
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
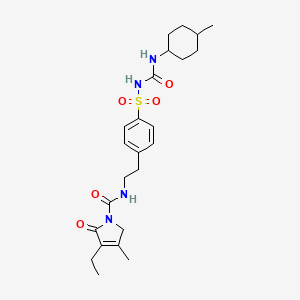
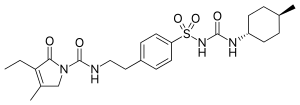
Glimepiride
- Molecular FormulaC24H34N4O5S
- Average mass490.616 Da
-
HOE 490UNII:6KY687524K
Glimepiride (original trade name Amaryl) is an orally available medium-to-long-acting sulfonylurea antidiabetic drug. It is sometimes classified as either the first third-generation sulfonylurea,[1] or as second-generation.[2]
Glimepiride is a Sulfonylurea. The chemical classification of glimepiride is Sulfonylurea Compounds.
Glimepiride is a long-acting, third-generation sulfonylurea with hypoglycemic activity. Compared to other generations of sulfonylurea compounds, glimepiride is very potent and has a longer duration of action. This agent is metabolized by CYP2C9 and shows peroxisome proliferator-activated receptor gamma (PPARgamma) agonistic activity.
Indications
Glimepiride is indicated to treat type 2 diabetes mellitus; its mode of action is to increase insulin production by the pancreas. It is not used for type 1 diabetes because in type 1 diabetes the pancreas is not able to produce insulin.[3]
Contraindications
Its use is contraindicated in patients with hypersensitivity to glimepiride or other sulfonylureas.
Adverse effects
Side effects from taking glimepiride include gastrointestinal tract (GI) disturbances, occasional allergic reactions, and rarely blood production disorders including thrombocytopenia, leukopenia, and hemolytic anemia. In the initial weeks of treatment, the risk of hypoglycemia may be increased. Alcohol consumption and exposure to sunlight should be restricted because they can worsen side effects.[3]
Pharmacokinetics

Two generic oral tablets of glimepiride, 2 mg each
Gastrointestinal absorption is complete, with no interference from meals. Significant absorption can occur within one hour, and distribution is throughout the body, 99.5% bound to plasma protein. Metabolism is by oxidative biotransformation, it is hepatic and complete. First, the medication is metabolized to M1 metabolite by CYP2C9. M1possesses about 1⁄3 of pharmacological activity of glimepiride, yet it is unknown if this results in clinically meaningful effect on blood glucose. M1 is further metabolized to M2metabolite by cytosolic enzymes. M2 is pharmacologically inactive. Excretion in the urine is about 65%, and the remainder is excreted in the feces.
Mechanism of action
Like all sulfonylureas, glimepiride acts as an insulin secretagogue.[4] It lowers blood sugar by stimulating the release of insulin by pancreatic beta cells and by inducing increased activity of intracellular insulin receptors.
Not all secondary sufonylureas have the same risks of hypoglycemia. Glibenclamide (glyburide) is associated with an incidence of hypoglycemia of up to 20–30%, compared to as low as 2% to 4% with glimepiride. Glibenclamide also interferes with the normal homeostatic suppression of insulin secretion in reaction to hypoglycemia, whereas glimepiride does not. Also, glibenclamide diminishes glucagon secretion in reaction to hypoglycemia, whereas glimepiride does not.[5]

Interactions
Nonsteroidal anti-inflammatory drugs (such as salicylates), sulfonamides, chloramphenicol, coumadin and probenecid may potentiate the hypoglycemic action of glimepiride. Thiazides, other diuretics, phothiazides, thyroid products, oral contraceptives, and phenytoin tend to produce hyperglycemia.
SYNTHESIS
EP 0031058; US 4379785, Arzneim-Forsch Drug Res 1988,38(8),1079

The condensation of 3-ethyl-4-methyl-3-pyrrolin-2-one (I) with 2-phenylethyl isocyanate (II) at 150 C gives 3-ethyl-4-methyl-2-oxo-N-(2-phenylethyl)-3-pyrrolin-1-carboxamide (III), which is sulfonated with chlorosulfonic acid at 40 C to yield the corresponding benzenesulfonyl chloride (IV). The reaction of (IV) with concentrated NH4OH affords the sulfonamide (V), which is finally condensed with 4-methylcyclohexyl isocyanate (VI) in acetone.
clip

CLIP

Following is one of the synthesis routes: 3-Ethyl-4-methyl-3-pyrrolin-2-one could be condensed (I) with 2-phenylethyl isocyanate (II) at 150 C to produce 3-ethyl-4-methyl-2-oxo-N-(2-phenylethyl)-3-pyrrolin-1-carboxamide (III), which is sulfonated with chlorosulfonic acid at 40 C to yield the corresponding benzenesulfonyl chloride (IV). The reaction of (IV) with concentrated NH4OH affords the sulfonamide (V), which is finally condensed with 4-methylcyclohexyl isocyanate (VI) in acetone.
CLIP
http://science24.com/paper/6906

PAPER
https://www.sciencedirect.com/science/article/pii/S073170850500378X

PATENT
https://www.google.com/patents/US20070082943
-
Glimepiride, according to U.S. Pat. No. 4,379,785 (EP 031058) issued to Hoechst is prepared via reaction of 4-[2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido) ethyl] benzene sulfonamide (IV) with trans-4-methylcyclohexyl isocyanate (VIII). U.S. Pat. No. 4,379,785 (EP 031058) (hereafter referred to as the ‘785 patent) discloses heterocyclic substituted sulfonyl ureas, particularly 3-Ethyl-2,5-dihydro-4-methyl-N-[2-[4-[[[[(trans-4-methyl cyclohexyl)amino]carbonyl]amino]sulfonyl]phenyl]ethyl]-2-oxo-1H-pyrrole-1-carboxamide i.e. Glimepiride (I). The ‘785 patent teaches the preparation of Glimepiride starting from 3-Ethyl-4-methyl-3-pyrolidine-2-one (II) and 2-phenyl ethyl isocyanate to give [2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido) ethyl] benzene (III). The [2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido) ethyl] benzene is converted to the 4-[2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido) ethyl] benzene sulfonamide (IV), by reacting with chlorosulphonic acid, followed by treatment with ammonia solution. This intermediate compound (IV) is then finally reacted with trans-4-methylcyclohexyl isocyanate (VIII) prepared from trans-4-methyl cyclohexylamine HCl (VII) to form Glimepiride.
-
[0004]Glimepiride can also be synthesized by reaction of N-[[4-[2-(3-ethyl-4-methyl-2-oxo-3-pyrroline-1-carboxamido)-ethyl]phenyl]sulphonyl]methylurethane (IX) with trans-4-methyl cyclohexyl amine (VII) as reported by R. Weyer, V. Hitzel in Arzneimittel Forsch 38, 1079 (1988).
-
[0005]trans-4Methylcyclohexyl isocyanate (VIII) is prepared from trans-4-methyl cyclohexyl amine HCl (VII), by phosgenation.
-
[0006]H. Ueda et. al., S.T.P Pharma Sciences, 13(4) 281-286, 2003 discloses a novel polymorph of Glimepiride, Form II obtained by recrystallisation from a solvent mixture of ethanol and water. It also discloses that earlier known form is Form I. Reported solvents for obtaining Form I are methanol, acetonitrile, chloroform, butyl acetate, benzene and toluene.
-
[0007]An alternative route is disclosed in WO03057131(Sun Pharmaceutical), where 3-ethyl-4-methyl-2,5-dihydro-N-(4-nitrophenyloxycarbonyl)-pyrrole-2-one is treated with 4-(2-aminoethyl)-benzene sulphonamide to obtain 4-[2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido) ethyl] benzene sulfonamide (IV) which was then converted to Glimepiride (I). However, nonavailability of raw material and the yield being poor, the process as described in U.S. Pat. No. 4,379,785 is preferred.
-
[0008]To obtain Glimepiride of highest purity, following intermediates should be of highest quality:
-
[0009]a) 4-[2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido) ethyl] benzene sulfonamide (IV) with lowest possible content of ortho and meta isomers.
-
[0010]b) Trans-4-methyl cyclohexyl amine (VII) and its respective isocyanate (VIII) should have lowest content of the cis isomer.
-
[0011]The preparation of the 4-[2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido) ethyl] benzene sulfonamide is well disclosed in the patent U.S. Pat. No. 4,379,785. It is prepared by condensation of 3-ethyl-4-methyl-3-pyrrolidine-2-one of Formula (II) with 2-phenyl ethyl isocyanate. The condensed product is then chlorosulphonated with chlorosulphonic acid followed by ammonolysis with liq. ammonia to give compound of Formula (IV). The purity is not well documented in the patents, and by following the patented process, ˜85 to 88% of desired para isomer is obtained. This is evident as the chlorosulphonation is ortho-para directing.
-
[0012]Hence, there is a need to develop purification process to maintain undesired ortho and meta isomers below 0.1%.
-
[0013]The other key intermediate trans-4-methylcyclohexyl amine HCl (VII) should preferably have lowest possible content of the cis isomer. The commonly used procedure is reduction of 4-methyl cyclohexanone oxime (V) with sodium in alcohol, preferably ethanol.
-
[0014]T. P. Johnston, et. al., J. Med. Chem., 14, 600-614 (1971); H. Booth, et. al., J. Chem. Soc (B) 1971, 1047-1050 and K. Ramalingam et. al., Indian Journal of Chem Vol. 40, 366-369 (April 1972) all report the abovementioned reduction. The amine obtained via this process typically contains between 8 to 10% of the cis isomer. However, use of high excess sodium metal (25 eqv.) for reduction makes process commercially and environmentally unviable. Also, the purification of trans amine from the mixture via the distillation is very difficult as the boiling points differ only by about 2° C. Also there is an inherent drawback of said free amine as, it immediately forms carbonate salt. Further purification of the amine to reduce the cis content via crystallization of its salt is not sufficiently documented. Prior art describes purification of crude trans-4-methylcyclohexylamine HCl by crystallization of its hydrochloride but the yield and purity are not sufficiently discussed. A description of such purification is provided in J. Med. Chem, 14, 600-614 (1971), wherein trans-4-methylcyclohexylamine HCl is obtained by triple crystallization in acetonitrile of the crude hydrochloride (m.p. 260° C.) in 27% yield.
-
[0015]WO 2004073585 (Zentiva) describes a process for preparation of trans-4-methylcyclohexylamine HCl wherein the highlights of the invention are the use of sodium metal and purification via the pivalic acid salt. However drawbacks of the process are use of sodium metal, which is hazardous and pivalic acid which is expensive. The overall yield is ˜40%.
-
[0016]Thus considering the current stringent pharmacopieal requirements for cis content, there is a need for obtaining Glimepiride having cis impurity content well below 0.15% by a cost effective process.
-
[0017]Key factors in the production of Glimepiride are:
-
[0018]a) Substantial purity of trans-4-methyl cyclohexyl amine HCl (VII) with the lowest possible content of the cis isomer.
-
[0019]b) Substantial purity of 4-[2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido) ethyl] benzene sulfonamide (IV) with the lowest possible content of the ortho and meta isomer.
-
[0020]The purity of intermediate compound of Formula (IV) when prepared by the process disclosed in ‘785 patent, was found to be 82 to 85% by HPLC.
-
-
schemes I to III.



-
[0037]The purification of trans-4-methyl cyclohexylamine HCl (VII) is accomplished by using an appropriate solvent combination. The mixture of cis/ trans stereoisomers (i.e. 50:50) were dissolved in diluted methanol and the desired trans isomer is coprecipitated by adding acetone to it. The process is repeated with different proportions of the solvent mixture to get the trans-4-Methyl cyclohexylamine HCl (VII) >99.5% with cis isomer less than 0.15%. The overall yield from 4-methyl cyclohexanone is ˜30%. The purification has been achieved using a solvent mixture of alcohol and ketone. A preferred alcohol for dissolution is an aliphatic one wherein carbon chain may be preferably C1-C4. Preferably methanol is used to dissolve the crude trans-4-Methyl cyclohexylamine HCl. The ratio of substrate:methanol:acetone is fixed at 1:1.5:6 for achieving the desired purity. The cosolvent used for precipitation is an aliphatic ketone. The preferred ketone is acetone. The precipitation is carried out at a temperature between 20 to 50° C., preferably between 30 to 50° C. and most preferably at about 40° C. The addition of acetone is carried out over a period of 2 to 6 hrs, more preferably for about 2 to 4 hrs and most preferably in about 3 hrs. The compound thus obtained has a purity >95% by gas chromatography.
-
[0038]The enriched trans-4-Methyl cyclohexylamine HCl (VII) (>95%) is further purified using different proportions of the same solvent mixture. The enriched trans isomer is dissolved in alcohol and reprecipitated using an aliphatic ketone. The ratio of substrate:methanol:acetone ratio is fixed at 1:1.5:13.6 for obtaining purity greater than 99.8%.
-

-
- EXAMPLE 1trans-4-Methyl cyclohexylamine HCl (VII)
-
[0053]1.5 Kg of crude 4-Methyl cyclohexyloxime (V) was dissolved in 8.33 L Methanol. To this 0.15 Kg Raney nickel was added. Then the mixture was hydrogenated at 4-5 Kg/cm2 pressure at 50 to 55° C. After the absorption of H2 ceases, the reaction mass is cooled down and filtered. From resulting reaction mixture, methanol was distilled completely. Crude concentrated oil obtained is cooled to 15 to 20° C. to which methanolic hydrochloric acid (12 to 13%) is added slowly, when the product i.e. 4-Methylcyclohexylamine HCl precipitates out. The yield obtained 1.5 Kg of crude 4-methyl cyclohexylamine HCl (85%) with ˜50% content of trans isomer. The crude 4-Methyl cyclohexylamine HCl 1.5 Kg (wet) was further purified in methanol/acetone mixture. The crude 4-methyl cyclohexylamine HCl (1.5 Kg) was dissolved in 2.25 L of methanol at 25 to 30° C. Slowly started addition of 13.5 L of acetone over a period of 3 hrs. The trans-4-methyl cyclohexylamine HCl precipitated out. Yield 0.6 Kg. The purity achieved of trans isomer is >95%. The cis isomer at this stage is ˜2 to 3%.
-
[0054]The trans-4-methyl cyclohexylamine HCl (0.6 Kg) thus obtained is again taken in 0.9 L of methanol and is dissolved completely at 25 to 30° C. 8.1 L acetone is added slowly over a period of 3 hrs when pure trans isomer precipitates out completely. The purity achieved at this stage is >99.8% and cis isomer well below 0.15%. The yield thus obtained after the second purification is 0.48 Kg of trans-4-Methyl cyclohexylamine HCl (27.2% yield calculated on the starting oxime). Purity of the desired trans isomer is greater than 99.8% by G.C.
-
[0055]Melting point of the trans-4-methyl cyclohexylamine HCl thus obtained is 262° C. to 263° C.
-
EXAMPLE 2Preparation of 4-[2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido) ethyl] benzene sulfonamide (IV)
-
-
[0056]3-Ethyl-4-methyl-2,5-dihydro-1H-pyrrole-2-one (II) (1.0 Kg) and β-phenylethyl isocyanate (1.488 Kg) were mixed in anhydrous toluene (4.0 L) and refluxed for 4 hrs. The toluene was distilled off and hexane (8.0 L) was added to the reaction mixture at 50° C. The product precipitated is cooled to 0 to 5° C. to obtain the solid compound viz. 4-[2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido) ethyl] benzene (2.17 Kg). It was filtered & washed with 2.0 L of hexane.
-
[0057]To a cooled (15 to 25° C.) solution of chlorosulfonic acid (2.8 L), 4-[2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido)ethyl] benzene (2.0 Kg) was added in small portions over a period of 2 to 3 hrs. Further it was stirred for 30 min at this temperature and then temperature was gradually raised to 30 to 35° C. The reaction mass is stirred further for 2 hrs. The reaction mixture was then quenched into ice-water and stirred for 1 hr and filtered to obtain the product 4-[2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido)ethyl] benzene sulfonyl chloride (2.0 kg). To a cooled (15 to 20° C.) solution of diluted ammonia (1.4 L) 4-[2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido) ethyl] benzene sulfonyl chloride was added in small portion over 1 to 2 hrs. The reaction mixture was then heated to 70° C. for 2 hrs when ammonolysis is complete. The product converted is then stirred for 1 hr at R.T. and filtered and dried at 90 to 100° C. to obtain crude 4-[2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido) ethyl] benzene sulfonamide (2.2 Kg) having HPLC purity in the range of 82 to 88%. The crude compound 4-[2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido) ethyl] benzene sulfonamide (2.2 Kg) is then purified from mixture of organic solvents chosen from Methanol, Acetone & toluene.
-
EXAMPLE 3APurification of 4-[2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido)ethyl] benzene sulfonamide (IV)
-
-
[0058]1st Purification
-
[0059]In a reaction vessel containing Toluene (12.0 L), 4-[2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido) ethyl] benzene sulfonamide (2.0 Kg) was charged at 25 to 30° C. Slowly the temperature was raised to 60 to 65° C. and methanol (5.0 L) was added via the dosing tank slowly when the product dissolved completely. Refluxed it for 0.5 hr. Charcoalised and filtered the product in another reaction vessel. Distill off toluene/methanol mixture till total recovery about 65% under vacuum. White crystalline product precipitated out. After the recovery, cool the reaction mass to 15 to 20° C. The resulting crystallized solid product was filtered and washed two times with chilled acetone (about 2 L) each. The resulting product was dried at 90 to 100° C. in air oven till constant weight to obtain about 1.4 Kg of 4-[2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido)ethyl] benzene sulfonamide with greater than 95% HPLC purity.
-
EXAMPLE 3BPurification of 4-[2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido)ethyl] benzene sulfonamide (IV)
-
-
[0060]2nd Purification
-
[0061]In a reaction vessel containing Acetone (8.4 L), (1.4 Kg) of 1st purified 4-[2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido)ethyl] benzene sulfonamide was charged at 25 to 30° C. slowly and the temperature was raised to 55 to 60° C. Methanol was added (5.6 L) via the dose tank at this reflux temperature to dissolve it completely. Refluxed it for further 30 min. Distilled off acetone/ methanol mixture till total recovery about 65 to 70%. White crystalline product precipitated out. After the recovery slowly cooled the product to 15 to 20° C. The resultant solid product was filtered, washed two times with chilled acetone (1.4 L) each. The 4-[2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido) ethyl] benzene sulfonamide was dried at 90 to 100° C. in air oven till constant weight to obtain about 1.12 Kg of 4-[2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido)ethyl] benzene sulfonamide (IV) with greater than 99.5% purity with other isomers i.e. ortho and meta well below 0.2% respectively.
-
EXAMPLE 4Preparation of 3-Ethyl-2,5-dihydro-4-methyl-N-[2-[4-[[[[(trans-4-methyl cyclohexyl)amino]carbonyl]amino]sulfonyl]phenyl]ethyl]-2-oxo-1 H-pyrrole-1-carboxamide (I).
-
-
[0062]In a reaction vessel containing (24.2 L) Acetone, 4-[2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido)ethyl] benzene sulfonamide (1.0 Kg) and potassium carbonate (0.46 Kg) was added and refluxed at about 55 to 60° C. for 1 hr. trans-4-Methyl-cyclohexyl isocyanate was obtained by method known in art from trans-4-methyl-cyclohexylamine. A solution of trans-4-methyl-cyclohexyl isocyanate (0.515 Kg) in toluene (5 L) was prepared and added to the above reaction mixture. This reaction mixture is refluxed for 12 hrs, then cooled. To this cooled reaction mass charge 27 L of water. The reaction mass was filtered and the pH was adjusted to 5.5 to 6.0 by adding acetic acid at about 20 to 25° C. The solid obtained was filtered and washed with water. The 3-Ethyl-2,5-dihydro-4-methyl-N-[2-[4-[[[[(trans-4-methyl cyclohexyl)amino]carbonyl]amino]sulfonyl]phenyl]ethyl]-2-oxo-1H-pyrrole-1-carboxamide (I) obtained is then dried at 90 to 100° C. till constant weight. Yield of the product is 86.3%.
-
EXAMPLE 5Purification of 3-Ethyl-2,5-dihydro-4-methyl-N-[2-[4-[[[[(trans-4-methyl cyclohexyl)amino]carbonyl]amino]sulfonyl]phenyl]ethyl]-2-oxo-1H-pyrrole-1-carboxamide (I)
-
[0063]In a reaction vessel containing 6.0 L methanol and 1.0 Kg crude Glimepiride, dry ammonia gas was purged at 20 to 25° C. till all Glimepiride dissolves and a clear solution is obtained. This homogeneous mass was then charcoalised, filtered and finally neutralized with Glacial acetic acid to pH 5.5 to 6.0, till the entire product precipitates out. The pure Glimepiride was then filtered and dried at 65° C. to 70° C. till constant weight. Yield obtained was ˜90%.
CLIP
- Journal of Pharmaceutical Sciences 100(11):4700-9
- DOI
- 10.1002/jps.22662

Magnified 1H NMR spectra of (a) glimepiride and its solid dispersions with hyperbranched polymers containing the (b) hydroxyl and (c) the tertiary amino functional groups.

Magnified 13C NMR spectra of (a) glimepiride and its solid dispersions with hyperbranched polymers containing (b) the hydroxyl and (c) the tertiary amino functional groups.

The difference spectra of the solid dispersions of glimepiride and the hyperbranched polymer containing (a) the hydroxyl groups and (b) the tertiary amino groups. The difference spectra were obtained by subtraction of the spectra of the pure hyperbranched polymers from the spectra of the solid dispersions. The ATR spectra of the pure hyperbranched polymers were recorded on samples that were prepared under the same conditions as solid dispersions, only without the presence of the glimepiride drug.
Patents
- US6150383
- US6211205
- US6303640
- US6329404
- US8071130
- US7538125
- US7700128
- US7358366
FDA Orange Book Patents
| FDA Orange Book Patents: 1 of 3 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 7358366 |
| Expiration | Apr 19, 2020. 7358366*PED expiration date: Oct 19, 2020 |
| Applicant | SB PHARMCO |
| Drug Application | |
| FDA Orange Book Patents: 2 of 3 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 8071130 |
| Expiration | Jun 8, 2028 |
| Applicant | TAKEDA PHARMS USA |
| Drug Application |
|
| FDA Orange Book Patents: 3 of 3 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 7700128 |
| Expiration | Jan 30, 2027 |
| Applicant | TAKEDA PHARMS USA |
| Drug Application |
|
CLIP
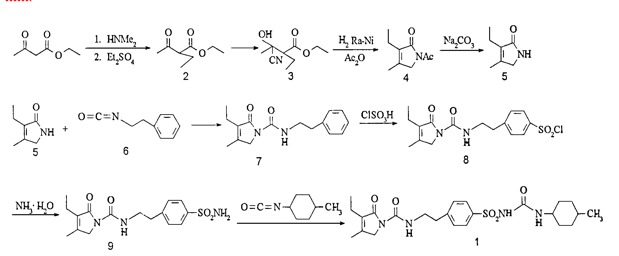
Journal of China Pharmaceutical University 1999 , 30(3):163 ~ 165
Ethyl acetoacetate (2) Preparation of literature more, such as with ethyl iodide or ethyl bromide as ethyl reagents will produce a double ethylation or oxyethylation, it is difficult to separate. We use dimethylamine and ethyl acetoacetate reaction enamine, then diethyl sulfate as ethylating agent, you can reduce the side reactions, product purity, the yield up to 80%. Preparation of cyanohydrin (3) Hydrochloric acid anhydrous literature, toxicity, difficult to operate, we use solid sodium cyanide and sodium bisulfite in the aqueous phase reaction, get 3, easy to operate. In the literature 1-acetyl-3- Ethyl-4-methyl-3-pyrrolin-2-one (4) was purified by high vacuum distillation and then hydrolyzed to give 3-ethyl- In the distillation of the product easy to loss, after the change to the crude hydrolysis, two-step yield of 33%. Reported in the literature 5 and phenethyl isocyanate (6) without solvent direct reaction of 3-ethyl-4-methyl-2-oxo-3-pyrroline-1 – N- (2 – phenethyl) A Amide (7), the experiment found that the reaction heat when the heating easy to red material, we add toluene as a solvent, the reaction is smooth, easy to post-treatment. 6 preparation, the general method is to use phosgene, but phosgene often Temperature of gas, highly toxic, difficult to operate, we use triphosgene instead of triphosgene as a yellow solid, easy to transport, weighing, laboratory convenience. We refer to the process of domestic glyburide, 7 chlorosulfonated, ammoniated sulfonamide (9), two-step yield of 77%. The last 9 reacts with trans-4-methylcyclohexylisocyanate to form glimepiride (1). Ethyl acetoacetate as the starting material, eight-step total yield of 11.5%.
Journal of China Pharmaceutical University 1999 , 30(3):163 ~ 165
Glimepi ride (1) trade name Amary l, chemical name 1- [4- [2- (3-ethyl-4-methyl-2-oxo-3-pyrroline-1-carboxamido ) Ethyl] phenylsulfonyl] -3- (trans-4-methylcyclohexyl) urea, a new sulfonylurea hypoglycemic agent developed by Hoechst AG in Germany and listed in the Netherlands and Switzerland in 1995, In 1996 the United States FDA approval
- 1 Campbell RK.Glimepiride :Role of a new sulfonylurea in the t reatment of type 2 diabetes mellitus .An n Pharmacother , 1998 , 32 :1044
- 2 Hans P , Joachim K .Synt hesis of oxyopsopy rrolecarboxyli c acid
and further investigations in the pyrrolone series.Ann Chem ,
1964 , 680 :60
- 3 Glimepiride.Dr ugs F ut , 1992 , 17(9):774
- 4 Corson BB, Dodge RA , Harris SA , et al .M andeli c acid.Org Syn , 1941 , Coll Vol 1 :329
- 5 M aurice WG , Roy VD, Brian I , et a l .A new synt hesis of iso- cyanates .J Chem Soc , Perkin Ⅰ , 1976 :141
- 6 天津医药工业研究所.糖尿病药物-优降糖的新合成法.医药工业, 1974 , 4 :11
- 7 Weyer R, Gei sen K , Hitzel V , et al .Heterocyclic subst ituted sul- f onyl ureas and their use .Ger O f fen , 1979 :2951135 A 1
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US4379785 * | Dec 17, 1980 | Apr 12, 1983 | Hoechst Aktiengesellschaft | Heterocyclic substituted sulfonyl ureas, and their use |
References
- Jump up^ Hamaguchi T, Hirose T, Asakawa H, et al. (December 2004). “Efficacy of glimepiride in type 2 diabetic patients treated with glibenclamide”. Diabetes Res. Clin. Pract. 66 Suppl 1: S129–32. doi:10.1016/j.diabres.2003.12.012. PMID 15563963.
- Jump up^ Davis SN (2004). “The role of glimepiride in the effective management of Type 2 diabetes”. J. Diabetes Complicat. 18 (6): 367–76. doi:10.1016/j.jdiacomp.2004.07.001. PMID 15531188.
- ^ Jump up to:a b “Glimepiride: MedlinePlus Drug Information”. nih.gov.
- Jump up^ Nissen SE, Nicholls SJ, Wolski K, et al. (April 2008). “Comparison of pioglitazone vs glimepiride on progression of coronary atherosclerosis in patients with type 2 diabetes: the PERISCOPE randomized controlled trial”. JAMA. 299 (13): 1561–73. doi:10.1001/jama.299.13.1561. PMID 18378631.
- Jump up^ Davis, Stephen N. (2005). “60. Insulin, oral hypoglycemic agents, and the pharmacology of the endocrine pancreas”. In Brunton, Laurence L.; Lazo, John S.; Parker, Keith L. (eds.). Goodman & Gilman’s The Pharmacological Basis of Therapeutics. New York: McGraw-Hill. p. 1636. ISBN 0-07-142280-3.
External links
- http://www.theodora.com/drugs/amaryl_tablets_sanofi_aventis.html
- http://www.rxlist.com/cgi/generic/glimepiride.htm
 |
|
| Clinical data | |
|---|---|
| Trade names | Amaryl |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a696016 |
| License data | |
| Pregnancy category |
|
| Routes of administration |
Oral (tablets) |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | 100% |
| Protein binding | >99.5% |
| Metabolism | Complete hepatic (1st stage through CYP2C9) |
| Biological half-life | 5–8 hours |
| Excretion | Urine (~60%), feces (~40%) |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| ECHA InfoCard | 100.170.771 |
| Chemical and physical data | |
| Formula | C24H34N4O5S |
| Molar mass | 490.617 g/mol |
| 3D model (JSmol) | |
///////////Amaryl, glimepiride, glymepiride, HOE 490
CCC1=C(CN(C1=O)C(=O)NCCC2=CC=C(C=C2)S(=O)(=O)NC(=O)NC3CCC(CC3)C)C


[…] https://newdrugapprovals.org/2018/02/05/glimepiride/ […]
LikeLike