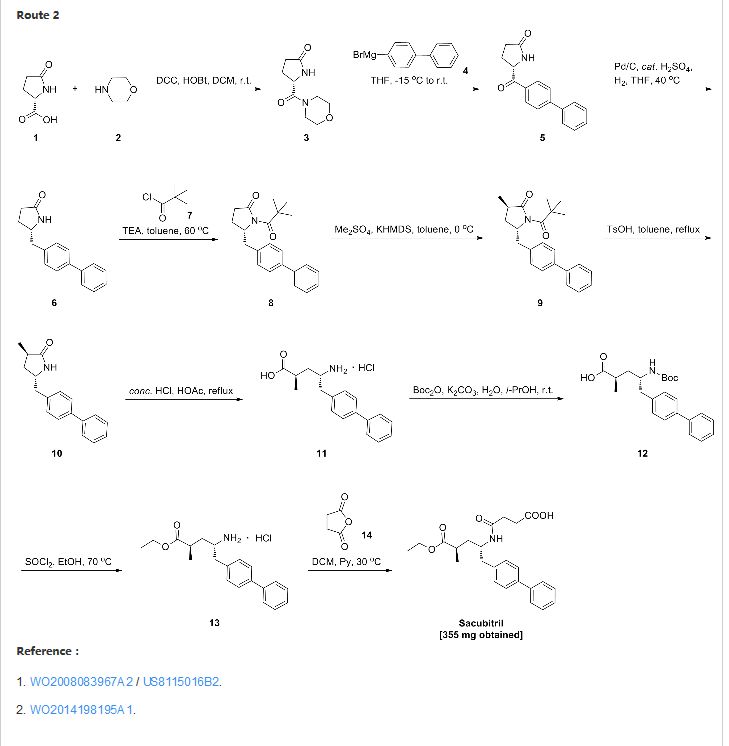
SYNTHESIS

WO-2008031567
http://www.google.com/patents/WO2008031567A1?cl=en
the following steps:


and optionally the following additional steps:


Example 1 :
(E)-(R)-5-biphenyl-4-yl-4-fert-butoxycarbonylamino-2-methylpent-2-enoic acid

(E)-(R)-5-Biphenyl-4-yl-4-tert-butoxycarbonylamino-2-methylpent-2-enoic acid ethyl ester (CAS# 149709-59-1) is hydrolysed using lithium hydroxide in ethanol to yield (£)-(f?)-5-biphenyl-4-yl-4-te/t-butoxycarbonylamino-2-methylpent-2-enoic acid as a white solid. δH (400 MHz; DMSO) 1.31 (9H1 s, (CH3J3), 1.59 (3H, s, 1- CH3), 2.68 (1H, dd, J 6.8, 13.2, 5-HA), 2.86 (1H, m, 5-HB), 4.44 (1H, m, 4-H), 6.51 (1H1 d, J 9.2, 3-H), 7.16 (1H, d, J 8.0, NH), 7.26 (2H, d, J 8.0, Ar-ortho- H(Ph)), 7.31 (1H, t, J 7.6, Ar-(Ph)-para-H), 7.40 (2H, t, J 8.0, Ar-(Ph)-metø-H), 7.54 (2H, d, J 8.0, Ar-mefa-H(Ph), 7.60 (2H, d, J 7.6, Ar-(Ph)-ort/vo-H), 12.26 (1H, s, CO2H); m/z (+ESI) 404 ([MNa]+, 17%), 382 ([MHf, 2), 326 (10), 264 (100), 167 (13).
Example 2:
(2/?,4S)-5-biphenyl-4-yl-4-fert-butoxycarbonylamino-2-methylpentanoic acid in crystalline form [2(i-a)]

2(i-a) To a suspension of (E)-(f?)-5-biphenyl-4-yl-4-te/t-butoxycarbonylamino-2- methylpent-2-enoic acid [2(ii-a)] (200 g, 524.3 mmol) in degassed ethanol (900 ml) at 40 °C a solution of diiodo(p-cymene)ruthenium(ll) dimer (0.052 g, 0.0524 mmol) and (αf?,αf?)-2,2>-bis(α-Λ/,Λ/-dimethylaminophenylmethyl)-(S,S)- 1 ,1′-bis[di(3,5-dimethyl-4-methoxyphenyl)phosphine]ferrocene (= Mandyphos SL-M004-1) (0.116 g, 0.110 mmol) is added in degassed ethanol (100 ml). The solution is degassed using vacuum and a pressure of 20 bar hydrogen applied. The mixture is stirred at 40 0C for 6 h. Vessel is then purged with nitrogen. Ethanol (700 ml) is removed by distillation, lsopropyl acetate (600 ml) is added. Solvent (600 ml) is removed by distillation, lsopropyl acetate (600 ml) is added. Solvent (600 ml) is removed by distillation, lsopropyl acetate (300 ml) is added and the solution is heated to reflux. Heptane fraction (1200 ml) is added and the mixture is cooled to room temperature. The solid is collected by filtration and washed with heptane fraction-isopropyl acetate 2 : 1 mixture (360 ml). The solid is dried overnight at 50 0C under 1-50 mbar vacuum to afford the title compound as a white/off-white solid [Ratio 2(i-a) : 2(i-b) 99 : 1, as determined by HPLC analysis]. Mpt 146-147 0C; δH (500 MHz; DMSO) 1.07 (3H1 d, J 7.0, 1-CH3), 1.34 (9H, s, (CH3)3), 1-38 (1H, m, 3-HA), 1.77 (1H, m, 3-HB), 2.43 (1H, m, 2-H), 2.70 (2H, d, J 7.0, 5-H)1 3.69 (1 H, m, 4-H), 6.74 (1 H, d, J 9.0, NH)1 7.27 (2H, d, J 8.0, Ar-ortA;o-H(Ph)), 7.36 (1H, t, J 7.0, Ar-(Ph)-para-H), 7.46 (2H, t, J 7.5, Ar-(Ph)- meta-H), 7.57 (2H, d, J 8.0, Ar-mefa-H(Ph), 7.64 (2H, d, J 7.5, Ar-(Ph)-orfΛo-H), 12.01 (1H, s, CO2H); δc (500 MHz, DMSO) 18.1 (1-CH3), 28.3 [(CH3)3], 35.9 (2- C), 37.9 (3-C), 40.7 (5-C), 50.0 (4-C), 77.4 [(C(CH3)3], 126.3, 126.5, 127.2, 128.9, 129.8 (Ar-CH), 137.7 (Ar-/pso-C(Ph)), 138.3 (Ar-para-C(Ph)), 140.1 (Ar- (Ph)-/pso-C), 155.2 (NCO), 177.2 (CO2H); mlz (+ESI) 406 ([MNa]+, 6%), 384 ([MH]+, 31 ), 328 (100), 284 (19); Found: [MH]+, 384.21691. C23H30NO4 requires MH 384.21693
PATENT
http://www.google.com/patents/EP0555175A1
Example 1….THE SODIUM SALT
To a solution of N-(3-carbo(t)butoxy-1-oxopropyl)-(4S)-(p-phenylphenylmethyl)-4-amino-2R-methylbutanoic acid ethyl ester (0.80 g) in 15 ml of CH2CI2 at room temperature are added 3 ml of trifluoroacetic acid. The mixture is stirred overnight and concentrated. The residue is dissolved in tetrahydrofuran (THF), and 6.5 ml of 1 N NaOH is added. The mixture is concentrated and triturated with ether. The solid can be recrystallized from methylene chloride-hexane to give sodium N-(3-carboxy-1-oxopropyl)-(4S)-(p-phenylphenylmethyl)-4-amino-2R-methyl butanoic acid ethyl ester melting at 159-160°C; [a]D20 = – 11.4° (methanol).
-
 SODIUM SALT
SODIUM SALT
The starting material is prepared as follows:
A solution of a-t-BOC-(R)-tyrosine methyl ester (5.9 g, 20 mmol) and pyridine (8 mL, 100 mmol) in methylene chloride (30 mL) is cooled to 0-5°C. Trifluoromethanesulfonic anhydride (4 mL, 23 mmol) is added at 0-5°C, and the resulting mixture is held for another 30 minutes. The reaction mixture is diluted with water (60 mL) and methylene chloride (100 mL), and washed sequentially with 0.5 N sodium hydroxide solution (1 x 50 mL), water (1 x 60 mL), 10% citric acid solution (2 x 75 mL) and water (1 x 60 mL). The organic phase is dried over MgS04 and concentrated to an oil. The oil is purified by column chromatography (silica gel, hexane/ethyl acetate, 2:1 to give methyl(R)-2-(t-butoxycarbonylamino)-3-[4-(trifluoromethylsulfonyloxy)phenyl]-propionate which crystallizes on standing; m.p. 46-48°C; [a]20 D-36.010 (c=1, CHCI3).
Nitrogen is passed through a suspension of (R)-2-(t-butoxycarbonylamino)-3-[4-(trifluoromethylsulfonyloxy)-phenyl]-propionate (1.75mmol), phenylboronic acid (3.5 mmol), anhydrous potassium carbonate (2.63 mmol) and toluene (17 mL) for 15 minutes. Tetrakis(triphenyiphosphine)paiiadium(0) is added, and the mixture is heated at 85-90° for 3 hours. The reaction mixture is cooled to 25°C, diluted with ethyl acetate (17 mL) and washed sequentially with saturated sodium bicarbonate (1 x 20 mL), water (1 x 20 mL), 10% citric acid (1 x 20 mL), water (1 x 20 mL) and saturated sodium chloride solution (1 x 20 mL). The organic phase is concentrated, and the residue is purified by column chromatography (silica gel, hexane/ethyl acetate 2:1) to yield methyl (R)-2-(t-butoxycarbonylamino)-3-(p-phenylphenyl)-propionate which can also be called N-(R)-t-butoxycarbonyl-(p-phenylphenyl)-alanine methyl ester.
To a solution of N-(R)-t-butoxycarbonyl-(p-phenylphenyl)-alanine methyl ester (6.8 g) in 60 ml of THF and 20 ml of methanol are added 20 ml of aqueous 1 N sodium hydroxide solution. The mixture is stirred for 1 h at room temperature and then acidified with 21 ml of 1 N hydrochloric acid. The aqueous solution is extracted 3x with ethyl acetate. The combined organic extracts are dried (MgS04), filtered and concentrated to give N-(R)-t-butoxycarbonyl-(p-phenylphenyl)-alanine, m.p. 98-99°C; [a]2°D -18.59° (c=1, methanol).
To a solution of N-(R)-t-butoxycarbonyl-(p-phenylphenyl)-alanine (4.8 g) in 70 ml of methylene chloride (CH2CI2) at 0°C with 1.65 g of N,O-dimethylhydroxylamine HCI, 1.7 g of triethylamine and 2.85 g of hydroxybenzotriazole are added 5.37 g of 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride. The mixture is stirred 17 h at room temperature. The mixture is concentrated taken up in ethyl acetate (EtOAc) and washed with saturated sodium bicarbonate, 1N HCI and brine, then dried (MgS04), filtered and concentrated to give N-(R)-t-butoxycarbonyl-(p-phenylphenyl)-alanine N,O-dimethyl hydroxylamine amide.
To a 0°C solution of N-(R)-t-butoxycarbonyl-(p-phenylphenyl)-alanine N,O-dimethyl hydroxylamine amide (5.2 g) in 250 ml of diethyl ether are added 0.64 g of lithium aluminum hydride. The reaction is stirred for 30 min. and quenched with aqueous potassium hydrogen sulfate. The mixture is stirred for additional 5 min., poured onto 1N HCI, extracted (3x) with EtOAc, dried (MgS04), filtered, and concentrated to give N-(R)-4-t-butoxycarbonyl-(p-phenylphenyl)-alanine carboxaldehyde as a colorless oil.
To a 0°C solution of N-(R)-t-butoxycarbonyl-(p-phenylphenyl)-alanine carboxaldehyde (4.4 g) in 200 ml of CH2CI2are added 10 g of carboethoxyethylidene phenyl phosphorane. The mixture is warmed to room temperature, stirred for 1 h, washed with brine, dried (MgS04), filtered and concentrated. The residue is chromatographed on silica gel eluting with (1:2) ether:hexane to give N-t-butoxycarbonyl-(4R)-(p-phenylphenylme- thyl)-4-amino-2-methyl-2-butenoic acid ethyl ester.
A solution of N-t-butoxycarbonyl-(4R)-(p-phenylphenylmethyl)-4-amino-2-methyl-2-butenoic acid ethyl ester (4.2 g) in 400 ml of ethanol is suspended with 2.0 g of 5% palladium on charcoal and then is hydrogenated at 50 psi for 6h. The catalyst is removed by filtration and the filtrate is concentrated to give N-t-butoxycarbonyl(4S)-(p-phenylphenylmethyl)-4-amino-2-methylbutanoic acid ethyl ester as a 80:20 mixture of diastereomers.
To the N-t-butoxycarbonyl(4S)-(p-phenylphenylmethyl)-4-amino-2-methylbutanoic acid ethyl ester (4.2 g) in 40 ml of CH2CI2 at 0°C is bubbled dry hydrogen chloride gas for 15 min. The mixture is stirred 2 h and concentrated to give (4S)-(p-phenylphenylmethyl)-4-amino-2-methylbutanoic acid ethyl ester hydrochloride as a 80:20 mixture of diastereomers.
To a room temperature solution of the above amine salt (3.12 g) in 15 ml of CH2CI2 and 15 ml of pyridine are added 13.5 g of succinic anhydride. The mixture is stirred for 17 h, concentrated, dissolved in ethyl acetate, washed with 1N HCI and brine, and dried (MgS04) to give N-(3-carboxy-1-oxopropyl)-(4S)-(p-phenylphenyl- methyl)-4-amino-2-methylbutanoic acid ethyl ester as a 80:20 mixture of diastereomers.
The above N-(3-carboxy-1-oxopropyl)-(4S)-(p-phenylphenylmethyl)-4-amino-2-methylbutanoic acid ethyl ester diastereomeric mixture (3.9 g) and N,N-dimethylformamide-di-t-butyl acetal (8.8 ml) are heated at 80°C in 40 ml of toluene for 2 h. The mixture is poured onto ice- 1N HCI, extracted with ether, chromatographed on silica gel eluting with (2:1) toluene:ethyl acetate to give N-(3-carbo(t)butoxy-1-oxopropyl)-(4S)-(p-phenylphe- nylmethyl)-4-amino-2R-methylbutanoic acid ethyl ester as the more polar material and the corresponding (S,S) diastereomer as the less polar material.
Example 2………THE ACID
To a solution of N-(3-carboxy-1-oxopropyl)-(4S)-(p-phenylphenylmethyl)-4-amino-(2R)-methylbutanoic acid ethyl ester (0.33 g) in 20 ml of (1:1) ethanol:tetrahydrofuran (THF) at room temperature are added 5 ml of 1 N sodium hydroxide solution (NaOH) and stirred for 17 h. The mixture is concentrated, dissolved in water and washed with ether. The aqueous layer is acidified with 1 N hydrochloric acid (HCI), extracted 3x with ethyl acetate (EtOAc), dried over magnesium sulfate (MgS04), filtered and concentrated. The residue is triturated with ether to yield N-(3-carboxy-1-oxopropyl)-(4S)-(p-phenylphenylmethyl)-4-amino-(2R)-methylbutanoic acid melting at 158-164°C, [α]D 20= -23.5° (methanol).
PATENT
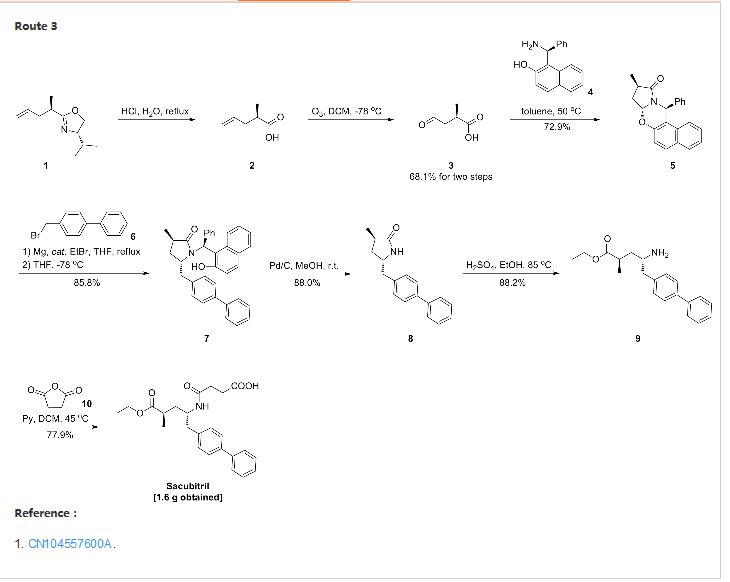
CN 104557600
http://www.google.com/patents/CN104557600A?cl=zh

United States Patent US5217996 and international patent W02008031567, W02010136474 and W02012025501 reported a synthetic route follows to the chiral amino alcohols as raw materials, oxidized to the aldehyde, Victoria ladder tin reaction, chiral hydrogenation and amidation condensation reaction to obtain the objective product.

In addition, the international patent TO2008083967, TO2011088797, TO2012025502 and TO2014198195 reported that another type of preparation. The route through the 2-oxo-proline as raw material, carboxyl activating biphenyl substituted carbonyl reduction, chiral methylation, ring-opening reaction and amide condensation reaction to obtain the objective product.


Example Eight:
in the reaction flask was added (2R, 4S) -2- methyl-4-amino -5- (l, P- biphenyl-4-yl) – pentanoic acid ethyl ester (VII) (1.55g, 5mmol ), Jie of pyridine (1.2g, 15mmol) and dichloromethane burning 25mL, stirring to dissolve, butyric anhydride (1.0g, 10mmol), was heated to 4〇-45 ° C, the reaction was stirred for 6 hours. Fill Gaudin anhydride (0. 5g, 5mmol), the reaction was continued for 4 hours and the end of the reaction by TLC. Concentrated under reduced pressure, the residue was recrystallized from ethyl acetate and n-hexane to give an off-white solid sand sacubitril Kubica song (I) L 6g, a yield of 77.9%;
1H NMR (CDCl3) S 7.51 (d, 2H), 7.46 ( d, 2H), 7.36 (m, 2H), 7. 27 (m, 1H), 7. 17 (d, 2H), 5. 72 (d, 1H), 4. 19 (brs, 1H), 4. 06 (q, 2H), 2. 87-2. 72 (m, 2H), 2. 62-2. 54 (m, 2H), 2. 49 (brs, 1H), 2. 43-2. 33 ( m, 2H), I 88 (m 1H), I 54-1 43 (m, 1H), I. 18 (t, 3H), l 10 (d, 3H);…..
FAB-MSm / z : 412 [M + H] +.
PATENT
http://www.google.com/patents/WO2014198195A1?cl=en
Example 7
(2 Standby

Acetyl chloride (1 mL) 0 ° C was added ethanol (10 mL), and at room temperature for 0.5 hours, the compound (3R, 5S) -5- biphenyl-4-methyl-1- (2,2- methyl-propionyl) -3-methyl pyrrolidone (520 mg, 1.49 mmol), the reaction was refluxed for 3 days. After cooling to room temperature, and concentrated. The reaction mixture was dissolved in 8 mL of dichloromethane and pyridine 1: 1 mixed solution, and then butyryl anhydride (223 mg, 2.23 mmol). 30 ° C overnight. LC-MS detection, a small amount of starting material remaining, fill Gading anhydride (75 mg, 0.74 mmol), continue to reflect four hours. Concentrated and reverse phase column chromatography to give a white foam solid (2R, 4S) -5- biphenyl-4-yl-4- (3-carboxy – propionylamino) -2-methyl – acetic acid ester a (355 mg, 58%) and white solid (2R, 4S) -5- biphenyl-4-yl-4- (3-carboxy – propionylamino) -2-methyl – pentanoic acid b ( 13 mg, 2.3%).
a: 1H MR (400 MHz, CDCl 3 ) [delta] 7.51 (d, = 7.8 Hz, 2H), 7.46 (d, = 7.8 Hz, 2H), 7.36 (t, J = 7.6 Hz, 2H), 7.27 (t, J = 7.2 Hz, IH), 7.17 (d, J = 7.9 Hz, 2H), 5.72 (d, J = 8.1 Hz, IH), 4.19 (brs, IH), 4.06 (q, J = 7.0 Hz, 2H) , 2.87-2.72 (m, 2H), 2.62-2.54 (m, 2H), 2.49 (brs, IH), 2.43-2.33 (m, 2H), 1.88 (ddd, = 13.2, 9.5, 3.9 Hz, IH), 1.54-1.43 (m, IH), 1.18 (t, = 7.0 Hz, 3H), 1.10 (d, = 7.2 Hz, 3H).
LC-MS: t R = 3.43 min; [M + H] +: 412.0.
……………………
Paper
JOURNAL OF MEDICINAL CHEMISTRY, vol. 38, no. 10, 1995, pages 1689-1700,
http://pubs.acs.org/doi/pdf/10.1021/jm00010a014
NOTE———–DIACID
(aR,yS)-y-[ (3-Carbo-1-oxopropyl)aminol-a-methyl- [l,l’-biphenyllpentanoic Acid (21a).
To the sodium salt of 19a (0.73 g, 1.68 mM) in 20 mL of THF:EtOH was added 1 N NaOH (5.0 mL, 5.0 “01). The reaction mixture was stirred overnight and then washed with ether. The aqueous layer was acidified with 1 N HCI, re-extracted with EtOAc (3 x 10 mL), dried (MgSO& and evaporated to dryness. The solid was recrystallized from ethanol to yield 435 mg of 21a DIACID OF SACUBITRIL
melting at 165-167 “C:
[a] D25~ -28.73 (c = 10.1 in MeOH);
‘H NMR, DMSOD6
PPM 12.0, (s, 2H), 7.75 (d, 1H), 7.62 (d, 2H), 7.55 (d, 2H), 7.45 (t, 2H), 7.32 (t, lH), 7.25 (d, 2H), 4.92 (m, lH), 2.70 (d, 2H), 2.35 (t, 3H), 2.25 (m, 2H), 1.75 (m, lH), 1.32 (m, lH), 1.03 (d, 3H).
Anal. (C22H25N05) C,H,N
Note diacid is sacubitrilat (LBQ657)
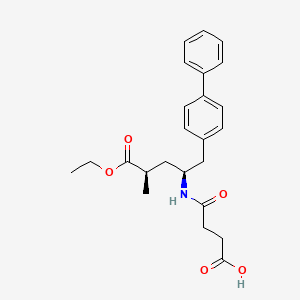
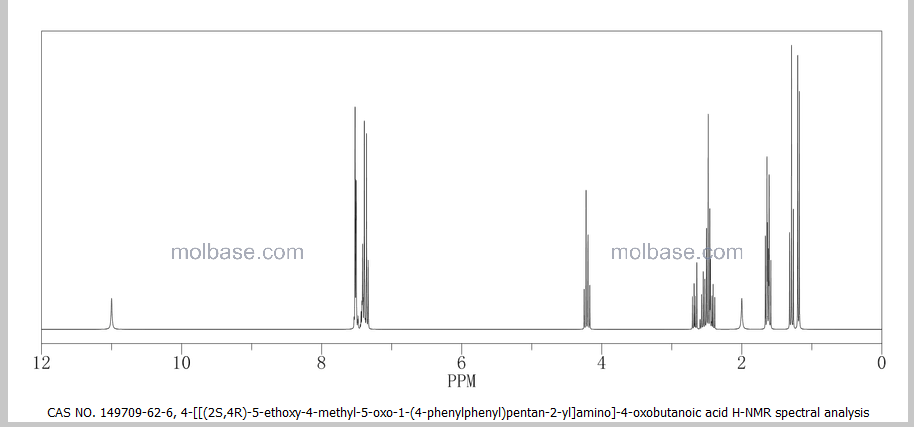
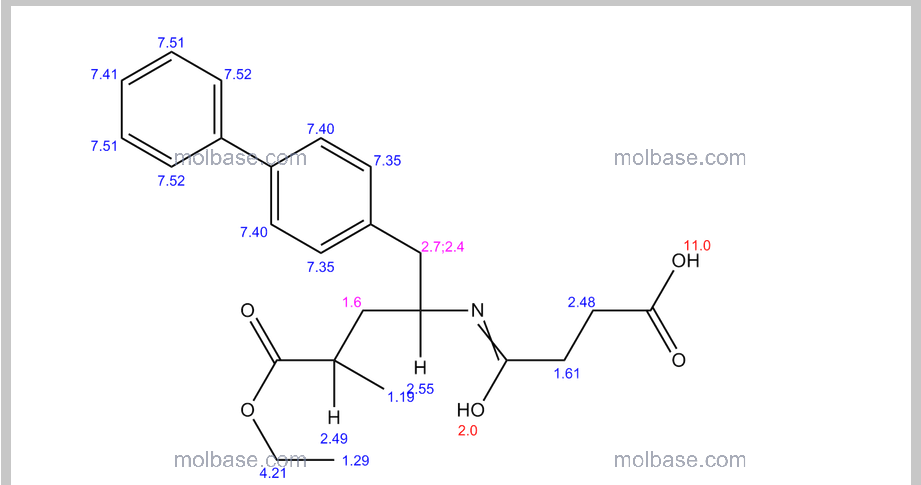
NMR PREDICT
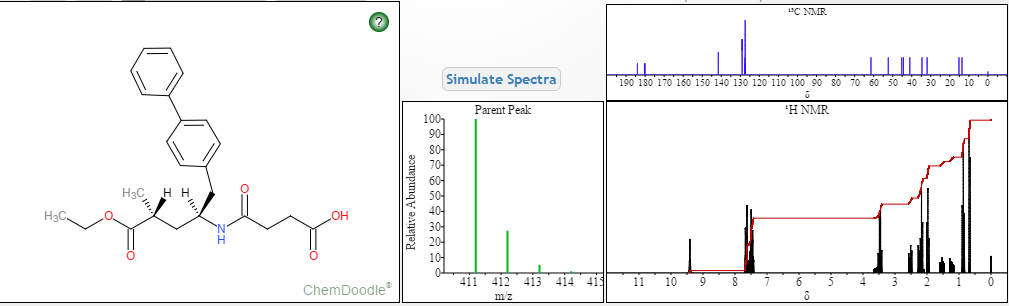
1H NMR PREDICT
13C NMR PREDICT
COSY PREDICT
…………….

NMR…..http://www.chemietek.com/Files/Line3/CHEMIETEK,%20AHU-377%20,%20Lot%2001,%20NMR-MeOD,%201.1.pdf
- Mol. Formula:C24H29NO5 ∙ C4H11NO3
- MW:532.6
- HPLC………http://www.chemietek.com/Files/Line2/CHEMIETEK,%20AHU-377%20,%20Lot%2001,%20HPLC.pdf
update………
PATENT
WO2016180275
Heart failure is a very high mortality syndrome, for patients with heart failure, so far no drug can significantly improve mortality and morbidity, and thus a new type of therapy is necessary. AHU-377 (CAS No. 149709-62-6) is an enkephalinase inhibitor, which is a prodrug ester groups can be lost through hydrolysis, converted to pharmaceutically active LBQ657, inhibit endorphin enzyme (NEP) the role of the main biological effects of NEP is to natriuretic peptides, bradykinin and other vasoactive peptide degradation failure. AHU-377 and angiotensin valsartan composition according to the molar ratio of 1 LCZ696. LCZ696 is an angiotensin receptor enkephalinase inhibitors, which can lower blood pressure, treat heart failure may become a new drug. Clinical data show, LCZ696 is more effective for the treatment of hypertension than valsartan alone.
Patents US 5,217,996 and US 5,354,892 reported the first synthesis of AHU-377, the synthetic route is as follows:
Reaction with unnatural D-tyrosine derivative as a substrate, more expensive, while the second step in the synthesis is necessary to use Pd-catalyzed Suzuki coupling reaction, whereby preparative route costs than the AHU-377 high.
Patent US 8,115,016 above routes also reported the departure from the pyroglutamate, through multi-step process for preparing a reaction AHU-377, which is more difficult methylation reaction, and the yield is not high. Patent US 8,580,974 also reported a carbonyl group of the a- introducing N, N- dimethyl enamine is converted to methyl, however, there are some problems in the route for constructing methyl chiral centers, are not suitable for scale-up synthesis route as follows:
About the latest AHU377 synthesis intermediates, Patent WO2014032627A1 reported using a Grignard reagent to react with epichlorohydrin, a quicker been important intermediates, synthetic route Compound AHU377 synthesized as follows:
However, the second step of the synthetic route use succinimide nitrogen atoms introduced by Mitsunobu reaction with hydrochloric acid hydrolysis to remove, then converted to Boc protected at the end of the synthesis process AHU377 Boc will have to take off protection, then any connection with succinic anhydride reaction product introduced into the structure of succinic acid portion, so that this method of atom economy and the economy of the steps are low.
Example 1
Synthesis of Compound 2
In inert atmosphere, a solution of three 500mL flask was added compound 1 (10g, 1eq), dissolved after 90mL THF, was added CuI (4.814g, 0.1eq), the system moves to the low temperature in the cooling bath to -20 ℃ when, biphenyl magnesium bromide dropwise addition, the internal temperature was controlled not higher than -10 ℃. Bi closed refrigeration drop, return to room temperature overnight. Completion of the reaction, the reaction solution was poured into saturated the NH 4 of Cl (10vol, 100 mL) was stirred at room temperature for 0.5h. Suction filtered, the filter cake was rinsed with a small amount of EA, and the filtrate was transferred to a separatory funnel carved, and the aqueous phase was extracted with EA (10vol × 2,100mL × 2) and the combined organic phases with saturated NaHC [theta] 3 , the NH 4 of Cl, each Brine 150mL (15vol) washed once, dried over anhydrous over MgSO 4 dried, suction filtered, and concentrated to give a white solid. Product obtained was purified by column 15.2g, yield 78%.
NMR data for the product are as follows:
1 the H NMR (400MHz, CDCl 3 ) [delta] 7.57 (D, J = 7.6Hz, 2H), 7.52 (D, J = 8.1Hz, 2H), 7.42 (T, J = 7.6Hz, 2H), 7.38-7.25 (m, 8H), 4.62-4.47 ( m, 2H), 4.09 (dd, J = 6.7,3.5Hz, 1H), 3.54 (dd, J = 9.5,3.5Hz, 1H), 3.43 (dd, J = 9.4 , 6.9Hz, 1H), 2.84 ( d, J = 6.6Hz, 2H), 2.38 (s, 1H).
Example 2
Synthesis of Compound 3
In an inert gas, at room temperature was added to the flask 500mL three Ph3P (18.54g, 2eq), 240mL DCM dissolution, butyryl diimide (of 6.44 g), compound 2 (15g), an ice-water bath cooling to 0 ℃ or so, was added dropwise DIAD (14mL) was complete, the reaction go to room temperature.Starting material the reaction was complete, the system was added to water (100 mL) quenched the reaction was stirred for 10min; liquid separation, the aqueous phase was extracted with DCM (100mL × 2), the combined organic phases with saturated Brine 100mL × 2), dried over anhydrous over MgSO 4 dried , filtration, spin dry to give a white solid; product was purified by column 15.4g, yield 82%.
NMR data for the product are as follows:
1 the H NMR (400MHz, CDCl 3 ) [delta] 7.56 (D, J = 7.4Hz, 2H), 7.49 (D, J = 8.0Hz, 2H), 7.42 (T, J = 7.6Hz, 2H), 7.37-7.30 (m, 3H), 7.27 ( d, J = 6.7Hz, 3H), 7.22 (d, J = 8.0Hz, 2H), 4.75 (s, 1H), 4.56 (d, J = 12.0Hz, 1H), 4.45 (d, J = 12.0Hz, 1H ), 4.06 (t, J = 9.6Hz, 1H), 3.70 (dd, J = 10.0,5.2Hz, 1H), 3.23 (dd, J = 13.8,10.3Hz, 1H) , 3.14-3.00 (m, 1H), 2.48 (d, J = 4.0Hz.4H).
Example 3
Synthesis of Compound 4
Protection of inert gas, at room temperature was added to the flask 1L three compound 3 (18.81g), 470mL EtOH was dissolved, was added Pd / C, replaced the H 2 three times, move heated on an oil bath at 60 ℃ reaction. Raw reaction was complete, the system was removed from the oil bath, the reaction solution was suction filtered through Celite and concentrated to give the crude product. It was purified by column pure 11.8g, a yield of 81.2%.
NMR data for the product are as follows:
1 the H NMR (400MHz, CDCl 3 ) [delta] 7.57 (D, J = 7.8Hz, 2H), 7.51 (D, J = 7.8Hz, 2H), 7.42 (T, J = 7.5Hz, 2H), 7.33 (T , J = 7.2Hz, 1H), 7.26 (d, J = 7.2Hz, 2H), 4.55 (d, J = 5.2Hz, 1H), 4.06-3.97 (m, 1H), 3.86 (dd, J = 12.0, 3.1Hz, 1H), 3.16 (dd , J = 8.1,2.9Hz, 2H), 2.58 (t, J = 7.0Hz, 4H), 1.26 (s, 2H).
Example 4
Synthesis of Compound 7
Protection of inert gas, at room temperature to a 25mL flask was added three Dess-Martin oxidant (767.7mg), 10mL DCM was dissolved, the system was cooled down to -10 deg.] C, was added 4 (500mg). Starting material the reaction was complete, to the system was added saturated NaHCO3 and Na2S2O3 each 5mL, quench the reaction stirred for 10min; aqueous phase was extracted with DCM (10mL × 3) and the combined organic phases with saturated NaHCO3, Brine 30mL each wash, dried over anhydrous MgSO4, filtration, spin dried to give the crude product used directly in the next reaction cast.
Example 5
Synthesis of Compound 8
Inert gas, at room temperature for three to 500mL flask 7 (497.5mg), 10mL DCM to dissolve an ice water bath to cool, added phosphorus ylide reagent (880.6mg), the system was removed from the ice water bath at room temperature. The reaction material completely stop the reaction, the system was added to water (5mL) to quench the reaction. Liquid separation, the aqueous phase was extracted with DCM (10mL × 2), organic phases were combined, washed with saturated Brine 20mL × 2, dried over anhydrous MgSO4, filtration, spin crude done. Product obtained was purified by column 563mg, 90% yield.
NMR data for the product are as follows:
1 the H NMR (400MHz, CDCl 3 ) δ7.60-7.53 (m, 2H), 7.51 (D, J = 8.1Hz, 2H), 7.42 (T, J = 7.6Hz, 2H), 7.33 (D, J = 7.3Hz, 1H), 7.23 (d , J = 8.1Hz, 2H), 7.13 (dd, J = 9.2,1.5Hz, 1H), 5.26 (td, J = 9.5,6.9Hz, 1H), 4.25-4.05 ( m, 2H), 3.40 (dd , J = 13.7,9.7Hz, 1H), 3.13 (dd, J = 13.8,6.7Hz, 1H), 2.53 (d, J = 2.2Hz, 4H), 1.85 (d, J = 1.4Hz, 3H), 1.30 ( t, J = 7.1Hz, 3H).
Example 6
Synthesis of Compound 9
Protection of inert gas, at room temperature to a 50mL flask was added three 8 (365mg, 1eq), 9mL of ethanol and stirred to dissolve, the system was replaced with hydrogen three times, was added Pd / C (25% w / w) at room temperature. The reaction material completely stop the reaction, the system was added to water (5mL) to quench the reaction. The reaction mixture was suction filtered through Celite and concentrated to give the crude product. Product was purified by column, yield 80.2%, purity 97.2%.
Example 7
Synthesis of Compound 10
Equipped with Compound 9 (100mg) acetic acid A reaction flask (9mL), hydrochloric acid (1mL). The reaction was heated oil bath at 80 deg.] C. The reaction material completely stop the reaction, the system was added to water (5mL) to quench the reaction. After saturated NaHCO3 and extracted with EA and concentrated to give crude product. Product obtained was purified by column 90mg, yield 84%.
NMR data for the product are as follows:
1 the H NMR (400MHz, CDCl 3 ) δ7.61-7.54 (m, 2H), 7.53-7.48 (m, 2H), 7.41 (dd, J = 10.5,4.9Hz, 2H), 7.31 (dd, J = 8.3 , 6.4Hz, 1H), 7.22 ( d, J = 8.2Hz, 2H), 5.93 (t, J = 9.7Hz, 1H), 4.34-4.00 (m, 3H), 2.91-2.71 (m, 2H), 2.68 -2.57 (m, 2H), 2.55 (ddd, J = 9.4,7.0,4.3Hz, 1H), 2.42 (dt, J = 13.3,6.8Hz, 2H), 1.97-1.74 (m, 1H), 1.64-1.46 (m, 1H), 1.23 ( td, J = 7.1,3.3Hz, 3H), 1.14 (dd, J = 7.1,3.9Hz, 3H)
Example 8
Synthesis of Compound 5
Example 8-1: The reaction flask was added compound 4 (1eq) was added water (2VOL), concentrated hydrochloric acid (2VOL), 110 ℃ reaction was heated in an oil bath overnight, complete conversion of starting material, the HPLC peak area 97%. 10% NaOH solution was added to adjust the pH to about 10, filtration products. Yield 85%.
Example 8-2: The reaction flask was added compound 4 (1eq) was added ethanol (5 vol), water (5 vol), potassium hydroxide (8 eq), was heated in an oil bath overnight at 110 ℃ reaction, complete conversion of the starting material, the HPLC peak area 99%. Water was added (5Vol), filtered to obtain the product. Yield 95%. Product was dissolved in toluene, was added ethanolic hydrochloric acid, the precipitated hydrochloride Compound 5.
NMR data for the product are as follows:
1 the H NMR (400MHz, of DMSO) [delta] 8.31 (S, 3H), 7.70-7.61 (m, 4H), 7.47 (T, J = 7.6Hz, 2H), 7.42-7.31 (m, 3H), 4.09 (the dq- , J = 42.6,7.1Hz, 1H), 3.62-3.51 (m, 1H), 3.50-3.41 (m, 1H), 3.11-3.00 (m, 1H), 2.95-2.84 (m, 1H), 1.30-1.10 (m, 1H).
EXAMPLE 9
Synthesis of Compound 6
To the reactor was added compound 5, was added absolute ethanol (3vol). Temperature of the outer set 30 ℃ heating, stirring was continued after the temperature reached 25 ℃ 20min. Was added 30% NaOH aqueous solution (1.1eq). External temperature 65 ℃ heating provided, after the internal temperature reached 60 deg.] C was slowly added (of Boc) 2 O (1.1 eq). Stirring 0.5h, reaction monitoring. After completion of the reaction, water was added slowly dropwise (8vol), turn off the heating and natural cooling. The system temperature was lowered to 25 deg.] C and continue stirring for 2h. Filter cake at 50 ℃ blast oven drying to obtain the product.
NMR data of the product are as follows:
1 the H NMR (400MHz, CDCl 3 ) δ7.61-7.50 (m, 4H), 7.61-7.50 (m, 4H), 7.46-7.39 (m, 2H), 7.48-7.38 (m, 2H), 7.38-7.23 (m, 3H), 7.37-7.26 ( m, 3H), 4.82 (d, J = 7.9Hz, 1H), 4.82 (d, J = 7.9Hz, 1H), 3.91 (s, 1H), 3.70 (d, J = 11.0Hz, 1H), 3.77-3.54 (m, 2H), 3.65-3.47 (m, 1H), 2.88 (d, J = 7.0Hz, 2H), 2.88 (d, J = 7.0Hz, 2H), 2.51 (s, 1H), 2.51 (s, 1H), 1.42 (s, 9H), 1.42 (s, 9H).
Synthesis of Intermediate Compound 6 to Compound 10, i.e., the AHU-377, a synthetic route in the background of the present invention, the cited patent application WO2014032627A1 loaded in detail, not in this repeat.
Example 10
Synthesis of Compound 2
Benzyl glycidyl ether preparation (50g) in THF (200mL) was added. Under inert gas protection, the biphenyl magnesium bromide (365mmol) was added to THF (1020mL) was added the reaction flask is placed in a low temperature bath -40 ℃ cooling. Cuprous iodide (O.leq) when the internal temperature dropped to -9 ℃. Continued to decrease the temperature of -23 ℃ dropwise addition of benzyl glycidyl ether in THF was added dropwise to control the internal temperature process of not higher than -15 deg.] C, 47 min when used, the addition was completed the cooling off the reaction was stirred overnight. The cooling system to -20 ℃ quenched with 1N HCl aqueous solution, <10 ℃ Go stirred 30min at room temperature. Liquid separation, the aqueous phase was extracted with THF, the combined THF phases. Respectively saturated ammonium chloride (250mL), saturated brine (250mL) washed. Rotary evaporation to remove THF, and water (200 mL) Continue rotary evaporation 1h, cool to precipitate a solid. Suction crude. Crude n-heptane was added 2Vol beating, suction filtration to obtain the product in a yield of 90 ~ 95%, HPLC peak area 94%. In another column purification was pure, columned yield 88.6%, HPLC 99.1%.
Example 11
Synthesis of Compound 3
Preparation Example 9, said compound taking the embodiment 2 (5g) added to the reaction flask, the reaction flask was added toluene (80mL), phthalimide (2.55 g of) and triphenylphosphine (5.35g of), the nitrogen was replaced protection. An ice-salt bath cooling to -5 deg.] C, was added dropwise DIAD (4.12g), dropwise addition was exothermic, the temperature was raised to 5 ℃. The reaction was continued 1h sampling HPLC test material substantially complete reaction. Join 12g silica spin column done to collect the product (including DIEA derivative).
Example 12
Synthesis of Compound 11
Compound 3 (3g) was added to the reaction flask embodiment taken in Preparation Example 10, was added ethanol (30 mL), with stirring. Was added hydrazine hydrate (2g) was heated in an oil bath reflux 1h, when supplemented with 20mL ethanol was stirred difficulties, the reaction was continued to 2.5h, HPLC showed the starting material the reaction was complete. Add EA / H2O 100mL each liquid separation, the EA phase was washed with water (100mL) and the combined organic phases were washed with water (100mL) and saturated brine (100mL) washed. Anhydrous magnesium sulfate and filtered spin column was done product 1.88g, yield 88%, HPLC 94%.
NMR data of the product are as follows:
1 the H NMR (400MHz, of DMSO) [delta] 7.64 (D, J = 7.2Hz, 2H), 7.57 (D, J = 8.1Hz, 2H), 7.45 (T, J = 7.6Hz, 2H), 7.39-7.32 ( m, 5H), 7.29 (d , J = 8.1Hz, 3H), 4.55-4.43 (m, 2H), 3.38-3.23 (m, 3H), 3.18-3.10 (m, 1H), 2.82-2.74 (m, 1H), 2.61-2.52 (m, 1H ).
Example 13
Synthesis of Compound 11
To the toluene solution of the compound 2 was added phthalimide (1.1 eq), triphenylphosphine (1.3 eq) with stirring. External bath set -10 ℃, to cool the system, the internal temperature dropped to 0 ~ 5 ℃, start dropping DIAD (1.3eq), control the internal temperature -5 ~ 5 ℃. Completion of the dropwise addition, the cooling bath was turned off outside the reaction was stirred at room temperature. The reaction was stirred for 1 to 4 hours. The reaction solution to give compound 3, administered directly in the next reaction. To the above reaction mixture was added hydrazine hydrate (6 eq), heated to 70 ~ 80 ℃, to complete the reaction, filtered hot, the filtrate. Aqueous sodium hydroxide solution (20vol 10%) was stirred for 0.5h, allowed to stand for liquid separation from toluene phase. Water was added (20vol) was stirred for 0.5h, allowed to stand for liquid separation from toluene phase. The toluene phase was added hydrochloric acid (20vol, 3N), stirred for 0.5h, to form a solid precipitate. Filtration and drying to obtain a product, i.e. compound 11, the hydrochloride salt, yield 60% in two steps.
NMR data of the product are as follows:
1 the H NMR (400MHz, of DMSO) [delta] 8.46 (S, 3H), 7.63 (dd, J = 16.4,7.7Hz, 4H), 7.47 (T, J = 7.6Hz, 2H), 7.42-7.22 (m, 8H ), 4.56 (d, J = 12.1Hz, 1H), 4.48 (d, J = 12.1Hz, 1H), 3.58 (d, J = 7.9Hz, 2H), 3.47 (dd, J = 10.9,6.3Hz, 1H ), 3.11 (dd, J = 13.5,4.9Hz, 1H), 2.92 (dd, J = 13.4,9.1Hz, 1H).
Example 14
Synthesis of Compound 12
Weigh Compound 11 (1.38g) was added to the reaction flask. To the reaction flask plus DCM (14ml) and Et3N (462mg, 0.73ml). Weighed (of Boc) 2O (1.23 g of) was added to DCM (5ml) was dissolved. Room temperature (8 ℃), a solution (of Boc) 2 DCM solution O was added dropwise to the reaction, (2ml) rinsed with DCM. The reaction mixture was stirred at room temperature, detected by HPLC, the reaction ends 4h. Reaction mixture was washed (15ml) 3 times with Brine (15ml) The reaction solution was washed 1 times. Inorganic sulfate, concentrated and purified by column PE:EA = 15:1 give product 560mg, yield 30.8%, HPLC 99.92%.
NMR data of the product are as follows:
1 the H NMR (400MHz, CDCl 3 ) [delta] 7.57 (D, J = 7.6Hz, 2H), 7.49 (D, J = 7.4Hz, 2H), 7.43 (T, J = 7.3Hz, 2H), 7.39-7.28 (m, 5H), 7.24 ( d, J = 9.0Hz, 3H), 5.00-4.80 (br, 1H), 4.51 (q, J = 11.8Hz, 2H), 4.08-3.85 (br, 1H), 3.43 ( d, J = 2.9Hz, 2H) , 3.02-2.77 (m, 2H), 1.42 (s, 9H).
Example 15
Synthesis of Compound 6
Weigh Compound 12 (250mg) and methanol (9ml) was added to the reaction flask. Added Pd / C (138mg, 1 / 4w / w, water content 55%). The H 2replaced 3 times, 50 ℃ stirred and heated. HPLC detection reaction, the reaction end 30h. Filtered off Pd / C, 40 ℃ concentrated under reduced pressure to remove methanol. PE:EA = 3:1 florisil column to give the product 196mg, 100% yield, 99.34% purity.
NMR data of the product are as follows:
1 the H NMR (400MHz, CDCl 3 ) δ7.61-7.50 (m, 4H), 7.61-7.50 (m, 4H), 7.46-7.39 (m, 2H), 7.48-7.38 (m, 2H), 7.38-7.23 (m, 3H), 7.37-7.26 ( m, 3H), 4.82 (d, J = 7.9Hz, 1H), 4.82 (d, J = 7.9Hz, 1H), 3.91 (s, 1H), 3.70 (d, J = 11.0Hz, 1H), 3.77-3.54 (m, 2H), 3.65-3.47 (m, 1H), 2.88 (d, J = 7.0Hz, 2H), 2.88 (d, J = 7.0Hz, 2H), 2.51 (s, 1H), 2.51 (s, 1H), 1.42 (s, 9H), 1.42 (s, 9H).
Method for preparing the AHU-377, characterized by comprising the steps of: (a) Compound (1) S- benzyl glycidyl ether and biphenyl Grignard reagent produced by the reaction of the compound (2) in an organic solvent; ( b) compound (2) with a succinimide or phthalimide Mitsunobu reaction occurs in an organic solvent to form a compound (3); (C) compound (3) in an organic solvent in the role of a catalyst under removal debenzylation protected form compound (4); (D) compound (4) with an oxidizing agent oxidation reaction occurs in an organic solvent to form a compound (7); (E) compound (7) with a phosphorus ylide reagent in an organic solvent to give the compound (8); (F.) compound (8) in an organic solvent in the selective catalytic hydrogenation of the compound (9); and (g) of the compound (9) in an organic solvent in the hydrolysis reaction of the amide compound occurs in the presence of an acid ( 10), i.e., AHU-377;
References
- John J.V. McMurray, Milton Packer, Akshay S. Desai, et al. for the PARADIGM-HF Investigators and Committees (August 30, 2014).“Angiotensin–Neprilysin Inhibition versus Enalapril in Heart Failure”. N Eng J Med 371. doi:10.1056/NEJMoa1409077.
- Solomon, SD. “HFpEF in the Future: New Diagnostic Techniques and Treatments in the Pipeline”. Boston. p. 48. Retrieved 2012-01-26.
- Gu, J.; Noe, A.; Chandra, P.; Al-Fayoumi, S.; Ligueros-Saylan, M.; Sarangapani, R.; Maahs, S.; Ksander, G.; Rigel, D. F.; Jeng, A. Y.; Lin, T. H.; Zheng, W.; Dole, W. P. (2009). “Pharmacokinetics and Pharmacodynamics of LCZ696, a Novel Dual-Acting Angiotensin Receptor-Neprilysin Inhibitor (ARNi)”. The Journal of Clinical Pharmacology 50 (4): 401–414. doi:10.1177/0091270009343932.PMID 19934029.
- Schubert-Zsilavecz, M; Wurglics, M. “Neue Arzneimittel 2010/2011.” (in German)


| WO2004085378A1 * | Mar 15, 2004 | Oct 7, 2004 | Joseph D Armstrong Iii | Process for the preparation of chiral beta amino acid derivatives by asymmetric hydrogenation |
| WO2006057904A1 * | Nov 18, 2005 | Jun 1, 2006 | Merck & Co Inc | Stereoselective preparation of 4-aryl piperidine amides by asymmetric hydrogenation of a prochiral enamide and intermediates of this process |
| WO2006069617A1 * | Dec 5, 2005 | Jul 6, 2006 | Dsm Fine Chem Austria Gmbh | Process for transition metal-catalyzed asymmetric hydrogenation of acrylic acid derivatives, and a novel catalyst system for asymmetric transition metal catalysis |
| US5217996 * | Jan 22, 1992 | Jun 8, 1993 | Ciba-Geigy Corporation | Biaryl substituted 4-amino-butyric acid amides |
NON-PATENT CITATIONS
| Reference | ||
|---|---|---|
| 1 | * | KSANDER, GARY M. ET AL: “Dicarboxylic Acid Dipeptide Neutral Endopeptidase Inhibitors” JOURNAL OF MEDICINAL CHEMISTRY, vol. 38, no. 10, 1995, pages 1689-1700, XP002340280 cited in the application |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
4-{[(2S,4R)-1-(4-Biphenylyl)-5-ethoxy-4-methyl-5-oxo-2-pentanyl]amino}-4-oxobutanoic acid
|
|
| Clinical data | |
| Legal status |
|
| Identifiers | |
| CAS Registry Number | 149709-62-6 |
| ATC code | None |
| PubChem | CID: 9811834 |
| ChemSpider | 7987587 |
| Synonyms | AHU-377; AHU377 |
| Chemical data | |
| Formula | C24H29NO5 |
| Molecular mass | 411.49 g/mol |
Relevant Clinical Literature
UK Guidance
Regulatory Literature
Other Literature
|
|
| Systematic (IUPAC) name | |
|---|---|
|
4-{[(2S,4R)-1-(4-Biphenylyl)-5-ethoxy-4-methyl-5-oxo-2-pentanyl]amino}-4-oxobutanoic acid
|
|
| Identifiers | |
| CAS Number | 149709-62-6 |
| ATC code | None |
| PubChem | CID: 9811834 |
| ChemSpider | 7987587 |
| Synonyms | AHU-377; AHU377 |
| Chemical data | |
| Formula | C24H29NO5 |
| Molecular mass | 411.49 g/mol |
Message
Comwinchem <comwinchem@foxmail.com>
Date: 1 September 2016 at 15:16
Subject: LCZ696 (SACUBITRIL+VALSARTAN)/ Changzhou Comwin Fine Chemicals Co,. Ltd
To: amcrasto <amcrasto@gmail.com>
Dear SirHow do you do! Sincerely hope my email will bring you more LCZ696 (SACUBITRIL+VALSARTAN) possibilities.I am Wang Zhuo of ComWin from China, and in charge of LCZ696 (SACUBITRIL+VALSARTAN) global marketing.
Date: 1 September 2016 at 15:16
Subject: LCZ696 (SACUBITRIL+VALSARTAN)/ Changzhou Comwin Fine Chemicals Co,. Ltd
To: amcrasto <amcrasto@gmail.com>
Dear SirHow do you do! Sincerely hope my email will bring you more LCZ696 (SACUBITRIL+VALSARTAN) possibilities.I am Wang Zhuo of ComWin from China, and in charge of LCZ696 (SACUBITRIL+VALSARTAN) global marketing.
LCZ696 (sacubitril/Valsartan) is a combination drug for use in heart failure developed by Novartis. It consists of valsartan and sacubitril, in a 1:1 mixture by molecule count. It was approved by US FDA in July 2015.
Sacubitril (Hemicalcium) is a neprilysin inhibitor, We have developed this project since 2nd half of 2014. At present, some intermediates are in commercial scale, and some are in pilot production.
We will put our most focus on this project from 2nd half of this year. Certainly we will file DMF for Sacubitril and make submission to regulartory market. We have sold Sacbutitril to some EU customers for evaluation purpose, such as Teva, Chemo. Also, we are doing the development of LCZ696 (the final API). It’s co-crystallized valsartan and sacubitril, in a one-to one molar ratio. One LCZ696 complex consists of six valsartan anions, six sacubitril anions, 18 sodium cations, and 15 molecules of water. Now we have the sample of LCZ696 in kilogram grade.
Best Regards
Wang Zhuo
Sales Executive
Changzhou ComWin Fine Chemicals Co.,Ltd.
24th Floor, Jiaye International Commercial Plaza
99 Yanling West Road, Changzhou
Jiangsu 213003 China
Tel: 0086 519 8663 2882
Fax: 0086 519 8661 3190
email: wang.zhuo@comwin-china.com
www.comwin-china.com


सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये। औकात बस इतनी देना, कि औरों का भला हो जाये।
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE


Join me on twitter

 LIONEL MY SON
LIONEL MY SONHe was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, Now I keep him as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son happy

सुकून उतना ही देना प्रभू, जितने से
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
////// antihypertensive drug, Heart Failure, Sacubitril, AHU 377
CCOC(=O)[C@H](C)C[C@@H](Cc1ccc(cc1)c2ccccc2)NC(=O)CCC(=O)O
CCOC(=O)[C@H](C)C[C@@H](CC1=CC=C(C=C1)C2=CC=CC=C2)NC(=O)CCC(=O)O
