
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
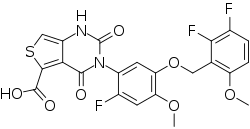
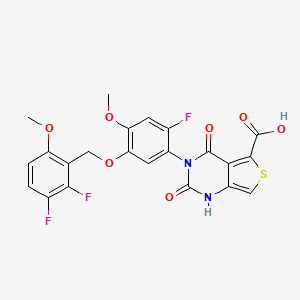
LINZAGOLIX
CAS 935283-04-8
C22H15F3N2O7S
- Hormone Antagonists
3-[5-[(2,3-difluoro-6-methoxyphenyl)methoxy]-2-fluoro-4-methoxyphenyl]-2,4-dioxo-1H-thieno[3,4-d]pyrimidine-5-carboxylic acid
- WHO 10711
- Treatment of Endometriosis Pain and Uterine Myoma-Associated Heavy Menstrual Bleeding
- OriginatorKissei Pharmaceutical
- DeveloperKissei Pharmaceutical; ObsEva
- Class2 ring heterocyclic compounds; Antihormones; Antineoplastics; Carboxylic acids; Fluorinated hydrocarbons; Ketones; Pyrimidines; Small molecules; Thiophenes
- Mechanism of ActionLHRH receptor antagonists
- PreregistrationUterine leiomyoma
- Phase IIIEndometriosis
- Phase IIAdenomyosis
- 22 Nov 2021FDA assigns PDUFA action date of (13/09/2022) for linzagolix for Uterine leiomyoma
- 22 Nov 2021The US FDA accepts NDA for linzagolix for Uterine leiomyoma for review
- 20 Oct 2021Efficacy and adverse events data from a phase II trial in Adenomyosis presented at the American Society for Reproductive Medicine (ASRM) 2021 Scientific Congress & Expo


Linzagolix choline
CAS#: 1321816-57-2 (choline)
Chemical Formula: C27H28F3N3O8S
Exact Mass: 611.1549
Molecular Weight: 611.58
Linzagolix is an orally bioavailable gonadotropin-releasing hormone (GnRH or LHRH) receptor antagonist, with potential hormone production inhibitory activity. Upon oral administration of linzagolix, this agent competes with GnRH for receptor binding and inhibits GnRH receptor signaling in the anterior pituitary gland, thereby inhibiting the secretion and release of luteinizing hormone (LH) and follicle stimulating hormone (FSH). In males, the inhibition of LH secretion prevents the release of testosterone. As a result, this may relieve symptoms associated with hormonally dependent disease states such as hormone-dependent prostate cancer. In women, this prevents the production of estrogen by the ovaries and may relieve symptoms from sex-hormone dependent diseases, such as pain associated with endometriosis, heavy menstrual bleeding or uterine fibroids.
Linzagolix (INN; developmental code names KLH-2109, OBE-2109; tentative brand name Yselty) is a small-molecule, non-peptide, orally active gonadotropin-releasing hormone antagonist (GnRH antagonist) which is under development by Kissei Pharmaceutical and ObsEva for the treatment of uterine fibroids, endometriosis, and adenomyosis.[1][3][2] As of December 2020, it is under review for approval for uterine fibroids, is in phase III clinical trials for endometriosis, and is in phase II clinical studies for adenomyosis.[1]
Estrogen-dependent disorders represent a challenging class of diseases that have a high incidence in the general population and are often associated with particularly severe symptomology. Uterine fibroids, for example, also referred to as leiomyomata, are among the most common benign tumors in women. Symptoms associated with uterine fibroids commonly include heavy or prolonged menstrual bleeding, pelvic pressure and pelvic organ compression, back pain, and adverse reproductive outcomes. Heavy menstrual bleeding may lead to iron deficiency anemia, a key symptom of uterine fibroids and the leading cause of surgical interventions that may include hysterectomy. Endometriosis is another estrogen-dependent gynecological condition, characterized by the presence of endometrial-like tissue outside the uterus.
Additional examples of estrogen-dependent diseases include adenomyosis and rectovaginal endometriosis, which are particularly severe endometrial growth disorders characterized by the invasion of endometrial tissue into the uterine myometrium and rectovaginal zones, respectively. The term adenomyosis or uterine adenomyosis is used to describe the presence of both endometrial glands and stroma deep within the myometrium. This condition is associated with hypertrophy and hyperplasia of the subjacent muscle cells, which may ultimately result in an altered size and globulous morphology of the uterus. Due to the severity of this disorder, one of the key symptoms is strong menstrual and even non-menstrual pelvic pain with abnormal uterine bleeding. Like adenomyosis, rectovaginal endometriosis patients present with a variety of pain symptoms including dysmenorrhea, dyspareunia, chronic pelvic pain, dysuria, and dyschezia. Treatment options for rectovaginal endometriosis are limited. Since medical therapies are either ineffective or have considerable side effects, rectovaginal endometriosis patients often undergo surgical procedures to reduce the endometrial node, and may even be subject to resection of the bowel if the node infiltrates the rectal or sigmoidal wall.

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
Obseva Announces U.S. FDA Acceptance of New Drug Application for Linzagolix
November 22, 2021 01:05 ET | Source: ObsEva SA………. https://www.globenewswire.com/news-release/2021/11/22/2338610/0/en/Obseva-Announces-U-S-FDA-Acceptance-of-New-Drug-Application-for-Linzagolix.html
FDA Accepts NDA for Linzagolix for the Management of Heavy Menstrual Bleeding Associated with Uterine Fibroids
GENEVA, Switzerland November 22, 2021 – Obseva SA (NASDAQ: OBSV; SIX: OBSN), a biopharmaceutical company developing and commercializing novel therapies to improve women’s reproductive health, today announced that the New Drug Application (NDA) for linzagolix for the management of heavy menstrual bleeding associated with uterine fibroids in premenopausal women has been accepted for review by the United States Food and Drug Administration (FDA). The submission is based on data from the two Phase 3 PRIMROSE trials. Linzagolix has a differentiated profile and if approved, would be the first and only GnRH receptor antagonist with flexible dosing options for uterine fibroids, including a low dose option to address the needs of women who cannot or do not want to take hormones.1,4 The FDA set a target action date of September 13, 2022 for this NDA under the Prescription Drug User Fee Act (PDUFA).
“Today marks an important milestone not only in the linzagolix clinical development process, but for Obseva as a company, and most importantly, the millions of women living with uterine fibroids throughout the US. Linzagolix is a significant innovation in the field of women’s health – an area that is consistently underinvested in – and we are incredibly excited about the potential of bringing this important treatment to market” said Brian O’Callaghan, CEO of Obseva. “We are encouraged by our positive Phase 3 PRIMROSE results. If approved, we believe linzagolix will address a significant unmet need in offering a more individualized treatment option for a broader range of women.”
The Phase 3 PRIMROSE trials of linzagolix (PRIMROSE 1: US; n=574 and PRIMROSE 2: Europe and US; n=535) investigated the efficacy and safety of two dosing regimens, 100mg once daily and 200mg once daily, alone or in combination with hormonal ABT (1 mg estradiol and 0.5 mg norethisterone acetate) for the treatment of heavy menstrual bleeding associated with uterine fibroids. The NDA submission comprises positive 24-week treatment results from both studies, as well as supportive results from Week 52 and the 76-week post-treatment follow-up.
“Uterine fibroids can have a devastating impact on women’s day-to-day life. With its unique dosing options, linzagolix has the potential to significantly advance medical options for women,” stated Elizabeth Garner, MD, MPH, Chief Medical Officer of Obseva. “A dosing option without hormonal ABT would be welcomed by the significant number of women who either have contraindications to or a personal preference to avoid the use of estrogen-based therapies, while also providing a dosing option for women in whom hormonal ABT is indicated.”
The linzagolix marketing authorization application (MAA) was validated by the European Medicine Agency (EMA) with an approval recommendation from the Committee for Medicinal Products for Human Use (CHMP) expected in Q4 2021. Obseva announced previously that the company has entered into a partnership with Syneos Health to support commercialization of linzagolix in the US and EU.
About Linzagolix
Linzagolix is a novel, once daily, oral GnRH receptor antagonist with a potentially best-in-class profile1,2,3. Linzagolix is the subject of submitted marketing authorization applications for the treatment of heavy menstrual bleeding associated with uterine fibroids and is currently in late-stage clinical development for the treatment of pain associated with endometriosis. Obseva licensed linzagolix from Kissei in late 2015 and retains worldwide commercial rights, excluding Asia, for the product. Linzagolix is not currently approved anywhere in the world.
About the Phase 3 PRIMROSE Program in Uterine Fibroids
PRIMROSE 1 & 2 were prospective, randomized, parallel group, double-blind, placebo-controlled Phase 3 studies that investigated the efficacy and safety of two dosing regimens of linzagolix, 100 mg and 200 mg once daily, alone and in combination with hormonal ABT (1 mg estradiol and 0.5 mg norethisterone acetate) for the treatment of heavy menstrual bleeding associated with uterine fibroids. PRIMROSE 1 was conducted in the United States and enrolled 574 women. PRIMROSE 2 was conducted in Europe and the United States and enrolled 535 women. Both trials comprised a 52-week treatment period followed by a 6-month post treatment follow-up period. Additional information can be found here.
About Uterine Fibroids
Uterine fibroids are common benign tumors of the muscular tissue of the uterus which affect women of childbearing age and can vary in size from undetectable to large bulky masses. Few long-term medical treatments are available, and as a result, approximately 300,000 hysterectomies are performed for uterine fibroids every year in the US.
The symptoms of uterine fibroids are wide-ranging and include heavy menstrual bleeding, anemia, pelvic pressure and bloating, urinary frequency and pain that can be extremely debilitating with a significant impact on quality of life. These symptoms can also have an impact on mental health, creating the additional burden of anxiety and distress.
About Obseva
Obseva is a biopharmaceutical company built to address some of the most challenging unmet needs in women’s health – an under-researched, under-invested field of medicine. With deep expertise in clinical development, Obseva is passionate about the pursuit of advances that benefit women and their health and the importance of delivering truly meaningful innovation in this space. Through strategic in-licensing and disciplined drug development, Obseva has established a late-stage clinical pipeline with development programs focused on new therapies for the treatment of uterine fibroids, endometriosis, and preterm labor. Obseva is listed on the Nasdaq Global Select Market and is traded under the ticker symbol “OBSV” and on the SIX Swiss Exchange where it is traded under the ticker symbol “OBSN”. For more information, please visit http://www.ObsEva.com.
About Kissei
Kissei is a Japanese pharmaceutical company with approximately 70 years of history, specialized in the field of urology, kidney-dialysis and unmet medical needs. Silodosin is a Kissei product for the treatment of the signs and symptoms of benign prostatic hyperplasia which is sold worldwide through its licensees. KLH-2109/OBE2109 is a new chemical entity discovered by Kissei R&D.

……………………………

PATENT
WO 2007046392
https://patents.google.com/patent/WO2007046392A1/en
PATENT
WO 2014042176
https://patents.google.com/patent/WO2014042176A1/en

(Process 1)
Compound (D) can be produced by reacting compound (B) or a salt thereof with compound (C) in the presence of a base in a solvent. Examples of the solvent include halogen solvents such as dichloromethane, cyclic ethers such as tetrahydrofuran, 2-methyltetrahydrofuran, and tetrahydropyran, amide solvents such as N, N-dimethylformamide, aromatic hydrocarbon solvents such as toluene, A nitrile solvent such as acetonitrile, an ester solvent such as ethyl acetate, or a mixed solvent thereof and a mixed solvent thereof and water are preferable, and a mixed solvent of tetrahydrofuran and water is preferable. Examples of the base include organic bases such as triethylamine and pyridine, and inorganic bases such as sodium hydrogen carbonate, potassium hydrogen carbonate, cesium carbonate, sodium carbonate, and potassium carbonate, preferably triethylamine, sodium hydrogen carbonate, or potassium carbonate Is mentioned. The equivalent of the base may be an equivalent amount capable of neutralizing the salt and neutralizing the acid generated by the reaction. The equivalent of (C) can be used in an amount of 0.8 to 1.1 equivalents relative to (B), preferably 1.0 equivalent. The reaction temperature is usually 0 to 30 ° C., and the reaction time is usually 0.5 to 3 hours, although it varies depending on the raw material used, the solvent, the reaction temperature and the like. Examples of the salt of the compound (B) include a salt with an inorganic acid, a salt with an organic acid, a salt with an acidic amino acid, and the like. Examples of the salt with an inorganic acid include salts with hydrochloric acid, hydrobromic acid, nitric acid, sulfuric acid, phosphoric acid and the like. Examples of salts with organic acids include formic acid, acetic acid, trifluoroacetic acid, fumaric acid, oxalic acid, tartaric acid, maleic acid, citric acid, succinic acid, malic acid, methanesulfonic acid, benzenesulfonic acid, p-toluene And salts with sulfonic acid and the like. Examples of salts with acidic amino acids include salts with aspartic acid, glutamic acid and the like. Among these salts, salts with hydrochloric acid and methanesulfonic acid are preferable. Compound (C) used in Scheme 1 may be a commercially available product, or can be produced according to a known method or a method analogous thereto. Compound (D) may be isolated before the next step, but it can also be used in the next step without isolation.(Process 2)
Compound (F) can be produced by reacting compound (D) with compound (E) or a salt thereof in a solvent in the presence or absence of a base. Examples of the solvent include cyclic ethers such as tetrahydrofuran, 2-methyltetrahydrofuran, tetrahydropyran, amide solvents such as N, N-dimethylformamide, aromatic hydrocarbon solvents such as toluene, nitrile solvents such as acetonitrile, An ester solvent such as ethyl acetate or a mixed solvent thereof and a mixed solvent thereof with water, and the like are preferable, and a mixed solvent of tetrahydrofuran and water is preferable. Examples of the base include organic bases such as N, N-dimethylaminopyridine, triethylamine, N-methylpyrrolidine, N-methylmorpholine, diisopropylethylamine, and preferably N, N-dimethylaminopyridine, triethylamine and the like. . The equivalent of the base can be used in an amount of 0.1 to 2.0 equivalents relative to the compound (E), preferably 0.1 to 0.5 equivalents (provided that when a salt of the compound (E) is used, Further base necessary for neutralization is required). The reaction temperature is from room temperature to 60 ° C., and the reaction time is usually from 1 to 24 hours, although it varies depending on the raw material used, the solvent, the reaction temperature, and the like. Examples of the salt of compound (E) include a salt with an inorganic acid, a salt with an organic acid, a salt with an acidic amino acid, and the like. Examples of the salt with an inorganic acid include salts with hydrochloric acid, hydrobromic acid, nitric acid, sulfuric acid, phosphoric acid and the like. Examples of salts with organic acids include formic acid, acetic acid, trifluoroacetic acid, fumaric acid, oxalic acid, tartaric acid, maleic acid, citric acid, succinic acid, malic acid, methanesulfonic acid, benzenesulfonic acid, p-toluene And salts with sulfonic acid and the like. Examples of salts with acidic amino acids include salts with aspartic acid, glutamic acid and the like. Compound (F) may be isolated before the next step, but it can also be used in the next step without isolation.(Process 3)
The intramolecular cyclization and hydrolysis reaction in this step can be performed simultaneously or separately.
(Step 3-1)
Compound (A) can be produced by subjecting compound (F) to intramolecular cyclization and hydrolysis in the presence of a base in a solvent. Examples of the solvent include cyclic ethers such as tetrahydrofuran, 2-methyltetrahydrofuran and tetrahydropyran, lower alcohols such as methanol, ethanol and 2-propanol, amide solvents such as N, N-dimethylformamide, and nitriles such as acetonitrile. Examples thereof include a solvent and the like or a mixed solvent of a mixed solvent thereof and water, and a mixed solvent of tetrahydrofuran / methanol / water is preferable. Examples of the base include inorganic bases such as sodium hydroxide, potassium hydroxide, lithium hydroxide and sodium hydride, and metal alkoxides such as sodium methoxide and potassium tert-butoxide, preferably lithium hydroxide and sodium And methoxide. The base can be used in an amount of 3.0 to 6.0 equivalents, preferably 4.0 to 4.5 equivalents, relative to compound (F). The reaction temperature is usually from 0 to 20 ° C., and the reaction time is usually from 1 to 10 hours, although it varies depending on the raw material used, solvent, reaction temperature and the like.
(Step 3-2)
When isolating compound (G), compound (G) can be produced by subjecting compound (F) to an intramolecular cyclization reaction in a solvent in the presence of a base. Examples of the solvent include cyclic ethers such as tetrahydrofuran, 2-methyltetrahydrofuran and tetrahydropyran, lower alcohols such as methanol, ethanol and 2-propanol, amide solvents such as N, N-dimethylformamide, and nitriles such as acetonitrile. Examples thereof include a solvent and the like or a mixed solvent thereof, and a mixed solvent of tetrahydrofuran / methanol is preferable. Examples of the base include inorganic bases such as sodium hydroxide, potassium hydroxide, lithium hydroxide or sodium hydride, metal alkoxides such as sodium methoxide and potassium tert-butoxide, and lithium hydroxide, sodium methoxide and the like. preferable. The base can be used in an amount of 0.1 to 1.5 equivalents, preferably 1.0 to 1.1 equivalents, relative to compound (F). The reaction temperature is usually from 0 to 20 ° C., and the reaction time is usually from 1 to 10 hours, although it varies depending on the raw material used, solvent, reaction temperature and the like.
(Step 3-3)
The hydrolysis reaction in this step can be performed by the same method as in step 3-1 or a method analogous thereto.(Process 4)
Compound (A) can be converted to a salt thereof by a conventional method. Examples of such salts include inorganic salts such as sodium salt, potassium salt, calcium salt, magnesium salt, triethylamine, diisopropylamine, N, N′-dibenzylethylenediamine, ethanolamine, (2-hydroxyethyl) trimethylammonium. (Hereinafter referred to as choline), addition salts with organic bases such as N-methylglucamine, arginine, lysine and the like, and choline salts are preferred. Examples of the reagent used for conversion to the choline salt include choline hydroxide, choline bicarbonate, choline chloride and choline acetate.Here, the compound (B) and the salt thereof used in the above-mentioned scheme 1 are commercially available, or manufactured by the method described in a) to c), the method described in the reference examples, or a method analogous thereto. Can do.
a) JP-A 64-29373
b) Synthetic Communications, 32, 2565 (2002)
c) Synthesis, 200 (1977)Further, the compound (E) or a salt thereof used in the scheme 1 can be produced by the method described in Patent Document 1, the method described in Reference Examples, or a method analogous thereto.The compound obtained in the production process in the present specification includes hydrates or solvates thereof, and any of them can be used. Furthermore, the compound obtained in the production process in the present specification may have tautomers and / or geometric isomers, any of which can be used, and also a mixture thereof. be able to.By the production method of the present invention, the compound (A) useful as a pharmaceutical product or a salt thereof can be obtained in high yield and high purity through the compound (D) which is a production intermediate.The content of the present invention will be described in more detail by the following examples, but the present invention is not limited to the content.Reference example 1
Dimethyl 4-oxothiolane-2,3-dicarboxylate methylthioglycolate (15.0 g), tetrahydrofuran (45 g), piperidine (0.361 g) in a reaction mixture at room temperature with dimethyl maleate (21.4 g) in tetrahydrofuran (30 g) The solution was added. To the reaction mixture was added 20% sodium methoxide in methanol (43 g) at 55 ° C. under a nitrogen atmosphere. The reaction mixture was stirred at reflux for 3 hours. Diisopropyl ether (105 g) and acetic acid (0.85 g) were added to the reaction mixture at 45-50 ° C., and then cooled. The suspension was filtered to obtain wet crystals (43.3 g) of sodium salt of dimethyl 4-oxothiolane-2,3-dicarboxylate. The wet crystals were added to a mixture of 85% phosphoric acid (9.8 g), water (20 g) and ethyl acetate (150 g) at room temperature, and the aqueous layer was removed. The obtained organic layer was washed with 10% brine and then dried over anhydrous magnesium sulfate. The drying agent was removed by filtration, and the filtrate was concentrated under reduced pressure to obtain the title compound (22.7 g).Reference example 2
Dimethyl 4- (hydroxyimino) thiolane-2,3-dicarboxylate Dimethyl 4-oxothiolane-2,3-dicarboxylate (10.0 g), pyridine (5.44 g), hydroxylamine hydrochloride (3.34 g) Was stirred at 50 ° C. for 1 hour. Ethyl acetate and 7% aqueous phosphoric acid solution were added to the reaction mixture at room temperature, and the aqueous layer was removed. The obtained organic layer was washed with 5% sodium bicarbonate water and 10% brine. The organic layer was dried over anhydrous sodium sulfate. After removing the desiccant by filtration, the filtrate was concentrated under reduced pressure to obtain the title compound (10.4 g).Reference example 3
4-Aminothiophene-2,3-dicarboxylic acid dimethyl hydrochloride 4- (hydroxyimino) thiolane-2,3-dicarboxylate (10.4 g) in acetic acid (32 g) solution in 4N-hydrogen chloride / ethyl acetate solution ( 120 g) was added at room temperature. The reaction mixture was stirred at room temperature for 8 hours. After filtering the suspension, the obtained solid was dried to obtain the title compound (9.42 g).Reference example 4
4-Aminothiophene-2,3-dicarboxylic acid dimethyl methanesulfonate To a solution of methanesulfonic acid (80.0 g) in ethyl acetate (900 g), dimethyl 4- (hydroxyimino) thiolane-2,3-dicarboxylate (97. 1 g) of ethyl acetate (500 g) was added at 65-75 ° C. The reaction mixture was stirred at the same temperature for 2 hours. Methyl isobutyl ketone (100 g) was added at 45-50 ° C. and cooled to room temperature. After filtering the suspension, the obtained solid was dried to obtain the title compound (102 g).Reference Example 5
1,2-difluoro-3-[(4-fluoro-2-methoxyphenoxy) methyl] -4-methoxybenzene sodium borohydride in a solution of 2,3-difluoro-6-methoxybenzaldehyde (150 g) in toluene (900 g) (13.2 g) of 0.1N sodium hydroxide aqueous solution (180 g) was added at 35 to 39 ° C. The reaction mixture was stirred at the same temperature for 5 hours. After cooling the reaction mixture to room temperature, the aqueous layer was removed. The obtained organic layer was washed with 20% brine to obtain a toluene solution of 2,3-difluoro-6-methoxybenzyl alcohol. To this solution was added concentrated hydrochloric acid (610 g) at room temperature. The reaction mixture was stirred at 38-43 ° C. for 5 hours. After cooling the reaction mixture to room temperature, the aqueous layer was removed. The obtained organic layer was washed with water and 20% brine to obtain a toluene solution of 3- (chloromethyl) -1,2-difluoro-4-methoxybenzene. To this solution, 4-fluoro-2-methoxyphenol (125 g) and tetrabutylammonium bromide (56.2 g) were added at room temperature. A 25% aqueous sodium hydroxide solution (170 g) was added to the reaction mixture at 60 to 63 ° C., and the mixture was stirred at the same temperature for 4 hours. Water was added to the reaction mixture and the aqueous layer was removed. The obtained organic layer was washed with water and concentrated under reduced pressure. The residue was dissolved in 2-propanol and water was added. After filtering the suspension, the obtained solid was dried to obtain the title compound (232 g).Reference Example 6
1,2-difluoro-3-[(4-fluoro-2-methoxy-5-nitrophenoxy) methyl] -4-methoxybenzene 1,2-difluoro-3-[(4-fluoro-2-methoxyphenoxy) methyl ] To a solution of 4-methoxybenzene (158 g) in acetic acid (1200 g) was added 60% nitric acid (72.2 g) at 59-62 ° C., and the mixture was stirred at the same temperature for 2 hours. Water (1200 g) was added to the suspension at 15 to 19 ° C., and the mixture was stirred at the same temperature for 1 hour. After filtering the suspension, the obtained solid was washed with water to obtain wet crystals of the title compound (190 g, Net amount 168 g).Reference Example 7
2-Fluoro-5-[(2,3-difluoro-6-methoxyphenyl) methoxy] -4-methoxyaniline Raney nickel (2.5 g), ethyl acetate (180 g), 1,2-difluoro-3-[(4 -Fluoro-2-methoxy-5-nitrophenoxy) methyl] -4-methoxybenzene wet crystal (10.9 g, Net amount 10.0 g) was stirred at room temperature under a hydrogen atmosphere for 4 hours. The catalyst was removed by filtration, and the filtrate was concentrated under reduced pressure. The residue was dissolved with methanol and water was added. After filtering the suspension, the obtained solid was dried to obtain the title compound (7.97 g).Example 1
4- (phenoxycarbonylamino) thiophene-2,3-dicarboxylic acid dimethyl potassium carbonate (17.1 g), water (90 g), tetrahydrofuran (150 g) and 4-aminothiophene-2,3-dicarboxylic acid dimethyl hydrochloride (30 0.06) was added phenyl chloroformate (18.6 g) at 6-13 ° C. The reaction mixture was stirred at 12-13 ° C. for 30 minutes, and then the aqueous layer was removed. To the obtained organic layer, tert-butyl methyl ether was added and washed with 20% brine. The obtained organic layer was concentrated under reduced pressure. The residue was dissolved with diisopropyl ether and n-hexane was added. After filtering the suspension, the obtained solid was dried to obtain the title compound (37.0 g).
1 H-NMR (DMSO-d 6 ) δ ppm: 3.82 (3H, s), 3.82 (3H, s), 7.13-7.30 (3H, m), 7.40-7.46 (2H, m), 7.80 (1H, s ), 10.24 (1H, s)Example 2
4- {3- [2-Fluoro-5- (2,3-difluoro-6-methoxybenzyloxy) -4-methoxyphenyl] ureido} dimethyl thiophene-2,3-dicarboxylate 2-fluoro-5-[( 2,3-difluoro-6-methoxyphenyl) methoxy] -4-methoxyaniline (7.70 g), dimethyl 4- (phenoxycarbonylamino) thiophene-2,3-dicarboxylate (8.65 g), triethylamine (0. 37 g) and tetrahydrofuran (80 mL) were stirred at room temperature for 24 hours. The reaction mixture was concentrated under reduced pressure. Ethyl acetate and methanol were added to the residue. After filtering the suspension, the obtained solid was dried to obtain the title compound (12.0 g).
1 H-NMR (DMSO-d 6 ) δ ppm: 3.71 (3H, s), 3.82 (3H, s), 3.83 (3H, s), 3.89 (3H, s), 5.00 (2H, d, J = 1.6 Hz), 6.87-6.93 (1H, m), 7.00 (1H, d, J = 12.8Hz), 7.41-7.50 (1H, m), 7.75 (1H, d, J = 8.0Hz), 7.94 (1H, s ), 8.82 (1H, s), 8.95 (1H, s)Example 3
3- [2-Fluoro-5- (2,3-difluoro-6-methoxybenzyloxy) -4-methoxyphenyl] -2,4-dioxo-1,2,3,4-tetrahydrothieno [3,4 d] methyl pyrimidine-5-carboxylate 4- {3- [2-fluoro-5- (2,3-difluoro-6-methoxybenzyloxy) -4-methoxyphenyl] ureido} thiophene-2,3-dicarboxylic acid A methanol solution (3.48 g) of 28% sodium methoxide was added to a suspension of dimethyl (10.0 g) in tetrahydrofuran (40 g), stirred at room temperature for 3 hours, and acetic acid (1.30 g) was added. The reaction mixture was concentrated under reduced pressure. Methanol was added to the residue, and water was further added. After filtering the suspension, the obtained solid was dried to obtain the title compound (8.58 g).
1 H-NMR (DMSO-d 6 ) δ ppm: 3.79 (3H, s), 3.81 (3H, s), 3.84 (3H, s), 4.95 (2H, s), 6.88-6.94 (1H, m), 7.08 (1H, d, J = 11.6Hz), 7.19-7.23 (2H, m), 7.44-7.53 (1H, m), 11.62 (1H, s)Example 4
4- (phenoxycarbonylamino) thiophene-2,3-dicarboxylate potassium carbonate (9.38 kg), water (49 kg), tetrahydrofuran (82 kg), dimethyl 4-aminothiophene-2,3-dicarboxylate hydrochloride (16 4 kg) of the reaction mixture was stirred for 40 minutes, and then phenyl chloroformate (10.1 kg) was added at 11-21 ° C. The reaction mixture was stirred for 30 minutes, and then the aqueous layer was removed to obtain a tetrahydrofuran solution of the title compound.Example 5
4- {3- [2-Fluoro-5- (2,3-difluoro-6-methoxybenzyloxy) -4-methoxyphenyl] ureido} dimethyl thiophene-2,3-dicarboxylate 4-obtained in Example 4 To a tetrahydrofuran solution of dimethyl (phenoxycarbonylamino) thiophene-2,3-dicarboxylate, 2-fluoro-5-[(2,3-difluoro-6-methoxyphenyl) methoxy] -4-methoxyaniline (17.0 kg), Tetrahydrofuran (8.5 kg) and triethylamine (1.1 kg) were added, and the mixture was stirred at 50 ° C. for 3.5 hours to obtain a tetrahydrofuran solution of the title compound.Example 6
3- [2-Fluoro-5- (2,3-difluoro-6-methoxybenzyloxy) -4-methoxyphenyl] -2,4-dioxo-1,2,3,4-tetrahydrothieno [3,4 d] pyrimidine-5-carboxylic acid tetrahydrofuranate 4- {3- [2-fluoro-5- (2,3-difluoro-6-methoxybenzyloxy) -4-methoxyphenyl] ureido} obtained in Example 5 Methanol (41 kg) and water (47 kg) are added to a tetrahydrofuran solution of dimethyl thiophene-2,3-dicarboxylate, a 7.3% lithium hydroxide aqueous solution (80.1 kg) is added at 11 to 13 ° C., and 90 ° C. at 11 ° C. Stir for minutes. Acetic acid (11.4 kg) was added to the reaction mixture at 9 to 16 ° C., and acetic acid (13.0 kg) was further added at 29 to 31 ° C. Seed crystals were added to the reaction mixture, and the mixture was stirred at the same temperature for 30 minutes. Water (34 kg) was added to the suspension and stirred at 30 ° C. for 40 minutes. The suspension was stirred at 4-9 ° C. for 90 minutes. After the suspension was filtered, the obtained solid was washed with a mixed solution of methanol (54 kg) and water (68 kg) to give wet crystals of the title compound (31.64 kg, Net amount (compound (A) free form equivalent)) 26 0.7 kg) was obtained.
A part of the wet crystals of the title compound was dried under reduced pressure at an external temperature of 60 ° C., and 1 H-NMR, HPLC and powder X-ray diffraction were measured on the obtained dried crystals of the title compound.
1 H-NMR (DMSO-d 6 ) δ ppm: 1.68-1.82 (3H, m), 3.53-3.65 (3H, m), 3.80 (3H, s), 3.81 (3H, s), 4.94-4.98 (2H , m), 6.87-6.94 (1H, m), 7.13 (1H, d, J = 11.2Hz), 7.25 (1H, d, J = 7.2Hz), 7.39 (1H, s), 7.43-7.52 (1H, m), 11,99 (1H, s), 14.53 (1H, s)
PATENT
WO 2020089190
https://patents.google.com/patent/WO2020089190A2/enFor example, the GnRH antagonist may be 3-[2-fluoro-5-(2,3-difluoro-6-methoxybenzyloxy)4- methoxyphenyl]-2,4-dioxo-1 ,2,3,4- tetrahydrothieno [3,4d]pyrimidine-5-carboxylic acid, or a pharmaceutically acceptable salt thereof. The salt may be, for instance, the choline salt thereof, represented by formula (Via), below.
Compound (VI) and pharmaceutically acceptable salts thereof, such as the choline salt thereof (compound (Via)), can be synthesized, for example, using the methodology described in WO 2014/042176, the disclosure of which is incorporated herein by reference in its entirety. An exemplary synthetic scheme that may be used for the preparation of compound (VI) and the choline salt thereof is shown in Scheme 1 , below.Scheme 1 . Exemplary preparation of compound (VI) and the choline salt thereof




wherein Ri and R are each independently C alkoxy groups; LG is a nucleofugal leaving group, such as chlorine or bromine, among others; R represents an optional substituent, such as halogen, acyl group, C alkyl group, or a nitro substituent; DMAP denotes A/-dimethylaminopyridine; and TEA denotes trimethylamine.Crystalline compound (Via) has been characterized spectroscopically, for instance, in US Patent No. 9,169,266, the disclosure of which is incorporated herein by reference in its entirety. The foregoing crystalline form has been shown to exhibit characteristic X-ray powder diffraction peaks at about 7.10 2Q, about 11 .5° 2Q, about 19.4° 2Q, about 21 .5° 2Q, about 22.0° 2Q, about 22.6° 2Q, about 23.5° 2Q, and about 26.2° 2Q. Additionally, this crystalline form exhibits 13C solid-state nuclear magnetic resonance (NMR) peaks centered at about 55.5 ppm, about 57.1 ppm, about 58.7 ppm, about 69.8 ppm, about 98.1 ppm, about 110.3 ppm, about 1 1 1 .6 ppm, about 113.7 ppm, about 1 18.0 ppm, about 145.3 ppm, about 149.8 ppm, and about 155.8 ppm. This crystalline form further exhibits 19F solid-state NMR peaks centered at about -151.8 ppm, -145.2 ppm, and -131 .6 ppm.Compound (VI), as well as pharmaceutically acceptable salts thereof, such as the choline salt thereof, exhibit a high affinity for human GnRH receptor (27.4 nM). Using the compositions and methods described herein, a patient that is presenting with or has been diagnosed as having, adenomyosis or rectovaginal endometriosis may be administered a compound of formula (VI), or a pharmaceutically acceptable salt thereof, such as the choline salt thereof, to treat the disease or ameliorate one or more symptoms of the disease. Exemplary doses of compound (VI) and pharmaceutically acceptable salts thereof, such as the choline salt thereof, include doses of from 25 mg to 500 mg daily, such as doses of 100 mg per day and 200 mg per day. Additional dosing information is provided below.3-Aminoalkyl pyrimidine-2, 4(1 H,3H)-dionesAdditional GnRH antagonists that may be used in conjunction with the compositions and methods described herein include optionally substituted 3-aminoalkyl pyrimidine-2, 4(1 H,3H)-dione derivatives, such as compounds represented by formula (VII)

PATENTWO 2021023876https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021023876&_cid=P11-KWFRM2-91270-1
In some embodiments, the compound is the choline salt of the compound represented by formula (VI), choline 3- [2-fluoro-5- (2,3-difluoro-6-methoxybenzyloxy) 4-methoxyphenyI] -2,4- dioxo-1,2,3,4-
tetrahydrothieno [3,4d] pyrimidine-5-carboxylate. It is to be understood that references herein to a compound represented by formula (VI) specifically include the choline salt of compound (VI), which is represented by formula (VIa), below.
In some embodiments, the choline 3- [2-fluoro-5- (2,3-difluoro-6-methoxybenzyloxy) 4-methoxyphenyI] -2,4-dioxo-1,2,3,4- tetrahydrothieno [3,4d ] pyrimidine-5-carboxylate is in a crystalline state.
PATENT
WO 2021023877
References
- ^ Jump up to:a b c “Linzagolix – Kissei Pharmaceutical/ObsEva – AdisInsight”.
- ^ Jump up to:a b Ezzati M, Carr BR (2015). “Elagolix, a novel, orally bioavailable GnRH antagonist under investigation for the treatment of endometriosis-related pain”. Womens Health (Lond). 11 (1): 19–28. doi:10.2217/whe.14.68. PMID 25581052.
- ^ Chodankar, Rohan; Allison, Jennifer (2018). “New Horizons in Fibroid Management”. Current Obstetrics and Gynecology Reports. 7 (2): 106–115. doi:10.1007/s13669-018-0242-6. ISSN 2161-3303.
External links
| Clinical data | |
|---|---|
| Trade names | Yselty |
| Other names | KLH-2109; OBE-2109 |
| Routes of administration | By mouth[1][2] |
| Drug class | GnRH modulator; GnRH antagonist; Antigonadotropin |
| ATC code | None |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 935283-04-8 |
| PubChem CID | 16656889 |
| ChemSpider | 17590169 |
| UNII | 7CDW97HUEX |
| KEGG | D11608 |
| ChEMBL | ChEMBL3668014 |
| Chemical and physical data | |
| Formula | C22H15F3N2O7S |
| Molar mass | 508.42 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
////////LINZAGOLIX, Hormone Antagonists, WHO 10711, KLH-2109, KLH 2109, OBE-2109, OBE 2109

NEW DRUG APPROVALS
ONE TIME
$10.00


{kind=link}
{kind=link}