
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
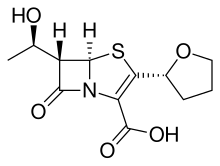
- Molecular FormulaC12H15NO5S
- Average mass285.316 Da
Faropenem
7086
(+)-(5R,6S)-6-((1R)-1-Hydroxyethyl)-7-oxo-3-((2R)-tetrahydro-2-furyl)-4-thia-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic Acid
(5R,6S)-6-[(1R)-1-Hydroxyethyl]-7-oxo-3-[(2R)-tetrahydro-2-furanyl]-4-thia-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid
(5R,6S)-6-[(1R)-1-Hydroxyethyl]-7-oxo-3-[(2R)-tetrahydrofuran-2-yl]-4-thia-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid
106560-14-9[RN]
4-Thia-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid, 6-[(1R)-1-hydroxyethyl]-7-oxo-3-[(2R)-tetrahydro-2-furanyl]-, (5R,6S)-
6α-[(R)-1-hydroxyethyl]-2-[(R)-tetrahydrofuran-2-yl]pen-2-em-3-carboxylic acid
4-Oxofenretinide
4-Oxo-N-(4-hydroxyphenyl)retinamide
6α-[(1R)-1-hydroxyethyl]-2-[(2R)-tetrahydrofuran-2-yl]-2,3-didehydropenam-3-carboxylic acid
7305146 [Beilstein]
FaropenemCAS Registry Number: 106560-14-9
CAS Name: (5R,6S)-6-[(1R)-1-Hydroxyethyl]-7-oxo-3-[(2R)-tetrahydro-2-furanyl]-4-thia-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid
Additional Names: fropenem; (5R,6S,8R,2¢R)-2-(2¢-tetrahydrofuryl)-6-hydroxyethylpenem-3-carboxylate
Molecular Formula: C12H15NO5S
Molecular Weight: 285.32
Percent Composition: C 50.51%, H 5.30%, N 4.91%, O 28.04%, S 11.24%
Literature References: Orally active, b-lactamase stable, penem antibiotic.Prepn: M. Ishiguro et al.,EP199446; eidem,US4997829 (1986, 1991 both to Suntory); eidem,J. Antibiot.41, 1685 (1988).Pharmacokinetics: A. Tsuji et al.,Drug Metab. Dispos.18, 245 (1990). In vitro antimicrobial spectrum: J. M. Woodcock et al.,J. Antimicrob. Chemother.39, 35 (1997). b-Lactamase stability: A. Dalhoff et al., Chemotherapy (Basel)49, 229 (2003).HPLC determn in plasma: R. V. S. Nirogi et al., Arzneim.-Forsch.55, 762 (2005). Clinical trial in urinary tract infections: S. Arakawa et al.,Nishinihon J. Urol.56, 300 (1994); in bacterial sinusitis: R. Siegert et al., Eur. Arch. Otorhinolaryngol.260, 186 (2003).
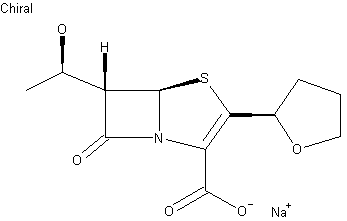
Derivative Type: Sodium salt
CAS Registry Number: 122547-49-3
Additional Names: Furopenem
Manufacturers’ Codes: ALP-201; SUN-5555; SY-5555; WY-49605
Trademarks: Farom (Daiichi)
Molecular Formula: C12H15NNaO5S
Molecular Weight: 308.31
Percent Composition: C 46.75%, H 4.90%, N 4.54%, Na 7.46%, O 25.95%, S 10.40%
Properties: [a]D22 +60° (c = 0.10).
Optical Rotation: [a]D22 +60° (c = 0.10)
Derivative Type: Daloxate
CAS Registry Number: 141702-36-5
CAS Name: (5R,6S)-6-[(1R)-1-Hydroxyethyl]-7-oxo-3-[(2R)-tetrahydro-2-furanyl]-4-thia-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid (5-methyl-2-oxo-1,3-dioxol-4-yl)methyl ester
Additional Names: faropenem medoxomil
Manufacturers’ Codes: Bay-56-6854; SUN-208
Trademarks: Orapem (Replidyne)
Molecular Formula: C17H19NO8S
Molecular Weight: 397.40
Percent Composition: C 51.38%, H 4.82%, N 3.52%, O 32.21%, S 8.07%
Literature References: Prepn: H. Iwata et al., WO9203442; eidem, US5830889 (1992, 1998 both to Suntory).
Properties: Pale yellow crystals.
Therap-Cat: Antibacterial (antibiotics).
Keywords: Antibacterial (Antibiotics); ?Lactams; Penems.
Faropenem is an orally active beta-lactam antibiotic belonging to the penem group.[1] It is resistant to some forms of extended-spectrum beta-lactamase.[2] It is available for oral use.[3]

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
Forms
Faropenem was developed by Daiichi Asubio Pharma, which markets it in two forms.
- The sodium salt faropenem sodium, available under the trade name Farom, has been marketed in Japan since 1997. (CID 636379 from PubChem)
- The prodrug form faropenem medoxomil[4] (also known as faropenem daloxate) has been licensed from Daiichi Asubio Pharma by Replidyne, which plans to market it in conjunction with Forest Pharmaceuticals. The trade name proposed for the product was Orapem, but company officials recently announced this name was rejected by the FDA.[5]
Clinical use
As of 8 September 2015, Faropenem has yet to receive marketing approval in the United States, and was submitted for consideration by the United States Food and Drug Administration (FDA) on 20 December 2005. The new drug application dossier submitted included these proposed indications:
- acute bacterial sinusitis
- community-acquired pneumonia
- acute exacerbations of chronic bronchitis
- uncomplicated skin and skin structure infections
- urinary tract infections
History
The FDA refused to approve faropenem, an antibiotic manufactured by Louisville-based Replidyne. The FDA said the drug was “nonapprovable”, but did not refer to specific safety concerns about the product. The company will have to conduct new studies and clinical trials, lasting an estimated two more years, to prove the drug treats community-acquired pneumonia, bacterial sinusitis, chronic bronchitis, and skin infections.[citation needed]
In India it is available as Farobact 200/300ER CIPLA.
PATENT
https://patents.google.com/patent/WO2008035153A2/enFaropenem is an orally active β-lactam antibiotic belonging to the penem group. Faropenem is chemically known as 6-(l-hydroxyethyl)-7-oxo-3-(oxolan-2-yl)-4-thia-l-azabicyclo[3.2.0]hept-2-ene-2-carboxylicacid. The known forms of Faropenem are Faropenem sodium and the prodrug form, FaropenemMedoxomil (also known as Faropenem Daloxate). In view of the importance of the compound of the formula (I), several synthetic procedures to prepare the compound have been reported.US 4,997,829 provides process for the preparation of faropenem according to the following scheme. The process is exemplified with the allyl protected carboxyl group. One of the process involves the reaction of A- acetoxyazetidinone with tetrahydrothiofuroic acid, condensation with allyl glyoxalate in refluxing benzene, chlorination with thionyl chloride, reaction of triphenylphosphine with lutidine in hot THF, cyclization in refluxing toluene, deprotection of silyl protecting group with tetrabutylammonium fluoride, treating with triphenylphosphine and, treating with sodium 2-ethylhexanoate and (PP^)4Pd to result faropenem sodium. The process exemplified utilizes benzene as solvent, which is not environmentally acceptable. Tetrabutylammonium fluoride was used as desilylating agent that is expensive. Even though the description teaches that optically active compounds can be employed, the examples utilized the dl-compound of tetrahydrothiofuroic acid further requiring resolution.

Methods are provided for the synthesis of series of penem compounds in J Antibiotics 1988, 41(11), 1685-1693. The provided methods utilize sulfonylazetidinone as the starting materials. As one of the procedures gives lesser yield, another procedure was adopted which uses silver salts.Japanese patent, JP2949363 describes a process for deallylation and salt formation with an alkali metal salt of carboxylic acid in the presence of a catalytic amount of palladium complex for the preparation of faropenem.EP410727 describes a process for removing allyl group from a penem compound using cyclic 1,3-diketone such as dimedone.The yield and quality of the final product is always less in the above prior art methods. With the continued research, the present inventors have undertaken extensive studies for developing a process for the preparation of compound of formula (I), which is commercially viable, involves simple techniques such as crystallizations, with improved yields and quality of the product, and with lesser reaction time. None of the prior art suggests or teaches the techniques provided herein.The process is shown in Scheme-I as given below:

One-pot process for the preparation of Faropenem sodium:Sodium salt of R(+)-tetrahydrofuran-2-thiocarboxylic acid (67 g) in aqueous acetone was added slowly to a solution of AOSA (100 g) in acetone (200 mL) and stirred for 3 h at pH 8.0 to 8.5 using sodium bicarbonate solution.After completion of the reaction, the product was extracted with toluene. The combined toluene layer was washed with saturated sodium bicarbonate solution and brine solution. Toluene was removed under vacuum completely and the mass obtained, 3-(l’-tert-butyldimethylsilyloxyethyl)-4-(2′- tetrahydrofuranoylthio)-2-azetidinone was directly taken for next step.3-(r-tert-Butyldimethylsilyloxyethyl)-4-(2′-tetrahydrofuranoylthio)-2- azetidinone obtained was dissolved in toluene (1000 mL) and cooled to -10 to -5 °C under nitrogen. Triethylamine (124 mL) was added to it followed by allyl oxalyl chloride (82 g) at -10 to- 5 0C for 2 h. After completion of the reaction, cold water was added to the mass and washed with dilute hydrochloric acid and sodium bicarbonate solution. Toluene layer was separated and washed with purified water. The toluene layer containing compound of formula (VI) was concentrated under vacuum at 50 to 60 °C and taken for next step as such.Compound of formula (VI) (150 g) was dissolved in triethyl phosphite (150 mL), heated to 60 0C and stirred under nitrogen atmosphere. Toluene (3000 mL) was added, heated to 100 to 110 °C and stirred for 20- 24 h. Toluene was distilled under vacuum completely. Product obtained, allyl (1 ‘R,2″R,5R,6S)-6-(l 5-tert-butyldimethylsilyloxyethyl)-2-(2″-tetrahydrofuranyl) penem-3-carboxylate (VII) was directly taken for next step.Compound (VII) obtained was dissolved in DMF (700 mL) at 30 °C.Ammonium hydrogen difluoride (80 g) and NMP (210 mL) were added and stirred at room temperature for 25 to 35 h. The reaction mass was quenched into a mixture of water-ethyl acetate and stirred at room temperature. The ethyl acetate layer was separated and the aqueous layer extracted with ethyl acetate. ■ The combined ethyl acetate layer was washed with water followed by saturated sodium bicarbonate solution. The ethyl acetate layer was charcoal treated. The ethyl acetate layer containing allyl (l’R,2″R,5R,6S)-6-(l’-hydroxyethyl)-2-(2″- tetrahydrofuranyl)penem-3-carboxylate (XII) was partially distilled and taken for the next step.The ethyl acetate layer containing compound of formula (XII), Pd/C, sodium bicarbonate and purified water (1000 mL) were taken in an autoclave and maintained 5 to 10 kg pressure of hydrogen gas for 2-5 h. After completion of the reaction the Pd/C was filtered off and ethyl acetate layer separated. The pH of the mass was adjusted to 1.5 and extracted with ethyl acetate. The aqueous layer was extracted again with ethyl acetate twice. The combined ethyl acetate layer was carbon treated. Sodium-2-ethylhexanoate in ethyl acetate was added slowly and stirred. The precipitated title compound was filtered under vacuum, washed with acetone and dried. Dry weight of the product: 65-75 g.Example 9Purification of Faropenem sodiumCrude Faropenem sodium (50 g) was dissolved in purified water (200 mL) at 25-30 0C. The solution was charcoalised. Acetone (1500 mL) was added. The reaction mass was stirred further for 10 min. The precipitated solid was cooled to 0 —2 °C then filtered, washed with acetone and dried at room temperature. Weight of pure Faropenem sodium is 43 to 46 g (Purity 99.95%).Example 9aPurification of Faropenem sodiumCrude Faropenem sodium (50 g) was dissolved in purified water (200 mL) at 25-30 °C. Acetone (150O mL) was added. The reaction mass was stirred further for 10 min. The precipitated solid was cooled to 0-2 °C then filtered, washed with acetone and dried at room temperature. Weight of pure Faropenem sodium is 43 to 46 g (Purity 99.95%).
PATENT
https://patents.google.com/patent/CN103880864B/enFaropenem sodium is developed by Japanese Suntory companies, and first penemss antibiosis in listing in 1997 Element, it are similar to the several carbapenem antibiotics for listing, strong with has a broad antifungal spectrum, antibacterial activity, to beta-lactamase Stably, the features such as also having good action to extended spectrumβ-lactamase producing strains, citrobacter, enterococcus and anaerobe etc.. It is first orally active, penems antibiotics stable to beta-lactamase in the world so far.Its structural formula As follows:
Report about Faropenem sodium preparation method is a lot, mainly has several as follows:1st, J. Antibiotics 1988, the method that reports in 41,1685, see below row reaction equation:
Acyl group substitution reaction is carried out in the basic conditions with 4-AA and three beneze methane thiols and obtains thio trityl as protecting group Aza cyclo-butanone, then when 2-TETRAHYDROFUROYL chlorine is connected with lactams, using silver nitrate as condensing agent, but nitric acid Silver is expensive, and cost is too high, while the silver chloride for generating is difficult to filter, is not suitable for large-scale production.2nd, the classical preparation method of United States Patent (USP) US4997829 report:There is acyl with (R) tetrahydrofuran -2- thiocarboxylic acids Base substitution reaction generates thioesters, then through condensation, chlorine replacement, intramolecular Witting cyclization, slough hydroxyl protecting group and carboxylic Base protection group obtains product, and this synthetic route yield is very low, while side chain is thio-compoundss, abnormal smells from the patient is extremely smelly, and prepares complexity, There is-fixed harm to human body and environment.It is also required in chloro building-up process using pungent thionyl chloride, these factors are all It is unfavorable for industrialized production
3rd, the method that reports in Chinese patent CN1314691 is as follows:
Said method route is shorter, is produced using one kettle way, more convenient.But said method is related to some other salt such as acetate using heavy metal palladium in last operation The deprotecting regent of compound and triphenyl phosphorus together as pi-allyl, metal palladium reagent is expensive, while triphenyl phosphorus are most More difficult removing in step afterwards, increases operation difficulty, affects product quality.Allyloxy is used easily to produce as protection group simultaneously A kind of double bond olefinic polymerization species impurity of life, affects product quality, reduces yield.Embodiment one(R) tetrahydrofuran -2- thiocarboxylic acids (198g, 1.5 mol) are put in 3L reaction bulbs, plus 1 mol/L hydrogen-oxygens Change sodium body lotion (I.5 L) to be adjusted at 5 DEG C of pH 9- 10,0-, Deca 4AA(287g, 1. 0mo l) acetone (1 L) Solution, drop are finished, and are adjusted to pH 8 or so, 2 h of room temperature reaction with 1 mol/L sodium hydroxide. and add water (500 ml) dilution, second Acetoacetic ester (600 ml x3) is extracted, and merges organic layer, successively with 5 % sodium bicarbonate solutions (300 ml x 2) and water (300 m1 x 2) is washed, and anhydrous sodium sulphate is dried, and is filtered, and filtrate concentrates, and obtains pale yellow oil (about 360 g), directly Input the next step.Embodiment twoThe mixing of concentrated solution as obtained above, triethylamine (l70g, 1.7 mol) and dichloromethane (1.5 L), 0-5 DEG C Deca chlorine oxalic acid is finished to p-Nitrobenzyl (414.1 g, 1 .7 mo l), drop, and equality of temperature reacts 2 h, and add water (1 L) dilution, Extracted with dichloromethane (500 ml x 4), merge organic layer, molten with water (300m1 x 2) and 5 % sodium bicarbonate successively Liquid (300 m1 x 2) is washed, anhydrous sodium sulfate drying, is filtered, and concentration obtains pale yellow oil (about 530g), direct plunges into The next step..Embodiment threeAbove-mentioned gained grease, dimethylbenzene (4L) and NSC 5284 (500ml) are mixed, heating reflux reaction 5h , reduce pressure and boil off dimethylbenzene and NSC 5284, residue ethyl acetate-hexane (1:5,1 L) recrystallization, obtain yellowish Color solid (334.3g, 61%, in terms of 4AA).Example IVAbove-mentioned solid (0.60 mol of 330g.) is dissolved in methanol (2 L), adds 1.0M hydrochloric acid (0.4 L), adds palladium carbon (15.0 g), hydrogen is passed through, 40 DEG C of stirrings, response time are 16 h, and the pressure of system is 4atm, after reaction terminates, crosses and filters Catalyst is removed, is concentrated.Embodiment fiveThe product obtained after above-mentioned concentration is dissolved in tetrahydrofuran 600ml, the 2 ethyl hexanoic acid sodium of 100.0g is added Tetrahydrofuran(200ml)And water(200 ml)Mixed solution, 2 h are stirred at room temperature, have faint yellow solid generate, filter, be method Faropenem crude product 147.0g.Embodiment sixBy above-mentioned solid deionized water(2200ml)Acetone is slowly added under dissolved solution, stirring to start to become to solution Muddiness, when about adding acetone 750ml, solution starts to become cloudy, and stops adding, and continues stirring and allows its crystallize overnight, sucking filtration, acetone Washing, dries, and obtains the Faropenem sodium fine work 125.0g of white.
Syn
AU 8654460; EP 0199446; JP 1994128267; US 4997829
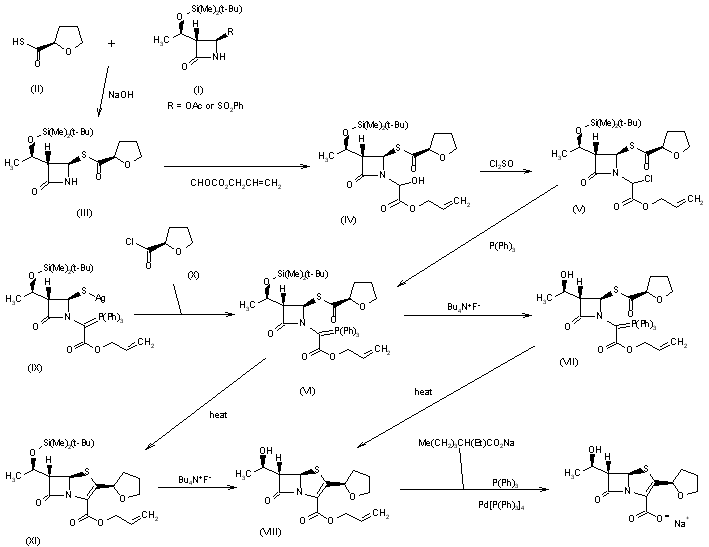
This compound is prepared by several related ways: 1) The reaction of silylated azetidinone (I) with tetrahydrofuran-2-thiocarboxylic acid (II) by means of NaOH in THF – water gives the azetidinone thioester (III), which is condensed with allyl glyoxylate in refluxing benzene yielding the hydroxyester (IV). The reaction of (IV) with SOCl2 affords the chloroester (V), which by reaction with triphenylphosphine by means of lutidine in hot THF is converted into the phosphoranylidene derivative (VI). The elimination of the silyl protecting group of (VI) with tetrabutylammonium fluoride gives the azetidinone (VII), which is cyclized in refluxing toluene yielding the (5R,6S)-6-[1(R)-hydroxyethyl]-2-[2(R)-tetrahydrofuryl]penem-3-carboxyli c acid allyl ester (VIII). Finally, this compound is hydrolyzed with triphenylphosphine, sodium 2-ethylhexanoate and Pd-tetrakis(triphenylphosphine). 2) The condensation of the silver salt of protected azetidinone (IX) with tetrahydrofuran-2(R)-carbonyl chloride (X) also yields the phosphoranylidene salt (VI). 3) Phosphoranylidene ester (VI) can also be cyclized first in refluxing benzene yielding the silylated penem ester (XI), which is deprotected with tetrabutylammonium fluoride to (VIII). 4) The hydrolysis of allyl ester (VIII) to the final product can also be performed with paladium tetrakis(triphenylphosphine) and sodium 4-(methoxycarbonyl)-5,5-dimethylcyclohexane-1,3-dione enolate in several different solvents such as methyl acetate, ethylacetate, tetrahydrofuran, dioxane, sec-butanol, acetonitrile, acetone, 2-butanone, 1,2-dichloroethane, chlorobenzene, toluene, or ethylene glycol dimethyl ether. 5) The preceding hydrolysis can also be performed with triphenylphosphine and paladium tetrakis(triphenylphosphine) with sodium propionate, sodium acetate or sodium lactate in tetrahydrofuran or acetone.
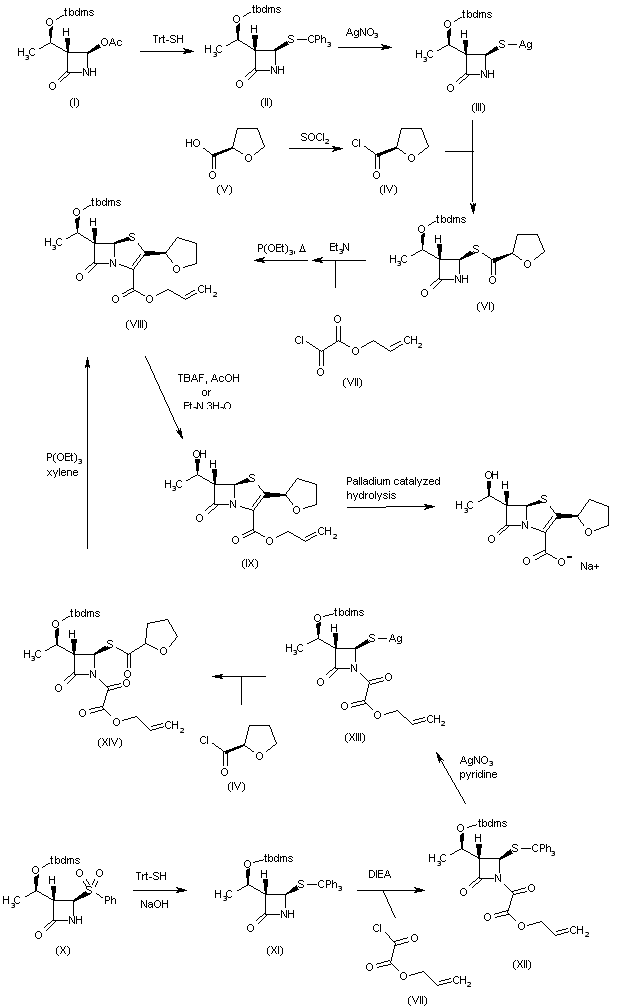
Treatment of the silylated azetidinone (I) with tritylmercaptan affords the tritylsulfanyl-azetidinone (II), which is converted into the silver salt (III) by reaction with AgNO3. Compound (III) is coupled with tetrahydrofuran-2(R)-carbonyl chloride (IV) — obtained by treatment of carboxylic acid (V) with thionyl chloride — providing the azetidinone thioester (VI). Coupling of azetidinone (VI) with allyl oxalyl chloride (VII) in CH2Cl2 by means of Et3N, followed by intramolecular Wittig cyclization by means of triethyl phosphite in refluxing xylene, affords penem (VIII). Alternatively, compound (VIII) can also be obtained as follows: Substitution of phenyl sulfonyl group of azetidinone (X) by tritylmercaptan by means of NaOH in acetone/water provides tritylsulfanyl-azetidinone (XI), which is condensed with allyl oxalyl chloride (VII) by means of DIEA in CH2Cl2 to give the oxalyl amide (XII). Compound (XII) is then treated with AgNO3 and pyridine in acetonitrile, providing the silver mercaptide (XIII), which is acylated with tetrahydrofuran-2(R)-carbonyl chloride (IV) in acetonitrile to afford the penem precursor (XIV). Penem (VIII) is obtained by intramolecular Wittig cyclization of (XIV) with P(OEt)3 in refluxing xylene. Finally, faropenem sodium can be obtained by removal of the tbdms protecting group of (VIII) by means of either Et3N tris(hydrogen fluoride) in ethyl acetate or tetrabutylammonium fluoride (TBAF) and HOAc in THF to give compound (IX). This is followed by allyl ester group removal of (IX), which can be performed under several different conditions: i) triphenylphosphine, sodium 2-ethylhexanoate and palladium tetrakis(triphenylphosphine); ii) palladium tetrakis(triphenylphosphine) and sodium 4-(methoxycarbonyl)-5,5-dimethylcyclohexane-1,3-dione enolate in several different solvents such as methyl acetate, ethyl acetate, tetrahydrofuran, dioxane, sec-butanol, acetonitrile, acetone, 2-butanone, 1,2-dichloroethane, chlorobenzene, toluene or ethylene glycol dimethyl ether; iii) triphenylphosphine and palladium tetrakis(triphenylphosphine) with sodium propionate, sodium acetate or sodium lactate in tetrahydrofuran or acetone; or iv) palladium acetate in the presence of P(OBu)3 and sodium propionate in THF.
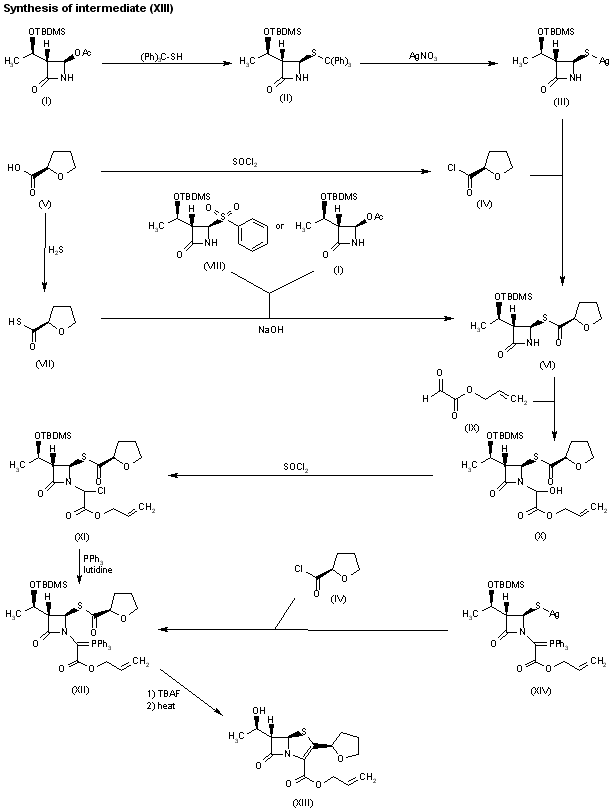
Treatment of the silylated azetidinone (I) with tritylmercaptan affords the tritylsulfanylazetidinone (II), which by reaction with AgNO3 is converted into the silver salt (III). Compound (III) is coupled with tetrahydrofuran-2(R)-carbonyl chloride (IV) ?obtained by treatment of carboxylic acid (V) with thionyl chloride ?to provide the azetidinone thioester (VI). Alternatively, compound (VI) can be obtained by condensation of tetrahydrofuran-2(R)-thiocarboxylic S-acid (VII) ?obtained by treatment of carboxylic acid (V) with hydrogen sulfide ?with silylated azetidinones (I) or (VIII) by means of NaOH in THF/water. Condensation of azetidinone thioester (VI) with allyl glyoxylate (IX) in refluxing benzene gives the hydroxy ester (X), which is treated with SOCl2 to yield the chloro ester (XI). Reaction of compound (XI) with triphenylphosphine and lutidine in hot THF provides the phosphoranylidene derivative (XII), which is converted into (5R,6S)-6-[1(R)-hydroxyethyl]-2-[2(R)-tetrahydrofuryl]penem-3-carboxylic acid allyl ester, faropenem allyl ester (XIII) by removal of the silyl protecting group with tetrabutylammonium fluoride, followed by cyclization in refluxing toluene. Compound (XII) can also be obtained by condensation of the silver salt of protected azetidinone (XIV) with tetrahydrofuran-2(R)-carbonyl chloride (V).
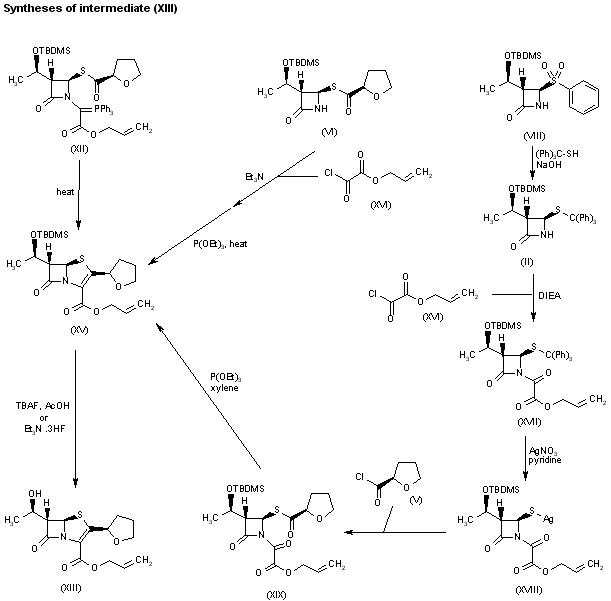
Alternatively, faropenem allyl ester (XIII) can also be prepared by cyclization of compound (XII) in refluxing benzene to yield silylated penem allyl ester (XV), which is then deprotected with either tetrabutylammonium fluoride in AcOH or triethylamine tris(hydrogen fluoride) in methyl isobutyl ketone or toluene. Penem (XV) can also be synthesized by several related ways: a) By coupling of azetidinone (VI) with allyl oxalyl chloride (XVI) in CH2Cl2 by means of Et3N, followed by intramolecular Wittig cyclization by means of triethyl phosphite in refluxing xylene. b) Substitution of phenyl sulfonyl group of azetidinone (VIII) by tritylmercaptan by means of NaOH in acetone/water provides tritylsulfanyl-azetidinone (II), which is condensed with allyl oxalyl chloride (XVI) by means of DIEA in CH2Cl2 to give the oxalyl amide (XVII). Compound (XVII) is then treated with AgNO3 and pyridine in acetonitrile to provide the silver mercaptide (XVIII), which is acylated with tetrahydrofuran-2(R)-carbonyl chloride (IV) in acetonitrile to afford the penem precursor (XIX). Finally, compound (XV) is obtained by intramolecular Wittig cyclization of (XX) with P(OEt)3 in refluxing xylene.
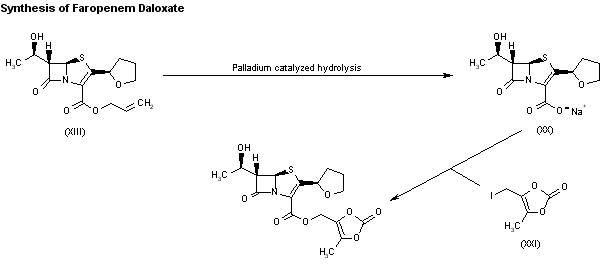
Hydrolysis of faropenem allyl ester (XIII) to faropenem sodium (XX) can be performed under several different conditions: i) triphenylphosphine, sodium 2-ethylhexanoate and palladium tetrakis(triphenylphosphine); ii) palladium tetrakis(triphenylphosphine) and sodium 4-(methoxycarbonyl)- 5,5-dimethylcyclohexane-1,3-dione enolate in several different solvents such as methyl acetate, ethyl acetate, tetrahydrofuran, dioxane, sec-butanol, acetonitrile, acetone, 2-butanone, 1,2-dichloroethane, chlorobenzene, toluene, or ethylene glycol dimethyl ether; iii) triphenylphosphine and palladium tetrakis(triphenylphosphine) with sodium propionate, sodium acetate or sodium lactate in tetrahydrofuran or acetone; and iv) palladium acetate in the presence of P(OBu)3 and sodium propionate in THF. Finally, faropenem daloxate can be directly obtained from faropenem sodium (XX) by esterification with 4-(iodomethyl)-5-methyl-1,3-dioxol-2-one (XXI) in DMF.
PATENT
https://patents.google.com/patent/CN103059046A/enFaropenem (Faropenem), chemistry (5R, 6S)-6-[(1R)-hydroxyethyl by name]-2-[(2R)-and tetrahydrofuran (THF)] penem-3-carboxylic acid list sodium salt, by the first exploitation listing in 1997 years of Japanese Suntory company.This medicine is a kind of atypical beta-lactam penems antibiotics, has very strong anti-microbial activity, especially to the anti-microbial activities of the anerobes such as the gram positive organisms such as golden Portugal bacterium, penicillin-fast streptococcus pneumoniae, streptococcus faecium and bacteroides fragilis apparently higher than existing cynnematin, anti-gram-negative bacteria is active similar to oral cephalosporin, and is stable to various β-lactamases.Various clinical studyes show that this medical instrument has clinical effectiveness good, safe, the advantage that renal toxicity and neurotoxicity are little.Its structural formula is as follows:
For synthesizing of Faropenem, existing many reports in the prior art, for example CN101125857A has reported following synthetic route:
Take (3R, 4R)-3-[(R)-1-tert-butyl dimethyl silica ethyl]-4-[(R)-and acetoxyl group] nitrogen heterocyclic din-2-ketone is as starting raw material, and warp gets intermediate compound I with R-(+)-sulfo-tetrahydrofuran (THF)-2-formic acid condensation; Intermediate compound I is carried out acylation reaction with monoene propoxy-oxalyl chloride under the catalysis of alkali, get intermediate II; Intermediate II cyclization under the effect of triethyl-phosphite gets intermediate III; Intermediate III is sloughed hydroxyl protecting group through the effect of tetrabutylammonium, gets intermediate compound IV; Intermediate compound IV decarboxylize protecting group under [four (triphenylphosphine)] palladium and triphenylphosphine effect gets Faropenem.Find that after deliberation the method for the present synthetic Faropenem of reporting is all similar with the disclosed method of above-mentioned CN101125857A, all need remove in two steps the protecting group of hydroxyl and carboxyl, reaction scheme is longer.When removing above-mentioned protecting group, need to use a large amount of tetrabutylammonium and [four (triphenylphosphine)] palladium and triphenylphosphine; these reagent costs are high, toxicity is large; be unfavorable for large industrial production; and can introduce the heavy metal palladium; so that the heavy metal remnants in the Faropenem exceed standard, be not suitable for the production of bulk drug.And when adopting aforesaid method deprotection base, the yield in per step only can reach 60%-75%, has further increased production cost.Embodiment 6The preparation of FaropenemWith intermediate 3(364.5g, 0.8mol) use the 700mL acetic acid ethyl dissolution, to open and stir, 0 ℃ of lower dropping with the 36g trifluoroacetic acid after the dilution of 100mL ethyl acetate dripped off in 1 hour, 0 ℃ of lower reaction 2h that continues.Stopped reaction stirs the sodium bicarbonate aqueous solution of lower dropping 5%, until reaction solution pH is neutral.Emit water layer from the reactor lower end, discard.In reactor, add gradually the ethanolic soln of sodium bicarbonate, until till no longer including solid and separating out.Suction filtration, filter cake gets white solid powder 230g(productive rate 93.7% with acetone-water (10:3, v/v) recrystallization), M.P. 163-164 ℃, detect through HPLC, purity is 99.8%Reference examples 1(5R, 6S)-6-[(R)-1-hydroxyethyl]-2-[(R)-and the 2-TETRAHYDROFUROYL sulfenyl] preparation of penem-3-carboxylic acid propyleneWith (5R, 6S)-6-[(R)-the 1-tert-butyl dimethyl silica ethyl]-2-[(R)-and the 2-TETRAHYDROFUROYL sulfenyl] penem-3-carboxylic acid propylene (150g, 0.342mol) and ammonium bifluoride (59.5g, 1.025mmol) add successively among the 400mL DMF, 55~60 ℃ were reacted 5 hours, stopped reaction, suction filtration, filtrate adds water 800ml, uses ethyl acetate extraction, and organic phase is washed with 5% sodium hydrogen carbonate solution, anhydrous sodium sulfate drying, concentrated, gained incarnadine oily matter gets yellow solid 73g through the petrol ether/ethyl acetate recrystallization, yield 66%.Reference examples 2The preparation of Faropenem(the 5R that reference examples 1 is prepared, 6S)-6-[(R)-the 1-hydroxyethyl]-2-[(R)-and the 2-TETRAHYDROFUROYL sulfenyl] penem-3-carboxylic acid propylene (73g, 0.224mol), 6.5g triphenylphosphine, 6.5g [four (triphenylphosphine)] palladium adds among the 500mL methylene dichloride l successively, the ethyl acetate solution that adds the 2 ethyl hexanoic acid sodium preparation of 500mL 0.5M, stirring at room 1 hour, stopped reaction adds 15mL water in reaction solution, stir 30min, suction filtration, this solid is dissolved in the 100mL water again, adds decolorizing with activated carbon 30min, filter, filtrate adds in the 500mL acetone, place crystallization, get Faropenem 66g, yield 96%.Find that by contrast the total recovery that two steps of reference examples remove hydroxyl and carboxyl-protecting group only has about 63.4%, and single stage method of the present invention removes the yield of hydroxyl and carboxyl-protecting group and can reach more than 90%.Preparation method of the present invention can the one-step removal hydroxyl and carboxyl on protecting group, shortened the production cycle, the deprotecting regent cost is low, toxicity is little, can not cause heavy metal remaining, and have higher reaction yield, is fit to very much the industrial production of raw material medicine.
Patent
Publication numberPriority datePublication dateAssigneeTitleCN1939924A *2006-09-082007-04-04鲁南制药集团股份有限公司Industrial production of Fallopeinan sodiumWO2008035153A2 *2006-08-022008-03-27Orchid Chemicals & Pharmaceuticals LimitedProcess for the preparation of beta-lactam antibioticCN103059046A *2013-01-282013-04-24苏州二叶制药有限公司Preparation method of faropenemFamily To Family CitationsCN100522975C *2007-08-232009-08-05东北制药集团公司沈阳第一制药厂Method for preparing faropenemPublication numberPriority datePublication dateAssigneeTitleCN1884284A *2005-06-212006-12-27浙江金华康恩贝生物制药有限公司Process for the preparation of sodium faropenemCN1939924A *2006-09-082007-04-04鲁南制药集团股份有限公司Industrial production of Fallopeinan sodiumCN101125857A *2007-08-232008-02-20东北制药集团公司沈阳第一制药厂Method for preparing faropenemWO2008035153A2 *2006-08-022008-03-27Orchid Chemicals & Pharmaceuticals LimitedProcess for the preparation of beta-lactam antibiotic
Publication numberPriority datePublication dateAssigneeTitle
EP0410727A1 *1989-07-261991-01-30Suntory LimitedProcesses for removing allyl groupsUS4997829A *1985-03-091991-03-05Suntory LimitedPenem compounds, and use thereofEP0574940A1 *1992-06-181993-12-22Tanabe Seiyaku Co., Ltd.Method for removing the protecting group for carboxyl groupWO2007039885A1 *2005-10-052007-04-12Ranbaxy Laboratories LimitedA process for the preparation of faropenemFamily To Family Citations
Publication numberPriority datePublication dateAssigneeTitleCN102964357A *2012-11-112013-03-13苏州二叶制药有限公司Faropenem sodium and tablet thereofCN103059046A *2013-01-282013-04-24苏州二叶制药有限公司Preparation method of faropenemCN103880864A *2014-03-252014-06-25江苏正大清江制药有限公司Method for synthesizing faropenem sodiumCN104086516A *2014-07-182014-10-08成都樵枫科技发展有限公司Synthetic method of R-(+)-sulfotetrahydrofuran-2-formic acidCN101941981B *2009-07-032015-01-21湖南华纳大药厂有限公司Catalyst composition and method for preparing faropenem sodiumCN106860405A *2015-12-142017-06-20山东新时代药业有限公司A kind of faropenem sodium granules and preparation method thereofCN108840877A *2018-06-122018-11-20赤峰迪生药业有限责任公司A kind of preparation method of oxygen cephalosporin intermediate
References
- ^ Critchley IA, Brown SD, Traczewski MM, Tillotson GS, Janjic N (December 2007). “National and regional assessment of antimicrobial resistance among community-acquired respiratory tract pathogens identified in a 2005-2006 U.S. Faropenem surveillance study”. Antimicrob. Agents Chemother. 51 (12): 4382–9. doi:10.1128/AAC.00971-07. PMC 2168020. PMID 17908940.
- ^ Mushtaq S, Hope R, Warner M, Livermore DM (May 2007). “Activity of faropenem against cephalosporin-resistant Enterobacteriaceae”. J. Antimicrob. Chemother. 59 (5): 1025–30. doi:10.1093/jac/dkm063. PMID 17353220.
- ^ Milazzo I, Blandino G, Caccamo F, Musumeci R, Nicoletti G, Speciale A (March 2003). “Faropenem, a new oral penem: antibacterial activity against selected anaerobic and fastidious periodontal isolates”. J. Antimicrob. Chemother. 51 (3): 721–5. doi:10.1093/jac/dkg120. PMID 12615878.
- ^ Gettig JP, Crank CW, Philbrick AH (January 2008). “Faropenem medoxomil”. Ann Pharmacother. 42 (1): 80–90. doi:10.1345/aph.1G232. PMID 18094341. Archived from the original on 2013-02-03.
- ^ (Q1 06 Investor Conf Call)(CID 6918218 from PubChem)
External links
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| Routes of administration | Oral |
| ATC code | J01DI03 (WHO) |
| Identifiers | |
| CAS Number | 106560-14-9 |
| PubChem CID | 65894 |
| ChemSpider | 59303 |
| UNII | F52Y83BGH3 |
| ChEBI | CHEBI:51257 |
| ChEMBL | ChEMBL556262 |
| CompTox Dashboard (EPA) | DTXSID0046430 |
| Chemical and physical data | |
| Formula | C12H15NO5S |
| Molar mass | 285.31 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
///////////Faropenem, ALP-201, SUN-5555, SY-5555, WY-49605, ANTIBACTERIALS, DIICHI, Daiichi Asubio Pharma

NEW DRUG APPROVALS
ONE TIME
$10.00


{kind=link}