
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
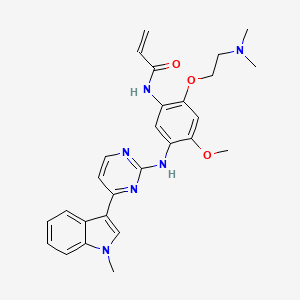
BPI-7711, Rezivertinib
1835667-12-3
C27H30N6O3, 486.576
N-[2-[2-(dimethylamino)ethoxy]-4-methoxy-5-[[4-(1-methylindol-3-yl)pyrimidin-2-yl]amino]phenyl]prop-2-enamide
Beta Pharma in collaboration Chinese licensee CSPC Pharmaceuticals Group , is developing BPI-7711
In June 2021, this drug was reported to be in phase 3 clinical development.
APPROVALS 2024, CHINA 2024
- OriginatorBeta Pharma
- ClassAmides; Amines; Antineoplastics; Indoles; Phenyl ethers; Pyrimidines; Small molecules
- Mechanism of ActionEpidermal growth factor receptor antagonists
- Phase IIINon-small cell lung cancer
- 30 Dec 2020Chemical structure information added
- 09 Apr 2020Beta Pharma initiates a phase I trial for Non-small cell lung cancer (In volunteers) in China (PO) (NCT04135833)
- 25 Mar 2020Beta Pharma completes a phase I pharmacokinetic trial for Non-small cell lung cancer (In volunteers) in China (NCT04135820)
N-(2-(2-(Dimethylamino)ethoxy)-4-methoxy-5-((4-(1-methyl-1H-indol-3-yl)-2-pyrimidinyl)amino)phenyl)-2-propenamideThe epidermal growth factor receptor (EGFR, Herl, ErbB l) is a principal member of the ErbB family of four structurally-related cell surface receptors with the other members being Her2 (Neu, ErbB2), Her3 (ErbB3) and Her4 (ErbB4). EGFR exerts its primary cellular functions though its intrinsic catalytic tyrosine protein kinase activity. The receptor is activated by binding with growth factor ligands, such as epidermal growth factor (EGF) and transforming growth factor-alpha (TGF-a), which transform the catalytically inactive EGFR monomer into catalytically active homo- and hetero- dimers. These catalytically active dimers then initiate intracellular tyrosine kinase activity, which leads to the autophosphorylation of specific EGFR tyrosine residues and elicits the downstream activation of signaling proteins. Subsequently, the signaling proteins initiate multiple signal transduction cascades (MAPK, Akt and JNK), which ultimately mediate the essential biological processes of cell growth, proliferation, motility and survival.EGFR is found at abnormally high levels on the surface of many types of cancer cells and increased levels of EGFR have been associated with advanced disease, cancer spread and poor clinical prognosis. Mutations in EGFR can lead to receptor overexpression, perpetual activation or sustained hyperactivity and result in uncontrolled cell growth, i.e. cancer. Consequently, EGFR mutations have been identified in several types of malignant tumors, including metastatic lung, head and neck, colorectal and pancreatic cancers. In lung cancer, mutations mainly occur in exons 18 to 21, which encode the adenosine triphosphate (ATP)-binding pocket of the kinase domain. The most clinically relevant drug- sensitive EGFR mutations are deletions in exon 19 that eliminate a common amino acid motif (LREA) and point mutations in exon 21, which lead to a substitution of arginine for leucine at position 858 (L858R). Together, these two mutations account for nearly 85% of the EGFR mutations observed in lung cancer. Both mutations have perpetual tyrosine kinase activity and as a result they are oncogenic. Biochemical studies have demonstrated that these mutated EGFRs bind preferentially to tyrosine kinase inhibitor drugs such as erlotinib and gefitinib over adenosine triphosphate (ATP).Erlotinib and gefitinib are oral EGFR tyrosine kinase inhibitors that are first line monotherapies for non-small cell lung cancer (NSCLC) patients having activating mutations in EGFR. Around 70% of these patients respond initially, but unfortunately they develop resistance with a median time to progression of 10-16 months. In at least 50% of these initially responsive patients, disease progression is associated with the development of a secondary mutation, T790M in exon 20 of EGFR (referred to as the gatekeeper mutation). The additional T790M mutation increases the affinity of the EGFR kinase domain for ATP, thereby reducing the inhibitory activity of ATP- competitive inhibitors like gefitinib and erlotinib.Recently, irreversible EGFR tyrosine kinase inhibitors have been developed that effectively inhibit the kinase domain of the T790M double mutant and therefore overcome the resistance observed with reversible inhibitors in the clinic. These inhibitors possess reactive electrophilic functional groups that react with the nucleophilic thiol of an active-site cysteine. Highly selective irreversible inhibitors can be achieved by exploiting the inherent non-covalent selectivity of a given scaffold along with the location of a particular cysteine residue within the ATP binding site. The acrylamide moieties of these inhibitors both undergo a Michael reaction with Cys797 in the ATP binding site of EGFRT790M to form a covalent bond. This covalent mechanism is thought to overcome the increase in ATP affinity of the T790M EGRF double mutant and give rise to effective inhibition. However, these inhibitors may cause various undesired toxicities. Therefore, development of new inhibitors for treatment of various EGFR-related cancers is still in high demand.
PatentCN201580067776) N-(2-(2-(dimethylamino)ethoxy)-4-methoxy-5-((4-(1-methyl-1H- Indol-3-yl)pyrimidin-2-yl)amino)phenyl)acrylamide (compound of formula I) can be prepared by the following synthetic route:
PATENT
https://patents.google.com/patent/WO2016094821A2/enExample 1N-(2-(2-(Dimethylamino)ethoxy)-4-methoxy-5-((4-(l-methyl-lH-indol-3- yl)pyrimidin-2-yl)amino)phenyl)acrylamide (1) Sche

N-(4-(2-(Dimethylamino)ethoxy)-2-methoxy-5-nitrophenyl)-4-(l-methyl-lH- indol-3-yl)pyrimidin-2-amine (Scheme 1, Intermediate B). To a slurry of NaH (30 mmol, 60% oil dispersion prewashed with hexanes) and 50 mL of 1,4-dioxane was added 2-dimethylaminoethanol (27 mmol, 2.7 mL) dropwise with stirring under N2. After stirring for 1 h, a slurry of A (5.4 mmol) in 50 mL of 1,4-dioxane was added portion-wise over 15 min under a stream of N2. The resulting mixture was stirred overnight, then poured into water and the solid was collected, rinsed with water, and dried under vacuum to yield 2.6 g of product as a yellow solid. A purified sample was obtained from chromatography (silica gel; CH2C12-CH30H gradient). 1H NMR (300 MHz, DMSO) δ 2.26 (s, 6H), 2.70 (t, 2H, J = 6 Hz), 3.87 (s, 3H), 4.01 (s, 3H), 4.32 (t, 2H, J = 6 Hz), 7.00-7.53 (m, 5H), 8.18-8.78 (m, 5H); C24H26N604 m/z MH+ 463.4-(2-(Dimethylamino)ethoxy)-6-methoxy-Nl-(4-(l-methyl-lH-indol-3- yl)pyrimidin-2-yl)benzene-l,3-diamine (Scheme 1, Intermediate C). A suspension of 2.6 g of Intermediate B, 1.6 g of Fe°, 30 mL of ethanol, 15 mL of water, and 20 mL of cone. HC1 was heated to 78 °C for 3 h. The solution was cooled to room temperature, adjusted to pH 10 with 10% NaOH (aq) and diluted with CH2C12. The mixture was filtered through Dicalite, and the filtrate layers were separated. The aqueous phase was extracted with CH2C12 twice, and the combined organic extracts were dried over Na2S04 and concentrated. Column chromatography (silica gel, CH2Cl2-MeOH gradient) afforded 1.2 g of Intermediate C as a solid. C24H28N602 m/z MH+ 433.N-(2-(2-(Dimethylamino)ethoxy)-4-methoxy-5-((4-(l-methyl-lH-indol-3- yl)pyrimidin-2-yl)amino)phenyl)acrylamide (1). To a solution of Intermediate C (2.8 mmol) in 50 mL of THF and 10 mL of water was added 3-chloropropionychloride (2.8 mmol) dropwise with stirring. After 5 h of stirring, NaOH (28 mmol) was added and the mixture was heated at 65°C for 18 h. After cooling to room temperature, THF was partially removed under reduced pressure, and the mixture was extracted with CH2C12, dried over Na2S04, and concentrated. Chromatography of the crude product (silica gel, CH2Cl2-MeOH) afforded 0.583 g of Example 1 as a beige solid. 1H NMR (300 MHz, DMSO) δ 2.28 (s, 6H), 2.50-2.60 (m, 2H), 3.86 (s, 3H), 3.90 (s, 3H), 4.19 (t, 2H, = 5.5 Hz), 5.73-5.77 (m, IH), 6.21-6.27 (m, IH), 6.44-6.50 (m, IH), 6.95 (s, IH), 7.11-7.53 (overlapping m, 3H), 7.90 (s, IH), 8.27-8.30 (overlapping m, 3H), 8.55 (s, IH), 8.84 (s, IH), 9.84 (s, IH) ppm; C27H30N6O3 m/z MH+ 487
PATENT WO2021115425
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021115425&tab=FULLTEXT&_cid=P20-KQN9F3-73566-1Epidermal growth factor receptors (EGFR, Her1, ErbB1) are the main members of the ErbB family of four structurally related cell surface receptors, and the other members are Her2 (Neu, ErbB2), Her3 (ErbB3) and Her4 (ErbB4). EGFR exerts its main cellular functions through its inherent catalytic tyrosine protein kinase activity. The receptor is activated by binding to growth factor ligands, such as epidermal growth factor (EGF) and transforming growth factor-α (TGF-α). The catalytically inactive EGFR monomer is transformed into a catalytically active homopolymer and Heterodimer. These catalytically active dimers then initiate intracellular tyrosine kinase activity, which leads to autophosphorylation of specific EGFR tyrosine residues and elicits downstream activation of signaling proteins. Subsequently, the signal protein initiates multiple signal transduction cascades (MAPK, Akt, and JNK), which ultimately regulate the basic biological processes of cell growth, proliferation, motility, and survival.
EGFR has been found to have abnormally high levels on the surface of many types of cancer cells, and elevated EGFR levels have been associated with advanced disease, cancer spread, and poor clinical prognosis. Mutations in EGFR can lead to overexpression of the receptor, permanent activation or continuous hyperactivity, leading to uncontrolled cell growth, which is cancer. Therefore, EGFR mutations have been identified in several types of malignant tumors, including metastatic lung cancer, head and neck cancer, colorectal cancer, and pancreatic cancer. In brain cancer, mutations mainly occur in exons 18-21, which encode the adenosine triphosphate (ATP)-binding pocket of the kinase domain. The most clinically relevant drug-sensitive EGFR mutations are deletions in exon 19 and point mutations in exon 21. The former eliminates a common amino acid motif (LREA), and the latter results in position 858 (L858R). The arginine is replaced by leucine. Together, these two mutations account for nearly 85% of the EGFR mutations observed in lung cancer. Both mutations have permanent tyrosine kinase activity, so they are carcinogenic. In at least 50% of patients who initially responded to current therapies, the progression of the disease is related to the development of a secondary mutation, T790M (also known as the goalkeeper mutation) in exon 20 of EGFR.
BPI-7711 is a third-generation EGFR-TKI compound developed by Beida Pharmaceuticals and disclosed in International Patent No. WO2017/218892. It is the N-(2-(2-(dimethylamino) )Ethoxy)-4-methoxy-5-((4-(1-methyl-1H-indol-3-yl)pyrimidin-2-yl)amino)phenyl)acrylamide methanesulfonic acid salt:
Need to develop improved properties containing N-(2-(2-(dimethylamino)ethoxy)-4-methoxy-5-((4-(1-methyl-1H-indole-3 -Yl)pyrimidin-2-yl)amino)phenyl)acrylamide pharmaceutically acceptable salt, in particular the pharmaceutical composition of BPI-7711 and its use, and the preparation of said pharmaceutical composition suitable for large-scale production method.
PATENT
WO2021061695 , for another filing, assigned to Beta Pharma, claiming a combination of an EGFR inhibitor (eg BPI-7711) and a CDK4/6 inhibitor, useful for treating cancer.
PATENT
WO-2021121146
Novel crystalline polymorphic form A of rezivertinib – presumed to be BPI-7711 – useful for treating diseases mediated by EGFR mutations eg lung cancer, preferably non-small cell lung cancer (NSCLC).Epidermal growth factor receptor (EGFR) is a type of transmembrane receptor tyrosine kinase in the human body. The activation (ie phosphorylation) of this kinase is of great significance to the inhibition of tumor cell proliferation, angiogenesis, tumor invasion, metastasis and apoptosis. EGFR kinase is involved in the disease process of most cancers, and these receptors are overexpressed in many major human tumors. Overexpression, mutations, or high expression of ligands associated with these family members can lead to some tumor diseases, such as non-small cell lung cancer, colorectal cancer, breast cancer, head and neck cancer, cervical cancer, bladder cancer, and thyroid. Cancer, stomach cancer, kidney cancer, etc.
In recent years, epidermal growth factor receptor tyrosine kinase has become one of the most attractive targets in current anti-tumor drug research. In 2003, the US FDA approved the first epidermal growth receptor tyrosine kinase inhibitor (EGFR-TKI) drug (gefitinib) for the treatment of advanced non-small cell lung cancer (NSCLC). Development of a generation of EGFR inhibitors. Numerous clinical trials have confirmed that for patients with EGFR-positive non-small cell lung cancer, the therapeutic effect of molecular targeted drugs is significantly better than traditional chemotherapy.
Although the first-generation EGFR-inhibiting targeted drugs responded well to the initial treatment of many non-small cell lung cancer (NSCLC) patients, most patients will eventually develop disease progression due to drug resistance (such as EGFR secondary T790M mutation). The emergence of drug resistance is caused by various mechanisms based on the mutations in the original EGFR pathway activity. In the drug resistance research on the first generation of EGFR inhibitors, the research frontier is the irreversible third generation EFGR inhibitor.
But so far, the third-generation EGFR inhibitors worldwide, in addition to AstraZeneca O’Higgins imatinib developed, there is no other effective against T790M resistance mutations in patients with drug approved for clinical use; Several drug candidates for the T790M mutation are in clinical development. The chemical structure of this third-generation EGFR inhibitor is completely different from that of the first-generation. The main difference from the first-generation EGFR inhibitors is that they both use a highly selective core structure to replace the low-selective aminoquinoline core structure of the first and second-generation EGFR-TKIs. Compared with wild-type EGFR, these third-generation compounds are highly specific and selective for the T790M mutation after EGFR positive resistance.
Chinese Patent Application No. CN201580067776.8 discloses a compound of the following formula I, which also belongs to the third-generation EGFR-TKI class of small molecule targeted drugs. The compound has a high inhibitory effect on non-small cell lung cancer (NSCLC) cells with single-activity mutation and T790M double-mutant EGFR, and its effective inhibitory concentration is significantly lower than the concentration required to inhibit the activity of wild-type EGFR tyrosine kinase. It has good properties, low side effects and good safety.
Chinese Patent Application No. CN201780050034.3 also discloses various salts and corresponding crystal forms of the compound of the above formula I. Example 2 discloses two crystal forms of the methanesulfonate of the compound of formula I, 2A and 2B, respectively.In the following examples, the “room temperature” can be 15-25°C.[0041](1) N-(2-(2-(Dimethylamino)ethoxy)-4-methoxy-5-((4-(1-methyl-1H-indol-3-yl)pyrimidine -2-yl)amino)phenyl)acrylamide (compound of formula I)[0042]
[0043]Known (for example, see CN201580067776.8) N-(2-(2-(dimethylamino)ethoxy)-4-methoxy-5-((4-(1-methyl-1H- Indol-3-yl)pyrimidin-2-yl)amino)phenyl)acrylamide (compound of formula I) can be prepared by the following synthetic route:[0044]
[0045]Step 1-Preparation of Intermediate J:[0046]
[0047]Preparation: In a 10L reaction flask, add 6L of anhydrous tetrahydrofuran solvent, protected by nitrogen, and cool to 0°C. While stirring, slowly add 101 g of sodium hydride (101 g, 2.52 mol), and the internal temperature does not exceed 10° C., and add 234 g of dimethylaminoethanol (234 g, 2.62 mol). After the addition, the temperature is adjusted to room temperature to prepare a sodium alkoxide solution.[0048]In a 30L reaction flask, add N-(4-fluoro-2-methoxy-5-nitrophenyl)-4-(1-methyl-1H-indol-3-yl)-2-pyrimidinamine ( Starting material B) (430g, 1.10mol), then add 9L of tetrahydrofuran, start stirring, dissolve it, control the temperature at 10±10°C, slowly add the prepared sodium alkoxide solution dropwise. Control the temperature at 10±10℃ and keep it for 5.0h. When the raw material content is ≤0.5%, the reaction ends. Control the temperature at 10±10°C, slowly add 3% hydrochloric acid solution dropwise, adjust the pH of the solution to 6-7, stir for 1.5h and then stand for stratification, separate the organic phase, and concentrate to 15-20L. After cooling to 20±5°C, 4.3 kg of water was slowly added dropwise, filtered, and dried to obtain 497 g of yellow powder intermediate J with a yield of 98.0% and an HPLC purity of 99.3%. MS m/z: 463.2 [M+1].[0049]Nuclear magnetic data: 1 HNMR (d 6 -DMSO): δ ppm: 8.78 (s, 1H); 8.42-8.28 (m, 3H); 8.16 (s, 1H); 7.53 (d, 1H, J = 8.28); 7.29- 7.20 (m, 2H); 7.13-7.07 (m, 1H); 7.01 (s, 1H); 4.33 (t, 2H, J = 5.65); 4.02 (s, 3H); 3.88 (s, 3H); 2.71 ( t, 2H, J = 5.77); 2.27 (s, 6H).[0050]Step 2-Preparation of Intermediate K:[0051]
[0052]Preparation: Add 5L of tetrahydrofuran and Intermediate J (350g, 108mmol) to a 10L hydrogenation reactor, add 17.5g of wet palladium charcoal, replace the hydrogenation reactor with hydrogen, adjust the pressure value to 0.2MPa, control the temperature at 25°C, and keep the temperature for reaction. At 9h, HPLC monitors the progress of the reaction, and stops the reaction when the substrate is ≤0.5%. Filter, concentrate the filtrate under reduced pressure until the solvent volume is about 2L, adjust the internal temperature to room temperature, slowly add 4L n-heptane dropwise within 4-7 hours, filter and dry the solid under reduced pressure to obtain 285g of white powder intermediate K The yield was 86%, and the HPLC purity was 99.60%. MS m/z: 433.3 [M+1].
Nuclear magnetic data: 1 HNMR (CDCl 3 ): δ ppm: 8.42 (d, 1H, J = 7.78), 8.28 (s, 1H), 8.26-8.23 (m, 1H), 7.78 (s, 1H), 7.51 (d, 1H,J=8.28),7.41(s,1H),7.26-7.23(m,1H),7.19- 7.11(m,2H),6.72(s,1H), 4.38(br,2H),4.06(t, 2H,J=5.77), 3.88(s,3H), 3.75(s,3H), 2.63(t,2H,J=5.77), 2.26(s,6H).
Step 3-Preparation of compound of formula I:
Add 250 mL of anhydrous tetrahydrofuran solvent and Intermediate K (14 g, 32 mmol) to the reaction flask and stir, cool to 0-5° C., add 10% hydrochloric acid (12 ml), and stir for 20 minutes. At 0-5°C, slowly drop 3-chloropropionyl chloride (5.6 g, 45 mmol) into the reaction flask. Stir for 3 hours, after sampling test (K/(U+K)≤0.5%) is qualified, add 36% potassium hydroxide aqueous solution (75ml, 480mmol), heat to 23-25°C, and stir for 12 hours. Raise the temperature to 50-60°C and stir for 4 hours. After the sampling test (U/(U+L)≤0.1%) is qualified, stand still for liquid separation. Separate the organic phase, wash with 10% brine three times, dry, filter, and concentrate the organic phase to 150 ml. The temperature was raised to 40° C., 150 ml of n-heptane was slowly added dropwise, and the temperature was lowered to room temperature to precipitate crystals. Filtered and dried to obtain 10.71 g of light brown solid (compound of formula I), yield 68%, HPLC purity: 99.8% (all single impurities do not exceed 0.15%). MS m/z: 487.3 [M+1].[0057]Nuclear magnetic data (Figure 1): 1 HNMR (d 6 -DMSO): δppm: 9.84 (s, 1H), 8.90 ~ 8.82 (m, 1H), 8.32-8.25 (m, 2H), 7.89 (s, 1H) ,7.51(d,1H,J=8.25), 7.27~7.10(m,1H), 6.94(s,1H), 6.49(dd,1H,J=16.88,10.13), 6.25(dd,1H,J=16.95 ,1.81),5.80~5.75(m,1H),4.19(t,2H,J=5.57),3.88(d,6H,J=14.63,6H),3.34(s,3H),2.58(d,2H, J=5.5), 2.28 (s, 6H).
(2) N-(2-(2-(Dimethylamino)ethoxy)-4-methoxy-5-((4-(1-methyl-1H-indol-3-yl)pyrimidine -2-yl)amino)phenyl)acrylamide methanesulfonate (Form A) preparation
Example 1
The compound of formula I (3 g, 6.1 mmol) was dissolved in 24 ml of dimethyl sulfoxide DMSO solvent, the temperature was raised to 65° C., and the mixture was stirred and dissolved. Add an equivalent amount of methanesulfonic acid (0.59 g, 6.1 mmol) to the system. The temperature was lowered to 50°C, and 12ml of isopropyl acetate IPAc was slowly added. Stir at 50°C for 1 hour, then lower the temperature to 15°C. 21ml IPAc was added in 4 hours. The solution was stirred and crystallized at 15°C, filtered under reduced pressure, the filter cake was washed with isopropyl acetate, and washed with acetone to reduce the residual DMSO solvent. Blow drying at 50°C (or vacuum drying at 50°C) to obtain 3.16 g of a pale yellow solid (crystal form A). HPLC purity is 100%, yield is 88%, DMSO: <100ppm; IPAc: <100ppm. MS m/z: 487.2 [M+1-MsOH]. Melting point: 242-244°C.
Nuclear magnetic data (figure 2): 1 HNMR(d 6 -DMSO): δppm: 9.57(brs,1H), 9.40(s,1H), 8.71(s,1H), 8.48(s,1H), 8.32(d ,1H,J=7.9),8.29(d,1H,J=5.3),7.96(s,1H),7.51(d,1H,J=8.2),7.23(ddd,1H,J=7.9,7.1,0.8 ), 7.19 (d, 1H, J = 5.4), 7.15 (ddd, 1H, J = 7.8, 7.3, 0.5), 6.94 (s, 1H), 6.67 (dd, 1H, J = 16.9, 10.2), 6.27 ( dd, 1H, J = 16.9, 1.8), 5.57 (dd, 1H, J = 16.9, 1.7), 4.44 (t, 2H, J = 4.6), 3.89 (s, 3H), 3.88 (s, 3H), 3.58 (t, 2H, J=4.6), 2.93 (s, 6H), 2.39 (s, 3H).
After testing, the powder X-ray diffraction pattern of crystal form A obtained in this example has diffraction angle 2θ values of 11.06±0.2°, 12.57±0.2°, 13.74±0.2°, 14.65±0.2°, 15.48±0.2°, 16.58±0.2°, 17.83±0.2°, 19.20±0.2°, 19.79±0.2°, 20.88±0.2°, 22.05±0.2°, 23.06±0.2°, 24.23±0.2°, 25.10±0.2°, 25.71±0.2°, 26.15±0.2°, 27.37±0.2°, 27.42±0.2° has a characteristic peak; its XRPD spectrum is shown in Figure 3 and the attached table, DSC diagram is shown in Figure 4, TGA diagram is shown in Figure 5, and infrared spectrum IR diagram is shown in Figure 6. Show.
Example 2
[0066]The compound of formula I (28.25 g, 58.1 mmol) was dissolved in 224 ml of dimethyl sulfoxide DMSO solvent, the temperature was raised to 15-35° C., and the mixture was stirred to clear. 0.97 equivalents of methanesulfonic acid (5.4 g, 0.97 mmol) were added to the system in batches. Slowly add 448 ml of methyl isobutyl ketone (MIBK). Stir for 1 hour, then lower the temperature to 10-15°C. The solution was reacted with salt formation at 10-15°C, sampled, and HPLC detected the residue of the compound of formula I in the mother liquor (≤0.4%). After the reaction was completed, vacuum filtration was performed to obtain 32 g of the crude methanesulfonate of the compound of formula I.Add 3g of the crude methanesulfonate of the compound of formula I into 24ml of dimethyl sulfoxide DMSO solvent, stir to clear at 65°C, cool down, slowly add 48ml of methyl isobutyl ketone (MIBK) dropwise, stir and crystallize 6-8 After hours, vacuum filtration, drying at 60° C. (or 60° C. vacuum drying) to obtain the target crystal form A. Melting point: 242-244°C. The XRPD pattern of the crystal form is consistent with Figure 3 (Figure 7), and all characteristic peaks are within the error range.
SYN
European Journal of Medicinal Chemistry 291 (2025) 117643
Rezivertinib, also known as BPI-7711, is a third-generation epidermal growth factor receptor (EGFR) TKI, developed by Beta Pharm. Rezivertinib selectively targets both EGFR-sensitizing mutations
and the T790 M resistance mutation, thereby addressing resistance mechanisms associated with first- and second-generation EGFR-tyrosine kinase inhibitors. In 2024, the NMPA approved Rezivertinib mesylate capsules (trade name: Ruibida) for the treatment of adult patients with locally advanced or metastatic NSCLC who have progressed during or after EGFR-TKI therapy and have confirmed EGFR T790 M mutation-positive status. Rezivertinib exerts its antitumor activity by forming covalent bonds with mutant EGFR, particularly the T790 M mutation, which effectively blocks the downstream signaling pathways responsible for promoting tumor cell proliferation and survival [21]. The mechanism of Rezivertinib effectively inhibits tumor growth in patients harboring T790M-mediated resistance to first- and second-generation EGFR-TKIs. In a Phase IIb clinical trial (NCT03812809), Rezivertinib demonstrated significant clinical efficacy among patients with EGFR T790 M mutation-positive NSCLC who had experienced disease progression following prior EGFR-TKI therapy. The trial reported an ORR of
64.6 % and a median PFS of 12.2 months, highlighting its potent antitumor activity in this specific patient cohort. In terms of safety, Rezivertinib exhibited a favorable tolerability profile [22]. The most
frequently observed treatment-related adverse events were rash, diarrhea, and elevated liver enzymes, predominantly of mild to moderate severity (grade 1 or 2). No dose-limiting toxicities were noted, and its safety profile aligned with those of other third-generation EGFR-TKIs.
The synthesis of Rezivertinib, illustrated in Scheme 5, initiates with nucleophilic substitution reaction between Rezi-001 and Rezi-002,affording Rezi-003 [23]. Fe-mediated reduction of Rezi-003 yields
Rezi-004, followed by amidation with Rezi-005 to deliver Rezivertinib [20] J.J. Cui, E.W. Rogers, Preparation of Fluorodimethyltetrahydroethenopyrazolobenzoxatriazacyclotridecinone
Derivatives for Use as Antitumor Agents, 2017. US20180194777A1.
[21] Y. Shi, Y. Zhao, S. Yang, J. Zhou, L. Zhang, G. Chen, J. Fang, B. Zhu, X. Li, Y. Shu,
J. Shi, R. Zheng, D. Wang, H. Yu, J. Huang, Z. Zhuang, G. Wu, L. Zhang, Z. Guo,
M. Greco, X. Li, Y. Zhang, Safety, efficacy, and pharmacokinetics of rezivertinib
(BPI-7711) in patients with advanced NSCLC with EGFR T790M mutation: a phase
1 dose-escalation and dose-expansion study, J. Thorac. Oncol. 17 (2022) 708–717.

//////////// BPI-7711, BPI 7711, rezivertinib, phase 3, CHINA 2024, APPROVALS 2024



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
CN1C=C(C2=CC=CC=C21)C3=NC(=NC=C3)NC4=CC(=C(C=C4OC)OCCN(C)C)NC(=O)C=C
NEW DRUG APPROVALS
one time
$10.00

