
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
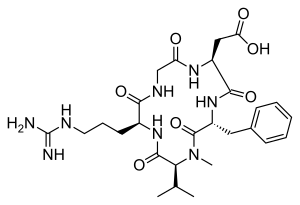
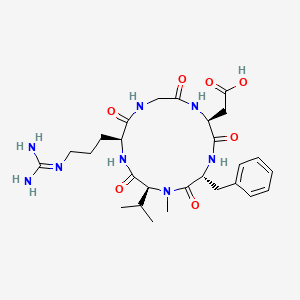
| IUPAC Condensed | cyclo[Arg-Gly-Asp-D-Phe-N(Me)Val] |
|---|---|
| HELM | PEPTIDE1{R.G.D.[dF].[meV]}$PEPTIDE1,PEPTIDE1,5:R2-1:R1$$$ |
| IUPAC | cyclo[L-arginyl-glycyl-L-alpha-aspartyl-D-phenylalanyl-N-methyl-L-valyl] |
CILENGITIDE
- Molecular FormulaC27H40N8O7
- Average mass588.656 Da
2-[(2S,5R,8S,11S)-5-benzyl-11-[3-(diaminomethylideneamino)propyl]-7-methyl-3,6,9,12,15-pentaoxo-8-propan-2-yl-1,4,7,10,13-pentazacyclopentadec-2-yl]acetic acid188968-51-6[RN]
4EDF46E4GI
7823
циленгитид
سيلانجيتيد
西仑吉肽
EMD 121974, EMD-121974, UNII-4EDF46E4GI
Cilengitide has been in phase III clinical trials by Merck Serono and NCI for the treatment of glioblastoma multiforme. However, this research has been discontinued.
Cilengitide was originally developed by Merck KGaA in collaboration with the Technical University of Munich, then received orphan drug designation from FDA for the treatment of glioma in 2005.
Cilengitide (EMD 121974) is a molecule designed and synthesized at the Technical University Munich in collaboration with Merck KGaA in Darmstadt. It is based on the cyclic peptide cyclo(-RGDfV-), which is selective for αv integrins, which are important in angiogenesis (forming new blood vessels), and other aspects of tumor biology. Hence, it is under investigation for the treatment of glioblastoma, where it may act by inhibiting angiogenesis, and influencing tumor invasion and proliferation.[1][2]
The European Medicines Agency has granted cilengitide orphan drug status.[3]
Cilengitide seems to function by inhibiting the FAK/src/AKT pathway and inducing apoptosis in endothelial cells.[4] Preclinical studies in mice of cilengitide were able to demonstrate efficacious tumor regression.[4]
In a rat xenograft model, cilengitide was able to potentiate the cytotoxic effects of radiation when cilengitide was administered prior to radiation therapy.[5] When combined with radiation, inhibition of integrin expression by cilengitide synergistically improves the cytotoxic effects of ionizing radiation for glioblastoma.[5]
Clinical trials
Phase II studies were able to demonstrate that cilengitide as a potential monotherapy in patients with recurrent glioblastoma[6] with high intratumor drug levels when 2000 mg of cilengitide is given twice weekly.[7]
Cilengitide is well tolerated, in combination with radiation and temozolomide, at a dose of 2000 mg in patients with newly diagnosed glioblastoma, regardless of MGMT promoter status.[8] In a phase I/IIa study, the addition of cilengitide to the standard of care for newly diagnosed glioblastoma (surgical resection followed by temozolomide and radiation therapy) improves progression-free survival and overall survival in patients with MGMT promoter methylation.[9]
However, in a subsequent study, cilengitide does not seem to alter the pattern of glioblastoma progression,[10]
and in an EORTC phase III randomized, controlled, multicenter clinical trial, consisting of over 500 patients in 23 countries, the addition of cilengitide to the standard of care did not improve overall survival in patients with newly diagnosed glioblastoma and methylated MGMT promoter status [11] A phase II study, the CORE trial, is currently being conducted in patients with newly diagnosed glioblastoma and unmethylated MGMT promoter status.[12]

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
SYN
Angewandte Chemie, International Edition, 55(4), 1540-1543; 2016

SYN
Chemistry – A European Journal, 16(18), 5385-5390, S5385/1-S5385/36; 2010
Reference:1. WO0047228A1 / US7115261B1.
2. US6001961A.Route 2
Reference:1. CN102731627A.PATENTWO/2021/224234ANTIVIRAL USE OF CILENGITIDEhttps://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021224234&_cid=P20-KW0M52-85135-1
PATENThttps://patents.google.com/patent/CN102731627A/enEMD121974 (Cilengitide), the Chinese another name: ring (L-arginyl glycyl-L-aspartoyl-D-phenylalanyl-N-methyl-L-valyl) is an a kind of new classification cancer therapy drug of synthetic.Merkel company discovers that EMD121974 amalgamation radiotherapy (merging to reach assists TM to add radiotherapy) possibly prolong lifetime; Simultaneously integrate plain supressor antitumor drug as first; Got into the III clinical trial phase, its important mechanism is to grow targeting that the blood supply structure of nutrition, the growth of promotion cancer cell is provided in tumour and for tumour through line artery.The EMD121974 molecular formula is: C 27H 40N 8O 7, have following structure:
The preparation method of cyclic peptide mainly contains liquid phase synthesis process, solid phase synthesis precursor peptide cyclization process, process for solid phase synthesis in liquid phase at present; Wherein preceding two kinds of synthesis techniques all are the cyclisation in liquid phase of synthetic precursor peptide, and this method needs reactant in extremely rare solvent, to react (10 -3~10 -4Mol/L), and intermolecular be prone to react generation line style or cyclic polymer, greatly reduced the cyclisation yield, bring trouble for follow-up purifying, and in large-scale production, produce a large amount of waste liquids, be unfavorable for suitability for industrialized production.In conjunction with the structure of EMD121974, utilize the false rare principle of benefit of solid phase, developed a kind of efficient cyclization reaction, the cyclisation time shortens to 20%~30% of liquid phase cyclisation, and the 2%-8% of solvent as liquid phase used in reaction.Embodiment 1The preparation of Fmoc-L-Asp (OtBu)-Wang ResinThe Wang Resin that takes by weighing the 10g substitution degree and be 0.5mmol/g joins in the reactor drum, adds an amount of DCM, and swelling 30min takes out DCM; 6.17g Fmoc-L-Asp-OtBu, DIC 2.40ml, HOBT2.1g are dissolved among the 30ml DMF; At 0-5 ℃ of activation 15min, activation solution is joined in the reactor drum that contains Wang Resin, behind the reaction 10min; Add DMAP 0.18g again, at 0~30 ℃ of reaction 1~5h.After reaction finishes, add sealing Wang Resin unreacted hydroxylation reagent diacetyl oxide 1ml and pyridine 0.5ml, behind the capping 1h, DMF, DCM, the CH of 80ml used in washing successively 3OH, DMF washing 2,1,1,2 times, each 1min.Through detecting, obtain the Fmoc-L-Asp that substitution degree is 0.47mmol/g (OtBu)-Wang Resin.Embodiment 2The EMD121974 precursor:The preparation of A-Wang Resin (Fmoc-D-Phe-N-Me-L-Val-L-Arg (Mtr)-Gly-L-Asp (OtBu)-Wang Resin)Fmoc-L-Asp (OtBu)-Wang Resin is joined in the reactor drum, behind DMF swelling 30min, take out solvent, the piperidines-DMF that adds 80ml 25% reacts 5min, and 80ml DMF washs 1 time (3min), and the piperidines-DMF that adds 80ml 25% reacts 15min; DMF, DCM, the CH of 80ml used in washing successively 3OH, DMF washing 2,1,1,2 times, each 1min; With 4.45g Fmoc-Gly-OH, 5.68g HBTU, 2.03g HOBt, be dissolved among the DMF of 30ml, dissolve the back and added DIEA 2.45ml; 0~5 ℃ of activation 15min; Activation solution is joined in the above-mentioned reactor drum, and behind reaction 1-3h under 0~30 ℃, reaction end detects with ninhydrin method.Adopt aforesaid method coupling Fmoc-L-Arg (Mtr)-OH, Fmoc-N-Me-L-Val, Fmoc-D-Phe-OH successively, finally obtain Fmoc-D-Phe-N-Me-L-Val-L-Arg (Mtr)-Gly-L-Asp (OtBu)-Wang Resin.Embodiment 3EMD121974 precursor peptide: the preparation of B-Wang Resin (D-Phe-N-Me-L-Val-L-Arg (Mtr)-Gly-L-Asp-Wang Resin)With volume ratio is that piperidines-DMF of 25% is the Fmoc deprotection agent of Fmoc-D-Phe-N-Me-L-Val-L-Arg (Mtr)-Gly-L-Asp (OtBu)-Wang Resin; Add piperidines-DMF 80ml of 25% first time; Reaction 5min, 80ml DMF washs 1 time (3min), adds piperidines-DMF 80ml of 25% for the second time; Behind the reaction 15min, DMF, DCM, the CH of 80ml used in washing successively 3OH, DMF washing 2,1,1,2 times, each 1min gets D-Phe-N-Me-L-Val-L-Arg (Mtr)-Gly-L-Asp (OtBu)-Wang Resin after washing finishes.80% the PhOH-DCM solution that adds volume ratio and be 100ml takes off OtBu with the TFA of catalytic amount, reacts 8h; DMF, DCM, the CH of 80ml used in washing successively 3OH, DMF washing 2,1,1,2 times, each 1min gets D-Phe-N-Me-L-Val-L-Arg (Mtr)-Gly-L-Asp-Wang Resin.Embodiment 4The preparation of EMD121974-Wang Resin (Cyclo (D-Phe-N-Me-L-Val-L-Arg-Gly-L-Asp)-Wang Rsin)In above-mentioned reactor drum, add cyclization reagent 3.9g DPPA, 2.5ml DIEA (reactant cyclization reagent amount of substance ratio is 1: 3), at 10~40 ℃ of reaction 3h, the multiple cyclization reagent reaction 3~5h (reaction end detects with ninhydrin method) that throws once above-mentioned equivalent; DMF, DCM, the CH of 80ml used in washing successively 3OH washing 2,1,3 times, each 3min gets Cyclo (D-Phe-N-Me-L-Val-L-Arg-Gly-L-Asp)-Wang Rsin.Embodiment 5The preparation of EMD121974 (Cyclo (D-Phe-N-Me-L-Val-L-Arg-Gly-L-Asp))In above-mentioned reactor drum, add the TFA/H of lytic reagent 120ml again 2Behind O/TlS (volume ratio is 95: 2.5: 2.5) the reaction 3h, suction filtration is removed resin, and filtrating slowly joins in the no water-ice ether; Static 2-5h, high speed centrifugation obtain thick peptide, prepare through high-pressure liquid phase; Lyophilize gets smart EMD121974; Its purity>99.5%, single impurity<0.2%, total recovery reaches 63%.Choosing substitution degree in the present embodiment is the Wang Resin of 0.5mmol/g, and can also choose substitution degree is the arbitrary Wang Resin and Fmoc-L-Asp-OtBu prepared in reaction Fmoc-L-Asp (the OtBu)-Wang Resin of 0.4~0.9mmol/g scope.All can realize technical scheme of the present invention, and obtain technique effect of the present invention.Above content is an EMD121974 and become one of best preferred version of route; And to further explain that the present invention did; But can not assert that practical implementation of the present invention is only limited to these explanations; Under the prerequisite that does not break away from the present invention’s design, can also make some simple deductions and replacement, all should be regarded as protection domain of the present invention.
CLIPhttps://www.eurekaselect.net/article/2607Cilengitide, a cyclic RGD pentapeptide, is currently in clinical phase III for treatment of glioblastomas and in phase II for several other tumors. This drug is the first anti-angiogenic small molecule targeting the integrins αvβ3, αvβ5 and α5β1. It was developed by us in the early 90s by a novel procedure, the spatial screening. This strategy resulted in c(RGDfV), the first superactive αvβ3 inhibitor (100 to 1000 times increased activity over the linear reference peptides), which in addition exhibited high selectivity against the platelet receptor αIIbβ3. This cyclic peptide was later modified by N-methylation of one peptide bond to yield an even greater antagonistic activity in c(RGDf(NMe)V). This peptide was then dubbed Cilengitide and is currently developed as drug by the company Merck-Serono (Germany). This article describes the chemical development of Cilengitide, the biochemical background of its activity and a short review about the present clinical trials. The positive anti-angiogenic effects in cancer treatment can be further increased by combination with “classical” anti-cancer therapies. Several clinical trials in this direction are under investigation.
CLIPJournal of Protein Chemistry

Schematic of the one-step chemoenzymatic synthesis of cilengitide using wild-type Mcy TE. (1) The chemically synthesised (SPPS, solid-phase peptide synthesis) mimetic substrate was condensed with benzyl mercaptane to produce pentapeptide thioester (pentapeptide-BMT). (2) Models of the substrate-O-TE acyl enzyme intermediate are marked with brackets (protein data bank, 1JMK). (3) Mechanism of TE domain catalysis: a pentapeptide -O-TE acyl-enzyme intermediate is formed by transfer of the peptidyl chain from the phosphopantethiene of the terminal peptidyl carrier protein (PCP), which was substituted by benzyl mercaptane, to the active site serine of the TE domain. For hydrolyzing TE domains, the intermediate is captured by water, generating the linear peptide; for cyclizing TE domains, an intramolecular nucleophile captures the intermediate, resulting in “cilengitide”
PATENTWO 9745447
WO 9745137
DE 19534177
WO 2000053627
WO 2000047228
US 20040063790
WO 2009124754
WO 2011079015
WO 2011069629
WO 2011144756WO 2016059622
PATENTWO 2012062777https://patents.google.com/patent/WO2012062777A1/enSynthesis of cyclic peptidesCyclo[-Arg-Gly-Asp- 6 or 7 -Phe-Val-Ala-] (1 and 2). Resin loading. 2- chlorotrityl chloride-resin ( 1 50 m g , 1 .5m m ol/g ) was p laced i n a 20 m l polypropylene syringe fitted with a polyethylene filter disk. The resin was then washed with CH2CI2 (5 χ 0.5 min), and a solution of Fmoc-L-Gly-OH (334 mg, 1 .125 mmol, 5 equiv) and DIEA (239 μΙ_, 6.25 equiv) in CH2CI2 (2.5 ml_) was added. The mixture was then stirred for 15 min. Extra DIEA (239 μΙ_, total 12.5 mmol) was added, and the mixture was stirred for an additional 45 min. The reaction was stopped by adding 3 χ DCM/ MeOH/ DIEA (85: 10:5) and stirring for 1 0 m in. The Fmoc-L-Gly-O-resin product was subjected to the following washings/treatments with CH2CI2 (3 χ 0.5 min), DMF (3 χ 0.5 min), piperidine and DMF (5 χ 0.5 min). The loading was 0.50 mmol/g, as calculated by Fmoc determination.Peptide coupling. Fmoc-L-Arg(Pbf)-OH (243 mg, 0.375 mmol, 5 equiv), Fmoc- L-Ala-OH (1 17 mg, 0.375 mmol, 5 equiv), Fmoc-L-Val-OH ( 127 mg, 0.375 mmol, 5 equiv) and Fmoc- L-Phe-OH ( 145 mg, 0.375 mmol, 5 equiv) were added sequentially to the above obtained H-L-Gly-O-resin using HCTU (155 mg, 0.375 mmol, 5 equiv), HOBt (50 mg, 0.375 mmol, 5 equiv) and DIEA (127 μΙ_, 0.75 mmol, 10 equiv) in DMF (2.5 ml_). In all cases, after 90 min of coupling, the ninhydrin test was negative. Removal of Fmoc group and washings were performed as described in general procedures. /V-Alloc-thiazole 6 or 7 (92 mg, 0.375 mmol, 5 equiv) was coupled with HATU (143 mg, 0.375 mmol, 5 equiv), HOAt (51 mg, 0.375 mmol, 5 equiv) and DIEA (127 μΙ_, 0.75 mmol, 10 equiv) for 90 min. This coupling was repeated twice in the same conditions. The Alloc group of the peptide resin was removed with Pd (PPh3)4 (9 mg, 0.0075 mmol, 0.1 equiv) in the presence of PhSiH3 (92.5 μΙ_, 0.75 mmol, 10 equiv) in DCM for 20 min. This deprotection was repeated three times in the same conditions. After washing, the resin was treated with dry THF (2ml_) for 15 min. Meanwhile, Fmoc-L-Asp(tBu)-OH (154 mg, 0.375 mmol, 5 equiv) was added to a 68 mM solution of triphosgene in dry THF (1 .15 equiv). Sym-collidine (99.5 μΙ_, 0.75 mmol, 10 equiv) was added to the clear solution, upon which a precipitate of collidinium chloride was formed. DIEA (102 μΙ_, 0.6 mmol, 8 equiv) was added to the resin, immediately followed by addition of the suspension. This coupling was repeated four times in the same conditions. The reaction mixture was stirred at 50 °C during 48 h.Peptide cleavage. Following Fmoc deprotection, the peptidyl-resin was treated with TFA-CH2CI2 (1 :99) (5 χ 30 s). The filtrate was collected on H20 (4 ml_) and the H20 was partially removed under reduced pressure. MeCN was then added to dissolve solid that formed during the removal of H20, and the solution was lyophilized to give 12 mg and 10 mg of the linear compounds 28 and 29 respectively with a purity of > 91 % as checked by HPLC (Column A, Rt 7.43 min and Rt 7.38 min respectively, linear gradient 35%-40% ACN in 15 min.)], which was used without further purification. MALDI-TOF-MS calculated for C50H71 N11 O13S2 1098.29; found mlz 1099.29 [M + H]+, 1 121 .28 [M + Na]+, 1 137.39 [M + K]+.Synthesis in solution. Cyclization. The protected linear peptides 28 and 29 were dissolved in DMF (1 L, 10“4 M), and HOAt (9.6 mg, 0.07 mmol, 5 equiv), DIPEA (24 μΙ_, 0.14 mmol, 10 equiv), and PyAOP (36.6 mg, 0.07 mmol, 5 equiv) were added. The mixture was stirred for 24 h at room temperature, and the course of the cyclization step was then checked by HPLC (Column A, Rt 1 1 -67 min and Rt 10.70 min respectively, linear gradient 45%-55% ACN in 15 min.). The solvent was removed by evaporation under reduced pressure and the protected cycle 30 and 31 were used in the next step without further purification. MALDI-TOF-MS calculated for C50H69N11 O12S2 1080.28; found mlz 1081 .28 [M + H]+, 1 103.27 [M + Na]+, 1 1 19.38 [M + K]+.Side chain deprotection. The protected cyclopeptides 30 and 31 (14.7 mg, 19.04 pmol) were treated with TFA-H20 (95: 5) during 1 h. The solvent was removed by evaporation under reduced pressure.Peptide purification. The crude product was purified by HPLC (Symmetry C8 5 μη-Ί, 30 mm x 100 mm), gradient of MeCN (30% to 75% in 15 min) MeCN (+0.05% TFA) in water (+0.05% TFA), 20 mL/min, detection at 220 nm, to give the cyclopeptides 1 and 2 (4.5 mg, 5.8 pmol and 6.5 mg, 8.37 pmol, 7.7% and 12% yield respectively). The products were characterized by HPLC (Rt 8.99 min, and Rt 8.02 min Column A, respectively, linear gradient 0%-100% ACN in 1 5 min. ) and by MALDI-TOF-MS: calculated for C33H45N11 O9S 771 .84; found mlz 772.84 [M + H]+, 794.83 [M + Na]+, 810.94 [M + K]+.Cyc/o-[Arg-Gly-Asp-Thz1X-] (3). General procedure for cyclopeptide synthesis. Solid phase synthesis: The synthesis of the linear peptide H- Asp(tBu)-XX-Arg(Pbf)-Gly-OH was performed using Fmoc-based solid phase peptide synthesis with 2-chlorotrityl chloride resin (2.0 g, 3.2 mmol).Resin loading: Fmoc-Gly-OH (594 mg, 2.0 mmol) was attached to the resin with DIPEA in DCM at room temperature for 1 .5 h. The remaining trityl groups were capped adding 0.5 mL of MeOH for 30 min. After that, the resin was filtered and washed with DCM (2x), DMF (2x). The loading of the resin was determined by titration of the Fmoc group (Chan WC and White PD. Fmoc Solid Phase Peptide Synthesis. Oxford University Press: New York, 2000). The final loading was 2.0 mmol/g. The Fmoc group was eliminated by treatment with 20% piperidine in DMF (2X10 min). The resin was washed with DMF (3x), DCM (3x). Peptide coupling: Fmoc-Arg(Pbf)-OH (5.19 g, 8.0 mmol), DIPCDI (1.23 mL, 8.0 mmol) and HOBt (1.08 g, 8.0 mmol) were dissolved in DMF and added to the resin for 1 .5 h. The end of the coupling was monitored by ninhydrin test (free amine group) (Kaiser E et al. Anal Biochem 1970, 34:595-598). The resin was filtered and washed with DMF (3X) and DCM (3X). The Fmoc group was eliminated with 20 % piperidine in DMF (2X10 min).The coupling of the thiazole module was carried out with 8 (1 .14 g, 3.0 mmol), PyAOP (1 .56 g, 3.0 mmol) and DIPEA (1 .02 mL, 6.0 mmol) in DMF for 1 .5 h. The completion of the reaction was checked with the ninhydrin test. Finally the deprotection of the amine and coupling of the Fmoc-Asp(‘Bu)-OH were carried out under the same conditions of the second amino acid.Peptide cleavage: The resin bound peptide was treated with 2% TFA in DCM (6 x 30 sec.) The resin was washed with DCM and the combined solution was evaporated under vacuum with Et20 several times, furnishing the linear peptide 32 as a white solid. The peptide was used for the next step without purification.H PLC (gradient 20 to 80% of CH3CN in 1 5 m in): tR= 8.33 min. HPLC-MS (ES(+)): m/z 795.3.Synthesis in solution. Cyclization: The product 32 (200 mg, 0.251 mmol) was dissolved in anhydrous DMF (50 mL, 5 mM), PyAOP (262 mg, 0.503 mmol) and DIPEA (213 μί, 1 .255 mmol) were added. The reaction was monitored by HPLC. Once the reaction was finished, the DMF was evaporated under vacuum. The crude was dissolved in AcOEt and the solution was washed with NH4CISat and Na2CO3 sat. The organic layer was collected, dried over Na2SO4, filtered and concentrated under vacuum. The peptide was purified by flash chromatography (CHCIs/MeOH 8:2) furnishing the protected cyclic peptide 33 as a white solid (1 56 mg, XX%). HPLC (gradient 40 to 90% of CH3CN in 1 5 min): tR= 8.86 min. HPLC-MS (ES(+)): m/z 778.2Side chain deprotection: The protected peptide 33 (125 mg, XX mmol), was treated with 25 mL of a solution of TFA H2O (95:5). After 3 h, the solvent was evaporated under vacuum and the residue was precipitated with Et2O (4X). The Et2O solution was discarded and the white solid was lyophilized to afford 3 55 mg (XX%).
Peptide purification. The end product 3 was dissolved in 5 ml MilliQ water and it was filtered through a 0.2 pm filter. The cyclic peptide was purified by semipreparative RP-HPLC using acetronitrile (0.05% TFA)/water (0.1 % TFA). The HPLC sample was vacuum concentred and transformed into the hydrochloride salt lyophilized with water with 0.05% HCI.1H-NMR (500 MHz, H20:D20-d2 9: 1 , 278 K): δ = 9.29 (t, NH Gly), 9.20 (d, J = 7.24 Hz, NH Asp), 8.90 (t, J = 5.89/5.89 Hz, NH Thz), 8.46 (d, J = 8.93 Hz, NH Arg), 7.79 (s, CH Thz), 7.22 (t, J = 5.39/5.39 Hz, ΝΗε Arg), 4.75 (m, CHa Arg), 4.63 (m, CHa Asp), 4.04 (dd, J = 3.35/14.90 Hz, CHa Gly), 3.82 (dd, J = 6.69/14.96 Hz, CHa Gly), 3.17 (m, CH25 Arg), 2.89 (m, CH2p Asp), 1 .92 (m, CH p Arg), 1 .82 (m, CHP Arg), 1 .63 (m, CH2 Arg). HPLC (gradient 0 to 20% of CH3CN in 15 min): tR= 10.52 m in. HRMS (E IS) m/z calculated 468.1540

found 469.16099 (M+H)+.Cyc/o-[Arg-Gly-Asp-Thz2X-] (4). The cyclopeptide 4 was prepared according to the process followed for 3 and using bithiazole 9 (XX mg, YY mmol) instead of 8. The linear peptide 34: HPLC (gradient 0 to 100% CH3CN in 15 min.): tR = 10.34 min, HPLC-MS (ES(+)): m/z 877.81 . The protected peptide 35: HPLC (gradient 0 to 100% CH3CN in 15 min.): tR = 13.91 min, HPLC-MS (ES(+)): m/z 860.54. The final peptide 4: 1H-NMR (500 MHz, H20:D20-d2 9: 1 , 298 K): δ = 8.93 (sbroad, NH Gly), 8.82 (d, J = 7.62 Hz, NH Asp), 8.75 (t, J = 5.69/5.69 Hz, NH Thz), 8.51 (d, J = 7.62 Hz, NH Arg), 8.05 (s, CH Thz1), 7.50 (s, CH Thz2), 7.19 (t, J = 5.38/5.38 Hz, ΝΗε Arg), 4.13 (dd, J = 5.82/14.24 Hz, CH Gly), 3.87 (dd, J = 5.96/15.69 Hz, CH Gly), 3.21 (m , CH25 Arg), 2.94 (m, CH2p Asp), 1 .95 (m , CHP Arg), 1 .87 (m , CHP Arg), 1 .68 (m , CH2y Arg). HPLC (gradient 1 0 to 25% of CH3CN in 1 5 m in): tR = 8.73 min. HRMS (EIS) m/z calculated 551 .1369 (C2oH25N906S2) found 552.14392 (2M+2H)+.Cyc/o-[Arg-Gly-Asp-Thz3X-] (5). The cyclopeptide 5 was prepared according to the process for 3 and using trithiazole 10 (XX mg, YY mmol) instead of 8. The linear peptide 36: HPLC (gradient 20 to 80% of CH3CN in 15 min.): tR = 7.60 min, HPLC-MS (ES(+)): m/z 961 .23. The protected peptide 37: HPLC (gradient 20 to 80% of CH3CN in 15 m in. ): tR = 1 3.13 min, HPLC-MS (ES(+)): m/z 944.3. The final peptide 5: HPLC (gradient 10 to 30% CH3CN in 15 m in): tR = 8.26 m in. HRMS (E IS) m/z calculated 634.1 1 99 (C23H26N10O6S3) found 635.12683 (2M+2H)+. 1H-NMR (500 MHz, DMSO-d6 298 K): δ = 9.21 (t, J = 5.4, NH Gly), 8.72 (m, NH Asp + NH Thz), 8.37 (s, CH Thz1), 7.96 (d, J = 9.2, NHa Arg), 7.77 (s, CH Thz2), 7.68 (t, J = 6.0, ΝΗε Arg), 7.23 (s, CH Thz3), 4.83 (dd, J = 14.3, 8.5, CHa Arg), 4.72 (dd, J = 16.3, 6.6, CH Thz), 4.59 (m, CH Thz + CHa Asp), 3.89 (d, J = 1 1 .5, CH Gly), 3.59 (d, J = 9.7, CH Gly), 3.13 (dd, J = 12.6, 6.3, CH25 Arg), 2.81 (dd, J = 16.3, 4.3, CHP Asp), 2.58 (dd, J = 16.5, 8.7, CHP Asp), 1 .82 (m, CHP Arg), 1 .71 (m, CHP Arg), 1 .49 (m, CH2y Arg).Cilengitide. The cilengitide was prepared according to the method described in Dechantsreiter MA et al. (J Med Chem 1999, 42:3033-3040). 1H- NMR (500 MHz, H20:D20-d2 9: 1 , 298 K): δ = 8.55 (d, J = 8.06 Hz, NH Asp), 8.37 (d, J = 7.28 Hz, NH Arg), 8.13 ( d, J = 9.19 Hz, NH Phe), 7.97 (m, NH Gly), 7.34 (m, 2H, C6H5 Phe), 7.26 (m, 3H, C6H5 Phe), 7.22 (t, J = 5.53/5.53 Hz, ΝΗε Arg), 5.19 (dd, J = 8.58/16.02 Hz, CHa Phe), 4.56 (dd, J = 7.45/- Hz, CHa Asp), 4.34 (d, J = 10.89 Hz, CHa MeVal), 4.12 (dd, J = 7.80/14.63 Hz, CH Gly), 3.95 (dd, J = 6.84/15.33 Hz, CHa Arg), 3.54 (dd, J = 3.37/14.60 Hz, CH Gly), 3.20 (m , CH25 Arg), 3.02 (m, CH2p Phe), 2.88 (s, CH3 MeVal), 2.84 (dd, J = 7.26/16.68 Hz, CHP Asp), 2.63 (dd, J = 7.60/16.54 Hz, CHP Asp), 2.06 (m, CHP Val), 1 .91 (m, CH2p Arg), 1 .57 (m, CH2 Asp), 0.88 (d, J = 6.55 Hz, CH3 Val1), 0.56 (d, J = 6.49 Hz, CH3 Val2).
PAPERJournal of medicinal chemistry (1999), 42(16), 3033-40.Peptide Science (2001), Volume Date2000, 37th, 249-250. Current opinion in investigational drugs (London, England : 2000) (2003), 4(6), 741-5. Journal of medicinal chemistry (2005), 48(24), 7675-87.Peptide Science (2006), 43rd, 215-216Angewandte Chemie, International Edition (2010), 49(15), 2732-2737, S2732/1-S2732/53.Accounts of Chemical Research (2017), 50(7), 1541-1556.
References
- ^ Burke PA, DeNardo SJ, Miers LA, Lamborn KR, Matzku S, DeNardo GL (August 2002). “Cilengitide targeting of alpha(v)beta(3) integrin receptor synergizes with radioimmunotherapy to increase efficacy and apoptosis in breast cancer xenografts”. Cancer Research. 62 (15): 4263–72. PMID 12154028.
- ^ Goodman SL, Hölzemann G, Sulyok GA, Kessler H (February 2002). “Nanomolar small molecule inhibitors for alphav(beta)6, alphav(beta)5, and alphav(beta)3 integrins”. Journal of Medicinal Chemistry. 45 (5): 1045–51. doi:10.1021/jm0102598. PMID 11855984.
- ^ Spreitzer H (October 27, 2008). “Neue Wirkstoffe – Cilengitide”. Österreichische Apothekerzeitung (in German) (22/2008): 1136–7.
- ^ Jump up to:a b Yamada S, Bu XY, Khankaldyyan V, Gonzales-Gomez I, McComb JG, Laug WE (December 2006). “Effect of the angiogenesis inhibitor Cilengitide (EMD 121974) on glioblastoma growth in nude mice”. Neurosurgery. 59 (6): 1304–12, discussion 1312. doi:10.1227/01.NEU.0000245622.70344.BE. PMID 17277694. S2CID 19861713.
- ^ Jump up to:a b Mikkelsen T, Brodie C, Finniss S, Berens ME, Rennert JL, Nelson K, Lemke N, Brown SL, Hahn D, Neuteboom B, Goodman SL (June 2009). “Radiation sensitization of glioblastoma by cilengitide has unanticipated schedule-dependency”. International Journal of Cancer. 124 (11): 2719–27. doi:10.1002/ijc.24240. PMID 19199360.
- ^ Reardon DA, Fink KL, Mikkelsen T, Cloughesy TF, O’Neill A, Plotkin S, et al. (December 2008). “Randomized phase II study of cilengitide, an integrin-targeting arginine-glycine-aspartic acid peptide, in recurrent glioblastoma multiforme”. Journal of Clinical Oncology. 26 (34): 5610–7. CiteSeerX 10.1.1.688.8987. doi:10.1200/JCO.2008.16.7510. PMID 18981465.
- ^ Gilbert MR, Kuhn J, Lamborn KR, Lieberman F, Wen PY, Mehta M, Cloughesy T, Lassman AB, Deangelis LM, Chang S, Prados M (January 2012). “Cilengitide in patients with recurrent glioblastoma: the results of NABTC 03-02, a phase II trial with measures of treatment delivery”. Journal of Neuro-Oncology. 106 (1): 147–53. doi:10.1007/s11060-011-0650-1. PMC 4351869. PMID 21739168.
- ^ Nabors LB, Mikkelsen T, Hegi ME, Ye X, Batchelor T, Lesser G, Peereboom D, Rosenfeld MR, Olsen J, Brem S, Fisher JD, Grossman SA (November 2012). “A safety run-in and randomized phase 2 study of cilengitide combined with chemoradiation for newly diagnosed glioblastoma (NABTT 0306)”. Cancer. 118 (22): 5601–7. doi:10.1002/cncr.27585. PMC 3423527. PMID 22517399.
- ^ Stupp R, Hegi ME, Neyns B, Goldbrunner R, Schlegel U, Clement PM, et al. (June 2010). “Phase I/IIa study of cilengitide and temozolomide with concomitant radiotherapy followed by cilengitide and temozolomide maintenance therapy in patients with newly diagnosed glioblastoma” (PDF). Journal of Clinical Oncology. 28(16): 2712–8. doi:10.1200/JCO.2009.26.6650. PMID 20439646.
- ^ Eisele G, Wick A, Eisele AC, Clément PM, Tonn J, Tabatabai G, et al. (March 2014). “Cilengitide treatment of newly diagnosed glioblastoma patients does not alter patterns of progression”(PDF). Journal of Neuro-Oncology. 117 (1): 141–5. doi:10.1007/s11060-014-1365-x. PMID 24442484. S2CID 21636884.
- ^ Merck Group. “Phase III Trial of Cilengitide Did Not Meet Primary Endpoint in Patients With Newly Diagnosed Glioblastoma, Date accessed: 3/24/2014.”
- ^ ASCO Meeting Library. [1] “Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma and methylated O6-methylguanine-DNA methyltransferase (MGMT) gene promoter: Key results of the multicenter, randomized, open-label, controlled, phase III CENTRIC study, Date accessed: 3/24/2014
| Names | |
|---|---|
| IUPAC name2-[(2S,5R,8S,11S)-5-benzyl-11-{3-[(diaminomethylidene)amino]propyl}-7-methyl-3,6,9,12,15-pentaoxo-8-(propan-2-yl)-1,4,7,10,13-pentaazacyclopentadecan-2-yl]acetic acid | |
| Identifiers | |
| CAS Number | 188968-51-6 |
| 3D model (JSmol) | Interactive image |
| ChEMBL | ChEMBL429876 |
| ChemSpider | 154046 |
| IUPHAR/BPS | 6597 |
| KEGG | D03497 |
| MeSH | Cilengitide |
| PubChem CID | 176873 |
| UNII | 4EDF46E4GI |
| CompTox Dashboard (EPA) | DTXSID9044035 |
| showInChI | |
| showSMILES | |
| Properties | |
| Chemical formula | C27H40N8O7 |
| Molar mass | 588.656 g/mol |
| Density | 1.417 g/mL |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
| Infobox references |
/////////CILENGITIDE, циленгитид , سيلانجيتيد ,西仑吉肽 , PHASE 3, EMD 121974, EMD-121974, UNII-4EDF46E4GI, orphan drug , MERCK, glioblastoma,
CC(C)C1C(=O)NC(C(=O)NCC(=O)NC(C(=O)NC(C(=O)N1C)CC2=CC=CC=C2)CC(=O)O)CCCN=C(N)N

NEW DRUG APPROVALS
ONE TIME
$10.00


{kind=link}
{kind=link}