

Candidate: TAK-981



| Patent ID | Date | Patent Title |
|---|---|---|
| US2015182533 | 2015-07-02 | 5-HT3 RECEPTOR ANTAGONISTS |
| US2014024644 | 2014-01-23 | 5-HT3 RECEPTOR ANTAGONISTS |
PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
Home » Posts tagged 'TAKEDA'
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |

TAK-981
C25 H28 Cl N5 O5 S2, 578.103
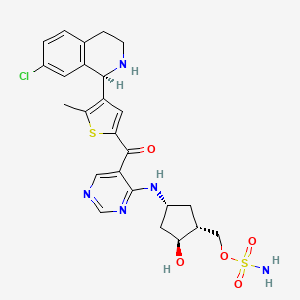
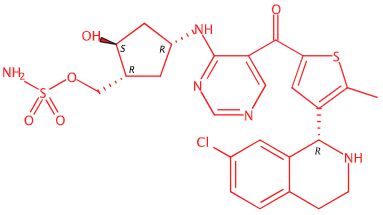
[(1R,2S,4R)-4-[(5-[4-[(1R)-7-Chloro-1,2,3,4-tetrahydroisoquinolin-1-yl]-5-methylthiophene-2-carbonyl]pyrimidin-4-yl)amino]-2-hydroxycyclopentyl]methyl sulfamate
[(1R,2S,4R)-4-[[5-[4-[(1R)-7-Chloro-1,2,3,4-tetrahydroisoquinolin-1-yl]-5-methyl-thiophene-2-carbonyl]pyrimidin-4-yl]amino]-2-hydroxy-cyclopentyl]methyl sulfamate
Sulfamic acid, [(1R,2S,4R)-4-[[5-[[4-[(1R)-7-chloro-1,2,3,4-tetrahydro-1-isoquinolinyl]-5-methyl-2-thienyl]carbonyl]-4-pyrimidinyl]amino]-2-hydroxycyclopentyl]methyl ester
| CAS 1858276-04-6 FREE
CAS 1858279-63-6 HYDRATE |
|
| MW | 578.103 |
Takeda is evaluating TAK-981, a SUMO-Activating Enzyme (SAE) inhibitor, in early clinical trials for the treatment of adult patients with advanced or metastatic solid tumors or with relapsed or refractory lymphomas.
Small ubiquitin-like modifier (SUMO) is a member of the ubiquitin-like protein (Ubl) family that is covalently conjugated to cellular proteins in a manner similar to Ub-conjugation (Kerscher, O., Felberbaum, R., and Hochstrasser, M. 2006. Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu Rev Cell Dev Biol. 22: 159-80). Mammalian cells express three major isoforms: SUMO l , SUM02 and SUM03. SUM02 and SUM03 share -95% amino acid sequence homology but have -45% sequence homology with SUMO l (Kamitani, T., Kito, K., Nguyen, H. P., Fukuda-Kamitani, T., and Yeh, E. T. 1998. Characterization of a second member of the sentrin family of ubiquitin-like proteins. J Biol Chem. 273( 18): 1 1349-53). SUMO proteins can be conjugated to a single lysine residue of a protein (monosumoylation) or to a second SUMO protein that is already conjugated to a protein forming a SUMO chain (polysumoylation). Only SUM02/3 can form such chains because they possess internal consensus SUMO modification sites (Tatham, M. H., Jaffray, E., Vaughan, O. A., Desterro, J. M., Botting, C. H., Naismith, J. H., Hay, R. T. 2001. Polymeric chains of SUMO-2 and SUM 0-3 are conjugated to protein substrates by SAE1/SAE2 and Ubc9. J Biol Chem. 276(38):35368-74). An additional isoform, SUM04, is found in kidney, lymph node and spleen cells, but it is not known whether SUM04 can be conjugated to cellular proteins.
[0003] SUMO l , SUM02 and SUM03 are activated in an ATP-dependent manner by the SUMO-activating enzyme (SAE). SAE is a heterodimer that consists of SAE 1 (SUMO-activating enzyme subunit 1) and SAE2 (UBA2). SAE, like other El activating enzymes, uses ATP to adenylate the C-terminal glycine residue of SUMO. In a second step, a thioester intermediate is then formed between the C-terminal glycine of SUMO and a cysteine residue in SAE2. Next, SUMO is transferred from the El to the cysteine residue of the SUMO conjugating enzyme (E2), UBC9. Unlike the Ub pathway that contains many E2 enzymes, Ubc9 is currently the only known conjugating enzyme for SUMO and functions with SUMOl , SUM02 and SUM03 proteins. SUMO proteins are then conjugated to the target protein, either directly or in conjunction with an E3 ligase, through isopeptide bond formation with the epsilon amino group of a lysine side chain on a target protein. Several SUMO E3 ligases, including PIAS (protein inhibitor of activated signal transducer and activator of transcription protein) proteins and Ran-binding protein 2 (RanBP2), and polycomb 2 (Pc2), have been identified (Johnson, E. S., and Gupta, A. A. 2001. An E3-like factor that promotes SUMO conjugation to the yeast septins. Cell. 106(6):735-44; Pichler, A., Gast, A., Seeler, J. S., Dejean, A.; Melchior, F. 2002. The nucleoporin RanBP2 has SUMOl E3 ligase activity. Cell. 108(1): 109-20; Kagey, M. H., Melhuish, T. A., and Wotton, D. 2003. The polycomb protein Pc2 is a SUMO E3. Cell. 1 13(1): 127- 37). Once attached to cellular targets, SUMO modulates the function, subcellular localization, complex formation and/or stability of substrate proteins (Miiller, S., Hoege, C, Pyrowolakis, G., and Jentsch, S. 2001. SUMO, ubiquitin’s mysterious cousin. Nat Rev Mol Cell Biol. 2(3):202-10). SUMO- conjugation is reversible through the action of de-sumoylating enzymes called SENPs (Hay, R. T. 2007. SUMO-specific proteases: a twist in the tail. Trends Cell Biol. 17(8):370-6) and the SUMO proteins can then participate in additional conjugation cycles.
[0004] SAE-initiated SUMO-conjugation plays a major role in regulating diverse cellular processes, including cell cycle regulation, transcriptional regulation, cellular protein targeting, maintenance of genome integrity, chromosome segregation, and protein stability (Hay, R. T. 2005. SUMO: a history of modification. Mol Cell. 18( 1): 1 -12; Gill, G. 2004. SUMO and ubiquitin in the nucleus: different functions, similar mechanisms? Genes Dev. 18(17):2046-59). For example, SUMO- conjugation causes changes in the subcellular localization of RanGAPl by targeting it to the nuclear pore complex (Mahajan, R., Delphin, C., Guan, T., Gerace, L., and Melchior, F. 1997. A small ubiquitin-related polypeptide involved in targeting RanGAPl to nuclear pore complex protein RanBP2. Cell. 88(1):97- 1070). Sumoylation counteracts ubiquitination and subsequently blocks the degradation of Ι Β, thereby negatively regulating NF-κΒ activation (Desterro, J. M., Rodriguez, M. S., Hay, R. T. 1998. SUMO- 1 modification of IkappaB alpha inhibits NF-kappaB activation. Mol Cell. 2(2):233-9). Sumoylation has been reported to play an important role in transcription exhibiting both repressive and stimulatory effects. Many of the transcriptional nodes that are modulated play important roles in cancer. For example, sumoylation stimulates the transcriptional activities of transcription factors such as p53 and HSF2 (Rodriguez, M. S., Desterro, J. M., Lain, S., Midgley, C. A., Lane, D. P., and Hay, R. T. 1999. SUMO- 1 modification activates the transcriptional response of p53. EMBO J. 18(22):6455-61 ; Goodson, M. L., Hong, Y., Rogers, R., Matunis, M. J., Park-Sarge, O. K., Sarge, K. D. 2001. Sumo- 1 modification regulates the DNA binding activity of heat shock transcription factor 2, a promyelocytic leukemia nuclear body associated transcription factor. J Biol Chem. 276(21 ): 18513-8). In contrast, SUMO-conjugation represses the transcriptional activities of transcription factors such as LEF (Sachdev, S., Bruhn, L., Sieber, H., Pichler, A., Melchior, F., Grosschedl, R. 2001. PIASy, a nuclear matrix-associated SUMO E3 ligase, represses LEF1 activity by sequestration into nuclear bodies. Genes Dev. 15(23):3088- 103) and c-Myb (Bies, J., Markus, J., and Wolff, L. 2002. Covalent attachment of the SUMO- 1 protein to the negative regulatory domain of the c-Myb transcription factor modifies its stability and transactivation capacity. / Biol Chem. 277( 1 1):8999-9009). Thus, SUMO-conjugation controls gene expression and growth control pathways that are important for cancer cell survival.
[0005] Altered expression of SAE pathway components have been noted in a variety of cancer types: (Moschos, S. J., Jukic, D. M., Athanassiou, C., Bhargava, R., Dacic, S., Wang, X., Kuan, S. F., Fayewicz, S. L., Galambos, C., Acquafondata, M., Dhir, R., and Becker, D. 2010. Expression analysis of Ubc9, the single small ubiquitin-like modifier (SUMO) E2 conjugating enzyme, in normal and malignant tissues. Hum Pathol. 41(9): 1286-980); including multiple myeloma (Driscoll, J. J., Pelluru, D., Lefkimmiatis, K., Fulciniti, M., Prabhala, R. H., Greipp, P. R., Barlogie, B., Tai, Y. T., Anderson, K. C, Shaughnessy, J. D. Jr., Annunziata, C. M., and Munshi, N. C. 2010. The sumoylation pathway is dysregulated in multiple myeloma and is associated with adverse patient outcome. Blood. 1 15(14):2827-34); and breast cancer (Chen, S. F., Gong, C, Luo, M., Yao, H. R., Zeng, Y. J., and Su, F. X. 201 1. Ubc9 expression predicts chemoresistance in breast cancer. Chin J Cancer. 30(9):638-44), In addition, preclinical studies indicate that Myc-driven cancers may be especially sensitive to SAE inhibition (Kessler, J. D., Kahle, K. T., Sun, T., Meerbrey, K. L., Schlabach, M. R., Schmitt, E. M., Skinner, S. O., Xu, Q., Li, M. Z., Hartman, Z. C, Rao, M., Yu, P., Dominguez-Vidana, R., Liang, A. C, Solimini, N. L., Bernardi, R. J., Yu, B., Hsu, T., Golding, I., Luo, J., Osborne, C. K., Creighton, C. J., Hilsenbeck, S. G., Schiff, R., Shaw, C. A., Elledge, S. J., and Westbrook, T. F. 2012. A SUMOylation-dependent transcriptional subprogram is required for Myc-driven tumorigenesis. Science. 335(6066):348-53; Hoellein, A., Fallahi, M., Schoeffmann, S., Steidle, S., Schaub, F. X., Rudelius, M., Laitinen, I., Nilsson, L., Goga, A., Peschel, C, Nilsson, J. A., Cleveland, J. L., and Keller, U. 2014. Myc-induced SUMOylation is a therapeutic vulnerability for B-cell lymphoma. Blood. 124( 13):2081 -90). Since SUMO-conjugation regulates essential cellular functions that contribute to the growth and survival of tumor cells, targeting SAE could represent an approach to treat proliferative disorders such as cancer.
[0006] SAE inhibitors may also be applicable for the treatment of other diseases and conditions outside of oncology. For example, SUMO modifies proteins that play important roles in neurodegenerative diseases (Steffan, J. S., Agrawal, N., Pallos, J., Rockabrand, E., Trotman, L. C, Slepko, N., Hies, K., Lukacsovich, T., Zhu, Y. Z., Cattaneo, E., Pandolfi, P. P., Thompson, L. M., Marsh, J. L. 2004. SUMO modification of Huntington and Huntington’s disease pathology. Science. 304(5667): 100-4); Dorval, V., and Fraser, P. E. 2006. Small ubiquitin-like modifier (SUMO) modification of natively unfolded proteins tau and alpha-synuclein. J Biol Chem. 281 ( 15):9919-24; Ballatore, C, Lee, V. M., and Trojanowski, J. Q. 2007. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat Rev Neurosci. 8(9):663-72). Sumoylation also has been reported to play important role in pathogenic viral infection, inflammation and cardiac function (Lee, H. R., Kim, D. J., Lee, J. M., Choi, C. Y., Ahn, B. Y., Hayward, G. S., and Ahn, J. H. 2004. Ability of the human cytomegalovirus ΓΕ1 protein to modulate sumoylation of PML correlates with its functional activities in transcriptional regulation and infectivity in cultured fibroblast cells. / Virol. 78(12):6527-42; Liu, B., and Shuai, K. 2009. Summon SUMO to wrestle with inflammation. Mol Cell. 35(6):731-2; Wang, J., and Schwartz, R. J. 2010. Sumoylation and regulation of cardiac gene expression. Circ Rei. l07( l): 19-29). [0007] It would be beneficial therefore to provide new SAE inhibitors that possess good therapeutic properties, especially for the treatment of proliferative, inflammatory, cardiovascular and neurodegenerative disorders.
PATENT
WO 2016004136

https://patents.google.com/patent/WO2016004136A1/en
Example 133: [(lR,2S,4R)-4-[[5-[4-[(lR)-7-Chloro-l,2,3,4-tetrahydroisoquinolin-l-yl]-5-methyl- thiophene-2-carbonyl]pyrimidin-4-yl]amino]-2-hydroxy-cyclopentyl]methyl sulfamate I-263a

Step 1: 7-Chloro-l-[5-(l,3-dioxolan-2-yl)-2-methyl-3-thienyl]-l,2,3,4-tetrahydroisoquinoline
[00714] An oven-dried 2-neck 250 mL round bottom flask under nitrogen was charged with THF (40 mL) and cooled to -74 °C . Added 2.50 M ra-BuLi in hexane (6.92 mL, 17.3 mmol). Added a solution of Int-1 (4.00 g, 16.0 mmol) in THF (60 mL) slowly keeping the internal temperature less than -70 °C . Stirred with cooling 5 min. A second oven-dried 250 mL round bottom flask under nitrogen was charged with THF (60 mL) and Int-50 (2.04 g, 12.4 mmol) and the resulting solution was cooled to 0 °C . Added boron trifluoride diethyl ether complex ( 1.71 mL, 13.6 mmol) slowly and cooled to -30 °C . The contents of the first flask were transferred via cannula to the second flask. Reaction was quenched with saturated aqueous NaHC03 and warmed to rt. Water was added, and the mixture was extracted three times with EtOAc. Combined organic portions were washed with brine, dried over anhydrous Na2S04, filtered, and concentrated in vacuo. Residue was purified via flash column chromatography eluting with a hexane / EtOAc gradient (0 to 100% EtOAc) to afford the title compound as a white solid ( 1.88g, 45%). Ή NMR (400 MHz, Chloroform-d) δ 7.17 – 7.01 (m, 2H), 6.83 – 6.61 (m, 2H), 5.92 (s, 1H), 5.09 (s, 1H), 4.17 – 4.04 (m, 2H), 4.03 – 3.92 (m, 2H), 3.37 – 3.25 (m, 1H), 3.13 – 2.91 (m, 2H), 2.82 – 2.69 (m, 1H), 2.46 (s, 3H). LCMS: (AA) M+l 336.1
Step 2: ieri-Butyl 7-chIoro-l-[5-(l,3-dioxolan-2-yl)-2-methyl-3-thienyl]-3,4-dihydroisoquinoIine -2(lH)-carboxyIate [00715] A 50 mL round bottom flask under nitrogen was charged with 7-chloro-l -[5-(l ,3-dioxolan-2- yl)-2-methyl-3-thienyl]- l ,2,3,4-tetrahydroisoquinoline (5.67 g, 16.9 mmol) and DCM ( 100 mL), to which was added triethylamine (4.71 mL, 33.8 mmol), di-ieri-butyldicarbonate (4.61 g, 21.1 mmol), and N,N-dimethylaminopyridine (23 mg, 0.18 mmol). Reaction was stirred for 1 h at rt and then poured into saturated NaHC03 solution. Mixture was extracted three times with DCM, and the combined organic portions were washed with brine, dried over Na2S04, filtered, and concentrated in vacuo. The residue was subjected to flash column chromatography eluting with a hexane / EtOAc gradient to afford 6.96g (95%) of the title compound. LCMS: (AA) M+ l 436.1
Step 3: tert-Butyl 7-chloro-l-(5-formyl-2-methyl-3-thienyl)-3,4-dihydroisoquinoline -2(1H)- carboxylate
[00716] A 1 L round bottom flask was charged with ferf-butyl 7-chloro-
1 -[5-( 1 ,3-dioxolan-2-yl)-2-methyl-3-thienyl]-3 ,4-dihydroisoquinoline-2( 1 H)-carboxylate (7.30 g, 16.7 mmol), methanol (200 mL), and water (20 mL), to which was added a solution of 12M HC1 (4.00 mL, 130 mmol) in methanol (200 mL), and the reaction was stirred at rt for 1 h. Reaction was quenched via addition of 50mL of saturated NaHC03 and stirred for 5 min. Methanol was removed in vacuo, and the resulting aqueous mixture was extracted three times with EtOAc, and then the combined organic layers were washed with brine, dried over anhydrous Na2S04 and concentrated in vacuo. The residue was subjected to flash column chromatography eluting with a hexane / EtOAc gradient to afford the title compound (4.55g, 70%). Ή NMR (400 MHz, Chloroform-d) δ 9.67 (s, 1 H), 7.27 – 7.15 (m, 2H), 7.12 (s, 1 H), 6.98 – 6.94 (m, 1 H), 6.34 (m, l H), 4.15 (s, 1 H), 3.18 – 3.06 (m, 1 H), 3.05 – 2.93 (m, 1H), 2.82 – 2.73 (m, 1 H), 2.69 (s, 3H), 1.50 (s, 9H). LCMS: (AA) M+Na 414.2
Step 4: tert-Butyl 7-chIoro-l-{5-[(4-chloropyrimidin-5-yl)(hydroxy)methyI]-2-methyl-3-thienyl}- 3,4-dihydroisoquinoline-2(lH)-carboxylate
[00717] An oven-dried 500 mL 3-neck round bottom flask under nitrogen was charged with 4-chloro- 5-iodopyrimidine (4.08 g, 17.0 mmol) and 2-methyltetrahydrofuran ( 150 mL). An addition funnel containing a solution of rert-butyl 7-chloro- l -(5-formyl-2-methyl-3-thienyl)-3,4- dihydroisoquinoline-2(l H)-carboxylate (4.75 g, 12.1 mmol) in 2-methyltetrahydrofuran (50 mL) was attached, and the contents of the reaction flask were cooled to -75 °C . 2.50 M n-BuLi in hexane ( 14.1 mL, 35.2 mmol) was added in small portions keeping the internal temperature less than -70 °C , at which point the contents of addtion funnel were added in a single portion. Upon completion of addition, the reaction was quenched by adding 20 mL of saturated NaHC03 in small portions and warmed to rt. The aqueous mixture was extracted three times with EtOAc, and then the combined organic layers were washed with brine, dried over anhydrous Na2S04 and concentrated in vacuo. The residue was subjected to flash column chromatography eluting with a hexane / EtOAc gradient to afford the title compound (4.85g, 79%). LCMS: (AA) M+Na 528.1
Step 5: tert-Butyl 7-chloro-l-{5-[(4-chloropyrimidin-5-yl)(hydroxy)methyl]-2-methyl-3-thienyl}- 3,4- dihydroisoquinoline-2(lH)-carboxylate
[00718] A 1 L round bottom flask was charged with fe/Y-butyl 7-chloro- l – { 5-[(4-chloropyrimidin-5- yl)(hydroxy)methyl]-2-methyl-3-thienyl}-3,4-dihydroisoquinoline-2(l H)-carboxylate (4.85 g, 9.58 mmol) and DCM (300 mL). Manganese (IV) oxide (14.2 g, 163 mmol) was added and the reaction was stirred at rt for 18 h. Mixture was filtered through Celite, and the filter cake was rinsed with hot EtOAc. Filtrate was concentrated in vacuo to afford the title compound (4.47g , 93%). Ή NMR (400 MHz, Chloroform-d) δ 9.09 (s, 1 H), 8.70 (s, 1 H), 7.24 – 7.16 (m, 1 H), 7.16
– 7.07 (m, 1 H), 7.00 – 6.90 (m, 2H), 6.32 (s, 1 H), 4.28 – 3.97 (m, 1H), 3.14 – 2.89 (m, 2H), 2.78
– 2.65 (m, 4H), 1 .53 – 1.43 (m, 9H).
Step 6: tert-Butyl (lR)-7-chloro-l-[5-[4-[[(lR,3R,4S)-3-(hydroxymethyl)-4-triisopropylsiIyloxy- cyclopentyl]amino]pyrimidine-5-carbonyl]-2-methyl-3-thienyl]-3,4-dihydro-lH-isoquinoline-2- carboxylate
[00719] A 1 L round bottom flask under nitrogen was charged with iert-butyl 7-chloro- l – { 5-[(4- chloropyrimidin-5-yl)carbonyI]-2-methyl-3-thienyl }-3,4-dihydroisoquinoline-2( l H)-carboxylate (4.47 g, 8.86 mmol), DMF (20.0 mL, 258 mmol), Int-259 (3.06 g, 10.6 mmol), and triethylamine (3.09 mL, 22.2 mmol) and the mixture was stirred at rt for 18 h. Reaction mixture was poured into water and saturated NaHC03, and then extracted three times with EtOAc, and then the combined organic layers were washed with brine, dried over anhydrous Na2S04 and concentrated in vacuo. The residue was subjected to flash column chromatography eluting with a 70/30 to 60/40 hexane/EtOAc gradient to afford 0.56g of first-eluting diastereomer 1 (not pictured), 4.3 l g of a mixture of diastereomers, and 1.1 lg ( 17%) of second-eluting diastereomer 2 (the title compound). The mixture of diastereomers thus obtained was resubjected to the described chromatography conditions two additional times to afford a total of 2.62 g of the desired diastereomer. Ή NMR (400 MHz, Methanol-d4) δ 8.54 – 8.46 (m, 2H), 7.27 – 7.19 (m, 2H), 7.09 – 6.99 (m, 2H), 6.37 (s, 1H), 4.87 – 4.75 (m, 1H), 4.38 – 4.29 (m, 1H), 4.20 – 4.09 (m, 1H), 3.66 – 3.52 (m, 2H), 3.28- 3.14 (m, 2H), 3.02 – 2.89 (m, 1 H), 2.89 – 2.78 (m, 1 H), 2.68 (s, 3H), 2.54 – 2.41 (m, 1 H), 2.22 – 2.09 (m, 2H), 1.86 – 1.73 (m, 1H), 1.50 (s, 8H), 1.39 – 1.23 (m, 2H), 1.15 – 1.04 (m, 20H).
LCMS: (AA) M+ 1 755.3
Step 7: tert-Butyl (lR)-7-chloro-l-[2-methyl-5-[4-[[(lR,3R,4S)-3-(sulfamoyloxymethyl)-4- triisopropylsilyloxy-cyclopentyl]amino]pyrimidine-5-carbonyl]-3-thienyl]-3,4-dihydro-lH- isoquinoline-2-carboxylate [00720] A solution of ie/t-butyl (lR)-7-chloro-l-[5-[4-[[( lR,3R,4S)-3-(hydroxymethyl)-4- triisopropylsilyloxy-cyclopentyl]amino]pyrimidine-5-carbonyl]-2-methyl-3-thienyl]-3,4-dih lH-isoquinoline-2-carboxylate (2.46 g, 3.26 mmol) in 2-methyltetrahydrofuran (25 mL), and DMF (25 mL) was cooled to 0 °C. Triethylamine ( 1.82 mL, 13.0 mmol) and chlorosulfonamide (1.50 g, 13.0 mmol) were added and the reaction was stirred for 10 min. Added methanol (0.53 mL, 13.0 mmol) and stirred for 15 min. Reaction mixture was poured into saturated NaHC03, extracted three times with EtOAc, and then the combined organic layers were washed with brine, dried over anhydrous Na2S04 and concentrated in vacuo. The residue was subjected to flash column chromatography eluting with a hexane / EtOAc gradient to afford the title compound (2.41g, 89%). Ή NMR (400 MHz, Methanol-d4) δ 8.58 – 8.45 (m, 2H), 7.29 – 7.17 (m, 2H), 7.1 1 – 6.98 (m, 2H), 6.36 (s, 1 H), 4.84 – 4.73 (m, 1H), 4.44 – 4.33 (m, 1H), 4.21 – 4.08 (m, 4H), 3.27- 3.17 (m, 1 H),3.02 – 2.89 (m, 1 H), 2.88 – 2.78 (m, 1 H), 2.67 (s, 3H), 2.57 – 2.47 (m, 1 H), 2.41 – 2.30 (m, 1 H), 2.23 – 2.13 (m, 1 H), 1.87- 1.78 (m, 1 H), 1.50 (s, 9H), 1.43 – 1 .33 (m, 1 H), 1 .17 – 1.04 (m, 20H). LCMS: (AA) M+l 834.3
Step 8: [(lR,2S,4R)-4-[[5-[4-[(lR)-7-Chloro-l,2,3,4-tetrahydroisoquinolin-l-yl]-5-methyl- thiophene-2-carbonyl]pyrimidin-4-yI]aniino]-2-hydroxy-cyclopentyl]methyl sulfamate
[00721] A solution of f«?r/-butyl ( l R)-7-chloro- l -[2-methyl-5-[4-[[( l R,3R,4S)-3-
(sulfamoyloxymethyl)-4-triisopropylsilyloxy-cyclopentyl]amino]pyrimidine-5-carbonyl]-3- thienyl]-3,4-dihydro- l H-isoquinoline-2-carboxylate (2.41 g, 2.89 mmol) in CH3CN ( 10 mL) was cooled in an ice bath to + 1 °C . Phosphoric acid ( 10 mL, 200 mmol) was added dropwise and the reaction was stirred with ice bath cooling for 60 min. The mixture was warmed to rt and stirred for an additional 3 h. Reaction was poured into a stirring mixture of 50 mL water and 50 mL EtOAc, and the the pH was adjusted to ~9 by slowly adding 200 mL of saturated NaHC03 with stirring. Resulting aqueous mixture was extracted three times with EtOAc, and then the combined organic layers were washed with brine, dried over anhydrous Na2S04 and concentrated in vacuo. The residue was subjected to flash column chromatography eluting with a gradient that began with 100% DCM and increased in polarity to 80% DCM / 20% methanol / 2% ammonium hydroxide gradient to afford the title compound (1.50 g, 90%). Ή NMR (400 MHz, Methanol-d4) δ 8.61 (s, 1H), 8.52 (s, 1 H), 7.27 (s, 1 H), 7.18 – 7.13 (m, 2H), 6.73 – 6.68 (m, 1 H), 5.23 (s, 1H), 4.81 – 4.70 (m, 1 H), 4.26 – 4.10 (m, 3H), 3.29 – 3.23 (m, 2H), 3.1 1 – 2.96 (m, 2H), 2.87 – 2.76 (m, 1H), 2.60 (s, 3H), 2.55 – 2.42 (m, 1 H), 2.33 – 2.19 (m, 1H), 2.18 – 2.07 (m, 1H), 1.95 – 1.81 (m, 1H), 1.47 – 1.35 (m, 1 H). LCMS: (AA) M+l 580.0
CLIP
Candidate: TAK-981

Presenter: Steven Paul Langston, associate director at Takeda Pharmaceuticals International
Target: Sumo activating enzyme
Disease: Solid tumors
Reporter’s notes: Langston gave the last talk of the morning session, placing him in the “precarious position of being between you and lunch,” he said. Takeda acquired this drug development program, falling under the umbrella of immuno-oncology, along with Millenium Pharmaceuticals in 2008. The team targeted a pathway known as SUMOylation, a protein post translation modification that is implicated in a number of cellular processes including immune response. In SUMOylation, enzymes attach a small protein to another protein. They found that inhibiting this pathway activates a type I interferon response in immune cells. How the molecule, TAK-981, inhibits this pathway is quite complicated, Langston said. TAK-981 forms an adduct with a small ubiquitin like modifier (SUMO) to inhibit a SUMO activating enzyme that catalyzes SUMOylation. While the synthesis of TAK-981 is fairly short, it requires a nonideal chiral chromatography separation after the first step. TAK-981 is in Phase I clinical trials as an intravenous infusion for patients with metastatic solid tumors or lymphomas.
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US2018311239 | HETEROARYL COMPOUNDS USEFUL AS INHIBITORS OF SUMO ACTIVATING ENZYME | 2018-03-16 | |
| US9962386 | HETEROARYL COMPOUNDS USEFUL AS INHIBITORS OF SUMO ACTIVATING ENZYME | 2017-04-17 | |
| US9683003 | HETEROARYL COMPOUNDS USEFUL AS INHIBITORS OF SUMO ACTIVATING ENZYME | 2015-06-30 | 2016-01-14 |
//////////TAK-981, TAK 981, Phase I, Lymphoma, Solid tumours, TAKEDA,
Cc3sc(cc3[C@@H]1NCCc2ccc(Cl)cc12)C(=O)c5cncnc5N[C@@H]4C[C@H](COS(N)(=O)=O)[C@@H](O)C4
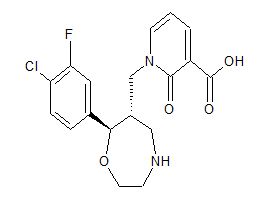
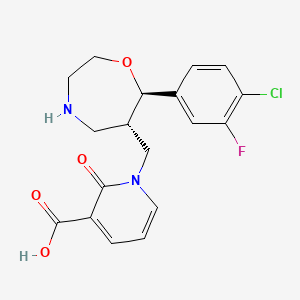
1-{[(6S,7R)-7-(4-Chloro-3-fluorophenyl)-1,4-oxazepan-6-yl]methyl}-2-oxo-1,2-dihydro-3-pyridinecarboxylic acid
CAS 1372185-97-1
CAS 1372180-09-0 hydrochloride
Peripherally selective noradrenaline reuptake inhibitor
 1-([(6S,7R)-7-(4-Chloro-3-fluorophenyl)-1,4-oxazepan-6-yl]methyl]-2-oxo-1,2-dihydropyridine-3-carboxylic acid monohydrochloride
1-([(6S,7R)-7-(4-Chloro-3-fluorophenyl)-1,4-oxazepan-6-yl]methyl]-2-oxo-1,2-dihydropyridine-3-carboxylic acid monohydrochloride
3-Pyridinecarboxylic acid, 1-[[(6S,7R)-7-(4-chloro-3-fluorophenyl)hexahydro-1,4-oxazepin-6-yl]methyl]-1,2-dihydro-2-oxo-, hydrochloride (1:1)
1-{[(6S,7R)-7-(4-Chloro-3-fluorophenyl)-1,4-oxazepan-6-yl]methyl}-2-oxo-1,2-dihydropyridine-3-carboxylic Acid Hydrochloride (1:1) (1·HCl)
TAKEDA PHARMACEUTICAL COMPANY LIMITED [JP/JP]; 1-1, Doshomachi 4-chome, Chuo-ku, Osaka-shi, Osaka 5410045 (JP)
ISHICHI, Yuji; (JP).
YAMADA, Masami; (US).
KAMEI, Taku; (JP).
FUJIMORI, Ikuo; (US).
NAKADA, Yoshihisa; (JP).
YUKAWA, Tomoya; (JP).
SAKAUCHI, Nobuki; (JP).
OHBA, Yusuke; (JP).
TSUKAMOTO, Tetsuya; (JP)
Paper

A practical synthesis of a peripherally selective noradrenaline reuptake inhibitor that has a chiral 6,7-trans-disubstituted-1,4-oxazepane as a new class of scaffold is described. The amino alcohol possessing the desired stereochemistry was obtained with excellent dr and ee, starting from a commercially available aldehyde via a Morita–Baylis–Hillman reaction, Michael addition, isolation as maleic acid salt, reduction, and diastereomeric salt formation with (+)-10-camphorsulfonic acid. The desired single stereoisomer obtained at an early stage of the synthesis was used for seven-membered ring formation in fully telescoped processes, providing the chiral 6,7-trans-disubstituted-1,4-oxazepane efficiently. In addition to controls of dr and ee of the chiral 1,4-oxazepane, and control of N,O-selectivity in SN2 reaction of the intermediate mesylate with a pyridone derivative, finding appropriate intermediates that were amenable to isolation and upgrade of purity enabled a practical chiral HPLC separation-free, column chromatograph-free synthesis of the drug candidate with excellent chemical and optical purities in a higher overall yield.


PATENT
https://www.google.com/patents/WO2012046882A1?cl=zh
PAPER
Volume 24, Issue 16, 15 August 2016, Pages 3716–3726
http://www.sciencedirect.com/science/article/pii/S0968089616304382
Peripheral-selective inhibition of noradrenaline reuptake is a novel mechanism for the treatment of stress urinary incontinence to overcome adverse effects associated with central action. Here, we describe our medicinal chemistry approach to discover a novel series of highly potent, peripheral-selective, and orally available noradrenaline reuptake inhibitors with a low multidrug resistance protein 1 (MDR1) efflux ratio by cyclization of an amide moiety and introduction of an acidic group. We observed that the MDR1 efflux ratio was correlated with the pKa value of the acidic moiety. The resulting compound 9exhibited favorable PK profiles, probably because of the effect of intramolecular hydrogen bond, which was supported by a its single-crystal structure. The compound 9, 1-{[(6S,7R)-7-(4-chloro-3-fluorophenyl)-1,4-oxazepan-6-yl]methyl}-2-oxo-1,2-dihydropyridine-3-carboxylic acid hydrochloride, which exhibited peripheral NET-selective inhibition at tested doses in rats by oral administration, increased urethral resistance in a dose-dependent manner.

REFERNCES
(a) Ishichi, Y.; Yamada, M.; Kamei, T.; Fujimori, I.; Nakada, Y.; Yukawa, T.; Sakauchi, N.; Ohba, Y.; Tsukamoto, T. WO 2012/046882 A1, Apr 12, 2012.
(b) Fujimori, I.; Yukawa, T.; Kamei, T.; Nakada, Y.; Sakauchi, N.; Yamada, M.; Ohba, Y.; Takiguchi, M.; Kuno, M.; Kamo, I.; Nakagawa, H.; Hamada, T.; Igari, T.; Okuda, T.; Yamamoto, S.; Tsukamoto, T.; Ishichi, Y.; Ueno, H. Bioorg. Med. Chem. 2015, 23, 5000– 5014 DOI: 10.1016/j.bmc.2015.05.017
(c) Yukawa, T.; Fujimori, I.; Kamei, T.; Nakada, Y.; Sakauchi, N.; Yamada, M.; Ohba, Y.; Ueno, H.; Takiguchi, M.; Kuno, M.; Kamo, I.; Nakagawa, H.; Fujioka, Y.; Igari, T.; Ishichi, Y.; Tsukamoto, T. Bioorg. Med. Chem. 2016, 24, 3207– 3217 DOI: 10.1016/j.bmc.2016.05.038
(d) Yukawa, T.; Nakada, Y.; Sakauchi, N.; Kamei, T.; Yamada, M.; Ohba, Y.; Fujimori, I.; Ueno, H.; Takiguchi, M.; Kuno, M.; Kamo, I.; Nakagawa, H.; Fujioka, Y.; Igari, T.; Ishichi, Y.; Tsukamoto, T. Bioorg. Med. Chem. 2016, 24, 3716– 3726 DOI: 10.1016/j.bmc.2016.06.014
//////////////////1372185-97-1, 1372180-09-0, Peripherally selective, noradrenaline reuptake inhibitor, TAKEDA
O=C(O)C3=CC=CN(C[C@@H]1CNCCO[C@H]1c2ccc(Cl)c(F)c2)C3=O
“NEW DRUG APPROVALS” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This is a compilation for educational purposes only. P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent

TAK-058 , ENV-8058
5-HT 3 receptor antagonist
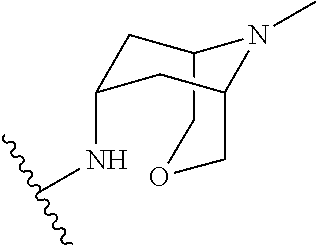
1-(1-methyl-1H-pyrazol-4-yl)-N-((1R,5S,7S)-9-methyl-3-oxa-9-azabicyclo[3.3.1]nonan-7-yl)-1H-indole-3-carboxamide
l-(l-methyl-lH-pyrazol-4-yl)-N-((lR,5 .7S)-9-methyl-3-oxa-9-azabicyclo[3.3.11nonan-7-yl)-lH-indole-3-carboxamide
1-(1-methyl-1H- pyrazol-4-yl)-N- ((1R,5S,7S)- 9-methyl-3- oxa-9-azabicyclo [3.3.1]nonan-7- yl)-1H-indole-3- carboxamide, 2,2,2- trifluoroacetic acid salt
N-(9-methyl-3-oxa-9-azabicyclo[3.3.1]nonan-7-yl)-1-(1-methylpyrazol-4-yl)indole-3-carboxamide
| Molecular Formula: | C21H25N5O2 |
|---|---|
| Molecular Weight: | 379.4555 g/mol |
https://clinicaltrials.gov/ct2/show/NCT02153099
Phase I Schizophrenia
| Latest Stage of Development | Phase I |
| Standard Indication | Schizophrenia |
| Indication Details | Treat schizophrenia |

1 -( 1 -methyl- 1 H-pyrazol-4-yl)-N-((lR,5S,7S)-9-methyl-3-oxa-9-azabicyclo[3.3.1]nonan-7-yl)-lH-indole-3-carboxamide, free base, which is an antagonist of the 5-HT3 receptor. 1 -(1 -Methyl- 1 H-pyrazol-4-yl)-N-((lR,5S,7S)-9-methyl-3-oxa-9-azabicyclo[3.3.1]nonan-7-yl)-lH-indole-3-carboxamide, 2,2,2-trifluoroacetic acid salt, is disclosed in PCT Publication No. WO
2014/014951, published January 23, 2014.
1-(1-methyl-1H-pyrazol-4-yl)-N-((1R,5S,7S)-9-methyl-3-oxa-9-azabicyclo[3.3.1]nonan-7-yl)-1H-indole-3-carboxamide a 5-HT3 receptor antagonist, useful for treating anxiety, depression, eating disorder, schizophrenia, cognitive dysfunction, Parkinson’s disease, Huntington’s Chorea, presenile dementia, Alzheimer’s disease and atherosclerosis.
This compound was originally claimed in WO2014014951, Takeda, following its acquisition of Envoy Therapeutics, is developing TAK-058 (ENV-8058), a 5-HT3 receptor antagonist, as an oral solution for treating schizophrenia, especially cognitive impairment associated with schizophrenia.
In July 2015, the drug was listed as being in phase I development. TAK-058 may have emerged from a schizophrenia therapy program which used Envoy’s bacTRAP translational profiling technology to identify a protein target in the brain.
PATENT
Example 5
Synthesis of l-(l-methyl-lH-pyrazol-4-yl)-N-((lR,5 .7S)-9-methyl-3-oxa-9-azabicyclo[3.3.11nonan-7-yl)-lH-indole-3-carboxamide. 2.2.2-trifluoroacetic acid salt
Step 1 : methyl 1-(1 -methyl- lH-pyrazol-4-yl)-lH-indole-3-carboxylate. TFA
To a sealed tube was added copper(I) iodide (65.2 mg, 0.342 mmol), methyl 1H-indole-3-carboxylate (200 mg, 1.142 mmol) and potassium phosphate (509 mg, 2.397 mmol), then the reaction vessel was evacuated and purged with nitrogen (3x). Next, 4-bromo-l-methyl-lH-pyrazole (184 mg, 1.142 mmol) and (lR,2R)- ,N2-dimethylcyclohexane-l,2-diamine (109 μΐ, 0.685 mmol) were added, followed by toluene (1 142 μΐ). The reaction tube was evacuated and purged with nitrogen, then sealed and heated at 1 10 °C for 24 h. HPLC purification provided the title compound as a colorless oil.
Step 2: 1-(1 -methyl- lH-pyrazol-4-yl)-lH-indole-3-carboxylic acid hydrochloride
To a solution of methyl 1-(1 -methyl- lH-pyrazol-4-yl)-lH-indole-3-carboxylate, TFA
(3.5 mg, 9.48 μιηοΐ) in MeOH (95 μΐ) was added a solution of aq. KOH (33.2 μΐ, 0.066 mmol, 2 M). The reaction mixture was stirred at RT overnight, then acidified with IN HC1.
The solvent was evaporated under reduced pressure and the residue was dried under vacuum overnight. The title compound was used without further purification.
Step 3 : l-(l-methyl-lH-pyrazol-4-yl)-N-((lR,5 .7S)-9-methyl-3-oxa-9-azabicyclor3.3.11nonan-7-yl)-lH-indole-3-carboxamide, 2,2,2-trifluoroacetic acid salt
To a mixture of 1-(1 -methyl- lH-pyrazol-4-yl)-lH-indole-3-carboxylic acid hydrochloride (2.6 mg, 9.36 μιηοΐ) in DMF (187 μΐ) was added HATU (4.27 mg, 0.01 1 mmol) and DIPEA (8.18 μΐ, 0.047 mmol). After the reaction mixture was stirred at RT for 15 min, (lR,5S,7S)-9-methyl-3-oxa-9-azabicyclo[3.3.1]nonan-7-amine, TFA (3.04 mg, 0.01 1 mmol) was added and stirring was continued for 2 h. HPLC purification afforded the title compound as a white solid. MS (ESI, pos. ion) m/z: 380.30 (M+l).
PATENT
EXAMPLE 1 : l-(l-methyl-lH-pyrazol-4-yl)-N-((lR,5S,7S)-9-methyl-3-oxa-9-azabicyclo[3.3.1 ]nonan-7-yl)- lH-indole-3-carboxamide
l-(l-Methyl-lH-pyrazol-4-yl)-lH-indole-3-carboxylic acid (128.7 g, 0.53 mol,) and anhydrous THF (645 mL) was heated to about 43°C. Oxalyl chloride (137.7 g, 92 mL, 1.08 mol) was added dropwise between 40 and 50°C. Gas evolution ceased in approximately 30 minutes. The resulting suspension was stirred for 2 hours at 50°C, allowed to cool to room temperature, and then stirred overnight. The suspension was diluted with heptane (1.5 L), stirred for 10 minutes, and allowed to settle. The supernatant was removed. The addition of heptane (1.5 L), followed by stirring, settling, and decanting was repeated two more times.
The resulting suspension was diluted with anhydrous THF (645 mL) and the ratio between THF and heptane was determined by NMR to be 3:2. The reaction mixture was cooled to 5°C and to the mixture was added DIPEA base (138 g, 1.07 mol) at such a rate that the temperature did not exceed 20°C. Next (li?,55*,7S)-9-methyl-3-oxa-9-azabicyclo[3.3.1]nonan-7-amine (101.4 g, 0.63 mol) in 500 mL of anhydrous THF was added. The reaction mixture was warmed to ambient temperature and stirred at 20 to 23°C overnight to give a suspension.
The suspension was filtered and the cake was dissolved in IN HC1 (2.6 L). The aqueous layer was washed with EtOAc (3 x 2.6 L). The aqueous layer was cooled to 5°C and was basified to pH 12 with aqueous potassium hydroxide (230 g) solution in water (500 mL). The mixture was stirred at 5 to 10°C overnight to give a solid. The product was filtered, washed with water (2 x 1.2 L), followed by MTBE (2 x 1.2 L), and then dried to give 128 g (64%) of the (crude) title compound.
Patent
https://www.google.co.in/patents/US20140024644
1-(1-methyl-1H- pyrazol-4-yl)-N- ((1R,5S,7S)- 9-methyl-3- oxa-9-azabicyclo [3.3.1]nonan-7- yl)-1H-indole-3- carboxamide, 2,2,2- trifluoroacetic acid salt
Synthetic Procedures Reference 1 Synthesis of (1R,5S,7S)-tert-butyl 7-hydroxy-3-oxa-9-azabicyclo[3.3.1]nonane-9-carboxylate

Example 4 Synthesis of N-((1R,5S,7S)-9-methyl-3-oxa-9-azabicyclo[3.3.1]nonan-7-yl)-1-(1H-pyrazol-4-yl)-1H-indole-3-carboxamide, 2,2,2-trifluoroacetic acid salt

Example 5 Synthesis of 1-(1-methyl-1H-pyrazol-4-yl)-N-((1R,5S,7S)-9-methyl-3-oxa-9-azabicyclo[3.3.1]nonan-7-yl)-1H-indole-3-carboxamide, 2,2,2-trifluoroacetic acid salt

Step 1: methyl 1-(1-methyl-1H-pyrazol-4-yl)-1H-indole-3-carboxylate, TFA
Step 2: 1-(1-methyl-1H-pyrazol-4-yl)-1H-indole-3-carboxylic acid hydrochloride
Step 3: 1-(1-methyl-1H-pyrazol-4-yl)-N-((1R,5S,7S)-9-methyl-3-oxa-9-azabicyclo[3.3.1]nonan-7-yl)-1H-indole-3-carboxamide, 2,2,2-trifluoroacetic acid salt
| 15 |
 |
 |
TFA |
| 379.456 MW | 380.30 MS +1
|
/////////TAK-058 , ENV-8058, phase I, takeda, 5-HT 3 receptor antagonist, Envoy Therapeutics, Inc., Phase I, Schizophrenia
C12CC(CC(N1C)COC2)NC(c4c3ccccc3n(c4)c5cnn(c5)C)=O
CN1C=C(C=N1)N2C=C(C3=CC=CC=C32)C(=O)NC4CC5COCC(C4)N5C
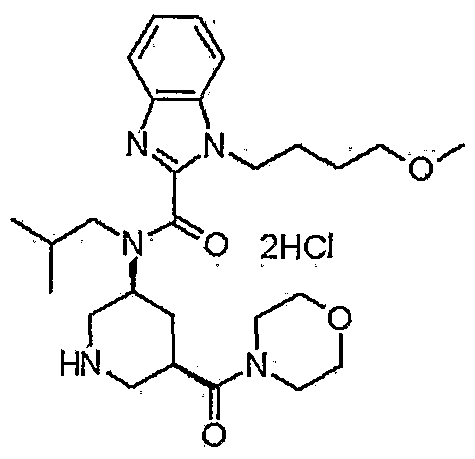
TAK 272
C27 H41 N5 O4 . Cl H, 536.106
CAS.1202269-24-6. MonoHCl
1202265-90-4 DIHCL
Base cas…1202265-63-1
Metanesulfonate…1202266-34-9
Takeda Pharmaceutical Company Limited, INNOVATOR
see……….http://www.allfordrugs.com/2015/10/21/tak-272-for-hypertension-takedas-next-sartan/
1-(4-methoxybutyl)-N-(2-methylpropyl)-N-[(3S,5R)-5-(morpholin-4-ylcarbonyl)-piperidin-3-yl]-1H-benzimidazole-2-carboxamide
1- (4-methoxybutyl) -N- (2-methylpropyl) -N- [ (3S, 5R) -5- (morpholin-4-ylcarbonyl) piperidin-3-yl] -lH-benzimidazole-2-carboxamide dihydrochloride
N-Isobutyl-1-(4-methoxybutyl)-N-[5(R)-(morpholin-4-ylcarbonyl)piperidin-3(S)-yl]-1H-benzimidazole-2-carboxamide hydrochloride
1- (4-methoxybutyl) -N- (2- methylpropyl) -N – [(3S, 5R) -5- (morpholin-4-ylcarbonyl) piperidine-3 – yl] -1H- benzimidazole-2-carboxamide hydrochloride,
![]()
The compound is used as renin inhibitor for treating diabetic nephropathy and hypertension
Takeda’s TAK-272, was reported to be in phase II in October 2015), an oral renin inhibitor, for treating diabetic nephropathy and hypertension

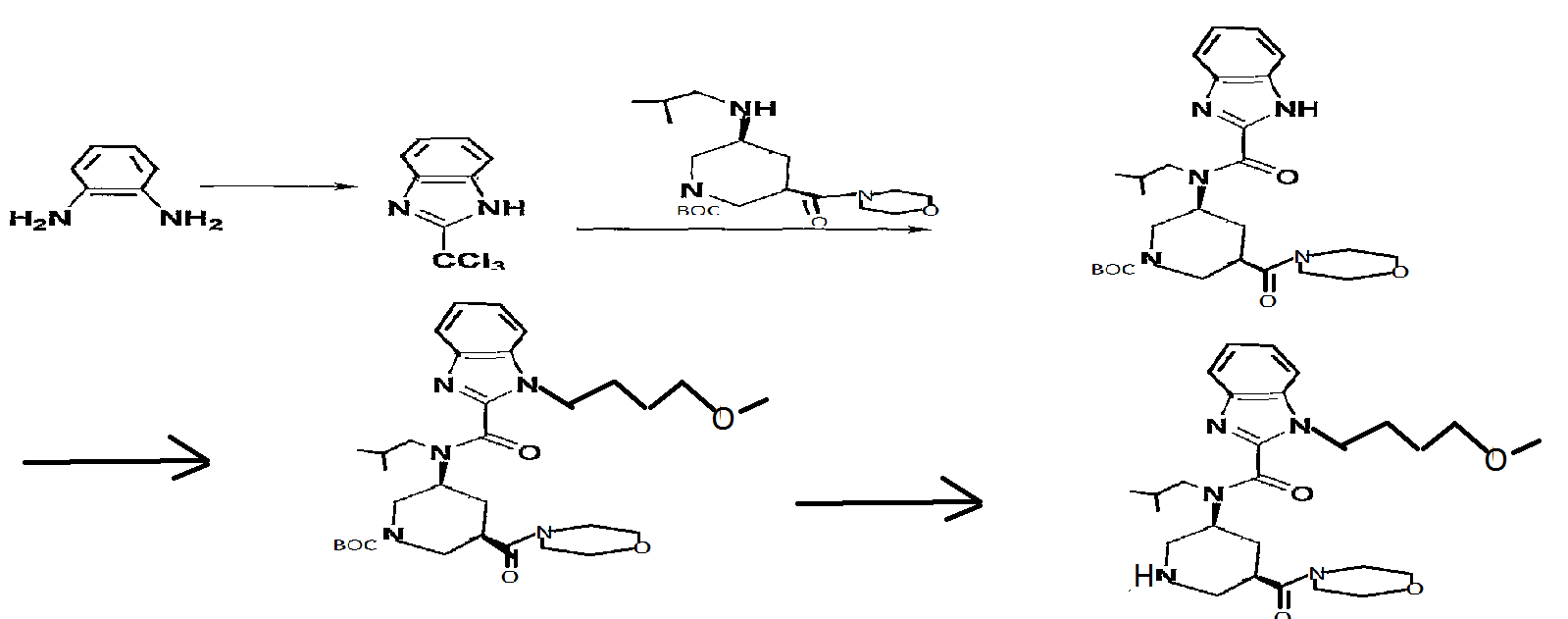
In the above method, the acid anhydride (BANC) from chiral dicarboxylic acid monoester ((-) – BMPA) were synthesized and then the carboxylic acid after conversion and hydrolysis reaction of the Z amine by the Curtius rearrangement of the carboxylic acid (BAPC) and it was then performs amidation by the condensation reaction with the amine (morpholine), is synthesized heterocyclic amide compound (BMPC). Further, Patent Document 2, the preparation of compounds useful as synthetic intermediates of the above heterocyclic compounds are disclosed.
(Wherein each symbol is as described in Patent Document 2.)
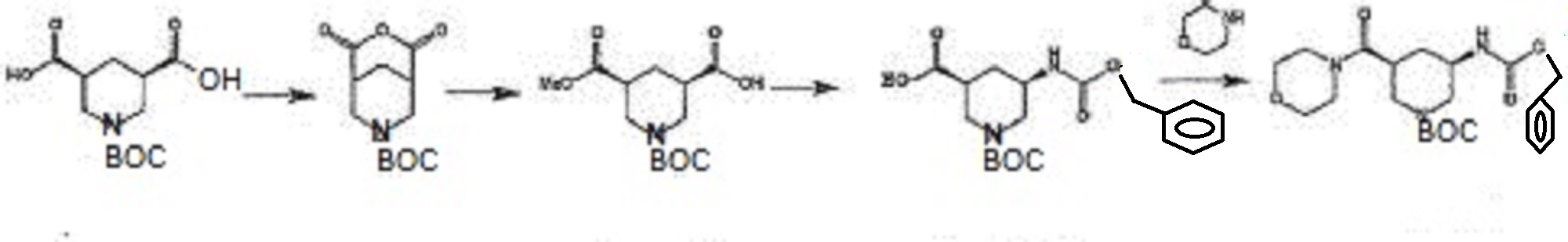
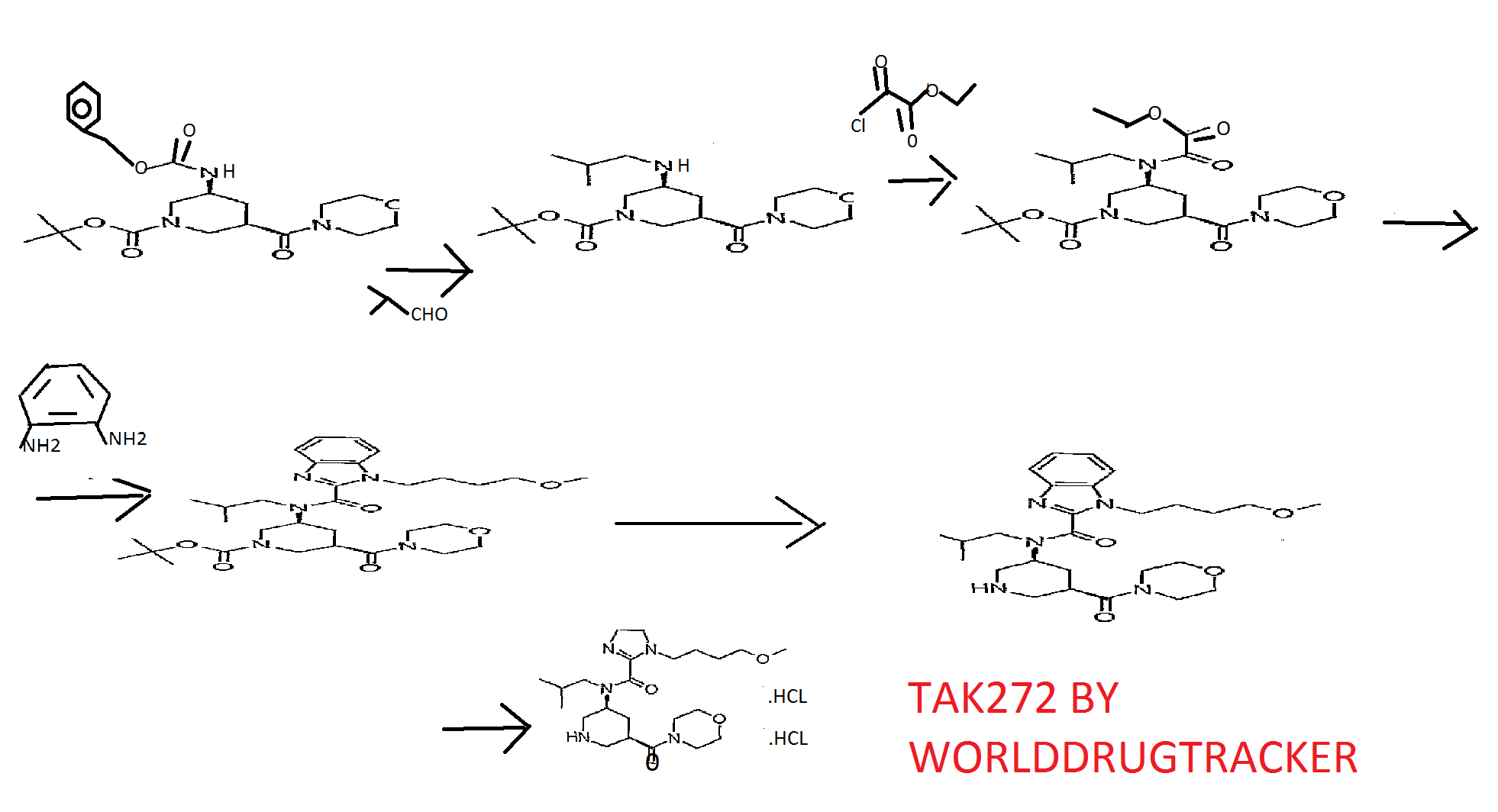
WO2009154300
https://www.google.co.in/patents/WO2009154300A2?cl=en
INTERMEDIATES FOR CONSTRUCTION

USE THIS ONE




Reference Example 31 tert-butyl (3S,5R)-3-[{ [1- (4-methoxybutyl) -lH-benzimidazol-2- yl] carbonyl} (2-methylpropyl) amino] -5- (morpholin-4- ylcarbonyl)piperidine-l-carboxylate and 1- (4-methoxybutyl) -N-
(2-methylpropyl) -N- [ (3S, 5R) -5- (morpholin-4- ylcarbonyl)piperidin-3-yl]-lH-benzimidazole-2-carboxamide
tert-Butyl (3S, 5R) -3-{ [ ( {2- [ (4- methoxybutyl) amino] phenyl}amino) (oxo) acetyl] (2- methylpropyl) amino} -5- (morpholin-4-ylcarbonyl) piperidine-1- carboxylate (9.11 g) was dissolved in acetic acid (50 ml), and the mixture was stirred at 😯0C for 15 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure, the residue was diluted with aqueous sodium bicarbonate, and the mixture was extracted with ethyl acetate. The extract was washed with saturated brine, and dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure. The residue was subjected to basic silica gel column chromatography, and a fraction eluted with ethyl acetate was concentrated under reduced pressure to give tert- butyl (3S, 5R) -3- [ { [1- (4-methoxybutyl) -lH-benzimidazol-2- yl] carbonyl } (2-methylpropyl) amino] -5- (morpholin-4- ylcarbonyl)piperidine-l-carboxylate (5.85 g) , and a fraction eluted with ethyl acetate-methanol (85:15) was concentrated under reduced pressure to give 1- (4-methoxybutyl) -N- (2- methylpropyl) -N- [ (3S, 5R) -5- (morpholin-4-ylcarbonyl) piperidin- 3-yl] -lH-benzimidazole-2-carboxamide (580 mg) . [0424] tert-butyl (3S,5R)-3-[{ [1- (4-methoxybutyl) -lH-benzimidazol-2- yl] carbonyl} (2-methylpropyl) amino] -5- (morpholin-4- ylcarbonyl ) piperidine-1-carboxylate 1H-NMR (CDCl3) δ 0.63-0.80 (2H, m) , 0.89-1.07 (4H, m) , 1.41- 1.59 (9H, m) , 1.59-1.80 (2H, m) , 1.87-2.23 (4H, m) , 2.30-2.98 (3H, m) , 3.21-3. 46 ( 6H, m) , 3.49-3. 91 (1OH, m) , 3. 95-4 . 47 (5H, m) , 7 . 18-7 . 51 (3H, m) , 7. 56-7 . 84 ( IH, m) .
MS (ESI+, m/e) 600 (M+l )
1- (4-methoxybutyl) -N- (2-methylpropyl) -N- [ (3S, 5R) -5- (morpholin- 4-ylcarbonyl)piperidin-3-yl] -lH-benzimidazole-2-carboxamide BASE
1H-NMR (CDCl3) δ 0.64-0.74 (2H, m) , 0.95-1.07 (4H, m) , 1.43-
1.74 (3H, m) , 1.84-2.41 (4H, m) , 2.48-2.67 (IH, m) , 2.67-3.01
(3H, m), 3.03-3.44 (8H, m) , 3.47-3.78 (9H, m) , 4.06-4.46 (3H, m) , 7.28-7.47 (3H, m) , 7.62-7.81 (IH, m) . MS (ESI+, m/e) 500 (M+l)
Example 10
1- (4-methoxybutyl) -N- (2-methylpropyl) -N- [ (3S, 5R) -5- (morpholin-
4-ylcarbonyl) piperidin-3-yl] -lH-benzimidazole-2-carboxamide dihydrochloride
tert-Butyl (3S,5R)-3-[{ [1- (4-methoxybutyl) -IH- benzimidazol-2-yl] carbonyl} (2-methylpropyl) amino] -5-
(morpholin-4-ylcarbonyl)piperidine-l-carboxylate (5.85 g) was dissolved in methanol (20 ml) , 4M hydrogen chloride-ethyl acetate (20 ml) was added, and the mixture was stirred at room temperature for 15 hr. The reaction mixture was concentrated, and the residue was diluted with aqueous sodium bicarbonate, and the mixture was extracted with ethyl acetate. The extract was washed with saturated brine, and dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure. The residue was subjected to basic silica gel column chromatography, and a fraction eluted with ethyl acetate- methanol (9:1) was concentrated under reduced pressure to give 1- (4-methoxybutyl) -N- (2-methylpropyl) -N- [ (3S, 5R) -5- (morpholin- 4-ylcarbonyl) piperidin-3-yl] -lH-benzimidazole-2-carboxamide (4.40 g) . The obtained 1- (4-methoxybutyl) -N- (2-methylpropyl) – N- [ (3S, 5R) -5- (morpholin-4-ylcarbonyl) piperidin-3-yl] -IH- benzimidazole-2-carboxamide (2.20 g) was dissolved in ethyl acetate (20 ml) , 4M hydrogen chloride-ethyl acetate (5 ml) and methanol (20 ml) were added, and the mixture was stirred at room temperature for 5 min. The reaction mixture was concentrated under reduced pressure to give the object product (2.52 g).
dihydrochloride
1H-NMR (DMSO-d6) δ 0.63-0.76 (2H, m) , 0.85-1.00 (4H, m) , 1.40-
1.60 (2H, m) , 1.68-1.89 (2H, m) , 1.93-2.17 (2H, m) , 2.20-2.44
(2H, m) , 2.81-3.81 (2OH, m) , 4.19-4.39 (3H, m) , 7.23-7.46 (2H, m) , 7.57-7.81 (2H, m) , 8.38-9.77 (2H, m) .
MS (ESI+, m/e) 500 (M+l)
Example 252
1- ( 4-methoxybutyl ) -N- ( 2-methylpropyl ) -N- [ ( 3S 1. 5R) -5- (morpholin- 4-ylcarbonyl ) piperidin-3-yl ] -lH-benzimidazole-2-carboxamide methanesulfonate

l-(4-Methoxybutyl) -N- (2-methylpropyl) -N- [ (3S,5R)-5- (morpholin-4-ylcarbonyl) piperidin-3-yl] -lH-benzimidazole-2- carboxamide (208 mg) was dissolved in ethyl acetate (2 ml) , a solution of methanesulfonic acid (40 μl) in ethyl acetate (1 ml) was added at 75°C, hexane (1 ml) was added, and the mixture was heated under reflux and stood at room temperature overnight. The precipitated crystals were collected by filtration, and dried at 7O0C for 3 hr to give the object product (158 mg) . MS (ESI+, m/e) 500 (M+l) melting point : 144.40C
Example 32
methyl (3R, 5S)-5-[{ [1- (4-methoxybutyl) -lH-benzimidazol-2- yl] carbonyl} (2-methylpropyl) amino] piperidine-3-carboxylate dihydrochloride [0675]

MS (ESI+, m/e) 445 (M+l)
Example 33
(3R, 5S) -5- [ { [1- (4-methoxybutyl) -lH-benzimidazol-2- yljcarbonyl} (2-methylpropyl) amino] piperidine-3-carboxylic acid dihydrochloride

MS (ESI+, m/e) 431 (M+l)
Reference Example 29
{ [ ( 3S , 5R) -1- (tert-butoxycarbonyl ) -5- (morpholin-4- ylcarbonyl ) piperidin-3~yl ] ( 2-itιethylpropyl ) amino } (oxo ) acetic acid
To a solution of tert-butyl (3S,5R)~3-{ [ethoxy (oxo) acetyl] (2-methylpropyl) amino}-5- (morpholin-4- ylcarbonyl) piperidine-1-carboxylate (10.3 g) in ethanol (40 ml) was added 2M aqueous sodium hydroxide solution (22 ml) , and the mixture was stirred at room temperature for 6 hr. The reaction mixture was adjusted to pH 7 with IM hydrochloric acid, and extracted with ethyl acetate. The extract was washed with saturated brine, and dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure to give the object product (10.3 g) .
1H-NMR (CDCl3) δ 0.78-0.99 (6H, m) , 1.37-1.52 (9H, m) , 1.79- 2.16 (3H, m) , 2.38-3.86 (14H, m) , 3.93-4.43 (2H, m) . MS (ESI+, m/e) 442 (M+l)
Reference Example 28
tert-butyl (3S, 5R) -3-{ [ethoxy (oxo) acetyl] (2- methylpropyl ) amino } -5- (morpholin-4-ylcarbonyl) piperidine-1- carboxylate
To a solution of tert-butyl (3S, 5R) -3- [ (2- methylpropyl) amino] -5- (morpholin-4-ylcarbonyl) piperidine-1- carboxylate (9.24 g) and diisopropylethylamine (10.5 ml) in DMA (100 ml) was added dropwise ethyl chloroglyoxylate (3.4 ml) at 0°C. The reaction mixture was stirred at room temperature for 15 hr, and the reaction mixture was concentrated. An aqueous sodium bicarbonate solution was added to the residue, and the mixture was extracted with ethyl acetate. The extract was washed with saturated brine, and dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography, and a fraction eluted with ethyl acetate was concentrated under reduced pressure to give the object product (10.3 g) . 1H-NMR (CDCl3) δ 0.84-1.00 (6H, m) , 1.37 (3H, q) , 1.42-1.53 (9H, m) , 1.80-2.19 (3H, m) , 2.26-2.42 (IH, m) , 2.59-2.96 (IH, in) , 2.97-3.30 (3H, m) , 3.37-3.92 (9H, m) , 4.01-4.26 (2H, m) , 4.26- 4.40 (2H, m) . MS (ESI4-, m/e) 470 (M+l) “
Reference Example 22 tert-butyl (3S, 5R) -3- [ (2-methylpropyl) amino] -5- (morpholin-4- ylcarbonyl)piperidine-l-carboxylate
[0369] tert-Butyl (3S,5R)-3-{ [ (benzyloxy) carbonyl] aminoJ-5- (morpholin-4-ylcarbonyl)piperidine-l-carboxylate (58 g) and palladium (II) hydroxide-carbon (5 g) were suspended in methanol (400 ml) and the mixture was stirred under a hydrogen atmosphere (1 atom) at room temperature for 16 hr. The palladium catalyst was filtered off, and the filtrate was concentrated under reduced pressure. The obtained residue and acetic acid (8.8 ml) were dissolved in methanol (400 ml), 2- methylpropanal (14.0 ml) was added, and the mixture was stirred at room temperature for 1 hr. Sodium triacetoxyborohydride (40.4 g) was added to the reaction mixture, and the mixture was stirred at room temperature for 2 hr. The reaction mixture was concentrated under reduced pressure, and the concentrate was basified with 3.5M aqueous potassium carbonate solution, and the mixture was extracted with ethyl acetate. The extract was washed with saturated brine, and dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure. The residue was subjected to basic silica gel column chromatography, and a fraction eluted with ethyl acetate-hexane (1:5) – ethyl acetate-hexane (1:1) was concentrated under reduced pressure to give the object product (33.3 g) .
1H-NMR (CDCl3) δ: 0.90 (6H, d) , 1.46 (9H, s) , 1.54 (IH, d) , 1.69 (IH, dt), 1.96-2.12 (2H, m) , 2.23-2.37 (IH, m) , 2.47 (3H, d) , 2.66 (IH, d) , 3.61 (IH, br s) , 3.55 (2H, d) , 3.69 (5H, ddd) , 4.01-4.46 (2H, m) .
Example 6 1-tert-butyl 3-methyl (3R, 5S) -5-aminopiperidine-l, 3- dicarboxylate [0318]

(3S, 5R) -1- (tert-Butoxycarbonyl) -5-(methoxycarbonyl)piperidine-3-carboxylic acid (2.83 g) was suspended in toluene (36 ml), diphenylphosphoryl azide (2.60 ml) and triethylamine (1.70 ml) were added, and the mixture was stirred at 100°C for 1 hr. The reaction mixture was cooled to room temperature, benzyl alcohol (1.53 ml) and triethylamine (7.00 ml) were added and the mixture was stirred at 80°C for 3 hr. The reaction mixture was concentrated, the residue was dissolved in ethyl acetate, and the solution was washed with water, 0.5M hydrochloric acid, saturated aqueous sodium hydrogen carbonate and saturated brine in this order, and dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography, and a fraction eluted with ethyl acetate-hexane (1:3 – 3:1) was concentrated under reduced pressure. The obtained residue was dissolved in methanol (60 ml), 10% palladium carbon (50% in water) (150 mg) was added and the mixture was stirred under a hydrogen pressurization (5 atom) at ambient temperature and normal pressure for 5 hr. The catalyst was filtered off, and the filtrate was concentrated under reduced pressure to give the object product (1.83 g) as an oil.
1H-NMR (CDCl3) δ 1.22-1.43 (4H, m) , 1.46 (9H, s), 2.27-2.79 (4H, m) , 3.70 (3H, s) , 4.13 (2H, br s) [0320] In the same manner as in the method shown in Reference Example 6, the following compound (Reference Example 7) was obtained.
Reference Example 8
1-tert-butyl 3-methyl (3R, 5S) -5- [ (2- methylpropyl) amino] piperidine-1, 3-dicarboxylate [0325]

1-tert-Butyl 3-methyl (3R, 5S) -5-aminopiperidine-l, 3- dicarboxylate (1.83 g) , isobutyraldehyde (0.78 ml) and acetic acid (0.49 ml) were dissolved in methanol (50 ml), and the mixture was stirred at room temperature for 30 min. Sodium triacetoxyborohydride (3.80 g) was added to the reaction mixture, and the mixture was stirred at room temperature for 7 hr. The reaction mixture was concentrated under reduced pressure, the concentrate was basified with aqueous sodium bicarbonate, and extracted with ethyl acetate. The extract was washed with water and saturated brine, and dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography, and a fraction eluted with ethyl acetate-hexane (1:1) – ethyl acetate 100% – ethyl acetate- methanol (9:1) was concentrated under reduced pressure to give the object product (1.42 g) as an oil.
1H-NMR (CDCl3) δ 0.90 (6H, d) , 1.22-1.38 (3H, m) , 1.46 (9H, s) , 1.69 (IH, dt), 2.23-2.39 (2H, m) , 2.44-2.59 (IH, m) , 2.47 (2H, d) , 2.74 (IH, br s) , 3.69 (3H, s) , 4.18-4.34 (2H, m)
Reference Example 27
N- (4-methoxybutyl) benzene-1, 2-diamine

To a solution of phenylenediamine (10.8 g) and 4- methoxybutyl methanesulfonate (9.11 g) in acetonitrile (100 ml) was added potassium carbonate (20.7 g) , and the mixture was stirred heated under reflux for 15 hr. Water was added to the reaction mixture, and the mixture was extracted twice with ethyl acetate. The extract was washed with saturated brine, and dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography, and a fraction eluted with ethyl acetate-hexane (35:65) was concentrated under reduced pressure to give the object product (5.44 g) . 1H-NMR (CDCl3) δ 1.67-1.82 (4H, m) , 3.13 (2H, t) , 3.24-3.39 (6H, m) , 3 . 38 -3 . 50 ( 2H, m) , 6 . 62 – 6 . 74 ( 3H, m) , 6 . 81 ( IH, in) . MS ( ESI+ , m/e ) 195 (M+l )
Reference Example 146 tert-butyl (3S, 5R) -3- [ { [1- (4-methoxybutyl) -lH-benzimidazol-2- yl]carbonyl} (2-methylpropyl) amino] -5- (morpholin-4- ylcarbonyl)piperidine-l-carboxylate

A solution of tert-butyl (3S, 5R) -3- [ (lH-benzimidazol-2- ylcarbonyl) (2-methylpropyl) amino] -5- (morpholin-4- ylcarbonyl)piperidine-l-carboxylate (200 mg) , 4-itιethoxybutyl methanesulfonate (107 mg) and cesium carbonate (254 mg) in N,N-dimethylacetamide (5 ml) was stirred at 60°C for 15 hr. After cooling to room temperature, the reaction mixture was diluted with water and extracted with ethyl acetate (10 ml*2) . The extract was washed with saturated brine, and dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography, and a fraction eluted with ethyl acetate-hexane (5:95 – 3:7) was concentrated under reduced pressure to give the object product (190 mg) . 1H-NMR (CDCl3) δ 0.63-0.80 (2H, m) , 0.89-1.07 (4H, m) , 1.41- 1.59 (9H, m) , 1.59-1.80 (2H, m) , 1.87-2.23 (4H, m) , 2.30-2.98 (3H, m) , 3.21-3.46 (6H, m) , 3.49-3.91 (1OH, m) , 3.95-4.47 (5H, m) , 7.18-7.51 (3H, m) , 7.56-7.84 (IH, m) . MS (ESI+, m/e) 600 (M+l)
ALTERNATE METHOD IN THIS PATENT



Reference Example 61
2- (trichloromethyl) -lH-benzimidazole

O-Phenylenediamine (25 g) was dissolved in acetic acid (750 ml), and methyl 2, 2, 2-trichloroacetimidate (28.5 ml) was added dropwise over 15 min. After stirring at room temperature for 1 hr, the reaction mixture was concentrated to about 150 ml, and poured into water (1500 ml) . The precipitated crystals were collected by filtration, washed with water (1000 ml) and suspended in toluene (500 ml) . The solvent was evaporated under reduced pressure. The residue was again suspended in toluene (500 ml) and the solvent was evaporated under reduced pressure. The residue was dried under reduced pressure to give the object product (51.8 g) . 1H-NMR (CDCl3) δ 7.31-7.45 (2H, m) , 7.49-7.55 (IH, m) , 7.89 (IH, d) , 9 . 74 ( IH, br s )
Reference Example 64
1-tert-butyl 3-methyl (3R, 5S) -5- [ (lH-benzimidazol-2- ylcarbonyl) (2-methylpropyl) amino] piperidine-1, 3-dicarboxylate

2- (Trichloromethyl) -lH-benzimidazole (19 g) and 1-tert- butyl 3-methyl (3R, 5S) -5- [ (2-methylpropyl) amino] piperidine- 1,3-dicarboxylate (25 g) were dissolved in THF (1200 ml), sodium hydrogen carbonate (67 g) and water (600 ml) were added, and the mixture was stirred at room temperature for 1 hr and at 5O0C for 1 hr. After evaporation of the solvent, the residue was extracted 3 times with ethyl acetate (700 ml) . The extract was washed successively with 10%-aqueous citric acid solution (500 ml) and brine, and dried over anhydrous sodium sulfate. The solvent was evaporated under reduced pressure.
The residue was dissolved in ethyl acetate (1000 ml), subjected to basic silica gel column chromatography, and a fraction eluted with ethyl acetate was concentrated under reduced pressure to give the object product (30.6 g) .
1H-NMR (CDCl3) δ 0.78-1.09 (6 H, m) , 1.17-1.55 (9 H, m) , 1.77-2.95 (5 H, m) , 3.11-3.79 (6 H, m) , 3.99-4.73 (4 H, m) , 7.24- 7.41 (2 H, m) , 7.45-7.59 (1 H, m) , 7.72-7.88 (1 H, m) , 10.66-10.98 (1 H, m)MS (ESI+, m/e) 459 (M+l)
Reference Example 69
1-tert-butyl 3-methyl (3R, 5S) -5- [ { [1- (4-methoxybutyl) -IH- benzimidazol-2-yl] carbonyl} (2-methylpropyl) amino] piperidine-1 , 3-dicarboxylate

1-tert-Butyl 3-methyl (3R, 5S) -5- [ (lH-benzimidazol-2- ylcarbonyl) (2-methylpropyl) amino] piperidine-1, 3-dicarboxylate (30 g) and 4-methoxybutyl methanesulfonate (12.5 g) were dissolved in DMA (600 ml), cesium carbonate (32 g) was added, and the mixture was stirred at 70°C for 12 hr. The reaction mixture was poured into ice water (1000 ml), and the mixture was extracted twice with ethyl acetate (1000 ml) . The extract was washed with brine, and dried over anhydrous sodium sulfate. The solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography, and a fraction eluted with ethyl acetate-hexane (1:4 – 1:1) was concentrated under reduced pressure to give the object product (28.7 g) .
1H-NMR (CDCl3) δ 0.76 (4H, d) , 1.01 (2H, d) , 1.30-1.52 (9H, m) , 1.58-2.07 (4H, m) , 2.10-2.93 (4H, m) , 3.27-3.75 (12H, m) , 4.06-4.57 (5H, m) , 7.26-7.48 (3H, m) , 7.79 (IH, d) MS (ESI+, m/e) 545 (M+l)
Example 71
1- (4-methoxybutyl) -N- (2-methylpropyl) -N- [ (3S, 5R) -5- (morpholin- 4-ylcarbonyl) piperidin-3-yl] -lH-benzimidazole-2-carboxamide

tert-Butyl (3S, 5R) -3- [{ [1- (4-methoxybutyl) -IH- benzimidazol-2-yl] carbonyl} (2-methylpropyl) amino] -5- (morpholin-4-ylcarbonyl)piperidine-l-carboxylate (5.85 g) was dissolved in methanol (20 ml) , 4M hydrogen chloride-ethyl acetate (20 ml) was added, and the mixture was stirred at room temperature for 15 hr. The reaction mixture was concentrated, the residue was diluted with aqueous sodium bicarbonate,…and, the mixture was extracted with ethyl acetate. The extract was washed with saturated brine, and dried over anhydrous sodium sulfate. The solvent was evaporated under reduced pressure. The residue was subjected to basic silica gel column chromatography, and a fraction eluted with ethyl acetate- methanol (9:1) was concentrated under reduced pressure to give the object product (4.40 g) . MS (ESI+, m/e) 500 (M+l)
Example 101
1- (5-methoxypentyl) -N- (2-methylpropyl) -N- [ (3S, 5R) -5- (morpholin-4-ylcarbonyl) piperidin-3-yl] -lH-benzimidazole-2- carboxamide dihydrochloride

[1144] tert-Butyl (3S, 5R) -3- [ { [1- (5-methoxypentyl) -IH- benzimidazol-2-yl] carbonyl} (2-methylpropyl) amino] -5- (morpholin-4-ylcarbonyl)piperidine-l-carboxylate (123 mg) was dissolved in 4M hydrogen chloride-ethyl acetate (5 ml) , and the mixture was stirred at room temperature for 3 hr. The reaction mixture was concentrated, and the residue was subjected to reversed-phase preparative HPLC and the eluted fraction was concentrated under reduced pressure. The residue was diluted with aqueous sodium bicarbonate, and the mixture was extracted with ethyl acetate. The extract was washed with saturated brine, and dried over anhydrous sodium sulfate. 4M Hydrogen chloride-ethyl acetate (1 ml) was added and the mixture was stirred for 5 min. The solvent was evaporated under reduced pressure to give the object product (76 mg) . MS (ESI+, m/e) 514 (M+l)
PATENT
WO2013122260
http://www.google.co.in/patents/WO2013122260A1?cl=en
PATENT
WO 2011158880
http://www.google.co.in/patents/WO2011158880A1?cl=en
Reference Example 1
1- (4-methoxybutyl) -N- (2- methylpropyl) -N – [(3S, 5R) -5- (morpholin-4-ylcarbonyl) piperidin-3-yl] -1H- benzimidazole -2 – carboxamide hydrochloride (A-type crystal)
tert- butyl (3S, 5R) -3 – [{[1- (4- methoxy-butyl) -1H- benzimidazol-2-yl] carbonyl} (2-methylpropyl) amino] -5- (morpholin-4- ylcarbonyl) was suspended dissolved piperidine-1-carboxylate The (300g) in 3N- hydrochloric acid water (1200mL) and Ethyl acetate (60mL), and stirred over 3 h at 25 ~ 35 ℃. After completion of the reaction, it was added ethyl acetate (2400mL) in the same temperature. After the addition, it was added 25% aqueous ammonia (600mL) with cooling. After the addition stirring and extracted the organic layer of 5% aqueous ammonia (600mL) was added and stirred. After stirring, the resulting organic layer it was concentrated until the solvent no longer distilled off. After concentrated, dissolved with ethyl acetate (1500mL), and transferred to solution to the crystallizer vessel, and washed with ethyl acetate (750mL). After washing, it was raised in stirring under 45 ~ 55 ℃. After raising the temperature, at the same temperature 4N- hydrogen chloride – it was dropped ethyl acetate (131.3mL). After dropping, it was to dissolve the precipitate at the same temperature. After dissolution confirmation, it was added heptane (750mL) at 40 ~ 50 ℃, after the addition, then cooled to 25 ~ 35 ℃. After cooling, the addition of A-type crystals of the seed crystals (300mg) which was obtained according to the method described in Example 265 of WO2009 / 154300, and stirred for 30 minutes or more. After stirring, the temperature was raised to 40 ~ 45 ℃, it was dropped heptane (1500mL). After the completion of the dropping, it was stirred at the same temperature. Then gradually cooled to 5 ℃ below, followed by stirring at the same temperature for 1 hour. After stirring, ethyl acetate and filtered crystals – heptane: washed with (1 1,600mL), to obtain a wet crystal. The obtained wet crystals dried under reduced pressure at 50 ℃, 1- (4- methoxybutyl) -N- (2- methylpropyl) -N – [(3S, 5R) -5- (morpholin-4-yl carbonyl) piperidin-3-yl] -1H- obtained a crystalline powder of benzimidazole-2-carboxamide hydrochloride (A-type crystal, 198.82g, 74.1% yield). FINAL PRODUCT
TERT BUTYL DERIVATIVE, N-1
Reference Example 4
tert- butyl (3S, 5R) -3 – [{[1- (4- methoxy-butyl) -1H- benzoimidazol-2-yl] carbonyl} (2-methylpropyl) amino] -5- (morpholin-4- ylcarbonyl) piperidine-1-carboxylate 1)
o- nitro aniline (50.0g, 0.362mol), tetrabutylammonium bromide (58.3g, 0.181mol), potassium bromide (43.1g, 0.362mol) in toluene (500mL ) and it was added. At a temperature of 20 ~ 30 ℃ 1- chloro-4-methoxy-butane (66.6g, 0.543mol) and, I was added to 50w / v% sodium hydroxide solution (145mL, 1.81mol). The reaction was heated to a temperature 85 ~ 95 ℃, and stirred for 6 hours. After cooling to a temperature 20 ~ 30 ℃, the reaction mixture water (250mL), 1N- aqueous hydrochloric acid (250mL × 2), 5w / v% aqueous solution of sodium bicarbonate (250mL), it was washed successively with water (250mL). After concentration under reduced pressure the organic layer to Contents (250mL), was added toluene (100mL), was obtained
N- (4- methoxy-butyl) -2-nitroaniline in toluene (350mL, 100% yield).
1 H-NMR (300MHz, CDCl 3) δ 1.64-1.89 (m, 4H), 3.25-3.39 (m, 2H), 3.35 (s, 3H), 3.44 (t, J = 6.1 Hz, 2H), 6.63 ( ddd, J = 8.5, 6.9, 1.2 Hz, 1H), 6.86 (dd, J = 8.5, 1.2 Hz, 1H), 7.43 (ddd, J = 8.5, 6.9, 1.5 Hz, 1H), 8.07 (br s, 1H ), 8.17 (dd, J = 8.5, 1.5 Hz, 1H).
2) N- (4-methoxy-butyl) -2-10 percent in nitroaniline of toluene solution (350mL) Pd / C (K-type, 50% water-containing product) (10.0g) and toluene (100mL) it was added. Hydrogen pressure of 0.1MPa, it was stirred for 3 hours at a temperature of 20 ~ 30 ℃. A stream of nitrogen, the catalyst was filtered, I was washed with toluene (100mL). After the water in the filtrate was separated off and adding magnesium sulfate (25.0g) at a temperature 20 ~ 30 ℃, and stirred at the same temperature for 30 minutes. Filtered over magnesium sulfate, washed with toluene (100mL), was obtained N- (4- methoxybutyl) -o- toluene solution of phenylenediamine (100% yield).
1 H NMR (500 MHz, CDCl 3) δ1.67-1.78 (m, 4H), 3.12-3.14 (m, 2H), 3.32 (br, 3H), 3.35 (s, 3H), 3.41-3.47 (m, 2H), 6.63-6.69 (m, 2H), 6.69-6.74 (m, 1H), 6.82 (td, J = 7.57, 1.58 Hz, 1H).
3) N- (4- methoxy-butyl) -o- After the toluene solution of phenylenediamine cooled to a temperature 0 ~ 10 ℃, acetic acid (65.2g, 1.09mol) and 2,2,2 trichloroacetimide acid methyl ( 70.3g, 0.398mol) and I were added. After stirring for 30 minutes at a temperature 0 ~ 10 ℃, it was stirred for 3 hours at a temperature of 20 ~ 30 ℃. The reaction was 5w / v% saline (250mL), 2N- aqueous hydrochloric acid / 5w / v% sodium chloride solution: a mixture of (1 1) (250mL × 2), 5w / v% aqueous solution of sodium bicarbonate (250mL), 5w / v It was washed successively with% saline solution (250mL). A stream of nitrogen, was added magnesium sulfate (25.0g) to the organic layer at a temperature 20 ~ 30 ℃, and stirred at the same temperature for 30 minutes. Filtered magnesium sulfate, and washed with toluene (100mL). The filtrate was concentrated under reduced pressure and the amount of contents (150mL). Stir the concentrated solution at a temperature 20 ~ 30 ℃, was allowed to precipitate crystals, was added dropwise heptane (750mL). The crystals bleeding is heated to a temperature 40 ~ 50 ℃, after stirring for 30 min, cooled to a temperature 0 ~ 10 ℃, and the mixture was stirred at the same temperature for 2 hours.The precipitated crystals were collected by filtration, toluene – heptane: was washed with (1 5,150 mL). And dried under reduced pressure at 40 ℃, it was obtained 1- (4-methoxy-butyl) -2-fine brown crystals of trichloromethyl -1H- benzimidazole (96.5g, 82.9% yield from o- nitroaniline).
1 H-NMR (300MHz, CDCl 3) δ: 1.68-1.85 (m, 2H), 1.99-2.17 (m, 2H), 3.37 (s, 3H), 3.48 (t, J = 6.1 Hz, 2H), 4.50 -4.65 (m, 2H), 7.27-7.49 (m, 4H), 7.82-7.93 (m, 1H).
. Anal Calcd for C 13 H 15 Cl 3 N 2 O:. C, 48.55; H, 4.70; N, 8.71; Cl, 33.07 Found: C, 48.30; H, 4.61; N, 8.74; Cl, 33.30.
4) pyridine-3,5-dicarboxylic acid (110g, 0.66mol), it was dropped methanol (660 mL) mixture of concentrated sulfuric acid at a temperature of 50 ℃ or less of (226.0g, 2.30mol). Thereafter, the mixture was stirred and heated to a temperature 55 ~ 65 ℃ 7 hours. The reaction was the temperature 40 ~ 50 ℃, was added water (220mL). And further dropping temperature 40-50 5% aqueous ammonia at ℃ (about 1.10L) was adjusted to pH8.0 ~ 8.5. After stirring at a temperature 40 ~ 50 ℃ 30 minutes and stirred for 1 hour and cooled to a temperature 0 ~ 10 ℃. Was collected by filtration precipitated crystals, methanol – water (1: 3,165mL), and washed successively with water (440mL). To obtain a white crystalline powder pyridine-3,5-dicarboxylic acid dimethyl and dried under reduced pressure at 50 ℃ (105.0g, 82.0% yield).
1 H-NMR (300 MHz, CDCl 3) δ 4.00 (s, 6H), 8.87 (s, 1H), 9.37 (s, 2H).
. Anal Calcd for C 9 H 9 NO 4:. C, 55.39; H, 4.65; N, 7.18; O, 32.79 Found: C, 55.42; H, 4.65; N, 7.16.
5) 1 L autoclave pyridine-3,5-dicarboxylic acid dimethyl (100g, 0.51mol) and was charged with dimethylacetamide (400mL), temperature 30 ℃ below with trifluoroacetic acid (59.2mL, after dropping the 0.77mol), 10% Pd-C (PE-type) the (20.0g) it was added. Hydrogen pressure of 0.5 ~ 0.7MPa, it was stirred for 12 hours at a temperature of 55 ~ 65 ℃. The catalyst was filtered off, it was washed with dimethylacetamide (50mL × 2). Triethylamine and the combined filtrates at a temperature 20 ~ 30 ℃ (77.8g, 0.77mol) was added dropwise, and adjusted to pH9.0 ~ 10.0. Temperature 30 ~ 40 ℃ by di -tert- butyl (134g, 0.614mol) was added dropwise and stirred at the same temperature for 2 hours. After the reaction mixture as a 20 ~ 30 ℃, it was added ethyl acetate (600mL), washed with water (900mL). The aqueous layer it was re-extracted with ethyl acetate (400mL). The combined organic layers 5w / v% citric acid -10w / v% sodium chloride solution (600mL), 3% aqueous sodium bicarbonate (600mL), and washed successively with water (600mL). Contents The organic layer (200mL) until it was concentrated under reduced pressure, methanol (250mL) was added to the concentrated solution, and then concentrated under reduced pressure until Contents (200mL). The addition of methanol (250mL) again concentrate, After concentration under reduced pressure until Contents (200mL), was added methanol (2.40L). The solution in water (18.5g, 1.03mol), cesium carbonate (417g, 1.28mol) was added and stirred for about 24 hours at a temperature 55 ~ 65 ℃. The reaction solution was the temperature 20 ~ 30 ℃, concentrated to Contents (700mL), it was added tetrahydrofuran (500mL). The solution temperature at 15 ~ 35 ℃ 2N- hydrochloric acid solution (1.28L, 2.56mol) was added dropwise and adjusted to pH3.0 ~ 3.5, and the mixture was stirred for 30 minutes at a temperature 20 ~ 30 ℃. Extracted with ethyl acetate (750mL × 2), and the organic layer was washed with 10w / v% aqueous sodium chloride solution (500mL × 3). Contents The organic layer (300mL) until it was concentrated under reduced pressure, to obtain a weight content by adding ethyl acetate (650mL).Heating the concentrate to a temperature of 55 ~ 65 ℃, it was added dropwise heptane (500mL). It cooled to a temperature 20 ~ 30 ℃ and stirred for 1 hour. The precipitated crystals were collected by filtration, ethyl acetate – heptane: was washed with (1 1,120mL). Dried under reduced pressure at 50 ℃ 1- (tert- butoxycarbonyl) to give a white crystalline powder of piperidine-3,5-dicarboxylic acid (113.3g, 80.9% yield).
1 H-NMR (300 MHz, DMSO-d 6) δ 1.40 (s, 9H), 1.44-1.61 (m, 1H), 2.21-2.26 (m, 1H), 2.31-2.41 (m, 2H), 4.10- 4.12 (m, 2H).
. Anal Calcd for C 12 H 19 NO 6:. C, 52.74; H, 7.01; N, 5.13; O, 35.13 Found: C, 52.96; H, 6.99; N, 5.39.
6) Under a nitrogen stream, 1- (tert- butoxycarbonyl) piperidine-3,5-dicarboxylic acid (5.00g, 18.3mmol) was suspended in tetrahydrofuran (10.0mL), trifluoroacetic acid anhydride at a temperature 20 ~ 30 ℃ It was dropping things (3.80mL, 27.5mmol). After the completion of the dropping, it was stirred for 1 hour at a temperature of 20 ~ 30 ℃. It was added dropwise heptane (20.0mL) at a temperature 20 ~ 30 ℃ the reaction solution, and stirred for 3 hours then cooled to a temperature 0 ~ 10 ℃. The precipitated crystals were collected by filtration, and washed with heptane (3.00mL). Dried under reduced pressure at 40 ℃ 2,4- dioxo-3-oxa-7-azabicyclo [3,3,1] white crystalline powder of nonane-7-carboxylic acid tert- butyl was obtained (4.03g, yield 86.1%).
1 H-NMR (300 MHz, CDCl 3) δ 1.43 (s, 9H), 1.93-1.99 (m, 1H), 2.40-2.46 (m, 1H), 3.06-3.11 (m, 4H), 4.50-4.54 ( m, 2H).
. Anal Calcd for C 12 H 17 NO 5:. C, 56.46; H, 6.71; N, 5.49; O, 31.34 Found: C, 56.51; H, 6.63; N, 5.69.
7) Under a nitrogen stream, quinidine (69.9g, 0.215mol) and was charged with tetrahydrofuran (200mL), and cooled to a temperature -5 ~ 5 ℃. At the same temperature 2,4-dioxo-3-oxa-7-azabicyclo [3,3,1] nonane-7-carboxylic acid tert- butyl (50.0g, 0.196mol) was added and washed with tetrahydrofuran (50.0mL) crowded. Temperature -5 ~ 5 methanol at ℃ (9.41g, 0.29 4mol) was added dropwise, and the mixture was stirred for 2 hours at a temperature -5 ~ 5 ℃. Ethyl acetate (350mL) to the reaction mixture, was by adding minute solution 20w / v% citric acid aqueous solution (250mL). The aqueous layer it was re-extracted with ethyl acetate (125mL × 2). The organic layers were combined 20w / v% aqueous solution of citric acid (250mL), I was washed successively with water (250mL × 2). The organic layer it was concentrated under reduced pressure. To the residue ethanol (100mL) was added ethyl acetate (450mL) was heated to a temperature 60 ~ 70 ℃, (R) – was added phenethylamine (23.7g, 0.196mol). Temperature 50-60 for one hour at ℃, 1 hour at a temperature of 20 ~ 30 ℃, it was stirred for 1 hour at a temperature of -5 ~ 5 ℃. The precipitated crystals were collected by filtration, ethanol – ethyl acetate: and washed with (2 9,100mL). And dried under reduced pressure at 50 ℃ (3S, 5R) -1- (tert- butoxycarbonyl) -5- (methoxycarbonyl) piperidin-3 to give a white crystalline powder of the carboxylic acid (1R) -1- phenylethylamine salt It was (55.7g, 69.6% yield).
1 H-NMR (300 MHz, DMSO-d 6) δ 1.42 (s, 9H), 1.43-1.51 (m, 3H), 2.06-2.14 (m, 1H), 2.21-2.26 (m, 1H), 2.39- 2.44 (m, 1H), 2.52-2.53 (m, 1H), 2.57 (br s, 2H), 3.64 (s, 3H), 4.12 (br s, 2H), 4.19-4.26 (m, 1H), 7.30- 7.40 (m, 3H), 7.45-7.48 (m, 2H).
. Anal Calcd for C 21 H 32 N 2 O 6:. C, 61.75; H, 7.90; N, 6.86; O, 23.50 Found: C, 61.54; H, 7.77; N, 6.86.
8) (3S, 5R) -1- (tert- butoxycarbonyl) -5- (methoxycarbonyl) piperidine-3-carboxylic acid (1R) -1- phenylethylamine salt (20.0g, 49.0mmol), methanol (20mL) and it was charged with water (80mL). Temperature 20-30 citric acid at ℃ (11.3g, 58.8mmol) was added dropwise a solution prepared by dissolving in water (20.0mL), and the mixture was stirred 1.5 hours at the same temperature. The precipitated crystals were collected by filtration and washed with water (60mL). And dried under reduced pressure at 50 ℃ (3S, 5R) -1- (tert- butoxycarbonyl) -5- give a white crystalline powder (methoxycarbonyl) piperidine-3-carboxylic acid (13.5g, 96.1% yield ).
1 H-NMR (300 MHz, CDCl 3) δ 1.40 (s, 9H), 1.46-1.59 (m, 1H), 2.22-2.27 (m, 1H), 2.37-2.45 (m, 2H), 2.63-2.73 ( m, 2H), 3.63 (s, 3H), 4.14 (br s, 2H), 12.51 (br s, 1H).
. Anal Calcd for C 13 H 21 NO 6:. C, 54.35; H, 7.37; N, 4.88; O, 33.41 Found: C, 54.14; H, 7.28; N, 4.85.
9) Under a nitrogen stream, (3S, 5R) -1- (tert- butoxycarbonyl) -5- (methoxycarbonyl) piperidine-3-carboxylic acid (30.0g, 104mmol), triethylamine (31.7g, 313mmol) and toluene ( It was charged with 180mL). Diphenylphosphorylazide at a temperature of 15 ~ 35 ℃ (28.7g, 313mmol) I was dropped a toluene (30.0mL) solution. After stirring at a temperature 30 ± 5 ℃ 30 minutes, and the mixture was stirred and heated to a temperature 65 ~ 75 ℃ 30 minutes. Temperature 60 ~ 70 ℃ in the benzyl alcohol (12.4g, 115mmol) it was dropped. To a temperature 80 ~ 90 ℃ was stirred and heated for 3 hours. The reaction mixture was cooled to a temperature 20 ~ 30 ℃, sodium nitrite (7.20g, 104mmol) and after stirring was added a solution prepared by dissolving in water (150mL) 1 hour, the aqueous layer was separated. The organic layer 5w / v% aqueous sodium bicarbonate solution (150mL), 20w / v% aqueous citric acid solution (150mL), washed successively with 5w / v% aqueous sodium chloride solution (150mL), the organic layer was concentrated under reduced pressure. The residue methanol (60.0mL) was added and concentrated under reduced pressure to. The more we went once in the same manner.To the residue was added methanol and the content amount of the (90.0g). Temperature 15 ~ 35 ℃ 2N- aqueous sodium hydroxide (62.6mL, 125mmol) was added and stirred for 1 hour at a temperature 30 ± 5 ℃. Temperature 20 ~ 30 ℃ in methanol (120mL), was added to 20w / v% aqueous citric acid solution (300mL), it was a pH3.0 ~ 3.5. After stirring for 30 minutes at a temperature 50 ~ 60 ℃, cooled to a temperature 20 ~ 30 ℃ and stirred for 1 hour. It was stirred for 1 hour at the temperature 0 ~ 10 ℃. The precipitated crystals were collected by filtration, and washed with water (90.0mL). And dried under reduced pressure at 50 ℃ (3R, 5S) -5 – {[(benzyloxy) carbonyl] amino} -1- (tert- butoxycarbonyl) to yield a white crystalline powder piperidine-3-carboxylic acid (35.0 g, 88.6% yield).
1 H-NMR (300 MHz, DMSO-d 6) δ 1.41 (s, 9H), 2.11 (d, J = 12.4 Hz, 1H), 2.40-2.48 (m, 4H), 2.62 (br s, 1H), 4.08 (t, J = 14.4 Hz, 2H), 5.04 (s, 2H), 7.31-7.41 (m, 5H), 12.53 (br s, 1H).
. Anal Calcd for C 19 H 26 N 2 O 6:. C, 60.30; H, 6.93; N, 7.40; O, 25.37 Found: C, 60.03; H, 6.99; N, 7.41.
10) Under a nitrogen stream, (3R, 5S) -5 – {[(benzyloxy) carbonyl] amino} -1- (tert- butoxycarbonyl) piperidine-3-carboxylic acid (30.0g, 79.3mmol), morpholine (7.60 g, 87.2mmol), 1- hydroxybenzotriazole monohydrate (2.43g, it was charged with 15.9mmol) and dimethylacetamide (90.0mL). Hydrochloride 1-ethyl at a temperature 20 ~ 30 ℃ -3- (3- dimethylaminopropyl) carbodiimide (16.7g, 87.1mmol) after addition and stirred for 1 hour at a temperature 45 ~ 55 ℃. Temperature 45 ~ 55 ℃ with tetrahydrofuran (90.0mL), sequentially dropwise addition of water (210mL), and stirred for 1 hour. After stirring for 1 hour and cooled to a temperature 20 ~ 30 ℃, were collected by filtration the precipitated crystals, tetrahydrofuran – water: washing with (1 3,120mL). And dried under reduced pressure at 50 ℃ tert- butyl piperidine -1- (3S, 5R) -3 – a white crystalline powder of {[(benzyloxy) carbonyl] amino} -5 (morpholin-4-yl-carbonyl) carboxylate It was obtained (32.7g, 92.3% yield).
1 H-NMR (300 MHz, DMSO-d 6) δ 1.41 (s, 9H), 1.49-1.57 (m, 1H), 1.87 (d, J = 12.3 Hz, 1H), 2.43 (br s, 1H), 2.63-2.71 (m, 1H), 2.79-2.83 (m, 1H), 3.37-3.54 (m, 9H), 3.89 (d, J = 11.5 Hz, 1H), 4.06 (br s, 1H), 5.03 (s , 2H), 7.30-7.38 (m, 5H).
. Anal Calcd for C 23 H 33 N 3 O 6:. C, 61.73; H, 7.43; N, 9.39; O, 21.45 Found: C, 61.59; H, 7.50; N, 9.43.
11) tert- Butyl piperidin -1- (3S, 5R) -3 – {[(benzyloxy) carbonyl] amino} -5- (morpholin-4-ylcarbonyl) carboxylate (30.0g, 67.0mmol), isobutyraldehyde (7.25g, 101mmol), it was charged with 10% Pd-C (PE type) (1.50g) and methanol (240mL).Hydrogen pressure of 0.2 ~ 0.3MPa, it was stirred for 4 hours at a temperature of 20 ~ 30 ℃. The catalyst is filtered off and washed with methanol (60.0mL). The filtrate was concentrated under reduced pressure, ethyl acetate was added (60.0mL), and concentrated under reduced pressure again. The residue ethyl acetate was added, followed by the amount of contents (360mL). Temperature 45-55 succinate by heating to ℃ (7.90g, 67.0mmol) was added. After stirring for 1 hour at a temperature 45 ~ 55 ℃, cooled to a temperature 20 ~ 30 ℃, and stirred for 1 hour. The precipitated crystals were collected by filtration, and washed with ethyl acetate (90.0mL). And dried under reduced pressure at 50 ℃ tert- butyl (3S, 5R) -3 – [(2- methyl-propyl) amino] -5- (morpholin-4-yl-carbonyl) piperidine – 1-carboxylate white crystals of alert succinate got sex powder (30.2g, 92.5% yield).
1 H-NMR (300 MHz, D 2 O) δ 1.02 (s, 3H), 1.04 (s, 3H), 1.47 (s, 9H), 1.97-2.09 (m, 2H), 2.26-2.30 (m, 1H ), 2.55 (s, 4H), 2.99 (d, J = 7.0 Hz, 2H), 3.23 (br s, 1H), 3.39-3.45 (m, 2H), 3.53-3.80 (m, 10H), 3.82-3.93 (br s, 1H).
. Anal Calcd for C 23 H 41 N 3 O 8:. C, 56.66; H, 8.48; N, 8.62; O, 26.25 Found: C, 56.48; H, 8.46; N, 8.39.
12) tert- Butyl (3S, 5R) -3 – [(2- methylpropyl) amino] -5- (morpholin-4-ylcarbonyl) piperidine – 1 – carboxylate succinate (30.3g, 62.2mmol), acetonitrile (60.0mL) and, it was charged with water (40.0mL). Then after stirring was added potassium carbonate (34.4g, 0.249mmol) 10 minutes, 1- (4-methoxybutyl) -2-trichloromethyl -1H- benzimidazole (20.0g, 62.2mmol) was added. After stirring for 2 hours at a temperature of 70 ~ 80 ℃, it was added dimethyl sulfoxide (15.0mL), and the mixture was stirred for 6 hours at a temperature 70 ~ 80 ℃. After cooling the reaction mixture to a temperature 20 ~ 30 ℃, water (120mL), it was separated and by adding toluene (240mL). The organic layer 10w / v% sodium chloride solution (100mL), 10w / v% aqueous solution of citric acid (100mL), it was washed sequentially with 10w / v% sodium chloride solution (100mL). The organic layer of activated carbon Shirasagi A a (1.0g) was added, and the mixture was stirred for 30 minutes at a temperature 20 ~ 30 ℃. Activated carbon was filtered, washed with toluene (40.0mL), and concentrated under reduced pressure of the filtrate to 110 mL. By heating to a temperature 35 ~ 45 ℃ was added dropwise heptane (280mL). At a temperature 35 ~ 45 ℃ tert- butyl (3S, 5R) -3 – [{[1- (4- methoxy-butyl) -1H- benzoimidazol-2-yl] carbonyl} (2-methylpropyl) amino] -5 – and the mixture was stirred for 1 hour at (morpholin-4-ylcarbonyl) piperidine-1-carboxylate was added to the same temperature the crystals (10mg) of the acrylate. Heptane (140mL) was stirred and added dropwise to 30 minutes at a temperature 35 ~ 45 ℃. It was cooled to a temperature 20 ~ 30 ℃ and stirred for 2 hours. The precipitated crystals were collected by filtration, toluene – heptane: was washed with (1 5,40.0mL). And dried under reduced pressure at 50 ℃ tert- butyl (3S, 5R) -3 – [{[1- (4- methoxy-butyl) -1H- benzoimidazol-2-yl] carbonyl} (2-methylpropyl) amino] – 5- (morpholin-4-ylcarbonyl) piperidine-1-carboxylate was obtained a pale yellowish crystalline powder of alert (27.7g, 74.2% yield).
1 H-NMR (300 MHz, CDCl 3) δ 0.68-0.80 (m, 3H), 0.96-1.08 (m, 3H), 1.31 (br s, 5H), 1.49 (s, 4H), 1.61-1.71 (m , 2H), 1.71 (br s, 0.5H), 1.92-2.05 (m, 3H), 2.05-2.24 (m, 2H), 2.45 (br s, 1H), 2.60 (br s, 1H), 2.72-2.96 (m, 2H), 3.26-3.35 (m, 3H), 3.35-3.47 (m, 2H), 3.47-3.73 (m, 10H), 4.02-4.26 (m, 2H), 4.26-4.34 (m, 1H) , 4.34-4.47 (m, 0.5H), 7.25-7.29 (m, 1H), 7.29-7.41 (m, 1H), 7.41-7.53 (m, 1H), 7.64 (br s, 0.5H), 7.79 (d , J = 8.2 Hz, 0.5H).
. Anal Calcd for C 32 H 49 N 5 O 6:. C, 64.08; H, 8.23; N, 11.68; O, 16.01 Found: C, 63.82; H, 8.12; N, 11.64.
PATENT
TAKEDA PHARMACEUTICAL COMPANY LIMITED [JP/JP]; 1-1, Doshomachi 4-chome, Chuo-ku, Osaka-shi, Osaka 5410045 (JP)
Provided is a method for producing a synthetic intermediate of a heterocyclic compound having a renin inhibitory activity and effective as a prophylactic or therapeutic drug against diabetic renal disease, hypertension, and the like. A method for producing a compound represented by formula (III-1a), (III-1b), (III-1c), and/or (III-1d) [where the symbols in the formulas are as defined in the description], or a salt thereof, said method characterized in that a compound represented by formula (Ia) or (Ib) [where the symbols in the formulas are as defined in the description] or a salt thereof is reacted with a compound represented by formula (II) [where the symbols in the formula are as defined in the description] or a salt thereof in the presence of an aluminum compound and a chiral amine compound.
In the above method, the acid anhydride (BANC) from chiral dicarboxylic acid monoester ((-) – BMPA) were synthesized and then the carboxylic acid after conversion and hydrolysis reaction of the Z amine by the Curtius rearrangement of the carboxylic acid (BAPC) and it was then performs amidation by the condensation reaction with the amine (morpholine), is synthesized heterocyclic amide compound (BMPC). Further, Patent Document 2, the preparation of compounds useful as synthetic intermediates of the above heterocyclic compounds are disclosed.[Formula 3]

(Wherein each symbol is as described in Patent Document 2.)
| WO2010150840A1 | 24 Jun 2010 | 29 Dec 2010 | Dainippon Sumitomo Pharma Co., Ltd. | N-substituted-cyclic amino derivative |
| WO2011158880A1 | 15 Jun 2011 | 22 Dec 2011 | Takeda Pharmaceutical Company Limited | Crystal of amide compound |
| WO2012062687A1 * | 7 Nov 2011 | 18 May 2012 | F. Hoffmann-La Roche Ag | Triazole derivatives and their use for neurological disorders |
| WO2013122260A1 | 14 Feb 2013 | 22 Aug 2013 | Takeda Pharmaceutical Company Limited | Tablet |
| CN103221402B * | 7 Nov 2011 | 17 Jun 2015 | 霍夫曼-拉罗奇有限公司 | 三唑衍生物及其用于神经障碍的用途 |
| US8329691 | 14 Oct 2008 | 11 Dec 2012 | Takeda Pharmaceutical Company Limited | Amide compounds and use of the same |
| US8389511 | 19 Dec 2008 | 5 Mar 2013 | Dainippon Sumitomo Pharma Co., Ltd. | Bicyclic heterocyclic derivative |
| US8658639 | 24 Jun 2010 | 25 Feb 2014 | Dainippon Sumitomo Pharma Co., Ltd | N-substituted-cyclic amino derivative |
| US8742097 | 2 Nov 2011 | 3 Jun 2014 | Hoffmann-La Roche Inc. | Triazole compounds I |
| US9018374 | 15 Jun 2011 | 28 Apr 2015 | Takeda Pharmaceutical Company Limited | Crystal of amide compound |
| US9090601 | 28 Jan 2010 | 28 Jul 2015 | Millennium Pharmaceuticals, Inc. | Thiazole derivatives |
///////////TAK 272, Hypertension

Millennium Pharmaceuticals, Inc. INNOVATOR
Millennium Pharmaceuticals, Inc., a subsidiary of Takeda Pharmaceutical Company Limited,
MLN4924, MLN 4924-003, TAK-924
905579-51-3 BASE
1160295-21-5 HcL
A potent and selective inhibitor of NAE. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. The ubiquitin-proteasome pathway mediates the destruction of unwanted proteins.
(((1S,2S,4R)-4-{4-[(S)-2,3-Dihydro-1H-inden-1-ylamino]-7H-pyrrolo[2,3-d]pyrimidin-7-yl}-2-hydroxycyclopentyl)methyl sulfamate hydrochloride) (pevonedistat), a novel NEDD8-activating enzyme (NAE) inhibitor, has demonstrated in vitro cytotoxic activity against a variety of human malignancies and is currently being developed by Takeda Pharmaceuticals Company Limited as a clinical candidate for the treatment of cancer
In 2011, orphan drug designation was assigned to MLN-4924 for the treatment of MDS and for the treatment of acute myelogenous leukemia.
PHASE 1…….CANCER SOLID TUMOR

………………….
PATENT
http://www.google.com/patents/US20120330013

preparing a compound represented by the following formula 1 by reacting the compound of formula 11 with TFA (step 9):


The retrosynthetic analysis of MLN4924 (1), as the final desired nucleoside, is shown in the following.

MLN 4924 (1) can be synthesized by condensing cyclic sulfate 3 as the glycosyl donor with a purine base. The glycosyl donor 3 can be produced from diol 4, which in turn can be obtained from cyclopentanone 5 via a stereoselective reduction and a regioselective cleavage of the isopropylidene moiety. The cyclopentanone 5 can be synthesized from cyclopentenone 6 by stereoselective reduction. The intermediate cyclopentenone 6 can be easily derived from D-ribose according to our previously published procedure (Jeong, L. S. et al., J. Org. Chem. 2004, 69, 2634-2636).
The synthetic route for the glycosyl donor 3 is shown in the following scheme 1.

Example 1 Preparation of MLN4924 Step 1: Preparation of 6-(tert-butyl-diphenyl-silanyloxymethyl)-2,2-dimethyl-tetrahydro-cyclopenta[1,3]dioxol-4-one (Compound 5)

To a suspension of the compound 6 (20.0 g, 47.1 mmol) in methanol (400 ml) was added 10% palladium on activated carbon (1.0 g), and the mixture was stirred at room temperature overnight under H2 atmosphere. After filtration of the reaction mixture, the solvent was removed and the residue was dissolved in methylene chloride and then filtered through short pad silica gel. Then, the solvent was evaporated to give the compound 5 (20.1 g, 100%) as a colorless syrup.
[α]20 D −28.32 (c 1.49, MeOH); HR-MS (ESI): m/z calcd for C25H32NaO4Si [M+Na]+ 447.1968, Found 447.1956; 1H NMR (400 MHz, CDCl3) δ 7.69 (m, 4H), 7.40 (m, 6H), 4.84 (t, J=4.4 Hz, 1H), 4.22 (dd, J=1.2, 4.8 Hz, 1H), 3.96 (dd, J=8.0, 10.0 Hz, 1H), 3.82 (dd, J=6.8, 10.0 Hz, 1H), 2.37 (m, 1H), 2.30 (ddd, J=1.2, 8.4, and 18.4 Hz, 1H), 2.20 (ddd, J=1.2, 12.0, and 18.4 Hz, 1H), 1.37 (s, 3H), 1.35 (s, 3H), 1.06 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 112.6, 80.5, 77.6, 77.2, 76.9, 63.6, 38.1, 36.9, 27.1, 27.02, 27.01, 25.3, 19.5; Anal. Calcd for C25H32O4Si: C, 70.72; H, 7.60. Found: C, 70.79; H, 7.75.
Step 2: Preparation of 6-(tert-butyl-diphenyl-silanyloxymethyl)-2,2-dimethyl-tetrahydro-cyclopenta[1,3]dioxol-4-ol (Compound 7)

To a suspension of the compound 5 (20.1 g, 47.1 mmol) in methanol (500 ml) were added sodium borohydride (2.17 g, 57.4 mmol) and cerium (III) chloride heptahydrate (21.3 g, 57.2 mmol) at 0° C., and the mixture was stirred at room temperature for 30 min. After the solvent was removed, the residue was partitioned between ethyl acetate and water. The organic layer was then washed with brine, dried with anhydrous MgSO4, filtered, and evaporated. The residue was purified by silica gel column chromatography (hexane/ethyl acetate=5/1) to give the compound 7 (20.86 g, 98%) as a colorless syrup.
[α]20 D +34.55 (c 0.55, MeOH); HR-MS (ESI): m/z calcd for C25H34NaO4Si [M+Na]+: 449.2124; Found: 449.2110; 1H NMR (400 MHz, CDCl3) δ 7.69 (m, 4H), 7.39 (m, 6H), 4.62 (t, J=5.6 Hz, 1H), 4.44 (t, J=5.6 Hz, 1H), 3.89 (dd, J=6.0, 7.6 Hz, 1H), 3.84 (m, 1H), 3.68 (dd, J=6.4, 10.0 Hz, 1H), 1.91 (m, 2H), 1.26 (m, 1H), 1.42 (s, 3H), 1.33 (s, 3H), 1.05 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 135.9, 135.8, 134.2, 134.1, 129.8, 129.7, 127.8, 127.7, 110.6, 79.4, 78.9, 77.6, 77.2, 76.9, 72.5, 62.9, 41.6, 33.4, 27.0, 25.9, 27.0, 25.9, 24.4, 19.5; Anal. Calcd for C25H34O4Si: C, 70.38; H, 8.03. Found: C, 70.41; H, 8.08.
Step 3: Preparation of 3-tert-butoxy-4-(tert-butyl-diphenyl-silanyloxymethyl)-cyclopentane-1,2-diol (Compound 4)

To a solution of the compound 7 (20.86 g, 47.12 mmol) in methylene chloride was added trimethylaluminum (2.0 M in toluene, 132.1 ml) at 0° C., and the mixture was stirred at room temperature for 2 days. The mixture was cooled to 0° C., slowly quenched with an aqueous saturated ammonium chloride solution, filtered, and evaporated. The residue was partitioned between ethyl acetate and water. The organic layer was washed with brine, dried with anhydrous MgSO4, filtered, and evaporated. The residue was purified by silica gel column chromatography (hexane/ethyl acetate=2/1) to give the compound 4 (13.42 g, 62%) as a colorless syrup.
[α]20 D +3.30 (c 0.55, MeOH); HR-MS (ESI): m/z calcd for C26H38NaO4Si [M+Na]+: 465.2437; Found: 465.2423; 1H NMR (400 MHz, CDCl3) δ 7.70 (m, 4H), 7.41 (m, 6H), 4.05 (dd, J=4.4, 7.2 Hz, 1H), 3.93 (m, 1H), 3.72 (m, 2H), 3.59 (dd, J=3.6, 12.0 Hz, 2H), 2.70 (d, J=20.8 Hz, 1H), 2.10 (m, 2H), 1.60 (m, 1H), 1.20 (s, 9H), 1.06 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 135.9, 133.5, 130.0, 129.9, 127.9, 127.9, 77.6, 77.2, 76.9, 74.9, 73.8, 72.7, 72.1, 63.3, 42.1, 34.0, 28.5, 27.0, 19.4; Anal. Calcd for C26H38O4Si: C, 70.55; H, 8.65. Found: C, 70.61; H, 8.70.
Step 4: Preparation of (4-tert-butoxy-2,2-dioxo-tetrahydro-2-yl-6-cyclopenta[1,3,2]-dioxathiol-5-ylmethoxy)-tert-butyl-diphenyl-silane (Compound 3)

To a solution of the compound 4 (13.42 g, 30.3 mmol) in methylene chloride were added triethyl amine (14.5 ml, 101.0 mmol) and thionyl chloride (3.7 ml, 47.4 mmol) at 0° C., and the reaction mixture was stirred at 0° C. for 10 minutes. The reaction mixture was partitioned between methylene chloride and water. The organic layer was washed with brine, dried with anhydrous MgSO4, filtered, and evaporated. The residue was purified by silica gel column chromatography (hexane/ethyl acetate=6/1) to give the cyclic sulfite (14.37 g, 97%) as a white foam.
[α]20 D +20.00 (c 0.05, MeOH); HR-MS (ESI): m/z calcd for C26H36NaO5SSi [M+Na]+: 511.1950; Found: 511.1929; 1H NMR (400 MHz, CDCl3) δ 7.64 (m, 4H), 7.40 (m, 6H), 5.23 (m, 1H), 5.04 (dd, J=4.4, 6.0 Hz, 1H), 4.01 (t, J=4.8 Hz, 1H), 3.68 (dd, J=3.6, 10.4 Hz, 1H), 3.56 (dd, J=8.0, 10.4 Hz, 1H), 2.07 (m, 2H), 1.96 (m, 1H), 1.14 (s, 9H), 1.05 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 135.8, 135.7, 133.9, 133.8, 129.9, 129.9, 127.9, 127.8, 85.7, 83.2, 77.6, 77.2, 76.9, 75.0, 71.1, 62.7, 44.7, 31.4, 28.5, 27.1, 19.4; Anal. Calcd for C26H36O5SSi: C, 63.90; H, 7.42; S, 6.56. Found: C, 63.94; H, 7.45; S, 6.61.
To a solution of the cyclic sulfite obtained above (14.37 g, 29.4 mmol) in the mixture of carbon tetrachloride, acetonitrile and water (1:1:1.5, 210 ml) were added sodium metaperiodate (18.56 g, 56.4 mmol) and ruthenium chloride (1.72 g, 8.25 mmol), and the reaction mixture was stirred at room temperature for 10 minutes. The reaction mixture was partitioned between methylene chloride and water. The organic layer was washed with brine, dried with anhydrous MgSO4, filtered, and evaporated. The residue was purified by silica gel column chromatography (hexane/ethyl acetate=4/1) to give the compound 3 (13.36 g, 90%) as a white solid.
mp 101-104° C.; [α]20 D −80.00 (c 0.05, MeOH); HR-MS (ESI): m/z calcd for C26H36NaO6SSi [M+Na]+: 527.1900; Found: 527.1881; 1H NMR (400 MHz, CDCl3) δ 7.64 (m, 4H), 7.41 (m, 6H), 5.13 (m, 1H), 4.83 (dd, J=4.4, 6.8 Hz, 1H), 4.13 (t, J=4.0 Hz, 1H), 3.92 (dd, J=6.4, 10.4 Hz, 1H), 3.69 (dd, J=5.2, 10.4 Hz, 1H), 2.11 (m, 2H), 2.02 (m, 1H), 1.15 (s, 9H), 1.05 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 135.7, 135.0, 133.8, 133.7, 130.0, 128.0, 127.9, 83.5, 82.2, 77.6, 77.2, 76.9, 75.4, 70.4, 70.4, 62.2, 43.9, 31.3, 28.2, 27.1, 26.8, 19.4; Anal. Calcd for C26H36O6SSi: C, 61.87; H, 7.19; S, 6.35. Found: C, 61.91; H, 7.14; S, 6.30.
Step 5: Preparation of 2-tert-butoxy-3-(tert-butyl-diphenyl-silanyloxymethyl)-5-[4-(indan-1-ylamino)-pyrrolo[2,3-d]pyrimidin-7-yl]-cyclopentanol (Compound 8)

A suspension of N6-indanyl-7-deazaadenine (8.80 g, 35.2 mmol), sodium hydride (1.38 g, 45.7 mmol) and 18-crown-6 (9.11 g, 45.7 mmol) in THF (200 ml) was stirred at 80° C. To the reaction mixture was added a solution for the compound 3 (13.36 g, 26.5 mmol) in THF (150 ml), and the stirring was continued at 80° C. overnight. The reaction mixture was cooled down to 0° C., and conc. HCl was added slowly until pH reaches 1-2. Then the reaction mixture was further stirred at 80° C. for 2 hours. After neutralized with saturated aqueous NaHCO3 solution, the reaction mixture was partitioned between ethyl acetate and water. The organic layer was washed with brine, dried with anhydrous MgSO4, filtered, and evaporated. The residue was purified by silica gel column chromatography (hexane/ethyl acetate=2/1) to give the compound 8 (11.62 g, 65%) as a white foam.
UV (CH2Cl2) λmax 272.5 nm; [α]20 D −8.89 (c 0.45, MeOH); HR-MS (ESI): m/z calcd for C41H51N4O3Si [M+H]+: 675.3730; Found: 675.3717; 1H NMR (400 MHz, CDCl3) δ 8.38 (s, 1H), 7.70 (m, 4H), 7.41 (m, 6H), 6.92 (d, J=3.6 Hz, 1H), 6.29 (d, J=3.2 Hz, 1H), 5.91 (dd, J=7.6, 14.8 Hz, 1H), 5.14 (br d, J=6.8 Hz, 1H), 4.77 (m, 1H), 4.36 (t, J=6.0 Hz, 1H), 4.22 (dd, J=5.2, 10.8 Hz, 1H), 3.84 (dd, J=5.6, 10.4 Hz, 1H), 3.73 (dd, J=8.4, 10.4 Hz, 1H), 3.37 (d, J=5.6 Hz, 1H), 3.06 (m, 1H), 2.95 (m, 1H), 2.75 (m, 1H), 2.75 (m, 1H), 2.58 (m, 1H), 2.38 (m, 1H), 2.15 (m, 1H), 1.98 (m, 1H), 1.65 (s, 1H), 1.55 (s, 1H), 1.16 (s, 9H), 1.07 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 156.4, 151.8, 150.3, 144.1, 143.8, 135.9, 134.0, 129.9, 128.2, 127.9, 127.9, 127.0, 125.1, 124.4, 123.3, 103.8, 97.4, 77.8, 77.6, 77.2, 76.9, 74.9, 72.4, 63.5, 62.1, 56.3, 43.9, 34.9, 30.5, 30.5, 28.5, 27.2, 19.5; Anal. Calcd for C41H50N4O3Si: C, 72.96; H, 7.47; N, 8.30. Found: C, 73.01; H, 7.45; N, 8.36.
Step 6: Preparation of {7-[3-tert-butoxy-4-(tert-butyl-diphenyl-silanyloxymethyl)-cyclopentyl]-7H-pyrrolo[2,3-d]pyrimidin-4-yl}-indan-1-yl-amine (Compound 9)

To a solution of the compound 8 (11.62 g, 17.2 mmol) in methylene chloride (300 ml) were added N,N-dimethylaminopyridine (5.64 g, 51.6 mmol) and phenyl chlorothionocarbonate (4.3 ml, 34.4 mmol), and the reaction mixture was stirred at room temperature overnight. After the solvent was removed, the residue was purified by silica gel column chromatography (hexane/ethyl acetate=6/1) to give the thiocarbonate (13.82 g, 99%) as a white foam.
UV (MeOH) λmax 271.50 nm; [α]20 D +10.00 (c 0.15, MeOH); HR-MS (ESI): m/z calcd for C48H55N4O4SSi [M+H]+: 811.3713; Found: 811.3687; 1H NMR (400 MHz, CDCl3) δ 8.36 (s, 1H), 7.61 (dd, J=1.6, 7.6 Hz, 4H), 7.34 (m, 5H), 7.26 (m, 4H), 7.18 (m, 6H), 6.86 (s, 1H), 6.25 (d, J=3.2 Hz, 1H), 6.00 (dd, J=3.2, 8.4 Hz, 1H), 5.83 (d, J=6.8 Hz, 1H), 5.19 (m, 1H), 5.07 (br s, 1H), 4.48 (t, J=3.6 Hz, 1H), 3.82 (dd, J=7.2, 10.4 Hz, 1H), 3.52 (dd, J=7.2, 10.0 Hz, 1H), 2.99 (m, 1H), 2.88 (m, 2H), 2.69 (m, 2H), 2.18 (dd, J=11.2, 13.6 Hz, 1H), 1.94 (m, 2H), 1.12 (s, 9H), 0.98 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 194.9, 153.5, 152.1, 143.9, 135.9, 135.8, 134.1, 129.9, 129.6, 128.3, 127.9, 127.0, 126.7, 125.1, 124.6, 123.2, 122.0, 87.9, 77.6, 77.2, 76.9, 74.6, 70.4, 63.5, 57.3, 42.8, 35.0, 30.7, 30.5, 29.9, 28.7, 27.1, 19.4; Anal. Calcd for C48H54N4O4SSi: C, 71.08; H, 6.71; N, 6.91; S, 3.95. Found: C, 71.14; H, 6.75; N, 6.95; S, 4.01.
To a solution of the thiocarbonate obtained above (13.82 g, 17.0 mmol) in toluene (200 ml) were added tri-n-butyltinhydride (9.4 ml, 34.1 mmol) and 2,2′-azo-bis-isobutyronitrile (4.32 g, 26.3 mmol), and the reaction mixture was stirred at 110° C. for 1 hour. After the mixture was cooled down, the solvent was removed. The resulting residue was purified by silica gel column chromatography (hexane/ethyl acetate=3/1) to give the compound 9 (9.21 g, 82%) as a white foam.
UV (MeOH) λmax 272.50 nm; [α]20 D −10.00 (c 0.20, MeOH); HR-MS (ESI): m/z calcd for C41H51N4O2Si [M+H]+: 659.3781; Found: 659.3757; 1H NMR (400 MHz, CDCl3) δ 8.41 (s, 1H), 7.69 (m, 4H), 7.41 (m, 6H), 7.29 (m, 2H), 7.23 (m, 2H), 6.92 (d, J=3.6 Hz, 1H), 6.31 (d, J=3.6 Hz, 1H), 5.90 (dd, J=7.2, 14.8 Hz, 1H), 5.38 (m, 1H), 5.15 (br s, 1H), 4.33 (dd, J=5.2, 8.4 Hz, 1H), 3.88 (dd, J=6.4, 10.0 Hz, 1H), 3.68 (dd, J=7.2, 10.4 Hz, 1H), 3.05 (m, 1H), 2.96 (dd, J=7.6, 15.6 Hz, 1H), 2.76 (m, 1H), 2.45 (d, J=5.2 Hz, 1H), 2.29 (m, 2H), 2.06 (m, 1H), 1.95 (m, 2H), 1.55 (s, 1H), 1.13 (s, 9H), 1.06 (s, 9H);13C NMR (100 MHz, CDCl3) δ 156.3, 151.9, 144.1, 143.9, 135.9, 135.8, 134.3, 129.8, 128.2, 127.8, 127.0, 125.1, 124.6, 121.8, 77.6, 77.2, 76.7, 73.5, 72.2, 63.6, 56.4, 52.8, 46.8, 42.8, 34.9, 34.5, 30.5, 28.6, 27.2, 28.7, 19.4; Anal. Calcd for C41H50N4O2Si: C, 74.73; H, 7.65; N, 8.30. Found: C, 74.79; H, 7.61; N, 8.25.
Step 7: Preparation of 2-tert-butoxy-4-[4-(indan-1-ylamino)-pyrrolo[2,3-d]pyrimidin-7-yl]-cyclopentanol (Compound 10)

To a solution of the compound 9 (9.21 g, 13.97 mmol) in the mixture of THF and pyridine (1:1, 160 ml) was added dropwise pyridine hydrofluoride (18.42 ml, 190.0 mmol) at 0° C., and the reaction mixture was stirred at room temperature for 1 hour. The mixture was neutralized with saturated aqueous NaHCO3 solution and partitioned between ethyl acetate and water. The organic layer was washed with brine, dried with anhydrous MgSO4, filtered, and evaporated. Then, the residue was purified by silica gel column chromatography (hexane/ethyl acetate=1/3) to give the compound 10 (5.63 g, 99%) as a white foam.
UV (MeOH) λmax 273.00 nm; [α]20 D −6.36 (c 1.10, MeOH); HR-MS (ESI): m/z calcd for C25H33N4O2 [M+H]+: 421.2604; Found: 421.2599; 1H NMR (400 MHz, CDCl3) δ 8.34 (s, 1H), 7.30 (d, J=7.6 Hz, 1H), 7.22 (d, J=7.2 Hz, 2H), 7.15 (t, J=6.8 Hz, 1H), 6.88 (d, J=3.2 Hz, 1H), 6.23 (d, J=3.6 Hz, 1H), 5.83 (dd, J=7.2, 15.2 Hz, 1H), 5.28 (m, 1H), 5.06 (m, 1H), 4.47 (dd, J=5.6, 10.4 Hz, 1H), 3.78 (m, 1H), 3.70 (m, 1H), 3.24 (t, J=5.2 Hz, 1H), 2.98 (m, 1H), 2.87 (m, 1H), 2.68 (m, 1H), 2.46 (m, 1H), 2.37 (m, 2H), 1.93 (m, 2H), 1.18 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 156.2, 151.8, 147.9, 143.9, 143.9, 128.3, 126.9, 125.1, 124.5, 121.9, 97.7, 77.6, 77.2, 76.9, 75.5, 74.9, 63.4, 56.4, 53.8, 44.2, 42.2, 34.9, 33.2, 30.5, 28.6; Anal. Calcd for C25H32N4O2: C, 71.40; H, 7.67; N, 13.32. Found: C, 71.46; H, 7.60; N, 13.35.
Step 8: Preparation of sulfamic acid 2-tert-butoxy-4-[4-(indan-1-ylamino)-pyrrolo[2,3-d]pyrimidin-7-yl]-cyclopentylmethyl ester (Compound 11)

Preparation of 2.0 M solution of chlorosulfonamide in acetonitrile: Formic acid (14.15 ml, 166.0 mmol) was added dropwise to chlorosulfonyl isocyanate (32.0 ml, 162.5 mmol) under nitrogen atmosphere at 0° C. When the addition was completed, the mixture was solidified. To the mixture was added acetonitrile (61.3 ml), and the resulting solution was left to stand under nitrogen source at room temperature overnight.
To a solution of the compound 10 (5.63 g, 13.83 mmol) and triethyl amine (9.7 ml, 0.74 mmol) in acetonitrile (278 ml) was added 2.0 M solution of chlorosulfonamide in acetonitrile (13.83 ml, 27.76 mmol) at 0° C., and the reaction mixture was stirred at room temperature for 45 minutes. Additional 2.0 M chlorosulfonamide solution in acetonitrile (13.83 ml, 27.76 mmol) was added and the mixture was stirred at room temperature for 15 minutes. The reaction was quenched with methanol, and the solvent was removed. The residue was purified by silica gel column chromatography (methylene chloride/methanol=20/1) to give the compound 11 (6.37 g, 92%) as a white foam.
UV (MeOH) λmax 273.00 nm; [α]20 D −18.00 (c 0.50, MeOH); HR-MS (ESI): m/z calcd for C25H34N5O4S [M+H]+: 500.2332; Found: 500.2331; 1H NMR (400 MHz, CDCl3) δ 8.38 (s, 1H), 7.36 (d, J=7.2 Hz, 1H), 7.29 (d, J=7.2 Hz, 1H), 7.22 (m, 2H), 6.95 (d, J=3.6 Hz, 1H), 6.31 (d, J=3.2 Hz, 1H), 5.89 (d, J=6.4 Hz, 1H), 5.10 (s, 2H), 4.41 (m, 2H), 4.26 (m, 1H), 3.05 (m, 1H), 2.94 (m, 1H), 2.76 (m, 2H), 2.27 (m, 3H), 2.06 (m, 1H), 1.97 (m, 1H), 1.76 (br s, 1H); 13C NMR (100 MHz, CDCl3) δ 156.4, 151.9, 149.9, 143.9, 143.8, 128.3, 126.9, 125.1, 124.5, 121.9, 121.9, 103.5, 97.9, 77.4, 77.2, 76.9, 74.3, 71.9, 71.3, 56.4, 53.1, 49.0, 42.3, 34.9, 34.3, 30.5, 28.6; Anal. Calcd for C25H33N5O4S: C, 60.10; H, 6.66; N, 14.02; S, 6.42. Found: C, 60.15; H, 6.71; N, 13.98; S, 6.39.
Step 9: Preparation of sulfamic acid 2-hydroxy-4-[4-(indan-1-ylamino)-pyrrolo[2,3-d]pyrimidin-7-yl]-cyclopentylmethyl ester (Compound 1)

A solution of the compound 11 (6.37 g, 12.72 mmol) in 70% trifluoroacetic acid (149.24 ml) was stirred at room temperature for 2 hours. The solvent was removed and the residue was purified by silica gel column chromatography (hexane/ethylene acetate=1/2) to give the compound 1 (5.08 g, 90%) as a white foam. BASE
UV (MeOH) λmax 279.50 nm; [α]20 D −6.41 (c 2.34, MeOH);
HR-MS (ESI): m/z calcd for C21H26N5O4S [M+H]+: 444.1705; Found: 444.1706;
1H NMR (400 MHz, CD3OD) δ 8.17 (d, J=1.6 Hz, 1H), 7.25 (m, 2H), 7.18 (m, 2H), 6.64 (d, J=3.6 Hz, 1H), 5.86 (t, J=7.6 Hz, 1H), 5.46 (m, 1H), 4.49 (d, J=2.8 Hz, 1H), 3.07 (m, 1H), 2.92 (m, 1H), 2.80 (m, 1H), 2.64 (m, 1H), 2.35 (m, 1H), 2.25 (m, 2H), 2.03 (m, 2H);
13C NMR (100 MHz, CD3OD) δ 152.1, 145.3, 144.6, 128.8, 127.6, 125.7, 125.2, 122.6, 100.5, 73.1, 70.9, 56.9, 54.0, 44.8, 43.6, 34.9, 34.6, 31.1;
Anal. Calcd for C21H25N5O4S: C, 56.87; H, 5.68; N, 15.79; S, 7.23. Found: C, 56.91; H, 5.73; N, 15.82; S, 7.26.
…………………….
http://www.google.com/patents/WO2010132110A1?cl=en
((lS,2S,4R)-4-{4-[(lS)-2,3-dihydro-lH-inden-l-ylamino]-7H-pyrrolo[2,3-d]pyrimidin-7-yl }-2-hydroxycyclopentyl)methyl sulfamate (//) is described in Intl. App. Pub. No. WO 07/092213, U.S. App. Pub. No. 2007/0191293, and U.S. App. Pub. No. 2009/0036678. The potassium salt of ((lS,2S,4R)-4-{4-[( 1 S)-2,3-dihydro- 1 H-inden- 1 -ylamino]-7H-pyrrolo[2,3-d]pyrimidin-7-yl } -2-hydroxycyclopentyl)methyl sulfamate is disclosed in Intl. App. Pub. No. WO 07/092213 and U.S. App. Pub. No. 2007/0191293.

(H)
((lS,2S,4R)-4-{4-[(lS)-2,3-dihydro-lH- inden-l-ylamino]-7H-pyπOlo[2,3-d]pyrimidin-7-yl}-2-hydroxycyclopentyl)methyl sulfamate (/):

Step 3: Synthesis of ((lS,2S.4R)-4-(4-r(lS)-2,3-dihydro-lH-inden-l-ylaminol-7H-pyrrolor2.3-dlpyrimidin-7-yl}-2-hvdroxycvclopentyl)methyl sulfamate hydrochloride Form 1
[0158] A reactor was charged with ((lS,2S,4R)-4-{4-[(lS)-2,3-dihydro-lH-inden-l-ylarnino]-7H-pyrrolo[2,3-d]pyrimidin-7-yl }-2-hydroxycyclopentyl)methyl sulfamate (13.4 Kg, 30.2 mol) and 200-proof ethanol (106.2 Kg). The mixture was heated to reflux to afford a clear solution. The mixture was cooled to 50 ± 5 0C and passed through a cartridge filter. 200 proof ethanol (8.9 Kg) was used to rinse the filter. 1.27M hydrogen chloride in ethanol (10.2 Kg) was added via a cartridge filter at a rate to maintain a temperature of 50 ± 5 0C. The mixture was then seeded with Form 1 (67 g). Further 1.27M HCl (10.2 Kg) was added via a cartridge filter at a rate to maintain a temperature of 50 ± 5 0C. The mixture was then stirred at 50 ± 5 0C for about 3 hours. The mixture was then cooled to 20 ± 5 0C over about 3 hours and then stirred for about 2.5 hours. The solid product was then isolated by filtration and washed with 200-proof ethanol (I x 20.4 Kg and 1 x 21.2 Kg). The solids were dried by aspiration on the filter until no supernatant was seen to be collected, and then further dried under reduced pressure at <30 0C to afford the title compound (12.2 Kg) as a white solid determined to be Form 1 by XRPD. IH NMR (300MHz, DMSO, δ): 9.83 (s, IH), 8.34 (s, IH), 7.62 (s, IH), 7.44 (s, 2H), 7.30 (m, 3H), 7.22 (t, IH), 7.07 (s, IH), 5.86 (dd, IH), 5.42 (m, IH), 4.32 (m, IH), 4.21 (dd, IH), 4.02 (dd, IH), 3.04 (m, IH), 2.88 (m, IH), 2.67 (m, 2H), 2.15 (m, 2H), 2.08 (m, 2H), 1.94 (m, IH). XRPD data for Form 1 is shown in FIGURE 1 and Table 1; DSC data is shown in FIGURE 2, and TGA data for Form 1 is shown in FIGURE 3.
…………..
http://www.google.com/patents/WO2007092213A2?cl=en
Example 70: Diastereoisomeric mixture of (lS/2R/4R)-4-{4-[(lS)-2/3-dihydro-lH-inden-l- ylaimnol-ZH-pyrrolop^-dlpyxirnidin-Z-ylJ^-hydroxycyclopentyl s ulf amate and (lRf2S/4S)-4-{4-[(lS)-2,3-dihydro-lH-inden-l-ylaminol-7H-pyrrolo[2,3d]- pyrimidin-7-yl}-2-hydroxycyclopentyl sulfamate (Compounds 1-77 and 1-78)

Step a: Cyclopent-3-en-l-yl methanesulfonate
[0335] 3-Cydopentene-l-ol (0.500 g, 5.94 mmol) was stirred in DCM (95 mL).
Pyridine (2.40 mL), N,N-dimethylaminopyridine (0.10 g, 1.00 mmol) and methanesulfonyl chloride (0.690 mL, 8.92 mmol) were added, and the reaction mixture was stirred at 350C for 4 h. N,N-Dimethylarrιinopyridirιe (0.14 g, 1.2 mmol) and methanesulfonyl chloride (0.69 mL, 8.92 mmol) were added, and the reaction was stirred overnight. TLC indicated complete conversion. The reaction mixture was cooled and concentrated. The residue was purified by silica gel chromatography, eluting with DCM, to afford the title compound as a clear oil (0.660 g, 68%).
Step b: 7-Cyclopent-3-en-l-yl-N-r(lSV2,3-dihydro-lH-inden-l-yn-7H-pyrrolor2,3-rfl- pyrmτidin-4-arnine
[0336] N-[(lS)-2,3-DihydrcHlH-mden-l-yl]-7H-pyrrolo[2/3-d]p3αimidin-4-amine (1.32 g, 5.29 mmol) was azeotroped with toluene and placed under high vacuum for 30 min. N,N-Dimethylformamide (17.7 mL) was added, followed by cesium carbonate (1.99 g, 6.10 mmol). The mixture was stirred at 700C for 10 min. Cyclopent-3-en-l-yl methanesulfonate (0.660 g, 4.07 mmol) in N,N-dimethylformarnide (12.6 mL) was added dropwise. The reaction mixture was heated to 1100C for 1 h. The reaction mixture was cooled, quenched with brine and diluted with H2O. The aqueous layer was extracted with EtOAc (3x), washed with H2O and brine, dried (Na2SO4), filtered, and concentrated. The residue -was purified by via silica gel chromatography, eluting with a gradient of 0 to 5% MeOH in DCM followed by 25 to 50% EtOAc in hexanes, to afford the title compound as a pale brown solid (0.684 g, 53%). LC/MS: R1 = 1.38 min, ES+ 317 (FA standard). Step c: (lR,2S,45)-4-{4-r(lS)-2,3-dihydro-lH-inden-l-ylaininol-7H-pyrrolof2.3- rf1pyrimidin-7-yl}cyclopentane-l,2-diol
[0337] 7-Cyclopent-3-en-l-yl-N-[(lS)-2^-dihyrdo-lH-inden-l-yl]-7H-pyrrolo[2,3- d]pyτimidin-4-amine (0.312 g, 0.986 mmol) was stirred in tert-butyl alcohol (4.9 mL) and H2O (4.9 mL). AD-mix-α (Sigma- Aldrich, 1.4 g) was added, and the suspension was stirred at rt overnight. TLC indicated complete conversion. The reaction was quenched with sodium sulfite (1.48 g, 11.7 mmol), and the mixture was stirred for 5 h. The reaction mixture was diluted with EtOAc and H2O, and the aqueous layer was extracted with EtOAc (2x). The organic layer was dried (Na2SO4), filtered, and concentrated. The residue was purified via silica gel chromatography, eluting with EtOAc, to afford the title compound as a white solid (0.190 g, 55%).
Step d: Diastereoisomeric mixture of (lS,2R,4R)-4-{4-r(15)-23-dihydro-lH-inden-l- ylarninoi^jH-pyrrolofΣ^dlpyrirnidin-y-yll-l-hydroxycyclopentyl sulfamate and (lR,2S,4S)-4-{4-iαSV2,3-dihydro-lH-inden-l-ylarninol-7H-pyrrolor2,3- rf1pyrimidm-7-yl)-2-hydroxycyclopenryl sulfamate (Compounds 1-77 and 1-78)
[0338] (lR,2S,4S)-4-{4-[(lS)-2,3-Dihydro-lH-inden-l-ylarnino]-7H-pyrrolo[2/3- d]pyrimidin-7-ylJcyclopentane-l,2-diol (0.080 g, 0.23 mmol) was azeotroped with toluene and then was dissolved in anhydrous acetonitrile (2.3 mL). Pyridine (0.0369 mL, 0.458 mmol) was added. The reaction mixture was cooled to 00C, and a 2N solution of chlorosulfonamide in acetonitrile (0.144 mL) was added dropwise. The reaction was stirred for 1 h, and then additional 2N chlorosulfonamide in acetonitrile (0.028 mL) was added. After 30 min, additional 2N chlorosulfonamide in acetonitrile (0.0342 mL) was added, and the reaction mixture was stirred for 2 h. The reaction was quenched with methanol, and the mixture was concentrated in vacuo. The residue was purified by preparative thin layer chromatography using DCM:AcCN:MeOH (50:45:5). The relevant band was cut, washed with acetone, filtered, and concentrated to give a mixture of diastereomers as a white solid. (11 mg, 11%). 1H NMR (CDCl3, 400 NMR, δ): 8.36-8.27 (m, IH); 7.38-7.09 (m, 5H); 6.90-6.80 (m, IH); 6.36- 6.20 (m, IH); 5.95-5.76 (m, IH); 5.51-5.22 (m, 2H); 4.83-4.68 (m, IH); 3.87-3.72 (m, IH); 3.12- 2.83 (m, 2H); 2.75-2.53 (m, IH); 2.50-2.14 (m, 2H); 2.08-1.79 (m, 2H) ppm. LC/MS: R, = 1.16 min, ES+ 430 (FA standard).
…………
WO 2012061551
http://www.google.im/patents/WO2012061551A1?cl=en
The compound ((lS,2S,4R)-4-(4-((lS)-2,3-dihydro-lH-inden-l-ylamino)-7H-pyrrolo[2,3-d]- pyrimidin-7-yl)-2-hydroxycyclopentyl)methyl sulfamate:

also known as MLN4924, is an inhibitor of NEDD8-activating enzyme (NAE). Inhibition of NAE has been shown to induce cancer cell death and inhibit the growth of tumors in xenograft models. See, e.g., T.A. Soucy et al., Nature, 2009, 458, 732-737; T.A. Soucy ei al., Clin. Cancer Res., 2009, 15 (12), 3912-3916; and J.E. Brownell et al., Mol. Cell., 2010, 37 (1), 102-111, each of which is hereby incorporated by reference herein in its entirety. MLN4924, pharmaceutical compositions of MLN4924, processes for its synthesis, and polymorphic forms have been described previously. See, e.g., US Patent Appl. Nos. 11/700,614 (Publ. No. 2007/0191293), 12/221,399 (Publ. No. 2009/0036678) and 12/779,331 (Publ. No. 2011/0021544),
……………

A practical synthesis of a novel NEDD8-activating enzyme (NAE) inhibitor pevonedistat (MLN4924) is described. Key steps include an enantioselective synthesis of an amino-diol cyclopentane intermediate containing three chiral centers and a novel, regioselective sulfamoylation using N-(tert-butoxycarbonyl)-N-[(triethylenediammonium)sulfonyl]azanide. The linear process, involving six solid isolations, has been carried out in multiple cGMP productions on 15–30 kg scale to produce pevonedistat in 98% (a/a) chemical purity and 25% overall yield.

((1S,2S,4R)-4-(4-(((S)-2,3-Dihydro-1H-inden-1-yl)amino)-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-2-hydroxycyclopentyl)methyl Sulfamate (1)
((1S,2S,4R)-4-(4-(((S)-2,3-Dihydro-1H-inden-1-yl)amino)-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-2-hydroxycyclopentyl)methyl Sulfamate·Hydrochloride (Pevonedistat)

MLN4924 (1), which is in clinical trials as an anticancer agent, was stereoselectively synthesized from d-ribose via a route involving stereoselective reduction, regioselective cleavage of an isopropylidene moiety, and selective displacement of a cyclic sulfate moiety as key steps.
Sulfamic Acid 2-Hydroxy-4-[4-(indan-1-ylamino)pyrrolo[2,3-d]pyrimidin-7-yl]cyclopentylmethyl Ester (1) BASE



| WO2012061551A1 * | Nov 3, 2011 | May 10, 2012 | Millennium Pharmaceuticals, Inc. | Administration of nedd8-activating enzyme inhibitor |
| WO2013028832A2 * | Aug 23, 2012 | Feb 28, 2013 | Millennium Pharmaceuticals, Inc. | Inhibitors of nedd8-activating enzyme |
| WO2013028832A3 * | Aug 23, 2012 | May 2, 2013 | Millennium Pharmaceuticals, Inc. | Inhibitors of nedd8-activating enzyme |
| US8809356 | Aug 23, 2012 | Aug 19, 2014 | Millennium Pharmaceuticals, Inc. | Inhibitors of NEDD8-activating enzyme |
| Reference | ||
|---|---|---|
| 1 | * | Lee, et. al., Journal of Organic Chemistry (2011), 76(9), 3557-3561. |
1H NMR PREDICT

13 C NMR


//////////Pevonedistat, MLN4924, Millennium Pharmaceuticals, TAKEDA, TAK-924 , PHASE 1, orphan drug designation
Takeda’s flagship experimental cancer drug ixazomib is a giant leap closer to being filed with regulatory authorities around the globe for multiple myeloma, after turning in a solid performance in late-stage trials.
by

Rainer studied chemistry and economics in Germany (Bonn, Marburg) and did postdoctoral work at Stanford University (CA, USA). He held various positions (R&D, Market Research, Marketing, Strategic Planning, Sourcing, etc) while working at Rütgers, Novartis, Syngenta, SK, Clariant, Archimica. His international background (he worked out of Belgium, Germany, Italy, Switzerland and the USA) combined with broad experience gained in China and India (he audited more than 200 companies) is the foundation of CAP INTELLIGENCE.
Dr Rainer Steinbach of CAP Intelligence profiles Japan’s most global drugs firm
Takeda is the largest Asian pharmaceuticals company. The company started as early as 1977 to establish major co-operations with Western firms. As a result, it has the most global orientation amongst Japanese pharmaceutical companies. The latest major acquisition, Nycomed, is part of this globalisation strategy.
History
Takeda dates back to 1781, when Chobei Takeda started selling Japanese and Chinese traditional medicines. In 1895, the firm started the first production of pharmaceuticals in Osaka. Research activities started in 1914 and in 1944 fermentation activities were added.
In 1981, the antibiotics Takesulin and Pansporin were launched in Japan. In 1985, Takeda formed TAP Pharmaceuticals, a 50:50 joint venture (JV) in the US with Abbott Laboratories. TAP began marketing the prostate cancer treatment leuprorelin (Lupron) in the same year. 1997 saw the launch in Europe of candesartan celexetil (Blopress/Kensen), an anti-hypertensive agent which is also marketed by AstraZeneca.
In 2008, Takeda acquired Millennium Pharmaceutical of Cambridge, Massachusetts, an oncology research specialist, for $8.8 billion. Larger still was the acquisition of Swiss company Nycomed for €9.6 billion ($13.3 billion) in 2011, not including Nycomed’s US-based dermatological business. Nycomed had itself grown substantially by acquiring the pharmaceuticals interests of Altana in Germany and Bradley Pharmaceuticals in 2007.
The acquired parts of Nycomed had revenues of about €2.84 billion in 2011 and a workforce of about 11,800 employees, plus production locations in 11 countries worldwide. Its revenues were mainly in Europe (48%), Russia (17%) and Latin America (13%), plus other emerging markets. The acquisition also gained Takeda access to romiflumast (Daliresp), a new drug against chronic obstructive pulmonary disease.
In 2012, Takeda acquired URL Pharma, a privately owned company headquartered in Philadelphia and employing about 500, for an upfront payment of $800 million and future performance-based contingent earn out payments. URL’s 2011 revenues amounted of nearly $600 million, over two thirds coming from colchicine (Colcrys), which is used to treat and prevent gout flares. URL Pharma was sold to Sun Pharma in January 2013.

Table 1 – Locations of Takeda sites
Structure
Takeda is a public share company that is listed at the Tokyo and Osaka stock exchange, with the ticker symbol 4502. The main shareholders are financial institutions (33%), foreign investors (30%), some 280,000 individuals (27%) security companies and others (10%). The three biggest single shareholders are Nippon Life Insurance, with 7.1%, Japan Trustee Services Bank (4.4%) and the Master Trust Bank of Japan (4.3%).
The company is headquartered in Osaka, with its European headquarters in London and the American one headquarters Deerfield, Illinois. It has 17 manufacturing sites and three JV manufacturing sites, the most important of which are listed in Table 1. It employs a global workforce of about 30,500. Regional data about this are not published, apart from in Japan itself. CAP Intelligence estimates the workforce split as 31% in Japan, 30% in Europe, 25% in the USA and 14% in the rest of the world.

As of today, the Takeda Group has 61 consolidated companies and 14 affiliates. Major subsidiaries include: Takeda Nycomed Pharmaceuticals, Takeda Europe Holdings (Amsterdam), Takeda USA Holdings (New York), Millennium Chemicals (Cambridge, Massachusetts), Nihon Pharmaceutical, Wako Pure Chemicals and Mizuzawa Industrial Chemicals (all Japan) and Tianjin Takeda Pharmaceuticals (China).
Takeda’s financial year starts on April 1 and ends on March 31. The company’s financial results are reported in Yen, but are given here in US dollars for ease of comparison with other profiled firms. Figure 1 shows revenues, EBITDA, operating income and net earnings for the years since 2002.
In the year to March 2013, earnings fell by 13.3% to $16.57 billion, but EBITDA was up by 6.1% to $5.52 million and operating income was 27.4% up to $4.28 billion while earnings more than doubled to $3.48 million. Consequently, the net profit margin shot up from the 8% mark in the previous two years to 21.2%.

Figure 1 – Takeda’s revenues & profits ($ billion), 2011-2013 fiscal years
Main activities
Takeda is overwhelmingly focused on ethical products, which account for 90% of its revenues. Within this, cardiovascular and metabolic therapies account for 74%, followed by oncology with 13.5% and inflammatory with the other 2.5%. The remainder of its revenues is split between consumer healthcare products, including cold remedies and vitamin-containing products (4%), and others (6%). The company’s five best selling products account for close to $10 billion in sales, more than half of the total (Figure 2).
Sales are 47% in Japan, 23% in North America, 16% in Europe, 4% each in the rest of Asia, Russia and the CIS and Latin America and 2% in the rest of the world. As the Japanese market will not deliver major growth opportunities, the company’s objective is to be strongly present in emerging markets, especially China and Russia. Sales and marketing efforts have been intensified to this end. In fiscal 2012, Takeda all started an e-commerce website for direct selling in Japan called the Takeda Online Shop as part of the consumer healthcare business.
Takeda’s stated vision is “to embody global pharmaceutical leadership through innovation, culture, and growth, guided by an unwavering commitment to significantly improve the lives of patients”. Its strategy includes:
Takeda is one of the few Japanese pharmaceuticals companies that have a truly global presence. Because of this, in addition to its Japanese competitors, such as Astellas, Eisai, Mitsubishi, Otsuka, Shionogi, Taiho and Teijin, it also competes with all the major international companies, including generics companies. The strongest competitors in its main area of cardiovascular and metabolic drugs are AstraZeneca, Bayer, Bristol-Myers Squibb (BMS), Boehringer Ingelheim (BI), Daiichi Sankyo, Dainippon Sumitomo, Merck & Co., Novartis, Pfizer and Sanofi.

Table 2 – Takeda’s leading brands by sales, 2010-2013 fiscal years
R&D structure
About 6,000 Takeda employees work in R&D. In 2012-2013, the company invested more than $3.9 billion in R&D, almost 21% of total revenues. It has R&D sites in: Osaka and Fujisawa in Japan; Palo Alto, San Diego, Deerfield, Cambridge, Bozeman and Fort Collins in the US; Cambridge and London, UK; Roskilde, Denmark; Konstanz, Germany; Singapore; Guangzhou, China; and, Sao Jerônimo in Brazil.
Following the opening of the new drug discovery research centre, the Shonan Research Centre in Osaka, a new R&D structure was implemented in early 2011, creating ‘Drug Discovery Units (DDUs)’, with research functions around each of the four core research activities of metabolic diseases, oncology, CNS-related diseases and inflammatory diseases.
In addition, R&D alliances continue to form a key part of Takeda’s strategy. This has included alliances with Advinus Therapeutics in India, Seattle Genetics, Sage Bionetworks, Xoma and Zinfandel Pharma in the US and BC Cancer Agency in Canada. The company has stated that R&D expenditure over the next three years will be divided as follows between different therapeutic areas: oncology 31%, cardiovascular and metabolic 27%, CNS 14%, immunology and respiratory 12%, general medicine and vaccines 16%.
Clinical development
As of July 2013, Takeda had more than 40 products in clinical development, with the main emphasis on cardiovascular and metabolic indications and oncology. These comprised 14 compounds in Phase I, six (all NMEs) in Phase II and 12 (including seven NMEs) in Phase III.
Eight products, including three NMEs, had been submitted for approval and submissions had been filed for: vedolizumab, a monoclonal antibody developed to treat Crown’s disease and ulcerative colitis (Figure 2a); vortioxetine an anti-depressant co-developed with Lundbeck to treat generalised anxiety disorder; and, BLB-750, a vaccine developed to prevent pandemic influenza. Amongst the developmental drugs in Phase III are:

Figure 3 – Pipeline drugs at Takeda
Key products
The main market products from Takeda have already been listed above in Table 2. The five most important by sales in the most recent fiscal year are as follows in alphabetical order, with the generic name first and the brand name in brackets after. Further information about the rest of the portfolio is available from CAP Intelligence.
Bortezomib (Velcade, Figure 3a) belongs to the class of targeted intra-cellular tumour therapeutics. It was the first therapeutic protease inhibitor ever approved and was originally developed by Myogenics, a company that was sold to Leukosite. This firm was in turn acquired by Millennium, which ultimately became part of Takeda.
Amongst others, bortezomib is approved against multiple myeloma and mantle cell lymphoma. Chemically, it is an N-protected dipeptide. The protection group contains a boron atom which binds the catalytic site of the 26S proteasome that regulates protein expression. Bortezomib is co-marketed with Johnson & Johnson (J&J) under the same trade name Velcade. Pharmstandard markets it in Russia.
Depending on the specific indication multiple myeloma, competing drugs include: other targeted tumour therapeutics, such as lenalidomide (Revlimid), pomalidomide (Pomast) and thalidomide (Thalidomide), all by Celgene; enzyme inhibitors, such as carfilzomib (Kyprolix by Onyx); and, topomerase inhibitors, such as doxorubicin (Doxil/Caelyx) by J&J.
Candesartan (Blopress/Kensen, Figure 3b) belongs to the class of angiotensin II receptor antagonists (ARBs) or ‘sartans’, which are chemically 2-tetrazoylbiphenyl derivatives. The drug is used for treatment of hypertension (high blood pressure). Depending on the specific indication, competing drugs include:

Figure 3 – Key market products by Takeda
Lansoprazole (Takepron/Ogast/Lansox, Figure 4c) belongs to the sub-group or proton pump inhibitors (PPIs) or ‘prazoles’ in the class of drugs for acid-related disorders. PPIs reduce acid secretion by inhibiting the enzyme ATPase in gastric parientel cells.
Lansoprazole is used to treat stomach ulcers, peptic ulcers and gastroesophagal reflux. The originator drug is marketed by Takeda but is now generic, being marketed as Lansul and Lansoptol by Krka, Lansopran by Sawai and Opiren by Almirall and as an over-the-counter drug by Novartis under the name Prevacid 24H. Depending on the specific indication, lansoprazole competes with:
Leuprorelin (Leuplin/Enatoe, Figure 3d) is an analogue to the gonadotropin-releasing hormone (GnRH) and acts as agonist at pituitary GnRH receptors. It regulates down the secretion of gonadotropins-luteinizing hormones (LHs) and follicle-stimulating hormones (FSHs), reducing estradiol and testosterone levels in both sexes.
Leuprolin is marketed by various companies, such as Eligard by Sanofi and Astellas and Vidadur by Bayer. Competing GnRH medications include goserelin (Zalodex by AstraZeneca), buserelin (Suprefact by Sanofi), histrelin (Vantas and Supprelin by Elan), triptorelin (Decapentyl by Ipsen, Gonapeptyl by Ferring, Trelstar by Watson), deslorelin (Ovuplant by Peptech) and nafarelin (Synarel by Pfizer).
Pioglitazone (Actos/Glustin/Zanctos, Figure 3e) belongs to the sub group of insulin sensitisers in the class of anti-diabetic drugs. These work against the core problem of Type II diabetes, insulin resistance. In India, the drug is marketed by Zydus Cadila. Depending on the specific indication, competing drugs include:
http://www.specchemonline.com/articles/view/takeda-pharmaceutical#.U6fUjUCs_yV
Contact:
Dr Rainer Steinbach
CEO
CAP Intelligence
Tel: +49 231 73 56 84
E-mail: rainer.steinbach@cap-intelligence.de
Website: http://www.cap-intelligence.de

Farnesoid X receptor (FXR, NRIH4) plays a major role in the control of cholesterol metabolism. This suggests that antagonizing the transcriptional activity of FXR is a potential means to treat cholestasis and related metabolic disorders. Here we describe the synthesis, biological evaluation, and structure–activity relationship (SAR) studies of trisubstituted-pyrazol carboxamides as novel and potent FXR antagonists. One of these novel FXR antagonists, 4j has an IC50 of 7.5 nM in an FXR binding assay and 468.5 nM in a cell-based FXR antagonistic assay. Compound 4j has no detectable FXR agonistic activity or cytotoxicity. Notably, 4j is the most potent FXR antagonist identified to date; it has a promising in vitro profile and could serve as an excellent chemical tool to elucidate the biological function of FXR.
Original Research Article
Pages 2919-2938
Donna D. Yu, Wenwei Lin, Barry M. Forman, Taosheng Chen
Tadataka Yamada, M.D., Chief Medical & Scientific Officer of Takeda

TAKEDA US CHICAGO OFFICE
TAKEDA PIPELINE SEE LINKS BELOW
1 https://www.takeda.com/investor-information/annual/files/ar2013_10_en.pdf
2. http://www.takeda.com/research/files/pipeline_20131031_en.pdf
3 http://www.takeda.com/research/pipeline/
Takeda’s top executives had frequently pointed to TAK-875 as one of their best shots at coming up with an important new approach to treating diabetes. The drug is designed to spur insulin secretion in the pancreas and Takeda had confidently projected an approval in Japan in 2015 with a follow-up approval in the big U.S. market a year or two later.
The termination of the high-profile program caused some anxiety among investors. Takeda’s shares plunged 8% on the loss as analysts wondered how the pharma company could counter the loss of Actos, a $3.7 billion drug that accounted for about a quarter of its revenue in 2011.
Takeda won an approval on a trio of DPP-4 diabetes drugs–Nesina (alogliptin) and two combos with alogliptin, dubbed Oseni and Kazano–at the beginning of the year. But Takeda suffered some big delays in gaining acceptance, a common fate in this field, where regulators are particularly cautious about new drugs. And Merck had already solidified its lead in the DPP-4 market with Januvia whileOnglyza trailed closely behind it. Takeda had hoped that a combination of TAK-875 and Januvia could help regain some lost market territory–but that dream has clearly vanished as well.
| January 27, 2014 | |
| January 22, 2014 | |
| January 17, 2014 | |
| January 14, 2014 | |
| January 10, 2014 |
2013
| December 27, 2013 | ||
| December 25, 2013 | ||
| December 25, 2013 | ||
| December 24, 2013 | ||
| December 20, 2013 | ||
| December 20, 2013 | ||
| December 19, 2013 | ||
| December 10, 2013 | ||
| December 10, 2013 | ||
| December 10, 2013 | ||
| December 10, 2013 | ||
| December 9, 2013 | ||
| December 5, 2013 | ||
| December 4, 2013 | ||
| November 30, 2013 | ||
| November 21, 2013 | ||
| November 19, 2013 | ||
| November 14, 2013 | ||
| November 12, 2013 | ||
| November 12, 2013 | ||
| October 21, 2013 | ||
| October 7, 2013 | ||
| October 2, 2013 | ||
| October 1, 2013 |
| September 26, 2013 | ||
| September 26, 2013 | ||
| September 24, 2013 | ||
| September 20, 2013 | ||
| September 20, 2013 | ||
| September 13, 2013 | ||
| September 13, 2013 | ||
| September 5, 2013 | ||
| September 2, 2013 | ||
| August 27, 2013 | ||
| August 27, 2013 | ||
| August 27, 2013 | ||
| August 22, 2013 | ||
| August 13, 2013 | ||
| August 1, 2013 | ||
| July 31, 2013 | ||
| July 31, 2013 | ||
| July 31, 2013 | ||
| July 30, 2013 | ||
| July 29, 2013 | ||
| July 26, 2013 | ||
| July 19, 2013 | ||
| July 19, 2013 | ||
| July 19, 2013 | ||
| July 18, 2013 | ||
| July 10, 2013 | ||
| July 1, 2013 |
CLIPPED
Takeda isn’t quite in the top 10 among global drugmakers, but the company boasts the 7th-largest pipeline in the industry, according to its presentation at the conference. Yamada noted that 31% of the pipeline assets are in late-stage trials. Millennium is leading development of three late-stage contenders, TAK-700 for prostate cancer, MLN9708 for multiple myeloma and MLN0002 for ulcerative colitis andCrohn’s disease.
In an effort to revive its diabetes franchise, Takeda is in the final stage of development for a first-of-a-kind GPR40 agonist called TAK-875, designed to provide glucose-dependent insulin secretion.
With a rich late-stage pipeline at Takeda, Yamada wants the company to focus on growing its ranks of earlier-stage drug candidates. To do this the company has landed a variety of deals, including the purchase of Intellikine for $310 million to acquire anti-cancer drugs and more recently the acquisition of Envoy Therapeutics last year for $140 million.
Takeda has formed a New Frontier Science group to scout out the hottest research in academia and elsewhere and form collaborations with scientists behind those innovations. At the J.P. Morgan conference, Yamada said, he was attending many meetings with members of the biotech community.

Takeda Pharmaceutical Company Limited (武田薬品工業株式会社 Takeda Yakuhin Kōgyō Kabushiki-gaisha?) is the largest pharmaceutical company in Japan and Asia and a top 15 pharmaceutical company. The company has over 30,000 employees worldwide and achieved $16.2 billion USD in revenue during the 2012 fiscal year.[1] The company is focused on metabolic disorders, gastroenterology, neurology, inflammation, as well asoncology through its independent subsidiary, Millennium: The Takeda Oncology Company.[2] Its headquarters is located in Chuo-ku, Osaka, and it has an office in Nihonbashi, Chuo, Tokyo.[3][4] In January 2012, Fortune Magazine ranked the Takeda Oncology Company as one the 100 best companies to work for in the United States.
Takeda Pharmaceuticals was founded on June 12, 1781 and was incorporated on January 29, 1925.
Takeda’s Japanese logo
In 1977, Takeda first entered the U.S. pharmaceutical market by developing a joint venture with Abbott Laboratories called TAP Pharmaceuticals.[5]Through TAP Pharmaceuticals, Takeda and Abbott launched the blockbusters Lupron (leuprolide) in 1985 and Prevacid (lansoprazole) in 1995.
One of the firm’s mainstay drugs is Actos, a compound in the thiazolidinedione class of drugs used in the treatment of type 2 diabetes. Launched in 1999, Actos has become the best-selling diabetes drug in the world with $4 billion USD in sales during the 2008 fiscal year.[6]
In February 2005, Takeda announced its acquisition of San Diego, California-based Syrrx, a company specializing in high-throughput X-ray crystallography, for $270 million.[7]
In February 2008, Takeda acquired the Japanese operations of Amgen and rights to a dozen of the California biotechnology company’s pipeline candidates for the Japanese market.[8]
In March 2008, Takeda and Abbott Laboratories announced plans to conclude their 30-year old joint venture, TAP Pharmaceuticals, that had over $3 billion in sales in its final year. The split resulted in Abbott acquiring U.S. rights to Lupron and the drug’s support staff. On the other hand, Takeda received rights to Prevacid and TAP’s pipeline candidates. The move also increased Takeda’s headcount by 3,000 employees.[9]
In April 2008, Takeda announced that it was acquiring Millennium Pharmaceuticals of Cambridge, Massachusetts, a company specializing in cancerdrug research, for $8.8 billion. The acquisition brought in Velcade, a drug indicated for hematological malignancies, as well as a portfolio of pipeline candidates in the oncology, inflammation, and cardiovascular therapeutic areas. Millennium now operates as an independent subsidiary, serving as the global center of excellence in oncology under its new name: “Millennium: The Takeda Oncology Company.” [10]
In May 2008, the company licensed non-exclusively the RNAi technology platform developed by Alnylam Pharmaceuticals, creating a potentially long-term partnership between the companies.[11]
On May 19, 2011, Takeda Pharmaceutical and Nycomed announced that Takeda will acquire Nycomed for € 9.6 billion. The acquisition was completed by September 30, 2011.[12]
On April 11, 2012, Takeda Pharmaceutical and URL Pharma announced that Takeda will acquire URL Pharma for $800 million. The acquisition is expected to be completed within 60 days.
On 25 May 2012, Takeda announced the purchase of Brazilian pharmaceutical company Multilab by R$ 540 million.[13]

Takeda Midosuji Building, headquarters of Takeda Pharmaceutical Company, inChuo-ku, Osaka, Japan
Takeda operates two primary bases in Japan in Osaka and Tokyo. Its United States subsidiary is based in Deerfield, Illinois, and all Global Operations outside of Japan and U.S. are based in Opfikon (Zurich), Switzerland. The company maintains research & development sites in Osaka and Tsukuba, Japan; San Diego andSan Francisco, United States; Cambridge, United Kingdom; and Singapore.[14]
The company has manufacturing facilities in Japan, China, Indonesia, Italy, and Ireland.[15] Following the Nycomed acquisition, the Takeda manufacturing sites have been extended with facilities in Argentina,Austria,Belgium,Brazil,Denmark, Estonia,Germany,Mexico,Norway and Poland. Takeda has overseas marketing presences in the U.S., UK, France, Italy, Germany, Austria, Switzerland, Spain, China, Taiwan, Philippines, Thailand, Indonesia, and Singapore. It has recently[when?] announced its first foray into Canada, Portugal, Spain, Mexico, and Ireland.[15]

AT INDONESIA
Some of the key products that Takeda produces on behalf of partners include:[16]
![]() AT UK
AT UK
 |
|
| Native name | 武田薬品工業株式会社 |
|---|---|
| Type | Public KK |
| Traded as | |
| Industry | Pharmaceuticals |
| Founded | Doshomachi, Osaka, Japan (June 12, 1781) |
| Headquarters | 1-1, Doshomachi Yonchome,Chuo-ku, Osaka, Japan |
| Key people | Yasuchika Hasegawa (President & CEO) |
| Revenue | |
| Operating income | |
| Net income | |
| Total assets | |
| Total equity | |
| Employees | 30,481 (2012) |
| Website | takeda.com (Global website) |
References:
|
|

CMC CENTRE
The Chemistry, Manufacturing and Controls (CMC) Center is a global organization responsible for overall R&D activities ranging from chemical information on development candidates to the processes leading to “manufacturing” of pharmaceutical products.
The main sites are located in Osaka and consist of the following laboratories: the Chemical Development Laboratories in charge of R&D for developing the manufacturing methods of active pharmaceutical ingredients and the manufacturing of drug substances for clinical samples; the Pharmaceutical Technology R&D Laboratories in charge of R&D for dosage forms, manufacturing and packaging, as well as manufacturing of clinical samples; and the Analytical Development Laboratories in charge of R&D for the development of analytical methods and stability studies of clinical samples. In addition, Hikari Bio-Manufacturing Technology Laboratories is located in Hikari (Yamaguchi) and this is where antibody drug substances are manufactured.
As for overseas sites, the Cambridge Biologics CMC Group (Massachusetts) and the Chicago Pharmaceutical Science Group (Illinois) are located in the USA, while the CMC Center Europe is mainly located in Roskilde, Denmark. All research and development activities at Takeda are promoted with the cooperation of these sites.

CAS#: 1201902-80-8
Synonym: Ixazomib; MLN-9708.
IUPAC/Chemical name:
4-(carboxymethyl)-2-((R)-1-(2-(2,5-dichlorobenzamido)acetamido)-3-methylbutyl)-6-oxo-1,3,2-dioxaborinane-4-carboxylic acid
UPDATES AT THE BOTTOM OF PAGE
CAMBRIDGE, Mass., May 23, 2013 – Takeda Pharmaceutical Company Limited (TSE:4502) today announced the initiation of an international phase 3 clinical trial evaluating once a week MLN9708 in combination with lenalidomide and dexamethasone in patients with newly diagnosed multiple myeloma who are not candidates for transplant. The multi-center study with MLN9708, an investigational, oral proteasome inhibitor, will be conducted in Europe and North America.———————-READ MORE AT
http://www.pharmalive.com/takeda-begins-phase-iii-trial-of-multiple-myeloma-drug
Description of Ixazomib: ixazomib is an orally bioavailable second generation proteasome inhibitor (PI) with potential antineoplastic activity. Ixazomib inhibits the activity of the proteasome, blocking the targeted proteolysis normally performed by the proteasome, which results in an accumulation of unwanted or misfolded proteins; disruption of various cell signaling pathways may follow, resulting in the induction of apoptosis. Compared to first generation PIs, second generation PIs may have an improved pharmacokinetic profile with increased potency and less toxicity. Proteasomes are large protease complexes that degrade unneeded or damaged proteins that have been ubiquinated
MLN9708 is an investigational proteasome inhibitor that, compared with bortezomib, has improved pharmacokinetics, pharmacodynamics, and antitumor activity in preclinical studies. MLN9708 rapidly hydrolyzes to MLN2238, the biologically active form. MLN9708 has a shorter proteasome dissociation half-life and improved pharmacokinetics, pharmacodynamics, and antitumor activity compared with bortezomib.MLN9708 has a larger blood volume distribution at steady state, and analysis of 20S proteasome inhibition and markers of the unfolded protein response confirmed that MLN9708 has greater pharmacodynamic effects in tissues than bortezomib. MLN9708 showed activity in both solid tumor and hematologic preclinical xenograft models, and we found a correlation between greater pharmacodynamic responses and improved antitumor activity. Moreover, antitumor activity was shown via multiple dosing routes, including oral gavage. Taken together, these data support the clinical development of MLN9708 for both hematologic and solid tumor indications. (source: Cancer Res. 2010 Mar 1;70(5):1970-80. Epub 2010 Feb 16.).
| References |
1: Mullard A. Next-generation proteasome blockers promise safer cancer therapy. Nat Med. 2012 Jan 6;18(1):7. doi: 10.1038/nm0112-7a. PubMed PMID: 22227650.
2: Anderson KC. The 39th David A. Karnofsky Lecture: bench-to-bedside translation of targeted therapies in multiple myeloma. J Clin Oncol. 2012 Feb 1;30(4):445-52. Epub 2012 Jan 3. PubMed PMID: 22215754.
3: Appel A. Drugs: More shots on target. Nature. 2011 Dec 14;480(7377):S40-2. doi: 10.1038/480S40a. PubMed PMID: 22169800.
4: Lee EC, Fitzgerald M, Bannerman B, Donelan J, Bano K, Terkelsen J, Bradley DP, Subakan O, Silva MD, Liu R, Pickard M, Li Z, Tayber O, Li P, Hales P, Carsillo M, Neppalli VT, Berger AJ, Kupperman E, Manfredi M, Bolen JB, Van Ness B, Janz S. Antitumor activity of the investigational proteasome inhibitor MLN9708 in mouse models of B-cell and plasma cell malignancies. Clin Cancer Res. 2011 Dec 1;17(23):7313-23. Epub 2011 Sep 8. PubMed PMID: 21903769.
5: Chauhan D, Tian Z, Zhou B, Kuhn D, Orlowski R, Raje N, Richardson P, Anderson KC. In vitro and in vivo selective antitumor activity of a novel orally bioavailable proteasome inhibitor MLN9708 against multiple myeloma cells. Clin Cancer Res. 2011 Aug 15;17(16):5311-21. doi: 10.1158/1078-0432.CCR-11-0476. Epub 2011 Jun 30. PubMed PMID: 21724551; PubMed Central PMCID: PMC3156932.
6: Kupperman E, Lee EC, Cao Y, Bannerman B, Fitzgerald M, Berger A, Yu J, Yang Y, Hales P, Bruzzese F, Liu J, Blank J, Garcia K, Tsu C, Dick L, Fleming P, Yu L, Manfredi M, Rolfe M, Bolen J. Evaluation of the proteasome inhibitor MLN9708 in preclinical models of human cancer. Cancer Res. 2010 Mar 1;70(5):1970-80. Epub 2010 Feb 16. Erratum in: Cancer Res. 2010 May 1;70(9):3853. Hales, Paul [added]. PubMed PMID: 20160034.
7: Dick LR, Fleming PE. Building on bortezomib: second-generation proteasome inhibitors as anti-cancer therapy. Drug Discov Today. 2010 Mar;15(5-6):243-9. Epub 2010 Jan 29. Review. PubMed PMID: 20116451.8: Marblestone JG. Ubiquitin Drug Discovery & Diagnostics 2009 – First Annual Conference. IDrugs. 2009 Dec;12(12):750-3. PubMed PMID: 19943215.

http://www.cancernetwork.com/conference-reports/ash2012/content/article/10165/2119611
Nasopharyngeal cancer is a sub-type of head and neck cancer that arises from the epithelial cells that cover the surface and line the nasopharynx. The incidence of nasopharyngeal cancer has been reported at approximately 0.5 to 2 new cases per year per 100,000 in Europe and the USA. Rottey et ah, Curr. Opin. Oncol., 23(3): 254-258 (201 1). There are three subtypes of nasopharyngeal cancer recognized in the World Health Organization (WHO) classification: (i) Type 1 – squamous cell carcinoma, typically found in the older adult population; (ii) Type 2 non-keratinizing carcinoma; and (iii) Type 3 – undifferentiated carcinoma. Treatment for nasopharyngeal cancer often involves radiotherapy and/or chemotherapy. There remains a continuing need for new and improved treatments for patients with nasopharyngeal cancer. There remains a further need to identify nasopharyngeal patients most likely to benefit from treatment with a proteasome inhibitor.
Proteasome inhibition represents an important new strategy in cancer treatment. King et al. , Science 274: 1652-1659 ( 1996), describes an essential role for the ubiquitin-proteasome pathway in regulating cell cycle, neoplastic growth and metastasis. The authors teach that a number of key regulatory proteins, including cyclins, and the cyclin-dependent kinases p21 and p27K,P ! , are temporally degraded during the cell cycle by the ubiquitin-proteasome pathway. The ordered degradation of these proteins is required for the cell to progress through the cell cycle and to undergo mitosis.
The proteasome inhibitor VELCADE© (bortezomib; N-2-pyrazinecarbonyl-L -phenylalanine -L- leucineboronic acid) is the first proteasome inhibitor to achieve regulatory approval. Mitsiades et ai, Current Drug Targets, 7: 1341 (2006), reviews the clinical studies leading to the approval of bortezomib for the treatment of multiple myeloma patients who have received at least one prior therapy. Fisher et ai , J. Clin. Oncol, 30:4867, describes an international multi-center Phase II study confirming the activity of bortezomib in patients with relapsed or refractory mantle cell lymphoma. Ishii et al, Anti-Cancer Agents in Medicinal Chemistry, 7:359 (2007), and Roccaro et al., Curr. Pharm. Biotech., 7: 1341 (2006), discuss a number of molecular mechanisms that may contribute to the antitumor activities of bortezomib. The proteasome inhibitor MLN9708 [2,2′-{2-[(lR)- l -( {[(2,5-dichlorobenzoyl)amino]acetyl}amino)-3- methylbutyl]-5-oxo-l,3,2-dioxaborolane-4,4-diyl}diacetic acid] is currently undergoing clinical evaluation for hematological and solid cancers. MLN9708 is a citrate ester which rapidly hydrolyzes to the active form [(lR)-l -({[(2,5-dichlorobenzoyl)amino]acetyl}amino)-3-methylbutyl]boronic acid (MLN2238) on exposure to aqueous solution or plasma. MLN9708 has demonstrated anti-tumor activity in a range of hematological and solid tumor xenograft models (Kupperman et al. (2010) Cancer Res. 70: 1970- 1980),
Summary
The invention relates to the discovery that patients with nasopharyngeal cancer respond to treatment with MLN9708. In one aspect, the invention relates to the discovery of the increased expression of Nuclear Factor Kappa-B RelA 65,000 dalton subunit (NFKB p65) in biological samples comprising cells obtained from patients with nasopharyngeal cancer and responsive to MLN9708.
Accordingly, the invention features treating nasopharyngeal cancer patients withMLN9708 if a sample from the patient demonstrates an elevated expression of NFKB p65.
PATENT

or a pharmaceutically acceptable salt or a pharmaceutical composition or a boronic acid anhydride thereof.
[048| The compound of formula (II), [( l R)-l -( } [(2,5-dichlorobenzoyl)amino]acetyl} amino)-3- methylbutyljboronic acid (MLN2238) is disclosed in Olhava and Danca, U .S. Patent No. 7,442,830, herein incorporated by reference in its entirety. [049] In some other embodiments, Z and Z together form a moiety derived from a compound having at least two hydroxyl groups separated by at least two connecting atoms in a chain or ring, said chain or ring comprising carbon atoms and, optionally, a heteroatom or heteroatoms which can be N, S, or O, wherein the atom attached to boron in each case is an oxygen atom.
In certain embodiments, wherein the alpha-hydroxy carboxylic acid or beta-hydroxy carboxylic acid is citric acid, the compound of formula (I) is characterized by formula (III-A) or (III-B):

(III-B), or a mixture thereof or a pharmaceutical composition thereof.
[054] In certain embodiments, wherein the alpha-hydroxy carboxylic acid or beta-hydroxy carboxylic acid is citric acid, the compound of formula (I) is characterized by formula (III-A):

or a pharmaceutical composition thereof.
[055] The compound of formula (III-A), 2,2′- {2-[( l i?)- l -( { [(2,5-dichlorobenzoyl)amino]acetyl } amino)- 3-methylbutyl]-5-oxo- l ,3,2-dioxaborolane-4,4-diyl} diacetic acid (MLN9708) is disclosed in Elliott et al. , WO 09/ 154737, herein incorporated by reference in its entirety
PATENT
http://www.google.com/patents/WO2009154737A1?cl=en
Example 1: Synthesis of 4-(/?,S)-(carboxymethyl)-2-( (R)-I -(2-(2,5- dichlorobenzamido)acetamido)-3-methylbutyl)-6-oxo-l,3,2-dioxaborinane-4- carboxylic acid (1-1)

Step l: 2,5-r(dichlorobenzoyI)aminolacetic acid
[0310] To a mixture of NaOH (12 g, 300 mmol) and glycine (18 g, 239 mmol) in water (120 mL) was added dropwise over 45 min a solution of 2,5-dichlorobenzoyl chloride (10 g, 48 mmol) in THF (15 mL) keeping the internal temperature below about 25 0C. After 1 h, the mixture was acidified with 2.0 M HCl (125 mL) keeping the internal temperature below about 5 0C. The resulting precipitate was collected by vacuum filtration. The crude product was recrystallized from water to give 2,5-[(dichlorobenzoyl)amino]acetic acid as a white, crystalline solid (6.1 g, 52%). mp 173.3 0C. 1H NMR (300 MHz, DMSOd6, δ): 12.72 (bs, IH), 8.89 (t, J = 6.0 Hz, IH), 7.54 (m, 2H), 7.48 (m, IH), 3.93 (d, J = 6.0 Hz). 13C NMR (75 MHz, DMSO-Ci6, δ): 41.6, 129.3, 129.6, 131.4, 132.2, 138.2, 171.4, 165.9. MS (ni/z): [M+H] calculated for C9H8Cl2NO3, 248.0; found, 248.0; [M+Na] calculated for C9H7Cl2NNaO3, 270.0; found 270.2.
2,5-[(dichlorobenzoyl)amino]acetic acid was also be prepared via the following procedure: To a mixture of glycine (21.5 g, 286 mmol) in water (437 mL), was added 2.0 M NaOH (130 mL) and the resulting solution was cooled to 0 0C. A solution of 2,5-dichlorobenzoyl chloride (50.0 g, 239 mmol) in THF (75 mL) was added dropwise at such a rate that the internal temperature was maintained at 0 ± 1 0C. During the addition, the pH was controlled at 11.0 ± 0.2 using a pH controller titrated with 2.0 M NaOH. After complete addition, the mixture was stirred at 0 ± 1 0C for an additional 2 h. The mixture was then acidified with 2.0 M HCl (176 mL) to a final pH of 2.5. The resulting precipitate was collected by filtration, washed with cold water (125 mL), and dried at 45 0C in a vacuum oven to afford 2,5-[(dichlorobenzoyl)amino]acetic acid as a white solid (57.6 g, 97.3%). Step 2: 2,5-dichloro-N-f2-(( (lR’)-3-niethyl-l-r(3aS,4S.6S.7aR)-3a,5,5-trimethylhexahvdro-
4,6-methano-l,3,2-benzodioxaborol-2-yllbutyl }amino)-2-oxoethvπbenzamide
To a solution of 2,5-[(dichlorobenzoyl)amino]acetic acid (6.10 g, 24.6 mmol) and TBTU (8.34 g, 26.0 mmol) in DMF (40 mL) with an internal temperature below about 5 0C was added (IR)- 3-methyl-l-[(3aS,4S,6S,7aR)-3a,5,5-trimethylhexahydro-4,6-methano-l,3,2-benodioxaborol-2- yl]butan-l-amine»TFA (9.35 g, 24.7 mmol). DIPEA (13 mL, 75 mmol) was then added dropwise over 2 h keeping the internal temperature below about 5 0C. After 40 min, the mixture was diluted with EtOAc (90 mL), washed with 5% NaCl (150 mL), twice with 10% NaCl (2 x 40 mL), once with 2% K2CO3 (1 x 40 mL), once with 1% H3PO4 (1 x 40 mL), and once with 10% NaCl (1 x 40 mL). The resulting organic layer was concentrated to a thick oil, diluted with heptane (40 mL) and evaporated to yield 2,5-dichloro-N-[2-({ (lR)-3-methyl-l-[(3aS,4S,6S,7aR)-3a,5,5- trimethylhexahydro-4,6-methano-l ,3,2-benzodioxaborol-2-yl]butyl }amino)-2-oxoethyl]benzamide as a white solid which was used in the next step without purification.
Step 3: N,N\N’Wboroxin-2A6-triyltrisir(lR)-3-methylbutane-l J-diyllimino(2-oxoethane- 2,l-diyl)^ ^tris(2,5-dichlorobenzamide)
To a solution of 2,5-dichloro-N-[2-({(lR)-3-methyl-l-[(3aS,4S,6S,7aR)-3a,5,5- trimethylhexahydro-4,6-methano-l,3,2-benzodioxaborol-2-yl]butyl }amino)-2-oxoethyl]benzamide (12.2 g, 24.6 mmol) in methanol/hexane (1 :1) (250 mL) were added IN HCl (30 mL, 30 mmol) and (2-methylpropyl)boronic acid (6.5 g, 64 mmol). The reaction mixture was allowed to stir overnight. The phases were separated and the methanol layer was washed twice with additional heptane (2 x 55 mL). The resulting organic layer was concentrated to about 10 mL and partitioned between 2.0M NaOH (30 mL) and DCM (25 mL). The DCM layer was washed once with additional 2.0M NaOH (5 mL). The basic aqueous layers were then combined, washed twice with DCM (2 x 25 mL) and acidified with IM HCl (60 mL). The resulting mixture was diluted with DCM (40 mL), the layers were separated, and the resulting aqueous layer was washed three times with DCM (3 x 10 mL). The combined DCM extracts were dried over MgSO4 (25 g) and evaporated to a thick oil. The product was precipitated with heptane (50 mL) and collected by filtration to yield N,N’,N”-{boroxin-2,4,6- -riyltris[[(lR)-3-methylbutane-l,l-diyl]imino(2-oxoethane-2,l-diyl)] }tris(2,5-dichlorobenzamide) as a white solid (6.6 g, 74%). 1H NMR (300 MHz, DMSO-Cl6, δ): 8.93 (t, J – 6.0 Hz, IH), 8.68 (bs, IH), 7.63 (m, IH), 7.52 (m, 2H), 4.00 (d, J = 6.0 Hz, 2H), 2.62 (m, IH), 1.59 (m, IH), 1.33 (m, IH), 1.24 (m, IH), 0.81 (d, / = 5.9 Hz, 6H). 13C NMR (125 MHz, DMSO-Cl6, δ): 23.2, 25.8, 40.1, 40.7, 43.0, 129.0, 130.0, 131.0, 137.5, 165.0, 172.5. MS (m/z) in CH3CN: [M+H] calculated for C42H52B3Cl6N6O9, 1027.2; found, 1027.3; [M+Na] calculated for C42H51B3Cl6N6NaO9, 1049.2; found 1049.5.
Step 4: 4-(/?.S)-(carboxymethyl)-2-((/?)-l-(2-(2,5-dichlorobenzamido)acetamido)-3- methylbutyl)-6-oxo-l,3,2-dioxaborinane-4-carboxylic acid (1-1)
Form 1: To a solution of citric acid (2.75 g, 14.3 mmol) in EtOAc (85 mL) with an internal temperature of about 74 0C was added N,N’,N”-{boroxin-2,4,6-triyltris[[(lR)-3-methylbutane-l,l- diyl]imino(2-oxoethane-2,l-diyl)] }tris(2,5-dichlorobenzamide) (5.00 g, 4.87 mmol) as a solid. The solution was cooled uncontrolled until the internal temperature was about 25 0C and the mixture was stirred overnight. The resulting precipitate was collected by filtration to yield 2,2′-{2-[(lR)-l-({ [(2,5- dichlorobenzoyl)amino]acetyl }amino)-3-methylbutyl]-5-oxo-l,3,2-dioxaborolane-4,4-diyl}diacetic acid Form 1 as a crystalline solid (6.65 g, 88 %). 1H NMR (500 MHz, DMSOd6, δ 110 0C): 10.08 (s, IH), 8.69 (s, IH), 7.61 (s, IH), 7.52 (d, J = 1.3 Hz, 2H), 4.26 (d, J = 5.5 Hz, 2H), 2.70 (q, J = 14.5 Hz, 4H), 2.70 (bs, IH), 1.72 (sept, J – 6.5 Hz, IH), 1.42 (ddd, J = 5.2 Hz, J = 8.6 Hz, J = 13.9 Hz, IH), 1.28 (ddd, J = 5.3, J = 9.4 Hz, J = 14.3 Hz, IH), 0.91 (dd, J = 3.3 Hz, J = 6.6 Hz, 6H). MS (m/z) in CH3CN: [M+Na] calculated for C20H23BCl2N2NaO9, 539.1; found, 539.1.
Ixazomib citrate [USAN]
1,3,2-Dioxaborolane-4,4-diacetic acid, 2-[(1R)-1-[[2-[(2,5-dichlorobenzoyl)amino]acetyl]amino]-3-methylbutyl]-5-oxo- [ACD/Index Name]
1,3,2-Dioxaborolane-4,4-diacetic acid,2-[(1R)-1-[[2-[(2,5-dichlorobenzoyl)amino]acetyl]amino]-3-methylbutyl]-5-oxo-
1239908-20-3 [RN]
2,2′-{2-[(1R)-1-{[N-(2,5-Dichlorbenzoyl)glycyl]amino}-3-methylbutyl]-5-oxo-1,3,2-dioxaborolan-4,4-diyl}diessigsäure [German] [ACD/IUPAC Name]
2,2′-{2-[(1R)-1-{[N-(2,5-dichlorobenzoyl)glycyl]amino}-3-methylbutyl]-5-oxo-1,3,2-dioxaborolane-4,4-diyl}diacetic acid [ACD/IUPAC Name]
2-[(1R)-1-[[2-[(2,5-dichlorobenzoyl)amino]acetyl]amino]-3-methylbutyl]-5-oxo-1,3,2-dioxaborolane-4,4-diacetic acid
2-[4-(carboxymethyl)-2-[(1R)-1-[[2-[(2,5-dichlorobenzoyl)amino]acetyl]amino]-3-methyl-butyl]-5-oxo-1,3,2-dioxaborolan-4-yl]acetic acid
Acide 2,2′-{2-[(1R)-1-{[N-(2,5-dichlorobenzoyl)glycyl]amino}-3-méthylbutyl]-5-oxo-1,3,2-dioxaborolane-4,4-diyl}diacétique [French] [ACD/IUPAC Name]
MLN9708
Ixazomib (trade name Ninlaro) is a drug for the treatment of multiple myeloma, developed by Takeda Pharma. It acts as aproteasome inhibitor and has orphan drug status in the US. In November 2015, the U.S. Food and Drug Administration approved ixazomib for use in combination with lenalidomide and dexamethasone for the treatment of multiple myeloma after at least one prior therapy.[2]
Ixazomib is a peptide analogue that reversibly inhibits the protein proteasome subunit beta type-5 (PSMB5), which is part of the 20Sproteasome complex.[3]

Ixazomib citrate—a prodrug for ixazomib
NINLARO Provides a New Option for Patients Living with Multiple Myeloma Who Have Received at Least One Prior Therapy
Cambridge, Mass. and Osaka, Japan, November 20, 2015 – Takeda Pharmaceutical Company Limited (TSE: 4502) today announced that the U.S. Food and Drug Administration (FDA) has approved NINLARO®(ixazomib) capsules, the first and only oral proteasome inhibitor, indicated in combination with lenalidomide and dexamethasone for the treatment of patients with multiple myeloma who have received at least one prior therapy. NINLARO is a once-weekly pill. More information is available at www.NINLARO.com.
Takeda submitted a New Drug Application for NINLARO to the FDA in July 2015, and in September NINLARO was granted Priority Review status with a PDUFA date of March 10, 2016, reflecting the profound and continuing unmet need for new treatments for multiple myeloma, a devastating, relapsing and incurable rare cancer.
“With the approval of NINLARO, we can now offer patients a once-weekly oral proteasome inhibitor as part of a highly active triplet therapy,” said Paul Richardson, M.D., Clinical Program Leader and Director of Clinical Research, Jerome Lipper Multiple Myeloma Center Institute Physician at Dana-Farber Cancer Institute, and investigator for TOURMALINE-MM1, the pivotal Phase 3 trial on which today’s approval is based. “We, as investigators of the TOURMALINE-MM1 trial, felt it was vital to conduct a comprehensive ‘real world’ evaluation of this combination that included some of the most common patient types in the relapsed/refractory multiple myeloma setting, such as older patients, patients with moderate renal impairment, light chain disease, and high risk cytogenetics. Further, we treated patients until disease progression to determine the sustainability of NINLARO in treating their relapsed/refractory disease. The TOURMALINE-MM1 data demonstrate convincingly that oral NINLARO-based triplet treatment is effective at extending progression-free survival, over and above the clinical benefit seen with lenalidomide and dexamethasone, with a tolerable safety profile.”
“We introduced the first proteasome inhibitor for multiple myeloma, VELCADE, into clinical research approximately 20 years ago. Since that time, we’ve significantly advanced scientific understanding of this rare cancer, culminating in the introduction of NINLARO,” said Andy Plump, M.D., Ph.D, Takeda Chief Medical and Scientific Officer. “NINLARO is an entirely new molecule that offers the efficacy of this proteasome inhibitor in a convenient once-weekly pill with a tolerable safety profile. Takeda is delighted to bring this significant innovation to multiple myeloma patients today, and we continue to examine the potential of NINLARO through a robust clinical development program.”
Dr. Brian Durie, Chairman of the International Myeloma Foundation, said, “The IMF is pleased by the approval of ixazomib. This opens the door for a fully oral proteasome inhibitor-based triplet combination therapy. Having worked in multiple myeloma for decades, I’ve seen notable progress, yet significant unmet needs remain. With today’s approval, we now have another attractive option for many patients living with multiple myeloma.”
The FDA approval of NINLARO is based on results from the TOURMALINE-MM1 Phase 3 clinical trial, the first double-blind, placebo-controlled trial with a proteasome inhibitor. TOURMALINE-MM1 is the first of five ongoing Phase 3 clinical trials with study results available. The TOURMALINE program has enrolled approximately 3,000 patients to date in 40 countries. Data from the NINLARO Phase 3 TOURMALINE-MM1 pivotal trial will be presented at the upcoming 57th Annual Meeting of the American Society of Hematology on December 7, 2015.
“The approval of ixazomib offers a much-needed additional option in the multiple myeloma treatment landscape. It is developments such as these that help us to better understand the disease and provide continued hope for patients,” said Kathy Giusti, Founder and Executive Chairman of the Multiple Myeloma Research Foundation (MMRF). “A cancer diagnosis today is different from what it was just a few years ago and it’s exciting to see continued progress. As a patient, I understand the urgent need for advancing research through partnerships that bring new treatment options, as we’ve done with Takeda.”
“NINLARO is a first-of-its-kind innovation that is supported by a global development program, unprecedented for us at Takeda Oncology, and we would like to express our immense appreciation for all patients involved for their incredible strength and invaluable participation. The introduction of NINLARO marks an important step forward, as its efficacy and safety profile – coupled with its completely oral administration – potentially can reduce some logistical burdens, and help enable patients to reap the full benefits of this sustainable therapy,” explained Christophe Bianchi, M.D., President, Takeda Oncology. “As part of our unwavering 20-year commitment, Takeda will continue to pursue advances for these patients, and we look forward to introducing and expanding access to NINLARO in other markets around the world.”
About the TOURMALINE-MM1 Trial
TOURMALINE-MM1 is an international, randomized, double-blind, placebo-controlled clinical trial of 722 patients, designed to evaluate NINLARO plus lenalidomide and dexamethasone compared to placebo plus lenalidomide and dexamethasone in adult patients with relapsed and/or refractory multiple myeloma. Results showed NINLARO is effective in extending Progression Free Survival (PFS) and has a manageable safety profile. The trial achieved its primary endpoint and demonstrated a clinically meaningful and statistically significant prolongation in PFS at this analysis, which showed that patients treated in the NINLARO arm lived without their disease worsening for a significantly longer time compared to patients in the control arm. Patients continue to be treated to progression in this trial and will be evaluated for long term outcomes.
In the TOURMALINE-MM1 trial, the most common adverse reactions (≥20%) in patients receiving NINLARO included diarrhea, constipation, thrombocytopenia, peripheral neuropathy, nausea, peripheral edema, vomiting and back pain. Serious adverse reactions reported in ≥2% patients included thrombocytopenia (2%) and diarrhea (2%).
Efficacy and safety data were reviewed by an Independent Data Monitoring Committee (IDMC), who recommended the study be continued in blinded fashion to allow further maturation of long term outcomes, including overall survival (OS) and long-term safety.
About NINLARO (ixazomib) capsules
NINLARO (ixazomib) is the first and only oral proteasome inhibitor indicated in combination with lenalidomide and dexamethasone for the treatment of patients with multiple myeloma who have received at least one prior therapy. NINLARO is administered orally, once-weekly on days 1, 8, and 15 of a 28-day treatment cycle. NINLARO is currently under review by the European Medicines Agency (EMA) and was granted an accelerated assessment by the Committee for Medicinal Products for Human Use (CHMP). NINLARO also received Breakthrough Therapy status by the U.S. FDA for relapsed or refractory systemic light-chain (AL) amyloidosis, a related ultra orphan disease, in 2014.
The TOURMALINE clinical development program further reinforces Takeda’s ongoing commitment to developing innovative therapies for people living with multiple myeloma worldwide and the healthcare professionals who treat them. Five global Phase 3 trials are ongoing:
In addition to the TOURMALINE program, a large number of investigator initiated studies are evaluating ixazomib for patients globally.
For additional information on the ongoing Phase 3 studies please visit www.clinicaltrials.gov. To learn more about NINLARO, please visit www.NINLARO.com or call 1-844-N1POINT (1-844-617-6468).
![]()
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
N2-(2,5-Dichlorobenzoyl)-N-[(1R)-1-(dihydroxyboryl)-3-methylbutyl]glycinamide
|
|
| Clinical data | |
| Trade names | Ninlaro |
| AHFS/Drugs.com | entry |
| Legal status |
|
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Bioavailability | 58%[1] |
| Protein binding | 99% |
| Metabolism | hepatic, CYP3A4 (42%),CYP1A2 (26%) and others |
| Biological half-life | 9.5 days |
| Excretion | urine (62%), feces (22%) |
| Identifiers | |
| CAS Number | 1072833-77-2 |
| ATC code | L01XX50 |
| PubChem | CID 25183872 |
| ChemSpider | 25027391 |
| UNII | 71050168A2 |
| KEGG | D10130 |
| ChEBI | CHEBI:90942 |
| Synonyms | MLN2238 |
| Chemical data | |
| Formula | C14H19BCl2N2O4 |
| Molar mass | 361.03 g·mol−1 |

//////////
see….http://apisynthesisint.blogspot.in/2016/02/takedas-ixazomib-multiple-myeloma-drug.html
