
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

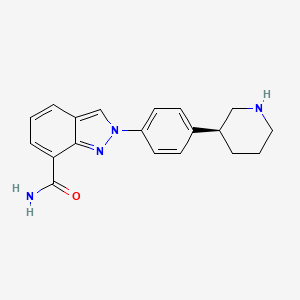
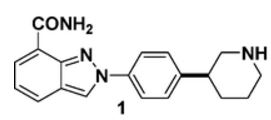
MK-4827,(S)-2-(4-(piperidin-3-yl)phenyl)-2H-indazole-7-carboxaMide
1038915-60-4 CAS, free form
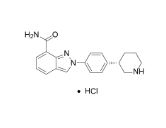
1038915-64-8 CAS HYDROCHLORIDE
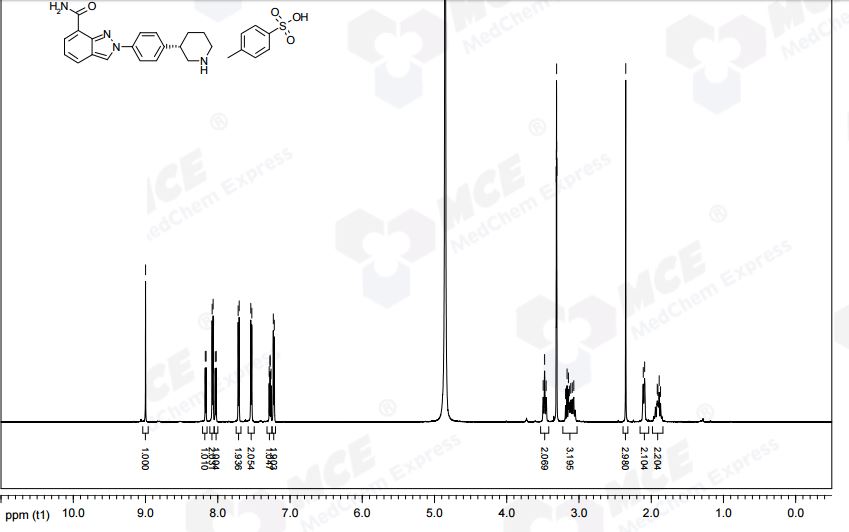
1613220-15-7 cas TOSYLATE MONOHYDRATE

| 1038915-73-9 TOSYLATE |

MK-4827(Niraparib) tosylate is a selective inhibitor of PARP1/PARP2 with IC50 of 3.8 nM/2.1 nM; with great activity in cancer cells with mutant BRCA-1 and BRCA-2; >330-fold selective against PARP3, V-PARP and Tank1.
IC50 value: 3.8 nM/2.1 nM( PARP1/2) [1]
Target: PARP1/2
in vitro: MK-4827 displays excellent PARP 1 and 2 inhibition with IC(50) = 3.8 and 2.1 nM, respectively, and in a whole cell assay, it inhibits PARP activity with EC(50) = 4 nM and inhibits proliferation of cancer cells with mutant BRCA-1 and BRCA-2 with CC(50) in the 10-100 nM range [1].
in vivo: MK-4827 is well tolerated in vivo and demonstrates efficacy as a single agent in a xenograft model of BRCA-1 deficient cancer [1]. In addition, MK-4827 strongly enhances the effect of radiation on a variety of human tumor xenografts, both p53 wild type and p53 mutant. The enhancement of radiation response is observed in clinically relevant radiation-dose fractionation schedules. The therapeutic window during which time MK-4827 interacts with radiation lasts for several hours. These biological attributes make translation of this therapeutic combination treatment feasible for translation to the treatment of a variety of human cancers [2].
MERCK
![]()
TESARO
![]()
An inhibitor of poly (ADP-ribose) polymerase (PARP) with potential antineoplastic activity. PARP Inhibitor MK4827 inhibits PARP activity, enhancing the accumulation of DNA strand breaks and promoting genomic instability and apoptosis. The PARP family of proteins detect and repair single strand DNA breaks by the base-excision repair (BER) pathway. The specific PARP family member target for PARP inhibitor MK4827 is unknown. (NCI Thesaurus)
Niraparib (originally MK-4827)[1] is an orally active[2] small molecule PARP inhibitor being developed (by Tesaro) to treat ovarian cancer.
It is an inhibitor of PARP1 and PARP2.[3]
Niraparib is due to be submitted for FDA approval (for maintenance therapy in ovarian cancer) later in 2016.[4]
Chemically, MK-4827 is C19H20N4O[5] (ignoring a possible tosylate group).[6]
A 2012 study found that PARP inhibitors exhibit cytotoxic effects not based solely on their enzymatic inhibition of PARP, but by their trapping of PARP on damaged DNA, and the strength of this trapping activity was ordered niraparib >> olaparib >> veliparib.[7]
MEDICINAL CHEMISTRY APPROACH

The Medicinal Chemistry approach to compound 1 is shown in Scheme ABOVE. The racemic piperidine 2 was accessed by reduction of the 3-aryl pyridine 3 and then resolved by salt formation with tartaric acid. Protection of the piperidine nitrogen in enantiomerically upgraded piperidine 2 and condensation with aldehyde 4 afforded imine 5 which, after displacement of the nitro group with sodium azide, underwent a thermally promoted cyclisation to afford the 2-aryl indazole 6. Conversion of the ester functionality to a primary amide and deprotection afforded the active pharmaceutical ingredient (API) as the hydrochloride salt. A final chiral HPLC purification was then required to upgrade the enantiomeric purity to >98% ee, followed by lyophilization to give the desired compound 1 as an amorphous HCl salt.

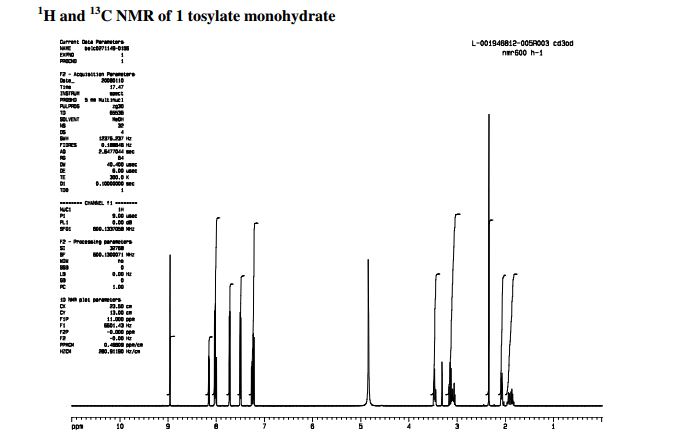
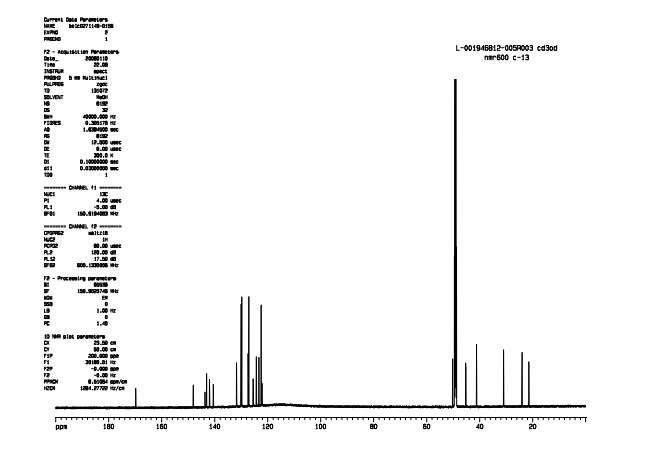
 NMR CD3OD
NMR CD3OD
Clinical trials
It has undergone a phase III trial for ovarian cancer.[8] It is reported that the primary endpoint (progression-free survival, PFS) was met.[4] Patients with and without a BRCA mutation both showed longer PFS.[4]
As of June 2016 seven clinical trials have been registered for MK-4827.[9]
PAPER
http://pubs.acs.org/doi/abs/10.1021/op400233z
Process Development of C–N Cross-Coupling and Enantioselective Biocatalytic Reactions for the Asymmetric Synthesis of Niraparib
Abstract

Process development of the synthesis of the orally active poly(ADP-ribose)polymerase inhibitor niraparib is described. Two new asymmetric routes are reported, which converge on a high-yielding, regioselective, copper-catalyzed N-arylation of an indazole derivative as the late-stage fragment coupling step. Novel transaminase-mediated dynamic kinetic resolutions of racemic aldehyde surrogates provided enantioselective syntheses of the 3-aryl-piperidine coupling partner. Conversion of the C–N cross-coupling product to the final API was achieved by deprotection and salt metathesis to isolate the desired crystalline salt form.
PAPER
http://pubs.acs.org/doi/full/10.1021/op2000783
Development of a Fit-for-Purpose Large-Scale Synthesis of an Oral PARP Inhibitor
Abstract

Compound (1) a poly(ADP-ribose)polymerase (PARP) inhibitor has been made by a fit-for-purpose large-scale synthesis using either a classical resolution or chiral chromatographic separation. The development and relative merits of each route are discussed, along with operational improvements and extensive safety evaluations of potentially hazardous reactions.
 as tosylate H20
as tosylate H20
1613220-15-7 cas
Free form 1038915-60-4
- 2-[4-(3S)-3-Piperidinylphenyl]-2H-indazole-7-carboxamide
- Niraparib
- Jones, Philip; Journal of Medicinal Chemistry 2009, V52(22), P7170-7185
- MK-4827
(S)-2-(4-(Piperidin-3-yl)phenyl)-2H-indazole-7-carboxamide Tosylate Monohydrate 1
Discovery of 2-{4-[(3S)-Piperidin-3-yl]phenyl}-2H-indazole-7-carboxamide (MK-4827): A Novel Oral Poly(ADP-ribose)polymerase (PARP) Inhibitor Efficacious in BRCA-1 and -2 Mutant Tumors
Abstract

We disclose the development of a novel series of 2-phenyl-2H-indazole-7-carboxamides as poly(ADP-ribose)polymerase (PARP) 1 and 2 inhibitors. This series was optimized to improve enzyme and cellular activity, and the resulting PARP inhibitors display antiproliferation activities against BRCA-1 and BRCA-2 deficient cancer cells, with high selectivity over BRCA proficient cells. Extrahepatic oxidation by CYP450 1A1 and 1A2 was identified as a metabolic concern, and strategies to improve pharmacokinetic properties are reported. These efforts culminated in the identification of 2-{4-[(3S)-piperidin-3-yl]phenyl}-2H-indazole-7-carboxamide 56 (MK-4827), which displays good pharmacokinetic properties and is currently in phase I clinical trials. This compound displays excellent PARP 1 and 2 inhibition with IC50 = 3.8 and 2.1 nM, respectively, and in a whole cell assay, it inhibited PARP activity with EC50 = 4 nM and inhibited proliferation of cancer cells with mutant BRCA-1 and BRCA-2 with CC50 in the 10−100 nM range. Compound 56 was well tolerated in vivo and demonstrated efficacy as a single agent in a xenograft model of BRCA-1 deficient cancer.
PATENT
https://www.google.com/patents/WO2014088983A1?cl=en

EXAMPLE 1
The following Example 1 describes synthesis of the compound 2-{4-[(3S)-Piperidin enyl}-2H-indazole-7-carboxamide:
2-{4-[(3S)-Piperidin-3-yl]phenyl}-2H-indazole-7-carboxamide tosylate monohydrate 1
Scheme
1.1 Acylation

2- crystalline
10
A mixture of succinic anhydride 1 (110 g) and bromobenzene (695 mL) was cooled to below 5°C then added A1C13 (294 g). The slurry was allowed to warm to RT and then aged until the reaction was complete judged by HPLC. The reaction mixture was then transferred slowly into a cold HC1 solution resulting in the formation of a white precipitate. The white slurry was filtered through a fritted funnel rinsing with H20. To the off-white product was added MTBE and extracted with aq. NaOH. The aqueous layer was cooled in an ice bath. Concentrated HC1 was added drop wise to adjust the solution pH to 1 , resulting in the formation of a white slurry. The slurry was collected on a fritted funnel, rinsed with H20, and dried under vacuum with a N2 sweep at RT to give the target compound (265 g, 93% corrected yield) as a white powder.
1.2 Esterification

A mixture of the acid 2 (205 g), IPA (4 L) and cone. H2S04 (2.13 mL / 3.91 g) was heated to a gentle reflux until the reaction was complete judged by HPLC. The solution was then cooled to RT and concentrated to a volume of 350-400 mL. The residue was dissolved in
MTBE (1.2 L), washed with aq. Na2C03 followed by water. After dried over MgS04 , the filtrate was solvent-switched into heptane. The slurry was then filtered, and the cake was washed with cold heptane. After drying under vacuum, the target compound (223.5 g, 93% corrected yield) was obtained as a white powder.
1.3 Epoxidation

A mixture of Me3SOI (230 g) and DMSO (300 mL) was added KOt-Bu (113 g) followed by DMSO (300 mL). The mixture was aged for a further 1.5 hr. In a separate flask, ketone 3 (230 g) was dissolved in a mixture of THF (250 mL) and DMSO (150 mL), and the resulting solution was added drop wise to the ylide solution. The mixture was aged for 2 hr at RT, added hexanes (1 L), and then quenched by the addition of ice-water (600 mL). The layers were cut, and the organic layer was washed with water then with brine. The slightly cloudy yellow organic layer was dried over Na2S04 and filtered through a fritted funnel. Product solution assay was 176.1 g (76%> assay yield). This solution was carried forward into the rearrangement step. 1.4 Epoxide rearrangement and bisulfite formation

5 – not isolated

A solution of crude epoxide 4 (assay 59.5 g) in hexanes was solvent switched into PhMe, and added ZnBr2 (10.7 g). When the rearrangement was complete judged by HPLC, the slurry was filtered through a fritted funnel. The clear filtrate was washed with 10% aq. NaCl and then stirred with a solution of sodium bisulfite (NaHS03, 24.7 g) in H20 (140 mL) vigorously at RT for 3 hr. The cloudy aqueous layer was separated and washed with heptanes. By 1H-NMR assay, the aqueous solution contained 71.15 g bisulfite adduct 6 (30.4 wt % solution, 90%) yield from crude epoxide 4). This solution was used directly in the subsequent transaminase step.
1.5 Transaminase DKR

45 C, inert, 40-46 hrs 7
100 g/L as 17.16 wt % aq solution 99.3% ee
85-87% yield
To a cylindrical Labfors reactor was charged pyridoxal-5 -phosphate (1.4 g, 5.66 mmol), 452 ml 0.2 M borate buffer pH 10.5 containing 1M iPrNH2, 52 g transaminase (SEQ ID NO: 180), and 75 ml DMSO, and the resulting mixture was warmed to 45°C. The pH was controlled at pH 10.5 using 8 M aq iPrNH2. To this was added dropwise a mixture of 17.16 wt% aq solution of ester bi-sulfite 6 (147.2 g, 353 mmol) and 219 ml DMSO under N2 atmosphere. When the reaction was complete judged by HPLC, the reaction mixture was cooled and extracted with 1 volume of 3:2 IPA:IPAc. The aq/rag layer was extracted again with 1 volume of 3:7 IPATPAc. The organic layer was washed with brine at pH >9. Assay yield in solution was 78 g (87%); 99.3% ee. After dried over MgS04, and filtered through a fritted funnel, the crude solution was concentrated under vacuum flushing with IP Ac to remove IPA. The resulting slurry was concentrated to a final volume of -200 mL, cool to below 0°C, and filtered to collect the solid. The cake was washed with ice-cold IPAc and dried at RT under vacuum to give the desired product (84% corrected yield, 99.3 LCAP) as a white powder. 1.6. Reduction of amide

(S)-3-(4-bromophenyl)piperidine
The lactam 7 can be reduced to form the i eridine 8 as described below:

7 – crystalline
A mixture of lactam 7 (10.25 g at 97.5 wt %) in THF (100 mL) was cooled to < 10°C, and added NaBH4 (4.47 g). EtOH (6.89 mL) was then added slowly over 20 min. The slurry was aged for an additional 1 hr at 2°C after which BF3 THF (13.03 mL) was added over 1 hr. The slurry was slowly warmed to RT and aged until complete conversion judged by HPLC. The reaction was then cooled to < 5°C then slowly quenched with MeOH (7.96 mL), added HC1 (9.69 mL), then the reaction was heated to 45°C until decomplexation of product-borane complex was complete, as indicated by LC assay. The reaction was cooled, diluted with IPAc (75 mL) and water (80 mL), and then pH was adjusted with aqueous NH4OH to pH 8. The organic layer was separated, added 75 mL water, then pH adjusted to 10.5 with 50 wt % NaOH. The layers were separated and the organic layer was washed with brine. After solvent-switched to IPAc, LC Assay yield was 9.1g; 95.9%.
1.7 Tosylate salt formation The tosylate salt of the piperidine 8 can be formed as described below:

The crude piperidine 8 free base in IPA was heated to ~40°C. TsOH H20 solids was added portion-wise. The slurry was warmed to 50°C and held at that temperature for 2 h, and then slowly cooled to RT and aged overnight. Supernatant concentration was measured to be 2.5 g/ml (free base concentration). The solids were filtered and washed with IP Ac (3×15 mL) and dried at RT. Isolated solides: 14.85 g, 96% corrected isolated yield.
1.8 Boc protection
The piperi ine 8 tosylate salt can be protected as described below:

To a stirred slurry of the tosylate salt of piperidine 8 (25.03 g, 60.6 mmol) in MTBE (375 ml) was added NaOH (aq. 1.0 N, 72.7 ml, 72.7 mmol) at RT. To the mixture, (BOC)20 (13.36 ml, 57.6 mmol) was added slowly over 3 min. The resulting mixture was stirred for 4.5 hr at RT, and then the aqueous layer was separated. The MTBE layer was washed with water (100 ml X 2). The organic layer was filtered, and DMAC (100 ml) was added to the filtrate and
concentrated under vacuum. Product assay: 21.86 g, quantitative yield.
1.9 Terf-Butylamide Formation

N-(tert-butyl)- 1 H-indazole-7-carboxamide

10 11
Indazole-7-carboxylic acid 10 (50.3 g, 295 mmol) was dissolved in DMF, and added CDI (59.1 g, 354 mmol) at RT. After 1.5hr, tert-butylamine (62.5 ml, 589 mmol) was added to the reaction mixture. The resulting reaction mixture was warmed to 40 °C until complete
conversion, then cooled to RT. Water (600 ml) was added dropwise causing the mixture to form a thick slurry. Solid was collected by filtration and washed with 10% DMF in water (250 ml) followed by water. The solid was dried under vacuum. Beige solid: 55.31 g, 86%> isolated yield.
1.10 Carbon-Nitrogen Coupling

(S)-tert-butyl 3-(4-(7-(tert-butylcarbamoyl)-2H-indazol-2-yl)phenyl)piperidine- 1 -carboxylate

A mixture of the protected piperidine 9 (113 g, 18.23 wt%, 60.6 mmol) in DMAc (160 mL), compound 11 (13.82 g, 63.6 mmol), and K2CO3 (25.6 g, 182 mmol) was degassed by bubbling nitrogen. To the mixture was added CuBr (0.444 g, 3.03 mmol) and 8- hydroxyquinoline 12 (0.889 g, 6.06 mmol), and the resulting mixture was warmed to 110°C until complete conversion. The reaction mixture was then cooled, filtered through a pad of Celite, and rinsed with DMAc (100 ml). The filtrate was warmed to 35°C and added citric acid aqueous solution (10%) dropwise to form a light green slurry. After cooled to room temperature, the slurry was filtered, and the cake was washed with DMAc/Water (2/1, 150ml) followed by copious amount of water. The solid was dried under vacuum with nitrogen. Net weight: 27.24g. LC assay: 26.77g, 98.3 wt %. Assay yield: 93.6%.
1.11 Double deprotection

To compound 13 (20.0 g, 41.2 mmol) was added MSA (60 ml) and o-xylene (40 ml), and the the reaction mixture was warmed to 40°C until the complete conversion judged by HPLC. The reaction mixture was cooled to RT and added water (140 ml) slowly maintaining the temperature < 25°C. When the water addition was completed, the organic layer was removed, and the aq. layer was washed with toluene. The aqueous layer was filtered through a glass funnel, and the filtrate was added an aqueos solution of TsOH (11.77g in 23.5 ml) slowly at RT causing a thick slurry to form. Solid was collected by filtration, washed with water, and dried under vacuum. The titled compound was obtained as a white powder. Net weight: 20.6 g. LC assay: 20.0 g, 97.3 wt %. Assay yield: 95.2%.
EXAMPLE 2
The following Example 2 describes synthesis of the trifluoromethylacetate salt of compound 2-{4-[(3S)-Piperidin-3-yl]phenyl}-2H-indazole-7-carboxamide:
2.1 Cumylamide Formation

N-(2-phenylpropan-2-yl)- 1 H-indazole-7-carboxamide

10 1 5
10
To the indazole-7-carboxylic acid 10 (400 mg, 2.47 mmol) in tetrahydrofuran (9.9 mL), was sequentially added HATU (1.13 g, 2.96 mmol), DIPEA (2.15 mL, 12.3 mmol), and cumylamine (500 mg, 3.70 mmol) at 50°C. The reaction was stirred overnight before being concentrated and loaded directly onto a silica column, eluting with 10-30% EtOAc/hexane. The product was collected and concentrated to afford the desired product as a colorless solid (557 mg, 81% yield).
2.2 Carbon-Nitrogen Coupling

-butyl 3-(4-(7-((2-phenylpropan-2-yl)carbamoyl)-2H-indazol-2-yl)phenyl)piperidine- carboxylate

15 16
A sealed vial containing the indazole-7-carboxamide 15 (50 mg, 0.18 mmol), copper(I) iodide (2.6 mg, 0.014 mmol), potassium phosphate tribasic (80 mg, 0.38 mmol), and aryl bromide 9 (73.1 mg, 0.215 mmol) was evacuated and backfilled with argon (x3). Trans-N,N’- dimethylcyclohexane-l,2-diamine (11.3 μΐ,, 0.072 mmol), and toluene (179 μΐ) were then added successively and the sealed vial was heated at 110 °C overnight. The vial was then cooled and toluene (0.30 mL) was added to the slurry. Crude LC/MS indicated >20: 1 selectivity for the desired indazole isomer. The crude product was purified by loading directly onto a Biotage Snap 10G silica column, eluting with 5-50% EtOAc/hexane. The product was collected and concentrated to afford the desired product as a colorless solid (78 mg, 81% yield).
2.3 Double deprotection
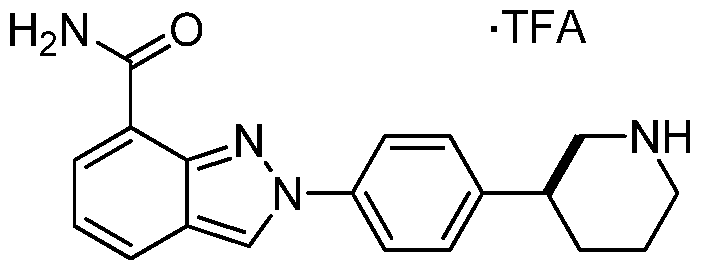
(5)-2-(4-(piperidin-3-yl)phenyl)-2H-indazole-7-carboxamide trifluoromethylacetate salt

16 17
To the piperidine-l-carboxylate 16 (45 mg, 0.084 mmol), was added triethylsilane (267 μί, 1.67 mmol) and TFA (0.965 mL, 12.5 mmol) at 25°C. The reaction was stirred for 4 hours and the reaction was concentrated in vacuo, and purified by mass triggered reverse phase HPLC (acetonitrile: water, with 0.1% TFA modifier). Lyphilization gave the desired product as the TFA salt and as a white solid (31 mg, 85% yield). HRMS (ESI) calc’d for Ci9H2iN40 [M+H]+: 321.1710, found 321.1710.
EXAMPLE 3
Following the conditions used in sections 2.1 and 2.2 of Example 2, this Example 3 shows regioselective N2 arylation of compound 9 using various amide protecting groups. The indazole-7-carboxylic acid 10 was reacted with various amines to generate a protected amide.
The amide protecting groups are indicated by the R group in Table 2. The amide coupling yield is provided in Table 2. The Cu-mediated carbon-nitrogen coupling of this indazole to compound 9 was then tested to determine if regioselective N2 arylation was possible. The arylation yield is also provided in Table 2. The data shows that various amide protecting groups on the indazole intermediate are suitable to generate efficient regioselective N2 arylation of compound 9.


The present invention relates to amide substituted indazoles which are inhibitors of the enzyme poly(ADP-ribose)polymerase (PARP), previously known as poly(ADP-ribose)synthase and poly(ADP-ribosyl)transferase. The compounds of the present invention are useful as monotherapies in tumors with specific defects in DNA-repair pathways and as enhancers of certain DNA-damaging agents such as anticancer agents and radiotherapy. Further, the compounds of the present invention are useful for reducing cell necrosis (in stroke and myocardial infarction), down regulating inflammation and tissue injury, treating retroviral infections and protecting against the toxicity of chemotherapy.
Poly(ADP-ribose) polymerase (PARP) constitute a super family of eighteen proteins containing PARP catalytic domains (Bioessays (2004) 26:1148). These proteins include PARP-1, PARP-2, PARP-3, tankyrase-1, tankyrase-2, vaultPARP and TiPARP. PARP-I, the founding member, consists of three main domains: an amino (N)-terminal DNA-binding domain (DBD) containing two zinc fingers, the automodification domain, and a carboxy (C)-terminal catalytic domain.
PARP are nuclear and cytoplasmic enzymes that cleave NAD+ to nicotinamide and ADP-ribose to form long and branched ADP-ribose polymers on target proteins, including
topoisomerases, histones and PARP itself (Biochem. Biophys. Res. Commun. (1998) 245:1-10).
Poly(ADP-ribosyl)ation has been implicated in several biological processes, including DNA repair, gene transcription, cell cycle progression, cell death, chromatin functions and genomic stability.
The catalytic activity of PARP-I and PARP-2 has been shown to be promptly stimulated by DNA strand breakages (see Pharmacological Research (2005) 52:25-33). In response to DNA damage, PARP-I binds to single and double DNA nicks. Under normal physiological conditions there is minimal PARP activity, however, upon DNA damage an immediate activation of PARP activity of up to 500-fold occurs. Both PARP-I and PARP-2 detect DNA strand interruptions acting as nick sensors, providing rapid signals to halt transcription and recruiting the enzymes required for DNA repair at the site of damage. Since radiotherapy and many chemotherapeutic approaches to cancer therapy act by inducing DNA damage, PARP inhibitors are useful as chemo- and radiosensitizers for cancer treatment. PARP inhibitors have been reported to be effective in radio sensitizing hypoxic tumor cells (US 5,032,617, US
5,215,738 and US 5,041,653).
Most of the biological effects of PARP relate to this poly (ADP-ribosyl)ation process which influences the properties and function of the target proteins; to the PAR oligomers that, when cleaved from poly(ADP-ribosyl)ated proteins, confer distinct cellular effects; the physical association of PARP with nuclear proteins to form functional complexes; and the lowering of the cellular level of its substrate NAD+ (Nature Review (2005) 4:421-440).
Besides being involved in DNA repair, PARP may also act as a mediator of cell death. Its excessive activation in pathological conditions such as ischemia and reperfusion injury can result in substantial depletion of the intercellular NAD+, which can lead to the impairment of several NAD+ dependent metabolic pathways and result in cell death (see Pharmacological Research (2005) 52:44-59). As a result of PARP activation, NAD+ levels significantly decline. Extensive PARP activation leads to severe depletion OfNAD+ in cells suffering from massive DNA damage. The short half-life of poly(ADP-ribose) results in a rapid turnover rate, as once poly(ADP-ribose) is formed, it is quickly degraded by the constitutively active poly(ADP-ribose) glycohydrolase (PARG). PARP and PARG form a cycle that converts a large amount OfNAD+ to ADP-ribose, causing a drop OfNAD+ and ATP to less than 20% of the normal level. Such a scenario is especially detrimental during ischemia when deprivation of oxygen has already drastically compromised cellular energy output. Subsequent free radical production during reperfusion is assumed to be a major cause of tissue damage. Part of the ATP drop, which is typical in many organs during ischemia and reperfusion, could be linked to NAD+ depletion due to poly(ADP-ribose) turnover. Thus, PARP inhibition is expected to preserve the cellular energy level thereby potentiating the survival of ischemic tissues after insult. Compounds which are inhibitors of PARP are therefore useful for treating conditions which result from PARP mediated cell death, including neurological conditions such as stroke, trauma and Parkinson’s disease.
PARP inhibitors have been demonstrated as being useful for the specific killing of BRCA-I and BRCA-2 deficient tumors {Nature (2005) 434:913-916 and 917-921; and Cancer Biology & Therapy (2005) 4:934-936).
PARP inhibitors have been shown to enhance the efficacy of anticancer drugs
{Pharmacological Research (2005) 52:25-33), including platinum compounds such as cisplatin and carboplatin {Cancer Chemother Pharmacol (1993) 33:157-162 and MoI Cancer Ther (2003) 2:371-382). PARP inhibitors have been shown to increase the antitumor activity of
topoisomerase I inhibitors such as Irinotecan and Topotecan (MoI Cancer Ther (2003) 2:371-382; and Clin Cancer Res (2000) 6:2860-2867) and this has been demonstrated in in vivo models (J Natl Cancer Inst (2004) 96:56-67).
PARP inhibitors have been shown to restore susceptibility to the cytotoxic and antiproliferative effects of temozolomide (TMZ) (see Curr Med Chem (2002) 9:1285-1301 and Med Chem Rev Online (2004) 1:144-150). This has been demonstrated in a number of in vitro models (Br J Cancer (1995) 72:849-856; Br J Cancer (1996) 74:1030-1036; MoI Pharmacol (1997) 52:249-258; Leukemia (1999) 13:901-909; GUa (2002) 40:44-54; and Clin Cancer Res (2000) 6:2860-2867 and (2004) 10:881-889) and in vivo models (Blood (2002) 99:2241-2244; Clin Cancer Res (2003) 9:5370-5379 and J Natl Cancer Inst (2004) 96:56-67). PAPR inhibitors have also been shown to prevent the appearance of necrosis induced by selective Λ3 -adenine methylating agents such as MeOSC>2(CH2)-lexitropsin (Me-Lex) {Pharmacological Research (2005) 52:25-33).
PARP inhibitors have been shown to act as radiation sensitizers. PARP inhibitors have been reported to be effective in radiosensitizing (hypoxic) tumor cells and effective in preventing tumor cells from recovering from potentially lethal {Br. J. Cancer (1984) 49(Suppl. VI):34-42; and Int. J. Radial Bioi. (1999) 75:91-100) and sub-lethal {Clin. Oncol. (2004) 16(l):29-39) damage of DNA after radiation therapy, presumably by their ability to prevent DNA strand break rejoining and by affecting several DNA damage signaling pathways.
PARP inhibitors have also been shown to be useful for treating acute and chronic myocardial diseases (see Pharmacological Research (2005) 52:34-43). For instance, it has been demonstrated that single injections of PARP inhibitors have reduced the infarct size caused by ischemia and reperfusion of the heart or skeletal muscle in rabbits. In these studies, a single injection of 3-amino-benzamide (10 mg/kg), either one minute before occlusion or one minute before reperfusion, caused similar reductions in infarct size in the heart (32-42%) while 1,5-dihydroxyisoquinoline (1 mg/kg), another PARP inhibitor, reduced infarct size by a comparable degree (38-48%). These results make it reasonable to assume that PARP inhibitors could salvage previously ischemic heart or reperfusion injury of skeletal muscle tissue {PNAS (1997) 94:679-683). Similar findings have also been reported in pigs {Eur. J. Pharmacol. (1998) 359:143-150 and Ann. Thorαc. Surg. (2002) 73:575-581) and in dogs (Shock. (2004) 21:426-32). PARP inhibitors have been demonstrated as being useful for treating certain vascular diseases, septic shock, ischemic injury and neurotoxicity {Biochim. Biophys. Actα (1989) 1014:1-7; J Clin. Invest. (1997) 100: 723-735). Oxygen radical DNA damage that leads to strand breaks in DNA, which are subsequently recognized by PARP, is a major contributing factor to such disease states as shown by PARP inhibitor studies (J Neurosci. Res. (1994) 39:38-46 and PNAS (1996) 93:4688-4692). PARP has also been demonstrated to play a role in the
pathogenesis of hemorrhagic shock {PNAS (2000) 97:10203-10208).
PARP inhibitors have been demonstrated as being useful for treatment of inflammation diseases (see Pharmacological Research (2005) 52:72-82 and 83-92).
It has also been demonstrated that efficient retroviral infection of mammalian cells is blocked by the inhibition of PARP activity. Such inhibition of recombinant retroviral vector infections has been shown to occur in various different cell types (J Virology, (1996)
70(6): 3992-4000). Inhibitors of PARP have thus been developed for use in anti- viral therapies and in cancer treatment (WO 91/18591).
In vitro and in vivo experiments have demonstrated that PARP inhibitors can be used for the treatment or prevention of autoimmune diseases such as Type I diabetes and diabetic complications {Pharmacological Research (2005) 52:60-71).
PARP inhibition has been speculated as delaying the onset of aging characteristics in human fibroblasts {Biochem. Biophys. Res. Comm. (1994) 201(2):665-672 and Pharmacological Research (2005) 52:93-99). This may be related to the role that PARP plays in controlling telomere function (Nature Gen., (1999) 23(l):76-80).
The vast majority of PARP inhibitors to date interact with the nicotinamide binding domain of the enzyme and behave as competitive inhibitors with respect to NAD+(Expert Opin. Ther. Patents (2004) 14:1531-1551). Structural analogues of nicotinamide, such as benzamide and derivatives were among the first compounds to be investigated as PARP inhibitors.
However, these molecules have a weak inhibitory activity and possess other effects unrelated to PARP inhibition. Thus, there is a need to provide potent inhibitors of the PARP enzyme.
Structurally related PARP inhibitors have previously been described. WO 1999/59973 discloses amide substituted benzene rings fused to 5 membered heteroaromatic rings;
WO2001/85687 discloses amide substituted indoles; WO 1997/04771, WO 2000/26192, WO 2000/32579, WO 2000/64878, WO 2000/68206, WO 2001/21615, WO 2002/068407, WO 2003/106430 and WO 2004/096793 disclose amide substituted benzo imidazoles; WO
2000/29384 discloses amide substituted benzoimidazoles and indoles; and EP 0879820 discloses amide substituted benzoxazoles.
It has now surprisingly been discovered that amide substituted indazoles of the present invention exhibit particularly high levels of inibition of the activity of poly(ADP-ribose)polymerase (PARP). Thus the compounds of the present invention are particularly useful as inhibitors of PARP-I and/or PARP-2. They also show particularly good levels of cellular activity, demonstrating good anti-proliferative effects in BRCAl and BRCA2 deficient cell lines.
The present invention provides compounds of formula I:

Scheme 1
A procedure to synthesize derivatives of those compounds of this invention is shown in scheme 1, whereby the substituted 2H-indazoles are prepared using a synthetic route similar to that described in WO 2005/066136. Following initial conversion of the 2-nitro-3-methyl-benzoic acid derivative into the corresponding ester, radical bromination of the methyl group using reagents like N-bromosuccinimide and benzoyl peroxide yields the key benzyl bromide derivative. Oxidation of this benzylic bromide to the corresponding benzaldehyde can be accomplished for instance using 7V-methylmorpholine-7V-oxide and molecular sieves. Following the condensation of the aldehyde with an amine, ring closure can be accomplished by treating the key intermediate with sodium azide at elevated temperature to introduce the final nitrogen atom and the resultant extrusion of nitrogen to furnish the indazole ring. A base such as lutidine can also be added to this reaction. Final conversion of the ester to the primary amide yields the desired derivatives. This can be accomplished either by heating the ester in an ammonia solution or by conversion to the corresponding carboxylic acid and then amide coupling.

Rx = C1-6alkyl
Oxidation
e.g. NMMO, mol sieves


NH3, THF or MeOH,
700C sealed tube, or
NaOH or KOH, NH3, HATU
or TBTU, DIPEA, DMF, RT
Scheme 1
Scheme 2
A variation of schemes 1 is shown below in scheme 2 and allows the introduction of substituents onto the indazole cores. When the required nitrobenzoic acid derivatives are not commercial available they can be prepared through nitration of the corresponding benzoic acid derivatives, for instance using potassium nitrate in concentrated sulphuric acid. Synthetic manipulations as decribed above allow the formation of the corresponding aniline which can either be cyclised to the indazole by firstly acetylation of the indazole and cyclisation with sodium nitrite in concentrated HCl acid at O0C. Alternatively, the aniline can be diazonitised with nitrosium tetrafluoroborate and the corresponding diazonium tetrafluoroborate salt decomposed at elevated temperatures to the corresponding dilfluorobenzene derivative by a Schiemann reaction
(Caution). Following the synthetic sequence as described in scheme 1 allows oxidation of the benzylic methyl group to the corresponding aldehyde and elaboration of the desired indazole derivatives by coupling with a (hetero)anilide and cyclisation with sodium azide.
Nitration Esterifi cation
KNO3, cone. e.g. AcCI, MeOH,



Reduction
H2, Pd/C

Scheme 2 Scheme 3
An alternative procedure involves functionalisation of the indazole at a late stage as shown in scheme 3. Here the indazole ester is first converted to the corresponding carboxamide and the subjected to nucleophilic aromatic substitution of the appropriate fluoro(hetero)aromatic bromide. This allows the preparation of a bromide derivative that can be cross coupled under Suzuki coupling conditions, for instance using tri(tert-butyl)phosphine and Pd2(dba)3 as catalysts in the presence of a base, such as sodium carbonate. Conversion to the desied piperidine moiety is then accomplished by a Fowler reaction using an acyl chloride, such as CBz-Cl and a reducing agent such as NaBH4. Final hydrogenation reaction can yield the corresponding piperidine derivatives.

Suzuki coupling

Scheme 3
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US8071623 * | Jan 8, 2008 | Dec 6, 2011 | Instituto Di Ricerche Di Biologia Molecolare P. Angeletti Spa | Amide substituted indazoles as poly(ADP-ribose)polymerase(PARP) inhibitors |
| US8129377 * | Sep 29, 2005 | Mar 6, 2012 | Mitsubishi Tanabe Pharma Corporation | 6-(pyridinyl)-4-pyrimidone derivates as tau protein kinase 1 inhibitors |
| US20100286203 * | Jan 8, 2009 | Nov 11, 2010 | Foley Jennifer R | Pharmaceutically acceptable salts of 2–2h-indazole-7-carboxamide |
| Reference | ||
|---|---|---|
| 1 | * | CHUNG ET AL.: “Process Development of C-N Cross-Coupling and Enantioselective Biocatalytic Reactions for the Asymmetric Synthesis of Niraparib.“, ORGANIC PROCESS RESEARCH & DEVELOPMENT, vol. 18, no. 1, 2014, pages 215 – 227, XP055263728 |
| 2 | * | JONES ET AL.: “Discovery of 2-(4-[(3S)-Piperidin-3-yl]phenyl}-2H-indazole-7-carboxamide ( MK -4827): A Novel Oral Poly(ADP-ribose)polymerase (PARP) Inhibitor Efficacious in BRCA-1 and -2 Mutant Tumors.“, JOURNAL OF MEDICINAL CHEMISTRY, vol. 52, no. 22, 2009, pages 7170 – 7185, XP055263725 |
| 3 | * | WALLACE ET AL.: “Development of a Fit-for-Purpose Large-Scale Synthesis of an Oral PARP Inhibitor.“, ORGANIC PROCESS RESEARCH & DEVELOPMENT, vol. 15, no. 4, 2011, pages 831 – 840, XP055263721 |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2016025359A1 * | Aug 10, 2015 | Feb 18, 2016 | Merck Sharp & Dohme Corp. | Processes for the preparation of a bace inhibitor |
References
- Jump up^ “PARP Inhibitors in Oncology. Chemosensitizers or Single-Agent Therapeutics?” (PDF). July 2009.
- Jump up^ A Phase III Trial of Niraparib Versus Physician’s Choice in HER2 Negative, Germline BRCA Mutation-positive Breast Cancer Patients (BRAVO)
- Jump up^ “PARP inhibitor, MK-4827, shows anti-tumor activity in first trial in humans”. 17 Nov 2010.
- ^ Jump up to:a b c Tesaro’s PARP ovarian cancer drug hits PhIII goal; prepares to file. June 2016
- Jump up^ MK-4827 @ pubchem
- Jump up^ MK-4827
- Jump up^ Murai, Junko, et al. “Trapping of PARP1 and PARP2 by clinical PARP inhibitors.” Cancer research 72.21 (2012): 5588-5599.
- Jump up^ AbbVie takes PARP inhibitor into third phase III trial. June 2014
- Jump up^ Niraparib studies
Further reading
 |
|
| Clinical data | |
|---|---|
| Routes of administration |
By mouth |
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | 1038915-60-4 |
| PubChem (CID) | 24958200 |
| ChemSpider | 24531930 |
| UNII | HMC2H89N35 |
| ChEMBL | CHEMBL1094636 |
| Chemical and physical data | |
| Formula | C19H20N4O |
| Molar mass | 320.394 g/mol |
| 3D model (Jmol) | Interactive image |


[…] NIRAPARIB […]
LikeLike