
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

Millennium Pharmaceuticals, Inc. INNOVATOR
Millennium Pharmaceuticals, Inc., a subsidiary of Takeda Pharmaceutical Company Limited,
MLN4924, MLN 4924-003, TAK-924
905579-51-3 BASE
1160295-21-5 HcL
A potent and selective inhibitor of NAE. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. The ubiquitin-proteasome pathway mediates the destruction of unwanted proteins.
(((1S,2S,4R)-4-{4-[(S)-2,3-Dihydro-1H-inden-1-ylamino]-7H-pyrrolo[2,3-d]pyrimidin-7-yl}-2-hydroxycyclopentyl)methyl sulfamate hydrochloride) (pevonedistat), a novel NEDD8-activating enzyme (NAE) inhibitor, has demonstrated in vitro cytotoxic activity against a variety of human malignancies and is currently being developed by Takeda Pharmaceuticals Company Limited as a clinical candidate for the treatment of cancer
In 2011, orphan drug designation was assigned to MLN-4924 for the treatment of MDS and for the treatment of acute myelogenous leukemia.
PHASE 1…….CANCER SOLID TUMOR

………………….
PATENT
http://www.google.com/patents/US20120330013

preparing a compound represented by the following formula 1 by reacting the compound of formula 11 with TFA (step 9):


The retrosynthetic analysis of MLN4924 (1), as the final desired nucleoside, is shown in the following.

MLN 4924 (1) can be synthesized by condensing cyclic sulfate 3 as the glycosyl donor with a purine base. The glycosyl donor 3 can be produced from diol 4, which in turn can be obtained from cyclopentanone 5 via a stereoselective reduction and a regioselective cleavage of the isopropylidene moiety. The cyclopentanone 5 can be synthesized from cyclopentenone 6 by stereoselective reduction. The intermediate cyclopentenone 6 can be easily derived from D-ribose according to our previously published procedure (Jeong, L. S. et al., J. Org. Chem. 2004, 69, 2634-2636).
The synthetic route for the glycosyl donor 3 is shown in the following scheme 1.

Example 1 Preparation of MLN4924 Step 1: Preparation of 6-(tert-butyl-diphenyl-silanyloxymethyl)-2,2-dimethyl-tetrahydro-cyclopenta[1,3]dioxol-4-one (Compound 5)

To a suspension of the compound 6 (20.0 g, 47.1 mmol) in methanol (400 ml) was added 10% palladium on activated carbon (1.0 g), and the mixture was stirred at room temperature overnight under H2 atmosphere. After filtration of the reaction mixture, the solvent was removed and the residue was dissolved in methylene chloride and then filtered through short pad silica gel. Then, the solvent was evaporated to give the compound 5 (20.1 g, 100%) as a colorless syrup.
[α]20 D −28.32 (c 1.49, MeOH); HR-MS (ESI): m/z calcd for C25H32NaO4Si [M+Na]+ 447.1968, Found 447.1956; 1H NMR (400 MHz, CDCl3) δ 7.69 (m, 4H), 7.40 (m, 6H), 4.84 (t, J=4.4 Hz, 1H), 4.22 (dd, J=1.2, 4.8 Hz, 1H), 3.96 (dd, J=8.0, 10.0 Hz, 1H), 3.82 (dd, J=6.8, 10.0 Hz, 1H), 2.37 (m, 1H), 2.30 (ddd, J=1.2, 8.4, and 18.4 Hz, 1H), 2.20 (ddd, J=1.2, 12.0, and 18.4 Hz, 1H), 1.37 (s, 3H), 1.35 (s, 3H), 1.06 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 112.6, 80.5, 77.6, 77.2, 76.9, 63.6, 38.1, 36.9, 27.1, 27.02, 27.01, 25.3, 19.5; Anal. Calcd for C25H32O4Si: C, 70.72; H, 7.60. Found: C, 70.79; H, 7.75.
Step 2: Preparation of 6-(tert-butyl-diphenyl-silanyloxymethyl)-2,2-dimethyl-tetrahydro-cyclopenta[1,3]dioxol-4-ol (Compound 7)

To a suspension of the compound 5 (20.1 g, 47.1 mmol) in methanol (500 ml) were added sodium borohydride (2.17 g, 57.4 mmol) and cerium (III) chloride heptahydrate (21.3 g, 57.2 mmol) at 0° C., and the mixture was stirred at room temperature for 30 min. After the solvent was removed, the residue was partitioned between ethyl acetate and water. The organic layer was then washed with brine, dried with anhydrous MgSO4, filtered, and evaporated. The residue was purified by silica gel column chromatography (hexane/ethyl acetate=5/1) to give the compound 7 (20.86 g, 98%) as a colorless syrup.
[α]20 D +34.55 (c 0.55, MeOH); HR-MS (ESI): m/z calcd for C25H34NaO4Si [M+Na]+: 449.2124; Found: 449.2110; 1H NMR (400 MHz, CDCl3) δ 7.69 (m, 4H), 7.39 (m, 6H), 4.62 (t, J=5.6 Hz, 1H), 4.44 (t, J=5.6 Hz, 1H), 3.89 (dd, J=6.0, 7.6 Hz, 1H), 3.84 (m, 1H), 3.68 (dd, J=6.4, 10.0 Hz, 1H), 1.91 (m, 2H), 1.26 (m, 1H), 1.42 (s, 3H), 1.33 (s, 3H), 1.05 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 135.9, 135.8, 134.2, 134.1, 129.8, 129.7, 127.8, 127.7, 110.6, 79.4, 78.9, 77.6, 77.2, 76.9, 72.5, 62.9, 41.6, 33.4, 27.0, 25.9, 27.0, 25.9, 24.4, 19.5; Anal. Calcd for C25H34O4Si: C, 70.38; H, 8.03. Found: C, 70.41; H, 8.08.
Step 3: Preparation of 3-tert-butoxy-4-(tert-butyl-diphenyl-silanyloxymethyl)-cyclopentane-1,2-diol (Compound 4)

To a solution of the compound 7 (20.86 g, 47.12 mmol) in methylene chloride was added trimethylaluminum (2.0 M in toluene, 132.1 ml) at 0° C., and the mixture was stirred at room temperature for 2 days. The mixture was cooled to 0° C., slowly quenched with an aqueous saturated ammonium chloride solution, filtered, and evaporated. The residue was partitioned between ethyl acetate and water. The organic layer was washed with brine, dried with anhydrous MgSO4, filtered, and evaporated. The residue was purified by silica gel column chromatography (hexane/ethyl acetate=2/1) to give the compound 4 (13.42 g, 62%) as a colorless syrup.
[α]20 D +3.30 (c 0.55, MeOH); HR-MS (ESI): m/z calcd for C26H38NaO4Si [M+Na]+: 465.2437; Found: 465.2423; 1H NMR (400 MHz, CDCl3) δ 7.70 (m, 4H), 7.41 (m, 6H), 4.05 (dd, J=4.4, 7.2 Hz, 1H), 3.93 (m, 1H), 3.72 (m, 2H), 3.59 (dd, J=3.6, 12.0 Hz, 2H), 2.70 (d, J=20.8 Hz, 1H), 2.10 (m, 2H), 1.60 (m, 1H), 1.20 (s, 9H), 1.06 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 135.9, 133.5, 130.0, 129.9, 127.9, 127.9, 77.6, 77.2, 76.9, 74.9, 73.8, 72.7, 72.1, 63.3, 42.1, 34.0, 28.5, 27.0, 19.4; Anal. Calcd for C26H38O4Si: C, 70.55; H, 8.65. Found: C, 70.61; H, 8.70.
Step 4: Preparation of (4-tert-butoxy-2,2-dioxo-tetrahydro-2-yl-6-cyclopenta[1,3,2]-dioxathiol-5-ylmethoxy)-tert-butyl-diphenyl-silane (Compound 3)

To a solution of the compound 4 (13.42 g, 30.3 mmol) in methylene chloride were added triethyl amine (14.5 ml, 101.0 mmol) and thionyl chloride (3.7 ml, 47.4 mmol) at 0° C., and the reaction mixture was stirred at 0° C. for 10 minutes. The reaction mixture was partitioned between methylene chloride and water. The organic layer was washed with brine, dried with anhydrous MgSO4, filtered, and evaporated. The residue was purified by silica gel column chromatography (hexane/ethyl acetate=6/1) to give the cyclic sulfite (14.37 g, 97%) as a white foam.
[α]20 D +20.00 (c 0.05, MeOH); HR-MS (ESI): m/z calcd for C26H36NaO5SSi [M+Na]+: 511.1950; Found: 511.1929; 1H NMR (400 MHz, CDCl3) δ 7.64 (m, 4H), 7.40 (m, 6H), 5.23 (m, 1H), 5.04 (dd, J=4.4, 6.0 Hz, 1H), 4.01 (t, J=4.8 Hz, 1H), 3.68 (dd, J=3.6, 10.4 Hz, 1H), 3.56 (dd, J=8.0, 10.4 Hz, 1H), 2.07 (m, 2H), 1.96 (m, 1H), 1.14 (s, 9H), 1.05 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 135.8, 135.7, 133.9, 133.8, 129.9, 129.9, 127.9, 127.8, 85.7, 83.2, 77.6, 77.2, 76.9, 75.0, 71.1, 62.7, 44.7, 31.4, 28.5, 27.1, 19.4; Anal. Calcd for C26H36O5SSi: C, 63.90; H, 7.42; S, 6.56. Found: C, 63.94; H, 7.45; S, 6.61.
To a solution of the cyclic sulfite obtained above (14.37 g, 29.4 mmol) in the mixture of carbon tetrachloride, acetonitrile and water (1:1:1.5, 210 ml) were added sodium metaperiodate (18.56 g, 56.4 mmol) and ruthenium chloride (1.72 g, 8.25 mmol), and the reaction mixture was stirred at room temperature for 10 minutes. The reaction mixture was partitioned between methylene chloride and water. The organic layer was washed with brine, dried with anhydrous MgSO4, filtered, and evaporated. The residue was purified by silica gel column chromatography (hexane/ethyl acetate=4/1) to give the compound 3 (13.36 g, 90%) as a white solid.
mp 101-104° C.; [α]20 D −80.00 (c 0.05, MeOH); HR-MS (ESI): m/z calcd for C26H36NaO6SSi [M+Na]+: 527.1900; Found: 527.1881; 1H NMR (400 MHz, CDCl3) δ 7.64 (m, 4H), 7.41 (m, 6H), 5.13 (m, 1H), 4.83 (dd, J=4.4, 6.8 Hz, 1H), 4.13 (t, J=4.0 Hz, 1H), 3.92 (dd, J=6.4, 10.4 Hz, 1H), 3.69 (dd, J=5.2, 10.4 Hz, 1H), 2.11 (m, 2H), 2.02 (m, 1H), 1.15 (s, 9H), 1.05 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 135.7, 135.0, 133.8, 133.7, 130.0, 128.0, 127.9, 83.5, 82.2, 77.6, 77.2, 76.9, 75.4, 70.4, 70.4, 62.2, 43.9, 31.3, 28.2, 27.1, 26.8, 19.4; Anal. Calcd for C26H36O6SSi: C, 61.87; H, 7.19; S, 6.35. Found: C, 61.91; H, 7.14; S, 6.30.
Step 5: Preparation of 2-tert-butoxy-3-(tert-butyl-diphenyl-silanyloxymethyl)-5-[4-(indan-1-ylamino)-pyrrolo[2,3-d]pyrimidin-7-yl]-cyclopentanol (Compound 8)

A suspension of N6-indanyl-7-deazaadenine (8.80 g, 35.2 mmol), sodium hydride (1.38 g, 45.7 mmol) and 18-crown-6 (9.11 g, 45.7 mmol) in THF (200 ml) was stirred at 80° C. To the reaction mixture was added a solution for the compound 3 (13.36 g, 26.5 mmol) in THF (150 ml), and the stirring was continued at 80° C. overnight. The reaction mixture was cooled down to 0° C., and conc. HCl was added slowly until pH reaches 1-2. Then the reaction mixture was further stirred at 80° C. for 2 hours. After neutralized with saturated aqueous NaHCO3 solution, the reaction mixture was partitioned between ethyl acetate and water. The organic layer was washed with brine, dried with anhydrous MgSO4, filtered, and evaporated. The residue was purified by silica gel column chromatography (hexane/ethyl acetate=2/1) to give the compound 8 (11.62 g, 65%) as a white foam.
UV (CH2Cl2) λmax 272.5 nm; [α]20 D −8.89 (c 0.45, MeOH); HR-MS (ESI): m/z calcd for C41H51N4O3Si [M+H]+: 675.3730; Found: 675.3717; 1H NMR (400 MHz, CDCl3) δ 8.38 (s, 1H), 7.70 (m, 4H), 7.41 (m, 6H), 6.92 (d, J=3.6 Hz, 1H), 6.29 (d, J=3.2 Hz, 1H), 5.91 (dd, J=7.6, 14.8 Hz, 1H), 5.14 (br d, J=6.8 Hz, 1H), 4.77 (m, 1H), 4.36 (t, J=6.0 Hz, 1H), 4.22 (dd, J=5.2, 10.8 Hz, 1H), 3.84 (dd, J=5.6, 10.4 Hz, 1H), 3.73 (dd, J=8.4, 10.4 Hz, 1H), 3.37 (d, J=5.6 Hz, 1H), 3.06 (m, 1H), 2.95 (m, 1H), 2.75 (m, 1H), 2.75 (m, 1H), 2.58 (m, 1H), 2.38 (m, 1H), 2.15 (m, 1H), 1.98 (m, 1H), 1.65 (s, 1H), 1.55 (s, 1H), 1.16 (s, 9H), 1.07 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 156.4, 151.8, 150.3, 144.1, 143.8, 135.9, 134.0, 129.9, 128.2, 127.9, 127.9, 127.0, 125.1, 124.4, 123.3, 103.8, 97.4, 77.8, 77.6, 77.2, 76.9, 74.9, 72.4, 63.5, 62.1, 56.3, 43.9, 34.9, 30.5, 30.5, 28.5, 27.2, 19.5; Anal. Calcd for C41H50N4O3Si: C, 72.96; H, 7.47; N, 8.30. Found: C, 73.01; H, 7.45; N, 8.36.
Step 6: Preparation of {7-[3-tert-butoxy-4-(tert-butyl-diphenyl-silanyloxymethyl)-cyclopentyl]-7H-pyrrolo[2,3-d]pyrimidin-4-yl}-indan-1-yl-amine (Compound 9)

To a solution of the compound 8 (11.62 g, 17.2 mmol) in methylene chloride (300 ml) were added N,N-dimethylaminopyridine (5.64 g, 51.6 mmol) and phenyl chlorothionocarbonate (4.3 ml, 34.4 mmol), and the reaction mixture was stirred at room temperature overnight. After the solvent was removed, the residue was purified by silica gel column chromatography (hexane/ethyl acetate=6/1) to give the thiocarbonate (13.82 g, 99%) as a white foam.
UV (MeOH) λmax 271.50 nm; [α]20 D +10.00 (c 0.15, MeOH); HR-MS (ESI): m/z calcd for C48H55N4O4SSi [M+H]+: 811.3713; Found: 811.3687; 1H NMR (400 MHz, CDCl3) δ 8.36 (s, 1H), 7.61 (dd, J=1.6, 7.6 Hz, 4H), 7.34 (m, 5H), 7.26 (m, 4H), 7.18 (m, 6H), 6.86 (s, 1H), 6.25 (d, J=3.2 Hz, 1H), 6.00 (dd, J=3.2, 8.4 Hz, 1H), 5.83 (d, J=6.8 Hz, 1H), 5.19 (m, 1H), 5.07 (br s, 1H), 4.48 (t, J=3.6 Hz, 1H), 3.82 (dd, J=7.2, 10.4 Hz, 1H), 3.52 (dd, J=7.2, 10.0 Hz, 1H), 2.99 (m, 1H), 2.88 (m, 2H), 2.69 (m, 2H), 2.18 (dd, J=11.2, 13.6 Hz, 1H), 1.94 (m, 2H), 1.12 (s, 9H), 0.98 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 194.9, 153.5, 152.1, 143.9, 135.9, 135.8, 134.1, 129.9, 129.6, 128.3, 127.9, 127.0, 126.7, 125.1, 124.6, 123.2, 122.0, 87.9, 77.6, 77.2, 76.9, 74.6, 70.4, 63.5, 57.3, 42.8, 35.0, 30.7, 30.5, 29.9, 28.7, 27.1, 19.4; Anal. Calcd for C48H54N4O4SSi: C, 71.08; H, 6.71; N, 6.91; S, 3.95. Found: C, 71.14; H, 6.75; N, 6.95; S, 4.01.
To a solution of the thiocarbonate obtained above (13.82 g, 17.0 mmol) in toluene (200 ml) were added tri-n-butyltinhydride (9.4 ml, 34.1 mmol) and 2,2′-azo-bis-isobutyronitrile (4.32 g, 26.3 mmol), and the reaction mixture was stirred at 110° C. for 1 hour. After the mixture was cooled down, the solvent was removed. The resulting residue was purified by silica gel column chromatography (hexane/ethyl acetate=3/1) to give the compound 9 (9.21 g, 82%) as a white foam.
UV (MeOH) λmax 272.50 nm; [α]20 D −10.00 (c 0.20, MeOH); HR-MS (ESI): m/z calcd for C41H51N4O2Si [M+H]+: 659.3781; Found: 659.3757; 1H NMR (400 MHz, CDCl3) δ 8.41 (s, 1H), 7.69 (m, 4H), 7.41 (m, 6H), 7.29 (m, 2H), 7.23 (m, 2H), 6.92 (d, J=3.6 Hz, 1H), 6.31 (d, J=3.6 Hz, 1H), 5.90 (dd, J=7.2, 14.8 Hz, 1H), 5.38 (m, 1H), 5.15 (br s, 1H), 4.33 (dd, J=5.2, 8.4 Hz, 1H), 3.88 (dd, J=6.4, 10.0 Hz, 1H), 3.68 (dd, J=7.2, 10.4 Hz, 1H), 3.05 (m, 1H), 2.96 (dd, J=7.6, 15.6 Hz, 1H), 2.76 (m, 1H), 2.45 (d, J=5.2 Hz, 1H), 2.29 (m, 2H), 2.06 (m, 1H), 1.95 (m, 2H), 1.55 (s, 1H), 1.13 (s, 9H), 1.06 (s, 9H);13C NMR (100 MHz, CDCl3) δ 156.3, 151.9, 144.1, 143.9, 135.9, 135.8, 134.3, 129.8, 128.2, 127.8, 127.0, 125.1, 124.6, 121.8, 77.6, 77.2, 76.7, 73.5, 72.2, 63.6, 56.4, 52.8, 46.8, 42.8, 34.9, 34.5, 30.5, 28.6, 27.2, 28.7, 19.4; Anal. Calcd for C41H50N4O2Si: C, 74.73; H, 7.65; N, 8.30. Found: C, 74.79; H, 7.61; N, 8.25.
Step 7: Preparation of 2-tert-butoxy-4-[4-(indan-1-ylamino)-pyrrolo[2,3-d]pyrimidin-7-yl]-cyclopentanol (Compound 10)

To a solution of the compound 9 (9.21 g, 13.97 mmol) in the mixture of THF and pyridine (1:1, 160 ml) was added dropwise pyridine hydrofluoride (18.42 ml, 190.0 mmol) at 0° C., and the reaction mixture was stirred at room temperature for 1 hour. The mixture was neutralized with saturated aqueous NaHCO3 solution and partitioned between ethyl acetate and water. The organic layer was washed with brine, dried with anhydrous MgSO4, filtered, and evaporated. Then, the residue was purified by silica gel column chromatography (hexane/ethyl acetate=1/3) to give the compound 10 (5.63 g, 99%) as a white foam.
UV (MeOH) λmax 273.00 nm; [α]20 D −6.36 (c 1.10, MeOH); HR-MS (ESI): m/z calcd for C25H33N4O2 [M+H]+: 421.2604; Found: 421.2599; 1H NMR (400 MHz, CDCl3) δ 8.34 (s, 1H), 7.30 (d, J=7.6 Hz, 1H), 7.22 (d, J=7.2 Hz, 2H), 7.15 (t, J=6.8 Hz, 1H), 6.88 (d, J=3.2 Hz, 1H), 6.23 (d, J=3.6 Hz, 1H), 5.83 (dd, J=7.2, 15.2 Hz, 1H), 5.28 (m, 1H), 5.06 (m, 1H), 4.47 (dd, J=5.6, 10.4 Hz, 1H), 3.78 (m, 1H), 3.70 (m, 1H), 3.24 (t, J=5.2 Hz, 1H), 2.98 (m, 1H), 2.87 (m, 1H), 2.68 (m, 1H), 2.46 (m, 1H), 2.37 (m, 2H), 1.93 (m, 2H), 1.18 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 156.2, 151.8, 147.9, 143.9, 143.9, 128.3, 126.9, 125.1, 124.5, 121.9, 97.7, 77.6, 77.2, 76.9, 75.5, 74.9, 63.4, 56.4, 53.8, 44.2, 42.2, 34.9, 33.2, 30.5, 28.6; Anal. Calcd for C25H32N4O2: C, 71.40; H, 7.67; N, 13.32. Found: C, 71.46; H, 7.60; N, 13.35.
Step 8: Preparation of sulfamic acid 2-tert-butoxy-4-[4-(indan-1-ylamino)-pyrrolo[2,3-d]pyrimidin-7-yl]-cyclopentylmethyl ester (Compound 11)

Preparation of 2.0 M solution of chlorosulfonamide in acetonitrile: Formic acid (14.15 ml, 166.0 mmol) was added dropwise to chlorosulfonyl isocyanate (32.0 ml, 162.5 mmol) under nitrogen atmosphere at 0° C. When the addition was completed, the mixture was solidified. To the mixture was added acetonitrile (61.3 ml), and the resulting solution was left to stand under nitrogen source at room temperature overnight.
To a solution of the compound 10 (5.63 g, 13.83 mmol) and triethyl amine (9.7 ml, 0.74 mmol) in acetonitrile (278 ml) was added 2.0 M solution of chlorosulfonamide in acetonitrile (13.83 ml, 27.76 mmol) at 0° C., and the reaction mixture was stirred at room temperature for 45 minutes. Additional 2.0 M chlorosulfonamide solution in acetonitrile (13.83 ml, 27.76 mmol) was added and the mixture was stirred at room temperature for 15 minutes. The reaction was quenched with methanol, and the solvent was removed. The residue was purified by silica gel column chromatography (methylene chloride/methanol=20/1) to give the compound 11 (6.37 g, 92%) as a white foam.
UV (MeOH) λmax 273.00 nm; [α]20 D −18.00 (c 0.50, MeOH); HR-MS (ESI): m/z calcd for C25H34N5O4S [M+H]+: 500.2332; Found: 500.2331; 1H NMR (400 MHz, CDCl3) δ 8.38 (s, 1H), 7.36 (d, J=7.2 Hz, 1H), 7.29 (d, J=7.2 Hz, 1H), 7.22 (m, 2H), 6.95 (d, J=3.6 Hz, 1H), 6.31 (d, J=3.2 Hz, 1H), 5.89 (d, J=6.4 Hz, 1H), 5.10 (s, 2H), 4.41 (m, 2H), 4.26 (m, 1H), 3.05 (m, 1H), 2.94 (m, 1H), 2.76 (m, 2H), 2.27 (m, 3H), 2.06 (m, 1H), 1.97 (m, 1H), 1.76 (br s, 1H); 13C NMR (100 MHz, CDCl3) δ 156.4, 151.9, 149.9, 143.9, 143.8, 128.3, 126.9, 125.1, 124.5, 121.9, 121.9, 103.5, 97.9, 77.4, 77.2, 76.9, 74.3, 71.9, 71.3, 56.4, 53.1, 49.0, 42.3, 34.9, 34.3, 30.5, 28.6; Anal. Calcd for C25H33N5O4S: C, 60.10; H, 6.66; N, 14.02; S, 6.42. Found: C, 60.15; H, 6.71; N, 13.98; S, 6.39.
Step 9: Preparation of sulfamic acid 2-hydroxy-4-[4-(indan-1-ylamino)-pyrrolo[2,3-d]pyrimidin-7-yl]-cyclopentylmethyl ester (Compound 1)

A solution of the compound 11 (6.37 g, 12.72 mmol) in 70% trifluoroacetic acid (149.24 ml) was stirred at room temperature for 2 hours. The solvent was removed and the residue was purified by silica gel column chromatography (hexane/ethylene acetate=1/2) to give the compound 1 (5.08 g, 90%) as a white foam. BASE
UV (MeOH) λmax 279.50 nm; [α]20 D −6.41 (c 2.34, MeOH);
HR-MS (ESI): m/z calcd for C21H26N5O4S [M+H]+: 444.1705; Found: 444.1706;
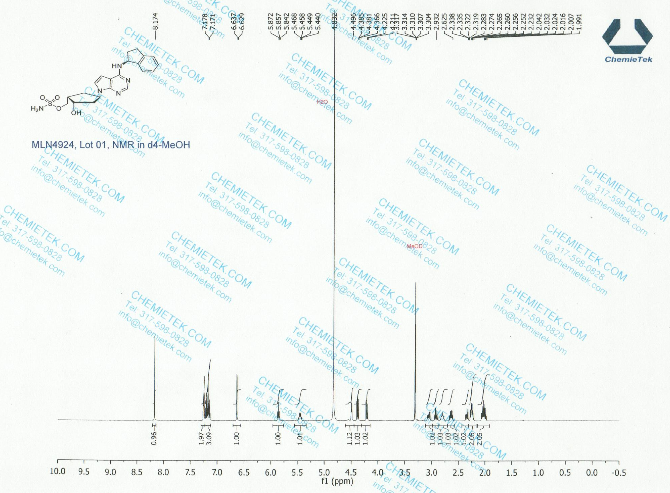
1H NMR (400 MHz, CD3OD) δ 8.17 (d, J=1.6 Hz, 1H), 7.25 (m, 2H), 7.18 (m, 2H), 6.64 (d, J=3.6 Hz, 1H), 5.86 (t, J=7.6 Hz, 1H), 5.46 (m, 1H), 4.49 (d, J=2.8 Hz, 1H), 3.07 (m, 1H), 2.92 (m, 1H), 2.80 (m, 1H), 2.64 (m, 1H), 2.35 (m, 1H), 2.25 (m, 2H), 2.03 (m, 2H);
13C NMR (100 MHz, CD3OD) δ 152.1, 145.3, 144.6, 128.8, 127.6, 125.7, 125.2, 122.6, 100.5, 73.1, 70.9, 56.9, 54.0, 44.8, 43.6, 34.9, 34.6, 31.1;
Anal. Calcd for C21H25N5O4S: C, 56.87; H, 5.68; N, 15.79; S, 7.23. Found: C, 56.91; H, 5.73; N, 15.82; S, 7.26.
…………………….
http://www.google.com/patents/WO2010132110A1?cl=en
((lS,2S,4R)-4-{4-[(lS)-2,3-dihydro-lH-inden-l-ylamino]-7H-pyrrolo[2,3-d]pyrimidin-7-yl }-2-hydroxycyclopentyl)methyl sulfamate (//) is described in Intl. App. Pub. No. WO 07/092213, U.S. App. Pub. No. 2007/0191293, and U.S. App. Pub. No. 2009/0036678. The potassium salt of ((lS,2S,4R)-4-{4-[( 1 S)-2,3-dihydro- 1 H-inden- 1 -ylamino]-7H-pyrrolo[2,3-d]pyrimidin-7-yl } -2-hydroxycyclopentyl)methyl sulfamate is disclosed in Intl. App. Pub. No. WO 07/092213 and U.S. App. Pub. No. 2007/0191293.

(H)
((lS,2S,4R)-4-{4-[(lS)-2,3-dihydro-lH- inden-l-ylamino]-7H-pyπOlo[2,3-d]pyrimidin-7-yl}-2-hydroxycyclopentyl)methyl sulfamate (/):

Step 3: Synthesis of ((lS,2S.4R)-4-(4-r(lS)-2,3-dihydro-lH-inden-l-ylaminol-7H-pyrrolor2.3-dlpyrimidin-7-yl}-2-hvdroxycvclopentyl)methyl sulfamate hydrochloride Form 1
[0158] A reactor was charged with ((lS,2S,4R)-4-{4-[(lS)-2,3-dihydro-lH-inden-l-ylarnino]-7H-pyrrolo[2,3-d]pyrimidin-7-yl }-2-hydroxycyclopentyl)methyl sulfamate (13.4 Kg, 30.2 mol) and 200-proof ethanol (106.2 Kg). The mixture was heated to reflux to afford a clear solution. The mixture was cooled to 50 ± 5 0C and passed through a cartridge filter. 200 proof ethanol (8.9 Kg) was used to rinse the filter. 1.27M hydrogen chloride in ethanol (10.2 Kg) was added via a cartridge filter at a rate to maintain a temperature of 50 ± 5 0C. The mixture was then seeded with Form 1 (67 g). Further 1.27M HCl (10.2 Kg) was added via a cartridge filter at a rate to maintain a temperature of 50 ± 5 0C. The mixture was then stirred at 50 ± 5 0C for about 3 hours. The mixture was then cooled to 20 ± 5 0C over about 3 hours and then stirred for about 2.5 hours. The solid product was then isolated by filtration and washed with 200-proof ethanol (I x 20.4 Kg and 1 x 21.2 Kg). The solids were dried by aspiration on the filter until no supernatant was seen to be collected, and then further dried under reduced pressure at <30 0C to afford the title compound (12.2 Kg) as a white solid determined to be Form 1 by XRPD. IH NMR (300MHz, DMSO, δ): 9.83 (s, IH), 8.34 (s, IH), 7.62 (s, IH), 7.44 (s, 2H), 7.30 (m, 3H), 7.22 (t, IH), 7.07 (s, IH), 5.86 (dd, IH), 5.42 (m, IH), 4.32 (m, IH), 4.21 (dd, IH), 4.02 (dd, IH), 3.04 (m, IH), 2.88 (m, IH), 2.67 (m, 2H), 2.15 (m, 2H), 2.08 (m, 2H), 1.94 (m, IH). XRPD data for Form 1 is shown in FIGURE 1 and Table 1; DSC data is shown in FIGURE 2, and TGA data for Form 1 is shown in FIGURE 3.
…………..
http://www.google.com/patents/WO2007092213A2?cl=en
Example 70: Diastereoisomeric mixture of (lS/2R/4R)-4-{4-[(lS)-2/3-dihydro-lH-inden-l- ylaimnol-ZH-pyrrolop^-dlpyxirnidin-Z-ylJ^-hydroxycyclopentyl s ulf amate and (lRf2S/4S)-4-{4-[(lS)-2,3-dihydro-lH-inden-l-ylaminol-7H-pyrrolo[2,3d]- pyrimidin-7-yl}-2-hydroxycyclopentyl sulfamate (Compounds 1-77 and 1-78)

Step a: Cyclopent-3-en-l-yl methanesulfonate
[0335] 3-Cydopentene-l-ol (0.500 g, 5.94 mmol) was stirred in DCM (95 mL).
Pyridine (2.40 mL), N,N-dimethylaminopyridine (0.10 g, 1.00 mmol) and methanesulfonyl chloride (0.690 mL, 8.92 mmol) were added, and the reaction mixture was stirred at 350C for 4 h. N,N-Dimethylarrιinopyridirιe (0.14 g, 1.2 mmol) and methanesulfonyl chloride (0.69 mL, 8.92 mmol) were added, and the reaction was stirred overnight. TLC indicated complete conversion. The reaction mixture was cooled and concentrated. The residue was purified by silica gel chromatography, eluting with DCM, to afford the title compound as a clear oil (0.660 g, 68%).
Step b: 7-Cyclopent-3-en-l-yl-N-r(lSV2,3-dihydro-lH-inden-l-yn-7H-pyrrolor2,3-rfl- pyrmτidin-4-arnine
[0336] N-[(lS)-2,3-DihydrcHlH-mden-l-yl]-7H-pyrrolo[2/3-d]p3αimidin-4-amine (1.32 g, 5.29 mmol) was azeotroped with toluene and placed under high vacuum for 30 min. N,N-Dimethylformamide (17.7 mL) was added, followed by cesium carbonate (1.99 g, 6.10 mmol). The mixture was stirred at 700C for 10 min. Cyclopent-3-en-l-yl methanesulfonate (0.660 g, 4.07 mmol) in N,N-dimethylformarnide (12.6 mL) was added dropwise. The reaction mixture was heated to 1100C for 1 h. The reaction mixture was cooled, quenched with brine and diluted with H2O. The aqueous layer was extracted with EtOAc (3x), washed with H2O and brine, dried (Na2SO4), filtered, and concentrated. The residue -was purified by via silica gel chromatography, eluting with a gradient of 0 to 5% MeOH in DCM followed by 25 to 50% EtOAc in hexanes, to afford the title compound as a pale brown solid (0.684 g, 53%). LC/MS: R1 = 1.38 min, ES+ 317 (FA standard). Step c: (lR,2S,45)-4-{4-r(lS)-2,3-dihydro-lH-inden-l-ylaininol-7H-pyrrolof2.3- rf1pyrimidin-7-yl}cyclopentane-l,2-diol
[0337] 7-Cyclopent-3-en-l-yl-N-[(lS)-2^-dihyrdo-lH-inden-l-yl]-7H-pyrrolo[2,3- d]pyτimidin-4-amine (0.312 g, 0.986 mmol) was stirred in tert-butyl alcohol (4.9 mL) and H2O (4.9 mL). AD-mix-α (Sigma- Aldrich, 1.4 g) was added, and the suspension was stirred at rt overnight. TLC indicated complete conversion. The reaction was quenched with sodium sulfite (1.48 g, 11.7 mmol), and the mixture was stirred for 5 h. The reaction mixture was diluted with EtOAc and H2O, and the aqueous layer was extracted with EtOAc (2x). The organic layer was dried (Na2SO4), filtered, and concentrated. The residue was purified via silica gel chromatography, eluting with EtOAc, to afford the title compound as a white solid (0.190 g, 55%).
Step d: Diastereoisomeric mixture of (lS,2R,4R)-4-{4-r(15)-23-dihydro-lH-inden-l- ylarninoi^jH-pyrrolofΣ^dlpyrirnidin-y-yll-l-hydroxycyclopentyl sulfamate and (lR,2S,4S)-4-{4-iαSV2,3-dihydro-lH-inden-l-ylarninol-7H-pyrrolor2,3- rf1pyrimidm-7-yl)-2-hydroxycyclopenryl sulfamate (Compounds 1-77 and 1-78)
[0338] (lR,2S,4S)-4-{4-[(lS)-2,3-Dihydro-lH-inden-l-ylarnino]-7H-pyrrolo[2/3- d]pyrimidin-7-ylJcyclopentane-l,2-diol (0.080 g, 0.23 mmol) was azeotroped with toluene and then was dissolved in anhydrous acetonitrile (2.3 mL). Pyridine (0.0369 mL, 0.458 mmol) was added. The reaction mixture was cooled to 00C, and a 2N solution of chlorosulfonamide in acetonitrile (0.144 mL) was added dropwise. The reaction was stirred for 1 h, and then additional 2N chlorosulfonamide in acetonitrile (0.028 mL) was added. After 30 min, additional 2N chlorosulfonamide in acetonitrile (0.0342 mL) was added, and the reaction mixture was stirred for 2 h. The reaction was quenched with methanol, and the mixture was concentrated in vacuo. The residue was purified by preparative thin layer chromatography using DCM:AcCN:MeOH (50:45:5). The relevant band was cut, washed with acetone, filtered, and concentrated to give a mixture of diastereomers as a white solid. (11 mg, 11%). 1H NMR (CDCl3, 400 NMR, δ): 8.36-8.27 (m, IH); 7.38-7.09 (m, 5H); 6.90-6.80 (m, IH); 6.36- 6.20 (m, IH); 5.95-5.76 (m, IH); 5.51-5.22 (m, 2H); 4.83-4.68 (m, IH); 3.87-3.72 (m, IH); 3.12- 2.83 (m, 2H); 2.75-2.53 (m, IH); 2.50-2.14 (m, 2H); 2.08-1.79 (m, 2H) ppm. LC/MS: R, = 1.16 min, ES+ 430 (FA standard).
…………
WO 2012061551
http://www.google.im/patents/WO2012061551A1?cl=en
The compound ((lS,2S,4R)-4-(4-((lS)-2,3-dihydro-lH-inden-l-ylamino)-7H-pyrrolo[2,3-d]- pyrimidin-7-yl)-2-hydroxycyclopentyl)methyl sulfamate:

also known as MLN4924, is an inhibitor of NEDD8-activating enzyme (NAE). Inhibition of NAE has been shown to induce cancer cell death and inhibit the growth of tumors in xenograft models. See, e.g., T.A. Soucy et al., Nature, 2009, 458, 732-737; T.A. Soucy ei al., Clin. Cancer Res., 2009, 15 (12), 3912-3916; and J.E. Brownell et al., Mol. Cell., 2010, 37 (1), 102-111, each of which is hereby incorporated by reference herein in its entirety. MLN4924, pharmaceutical compositions of MLN4924, processes for its synthesis, and polymorphic forms have been described previously. See, e.g., US Patent Appl. Nos. 11/700,614 (Publ. No. 2007/0191293), 12/221,399 (Publ. No. 2009/0036678) and 12/779,331 (Publ. No. 2011/0021544),
……………

A practical synthesis of a novel NEDD8-activating enzyme (NAE) inhibitor pevonedistat (MLN4924) is described. Key steps include an enantioselective synthesis of an amino-diol cyclopentane intermediate containing three chiral centers and a novel, regioselective sulfamoylation using N-(tert-butoxycarbonyl)-N-[(triethylenediammonium)sulfonyl]azanide. The linear process, involving six solid isolations, has been carried out in multiple cGMP productions on 15–30 kg scale to produce pevonedistat in 98% (a/a) chemical purity and 25% overall yield.

((1S,2S,4R)-4-(4-(((S)-2,3-Dihydro-1H-inden-1-yl)amino)-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-2-hydroxycyclopentyl)methyl Sulfamate (1)
((1S,2S,4R)-4-(4-(((S)-2,3-Dihydro-1H-inden-1-yl)amino)-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-2-hydroxycyclopentyl)methyl Sulfamate·Hydrochloride (Pevonedistat)

MLN4924 (1), which is in clinical trials as an anticancer agent, was stereoselectively synthesized from d-ribose via a route involving stereoselective reduction, regioselective cleavage of an isopropylidene moiety, and selective displacement of a cyclic sulfate moiety as key steps.
Sulfamic Acid 2-Hydroxy-4-[4-(indan-1-ylamino)pyrrolo[2,3-d]pyrimidin-7-yl]cyclopentylmethyl Ester (1) BASE



| WO2012061551A1 * | Nov 3, 2011 | May 10, 2012 | Millennium Pharmaceuticals, Inc. | Administration of nedd8-activating enzyme inhibitor |
| WO2013028832A2 * | Aug 23, 2012 | Feb 28, 2013 | Millennium Pharmaceuticals, Inc. | Inhibitors of nedd8-activating enzyme |
| WO2013028832A3 * | Aug 23, 2012 | May 2, 2013 | Millennium Pharmaceuticals, Inc. | Inhibitors of nedd8-activating enzyme |
| US8809356 | Aug 23, 2012 | Aug 19, 2014 | Millennium Pharmaceuticals, Inc. | Inhibitors of NEDD8-activating enzyme |
| Reference | ||
|---|---|---|
| 1 | * | Lee, et. al., Journal of Organic Chemistry (2011), 76(9), 3557-3561. |
1H NMR PREDICT

13 C NMR


//////////Pevonedistat, MLN4924, Millennium Pharmaceuticals, TAKEDA, TAK-924 , PHASE 1, orphan drug designation

