
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
Iguratimod
- Molecular FormulaC17H14N2O6S
- Average mass374.368 Da
- UNII-4IHY34Y2NVигуратимодإيغوراتيمود艾拉莫德
123663-49-0[RN]
3-Formylamino-7-methylsulfonylamino-6-phenoxy-4H-1-benzopyran-4-one
4IHY34Y2NV8176IGU
Methanesulfonamide, N-[3-(formylamino)-4-oxo-6-phenoxy-4H-1-benzopyran-7-yl]-
N-(3-Formamido-4-oxo-6-phenoxy-4H-chromen-7-yl)methanesulfonamide
product patent US4954518
Research Code:T-614
Trade Name:Iremod® / Kolbet® / Careram®
MOA:Nuclear factor NF-κB activation inhibitor
Indication:Rheumatoid arthritis
Status:Approved
Company:Simcere (Originator) , Taisho Toyama,EisaiSales:ATC Code:
Approved Countries or Area
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2012-06-29 | Marketing approval | Careram | Rheumatoid arthritis | Tablet, Film coated | 25 mg | Eisai | |
| 2012-06-29 | Marketing approval | Kolbet | Rheumatoid arthritis | Tablet, Film coated | 25 mg | Toyama Chemical, Taisho Toyama |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2011-08-15 | Marketing approval | 艾得辛/Iremod | Rheumatoid arthritis | Tablet, Film coated | 25 mg | Simcere |
Iguratimod was first approved by China Food and Drug Administration (CFDA) on August 15, 2011, then approved by Pharmaceuticals and Medicals Devices Agency of Japan (PMDA) on June 29, 2012. It was developed by Simcere and marketed as 艾得辛®/Iremod® by Simcere and as Kolbet® by Taisho Toyama and by Eisai in Japan.
Iguratimod is a nuclear factor NF-κB activation inhibitor used in the treatment of rheumatoid arthritis.
Iremod® is available as tablet for oral use, containing 25 mg of free Iguratimod, and the recommended dose is 25 mg once daily or 25 mg at a time, twice daily.
Iguratimod: (Iremod)-First approval: 2009
Iguratimod is a disease-modifying antirheumatic drug (DMARD) that was approved for use in rheumatoid arthritis (RA) patients in China and Japan in 2009
Toyama Chemical , Taisho Toyama Pharmaceutical , Eisai , Simcere and Tianjin Institute of Pharmaceutical Research have codeveloped and launched iguratimod, an inflammatory cytokine and IL-6 gene inhibiting compound that also inhibits immunoglobulin production in B cells. Iguratimod is indicated for the oral treatment of rheumatoid arthritis.
Iguratimod is an anti-inflammatory small molecule drug used for the treatment of rheumatoid arthritis, together with methotrexate in Japan and China.[1] As of 2015 the biological target was not known, but it prevents NF-κB activation and subsequently selectively inhibits COX-2 and several inflammatory cytokines.[1]
Adverse effects include elevated transaminases, nausea, vomiting, stomach pain; rashes, and itchiness.[1]
It is a derivative of 7-methanesulfonylamino-6-phenoxychromones and is a chromone with two amide groups; it was first published in 2000.[1][2] It was submitted for regulatory approval in Japan in 2003; the application was withdrawn in 2009, and it was resubmitted with additional data in 2011 and approved for marketing in Japan in 2012.[1] Eisai and Toyama Chemical market it in Japan.[3] Approval was obtained in China in 2011 by Simcere, independently of the Japanese originators.[1][4]
During discovery and development it was called T-614 and it is marketed under the names Careram and Kolbet.[5]
Syn
Indian Pat. Appl., 2014MU01507

SYN


Reference:1. US4954518.Route 2
Reference:1. Chem. Pharm. Bull. 2000, 48, 131-139.
2. Chin. J. New. Drugs. 2006, 15, 2042-2044.
3. Shanghai Chem. Ind. 2007, 32, 22-24.Route 3
Reference:1. CN1462748A.
2. Chem. Pharm. Bull. 2000, 48, 131-139.
SYN
AU 8823489; CH 679397; FR 2621585; GB 2210879; JP 1995267943; US 4954518
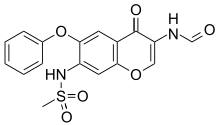
The reduction of 3-nitro-4-phenoxyphenol (I) with Fe, aqueous HCl gives 3-amino-4-phenoxyphenol (II), which is acylated with methanesulfonyl chloride by means of pyridine in dichloromethane affording 3-(methylsulfonamido)-4-phenoxyphenol (III). The condensation of (III) with 3-chloropropionic acid (IV) by means of NaOH in water gives 3-[3-(methylsulfonamido)-4-phenoxyphenoxy]propionic acid (V), which is cyclized by means of polyphosphoric acid at 65-70 C yielding 7-(methylsulfonamido)-6-phenoxy-3,4-dihydro-2H-1-benzopyran-4-one (VI). The bromination of (VI) with Br2 in CHCl3 affords 3-bromo-7-(methylsulfonamido)-6-phenoxy-3,4-dihydro-2H-1-benzopyran-4-one (VII), which is treated with sodium azide in DMF at 70-75 C giving 3-amino-7-(methylsulfonamido)-6-phenoxy-2H-1-benzopyran-4-one (VIII). Finally, this compound is formylated with formic acid in acetic anhydride.
SYN
Chem Pharm Bull 2000,48(1),131
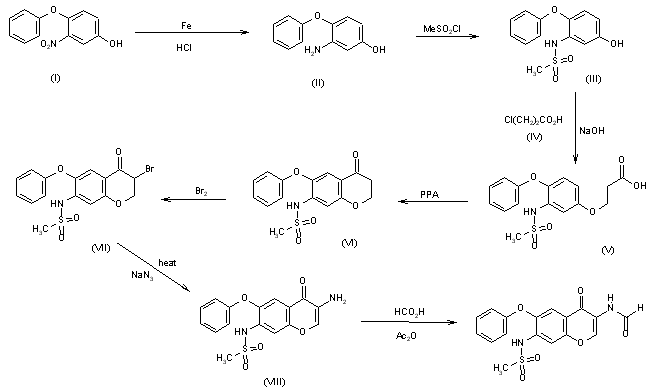
A preparative-scale synthetic route for T-614 has been reported: The reaction of 4-chloro-3-nitroanisole (I) with potassium phenolate in hot DMF gives 4-phenoxy-3-nitroanisole (II), which is reduced to the corresponding 3-amino compound (III) by treatment with Fe and HCl. The reaction of (III) with mesyl chloride in pyridine affords the sulfonamide (IV), which is acylated with 2-aminoacetonitrile (V) and AlCl3 in nitrobenzene/HCl giving the 2-aminoacetophenone (VI). Formylation of (VI) at the NH2 with acetic formic anhydride yields the formamide (VII), which is demethylated with AlCl3 and NaI in acetonitrile affording the phenol (VIII). Finally, this compound is cyclized with dimethylformamide dimethylacetal in DMF.
SYN
https://www.sciencedirect.com/science/article/abs/pii/S0968089614001230
Synthetic approaches to the 2012 new drugs
Hong X. Ding, … Christopher J. O’Donnell, in Bioorganic & Medicinal Chemistry, 2014

13 Iguratimod (Careram®, Iremod®)
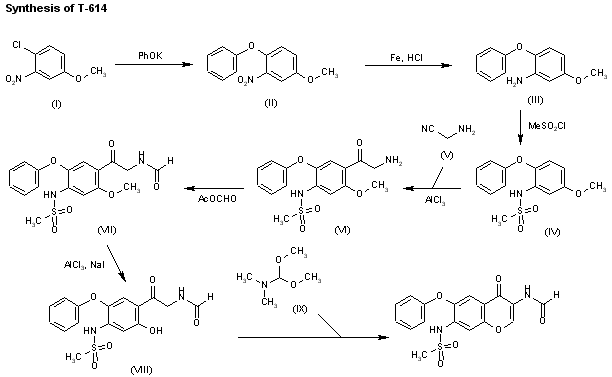
Iguratimod, which was discovered by Toyama Pharmaceuticals and jointly co-developed with Eisai in Japan, was approved by the PMDA (Pharmaceuticals and Medical Devices Agency) of Japan on June 29, 2012 for the treatment of rheumatoid arthritis.83 This drug was also independently developed by Simcere Pharmaceutical Group and is marketed as Iremod® in China. The drug exhibited inhibitory effects on granuloma inflammation, and was shown to be efficacious for the prevention of joint destruction in adjuvant arthritis.84,85 While several synthesis of iguratimod have been published,86 the most likely scale synthesis, which does not require chromatographic purification, is described in Scheme 14.87
The synthesis began with commercially available 3-nitro-4-chloro anisole (78) which was reacted with potassium phenoxide (generated from phenol and potassium t-butoxide at 110 °C) to provide the corresponding nitrophenyl ether which was subsequently reduced and sulfonylated to furnish sulfonamide 79. Next, this diphenyl ether was submitted to a Friedel–Crafts reaction with aminoacetonitrile hydrochloride which gave rise to aminomethylacetophenone 80 in 90% yield. This aminoketone was then formylated with formic trimethylacetic anhydride 81 at room temperature to afford formamide 82 in 91% yield, and this material was immediately subjected to O-demethylation conditions with aluminum trichloride and sodium iodide in acetonitrile to give the phenol 83 in 95% yield. Finally, treatment of the aminomethyl acetophenone phenol 83 with N,N-dimethylformamide dimethylacetal in DMF at low temperatures furnished iguratimod (XII) in 87% yield
83. Eisai and Toyama Chemical Receive Approval to Market Anti-rheumatic Agent Iguratimod in Japan, 2012, http://www.eisai.com/news/news201239.html, [Access Date: 2012-July-29].
84. Tanaka, K.; Shimotori, T.; Makino, S.; Aikawa, Y.; Inaba, T.; Yoshida, C.; Takano, S. Arzneim.-Forsch. 1992, 42, 935.
85. Tanaka, K.; Makino, S.; Shimotori, T.; Aikawa, Y.; Inaba, T.; Yoshida, C. Arzneim.-Forsch. 1992, 42, 945.
86. Takano, S.; Yoshida, C.; Inaba, T.; Tanaka, K.; Takeno, R.; Nagaki, H.; Shimotori, T.; Makino, S. US Patent 4954518 A, 1990.
87. Inaba, T.; Tanaka, K.; Takeno, R.; Nagaki, H.; Yoshida, C.; Takano, S. Chem. Pharm. Bull. 2000, 48, 131.
SYN

https://chemistry-europe.onlinelibrary.wiley.com/doi/abs/10.1002/slct.202003553A Convenient Synthesis of Iguratimod‐Amine Precursor via NHC‐Catalyzed Aldehyde‐Nitrile Cross Coupling ReactionNithya MurugeshProf. Ramasamy KarvembuDr. Seenuvasan VedachalamFirst published: 24 November 2020https://doi.org/10.1002/slct.202003553
A protocol for the synthesis of iguratimod‐amine precursor has been developed using N‐heterocyclic carbene (NHC)‐catalyzed aldehyde‐nitrile cross coupling reaction with overall atom efficiency of 71 %. The first step involves a nucleophilic aromatic substitution (SNAr) of 1‐chloro‐4‐methoxy‐2‐nitrobenzene (1) with phenol to produce 4‐methoxy‐2‐nitro‐1‐phenoxybenzene (2) which further undergoes nitro reduction followed by mesylation to produce N‐(5‐methoxy‐2‐phenoxyphenyl)methanesulfonamide (4). Furthermore, it was subjected to Vilsmeier‐Haack formylation and demethylation (using BBr3) to produce N‐(4‐formyl‐5‐hydroxy‐2‐phenoxyphenyl)methanesulfonamide (6). Subsequently, O‐alkylation followed by NHC‐catalyzed aldehyde‐nitrile cross coupling yields the amine precursor of iguratimod (8).
N-(3-Amino-4-oxo-6-phenoxy-4H-chromen-7-yl)methanesulfonamide (8):4=4. S. Vedachalam, J. Zeng, B. K. Gorityala, M. Antonio, X.-W. Liu, Org. Lett. 2010, 12, 352–355.
Compound 7 (290 mg, 0.83 mmol) and triazolium carbene catalyst (34 mg, 0.1245 mmol) were dissolved in dry CH2Cl2 under N2 atmosphere. To this, DBU (24.7 µL, 0.16 mmol) was added at room temperature, and the mixture was stirred for 12 h. After the completion of reaction, the reaction mixture was dried, and the residue was purified by column chromatography to yield compound 8. Yield: 200 mg, 70 %; m. p. 162 ℃; 1H NMR (500 MHz, DMSO-d6): δ 8.25 (s, 1H), 7.96 (s, 1H), 7.78 (s, 1H), 7.45 (t, J = 8.0 Hz, 2H), 7.25–7.21 (m, 3H), 7.16 (s, 1H), 4.91 (s, 2H), 3.23 (s, 3H); 13C NMR (125 MHz, DMSO-d6): δ 171.2, 155.0, 152.1, 150.4, 137.9, 133.0, 132.8, 131.0 (2C), 125.9, 122.4, 121.1 (2C), 117.2, 110.0, 39.0; FTIR (KBr): v 3423, 3347, 1620, 1592, 1487, 1342, 1210, 1155, 970, 757 cm-1 ; HR-MS (ESI): m/z calcd. for C16H15N2O5S 347.0702, found 347.0714 [M+H]+ …..https://chemistry-europe.onlinelibrary.wiley.com/action/downloadSupplement?doi=10.1002%2Fslct.202003553&file=slct202003553-sup-0001-misc_information.pdf
Patent
WO 2021020481
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021020481&tab=FULLTEXT&_cid=P11-KKXGLE-06477-1The following formula (1)
[Chemical 1]
Iguratimod (chemical name: N- [3- (formylamino) -4-oxo-6-phenoxy-4H-chromen-7-yl] methanesulfonamide), which is indicated by, has excellent anti-inflammatory and antipyretic and analgesic effects. It is a very useful compound as a therapeutic agent exhibiting anti-arthritis and anti-allergic effects (see Patent Document 1).
A plurality of synthetic routes are known for the method for producing iguratimod and its derivatives (hereinafter, may be referred to as iguratimod derivatives as a term meaning both iguratimod and its derivatives), and all of them use intermediates of iguratimod derivatives. This is a stepwise manufacturing method. The present inventors have studied the stepwise production of intermediates of iguratimod derivatives of formulas (II) to (XI) by the following synthetic route to synthesize the iguratimod derivatives represented by formula (XII). doing.
[Chemical formula 2]R 1 = hydroxyl protecting group, R 2 = amino protecting group, Ar = aromatic ring group which may have a substituent, X 2 = halogen atom.[Chemical 3]
Ms = methylsulfonyl group[Chemical 4]
Production Example 1
(Production of igratimodo: Patent Documents 3 and 4)
N, N-dimethylformamide 150 mL, N, N-dimethylformamide dimethyl acetal 40.9 g (N, N-dimethylformamide dimethyl acetal ) in a 1000 mL four-necked flask equipped with two stirring blades having a diameter of 10 cm ( 343 mmol) was added, and the mixture was cooled to 10 ° C. with stirring. 8.2 g (137 mmol) of glacial acetic acid and 50.0 g (137 mmol) of formylaminomethyl (2-hydroxy-4-methylsulfonylamino-5-phenoxyphenyl) ketone were sequentially added thereto, and the temperature was raised to 20 ° C. The reaction was carried out at the same temperature for 5 hours. 250 mL of methylene chloride was added to the reaction suspension, and 500 mL of water was added dropwise to the obtained solution. After adjusting the pH to 5 with a 10% aqueous hydrochloric acid solution, the mixture was stirred at 20 ° C. for 1 hour. The obtained precipitated crystals were separated, washed successively with 50 mL of methylene chloride, 50 mL of water and 50 mL of ethanol, and then dried at 50 ° C. for 12 hours. Next, the obtained crystals were dissolved in a mixed solvent of 7.7 g (137 mmol) of potassium hydroxide, 750 mL of water and 750 mL of acetone, and then neutralized with 2N hydrochloric acid water, and the obtained precipitated crystals were separated. Then, after washing with 50 mL of water, it was dried at 50 ° C. for 12 hours to obtain 42.8 g of iguratimod (iguratimod purity: 99.72%, N-methyl compound: 0.23%).
References
- ^ Jump up to:a b c d e f Tanaka K, Yamaguchi T, Hara M (May 2015). “Iguratimod for the treatment of rheumatoid arthritis in Japan”. Expert Review of Clinical Immunology. 11 (5): 565–73. doi:10.1586/1744666X.2015.1027151. PMID 25797025. S2CID 25134255.
- ^ Inaba T, Tanaka K, Takeno R, Nagaki H, Yoshida C, Takano S (January 2000). “Synthesis and antiinflammatory activity of 7-methanesulfonylamino-6-phenoxychromones. Antiarthritic effect of the 3-formylamino compound (T-614) in chronic inflammatory disease models”. Chemical & Pharmaceutical Bulletin. 48 (1): 131–9. doi:10.1248/cpb.48.131. PMID 10705489.
- ^ Bronson J, Dhar M, Ewing W, Lonberg N (2012). “Chapter Thirty-One – To Market, To Market—2011”. Annual Reports in Medicinal Chemistry. 47: 499–569. doi:10.1016/B978-0-12-396492-2.00031-X.
- ^ “Iguratimod – Simcere”. AdisInsight. Retrieved 27 May 2018.
- ^ “Iguratimod – Toyama Chemical”. AdisInsight. Retrieved 27 May 2018.
| Clinical data | |
|---|---|
| Trade names | Careram; Kolbet |
| Other names | T-614 |
| ATC code | None |
| Identifiers | |
| IUPAC name[show] | |
| CAS Number | 123663-49-0 |
| PubChem CID | 124246 |
| ChemSpider | 110694 |
| UNII | 4IHY34Y2NV |
| ChEMBL | ChEMBL2107455 |
| CompTox Dashboard (EPA) | DTXSID0048971 |
| ECHA InfoCard | 100.236.037 |
| Chemical and physical data | |
| Formula | C17H14N2O6S |
| Molar mass | 374.37 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES[hide]O=S(=O)(Nc3c(Oc1ccccc1)cc2c(O/C=C(\C2=O)NC=O)c3)C | |
| InChI[hide]InChI=1S/C17H14N2O6S/c1-26(22,23)19-13-8-15-12(17(21)14(9-24-15)18-10-20)7-16(13)25-11-5-3-2-4-6-11/h2-10,19H,1H3,(H,18,20)Key:ANMATWQYLIFGOK-UHFFFAOYSA-N |
////////////IGURATIMOD, UNII-4IHY34Y2NV , игуратимод , إيغوراتيمود , 艾拉莫德 , T-614, T 614, Kolbet, Careram, Rheumatoid arthritis, JAPAN 2012, CHINA 2011
CS(=O)(=O)NC1=C(C=C2C(=C1)OC=C(C2=O)NC=O)OC3=CC=CC=C3


{kind=link}
{kind=link}
{kind=link}
{kind=link}